
Single-atom alloy structure and unique bonding properties of Au104Ag40(PET)60 nanoclusters†
David
Morris
 a,
Xiangsha
Du
b,
Rongchao
Jin
b and
Peng
Zhang
*a
a,
Xiangsha
Du
b,
Rongchao
Jin
b and
Peng
Zhang
*a
aDepartment of Chemistry, Dalhousie University, Halifax B3H 4R2, Canada. E-mail: Peng.Zhang@Dal.ca
bDepartment of Chemistry, Carnegie Mellon University, Pittsburgh 15213, USA
First published on 5th August 2024
Abstract
The detailed characterization of AuAg alloy nanoclusters is essential to guide the discovery of species ideal for applications in various fields including catalysis and biomedicine. This work presents structural analysis of the Au104Ag40(PET)60 species through X-ray absorption spectroscopy (XAS). First, XAS fitting is utilized to model the distribution of Au and Ag atoms within the structure. Our proposed model assigns Ag atoms to the vertex sites of the second shell of the metal core, as well as the outermost staple sites. This distribution reveals Au104Ag40(PET)60 to be a Ag single-atom alloy. The proposed model shows outstanding agreement with the coordination number values derived from XAS. XAS near-edge analysis is employed to investigate the alloy bonding interactions between Au and Ag. Substantial d-electron transfer from Au to Ag is observed in this sample, beyond the magnitude of previously studied AuAg NCs. This work enhances the understanding of the structure–property relationship of AuAg alloy NCs, offering insights which can be applied to other large NCs and even NPs. These insights will in turn aid the discovery of new materials for use in various applications.
1. Introduction
Au nanoclusters (NCs) have been widely studied due to the many unique properties which arise from their ultrasmall size.1 These properties result in promising candidates for a wide variety of applications, in fields including catalysis, optics, and electronics.2–4 As the properties of a NC are dependent on structural features including shape, size, and atomic arrangement, detailed characterization is vital to establish a structure–property relationship and help guide the discovery of NC species with practical applications. To this end, atomically precise NCs, defined by their uniform, well-defined structure, have emerged as an area of focus due to their consistent and predictable properties.5–9Beyond monometallic NCs, introducing a second metal to form alloy NCs increases the compositional space, unlocking many new possibilities.10–12 By using atomically precise NCs as a starting template the properties can be carefully tuned through adjusting the exact dopant concentration of the second metal, which in turn modifies the composition of the final product. This high degree of control increases the feasibility of creating alloy NCs to be utilized for desirable applications.13 This includes classes of materials such as single-atom alloys, which are of interest due to their excellent catalytic activity and selectivity.14–19
One widely studied combination of metals is Au and Ag, owing to the synergistic effects between them resulting in enhanced catalytic performance, stability, and biocompatibility.10,11,20 AuAg alloy NCs have been successfully synthesized through methods including galvanic replacement and co-reduction.20 The exact atomic positions that each element occupies has been shown to have a significant impact on the properties expressed by the NC.21 Therefore, an accurate determination of these positions is vital to develop the structure–property relationship. Previous work has accomplished this for relatively large AuAg NCs including (AuAg)130 and (AuAg)267.21,22 These studies primarily relied on X-ray crystallography, which requires the synthesis of a high-quality single-crystal. This has proven to be very challenging due to the complex and flexible structures of NCs.23 X-ray crystallography data interpretation can be challenging for large NCs due to the high atomic number contrast between heavy metal atoms in the core and lighter atoms in the thiolate ligands, leading to uncertainty in the atomic identity and exact atomic positions.21,24 Other techniques also run into challenges for the analysis of large AuAg NCs. Imaging techniques struggle to observe the metal core as it is covered by the protecting organothiolate ligands, and while DFT calculations can provide useful information, the resources necessary to run these simulations increase significantly when considering NCs of increased size and multiple elements.5
A tool that has shown great success in the characterization of metal NCs is X-ray absorption spectroscopy (XAS).4,8,10,11,25,26 XAS allows for flexible sample preparation, circumventing the single-crystal requirement for X-ray crystallography. XAS provides element specific analysis, making it ideal for alloy samples containing multiple elements. X-ray absorption near-edge spectroscopy (XANES) provides insight into electronic properties, and extended X-ray absorption fine structure (EXAFS) yields detailed structural information including accurate values for the average coordination number (CN) and bond distances (R) of various interactions within the sample.27–29 All of these features makes XAS an ideal tool to provide insight into the structure–property relationship of metal alloy NCs.
This work focuses on the previously reported30 Au104Ag40 NC, utilizing a XAS-focused characterization strategy to gain an in-depth understanding of the structure of this sample. The previously reported crystal structure of Au144,24 the monometallic analogue of Au104Ag40, is used as a template to assign likely Ag locations, resulting in a model representing the exact structure of Au104Ag40. Our proposed model assigns 40 Ag atoms within the 144-metal atom structure, with 30 of them occupying the entirety of the staple sites and the remaining 10 occupying vertex sites of the second shell of the metal core. This model shows excellent agreement with the EXAFS fitting results, as well as consistent trends with other AuAg NCs of similar size.21,22 Excitingly, our findings indicate that Au104Ag40 is a single-atom Ag alloy, making it a promising candidate for applications such as catalysis. XANES analysis is utilized to probe the electronic properties of the sample, indicating d-electron transfer away from Au at a larger magnitude than what has been observed in other NCs of similar size.
2. Experimental methods
2.1 Reagents and materials
Tetrachloro Auric(III) acid (HAuCl4·3H2O, >99.99% metals basis, Aldrich), silver tetrafluoroborate (AgBF4, >99.99% metals basis, Aldrich), tetraoctylammonium bromide (TOAB, ≥98%, Fluka), 2-phenylethanethiol (PET, C8H9SH, 98%, Aldrich), sodium borohydride (NaBH4, Aldrich), methanol (MeOH, HPLC grade, ≥99.9%, Aldrich), dichloromethane (DCM, ACS reagent, ≥99.5%, Aldrich). All chemicals were used without further purification. Nanopure water was prepared using the Barnstead NANOpure Diamond system.2.2 Synthesis and mass spectrometry
Au104Ag40 NCs were synthesized following previously reported procedures.30 Briefly, 120 mg of HAuCl4·3H2O and 30 mg of AgBF4 were dissolved in 10 mL of methanol and vigorously stirred for 15 minutes. Under continued vigorous stirring, 190 mg of 2-phenylethanethiol in the presence of tetraoctylammonium bromide was added to the reaction mixture, followed by 114 mg of NaBH4 dissolved in 5 mL of cold Nanopure water. Black precipitate containing the NCs was precipitated out of solution. The remaining solution was stirred overnight, after which the supernatant was separated via centrifuge and discarded. The remaining precipitate was washed three times with methanol, then NCs were extracted using dichloromethane. The exact mass of the product was determined through electrospray ionization mass spectrometry (ESI-MS), collected using a Waters Q-TOF mass spectrometer equipped with Z-spray source.2.3 X-ray absorption spectroscopy
Au L3-edge and Ag K-edge XAS data was collected from the CLS@APS (Sector 20-BM) beamline at the Advanced Photon Source (operating at 7.0 GeV) in Argonne National Laboratory, Chicago, IL, USA. Solid powdered sample was packed into kapton tape and measured in fluorescence mode simultaneously with a metal foil reference for each element. All measurements were collected at room temperature under ambient pressure. EXAFS data was transformed and normalized into k-space (Fig. S1†) and R-space using the Athena program following conventional procedures.31 A k-range of 2.9–13.4 Å−1 for Au, and 3.4–10.5 Å−1 for Ag was used to obtain each Fourier-transformed EXAFS (FT-EXAFS) spectra with a k weighting of 2. Self-consistent multiple-scattering calculations were performed using the FEFF6 program to obtain the scattering amplitudes and phase-shift functions used to fit scattering paths within the Artemis program. In the fitting a R–window of 1.2–3.6 Å for Au and 1.5–3.6 Å for Ag was used. Au L3-edge and Ag K-edge data were simultaneously fitted, and various parameters were correlated together to minimize the number of independent variables and ensure a high fit quality. These correlations are clearly indicated in the relevant data tables.3. Results and discussion
3.1 Structural determination of Au104Ag40
Au104Ag40 NCs were synthesized following the previously reported procedure.30 The sample of interest in this work was synthesized using a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of AuIII/AgI. ESI-MS results (Fig. S2†) determine the most abundant species within this sample contains 40 Ag and 104 Au atoms. The well-defined peaks in the ESI-MS spectrum provide confidence in the purity of Au104Ag40. As such, we assume the identity of Au104Ag40 to be accurate, and that the shape and atomic positions of Au104Ag40 will be consistent with the Au144 structure determined through X-ray crystallography.24 Our approach is to use the structural parameters calculated from EXAFS fitting to determine the most likely locations that the 40 dopant Ag atoms will occupy within this 144-metal atom structure.
:1 molar ratio of AuIII/AgI. ESI-MS results (Fig. S2†) determine the most abundant species within this sample contains 40 Ag and 104 Au atoms. The well-defined peaks in the ESI-MS spectrum provide confidence in the purity of Au104Ag40. As such, we assume the identity of Au104Ag40 to be accurate, and that the shape and atomic positions of Au104Ag40 will be consistent with the Au144 structure determined through X-ray crystallography.24 Our approach is to use the structural parameters calculated from EXAFS fitting to determine the most likely locations that the 40 dopant Ag atoms will occupy within this 144-metal atom structure.
The metal atoms in Au144 can be separated into 4 categories, each of which forms a “shell” within the structure. Beginning at the innermost core of the NC, shell 1 contains 12 atoms arranged in icosahedral geometry, with the notable lack of a center atom resulting in a M–M CN of 11 for each site. Shell 2 contains 42 atoms, also in icosahedral geometry. The 12 vertices of shell 2 have a M–M CN of 11, while the remaining shell 2 atoms have a M–M CN of 12. Shell 3, which forms the surface of the NC, has a rhombicosidodecahedron shape and contains 60 atoms, each with a M–M CN of 9. Finally shell 4, which takes the form of a series of staple units, contains 30 atoms, each with a M–M CN of 4. The S atoms in the protective organothiolate ligands form bonds with M atoms in shells 3 and 4. Atoms in each shell will form M–M bonds with atoms in the shell below and above it, as well as bonds with other atoms within the same shell. The exception is shell 4, which will only form bonds with shell 3 atoms due to the long distance between shell 4 atoms. The Au144 bond distribution (Fig. S3†) shows a general trend of shorter M–M bonds with an approximate bond length ∼2.8 Å towards the core, and longer M–M bonds with an approximate bond length ∼3.0 Å towards the surface. With this understanding of the structure of Au144 established, EXAFS fitting was conducted on the Au104Ag40 to compare structural parameters to those observed in Au144.
Wavelet transform-EXAFS (WT-EXAFS) plots (Fig. 1) were used to observe the most intense features within the k-space and R-space, allowing for an appropriate fitting procedure to be chosen for the EXAFS fitting procedure. Intense features in the WT-EXAFS plot indicate distinct bonding interactions which should have separate fitting paths in the EXAFS fitting procedure.32 Feature A represents a Au–S interaction, and feature D represents a Ag–S interaction. As both Au and Ag show evidence of a significant bonding interaction with S atoms, it is concluded that both Au and Ag must occupy positions within shell 3 and/or shell 4. Two additional distinct maxima are observed in both the Au L3-edge (features B and C) and the Ag K-edge (features E and F). These features are within the region characteristic of M–M interactions. The presence of two distinct features indicates that there is a wide distribution of the Au–M and Ag–M bond lengths, consistent with what is observed in Au144 (Fig. S3†). As both Au and Ag show evidence of a wide range of M–M bond distances, it is likely that they are both distributed relatively evenly across the structure, ruling out the possibility of a core–shell type structure.
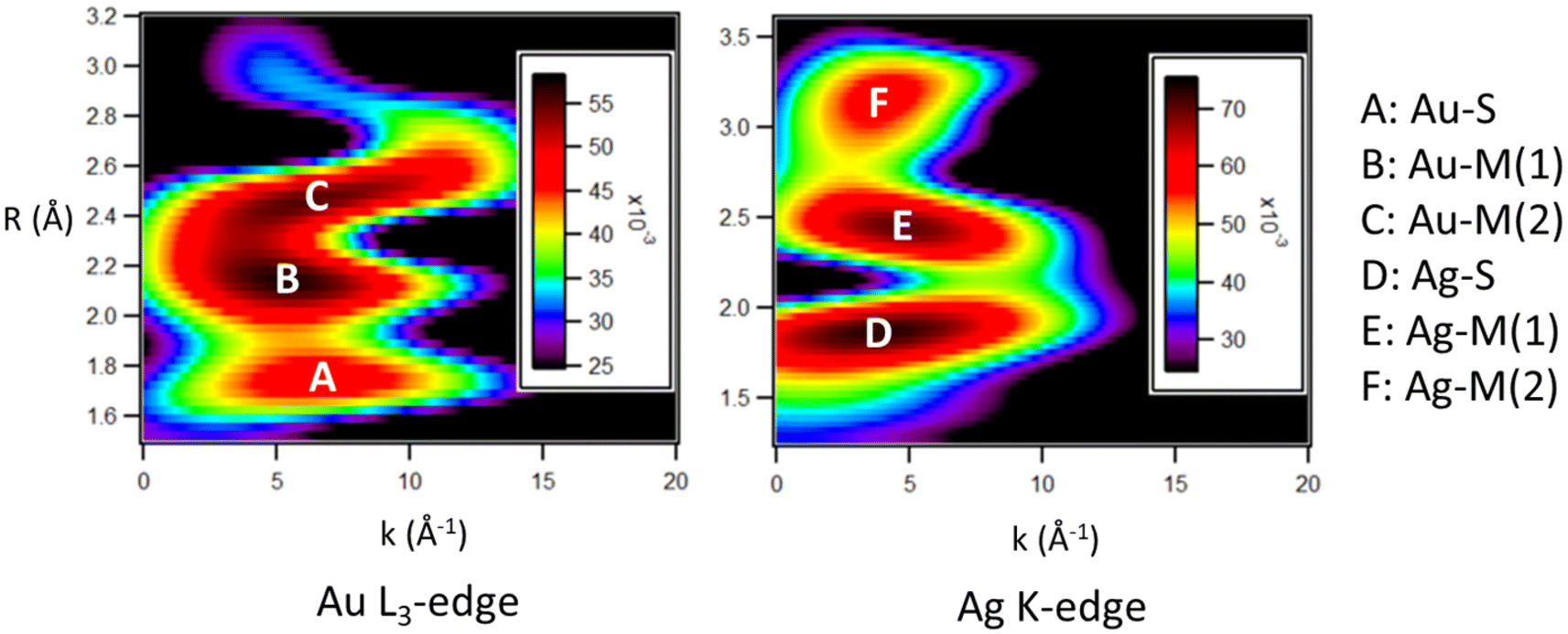 | ||
| Fig. 1 WT-EXAFS plots for Au104Ag40. Intense features are labelled according to the bond types they represent. In the Au L3-edge spectra: feature A represents Au–S, feature B represents the shorter distance Au–M, and feature C represents the longer distance Au–M. In the Ag K-edge: feature D represents Ag–S, feature E represents the shorter distance Ag–M, and feature F represents the longer distance Ag–M. | ||
Based on the features observed in the WT-EXAFS plots, a multi-shell fitting method for the M–M bonds is necessary to accurately model this system. The fitted FT-EXAFS spectra (Fig. S4†) and EXAFS fitting results (Table 1) allow for the average CN values of various bond types to be calculated. The most reliable fitting result utilizes four fitting paths for Au (Au–S, Au–Au, 2× Au–Ag) and three fitting paths for Ag (Ag–S, 2× Ag–Au). Interestingly, all fitting attempts which used a Ag–Ag fitting path gave a poor fit quality and infeasible values such as a negative CN. This result is strong evidence of a lack of Ag–Ag nearest-neighbor interactions in the sample, making Au104Ag40 a single-atom Ag alloy.
| Bond | CN | R (Å) | σ 2 (Å) × 10−3 | E 0 (eV) | R factor |
|---|---|---|---|---|---|
| Au–S | 0.9(2) | 2.30(1) | 5(2) | 7(1) | 0.0249 |
| Au–Au | 6(2) | 2.81(2) | 17(4) | ||
| Au–Ag | 1.0(9) | 2.84(4) | |||
| Au–Ag | 1.4(8) | 2.99(4) | |||
| Ag–S | 0.8(4) | 2.43(3) | 4(5) | 1(3) | 0.0249 |
| Ag–Au | 3(1) | 2.84(4) | |||
| Ag–Au | 3(1) | 2.99(4) |
Given the large proportion of Ag in this sample, there are very few possible configurations which will satisfy a Ag single-atom alloy distribution. The lack of nearest-neighbor Ag atoms is the main guiding principle for the proposed model, but there are also many other conditions which must be satisfied. The Au–Au fitting path, with a relatively large CN of 6 and relatively short bond distance of 2.81 Å, indicates a Au-rich metal core. The two distinct Au–Ag fitting paths indicate Au–Ag interactions are present within shells 1 and 2 (2.84 Å) and within shells 3 and 4 (2.99 Å). The Au–S and Ag–S CNs are roughly equal, providing insight into the elemental distribution of shells 3 and 4. Finally, the total Ag–M CN (6) is lower than that of Au (8.4). This finding further supports the innermost part of the structure being dominated by Au. It also indicates that shell 4, having a low M–M CN of 4, will be largely occupied by Ag.
Accounting for all these structural features, a model has been proposed for the exact element distribution of Au104Ag40 (Fig. 2). The CN values of this model show remarkable agreement with those calculated from EXAFS (Table 2), particularly for the alloy bonding interactions, providing high confidence in this assignment. Note that it is understandable to have a high degree of uncertainty for the M–S interactions due to the large NC size. In this model, all 30 of the shell 4 sites are occupied with Ag, and all 60 of the shell 3 sites are occupied with Au. This assignment satisfies a single-atom Ag distribution, a low overall Ag-M CN, and a roughly equal CN of Au–S and Ag–S. The staple layer being dominated by Ag has precedent in other AuAg NCs.21 Furthermore, the truncated geometry of the shell 3 surface layer has been observed in many monometallic Au NCs, as a way to reduce the high surface energy of Au, while monometallic Ag NCs have shown preference for a non-truncated geometry.33,34 Therefore, in the case of a surface shell with truncated shape as in Au104Ag40, Au showing a preference for occupying these shell 3 sites is expected.
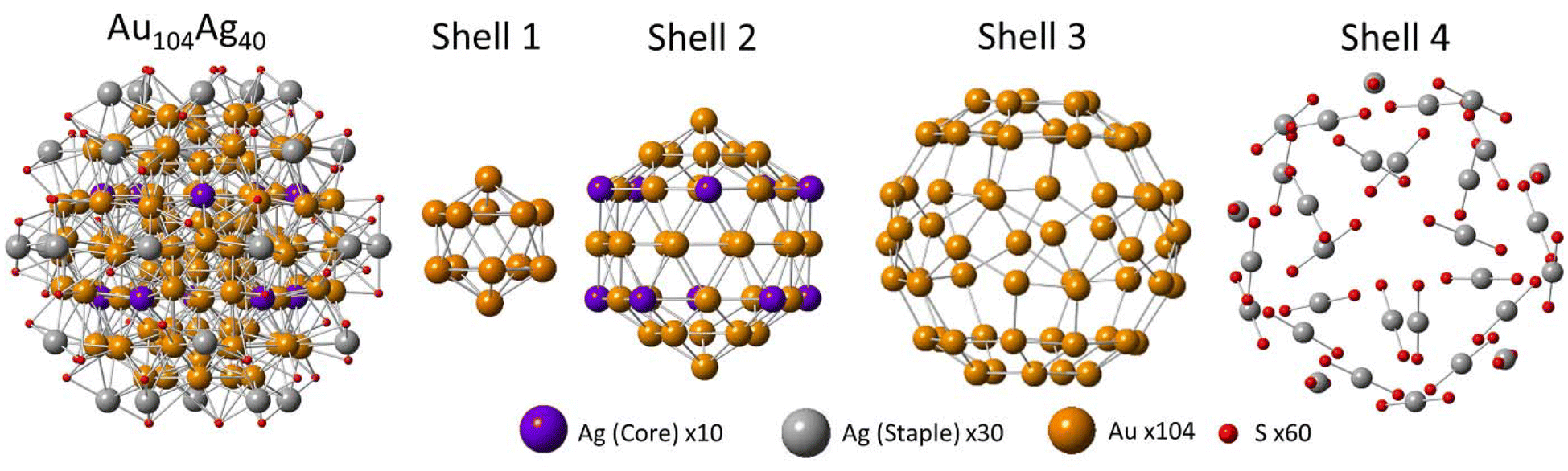 | ||
| Fig. 2 Proposed model of Au104Ag40. Each shell is individually shown for clarity purposes. | ||
| Bond | CN (EXAFS) | CN (model) |
|---|---|---|
| Au–S | 0.9(2) | 0.6 |
| Au–Au | 6(2) | 7.7 |
| Au–Ag | 1.0(9) | 1.0 |
| Au–Ag | 1.4(8) | 1.2 |
| Ag–S | 0.8(4) | 1.5 |
| Ag–Au | 3(1) | 2.8 |
| Ag–Au | 3(1) | 3.0 |
The remaining 10 Ag atoms have been assigned to the vertex sites of the icosahedron of shell 2. While other assignments of Ag within shell 2 satisfy the CN and bond distance values determined through EXAFS, this specific assignment is justified based on a similar distribution observed in the (AuAg)130 NC through X-ray crystallography.21 The top and bottom vertices of the shell 2 icosahedron were chosen to be the two vertices that are occupied by Au in our model. This is necessary to ensure a model containing exactly 40 Ag atoms, but also has precedent in (AuAg)130.21
To validate this model, the EXAFS fitting was refined with the CN values fixed according to the values in the model (Fig. S5† and Table 3). A high-quality fit is obtained using this fitting method, providing confidence in the proposed model. By fixing the CN values more independent variables can be utilized in the fitting procedure, allowing for the uncorrelation of σ2 values which was previously necessary to ensure a high-quality fit. This fitting method allows for further insight regarding the bond distance distribution of each fitting path in Au104Ag40. Notably the Au–Au and short-length Au–Ag bonds have a very large σ2 of 18 × 10−3 Å, indicating a wide distribution of these bond types in Au104Ag40.
| Bond | CN (fixed) | R (Å) | σ 2 (Å) × 10−3 | E 0 (eV) | R factor |
|---|---|---|---|---|---|
| Au–S | 0.6 | 2.28(1) | 2(1) | 5(1) | 0.0209 |
| Au–Au | 7.7 | 2.81(2) | 18(2) | ||
| Au–Ag | 1.0 | 2.85(2) | |||
| Au–Ag | 1.2 | 2.98(2) | 8(1) | ||
| Ag–S | 1.4 | 2.45(2) | 10(3) | 2(1) | 0.0209 |
| Ag–Au | 2.8 | 2.85(2) | 5(3) | ||
| Ag–Au | 3.0 | 2.98(2) | 6(3) |
3.2 Alloy bonding of Au104Ag40
Following the discovery of the single-atom Ag nature of Au104Ag40, we conducted an in-depth investigation into the alloy bonding interactions present within this sample. The 30 Ag atoms located within shell 4 are non-metallic in nature, making them of less interest in applications such as catalysis. We therefore focus our analysis on the 10 Ag atoms localized within shell 2.An in-depth analysis of this structure reveals several interesting features. Each of the Ag atoms has a CN of 11, with one shell 1, five shell 2, and five shell 3 neighbor atoms (Fig. 3). The EXAFS fitting results indicate Au–Ag bonds are on average slightly longer in length than Au–Au bonds. The lack of an innermost center atom in shell 1 provides the metal core of the NC with a higher degree of flexibility, allowing for the core to expand or contract as necessary to accommodate changes within the structure such as the introduction of dopant Ag atoms.
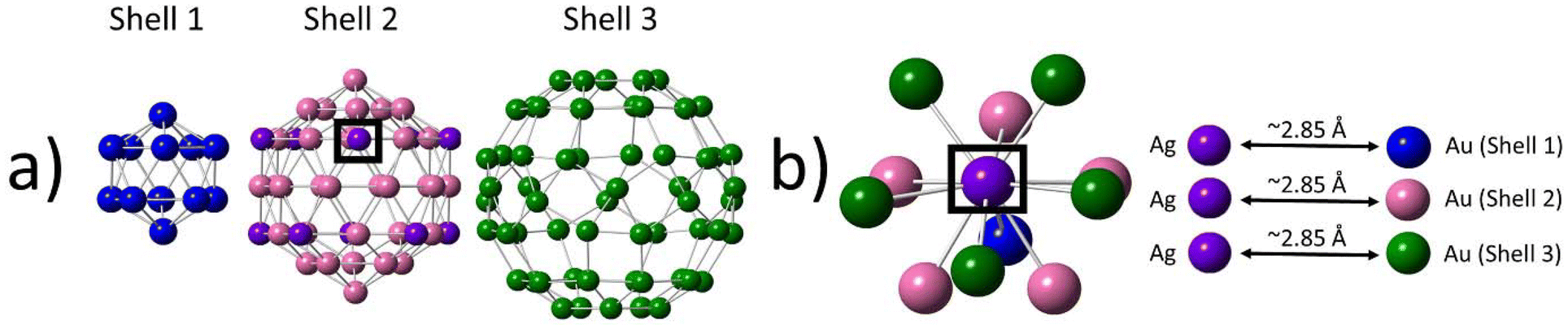 | ||
| Fig. 3 Local bonding environment of the single-atom Ag sites within shell 2. (a) Breakdown of shells 1, 2, and 3. (b) Nearest-neighbor coordination environment of a single-atom Ag site in shell 2. | ||
When considering Au104Ag40 as a whole, this model can be viewed as having a “sandwich” type distribution of Au atoms, with shells 1 and 3 consisting entirely of Au and surrounding the shell 2 layer where the single-atom Ag sites are located. Au104Ag40 approaches the upper limit of the number of Ag atoms which can be accommodated while still maintaining a single-atom alloy distribution. A high number of single-atom sites is of particular interest for applications such as catalysis, and this principle can potentially be applied to not only other AuAg NCs but also larger nanoparticles. Interestingly the final composition of this sample contained slightly less Ag than what was expected from the 2:1 Au:Ag loading, which may indicate that additional dopant Ag atoms were repelled from the structure in order to ensure the single-atom Ag arrangement is maintained. This discrepancy indicates that Au NCs are a good template structure for the creation of single-atom Ag alloys.
Direct comparison with the similar sized (AuAg)130 (Fig. 4) reveals interesting similarities and differences. As (AuAg)130 has a roughly 1:2 ratio of Au:Ag, a single-atom Ag distribution is not possible. Despite this, the second shell shows a very similar distribution of Ag atoms to that of Au104Ag40, with Ag maintaining a single-atom alloy type distribution within shell 2. A similar phenomenon was also observed in the larger (AuAg)267, where the third shell (outermost core shell) also showed evidence of a single-atom alloy type Ag distribution.22 The similarities in the distribution of Ag atoms within shell 2 hold despite the differences in shape of this layer between (AuAg)130 and Au104Ag40. When viewing (AuAg)130 from the side (Fig. 4a and c) the second shell can be divided into 7 horizontal rows of atoms, with the atoms in each row being arranged in the shape of a pentagon as can be clearly observed in the top-down view (Fig. 4b and d). The pentagon of every row has the same orientation, with all the vertices aligned.
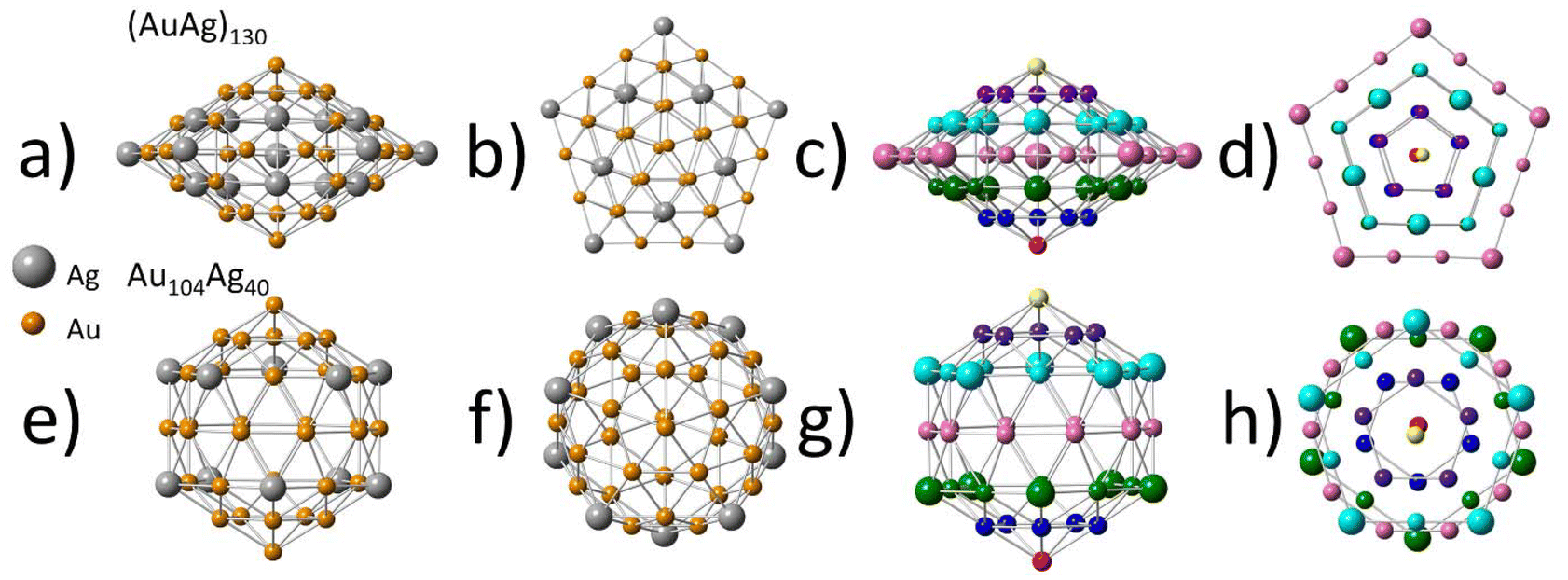 | ||
| Fig. 4 Shell 2 comparison between (AuAg)130 and Au104Ag40. (a) (AuAg)130 side view. (b) (AuAg)130 top-down view. (c) (AuAg)130 side view (colored). (d) (AuAg)130 top-down view (colored). (e) Au104Ag40 side view. (f) Au104Ag40 top-down view. (g) Au104Ag40 side view (colored). (h) Au104Ag40 top-down view (colored). Ag atoms are enlarged for clarity purposes. Colored view separates atoms by row to help highlight geometry from various viewpoints. In the top-down (colored) views, bonds between rows are omitted for clarity purposes. | ||
For Au104Ag40 the side view (Fig. 4e and g) also reveals 7 horizontal rows of atoms. While most of these rows maintain the same pentagon shape observed in (AuAg)130, the middle row instead has a decagon shape. From a top-down view (Fig. 4f and h) it is observed that rows in the top half have reverse orientation to rows in the bottom half. This mismatch results in shell 2 having a more rounded shape in Au104Ag40, and a more flat, spread-out shape in (AuAg)130. These changes in geometry are not enough to impact the bond distances, as EXAFS fitting indicates both (AuAg)130 and Au104Ag40 to have Au–Ag bond distances within uncertainty.
Interestingly, despite the 1:2 ratio of Au:Ag in (AuAg)130, this sample does not approach a single-atom Au type structure, with Au clustering in shell 2 and Ag spreading out across all 4 shells. This variation in the distribution between Au and Ag makes Ag the more likely candidate to form single-atom alloys at the appropriate composition. Comparison between (AuAg)130 and Au104Ag40 indicates that as the composition is varied to have a decreased amount of Ag, there will be less Ag atoms present within the innermost core (shell 1) and on the surface (shell 3) of the NC. Future study is necessary to confirm if this trend can be applied more broadly to AuAg NCs.
XANES was utilized to probe the electronic properties of the alloy bonding interaction. The Au L3-edge XANES spectra (Fig. 5a) of Au104Ag40 shows an increased white-line intensity compared to the metallic foil reference. There are two plausible explanations for the increase white-line intensity in Au104Ag40; the nanoscale effect, or a decrease in d-electron density given that the L3-edge represents an electronic transition to the valence d-orbital (Fig. 5b). As Au144 shows a similar white-line intensity to the metallic foil, the nanoscale effect can be ruled out and it can be concluded that a decrease in d-electron density is responsible. This finding indicates that Au donates d-electrons to Ag to form the alloying interaction, and in turn receives s/p-electron density back from Ag (Fig. 5c). Similar charge transfer has been previously observed in AuAg130.21
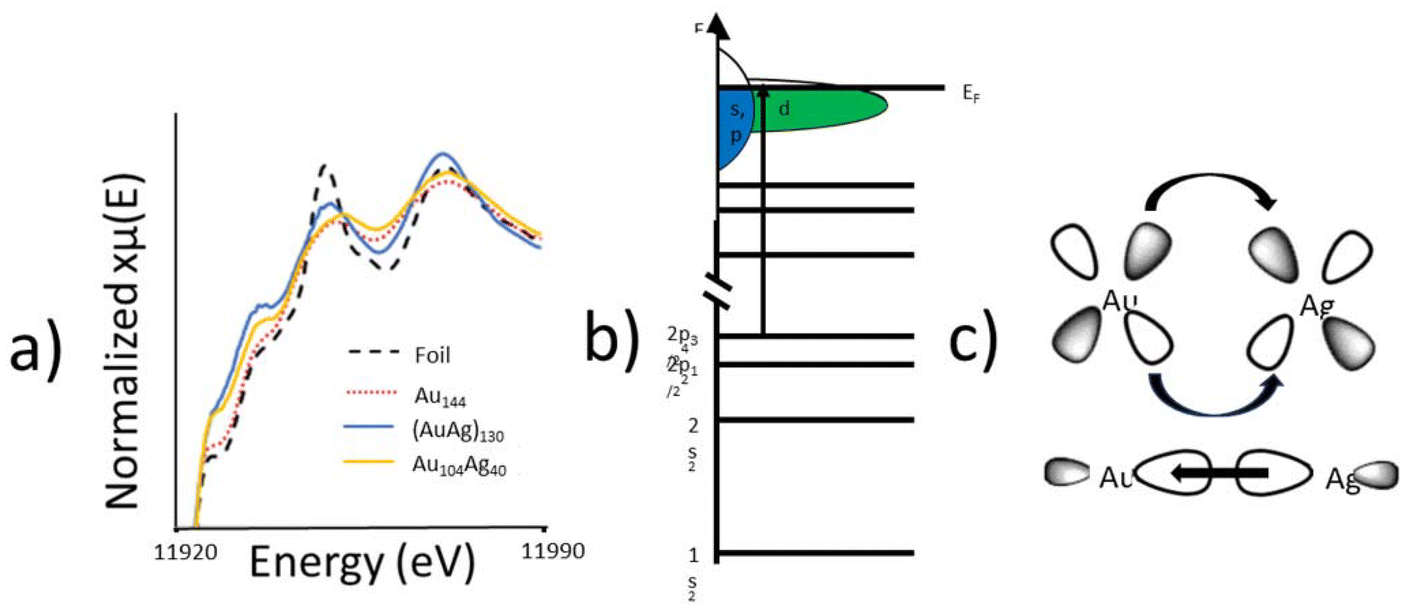 | ||
| Fig. 5 Electronic properties of Au104Ag40. (a) Au L3-edge XANES spectra for Au104Ag40 alongside metallic foil reference, Au144, and (AuAg)130. (b) Energy diagram illustrating the Au L3-edge electronic transition. (c) Schematic showing the electron transfer involved in the alloy interaction between Au and Ag atoms. Au donates d-electron density to Ag and receives s/p-electron density from Ag. | ||
To quantitatively evaluate the alloying phenomenon, XANES integration was used to calculate the d-hole (10 – number of d electrons) of Au for each of these samples (Table 4). The d-hole for Au144 is slightly smaller than for Au foil, as expected due to quantum size effects. Despite being of similar size, both (AuAg)130 and Au104Ag40 have a larger d-hole than the Au foil, arising from d-electron density being transferred to Ag from Au. Interestingly, the single-atom Ag structure of Au104Ag40 results in a substantially larger d-hole, due to Au–S interactions which are not present in (AuAg)130. The relative charge of the catalytically active site has been shown to play a key role in the catalytic performance of alloy NCs, making this observation of particular importance for future catalytic applications.35,36
| Sample | d-Hole |
|---|---|
| Au144 | 0.372 |
| Au foil | 0.400 |
| (AuAg)130 | 0.413 |
| Au104Ag40 | 0.673 |
4. Conclusions
XAS analysis has been used to identify the exact locations occupied by Au and Ag within the Au104Ag40 NC. In this model Ag occupies all 30 staples sites and 10 of the 12 vertices of the second shell of the metal core, resulting in a Ag single-atom alloy structure. The multi-step EXAFS fitting procedure provides a high degree of confidence for this model, showcasing how structural insight can be obtained for large AuAg NCs when single-crystal synthesis is not possible. Structural analysis revealed Au104Ag40 is an example of a single-atom Ag alloy, with Ag atoms located in a “sandwich” distribution between layers of Au atoms. XANES analysis provided insight into the alloy interaction between Au and Ag, revealing that Au donates d-electron density to Ag and receives s/p-electron density from Ag. This understanding of the charge transfer between elements is of particular interest in the context of future catalytic applications of AuAg alloy NCs. This work furthers understanding into the structure–property relationship and alloying behavior of large AuAg alloy NCs, which will be applied in future discovery of AuAg NCs for use in various applications.Author contributions
David Morris: conceptualization, methodology, investigation, XAS data collection, XAS data analysis, visualization, data curation, formal analysis, validation, writing – original draft, writing – review and editing. Xiangsha Du: sample preparation, mass spectrometry data collection, mass spectrometry data analysis, writing – review and editing. Rongchao Jin: supervision, writing – review and editing, resources, funding acquisition. Peng Zhang: supervision, writing – review and editing, resources, funding acquisition.Data availability
The data supporting this article has been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
P.Z. thanks the NSERC Canada Discovery Grant for funding. CLS@APS facilities (Sector 20-BM) at the Advanced Photon Source (APS) are supported by the U.S. Department of Energy (DOE), NSERC Canada, the University of Washington, the Canadian Light Source (CLS), and the APS. Use of the APS is supported by the DOE under Contract no. DEAC02-06CH11357. The CLS is financially supported by NSERC Canada, CIHR, NRC, and the University of Saskatchewan.References
- R. Jin, Nanoscale, 2010, 2, 343 RSC
.
- J. Xie, Y. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888 CrossRef CAS PubMed
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261 CrossRef CAS PubMed
- R. Yang, D. J. Morris, T. Higaki, M. J. Ward, R. Jin and P. Zhang, ACS Omega, 2018, 3, 14981 CrossRef CAS PubMed
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346 CrossRef CAS PubMed
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883 CrossRef CAS PubMed
- J. Liu, K. S. Krishna, Y. B. Losovyj, S. Chattopadhyay, N. Lozova, J. T. Miller, J. J. Spivey and C. S. S. R. Kumar, Chemistry, 2013, 19, 10201 CrossRef CAS PubMed
- D. M. Chevrier, R. Yang, A. Chatt and P. Zhang, Nanotechnol. Rev., 2015, 4, 193 CrossRef CAS
- X. Kang, X. Wei, S. Jin, Q. Yuan, X. Luan, Y. Pei, S. Wang, M. Zhu and R. Jin, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 18834 CrossRef CAS PubMed
- J. Liu, K. S. Krishna, C. Kumara, S. Chattopadhyay, T. Shibata, A. Dass and C. S. S. R. Kumar, RSC Adv., 2016, 6, 25368 RSC
- X. Zan, Q. Li, Y. Pan, D. J. Morris, P. Zhang, P. Li, H. Yu and M. Zhu, ACS Appl. Nano Mater., 2018, 1, 6773 CrossRef CAS
- Y. Yong, C. Li, X. Li, T. Li, H. Cui and S. Lv, J. Phys. Chem. C, 2015, 119, 7534 CAS
- Y. Chen, C. Zeng, D. R. Kauffman and R. Jin, Nano Lett., 2015, 15, 3603 CrossRef CAS PubMed
- X. Zhang, G. Cui, H. Feng, L. Chen, H. Wang, B. Wang, X. Zhang, L. Zheng, S. Hong and M. Wei, Nat. Commun., 2019, 10, 5812 CrossRef CAS PubMed
- T. Zhang, A. G. Walsh, J. Yu and P. Zhang, Chem. Soc. Rev., 2021, 50, 569 RSC
- P. N. Duchesne, Z. Y. Li, C. P. Deming, V. Fung, X. Zhao, J. Yuan, T. Regier, A. Aldalbahi, Z. Almarhoon, S. Chen, D. Jiang, N. Zheng and P. Zhang, Nat. Mater., 2018, 17, 1033 CrossRef CAS PubMed
- A. G. Walsh and P. Zhang, Adv. Mater. Interfaces, 2021, 8, 2001342 CrossRef CAS
- T. Zhang, Z. Chen, A. G. Walsh, Y. Li and P. Zhang, Adv. Mater., 2020, 32, 2002910 CrossRef CAS PubMed
- Z. Chen, A. G. Walsh and P. Zhang, Acc. Chem. Res., 2024, 57, 521 CAS
- X. Kang, Y. Li, M. Zhu and R. Jin, Chem. Soc. Rev., 2020, 49, 6443 RSC
- T. Higaki, C. Liu, D. J. Morris, G. He, T. Luo, M. Y. Sfeir, P. Zhang, N. L. Rosi and R. Jin, Angew. Chem., Int. Ed., 2019, 58, 18798 CrossRef CAS PubMed
- J. Yan, S. Malola, C. Hu, J. Peng, B. Dittrich, B. K. Teo, H. Hakkinen, L. Zheng and N. Zheng, Nat. Commun., 2018, 9, 3357 CrossRef PubMed
- P. Zhang, J. Phys. Chem. C, 2014, 118, 25291 CrossRef CAS
- N. Yan, N. Xia, L. Liao, M. Zhu, F. Jin and R. Jin, Sci. Adv., 2018, 4, eaat7259 CrossRef CAS PubMed
- G. Simms, J. D. Padmos and P. Zhang, J. Chem. Phys., 2009, 131, 214703 CrossRef PubMed
- H. Wang, J. T. Miller, M. Shakouri, C. Xi, T. Wu, H. Zhao and M. C. Akatay, Catal. Today, 2013, 207, 3 CrossRef CAS
- J. H. Sinfelt and G. D. Meitzner, Acc. Chem. Res., 1993, 26, 1 CrossRef CAS
- J. J. Rehr and R. C. Albers, Rev. Mod. Phys., 2000, 72, 621 CrossRef CAS
- T. K. Sham, S. J. Naftel and I. Coulthard, J. Appl. Phys., 1996, 79, 7134 CrossRef CAS
- X. Du, H. Ma, X. Zhang, M. Zhou, Z. Liu, H. Wang, G. Wang and R. Jin, Nano Res., 2022, 15, 8573 CrossRef CAS
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537 CrossRef CAS PubMed
- Z. Chen, D. M. Chevrier, B. E. Cohn, T. P. Bigioni and P. Zhang, J. Phys. Chem. C, 2023, 127, 20771 CrossRef CAS
- Y. Chen, C. Zeng, C. Liu, K. Kirschbaum, C. Gayathri, R. R. Gil, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2015, 137, 10076 CrossRef CAS PubMed
- L. D. Marks and L. Peng, J. Phys.: Condens. Matter, 2016, 28, 053001 CrossRef CAS PubMed
- S. Masuda, K. Sakamoto and T. Tsukuda, Nanoscale, 2024, 16, 4514 RSC
- Y. Liu, X. Chai, X. Cai, M. Chen, R. Jin, W. Ding and Y. Zhu, Angew. Chem., Int. Ed., 2018, 57, 9775 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02688h |
| This journal is © The Royal Society of Chemistry 2024 |