
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic studies of NOx reduction reactions involving copper complexes: encouragement of DFT calculations
Yohei
Kametani
 and
Yoshihito
Shiota
*
and
Yoshihito
Shiota
*
Institute for Materials Chemistry and Engineering and IRCCS, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: shiota@ms.ifoc.kyushu-u.ac.jp
First published on 7th November 2024
Abstract
The reduction of nitrogen oxides (NOx), which is mainly mediated by metalloenzymes and metal complexes, is a critical process in the nitrogen cycle and environmental remediation. This Frontier article highlights the importance of density functional theory (DFT) calculations to gain mechanistic insights into nitrite (NO2−) and nitric oxide (NO) reduction reactions facilitated by copper complexes by focusing on two key processes: the reduction of NO2− to NO by a monocopper complex, with special emphasis on the concerted proton–electron transfer, and the reduction of NO to N2O by a dicopper complex, which involves N–N bond formation, N2O2 isomerization, and N–O bond cleavage. These findings underscore the utility of DFT calculations in unraveling complicated reaction mechanisms and offer a foundation for future research aimed at improving the reactivity of transition metal complexes in NOx reduction reactions.
Introduction
In the denitrification process of the nitrogen cycle, nitrate (NO3−) is sequentially reduced to nitrogen gas (N2) according to the process NO3− → NO2− → NO → N2O → N2.1–3 Among these steps, the reduction of nitrite (NO2−) and nitric oxide (NO) to produce NO and nitrous oxide (N2O), respectively, is catalyzed by metalloenzymes.4 Inspired by these enzymes, diverse transition metal complexes that mediate NO2− and NO reductions have been reported. The electronic structure of such reactions, particularly those involving nitrosyl complexes (M–NO), is complicated due to the non-innocent nature of the NO ligand.5,6 Experimentally, the oxidation state of the NO ligand is estimated from the M–N–O angle and N–O stretching vibration values. In contrast, density functional theory (DFT) calculations provide direct information about the electron densities in molecules and atoms.7–12 Additionally, DFT calculations allow elucidating complicated reaction mechanisms such as concerted proton–electron transfer (CPET).13–18 Therefore, DFT calculations are highly recommended for the analysis of the reaction mechanisms of NO2−/NO reduction catalyzed by transition metal complexes. Here, we discuss two DFT studies on the copper-mediated reductions of NO2− and NO. The former focuses on the CPET in a copper-mediated NO2− reduction and quantitatively estimates the asynchronicity between proton transfer (PT) and electron transfer (ET) during CPET. The latter explores the complicated electronic state changes in a NO reduction reaction mediated by a dicopper complex.Reduction of NO2− to NO mediated by a monocopper complex using a phenol derivative
The reduction of NO2− to NO in the denitrification process is catalyzed by nitrite reductases (NiRs), including copper-containing and heme-containing NiRs.2–4,19–22 In addition, the NO2− reduction complements a physiological synthesis of NO, which functions as a signal transducer in immune response, vasodilation, and neurotransmission.23,24 Enzymes such as Mo-containing NiRs, xanthine oxidase, aldehyde oxidase, sulfite oxidase, and globins also perform the NO2− reduction as a NO reservoir.21,24–27Crystallographic studies have identified the mononuclear type 2 Cu catalytic active site in CuNiRs, where three histidines bind to the Cu center.19–22,28–30 This has inspired researchers to investigate the Cu-complex-mediated NO2− reduction. Woollard-Shore et al.31 synthesized, characterized and evaluated the catalytic properties of four Cu(II) complexes as potential agents for NO2 reduction. The crystallographic study revealed three distinct coordination modes: η1-NO2, η1-ONO, and η2-ONO. Chandra Maji et al.32 conducted an electrochemical study and suggested that the coordination mode is isomerized from η2-ONO to η1-NO2 during NO2− reduction by a Cu(II) complex. Kundu and coworkers have expanded the study of the phenol-mediated NO2− reduction using Cu(II) complexes.33–35 Among them, a reminiscent model of the red copper site in nitrosocyanin exhibited the NO-oxidase activity as well as the NiR activity.35 Cioncoloni et al.36 showed, based on kinetic and DFT studies, the involvement of CPET in the electrocatalytic NO2− reduction by two types of Cu complexes. However, differences of reactivity depending on the coordination mode of NO2− and detailed mechanism of CPET in the Cu-mediated NO2− reduction remain unclear.
This study aims to characterize the mechanism of the NO2− reduction by Cu(II) complexes, as reported by Kundu et al.33 They utilized a Cu(II) complex with a tripodal heteroditopic cryptand ligand (L) and phenol. The reaction of a phenol derivative (ArOH) with a Cu(II) nitrite complex ([CuII(L)(ONO)]+) forms a hydroxyl Cu(II) complex ([CuII(L)(OH)]+), NO, and a biphenol derivative, which was presumed to be formed via coupling with the corresponding phenoxyl radical (ArO˙). Mechanistic studies indicated that CPET occurred during the NO2− reduction. We recently reported a DFT study on the reaction mechanism of this NO2− reduction.37 DFT calculations were used to elucidate the mechanisms of the NO2− reduction in O- and N-coordination modes, highlighting the differences in the CPET process between these modes.
The X-ray crystallography study performed by Kundu et al. shows that NO2− is O-coordinated to Cu.33 However, DFT calculations suggest the formation of an N-coordinated Cu(II)–nitro complex (ICN) in addition to an O-coordinated Cu(II)–nitrite complex ([CuONO]+, ICO). Therefore, two reaction pathways from ICO (nitrite pathway) and ICN (nitro pathway) were examined. Both reaction pathways finally lead to the same product. In the nitrite pathway (Fig. 1), the reaction of ICO with ArOH forms the reactant complex RCO. The simultaneous H atom migration and O–N bond cleavage through transition state TSO produces a hydroxyl Cu(II) complex, ArO˙, and NO. The overall reaction in the nitrite pathway is endothermic by 17.8 kcal mol−1 with an activation energy of 25.2 kcal mol−1 for the H atom migration and O–N bond cleavage. Meanwhile, the nitro pathway involves three steps: H atom migration, N–O bond cleavage, and NO desorption, as shown in Fig. 2. The reaction begins with the association of ICN with ArOH to form the reactant complex RCN. Then, H atom migration from ArOH to NO2− occurs, leading to the HONO intermediate [Cu(HONO)]+ and ArO˙ (IM1N). N1–O2 bond cleavage forms the nitrosyl hydroxyl complex [Cu(NO)(OH)]+ (IM2N) having two spin states corresponding to CuINO+ and CuIINO0. The final step involves NO ligand dissociation. The rate-determining step, i.e., H atom migration, has an activation energy of 22.8 kcal mol−1. Thus, DFT calculations suggest that the nitrite pathway (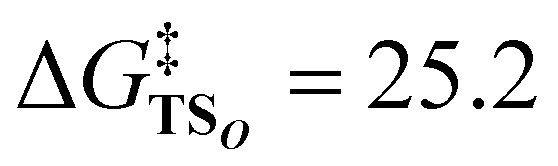 ) and the nitro pathway (
) and the nitro pathway (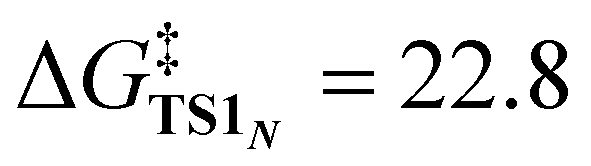 ) are comparable.
) are comparable.
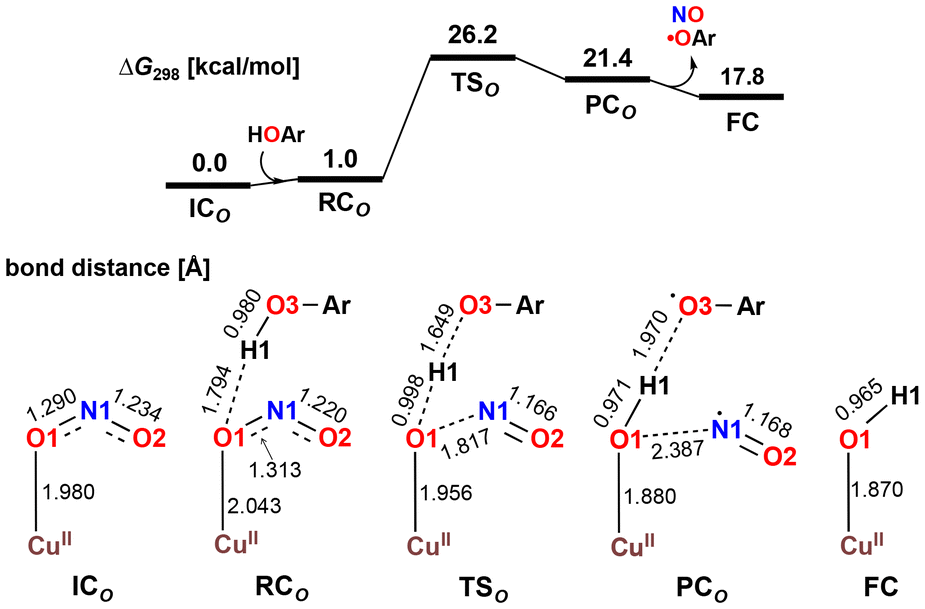 | ||
| Fig. 1 Free energy profile and optimized structures for the reduction of NO2− to NO in the nitrite pathway computed at the B3LYP-D3/6-31G**(Wachters-Hay for Cu) level of theory with the solvent effect of acetonitrile using the polarizable continuum model. Reproduced from ref. 37 with permission from the American Chemical Society, copyright 2023. | ||
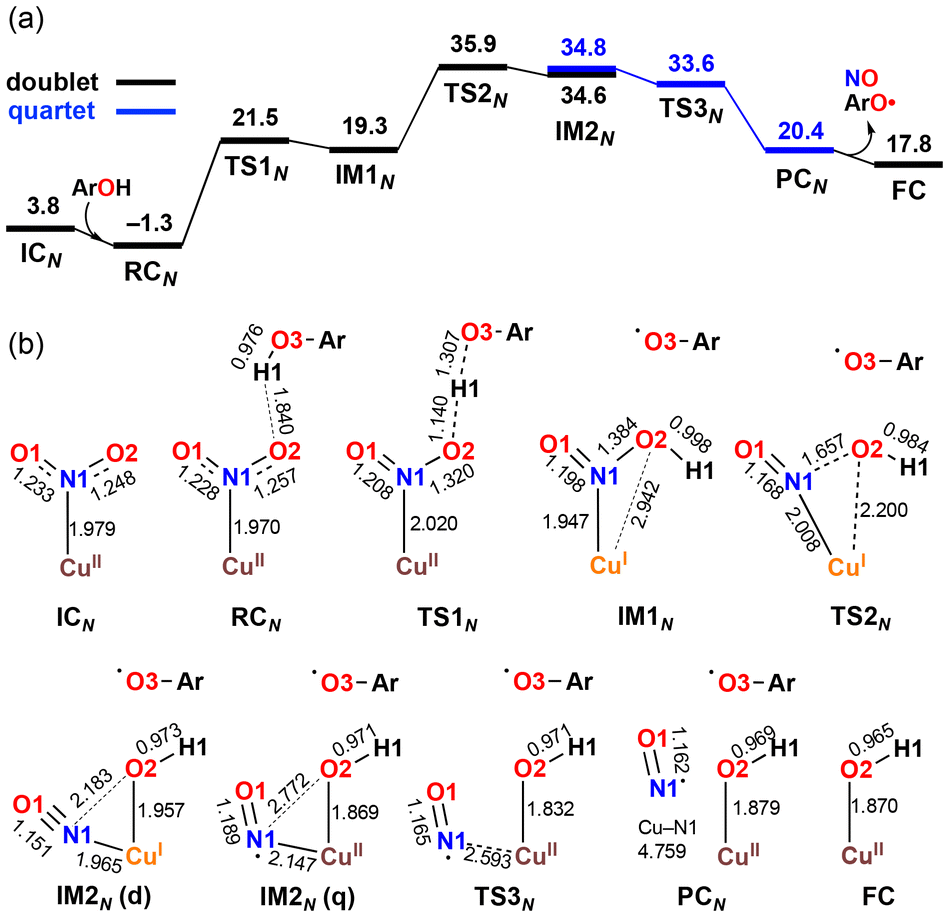 | ||
| Fig. 2 Free energy profile (a) and optimized structures (b) for the reduction of NO2− to NO at the B3LYP-D3/6-31G**(Wachters-Hay for Cu) level of theory with the solvent effect of acetonitrile using the polarizable continuum model. Free energy and bond distance are represented in kcal mol−1 and Å, respectively. Reproduced from ref. 37 with permission from the American Chemical Society, copyright 2023. | ||
The above mentioned approach allowed tracing changes on the potential energy surface by connecting discrete points of stable structures and transition states; however, further insights can be obtained by using intrinsic reaction coordinate (IRC) calculations to trace continuous changes in electronic energies, geometries, and electronic configurations, particularly regarding the CPET mechanism. To better understand the CPET-derived NO2− reduction, we carefully examined the electronic state changes associated with the structural changes derived from the IRC analysis, revealing the PT and ET behavior along the reaction coordinate. Over the past decade, a mechanism involving the concerted but asynchronous movement of a proton and an electron (i.e., asynchronous CPET) has been proposed.38–43 Given the existence of multiple CPET mechanisms, a detailed analysis requires information on the sequential changes in charge and structure along the reaction path. To this aim, IRC calculations, which can determine the reaction pathway itself, are highly valuable. We recently introduced indices to spotlight the asynchronicity of PT and ET in a CPET reaction, which we called PT point and ET point, respectively. The PT and ET points designate the location of the middle progression of PT and ET in the reaction coordinate as depicted in Fig. 3. In particular, the PT point is the location where the two O–H bonds are of the same length and the ET point is the location where 50% of the total change in the spin density of the atoms involved in ET occurs. Calculating the gap between PT and ET points (Δs) allows evaluating quantitatively their asynchronicity and classifying the type of CPET (Fig. 3(b)).
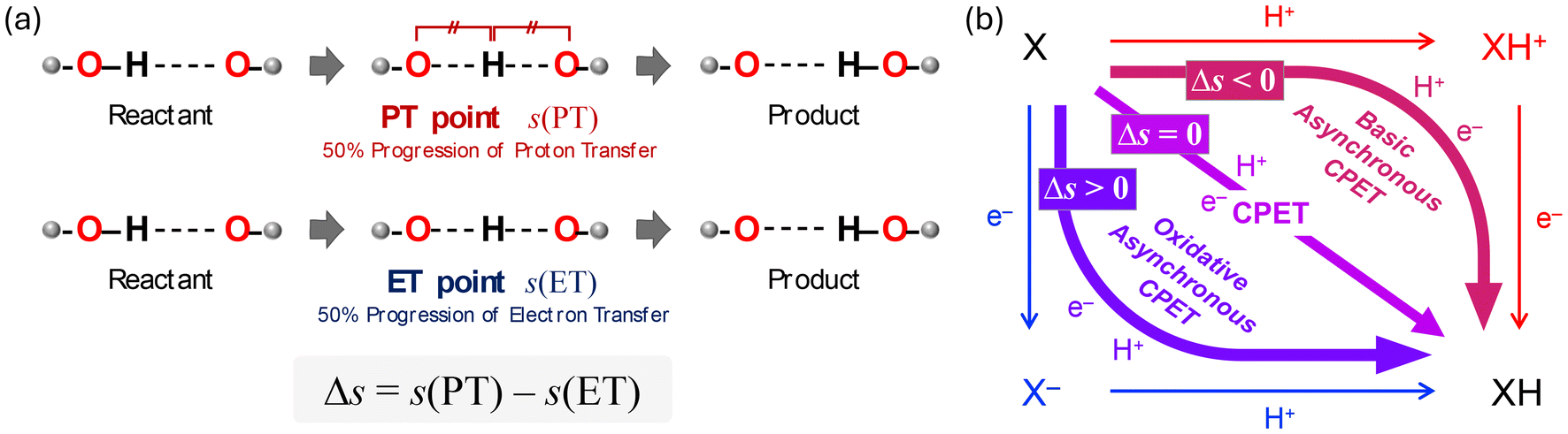 | ||
| Fig. 3 (a) Conceptual representation of the definition of proton transfer (PT) point and electron transfer (ET) point. (b) Application of PT and ET points to the square scheme. When Δs < 0, PT occurs prior to ET (basic-asynchronous CPET). When Δs = 0, PT and ET proceed simultaneously (synchronous CPET). When Δs > 0, ET occurs before PT (oxidative-asynchronous CPET). | ||
The IRC calculation results shown in Fig. 4 revealed changes in bond distance and spin density as a function of the reaction coordinate (s), where a reaction coordinate of s = 0.0 corresponds to a transition state. The values of two O–H distances, one N–O distance, and the spin densities for the Cu, N1, O1, O2, and O3 atoms are shown. In the PT point, the two O–H bonds are of equal length, and the O–H distances change before and after PT. Meanwhile, in the ET point, 50% of the total change in spin density for the atoms involved in ET occurs. The results for the reaction from RCO to PCOviaTSO in the nitrite pathway are shown in Fig. 4(a) and (c). As depicted in Fig. 4(a), the increase in the O3–H1 distance and the decrease in the O1–H1 distance, which correspond to the migration of the H1 atom from the O3 atom to the O1 atom, is nearly complete before reaching the transition state (s = 0.0). However, the O1–N1 distance consistently increases throughout the reaction. Here, the PT point is defined as the intersection between the two plots of the O1–H1 and O3–H1 distances, which was determined to be s(PT) = −1.72. Fig. 4(c) shows that the α-spin emerges on the O3 atom, whereas the β-spin appears on the N1 and O2 atoms. The ET point is defined as the 50% completion point of the spin-density change for the N1 atom, which was identified as s(ET) = −0.07. Consequently, Δs = s(PT) − s(ET) = −1.65, indicating that PT occurs earlier than ET. These computational results indicate that TSO represents a basic-asynchronous CPET, with “basic” referring to the dominance of the acid–base character.43–45
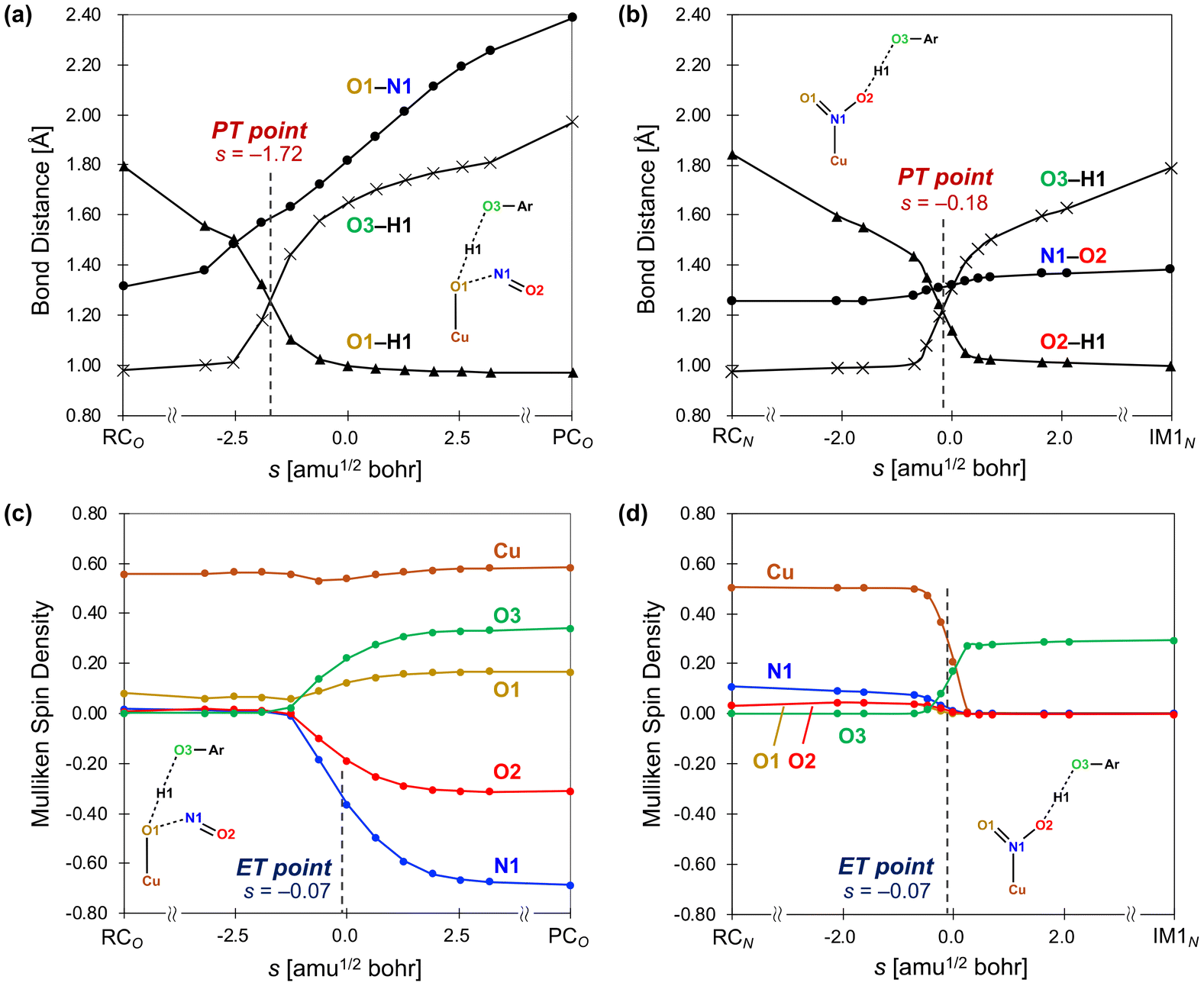 | ||
| Fig. 4 Intrinsic coordinate plots of the changes in structures and spin densities from RCO to PCOviaTSO in the nitrite pathway (a and c) and from RCN to IM1NviaTS1N in the nitro pathway (b and d) as a function of the reaction coordinate s [amu1/2 bohr]. The transition state is s = 0.0. The proton transfer (PT) point is defined as the intersection between the two O–H distances (O1–H1 and O3–H1 in (a) and O2–H1 and O3–H1 in (b)). The electron transfer (ET) point is defined as the point where the spin density for the N1 (Cu) atom reaches the midpoint of RCO and PCO in (c) (RCN and IM1N in (d)). Reproduced from ref. 37 with permission from the American Chemical Society, copyright 2023. | ||
Fig. 4(b) and (d) illustrate the results for the reaction from RCN to IM1NviaTS1N in nitro pathway. As shown in Fig. 4(b), the increase in the O3–H1 distance and the decrease in the O2–H1 distance, corresponding to the migration of the H1 atom from the O3 atom to the O2 atom, progress considerably near s = 0. The O1–N1 distance also increases substantially near s = 0; however, this change is minimal and can be attributed to the elongation of the N–O bond from a double bond to a single bond. In this case, the PT point, which is defined as the intersection between the two plots of the O2–H1 and O3–H1 distances, was determined to be s(PT) = −0.18. Fig. 4(d) shows an increase and a decrease in the spin densities on the Cu atom and the O3 atom, respectively. The ET point, defined as the 50% completion point of the spin-density change for the Cu atom, was identified as s(ET) = −0.07. Consequently, a gap of Δs = −0.11 exists between PT and ET, indicating that both processes progress almost synchronously. According to the IRC results, the reaction step viaTS1N was classified as a synchronous CPET. These characterizations of CPET are consistent with a previous work. Bím et al.46 introduced the asynchronicity factor based on reduction potential and acidity constant, proposing an inverse correlation between asynchronicity and the imaginary frequency of the transition state of CPET. In our calculations, we also identified this inverse correlation; TSO with a smaller imaginary frequency (300i cm−1) is characterized as basic-asynchronous CPET while TS1N with a larger imaginary frequency (1461i cm−1) is classified as synchronous CPET.
The concept of PT and ET points allows evaluating simple reactions involving the transfer of one proton and one electron. However, it is expected to be applicable to the elucidation of more complex reaction mechanisms in biological systems, such as ion channels, which involve the transfer of multiple protons and electrons.
DFT study on the reduction of NO to N2O catalyzed by a dicopper complex
The second topic of this Frontier article is the reduction of NO to N2O, which is crucial in the processes of denitrification and catalytic flue gas purification. This reaction, which is catalyzed by NO reductases (NOR) and flavodiiron NO reductases, involves the formation of a hyponitrite (N2O22−) intermediate via N–N bond formation.4,47,48 However, details of the N–N bond formation, N–O bond cleavage, the possible intermediate species, and the precise timing of the metal redox shuttling remain to be elucidated. This knowledge will provide important insights into the design of NOR-related therapeutics and improved NOx purification systems.Theoretical studies on transition-metal-complex-mediated NO reduction reactions are limited. We previously reported the investigation of the mechanism of NO reduction reactions catalyzed by a dinuclear Ru complex49 and Cu–ZSM-550 using DFT calculations. Recently, we focused on the experimental study by Tao et al.51 and theoretically elucidated the reaction mechanism of the NO reduction mediated by a dinuclear copper complex52 and the alkane hydroxylation performed using the oxo species produced after the reaction.53 Our DFT analysis revealed an interesting ET during the NO reduction reaction.
The dinuclear copper complex bearing a 1,2-bis(di(pyridin-2-yl)methoxy)benzene ligand (A) reported by Tao et al. was found to activate NO.51 The reaction of A with 3 equiv. of NO yields a (μ-oxo)(μ-nitrosyl) dicopper complex and N2O (Fig. 5). We investigated this reaction and proposed a mechanism for the reduction of NO to N2O by a dicopper complex. In this type of reaction, a μ-oxo complex is generally considered as an intermediate or a product. Therefore, the reaction was presumed to proceed via the reaction of the dicopper complex A with 2 equiv. of NO to form μ-oxo complex B followed by the coordination of another NO molecule to B to give the complex product (Fig. 5). In that study, we focused on the NO reduction to form N2O and B.
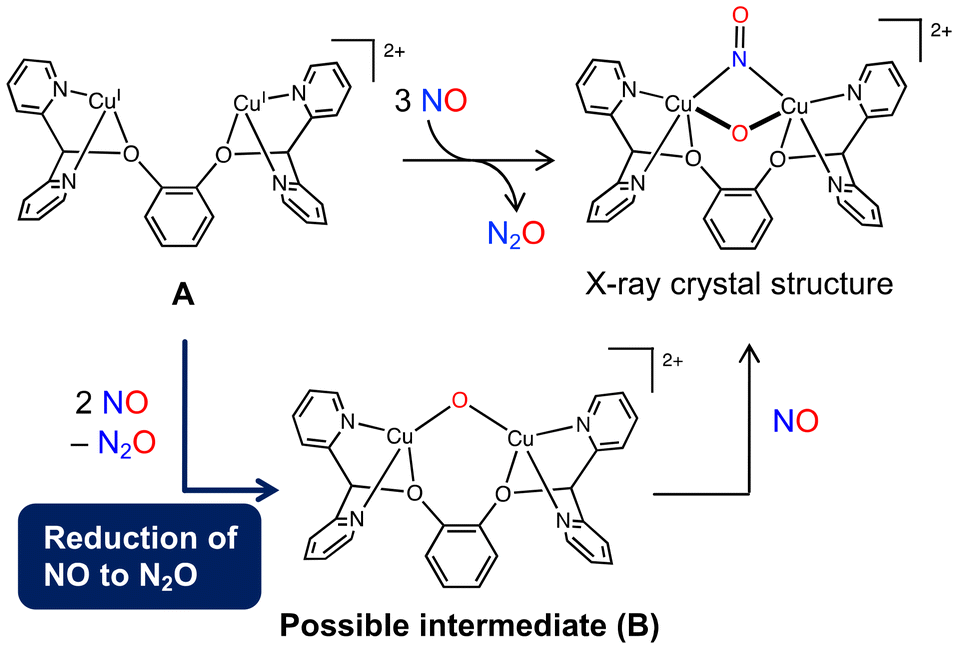 | ||
| Fig. 5 Possible reaction pathway of the NO reduction using dicopper complex A reported by Tao et al. Reproduced from ref. 52 with permission from the Royal Society of Chemistry, copyright 2022. | ||
The reaction mechanism and ET proposed on the basis of the calculations are depicted in Fig. 6. The reaction comprises three steps: (1) N–N bond formation, (2) isomerization of N2O2, and (3) N–O bond cleavage. In the initial complex, two NO molecules are coordinated to two Cu centers in an end-on manner. Initially, one electron from one Cu atom is transferred to NO, forming an intermediate with an N–N bond of 1.417 Å. The activation energy for this reaction is 21.8 kcal mol−1 and the formal charge of the N2O2 ligand is −1, which indicates that the coordination mode of the NO moiety changes from end-on to side-on. Subsequently, isomerization of the N2O2 ligand with rotation around the N–N bond occurs. In the transition state of this reaction, the N–N bond rotation barrier is lowered as the electrons of N2O2 temporarily return to the Cu atom, reducing the N–N bond order. This results in an activation energy of 14.0 kcal mol−1. Regarding the electron configuration, the closed-shell singlet of the Cu(I)Cu(I) moiety is most stable in the transition state. After the reaction, two electrons are transferred from one of the Cu atoms to N2O2, resulting in a Cu(I)Cu(III) configuration. However, due to their instability, charge transfer between the Cu atoms leads to a Cu(II)Cu(II) open-shell singlet or triplet as the ground state. The final step involves the cleavage of the bond between the bridged O atom and the adjacent N atom, yielding a N2O molecule. The activation energy of this process is 7.4 kcal mol−1, indicating that the N–O bond of the N2O2 ligand is easily broken. The overall reaction proceeds exothermically with an energy of 35.0 kcal mol−1. The rate-determining step is the first step of the N–N bond formation, and its activation energy is 21.8 kcal mol−1. During the NO reduction, the dicopper complex possesses various intermediates containing the cis- and trans-N2O2 isomers with different coordination modes to the Cu atoms. This flexibility of the dicopper complex allows adopting the most favored structures for each step in the NO reduction, i.e., N–N bond formation, N2O2 isomerization, and N–O bond cleavage. In addition to this structural flexibility, the frequent ET between the Cu atoms and the ligand, as illustrated in Fig. 6(b), facilitate the occurrence of the sequential reactions during the NO reduction. In a recent publication about the same dicopper complex, Tao et al. reported new kinetic data indicating the necessity of the third equivalent of NO in the reductive NO coupling.54 The paper suggested that the third NO facilitates the N–N bond formation to form the μ-oxo μ-nitroxyl complex [Cu2(NO)O] without through the μ-oxo complex [Cu2O]. This mechanism is strongly linked to the disproportionation of NO (3 NO → NO2 + N2O). In light of these findings, it would be beneficial for future studies to consider the third NO in the mechanistic study of NO reduction reactions.
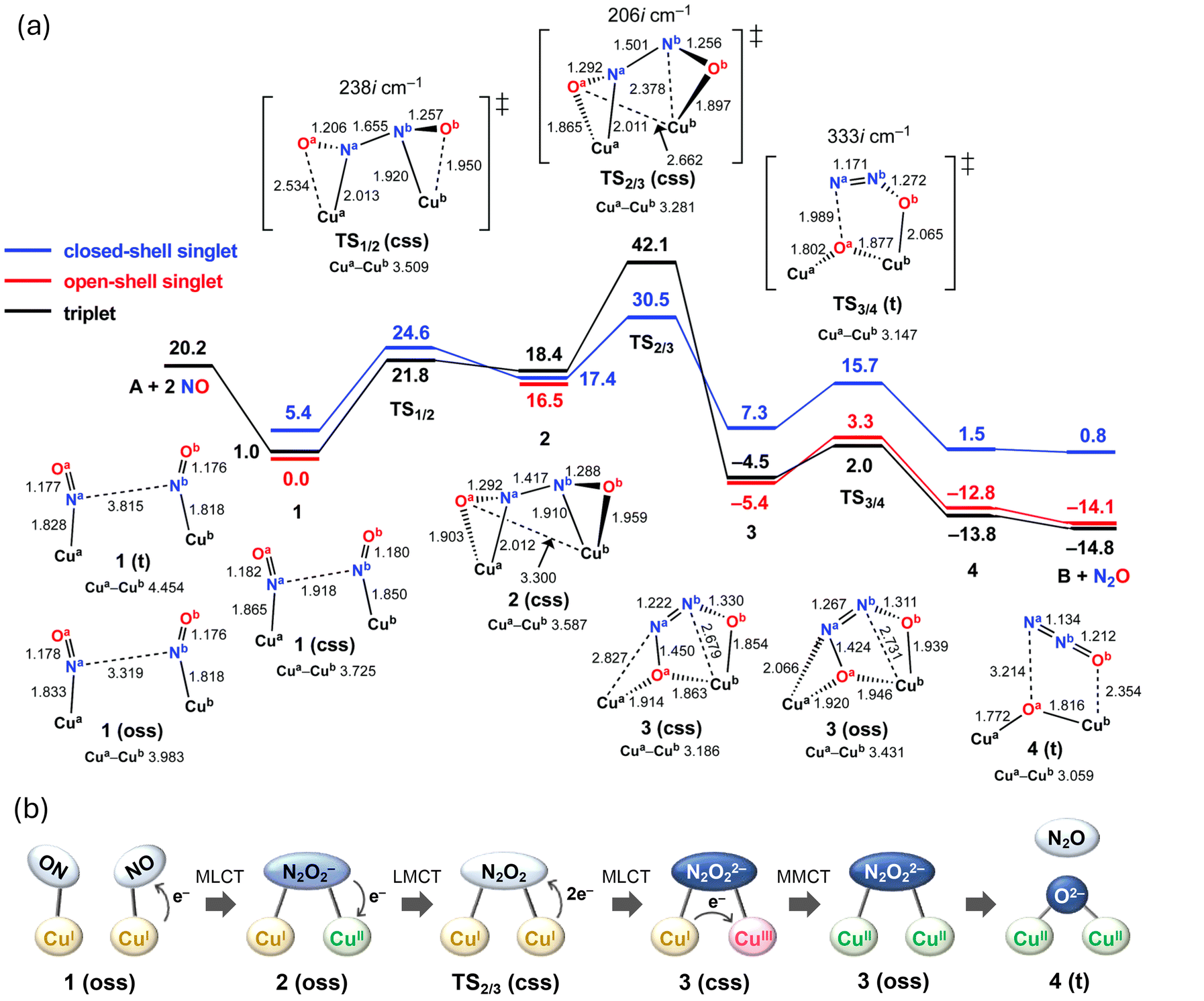 | ||
| Fig. 6 (a) Free-energy profile for the reduction of NO to N2O by dicopper complex A. Free energies are expressed in kcal mol−1. Energies and optimized geometries were calculated at the B3LYP-D3/D95**(Wachters-Hay for Cu) level of theory with the solvent effect of tetrahydrofuran using the polarizable continuum model. (b) Schematic representation of the changes in electronic states during the NO reduction by the dicopper complex. Reproduced from ref. 52 with permission from the Royal Society of Chemistry, copyright 2022. | ||
Conclusions
This Frontier article summarizes the mechanistic aspects of two important NOx reduction processes: NO reduction and NO2− reduction. First, the mechanism of the reduction of NO2− to NO mediated by a Cu(II) complex is discussed. According to geometry optimization, we identified two types of [CuNO2]+ initial complexes, i.e., an O-coordinated Cu(II)–nitrite complex (ICO) and an N-coordinated Cu(II)–nitro complex (ICN), which allowed proposing the nitrite and nitro reaction pathways, respectively. Given their energy profiles, the two pathways are comparable. In addition, to determine the exact nature of the CPET reactions in both pathways, we studied the changes in the geometric and electronic structures using an IRC analysis. Then, we introduced the PT and ET points as indices to evaluate the asynchronicity of PT and ET in a CPET reaction using the difference between the PT and ET points (Δs = s(PT) − s(ET), where the s value represents the location of PT and ET). In the nitrite pathway, Δs = −1.65, indicating that the PT point precedes the ET point, that is the reaction is a basic asynchronous CPET. In contrast, in the nitro pathway, the reaction is a synchronous CPET because the PT and ET points are close (Δs = −0.11). Although the two pathways lead to the same products, i.e., [Cu(II)OH]+, NO, and ArO˙, their CPET mechanisms differ substantially in terms of the asynchronicity of PT and ET. Thus, by tracking the continuous change in electronic structure via IRC analysis, we successfully described the CPET behavior in the NO2− reduction. The concept of PT and ET points will facilitate the characterization and understanding of CPET reactions.We also investigated the mechanism of the reduction of NO to N2O by a dicopper complex using DFT calculations. The computed results indicated that the reaction consists of three fundamental steps: (1) N–N bond formation, (2) isomerization of the N2O2 moiety, and (3) N–O bond cleavage. The calculated reaction mechanism predicts the initial coupling of two NO molecules. These results are consistent with experimental observations, where N2O is released in the presence of the dicopper complex. Additionally, the large binding energy of NO and Cu atoms in the reactant complex [Cu2(NO)2]2+ and the small binding energy of N2O in the product complex [Cu2(N2O)(μ-O)]2+ favor this catalytic cycle. Thus, DFT calculations allow following the frequent ET between Cu and the ligand in addition to structural changes in the reaction mechanism, providing a useful tool for the analysis of reaction pathways and revealing the details of the sequential N–N bond formation, isomerization of N2O2, and N–O bond cleavage reactions.
Author contributions
YK – Conceptualization, writing – original draft. YS – Conceptualization, writing – review and editing.Data availability
Data sharing is not applicable to this article as no new data were created or analyzed in this study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Japan Society for the Promotion of Science National Science (JSPS) KAKENHI Grant-in-Aid for JSPS Fellows JP22KJ2475.References
- D. E. Canfield, A. N. Glazer and P. G. Falkowski, Science, 2010, 330, 192–196 CrossRef.
- D. J. Richardson and N. J. Watmough, Curr. Opin. Chem. Biol., 1999, 3, 207–219 CrossRef.
- A. J. Timmons and M. D. Symes, Chem. Soc. Rev., 2015, 44, 6708–6722 RSC.
- I. M. Wasser, S. de Vries, P. Moënne-Loccoz, I. Schröder and K. D. Karlin, Chem. Rev., 2002, 102, 1201–1234 CrossRef.
- C. K. Jørgensen, Coord. Chem. Rev., 1966, 1, 164–178 CrossRef.
- W. Kaim, Inorg. Chem., 2011, 50, 9752–9765 CrossRef PubMed.
- G. Schenk, M. Y. M. Pau and E. I. Solomon, J. Am. Chem. Soc., 2004, 126, 505–515 CrossRef PubMed.
- R. G. Serres, C. A. Grapperhaus, E. Bothe, E. Bill, T. Weyhermüller, F. Neese and K. Wieghardt, J. Am. Chem. Soc., 2004, 126, 5138–5153 CrossRef PubMed.
- F. Roncaroli, M. Videla, L. D. Slep and J. A. Olabe, Coord. Chem. Rev., 2007, 251, 1903–1930 CrossRef.
- V. K. K. Praneeth, F. Paulat, T. C. Berto, S. D. George, C. Näther, C. D. Sulok and N. Lehnert, J. Am. Chem. Soc., 2008, 130, 15288–15303 CrossRef PubMed.
- M. Radon, E. Broclawik and K. Pierloot, J. Phys. Chem. B, 2010, 114, 1518–1528 CrossRef PubMed.
- M. A. Hough, J. Conradie, R. W. Strange, S. V. Antonyuk, R. R. Eady, A. Ghosh and S. S. Hasnain, Chem. Sci., 2020, 11, 12485–12492 RSC.
- J. M. Mayer, D. A. Hrovat, J. L. Thomas and W. T. Borden, J. Am. Chem. Soc., 2002, 124, 11142–11147 CrossRef PubMed.
- G. A. DiLabio and E. R. Johnson, J. Am. Chem. Soc., 2007, 129, 6199–6203 CrossRef PubMed.
- D. Usharani, D. C. Lacy, A. S. Borovik and S. Shaik, J. Am. Chem. Soc., 2013, 135, 17090–17104 CrossRef PubMed.
- T. Ishizuka, A. Watanabe, H. Kotani, D. C. Hong, K. Satonaka, T. Wada, Y. Shiota, K. Yoshizawa, K. Ohara, K. Yamaguchi, S. Kato, S. Fukuzumi and T. Kojima, Inorg. Chem., 2016, 55, 1154–1164 CrossRef PubMed.
- K. Koshiba, K. Yamauchi and K. Sakai, Angew. Chem., Int. Ed., 2017, 56, 4247–4251 CrossRef PubMed.
- M. J. Chalkley, T. J. Del Castillo, B. D. Matson and J. C. Peters, J. Am. Chem. Soc., 2018, 140, 6122–6129 CrossRef PubMed.
- B. A. Averill, Chem. Rev., 1996, 96, 2951–2964 CrossRef PubMed.
- S. Besson, M. G. Almeida and C. M. Silveira, Coord. Chem. Rev., 2022, 464, 214560 CrossRef CAS.
- L. B. Maia and J. J. G. Moura, Chem. Rev., 2014, 114, 5273–5357 CrossRef CAS.
- F. E. Dodd, J. Van Beeumen, R. R. Eady and S. S. Hasnain, J. Mol. Biol., 1998, 282, 369–382 CrossRef CAS.
- E. Culotta and D. E. Koshland, Science, 1992, 258, 1862–1865 CrossRef CAS PubMed.
- J. O. Lundberg, M. T. Gladwin and E. Weitzberg, Nat. Rev. Drug Discovery, 2015, 14, 623–641 CrossRef CAS.
- J. Wang, G. Keceli, R. Cao, J. T. Su and Z. Y. Mi, Redox Rep., 2017, 22, 17–25 CrossRef CAS PubMed.
- C. E. Sparacino-Watkins, J. Tejero, B. Sun, M. C. Gauthier, J. Thomas, V. Ragireddy, B. A. Merchant, J. Wang, I. Azarov, P. Basu and M. T. Gladwin, J. Biol. Chem., 2014, 289, 10345–10358 CrossRef CAS.
- M. T. Gladwin, R. Grubina and M. P. Doyle, Acc. Chem. Res., 2009, 42, 157–167 CrossRef CAS PubMed.
- J. W. Godden, S. Turley, D. C. Teller, E. T. Adman, M. Y. Liu, W. J. Payne and J. LeGall, Science, 1991, 253, 438–442 CrossRef CAS.
- R. R. Eady and S. S. Hasnain, Coord. Chem. Rev., 2022, 460, 214463 CrossRef.
- S. L. Rose, F. M. Ferroni, S. Horrell, C. D. Brondino, R. R. Eady, S. Jaho, M. A. Hough, R. L. Owen, S. V. Antonyuk and S. S. Hasnain, J. Mol. Biol., 2024, 436, 168706 CrossRef.
- J. G. Woollard-Shore, J. P. Holland, M. W. Jones and J. R. Dilworth, Dalton Trans., 2010, 39, 1576–1585 RSC.
- R. Chandra Maji, S. Mishra, A. Bhandari, R. Singh, M. M. Olmstead and A. K. Patra, Inorg. Chem., 2018, 57, 1550–1561 CrossRef PubMed.
- A. Mondal, K. P. Reddy, J. A. Bertke and S. Kundu, J. Am. Chem. Soc., 2020, 142, 1726–1730 CrossRef PubMed.
- S. Gupta, S. Arora, A. Mondal, S. C. E. Stieber, P. Gupta and S. Kundu, Eur. J. Inorg. Chem., 2022, 15, e202200105 CrossRef.
- B. S. Anju, N. R. Nair and S. Kundu, Angew. Chem., Int. Ed., 2023, 62, e202311523 CrossRef PubMed.
- G. Cioncoloni, I. Roger, P. S. Wheatley, C. Wilson, R. E. Morris, S. Sproules and M. D. Symes, ACS Catal., 2018, 8, 5070–5084 CrossRef.
- Y. Kametani, K. Ikeda, K. Yoshizawa and Y. Shiota, Inorg. Chem., 2023, 62, 13765–13774 CrossRef PubMed.
- J. W. Darcy, S. S. Kolmar and J. M. Mayer, J. Am. Chem. Soc., 2019, 141, 10777–10787 CrossRef.
- S. C. Coste, A. C. Brezny, B. Koronkiewicz and J. M. Mayer, Chem. Sci., 2021, 12, 13127–13136 RSC.
- R. Tyburski, T. F. Liu, S. D. Glover and L. Hammarström, J. Am. Chem. Soc., 2021, 143, 560–576 CrossRef.
- H. Kotani, H. Shimomura, K. Ikeda, T. Ishizuka, Y. Shiota, K. Yoshizawa and T. Kojima, J. Am. Chem. Soc., 2020, 142, 16982–16989 CrossRef PubMed.
- D. Bím, M. Maldonado-Domínguez, L. Rulísek and M. Srnec, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E10287–E10294 CrossRef.
- J. S. Zhang, Y. M. Lee, M. S. Seo, Y. Kim, E. Lee, S. Fukuzumi and W. Nam, Inorg. Chem. Front., 2022, 9, 3233–3243 RSC.
- M. K. Goetz and J. S. Anderson, J. Am. Chem. Soc., 2019, 141, 4051–4062 CrossRef.
- X. H. Chen, Y.-F. Yang and Y. B. She, Org. Chem. Front., 2024, 11, 1039–1049 RSC.
- D. Bím, M. Maldonado-Domínguez, L. Rulíšek and M. Srnec, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E10287–E10294 CrossRef.
- C. Varotsis, T. Ohta, T. Kitagawa, T. Soulimane and E. Pinakoulaki, Angew. Chem., Int. Ed., 2007, 46, 2210–2214 CrossRef.
- T. Hayashi, J. D. Caranto, D. A. Wampler, D. M. Kurtz and P. Moënne-Loccoz, Biochemistry, 2010, 49, 7040–7049 CrossRef.
- T. Suzuki, H. Tanaka, Y. Shiota, P. K. Sajith, Y. Arikawa and K. Yoshizawa, Inorg. Chem., 2015, 54, 7181–7191 CrossRef PubMed.
- P. K. Sajith, Y. Shiota and K. Yoshizawa, ACS Catal., 2014, 4, 2075–2085 CrossRef.
- W. Tao, J. K. Bower, C. E. Moore and S. Zhang, J. Am. Chem. Soc., 2019, 141, 10159–10164 CrossRef PubMed.
- Y. Kametani, T. Abe, K. Yoshizawa and Y. Shiota, Dalton Trans., 2022, 51, 5399–5403 RSC.
- T. Abe, Y. Kametani, K. Yoshizawa and Y. Shiota, Inorg. Chem., 2021, 60, 4599–4609 CrossRef PubMed.
- W. Tao, S. Carter, R. Trevino, W. Zhang, H. S. Shafaat and S. Zhang, J. Am. Chem. Soc., 2022, 144, 22633–22640 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |