
Engineering a molecular electrocatalytic system for energy-efficient ammonia production from wastewater nitrate†
Dean M.
Miller
 a,
Matthew J.
Liu
a,
Kristen
Abels
a,
Anna
Kogler
b,
Kindle S.
Williams
a and
William A.
Tarpeh
*abc
a,
Matthew J.
Liu
a,
Kristen
Abels
a,
Anna
Kogler
b,
Kindle S.
Williams
a and
William A.
Tarpeh
*abc
aDepartment of Chemical Engineering, Stanford University, Stanford, CA 94305, USA. E-mail: wtarpeh@stanford.edu; Tel: +1-650-497-1324
bDepartment of Civil and Environmental Engineering, Stanford University, Stanford, CA 94305, USA
cWoods Institute for the Environment, Stanford University, Stanford, California 94305, USA
First published on 24th June 2024
Abstract
Anthropogenic ammonia production has sustained exponential population growth but exacerbated wastewater nitrate pollution. Abundant nitrate can be refined to purified nitrogenous chemicals through the electrochemical nitrate reduction reaction (NO3RR). However, the dilute and impure composition of nitrate-bearing wastewaters presents barriers to practical electrocatalytic systems. We address these barriers in our investigation of the model ammonia-selective homogeneous molecular NO3RR catalyst Co(DIM) in real wastewater and reactive separations architectures. In this work, we elucidate catalysis inhibition mechanisms imposed by magnesium in real wastewaters that decrease nitrate conversion activity by 62.0%. These mechanisms informed our design of electrocatalyst-in-a-box (ECaB), a novel NO3RR reactive separation that exhibits the lowest reported energy consumption for purified wastewater-derived ammonia production (90.0 ± 2.7 kW h kg N−1). Engineering ECaB's subunit processes enhanced the rate of ammonia production by 20.4×. This work demonstrates a use-informed engineering approach that iterates between mechanistic insights and unit process-level performance of electrochemical wastewater refining systems in complex aqueous streams.
Broader contextWastewater refining generates valuable products from aqueous waste streams. While promising for chemical manufacturing, wastewaters are dilute and impure. These challenges have generally precluded the use of molecular catalysts for wastewater treatment. We evaluated the potential for a cobalt-centered molecular catalyst to convert wastewater nitrate into high-purity ammonia by systematically identifying wastewater constituents that interfered with catalytic activity. With these insights, we designed a novel electrochemical reactive separation process, electrocatalyst-in-a-box (ECaB), that achieved direct treatment of real municipal wastewater, recovery of purified ammonium sulfate, and reuse of homogeneous catalysts. This study advances energy-efficient distributed ammonia production, and provides a tunable platform for molecular electrocatalysts to interface with wastewater treatment. |
1. Introduction
Circular wastewater refining processes can responsibly manage aqueous waste streams and sustainably produce chemical commodities, including nitrogen compounds. Conventionally, nitrogenous commodities are produced from ammonia-nitrogen made using the Haber–Bosch (HB) process; this process is highly energy-efficient but contributes 1–2% of global carbon dioxide emissions.1 The majority of HB-nitrogen is used as fertilizer,2 but 24–54% of HB-nitrogen is discharged from anthropogenic processes as aqueous nitrate (NO3−) and ammonium (NH4+).3 Excess reactive nitrogen species in surface waters cause algal blooms that damage aquatic ecosystems and exacerbate climate change.4–6 Refining these fugitive nitrogen emissions into a tunable, diverse portfolio of products could simultaneously remediate harmful pollutants and generate 19 billion USD annually.3 The electrocatalytic NO3− reduction reaction (NO3RR) to ammonia can refine nitrate-rich wastewaters that contain 19–48 Tg NO3−–N globally per year.3 Electrified NO3RR processes will readily integrate with renewable and decentralized energy sources, which could enable ammonia production at sites of wastewater generation. This circular NO3− refining paradigm can improve sanitation access, expand nitrogen commodity access, and offset costs and emissions of industrial ammonia production.Despite the large overall flux of anthropogenic nitrogen discharges into the environment, most NO3− is contributed by dilute and impure wastewater sources.7 Both point sources (e.g., municipal wastewater) and nonpoint sources (e.g., agricultural runoff) generally contain less than 50 mg NO3−–N L−1 (3.6 mM).8–13 Municipal and agricultural wastewaters are also impure; they contain co-constituents (e.g., organic/inorganic ions, organic carbon, suspended solids) that compete for catalyst active sites, degrade catalysts over time, passivate electrode surfaces, and alter solution pH outside of catalyst operating ranges.14–16 In contrast to real wastewaters, NO3RR electrocatalysts are most heavily documented with NO3− concentrations above 100 mM (i.e., concentrated) in synthetic (i.e., pure) electrolytes.14,17–26 NO3− refining research must overcome the difference in the NO3− concentration and purity between catalysis investigations and real wastewaters to be relevant for wastewater treatment and chemical manufacturing.
Homogeneous molecular catalysts are promising for wastewater NO3− refining because their atomically precise reactivity could overcome both dilute and impure wastewater conditions;27 however, these catalysts are rarely explored for wastewater treatment because they require separation. The ligand structures of molecular catalysts promote high reactant and product selectivities that could enhance NO3RR faradaic efficiency (FE) and reaction rates to total ammonia nitrogen (TAN; the sum of NH4+–N(aq) and NH3–N(aq)) in dilute NO3− solutions. The metal complex 2,3-dimethyl-1,4,8,11-tetraazacyclotetradeca-1,3-diene (DIM) with a Co metal center, abbreviated as Co(DIM), yields TAN as its major product and suppresses the competitive hydrogen evolution reaction (HER) under aqueous conditions from pH 3.5 to 10.1.28 Co(DIM) is easily synthesized29 and exemplifies untapped benefits to employing molecular electrocatalysts for wastewater refining. The positively charged metal center of Co(DIM) facilitates binding of negatively-charged NO3−.28 Additionally, homogeneous electrocatalytic processes directly scale with volume30 (e.g., Fenton catalysts,31 porphyrins for PFAS remediation32) and can use economical carbonaceous electrode materials. Rational design principles for molecular electrocatalyst design, operation, and separation in wastewater refining processes are currently ill-defined because there are few use-informed investigations of homogeneous catalysis in real wastewaters contexts.
Likewise, rational design of wastewater refining unit processes is hampered by a dearth of electrochemical investigations that integrate reactions and separations, or reactive separations.3,33 Consequently, unit process performance (namely efficiencies, rates, and energy consumption) is difficult to benchmark against incumbent wastewater management processes. Reactive separations can overcome the challenges of dilute and impure wastewaters to enable NO3− extraction, NO3− conversion to TAN, and purified TAN recovery.3,33 However, there are few investigations of reactive separations in real wastewaters,34–36 and even fewer in dilute wastewaters.37 Our recent work demonstrated that homogeneous electrocatalysis in existing NO3RR reactive separation architectures can produce purified TAN ((NH4)2SO4(aq)).38 Elevating this preliminary demonstration to rationally-designed unit processes requires investigation of interfacial phenomena imposed by catalytically influential wastewater species.16,33
To address the rational design challenges imposed by dilute and impure NO3−, we investigated interfacial mechanisms that govern Co(DIM)-mediated NO3RR reactive separations performance in real wastewaters, and leveraged these insights toward engineering novel molecular catalysis systems. The major contribution of this work was to develop and interrogate molecular catalysis systems capable of converting wastewater nitrate into high-purity ammonia; which was enabled by several key advances. First, investigating catalysis (via electrolysis, amperometry, and spectroscopy) in a systematic suite of simulated and real wastewater electrolytes elucidated the mechanistic influences of inorganic ions on reactor performance. More specifically, we investigated the effect of bulk and interfacial ionic composition on activity and FE, and found that magnesium ions play a deterministic role in electrode fouling. Second, these mechanistic insights enabled rational design of a novel reactive separations unit process, electrocatalyst-in-a-box (ECaB). Our first ECaB iteration (proof-of-concept ECaB) exhibited sustained activity, high FE, and low energy consumption for several cycles to treat a real, NO3−-bearing wastewater: municipal secondary effluent. Third, proof-of-concept ECaB enabled long-term evaluation and benchmarking of the full unit process and its subunit processes against related NO3RR efforts and conventional nitrogen management (wastewater treatment and HB-ammonia production). Proof-of-concept ECaB also generated a high-purity TAN product with the lowest reported energy consumption (90.0 ± 2.7 kW h kg N−1) for any NO3RR reactive separation process to date. In a second, improved iteration (subunit engineered ECaB), we leveraged reactor design to improve subunit process rates toward scalable performance targets. Ultimately, our findings span from the microenvironment to the unit process scale and contribute to the informed design of catalysts, electrode and membrane interfaces, and electrochemical reactors. This study demonstrates a novel use-informed, iterative approach to wastewater electrocatalysis that guides the development of efficient and practical reactors for pollution remediation and circular chemical manufacturing.
2. Results & discussion
Because the primary NO3−-rich wastewaters are municipal secondary effluent and fertilizer runoff, NO3RR processes must operate with feed compositions of dilute NO3− (<4 mM), low conductivity (<2 mS cm−1), and water hardness (i.e., presence of divalent metal cations).37,38 We collected and conducted experiments in a representative secondary effluent (Table S1, ESI†) containing 120 mg NO3− L−1 (28 mg NO3−–N per L, 2.0 mM NO3−) and a matrix of anions, alkaline earth metal cations, suspended solids, and organic and inorganic carbon. Within this complex composition, we sought to uncover the promoting or inhibiting effects of specific wastewater constituents on Co(DIM)-mediated NO3RR (Section 2.1). Toward this goal, we assessed the NO3RR performance as a function of electrolyte composition (Section 2.1.1) and described generalizable interfacial mechanisms that influence Co(DIM)-mediated NO3RR behavior (Section 2.1.2). These insights informed the design and development of the novel reactive separations process ECaB (Section 2.2). Proof-of-concept ECaB (Section 2.2.1) demonstrated robust performance with real wastewater and enabled systematic comparisons to previous NO3RR investigations (Section 2.2.2). Finally, subunit engineered ECaB demonstrated improved subunit process rates toward tractable performance targets (Section 2.2.3).2.1. Effects of electrolyte composition on Co(DIM)-mediated NO3RR
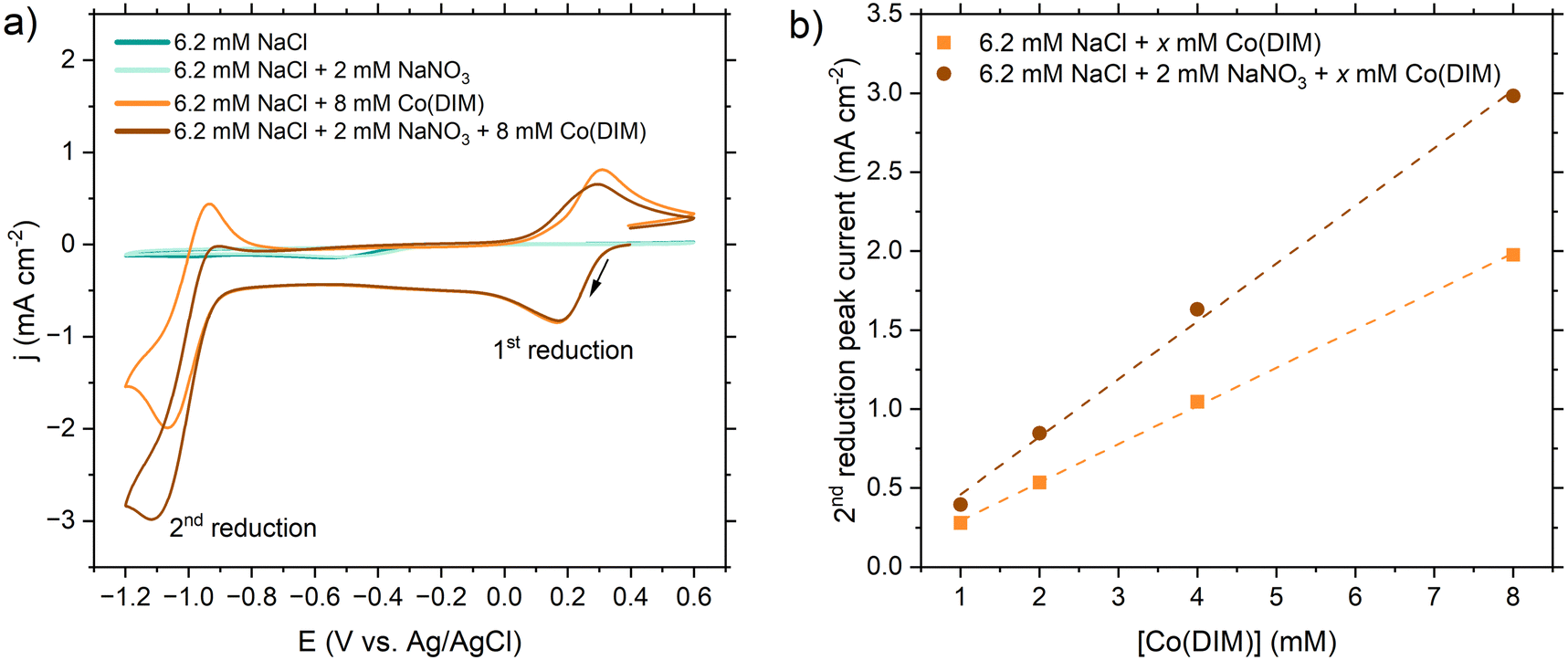 | ||
| Fig. 1 (a) Cyclic voltammograms collected in 6.2 mM NaCl (background electrolyte matching the Cl− concentration in real wastewater), 6.2 mM NaCl with 2 mM NaNO3 (background electrolyte with representative nitrate concentration), 6.2 mM NaCl with 8 mM Co(DIM) (background electrolyte with catalyst), and 6.2 mM NaCl with 2 mM NaNO3 and 8 mM Co(DIM) (background electrolyte with catalyst and representative nitrate concentrations). Working electrode: 5 mm GC disk. Counter electrode: 6.4 mm graphite rod. Reference electrode: Ag/AgCl (4.0 M KCl). Scan rate: 100 mV s−1. (b) Peak current density of the second reduction peak (∼−1.1 V vs. Ag/AgCl) of 6.2 mM NaCl with 8 mM Co(DIM) in the presence and absence of 2 mM NaNO3. Associated voltammograms are provided in Fig. S4 (ESI†). The excess factor, defined as the concentration of substrate (NO3−) divided by the concentration of catalyst (Co(DIM)), ranged from 2 to 200 previously. To address dilute real wastewaters, we minimized the excess factor with 8 mM Co(DIM) (the observed solubility limit at room temperature) to maximize NO3RR activity (excess factor = 0.25, Fig. 1b). | ||
 | ||
| Fig. 2 (a) Mass balance of aqueous nitrogen species as a function of time during two-chamber controlled-potential electrolysis (CPE) experiments at −1.05 V vs. Ag/AgCl performed in three electrolytes: 8 mM Co(DIM) in simplified wastewater, 8 mM Co(DIM) in simulated wastewater, and 8 mM Co(DIM) in real wastewater. Error bars represent ± one standard deviation from triplicate experiments (n = 3). For all three electrolytes, NO3−–N and TAN closed the nitrogen mass balance within ±3%. No TAN was detected after CPE without NO3− (Fig. S6, ESI†), confirming that the measured TAN was the result of the NO3RR. The closed mass balance and catalytic TAN formation support for NO3RR selectivity to TAN remained near 100% in all three electrolytes tested, consistent with previous investigations.28,38 (b) Cumulative TAN selectivity, TAN faradaic efficiency, and NO3− conversion for 8 hour CPE experiments. (c) Mg 1s XPS spectra of the glassy carbon plate cathode after CPE experiments. After CPE the electrode was rinsed with nanopure water and blow dried with N2 before XPS and SEM-EDS analysis. (d) SEM image and (e) Mg EDS map of the glassy carbon plate cathode after a CPE experiment in real wastewater, showing dispersed deposits of Mg on the surface. | ||
Whereas NO3RR activity was strongly influenced by wastewater impurities, the relatively low FETAN was primarily influenced by NO3− concentration. Under purely kinetic conditions,39 Co(DIM)-mediated NO3RR is first order in [Co(DIM)] and [NO3−], motivating the high catalyst concentrations employed in CVs and CPEs. However, purely kinetic conditions do not apply in our CPE experiments because the low excess factor imposed by the wastewater caused substrate consumption in the RDL, and consequently a significant amount of current was consumed by non-catalytic reductions of Co(DIM). Several control CPE experiments were performed to quantify non-catalytic charge allocation (i.e., FE). CPE at −0.75 VAg/AgCl (−0.99 V vs. first reduction, +0.25 V vs. second reduction) in simplified wastewater (6.2 mM NaCl + 2 mM NaNO3) served as a proxy for non-catalytic charge toward the first reduction of Co(DIM); CPE at −1.05 VAg/AgCl in a modified simplified wastewater (6.2 mM NaCl + 0 mM NaNO3) served as a proxy for non-catalytic charge toward the first and second reductions. The sum of charge associated with non-catalytic reductions of Co(DIM) accounts for 89.4% of charge passed in the simplified wastewater CPEs (Fig. S6 and S7, ESI†), reasonably closing the balance of FE. Notably, the non-catalytic charge passed (49.3% FE) exceeded the catalytic charge passed (41.4 ± 2.4% FE). The excess factor therefore significantly influences the ratio of activated Co(DIM) molecules that successfully perform NO3RR in the homogeneous phase.
Because only NO3− conversion was significantly changed as a function of electrolyte composition, we hypothesized that electrode fouling in simulated and real wastewater reduced the observed NO3RR rate by impeding Co(DIM) activation at the heterogeneous electrode–electrolyte interface. Visible deposits were present on the electrode after 8 hours for all three electrolytes (Fig. S8, ESI†), prompting spectroscopic and electrochemical characterization of the GC surface. Based on simulated wastewater exhibiting electrode fouling, we expected that hardness (e.g., Mg2+, Ca2+) and HCO3−/CO32− ions were precipitate components. XPS (Fig. 2c) and SEM/EDS (Fig. 2d and e) revealed magnesium on the GC surface for real and simulated wastewater. The pH required for precipitation of Mg(OH)2 from 1.5 mM Mg2+ is 9.8 (Table S2, ESI†). Because the bulk pH of all three catholytes remained below 9.8 (Fig. S9, ESI†), the interfacial pH in the RDL likely exceeded pH 9.8, which is common under reducing electrochemical conditions.40 No significant precipitation of Ca2+ was observed by EDS after real wastewater CPE (Fig. S10 and S11, ESI†), implying that the interface remained below pH 12.8 which is required to precipitate Ca(OH)2 from 1.9 mM Ca2+. We also compared the catalytic activity and charge transfer resistance of the deposits formed on GC cathodes in simplified and real wastewaters. In rinse tests, both deposits were less active than homogeneous Co(DIM) and were not selective for TAN or NO2− (Fig. S12 and S13, ESI†). Thus, we did not consider the Mg-containing deposits to be catalytic for NO3RR. The charge transfer resistance between the electrode surface and homogeneous Co(DIM) at the open circuit potential (OCP) was greater for the real deposit (Rct = 310 kΩ) than for the simplified deposit (Rct = 189.4 kΩ) and the pristine GC surface (Rct = 132.7 kΩ) (Fig. S14, ESI†). In conjunction with the insensitivity of TAN FE and selectivity to electrolyte composition (Fig. 2b), greater charge transfer resistance suggested that catalysis was heterogeneously inhibited by Mg deposition and that the homogeneous Co(DIM)-mediated NO3RR mechanism was unaffected by real wastewater impurities.
 | ||
| Fig. 3 (a) Current density vs. time of RDE CPEs −1.05 VAg/AgCl with suspected foulants manually dosed into the electrolyte at t = 10 min (no contaminant dose, 28 μL 1 M MgCl2, 30 μL 1 M NaHCO3, 38 μL 1 M CaCl2). Two experiment conditions were compared: the first experiment held −1.05 VAg/AgCl for the full 50 minutes (green trace), and the second experiment introduced a period of open circuit potential (OCP) from t = 20 min to t = 30 min. The counter electrode was a 6.4 mm graphite rod and the reference electrode was a Ag/AgCl (4.0 M KCl) electrode. The RDE was operated at 25 rotations per minute (RPM) because both catalytic current (for simplified wastewater) and inhibition (for real wastewater) were observed at this rotation rate (Section S4.1 and Fig. S15, ESI†). (b) Mg 1s XPS of the glassy carbon RDE electrode post Mg2+ dose CPE for both the uninterrupted CPE and the CPE with OCP period. | ||
Magnesium precipitation during the Co(DIM)-mediated electrocatalytic NO3RR is generalizable to reductive homogeneous electrocatalysis and to the NO3RR (heterogeneous or homogeneous) in real wastewaters. Similar precipitation processes have been observed in homogeneous CO2 reduction electrocatalysis,41 and water hardness (i.e., Mg2+, Ca2+) has been shown to decrease heterogeneous NO3− removal.16 In the secondary effluent used in this work (1.5 mM Mg2+), a negligible proportion of Mg2+ present was consumed to form a passivating cathode deposit (Fig. S22, ESI†). Other nitrate-rich wastewaters like polluted groundwaters, reverse osmosis brines, and ion exchange brines may contain higher Mg2+ concentrations (up to 84 mM),42 increasing the likelihood of forming precipitates. Mg2+ electrode fouling is likely a widespread barrier to implementation. A reactive separation system could conceivably perform uninhibited NO3RR directly in the native wastewater if divalent cations are repelled by near-surface confined Co(DIM) molecules. This insight motivates repulsive moieties at the electrode–electrolyte interface as a design principle for the microenvironment of homogeneous and heterogeneous catalyst systems. Electrode and catalyst design (e.g., heterogeneous molecular catalysts, single-atom catalysts, ionomer coatings) could protect the cathode from passivation, but would not address the low TAN FE induced by inherently low NO3− concentrations in real wastewaters. The remainder of this study addresses cathode protection and FETAN with reactive separations that selectively extract and up-concentrate NO3− (and reject Mg2+) from wastewater before catalysis.
2.2. Electrocatalyst-in-a-box (ECaB)
We mitigated cathode passivation in the NO3RR by pre-catalysis extraction of NO3− from wastewater via reactive separations. Extracting NO3− into a synthetic electrolyte for Co(DIM)-mediated NO3RR could be achieved by electrochemical separations such as electrodialysis,43 capacitive deionization,44 or electrosorption,37 but in low-conductivity NO3−-rich wastewaters, electrochemical extraction can dominate the total process energy consumption.33,37 This observation motivates low-energy extraction methods like Donnan membrane dialysis.45 Because Donnan equilibrium conditions are governed by electrochemical potentials (not concentrations) across the membrane, NO3− ions can diffuse against their concentration gradient by exchanging with high-activity receiver solution anions (e.g., Cl−). If the receiver solution volume is less than the feed solution (wastewater) volume, NO3− in the receiver solution can be substantially up-concentrated relative to the feed concentration. The resulting receiver solution is an ideal electrolyte for the NO3RR because of its high conductivity, high NO3− concentration, and absence of Mg2+. Donnan dialysis (DD) can also obviate separations challenges imposed by complex electrolyte compositions (e.g., high [Cl−], divalent ions, organics), like unselective ion extraction, high energy consumption, and high capital costs.45 In this section, we report a combined DD, NO3RR, and ammonia stripping system coined electrocatalyst-in-a-box (ECaB; Fig. 4a). | ||
| Fig. 4 (a) Electrocatalyst-in-a-box (ECaB) schematic. Wastewater–NO3− extraction (subunit process 1, orange) is achieved by Donnan dialysis (DD) in the extraction/recovery cell (left) where NO3− ions exchange with Cl− ions due to an electrochemical potential gradient across the anion exchange membrane (AEM). The catholyte recirculates between the extraction/recovery cell and the bipolar membrane (BPM)-separated electrocatalysis cell (right) where Co(DIM)-mediated NO3RR (subunit process 2, green) converts NO3− to TAN. As the catholyte basifies under reducing conditions, the majority of TAN exists as NH3 that is recovered (subunit process 3, blue) by diffusing across the gas-permeable membrane (GPM) into the acidic trap chamber. (b) NO3− and TAN concentrations and (c) nitrogen mass balance for electrocatalyst-in-a-box experiments. Error bars represent ± one standard deviation from triplicate experiments (n = 3). Detailed experimental parameters of ECaB tests can be found in the Methods section 4.2.3. Time periods where the electrochemical cell was held at open circuit (OCP; 0–24 h, 34–48 h, 58–72 h, 82–96 h) are indicated by a white background and time periods where the electrochemical cell was held at −1.05 VAg/AgCl (24–34 h, 48–58 h, 72–82 h) are indicated by a green background. Applied potentials were held for 10 h at a time to keep Co(DIM) in its stable operating pH regime.28,38 The dashed line in panel (b) indicates the drinking water limit of 10 mg NO3–N per L. | ||
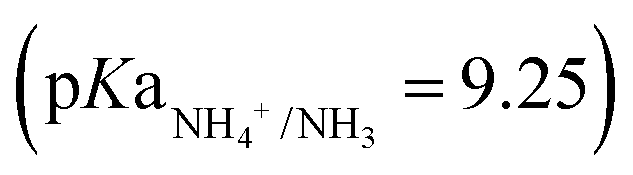 , which volatilized, crossed the GPM, and was recovered in the acid trap (0.1 M H2SO4). ECaB was recirculated overnight (34–48 h) at open circuit as the remaining TAN migrated across the GPM. At 48 h, the catholyte was adjusted back to pH 6 with ∼200 μL 10 wt% HCl to keep Co(DIM) in its stable operating pH regime28,38 before a second cycle of CPE (48–58 h) and OCP (58–72 h) began. ECaB experiments were performed for three cycles for a total of 96 h (30 cumulative hours of CPE) to demonstrate proof-of-concept. The total measured N masses (Fig. 4c) at 24 h, 48 h, 72 h, and 96 h accounted for 97.6 ± 1.4%, 88.7 ± 5.1%, 84.1 ± 2.7%, and 84.9 ± 1.8% of the influent wastewater NO3−–N. We hypothesize that binding of NH3 to Co(DIM) was the largest contributor to unclosed mass balances because the largest changes occurred during recirculation at OCP from 34–48 h and Co(DIM) in its Co(III) oxidation has a high NH3 binding capacity (at least 1.6 mM TAN/mM Co(DIM); Fig. S24, ESI†). Unlike many electrolysis approaches, the wastewater remained circumneutral for the duration of experiments (pH 7.07 ± 0.25 at t = 96 h, Fig. S23, ESI†), demonstrating potential for downstream water reuse. Combining membrane-based DD and ammonia stripping isolates the homogeneous Co(DIM) solution from the wastewater feed and the TAN product (Fig. S25, ESI†), enabling treated wastewater recovery, product TAN recovery, and homogeneous catalyst reuse. This catalyst reuse is similar to catalyst-in-a-cup48 (in which thermal homogenous catalysts are separated from solvents, reactants, and products in chemical production), inspiring the ECaB name.
, which volatilized, crossed the GPM, and was recovered in the acid trap (0.1 M H2SO4). ECaB was recirculated overnight (34–48 h) at open circuit as the remaining TAN migrated across the GPM. At 48 h, the catholyte was adjusted back to pH 6 with ∼200 μL 10 wt% HCl to keep Co(DIM) in its stable operating pH regime28,38 before a second cycle of CPE (48–58 h) and OCP (58–72 h) began. ECaB experiments were performed for three cycles for a total of 96 h (30 cumulative hours of CPE) to demonstrate proof-of-concept. The total measured N masses (Fig. 4c) at 24 h, 48 h, 72 h, and 96 h accounted for 97.6 ± 1.4%, 88.7 ± 5.1%, 84.1 ± 2.7%, and 84.9 ± 1.8% of the influent wastewater NO3−–N. We hypothesize that binding of NH3 to Co(DIM) was the largest contributor to unclosed mass balances because the largest changes occurred during recirculation at OCP from 34–48 h and Co(DIM) in its Co(III) oxidation has a high NH3 binding capacity (at least 1.6 mM TAN/mM Co(DIM); Fig. S24, ESI†). Unlike many electrolysis approaches, the wastewater remained circumneutral for the duration of experiments (pH 7.07 ± 0.25 at t = 96 h, Fig. S23, ESI†), demonstrating potential for downstream water reuse. Combining membrane-based DD and ammonia stripping isolates the homogeneous Co(DIM) solution from the wastewater feed and the TAN product (Fig. S25, ESI†), enabling treated wastewater recovery, product TAN recovery, and homogeneous catalyst reuse. This catalyst reuse is similar to catalyst-in-a-cup48 (in which thermal homogenous catalysts are separated from solvents, reactants, and products in chemical production), inspiring the ECaB name.
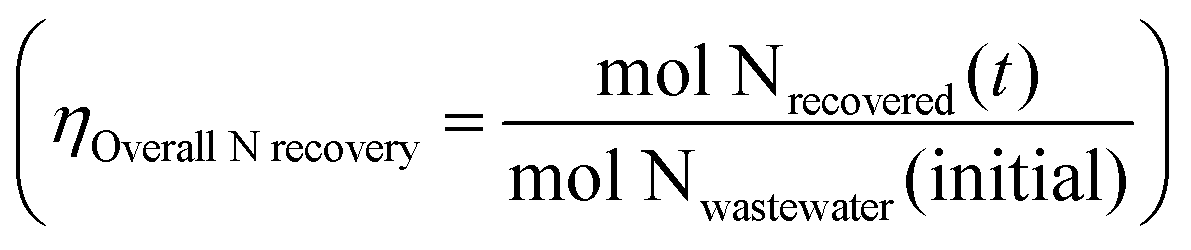 is the product of NO3− extraction efficiency, TAN yield, and TAN recovery efficiency (Section S5.1 and Fig. S26, ESI†). For ECaB, 98.1 ± 0.3% of wastewater NO3− was extracted by the end of the 96 h experiments, with 81.3 ± 3.3% extracted in the first 24 hours (Fig. 5a). 60.8 ± 1.1% of extracted NO3− was converted via Co(DIM)-mediated NO3RR and 91.0 ± 2.1% of the produced TAN was recovered in the trap. Overall, 42.6 ± 1.4% of influent wastewater NO3−–N was recovered as TAN. Proof-of-concept ECaB prevented cathode Mg fouling and enabled sustained NO3− conversion via selective NO3− extraction (Fig. S27–S29, ESI†). Despite having a more complex composition, ECaB with real wastewater outperformed CPE in simplified wastewater in terms of FETAN (Fig. 5b) and NO3− conversion rate (Fig. 5c). Additionally, Mg2+ was not detected in the catholyte nor on the GC surface by XPS (Fig. S30, ESI†) or EDS (Fig. S31, ESI†), reinforcing that Co(DIM)-mediated NO3RR activity in ECaB was not inhibited by cathode passivation. ECaB exemplifies a reactive separation process for treating real wastewater, and outperforms a simplified (i.e., ideal) system driven by insights into wastewater-induced inhibition.
is the product of NO3− extraction efficiency, TAN yield, and TAN recovery efficiency (Section S5.1 and Fig. S26, ESI†). For ECaB, 98.1 ± 0.3% of wastewater NO3− was extracted by the end of the 96 h experiments, with 81.3 ± 3.3% extracted in the first 24 hours (Fig. 5a). 60.8 ± 1.1% of extracted NO3− was converted via Co(DIM)-mediated NO3RR and 91.0 ± 2.1% of the produced TAN was recovered in the trap. Overall, 42.6 ± 1.4% of influent wastewater NO3−–N was recovered as TAN. Proof-of-concept ECaB prevented cathode Mg fouling and enabled sustained NO3− conversion via selective NO3− extraction (Fig. S27–S29, ESI†). Despite having a more complex composition, ECaB with real wastewater outperformed CPE in simplified wastewater in terms of FETAN (Fig. 5b) and NO3− conversion rate (Fig. 5c). Additionally, Mg2+ was not detected in the catholyte nor on the GC surface by XPS (Fig. S30, ESI†) or EDS (Fig. S31, ESI†), reinforcing that Co(DIM)-mediated NO3RR activity in ECaB was not inhibited by cathode passivation. ECaB exemplifies a reactive separation process for treating real wastewater, and outperforms a simplified (i.e., ideal) system driven by insights into wastewater-induced inhibition.
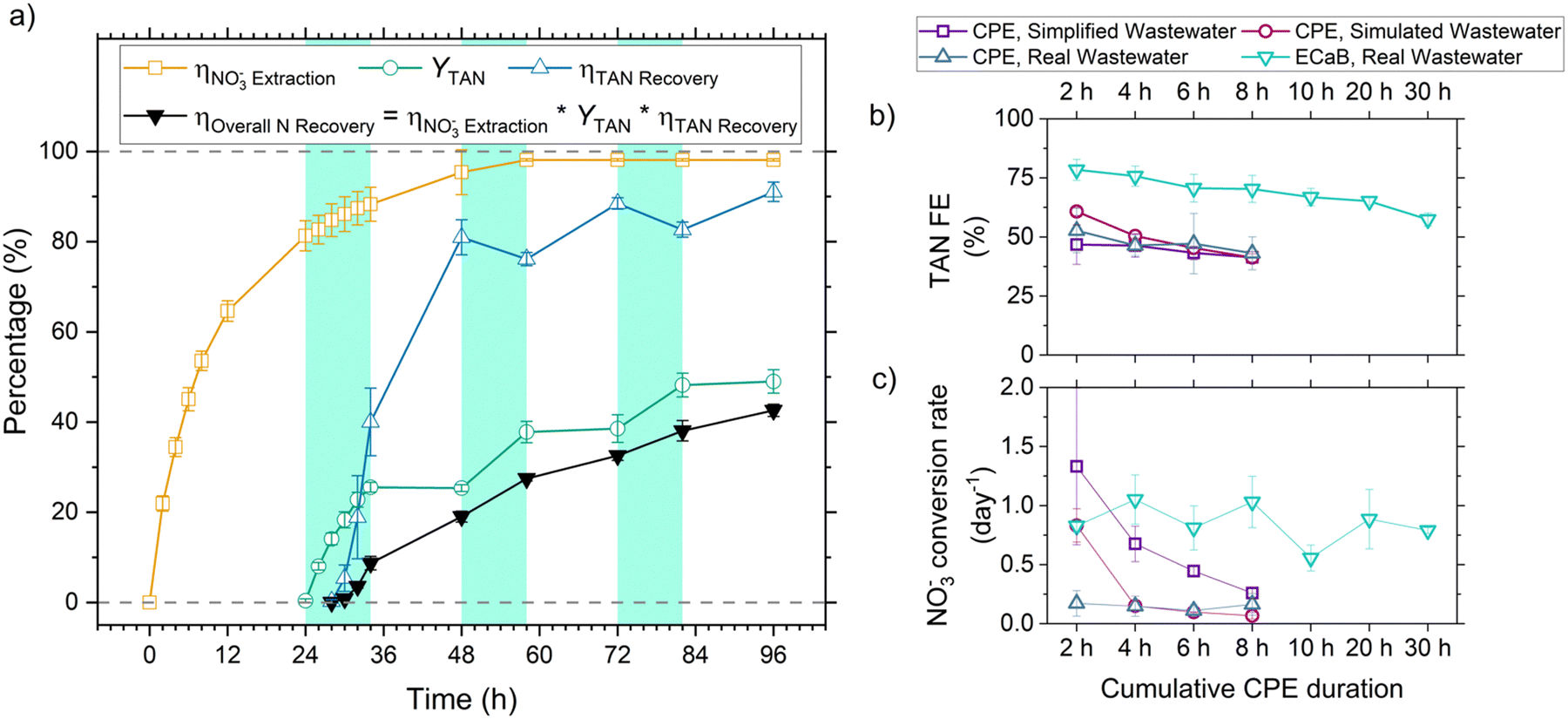 | ||
Fig. 5 (a) Cumulative NO3− extraction efficiency 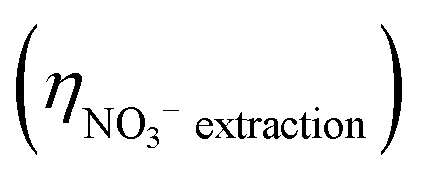 , TAN yield (YTAN), TAN recovery efficiency (ηTAN , TAN yield (YTAN), TAN recovery efficiency (ηTAN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) recovery), and overall nitrogen recovery efficiency (ηOverallNrecovery) during ECaB experiments. Error bars represent ± one standard deviation from triplicate experiments (n = 3). Detailed experimental parameters of ECaB tests can be found in the Materials & methods section 4.2.3. Time periods where the electrochemical cell was held at open circuit (OCP; 0–24 h, 34–48 h, 58–72 h, 82–96 h) are indicated by a white background and time periods where the electrochemical cell was held at −1.05 VAg/AgCl (24–34 h, 48–58 h, 72–82 h) are indicated by a green background. (b) Cumulative FETAN as a function of cumulative CPE duration. (c) Pseudo-instantaneous NO3− conversion rate (nitrate converted over one sample period divided by the preceding sample period nitrate concentration; eqn (S5.1.16), ESI†). Equations used to calculate efficiency and rate metrics can be found in Section S5.1 (ESI†). recovery), and overall nitrogen recovery efficiency (ηOverallNrecovery) during ECaB experiments. Error bars represent ± one standard deviation from triplicate experiments (n = 3). Detailed experimental parameters of ECaB tests can be found in the Materials & methods section 4.2.3. Time periods where the electrochemical cell was held at open circuit (OCP; 0–24 h, 34–48 h, 58–72 h, 82–96 h) are indicated by a white background and time periods where the electrochemical cell was held at −1.05 VAg/AgCl (24–34 h, 48–58 h, 72–82 h) are indicated by a green background. (b) Cumulative FETAN as a function of cumulative CPE duration. (c) Pseudo-instantaneous NO3− conversion rate (nitrate converted over one sample period divided by the preceding sample period nitrate concentration; eqn (S5.1.16), ESI†). Equations used to calculate efficiency and rate metrics can be found in Section S5.1 (ESI†). | ||
Proof-of-concept ECaB recovered a pure (NH4)2SO4 with record low energy consumption (90.0 ± 2.7 kW h kg N−1) for a NO3RR reactive separation process (Fig. 6), due to the selectivity and FETAN of Co(DIM)-mediated NO3RR in relatively dilute NO3− (≤5.8 ± 0.2 mM for all time points). Energy consumption is a major driver of wastewater treatment technology feasibility49 and is therefore a crucial metric to compare to related systems and to incumbent processes. Proof-of-concept ECaB demonstrates that its subunits (NO3− extraction, NO3− conversion, and TAN recovery; Fig. 6 symbol shape) consume a proportionate amount of energy in an integrated unit process format. The cumulative energy consumption was also achieved from real wastewater using reactive separations, demonstrating robust process capabilities (Fig. 6 symbol color). The cumulative energy consumption of proof-of-concept ECaB is within 3.6–7.1 times that of conventional centralized nitrogen management (12.7–25.1 kW h kg N−1), consisting of HB ammonia production (7.7–10.1 kW h kg N−1)50 and nitrification–denitrification (NDN) wastewater treatment (5–15 kW h kg N−1).51 Additionally, the cost of producing ammonium sulfate in ECaB is $2.70 kg N−1 and competitive with the retail price ($2.89 kg N−1) if electricity is considered the only operational expenditure (Section S5.1, ESI†).52 The low energy consumption and product cost in proof-of-concept ECaB are promising, but the long operational timescale (96 h) is limited by the relatively slow NO3− conversion subunit rate (Fig. S32, ESI†).
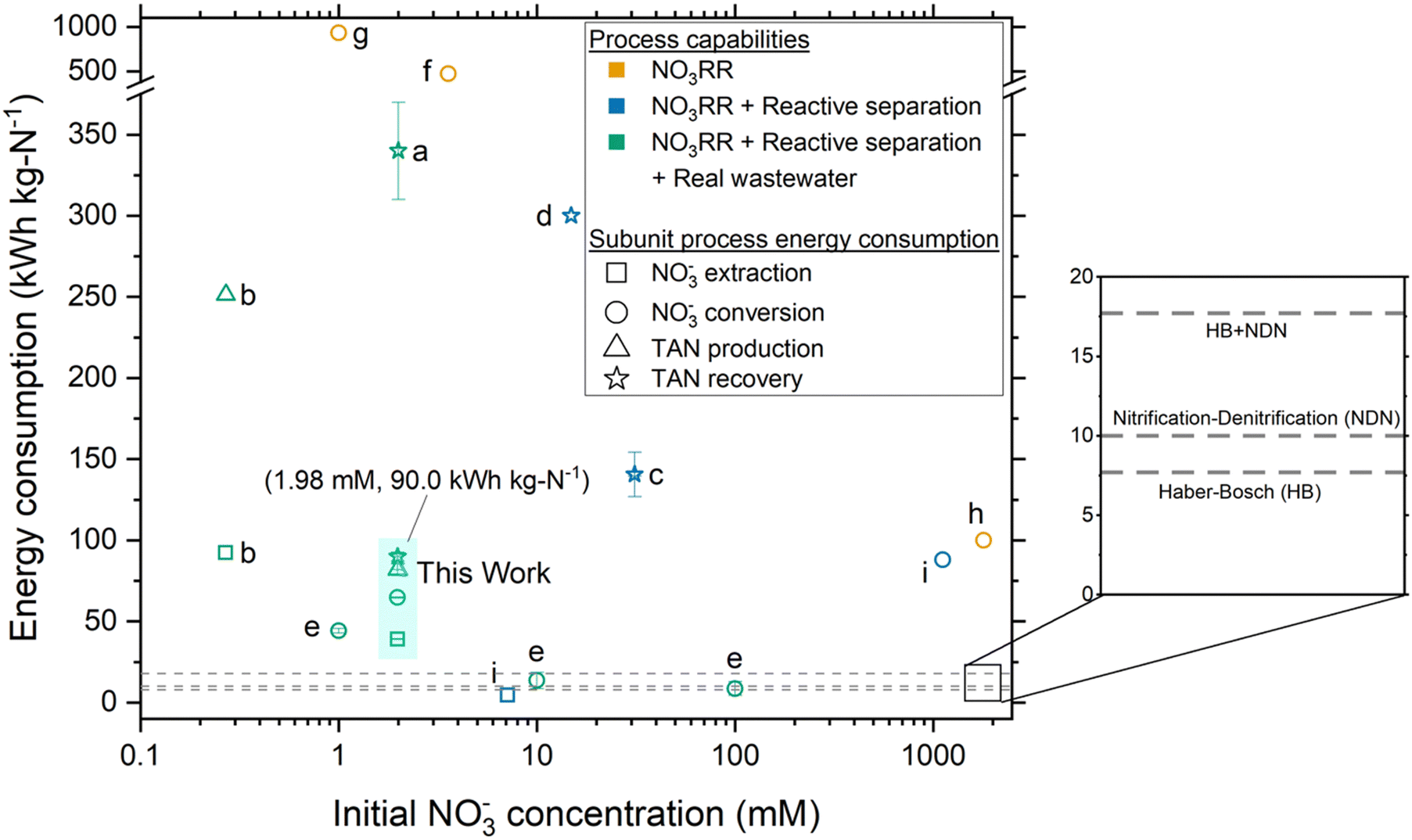 | ||
| Fig. 6 Energy consumption of NO3− extraction, NO3− conversion, TAN production, and TAN recovery subunit processes for reports of reactive separations in NO3RR literature. Error bars for this work represent ± one standard deviation from triplicate experiments (n = 3). Error bars were included as-reported for references a, c, and e. Fig. 6 shows ECaB energy consumption (kW h kg N−1) where the numerator is the same energy for all metrics and the denominator is the nitrogen mass associated with NO3− extraction, NO3− conversion, TAN production, and TAN recovery (Section S5.1, ESI†). Energy consumption values shown are therefore cumulative, such that a subunit process energy consumption is the sum of all preceding subunit processes (i.e., TAN recovery will always be the largest because it is the sum of all subunit processes). Equations used to calculate energy consumption are detailed in Section S5.1 (ESI†). For proof-of-concept ECaB, NO3− extraction and TAN recovery do not explicitly consume energy but are calculated as non-zero values here as the electrochemical energy consumed divided by the mass of nitrogen extracted or recovered, respectively. References for reported values from NO3RR studies in the literature are as follows: a,38 b,37 c,34 d,35 e,53 f,54 g,55 and h.56 Note that studies e, f, and g were focused on the removal of NO3− as N2. Tables S4–S7 (ESI†) provide a detailed comparison of ECaB to the NO3RR systems referenced in Fig. 6. The dashed lines indicate the energy consumption of Haber–Bosch (HB), nitrification–denitrification (NDN), and the summed energy consumption of HB + NDN. | ||
| Metric | Subunit | Proof-of-concept ECaB | Subunit engineered ECaB |
|---|---|---|---|
| Operation time (h) | NO3− extraction | 96 h | 24 h |
| TAN production | 30 h | 8 h | |
| TAN recovery | 72 h | 24 h | |
| Total | 96 h | 48 h | |
| Rate (μg N cm−2 h−1) | NO3− extraction | 10.4 ± 0.1 | 151.6 |
| TAN production | 15.7 ± 0.9 | 319.6 | |
| TAN recovery | 6.0 ± 0.2 | 45.8 | |
| Energy consumption (kW h kg N−1) | NO3− extraction | 0 (39.0 ± 0.2 considering all subunits) | 0 (48.2 considering all subunits) |
| TAN production | 81.9 ± 3.5 | 67.2 | |
| TAN recovery | 90.0 ± 2.7 | 295.3 | |
| Efficiency (%) | NO3− extraction (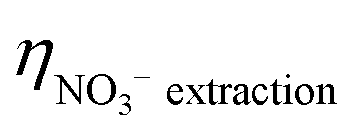 ) ) |
98.1 ± 0.3% | 67.5% |
| TAN production (YTAN) | 49.0 ± 2.6% | 82.5% | |
| TAN recovery (ηTANrecovery) |
91.0 ± 2.1% | 22.8% |
To quantify the performance gap between subunit engineered ECaB and operationally feasible systems, we derived scenario-based jTAN targets from a cost assessment of wastewater treatment coupled with fertilizer production. Cost was chosen as a basis for targets because it is the largest driver of wastewater treatment technology feasibility;49jTAN was chosen as a representative performance target because it influences cost57,58 and facilitates comparisons across electrochemical systems. ECaB's primary function (wastewater treatment) can be offset by its ability to recover chemical value, so we determined target jTAN values that would allow Co(DIM) in the ECaB system to be a net-zero cost over a ten year lifetime treating wastewater NO3− from a large wastewater treatment plant (1 × 106 galwastewater day−1)59 (Fig. 7a, Section S7.2, ESI†). Our assessment is conservative because it considered revenue from (NH4)2SO4 fertilizer but did not consider other financial incentives of ECaB in a wastewater treatment process, such as water recovery revenue, conventional nitrogen treatment costs, or avoided regulatory fines for environmental NO3− discharge. The calculated difference between target jTAN and our experimental jTAN describes the gap in reactor-level catalytic activity required for ECaB process feasibility. Using the same FETAN, ηNO3RR, and ηanode,membrane observed in subunit engineered ECaB, the required jTAN for the unit process to be a net-zero cost was 619 mA cm−2 (120× that of our experimental value; Fig. 7b). While this gap is large, it can be overcome by engineering anode cost (scenario B), thermodynamic performance (scenarios C–E), electrolyte identity (scenario F), and materials synthesis (scenarios G and H). Anode cost played the largest role in reducing the target jTAN due to the high cost of platinum-group metals; nickel-based anodes are under active investigation as lower-cost oxygen evolution anode materials.60,61 FETAN played the second largest role in reducing target jTAN and could be addressed by unit process-level development (e.g., enhancing NO3− up-concentration to its thermodynamic limit). Seemingly innocuous changes had large contributions to reducing target jTAN (e.g., using seawater as a catholyte reduced target jTAN by 33.8%). In the limit where all scenarios B–H are achieved and electricity is a free utility (i.e., scenario J), the target jTAN is 2.46× our subunit engineered ECaB jTAN. The dominant cost contribution to jTAN targets in scenarios A–H (Fig. 7c) was operational expenditures (OpEx, specifically electricity for NO3RR). Only in scenario I, when anode cost, FETAN, ηNO3RR, and ηanode,membrane were improved together, did capital expenditures (CapEx) outweigh OpEx. Meanwhile, Co(DIM) synthesis was a minority cost contributor in all scenarios, indicating that homogenous catalysts like Co(DIM) can be financially feasible and that efforts to reduce the overall system cost should prioritize engineering reaction potentials and FEs. Practically, our cost analysis highlights that overcoming limitations of reaction thermodynamics, electrolyte engineering, and materials synthesis can significantly reduce gaps between experimental and target jTAN of scalable systems.
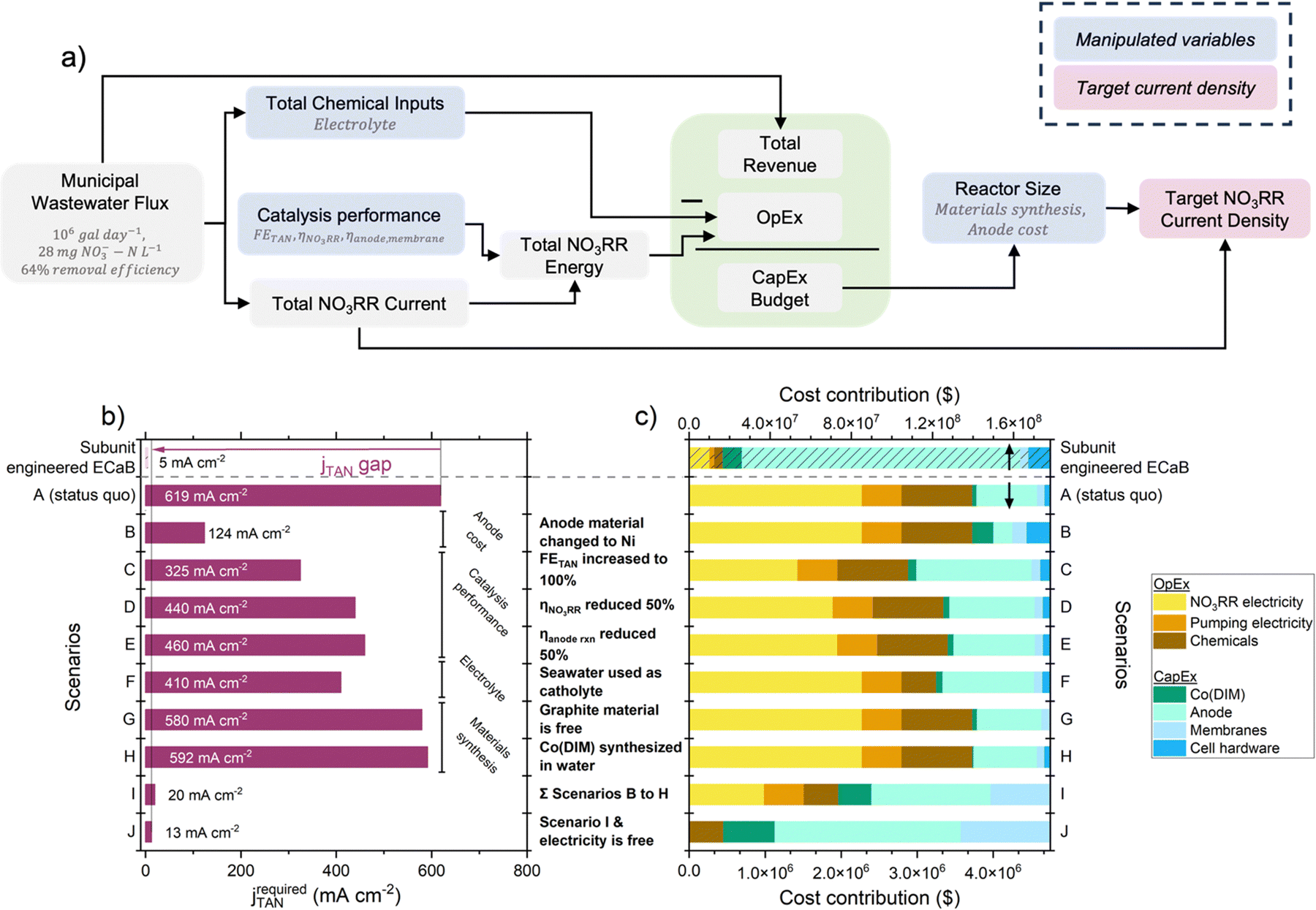 | ||
| Fig. 7 (a) Flow diagram for simplified cost assessment of Co(DIM)-mediated NO3RR in ECaB. The flux of wastewater NO3− from a large wastewater treatment plant (1 × 106 galwastewater day−1, 28 mg NO3−–N L−1 influent treated to 10 mg NO3−–N L−1 effluent = 64% removal efficiency to meet the EPA standard for drinking water)59 was used as a basis for the operating expenditures (OpEx; chemical costs, NO3RR electricity costs, and pumping electricity costs), capital expenditure (CapEx), and revenue of a scaled-up ECaB system. The difference between the net present values of revenue and expenditures was set to zero to determine the CapEx budget. The entire CapEx budget was spent on Co(DIM), anode material, membrane material, and cell hardware material, which defined the geometric surface area required. The total current was divided by the surface area to obtain jTAN. Detailed calculations are provided in Section S7.2 (ESI†). (b) TAN current density (jTAN; red) for experimental ECaB performance (this work, Co(DIM)-ECaB) and for scenario-specific performance targets. In all scenarios, the cost of electricity was ¢3 kW h−1.62 For Co(DIM)-ECaB and scenario A, FETAN was 62.9% and Ecell was 2.7 V (ηNO3RR was 0.905 V and ηanoderxn was 0.765, calculated from the pH-dependent equilibrium potentials of the NO3RR and OER). With improvements to catalysis thermodynamic performance, electrolytes, and materials synthesis, the target current density for Co(DIM)-ECaB to be a net zero cost is decreased by 31.0 times (from 619 mA cm−2 to 20.0 mA cm−2). If electricity becomes a free utility, this target can be further reduced to 13 mA cm−2. (c) Cost contribution of OpEx (chemical inputs, NO3RR electricity, and pumping electricity) and CapEx (Co(DIM), anodes, membranes, and cell hardware) to the overall system. | ||
In addition to converting wastewater NO3− to purified TAN at unprecedented energy efficiency, ECaB also provides a generalizable, modular platform for benchmarking electrocatalysis. For example, we performed Co(DIM)-mediated nitrite reduction in ECaB with a simplified wastewater feed containing NO2−, achieving a TAN yield of 71.0% in 2 hours of CPE (Fig. S39, ESI†). ECaB could similarly be used for CO2 reduction (homogeneous or heterogeneous) from HCO3−/CO32− feeds. The ECaB platform and our cost assessment could be adapted to other homogeneous and heterogeneous catalysts, electrodes, membranes, mass transport conditions, and operating parameters in specific wastewaters. Performing both extraction and recovery via membranes facilitates comparison of co-dependent reaction and separation rates and efficiencies (Tables S4–S7, ESI†).33 Volume reduction of NO3−-containing electrolytes by low-energy separations processes like DD could help remediate the majority of NO3− emissions contributed by high volume, dilute NO3− sources. The combination of subunit processes in ECaB driven by only one electrochemical power source will integrate well with distributed renewable power generation and storage.63 ECaB is therefore a promising and practical platform to investigate modular and on-site reactive separations.
3. Conclusions
In this study, we elucidated catalytically influential wastewater constituents (specifically Mg2+) and their effects on a homogeneous NO3RR catalyst's (Co(DIM)) performance to rationally design a novel reactive separations process: electrocatalyst-in-a-box (ECaB). Iteratively investigating reactor-scale performance and microenvironment-scale interfacial phenomena provided design principles for a system resilient to wastewater impurities, which we systematically investigated using simplified, simulated, and real wastewaters. The presence of Mg2+ in real NO3−-bearing secondary effluent inhibited NO3RR activity but did not affect NO3RR selectivity or FE. Mg deposition on the cathode surface was shown to be the major passivation mechanism, aligned with recent heterogeneous NO3RR reports of water hardness passivating cathodes.16 Heterogeneous electron transfer between the cathode and homogeneous Co(DIM) was slowed by the passivated surface, but the homogeneous NO3RR reaction was not observably affected. We showed that Co(DIM) can protect the cathode from inhibition by preventing specific adsorption of Mg2+. We also showed that anion-selective separations via DD enable cathode protection along with homogeneous catalyst reuse and treated water recovery. Combining DD and NO3RR within the ECaB process demonstrated promising wastewater NO3− extraction, NO3− conversion to TAN, and TAN recovery as a purified product. Proof-of-concept ECaB achieved 98.1 ± 0.3% NO3−–N removal from a real municipal secondary effluent and recovered a pure TAN product with the lowest reported energy consumption to date for an NO3RR system (90.0 ± 2.7 kW h kg N−1). Subunit engineered ECaB enabled a sustained jTAN of 5.14 mA cmgeometric−2 for 8 h of Co(DIM)-mediated NO3RR with low energy consumption maintained (67.2 kW h kg N−1 for TAN production). A cost assessment showed that subunit engineered ECaB jTAN must be improved by 120× to meet status-quo performance targets, but that tractable improvements to catalysts, electrolytes, and materials in ECaB systems can reduce targets to 2.46× the jTAN we achieved in subunit engineered ECaB.This work encourages the use-informed study of molecular catalysts and integrated unit process analysis for wastewater refining. The development of ECaB was informed entirely by the composition of a generalizable NO3−-rich wastewater, underscoring the imperative, insightful connections between systematic wastewater-based electrochemical research and the value propositions (i.e., pollutant removal and product generation) posed by a novel unit process. The developed ECaB architecture can also serve as a platform for comparative investigations of wastewaters, catalysts, and reactor operating conditions. Deployment feasibility for NO3RR reactive separations systems will be governed by analysis of efficiencies, rates, and energy consumption for extraction, conversion, and recovery unit processes.3 The study of integrated reactive separations that match the scale of wastewater–NO3− generation will accelerate use of wastewaters as feedstocks for electrified chemical production. Ultimately, this work innovates on incumbent centralized nitrogen management systems by producing commodities from impure, variable, and complex NO3−-rich wastewaters.
4. Methods
All chemicals were purchased as reagent grade and used as received. Nanopure water (resistivity: 18.2 MΩ cm) was used for all experiments and measurements unless stated otherwise. Before experiments, cation exchange membranes (CEM; CMI-7000, Membranes International Inc.) were stored in 0.1 M KClO4; anion exchange membranes (AEM; ASVN, Selemion) were stored in nanopure water; and bipolar membranes (BPM; FBM, Fumasep) were stored in 1 M NaCl. Hydrophobic gas-permeable membranes (GPM; CLARCOR QP 952 (polytetrafluoroethylene), CLARCOR Industrial Air) were used as received.4.1. Catalyst synthesis and characterization
The perchlorate salt of [Co(DIM)Br2]+ (Fig. 8) was prepared as detailed previously.291H NMR spectra (Varian Inova 600 at 600 MHz; Fig. S1, ESI†) and LC-MS spectra (Agilent 1260 HPLC with an Agilent 6460 Triple Quadrupole MS; Fig. S2, ESI†) of Co(DIM) confirmed the anticipated structure, molecular weight, and axial ligation of the catalyst.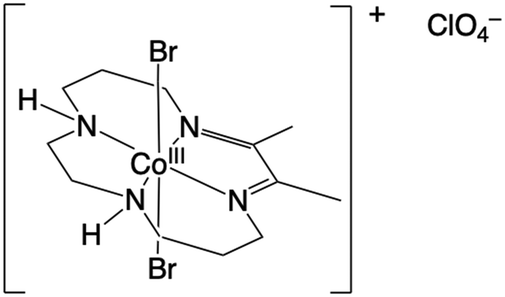 | ||
| Fig. 8 The structure of Co(DIM) in its synthesized, crystalline form. | ||
4.2. Electrochemical methods
A BioLogic VMP-300 potentiostat was used to control the potential applied to the working electrode versus the reference electrode. All electrochemical experiments were recorded using 85% IR compensation based on the ohmic resistance obtained via potentiostatic electrochemical impedance spectroscopy.The statistical significance of differences between the observed CPE performance in the three catholytes was compared using paired t-tests for equal means, where we rejected the null hypothesis (difference between CPE performance results from a normal distribution with mean equal to zero and unknown variance) if p < 0.05.
Rinse tests were performed to assess the catalytic activity of deposits on GC cathodes. The cell was then reconstructed with the deposit-containing GC electrode. An eight-hour CPE was performed (identical operation to the first CPE) with 50 mL simplified wastewater without dissolved Co(DIM) in the catholyte (i.e., 0 mM Co(DIM), 6.2 mM NaCl, 2 mM NaNO3).
4.3. Aqueous characterization
Aqueous ion speciation was quantified with cation chromatography (Na+, NH4+, K+, Ca2+, Mg2+) and anion chromatography (Cl−, NO2−, NO3−, SO4−, PO43−). Both cation chromatography (4 mM tartaric acid/2 mM oxalic acid eluent, SCS 1 column at 30 °C) and anion chromatography (4.5 mM carbonate/0.8 mM bicarbonate eluent, AS23-4 μm column at 30 °C) were performed with a dual Dionex ICS-6000 system (ThermoFisher Scientific). For ECaB, the high concentration of K+ in the catholyte made quantification of TAN by cation chromatography difficult, so flow injection analysis (indophenol method) was conducted with a SEAL AA500 AutoAnalyzer.38 For both cation chromatography and flow injection analysis, aliquots above pH 7 were acidified to prevent ammonia volatilization and facilitate accurate quantification of the measure of total ammonia nitrogen (TAN: sum of aqueous ammonium, NH4+, and aqueous ammonia, NH3). Inductively coupled plasma mass spectrometry (ICP-MS) was used to quantify the concentration of 59Co in each ECaB chamber at the initial and final time points. ICP-MS (ThermoFisher Scientific) was performed using a parallel flow nebulizer (Burgener PEEK Mira Mist) and a Peltier-cooled Scott-type double pass cyclonic spray chamber cooled to 2.7 °C.644.4. Electrode characterization
X-ray photoelectron spectroscopy (XPS) was performed using a Phi Versaprobe 3 with monochromatized Al Kα (1486 eV) radiation. Scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDS) characterization was performed on a Thermo Fisher Scientific Apreo S LoVac (5 kV, 5.0 pA) with a Bruker Quantax EBSD 400i integrated system containing an XFlash 6|60 SDD EDS detector. Raw EDS data were processed and plotted with Bruker's ESPRIT software.4.5. Efficiency, rate, and energy comparison with the literature
The scope of our comparison to precedent literature was constrained to literature studies that investigated reactive separations in real wastewaters. Reported values of efficiencies, rates, and energy consumption are shown for the four subunit processes of nitrate extraction (Table S4, ESI†), nitrate conversion by the NO3RR (Table S5, ESI†), TAN production by the NO3RR (Table S6, ESI†), and TAN recovery (Table S7, ESI†). Efficiencies, rates, and energy consumption were shown by Kogler and co-workers to be the most important operational metrics for wastewater treatment practitioners to make technology decisions for future systems.49Data availability
Data for this article are available at Stanford Digital Repository at https://doi.org/10.25740/sk954gp2933. These data include NMR and XPS spectra, aqueous nitrate and ammonia/ammonium mass balances for controlled-potential electrolysis and electrocatalyst-in-a-box (ECaB) experiments, electrochemical data used to calculate energy consumption for ECaB experiments, and worksheet and tabulated results of ECaB cost assessment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to several funders of this work, including the National Science Foundation EFRI program (Award 2132007), the Dreyfus Foundation (Camille Dreyfus Teacher-Scholar Award), the Sustainability Accelerator within the Doerr School of Sustainability at Stanford University, and the Chemical Engineering Department at Stanford University. D. M. M. acknowledges support from the National Aeronautics and Space Administration (NASA) Space Technology Graduate Research Opportunities fellowship (Award 80NSSC22K1191). M. J. L. acknowledges support from the Northern California Chapter of the Achievement Rewards for College Scientists (ARCS) Foundation. K. A. acknowledges support from the Stanford Graduate Fellowship. A.K. acknowledges support from the Stanford Interdisciplinary Graduate Fellowship. Part of this work was performed at the Stanford Nano Shared Facilities (SNSF), supported by the National Science Foundation under award ECCS-2026822. The authors thank Gaurav Kamat for the ICP-MS analysis, Jinyu Guo for the XRD analysis and for her thoughtful engagement with this work, and the Tarpeh Laboratory for their support, feedback, and inspiration.References
- C. Smith, A. K. Hill and L. Torrente-Murciano, Energy Environ. Sci., 2020, 13, 331–344 RSC.
- J. Lim, C. A. Fernández, S. W. Lee and M. C. Hatzell, ACS Energy Lett., 2021, 6, 3676–3685 CrossRef CAS.
- D. M. Miller, K. Abels, J. Guo, K. S. Williams, M. J. Liu and W. A. Tarpeh, J. Am. Chem. Soc., 2023, 145, 19422–19439 CrossRef CAS PubMed.
- J. Zhou, P. R. Leavitt, Y. Zhang and B. Qin, Water Res., 2022, 221, 118728 CrossRef CAS PubMed.
- J. Merder, T. Harris, G. Zhao, D. M. Stasinopoulos, R. A. Rigby and A. M. Michalak, Nat. Water, 2023, 1, 844–854 CrossRef.
- W. K. Dodds, W. W. Bouska, J. L. Eitzmann, T. J. Pilger, K. L. Pitts, A. J. Riley, J. T. Schloesser and D. J. Thornbrugh, Environ. Sci. Technol., 2009, 43, 12–19 CrossRef CAS PubMed.
- The Challenge of Tracking Nutrient Pollution 2300 Miles|U.S. Geological Survey, https://www.usgs.gov/news/challenge-tracking-nutrient-pollution-2300-miles, (accessed February 4, 2022).
- M. Lang, P. Li and X. Yan, Sci. Total Environ, 2013, 458–460, 238–245 CrossRef CAS PubMed.
- T. Kato, H. Kuroda and H. Nakasone, J. Hydrol., 2009, 368, 79–87 CrossRef CAS.
- B. Eghball and J. E. Gilley, J. Environ. Qual., 1999, 28, 1201–1210 CrossRef CAS.
- C. D. A. McLay, R. Dragten, G. Sparling and N. Selvarajah, Environ. Pollut., 2001, 115, 191–204 CrossRef CAS PubMed.
- R. K. Hubbard and J. M. Sheridan, J. Environ. Qual., 1983, 12, 291–295 CrossRef.
- Z.-H. Wang and S.-X. Li, in Advances in Agronomy, ed. D. L. Sparks, Academic Press, 2019, vol. 156, pp. 159–217 Search PubMed.
- J. M. McEnaney, S. J. Blair, A. C. Nielander, J. A. Schwalbe, D. M. Koshy, M. Cargnello and T. F. Jaramillo, ACS Sustainable Chem. Eng., 2020, 8, 2672–2681 CrossRef CAS.
- S.-E. Bae, K. L. Stewart and A. A. Gewirth, J. Am. Chem. Soc., 2007, 129, 10171–10180 CrossRef CAS PubMed.
- A. Atrashkevich, A. S. Fajardo, P. Westerhoff, W. S. Walker, C. M. Sánchez-Sánchez and S. Garcia-Segura, Water Res., 2022, 225, 119118 CrossRef CAS PubMed.
- F.-Y. Chen, Z.-Y. Wu, S. Gupta, D. J. Rivera, S. V. Lambeets, S. Pecaut, J. Y. T. Kim, P. Zhu, Y. Z. Finfrock, D. M. Meira, G. King, G. Gao, W. Xu, D. A. Cullen, H. Zhou, Y. Han, D. E. Perea, C. L. Muhich and H. Wang, Nat. Nanotechnol., 2022, 17, 759–767 CrossRef CAS PubMed.
- J. Li, G. Zhan, J. Yang, F. Quan, C. Mao, Y. Liu, B. Wang, F. Lei, L. Li, A. W. M. Chan, L. Xu, Y. Shi, Y. Du, W. Hao, P. K. Wong, J. Wang, S.-X. Dou, L. Zhang and J. C. Yu, J. Am. Chem. Soc., 2020, 142, 7036–7046 CrossRef CAS PubMed.
- J. Lim, C.-Y. Liu, J. Park, Y.-H. Liu, T. P. Senftle, S. W. Lee and M. C. Hatzell, ACS Catal., 2021, 11, 7568–7577 CrossRef CAS.
- P. Li, Z. Jin, Z. Fang and G. Yu, Energy Environ. Sci., 2021, 14, 3522–3531 RSC.
- Z.-Y. Wu, M. Karamad, X. Yong, Q. Huang, D. A. Cullen, P. Zhu, C. Xia, Q. Xiao, M. Shakouri, F.-Y. Chen, J. Y. (Timothy) Kim, Y. Xia, K. Heck, Y. Hu, M. S. Wong, Q. Li, I. Gates, S. Siahrostami and H. Wang, Nat. Commun., 2021, 12, 2870 CrossRef CAS PubMed.
- J. Li, M. Li, N. An, S. Zhang, Q. Song, Y. Yang and X. Liu, Proc. Natl. Acad. Sci., 2021, 118, e2105628118 CrossRef CAS PubMed.
- R. Jia, Y. Wang, C. Wang, Y. Ling, Y. Yu and B. Zhang, ACS Catal., 2020, 10, 3533–3540 CrossRef CAS.
- Y. Wang, W. Zhou, R. Jia, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 5350–5354 CrossRef CAS PubMed.
- Y. Wang, A. Xu, Z. Wang, L. Huang, J. Li, F. Li, J. Wicks, M. Luo, D.-H. Nam, C.-S. Tan, Y. Ding, J. Wu, Y. Lum, C.-T. Dinh, D. Sinton, G. Zheng and E. H. Sargent, J. Am. Chem. Soc., 2020, 142, 5702–5708 CrossRef CAS PubMed.
- G.-F. Chen, Y. Yuan, H. Jiang, S.-Y. Ren, L.-X. Ding, L. Ma, T. Wu, J. Lu and H. Wang, Nat. Energy, 2020, 5, 605–613 CrossRef CAS.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, eaar6611 CrossRef PubMed.
- S. Xu, D. C. Ashley, H.-Y. Kwon, G. R. Ware, C.-H. Chen, Y. Losovyj, X. Gao, E. Jakubikova and J. M. Smith, Chem. Sci., 2018, 9, 4950–4958 RSC.
- S. C. Jackels, Keith Farmery, E. Kent Barefield, N. J. Rose and D. H. Busch, Inorg. Chem., 1972, 11, 2893–2901 CrossRef CAS.
- D. J. Cole-Hamilton, Science, 2003, 299, 1702–1706 CrossRef CAS PubMed.
- K. Hu, P. Zhou, Y. Yang, T. Hall, G. Nie, Y. Yao, X. Duan and S. Wang, ACS ES&T Eng., 2022, 2, 110–120 Search PubMed.
- J. Sun, S. Jennepalli, M. Lee, A. Jones, D. M. O’Carroll, M. J. Manefield, M. Bhadbhade, B. Åkermark, B. Das and N. Kumar, Environ. Sci. Technol., 2022, 56, 7830–7839 CrossRef CAS PubMed.
- V. A. Niemann, P. Benedek, J. Guo, Y. Xu, S. J. Blair, E. R. Corson, A. C. Nielander, T. F. Jaramillo and W. A. Tarpeh, ACS Catal., 2023, 13, 6268–6279 CrossRef CAS.
- J. Gao, N. Shi, Y. Li, B. Jiang, T. Marhaba and W. Zhang, Environ. Sci. Technol., 2022, 56, 11602–11613 CrossRef CAS PubMed.
- J. Gao, N. Shi, X. Guo, Y. Li, X. Bi, Y. Qi, J. Guan and B. Jiang, Environ. Sci. Technol., 2021, 55, 10684–10694 CrossRef CAS PubMed.
- W. Zheng, L. Zhu, Z. Yan, Z. Lin, Z. Lei, Y. Zhang, H. Xu, Z. Dang, C. Wei and C. Feng, Environ. Sci. Technol., 2021, 55, 13231–13243 CAS.
- K. Kim, A. Zagalskaya, J. L. Ng, J. Hong, V. Alexandrov, T. A. Pham and X. Su, Nat. Commun., 2023, 14, 823 CrossRef CAS PubMed.
- M. J. Liu, D. M. Miller and W. A. Tarpeh, Environ. Sci. Technol. Lett., 2023, 10, 458–463 CrossRef CAS.
- J. M. Savéant and C. Costentin, Elements of molecular and biomolecular electrochemistry: an electrochemical approach to electron transfer chemistry, Wiley, Hoboken, NJ, Second edn, 2019 Search PubMed.
- J. Guo, P. Brimley, M. J. Liu, E. R. Corson, C. Muñoz, W. A. Smith and W. A. Tarpeh, ACS Sustainable Chem. Eng., 2023, 11, 7882–7893 CrossRef CAS.
- I. Bhugun, D. Lexa and J.-M. Savéant, J. Phys. Chem., 1996, 100, 19981–19985 CrossRef CAS.
- A. Pérez-González, A. M. Urtiaga, R. Ibáñez and I. Ortiz, Water Res., 2012, 46, 267–283 CrossRef PubMed.
- R. Mohammadi, D. L. Ramasamy and M. Sillanpää, Desalination, 2021, 498, 114726 CrossRef CAS.
- S. A. Hawks, M. R. Cerón, D. I. Oyarzun, T. A. Pham, C. Zhan, C. K. Loeb, D. Mew, A. Deinhart, B. C. Wood, J. G. Santiago, M. Stadermann and P. G. Campbell, Environ. Sci. Technol., 2019, 53, 10863–10870 CrossRef CAS PubMed.
- H. Chen, M. Rose, M. Fleming, S. Souizi, U. Shashvatt and L. Blaney, Chem. Eng. J., 2023, 455, 140522 CrossRef CAS.
- M. J. Pennino, S. G. Leibowitz, J. E. Compton, R. A. Hill and R. D. Sabo, Sci. Total Environ, 2020, 722, 137661 CrossRef CAS PubMed.
- D. B. Shah and G. A. Coulman, Biotechnol. Bioeng., 1978, 20, 43–72 CrossRef CAS.
- M. Janssen, C. Müller and D. Vogt, Green Chem., 2011, 13, 2247–2257 RSC.
- A. Kogler, M. Farmer, J. A. Simon, S. Tilmans, G. F. Wells and W. A. Tarpeh, ACS ES&T Eng., 2021, 1, 662–684 Search PubMed.
- G. Soloveichik, Nat. Catal., 2019, 2, 377–380 CrossRef CAS.
- D. Wu, X. Li and X. Li, ACS ES&T Water, 2021, 1, 796–807 Search PubMed.
- US Department of Agriculture, Pacific Northwest Production Cost Report (Bi-Weekly), https://usda.library.cornell.edu/concern/publications/h989sc11z?locale=en, (accessed June 5, 2024).
- P. Gayen, J. Spataro, S. Avasarala, A.-M. Ali, J. M. Cerrato and B. P. Chaplin, Environ. Sci. Technol., 2018, 52, 9370–9379 CrossRef CAS PubMed.
- J. Wang, S. Wang, Z. Zhang and C. Wang, J. Environ. Manage., 2020, 276, 111357 CrossRef CAS PubMed.
- H. Cheng, K. Scott and P. A. Christensen, Chem. Eng. J., 2005, 108, 257–268 CrossRef CAS.
- I. Katsounaros, M. Dortsiou and G. Kyriacou, J. Hazard. Mater., 2009, 171, 323–327 CrossRef CAS PubMed.
- N. Lazouski, A. Limaye, A. Bose, M. L. Gala, K. Manthiram and D. S. Mallapragada, ACS Energy Lett., 2022, 7, 2627–2633 CrossRef CAS.
- C. A. Fernandez and M. C. Hatzell, J. Electrochem. Soc., 2020, 167, 143504 CrossRef CAS.
- K. E. Kapo, M. Paschka, R. Vamshi, M. Sebasky and K. McDonough, Sci. Total Environ., 2017, 603–604, 445–452 CrossRef CAS PubMed.
- E. K. Volk, S. Kwon and S. M. Alia, J. Electrochem. Soc., 2023, 170, 064506 CrossRef CAS.
- Z.-Y. Wu, F.-Y. Chen, B. Li, S.-W. Yu, Y. Z. Finfrock, D. M. Meira, Q.-Q. Yan, P. Zhu, M.-X. Chen, T.-W. Song, Z. Yin, H.-W. Liang, S. Zhang, G. Wang and H. Wang, Nat. Mater., 2023, 22, 100–108 CrossRef CAS PubMed.
- W. J. Cole, B. A. Frew, P. J. Gagnon, J. Richards, Y. Sun, R. M. Margolis and M. A. Woodhouse, SunShot 2030 for Photovoltaics (PV): Envisioning a Low-cost PV Future, 2017.
- C. A. Fernández, O. Chapman, M. A. Brown, C. E. Alvarez-Pugliese and M. C. Hatzell, Environ. Sci. Technol., 2024, 58, 6964–6977 CrossRef PubMed.
- D. H. Marin, J. T. Perryman, M. A. Hubert, G. A. Lindquist, L. Chen, A. M. Aleman, G. A. Kamat, V. A. Niemann, M. B. Stevens, Y. N. Regmi, S. W. Boettcher, A. C. Nielander and T. F. Jaramillo, Joule, 2023, 7, 765–781 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary Fig. S1–S39, Tables S1–S7, and eqn S3.1.1–S7.2.1. See DOI: https://doi.org/10.1039/d4ee01727g |
| This journal is © The Royal Society of Chemistry 2024 |