
Molecular modulation of nickel–salophen organic frameworks enables the selective photoreduction of CO2 at varying concentrations†
Xiaohan
Yu‡
ab,
Mingzi
Sun‡
 c,
Tianran
Yan
ab,
Lin
Jia
ab,
Mingyu
Chu
ab,
Liang
Zhang
ab,
Wei
Huang
*ab,
Bolong
Huang
*ce and
Yanguang
Li
*abd
c,
Tianran
Yan
ab,
Lin
Jia
ab,
Mingyu
Chu
ab,
Liang
Zhang
ab,
Wei
Huang
*ab,
Bolong
Huang
*ce and
Yanguang
Li
*abd
aInstitute of Functional Nano & Soft Materials (FUNSOM), Soochow University, Suzhou, 215123, China. E-mail: weihuang@suda.edu.cn; yanguang@suda.edu.cn
bJiangsu Key Laboratory for Advanced Negative Carbon Technologies, Soochow University, Suzhou 215123, China
cDepartment of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong SAR 999077, China. E-mail: bhuang@polyu.edu.hk
dMacao Institute of Materials Science and Engineering (MIMSE), MUST-SUDA Joint Research Center for Advanced Functional Materials, Macau University of Science and Technology, Taipa 999078, Macau SAR, China
eResearch Centre for Carbon-Strategic Catalysis, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong SAR 999077, China
First published on 6th February 2024
Abstract
Photocatalytic CO2 reduction to value-added chemicals is appealing but challenging, especially under dilute CO2 conditions. Herein, we present a molecular modulation strategy for porous metal–salophen organic frameworks (M-SOFs), involving cooperative regulation of the catalytically active metal centers and their local coordination environments for selective photocatalytic CO2 reduction across a wide range of CO2 concentrations. The optimal Ni-SOF shows a remarkable photocatalytic CO production rate of 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 908 μmol h−1 g−1 and near-unity selectivity under a pure CO2 atmosphere, along with excellent structural stability. More impressively, it largely preserves the catalytic activity and selectivity even when exposed to dilute CO2 (5–20 vol%). Both experimental and theoretical analyses support that the specific Ni–N2O2 coordination environment in the Ni-SOF endows it with strong CO2 binding capacity. This, coupled with nanoporous skeletons, enhances local CO2 enrichment and facilitates its subsequent conversion at the catalytic centers, thereby leading to superior photocatalytic performances at various CO2 concentrations.
908 μmol h−1 g−1 and near-unity selectivity under a pure CO2 atmosphere, along with excellent structural stability. More impressively, it largely preserves the catalytic activity and selectivity even when exposed to dilute CO2 (5–20 vol%). Both experimental and theoretical analyses support that the specific Ni–N2O2 coordination environment in the Ni-SOF endows it with strong CO2 binding capacity. This, coupled with nanoporous skeletons, enhances local CO2 enrichment and facilitates its subsequent conversion at the catalytic centers, thereby leading to superior photocatalytic performances at various CO2 concentrations.
Broader contextSolar-driven CO2 conversion has been considered as an ideal approach for reducing atmospheric CO2 concentration while producing value-added chemicals and fuels. Over the past decades, tremendous progress has been made in the development of high-performance photocatalysts. Unfortunately, most of them are only operable under high-purity CO2 conditions. Direct photoreduction of dilute CO2 is appealing, given that industrial exhaust gases typically contain low concentrations of CO2; however, it remains a formidable challenge. The key obstacles lie in the inadequate CO2 capturing ability of the photocatalysts and unfavourable interactions between catalytic sites and CO2 molecules. Here, we demonstrate the unique capability of metal–salophen organic frameworks – with properly engineered metal centers and coordination environments – to facilitate the local enrichment and subsequent conversion of CO2. The optimal sample shows exceptional photocatalytic performance and near-unity CO2-to-CO selectivity across a wide range of CO2 concentrations. |
Introduction
Excessive CO2 emissions from fossil fuel combustion have been recognized as the dominant cause of global warming and climate change, posing a significant threat to our economic and environmental sustainability.1 Several strategies have been proposed to reduce the atmospheric CO2 concentration, either through physical sequestration or chemical conversion.2,3 Among them, solar-driven CO2 reduction to valuable chemicals (e.g., CO, CH4, CH3OH, and HCOOH) offers a particularly attractive approach by transforming solar energy into chemical energy while neutralizing CO2 emissions.4,5 Nevertheless, current investigations are primarily based on pure CO2 feedstocks. Given that a major source of CO2 emissions is industrial exhaust gases which contain relatively low CO2 concentrations typically in the range of 5–20%, the direct conversion of low-concentration CO2 is more practically relevant and highly desirable.6 Unfortunately, limited attempts so far have shown that diluted CO2 concentrations usually compromise the activity and/or selectivity.7 This is mainly attributed to inefficient CO2 capture and unfavorable binding interactions between catalytic sites and substrates.8 As a result, the rational design of photocatalysts with optimized catalytic centers and CO2 binding capacity is crucial.In nature, enzymes can catalyze complicated redox reactions with high activity and specificity towards target products.9 Their active sites are typically composed of metal cations bound to the amino acid residues of the proteins containing nitrogen (N), sulfur (S), and oxygen (O) atoms. For instance, galactose oxidase is a well-known mononuclear oxidoreductase, in which the Cu center is ligated by two pyrrolic N atoms and two phenolic O atoms.10 Such a Cu–N2O2 site can efficiently catalyze the two-electron oxidation of primary alcohols to corresponding aldehydes. Inspired by these biocatalysts, great efforts have been devoted to exploiting artificial enzymes in order to mimic the functions of natural molecules.11–14 Salen or salophen compounds are a type of Schiff-base ligands with planar tetradentate N2O2 sites, capable of accommodating various transition metal ions.15 By judiciously varying metal catalytic centers and ancillary organic ligands, it is possible to fine-tune their catalytic performances. Benefiting from their structural diversity and easy accessibility, metal–salen/salophen complexes have been widely used as enzyme mimics for different catalytic reactions.16,17 Nevertheless, their potential in photocatalytic CO2 reduction has not been explored. This may be partly attributed to the unsatisfactory long-term stability of molecular salen/salophen catalysts during photocatalytic reactions as a result of their aggregation or degradation.18
Immobilizing molecular catalysts onto suitable porous hosts offers a potential solution to the above issue. Previous investigations have demonstrated that porous skeletons could enhance the robustness of catalytic sites, and facilitate the enrichment of CO2 molecules in their vicinity.19,20 This is of great significance for durable and efficient CO2 conversion, particularly under diluted CO2 concentrations.
To this end, here we synthesize a new type of metal–salophen organic framework (M-SOF, M = Co, Ni, Cu, and Zn) by covalently incorporating metal–salophen nodes into porous architectures for the efficient and durable photoreduction of CO2 at varying concentrations. By tuning the predesigned organic precursors and metal species, the activity and CO selectivity of the resulting catalysts could be readily modulated. Of particular note is that the Ni-SOF containing Ni–N2O2 centers is the most photocatalytically active; across a wide range of CO2 concentrations (5–100 vol%), it exhibits an extremely high photocatalytic activity of up to 16908 μmol h−1 g−1 and near-unity selectivity for CO production. Experimental and theoretical studies show that the outstanding catalytic activity of the Ni-SOF originates from its porous frameworks and favorable binding towards CO2 and reaction intermediates.
Results and discussion
Fig. 1a schematically illustrates the synthetic procedure of Ni-SOFs via a metal-template-assisted Schiff-base reaction between salicylaldehyde and 1,3,5-tris(4-aminophenyl)triazine (TAPT) in N,N-dimethylformamide (DMF). During the synthesis, salicylaldehyde and nickel acetate were first mixed in DMF at room temperature to form a soluble O-coordinated Ni intermediate (Ni–O4), as evidenced by the change in solution color from light green to yellow.21 The subsequent addition of TAPT initiated the condensation reaction with the aldehyde functionalities of Ni–O4 at elevated temperatures and ultimately gave rise to an extended coordination polymer with Ni–N2O2 nodes. It is worth noting that such a stepwise synthetic procedure is crucial in order to achieve the desired metal–N2O2 coordination. TAPT is employed as the building block because of its nitrogen-rich composition, which would result in nitrogen-rich porous skeletons to facilitate CO2 capture. Other samples with different metal centers (Co-SOFs, Cu-SOFs, and Zn-SOFs) were synthesized by adding corresponding metal salts under otherwise identical conditions. To investigate the effect of coordination environments on photocatalysis, N-coordinated Ni–N4, and N/S-coordinated Ni–N2S2 were also synthesized by replacing salicylaldehyde with 2-pyridinecarboxaldehyde and 2-mercaptobenzaldehyde, respectively.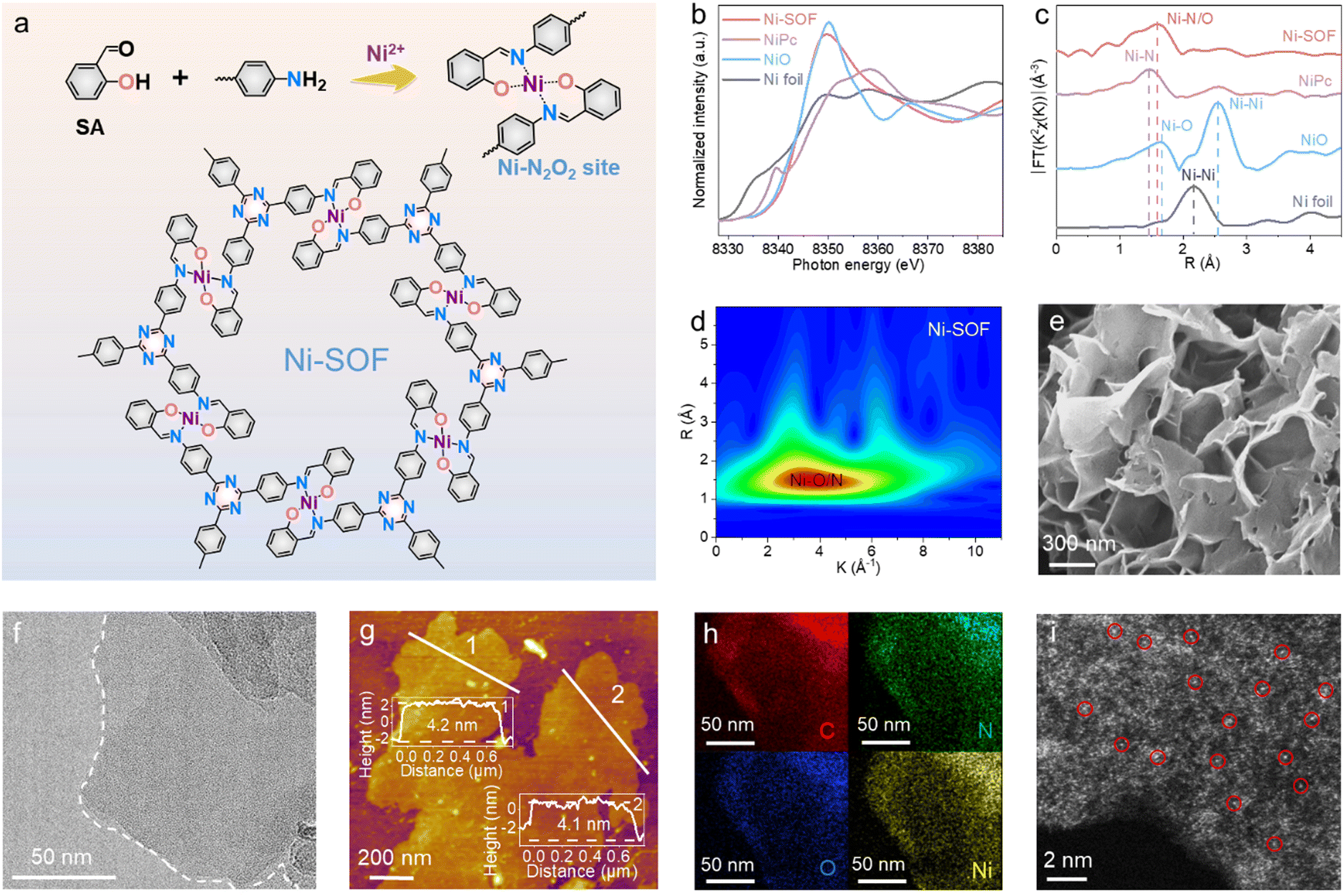 | ||
| Fig. 1 Synthesis and characterizations of Ni-SOFs. (a) Schematic illustration of the synthetic procedure of Ni-SOFs. (b) XANES and (c) EXAFS spectra of Ni-SOFs, NiPc, NiO and Ni foil. (d) Wavelet transform of the EXAFS spectra of Ni-SOFs. (e) SEM, (f) TEM, (g) AFM, (h) EDS elemental mapping and (i) HAADF-STEM images of Ni-SOFs. | ||
The chemical structures of our samples were first interrogated by Fourier transform infrared (FT-IR) and X-ray photoelectron spectroscopy (XPS). For the sake of clarity, the following discussion will be focused on Ni-SOF as a representative unless otherwise specified. From its FT-IR spectrum, the successful condensation between salicylaldehyde and TAPT is evidenced by the disappearance of the –NH2 stretching vibrations at 3209 and 3321 cm−1 as well as the emergence of a new peak at 1621 cm−1 attributed to –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (Fig. S1, ESI†).22 The presence of two peaks at 1361 and 1508 cm−1, characteristic of triazine, also signifies the incorporation of TAPT within the coordination networks.23 Moreover, two weak bands corresponding to Ni–O and Ni–N are observed at 589 and 458 cm−1, respectively.24 They suggest that the coordination nodes in Ni-SOF are formed through the interactions between Ni cations and surrounding imine/hydroxyl groups. Fig. S2 (ESI†) summarizes the XPS results of Ni-SOFs. The N 1s spectrum of the Ni-SOF could be deconvoluted into three peaks assigned to pyridinic N in the triazine unit (398.0 eV), CN (398.8 eV) and Ni–N (399.8 eV).22 Its O 1s spectrum shows two peaks at 528.3 and 531.6 eV, corresponding to Ni–O and C–O, respectively.25 The Ni center in the Ni-SOF is determined to be in the divalent state based on its Ni 2p spectrum.26
N (Fig. S1, ESI†).22 The presence of two peaks at 1361 and 1508 cm−1, characteristic of triazine, also signifies the incorporation of TAPT within the coordination networks.23 Moreover, two weak bands corresponding to Ni–O and Ni–N are observed at 589 and 458 cm−1, respectively.24 They suggest that the coordination nodes in Ni-SOF are formed through the interactions between Ni cations and surrounding imine/hydroxyl groups. Fig. S2 (ESI†) summarizes the XPS results of Ni-SOFs. The N 1s spectrum of the Ni-SOF could be deconvoluted into three peaks assigned to pyridinic N in the triazine unit (398.0 eV), CN (398.8 eV) and Ni–N (399.8 eV).22 Its O 1s spectrum shows two peaks at 528.3 and 531.6 eV, corresponding to Ni–O and C–O, respectively.25 The Ni center in the Ni-SOF is determined to be in the divalent state based on its Ni 2p spectrum.26
The electronic state and coordination environment of the Ni-SOF were further examined by synchrotron X-ray absorption spectroscopy (XAS). Fig. 1b illustrates its X-ray absorption near-edge structure (XANES) spectrum at the Ni K-edge. The white-line peak of Ni-SOFs is similar to that of NiO, corroborating the divalent state of Ni centers in Ni-SOFs. The corresponding Fourier transform extended X-ray absorption fine structure (EXAFS) spectrum of Ni-SOFs exhibits a prominent peak at 1.60 Å, which is a value between the bonding length of Ni–N in nickel phthalocyanine (NiPc) (1.46 Å) and that of Ni–O in NiO (1.65 Å) (Fig. 1c). This again supports the mixed Ni–N/O first-shell coordination.27 No Ni–Ni scattering is observed, confirming the atomic dispersion of Ni2+ cations in Ni-SOFs. Further evidence is obtained from the wavelet transform (WT) of the EXAFS spectra (Fig. 1d and Fig. S3, ESI†). The maximum WT position of Ni-SOFs apparently differs from those of Ni–N in NiPc and Ni–O in NiO, but partially overlaps with them.
To fully unveil the steric structure of Ni–N2O2 nodes in Ni-SOFs, a model molecule (denoted as Ni–salophen) was synthesized as its mimic and analyzed by single-crystal X-ray diffraction (XRD) measurement (Fig. S4 and Table S1, ESI†). It is shown to possess a four-coordinated Ni center with the first coordination sphere consisting of two phenolic O atoms and two imine N atoms. The bond length of Ni–O is calculated to be 1.83 Å, slightly shorter than that of Ni–N (1.91 Å) as a result of the higher electronegativity of O atoms. Two pairs of trans-coordinated O and N atoms around the central Ni atom form a nearly planar rhomboid geometry owing to the small differences in bond lengths and bond angles. It is believed that such an open metal center with exposed axial coordination is advantageous for CO2 binding and activation.28
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) imaging show that the Ni-SOF consists of crumpled and stacked nanosheets (Fig. 1e and f). Individual nanosheets are estimated to have a size ranging from 200 nm to 1 μm. From atomic force microscopy (AFM) height profiles at multiple locations, the nanosheet thickness is measured to be approximately 4 nm (Fig. 1g). Such an ultrathin nanosheet geometry is desirable for catalytic applications as it promotes the exposure of inner active sites and maximizes atom utilization. Energy dispersive X-ray spectroscopy (EDS) mapping under scanning transmission electron microscopy (STEM) reveals the uniform distribution of C, N, O and Ni on Ni-SOF nanosheets (Fig. 1h). The aberration-corrected high-angle annular dark-field STEM (HAADF-STEM) image shows discrete bright spots (some marked with red circles) corresponding to heavy Ni atoms, corroborating the atomic dispersion of Ni sites throughout the examined region (Fig. 1i). Moreover, the Ni content in Ni-SOFs is found to be 10.5 wt% as analyzed by inductively coupled plasma mass spectrometry (ICP-MS), which is in good agreement with the theoretical value (10.2 wt%). N2 isotherm sorption measurements show that the Ni-SOF has a specific surface area of 116 m2 g−1 (Fig. S5, ESI†). In addition to Ni-SOFs, the structures of other samples with different metal sites and coordination environments were also characterized, and results are summarized in the ESI† (Fig. S6–S8 and Table S2).
We evaluated the photocatalytic performance of our samples under pure CO2. The reactions were conducted in a mixed solution of acetonitrile and water under visible light irradiation (λ > 420 nm) using [Ru(bpy)3]Cl2 as the photosensitizer and triisopropanolamine (TIPA) as the hole scavenger. Gaseous products (e.g., CO and H2) were analyzed and quantified by gas chromatography (GC) based on their calibration curves (Fig. S9, ESI†). We explored different solvent ratios, catalyst concentrations, and hole scavengers (Fig. S10, ESI†). Under the optimized conditions, the Ni-SOF exhibits an almost linear accumulation of CO over time and yields a total of 67.6 μmol of CO after 4 h irradiation (Fig. 2a). This corresponds to an average mass-specific CO evolution rate of 16908 μmol h−1 g−1. Its maximum apparent quantum efficiency (AQE) toward CO production is 2.8% at 450 nm (Fig. S11, ESI†). The isotope experiment using 13CO2 provides evidence that CO stems from CO2 photoreduction rather than other organic substances or the catalyst itself (Fig. S12, ESI†). Control experiments also show that negligible CO is produced in the absence of light, CO2, Ni-SOF, photosensitizer or hole scavenger under otherwise identical conditions (Fig. 2b). In addition to CO, only a trace amount of H2 (1.3 μmol) is measured on Ni-SOFs, and no liquid products (such as formic acid or methanol) are detected in the reaction solution by nuclear magnetic resonance (NMR) spectroscopy (Fig. S13, ESI†). As a result, a high CO selectivity of 98% is achieved for our catalyst. It is worth highlighting that the great activity and selectivity measured for our Ni-SOF places it at the top of other organic competitors including covalent organic frameworks (COFs) and metal–organic frameworks (MOFs) as well as most inorganic counterparts (Fig. 2c and Table S3, ESI†).29–47 In particular, its mass-specific CO production rate is about 17.5 and 8.5 times larger than those of the state-of-the-art COF catalyst (Ni-TpBpy) and MOF catalyst (2D-Ni2TCPE), respectively.32,47 We are aware that higher mass-specific rates have been reported for some transition metal oxides, but their selectivity for carbonaceous products was generally lower than 80%.41
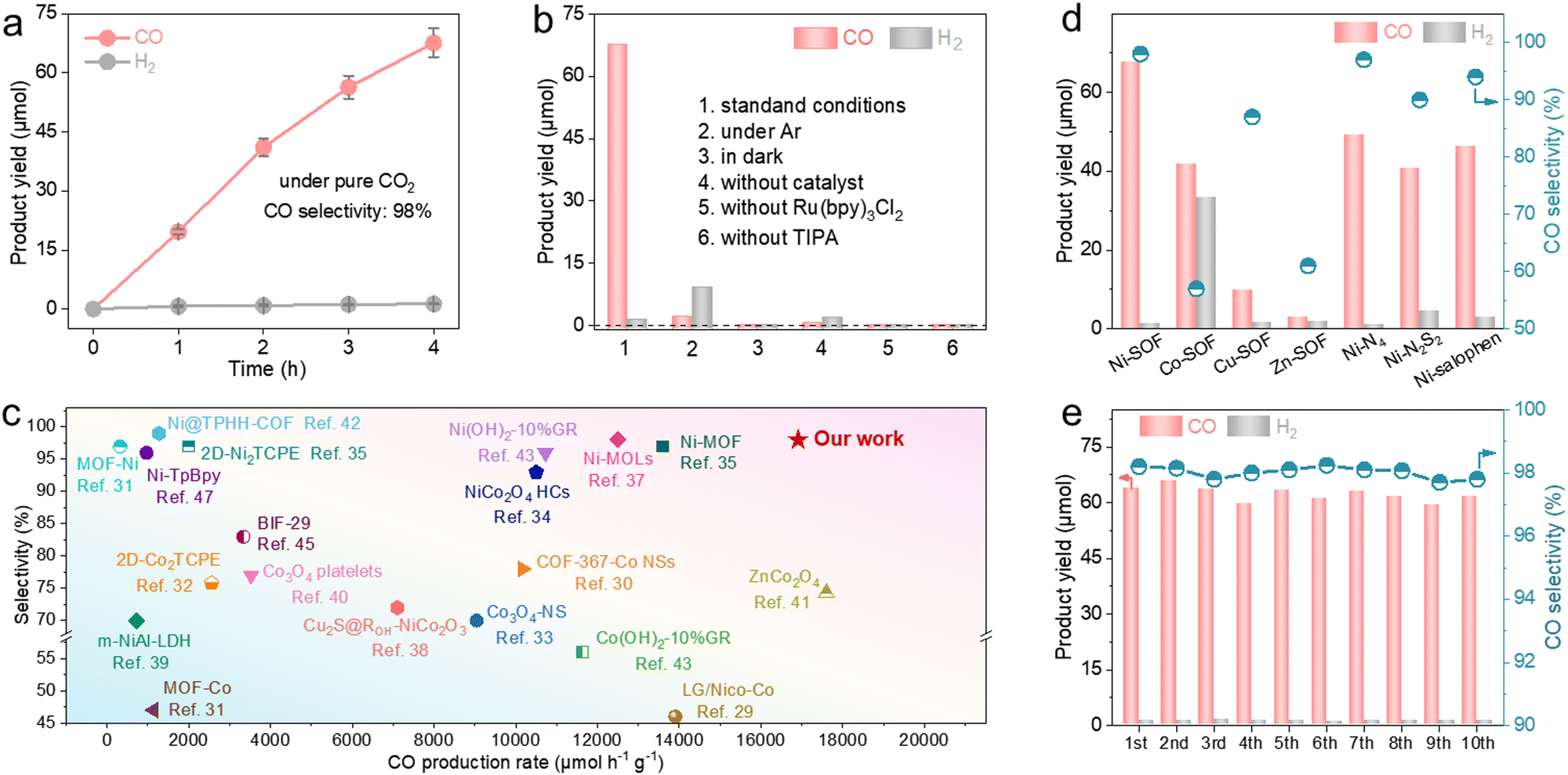 | ||
| Fig. 2 Photocatalytic performance of Ni-SOFs under pure CO2. (a) Time-dependent CO and H2 production on Ni-SOFs. (b) Control experiments conducted under different conditions. (c) Performance comparison of Ni-SOFs with other organic or inorganic catalysts in terms of the CO production rate and selectivity under pure CO2. (d) CO production and selectivity of different samples. (e) Cycling tests of Ni-SOFs. | ||
The catalytic performances of coordinated metal centers are greatly impacted by not only the metal identities but also their local chemical environments (Fig. 2d). Under identical reaction conditions, the Co-SOF shows a significantly decreased CO production rate of 10157.5 μmol h−1 g−1 and a higher H2 evolution activity of 8322.5 μmol h−1 g−1, leading to a low CO selectivity of 57%. Even though the Cu-SOF demonstrates a good CO selectivity (87%), its CO production rate is 7 times lower than that of Ni-SOFs. The Zn-SOF exhibits the lowest CO production rate of 695 μmol h−1 g−1 and a moderate CO selectivity of 61%. Moreover, the substitution of O atoms in the first coordination sphere of Ni-SOFs with less electronegative N or S (Ni–N4 and Ni–N2S2, respectively) leads to noticeable decreases in both the activity and selectivity, and their measured activities are found to correlate with the electronegativity of coordinating elements. The above results suggest the critical roles of metal centers and their coordination environments during CO2 photoreduction. The Ni-SOF featuring Ni–N2O2 active sites represents the optimal combination to enable active and selective CO2 photoreduction to CO. In addition, despite having identical Ni–N2O2 catalytic sites, the Ni-SOF is more active than Ni–salophen, presumably due to its porous microstructure which enriches local CO2 concentration.48
Stability is another important performance metric of photocatalysts. Here, we carried out long-term photocatalysis as shown in Fig. S14 (ESI†). The CO production rate of the Ni-SOF gradually decreases and reaches a plateau after 5 h. However, its activity can be completely restored when fresh [Ru(bpy)3]2+ is replenished. This observation suggests that the performance decay is due to the degradation of the photosensitizer rather than the Ni-SOF.49 In contrast, Ni–salophen experiences an irreversible activity loss after about 5 h. It underlines the evident advantage of metal–salophen organic frameworks with improved structural robustness and catalytic stability. The excellent stability of Ni-SOFs is also supported by the cycling experiment showing no noticeable performance loss after a total of 40 h illumination (Fig. 2e). In addition, the ICP-MS analysis of the reaction filtrate after the cycling test reveals a negligible Ni concentration (0.264 ppm), thus ruling out the possibility of Ni leaching out of Ni-SOFs during photocatalysis. Spectroscopic and microscopic analyses of the recovered Ni-SOF disclose no discernable structural changes (Fig. S15, ESI†).
Effective charge transfer from the photosensitizer to the catalytic sites is a prerequisite for initiating redox reactions. To probe the charge transfer behavior between [Ru(bpy)3]2+ and Ni-SOFs, steady-state and time-resolved photoluminescence (PL) spectroscopy studies were conducted. As shown in Fig. S16 (ESI†), the steady-state PL peak intensity of [Ru(bpy)3]2+ gradually decreases as the Ni-SOF concentration increases, while the TIPA concentration has no significant effect on the PL intensity. Corresponding Stern–Volmer plots show the much higher quenching efficiency of Ni-SOFs than TIPA. These results reveal that the electron transfer from the photosensitizer to the catalyst is the initial step of the photocatalytic cycle.32 The time-resolved PL spectrum of [Ru(bpy)3]2+ follows a single exponential decay with an average PL lifetime of 237 ns, consistent with literature results (Fig. 3a).50 In the presence of Ni-SOFs, a double-exponential decay is observed with fitted lifetimes of 0.49 ns and 225 ns (Fig. 3b). The former is attributed to the charge transfer between [Ru(bpy)3]2+ and Ni-SOFs. Compared to those measured in the presence of other M-SOFs (M = Co, Cu, and Zn), the shorter lifetime observed with Ni-SOFs at the early stage suggests more efficient electron transfer (Fig. S17, ESI†).51 Moreover, electron paramagnetic resonance (EPR) spectroscopy was conducted (Fig. 3c). Upon irradiation, a sharp EPR signal ascribed to Ni+ is observed at g = 2.06,52 while it becomes significantly attenuated upon CO2 introduction. Considering that Ni2+ is EPR-silent, this observation indicates that Ni2+ sites can readily accept electrons from the excited photosensitizer, and subsequently transfer them to CO2 to initiate the reduction reaction. The CO2 reduction process was then be tracked by in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS). Fig. 3d clearly shows the vibration bands from intermediates within a 30-minute irradiation period. The two peaks at 1611 and 1552 cm−1 are characteristic of the *COOH intermediate, and their peak intensity increases as the irradiation continues.53 In the meantime, a pronounced *CO adsorption peak at 2163 cm−1 is also observed, indicating a high *CO surface coverage under irradiation.54
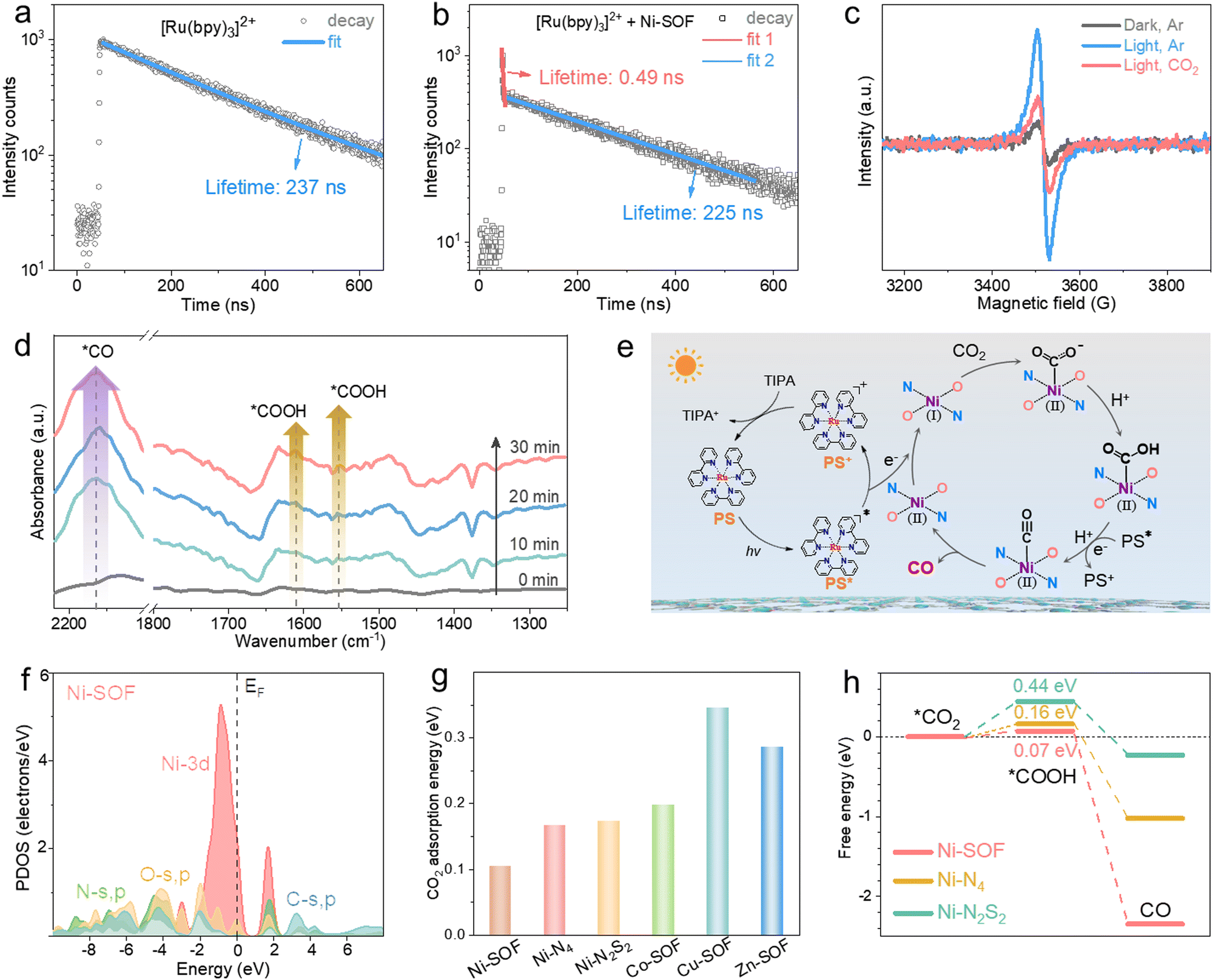 | ||
| Fig. 3 Charge transfer kinetics and possible reaction mechanism. (a) and (b) Time-resolved PL spectra of (a) [Ru(bpy)3]Cl2 and (b) [Ru(bpy)3]Cl2 + Ni-SOF in acetonitrile. (c) EPR spectra of Ni-SOFs under different conditions. (d) Operando DRIFTS spectra of Ni-SOFs under irradiation for different durations of time. (e) Schematic illustration of the proposed catalytic cycle. (f) PDOS spectra of Ni-SOFs. (g) CO2 adsorption energy on different catalysts. (h) Calculated free energy of CO2 photoreduction on Ni sites with different coordination environments. | ||
Based on the above spectroscopic results, a possible mechanism for CO2 photoreduction is proposed and illustrated in Fig. 3e. Upon visible light irradiation, [Ru(bpy)3]2+ (PS) is promoted to its excited state (PS*). Subsequently, photoexcited electrons are transferred to the Ni2+ sites of Ni-SOFs within a short time scale, generating catalytically active Ni+ centers. The oxidized photosensitizer (PS+) is then reduced back to its ground state by accepting electrons from TIPA. Electrons located on the Ni+ centers are further transferred to absorbed CO2, initiating the reduction reaction which involves the sequential formation of *COOH and *CO intermediates. At the end, *CO desorbs from the catalyst surface, thus stopping the reaction cycle.
To gain more insights into the catalytic mechanism, density functional theory (DFT) calculations were performed. The electronic distribution diagrams of Ni-SOFs, Ni–N4, and Ni–N2S2 near the Fermi level (EF) are depicted in Fig. S18 (ESI†). The maximum bonding orbital distribution is located closer to Ni in Ni-SOFs compared to those in Ni–N4 and Ni–N2S2, suggesting the electron-rich feature of Ni sites in Ni-SOFs. The electronic modulation of metal centers by the coordination environment is evident from the projected partial density of states (PDOS) analysis. As shown in Fig. 3f, the Ni-3d orbitals of Ni-SOFs exhibit a sharp peak near EF at Ev −0.88 eV (Ev = 0 eV). The s, p orbitals of O and N sites have good overlaps with the Ni-3d orbitals, indicating their strong binding interactions with central Ni atoms are necessary for the formation of a stable coordination configuration and efficient site-to-site electron transfer.55 By comparison, the metallic 3d orbitals of other catalysts (Ni–N4, Ni–N2S2, Co–N2O2, Cu–N2O2, and Zn–N2O2) are located at more negative positions related to their EF with obviously decreased overlaps with the s and p orbitals of surrounding heteroatoms (Fig. S19, ESI†). The d-band center and optical absorption comparisons also confirm the improved catalytic activity and optical properties of Ni-SOFs to promote the photocatalytic performances of CO2 reduction (Fig. S20, ESI†).
Furthermore, CO2 adsorption energies on different catalysts were calculated and compared (Fig. 3g). The Ni-SOF has the lowest adsorption energy, suggesting its strongest binding with CO2. This result is corroborated by the temperature programmed CO2 desorption (TPD) measurements showing that the Ni-SOF has the largest CO2 desorption capacity and highest desorption temperature (Fig. S21, ESI†). Favorable CO2 binding ability is believed to be conducive to CO2 capture and enrichment around the catalytic sites, thereby accelerating CO2 conversion.56 In addition, the free energy profiles of different catalysts during CO2 reduction were simulated. The conversion from *CO2 to *COOH is found to be the rate-determining step (RDS) for all the catalysts (Fig. 3h and Fig. S22, ESI†). Among them, the Ni-SOF shows the smallest energy barrier of 0.07 eV, indicating that the redox reaction on the Ni-SOF is the most favorable. Further conversion from *COOH to CO is exothermic and thereby spontaneous once *COOH is formed.
Encouraged by the great catalytic capacity and favorable CO2 binding of the Ni-SOF, we moved on to assess its catalytic performance at dilute CO2 concentrations. Fig. 4a schematically illustrates the local enrichment and subsequent photoreduction of diluted CO2 within the porous skeletons of Ni-SOFs. As demonstrated in Fig. 4b and Fig. S23 (ESI†), the Ni-SOF exhibits an exceptional catalytic performance at varying CO2 concentrations. Both the CO production rate and selectivity remain largely stable across a wide range of CO2 concentrations from 5 to 20 vol% in Ar. At a CO2 concentration as low as 2 vol%, the mass-specific CO production rate is measured to be 12100 μmol h−1 g−1, retaining 72% of the value achieved under pure CO2 as well as a high CO selectivity of 93%. Moreover, the AQE value at 10 vol% CO2 is 2.7% at 450 nm, which is very close to that obtained under pure CO2 (2.8%) (Fig. S24, ESI†). To the best of our knowledge, such high activity and selectivity have rarely been achieved before under dilute CO2 (Fig. 4c and Table S4, ESI†),37,43,57,58 exceeding those of other organic and inorganic candidates, even measured under pure CO2.34,42 The DRIFTS spectra of Ni-SOFs under pure and diluted (10 vol%) CO2 show two identical absorption bands at 2359 and 2341 cm−1, assigned to adsorbed CO2 with the end-on configuration (Fig. 4d).59 Importantly, their peak intensity and integrated areas are found to be largely independent of the CO2 concentration. This implies that Ni-SOFs can enrich local CO2 concentration when the feedstock becomes much diluted and may rationalize the exceptional photocatalytic activity measured under low-concentration CO2.
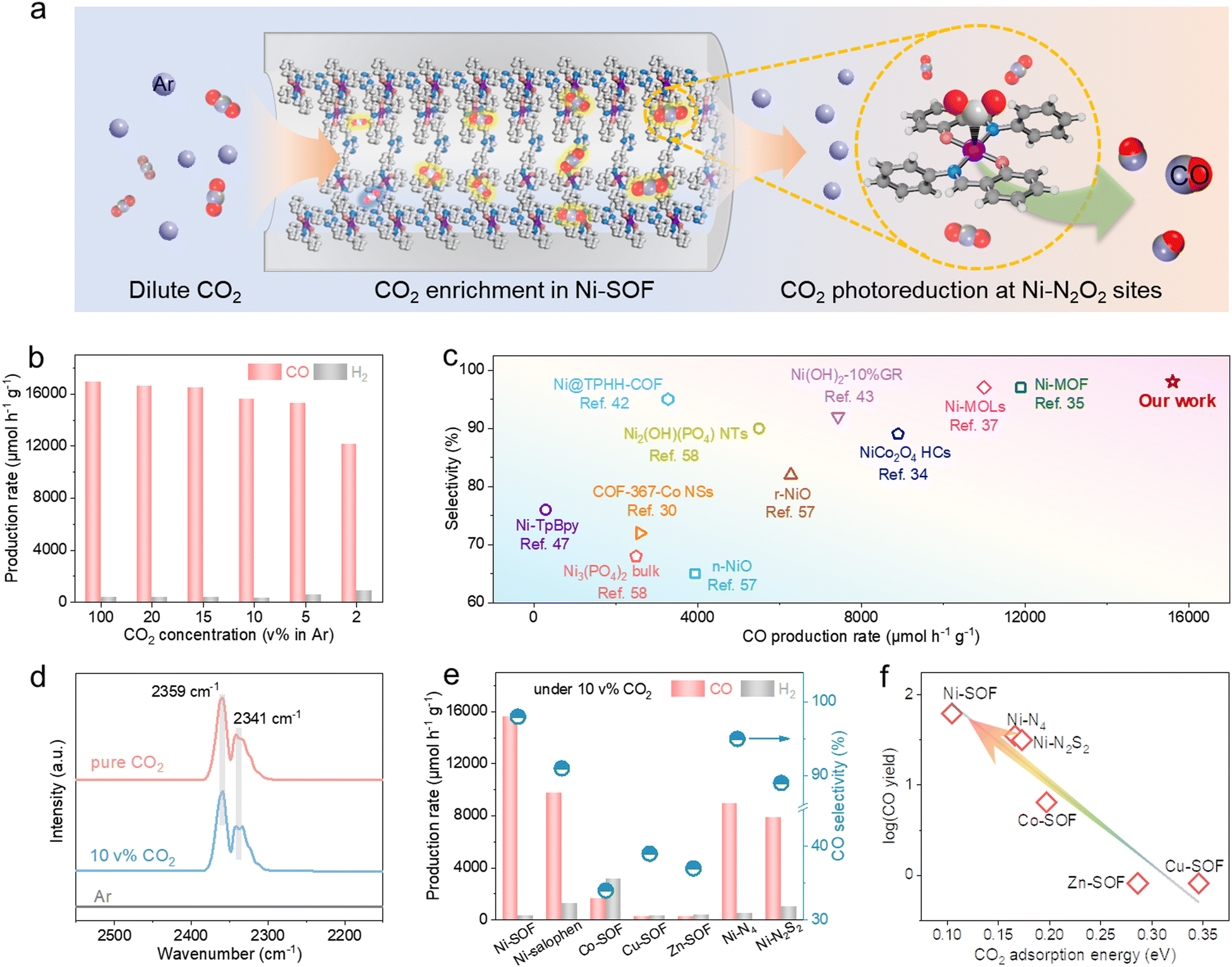 | ||
| Fig. 4 Performance evaluation of the catalysts under dilute CO2. (a) Schematic illustration of the enrichment and photoreduction of dilute CO2 in Ni-SOFs. (b) Photocatalytic activities of Ni-SOFs at various CO2 concentrations. (c) Performance comparison of Ni-SOFs with other organic or inorganic catalysts in terms of the CO production rate and selectivity under 10 vol% CO2 in Ar. (d) DRIFTS spectra of Ni-SOFs in pure Ar, pure CO2 or 10 vol% CO2 atmospheres. (e) CO production rate and selectivity of different catalysts under 10 vol% CO2. (f) Correlation between the logarithm of the CO production rate on different catalysts under 10 vol% CO2 and their CO2 adsorption energy. | ||
The great photocatalytic performance under low-concentration CO2 appears to be unique to Ni-SOFs. Ni–salophen presents considerably lower activity despite its similar coordination environment with Ni-SOFs (Fig. 4e). Replacing the central Ni with Co results in a decreased mass-specific activity of 2167 μmol h−1 g−1 (more than 4 times lower than that under pure CO2 on Co-SOFs) and low CO selectivity of 34%. More strikingly, CO2 photoreduction on Cu-SOFs and Zn-SOFs becomes almost quenched under the same conditions. Furthermore, both Ni–N4 and Ni–N2S2 exhibit about half of the activity of Ni-SOFs under 10 vol% CO2. We note that the measured activity here is roughly correlated with the CO2 binding energy predicted in Fig. 4f. The strongest binding affinity of Ni-SOF affords our catalyst with the most appealing activity by enriching CO2 local concentration and facilitating its subsequent conversion.
Conclusions
In summary, we synthesized a new class of M-SOF materials via facile metal-assisted Schiff-base reactions for efficient photocatalytic CO2 reduction. All the samples are composed of atomically dispersed metal sites and controlled local coordination environments, both of which could be precisely modulated via varying the predesigned organic moieties and metal species. The optimal sample, the Ni-SOF with Ni–N2O2 nodes, showed the highest CO production rate of 16908 μmol h−1 g−1 with an excellent selectivity up to 98%. Impressively, the Ni-SOF was able to maintain its photocatalytic performance across a wide range of CO2 concentrations (5–20 vol% in Ar) without reducing the selectivity. Experimental and theoretical studies revealed that the superior catalytic performance of Ni-SOFs mainly originated from the favorable d-band electronic modulation of the coordinated Ni atoms and optimal binding strength with the intermediates. In particular, the strong CO2 affinity of Ni-SOFs is identified as the key factor for the efficient conversion of diluted CO2. Our study demonstrates a promising strategy for rationalizing the chemical and electronic configurations of metal catalytic centers toward the direct utilization of diluted CO2 from industrial exhaust gas.
Author contributions
Y. L. and W. H. conceived the project and designed the experiments. X. Y. synthesized the catalysts and conducted the structure analysis and photocatalytic tests. B. H. and M. S. performed the theoretical calculations. T. Y. and L. Z. conducted the X-ray absorption measurements. L. J. carried out TEM characterization studies. M. C. performed TPD measurements. X. Y., M. S., W. H., B. H., and Y. L. co-wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support received from the National Key R&D Program of China (2021YFA1501101), the National Natural Science Foundation of China (22002100, U2002213, and 52161160331), the National Natural Science Foundation of China/Research Grant Council of Hong Kong Joint Research Scheme (N_PolyU502/21), the National Natural Science Foundation of China/Research Grants Council of Hong Kong Collaborative Research Scheme (CRS_PolyU504/22), the Natural Science Foundation of Jiangsu Province (BK20220027), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (20KJA430002), the Science and Technology Development Fund Macau SAR (0077/2021/A2), the Collaborative Innovation Center of Suzhou Nano Science and Technology, and the funding for Projects of Strategic Importance of The Hong Kong Polytechnic University (Project Code: 1-ZE2V), the Shenzhen Fundamental Research Scheme-General Program (JCYJ20220531090807017), the Natural Science Foundation of Guangdong Province (2023A1515012219) and Departmental General Research Fund of The Hong Kong Polytechnic University (Project Code: ZVUL). We thank the Shanghai Synchrotron Radiation Facility (beamline 11B) for the allocation of synchrotron beamtime, Dr Jie Xu for HAADF-STEM characterization and Chunpeng Wu for DRFITS measurements. B. H. is also grateful for the support provided by the Research Centre for Carbon-Strategic Catalysis (RC-CSC), the Research Institute for Smart Energy (RISE), and the Research Institute for Intelligent Wearable Systems (RI-IWEAR) of the Hong Kong Polytechnic University.Notes and references
- D. Campbell-Lendrum, T. Neville, C. Schweizer and M. Neira, Nat. Med., 2023, 29, 1631–1638 CrossRef CAS PubMed.
- C. J. Nielsen, H. Herrmann and C. Weller, Chem. Soc. Rev., 2012, 41, 6684–6704 RSC.
- J. Wu, Y. Huang, W. Ye and Y. Li, Adv. Sci., 2017, 4, 1700194 CrossRef PubMed.
- E. Gong, S. Ali, C. B. Hiragond, H. S. Kim, N. S. Powar, D. Kim, H. Kim and S.-I. In, Energy Environ. Sci., 2022, 15, 880–937 RSC.
- W. Huang, W. Luo and Y. Li, Mater. Today, 2020, 40, 160–172 CrossRef CAS.
- T. Nakajima, Y. Tamaki, K. Ueno, E. Kato, T. Nishikawa, K. Ohkubo, Y. Yamazaki, T. Morimoto and O. Ishitani, J. Am. Chem. Soc., 2016, 138, 13818–13821 CrossRef CAS PubMed.
- Y. Yamazaki, M. Miyaji and O. Ishitani, J. Am. Chem. Soc., 2022, 144, 6640–6660 CrossRef CAS PubMed.
- X. Wu, Y. Li, G. Zhang, H. Chen, J. Li, K. Wang, Y. Pan, Y. Zhao, Y. Sun and Y. Xie, J. Am. Chem. Soc., 2019, 141, 5267–5274 CrossRef CAS PubMed.
- S. Bierbaumer, M. Nattermann, L. Schulz, R. Zschoche, T. J. Erb, C. K. Winkler, M. Tinzl and S. M. Glueck, Chem. Rev., 2023, 123, 5702–5754 CrossRef CAS PubMed.
- A. John, M. M. Shaikh and P. Ghosh, Dalton Trans., 2008, 2815–2824 RSC.
- H. Huang, X. Jing, J. Deng, C. Meng and C. Duan, J. Am. Chem. Soc., 2023, 145, 2170–2182 CrossRef CAS PubMed.
- M. Vázquez-González, Z. Zhou, Y. Biniuri, B. Willner and I. Willner, Biochemistry, 2021, 60, 956–965 CrossRef PubMed.
- A. A. Shteinman, Catalysts, 2023, 13, 415 CrossRef CAS.
- Z. Wang, P. Yeary, X. Feng and W. Lin, J. Am. Chem. Soc., 2023, 145, 8647–8655 CAS.
- J. Zhang, L. Xu and W.-Y. Wong, Coord. Chem. Rev., 2018, 355, 180–198 CrossRef CAS.
- C. J. Whiteoak, G. Salassa and A. W. Kleij, Chem. Soc. Rev., 2012, 41, 622–631 RSC.
- S. Signorella, C. Palopoli and G. Ledesma, Coord. Chem. Rev., 2018, 365, 75–102 CrossRef CAS.
- C.-B. Li, Y. Chu, J. He, J. Xie, J. Liu, N. Wang and J. Tang, ChemCatChem, 2019, 11, 6324–6331 CrossRef CAS.
- V. Mouarrawis, R. Plessius, J. I. van der Vlugt and J. N. H. Reek, Front. Chem., 2018, 6, 623 CrossRef CAS PubMed.
- M. Lu, M. Zhang, J. Liu, Y. Chen, J.-P. Liao, M.-Y. Yang, Y.-P. Cai, S.-L. Li and Y.-Q. Lan, Angew. Chem., Int. Ed., 2022, 61, e202200003 CrossRef CAS PubMed.
- N. Zhao, K. Cai and H. He, Dalton Trans., 2020, 49, 11467–11479 RSC.
- H. Zhang, Z. Lin, P. Kidkhunthod and J. Guo, Angew. Chem., Int. Ed., 2023, 62, e202217527 CrossRef CAS PubMed.
- L. Chen, L. Wang, Y. Wan, Y. Zhang, Z. Qi, X. Wu and H. Xu, Adv. Mater., 2020, 32, 1904433 CrossRef CAS PubMed.
- S. Rai, A. Bajpai and S. Lokhandwala, J. Polym., 2013, 2013, 278576 Search PubMed.
- P. Thangasamy, S. Shanmuganathan and V. Subramanian, Nanoscale Adv., 2020, 2, 2073–2079 RSC.
- J. Wang, W. Zhu, F. Meng, G. Bai, Q. Zhang and X. Lan, ACS Catal., 2023, 13, 4316–4329 CrossRef CAS.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 CrossRef CAS.
- G. Yang, S. Li, N. Li, P. Zhang, C. Su, L. Gong, B. Chen, C. Qu, D. Qi, T. Wang and J. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202205585 CrossRef CAS PubMed.
- W. Liu, X. Li, C. Wang, H. Pan, W. Liu, K. Wang, Q. Zeng, R. Wang and J. Jiang, J. Am. Chem. Soc., 2019, 141, 17431–17440 CrossRef CAS PubMed.
- X.-K. Wang, J. Liu, L. Zhang, L.-Z. Dong, S.-L. Li, Y.-H. Kan, D.-S. Li and Y.-Q. Lan, ACS Catal., 2019, 9, 1726–1732 CrossRef CAS.
- H.-L. Zheng, S.-L. Huang, M.-B. Luo, Q. Wei, E.-X. Chen, L. He and Q. Lin, Angew. Chem., Int. Ed., 2020, 59, 23588–23592 CrossRef CAS PubMed.
- W. Chen, B. Han, C. Tian, X. Liu, S. Liang, H. Deng and Z. Lin, Appl. Catal., B, 2019, 244, 996–1003 CrossRef CAS.
- B. Han, J. Song, S. Liang, W. Chen, H. Deng, X. Ou, Y.-J. Xu and Z. Lin, Appl. Catal., B, 2020, 260, 118208 CrossRef CAS.
- B. Han, X. Ou, Z. Zhong, S. Liang, X. Yan, H. Deng and Z. Lin, Appl. Catal., B, 2021, 283, 119594 CrossRef CAS.
- K. Song, S. Liang, X. Zhong, M. Wang, X. Mo, X. Lei and Z. Lin, Appl. Catal., B, 2022, 309, 121232 CrossRef CAS.
- B. Han, X. Ou, Z. Deng, Y. Song, C. Tian, H. Deng, Y.-J. Xu and Z. Lin, Angew. Chem., Int. Ed., 2018, 57, 16811–16815 CrossRef CAS PubMed.
- L. Li, X. Dai, D.-L. Chen, Y. Zeng, Y. Hu and X. W. Lou, Angew. Chem., Int. Ed., 2022, 61, e202205839 CrossRef CAS PubMed.
- L. Tan, S.-M. Xu, Z. Wang, Y. Xu, X. Wang, X. Hao, S. Bai, C. Ning, Y. Wang, W. Zhang, Y. K. Jo, S.-J. Hwang, X. Cao, X. Zheng, H. Yan, Y. Zhao, H. Duan and Y.-F. Song, Angew. Chem., Int. Ed., 2019, 58, 11860–11867 CrossRef CAS PubMed.
- C. Gao, Q. Meng, K. Zhao, H. Yin, D. Wang, J. Guo, S. Zhao, L. Chang, M. He, Q. Li, H. Zhao, X. Huang, Y. Gao and Z. Tang, Adv. Mater., 2016, 28, 6485–6490 CrossRef CAS PubMed.
- S. Wang, Z. Ding and X. Wang, Chem. Commun., 2015, 51, 1517–1519 RSC.
- M. Dong, J. Zhou, J. Zhong, H.-T. Li, C.-Y. Sun, Y.-D. Han, J.-N. Kou, Z.-H. Kang, X.-L. Wang and Z.-M. Su, Adv. Funct. Mater., 2022, 32, 2110136 CrossRef CAS.
- K.-Q. Lu, Y.-H. Li, F. Zhang, M.-Y. Qi, X. Chen, Z.-R. Tang, Y. M. A. Yamada, M. Anpo, M. Conte and Y.-J. Xu, Nat. Commun., 2020, 11, 5181 CrossRef CAS PubMed.
- J.-W. Wang, L.-Z. Qiao, H.-D. Nie, H.-H. Huang, Y. Li, S. Yao, M. Liu, Z.-M. Zhang, Z.-H. Kang and T.-B. Lu, Nat. Commun., 2021, 12, 813 CrossRef CAS PubMed.
- H.-X. Zhang, Q.-L. Hong, J. Li, F. Wang, X. Huang, S. Chen, W. Tu, D. Yu, R. Xu, T. Zhou and J. Zhang, Angew. Chem., Int. Ed., 2019, 58, 11752–11756 CrossRef CAS PubMed.
- K. Niu, Y. Xu, H. Wang, R. Ye, H. L. Xin, F. Lin, C. Tian, Y. Lum, K. C. Bustillo, M. M. Doeff, M. T. M. Koper, J. Ager, R. Xu and H. Zheng, Sci. Adv., 2017, 3, e1700921 CrossRef PubMed.
- W. Zhong, R. Sa, L. Li, Y. He, L. Li, J. Bi, Z. Zhuang, Y. Yu and Z. Zou, J. Am. Chem. Soc., 2019, 141, 7615–7621 CrossRef CAS PubMed.
- W. Zhou, Q.-W. Deng, G.-Q. Ren, L. Sun, L. Yang, Y.-M. Li, D. Zhai, Y.-H. Zhou and W.-Q. Deng, Nat. Commun., 2020, 11, 4481 CrossRef CAS PubMed.
- J. Van Houten and R. J. Watts, J. Am. Chem. Soc., 1976, 98, 4853–4858 CrossRef CAS.
- Y. Su, Z. Song, W. Zhu, Q. Mu, X. Yuan, Y. Lian, H. Cheng, Z. Deng, M. Chen, W. Yin and Y. Peng, ACS Catal., 2021, 11, 345–354 CrossRef CAS.
- C. Gao, S. Chen, Y. Wang, J. Wang, X. Zheng, J. Zhu, L. Song, W. Zhang and Y. Xiong, Adv. Mater., 2018, 30, 1704624 CrossRef PubMed.
- M. Hartmann, N. Azuma and L. Kevan, J. Phys. Chem. C, 1995, 99, 10988–10994 CrossRef CAS.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- G. Qian, W. Lyu, X. Zhao, J. Zhou, R. Fang, F. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202210576 CrossRef CAS PubMed.
- S. Jiao, X. Fu and H. Huang, Adv. Funct. Mater., 2022, 32, 2107651 CrossRef CAS.
- Y. Wang, N.-Y. Huang, J.-Q. Shen, P.-Q. Liao, X.-M. Chen and J.-P. Zhang, J. Am. Chem. Soc., 2018, 140, 38–41 CrossRef CAS PubMed.
- W. Chen, X. Liu, B. Han, S. Liang, H. Deng and Z. Lin, Nano Res., 2021, 14, 730–737 CrossRef CAS.
- S. Liang, X. Liu, Z. Zhong, B. Han, X. Zhong, W. Chen, K. Song, H. Deng and Z. Lin, Nano Res., 2021, 14, 2558–2567 CrossRef CAS.
- P. D. C. Dietzel, R. E. Johnsen, H. Fjellvag, S. Bordiga, E. Groppo, S. Chavan and R. Blom, Chem. Commun., 2008, 5125–5127 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee04121b |
| ‡ These two authors contribute equally. |
| This journal is © The Royal Society of Chemistry 2024 |