
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWhite light and near-infrared emissions of Pr3+ ions in SiO2-LaF3 sol–gel nano-glass-ceramics
Natalia
Pawlik
 *a,
Tomasz
Goryczka
b,
Maciej
Zubko
bc,
Joanna
Śmiarowska
a and
Wojciech A.
Pisarski
a
*a,
Tomasz
Goryczka
b,
Maciej
Zubko
bc,
Joanna
Śmiarowska
a and
Wojciech A.
Pisarski
a
aUniversity of Silesia, Institute of Chemistry, Szkolna 9 Street, 40-007 Katowice, Poland. E-mail: natalia.pawlik@us.edu.pl
bUniversity of Silesia, Institute of Materials Engineering, 75. Pułku Piechoty 1A Street, 41-500 Chorzów, Poland
cUniversity of Hradec Králové, Department of Physics, Rokitanského 62, 50003, Hradec Králové, Czech Republic
First published on 1st February 2024
Abstract
In this work, we report the results of our investigations on the structural and luminescence properties of SiO2-LaF3:Pr3+ nano-glass-ceramics synthesized using the sol–gel method. Based on XRD, microscopic (TEM), and ATR-IR measurements, the crystallization of LaF3 nanocrystals favorably occupied by Pr3+ ions and overall transformations within the silicate sol–gel hosts dependent on heat-treatment conditions of the as-prepared amorphous xerogels were characterized. The fabricated oxyfluoride nano-glass-ceramics revealed the emissions within the greenish-blue (3P0,1 → 3H4, 3P0,1 → 3H5), reddish-orange (3P0,1 → 3H6, 1D2 → 3H4, 3P0 → 3F2,3), and NIR spectral scopes (1D2 → 3F4,1G4, 1G4 → 3H5, 3F3,4 → 3H4). Based on the luminescence spectra in the VIS range, the CIE chromaticity coordinates, correlated color temperatures (CCT), and color purities (CP) were calculated. The obtained results clearly indicate that the prepared Pr3+-doped sol–gel nano-glass-ceramics exhibit warm or neutral white light emissions with CCT values in the range from 2567 K to 3962 K. The lowest CP value was estimated at 12.8%, indicating that the fabricated samples are able to emit bright white light. Additionally, the NIR emissions cover E, S, C, and L bands, which are important for devices applicable in telecommunication technologies. For further characterization, the τ(3P0) and τ(1D2) decay times were estimated. It was established that the emissions from the 3P0 and the 1D2 excited states of Pr3+ ions, as well as the participation of cross-relaxation (CR) processes, are dependent on the size of crystallized LaF3 phase, distribution of optically active Pr3+ ions between amorphous and crystalline phase (determining the Pr3+–Pr3+ inter-ionic distances), and relative content of OH groups in the prepared sol–gel hosts.
Introduction
Trivalent praseodymium ion is one of the most promising rare-earth dopants, whose energy level diagrams and abundant metastable multiplets (i.e., 3P0–2, 1D2, and 1G4) enable the generation of luminescence across the broad spectral ranges from blue, green, and reddish-orange light up to near-infrared irradiation.1,2 Thus, such a tremendous spectrum of emitted wavelengths opens vast application possibilities for optical materials containing Pr3+ ions. For instance, the emission within the red or reddish-orange light area predisposes Pr3+-doped materials as essential parts of optical devices with great utility in LED technologies3–7 or as gain media for laser output at ∼645 nm.8–10 Additionally, the modifications in the composition of the host materials as well as in Pr3+ concentration allow for obtaining the appropriate combinations of blue, green, and reddish-orange luminescence, which could result in the generation of white-light emission, as described for LaAlO3:Pr3+ and YF3:Pr3+ nanocrystals or K3La(PO4)2:Pr3+ phosphors.11,12 The infrared luminescence of Pr3+-doped materials at ∼1.0 μm (1D2 → 3F3,4 transitions), ∼1.35 μm (1G4 → 3H5 transition), and ∼1.6 μm (3F3,4 → 3H4 transition), which covers NIR-II and NIR-III biological windows, make them interesting host matrices in the field of in vivo bioimaging.13–15 The above-mentioned infrared emissions cover E, S, C, and L bands belonging to the fifth optical telecommunication window; thus, Pr3+-doped materials are also considered as promising candidates for waveguide amplification.16,17 Additionally, since the de-excitation channels of the 3P0 and the 1D2 excited states, such as phonon-assisted cross-relaxation (CR) and multiphonon relaxation (MPR), reveal varied temperature-dependent behavior, Pr3+-doped materials could also be used as optical thermometers, as was described for LiYO2:Pr3+ (additionally adjusted by the phase transformation between α and β polymorphs) or La2MgTiO6:Pr3+.18,19 Another pathway relates to the involvement of excitations by ground-state absorption (GSA, λex = 444 nm) and excited-state absorption (ESA, λex = 525 nm) mechanisms on the 3P0 → 3H4 emission, as was performed for LaF3:Pr3+ crystals in the temperature range from 273.15 K to 573.15 K.20 Finally, Pr3+-doped luminescent thermometers could also be based on the intensity ratio of the 3P1 → 3H5 and 3P0 → 3H5 transitions. Indeed, the 3P1 state is thermally populated following the 3P0 excitation and governed by the Boltzmann statistics, as was described in the literature for YF3:Pr3+,21 LaF3:Pr3+,22,23 and LiYF4:Pr3+.23 Their usage could also extend the broad application area of Pr3+-doped materials as scintillators24,25 or dye-sensitized solar cells (DSSC).26 Thus, the presented vast fields of practical applications of Pr3+-doped optical materials make them highly needed for the development of modern optoelectronics.Among the various classes of optical materials doped with Pr3+ ions (e.g., glasses3–5,27,28 or crystals21–23), many research papers are devoted to oxyfluoride glass-ceramics (OxGCs). Indeed, oxyfluoride GCs are very attractive host materials because Pr3+ ions could be effectively insulated from the high-energetically oscillations of the glassy framework by their incorporation inside the low-phonon energy fluoride crystals; nevertheless, the glassy framework provides good chemical, mechanical, and thermal stability of the OxGCs. Most of the Pr3+-doped OxGC materials characterized and described in the current literature are focused on the preparation by high-temperature melt-quenching method, followed by the annealing of parent glasses at controlled and specified time and temperature conditions. Among them, OxGCs containing plentiful fluoride phases classified to AFx type (CaF2,29 SrF2,30,31 BaF2,32,33 LaF3,2,34–37 YF3,38 or PbF2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 39) and AMFx type (NaYF4,1 NaLaF4,25 BaYF5,40 LiGdF4/GdF3,41 NaGdF4,42 KYF443) have been reported. Interestingly, the spectroscopic behavior of Pr3+ ions could be successfully modulated by changing their local environment, i.e., glassy or crystalline. In this matter, C. Koepke et al.32 reported that Pr3+ photoluminescence in BaF2-based OxGCs is strictly correlated to their residing in oxide higher-field (O–Pr–O) or fluorine lower-field (F–Pr–F and/or O–Pr–F) sites. Further, for KYF4:Pr3+-based glass-ceramics, it was proven that the modification in a local field around Pr3+ from fully amorphous to crystalline (for the latter with parallel controll of the growth of fluoride crystals from 5 nm to 30 nm) resulted in emission color tuning from red via warm white, finally to cold white.43 The tunability of Pr3+ emission could also be triggered by modifications in their concentration, as presented by H. Lee et al.,2 who reported that LaF3:Pr3+-based OxGC materials could be used as color converters with a wide color gamut for blue LEDs emitting 450 nm wavelength, dependent on Pr3+ concentration (from 1 mol% to 5 mol%). In addition, the elongation in the heat-treatment time (from 1 hour to 10 hours) and elevation of temperature (from 700 °C to 800 °C) also allowed for successfully shifting the chromaticity coordinates of emitted light color from blue regions to white light. Hence, these peculiarities make the Pr3+-doped OxGC materials very attractive both from cognitive and utilitarian points of view.
39) and AMFx type (NaYF4,1 NaLaF4,25 BaYF5,40 LiGdF4/GdF3,41 NaGdF4,42 KYF443) have been reported. Interestingly, the spectroscopic behavior of Pr3+ ions could be successfully modulated by changing their local environment, i.e., glassy or crystalline. In this matter, C. Koepke et al.32 reported that Pr3+ photoluminescence in BaF2-based OxGCs is strictly correlated to their residing in oxide higher-field (O–Pr–O) or fluorine lower-field (F–Pr–F and/or O–Pr–F) sites. Further, for KYF4:Pr3+-based glass-ceramics, it was proven that the modification in a local field around Pr3+ from fully amorphous to crystalline (for the latter with parallel controll of the growth of fluoride crystals from 5 nm to 30 nm) resulted in emission color tuning from red via warm white, finally to cold white.43 The tunability of Pr3+ emission could also be triggered by modifications in their concentration, as presented by H. Lee et al.,2 who reported that LaF3:Pr3+-based OxGC materials could be used as color converters with a wide color gamut for blue LEDs emitting 450 nm wavelength, dependent on Pr3+ concentration (from 1 mol% to 5 mol%). In addition, the elongation in the heat-treatment time (from 1 hour to 10 hours) and elevation of temperature (from 700 °C to 800 °C) also allowed for successfully shifting the chromaticity coordinates of emitted light color from blue regions to white light. Hence, these peculiarities make the Pr3+-doped OxGC materials very attractive both from cognitive and utilitarian points of view.
Despite many undoubted advantages, there is a substantial drawback in the fabrication of OxGCs via the melt-quenching technique, which is related to the significant losses of fluorides during processing at high temperatures. Hence, such conditions could contribute to noticeable inhibition in the formation of fluoride crystal fraction, consequently resulting in a significant part of rare earth ions remaining inside the amorphous glassy host.44 This limitation, according to the risk of fluorides evaporation, could be overcome by the involvement of sol–gel synthesis as an alternative preparation route because the individual chemical reactions (i.e., hydrolysis of the metal/semi-metal alkoxide and (poly)condensation) are usually performed at relatively low temperatures. However, despite this unquestionable ascendancy, due to the small energy gap of Pr3+ between the 3P0 and the lower-lying 1D2 states (∼4080 cm−1),27 the luminescence of Pr3+––especially within the NIR scope––is very sensitive to high-vibrational OH groups, which are naturally created during aqueous sol–gel processing.45 Due to this limitation, studies on Pr3+ luminescence in oxyfluoride sol–gel materials are scarce and, as far as we know, there is only one paper published by the research team of J. J. Velázquez46 dedicated to studies of Pr3+ spectroscopy in OxGCs with 95SiO2-5LaF3:0.1 mol% Pr3+ composition. The paper describes the photoluminescence of singly-(Pr3+) and doubly-doped (Pr3+/Yb3+) systems within the visible light range. Additionally, it should also be noted that the luminescence of Pr3+ ions in NIR scope, according to OxGC materials, has not been explored in the literature, as far as we know. Therefore, due to the reasons mentioned above, an investigation of Pr3+ luminescence within both visible and NIR ranges in oxyfluoride sol–gel glass-ceramic systems seems to be highly meaningful and necessary.
The main concept of this research reported here was to prepare the series of Pr3+-doped oxyfluoride glass-ceramics containing LaF3 nanocrystals and investigate the impact of variable La3+:Pr3+ molar ratios as well as heat-treatment conditions (500 °C, 700 °C, and 900 °C) on the efficiency of host densification by the removal of OH groups (responsible for quenching luminescence of Pr3+) and efficiency of Pr3+ incorporation inside the fluoride crystal lattice. LaF3 was chosen due to the high similarity of La3+ and Pr3+ ionic radii, ensuring the efficient incorporation of Pr3+ inside the LaF3 crystal phase, as was stated in the literature.2,36 The ATR-IR and XRD techniques as well as TEM microscopy were used to determine the structural changes within the silicate sol–gel host and the crystallization of the LaF3:Pr3+ phase. The luminescence properties of the synthesized oxyfluoride glass-ceramic materials were characterized based on the excitation and emission spectra, along with the decay analysis from the 3P0 and 1D2 excited states. The results showed that the fabricated series of sol–gel Pr3+-doped nano-glass-ceramics could be potentially used as white light emitters or elements of amplifiers operating at E, S, C, and L bands belonging to the fifth optical telecommunication window.
Experimental
The series of xerogels singly-doped with Pr3+ ions were prepared using the sol–gel synthesis described elsewhere.47 All reagents were taken from Sigma Aldrich Chemical Company. The subsequent chemical reactions, which undergo during the sol–gel evolution, e.g., hydrolysis, condensation, and polycondensation of precursor (tetraethoxysilane, TEOS), were carried out in a solution of ethyl alcohol, deionized water, and acetic acid with molar ratio equals to 1:4:10:0.5. Parallelly, the solutions of La(CH3COO)3 and Pr(CH3COO)3 in deionized water and trifluoroacetic acid (TFA) were added dropwise to TEOS-based mixtures. The molar ratio of TFA:La3+:Pr3+ was set to 5:(1 − x):x, where x = 0.003, 0.006, 0.012, 0.03, 0.06, and 0.12. The as-prepared sols were dried at 35 °C for several weeks to form green-colored rigid xerogels. Afterward, the nano-glass-ceramic materials were obtained during controlled heat-treatment at selected temperatures (500 °C, 700 °C, and 900 °C). The prepared samples were denoted as GC1Prx–GC6Prx, where x refers to the temperature conditions of performed heat-treatment and 1–6 expresses the variable La3+:Pr3+ molar ratios in the following samples from the series.
The fabricated nano-glass-ceramics were characterized by X-ray diffraction analysis using an X'Pert Pro diffractometer supplied by PANalytical with CuKα radiation (λ = 1.54056 Å). The transmission electron microscopy (TEM) observations were performed using a JEOL high resolution (HR-TEM) JEM 3010 microscope working at 300 kV accelerating voltage and equipped with Gatan 2k × 2k Orius™ 833SC200D CCD camera and the Elite T Energy Dispersive Spectroscopy (EDS) silicon drift detector (SDD) from AMETEK EDAX. The sol–gel samples were suspended in isopropanol, and after ultrasonication for 10 minutes in an ultrasonic cleaner, the resulting materials were deposited on a Cu grid coated with amorphous carbon film standardized for TEM observations. Selected area electron diffraction (SAED) patterns were characterized using the ElDyf software (version 2.1). The structural characterization also included the infrared measurements; the experiment was performed with the use of a Nicolet iS50 ATR spectrometer, and the spectra were collected in attenuated total reflectance (ATR) configuration within the 4000–400 cm−1 as well as 500–300 cm−1 frequency range (64 scans, 0.1 cm−1 resolution). The ATR-IR spectra were decomposed using Fityk software (0.9.8 software, open-source (GPL2+)) based on the analysis of second derivatives, preceded by the appropriate smoothing of the spectra (using the Savitsky-Golay algorithm). The shape of the bands was described by the sum of the Gaussian and Lorentzian functions, and the coefficient of determination was 0.99. The luminescence measurements were performed on a Photon Technology International (PTI) Quanta-Master 40 (QM40) UV/VIS Steady State Spectrofluorometer supplied with a tunable pulsed optical parametric oscillator (OPO) pumped by the third harmonic of a Nd:YAG laser. The laser system was coupled with a xenon lamp, a double 200 mm monochromator, a multimode UV/VIS PMT detector (R928, PTI Model 914), and a Hamamatsu detector (H10330B-75). The excitation and emission spectra were recorded with a resolution of 0.5 nm. The luminescence decay curves were recorded by a PTI ASOC-10 (USB-2500) oscilloscope with an accuracy of ±0.1 μs. All structural and optical measurements were carried out at room temperature.
Results and discussion
ATR-IR characterization
The fabricated sol–gel materials, as porous species, could evolve to more condensed stages and create a more dense and continuous silicate network during the gradual elevation of heat-treatment temperature. These peculiarities of sol–gel processing, according to differential structure and texture of prepared materials, were verified based on ATR-IR measurements, whose results were presented in Fig. 1 and 2. To determine the impact of variable temperature conditions (500 °C, 700 °C, and 900 °C) on a dynamically formed sol–gel network, the spectra were recorded within the two frequency intervals: from 4000 cm−1 to 400 cm−1 (to observe the changes during the polycondensation of the silicate skeleton) and from 500 cm−1 to 300 cm−1 (to visualize the structural changes according to the crystallization of the LaF3 phase and incorporation of Pr3+ inside the fluoride crystallites). | ||
| Fig. 1 ATR-IR spectra recorded for individual sol–gel materials heat-treated at 500 °C (a), 700 °C (b), and 900 °C (c). The deconvolution of spectra for representative GC2Pr500, GC2Pr700, and GC2Pr900 nano-glass-ceramics in the range from 1400 cm−1 to 400 cm−1 region is also presented in (d)–(f), respectively. | ||
 | ||
| Fig. 2 The comparison of the ATR-IR spectra within the frequency range from 500 cm−1 to 300 cm−1 for representative GC2Pr500, GC2Pr700, and GC2Pr900 samples (the spectra of xerogel, as well as reference LaF3 and PrF3 are also given) (a). The juxtaposition of the ATR-IR spectra and localization of the La–F band for individual samples from each series: 500 °C (b), 700 °C (c), and 900 °C (d) are also presented. | ||
The comparison of the IR-ATR spectra, recorded in the frequency region from 4000 cm−1 to 400 cm−1 for all the samples from the individual series (GCPr500, GCPr700, and GCPr900), is presented in Fig. 1a–c. It should be stated that the shape of the recorded IR spectra for samples from the individual series is practically indistinguishable. These similarities in the spectral profiles clearly prove the formation of equivalent structures of silicate sol–gel host in each series, indicating their high reproducibility. Among the recorded IR signals, a weak and broad band at ∼3350 cm−1, assigned to vibrations of unreacted Si–OH groups, was identified. It is worth noticing that the band's intensities of the band are relatively low for all the prepared series of nano-glass-ceramics, indicating the residual amount of OH groups. Further, it was verified that the band slowly disappeared with the simultaneous elevation of heat-treatment temperature, as was shown in the following Fig. 1a–c, which continuously underwent polycondensation reaction within the silicate network. These speculations were also evaluated by the analysis of the spectral profile in the frequency range from 850 cm−1 to 1300 cm−1. Generally, a gradual disappearance of the shoulder at ∼955 cm−1 and a shift of the infrared signal from ∼1045 cm−1 (500 °C and 700 °C) to ∼1065 cm−1 (900 °C) could be observed. Therefore, to interpret the differences in the spectral profiles, deconvolution was implemented. It revealed the existence of the following oscillations inside the silicate sol–gel hosts: ∼440 cm−1 (Si–O–Si siloxane bridges), ∼560 cm−1 (4-fold siloxane rings), ∼620 cm−1 (6-fold siloxane rings), ∼730 cm−1 (Si–O bonds), ∼800 cm−1 (TO2 mode of Si–O–Si), ∼835 cm−1 (2-fold siloxane rings), ∼910 cm−1 (Q1 units, [Si(O1/2)O3]3−), ∼970 cm−1 (Q2 units, [Si(O1/2)2O2]2−), ∼1030 cm−1 (Q3 units, [Si(O1/2)3O]−), ∼1075 cm−1 (TO3 mode of Si–O–Si), ∼1160 cm−1 (Q4 units, [Si(O1/2)4]), and ∼1220 cm−1 (TO4 and/or LO3 modes of Si–O–Si).48 “O1/2” and “O” denote SiO4 tetrahedral units: the first of them corresponds to each oxygen atom involved in the formation of the Si–O–Si bridge, but the second to each non-bridging oxygen atom. It could be observed that the mutual intensities of individual components change while the annealing temperature increases. Particularly interesting is the change in the intensity of the deconvoluted component assigned to the oscillations inside Q3 units (∼1045 cm−1), which enhanced with elevation of treatment temperature from 500 °C to 700 °C and further reduced with the continuous elevation of temperature from 700 °C to 900 °C. As a result, the component located near ∼1075 cm−1 (TO3 mode of Si–O–Si bridges) determines the subtle but progressive overall position of the entire infrared signal toward higher frequencies, from ∼1045 cm−1 (500 °C and 700 °C) up to ∼1065 cm−1 (900 °C). Additionally, it is quite good visible that the participation of the component near ∼1155 cm−1 (Q4 units) gradually increases for nano-glass-ceramics fabricated at higher temperatures. Therefore, it could be concluded that increasing the annealing temperature of silicate xerogels favors the more efficient conversion of SiO4 units into Q3 and Q4 species without non-bridging oxygen atoms, proving the forming of condensed silicate sol–gel hosts. The conclusions mentioned above about the progressive polycondensation of sol–gel hosts could be additionally confirmed by a gradual shift of the signal assigned to the Si–O–Si bending vibration from 440 cm−1 (500 °C), through 444 cm−1 (700 °C), up to 451 cm−1 (900 °C). Similar results for the analogous SiO2-LaF3 sol–gel system were elaborated in the work by G. Gorni et al.49 Indeed, during the studied sol–gel evolution, the maximum of indicated signal noticeably shifted from 425 cm−1 (for xerogel) to higher wavenumbers of about 450 cm−1 for glass-ceramics prepared at 550 °C at various time intervals ranging from 1 minute to 80 hours. The maxima were compared with the location of the signal for SiO2 glass (∼460 cm−1). As was also stated in another paper,50 the observed shift is characteristic of silicate xerogels as they evolve toward more densified states with decreasing porosity. Moreover, according to the texture of sol–gel hosts, the shift of the signal assigned to the TO3 mode of Si–O–Si siloxane bridges with increasing treatment temperature is strictly attributed to structural modifications related to the change in the residual porosity of the sol–gel framework.48 Indeed, the pores induce deformations of Si–O–Si bridges that give rise to higher bridging angles and longer Si–O silanol bonds compared to bulk SiO2 glasses. Therefore, the changes in the porosity alter the appropriate modifications in Si–O–Si deformations; thus, shifts in the infrared signal are expected. This theory was justified by Almeida et al.51 and Parrill et al.,52 who proved that the maximum of the infrared signal corresponding to the TO3 mode shifts from 1073 cm−1 (500 °C) to 1083 cm−1 (900 °C) and from 1061 cm−1 (500 °C) to 1072 cm−1 (900 °C), respectively. Also, in our studies, such a shift of the component from 1069 cm−1 (500 °C) through 1075 cm−1 (700 °C) to 1083 cm−1 (900 °C) was denoted, confirming the polycondensation of the host parallelly with an elevation of heat-treatment temperature.
To determine the impact of heat-treatment conditions on the fabrication of the fluoride crystal phase, the ATR-IR spectra were also collected within the 500 cm−1–300 cm−1 frequency region to observe the vibrations originating from the La–F bond (Fig. 2). Indeed, highly visible bands with a maximum in the 360 cm−1–342 cm−1 range assigned to vibrations of fluoride crystal phase were recorded for nano-glass-ceramic samples from all the prepared series. To assess if the recorded infrared signal is associated with fluoride crystal fraction, the spectra of glass-ceramics fabricated at 500 °C, 700 °C, and 900 °C were compared with the spectra of the xerogel before heat-treatment, for which there is no band in this frequency region (Fig. 2a). The differences in the infrared spectra of xerogels and glass-ceramics could be explained by the thermal decomposition of lanthanum(III) trifluoroacetate and the further formation of a fluoride crystal fraction at about 300 °C, as shown in our previous work.53 Thus, the infrared spectra revealed the band ∼350 cm−1 only for annealed species. It is quite interesting that the frequency of La–F band was gradually shifted toward lower wavenumbers in the direction GC2Pr500 (358.7 cm−1) → GC2Pr700 (358.2 cm−1) → GC2Pr900 (355.6 cm−1) as heat-treatment temperature increases (Fig. 2a). To perform a more in-depth analysis of the changes in the band's frequency, the ATR-IR spectra were recorded for all glass-ceramic samples from individual series with varied La3+:Pr3+ molar ratio (Fig. 2b–d). Based on the obtained results, it could be observed that for samples from the series fabricated at 500 °C, the maximum of the La–F band was slightly changed (from 358.2 cm−1 for GC1Pr500 to 360.7 cm−1 for GC6Pr500). In the case of glass-ceramics produced at 700 °C, it was denoted that the tendency to increase in the La–F band's frequency from 357.2 cm−1 to 358.2 cm−1 up to reaching the La3+:Pr3+ molar ratio equaled 0.994:0.006 (GC2Pr700) and then to decrease with further growing concentration of Pr3+ ions (through 354.8 cm−1 for GC3Pr700 up to 352.8 cm−1 for GC6Pr700). Hence, any significant changes in the frequencies were observed, and the differences are greater than that for glass-ceramics fabricated at 500 °C. For samples annealed at 900 °C, the differences in the La–F band's frequencies parallelly with increasing concentration of Pr3+ ions are best visible, and a clear downward trend is observed from 357.0 cm−1 (GC1Pr900) to 342.3 cm−1 (GC6Pr900). Therefore, it seems to be reasonable to associate the observed differences in the frequencies of the La–F band with the efficiency of Pr3+ incorporation within the LaF3 crystal lattice. The considerations of the observed changes in the profiles of the recorded infrared spectra were made based on the data from the literature. Indeed, according to a paper by Kadlec et al.54 that concentrated on studying the structure of (BaF2)1−x(LaF3)x mixed crystals, the gradual entering of La3+ cations into the parent BaF2 crystal lattice with cubic symmetry was manifested by a change in the IR band frequencies. The authors reported that the progressive and significant changes in the profile of infrared spectra could be clearly observed for high x values (x > 0.13), where LaF3 formed continuously arranged clusters with tysonite symmetry within the cubic BaF2 parent crystal lattice. Indeed, the modifications in the (BaF2)1−x(LaF3)x crystals structure (influenced by high-doping level of La3+) are highly visible by the appearance of additional infrared signals as well as the shift of the band from 330 cm−1 (for x = 0, pure BaF2 crystal phase) to 402 cm−1 with growing amounts of LaF3 (x = 0.47, mixed BaF2–LaF3 crystals). Such a huge shift (Δ = 72 cm−1) in the maximum of the infrared signal is due to the significant difference in the ionic radius of Ba2+ (135 pm)55 and La3+ (122 pm),56 resulting in the modification of the lattice parameters and gradual changing of the force constants. Kovács et al.57 also studied an increase toward higher wavenumbers for selected rare earth trichlorides, i.e., CeCl3, NdCl3, SmCl3, GdCl3, and DyCl3. As a result of lanthanides contraction from Ce3+ to Dy3+, a highly visible increase in the vibrational frequency of the M–Cl bond was denoted from 328 cm−1 (CeCl3), through 333 cm−1 (NdCl3), 338 cm−1 (SmCl3), 343 cm−1 (GdCl3) up to 346 cm−1 (DyCl3). The observed shifts are not as radical as for (BaF2)1−x(LaF3)x mixed crystals, and we concluded that it is correlated with noticeable smaller differences in the ionic radius of trivalent rare earths than that for Ba2+/La3+ cations. Hence, in our experiment, we suppose that the indicated differences in the La–F band frequencies are strictly associated with the efficiency of Pr3+ incorporation inside the LaF3 crystal lattice. Due to the slightly smaller ionic radius of Pr3+ (118 pm) than of La3+ (122 pm),56 the increasing frequency should indicate the growing efficiency of Pr3+ entering into the LaF3 lattice. Indeed, this effect was clearly denoted for GC1Pr500–GC6Pr500 and GC1Pr700–GC2Pr700 samples. Unexpectedly, for the remaining GC3Pr700–GC6Pr700 and GC1Pr900–GC6Pr900 nano-glass-ceramic systems, the frequency of the infrared signal decreases, and the maximum is localized between the signal from the reference LaF3 (340.0 cm−1) and PrF3 phase (345.0 cm−1). In general, the results from XRD analysis, TEM microscopy, and luminescence characterization (presented in the next section) clearly demonstrate that the increase in the heat-treatment temperature and growing activator concentration favor the efficiency of Pr3+ incorporation inside the LaF3 nanocrystal lattice. It allows us to conclude that the initial shift of the IR signal to higher wavenumbers (GC1Pr500–GC6Pr500 and GC1Pr700–GC2Pr700) could be correlated with the incorporation of Pr3+ into the LaF3 phase at the dopant level, while for the remaining samples (GC3Pr700–GC6Pr700 and GC1Pr900–GC6Pr900), the IR signal “returns” to the frequency range close to the pure PrF3 phase; thus, we tentatively assumed that this observed nonlinear correlation and highly visible shift of the IR signals near frequency characteristic for PrF3 phase might be associated with the creation of PrF3 clusters inside the LaF3 lattice. Interestingly, the observed direction of shifting in the IR signal during the expected creation of PrF3 clusters is opposite to that of the (BaF2)1−x(LaF3)x mixed crystals.54 However, these differences could be correlated with the fact that both PrF3 and LaF3 are isostructural and crystallize in the hexagonal system; in the case of (BaF2)1−x(LaF3)x mixed crystals, the formation of LaF3 tysonite clusters within the cubic BaF2 parent lattice is responsible for causing significant changes in the symmetry and force constants, which could influence a different direction of change in the frequencies of IR signals.
XRD measurements
The XRD patterns recorded for fabricated Pr3+-doped sol–gel glass-ceramics are presented in Fig. 3. For all the prepared series, the diffraction lines are highly visible and were attributed to the hexagonal LaF3 phase crystallized in the P63cm space group (ICDD card no. 00-008-0461). Based on the broadening of the XRD lines, the average diameters (D) of the crystallized LaF3 phase were calculated using the Williamson-Hall theorem58where βhkl is the broadening of the (hkl) diffraction line, θ is the diffraction angle, λ is the X-ray wavelength, D is the average crystal size, and Z is the effective strain. The lattice strain and the crystallite size were deduced from the intercept of β
 cosθ/λ versus sinθ/λ. It was found that for all series of sol–gel samples, the LaF3 phase crystallized in the nanoscale, and the results of performed calculations are collected in Table 1. In general, analyzing the obtained data allows us to notice that the average crystallite sizes assessed for samples from GCPr500 and GCPr700 series are very similar (the range from 8.6 ± 0.1 nm to 13.5 ± 0.1 nm). In comparison, the treatment at 900 °C clearly contributed to an increase in the crystallite size (from 57.7 ± 0.3 nm to 81.7 ± 5.8 nm).
cosθ/λ versus sinθ/λ. It was found that for all series of sol–gel samples, the LaF3 phase crystallized in the nanoscale, and the results of performed calculations are collected in Table 1. In general, analyzing the obtained data allows us to notice that the average crystallite sizes assessed for samples from GCPr500 and GCPr700 series are very similar (the range from 8.6 ± 0.1 nm to 13.5 ± 0.1 nm). In comparison, the treatment at 900 °C clearly contributed to an increase in the crystallite size (from 57.7 ± 0.3 nm to 81.7 ± 5.8 nm).
 | ||
| Fig. 3 XRD patterns for the series of the fabricated sol–gel samples at: 500 °C (a), 700 °C (b), and 900 °C (c). | ||
| Oxyfluoride glass-ceramics | Lattice parameters [Å] | Crystallite size [nm] | |
|---|---|---|---|
| LaF3a | Sol–gel sample | ||
| a Refers to lattice parameters of pure LaF3 phase assigned to ICDD card no. 00-008-0461. | |||
| GC1Pr500 | a 0 = 7.184 | a 0 = 7.176(4) | 10.8 ± 0.1 |
| c 0 = 7.345(5) | |||
| GC2Pr500 | a 0 = 7.176(9) | 12.5 ± 0.2 | |
| c 0 = 7.346(8) | |||
| GC3Pr500 | a 0 = 7.178(1) | 8.6 ± 0.1 | |
| c 0 = 7.347(9) | |||
| GC4Pr500 | c 0 = 7.351 | a 0 = 7.184(3) | 9.4 ± 0.1 |
| c 0 = 7.348(1) | |||
| GC5Pr500 | a 0 = 7.185(8) | 9.7 ± 0.1 | |
| c 0 = 7.348(9) | |||
| GC6Pr500 | a 0 = 7.179(8) | 9.0 ± 0.1 | |
| c 0 = 7.347(9) | |||
| GC1Pr700 | a 0 = 7.184 | a 0 = 7.194(6) | 13.5 ± 0.1 |
| c 0 = 7.362(9) | |||
| GC2Pr700 | a 0 = 7.195(4) | 11.6 ± 0.1 | |
| c 0 = 7.361(7) | |||
| GC3Pr700 | a 0 = 7.195(3) | 9.5 ± 0.1 | |
| c 0 = 7.360(5) | |||
| GC4Pr700 | c 0 = 7.351 | a 0 = 7.192(8) | 9.7 ± 0.1 |
| c 0 = 7.358(8) | |||
| GC5Pr700 | a 0 = 7.185(2) | 10.2 ± 0.1 | |
| c 0 = 7.349(8) | |||
| GC6Pr700 | a 0 = 7.184(7) | 10.9 ± 0.1 | |
| c 0 = 7.349(9) | |||
| GC1Pr900 | a 0 = 7.184 | a 0 = 7.203(3) | 79.2 ± 1.0 |
| c 0 = 7.367(5) | |||
| GC2Pr900 | a 0 = 7.200(5) | 57.7 ± 0.3 | |
| c 0 = 7.364(1) | |||
| GC3Pr900 | a 0 = 7.198(8) | 81.7 ± 5.8 | |
| c 0 = 7.363(6) | |||
| GC4Pr900 | c 0 = 7.351 | a 0 = 7.197(4) | 68.8 ± 1.4 |
| c 0 = 7.361(5) | |||
| GC5Pr900 | a 0 = 7.195(5) | 70.0 ± 0.9 | |
| c 0 = 7.360(1) | |||
| GC6Pr900 | a 0 = 7.191(7) | 58.1 ± 0.2 | |
| c 0 = 7.352(7) | |||
Table 1 also summarizes the information about the calculated lattice parameters of the LaF3:Pr3+ phase for individual sol–gel samples. The crystal lattice parameters were determined using Le Bail method.59 Since the ionic radius of Pr3+ (r = 1.179 Å) is slightly smaller than that of La3+ (r = 1.216 Å),56 it was expected that Pr3+ co-doping––parallelly with the elevation of heat-treatment temperature––would result in a progressive decrease in the lattice parameters. Interestingly, according to the representative GC2Prx samples, the reverse trend was denoted, and the lattice parameters changed as follows: a0 = 7.179(9) Å and c0 = 7.346(8) Å (GC2Pr500), a0 = 7.195(4) Å and c0 = 7.361(7) Å (GC2Pr700), as well as a0 = 7.179(9) Å and c0 = 7.346(8) Å (GC2Pr900); thus, it could be clearly stated that the lattice parameters increased simultaneously with the increase in the annealing temperature. The observed trend might be correlated with the positive thermal expansion coefficients (along a (α⊥) and c axis (α∥) as well as the directionally averaged linear expansion coefficient (![[small alpha, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c2.gif) )) of the LaF3 fluoride phase, as was proved in the work by Korczak et al.,60 similar to that for other fluoride crystal phases, i.e., CaF2, SrF2, and BaF2.61 Such a direction of changes in the lattice parameters is consistent with the shift of infrared signals from 358.7 cm−1 (GC2Pr500), 358.2 cm−1 (GC2Pr700) to 355.6 cm−1 (GC2Pr900), as was demonstrated in Fig. 2a. The increase in the lattice parameters were also denoted for LaF3:Pr3+ nanocrystals prepared by hydrothermal (a0 = 7.201 Å, c0 = 7.432 Å) and co-precipitation (a0 = 7.197 Å, c0 = 7.375 Å) methods, as presented in a paper by M. S. Pudovkin et al.23
)) of the LaF3 fluoride phase, as was proved in the work by Korczak et al.,60 similar to that for other fluoride crystal phases, i.e., CaF2, SrF2, and BaF2.61 Such a direction of changes in the lattice parameters is consistent with the shift of infrared signals from 358.7 cm−1 (GC2Pr500), 358.2 cm−1 (GC2Pr700) to 355.6 cm−1 (GC2Pr900), as was demonstrated in Fig. 2a. The increase in the lattice parameters were also denoted for LaF3:Pr3+ nanocrystals prepared by hydrothermal (a0 = 7.201 Å, c0 = 7.432 Å) and co-precipitation (a0 = 7.197 Å, c0 = 7.375 Å) methods, as presented in a paper by M. S. Pudovkin et al.23
The modification in lattice parameters of the LaF3:Pr3+ nanocrystal phase is also correlated with the changes in the Pr3+ concentration, resulting from the variable La3+:Pr3+ molar ratio. In the case of samples fabricated at 500 °C, the lattice parameters remain almost constant (the deviations were denoted only for GC4Pr500 and GC5Pr500 samples; however, an increase in a0 and c0 parameters may result from the thermal expansion). For GC1Pr700-GC3Pr700 nano-glass-ceramic samples, similar lattice parameters were denoted, while from the GC4Pr700 sample, a more noticeable decrease in a0 and c0 parameter values was observed. Such parameter changes could suggest a particularly efficient Pr3+ incorporation into the LaF3 phase for GC4Pr700–GC6Pr700 sol–gel samples. The most remarkable differences in the lattice parameters were identified for nano-glass-ceramics fabricated during heat-treatment performed at 900 °C, suggesting the highest efficiency of Pr3+ entering into the LaF3 nanocrystal fraction compared with other series of sol–gel samples. In conjunction with the tentative conclusions from the analysis of the IR spectra according to the possibility of the PrF3 clusters formation (Fig. 2b–d), the observed changes in the lattice parameters for GC4Pr700–GC6Pr700 and GC1Pr900–GC6Pr900 samples seem to confirm this hypothesis.
TEM microscopy
The transmission electron microscopy (TEM) analyses were conducted for selected nano-glass-ceramics with low Pr3+ content (GC1Prx) and high Pr3+ concentration (GC6Prx) for each temperature level, i.e., 500 °C, 700 °C, and 900 °C. The resulting micrographs are illustrated in Fig. 4. The TEM investigations unequivocally delineated the nanocrystalline nature of the materials obtained during the controlled heat-treatment of the as-prepared xerogels at the proposed annealing conditions. The TEM micrographs recorded in both the bright and dark fields apparently show that all sol–gel samples comprise crystalline grains embedded within an amorphous host. According to the images from Fig. 4, it could be observed that following the elevation in the heat-treatment temperature, the crystalline phase becomes notably more prominent in size and crystallinity, particularly evidenced by the increase in crystallite sizes for nano-glass-ceramics obtained at 900 °C independent from Pr3+ concentration. In other words, the crystal sizes depend on the heat-treatment conditions, without any significant influence of the established La3+:Pr3+ molar ratio, as was evidenced from XRD analysis. Furthermore, the phase analysis conducted for the selected nano-glass-ceramics also agrees with the XRD results; the identification of the LaF3 nanophase with P63cm symmetry was confirmed through electron diffraction measurements. The acquired selected area electron diffraction (SEAD) patterns were indexed using phase data sourced from the ICDD database with lattice parameter values obtained from XRD analysis. The implemented heat-treatment process of the as-prepared xerogels led to the fluoride crystalline particles becoming more conspicuous, increasing in size, and transitioning into equiaxial nanoparticles. Moreover, it could be observed that these fluoride particles are dispersed more homogeneously within the amorphous sol–gel host parallelly with an elevation in heat-treatment temperature. Additional microscopic observations were carried out for the GC6Pr900 nano-glass-ceramic sample, and the results are collected in Fig. 5. The high-resolution (HR-TEM, Fig. 5c) microscopic image demonstrated the good quality of the formed LaF3 nanocrystals. The crystallization of the LaF3 phase inside the silicate sol–gel host was also confirmed by fast Fourier transform (FFT, Fig. 5d) analysis conducted on the acquired images, and the observed interplane distances correlate well with the theoretical values for the reference material. Furthermore, it should be also emphasized that in correlation with our previous assumptions regarding the efficient incorporation of Pr3+ ions inside LaF3 nanocrystals from ATR-IR and XRD analysis, the results from energy-dispersive X-ray spectroscopy (EDS, Fig. 5b) of the edge region presented in Fig. 5a confirmed that the nanocrystals consist primarily of lanthanum and fluorine, with praseodymium admixtures. Simultaneously, EDS analysis revealed that the amorphous part of the host matrix predominantly comprises silicon oxide, confirming the conclusions from ATR-IR spectra interpretation according to the polycondensation reaction of the sol–gel network.
 | ||
| Fig. 4 TEM observations of the GC1Prx (left side) and GC6Prx (right side) for the nano-glass-ceramics obtained during heat-treatment at 500 °C (a and b), 700 °C (c and d), and 900 °C (e and f). The right part of the columns presents corresponding dark field images and recorded SAED patterns. Red circles indicate theoretical Bragg positions. | ||
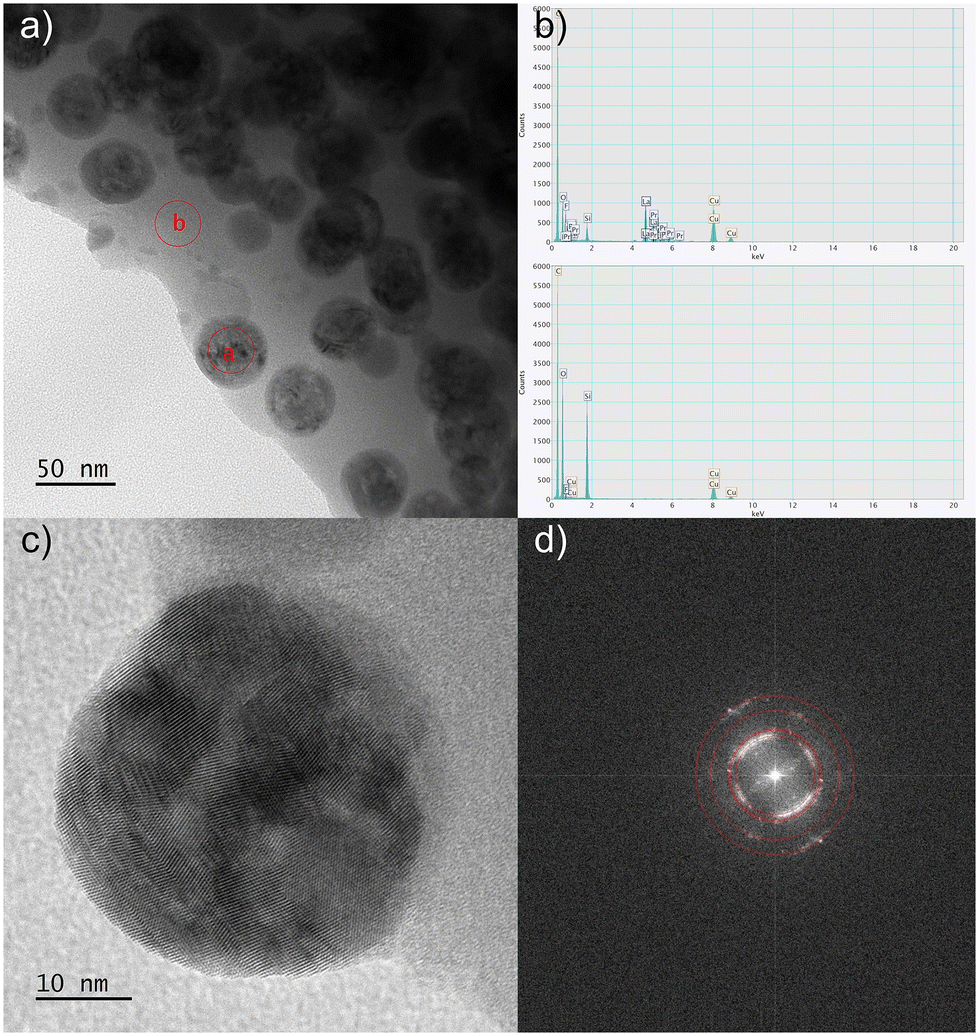 | ||
| Fig. 5 TEM observations of the GC6Pr900 nano-glass-ceramic sample: enlargement of the edge region showing the nanocrystals and the amorphous host regions (a) with red circles, which indicate the area used in the EDS analysis (b). HR-TEM image of the nanocrystals with visible interatomic planes (c) and corresponding FFT with the theoretical lattice distances marked by red rings (d) are also presented. | ||
Excitation spectra
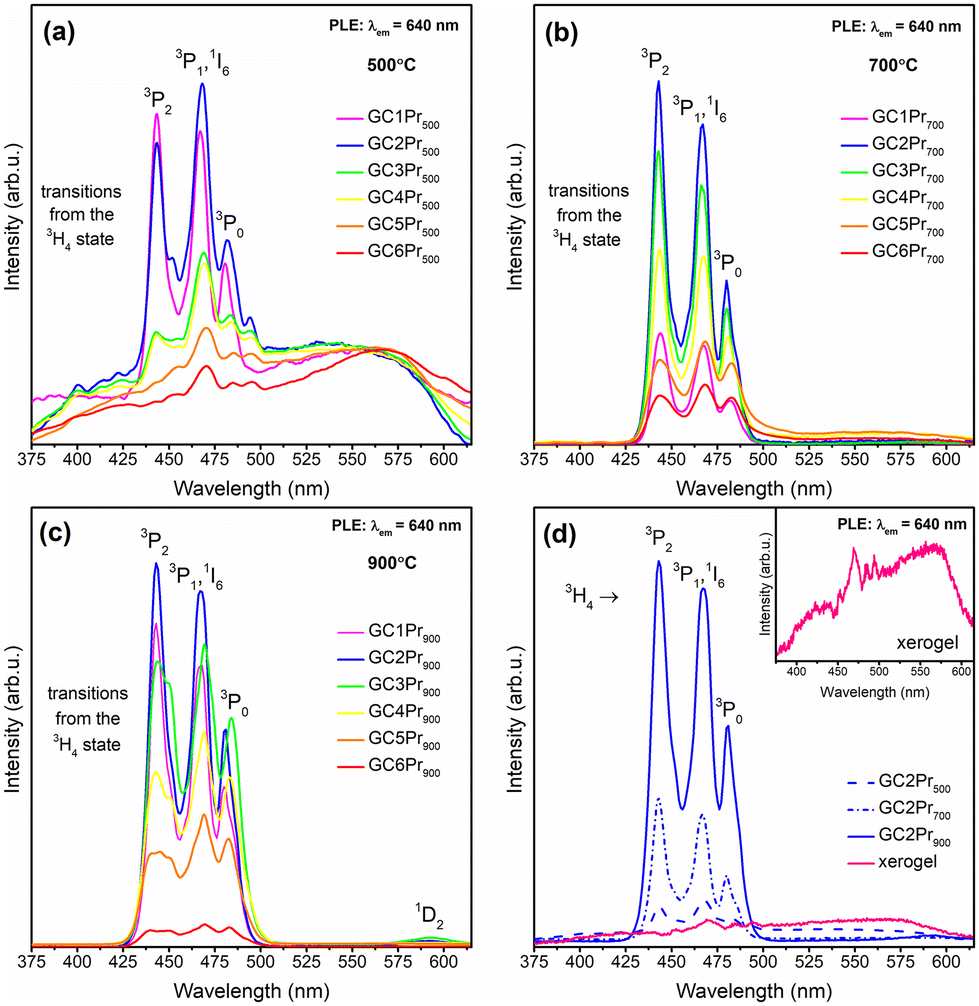
Fig. 6a–c illustrate the excitation spectra (PLE) for the individual series of Pr3+-doped oxyfluoride glass-ceramics fabricated at 500 °C, 700 °C and 900 °C, registered by collecting the red emission of the 3P0 → 3F2 transition at λem = 634 nm. The spectra consisted of 4f2 → 4f2 intra-configurational transitions of Pr3+ ions from the 3H4 ground state to the various excited 3PJ levels, appropriately labeled as the 3P2 (444 nm), 3P1 and 1I6 (467 nm), as well as 3P0 (480 nm). Moreover, the weak excitation band within the orange spectral range (592 nm), assigned to the 3H4 → 1D2 transition, was also recorded. It could be observed that for all the fabricated series, independent from the heat-treatment conditions, the intensities of individual excitation bands have grown with increasing Pr3+ content from La3+:Pr3+ molar ratio equal to 0.997:0.003 (GC1Prx) up to 0.994:0.006 (GC2Prx). However, exceeding this La3+:Pr3+ molar ratio resulted in a gradual decrease in the band intensities, suggesting that concentration quenching between neighboring Pr3+ ions (CQPr) takes place.
 | ||
| Fig. 6 Excitation spectra recorded for Pr3+-doped sol–gel nano-glass-ceramics fabricated at: 500 °C (a), 700 °C (b), and 900 °C (c). The comparison of excitation spectra for the as-prepared xerogels and GC2Prx nano-glass-ceramics with La3+:Pr3+ molar ratio equal 0.994:0.006 (d); the inset in (d) shows an enlargement of the PLE spectra recorded for the as-prepared xerogels. | ||
It should also be noted that for the series of sol–gel glass-ceramics obtained during annealing at 700 °C, the 3H4 → 3P2 excitation line at 444 nm is characterized by the highest intensity for GC1Pr700–GC4Pr700 samples, while with further increase in concentration of optically active dopant (GC5Pr700, GC6Pr700), the 3H4 → 3P1,1I6 as well as the 3H4 → 3P0 bands at 467 nm and 480 nm, respectively, became dominant. A similar tendency, according to the mutual alteration in the feeding of the 3P2, 3P1, 1I6, and 3P0 excited levels, was also denoted for samples from GC1–6Pr900 series, for which an increase in the intensities of the 3H4 → 3P1,1I6 and the 3H4 → 3P0 bands were observed from the GC3Pr900 sample. A detailed discussion on determining the effect of Pr3+ concentration on the excitation bands’ intensities was performed in the work.62 It was proven that the contribution of the 3H4 → 3P2 band's intensity according to the overall excitation spectrum gradually decreased parallelly with an increase in the Pr3+ concentration; additionally, it was denoted that the shape of the excitation bands became wider. It was assumed that such a spectroscopic behavior, characteristic for higher dopant concentration, could be explained by the reabsorption processes or by the location of the dopant ions in slightly different crystallographic sites of symmetry modified by the high Pr3+ concentration. Therefore, the observed changes in the intensities of individual excitation bands for samples from GC1–6Pr700 and GC1–6Pr900 series could suggest the high efficiency of Pr3+ ions embedding into a fluoride environment, especially according to our hypothesis about the possibility of forming PrF3 clusters within the LaF3 parent crystalline phase at high Pr3+ concentrations. Interestingly, for each nano-glass-ceramics fabricated at 500 °C (with the exception of GC1Pr500), the 3H4 → 3P1,1I6 band is more intense than the 3H4 → 3P2 one. However, it should be noted that the 3H4 → 3P1,1I6 band (∼468 nm) coincides with the strong background attributed to the photon recombinations from the defects inside the silicate sol–gel host (∼470 nm, similar to that shown for the as-prepared xerogels in the inset in Fig. 6d). According to the studies performed by Kłonkowski et al.,63 this effect could be significantly reduced after the thermal treatment at higher temperatures; therefore, it effectively disappeared for samples from the GC1–6Pr700 and GC1–6Pr900 series. Moreover, according to Fig. 6d, it could be observed that the intensities of whole excitation bands grow up in the following order of the representative samples: GC2Pr500 → GC2Pr700 → GC2Pr900, which could be explained by increasing the segregation of Pr3+ ions inside the LaF3 fluoride crystal phase with low-phonon energy. For the same reason, the 3H4 → 1D2 (592 nm) weak excitation band appeared only for glass-ceramics fabricated at 900 °C. Additionally, according to the inset in Fig. 6d, none of the PLE bands were recorded for the as-prepared xerogels, indicating a strong influence of OH groups on Pr3+ luminescence. Indeed, the observed quenching for xerogels could be explained by the occurrence of plentiful high-vibrational OH groups inside the porous silicate network, which was verified by us earlier by the presence of intense and broad band with a maximum >3000 cm−1 in the IR-ATR spectra.47,53
Emission spectra in VIS range and white light emission
The emission (PL) spectra of the individual series of Pr3+-doped glass-ceramic materials are presented in Fig. 7a–c. The spectra were recorded upon excitation at λex = 444 nm (3H4 → 3P2 transition) and showed several luminescence bands with maxima located within the greenish-blue and the reddish-orange light regions at 478 nm (3P1 → 3H4), 484 nm (3P0 → 3H4), 520 nm (3P1 → 3H5), 535 nm (3P0 → 3H5), 578 nm (3P1 → 3H6), 593 nm/598 nm (1D2 → 3H4), 608 nm (3P0 → 3H6), 633 nm/637 nm/648 nm (3P0 → 3F2), 668 nm (3P1 → 3F3), as was also illustrated in the energy level diagram (Fig. 8). The shape of the recorded emission bands is very similar to those of hexagonal LaF3:Pr3+ nanocrystals synthesized by the hydrothermal route.4 For each series, the greatest emission intensity was recorded for glass-ceramics with La3+:Pr3+ molar ratio of equal 0.994:0.006 (GC2Pr500, GC2Pr700, and GC2Pr900). From GC3Prx samples with higher content of Pr3+ ions, the luminescence started to quench, indicating the occurrence of energy transfer between neighboring Pr3+ ions in the host. The possible cross-relaxation (CR) channels involved the following transitions: {3P0 + 3H4} → {3H6 + 1D2} (1), {3P0 + 3H4} → {1G4 + 1G4} (2), {3P0 + 3H4} → {1D2 + 3H6} (3), {1D2 + 3H4} → {1G4 + 3F3} (4), {1D2 + 3H4} → {(3F3 + 3F4) + 1D2} (5), or via resonant energy transfer (RET) {1D2 + 3H4} → {3H4 + 1D2} (6).64 Similarly, as in the case of the excitation spectra, the intensities of emission bands gradually increase in the following order of representative samples: GC2Pr500 → GC2Pr700 → GC2Pr900 (Fig. 7d). Such a tendency could be associated with the growing tendency to incorporate Pr3+ ions inside LaF3 crystals and the removal of OH groups from the silicate sol–gel host. Moreover, Fig. 7d shows the comparison of PL spectra recorded for the as-prepared xerogels and GC2Prx nano-glass-ceramics. As could be seen, for the as-prepared xerogels, the luminescence of Pr3+ was not recorded, contrary to various types of Pr3+-doped glasses prepared by the conventional melt-quenching method, as presented in numerous papers published elsewhere.1,30,32,33,36,38,40–42 The observed quenching of Pr3+ emission in xerogels is strictly correlated with the occurrence of plentiful high-vibrational OH groups inside the silicate pores of the sol–gel network, which are responsible for the fast and efficient quenching of the Pr3+ luminescence from the emitting 3P0 and 1D2 levels, considering the relatively small 3P0–1D2 (∼4080 cm−1) and 1D2–1G4 (∼6570 cm−1) energy gaps. As was presented in the inset in Fig. 7d, upon excitation at λex = 444 nm, only a broad band originating from defects inside the sol–gel host was registered.63 Thus, the PL results clearly proved that only heat-treated samples revealed the characteristic 4f2–4f2 visible luminescence lines of Pr3+, and the appearance of emission characteristic for Pr3+ ions could be associated with their favorable cumulation inside precipitated fluoride nanocrystals as well as with the removal of OH groups.
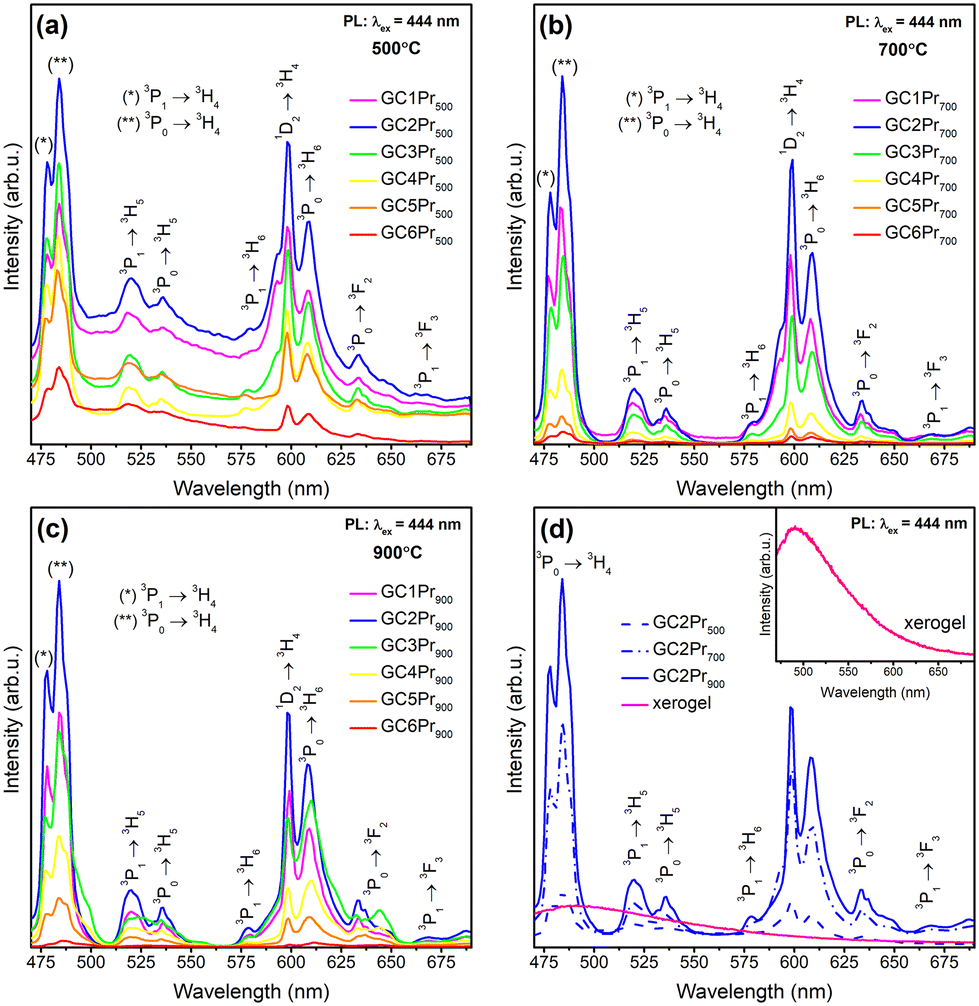 | ||
| Fig. 7 Emission spectra recorded within the VIS range for Pr3+-doped sol–gel samples prepared at: 500 °C (a), 700 °C (b), and 900 °C (c). The comparison of the emission spectra for the as-prepared xerogels and GC2Prx nano-glass-ceramics with La3+:Pr3+ molar ratio equal 0.994:0.006 (d); the inset in Fig. 5d shows an enlargement of the PL spectra recorded for the as-prepared xerogels. | ||
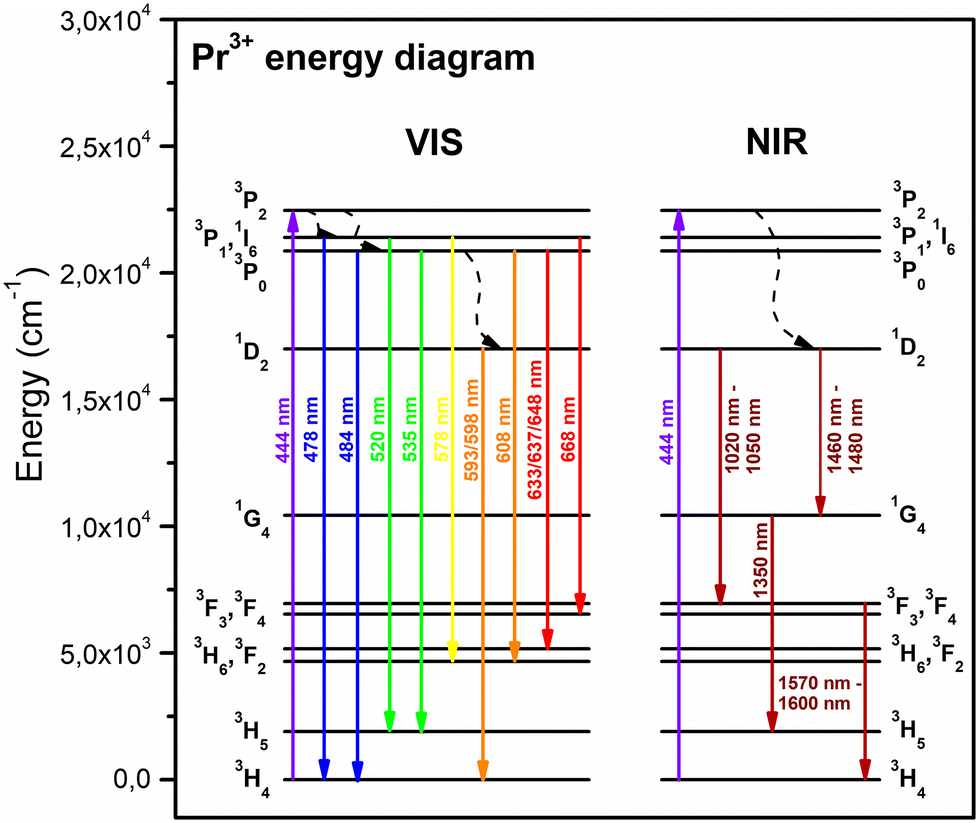 | ||
| Fig. 8 The energy level scheme of Pr3+ in the studied sol–gel samples. | ||
The further characterization of luminescent properties involved the analysis of emission intensities from the 3P0 as well as the 1D2 states of Pr3+. Indeed, the population of these two excited levels strongly depends on the concentration of Pr3+ ions3,8 and their distribution between the oxide amorphous host and the fluoride crystal lattice.65 Generally, according to the literature, at relatively low concentrations of Pr3+ ions, the multiphonon relaxation from the 3P2 state through the 3P1, 1I6, 3P0 up to the 1D2 level favors the orange luminescence assigned to the 1D2 → 3H4 transition. For higher dopant concentrations, the non-radiative energy transfer processes from excited Pr3+ to nearby unexcited Pr3+ ion become more efficient, and the luminescence associated with the 1D2 → 3H4 transition is gradually quenched by cross-relaxation (CR) mechanisms. Concentration quenching (CQPr) does not occur at low Pr3+ concentrations because the average distance among the neighboring dopants is relatively long; thus, the Pr3+–Pr3+ interaction is very weak. This effect was particularly well-observable in a paper by C. Y. Morassuti et al.3 concentrated on the characterization of Pr3+-doped calcium aluminosilicate glasses, for which the emissions from the 3P0 multiplet (into the lower-lying 3H4, 3H5, and 3F2,3,4 states) gradually increased with growing content of Pr3+ from 0.2 wt% Pr3+ up to 2.0 wt%; meanwhile, the luminescence from the 1D2 level was simultaneously quenched from 0.5 wt% Pr3+. Thus, the authors suggest that concentration quenching is strongly influenced by the cross-relaxation (CR) mechanisms between Pr3+ ions located in the immediate vicinity in the glassy host by involving the following transitions: {1D2 + 3H4} → {1G4 + 3F3} and {1D2 + 3H4} → {(3F3 + 3F4) + 1D2}. Similar results were denoted by F. Zhang et al.8 for TeO2–BaF2–NaF–PrF3 glasses, for which the orange emission from the 1D2 manifold was successfully quenched from 0.5 mol% Pr3+, while the luminescence from the 3P0 level started to quench beyond 1.5 mol% Pr3+. In the case of our experiment, as presented in Fig. 7, the emissions from the 3P0 and the 1D2 states are well-observed, and the intensity of the 1D2 → 3H4 band is higher for samples with lower concentrations of Pr3+ ions; however, for higher concentrations its intensity is reduced, suggesting the activation of cross-relaxation channels from the 1D2 manifold. Thus, these dependencies in the luminescence intensity as a function of Pr3+ concentration and heat-treatment conditions are graphically represented in Fig. 9. The luminescence intensity coefficients were calculated as ratios of the 3P0 → 3H4 band intensity to the sum of the 1D2 → 3H4 and 3P0 → 3H6 bands intensities (blue-to-reddish-orange emission, B/(RO)). For each series of prepared Pr3+-doped sol–gel materials, the calculated B/(RO)-ratio values gradually increase with growing Pr3+ ions concentration: from 0.66 (GC1Pr500) to 1.68 (GC6Pr500), from 0.87 (GC1Pr700) to 1.39 (GC6Pr700), and finally from 1.26 (GC1Pr900) to 1.65 (GC6Pr900). It could be observed that the B/(RO)-ratio values for samples with higher Pr3+ concentration from the series annealed at 500 °C are very close to those for samples from the series fabricated at 900 °C. However, we assumed that it is associated with a huge overlapping of characteristic emissions of Pr3+ with a background band from the silicate sol–gel host, which is highly visible for samples heat-treated at 500 °C. Hence, overlapping does not allow for a reliable assessment of the B/(RO) coefficients, especially for the GC4Pr500–GC6Pr500 samples.
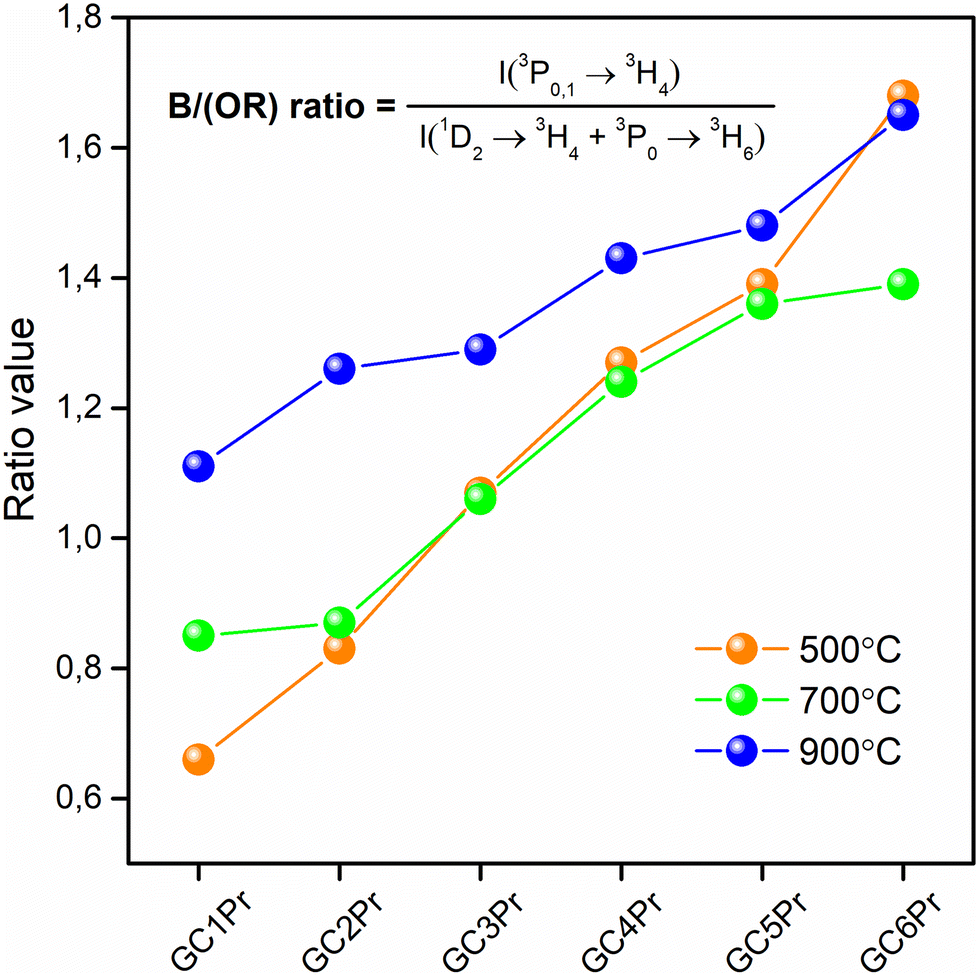 | ||
| Fig. 9 The B/(OR)-ratios calculated for the prepared Pr3+-doped sol–gel materials heat-treated at 500 °C, 700 °C, and 900 °C. | ||
It is also well-observed that the calculated B/(RO)-ratio values are generally higher as the annealing temperature increase from 500 °C to 900 °C. These differences in the B/(RO) coefficient could be explained by the vibration energies in the nearest framework around the Pr3+:LaF3 crystal lattice (340 cm−1) or the silicate sol–gel host (∼3350 cm−1 due to the presence of OH groups or ∼1050 cm−1 due to the Q3 [SiO4] units). The energy gap between the 3P0 and the 1D2 multiplets equals about ∼4080 cm−1,27 and it could be covered by 12 La–F phonons from the fluoride lattice, as well as by 4 or 1 phonon(s) of Q3 [SiO4] units or OH groups, respectively. As a consequence of the above-mentioned differences in vibrational energies, the 3P0 state is depopulated very quickly if Pr3+ ions are incorporated inside an amorphous silicate sol–gel host, and the excitation energy is transferred non-radiatively from the 3P0 multiplet to the lower-lying 1D2 state. The analysis of the available literature confirms the abovementioned considerations. For OxGCs-containing BaF2 nanocrystals, the calculated B/(RO)-ratio values are substantially higher (2.30 for the sample annealed at 600 °C/4 hours and 2.36 for the sample obtained during heat-treatment at 600 °C/24 hours) than for the parent amorphous glass (0.91).33 Therefore, the B/(RO)-ratio values increase with a growing degree of ceramization in the immediate vicinity around Pr3+ ions, confirming that an elevation in heat-treatment temperature promotes the cumulation of Pr3+ inside the LaF3 crystal phase.
Since the fabricated Pr3+-doped sol–gel nano-glass-ceramic materials emit greenish-blue and reddish-orange light (due to the 3P0,1 → 3H4–6, 1D2 → 3H4, 3P0 → 3F2, and 3P1 → 3F3 transitions), the chromaticity coordinates (x||y) were calculated and graphically presented in the CIE diagrams. The results are collected in Table 2, as well as Fig. 10 and 11. It should be noted that for the series of samples fabricated at 500 °C, the high background (associated with the photon recombinations from defects inside the silicate sol–gel hosts) does not allow for a faithful rendering of the actual color emitted by the samples. Thus, chromaticity coordinates were analyzed only for glass-ceramic samples fabricated at 700 °C and 900 °C. For the samples from the GC1Pr700–GC6Pr700 series (Fig. 10), the evaluated chromaticity coordinates shift slightly to red-tone color with the modification in the La3+:Pr3+ molar ratio from 0.997:0.003 (GC1Pr700) to 0.994:0.006 (GC2Pr700), as the content of Pr3+ ions increases. With the further growth of Pr3+ content (defined by the La3+:Pr3+ molar ratio from 0.988:0.012 (GC3Pr700) to 0.94:0.06 (GC5Pr700)), it is observed that the chromaticity coordinates shift and lie––especially for the GC4Pr700 sample––near the standard point for the illuminant. For the sample with the highest concentration of Pr3+ ions in the series (La3+:Pr3+ molar ratio equals 0.880:0.120), it is observed that the chromaticity coordinates shift into yellow-tone color. For the samples from GC1Pr900–GC6Pr900 series, the chromaticity coordinates shift from red-tone to yellow-tone color region with the increasing content of Pr3+ ions (simultaneously to decrease in the La3+:Pr3+ molar ratio from 0.997:0.003 to 0.88:0.1, Fig. 11). Nevertheless, similar to that in the case of nano-glass-ceramics prepared during heat-treatment at 700 °C, all samples from the GC1–6Pr900 series emit white light. The quality of generated white light by the individual sol–gel sample was examined by the correlated color temperature (CCT) using McCamy's analytical relation given by the below equation66
| CCT = −449n3 + 3525n2 − 6832n + 5520.33, |
in which (x, y) are the chromaticity coordinates for individual Pr3+-doped sample, (xi, yi) are the coordinates of standard white light (0.333||0.333), and (xd, yd) are the chromaticity coordinates of the dominant wavelength (determined from the straight line connecting the white light point and the (x, y) coordinates of a sample, as presented in the paper).73 Generally, the CP for standard white light equals 0%; thus, it is preferred if the CP values for the prepared Pr3+-doped sol–gel samples would be as low as possible. The calculated CP values for the individual samples in the prepared series are changed as follows: 33.3%, 24.0%, 12.8%, 16.2%, and 24.0% for GC1–6Pr700, as well as 24.5%, 21.5%, 36.6%, 26.6%, 28.7%, and 43.2% for GC1–6Pr900. The results clearly indicate that GC3Pr700 (CP = 12.8%) and GC4Pr700 (CP = 16.2%) samples exhibit bright white light, which could make them potential and interesting candidates for use in the development of white light emitters. Comparably low CP values (CP ≈ 10%, as for GC3Pr700 sample) were denoted for the Dy3+-doped CaSrAl2SiO7 phosphors prepared by high-temperature solid-state reaction.74
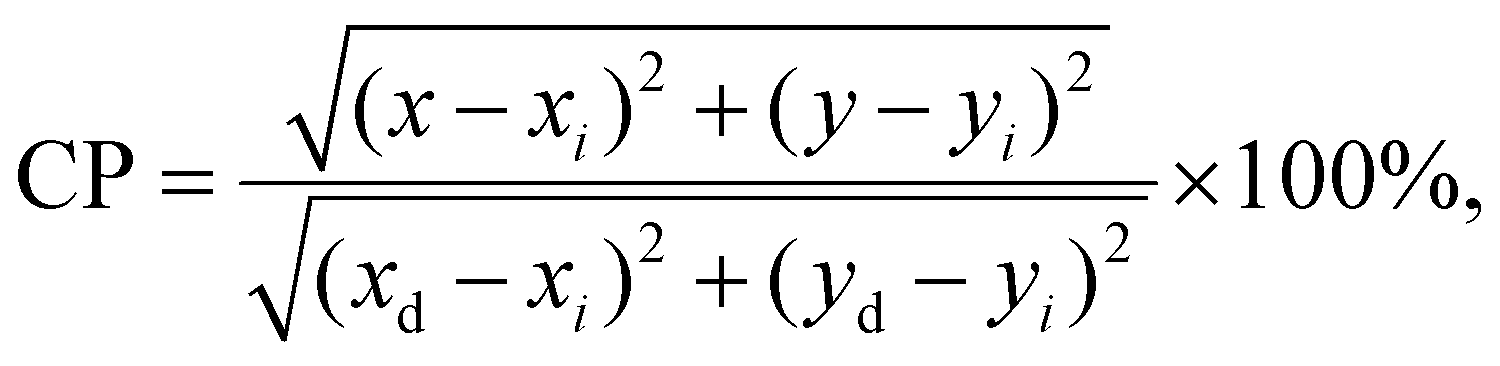
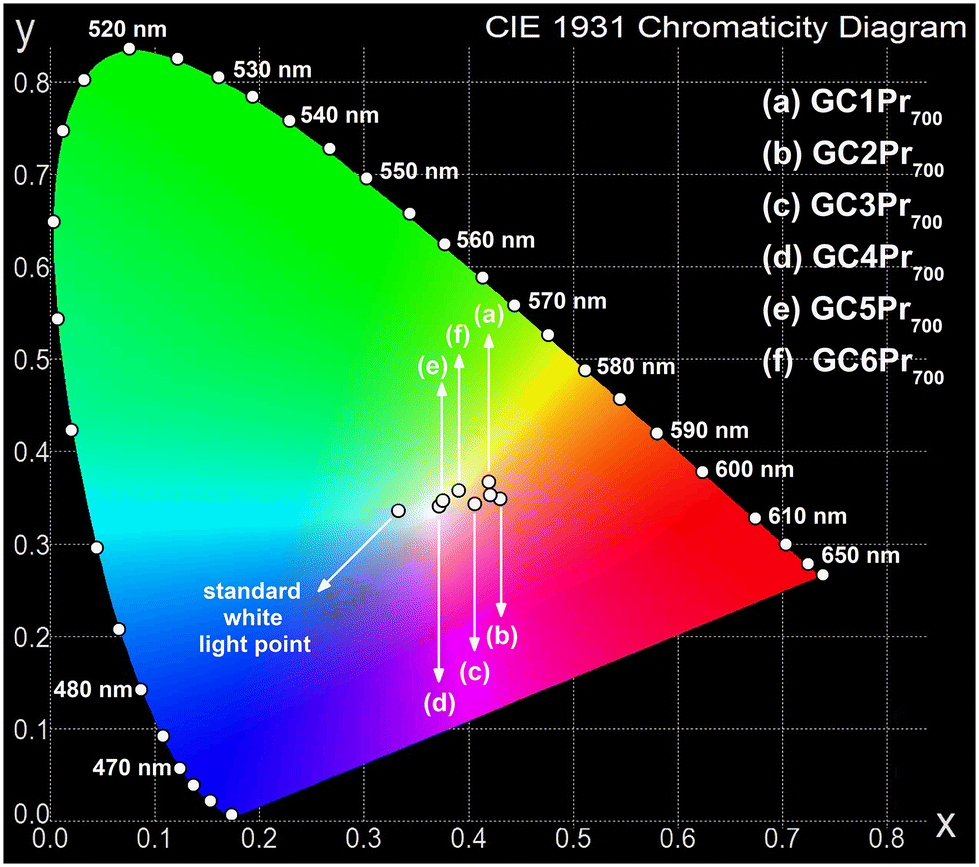 | ||
| Fig. 10 CIE chromaticity diagram with coordinates assigned for Pr3+-doped nano-glass-ceramics prepared at 700 °C. | ||
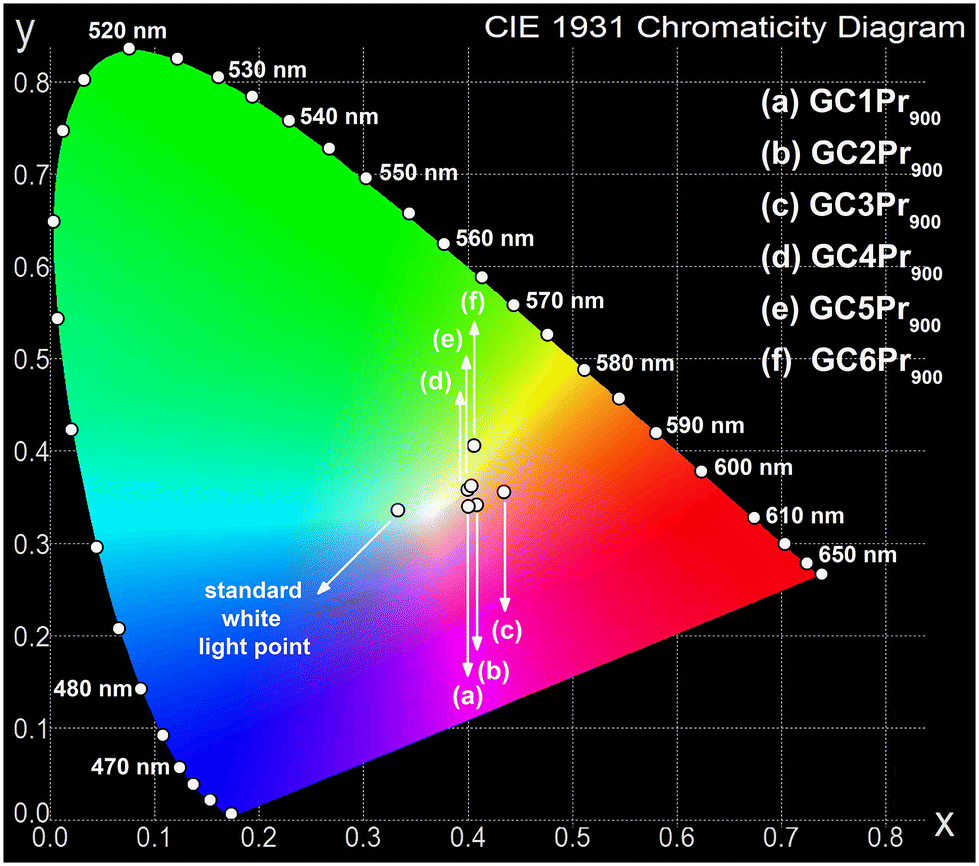 | ||
| Fig. 11 CIE chromaticity diagram with coordinates assigned for Pr3+-doped nano-glass-ceramics prepared at 900 °C. | ||
| Type of material/sample | CIE (x||y) | CCT [K]a | Ref. |
|---|---|---|---|
| a CCT was calculated using the following McCamy's formula: CCT = an3 + bn2 + cn + d. b a = −449, b = 3525, c = −6823, d = 5520.33. c a = −437, b = 3601, c = −6861, d = 5514.21. | |||
| YF3:1%Pr3+ | (0.441||0.390) | 2814b | 11 |
| BaWO4:0.01Pr3+ | (0.284||0.311) | 8680b | 26 |
| BaWO4:0.03Pr3+ | (0.291||0.325) | 7847b | |
| BaWO4:0.05Pr3+ | (0.320||0.340) | 6073b | |
| BaWO4:0.07Pr3+ | (0.297||0.337) | 7294b | |
| 60PbGeO3-20PbF2-20CdF2:0.5Pr3+ | (0.38||0.33) | 3623b | 70 |
| 60PbGeO3-20PbF2-20CdF2:0.75Pr3+ | (0.34||0.33) | 5153b | |
| β-NaYF4:0.1Pr3+ | (0.354||0.339) | 4563 | 71 |
| β-NaYF4:0.5Pr3+ | (0.323||0.338) | 5951 | |
| β-NaYF4:1.0Pr3+ | (0.307||0.335) | 6767 | |
| Lu1.94Pr0.06MoO6 | (0.315||0.312) | 6485c | 72 |
| Lu1.92Pr0.08MoO6 | (0.352||0.316) | 4499c | |
| GC1Pr700 | (0.419||0.364) | 2977b | This work |
| GC2Pr700 | (0.429||0.346) | 2581b | |
| GC3Pr700 | (0.405||0.340) | 3032b | |
| GC4Pr700 | (0.372||0.338) | 3962b | |
| GC5Pr700 | (0.375||0.344) | 3917b | |
| GC6Pr700 | (0.390||0.355) | 3577b | |
| GC1Pr900 | (0.408||0.339) | 2948b | |
| GC2Pr900 | (0.400||0.337) | 3124b | |
| GC3Pr900 | (0.434||0.353) | 2567b | |
| GC4Pr900 | (0.399||0.355) | 3343b | |
| GC5Pr900 | (0.402||0.359) | 3309b | |
| GC6Pr900 | (0.405||0.403) | 3608b | |
Emission spectra in the NIR region
The photoluminescence emission spectra inside the near-infrared (NIR) region for the synthesized Pr3+-doped oxyfluoride sol–gel materials were also recorded and are presented in Fig. 12. In the result of excitation using λex = 444 nm wavelength from blue-light scope, the emission bands for the transitions 1D2 → 3F4 (with maximum at ∼1020 nm for GC1–6Pr500 and GC1–6Pr700 series to ∼1050 nm for GC1–6Pr900 series), 1G4 → 3H5 (∼1350 nm), 1D2 → 1G4 (1460–1480 nm), and 3F3,4 → 3H4 (shifted from 1570 nm for GC1–6Pr500 series up to 1600 nm for GC1–6Pr900 series) were clearly observed. These three latter emissions overlap and form one less-resolved band. Such NIR emissions are essential for telecommunication applications, covering E, S, C, and L bands.16 | ||
| Fig. 12 Emission spectra recorded within the NIR range for Pr3+-doped sol–gel samples prepared at 500 °C (a), 700 °C (b), and 900 °C (c). The comparison of emission spectra for the as-prepared xerogels and GC2Prx nano-glass-ceramics with La3+:Pr3+ molar ratio equal to 0.994:0.006 is also presented (d). | ||
Similarly, as in the case of luminescence within the visible light scope, the modification in the La3+:Pr3+ molar ratio from 0.997:0.003 (GC1Prx) to 0.994:0.006 (GC2Prx) caused an increase in emission within the NIR region, while the further growing concentration of Pr3+ ions resulted in a substantial decrease in photoluminescence intensity. Additionally, as can be seen from Fig. 12d, the NIR emissions are enhanced with the gradual elevation in heat-treatment temperature of the parent xerogels from 500 °C up to 700 °C and 900 °C, clearly illustrating that the entry of Pr3+ into the LaF3 phase as well as the removal of residual OH groups from the sol–gel host are crucial factors determining the NIR luminescence. This hypothesis could be additionally confirmed by the fully quenched NIR luminescence for the as-prepared xerogels, as shown in Fig. 12d. Similar to the PL spectra recorded in the VIS range (Fig. 7d), the presence of high-vibrational OH groups in the porous structure of the as-prepared xerogels determines the effective share of non-radiative transitions from the emitting levels, i.e., 1D2, 1G4, and 3F3,4, strongly quenching the NIR luminescence.
Independent from heat-treatment temperature, it should also be pointed out that the NIR luminescence for the prepared GC4Prx nano-glass-ceramics is strongly inhibited, and for samples with the highest content of Pr3+ ions (GC5Prx and GC6Prx), the emission is wholly quenched. The mechanism, which is responsible for the observed photoluminescence behavior, is according to the cross-relaxation (CR) processes involving the following channels: {1D2 + 3H4} → {1G4 + (3F3,3F4)} and {1D2 + 3H4} → {(3F3,3F4) + 1G4}. Thus, the obtained results clearly indicate that NIR emissions are particularly sensitive to the shortening of the Pr3+–Pr3+ interionic distance. Indeed, for comparison, the luminescence from the 3P0, 3P1, and the 1D2 excited states in the VIS range is well-observable even for the GC5Prx and GC6Prx samples with the highest Pr3+ concentration. Additionally, it could be observed that for the samples from the series heat-treated at 500 °C and 700 °C, the intensity of the wide band in the NIR region with a maximum located near 1470 nm (1G4 → 3H5, 1D2 → 1G4, 3F3,4 → 3H4 transitions) is dominant compared with the luminescence recorded at ∼1050 nm (1D2 → 3F4 transition). Interestingly, a gradual change in the mutual intensities of these emissions is well-observable for samples with the same La3+:Pr3+ molar ratio but fabricated at varied temperature conditions. The most prominent change in the bands’ intensities was denoted for samples from the series prepared at 900 °C, especially for the GC3Pr900 material, for which the intensity of the 1D2 → 3F4 emission line noticeably increased in comparison with a wide band according to the 1G4 → 3H5, 1D2 → 1G4, 3F3,4 → 3H4 transitions. Therefore, such changes in the NIR luminescence profile of the studied sol–gel samples may indicate that the expected shortening in the Pr3+–Pr3+ distances promotes the {1D2 + 3H4} → {1G4 + (3F3,3F4)} cross-relaxation (CR) channel.
Luminescence decay analysis of the 3P0 and the 1D2 excited states of Pr3+
The further evaluation of the luminescence from Pr3+ ions in the series of prepared nano-glass-ceramic samples was carried out based on the analysis of the luminescence decay curves from 3P0 (λex = 444 nm, monitoring the 3P0 → 3H4 transition) as well as the 1D2 excited states (λex = 444 nm, monitoring the 1D2 → 1G4 transition). The resultant curves are presented in Fig. 13; all of them follow the second-order exponential nature, and the average lifetimes were calculated using the formula given belowin which A1 and A2 are percentage residual weighting factors, and τ1 and τ2 are decay components. The bi-exponential character of the recorded curves is associated with the distribution of Pr3+ ions either between a silicate amorphous sol–gel host and LaF3 nanocrystals, but it is also correlated with the involvement of cross-relaxation (CR) processes between neighboring Pr3+ ions if the critical interionic distance would be exceeded. It should also be noted that the lifetimes of the 1D2 state have been designated only for GC1Prx–GC3Prx glass-ceramics from each series. Although the NIR luminescence has been recorded for GC4Prx samples, but its intensity is very low; thus, the lifetimes would be calculated with a huge error. The resultant τavg(3P0):Pr3+ and τavg(1D2):Pr3+ average lifetimes, compared with the literature data for other Pr3+-doped oxyfluoride glass-ceramics, are collected in Tables 3 and 4, respectively. Generally, it should be noticed that the lifetimes of the 1D2 excited state are longer compared with those of the 3P0 level, and similar observations were also reported for Pr3+-doped OxGCs containing BaF2,33 PbF2,39 and LaF3 crystals.41
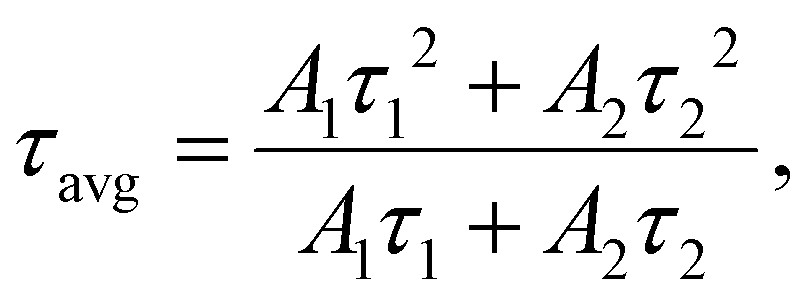
 | ||
| Fig. 13 Luminescence decay curves recorded for the 3P0 and the 1D2 states of Pr3+ ions in the fabricated nano-glass-ceramic samples. | ||
| Oxyfluoride glass-ceramics | Type of crystalline phase | τ(3P0) [μs] | Ref. |
|---|---|---|---|
| GC1Pr500 | LaF3 | 24.9 | This work |
| GC2Pr500 | 31.8 | ||
| GC3Pr500 | 21.5 | ||
| GC4Pr500 | 7.9 | ||
| GC5Pr500 | 6.7 | ||
| GC6Pr500 | 5.3 | ||
| GC1Pr700 | LaF3 | 42.6 | This work |
| GC2Pr700 | 52.9 | ||
| GC3Pr700 | 31.0 | ||
| GC4Pr700 | 12.6 | ||
| GC5Pr700 | 7.9 | ||
| GC6Pr700 | 5.4 | ||
| GC1Pr900 | LaF3 | 53.5 | This work |
| GC2Pr900 | 58.4 | ||
| GC3Pr900 | 40.0 | ||
| GC4Pr900 | 20.4 | ||
| GC5Pr900 | 8.7 | ||
| GC6Pr900 | 4.9 | ||
| 48.8TeO2-30ZnO-10YF3-10NaF-0.2Pr2O3 | NaYF4 | 24–62 | 1 |
| 41SiO2-10Al2O3-25.5LiF-23SrF2-0.5Pr2O3 | SrF2 | 12.0 | 30 |
| 67.7SiO2-14.9BaF2-12.9K2CO3-3La2O3-1.0Sb2O3-0.5Pr2O3 | BaF2 | 40–54 | 33 |
| 50GeO2-45PbO-5PbF2:2 mol% Pr3+ | PbF2 | 8.5 | 39 |
| 45SiO2-15Al2O3-10Na2O-24BaF2-6Y2O3:0.1 mol% Pr3+ | BaYF5 | 30.1 | 40 |
| 40SiO2-20Al2O3-20LiF-19GdF3-1.0PrF3 | LiGdF4/GdF3 | 57.0 | 41 |
| Oxyfluoride glass-ceramics | Type of crystalline phase | τ(1D2) [μs] | Ref. |
|---|---|---|---|
| GC1Pr500 | LaF3 | 106.0 | This work |
| GC2Pr500 | 127.4 | ||
| GC3Pr500 | 99.3 | ||
| GC4Pr500 | 35.5 | ||
| GC1Pr700 | LaF3 | 238.3 | This work |
| GC2Pr700 | 274.5 | ||
| GC3Pr700 | 151.6 | ||
| GC4Pr700 | 58.1 | ||
| GC1Pr900 | LaF3 | 174.8 | This work |
| GC2Pr900 | 195.3 | ||
| GC3Pr900 | 132.2 | ||
| GC4Pr900 | 49.2 | ||
| 67.7SiO2-14.9BaF2-12.9K2CO3-3La2O3-1.0Sb2O3-0.5Pr2O3 | BaF2 | 51–78 | 33 |
| 50GeO2-45PbO-5PbF2:2 mol% Pr3+ | PbF2 | 109.0 | 39 |
| SiO2-Al2O3-AlF3-Na2CO3-NaNO3-BaCO3-Ba(NO3)2-LaF3:Pr3+ (x = 0.01–0.5 mol%) | LaF3 | 525–25 | 41 |
From the analysis of the obtained data, it could be assumed that the τavg(3P0):Pr3+ lifetimes are prolonged with changing La3+:Pr3+ molar ratio from 0.997:0.003 (24.9 μs (500 °C), 42.6 μs (700 °C), 53.4 μs (900 °C)) to 0.994:0.006 (31.8 μs (500 °C), 52.9 μs (700 °C), 58.4 μs (900 °C)). A similar tendency was observed for the τavg(1D2):Pr3+ lifetimes, whose values were changed from 106.0 μs (500 °C), 238.3 μs (700 °C), and 174.5 μs (900 °C) (for samples with La3+:Pr3+ molar ratio of 0.997:0.003) to 127.4 μs (500 °C), 274.5 μs (700 °C), and 195.3 μs (900 °C) (for La3+:Pr3+ molar ratio of 0.994:0.006). Further changes in the La3+:Pr3+ molar ratios resulted in a progressive and strong reduction in the τavg(3P0):Pr3+ and τavg(1D2):Pr3+ decay times to values of about ∼5 μs (3P0) and 35.5–58.1 μs (1D2), respectively. Such optical behavior of the fabricated Pr3+-doped oxyfluoride sol–gel materials clearly indicates that a La3+:Pr3+ molar ratio of 0.994:0.006 determines the critical Pr3+–Pr3+ interionic distance, beyond which the concentration quenching process (CQPr) is well-observable in each of the prepared series.
According to the τavg(3P0):Pr3+ lifetimes, it could be stated that their values undergo a progressive prolongation for the individual samples with the same La3+:Pr3+ molar ratio along with the simultaneous elevation of the heat-treatment temperature (500 °C, 700 °C, and 900 °C) of the initial amorphous xerogels. This behavior clearly reveals the beneficial impact of the efficient migration of Pr3+ into the lattice of LaF3 nanocrystals characterized by low-phonon energy (∼340 cm−1) and the removal of OH groups from the sol–gel host on the photoluminescent properties. However, for the τavg(1D2):Pr3+ lifetimes, the observed tendency is slightly different because the longest decay times were denoted for the series of glass-ceramics heat-treated at 700 °C. In comparison with the samples from GC1–3Pr500 series, the lifetimes of the 1D2 level are noticeably extended for the GC1–3Pr700 materials, indicating the favorable influence of the partial elimination of OH groups, which are mainly responsible as quenching channels for suppressing the photoluminescence in the NIR region; the observed elongation of τavg(1D2):Pr3+ lifetimes is also caused by the more efficient incorporation of Pr3+ ions inside the LaF3 crystal lattice during annealing performed at 700 °C than at 500 °C. In contrast to the lifetimes from the 3P0 state, the further elevation in the heat-treatment temperature (900 °C) led to the shortening of the decay times from the 1D2 excited level, suggesting the progressive shortening of the Pr3+–Pr3+ distances, which is sufficient to quench the NIR luminescence. According to the differences mentioned above in the luminescence decay kinetics from the 3P0 and the 1D2 levels, it could be suggested that cross-relaxation (CR) processes occur in divergent ways for each of these two states. For this purpose, we compared the cross-relaxation efficiencies (ηCR) from the 3P0 and the 1D2 levels for the studied glass-ceramic samples based on estimations from the below equation.3
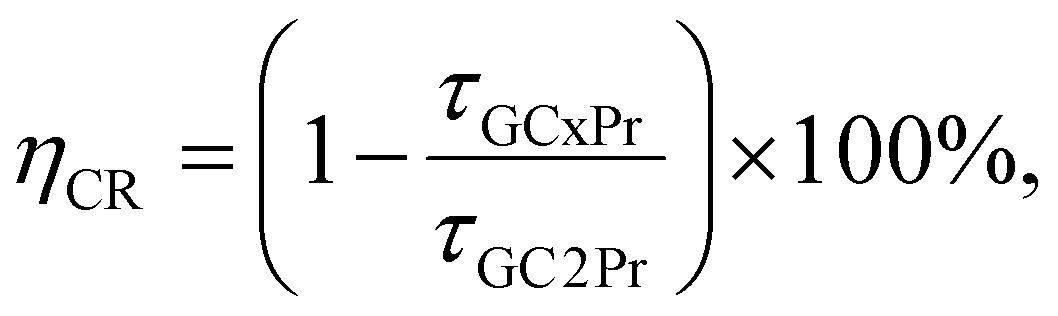 :Pr3+ 0.994:0.006 molar ratio, beyond which concentration quenching (CQPr) occurred. For the 3P0 level, the ηCR values are 32%, 75%, 79%, and 83% for GC3Pr500–GC6Pr500 samples, 41%, 76%, 85%, and 90% for GC3Pr700–GC6Pr700 series, as well as 32%, 65%, 85%, and 92% for GC3Pr900–GC6Pr900 glass-ceramics. Therefore, it could be observed that the ηCR(3P0) values for the samples with the same La3+:Pr3+ molar ratio but fabricated at different temperature conditions are quite similar; however, ηCR(3P0) ≥ 90% values were achieved only for samples from the series obtained at 700 °C and 900 °C. The ηCR values for the 1D2 state changed for GC3Prx as follows: 22% (GC3Pr500), 45% (GC3Pr700), and 32% (GC3Pr900). It is quite interesting that ηCR(3P0) > ηCR(1D2) for GC3Pr500, and ηCR(3P0) ≈ ηCR(1D2) for GC3Pr700 and GC3Pr900. Additionally, based on the NIR spectra (Fig. 12) and completely quenched luminescence for GC5Pr and GC6Pr samples from the series fabricated at 700 °C and 900 °C (it should be noted that for the GC5Pr500 sample, weak NIR emission is still detectable), we assumed that further differences in the ηCR(1D2) values would be highly visible, indicating that the luminescence from the 1D2 state is strongly sensitive to Pr3+–Pr3+ interionic distances in the studied sol–gel materials. Therefore, based on the performed luminescence measurements, we could suppose that the effect of cross-relaxation (CR) processes from the 1D2 excited state of Pr3+ on the overall luminescence – compared to the cross-relaxation (CR) processes from the 3P0 state – is particularly dependent on the heat-treatment temperature of initial xerogels, which influences the preferable cumulation of Pr3+ ions inside the LaF3 phase, resulting in the probability of formation of continuously arranged PrF3 clusters for samples fabricated at 700 °C and 900 °C (as was tentatively assumed from the performed ATR-IR and XRD measurements). In addition, it seems that the size of the precipitated fluoride nanocrystals obtained for individual series of sol–gel samples could also influence the involvement of CR processes, which might be potentially correlated with uniformity in Pr3+ distribution inside the fluoride crystal phase.
:Pr3+ 0.994:0.006 molar ratio, beyond which concentration quenching (CQPr) occurred. For the 3P0 level, the ηCR values are 32%, 75%, 79%, and 83% for GC3Pr500–GC6Pr500 samples, 41%, 76%, 85%, and 90% for GC3Pr700–GC6Pr700 series, as well as 32%, 65%, 85%, and 92% for GC3Pr900–GC6Pr900 glass-ceramics. Therefore, it could be observed that the ηCR(3P0) values for the samples with the same La3+:Pr3+ molar ratio but fabricated at different temperature conditions are quite similar; however, ηCR(3P0) ≥ 90% values were achieved only for samples from the series obtained at 700 °C and 900 °C. The ηCR values for the 1D2 state changed for GC3Prx as follows: 22% (GC3Pr500), 45% (GC3Pr700), and 32% (GC3Pr900). It is quite interesting that ηCR(3P0) > ηCR(1D2) for GC3Pr500, and ηCR(3P0) ≈ ηCR(1D2) for GC3Pr700 and GC3Pr900. Additionally, based on the NIR spectra (Fig. 12) and completely quenched luminescence for GC5Pr and GC6Pr samples from the series fabricated at 700 °C and 900 °C (it should be noted that for the GC5Pr500 sample, weak NIR emission is still detectable), we assumed that further differences in the ηCR(1D2) values would be highly visible, indicating that the luminescence from the 1D2 state is strongly sensitive to Pr3+–Pr3+ interionic distances in the studied sol–gel materials. Therefore, based on the performed luminescence measurements, we could suppose that the effect of cross-relaxation (CR) processes from the 1D2 excited state of Pr3+ on the overall luminescence – compared to the cross-relaxation (CR) processes from the 3P0 state – is particularly dependent on the heat-treatment temperature of initial xerogels, which influences the preferable cumulation of Pr3+ ions inside the LaF3 phase, resulting in the probability of formation of continuously arranged PrF3 clusters for samples fabricated at 700 °C and 900 °C (as was tentatively assumed from the performed ATR-IR and XRD measurements). In addition, it seems that the size of the precipitated fluoride nanocrystals obtained for individual series of sol–gel samples could also influence the involvement of CR processes, which might be potentially correlated with uniformity in Pr3+ distribution inside the fluoride crystal phase.
Conclusions
In this work, we have synthesized and investigated the series of oxyfluoride Pr3+-doped nano-glass-ceramics fabricated by sol–gel method and annealed at different temperatures, i.e., 500 °C, 700 °C, and 900 °C. Based on the performed structural measurements (XRD, ATR-IR) and TEM microscopy, it was proven that the gradual elevation of annealing temperature favors the densification of the silicate sol–gel host by the efficient removal of OH groups and crystallization of the LaF3 phase in the nanometer range, with preferable cumulated Pr3+ optically active ions. Additionally, the observed changes in the localization of the IR signal assigned to the vibrations of the fluoride crystal lattice allowed for the tentative assumption that the incorporation of Pr3+ ions inside the LaF3 phase is so effective that increasing the Pr3+ concentration in the samples from the series fabricated at 700 °C and 900 °C favors the formation of continuously arranged PrF3 clusters within the LaF3 parent crystal phase. All of the prepared LaF3:Pr3+-based oxyfluoride nano-glass-ceramic samples exhibit strong luminescence in the VIS range from greenish-blue to reddish-orange light, whose combination allows for successfully obtaining warm or neutral white light emissions with CCT values ranging from 2567 K to 3962 K. Additionally, the luminescence of Pr3+ ions within the NIR region was also recorded, and it covers E, S, C, and L bands belonging to the fifth optical telecommunication window. Based on the luminescence decay analysis, it was verified that the longest lifetimes of the 3P0 excited state were attributed to glass-ceramics fabricated by the controlled heat-treatment of initial xerogels at 900 °C, while the longest lifetimes of the 1D2 level was attributed to samples from the series annealed at 700 °C. Independently from the heat-treatment conditions, the longest decay times, both for the 3P0 and the 1D2 levels, were estimated for samples with La3+:Pr3+ molar ratio of 0.994:0.006, beyond which the concentration quenching (CQPr) process between neighboring Pr3+ ions takes place. Further analysis of the decay kinetics showed that the involvement of cross-relaxation (CR) channels from the 1D2 state is particularly sensitive to the heat-treatment conditions, which could be correlated with the size of the fluoride crystals and the possible creation of PrF3 clusters within the parent LaF3 fluoride crystal lattice. The obtained results suggest that the fabricated Pr3+-doped oxyfluoride sol–gel nano-glass-ceramics could be predisposed for applications as white light emitters and optical elements of NIR amplifiers.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research activities are co-financed by the funds granted under the Research Excellence Initiative of the University of Silesia in Katowice.References
- D. Rajesh, Opt. Mater., 2018, 86, 178 Search PubMed.
- H. Lee, W. J. Chung and W. B. Im, J. Lumin., 2021, 236, 118064 Search PubMed.
- C. Y. Morassuti, L. H. C. Andrade, J. R. Silva, M. L. Baesso, F. B. Guimarães, J. H. Rohling, L. A. O. Nunes, G. Boulon, Y. Guyot and S. M. Lima, J. Lumin., 2019, 210, 376 Search PubMed.
- T. Q. H. Tran, M. H. Hoang, T. A. T. Do, A. T. Le, T. H. Nguyen, T. D. Nguyen and M. T. Man, J. Lumin., 2021, 237, 118162 Search PubMed.
- N. Deopa, A. S. Rao Dr., S. k. Mahamuda, M. Gupta, M. Jayasimhadri, D. Haranath and G. Vijaya Prakash, J. Alloys Compd., 2017, 708, 911 Search PubMed.
- Z. He, C. Zhu, S. Huang and X. Wu, Vacuum, 2019, 159, 269 Search PubMed.
- Z. Chen, X. Wang, S. Pan and J. Pan, J. Lumin., 2021, 236, 118141 Search PubMed.
- F. Zhang, Z. Bi, A. Huang and Z. Xiao, J. Lumin., 2015, 160, 85 Search PubMed.
- W. Wang, J. Tian, N. Li, J. Dong, Q. Wang, Y. Xu, X. Xu, H. Tang, H. Lin, D. Li and J. Xu, J. Alloys Compd., 2021, 887, 161327 Search PubMed.
- M. Venkateswarlu, M. V. V. K. S. Prasad, K. Swapna, Sk. Mahamuda, A. S. Rao, A. M. Babu and D. Haranath, Ceram. Int., 2014, 40, 6261 Search PubMed.
- A. Jusza, L. Lipińska, M. Baran, A. Olszyna, A. Jastrzębska, M. Gil, P. Mergo and R. Piramidowicz, Opt. Mater., 2019, 95, 109247 Search PubMed.
- A. K. Bedyal, D. D. Ramteke, V. Kumar and H. C. Swart, Mater. Res. Bull., 2018, 103, 173 Search PubMed.
- L. R. R. Nunes, H. P. Labaki, F. J. Caixeta and R. R. Gonçalves, J. Lumin., 2022, 241, 118485 Search PubMed.
- M. Y. Tsang, P. Fałat, M. A. Antoniak, R. Ziniuk, S. J. Zelewski, M. Samoć, M. Nyk, J. Ou, T. Y. Ohulchanskyy and D. Wawrzyńczyk, Nanoscale, 2022, 14, 14770 Search PubMed.
- Y. Fan, P. Wang, Y. Lu, R. Wang, L. Zhou, X. Zheng, X. Li, J. A. Piper and F. Zhang, Nat. Nanotechnol., 2018, 13, 941 Search PubMed.
- L. F. Shen, B. J. Chen, H. Lin and E. Y. B. Pun, J. Alloys Compd., 2015, 622, 1093 Search PubMed.
- D. Wei, H. J. Seo, Y. Liu and X. Yang, J. Lumin., 2023, 253, 119488 Search PubMed.
- S. Wang, J. Zhang, Z. Ye, H. Yu and H. Zhang, Chem. Eng. J., 2021, 414, 128884 Search PubMed.
- R. Shi, L. Lin, P. Dorenbos and H. Liang, J. Mater. Chem. C, 2017, 5, 10737 Search PubMed.
- J. Stefanska, K. Maciejewska and L. Marciniak, New J. Chem., 2021, 45, 11898 Search PubMed.
- M. Runowski, P. Woźny, I. R. Martín, V. Lavín and S. Lis, J. Lumin., 2019, 214, 116571 Search PubMed.
- M. S. Pudovkin, O. A. Morozov, V. V. Pavlov, S. L. Korableva, E. V. Lukinova, Y. N. Osin, V. G. Evtugyn, R. A. Safiullin and V. V. Semashko, J. Nanomater., 2017, 3108586 Search PubMed.
- M. S. Pudovkin, S. L. Korableva, D. A. Koryakovtseva, E. V. Lukinova, A. V. Lovchev, O. A. Morozov and V. V. Semashko, J. Nanopart. Res., 2019, 21, 266 Search PubMed.
- J. P. Zuniga, S. K. Gupta, M. Pokhrel and Y. Mao, New J. Chem., 2018, 42, 9381 Search PubMed.
- E. Elsts, G. Krieke, U. Rogulis, K. Smits, A. Zolotarjovs, J. Jansons, A. Sarakovskis and K. Kundzins, Opt. Mater., 2016, 59, 130 Search PubMed.
- R. Kamal and H. Hafez, Novel, Opt. Mater., 2021, 121, 111646 Search PubMed.
- P. Singh and J. Joshi, J. Lumin., 2018, 203, 331 Search PubMed.
- M. Kuwik, C. K. Jayasankar, W. A. Pisarski and J. Pisarska, J. Alloys Compd., 2020, 842, 155801 Search PubMed.
- L. P. Naranjo, N. T. C. Oliveira, L. R. P. Kassab and C. B. de Araújo, J. Lumin., 2021, 238, 118225 Search PubMed.
- C. R. Kesavulu, C. S. Dwaraka Viswanath, S. Karki, P. Aryal, H. J. Kim and C. K. Jayasankar, Ceram. Int., 2018, 44, 1737 Search PubMed.
- T. Murakami and S. Tanabe, J. Ceram. Soc. Jpn., 2007, 115, 605 Search PubMed.
- C. Koepke, M. Środa, A. Marczewska, K. Wisniewski and P. Pichniarczyk, J. Lumin., 2016, 179, 139 Search PubMed.
- K. Biswas, A. D. Sontakke, J. Ghosh and K. Annapurna, J. Am. Ceram. Soc., 2010, 93, 1010 Search PubMed.
- H. Zheng, X.-J. Wang, S.-X. Qu, M. J. Dejneka and R. S. Meltzer, J. Lumin., 2006, 119–120, 153 Search PubMed.
- X.-J. Wang, S. H. Huang, R. Reeves, W. Wells, M. J. Dejneka, R. S. Meltzer and W. M. Yen, J. Lumin., 2001, 94–95, 229 Search PubMed.
- Y. Xu, X. Zhang, S. Dai, B. Fan, H. Ma, J.-L. Adam, J. Ren and G. Chen, J. Phys. Chem. C, 2011, 115, 13056 Search PubMed.
- H. Zheng, X.-J. Wang, M. J. Dejneka, W. M. Yen and R. S. Meltzer, J. Lumin., 2004, 108, 395 Search PubMed.
- Y. Yu and X. Li, Mater. Res. Bull., 2016, 73, 96 Search PubMed.
- B. Klimesz, G. Dominiak-Dzik, P. Solarz, M. Żelechower and W. Ryba-Romanowski, J. Alloys Compd., 2004, 382, 292 Search PubMed.
- M. Gu, Q.-C. Gao, S.-M. Huang, X.-L. Liu, B. Liu and C. Ni, J. Lumin., 2012, 132, 2531 Search PubMed.
- G. Lakshminarayana and J. Qiu, Phys. B, 2009, 404, 1169 Search PubMed.
- J. J. Velázquez, R. Balda, J. Fernández, G. Gorni, L. Pascual, G. Chen, M. Sundararajan, A. Durán and M. J. Pascual, J. Lumin., 2018, 193, 61 Search PubMed.
- Y. Sun, M. Wu, S. Tian, Z. Shi, Z. Lun, Q. Jiang, Y. Zhao, D. Chen, P. Xiong and Z. Yang, J. Mater. Chem. C, 2022, 10, 5266 Search PubMed.
- P. P. Fedorov, A. A. Luginina and A. I. Popov, J. Fluor. Chem., 2015, 172, 22 Search PubMed.
- A. E. Danks, S. R. Hall and Z. Schnepp, Mater. Horiz., 2016, 3, 91 Search PubMed.
- J. J. Velázquez, A. C. Yanes, J. del-Castillo, J. Méndez-Ramos and V. D. Rodríguez, J. Non-Cryst. Solids, 2010, 356, 1349 Search PubMed.
- N. Pawlik, B. Szpikowska-Sroka, T. Goryczka, M. Zubko, J. Lelątko and W. A. Pisarski, J. Eur. Ceram. Soc., 2019, 39, 5010 Search PubMed.
- P. Innocenzi, J. Non-Cryst. Solids, 2003, 316, 309 Search PubMed.
- G. Gorni, M. J. Pascual, A. Caballero, J. J. Velázquez, J. Mosa, Y. Castro and A. Durán, J. Non-Cryst. Solids, 2018, 501, 145 Search PubMed.
- C. Kinowski, M. Bouazaoui, R. Bechara, L. L. Hench, J. M. Nedelec and S. Turrell, J. Non-Cryst. Solids, 2001, 291, 143 Search PubMed.
- R. M. Almeida, H. C. Vasconcelos and L. M. Ilharco, SPIE Sol-gel Optics III, 1994, 2288, 678 Search PubMed.
- T. M. Parrill, J. Mater. Res., 1992, 7, 2230 Search PubMed.
- N. Pawlik, B. Szpikowska-Sroka, E. Pietrasik, T. Goryczka and W. A. Pisarski, J. Am. Ceram. Soc., 2018, 101, 4654 Search PubMed.
- F. Kadlec, P. Simon and N. Raimboux, J. Phys. Chem. Solids, 1998, 60, 861 Search PubMed.
- X. Chen and Y. Wu, J. Alloys Compd., 2020, 817, 153075 Search PubMed.
- P. D'Angelo, A. Zitolo, V. Migliorati, G. Chillemi, M. Duvail, P. Vitorge, S. Abadie and R. Spezia, Inorg. Chem., 2011, 50, 4572 Search PubMed.
- A. Kovács, R. J. M. Konings and A. S. Booij, Vib. Spectrosc., 1995, 10, 65 Search PubMed.
- S. Devesa, A. P. Rooney, M. P. Graça, D. Cooper and L. C. Costa, Mater. Sci. Eng., B, 2021, 263, 114830 Search PubMed.
- A. Le Bail, Powder Diffr., 2005, 20, 316 Search PubMed.
- W. Korczak and P. Mikołajczak, J. Cryst. Growth, 1983, 61, 601 Search PubMed.
- R. B. Roberts and G. K. White, J. Phys. C: Solid State Phys., 1986, 19, 7167 Search PubMed.
- J. Stefanska and L. Marciniak, Adv. Photonics Res., 2021, 2100070 Search PubMed.
- A. M. Kłonkowski, W. Wiczk, J. Ryl, K. Szczodrowski and D. Wileńska, J. Alloys Compd., 2017, 724, 649 Search PubMed.
- V. H. Rao, P. S. Prasad and K. S. Babu, Opt. Mater., 2020, 101, 109740 Search PubMed.
- M. Y. A. Yagoub, H. C. Swart and E. Coetsee, Mater. Res. Bull., 2017, 93, 170 Search PubMed.
- C. S. McCamy, Color Res. Appl., 1992, 17, 142 Search PubMed.
- D. Ramachari, L. Rama Moorthy and C. K. Jayasankar, Ceram. Int., 2014, 40, 11115 Search PubMed.
- I. Golasi, F. Salata, E. de Lieto Vollaro and A. Peña-García, Energy Build., 2019, 185, 112 Search PubMed.
- C. Liu, L. Sun, X. Jing, Y. Zhang, X. Meng, C. Jia and W. Gao, J. Build. Eng., 2022, 45, 103573 Search PubMed.
- A. S. Gouveia-Neto, N. P. S. M. Rios and L. A. Bueno, Opt. Mater., 2012, 35, 126 Search PubMed.
- W. Tang, B. Wang, Y. Chen, Y. Chen, J. Lin and Q. Zeng, Opt. Mater. Express, 2019, 9, 223 Search PubMed.
- D. Zeng, Y. Chen, K. Cai, C. Peng and S. Peng, J. Lumin., 2019, 206, 376 Search PubMed.
- Y.-F. Wu, Y.-T. Nien, Y.-J. Wang and I.-G. Chen, J. Am. Ceram. Soc., 2012, 95, 1360 Search PubMed.
- S. Sharma, N. Brahme, D.P. Bisen and P. Dewangan, Opt. Express, 2018, 26, 29495 Search PubMed.
| This journal is © The Royal Society of Chemistry 2024 |