DOI:
10.1039/D4DT02790F
(Perspective)
Dalton Trans., 2024,
53, 19058-19074
Amidinato silylene-based inorganic aromatic rings
Received
3rd October 2024
, Accepted 8th November 2024
First published on 13th November 2024
Abstract
Aromaticity is a key concept that underpins the behavior and applications of a wide range of chemical compounds. Its impact on stability, reactivity, biological functions, material properties, and environmental persistence underscores the importance of understanding and harnessing aromaticity in chemistry and materials sciences. We have been pioneers in the field of silylene chemistry and recently, our silylene molecules have been used to synthesize several inorganic aromatic ring compounds. Aromaticity in inorganic compounds is not commonly observed; hence, inorganic aromatic rings derived from silylene would further enhance our understanding of aromaticity and stability. Herein, we discuss the inorganic aromatic rings which have been synthesized from amidinato silylene.
 Saroj Kumar Kushvaha | Saroj Kumar Kushvaha earned his B.Sc. degree from the University of Lucknow in 2010 and his M.Sc. degree from DAVV Indore in 2012. He then worked as a chemistry lecturer from 2013 to 2016 and briefly as a junior researcher at the Defense Research and Development Organization (DRDO). In 2022, he received his Ph.D. from the Indian Institute of Technology Madras, Chennai. Currently, he is a post-doctoral researcher at Institut für Anorganische Chemie, Georg-August-Universität Göttingen, Germany. His research endeavors are mainly focused around main-group chemistry, particularly the chemistry of group 13 and group 14 elements. |
 Herbert W. Roesky | Herbert W. Roesky earned his doctorate from the Georg-August University Göttingen, Germany, in inorganic chemistry. After working at Du Pont in the United States, he returned to Göttingen to complete his habilitation. By 1971, he had become a professor at Johann Wolfgang Goethe University in Frankfurt am Main. In 1980, he moved back to the University of Göttingen, where he served as the director of the Institute for Inorganic Chemistry until 2004. He has been a visiting professor at several internationally reputed universities. Roesky is renowned for his groundbreaking research on fluorides involving both transition and main group elements. Currently, he focuses on various aspects of compounds with low-valent silicon. His prolific contributions include over 1350 peer-reviewed papers, articles, patents, and books in the fields of inorganic chemistry and materials sciences. He is also famous for his book Chemical Curiosities and demonstration of colourful chemical reactions. |
Introduction
Generally, silylenes are neutral divalent silicon compounds that are typically unstable and highly reactive intermediates due to their unsaturated nature. However, a broad range of stable and isolable silylenes have now been developed and are available to chemists.1,2 Silylenes are candidates for the chemical vapor deposition (CVD) process to deposit thin films of silicon-containing materials on substrates. This is particularly important in the semiconductor industry for the production of microelectronics and photovoltaic cells. Silylenes can also be used to modify the surface properties of materials, such as enhancing the adhesion, wettability, and chemical resistance of surfaces. This is useful in creating advanced materials with tailored surface characteristics for specific applications. Unlike carbenes, which can exist in either triplet or singlet ground states, the silicon atom in silylenes is in the singlet state and favors the (3s)2(3p)2 valence electron configuration because it is inherently reluctant to undergo s, p-orbital hybridization.3–5 Therefore, silylenes possess a singlet ground state with a non-bonding lone pair of electrons in the plane, affording a significant 3s-character. Consequently, the predominantly 3p out-of-plane orbital readily accepts electrons to comply with the “octet rule”. However, donor-stabilized silylenes have also been reported where the electron pair on the nitrogen atom of the amidinate group is donated to the empty p-orbital of the silicon atom. As a result, these silylenes lack an empty p-orbital and are therefore more stable than typical silylenes (NHSi).6,7 In 1986, Jutzi et al. made a groundbreaking advancement in silylene chemistry by isolating decamethylsilicocene ((η5-C5Me5)2Si:), the first stable compound containing a divalent silicon(II) atom.8 This discovery highlighted the necessity of electronic and steric saturation for synthesizing isolable silylenes. The first major breakthrough in silylene chemistry came in 1994 when West and Denk synthesized the first N-heterocyclic silylene (NHSi).9 Since the initial report of NHSi, the chemistry of these molecules has evolved in various directions, leading to several applications in the activation of small molecules,4,10–13 low-valent metal chemistry14 and preparation of polymers.15 Recently, a large number of reports have shown the uses of more stable silylenes such as amidinato-chloro-silylene in diverse fields of chemistry6,16–19 This type of amidinato-silylene has also been utilized to isolate several heterocyclic aromatic and homo-aromatic compounds.
Aromaticity is one of the most debated and important concepts that finds huge applications in organic chemistry as well as inorganic chemistry. Aromaticity is a highly debated topic of chemistry because of the widespread misunderstandings and disagreements regarding its definition and the aromatic nature of specific systems. Firstly, we will discuss some basic criteria for aromaticity.20 For a compound to be aromatic, the key criteria must be satisfied: (1) The molecule must be cyclic, meaning it has a ring structure. This allows for the delocalization of π electrons around the ring. (2) The molecule must be planar or nearly planar, allowing for the effective overlap of p-orbitals and thus the delocalization of π electrons across the ring. (3) The ring must have a continuous overlap of p-orbitals, meaning that every atom in the ring should have a p-orbital available for π electron delocalization. This typically involves alternating single and double bonds or a similar arrangement that provides continuous conjugation. (4) It must satisfy Hückel's rule. The molecule must have a specific number of π electrons that fit the formula 4n + 2, where n is a non-negative integer (0, 1, 2, 3, 4....). For example, benzene (C6H6) has 6 π electrons (n = 1), which satisfies Hückel's rule.21 (5) For four-membered heterocyclic (B/Si/C) rings to be aromatic, the ring should display a dissymmetric A2B2 arrangement and 2e should be present at 1,3-positions. Additionally, 2e should be able to delocalize through vacant orbitals of atoms at 1,4-positions. These atom arrangements are made possible by carefully designing molecules in such a way that the orbitals with matching symmetry and energy are made available to facilitate effective delocalization of electrons and covalent interaction.22a
As the above-listed criteria suggest, there may be numerous features of the molecules that describe the presence of aromaticity in a molecule; however, it is quite difficult to give a universal definition of aromaticity. In this regard, several attempts have been made to define aromaticity. However, the definition of aromaticity given by Chen et al. in 2005 is quite inclusive, which is worth quoting here “Aromaticity is a manifestation of electron delocalization in closed circuits, either in two or in three dimensions. This results in energy lowering, often quite substantial, and a variety of unusual chemical and physical properties. These include a tendency toward bond length equalization, unusual reactivity, and characteristic spectroscopic features”.22b,c
For twisted ring systems, the term Möbius aromaticity is used. Möbius aromaticity is a concept that extends the idea of aromaticity to systems that have a twisted ring structure, leading to the delocalization of both σ and π electrons. A Möbius aromatic system has a single twist, forming a Möbius strip-like structure. While Möbius aromaticity is often discussed in theoretical contexts, it has been reported in several systems representing a fascinating extension of aromaticity principles.22d
In planar cyclic structures, Baird's rule is used to estimate whether the lowest triplet state will exhibit aromatic properties. According to Baird's rule, the lowest triplet state of an annulene is aromatic if it contains 4n π-electrons and antiaromatic if it has (4n + 2) π-electrons, where n is a positive integer. This trend is the opposite of Hückel's rule, which applies to the ground state, usually the lowest singlet state (S0). As a result, Baird's rule is often referred to as the photochemical analogue of Hückel's rule.23a
Recently, the term spherical aromaticity has been used to describe the unusual stability of certain spherical compounds like fullerenes and polyhedral boranes.23b Fullerenes are a unique and interesting class of spherical molecules characterized by a conjugated π system, wherein each fullerene molecule consists of a closed network of interconnected hexagons and pentagons. Therefore, they have become a highly important class of compounds for studying their properties including stability and reactivity. In 2000, Hirsch and colleagues developed a rule to predict when a fullerene would exhibit aromaticity. According to Hirsch's rule, a fullerene would display aromatic properties if it contained 2(n + 1)2 π-electrons.23c This is because an aromatic fullerene must possess full icosahedral (or other appropriate) symmetry, requiring that its molecular orbitals be completely filled. This condition is satisfied only when there are exactly 2(n + 1)2 electrons, where n is a nonnegative integer. For example, buckminsterfullerene, with its 60 π-electrons, is non-aromatic since 60/2 = 30, which is not a perfect square. Subsequently, in 2011, Poater and coworkers expanded this rule to predict aromaticity in open-shell fullerene species. They determined that a fullerene would be aromatic if it had (2n2 + 2n + 1) π-electrons.23d This is based on the fact that a spherical species with a same-spin half-filled outer energy level and all inner levels fully filled can also be aromatic.
Although the concept of aromaticity holds huge importance in chemistry, primarily within the realm of organic compounds, its applicability, however, has also been extended to inorganic compounds.24–26 The inorganic analogues of benzene have been reported.27 In general, aromatic metal clusters have closed-shell electronic configurations and contain a cyclic arrangement of metal atoms with delocalized π electrons. These clusters exhibit special stability, electronic structure, and reactivity and have applications in fields such as catalysis, materials science, and electronics. Recent discoveries of aromatic and antiaromatic behavior in all-metal clusters have provided further directions into describing the chemical bonding, structures, and stability of transition-metal clusters and main-group clusters through the lens of aromaticity and antiaromaticity.20 Although organic molecules show σ- and π-aromaticity, transition metal clusters28 and main group clusters display more diverse types of aromaticity (σ, π, δ and ϕ).29,30 In σ-aromatic compounds, the electrons involved in sigma bonds are delocalized over the entire ring, resulting in a planar, cyclic structure with increased stability. This delocalization typically occurs in main group elements and transition metal complexes with cyclic arrangements of atoms connected by sigma bonds.
σ-Aromaticity is less common than π-aromaticity; however, it plays a crucial role in understanding the stability and reactivity of these compounds, as well as their applications in various fields of chemistry. Due to the presence of the d-orbital and possible electronic delocalization, transition metal clusters exhibit much more diverse chemistry and display a new type of aromaticity called δ-aromaticity.28 δ-Aromaticity is a relatively less common form of aromaticity observed in certain transition metal clusters and involves the delocalization of electrons in d-orbitals of transition metals. Metallabenzenes are another class of compounds where δ-aromaticity can be observed. These compounds are analogs of benzene in which one carbon atom is replaced by a transition metal atom.31 The metal atom can participate in the delocalized electron system using its d-orbitals. The π-aromaticity in inorganic systems has been so commonly observed. For example, Frenking et al. reported boron based small aromatic rings with π-aromaticity.32a Herein, Frenking et al. described computational and spectroscopic identification of [B3(NN)3]+ and [B3(CO)3]+ complexes, which feature the smallest π-aromatic system B3+. Similar cage aromatic compounds/charged aromatic clusters have also been reported, for example Al42− (characterized by SC-XRD),32b B3− (in the gas phase),32c [B3(NCy2)3]2− (characterized by SC-XRD)32detc. Among several reported aromatic systems, all metal aromatic cages would be really interesting to mention here. For instance, Al42− which was reported by Li et al. in 2001 in a series of bimetallic ionic systems with the chemical composition MAl4− (M = Li, Na, or Cu).32a The Al42− unit possesses square planar geometry and has two π electrons delocalized over entire four membered ring, thus conforming to Hückel's (4n + 2) π electron rule for aromaticity. Borazine, often referred to as “inorganic benzene”, is considered aromatic due to its isostructural similarity to benzene and their shared physical properties.33 In recent years, various anionic boron compounds exhibiting aromatic characteristics have been isolated by different research groups.34 Given the similarities between carbon and silicon, it was anticipated that silicon-substituted benzene would also demonstrate aromatic properties, which was also predicted theoretically and observed spectroscopically in the gas phase and in low-temperature matrices. Märkl and Schlosser came together, reporting the formation and NMR observation of 1,4-di-tert-butyl-2,6-bis(trimethylsilyl)-1-silabenzene.35 A significant breakthrough occurred when Tokitoh and coworkers synthesized a stable silabenzene by introducing the sterically bulky 2,4,6-tris[bis(trimethylsilyl)methyl]phenyl substituent at the silicon atom.36 Concurrently, Ando et al. reported the synthesis of 1,4-disila-(Dewar-benzene).37 Furthermore, Scheschkewitz and coworkers have made significant contributions in silicon based inorganic aromatic compounds.38–42 π-Aromaticity in inorganic systems has been scarcely reported; therefore, inorganic rings with π-aromaticity are highly desired.43–45
Some compounds display a special type of aromaticity called homoaromaticity, wherein a molecule exhibits aromatic stability despite having a disruption in delocalization, such as a single sp3-hybridized carbon or silicon atom, disrupting the continuous overlap of p-orbitals typically required for aromatic stabilization.3 The concept of homoaromaticity was first proposed by Winstein in 1959.46 Homoaromatic compounds retain delocalization of π-electrons over a bridged structure, allowing for some degree of aromatic stabilization. The overall stabilization is maintained through non-bonding interactions or through-bond coupling. The most well-known example of a homoaromatic compound is the homoaromatic cation (homotropylium ion), where a positive charge delocalizes over a seven-membered ring interrupted by a single methylene bridge47a or in cyclopentasilane derivatives wherein the delocalization is interrupted by an sp3-hybridized silicon atom.47b The cyclopentasilane ring with an sp3-hybridized silicon can still exhibit aromatic stabilization through non-bonding interactions or through-bond coupling among non-adjacent p-orbitals. The concept of homoaromaticity provides insights into the flexibility and breadth of the aromaticity concept, expanding the understanding of electron delocalization in non-traditional ring systems and its ability to explain stability and reactivity in interrupted conjugated systems makes it a valuable tool for chemists in various fields, enabling the design and synthesis of novel compounds with unique and useful properties.
The concept of aromaticity has constantly evolved since the discovery of the aromatic nature of benzene. Earlier, compounds were labeled aromatic if they display resemblance with benzene. Today, molecules are classified as aromatic if they possess two- or three-dimensional circuits of highly delocalized electrons, typically resulting in enhanced thermodynamic stability, equalized bond lengths, unique reactivity, and characteristic magnetic and spectroscopic properties.48 The so-called aromaticity criteria are derived from these characteristics, but none of them are free of ambiguities. Aromaticity in compounds is explained using different descriptors. These descriptors may have limitations. The most commonly used descriptors include the nucleus-independent chemical shifts (NICS) and induced ring current, e.g., current density analysis, the anisotropy of the induced current density (ACID) and gauge-including magnetically induced currents (GIMIC). Due to its simplicity and efficiency, NICS has become the most widely used index for evaluating the magnetic criterion of aromaticity and antiaromaticity. NICS is based on the absolute shielding (the negative value), that is, the manifestation of the induced magnetic field, at a certain location in space (or around a molecule). Initially, NICS values were used to be calculated at the geometric centers of the rings, which are generally denoted as NICS(0) values. However, later on, it was realized that the σ-frame influences these values. Therefore, it was suggested that the assessment of the aromaticity and antiaromaticity should be done by calculating NICS values at a distance from the plane of the ring. As a result, the NICS values are also calculated at 1.0 Å above the plane of the molecule, which are denoted as NICS(1). To gain deeper insights into electron delocalization, the out-of-plane component of NICS (i.e. NICSzz) is also calculated. Due to the minimal influence of σ-bonds on NICSzz values, NICS(1) and NICSzz are considered more reliable indicators than NICS(0). NICSzz not only distinguishes the electronic ring currents in aromatic, nonaromatic, and antiaromatic species but also provides a means to compare the strength of diatropic and paratropic currents across molecules with varying ring sizes or π electron counts, independent of σ–π separation techniques. To assess π delocalization above the ring center, isotropic chemical shifts, along with in-plane and out-of-plane components, are scanned and plotted. The NICS scan provides a broader picture of electron delocalization. A minimum in the NICSzz curve at a nonzero r value indicates aromatic π delocalization. For strongly aromatic molecules, the NICSzz curve continues to be in the negative region for a considerable distance.
The evaluation of aromaticity in four-membered or smaller rings is quite difficult because σ electron contributions and d electrons of heavy elements are not really negligible. Therefore, NICS values calculated for four-membered or small rings may not be reliable. NICS calculations were known to be an established method for identifying aromatic compounds with five or more; thus, utmost care should be taken while calculating and interpreting NICS values for small ring systems. Hence, the aromatic nature assessment of small ring systems is always accomplished by other methods such as ACID and GIMIC.
The current density analysis employing ACID and GIMIC methods are very common in assessing electron density in aromatic systems. ACID is a computational technique used to analyze electron delocalization and current density in molecules, which helps visualize how electron density flows in response to an external magnetic field. We know that the square of the wavefunction defines the total electron density; similarly, the ACID scalar field defines the density of delocalized electrons. The ACID method proved to be a highly versatile descriptor to analyze delocalization in ground-state molecules, excited states, transition states, organometallics, hyperconjugation and other through-bond and through-space interactions. In an ACID plot, the current density is represented as a 3D or 2D vector field, where the arrows show the direction and magnitude of electron flow in response to an applied magnetic field. The anisotropic current density highlights the regions of a molecule that exhibit circulating currents, often associated with aromatic rings, where electrons are more delocalized and hence exhibit characteristic ring currents. GIMIC specifically accounts for the gauge (reference point for phase) of the wavefunction, which ensures that the computed current densities are gauge-invariant and thus physically meaningful. The GIMIC method uses gauge-including atomic orbitals (GIAOs), which are modified orbitals that incorporate the magnetic vector potential to make the current density independent of the choice of origin. This enables more accurate and consistent results. GIMIC calculates current densities in a molecule by simulating the response of electron density to an external magnetic field. It provides detailed information on where and how current flows within the molecular structure, highlighting both intra- and inter-atomic currents. This current density can be visualized as a vector field or as ring currents around aromatic rings.
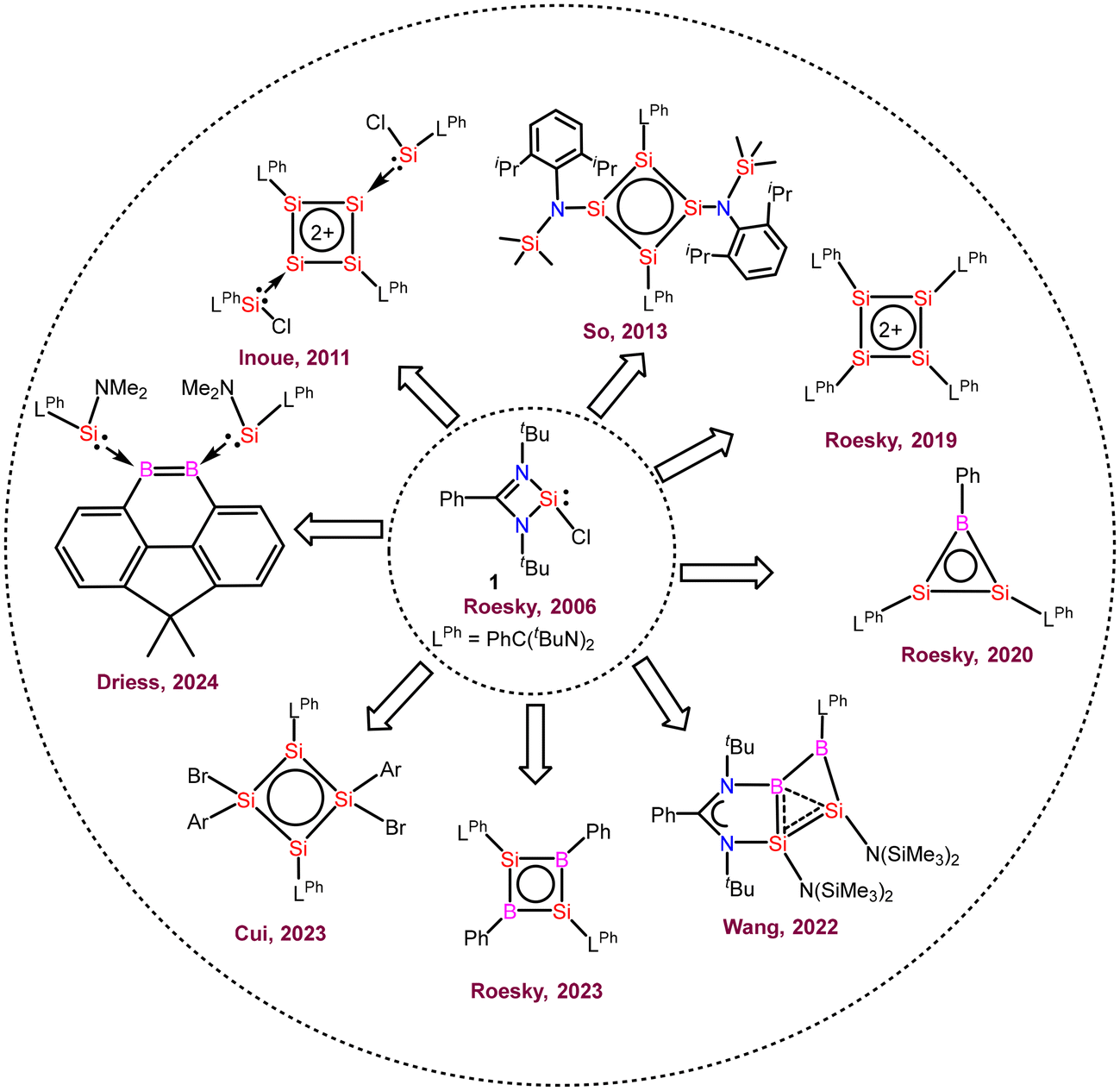
Herein, we will focus on the use of amidinato-silylenes in the preparation of aromatic and homo-aromatic ring compounds (Fig. 1) and the above-mentioned descriptors will be frequently used to describe aromatic and homoaromatic characteristics of the compounds synthesized from amidinato-silylenes.
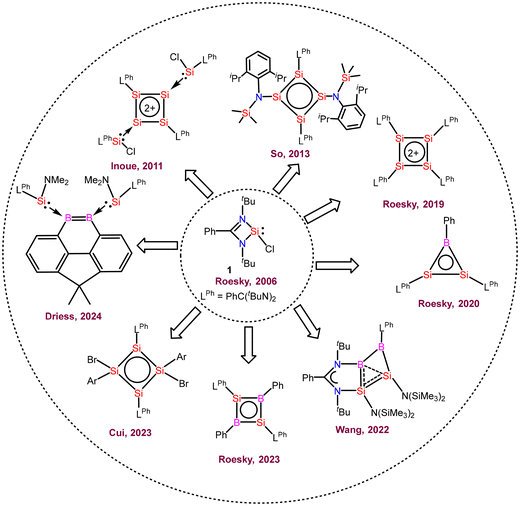 |
| | Fig. 1 Aromatic compounds prepared from amidinato-chlorosilylene. | |
Amidinato-silylenes and bis-silylenes
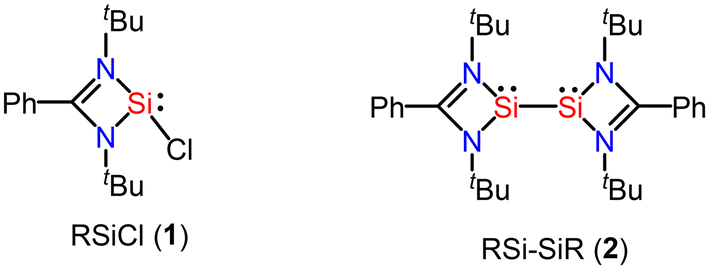
The amidinato-chlorosilylene RSiCl (1)6,7 was prepared by us in 2006 (Fig. 2). Subsequently, we made a small procedural change and we were able to isolate this silylene with highly improved yield in 2010. Furthermore, the interconnected amidinato-bis-silylene (2) was prepared by dehalogenative coupling in the presence of KC8 in 2009.49,50 The silylene and bis-silylene molecules have been subjected to huge research endeavours for more than one decade. Various research groups have used these molecules in various synthetic applications. These compounds have also been used for the synthesis of aromatic compounds. In some instances, interconnected amidinato-bis-silylenes have been used to prepare aromatic heterocyclic rings; however, spacer-separated bis-silylenes have not been employed to make aromatic compounds. Herein, we describe the use of silylene and interconnected bis-silylene molecules in the preparation of heterocyclic aromatic compounds. Notably, in these amidinato-silylenes, the silicon atom possesses five electrons in its valence shell. The extra one electron in the valence shell of this silylene makes it a unique molecule in terms of reactivity. When these silylene molecules are reacted with suitable substrates, they yield cyclic molecules. If empty p-orbitals are available in the neighbouring atoms of the silylene–silicon atom, the extra electron would be delocalized over the ring, resulting in the aromatic ring current. Utilizing this unique electronic property of silylene, several aromatic and homoaromatic compounds have been synthesized.
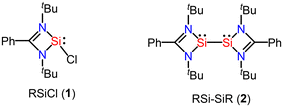 |
| | Fig. 2 Amidinato-chlorosilylenes and interconnected bis-silylene were reported by us. | |
Amidinato-supported 1,4-disilabenzene
Interconnected bis-silylenes have been proved to be very good precursors for the preparation of six-membered heterocyclic aromatic rings. The reaction of interconnected bis-silylene unsaturated compounds, generally, proceeds with the breakage of the Si(I)–Si(I) bond; as a result, various amidinato-supported 1,4-disilabenzene rings (3 and 4) have been synthesized by reacting amidinato-bis-silylene (2) with different alkynes.51 Dark red crystals of compound 3 were isolated with 30% yield (Scheme 1). The compound was thoroughly characterized with suitable techniques. The nearly planar six-membered ring containing 6 π-electrons resonates at δ = 18.05 ppm in the 29Si NMR spectrum. The compound 3 crystallizes in the trigonal space group P3121. The structure revealed that both silicon atoms have a distorted tetrahedral geometry (Fig. 3). The 1,4-disilabenzene ring is nearly planar, a key factor contributing to the enhanced stability of this compound. To determine whether the 1,4-disilabenzene rings in compounds 3 exhibit aromaticity, NICS values at 1 Å above the ring centers were calculated. Compound 3 has a NICS(1) value of −3.64, indicating it is slightly aromatic in nature. Additionally, the highest occupied molecular orbitals (HOMOs) with a nodal plane in the ring plane closely resemble the equivalent π-molecular orbitals (π-MOs) formed from carbon pz-orbitals in benzene in terms of shape and symmetry, although the energetic degeneracy of the HOMO is not preserved (Fig. 4). Later on, we substituted the phenyl group on the alkyne with less bulkier groups to prepare another compound with a central C4Si2 ring (4), to see if electronic delocalization is affected. The theoretical calculations reveal the very small π-electron delocalization over the central C4Si2 with a NICS(1) value of 1.62, wherein a positive NICS(1) value indicates the disruption of electron delocalization.52
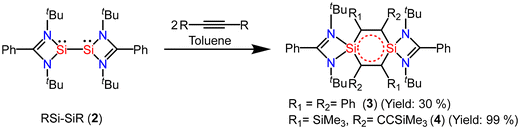 |
| | Scheme 1 Preparation of amidinato-supported 1,4-disilabenzenes (3 and 4). | |
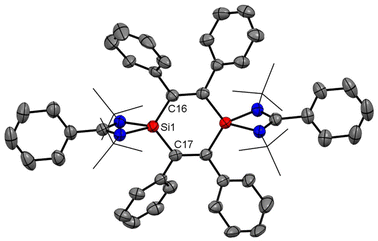 |
| | Fig. 3 The structure of 3 determined by single crystal XRD. Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. | |
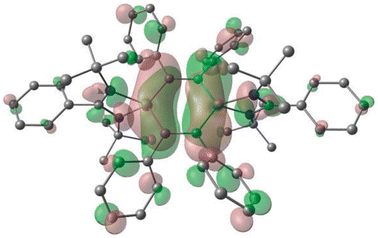 |
| | Fig. 4 HOMO of compound 3. Positive (green) and negative (red) isosurface representation at 0.02 a.u. Reproduced with permission from ref. 51. Copyright (2010) Royal Society of Chemistry. | |
Tetracyclic-silicon-aromatic rings
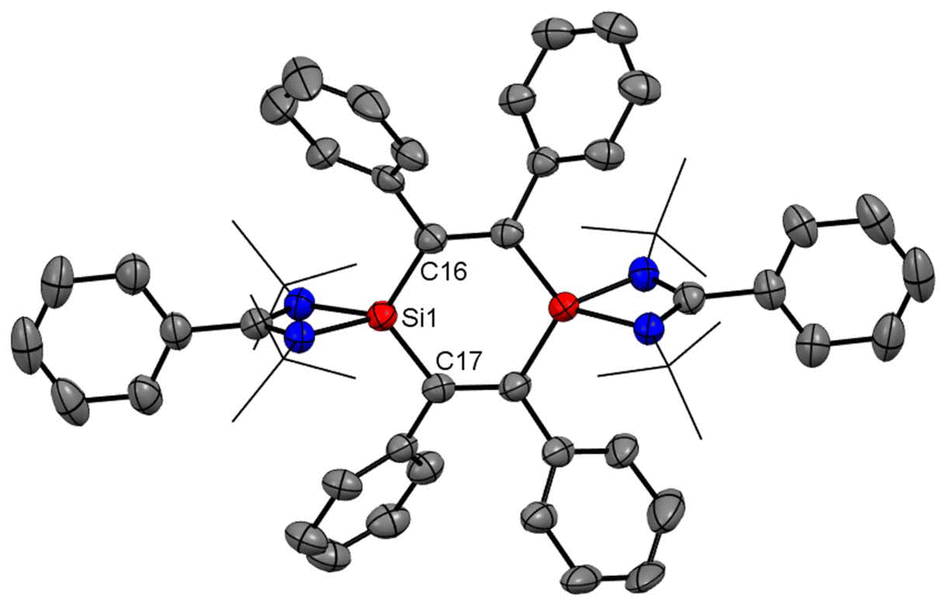
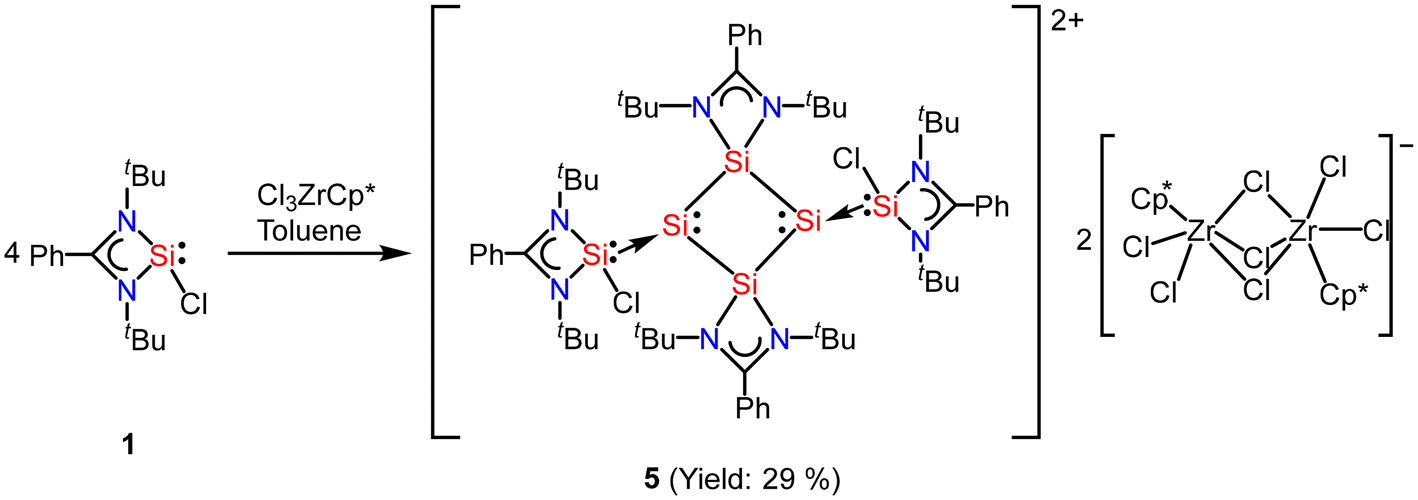
In most of the reports, the amidinato silylene-derived aromatic rings are four-membered cationic silicon cyclic rings and are connected to a bulky counter anion. Driess and co-workers reported the first donor-stabilized tetrasilacyclobutadiene dication species (5) that displays aromatic delocalization in a tetracyclic silicon ring.53 This unexpected compound (5) forms through a Lewis acid-assisted reaction involving amidinato chloro-silylene (1) and CpZrCl3 (Cp = pentamethylcyclopentadienyl) in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio (Scheme 2). Compound 5 was isolated as yellow crystals in 29% yield. Notably, the four-membered Si4 ring of compound 5 comprises of two N-donor-stabilized silylium units and two silylene-like units (Fig. 5). Notably, the nucleus independent chemical shift (NICS) of 5 showed negative values [NICS(1) = −3.8 and NICS(0) = −4.7 ppm], suggesting that compound 5 possesses an aromatic ring current due to the delocalization of π-electrons.
:2 molar ratio (Scheme 2). Compound 5 was isolated as yellow crystals in 29% yield. Notably, the four-membered Si4 ring of compound 5 comprises of two N-donor-stabilized silylium units and two silylene-like units (Fig. 5). Notably, the nucleus independent chemical shift (NICS) of 5 showed negative values [NICS(1) = −3.8 and NICS(0) = −4.7 ppm], suggesting that compound 5 possesses an aromatic ring current due to the delocalization of π-electrons.
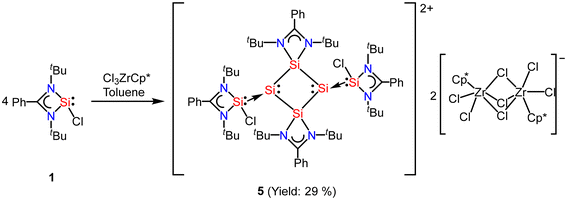 |
| | Scheme 2 Preparation of tetrasilacyclobutadiene dication species 5. | |
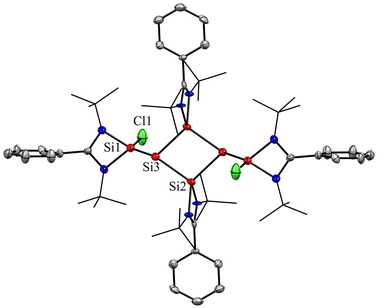 |
| | Fig. 5 Molecular structure of the dication of 5, determined by X-ray diffraction technique. Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. | |
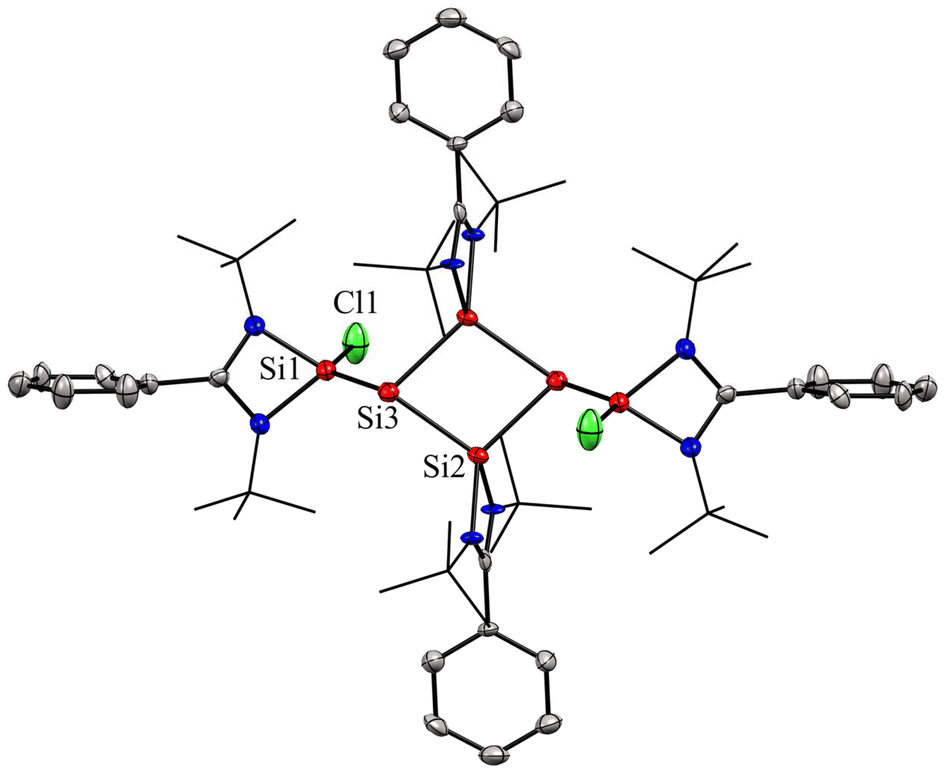
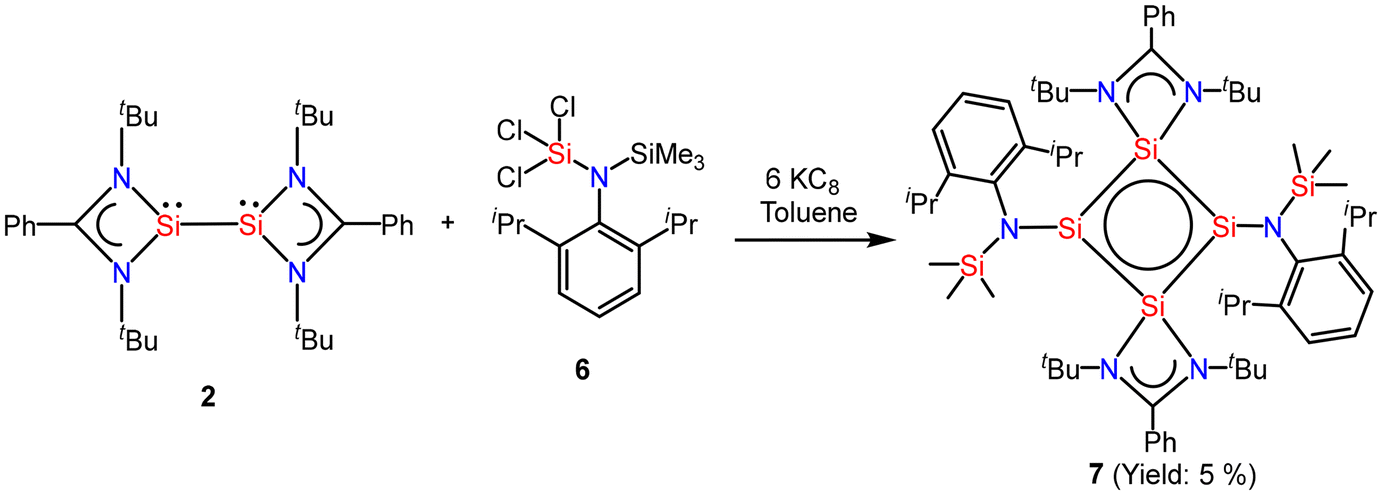
So and coworkers have also reported a four-membered Si4 ring compound (7), but with a completely different delocalization pattern of bonding electrons, in which all types of bonds in the molecule participate in delocalization.54 Compound 7 was synthesized by the reaction of amidinate bis-silylene (2) with two equivalents of amido trichlorosilane (6) and six equivalents of KC8 (Scheme 3, Fig. 7). This compound was isolated as a highly moisture sensitive brown crystalline solid in 5% yield. Compound 7 displays the cyclic and extensive delocalization of n, π, and σ-electrons in the Si4 ring affording a planar and rhombic shape.54 The 29Si{1H} NMR spectrum displays two distinct sharp singlets at δ: −70.8 and 32.2 ppm, corresponding to the three-coordinate and four-coordinate silicon atoms in the Si4 ring, respectively. Additionally, the 29Si{1H} NMR spectrum also features a sharp singlet at δ 1.41 ppm, which is associated with the SiMe3 group present in the molecule. The computational calculations reveal that the highest occupied molecular orbitals, HOMO, HOMO−1, and HOMO−3 delocalize around the Si4 ring in a cyclic fashion. The NICS(1) and NICS(0) values were found to be −10.07 ppm and −15.71 ppm which are comparable to those of benzene. Hence, these values support strong electron delocalization throughout the ring.
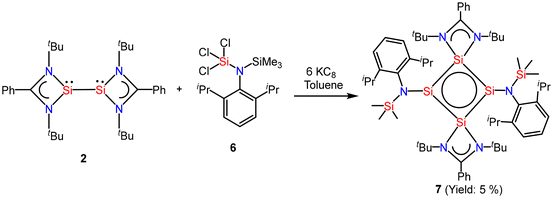 |
| | Scheme 3 Preparation of compound 7. | |
In compound 7, the delocalization is quite similar to the dismutational aromaticity observed in the Si4 ring of the tricyclic isomer of hexasilabenzene.41 This hexasilabenzene is a double intramolecular dismutational derivative of a Hückel aromatic compound, featuring silicon atoms in +2, +1, and 0 formal oxidation states. Dismutational aromaticity refers to a concept where an initially non-aromatic compound undergoes a dismutation (disproportionation) reaction, resulting in the formation of two products, one of which is aromatic. This concept is related to the idea that certain non-aromatic compounds can transform into more stable aromatic compounds through chemical reactions that redistribute electrons. However, compound 7 is not a Hückel aromatic compound, and all silicon atoms in its canonical forms 7A–E are formally in the +1-oxidation state (Fig. 6). Additionally, the delocalization in compound 7 differs from the 2π-electron delocalization found in the tetrasilacyclobutadiene dication.55
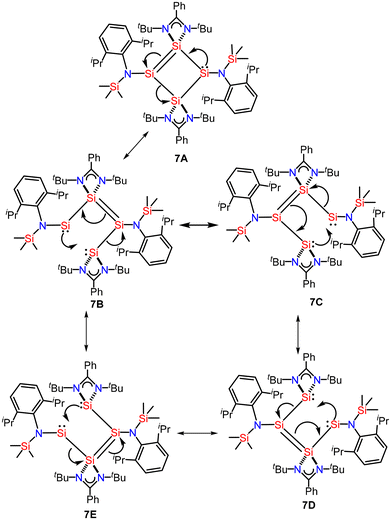 |
| | Fig. 6 The delocalization of two π, two σ, and two lone-pairs of electrons in canonical structures/resonating structures of 7. | |
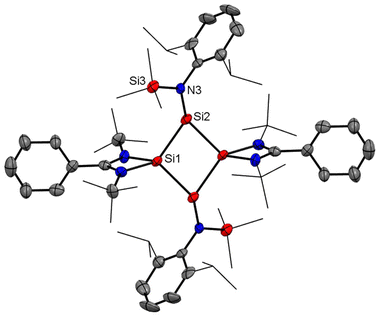 |
| | Fig. 7 Molecular structure of compound 7. Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. | |
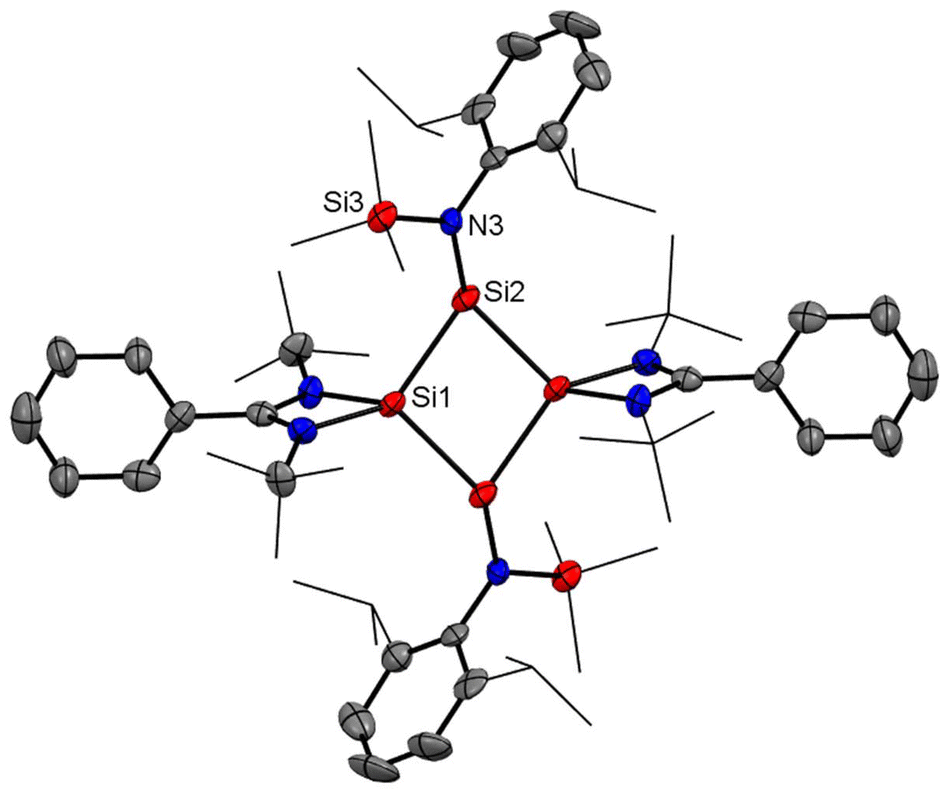
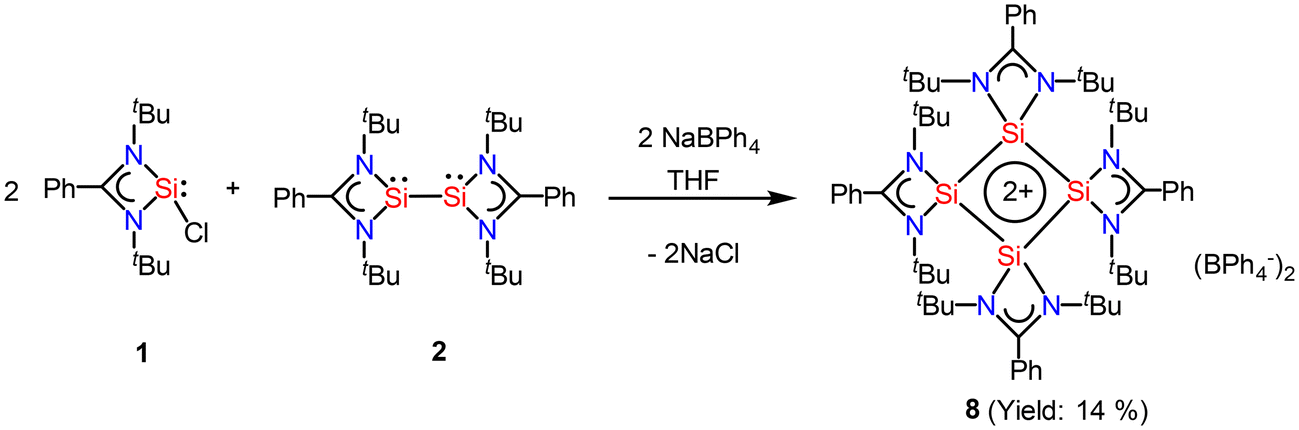
After eight years of synthesis of a tetrasilacyclobutadiene dication by Driess and co-workers, Roesky and coworkers went on to report another aromatic tetrasilacyclobutadiene dication (8) in 2019. Compound 8 was synthesized by reacting amidinato silylene (1) and amidinato bis-silylene (2) and NaBPh4 in a 2:1:1 molar ratio in THF (Scheme 4).56 Red colored crystals were isolated with 14% yield. The [Si4]2+ ring of 8 possesses a slightly puckered square-planar geometry as determined by single crystal X-ray diffraction (Fig. 8). Each silicon atom resides in a distorted tetrahedral arrangement bonded to a single bidentate amidinato ligand, which is positioned nearly perpendicular to the Si4 ring. The 29Si{1H} NMR spectrum of compound 8 produces a single resonance peak at −28.9 ppm indicating four equivalent silicon atoms in the molecule. Further insights were obtained by DFT calculations. The vertical HOMO/LUMO gap was calculated to be 2.28 eV. The lowest energy transition in the UV/vis spectrum, identified using time-dependent DFT, was 2.33 eV (532 nm), which aligns well with the experimental value of 465 nm and is attributed to the HOMO/LUMO transition (Fig. 9). The NICS(0) and NICS(1) values for compound 8 were calculated to be −17.4 and −12.7 ppm, respectively, confirming its aromatic nature. The higher (negative) NICS values of this compound compared to those of previously discussed compounds indicate the high diamagnetic ring current and better electron delocalization throughout the ring.
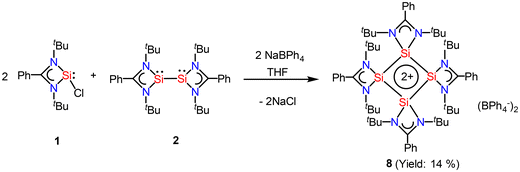 |
| | Scheme 4 Preparation of compound 8. | |
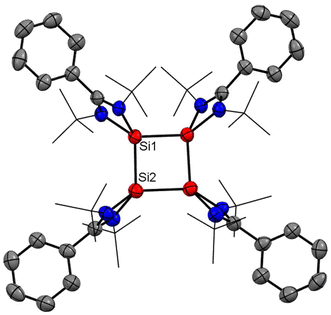 |
| | Fig. 8 Molecular structure of dication of 8. Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. | |
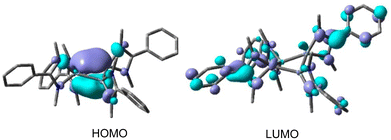 |
| | Fig. 9 HOMO and LUMO iso-surface plots of dication of 8 (values ±0.04). Reproduced with permission from ref. 56. Copyright (2019) American Chemical Society. | |
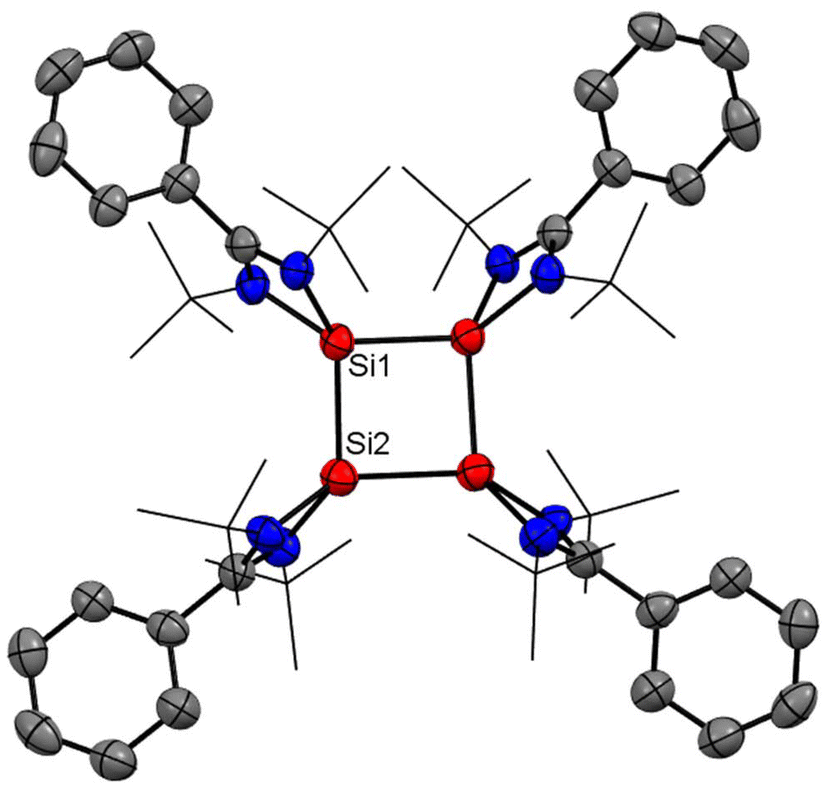
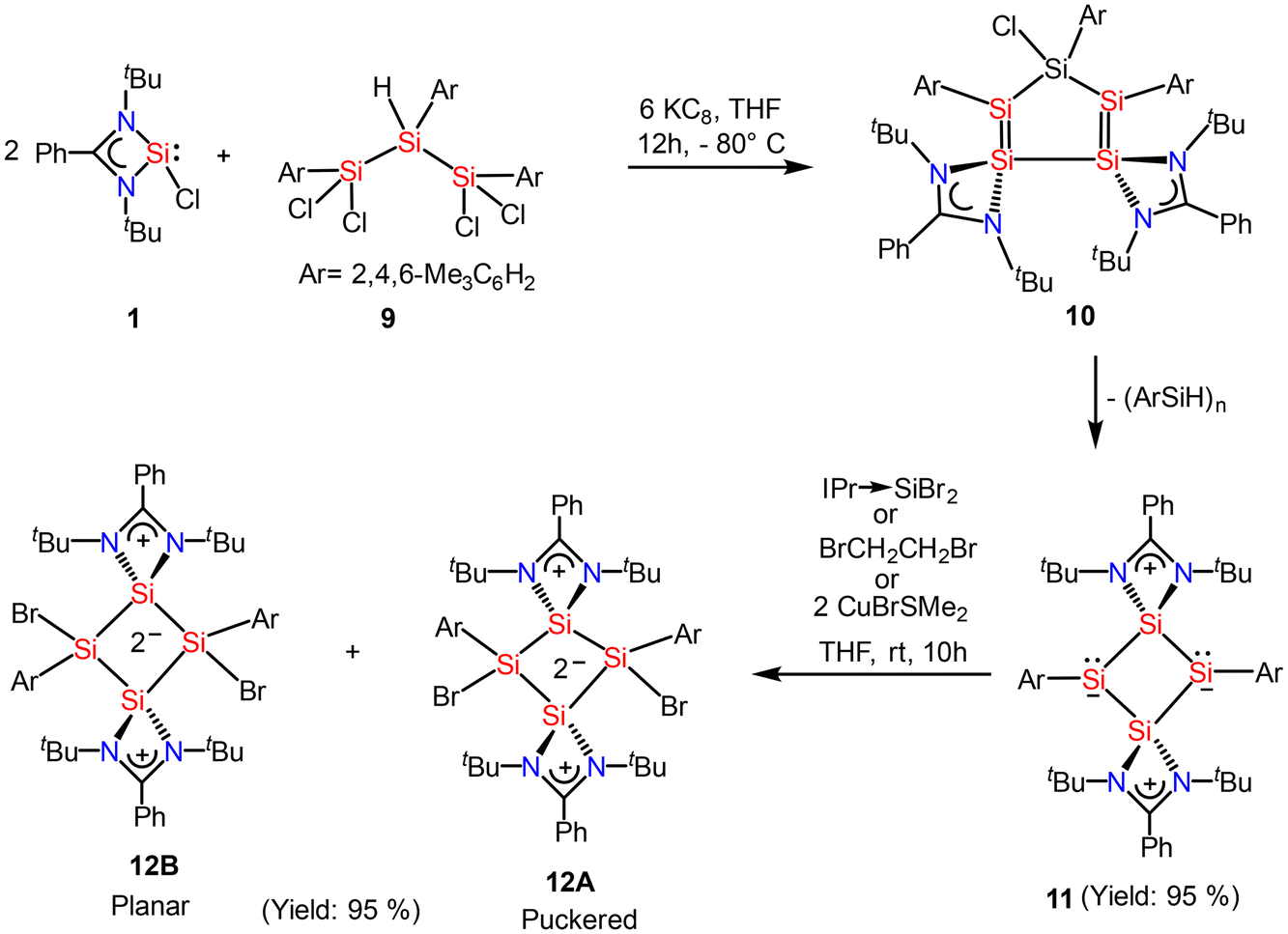
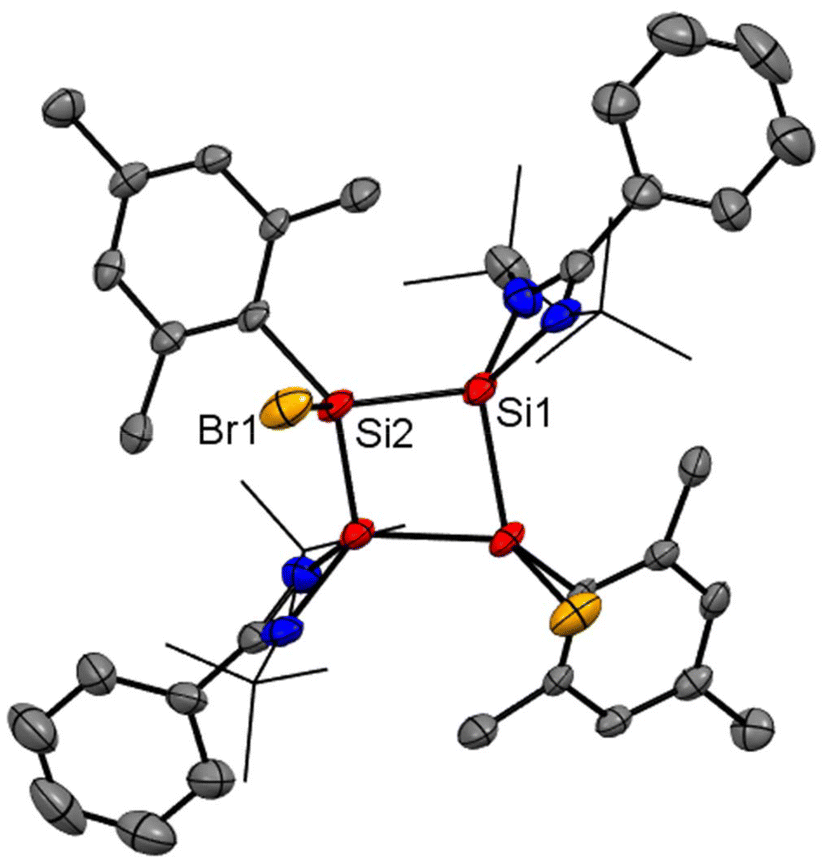
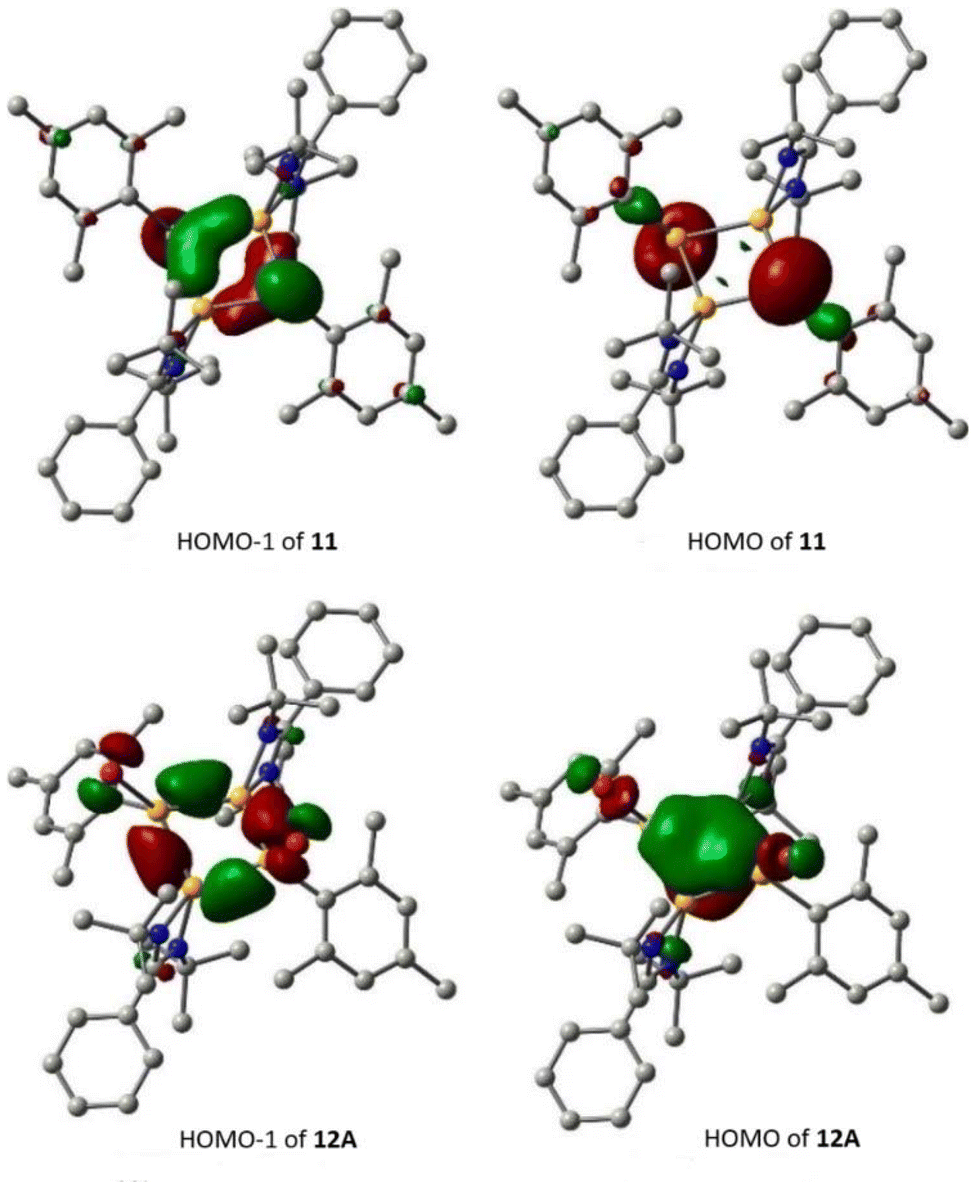
Recently, Cui and coworkers have isolated and well characterized a series of Si4 ring compounds (11, 12A and 12B) derived from an amidinate silylene.57 Among them some of the compounds are aromatic due to electronic delocalization enabled by the planar Si4 ring structure, and some of them are non-aromatic due to the ring strained/puckered structure, as we will discuss later in this section. For the synthesis of these compounds, amidinato silylene (1) and 1,2,3-triargio-1,1,3,3-tetrachlorotrisilane (9, Ar3Cl4HSi3) were mixed in THF, followed by addition of KC8. The stirring of the reaction mixture at −80° C for 12 h produces a tetrasilacyclobutadiene (LSiSiAr)2 compound (11) with 52% yield (Scheme 5, Fig. 10).57 Compound 11 upon halogenation with NHC-stabilized bromosilylene, produces neutral dihalo-substituted tetrasilacyclobutane-1,3-dilyls (12A and 12B). Upon characterization with single-crystal XRD, it was observed that two types of structures of 12A and 12B exist in the unit cell. Although both structures are the same by composition, their geometries are different. While 12A has a puckered ring structure with the two bromine groups in a cis configuration, 12B exhibits a planar, rhombic Si4 ring with two bromine atoms arranged in a trans configuration.
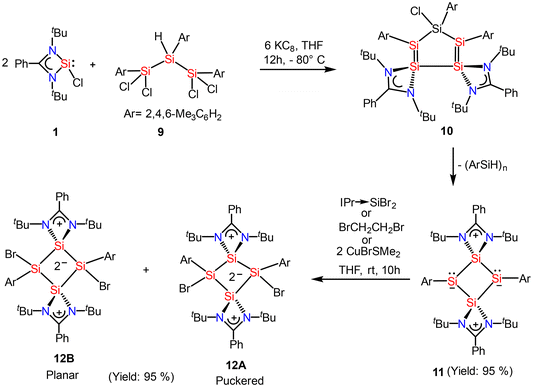 |
| | Scheme 5 Preparation of tetrasilacyclobutadiene, 11 and tetrasilacyclobutane-1,3-diyls, 12 (IPr = 1,3-bis(2,6-iPr2C6H3)-imidazole-2-ylidene). | |
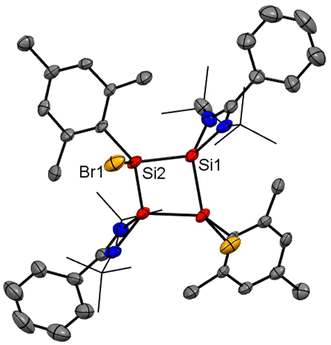 |
| | Fig. 10 Molecular structure of 12. Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. | |
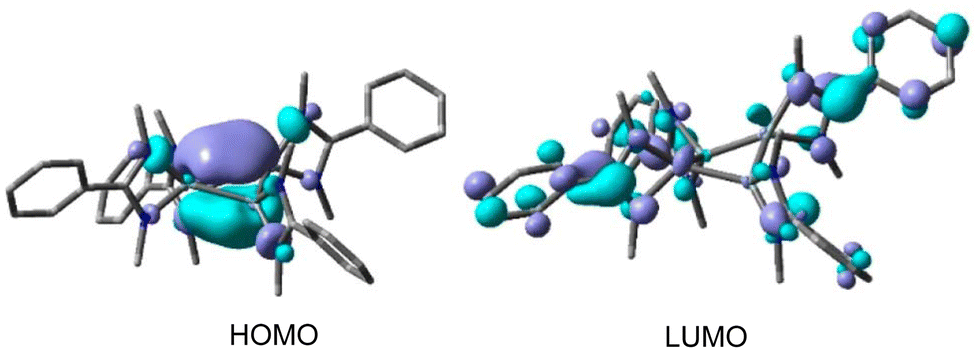
To investigate the electronic structures of the Si4 rings in these compounds, DFT calculations were carried out using the (U)B3LYP-D3/def2-SVP level of theory. The calculations revealed that the HOMOs of 12A and 12B primarily correspond to the π-type orbitals over the Si4 rings, with some additional contributions (Fig. 11). However, the HOMO−1s of 12A and 12B are predominantly characterized by Si–Si σ bonds. In contrast, the HOMO of 11 is mainly composed of the lone-pair orbitals of the silicon atoms, and its HOMO−1 consists of distorted π-type orbitals over the Si4 ring. These compounds are expected to display aromatic character. Compound 12B is expected to display a better aromatic character due to the planar Si4 ring structure which enables efficient electron delocalization. The presence of electron delocalization in these compounds was quantified by NICS calculations. The NICS(0) and NICS(1) values of 12A and 12B were found to be −11.8, 9.5, and −12.3, −9.2 ppm, respectively, which indicates that these rings possess aromatic electron delocalization. Additionally, the electron density of delocalized bonds (EDDB) has been employed to measure the aromaticity of the Si4 ring. The findings indicate that the EDDBSi4 values are similar to those of 2π-aromatic systems.58
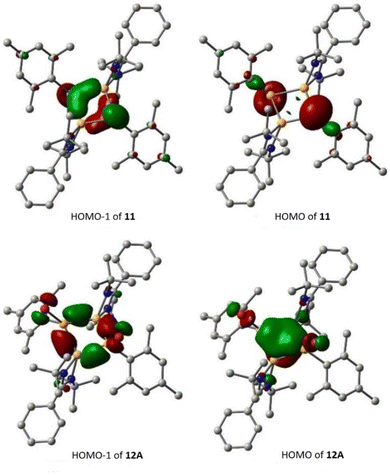 |
| | Fig. 11 Plots of the HOMO and HOMO−1 of 11 and 12A (contour value = 0.05). Reproduced with permission from ref. 57. Copyright (2023) American Chemical Society. | |
Heterocyclic aromatic rings
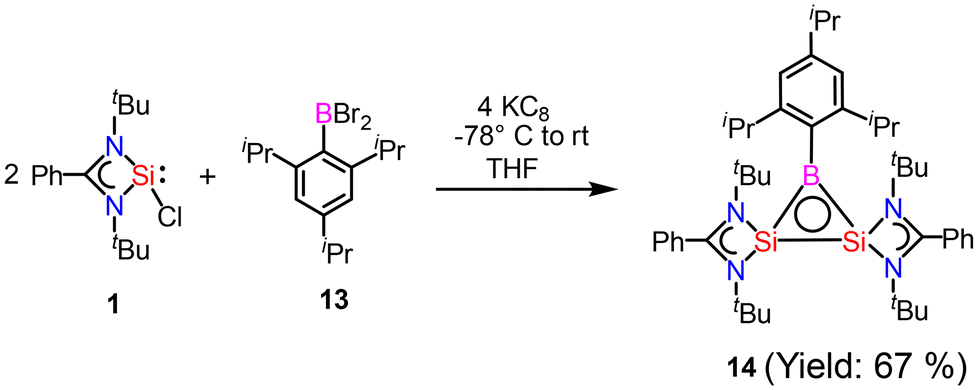
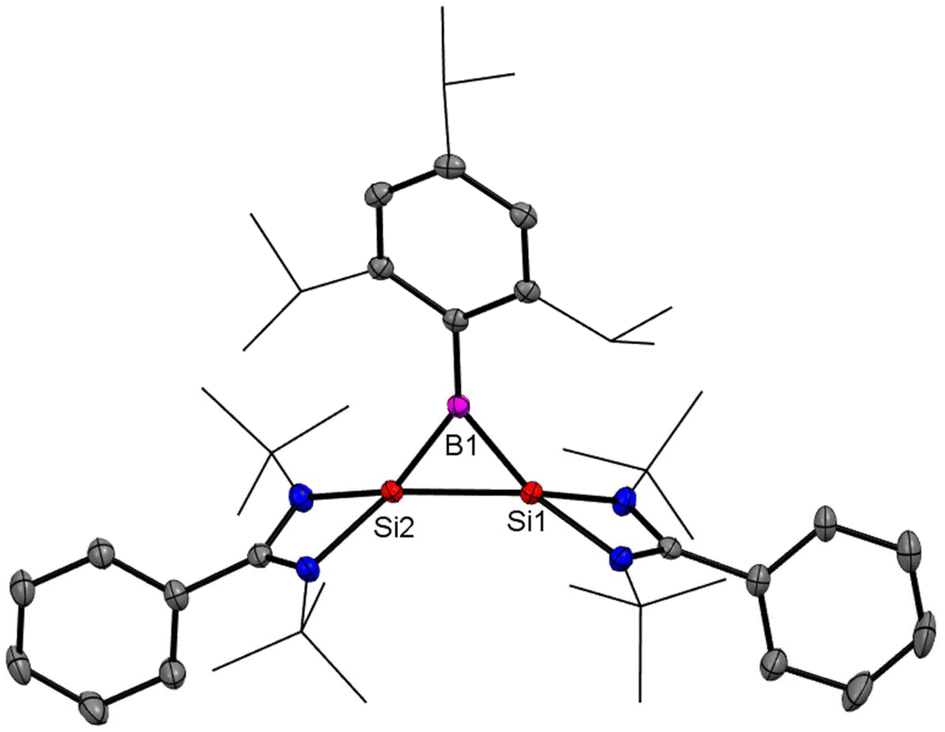
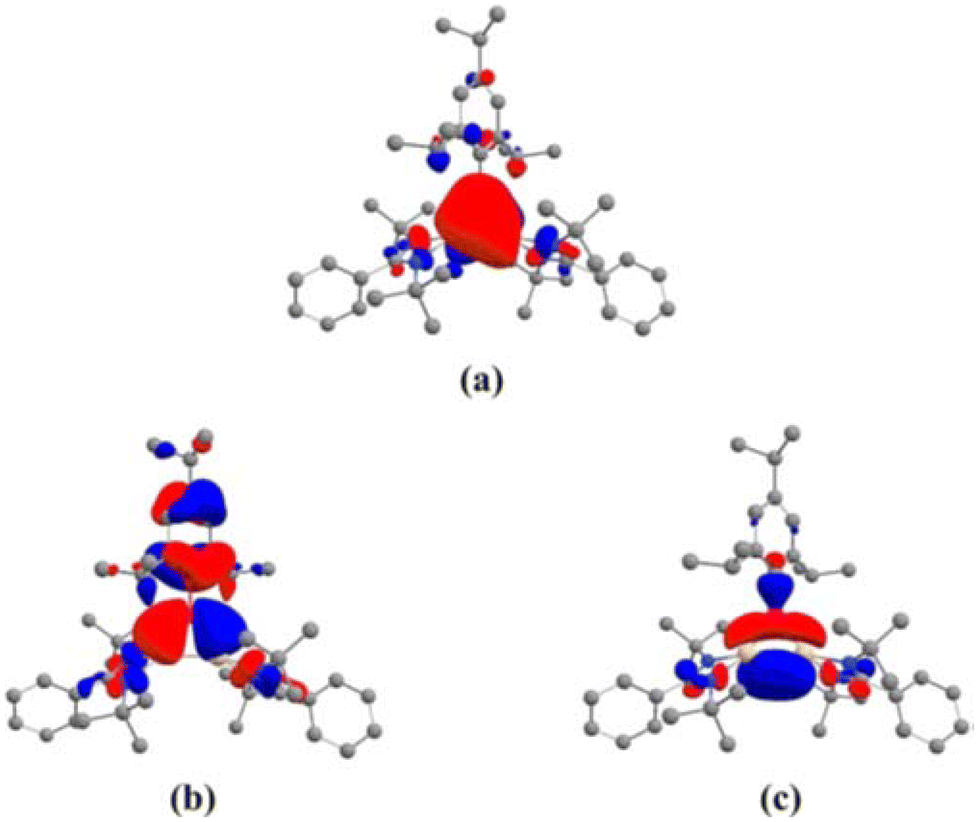
After reports of amidinato silylene-derived Si4 aromatic ring compounds, we thought to replace some of the silicon atoms of the Si4 ring with hetero-atoms such as group 13 elements. We were successful in replacing silicon atoms with boron to prepare new heterocyclic aromatic rings. In 2020, a neutral three-membered Si2B aromatic ring (14) was prepared by us. Amidinato silylene (1), dibromo(2,4,6-triisopropylphenyl) borane (13) and KC8 were mixed in THF in a 2:1:4 molar ratio at −78° C and the reaction mixture brought to room temperature slowly and stirred overnight (Scheme 6).59 Compound 14 was isolated as a red block shaped crystal in 67% yield. This compound was analyzed using NMR and various other methods. In the 11B NMR spectrum, the compound exhibits a singlet at 11.09 ppm, indicating it lies within the aromatic region for 11B NMR. The 29Si NMR spectrum shows a resonance at −71.03 ppm, which is upfield compared to the amidinato-silylene chloride, which resonates at 14.6 ppm. Compound 14 crystallizes in the monoclinic space group P21/c, with one molecule of 14 and one and a half toluene molecules in the asymmetric unit. The heterocycle forms an isosceles triangle, featuring identical Si–B bond lengths of 1.9190(13) and 1.9186(13) Å (Fig. 12). The amidinato ligands are oriented almost perpendicular to the three-membered Si2B ring. Additional structural and electronic details were obtained via DFT calculations. The HOMO is a delocalized π molecular orbital, primarily localized on the two Si and B atoms. Nitrogen's contribution to the σ* MOs of π symmetry is antibonding but minimal. The HOMO−1 and HOMO−11 are Walsh orbitals in the σ-plane, similar to those seen in the cyclopropenyl cation (Fig. 13). NBO analysis indicates double bond characteristic for the Si–Si and Si–B bonds, as suggested by Wiberg bond index values of 1.128 and 1.288, respectively. To quantify aromaticity, the NICSzz(1) value was calculated for the Si2B ring, which was found to be −8.1 ppm, indicating the aromatic nature of the ring. To assess π delocalization above the ring center of 14, isotropic chemical shifts, along with in-plane and out-of-plane components, were scanned and plotted. A minimum in the NICSzz curve at a nonzero r value indicates aromatic π delocalization. The NICSzz curve decreases from 0 Å, reaching a minimum between 0.8 Å and 1.4 Å, remaining in the negative region over a significant range. This behavior suggests π cloud delocalization above the three-membered rings (Fig. 14).
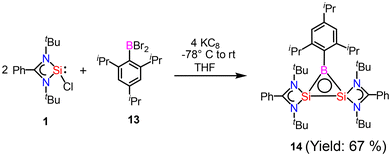 |
| | Scheme 6 Preparation of compound 14. | |
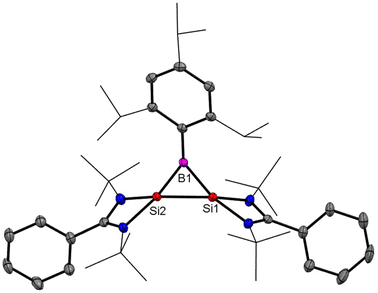 |
| | Fig. 12 Structure of 14 determined by single-crystal XRD. Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. | |
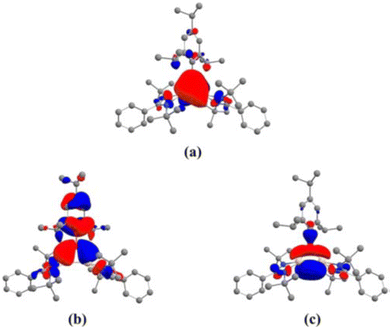 |
| | Fig. 13 (a) The delocalized π MO, HOMO, and Walsh orbitals (b) HOMO−1 and (c) HOMO−2 of 14. Reproduced with permission from ref. 59. Copyright (2023) Wiley-VCH GmbH. | |
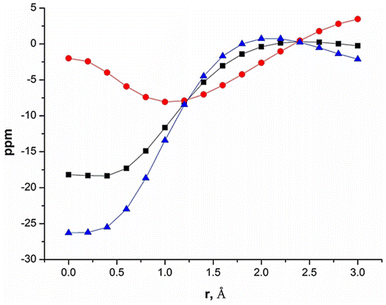 |
| | Fig. 14 NICS-curve scan for NICS-scan curves for the three-membered Si2B ring of disilaborirane, 14: (square) isotropic NICS; (dot) out-of-plane component; (triangle) in-plane component. Reproduced with permission from ref. 59. Copyright (2023) Wiley-VCH GmbH. | |
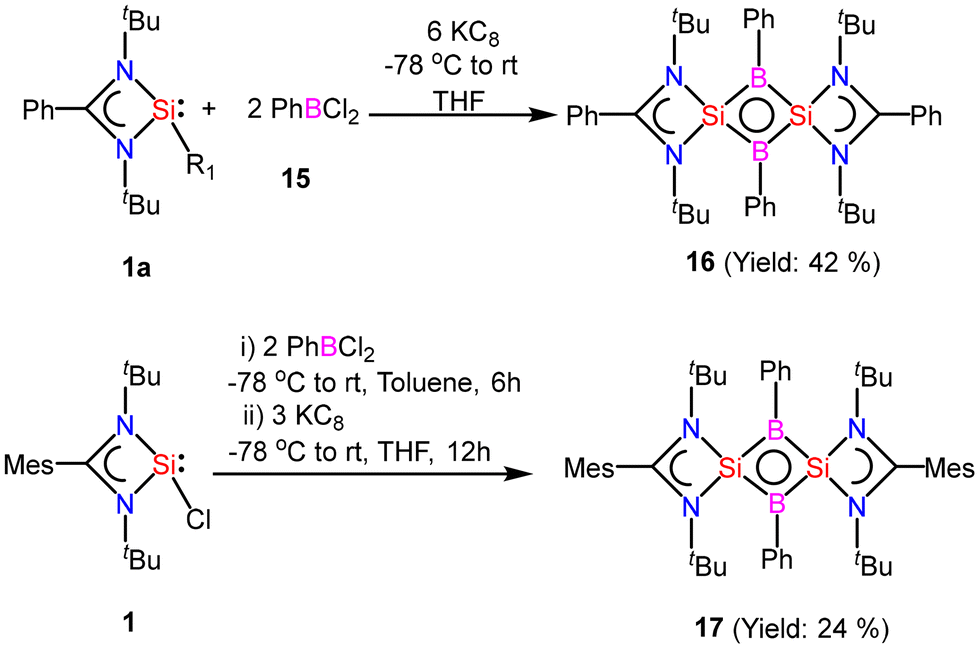
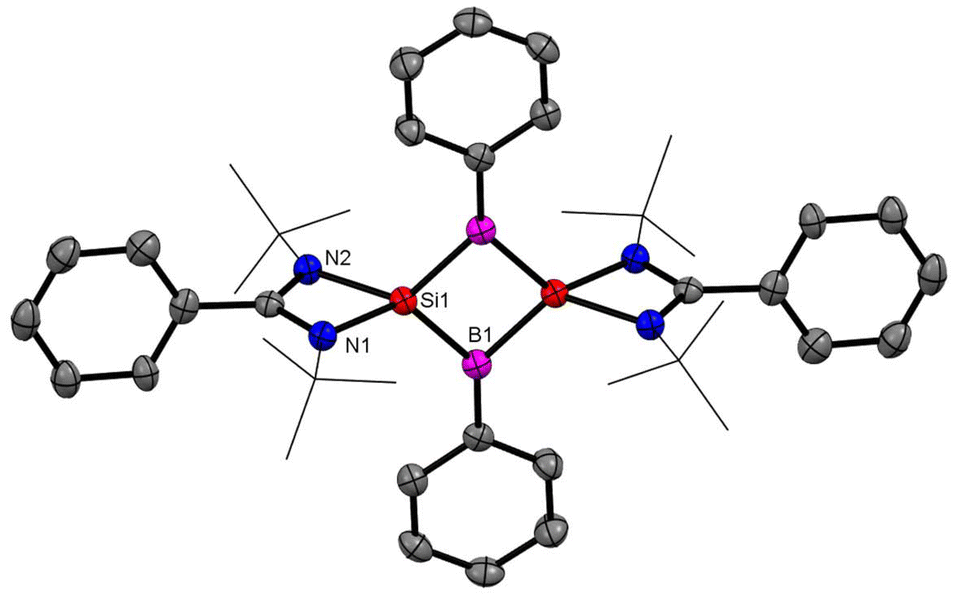
After successful introduction of a boron atom into the ring to obtain the Si2B aromatic ring, we thought to go further with the introduction of two boron atoms in the ring to make the Si2B2 ring. To approach the possibility of having a planar Si2B2 ring, we used a less bulky dichloro(phenyl)borane.60 The Si2B2 four-membered ring (16) was synthesized by reaction of amidinato-silylene (1a) and dichlorophenylborane (15), followed by reduction with KC8. The amidinato-silylene [(PhC(NtBu)2Si–R1)] (1a) and dichlorophenylborane were mixed in a 1:1 molar ratio in THF at −78 °C and stirred for seven hours at room temperature and subsequently mixed with 3 equivalents of KC8 at −78 °C and stirred overnight (Scheme 7).60,61 Red colored crystals were obtained from concentrated mother liquor with 42% yield. Compound 16 was characterized by single crystal XRD, NMR and mass spectrometry. The NMR 11B NMR spectrum of 16 exhibits a singlet at δ = −15.86 ppm. The 29Si NMR spectrum of 16 shows a resonance at 10.8 ppm. The mass spectrometry measurement of compound 16 exhibits a single molecular ion peak at m/z: 694.3 in the spectrum, validating the formation of compound 16. Single-crystal XRD revealed that compound 16 crystallizes in the space group C2/c with half a molecule in the asymmetric unit and the molecule has a planar Si2B2 ring structure with equal Si–B bond lengths of 1.946(2) Å and 1.947(2) Å. The bond lengths of the B–Si bond in the planar four-membered Si2B2 ring are comparable to the Si–B bond lengths (1.9186(13) and 1.9211(18) Å)19 of a previously reported Si2B neutral 2π aromatic ring (Fig. 15).
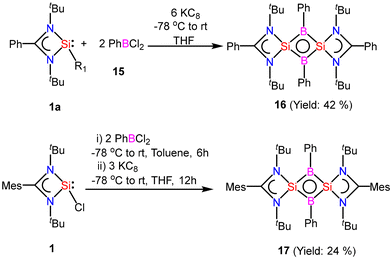 |
| | Scheme 7 Preparation of compounds 16 and 17 (R1: 3,5-diphenyl-1H-pyrazolyl). | |
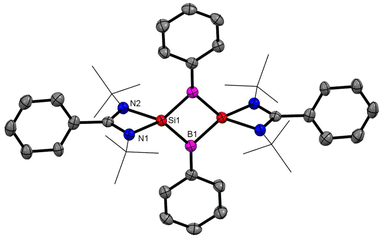 |
| | Fig. 15 The molecular structure of 16. Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. | |
Quantum mechanical calculations were carried out at the M06/def2-TZVPP//BP86-D3BJ/def2-TZVPP level of theory to understand the electronic and bonding characteristics of compound 16. DFT calculations revealed a singlet ground state of compound 16 with an adiabatic/vertical singlet–triplet energy gap of 28.7/24.0 kcal mol−1. Furthermore, CASSCF calculations were performed to understand if compound 16 has diradical character. The CASSCF (2, 2)/6-31G* calculation shows that the virtual MOs possess 0.13 electrons, which eliminates the possibility of a diradical ground state for 16. The unavailability of electrons in virtual MOs also suggests the delocalization of electrons. MO analysis shows that the HOMO of 16 is a symmetrical π molecular orbital and is delocalized over the entire Si2B2 ring. Negative charges on the boron atoms (−0.44e to −0.46e) obtained from NBO analysis clearly indicate a flow of charge from the amidinate-silicon to B, resulting in highly polarized Si–B bonds. Furthermore, the aromatic nature of 16 was confirmed by NICS values. The NICS values calculated at the center (NICS(0) = −6.2) and 1 Å above the ring (NICS(1) = −6.6) indicate aromatic delocalization. The NICSzz(1) value calculated for 16 is −3.1. The NICS values obtained for compound 16 are not as highly negative as we observe in common 6π-aromatic compounds, because, in compound 16, there are only two π-electron delocalizing in a four-membered ring. Additionally, aromatic delocalization was also reaffirmed by electron localization function (ELF) analysis, wherein, disynaptic basins generated from the ELF analysis of 16 V(Si, B) integrates to 2.35–2.40 electrons between the Si and B atoms, which suggests π-electron delocalization (Fig. 16). The substitution of phenyl with mesityl group on the amidinato moiety produces another aromatic compound 16, which displays better aromatic properties as supported by its higher NICS(1) values (−16.36 ppm).
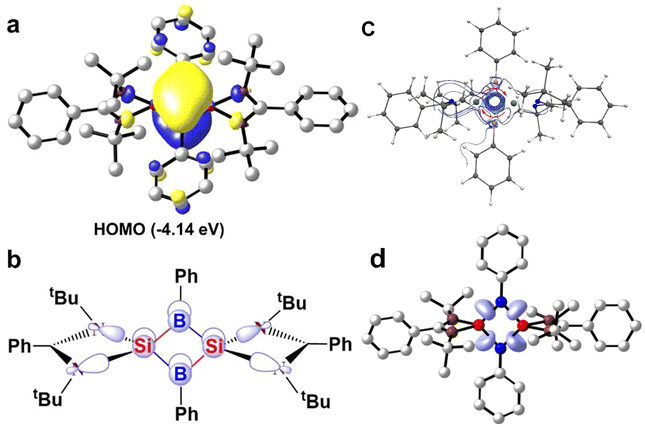 |
| | Fig. 16 (a) HOMO of 16 calculated at the M06/def2-TZVPP level of theory (contour value: 0.04). (b) Walsh orbital of 16 showing π-delocalization. (c) Streamline plot of the current density of the Si2B2 ring in 16. (d) A plot of ELF basins plotted between Si (red) and B (blue) atoms of 16. Reproduced with permission from ref. 60. Copyright (2023) American Chemical Society. | |
Since NICS values may be affected by neighboring σ-bonds, the current density was also calculated using the GIMIC program. The streamline plot of current density generated using the GIMIC program to visualize the direction of the magnetically induced ring current also suggests aromatic delocalization. It shows a net diatropic ring current of 1.6 nA T−1 around the Si2B2 ring, indicating that compound 16 exhibits aromatic delocalization, albeit to a much lesser extent than benzene. This difference is explained by the fact that benzene has six electrons contributing to the induced current, while compound 16 has only two, which are delocalized over four different centers. Additionally, the disynaptic basins from the electron localization function (ELF) analysis of 16 V(Si, B) show an integration of 2.3–2.40 electrons between the Si and B atoms, further supporting the presence of π-delocalization.
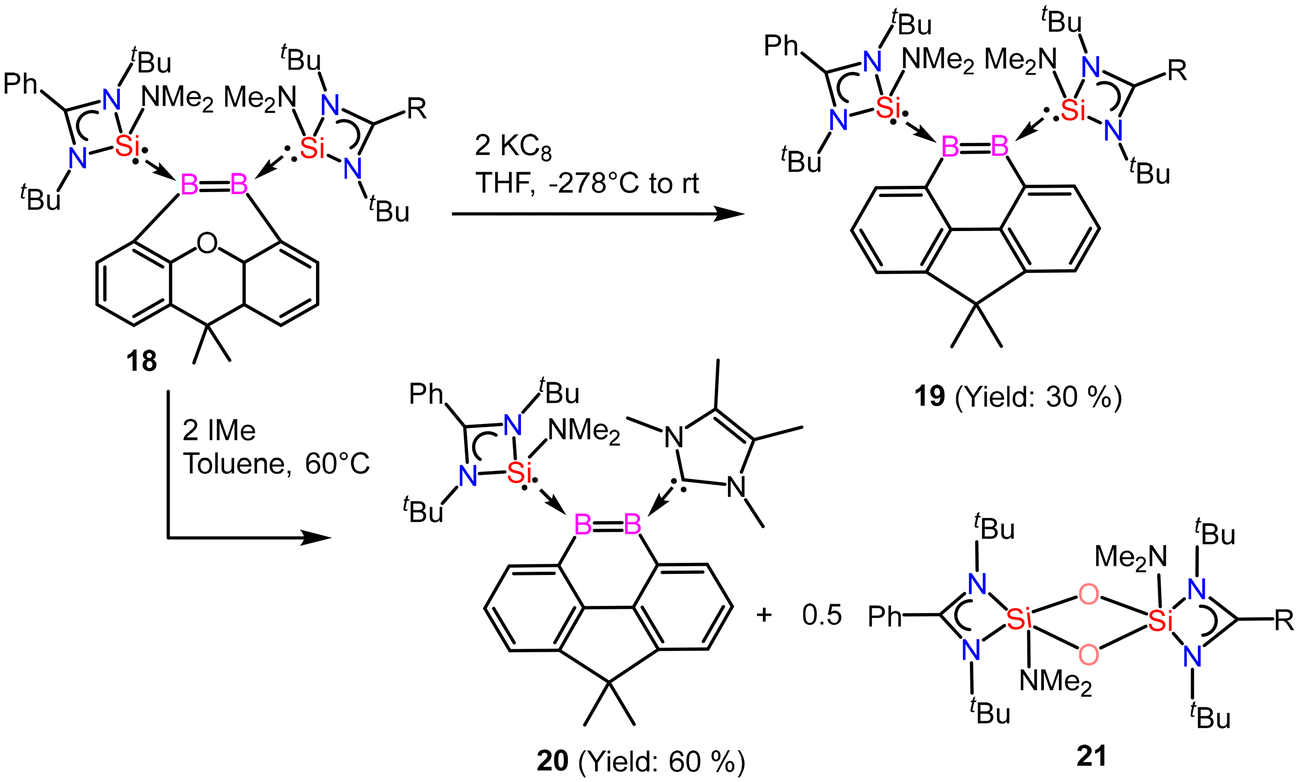
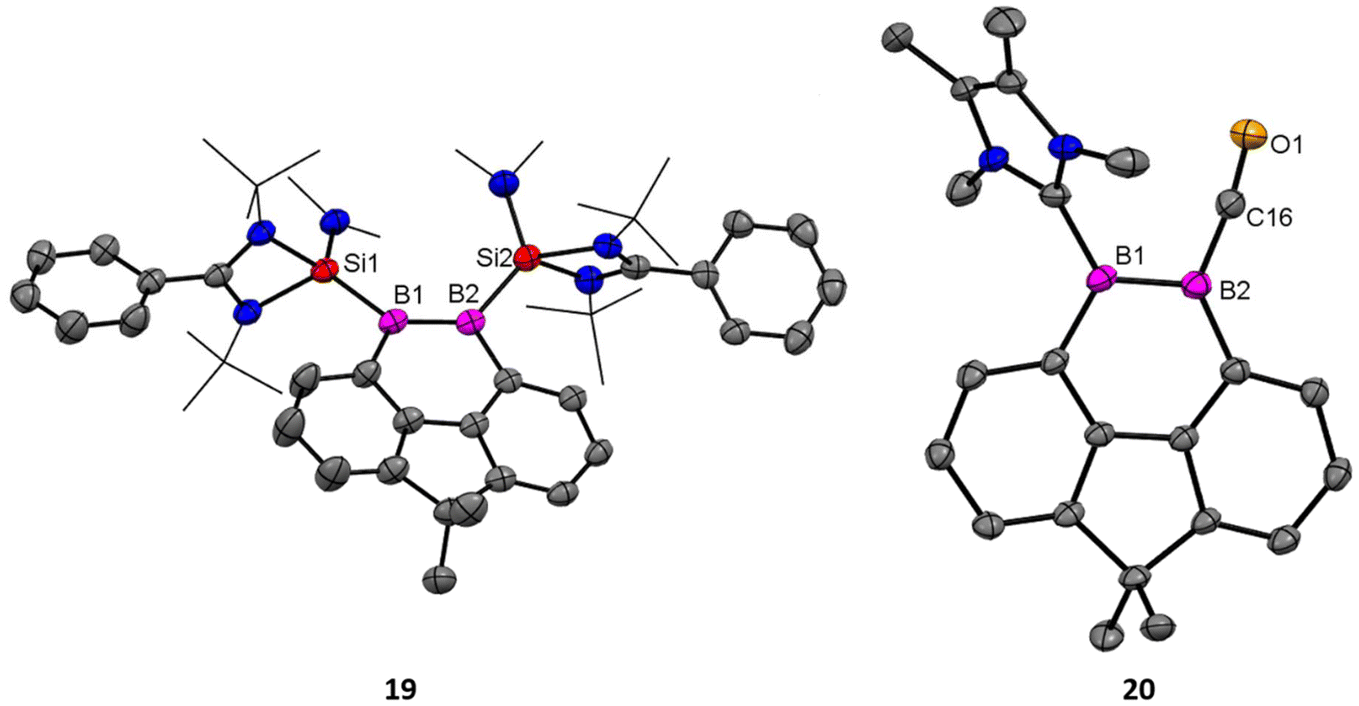
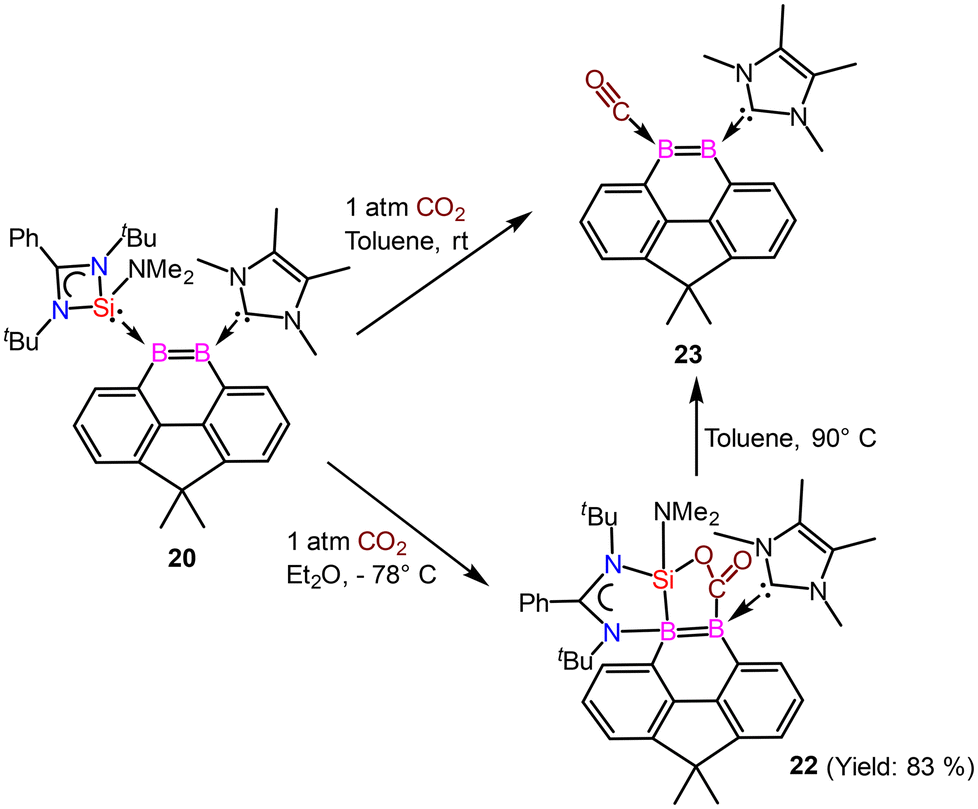
Driess and coworkers employed an amidinato-silylene to stabilize the boron–boron double bond and prepared boron based aromatic heterocyclic rings 19 and 20 from the deoxygenation reaction of 18 (Scheme 8).62 Exposure of 18 to 2 equivalents of KC8 in THF at room temperature for 2 hours yields the bis(NHSi)-stabilized 9,10-diboraphenanthrene derivative 19. Compound 19 was isolated with a 30% yield. The 11B{1H} NMR spectrum of compound 19 shows a signal at δ 29.2 ppm, while its 29Si{1H} NMR resonance appears at δ 17.7 ppm. Single-crystal X-ray diffraction (XRD) analysis shows that compound 19 features a 9,10-diboraphenanthrene core with a CMe2 linker at the 4,5-positions, where two NHSi ligands coordinate to two trigonal planar boron atoms. The B1–B2 bond length in 19 is 1.650(4) Å, with the elongation attributed to π-electron delocalization. When compound 18 was reacted with IMe as an alternative reducing agent in toluene at 60 °C for 12 hours, it produced the 9,10-diboraphenanthrene derivative 20 as a red compound (60% yield), along with the known byproduct 21. The 11B{1H} NMR spectrum of 20 exhibits two signals at δ 33.5 and 11.6 ppm. Single-crystal X-ray diffraction (XRD) analysis of 20 reveals a similar diboraphenanthrene framework, with one NHSi ligand from 19 replaced by IMe. The B–B bond length in 20 is 1.623(7) Å, comparable to that in 19 (1.650(4) Å). The aromatic character of 19 and 20 was stabilized by calculation of NICS values for these rings. The NICS values were obtained in the range of −9 to −10 ppm suggesting a significant ring current (Fig. 17).
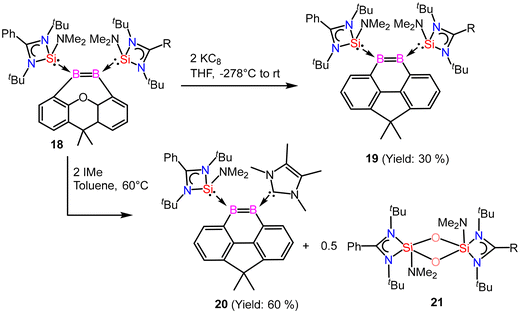 |
| | Scheme 8 Preparation of amidinato-silylene stabilized aromatic compounds (19 and 20) containing boron–boron double bond. | |
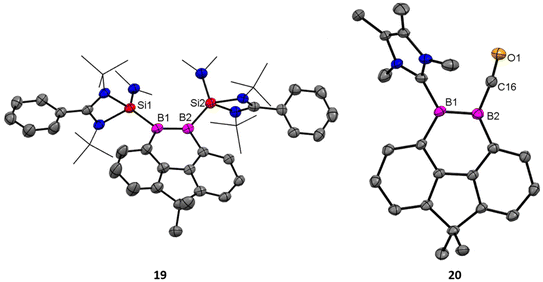 |
| | Fig. 17 Molecular structures of 19 and 20. Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. | |
The reactivity of compound 20 was examined with CO2 (Scheme 9). The NHSi ligand in 20 facilitates the monodeoxygenation of carbon dioxide (CO2) in toluene at room temperature, yielding the carbon monoxide-stabilized 9,10-diboraphenanthrene derivative 23via a silaoxadiborinanone intermediate, 22.
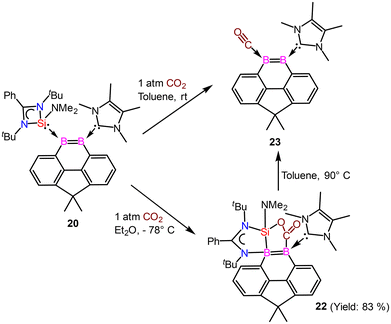 |
| | Scheme 9 Reactivity of compound 20 with CO2. | |
Homoaromatic rings
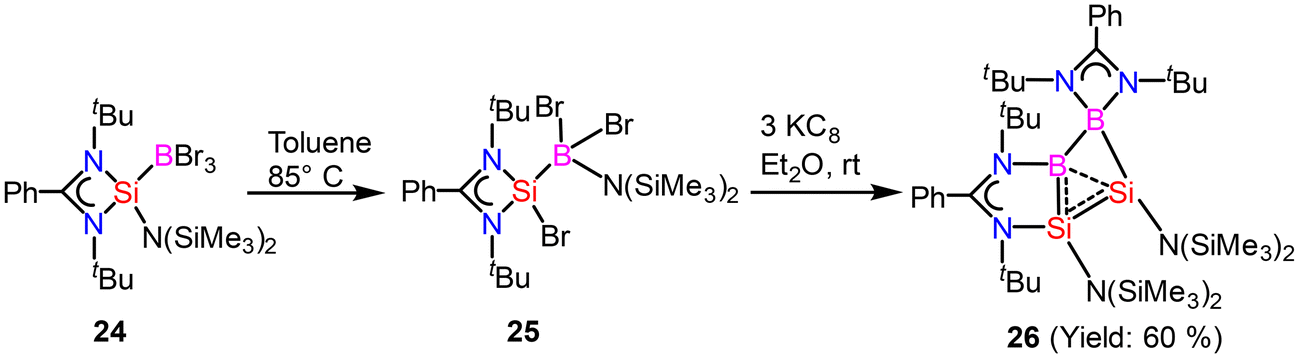
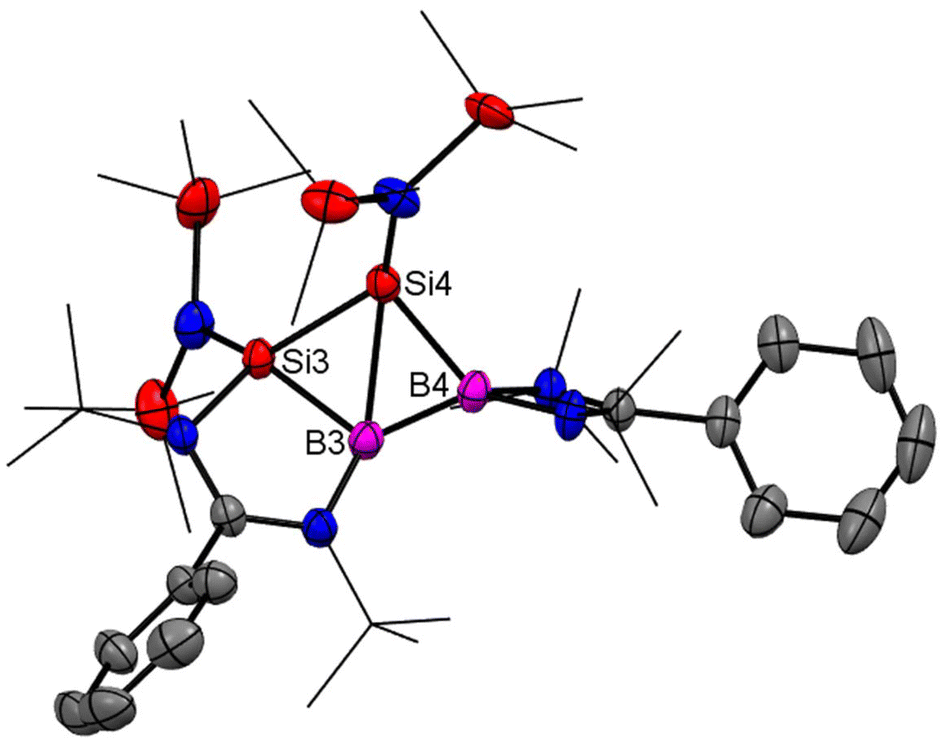
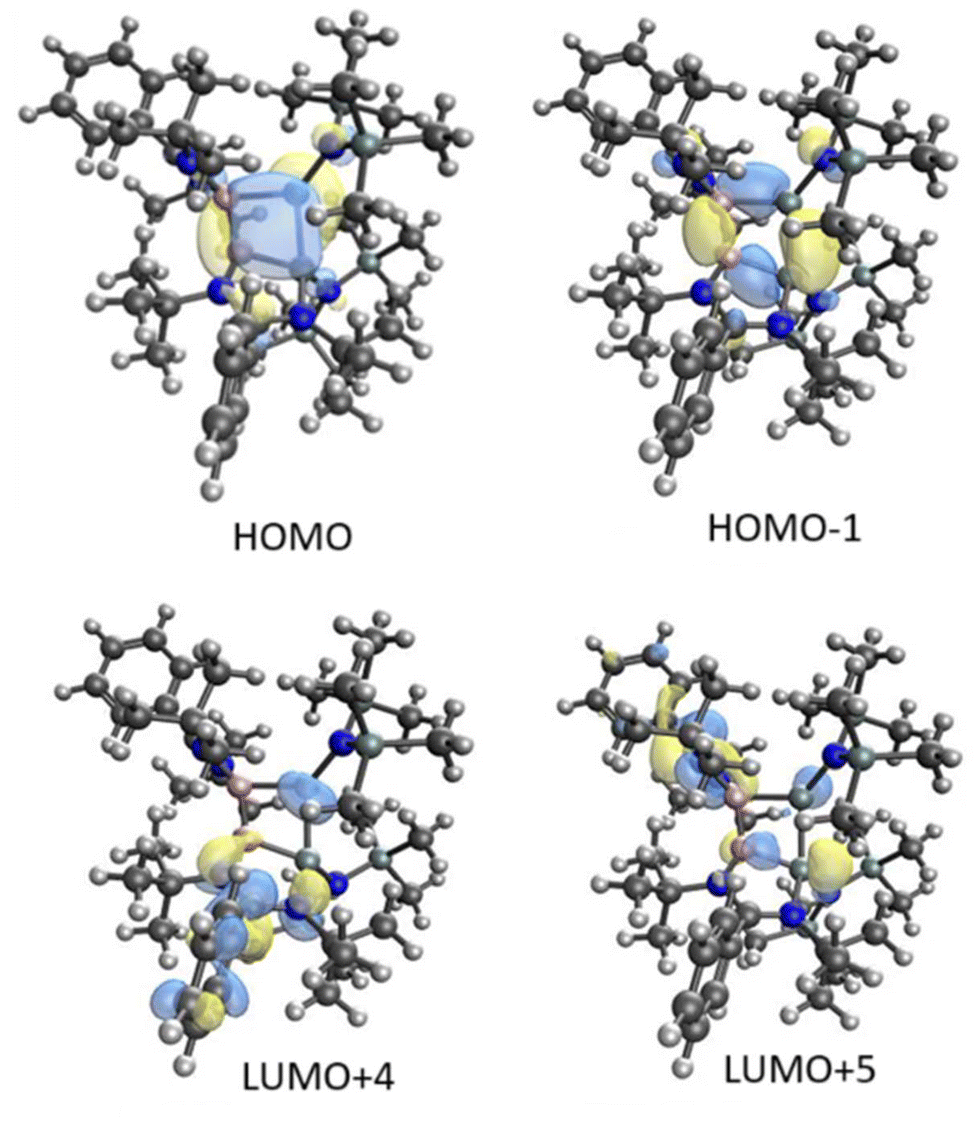
Homoaromaticity is a concept that extends the traditional definition of aromaticity by allowing for unique structural variations that still permit a degree of electronic delocalization and stabilization despite the presence of a saturated carbon/silicon atom or a group of atoms within the conjugated π-electron system. Compared to the vast array of aromatic compounds, isolated homoaromatic compounds remain quite uncommon. Additionally, the majority of these isolated homoaromatic species are either cationic or anionic, where the delocalization of charge provides an additional impetus for homoaromaticity. However, very few neutral homoaromatic compounds have been reported.63 Wang and coworkers prepared a formally neutral homoaromatic diboradisilacyclobutene (26) via the reduction of borylaminobromosilane (25) with KC8 (Scheme 10).64 Compound 26 represents the first example of a neutral boron and silicon mixed analogue of the homocyclopropenylium ion. Compound 26 was synthesized as per the procedure shown in Scheme 10. Single-crystal XRD analysis revealed that compound 26 possesses a folded four-membered Si2B2 ring and a remarkably short transannular B–Si distance of 2.306(3) Å. The most prominent structural characteristic of 26 is its puckered central B2Si2 ring, which has a folding angle of 26.2° between the B1–Si1–Si2 and B1–B2–Si2 planes. The homoaromatic nature of compound 26 was further validated by DFT calculations at the B3LYP level of theory. The calculations revealed that the HOMO mainly corresponds to the π-electron delocalization over the BSi2 group (Fig. 18). HOMO−1 is predominantly localized on the four-membered B2Si2 ring. Additionally, the atomic orbitals of B1, Si1, and Si2 contribute 23%, 11%, and 41% to the HOMO, respectively, while B2's atomic orbitals contribute only 4%. This suggests that the B2 atom does not participate in the aromatic delocalization and works as a disruption center for electronic conjugation. The orbitals from LUMO to LUMO+3 mainly correspond to the π* orbitals localized on the PhC(NtBu)2 groups. DFT calculations revealed that two π-electrons are delocalized over the BSi2 moiety (Fig. 19). Both the structural characteristics and DFT calculations demonstrate the notable 2π-homoaromatic nature of diboradisilacyclobutene. Furthermore, negative NICS(0) and NICS(1) values were found to be −19.1 and −13.9 ppm respectively, which support the pronounced homoaromatic characteristic of 26.
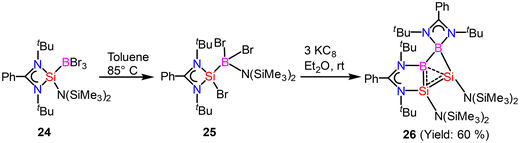 |
| | Scheme 10 Synthesis of homoaromatic compound 26. | |
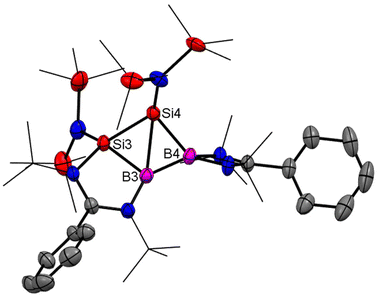 |
| | Fig. 18 Molecular structures of 26. Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. | |
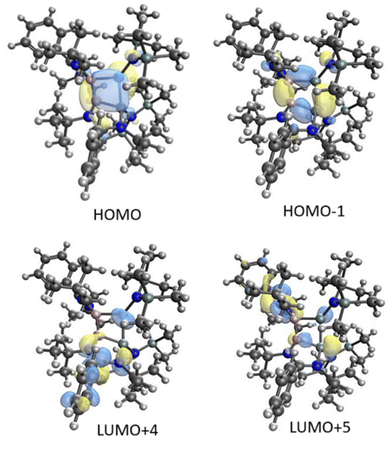 |
| | Fig. 19 Selected molecular orbitals calculated for 26. Reproduced with permission from ref. 64. Copyright (2022) American Chemical Society. | |
The comparison of NICS values of benzene with the compounds discussed in this review suggests that the benzene has a high degree of aromatic character (Table 1).65 The benzene molecule possesses quite high NICS values because it has six electrons which delocalize among six carbon centers; however, in the case of amidinate-silylene derived aromatic compounds, there are only two electrons delocalizing among four hetero-centers. Due to its stronger aromatic character, benzene is very much unreactive compared to the silicon based aromatic compounds.
Table 1 NICS values calculated for amidinato-silylene-derived aromatic compounds
| Compound |
NICS values (ppm) |
Ref. |
| NICS(0) |
NICS(1) or NICSZZ |
|
3
|
— |
−3.64 |
51
|
|
4
|
— |
1.62 |
52
|
|
5
|
−4.7 |
−3.8 |
53
|
|
7
|
−15.71 |
−10.07 |
54
|
|
8
|
−17.4 |
−12.7 |
56
|
|
12A
|
−11.8 |
9.5 |
57
|
|
12A
|
−12.3 |
−9.2 |
57
|
|
14
|
— |
−8.1 |
59
|
|
16
|
−6.2 |
−6.6 |
60
|
|
17
|
— |
−16.36 |
61
|
|
19
|
— |
−24.71 |
62
|
|
20
|
— |
−24.72 |
62
|
|
26
|
−19.1 |
−13.9 |
64
|
| Benzene |
−29.1 |
−31.2 |
65
|
Conclusion and future perspective
In conclusion, amidinato silylenes constitute a versatile class of silicon molecules that possess excellent electronic and structural properties. Due to their amphiphilic nature, these silylenes can be used in the synthesis of diverse classes of compounds including inorganic aromatic ring systems. These aromatic rings prepared from amidinao-silylenes are not as unreactive as benzene and other organic congeners. Notably, these rings have shown reactivity with small molecules and other reactive species. Presently, amidinato silylene-based silicon homoaromatic rings and silaborirane heterocyclic rings have been reported; however, heterocyclic aromatic rings with higher congeners of boron can also be prepared as predicted by density functional theory calculation.66 Hence, several hetero-aromatic compounds prepared using amidinato silylene and heavy group 13 and group 14 elements are expected to be reported in the future. These compounds hold a promising prospect due to their higher reactivity compared to organic congeners and unique electronic features. These compounds can also be used to prepare a series of potentially useful compounds for 2D materials and other applications.
Data availability
This is a review article. No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Conflicts of interest
There are no conflicts to declare.
References
- T. Imagawa, L. Giarrana, D. M. Andrada, B. Morgenstern, M. Nakamoto and D. Scheschkewitz, J. Am. Chem. Soc., 2023, 145, 4757–4764 CrossRef CAS PubMed.
- A. Saurwein, M. Nobis, S. Inoue and B. Rieger, Inorg. Chem., 2022, 61, 9983–9989 CrossRef CAS PubMed.
- J. Keuter, A. Hepp, A. Massolle, J. Neugebauer, C. Mück-Lichtenfeld and F. Lips, Angew. Chem., Int. Ed., 2022, 61, e202114485 CrossRef CAS PubMed.
- S. S. Sen, S. Khan, P. P. Samuel and H. W. Roesky, Chem. Sci., 2012, 3, 659–682 RSC.
- M. K. Bisai, T. Das, K. Vanka, R. G. Gonnade and S. S. Sen, Angew. Chem., Int. Ed., 2021, 60, 20706–20710 CrossRef CAS PubMed.
-
(a) S. S. Sen, H. W. Roesky, D. Stern, J. Henn and D. Stalke, J. Am. Chem. Soc., 2010, 132, 1123–1126 CrossRef CAS PubMed;
(b) S. K. Kushvaha, P. Kallenbach, S. M. N. V. T. Gorantla, R. Herbst-Irmer, D. Stalke, P. Parameswaran and H. W. Roesky, Chem. – Eur. J., 2023, e202303113 Search PubMed;
(c) S. K. Kushvaha, S. M. N. V. T. Gorantla, P. Kallenbach, R. Herbst-Irmer, D. Stalke, P. Parameswaran and H. W. Roesky, Dalton Trans., 2024, 53, 11410–11416 RSC.
- C. W. So, H. W. Roesky, J. Magull and R. B. Oswald, Angew. Chem., Int. Ed., 2006, 45, 3948–3950 CrossRef CAS PubMed.
- P. Jutzi, D. Kanne and C. Krüger, Angew. Chem., Int. Ed. Engl., 1986, 25, 164–164 CrossRef.
- M. Denk, R. Lennon, R. Hayashi, R. West, A. V. Belyakov, H. P. Verne, A. Haaland, M. Wagner and N. Metzler, J. Am. Chem. Soc., 1994, 116, 2691–2692 CrossRef CAS.
- C. Shan, S. Yao and M. Driess, Chem. Soc. Rev., 2020, 49, 6733–6754 RSC.
- S. Fujimori and S. Inoue, Eur. J. Inorg. Chem., 2020, 2020, 3131–3142 CrossRef CAS PubMed.
- M. Kira, Chem. Commun., 2010, 46, 2893–2903 RSC.
- M. Haaf, T. A. Schmedake and R. West, Acc. Chem. Res., 2000, 33, 704–714 CrossRef CAS PubMed.
-
(a) Y. Wang, M. Karni, S. Yao, A. Kaushansky, Y. Apeloig and M. Driess, J. Am. Chem. Soc., 2019, 141, 12916–12927 CrossRef CAS PubMed;
(b) Z. Hendi, M. K. Pandey, S. K. Kushvaha and H. W. Roesky, Chem. Commun., 2024, 60, 9483–9512 RSC.
- F. A. D. Herz, M. Nobis, D. Wendel, P. Pahl, P. J. Altmann, J. Tillmann, R. Weidner, S. Inoue and B. Rieger, Green Chem., 2020, 22, 4489–4497 RSC.
- R. S. Ghadwal, H. W. Roesky, S. Merkel, J. Henn and D. Stalke, Angew. Chem., Int. Ed., 2009, 48, 5683–5686 CrossRef CAS PubMed.
- M. Ghosh, S. Tothadi and S. Khan, Organometallics, 2021, 40, 3201–3210 CrossRef CAS.
- N. Parvin, S. Pal, J. Echeverría, S. Alvarez and S. Khan, Chem. Sci., 2018, 9, 4333–4337 RSC.
- C. Heinemann, T. Müller, Y. Apeloig and H. Schwarz, J. Am. Chem. Soc., 1996, 118, 2023–2038 CrossRef CAS.
- J. M. Mercero, A. I. Boldyrev, G. Merino and J. M. Ugalde, Chem. Soc. Rev., 2015, 44, 6519–6534 RSC.
-
(a) E. Hückel, Z. Phys., 1931, 70, 204–286 CrossRef;
(b) D. J. Klein and N. Trinajstic, J. Am. Chem. Soc., 1984, 106, 8050–8056 CrossRef CAS.
-
(a) Y. Matsuo and M. Maruyama, The chemistry of four-membered aromatics, Chem. Commun., 2012, 48, 9334–9342 RSC;
(b) Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed;
(c) G. Merino, M. Sola, I. Fernandez, C. Foroutan-Nejad, P. Lazzeretti, G. Frenking, H. L. Anderson, D. Sundholm, F. P. Cossio, M. A. Petrukhina, J. Wu, J. I. Wu and A. Restrepo, Chem. Sci., 2023, 14, 5569–5576 RSC;
(d) Z. S. Yoon, A. Osuka and D. Kim, Nat. Chem., 2009, 1, 113–122 CrossRef CAS PubMed.
-
(a) N. C. Baid, J. Am. Chem. Soc., 1972, 94, 4941–4948 CrossRef;
(b) M. Buehl and A. Hirsch, Chem. Rev., 2001, 101, 1153–1183 CrossRef CAS PubMed;
(c) A. Hirsch, Z. Chen and H. Jiao, Angew. Chem., Int. Ed., 2000, 39, 3915–3917 CrossRef CAS;
(d) J. Poater and M. Sola, Chem. Commun., 2011, 47, 11647–11649 RSC.
- A. N. Alexandrova, A. I. Boldyrev, H. J. Zhai and L. S. Wang, Coord. Chem. Rev., 2006, 250, 2811–2866 CrossRef CAS.
- A. I. Boldyrev and L. S. Wang, Chem. Rev., 2005, 105, 3716–3757 CrossRef CAS PubMed.
- C. A. Tsipis, Coord. Chem. Rev., 2005, 249, 2740–2762 CrossRef CAS.
- A. E. Seitz, M. Eckhardt, A. Erlebach, E. V. Peresypkina, M. Sierka and M. Scheer, J. Am. Chem. Soc., 2016, 138, 10433–10436 CrossRef CAS PubMed.
- D. Y. Zubarev, B. B. Averkiev, H. Zhai, L. Wang and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 257–267 RSC.
- H.-J. Zhai, B. B. Averkiev, D. Y. Zubarev, L.-S. Wang and A. I. Boldyrev, Angew. Chem., Int. Ed., 2007, 46, 4277–4280 CrossRef CAS PubMed.
- B. Peerless, Y. J. Franzke and S. Dehnen, Nat. Chem., 2002, 15, 347–356 CrossRef PubMed.
- I. Fernández and G. Frenking, Chem. – Eur. J., 2007, 13, 5873–5884 CrossRef PubMed.
-
(a) J. Jin, G. Wang, M. Zhou, D. M. Andrada, M. Hermann and G. Frenking, Angew. Chem., Int. Ed., 2016, 55, 2078–2082 CrossRef CAS PubMed;
(b) X. Li, A. E. Kuznetsov, H. F. Zhang, A. I. Boldyrev and L. S. Wang, Science, 2001, 291, 859–861 CrossRef CAS PubMed;
(c) A. N. Alexandrova, A. I. Boldyrev, H. J. Zhai, L. S. Wang, E. Steiner and P. W. Fowler, J. Phys. Chem. A, 2003, 107, 1359–1369 CrossRef CAS;
(d) T. Kupfer, H. Braunschweig and K. Radacki, Angew. Chem., Int. Ed., 2015, 54, 15084–15088 CrossRef CAS PubMed.
- K. Ota and R. Kinjo, Angew. Chem., Int. Ed., 2020, 59, 6572–6575 CrossRef CAS PubMed.
- H. Braunschweig, C. W. Chiu, K. Radacki and T. Kupfer, Angew. Chem., Int. Ed., 2010, 49, 2041–2044 CrossRef CAS PubMed.
- G. Märkl and W. Schlosser, Angew. Chem., Int. Ed. Engl., 1988, 27, 963–965 CrossRef.
- N. Tokitoh, K. Wakita, T. Matsumoto, T. Sasamori, R. Okazaki, N. Takagi, M. Kimura and S. Nagase, J. Chin. Chem. Soc., 2008, 55, 487–507 CrossRef CAS.
- Y. Kabe, K. Ohkubo, H. Ishikawa and W. Ando, J. Am. Chem. Soc., 2000, 122, 3775–3776 CrossRef CAS.
- D. Scheschkewitz, A. Ghaffari, P. Amseis, M. Unverzagt, G. Subramanian, M. Hofmann, P. V. R. Schleyer, H. F. Schaefer III, G. Geiseler, W. Massa and A. Berndt, Angew. Chem., Int. Ed., 2000, 39, 1272–1275 CrossRef CAS PubMed.
- C. Präsang, P. Amseis, D. Scheschkewitz, G. Geiseler, W. Massa, M. Hofmann and A. Berndt, Angew. Chem., Int. Ed., 2006, 45, 6745–6747 CrossRef PubMed.
- R. J. F. Berger, H. S. Rzepa and D. Scheschkewitz, Angew. Chem., Int. Ed., 2010, 49, 10006–10009 CrossRef CAS PubMed.
- K. Abersfelder, A. J. P. White, H. S. Rzepa and D. Scheschkewitz, Science, 2010, 327, 564–566 CrossRef CAS PubMed.
- C. B. Yildiz, K. I. Leszczyńska, S. González-Gallardo, M. Zimmer, A. Azizoglu, T. Biskup, C. W. M. Kay, V. Huch, H. S. Rzepa and D. Scheschkewitz, Angew. Chem., Int. Ed., 2020, 59, 15087–15092 CrossRef CAS PubMed.
- A. E. Kuznetsov, A. I. Boldyrev, X. Li and L.-S. Wang, J. Am. Chem. Soc., 2001, 123, 8825–8831 CrossRef CAS PubMed.
- X. Li, A. E. Kuznetsov, H. F. Zhang, A. I. Boldyrev and L. S. Wang, Science, 2001, 291, 859–861 CrossRef CAS PubMed.
- M. Tian, J. Zhang, L. Guo and C. Cui, Chem. Sci., 2021, 12, 14635–14640 RSC.
- S. Winstein, J. Am. Chem. Soc., 1959, 6523, 6524–6525 CrossRef.
-
(a) R. F. Childs, Acc. Chem. Res., 1984, 17, 347–352 CrossRef CAS;
(b) D. Scheschkewitz, M. Hofmann, A. Ghaffari, P. Amseis, C. Präsang, W. Mesbah, G. Geiseler, W. Massa and A. Berndt, J. Organomet. Chem., 2002, 646, 262–270 CrossRef CAS.
- G. Merino, M. Sola, I. Fernandez, C. Foroutan-Nejad, P. Lazzetti, G. Frenking, H. L. Anderson, D. Sundholm, F. Cossio, M. A. Petrukhina, J. Wu, J. I. Wu and A. Restredo, Chem. Sci., 2023, 14, 5569–5576 RSC.
- S. S. Sen, A. Jana, H. W. Roesky and C. Schulzke, Angew. Chem., Int. Ed., 2009, 48, 8536–8538 CrossRef CAS PubMed.
- S. S. Sen, S. Khan, S. Nagendran and H. W. Roesky, Acc. Chem. Res., 2012, 45, 578–587 CrossRef CAS PubMed.
- S. S. Sen, H. W. Roesky, K. Meindl, D. Stern, J. Henn, A. C. Stückl and D. Stalke, Chem. Commun., 2010, 46, 5873–5875 RSC.
- Y. Chen, J. Li, Y. Zhao, L. Zhang, G. Tan, H. Zhu and H. W. Roesky, J. Am. Chem. Soc., 2021, 143, 2212–2216 CrossRef CAS PubMed.
- S. Inoue, J. D. Epping, E. Irran and M. Driess, J. Am. Chem. Soc., 2011, 133, 8514–8517 CrossRef CAS PubMed.
- S. H. Zhang, H. W. Xi, K. H. Lim and C. W. So, Angew. Chem., Int. Ed., 2013, 52, 12364–12367 CrossRef CAS PubMed.
- Y. F. Yang, G. J. Cheng, J. Zhu, X. Zhang, S. Inoue and Y. D. Wu, Chem. – Eur. J., 2012, 18, 7516–7524 CrossRef CAS PubMed.
- X. Sun, T. Simler, R. Yadav, R. Ko and P. W. Roesky, J. Am. Chem. Soc., 2019, 141, 14987–14990 CrossRef CAS PubMed.
- Y. Li, S. Dong, J. Guo, Y. Ding, J. Zhang, J. Zhu and C. Cui, J. Am. Chem. Soc., 2023, 145, 21159–21164 CrossRef CAS PubMed.
- M. Solà, F. Feixas, J. O. C. Jiménez-Halla, E. Matito and J. Poater, Symmetry, 2010, 2, 1156–1179 CrossRef.
- S. K. Sarkar, R. Chaliha, M. M. Siddiqui, S. Banerjee, A. Münch, R. Herbst-Irmer, D. Stalke, E. D. Jemmis and H. W. Roesky, Angew. Chem., Int. Ed., 2020, 59, 23015–23019 CrossRef CAS PubMed.
- S. K. Kushvaha, P. Kallenbach, S. S. Rohman, M. K. Pandey, Z. Hendi, F. Rüttger, R. Herbst-Irmer, D. Stalke, P. Parameswaran and H. W. Roesky, J. Am. Chem. Soc., 2023, 145, 25523–25527 CrossRef CAS PubMed.
- J. Fan, L. Yue, C. Liu, B. Rao, G. Zhou, A. Li and B. Su, J. Am. Chem. Soc., 2024, 146, 39–44 CrossRef CAS PubMed.
- J. Fan, J. Xu, Q. Ma, S. Yao, L. Zhao, G. Frenking and M. Driess, J. Am. Chem. Soc., 2024, 146, 20458–20467 CrossRef CAS PubMed.
- T. Tran Ngoc, N. Grabicki, E. Irran, O. Dumele and J. F. Teichert, Nat. Chem., 2023, 15, 377–385 CrossRef CAS PubMed.
- Y. Zhang, L. Wu and H. Wang, J. Am. Chem. Soc., 2022, 144, 22446–22450 CrossRef CAS PubMed.
- A. Stanger, ChemPhysChem, 2023, 24, e202300080 CrossRef CAS PubMed.
- H. Sharma; and K. Vanka, Organometallics, 2024, 43(14), 1583–1592 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  and
Herbert W.
Roesky
and
Herbert W.
Roesky