
Poisoning resistance: challenges for hydrogen storage alloys toward engineering applications
Jing
Gu†
ab,
Zhendong
Yao†
 *acd,
Ge
Gao†
a,
Yijing
Wang
c,
Min
Liu
a,
Miaogen
Chen
b,
Chao
Li
a,
Meiqiang
Fan
a,
Xuezhang
Xiao
d and
Lixin
Chen
*d
*acd,
Ge
Gao†
a,
Yijing
Wang
c,
Min
Liu
a,
Miaogen
Chen
b,
Chao
Li
a,
Meiqiang
Fan
a,
Xuezhang
Xiao
d and
Lixin
Chen
*d
aCollege of Materials and Chemistry, China Jiliang University, Hangzhou 310018, China. E-mail: zhendongyao@foxmail.com
bKey Laboratory of Intelligent Manufacturing Quality Big Data Tracing and Analysis of Zhejiang Province, College of Science, China Jiliang University, Hangzhou 310018, China
cKey Laboratory of Advanced Energy Materials Chemistry, Nankai University, Tianjin 300071, China
dState Key Laboratory of Silicon and Advanced Semiconductor Materials, School of Materials Science and Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: lxchen@zju.edu.cn
First published on 14th June 2024
Abstract
The safety and stability of hydrogen storage alloys in the field of large-scale energy storage has now become a hot spot of attention for researchers. However, the surface poisoning phenomenon caused by impurity gas (O2, H2O, CO, etc.) contained in hydrogen is an unavoidable problem in engineering applications. On the one hand, in the process of “manufacturing – storage – transportation” of hydrogen, a small amount of impurity gas is easily mixed into it. On the other hand, producing ultra-high-purity hydrogen will significantly increase the cost of hydrogen. Thus, in the face of the impurity gas contained in hydrogen, the only solution is to improve the poisoning resistance of the hydrogen storage alloy. Therefore, researchers have carried out a lot of work on the poisoning as well as coping strategies for hydrogen storage alloys, and have made considerable progress. In order to overcome the influence of the poisoning effect in the engineering application, this review summarizes the interaction rules of poisoning behavior on the surface/interface of hydrogen storage alloys under impurity gases, and analyzes the effects of improving the anti-poisoning abilities of different modification methods carried on the bulk phase and surface/interface. Notably, the concept of constructing “hydrogen-permeable” and “impurity gas-barrier” layers is established based on the in-depth discussion of the anti-poisoning strategies. Finally, we propose rational directions for future anti-poisoning modification, hoping to promote the solution of poisoning to some extent.
1 Introduction
Hydrogen as an energy carrier and chemical feedstock could be a principal medium for replacing traditional fossil energy owing to its diverse sources, renewability and zero-carbon emissions.1–3 To usher in the age of hydrogen, developing safe and efficient ways to store and transport hydrogen is gaining attention. Various suitable carriers for indirectly storing hydrogen have been explored and modified in the field of chemical hydrogen storage, including hydrogen-containing compounds (ammonia, methanol, and natural gas, etc.) and substances with the ability to absorb hydrogen (hydrogen storage alloys).4,5 In terms of hydrogen-containing compounds, the on-site generation of COx-free hydrogen via ammonia reforming is relatively more suitable for hydrogen storage and transportation.6 However, the hydrogen–ammonia–hydrogen conversion usually requires severe conditions (high temperature and high pressure) and involves a certain amount of energy loss. Furthermore, the toxicity and corrosiveness of ammonia reduce its safety during storage and transportation.7,8 Thus, it is imperative to explore more effective and stable hydrogen storage substances to meet the needs of large-scale energy storage. As reported, storing hydrogen atoms via tetrahedral/octahedral gaps within alloys is a highly promising strategy for hydrogen storage.9 On the one hand, specific hydrogen storage alloys can achieve complete ultra-fast hydrogen absorption and desorption at room temperature without harsh conditions. On the other hand, most hydrogen storage alloys can undergo hundreds of reversible de/hydrogenation cycles without attenuating the hydrogen storage properties, which provides long-term stability and safety for engineering applications.10–12 According to the different alloy compositions, hydrogen storage alloys can be roughly divided into AB5-type (LaNi5), AB2-type (TiMn2), AB-type (TiFe), A2B-type (Zr2Ni), BCC-type (V-based Solid Solution) and Mg-based hydrides. Different types of hydrogen storage alloys possess specific advantages and, likewise, different obstacles in the industrialization process. For example, AB5-type alloys have stable cycling capacity but low gravimetric hydrogen density, and easily pulverize during cycles. Mg-based hydrides have attractive capacity but with high desorption temperature and low desorption pressure. Although the AB-type alloys are cost-effective, their activation conditions are rigorous and the plateau characteristics are poor. The above problems have more or less affected the commercial application of hydrogen storage alloys. Among them, a common problem of all hydrogen storage alloys is that “poisoning” will occur when they encounter impurity gases such as H2O, CO, O2, N2, CH4, SO2, CO2, etc., which will reduce or even cause complete loss of hydrogen ab/desorption activity.13 Since in the commercial environment it is difficult to meet the strict operational requirements of the laboratory, trace amounts of impurity gases will inevitably be mixed into the hydrogen flow. Consequently, surface poisoning is an unavoidable process in the long-term commercialization of hydrogen storage alloys.In the hydrogen storage community, research addressing the poisoning behavior of hydrogen storage materials is a relatively small field. Some of these works compare the poisoning degree of different impurity gases on hydrogen storage alloys, some reveal the interaction mechanism of impurity gases at the alloy interface, some explore the anti-poisoning modification law of hydrogen storage alloys, and some of them make outstanding contributions to the modification of anti-poisoning on hydrogen storage alloys. Therefore, there are few statistics for the relevant literature, as summarized in Fig. 1. It can be seen that most published articles on poisoning mainly focus on AB5 and AB alloys and MgH2 systems, which are relatively commercially and technologically mature hydrogen storage materials. On the other hand, the limited number of publications implies that the research field is far from saturated and that there is still plenty of potential to be explored.
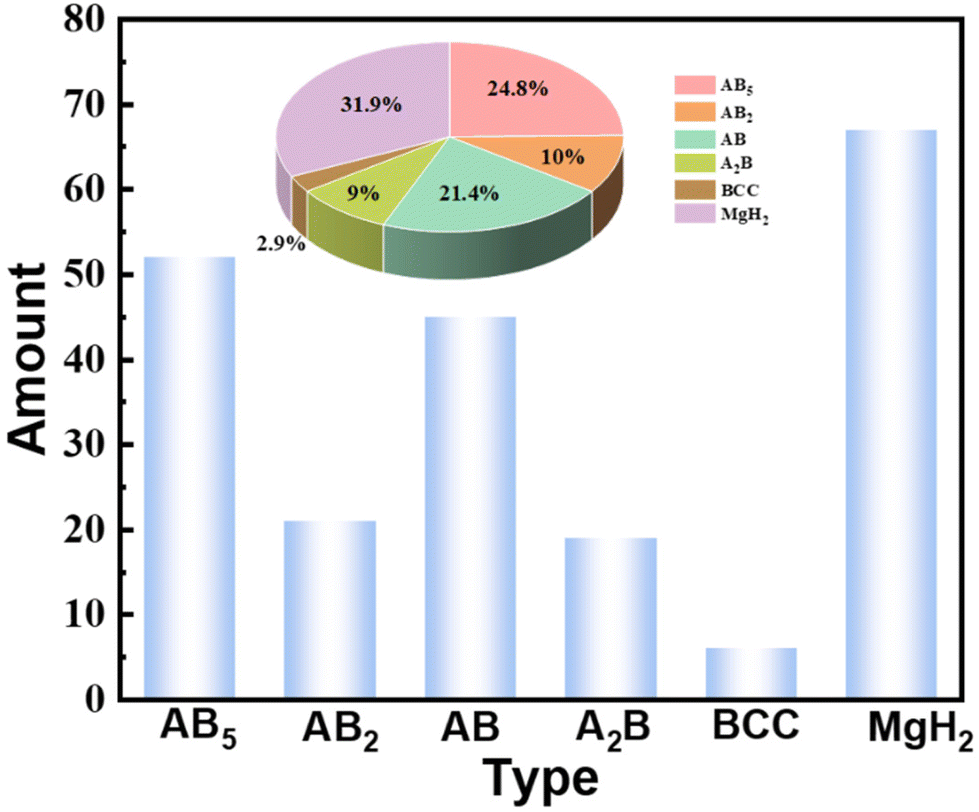 | ||
| Fig. 1 The number of articles related to poisoning for different materials and their corresponding proportions. | ||
In general, the poisoning behavior can be regarded as the interaction between the impurity gas and the surface followed by formation of a passivation layer that prevents the dissociation of H2 into H atoms or the diffusion of H atoms to the bulk phase, thus impairing the catalytic properties of the surface/interface and the hydrogen diffusion channel effect, respectively. As the concentration of gaseous impurity increases, the possibility of passivation layer formation increases, while the activation energy barrier for hydride formation also increases accordingly. Notably, the reaction with impurities is not limited to the surface but also involves the bulk phase, resulting in the continuous deterioration of hydrogen storage properties. In a nutshell, the reaction of hydrogen with the bulk phase requires a preliminary surface decomposition process, which is strongly influenced by gaseous impurities and thereby deactivates the surface, also known as surface poisoning.14 Reducing the concentration of polluting gases in industrial hydrogen streams is an appropriate strategy but not conducive to large-scale production.15 Therefore, to further enhance hydrogen storage, transportation and recovery, it is imperative to construct surface structures that selectively allow the entry of hydrogen and shield against impurity gas for long-term applications.
To date, extensive research has been conducted on the poisoning characteristics and anti-poisoning modification of various hydrogen storage alloys. Based on the available literature, the poisoning conditions of hydrogen storage materials can be divided into two types, which are defined here as online and offline poisoning, respectively. Online poisoning refers to the poisoning behavior of materials during the de/hydrogenation process under hydrogen atmosphere containing impurities. Therefore, the hydrogen-to-impurity ratio can be flexibly adjusted to determine the corresponding tolerance of the material to the impurity gas, to obtain more valuable and abundant experimental data. Comparatively, offline poisoning denotes the poisoning behavior of materials with pre-exposure to air or other environments before de/hydrogenation testing in pure hydrogen. Although the source of impurities is complex, it is demonstrated that offline poisoning has a certain reference value for the production, packaging and transportation of hydrogen storage alloys.
Additionally, the characterization of the composition, phase and structure of the poisoned samples is still of interest. Since gaseous impurities interact with solid metals mainly at the surface/interface, XPS (X-ray photoelectron spectroscopy), AES (Auger electron spectroscopy), TOF-SIMS (time-of-flight secondary ion mass spectrometry), GC (gas chromatography), IR (infrared spectrometry) and MS (mass spectrometry) characterization techniques are most commonly used in addition to the conventional XRD (X-ray diffraction), SEM (scanning electron microscope), TEM (transmission electron microscope) and SAED (selected area electron diffraction) microscopic analysis. In particular, XPS, which characterizes the near-surface regions of materials, is widely employed to determine the species of surface substances and the evolution of chemical valence states of elements.
Mostly the poisoning behavior of hydrogen storage alloys in mixture gas has been reported, albeit only focusing on a specific type of alloy, resulting in scattered studies without classifying and summarizing the poisoning commonness. Simultaneously, surface poisoning is a complex process involving many extrinsic factors (temperature, pressure, time, etc.). The types and concentrations of impurity gases and the reported hydrogen storage alloys are diverse, undoubtedly increasing the breadth and depth of poisoning research. By summarizing the current research work, the basic logic of poisoning and modification of hydrogen storage alloys is analyzed, in order to alleviate the poisoning effect. Hence, the purpose of this paper is to illustrate the poisoning behavior of different impurity gases on various alloys and metal hydrides, clarify the exact poisoning mechanism and discuss the subsequent anti-poisoning modification and regeneration approaches.
According to the relevant literature from 1978 to 2024 counted by the Web of Science Core Collection, the relationship between keywords obtained by frequency is depicted in Fig. 2. Since many related publications do not have the desired keywords or words in the thesis statement, the visualization diagram can only paint a rough overview of our work. The larger fonts and node sizes indicate that the word occurs frequently in the study. The clustering and connecting line colors also emphasize the degree of interconnection between various keywords. It can be seen that the activation and poisoning properties of hydrogen storage alloys are surface-dependent. Moreover, surface–interface-related studies are mainly concerned with the poisoning condition of materials and the purification of gases in the system. Descriptively, this review traces the poisoning behavior and surface–interface interaction mechanisms of alloys, magnesium and metal hydrides as main hydrogen storage materials through their intrinsic microstructure and surface–interface properties upon exposure to gaseous impurities. More improvement strategies for anti-poisoning abilities and regeneration conditions of alloys will be elucidated in the following sections.
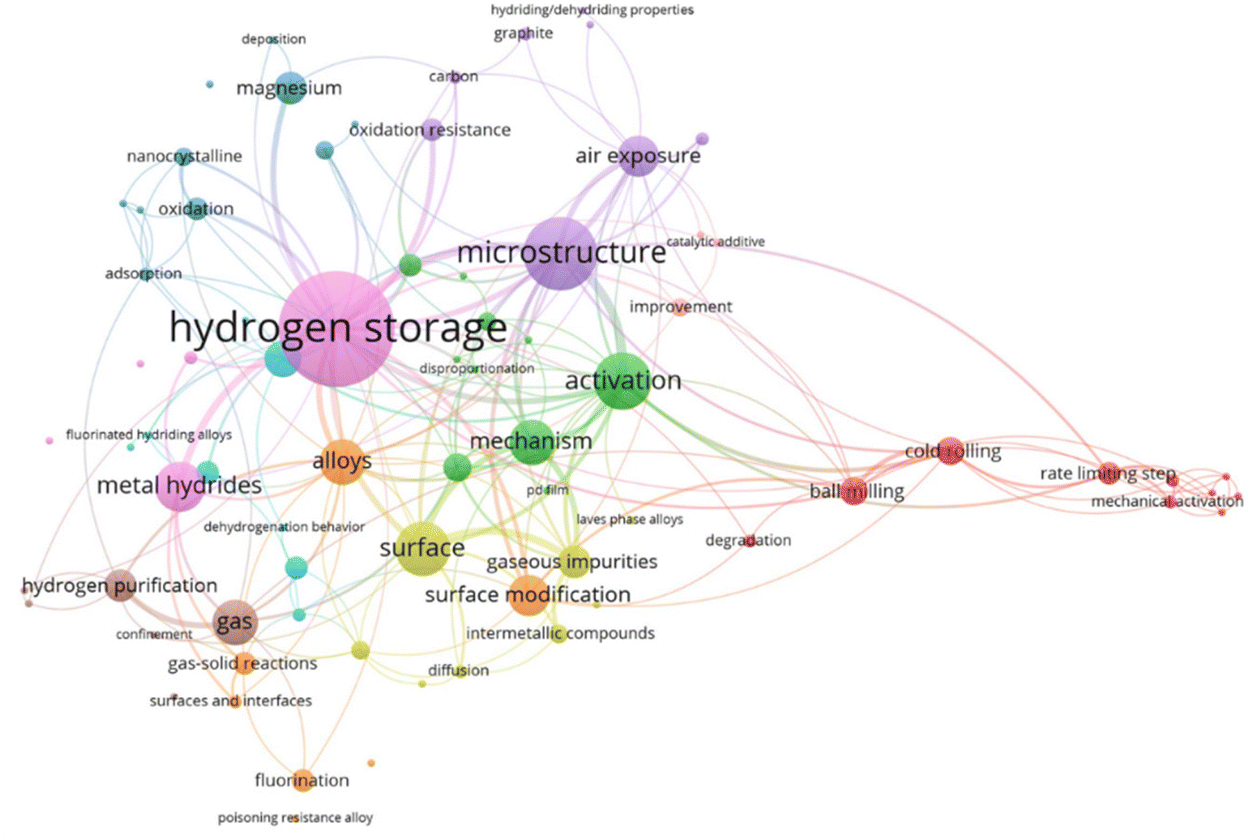 | ||
| Fig. 2 Keywords in the relevant literature from 1978 to 2024 counted by the Web of Science Core Collection. | ||
2 Various alloy–impurity interactions
Conventionally, the poisoning degree of the alloys depends not only on the nature of gas phase impurities, but also on the intrinsic properties and surface state of the alloy. Trace amounts of impurities can damage the hydrogen storage performance, usually in the form of degraded reaction rates or/and capacity. In this section, the typical metal hydrides are discussed, while the multicomponent alloys are classified to the subsequent modification part.2.1 AB5-type alloys
Regarding AB5 alloys, the most representative is LaNi5 which is easily activated under an ambient atmosphere, has fast cycling reversibility and is relatively insensitive to impurities. In the early 1980s, Goodell and Sandrock16 conducted extensive research on the poisoning behavior of LaNi5. The results showed that CO had the most significant poisoning effect. Afterward, it was found that CO chemisorbed on the surface to hinder the dissociation and penetration of hydrogen at low temperatures due to the higher adsorption activity of CO in comparison with H2.17 Other studies18,19 further pointed out that the strong adsorption tendency of active site Ni clusters to CO was the primary cause of surface deactivation. When the temperature exceeds 388 K, CO will undergo the following reaction:20,21| CO + 2H2 → CH4 + 1/2O2 | (1) |
Since CH4 is virtually innocuous to the hydrogen absorption performance, the subsequent analysis will focus on oxygen contamination. Interestingly, in the hydrogen atmosphere containing O2 and H2O, the hydrogen capacity decreased progressively during the first few cycles followed by a recovery, exhibiting a self-restore mechanism of the active surface.22 The oxygen-induced surface segregation that forms lanthanide oxide/hydroxide while keeping the catalytic Ni clusters on the subsurface may be responsible for the above retard–recovery phenomenon.23 Concretely, LaNi5 is initially subjected to a transient poisoning ascribed to the competitive adsorption of H2 and O2 on the surface. Later, the surface energy further decreases as oxygen chemisorption increases, resulting in the aggravated segregation and the recovery of hydrogen storage capacity.24 In fact, the metallic Ni cluster can only serve as a hydrogen dissociative catalyst to promote the kinetic property. The positive effect in the initial stage of poisoning is partly due to the surface reconstruction effect induced by the heat treatment during dehydrogenation,25 which partially cracks the oxide layer and slightly restores the hydrogen storage capacity. However, when the cycling number reaches a certain value, the hydrogenation capacity will still decay with the continuous consumption of the alloy, causing the decrease of bulk phase content and oxidation in the Ni-rich region.26,27 The dual effect was also determined by Han et al.28 using specific XPS analysis. It was concluded that the above-mentioned passivation is La(OH)3, which has a synergistic catalytic effect with Ni clusters. The corresponding reaction process can be described as follows:
| 2LaNi5 + 6O(adsorbed) + 6H(adsorbed) → 2La(OH)3 + 10Ni | (2) |
According to the report of Wallace et al.,29 LaNi5 combines with oxygen to generate La2O3, and then the oxide continues to transform into the aforementioned La(OH)3 upon contact with H2. Alternatively, Block et al.30 evaluated the cycling performance of LaNi5 under various impurity atmospheres. They showed that N2 and CH4 had no significant effect on LaNi5 in terms of hydrogen uptake rates. In addition, even 20 vol% CO2 is less harmful regarding hydrogen absorption capacity. In contrast, in the 1 vol% H2S mixture, the sample completely loses its hydrogen absorption ability after three cycles due to the high sulfur content on the surface. Similarly, Schweppe et al.31 experimentally observed the degradation extent of the impurities on the relative hydrogen absorption rate of LaNi5, viz N2 < CO2 < O2 < H2S < CO. Other impurities such as SO2 are also fatal poisons that can further improve the stability of LaNi5H6, leaving it unable to release hydrogen under conventional conditions.32 Furthermore, the poisoning behavior of other AB5-type binary alloys has been investigated under different atmospheres. A comprehensive comparison of the hydrogenation rate and capacity of the alloys in CO2/H2 co-flow and after pre-exposure to CO2 indicated that the anti-poisoning ability could be described as: LaCo5 ≪ LaNi5 < CaNi5. The first principles calculation demonstrated that the Ni element on the B side has stronger resistance to impurities than the Co element. Meanwhile, the Ni and the A-site Ca elements have a synergistic effect of reducing the CO2 adsorption energy.33 The mechanism is also applicable to O2 poisoning, as the Ni segregation observed on the CaNi5 surface is distinctly higher than that on the LaNi5 surface after air exposure,34,35 representing fewer impurities adsorbed on the Ni site, thus keeping it in a metallic state. From the perspective of thermodynamics, the formation enthalpy (absolute value) of Ca oxide is less than that of La oxide. Thus, the concentration of Ca oxide on the surface is relatively low, which can also indirectly verify the enrichment of catalytic nickel, leading to easier activation property.36
2.2 AB2-type alloys
AB2-type alloys have advantages of easy activation and low hysteresis effect but are poorly reversible and susceptible to impurity interference.37 Although AB2-type alloys are one of the most practical series of hydrogen storage materials available, there are few studies on the poisoning behavior of pure binary alloys. Most of the research focuses on multicomponent alloys, which will be recounted in the subsequent modification section. Previous studies claimed that 5 vol% CH4 only slightly hindered the adsorption kinetics of Ti1.2Mn1.8 and scarcely changed the reversible capacity. However, the deterioration was severe with prolonged cycles when CO was present in the hydrogen flow.38 Subsequently, Bernauer et al.39 proposed and outlined that the intrinsic effects of impurity gases on such materials can be divided into two categories, i.e., high concentrations of N2 and CH4 only negligibly reduce the hydrogen absorption kinetics, while low concentrations of CO, CO2, O2 and H2O could severely impair capacity and cycling stability. However, unlike the common A-side elemental segregation described above, Schlapbach et al.40 considered that two types of AB2 alloys (ZrMn2 and TiMn2−x) have the unique feature of B-side elemental segregation. Mn was enriched on the topmost surface and preferentially oxidized while Zr and Ti in the subsurface continued to form metal precipitates (also oxidized to some extent), observed by XPS technology. The lower surface energy and oxide formation enthalpy of Mn can largely account for this uncommon behavior. From a different theoretical point of view, Schülke et al.41 explained that the relatively minimal excess Gibbs free energy of Mn is responsible for its segregation tendency.2.3 AB-type alloys
AB-type TiFe alloy is of low cost but has relatively harsh in activation conditions and is sensitive to impurities. For example, air-exposed TiFe is easily deactivated under room temperature conditions due to the lack of surface segregation to limit the malignant oxidation of the surface.42–44 TiFe was completely poisoned after two cycles in 1 vol% CO2, and the poisoning effect of CO was more intense. Similarly, the concentration of 0.2 vol% H2S severely affected the hydrogenation performance of the alloy, resulting in a capacity decline of up to 25% per cycle. All three impurity gases mentioned above will seriously block the hydrogen uptake active sites, thus reducing the reactivity of the surface to hydrogen. In contrast, the poisoning effect of TiFe alloy under the CH4 atmosphere was not apparent because it did not passivate the surface. The cycling capacity reduction may be related to the lower H2 partial pressure in the presence of CH4.30 Moreover, Ti was gradually oxidized while Fe revealed its metallic state as the cycling proceeded in a 300 ppm O2 and H2O hydrogen mixture. It should be noted that no self-recovery phenomenon occurred, compared with LaNi5. The cycling capacity continued to decay mainly due to the insufficient catalytic activity of Fe or the denseness of the Ti oxide film.16 Shwartz et al.45 compared the evolution of surface composition and chemical valence state of TiFe exposed to O2 and H2O. It was found that surface Ti underwent two-step oxidation under the action of O2 to eventually generate TiO2, while only the first step oxidation (TiO) took place under H2O with a weak oxidation ability. Although thermodynamic analysis suggests that Fe in the bulk phase is prone to being oxidized, experimental results showed that Fe remained in a metallic state. This contradiction was mainly attributed to the fact that the surface reaction required a much higher enthalpy of formation than the bulk reaction under the induction of O2/H2O and the stronger oxidation resistance of the alloy (TiFe) relative to the metal (Ti and Fe). As a result, the valence transformation of the uppermost Fe was difficult.Additionally, AB-type ZrCo alloy is also in a severe situation in terms of gas contamination.46 Fortunately, the research showed that the reactivity of ZrCo with specific gaseous impurities is relatively low. The corresponding carbides/nitrides will be formed only at a fairly high temperature (above 400 °C).47 The hydrogenation enthalpy and entropy of ZrCo were almost unchanged after nitriding and carbonizing, indicating that the influence of impurities on thermodynamics is negligible.48 Glasbrenner et al.49 elucidated that zirconium compounds were more hazardous for hydrogen storage capacity than cobalt compounds due to the dominant role of Zr in the hydrogenation process. Therefore, the high reactivity of Zr with CH4, C2H4, O2 and N2 substantially reduces the hydrogen absorption capacity. Among them, N2 inhibits hydrogen uptake more strongly than CH4, and O2 is relatively less harmful.50,51 The preferential oxidation behavior of Zr compared with Co was also observed by Bredendiek-Kämper et al.52 The high oxygen solubility in Zr promoted the enrichment of ZrO2 on the surface. Conversely, the lower affinity of CO and CO2 for Zr allowed more uncontaminated Zr to react with hydrogen, resulting in insignificant capacity attenuation.49 However, a different view pointed out that the extremely low plateau pressure of ZrCo alloy at room temperature is the non-lethal factor of CO to the system, enabling a greater hydrogen uptake driving force.53 Kato et al.54 investigated the interference of hydride ZrCoHx on the catalytic activity of CO2 in the hydrogen mixture, since a higher desorption temperature (above 300 °C) facilitates the methanation of the released hydrogen atoms with CO2 on the surface. The reaction equation is as follows:
| CO2 + 4H2 → CH4 + 2H2O | (3) |
As illustrated by the parabolic-type curve in Fig. 3a and the linear relationship in Fig. 3b, the higher the hydrogen concentration in the hydride, the greater the flux of hydrogen atoms diffused from the bulk phase to the surface, resulting in more intense methane generation (Fig. 3b). Furthermore, Fig. 3c corroborates that methanation is temperature dependent and elaborates on the intrinsic mechanism. Initially, the lower temperature is insufficient to decompose hydrogen molecules in the gas and bulk phases. Meanwhile, CO2 and H2 adsorb on the surface to form formic acid (HCOOH) (Fig. 3c(a)). Hydrogen atoms then diffuse from the bulk phase to the surface and react with the formic acid at elevated temperatures to generate CH4 and H2O (Fig. 3c(b)), in agreement with eqn (3). Afterward, the excessive temperature leads to the dissociation of H2 within the gas mixture hitting the surface, allowing the hydrogen atoms in the gas mixture and hydride to participate in the reaction jointly and produce more products (Fig. 3c(c)). Eventually, the declined content of CH4 is mainly ascribed to the decrease of CO2 adsorption upon reaching a certain high temperature. Normally, hydrogen storage alloys undergo the hydrogenation process at room temperature rather than high temperature. As a result, considerable attention has been focused on the hydrogenation and poisoning resistance performance of ZrCo alloys under oxygen-containing atmospheres at room temperature. It has been confirmed that ZrCo is completely deactivated after three cycles under the 1 mol% O2 atmosphere (Fig. 3d), indicating its extremely oxygen-sensitive nature even at room temperature. Furthermore, ZrCo could quickly absorb saturated 2 wt% H2 under pure hydrogen, whereas only 0.26 wt% H2 could be obtained when 5 mol% O2 was introduced into the hydrogen, as illustrated in Fig. 3e. In particular, the material manifests a two-step hydrogenation process under 0.5 mol% O2, which is divided into a rapid hydriding stage and sluggish hydriding stage with an S-shaped kinetic curve (Curve 1).55 Subsequently, Jiapeng et al.56 observed a similar deterioration phenomenon of pure ZrCo, revealing the intrinsic mechanism of general physical blanket and chemical surface poisoning effects induced by various impurity gases. Specifically, the hydrogenation kinetic curves for ZrCo depicted in Fig. 3f demonstrated that active gases like CO2 initially have a chemical poisoning effect that severely impairs the hydrogenation kinetics. Afterward, ZrCo still achieves its saturated capacity with extended time since the intrinsic physical blanket effect of active CO2 dominates. Inactive gas N2, on the other hand, exhibits merely a general physical blanket effect, which damages hydrogenation kinetics without attenuating the ultimate hydrogenation capacity. A GC analysis was conducted to further elaborate and verify the proposed adsorption behavior, as shown in Fig. 3g. The extremely low signal of N2 in the empty reactor compared with H2 illustrates the uniform distribution of the gas mixture throughout the device (Fig. 3h). In contrast, the signal of N2 is more prominent than that of H2 in the reactor equipped with ZrCo. The intuitive physical blanket process is portrayed in Fig. 3i. Once the valve is opened, the selective absorption of H2 by the ZrCo material is responsible for the localized enrichment of the impurity gas and thus hinders the physical inward diffusion of H2. Since inert gases are only physically adsorbed to the alloy surface rather than impairing the hydrogenation activity, elimination strategies regarding the inherent physical blanket effect are well worth exploring to facilitate hydrogenation under inactive gas mixture. Notably, innovative approaches are discussed in section 3.3.4.
 | ||
| Fig. 3 (a) The formation of CH4 on ZrCoHx (x = 0, 0.1, 1.2, 2.9) in the flow of CO2, H2 (H2/CO2 = 5), and He. (b) The peak area of the ion current ratio m/z = 15 (CH3+) to m/z = 4 (He+) vs. the initial hydrogen concentration in ZrCoHx (x = 0, 0.1, 1.2, 2.9). (c) The schematic representation of the reduction of carbon dioxide on the ZrCoHx. Copyright 2016 Wiley; (d) cycling hydriding curves for ZrCo in 1 mol% O2. (e) Hydriding curves for ZrCo in pure hydrogen and hydrogen–oxygen mixed gas. Copyright 2024 Elsevier; (f) hydrogenation kinetic curves for ZrCo in 5 mol% N2/CO2. (g) GC analysis for the residual gas in reactor without/with ZrCo after introducing 5 mol% N2. (h–i) Schematic mixed gas concentration distribution for reactor without/with ZrCo. Copyright 2024 Elsevier.54–56 | ||
2.4 A2B-type alloys
As a typical A2B alloy, Mg2Ni is an ideal hydrogen storage medium with a relatively high hydrogen storage capacity (3.6 wt%). However, it suffers from difficulties in activation and poor hydrogen desorption kinetics. Similar to LaNi5, Mg2Ni has prominent recession–recovery characteristics under the CO/H2 environment at 300 °C, most likely related to MgO and Ni clusters.20 Later, Selvam et al.36 experimentally observed that both Mg2Ni and Mg2Cu exhibited surface segregation of Mg depending on the lower surface energy and higher oxygen affinity. The MgO produced by Mg coming into contact with oxygen prevents the large-scale oxidation of the metallic Ni/Cu. As reported, the poisoning resistance of Mg–Ni alloys could be notably improved with the increased Ni proportion in the whole system, further confirming the catalytic effect of Ni clusters on hydrogen uptake under the gas mixture.57 The XPS depth profile (Fig. 4a) obtained by Kato et al.58 under the CO2-containing H2 atmosphere can better explain the segregation of Mg/MgO. Meanwhile, the concentration of Ni on the fresh surface gradually increases with prolonged sputtering time, representing more metallic nickel on the subsurface. The TOF-SIMS image (Fig. 4b) further confirmed the correlation between the distribution of MgO and Ni. The uppermost layer was covered with a large amount of MgO and a small amount of Ni was unevenly distributed around it, corresponding to the green and yellow areas, respectively. This stacking relationship between layers is visually illustrated by the schematic diagram of Fig. 4d. Therein, determined by the XRD diffraction peaks of Mg2NiH4 after multiple hydrogen ab/desorption cycles in the inset of Fig. 4c, the MgO and Ni are disproportionation products of Mg2Ni/Mg2NiH4 in a mixed CO2/H2 gas owing to the selective oxidation of Mg. At the same time, the precipitated Ni clusters can compensate for the interference of the passivation layer with hydrogen and promote the methanation of CO2 to produce a less polluted gas. Moreover, the surface oxide layer of Mg2Cu is thicker than that of Mg2Ni by virtue of the brittle nature of Mg2Cu, which facilitates oxygen penetration and the migration of oxygen atoms.59 Yoshihiko et al.57 found that the influence of 5.07 vol% N2, 5 vol% CH4 or 1 vol% CO2 impurities on the thermodynamics of Mg2Ni alloy was negligible. The N2 and CH4 did not appear to form associated nitrogen/carbides, while CO2 was completely converted to carbonate groups. In contrast, Tran et al.60 found that 0.5 vol% N2 completely inhibited the hydrogenation of Mg2Ni because the material here was not pre-activated. Generally, H2O is considered to have at least moderate or limited negative effects, though it is relatively less poisonous for most alloys. Surprisingly, the work reported by Hampton et al.61 claimed that steam could essentially cause complete activation of Mg2Ni, allowing its initial hydrogen uptake. The percentage of hydrogen released increases with prolonged water exposure. The underlying mechanism of this water-induced activation of Mg2Ni will be discussed in section 3.3.4.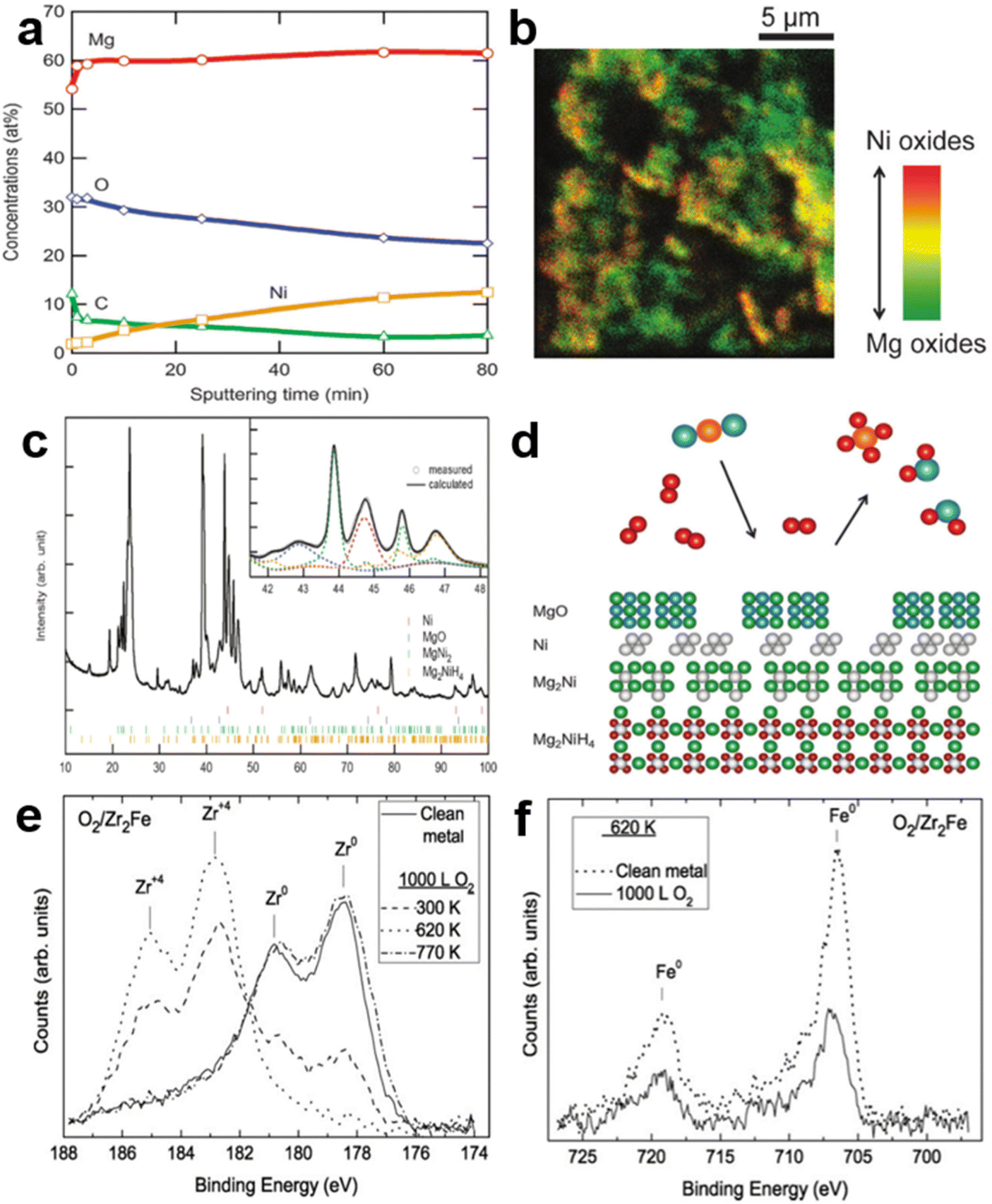 | ||
| Fig. 4 (a) The XPS and (b) TOF-SIMS of Mg2NiH4 after 5 hydrogen absorption/desorption cycles. (c) The XRD pattern after 18 cycles of the hydrogen absorption/desorption and the hydrogen desorption in CO2. (d) The disproportionated surface of Mg2NiH4. Copyright 2012 Royal Society of Chemistry; (e) Zr (3d) XPS spectra for clean Zr2Fe and for exposure to 1000 L oxygen at P = 10−5 Torr and various temperatures. (f) Fe (2p) XPS spectra for 347 °C, clean and 1000 L (P = 10−5 Torr), exposed to oxygen Zr2Fe. Copyright 2010 Elsevier.58,62 | ||
Zr2Fe, with extremely low hydrogen absorption equilibrium pressure, is considered as an ideal hydrogen isotope-trapping material. Since the purpose of hydrogen isotope capture is mainly to separate the scarce hydrogen isotope from the impurity atmosphere, the poisoning resistance of Zr2Fe alloy is crucial for its application.63 Fukada et al.64 discovered that the activated particle bed was efficient in removing hydrogen in N2 and Ar gas flows above 150 °C, while O2 above 640 ppm caused a complete loss of hydrogen-trapping activity in bulk. Similar results suggested that the operating conditions affected the gas removal efficiency. With temperature exceeding 278 °C, Zr2Fe adsorbed hydrogen isotope at an adequate rate in the presence of oxygen owing to the relatively low impurity content and the more active surfaces.65,66 The surface state evolution of Zr and Fe under an oxygen atmosphere was explicitly characterized by XPS, as shown in Fig. 4e and f. The adsorption and diffusion of oxygen were slow at 27 °C, corresponding to the coexistence of Zr0 and Zr4+. When the temperature rose to 347 °C, the oxidation gradually approached saturation while Fe remained metallic, meaning that oxygen was penetrating into the subsurface and bulk. Conversely, Zr showed only the metallic phase similar to the uncontaminated Zr2Fe at 487 °C, in which the elevated temperature was thought to promote oxide diffusion into the bulk phase and re-expose the active surface.62 This observation is consistent with the work of Cohen et al.,67 who suggested that Zr2Fe first induced the oxidation of Zr and the formation of elemental Fe under the oxygen atmosphere, followed by the oxidation of Fe. The high affinity of Zr for oxygen is the same as for Zr in ZrCo mentioned earlier. The complete reaction can be divided into the following two processes:
| Zr2Fe + 2O2 → 2ZrO2 + Fe | (4) |
| Zr2Fe + 11O2 → 8ZrO2 + 2Fe2O3 | (5) |
2.5 BCC-based solid solution alloys
Ti–V alloy is a widely studied BCC solid solution system that can form solid solutions at various Ti/V ratios.68 Relatively few investigations have been conducted on the poisoning aspects of pure Ti–V alloys. Suwarno et al.69 compared the cycling performance of two Ti–V alloys under different atmospheres. It was found that the sample could still absorb a considerable amount of hydrogen from the CO/H2 mixture in the initial cycle. However, compared with the Ar/H2 environment, the hydride was confined to the bulk phase due to the surface carbon/oxygen enrichment by the interaction with CO, resulting in an abrupt decrease in reversible hydrogen capacity in the second cycle. The cycling capacities of the two materials converged as the cycling continued, presumably because the surface-deposited layer limited further matrix oxidation. The surface contamination behavior assumed above can be confirmed by AES analysis. Under the pure hydrogen cycle, carbon and oxygen-containing products were present on the surface. Then, the O peak gradually diminished while the C peak disappeared as the ion sputtering extended. When cycled in Ar + 25%H2 + 1%CO, the C peak still existed after 690 s of sputtering (corresponding to 23 nm of surface removal), indicating that a large amount of CO-induced carbonization passivation covered the surface.2.6 MgH2
Mg has a high theoretical hydrogen storage capacity (7.6 wt%), good reversibility, cycling stability and meager cost. However, its strong oxidation tendency and impurity reactivity lead to activation difficulties and hydrogen storage performance attenuation, respectively. Low concentrations of impurities, such as a mixture of 85 ppm O2 and 8 ppm H2O, had a negligible effect on the Mg–H system, losing 8% capacity only after 478 cycles.70 Although the research demonstrated that oxygen reacted heavily with the Mg powders, it still allowed hydride formation at a reduced rate under 0.5 vol% O2, resulting in a 20% reduction in capacity. Moreover, the decay rate of N2 and CO on hydrogenation was relatively slow under the same conditions.71 It has been confirmed that the screening effect of CO2 is much more pronounced than O2 and N2. Thus, a concentration of 2 vol% CO2 caused an instantaneous loss of hydrogen absorption ability of Mg.72 The superficial phenomena alone are insufficient for a comprehensive evaluation. Thus, specific mechanisms are needed to explain the intrinsic poisoning behavior of these impurities. In recent years, most researchers have focused on the hydrogen ab/desorption performance of Mg/MgH2 after pre-exposure to air, which can be defined as the air stability of the material. For example, Friedrichs et al.73 analyzed the oxygen passivation behavior of MgH2 through in situ XPS and TEM. It was observed that the MgO and Mg(OH)x existed on the surfaces of Mg and MgH2, respectively. The Mg(OH)x content increased after air exposure at room temperature. In response to this phenomenon, Liu et al.74 further confirmed that MgO gradually converted to Mg(OH)2 by continuously absorbing H2O in the air. Radically, the Mg(OH)2 was responsible for surface passivation. Later, Jensen et al.75 discovered that the dehydrogenation kinetics of MgH2 slowed down upon contact with air due to the increase in the surface oxide layer that severely interfered with the outward diffusion of hydrogen, resulting in a significant increase in the dehydrogenation activation energy.In general, the alloy–impurity interactions of hydrogen storage alloys are surface/interface-dependent. The surface poisoning of the typical binary alloys (LaNi5, TiFe, Mg2Ni, Ti0.8V0.2, etc.) is mostly accompanied by A-side elemental segregation, whereas the AB2 alloy (Ti1.2Mn1.8, ZrMn2, etc.) has the unique feature of B-side elemental segregation. In most cases, hydrogen storage alloys of the same type have similar action rules under impurity gases. For example, the AB5-type alloys possess a relatively better anti-poisoning ability thanks to the subsurface enrichment of B-site elements, allowing the metallic active sites to promote hydrogen dissociation and penetration. However, the AB-type alloys and MgH2 exhibit high impurity reactivity and inferior poisoning resistance, attributed to the lack of subsurface segregation. Therefore, extensive modification strategies should be conducted to improve the anti-poisoning ability of binary alloys.
3 Modification of anti-poisoning ability
Over the past decades, much effort has been made to improve the tolerance of metal hydrides to impurities. The simplest method is to increase the hydrogen ab/desorption temperature, which can obviously suppress the poisoning tendency.20,76,77 However, a blindly elevated temperature is unrealistic for the practical application of hydrogen storage alloys. Therefore, a variety of reasonable modification methods are described below.3.1 Multiple alloying
Partial replacement of elements on either A or B side by neighboring elements to slow down the extent of poisoning of the alloy is a simple yet effective modification strategy, usually based on the typical metal hydrides introduced in Part 2. By doping the binary alloy with other elements, a new phase can be formed at the surface/interface to catalyse hydrogen absorption and block the impurity gas. For example, the partial substitution of Ni by Al enhances the oxidation resistance of LaNi5 alloy, since the substituted Al atoms reduce O2 adsorption by forming a thinner Al2O3 protective layer.27,28 The optimum Al doping content (in the range of 0.5–0.8) for long-term cycling stability of LaNi5−xAlx under a hydrogen mixture containing 100 ppm O2 has been derived by Nishimura et al.78 from a fitted curve of hydrogen uptake versus the number of cycles. Other doping elements, such as Ce, also substantially reduce the time required for complete dehydrogenation of CO-treated (La, Ce)Ni5 only because Ce elevates the plateau pressure of the system.79 Uchida et al.80,81 noted that, unlike Ce, Mn doping increased the sensitivity of LaNi4.5Mn0.5 to oxygen, which was reflected in the thickness of the generated MnO2 and Mn3O4 films (more than 100 nm), thus severely impeding the penetration of hydrogen into the bulk phase. In comparison with pure LaNi5, the Sn-substituted LaNi4.73Sn0.27 also exhibited poor hydrogen uptake capacity under a trace CO environment but showed a steady state after ten cycles without further deterioration.82 However, the pre-exposure of LaNi4.7Sn0.3 to O2 and H2O significantly enhanced the H2 reactivity due to the hydrogen-induced reduction of the deposited layer SnO2 under ambient atmosphere, allowing the highly catalytically active oxygen-deficient phase SnO2−x to promote the dissociation of hydrogen molecules.83In the aspect of poisoning resistance of AB2-type alloys, Yang et al.84 investigated the influence of doped elements Hf, Ti and Pd on ZrV2 at 0.5 vol% O2. It was found that the anti-poisoning order of the alloy was Zr(V0.9Pd0.1)2 > Zr0.9Hf0.1V2 > ZrV2, decaying to 80%, 69% and 19% of the original hydrogen storage capacity, respectively. Additionally, since metallic Ni always precipitates after air exposure and acts as an active surface, partial substitution of V by Ni is a rational strategy for improving the oxidation resistance of ZrV2 alloys.85 The situation is different under the CO2 atmosphere, where the addition of Ni reduces the hydrogen absorption properties of AB2-type (Ti0.515Zr0.485Mn1.2Cr0.8) alloys due to the relatively strong CO2–(AB2–Ni) bonds on the surface, while Fe and Co are suitable additives to reduce the CO2 sensitivity of the alloy.86 Maeda et al.87 found that the addition of trace amounts of La improved the cycling stability of AB2-type (Ti, Zr)(Ni, Mn, V, Fe)2.18 alloy in water-contaminated hydrogen, attributed to the selective oxidation of La. Since the diffraction peak of La oxide was not observed in the XRD pattern, the inherent antioxidant properties could not be clarified yet. Similarly, La or Mn was also added to other systems, giving the alloy a superior cycling stability than the original sample in hydrogen mixture, such as BCC-type V–Fe–Cr–Ti88 and AB-type Ti–Fe–Mn–Co.89
For AB-type alloys, most studies have focused on reducing the interference of air exposure to TiFe. Usually, Mn90 and Zr91,92 are considered as suitable dopants to promote surface segregation of TiFe, which effectively protects the surface particles from oxidation. Subsequently, Shwartz et al.45 observed that Fe remained in the metallic state even when exposed to a certain amount of oxygen, while Mn was readily oxidized. Similar oxidation behavior confirmed that Mn, as a sacrificial element, would be oxidized first to prevent further oxidation of Fe due to the lower formation enthalpy of Mn oxides.93,94 Then, Park et al.95,96 compared the first hydrogen absorption (activation) behavior of TiFe alloys after air exposure with different substituted elements. It was found that Mn- and Cr-doped alloys have a hydrogen-prone second phase MgZn2-type Mn/Cr-rich phase (depicted in the dotted line area of Fig. 5a and b), commonly referred to as the C14 Laves phase. Meanwhile, a large number of cracks in the Laves phase can be seen, confirming that hydrogen is first absorbed by the Laves phase, resulting in hydrogen absorption ability even after air-exposure for 12 h. Nonetheless, other Ni, Co and Cu-doped TiFe alloys with only a single TiFe phase are unable to absorb hydrogen. Hence, for TiFe-based alloys, the C14 Laves phase serves as an absorption medium and diffusion pathway that contributes to the hydrogen absorption properties of oxygen-contaminated alloys.97–99 Lv et al.100 further confirmed the validity of the C14 Laves phase, i.e., adding different levels of ZrMn2 could strikingly reduce the air sensitivity of TiFe. It is evident from the data shown in Fig. 5d that the increase in reaction rate and hydrogen capacity as well as the shortening of the incubation period are proportional to the ZrMn2 fraction. This correspondence is clearly related to the microstructure observed in Fig. 5c, where the bright phase (Zr/Mn-rich Laves phase) is more abundant and uniformly distributed with a reasonable increase in ZrMn2 fraction. The bright phase acts as a window for hydrogen penetration into the dark phase (TiFe main phase), giving rise to improved kinetics. Furthermore, air-exposure TiFe + 12 wt% ZrMn2 manifested similar kinetic properties compared with the counterpart under Ar (Fig. 5e), indicating that the ZrMn2 addition notably retards the oxidation trend. However, no C14 Laves phase was observed in the experiments of Modi et al.93 Instead, a new BCC primary phase and a better hydrogen-permeable FCC secondary phase emerged.
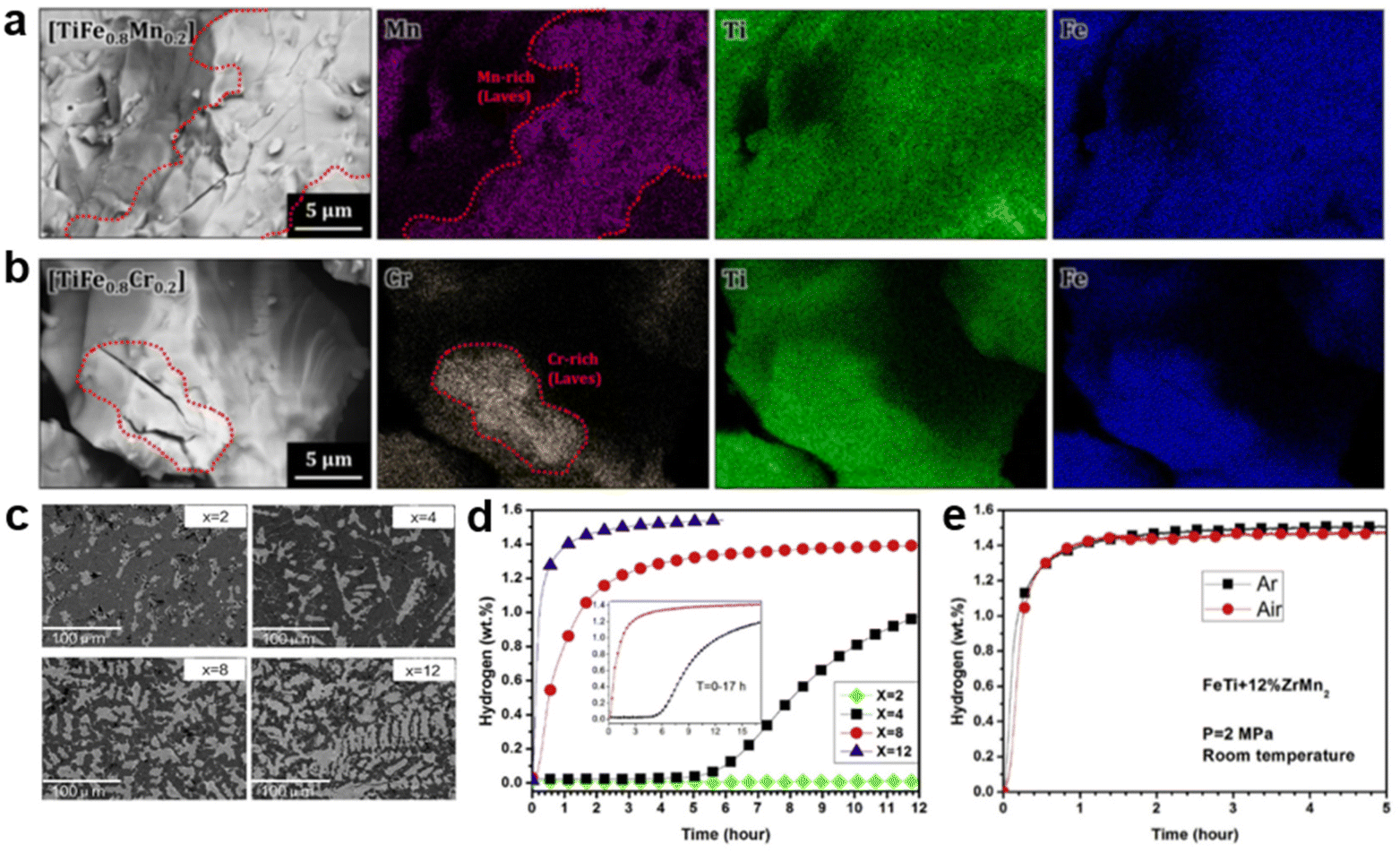 | ||
| Fig. 5 The morphologies of the (a) TiFe0.8Mn0.2 and (b) TiFe0.8Cr0.2 alloys after hydrogenation observed by FE-SEM. Copyright 2021 Elsevier; (c) backscattered electron micrographs of arc-melted TiFe + x wt% ZrMn2 (x = 2, 4, 8, 12) alloys. (d) First hydrogenation curves of TiFe + x wt% ZrMn2 (x = 2, 4, 8, 12) alloys at room temperature under 3 MPa hydrogen pressure. (e) The first hydrogenation of TiFe + 12 wt% ZrMn2 alloys at room temperature under 2 MPa hydrogen pressure. Copyright 2017 Elsevier.95,100 | ||
Mg2Ni and MgH2 are summed up as Mg-containing materials in this paragraph. It has been confirmed that prolonged air exposure to MgH2–5% VTiCr deteriorated the kinetics only slightly, with negligible degradation in reversibility and cycling stability.74 Dehouche et al.101 found that a nano Mg–V–Ti composite exhibited improved hydrogen absorption properties due to structural relaxation under long-term cycling conditions of 110 ppm H2O. However, the surface effects continued to aggravate the desorption properties. Tran et al.60 probed the surface interaction between Na-doped Mg2Ni and N2. The Na-doped Mg2Ni was significantly resistant to impurity compared with the pristine one at a concentration of 5 vol% N2, in that a Na-modified surface with positive charge characteristics could promote the selective adsorption of N2, thus retaining the active site of Ni for hydrogen adsorption and dissociation.
3.2 Catalytic doping
If the active sites on the surface are occupied by impurity gases, it is difficult for hydrogen molecules to decompose into hydrogen atoms to penetrate the bulk phase. Adding catalysts is another strategy to alleviate surface poisoning, primarily for Mg-based hydrides (MgH2 and Mg2NiH4). As mentioned in the previous section, pure Mg can severely deteriorate the hydrogen uptake/desorption kinetics upon air exposure. However, suitable catalysts and effective treatment can mitigate the oxidation degree. Several studies102,103 have shown that an appropriate amount of MgO is a suitable additive for the MgH2 system. Recently, Zhang et al.104 explored the relationship between the way of introducing MgO and its position in the Mg matrix. It was found that the doped-MgO can limit the grain size and avoid particle agglomeration, thus maintaining kinetic stability. Furthermore, the doped-MgO forms a cladding layer at a localised location, preventing impurity gas attacks and accelerating the de/hydrogenation process. In contrast, MgO formed through air exposure acted as a surface passivation layer that severely impaired the hydrogen storage properties. Xie et al.105 claimed that introducing CeO2 allowed the dehydrogenation of MgH2 after air exposure with almost no incubation period and rapid desorption at the initial stage, attributed to the unique intrinsic properties of CeO2. The CeO2 is uniformly distributed on the substrate surface to limit the formation of dense MgO films. Specifically, Ce is susceptible to oxidation upon air exposure. The formed CeO2 is unstable and converts to CeH2.73 after several cycles in pure hydrogen. Although the transition from CeO2 to CeH2.73 is thermodynamically unfavorable owing to a more negative formation enthalpy of CeO2 than CeH2.73, the migration of oxygen atoms in CeO2 is less limited by the Ce lattice and the incorporation of hydrogen in cerium oxide is thermodynamically spontaneous, which facilitates the phase transformation. CeO2 was then regenerated spontaneously on contact of CeH2.73 with oxygen. It is interesting that the oxide and hydride of cerium can be interconverted under air and hydrogen atmosphere, probably favoring the desired practical application. In fact, both Ce-containing substances have catalytic and nucleation sites that promote hydrogen decomposition/recombination and MgH2 formation, respectively. Moreover, the exceptionally high affinity of cerium for oxygen allows the faster production of CeO2 than MgO,106 thus explaining the superior antioxidant properties of MgH2–CeO2 and the stronger oxidation resistance in the Mg–Ni–Ce system with higher Ce content.Thermodynamically, the poisoning reaction of Mg2NiH4 in air at room temperature can be satisfied. The three reaction processes inferred by combining TEM, XPS and FTIR analysis are as follows:107
| Mg2NiH4 + 5/2O2 → 2MgO + NiO + 2H2 | (6) |
| Mg2NiH4 + 2H2O + 2CO → 2MgCO3 + Ni + 4H2 | (7) |
| Mg2NiH4 + 2H2O → 2Mg(OH)2 + Ni + 2H2 | (8) |
Afterward, Ma et al.108 compared the difference of TiH2 and/or graphite addition on the hydrogen storage performance of the composites. Surprisingly, the hydrogen uptake and release rates of all three air-exposure samples were instead enhanced compared with the uncontaminated samples, especially for Mg2NiH4–TiH2–graphite with synergistic catalytic effect, as shown in Fig. 6i and j. In addition, Mg2NiH4–TiH2 showed the best air stability and exhibited the least capacity loss. Subsequent TEM analysis revealed the mechanism by which this material has excellent air stability. Compared with Mg2NiH4–TiH2–graphite, 30-day air-exposed Mg2NiH4–TiH2 possessed a relatively thinner deposited layer (Fig. 6b and f) and less oxygen segregation (Fig. 6d and h). This exceptional performance is illustrated by the schematic diagram of Fig. 6k. The formation of the passivation layer and the TiH2@TiO2 slowed down the oxidation of the substrate. At the same time, TiH2@TiO2 and Ni-based products play a catalytic role. Therefore, these substances function together to improve air stability and kinetics. Recently, a heterophase composite (Mg2NiH4–MgNi2) was constructed in a Mg–Ni system, leading to superior anti-oxidation and kinetic performance. The relative content of Mg2NiH4 and MgNi2 can be modulated by varying the ratio of metallic Mg to Ni using the hydrogenation combustion synthesis (HCS) method. The in situ-formed MgNi2 can alleviate surface oxidation and act as a fast channel for hydrogen diffusion, thus improving the air stability and dehydrogenation kinetics, respectively.109
 | ||
| Fig. 6 TEM, HRTEM and element mapping images of (a–d) Mg2NiH4–TiH2 and (c–h) Mg2NiH4–TiH2–graphite after air exposure for 30 days. Isothermal hydrogen (i) desorption and (j) absorption reaction fraction curves at 230 °C for the as-milled Mg2NiH4–additive (TiH2, graphite and TiH2–graphite) samples before and after air exposure for 30 days. (k) Schematic illustration of air stability mechanism of Mg2NiH4–TiH2 during air exposure. Copyright 2022 Elsevier.108 | ||
Table 1 summarizes the most representative data on the hydrogen storage properties of various alloys/hydrides in impurity gases. Off-line poisoning and on-line poisoning are taken as two types. It should be noted that for comparison, the maximum hydrogen storage capacity refers to the hydrogen ab/desorption capacity under a pure hydrogen atmosphere at a specific temperature. Given the extremely slow de/hydrogenation rates of some alloys/hydrides under certain impurities, the initial capacity is the maximum hydrogen capacity that can be absorbed in a limited time or up to 95% saturation capacity.
| Alloy/hydride | Temperature (°C) | Impurity | C max (wt%) | C poisoned (wt%) | Ref. |
|---|---|---|---|---|---|
| C max denotes the maximum hydrogen storage capacity at a given temperature. Cpoisoned denotes the initial hydrogen storage capacity after poisoning. | |||||
| (1) Off-line poisoning | |||||
| LaNi4.7Al0.3 | RT | 48 h in air | 0.65 (H/A) | 0.60 (H/A) | 110 |
| TiCr2 | RT | 6 months in air | 1.3465 | 0.7119 | 111 |
| TiCr2 + 2 wt% Zr | RT | 48 h in air | 1.261 | 1.100 | 112 |
| TiCr2 + 4 wt% Zr | RT | 48 h in air | 1.315 | 1.182 | 112 |
| TiCr2 + 8 wt% Zr | RT | 48 h in air | 1.556 | 1.520 | 112 |
| TiCr2 + 8 wt% Mn | RT | 6 months in air | 1.3204 | 0.8417 | 111 |
| Zr0.9Ti0.1V2 | RT | 30 s in N2 | 3.90 (H/A) | 0.25 (H/A) | 113 |
| TiFe | 40 | 5 min in air | 0.730 | 0.18 | 114 |
| TiFe0.85Mn0.15 | RT | 24 h in air | 1.28 | 0.05 | 94 |
| TiFe0.8Mn0.2 | 20 | 1 h in air | — | 1.41 | 96 |
| TiFe0.8Cr0.2 | 20 | 1 h in air | — | 1.81 | 96 |
| TiFe0.9Ni0.1 | RT | 5 min in air | 0.923 | 0.20 | 114 |
| TiFe0.8Ni0.2 | 40 | 5 min in air | 1.092 | 0.29 | 114 |
| TiFe0.6Ni0.4 | 40 | 5 min in air | 0.87 | 0.20 | 114 |
| TiFe0.85Mn0.15Cr0.15 | 30 | 2 h in air | — | 1.6 | 115 |
| TiFe0.85Mn0.15Cr0.30 | 30 | 2 h in air | — | 0.15 | 115 |
| Ti0.515Zr0.485Mn1.2Cr0.8 | 20 | 0.5 h in CO2 | 1.69 | 1.55 | 86 |
| Ti0.515Zr0.485Mn1.2Cr0.8Fe0.1 | 20 | 0.5 h in CO2 | 1.58 | 1.50 | 86 |
| Ti0.515Zr0.485Mn1.2Cr0.8Co0.1 | 20 | 0.5 h in CO2 | 1.57 | 1.54 | 86 |
| Ti0.515Zr0.485Mn1.2Cr0.8Ni0.1 | 20 | 0.5 h in CO2 | 1.63 | 1.35 | 86 |
| TiFe + 8 wt% Zr | RT | 35 h in air | 1.70 | 1.30 | 91 |
| TiFe + 12 wt% Zr | RT | 35 h in air | 1.85 | 1.40 | 91 |
| TiFe + 16 wt% Zr | RT | 35 h in air | 1.90 | 1.79 | 91 |
| TiFe + 12 wt% (Zr + 2Cr) | RT | 3 h in air | 1.30 | 1.06 | 116 |
| MgH2 | T abs: 350 | 40 h in air | 5.6 | 4.2 | 74 |
| T des: 350 | 40 h in air | 5.35 | 4.0 | ||
| MgH2 | T des: 300 | 24 h in air | 6.5 | 1.5 | 117 |
| MgH2–5%VTiCr | T abs: 25 | 40 h in air | 3.17 | 1.22 | 74 |
| T des: 300 | 40 h in air | 5.55 | 2.30 | ||
| MgH2–CeO2 | T des: 300 | 168 h in air | 5.3 | 4.7 | 105 |
| MgH2–5%NbF5 | T des: 300 | 24 h in air | 6.8 | 5.0 | 117 |
| MgH2–5%Nb2O5 | T des: 300 | 24 h in air | 6.4 | 3.5 | 117 |
| Mg2NiH4 (0.5 h milled) | T des: 230 | 67 days in air | 3.10 | 2.88 | 107 |
| Mg2NiH4 (10 h milled) | T des: 230 | 67 days in air | 2.90 | 2.35 | 107 |
| Mg80Ni20Hx | T abs: 275 | 4 months in air | 3.5 | 3.1 | 118 |
| Mg80Ni20Hx | T des: 275 | 4 months in air | 3.7 | 3.3 | 118 |
| Mg–20Ni | T abs: 350 | 168 h in air | 5.9 | 2.7 | 106 |
| T des: 350 | 168 h in air | 6.1 | 1.6 | ||
| Mg–20Ce | T abs: 350 | 168 h in air | 6.0 | 5.4 | 106 |
| T des: 350 | 168 h in air | 6.0 | 5.7 | ||
| Mg–5Ni15Ce | T abs: 350 | 168 h in air | 5.8 | 5.0 | 106 |
| T des: 350 | 168 h in air | 6.0 | 5.4 | ||
| Mg–10Ni10Ce | T abs: 350 | 168 h in air | 5.7 | 4.5 | 106 |
| T des: 350 | 168 h in air | 6.0 | 5.2 | ||
| Mg2NiH4–TiH2 | T des: 230 | 1 month in air | 3.07 | 2.36 | 108 |
| Mg2NiH4–graphite | T des: 230 | 1 month in air | 3.03 | 1.78 | 108 |
| Mg2NiH4–TiH2–graphite | T des: 230 | 1 month in air | 2.93 | 1.14 | 108 |
| (2) On-line poisoning | |||||
| LaNi5 | 20 | 11 vol% CO | 1.39 | 0.49 | 33 |
| LaNi5 | 40 | 0.01 vol% CO | — | 1.38 | 82 |
| LaNi5 | 27 | 1 vol% H2S | 7.7 (H/A) | 2.8 (H/A) | 30 |
| LaNi4.73Sn0.27 | 40 | 0.01 vol% CO | — | 1.20 | 82 |
| LaNi4.9Al0.1 | 20 | 0.1 vol% CO | 1.09 | 0.18 | 18 |
| LaNi4.9Al0.1 | 50 | 0.1 vol% CO | 1.17 | 0.20 | 18 |
| LaNi4.9Al0.1 | 80 | 0.1 vol% CO | 1.08 | 0.72 | 18 |
| LaNi4.7Al0.3 | 30 | 0.03 vol% CO | 1.40 | 1.31 | 77 |
| LaNi4.7Al0.3 | 70 | 0.1 vol% CO | 1.18 | 0.95 | 119 |
| LaNi4.7Al0.3 | 80.2 | 0.1 vol% CO | 0.85 | 0.56 | 19 |
| LmNi4.8Al0.2 | 40 | 0.1 vol% H2S | 1.37 | 1.32 | 76 |
| LmNi4.8Al0.2 | 60 | 0.1 vol% H2S | 1.35 | 1.30 | 76 |
| LmNi4.8Al0.2 | 80 | 0.1 vol% H2S | 1.20 | 1.21 | 76 |
| LmNi4.8Al0.2 | 40 | 0.1 vol% CO | 1.37 | 0.65 | 76 |
| LmNi4.8Al0.2 | 60 | 0.1 vol% CO | 1.35 | 1.30 | 76 |
| LmNi4.8Al0.2 | 80 | 0.1 vol% CO | 1.20 | 1.15 | 76 |
| CaNi5 | 20 | 11 vol% CO | 1.50 | 1.13 | 33 |
| LaCo5 | 20 | 11 vol% CO | 0.93 | 0.49 | 33 |
| MmNi4.025Co0.4Mn0.275Al0.3 | 20 | 11 vol% CO | 1.25 | 0.54 | 33 |
| Ti1.2Mn1.8 | 27 | 0.5 vol% CO | 1.78 | 0.60 | 38 |
| Zr(V0.95Ni0.05)2 | 35 | 0.25 vol% O2 | 4.33 (H/A) | 3.79 (H/A) | 120 |
| Zr(V0.95Ni0.05)2 | 35 | 0.5 vol% O2 | 4.33 (H/A) | 3.3 (H/A) | 120 |
| Zr(V0.95Ni0.05)2 | 35 | 1 vol% O2 | 4.33 (H/A) | 2.4 (H/A) | 120 |
| Zr(V0.95Ni0.05)2 | 150 | 1 vol% O2 | 3.60 (H/A) | 2.23 (H/A) | 120 |
| Zr(V0.95Ni0.05)2 | 300 | 1 vol% O2 | 2.80 (H/A) | 2.56 (H/A) | 120 |
| Zr(V0.95Ni0.05)2 | 450 | 1 vol% O2 | 2.10 (H/A) | 1.82 (H/A) | 120 |
| Zr(V0.95Ni0.05)2 | 35 | 2 vol% O2 | 4.33 (H/A) | 0.07 (H/A) | 120 |
| Ti0.98Zr0.02V0.45Fe0.1Cr0.05Mn1.4 | 27 | 2.4 vol% CO | 3.15 | 0.10 | 38 |
| TiFe0.86Mn0.07Co0.07 + 4 wt% Mm | 25 | 0.025 vol% O2 | 1.76 | 1.58 | 89 |
| ZrCo | 25 | 5.1 vol% CO | 1.90 | 0.49 | 53 |
| ZrCo | 25 | 3.5 vol% CO, 1 vol% CH4, 0.2 vol% CO2, 0.2 vol% O2 | — | 0.73 | 121 |
| Zr70V25Ti5 | 25 | 5.51 vol% CO | 2.33 | 0.31 | 53 |
| Zr2Fe | 25 | 5.15 vol% CO | 1.88 | 0.21 | 53 |
| Zr0.8Ti0.5V1.7 | 25 | 0.1 vol% air | 2.86 | 2.86 | 122 |
| Zr0.8Ti0.5V1.7 | 25 | 1 vol% air | 2.86 | 0 | 122 |
| Zr0.8Ti0.5V1.7 | 180 | 1 vol% air | — | 0.70 | 122 |
| Zr0.8Ti0.5V1.7 | 340 | 1 vol% air | — | 1.55 | 122 |
| Ti0.9V0.1 | 425 | 1 vol% CO | 3.75 | 0.60 | 123 |
| V40Fe8Ti32Cr20 | 30 | 0.025 vol% O2 | 2.3 | 2.2 | 88 |
3.3 Surface modification
The fluoride layers mentioned above are insoluble in water and therefore can be generated stably on the surface. However, most of the AB and AB2 alloys form soluble fluorides, making it difficult to construct stable surface structures.134,135 In order to achieve better selective penetration of hydrogen, Liu et al.136 proposed to partially replace the AB2-based alloy with La to investigate the feasibility of fluorination. The results demonstrated that the fluorinated film containing La was successfully prepared on the surface of the AB2 alloy. The fluorinated alloy still maintained surface reactivity after 6 months of air exposure.
| 3Pd + 2ZrCo → Pd3Zr + ZrCo2 | (9) |
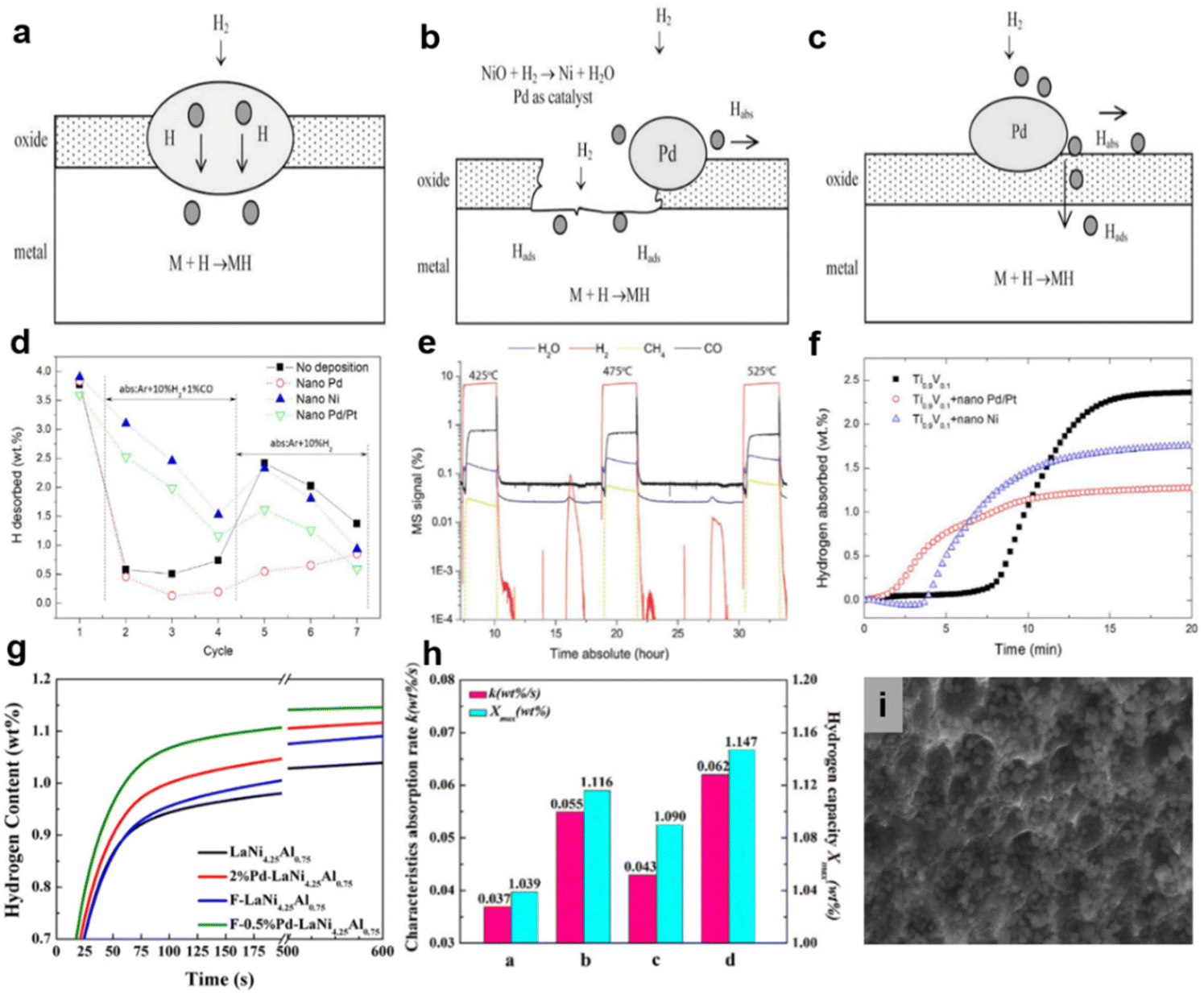 | ||
| Fig. 7 (a–c) Mechanism of Pd-treatment. Copyright 2009 Elsevier; (d) amount of hydrogen desorbed from the hydrides hydrogenated at various temperatures. (e) MS signal during hydrogenation cycle of Ti0.9V0.1 + nano Pd at 425 °C, 475 °C and 525 °C in Ar + 10% H2 + 1% CO. (f) Hydrogenation curves of Ti0.9V0.1 and nanoparticle-deposited Ti0.9V0.1 at 475 °C (cycle 6). Copyright 2017 Elsevier; (g) hydrogen absorption kinetics curves of the samples in impure hydrogen. (h) Parameters from Avrami Erofeev equation fitting of hydrogen absorption kinetics curves (a: LaNi4.25Al0.75, b: 2% Pd–LaNi4.25Al0.75, c: F–LaNi4.25Al0.75, d: F–0.5%Pd–LaNi4.25Al0.75). (i) SEM images of F–0.5% Pd–LaNi4.25Al0.75. Copyright 2016 Elsevier.123,143,147 | ||
 | ||
| Fig. 8 (a) Hydriding/dehydriding cycles of ZrCo and Pd@ZrCo in 1 mol% O2 (green line) and temperature–time curve with cycling (red line). (b) XRD patterns of samples treated with different methods. (c) The first hydriding kinetics of samples treated with different methods in 1 mol% O2. TEM images of (d) layer and (e) surface of the sample. (f) Comparison diagram of cycling stability for samples. (g) Structural diagram for samples treated with different methods. Copyright 2024 Elsevier; (h) illustrations depicting the structure of the rGO–Mg nanolaminates. Copyright 2016 Nature Publishing Group; (i) TPD curves of the two samples. Copyright 2016 Nature Publishing Group; (j) FESEM images of WA-Mg2NiH4. (k) HRTEM image of Ni@Mg(OH)2 nanosheets on the surface of WA-Mg2NiH4. (l–n) Schematic of VMHS promoting hydrogen desorption process of Mg2NiH4. Copyright 2021 Wiley.55,148,149 | ||
Originally, the relatively fast initial hydrogenation kinetics of the Pd-decorated sample confirms the effectiveness of Method-1 and Method-2 in resisting oxygen interference, as illustrated in Fig. 8c. Then, ten cycles were conducted and the capacity retention rate of each cycle was labeled in Fig. 8f. It is noteworthy that Method-3 owned the highest capacity retention of 90.38% after cycles, which warrants an in-depth investigation into the mechanism of its superior cycling stability under H2/O2 mixture. As observed in Fig. 8b, Method-3 simultaneously produces both Pd3Zr and Pd phases, whereas the other two thermal treatments are characterized by the presence of a single Pd-containing substance. Meanwhile, TEM results highlight the surface distribution of Pd and interface distribution tendency of Pd3Zr, as shown in Fig. 8d and e. The intrinsic phase transition mechanism on the anti-oxidation of ZrCo matrix during the three heat treatments is fully schematized in Fig. 8g. The structural evolution from Pd and ZrCo to Pd3Zr and ZrCo2 originating from Method-1 caused severe capacity decay due to the low hydrogen solubility of Pd3Zr. Although Method-2 can well maintain sufficient phase abundance of Pd to promote the hydrogen permeation and shield oxygen impurity, the ZrCo surface/interface is susceptible to deactivation by virtue of the detachment-prone nature of the Pd film. Inspired by the above strategies, Method-3 effectively constructs Pd/Pd3Zr@ZrCo coherent structures through two-stage thermal regulation. The protective effect of surface Pd and the structural stability of interface Pd3Zr improved the oxidation resistance and mitigated the hydrogen embrittlement of Pd-coated ZrCo, respectively. Hence, the Pd/Pd3Zr@ZrCo is capable of achieving ultrafast hydrogenation kinetics as well as superior cycling stability under oxygen-contaminated hydrogen atmospheres owing to the synergistic effect of the Pd and Pd3Zr.
| Alloy | Surface modification | Temperature (°C) | Impurity | C max (wt%) | C poisoned (wt%) | Ref. |
|---|---|---|---|---|---|---|
| F refers to fluorination. Cmax is the maximum hydrogen storage capacity after surface modification. Cpoisoned is the initial hydrogen storage capacity after poisoning. | ||||||
| LaNi4.7Al0.3 | 10% Pd coating | RT | 2 weeks in air | 0.67 (H/A) | 0.56 (H/A) | 143 |
| LaNi4.25Al0.75 | F | 30 | 0.1 vol% O2, 0.1 vol% N2 | — | 1.090 | 153 |
| LaNi4.25Al0.75 | 2% Pd coating | 30 | 0.1 vol% O2, 0.1 vol% N2 | — | 1.116 | 153 |
| LaNi4.25Al0.75 | F–0.5% Pd coating | 30 | 0.1 vol% O2, 0.1 vol% N2 | — | 1.147 | 153 |
| Zr70Fe5.4V24.6 | Pd coating | 30 | 1 vol% air | 5.0 (H/A) | 4.0 (H/A) | 137 |
| ZrV2 | Pd–Ag coating | 30 | 1 vol% air | 2.25 | 0.80 | 146 |
| Zr0.9Ti0.1V2 | Pd–Ag coating | 30 | 1 vol% air | 2.18 | 1.22 | 146 |
| Zr57V36Fe7 | Pd–Ag coating | 30 | 1 vol% air | 2.15 | 1.69 | 146 |
| ZrCoMm0.04 | F–(Pd–Ag) coating | 25 | 3.5 vol% CO, 1 vol% CH4, 0.2 vol% CO2, 0.2 vol% O2 | — | 0.88 | 121 |
| Ti0.9V0.1 | Pd coating | 425 | 1 vol% CO | — | 0.50 | 123 |
| Ti0.9V0.1 | Ni coating | 425 | 1 vol% CO | — | 1.52 | 123 |
| Ti0.9V0.1 | Pd/Pt coating | 425 | 1 vol% CO | — | 0.20 | 123 |
As mentioned above, inactive gases only have the general physical blanket effect owing to their local enrichment around the ZrCo powder and thus preventing inward hydrogen permeation. Once the enrichment of these impurity gases is mitigated, the material reobtains excellent hydrogenation kinetics. Recently, various innovative strategies for eliminating the general physical blanket effect have been explored thoroughly. As demonstrated in Fig. 9a, dynamical multiple-step hydrogenation can reach saturation capacity within 0.4 h, while static one-step hydrogenation yields only 0.46 wt% H2. Since the hydrogenation kinetics can be significantly enhanced by limiting the local enrichment of CH4, creating an addition flow may retard the formation of the destructive blanket layer. Subsequently, a further upgraded pump is connected throughout the system to facilitate the rapid flow of the gas mixture, as depicted in Fig. 9b. After introducing inactive mixed gases into the system, the static hydrogenation kinetic curve (purple line) of ZrCo shown in Fig. 9c indicates a prominent blanket hydrogenation stage, attaining only 0.4 wt% H2 in 0.2 h. Instead, the hydrogenation kinetics is obviously enhanced upon gas circulation (Fig. 9c-green line), and ZrCo reaches the theoretical capacity in 0.2 h. Afterward, the additional gas circulation is carried out once the blanket hydrogenation state is present, as illustrated in Fig. 9c-orange line. Intuitively, the blanket state is easily eliminated by gas circulation, allowing ZrCo to reach theoretical capacity as before. Generally, the elimination phenomena induced by gas disturbance are attributed to the transformation of the kinetic model from a diffusion-controlled process to an interface-controlled process, thus impairing the blanket passivation layer and facilitating the inward hydrogen adsorption and infiltration. Despite the gas circulation, other approaches were further proposed to probe the feasibility of eliminating the blanket effect. In a nutshell, increasing the gas tank volume, lowering the hydrogenation temperature and reducing the amount of loaded sample enormously favours the hydrogenation kinetics, as derived in Fig. 9d. Moreover, the influences of various engineering parameters on the amount of absorbed H2 when the system pressure reaches Pb (slow blanket hydrogenation stage) were optimized to eliminate the undesired physical blanket effect. The investigations shown in Fig. 9e reveal that elevated hydrogenation backpressure (P0) and storage tank volume (Vt) or reduced impurity gas concentration (x) are responsible for the superior hydrogen uptake.
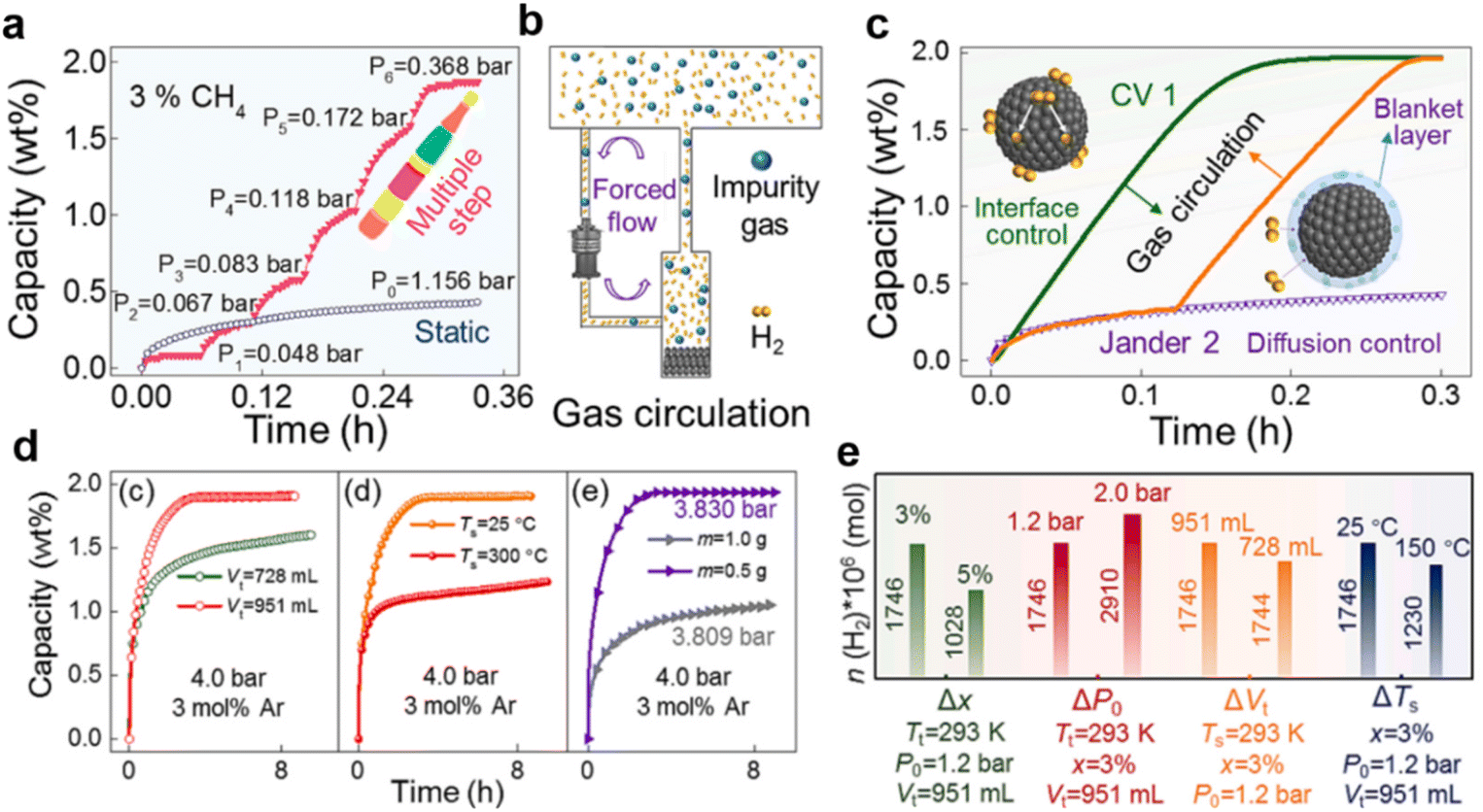 | ||
| Fig. 9 (a) Dynamical multiple-step and static hydrogenation kinetic curves for ZrCo in 3 mol% CH4 mixed gas; (b) schematic diagram of the gas circulation process; (c) influence of gas circulation on the hydrogenation kinetics of ZrCo in 1.2 bar 97 mol% H2 + 1 mol% He + 1 mol% N2 + 1 mol% CH4 quaternary mixed gas (purple line: static; orange line: static-circulation; green line: circulation); (d) influences of gas tank volume Vt, hydrogenation temperature Ts and sample amount m on the hydrogenation kinetics of ZrCo in 4 bar 3 mol% Ar mixed gas; (e) influences of various engineering parameters on the amount of absorbed H2 when the system pressure reaches Pb. Copyright 2024 Elsevier.56 | ||
This section discussed the enhanced anti-poisoning performance of hydrogen storage alloys that can be achieved via multiple alloying, catalytic doping, and surface modification to form a protective layer that permeates hydrogen and blocks impurity gases. Briefly, the multiple alloying possesses two-sidedness. Various factors determine the promotion or retardation effects of doped elements/compounds on the system. For instance, the high affinity of the dopant for impurities alleviates the poisoning tendency of the bulk phase. In particular, certain doped elements are capable of precipitating a second phase within the initial binary alloy, which favors hydrogen absorption. However, inappropriate doping elements can form excessively thick passivation layers or make the primitive binary alloy exhibit more pronounced interactions with impurity gases, which is not conducive to hydrogen dissociation and penetration.
Regarding catalytic doping, reasonable catalytic additives and mechanical treatments have been confirmed to promote the air stability of Mg-based hydrides. Since the catalyst is mainly compounded with the bulk phase by ball milling, it can be evenly distributed on the particle surface, acting as a kinetically preferred channel for hydrogen diffusion and an optimal sacrificial substance for impurity adsorption.
In addition, surface modifications such as fluorination treatment and metallic coatings can induce structural reconstruction of the material, which selectively shields against impurity gases and improves the catalytic activity of the surface. Moreover, many valid and innovative surface structures have been designed to enhance the poisoning resistance of alloys. However, the surface and bulk phase subjected to shrinkage and expansion during the long-term circulation may reduce the gas-selective permeability of the protective film, resulting in continuous exposure of fresh surfaces susceptible to secondary passivation. On the premise of constructing the gas-barrier membrane, improving the stability and self-restore ability of the surface structure to prolong cycling life is the exploration trend of surface modification.
4 Regeneration
Regeneration, also known as reactivation, has not received much attention in the last few decades but is still an essential practical property aimed at regaining the hydrogen absorption ability of contaminated samples. Assuming for now that the poisoning phenomenon cannot be avoided, regeneration of the hydrogen storage alloy under specific conditions is required to facilitate the extension of its service life and improve economic efficiency.4.1 Heat treatment
Since the poisoning reaction occurs mainly on the surface and subsurface, the composition and structure of the bulk phase are barely changed. Thus, in most cases, the poisoned alloy can be regenerated by heat treatment and circulation in pure hydrogen. Considering that a dynamic vacuum promotes the desorption of impurities adsorbed on the surface and the decomposition of passivation layers, heat treatment is usually combined with the dynamic vacuum. The ease of reactivation depends on the surface properties, the thickness and stability of the deposited layer, etc. For example, H2S-poisoned LaNi5 alloys can be reactivated by heating under dynamic vacuuming to about 427 °C and subsequently charged with hydrogen. However, the H2S poisoning is irreversible in the case of TiFe alloys. It is demanding to reactivate the sample even by intense heat treatment and pure hydrogen cycling, as the sulfur deposits covering the surface are difficult to remove.30 It can be inferred from the experiments of Zhang et al.113 that in most cases, alloys poisoned by gases such as N2, NH3, etc., are easier to regenerate because these impurities cover the surface by physical adsorption and are therefore easily released. Other impurities, like O2, CO, CO2, etc., come into contact with the surface to form a deposited barrier, thus a more severe heat treatment is required to reobtain the active surface. Suwarno et al.158 observed the regeneration behavior of CO-poisoned Ti–V alloy under a hydrogen mixture, as shown in Fig. 10a. The introduction of CO in cycle 9 caused an abrupt decrease in hydrogen desorption. Later, the capacity was partially recovered in cycles 12–15 due to the reintroduction of H2 and inert Ar. Subsequent cycles also followed this trend, indicating that the CO-induced surface state change had not yet fully penetrated the bulk phase. However, some materials will suffer a permanent capacity loss, ascribed to the formation of surface oxide films.159Fig. 10b illustrates the dependence of the cycling capacity on pressure and gas. The pressure had a minor influence on the hydrogen capacity (cycles 1–15), whereas 2 vol% CO caused a sudden degradation in capacity (cycles 16–18). Then, CO was removed from the gas stream after cycle 18, at which point the initial hydrogen capacity was restored by 80%.69 Furthermore, heat treatment conditions of various deactivated multicomponent alloys have been measured in large quantities. It can be seen from Fig. 10c that the Zr0.8Ti0.5V1.7 alloy takes only 6 min to reach a capacity of 1.24 wt% at 500 °C in 1 vol% air after a single pure hydrogen cycle, whereas the previously contaminated alloy takes more than 1 h to reach the same capacity. Also, flexible control of the heat treatment is necessary. When the alloy undergoes a heat treatment at 500 °C in the gas mixture, the final hydrogen uptake can reach 2.68 wt% as the temperature drops to 25 °C.160 The temperature dependence of the reactivation was again verified in Fig. 10d. Below 50 °C, the CO-contaminated LaNi4.3Al0.7 completely loses its hydrogen absorption capacity. With increasing temperature, the poisoning effect of CO becomes weaker during cycles. Up to 130 °C, the alloy maintains almost stable cycling properties,119 attributed to the high temperature driving the breakage of metal–carbon bonds and facilitating methanation reaction.76 However, the disadvantage is that CO promotes the formation of a hydrogen-deficient phase (LaNi5H3.8/LaNi4AlH4.1), leading to a continuous attenuation of the saturated hydrogen capacity.119 Park et al.161 traced the nature and evolution of oxide film on TiFe surface during heat treatment by in situ XPS. The results show that Ti has a strong oxidation driving force and that a critical temperature of about 400 °C is needed to reduce Ti oxides initially. In comparison, the onset reduction temperature of Fe oxides is 200 °C. Therefore, strict reactivation conditions for TiFe alloys are inevitable. The experiments of Zhu et al.162 demonstrated that the oxidized TiFe required at least seven repeated hydrogen ab/desorption cycles to be reactivated. However, the situation improved when performed on the multicomponent alloy. Fig. 10e verifies that with only a single treatment process by heating the oxidized alloy to 300 °C under 3 MPa hydrogen pressure, the TiFe0.85Mn0.15 began to reabsorb hydrogen at 30 °C and returned to 1.1 wt% hydrogen capacity due to the massive reduction of Fe and Mn oxides, followed by superior hydrogen uptake and release kinetics in the third cycle.93 In a follow-up study, Modi et al.115 established that adding Cr had certain deactivation resistance, resulting in a lower reactivation temperature (150 °C) under 3 MPa pure hydrogen for TiFe0.85Mn0.15Cr0.30. By cycling in impurity-pure hydrogen, Bouaricha et al.163 evaluated the recovery properties of MgH2–V 5 at%. It shows that the N2-and CO2-poisoned alloys can be restored to their initial state, while the O2-poisoned alloys are not affected by impurities in terms of kinetics. From the capacity point of view, the capacity lost in the N2 and O2 mixture could not be recovered, but it was partially recovered to 80% under the CO2 atmosphere. Shen et al.164 precisely determined the onset reactivation temperature of the poisoned BCC-type Ti–V-based alloy using thermal analysis. It was found that the peaks of various gases, except CO2 and H2O, started fluctuating around 170 °C (Fig. 10f), showing that the initial decomposition temperature of the passivation layer caused by various impurities was about 170 °C, followed by the strongest peak at about 220 °C. The above conclusion can also be confirmed by a similar trend in Fig. 10g. Additionally, PCT comparison curves (Fig. 10h) reveal that the plateau pressure and maximum hydrogen capacity of the poisoned samples heated at 600 °C for 2 h under vacuum are almost identical to their first activation counterpart, indicating that the hydrogen storage performance is basically recovered by effective reduction of the surface-deposited layer under such conditions. For a few materials, multiple alloying is not beneficial for regeneration properties. In the same environment, XPS analysis showed that the oxidized ZrV2 had been reduced to its initial state at 269 °C, while Zr(V0.1Fe0.9)2 was still in the 30% oxidized state when heated to 357 °C.165 Other investigation120 showed that 1 vol% CO-contaminated Zr(V0.95Ni0.05)2 required evacuation at 550 °C for 1 h to reactivate. However, for (Ti, Zr)(V, Fe, Cr, Mn)2 contaminated with only 0.1 vol% CO, the absorption performance was essentially unimproved even when the temperature rose to 60 °C. Reactivation by vacuum at 400 °C was necessary to slowly restore the hydrogen absorption capacity, although the cycling capacity continued to decay after reactivation due to surface deactivation induced by manganese segregation.38 The regeneration conditions of alloys after surface poisoning are listed in Table 3, where the heat treatment temperature is the determining factor for regeneration.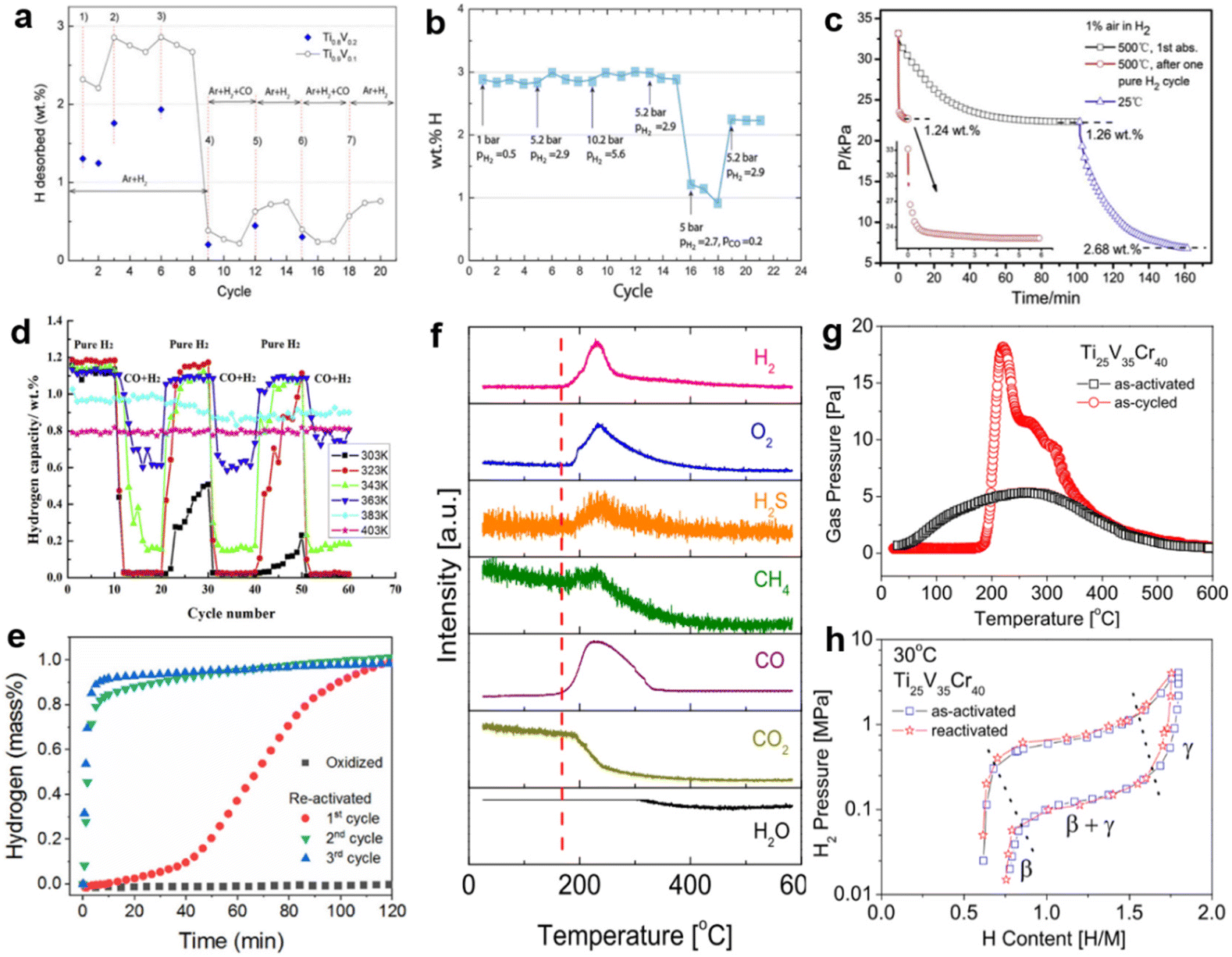 | ||
| Fig. 10 (a) Amount of hydrogen desorbed during the TPR cycles. Copyright 2016 Elsevier; (b) reversible hydrogen storage capacity of Ti0.9V0.1. Copyright 2019 Elsevier; (c) absorption curves for hydrogen containing 1 vol% air of Zr0.8Ti0.5V1.7. Copyright 2017 Elsevier; (d) hydrogen storage capacity of the material in 60 cycles under different temperatures. Copyright 2012 Elsevier; (e) hydrogen absorption of the oxidized and the heat-treated alloy. Copyright 2017 Elsevier; (f) in situ TPD spectra of the as-activated and as-cycled specimens. (g) In situ TPD-MS spectra of the as-cycled specimen. (h) P–C isotherm curves for the as-activated and reactivated Ti25V35Cr40 alloy. Copyright 2015 Elsevier.69,93,119,158,160,166 | ||
| Alloy | Impurity | Heat treatment (°C) | C regenerated (wt%) | Ref. |
|---|---|---|---|---|
| C regenerated denotes the initial hydrogen storage capacity after regeneration. | ||||
| (1) Off-line poisoning | ||||
| Zr0.9Ti0.1V2 | 30 s in O2 | 550 | 0.64 (H/A) | 113 |
| Zr0.9Ti0.1V2 | 30 s in N2 | 550 | 2.85 (H/A) | 113 |
| Zr0.9Ti0.1V2 | 1 min in O2 | 550 | 0.27 (H/A) | 113 |
| Zr0.9Ti0.1V2 | 1 min in N2 | 550 | 2.25 (H/A) | 113 |
| Zr0.9Ti0.1V2 | 3 min in O2 | 550 | 0.27 (H/A) | 113 |
| Zr0.9Ti0.1V2 | 3 min in N2 | 550 | 1.27 (H/A) | 113 |
| Ti0.96Zr0.04Mn1.43V0.45Fe0.08 | Several months in air | 120 | 2.6 (H/A) | 41 |
| TiFe | 5 min in air | 300 | 0.69 | 114 |
| TiFe0.85Mn0.15 | 2 h in air | 300 (3 MPa H2) | 1.0 | 93 |
| TiFe0.85Mn0.15 | 24 h in air | 400 | 1.18 | 94 |
| TiFe0.9Ni0.1 | 5 min in air | 300 | 0.72 | 114 |
| TiFe0.8Ni0.2 | 5 min in air | 300 | 0.68 | 114 |
| TiFe0.6Ni0.4 | 5 min in air | 300 | 1.09 | 114 |
| TiFe0.85Mn0.15Cr0.15 | 2 h in air | 300 (3 MPa H2) | 0.75 | 115 |
| TiFe0.85Mn0.15Cr0.30 | 2 h in air | 150 (3 MPa H2) | 0.4 | 115 |
| (2) On-line poisoning | ||||
| LaNi5 | 1 vol% H2S | 427 | 7.5 (H/A) | 30 |
| LmNi4.8Al0.2 | 0.1 vol% CO | 60 | 1.33 | 76 |
| Ti1.2Mn1.8 | 0.5 vol% CO | 30 | 1.72 | 38 |
| Ti0.98Zr0.02V0.45Fe0.1Cr0.05Mn1.4 | 2.4 vol% CO | 400 | 2.6 | 38 |
| Zr0.8Ti0.5V1.7 | 1 vol% air | 500 | 2.68 | 122 |
| V40Fe8Ti32Cr20 | 0.025 vol% O2 | 150 | 1.9 | 88 |
4.2 Ball milling
Once poisoned, the material requires an extremely long time or high pressure to initially absorb hydrogen without mechanical treatment. Ball milling can crack the passivation layer during the process and refine the grain and particle size.167 Manna et al.168 effectively reactivated the air-exposed TiFe alloy by ball milling. The material showed a noticeable improvement in hydrogenation capacity and kinetics, reaching 1.1 wt% capacity within 4 h. Empirically, reasonable control of the milling time is required to achieve the desired effect. It was experimentally observed that the short-term (0.5 h) milled Mg2NiH4 exhibited better hydrogen storage performance in comparison with the long-term milled (10 h) sample, which was probably associated with the relatively uniform particle size distribution of the 0.5 h milled sample. On the other hand, as prolonged milling time results in the increased specific surface area and active sites, the surface is more susceptible to off-line poisoning.107 The crystal structure and adsorption behavior of MgH2 were not affected by short-time ball milling under argon or air. However, a long period of ball milling under the atmosphere leads to capacity attenuation, although the kinetics are unaffected.169 In addition, the CO-contaminated (Mg(NH2)2 + 2LiH) system regained hydrogen desorption performance since the impurity passivation layers were crushed via ball milling, leading to enhanced kinetics and decreased dehydrogenation activation energy.170Overall, the most common and effective regeneration approach for poisoned alloy belongs to heat treatment. The heat treatment conditions of various deactivated hydrogen storage alloys are different due to the complicated interaction laws of the surface/interface. Normally, alloys poisoned by physisorbed gases (N2, CH4, He, etc.) are prone to reobtain the active surface and reach the saturated hydrogenation capacity after heat treatment. Nevertheless, a harsher heat treatment is required for alloys poisoned by chemisorbed impurity gases (O2, CO2, H2S, etc.). Ordinarily, it is tough to fully decompose the thermally stable deposition layers under experimental conditions. As a result, most reactivated alloys could not restore the saturated hydrogen storage capacity owing to the consumption of the bulk phase. Besides, the ease of reactivation depends on the structural composition of the alloys. The more sensitive the alloy is to impurity gases, the higher the temperature and longer the time required for regeneration. In addition to heat treatment, ball milling is applied to generate abundant active surfaces and re-exposed hydrogen-absorbing phases, which is a convenient way to restore hydrogen storage ability.
5 Conclusions and outlook
This review illustrates the poisoning behavior and mechanisms of various representative metal hydrides and, more importantly, the reasonable strategies for developing surface structures with resistance to impurity gases and designing effective regeneration cycles. In general, the poisoning of alloys by impurities can be attributed to two aspects: (1) slow kinetics, primarily due to the adsorption of impurities on surface active sites that hinder adsorption and dissociation of the hydrogen molecule, and (2) decay of hydrogen capacity, in the case of products generated with little or no contribution to the capacity. Normally, CO is the most drastic deactivator of the alloy system, and its concentration in the hydrogen stream needs to be strictly monitored and controlled. In particular, the activation or hydrogen desorption conditions of some alloys/hydrides are so severe that excessive temperatures will cause the methanation of CO and CO2 to produce CH4 and H2O in the gas mixture, which should be taken into account separately. Interestingly, for some specific Mg-based hydrides, a certain degree of atmospheric exposure is beneficial for the hydrogen storage properties owing to the H2O- and O2-induced catalytically active surfaces. Indeed, correlating the surface structure with the reaction property is essential to obtain appropriate anti-poisoning modifications. Depending on the different intrinsic characteristics of materials, attempts to alleviate the poisoning tendency include multiple alloying, catalytic doping and surface modification. The core of achieving an effective anti-poisoning system is to construct a “hydrogen-permeable” and “impurity gas-barrier” protective layer. Since the surface and bulk phase undergo repeated expansion/contraction as the cycle proceeds, the constructed layer should have certain anti-pulverization and self-recovery functions. Presently, except for a few water- and oxygen-friendly materials, no literature on complete anti-poisoning of hydrogen storage alloys has been reported, which is still an urgent demand for realizing solid hydrogen storage. Typically, the improvement of the anti-poisoning performance of most alloys is at the expense of hydrogen capacity. Meanwhile, it is difficult to reach the initial saturated hydrogen capacity after regeneration. Thus, more effective modification methods should be explored to keep the poisoning effects and hydrogen storage behavior in a tolerable range. Furthermore, the hydrogenation properties and potential mechanisms of some materials in the presence of gaseous impurities have not yet been clarified. It is hoped that a more comprehensive and in-depth understanding of poisoning and anti-poisoning behavior can be achieved in the future.Author contributions
Jing Gu, Zhendong Yao, Ge Gao: conceptualization, methodology, writing-original draft. Yijing Wang, Min Liu: resources, data curation. Miaogen Chen: visualization, investigation. Chao Li, Meiqiang Fan: visualization, supervision. Xuezhang Xiao, Lixin Chen: writing-reviewing and editing.Data availability
Data for this article, including figures and tables, are available at Royal Society of Chemistry.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work is supported by the Natural Science Foundation of Zhejiang Province (No. LQ24E010003), the Baima Lake Laboratory Joint Funds of the Zhejiang Provincial Natural Science Foundation of China (LBMHY24E060004, LBMHY24E060005).References
- R. Bhattacharyya and S. Mohan, Solid state storage of hydrogen and its isotopes: an engineering overview, Renewable Sustainable Energy Rev., 2015, 41, 872–883 CrossRef CAS
.
- Y. Aikai, Y. Kai, S. Mareen, D. Enkhtsetseg, L. Hang, Z. Shuo, Z. Qiu, E. Martin, S. Xingchen, S. Huimin, L. Qiongqiong, Y. Ruijie, M. Igor, P. Quanquan, I. Sylvio, W. Xingchao, M. Qianli, T. Frank, C. Jun and G. Olivier, Enhanced room-temperature Na+ ionic conductivity in Na4.92Y0.92Zr0.08Si4O12, eScience, 2023, 3, 100175 CrossRef
- L. Min, Y. Zhendong, G. Jing, L. Chao, H. Xu, Z. Liuting, H. Zengyang and F. Meiqiang, Issues and opportunities facing hydrolytic hydrogen production materials, Chem. Eng. J., 2023, 461, 141918 CrossRef
- H. Chuanqing, Y. Yingzhi, T. Xiaoyue, L. Zeyu, Z. Jin, Y. Chuanzhen, Y. Yong and Z. Rongbin, Hydrogen generation by ammonia decomposition over Co/CeO2 catalyst: Influence of support morphologies, Appl. Surf. Sci., 2020, 532, 147335 CrossRef
- L. Min, Y. Zhendong, L. Wenqing, Z. Jing, L. Chao, H. Xu, Z. Jiaguang, Z. Liuting and F. Meiqiang, Local high concentration alkali field accelerated silicon hydrolysis for hydrogen production, Chem. Eng. J., 2023, 475, 146201 CrossRef
- H. Chuanqing, Y. Yingzhi, Y. Jinmei, Y. Yue, W. Dashan, H. Feiyang, W. Xuewen, Z. Rongbin and F. Gang, Ru/La2O3 catalyst for ammonia decomposition to hydrogen, Appl. Surf. Sci., 2019, 476, 928–936 CrossRef
- H. Hu, Z. Tu, X. Yan, X. Chen, L. Zhou, Z.-H. Lu, R. Ye, G. Feng and R. Zhang, Effects of Organic Compounds on Ni/AlLaCe Catalysts for Ammonia Decomposition to Hydrogen, Ind. Eng. Chem. Res., 2024, 63, 3910–3920 CrossRef CAS
- L. Wenqing, Y. Zhendong, G. Jing, C. Yongfu, H. Zengyang, T. Guoping, L. Min, L. Chao, C. Miaogen, Z. Jiaguang, Z. Liuting and F. Meiqiang, Novel flame-retardant aluminum-based hydrolyzed material, Int. J. Hydrogen Energy, 2024, 66, 387–395 CrossRef
- J. Qi, X. Huang, X. Xiao, X. Zhang, P. Zhou, S. Zhang, R. Li, H. Kou, F. Jiang, Y. Yao, J. Song, X. Feng, Y. Shi, W. Luo and L. Chen, Isotope engineering achieved by local coordination design in Ti-Pd co-doped ZrCo-based alloys, Nat. Commun., 2024, 15, 2883 CrossRef CAS
- Z. Yao, Z. Liang, X. Xiao, J. Qi, J. He, X. Huang, H. Kou, W. Luo, C. Chen and L. Chen, Achieving excellent cycle stability in Zr–Nb–Co–Ni based hydrogen isotope storage alloys by controllable phase transformation reaction, Renewable Energy, 2022, 187, 500–507 CrossRef CAS
- L. Ouyang, F. Liu, H. Wang, J. Liu, X.-S. Yang, L. Sun and M. Zhu, Magnesium-based hydrogen storage compounds: A review, J. Alloys Compd., 2020, 832, 154865 CrossRef CAS
- Z.-M. Cao, P.-P. Zhou, X.-Z. Xiao, L.-J. Zhan, Z.-F. Jiang, S.-M. Wang, L.-J. Jiang and L.-X. Chen, Development of Ti0.85Zr0.17 (Cr-Mn-V) 1.3Fe0.7-based Laves phase alloys for thermal hydrogen compression at mild operating temperatures, Rare Met., 2022, 41, 2588–2594 CrossRef CAS
- F. Manchester and D. Khatamian, Mechanisms for Activation of Intermetallic Hydrogen Absorbers, Mater. Sci. Forum, 1988, 31, 261–296 CAS
- F. Cuevas and M. Latroche, Intermetallic alloys as hydrogen getters, J. Alloys Compd., 2022, 905, 164173 CrossRef CAS
- G. Bernardo, T. Araújo, T. da Silva Lopes, J. Sousa and A. Mendes, Recent advances in membrane technologies for hydrogen purification, Int. J. Hydrogen Energy, 2020, 45, 7313–7338 CrossRef CAS
- G. Sandrock and P. Goodell, Surface poisoning of LaNi5, FeTi and (Fe, Mn) Ti by O2, Co and H2O, J. Less-Common Met., 1980, 73, 161–168 CrossRef CAS
- H. Sakaguchi, T. Tsujimoto and G.-y. Adachi, The confinement of hydrogen in LaNi5 by poisoning of the hydride surface, J. Alloys Compd., 1995, 223, 122–126 CrossRef CAS
- S. Han, X. Zhang, S. Shi, H. Tanaka, N. Kuriyama, N. Taoka, K. Aihara and Q. Xu, Experimental and theoretical investigation of the cycle durability against CO and degradation mechanism of the LaNi5 hydrogen storage alloy, J. Alloys Compd., 2007, 446, 208–211 CrossRef
- X.-L. Wang, K. Iwata and S. Suda, Effects of carbon monoxide on the hydriding reactions of the untreated and fluorinated LaNi4.7Al0.3 alloys, J. Alloys Compd., 1995, 231, 829–834 CrossRef CAS
- F. Eisenberg and P. Goodell, Cyclic response of reversible hydriding alloys in hydrogen containing carbon monoxide, J. Less-Common Met., 1983, 89, 55–62 CAS
- G. Sandrock and P. Goodell, Cyclic life of metal hydrides with impure hydrogen: overview and engineering considerations, J. Less-Common Met., 1984, 104, 159–173 CrossRef CAS
- H. Siegmann, L. Schlapbach and C. Brundle, Self-Restoring of the Active Surface in the Hydrogen Sponge LaNi5, Phys. Rev. Lett., 1978, 40, 972 CrossRef CAS
- T. Von Waldkirch and P. Zürcher, Surface segregation in LaNi5 induced by oxygen, Appl. Phys. Lett., 1978, 33, 689–691 CrossRef CAS
- L. Schlapbach, A. Seiler, F. Stucki and H. Siegmann, Surface effects and the formation of metal hydrides, J. Less-Common Met., 1980, 73, 145–160 CrossRef CAS
- J. Bi, P. Zhou, X. Xiao, Z. Liang, J. Qi, Y. Zhang, H. Kou, T. Tang and L. Chen, Achieving excellent CO2 poisoning resistance of ZrCo hydrogen isotope storage material by surface reconstruction strategy, J. Alloys Compd., 2023, 954, 170220 CrossRef CAS
- Y. Xin-Nan and L. Schlapbach, Surface properties of chemically prepared LaNi5 and its oxidation and poisoning by O2 and CO, Int. J. Hydrogen Energy, 1988, 13, 429–432 CrossRef
- P. Goodell, Cycling hydriding response of LaNi5 in hydrogen containing oxygen as a minor impurity, J. Less-Common Met., 1983, 89, 45–54 CrossRef CAS
- J. I. Han and J.-Y. Lee, Influence of oxygen impurity on the hydrogenation properties of LaNi5, LaNi4.7Al0.3 and MmNi4.5Al0.5 during long-term pressure-induced hydriding-dehydriding cycling, J. Less-Common Met., 1989, 152, 329–338 CrossRef CAS
- W. Wallace, R. Karlicek Jr. and H. Imamura, Mechanism of hydrogen absorption by LaNi5, J. Phys. Chem., 1979, 83, 1708–1712 CrossRef CAS
- F. Block and H.-J. Bahs, Investigation of selective absorption of hydrogen by LaNi5 and FeTi, J. Less-Common Met., 1983, 89, 77–84 CrossRef CAS
- F. Schweppe, M. Martin and E. Fromm, Hydrogen absorption of LaNi5 powders precovered with O2, CO, H2S, CO2 or N2, J. Alloys Compd., 1997, 253, 511–514 CrossRef
- S. Myhra, E. Kisi and E. Gray, A surface analytical study of SO2 stabilisation of LaNi5Hx surfaces, J. Alloys Compd., 1995, 224, 305–315 CrossRef CAS
- N. Hanada, T. Nakagawa, H. Asada, M. Ishida, K. Takahashi, S. Isobe, I. Saita, K. Asano, Y. Nakamura and A. Fujisawa, Dependence of constituent elements of AB5 type metal hydrides on hydrogenation degradation by CO2 poisoning, J. Alloys Compd., 2015, 647, 198–203 CrossRef CAS
- P. Selvam, B. Viswanathan and V. Srinivasan, XPS studies of the surface properties of CaNi5, J. Electron Spectrosc. Relat. Phenom., 1989, 49, 203–211 CrossRef CAS
- P. Selvam, B. Viswanathan and V. Srinivasan, Evidence for the formation of surface carbonates on some hydrogen storage intermetallic compounds: an XPS study, Int. J. Hydrogen Energy, 1990, 15, 133–137 CrossRef CAS
- P. Selvam, B. Viswanathan, C. Swamy and V. Srinivasan, Surface properties and their consequences on the hydrogen sorption characteristics of certain materials, J. Less-Common Met., 1990, 163, 89–108 CrossRef CAS
- Z. Yao, L. Liu, X. Xiao, C. Wang, L. Jiang and L. Chen, Effect of rare earth doping on the hydrogen storage performance of Ti1.02Cr1.1Mn0.3Fe0.6 alloy for hybrid hydrogen storage application, J. Alloys Compd., 2018, 731, 524–530 CrossRef CAS
- F. Block and H.-J. Bahs, Selective absorption of hydrogen by Ti-Mn-based alloys from gas mixtures containing CO or CH4, J. Less-Common Met., 1984, 104, 223–230 CrossRef CAS
- O. Bernauer, J. Töpler, D. Noreus, R. Hempelmann and D. Richter, Fundamentals and properties of some Ti/Mn based laves phase hydrides, Int. J. Hydrogen Energy, 1989, 14, 187–200 CrossRef CAS
- L. Schlapbach, Surface properties of hydride-forming AB2 compounds, J. Less-Common Met., 1983, 89, 37–43 CrossRef CAS
- M. Schülke, H. Paulus, M. Lammers, G. Kiss, F. Réti and K.-H. Müller, Influence of surface contaminations on the hydrogen storage behaviour of metal hydride alloys, Anal. Bioanal. Chem., 2008, 390, 1495–1505 CrossRef PubMed
- L. Schlapbach, A. Seiler and F. Stucki, Surface segregation in FeTi and its catalytic effect on the hydrogenation, Mater. Res. Bull., 1978, 13, 697–706 CrossRef CAS
- G. Busch, L. Schlapbach, F. Stucki, P. Fischer and A. Andresen, Hydrogen storage in FeTi: Surface segregation and its catalytic effect on hydrogenation and structural studies by means of neutron diffraction, Int. J. Hydrogen Energy, 1979, 4, 29–39 CrossRef CAS
- L. Schlapbach, A. Seiler and F. Stucki, Surface segregation in FeTi and its catalytic effect on the hydrogenation II: AES and XPS studies, Mater. Res. Bull., 1978, 13, 1031–1037 CrossRef CAS
- A. Shwartz, N. Shamir, N. Froumin, S. Zalkind, I. Edry, A. Haim and M. Mintz, Initial oxidation of TiFe1−xMnx (x = 0–0.3) by low dose exposures to H2O and O2, J. Alloys Compd., 2014, 610, 6–10 CrossRef CAS
- Z. Liang, X. Xiao, J. Qi, H. Kou and L. Chen, ZrCo-based hydrogen isotopes storage alloys: a review, J. Alloys Compd., 2022, 932, 167552 CrossRef
- R.-D. Penzhorn, M. Devillers and M. Sirch, Storage of tritium in ZrCo alloy: Effect of pre-exposure to impurities relevant to the fusion fuel cycle, J. Nucl. Mater., 1991, 179, 863–866 CrossRef
- M. Devillers, M. Sirch and R.-D. Penzhorn, Hydrogen isotopes in pure and nitrided ZrCo, Z. Phys. Chem., 1989, 164, 1355–1360 CrossRef
- H. Glasbrenner, H. Klewe-Nebenius, M. Bruns, G. Pfennig, R.-D. Penzhorn and H. Joachim Ache, Surface analytical investigation of the tritium getter ZrCo after exposure to various gases, Microchim. Acta, 1992, 107, 207–217 CrossRef CAS
- E. Willin, M. Sirch, R.-D. Penzhorn and M. Devillers, Metal getters for tritium storage, Fusion Technol., 1988, 14, 756–763 CrossRef CAS
- R.-D. Penzhorn and M. Sirch, Evaluation of ZrCo and other getters for tritium handling and storage, J. Nucl. Mater., 1990, 170, 217–231 CrossRef CAS
- S. Bredendiek-Kämper, H. Klewe-Nebenius, G. Pfennig, M. Bruns, M. Devillers and H. Ache, Surface analytical characterization of the hydrogen getter material ZrCo, Fresenius’ Z. Anal. Chem., 1989, 335, 669 CrossRef
- J. Prigent, M. Latroche, E. Leoni and V. Rohr, Hydrogen trapping properties of Zr-based intermetallic compounds in the presence of CO contaminant gas, J. Alloys Compd., 2011, 509, S801–S803 CrossRef CAS
- S. Kato, S. K. Matam, P. Kerger, L. Bernard, C. Battaglia, D. Vogel, M. Rohwerder and A. Züttel, The origin of the catalytic activity of a metal hydride in CO2 reduction, Angew. Chem., Int. Ed., 2016, 55, 6028–6032 CrossRef CAS
- Z. Yajie, Z. Panpan, X. Xuezhang, B. Jiapeng, Z. Xinyi, K. Huaqin, H. Xu, T. Tao and C. Lixin, Superior oxygen-resistance and intrinsic mechanisms of coherent Pd/Pd3Zr@ZrCo structure with excellent cycling durability, Chem. Eng. J., 2024, 479, 147660 CrossRef
- B. Jiapeng, Z. Panpan, X. Xuezhang, Z. Yajie, K. Huaqin, T. Tao and C. Lixin, General impurity gas blanket effect mechanism and elimination strategies for hydrogen storage materials, Chem. Eng. J., 2024, 481, 148517 CrossRef
- I. Yoshihiko, O. Shuichiro and A. Etsuo, Effect of impurities and air exposure on hydrogen absorbing properties of Mg-Ni alloys, J. Less-Common Met., 1986, 120, 163–169 CrossRef
- S. Kato, A. Borgschulte, D. Ferri, M. Bielmann, J.-C. Crivello, D. Wiedenmann, M. Parlinska-Wojtan, P. Rossbach, Y. Lu and A. Remhof, CO2 hydrogenation on a metal hydride surface, Phys. Chem. Chem. Phys., 2012, 14, 5518–5526 RSC
- P. Selvam, B. Viswanathan and V. Srinivasan, XPS and XAES studies on hydrogen storage magnesium-based alloys, Int. J. Hydrogen Energy, 1989, 14, 899–902 CrossRef CAS
- X. Q. Tran, S. D. McDonald, Q. Gu, X. F. Tan and K. Nogita, Effect of impurity N2 concentration on the hydriding kinetics of Na-doped Mg–Ni alloys, Int. J. Hydrogen Energy, 2017, 42, 366–375 CrossRef CAS
- M. D. Hampton, R. Juturu and J. Lomness, The activation of Mg2Ni for initial hydrogen uptake by treatment with water vapor, Int. J. Hydrogen Energy, 1999, 24, 981–988 CrossRef CAS
- S. Zalkind, M. Nahmani and N. Shamir, The interaction of Zr2Fe surface with O2 and H2O at the temperature range 300–770 K, J. Alloys Compd., 2010, 501, 221–226 CrossRef CAS
- C. Boffito, F. Doni and L. Rosai, The properties of some zirconium-based gettering alloys for hydrogen isotope storage and purification, J. Less-Common Met., 1984, 104, 149–157 CrossRef CAS
- S. Fukada, K. Tokunaga and M. Nishikawa, Recovery of low-concentration hydrogen from different gas streams with Zr2Fe particle beds, Fusion Eng. Des., 1997, 36, 471–478 CrossRef CAS
- J. Klein and J. Wermer, Tritium stripping in a nitrogen glove box using palladium/zeolite and SAES St 198, Fusion Technol., 1995, 28, 1532–1539 CrossRef CAS
- D. W. James and G. A. Morgan, Evaluation of the effects of impurities on SAES ST198 hydrogen gettering, Fusion Sci. Technol., 2017, 71, 321–325 CrossRef
- D. Cohen, M. Nahmani, G. Rafailov, S. Attia, Z. Shamish, M. Landau, J. Merchuk and Y. Zeiri, Oxidation mechanism of porous Zr2Fe used as a hydrogen getter, Appl. Radiat. Isot., 2016, 107, 47–56 CrossRef CAS
- S. Kumar, A. Jain, T. Ichikawa, Y. Kojima and G. Dey, Development of vanadium based hydrogen storage material: A review, Renewable Sustainable Energy Rev., 2017, 72, 791–800 CrossRef CAS
- S. Suwarno, J. Solberg, B. Krogh, S. Raaen and V. Yartys, High temperature hydrogenation of Ti–V alloys: The effect of cycling and carbon monoxide on the bulk and surface properties, Int. J. Hydrogen Energy, 2016, 41, 1699–1710 CrossRef CAS
- B. Vigeholm, J. Kjøller, B. Larsen and A. S. Pedersen, Effect of oxygen on the Mg-H reaction, J. Less-Common Met., 1984, 104, 141–148 CrossRef CAS
- A. S. Pedersen and B. Larsen, The storage of industrially pure hydrogen in magnesium, Int. J. Hydrogen Energy, 1993, 18, 297–300 CrossRef CAS
- A. S. Pedersen, V. Bj, J. Kjøller and B. Larsen, The effect of cycling in impure hydrogen on the hydrogen capacity of magnesium powder, Int. J. Hydrogen Energy, 1987, 12, 765–771 CrossRef CAS
- O. Friedrichs, J. Sánchez-López, C. López-Cartes, M. Dornheim, T. Klassen, R. Bormann and A. Fernández, Chemical and microstructural study of the oxygen passivation behaviour of nanocrystalline Mg and MgH2, Appl. Surf. Sci., 2006, 252, 2334–2345 CrossRef CAS
- H. Liu, P. Sun, R. C. Bowman Jr., Z. Z. Fang, Y. Liu and C. Zhou, Effect of air exposure on hydrogen storage properties of catalyzed magnesium hydride, J. Power Sources, 2020, 454, 227936 CrossRef CAS
- T. R. Jensen, A. Andreasen, T. Vegge, J. W. Andreasen, K. Ståhl, A. S. Pedersen, M. M. Nielsen, A. M. Molenbroek and F. Besenbacher, Dehydrogenation kinetics of pure and nickel-doped magnesium hydride investigated by in situ time-resolved powder X-ray diffraction, Int. J. Hydrogen Energy, 2006, 31, 2052–2062 CrossRef CAS
- H. Lin, K. Lin, C. Sung and K. Wu, Characteristics of activation and anti-poisoning in an LmNi4.8Al0.2 hydrogen storage alloy, Int. J. Hydrogen Energy, 2007, 32, 2494–2500 CrossRef CAS
- Q. Wan, P. Li, Y. Li, F. Zhai, W. Zhang, L. Cui, A. A. Volinsky and X. Qu, CO impurities effect on LaNi4.7Al0.3 hydrogen storage alloy hydrogenation/dehydrogenation properties, Bull. Mater. Sci., 2014, 37, 837–842 CrossRef CAS
- K. Nishimura, K. Sato, Y. Nakamura, C. Inazumi, K. Oguro, I. Uehara, S. Fujitani and I. Yonezu, Stability of LaNi5−xAlx alloys (x = 0∼ 0.5) during hydriding and dehydriding cycling in hydrogen containing O2 and H2O, J. Alloys Compd., 1998, 268, 207–210 CrossRef CAS
- S. Corré, M. Bououdina, D. Fruchart and G.-y. Adachi, Stabilisation of high dissociation pressure hydrides of formula La1−xCexNi5 (x = 0–0.3) with carbon monoxide, J. Alloys Compd., 1998, 275, 99–104 CrossRef
- H. Uchida, Surface processes of H2 on rare earth based hydrogen storage alloys with various surface modifications, Int. J. Hydrogen Energy, 1999, 24, 861–869 CrossRef CAS
- H. Uchida, S. Seki and S. Seta, Effect of surface contamination on the hydriding behaviors of LaNi4.5Al0.5, LaNi2.5Co2.5 and LaNi4.5Mn0.5, J. Alloys Compd., 1995, 231, 403–410 CrossRef CAS
- E. M. Borzone, M. V. Blanco, G. O. Meyer and A. Baruj, Cycling performance and hydriding kinetics of LaNi5 and LaNi4.73Sn0.27 alloys in the presence of CO, Int. J. Hydrogen Energy, 2014, 39, 10517–10524 CrossRef CAS
- M. Sato, H. Uchida, M. Stange, V. A. Yartys, S. Kato, Y. Ishibashi, M. Terashima, R. Yamakawa and H.-H. Uchida, H2 reactivity on the surface of LaNi4.7Sn0.3, J. Alloys Compd., 2005, 402, 219–223 CrossRef CAS
- S. Yang, R. Fang, G. Yang, L. Lv, X. Han, W. Liu, X. He and P. Zhu, Influence of Element Substitutions on the Poisoning Behavior of ZrV2 Alloy Via Theoretical and Experimental Study, Nucl. Sci. Tech., 2023, 34, 113 CrossRef CAS
- T. Wu, X. Xue, T. Zhang, Y. Zhang, H. Kou and J. Li, Poisoning effect of oxygen on hydrogenation performance of a Zr-V-Ni Laves phase alloy, Int. J. Hydrogen Energy, 2016, 41, 19114–19122 CrossRef CAS
- N. Hanada, H. Asada, T. Nakagawa, H. Higa, M. Ishida, D. Heshiki, T. Toki, I. Saita, K. Asano and Y. Nakamura, Effect of CO2 on hydrogen absorption in Ti-Zr-Mn-Cr based AB2 type alloys, J. Alloys Compd., 2017, 705, 507–516 CrossRef CAS
- T. Maeda, T. Fuura, I. Matsumoto, Y. Kawakami and M. Masuda, Cyclic stability test of AB2 type (Ti, Zr)(Ni, Mn, V, Fe)2.18 for stationary hydrogen storage in water contaminated hydrogen, J. Alloys Compd., 2013, 580, S255–S258 CrossRef CAS
- U. Ulmer, D. Oertel, T. Diemant, C. Bonatto Minella, T. Bergfeldt, R. Dittmeyer, R. J. r. Behm and M. Fichtner, Performance improvement of V–Fe–Cr–Ti solid state hydrogen storage materials in impure hydrogen gas, ACS Appl. Mater. Interfaces, 2018, 10, 1662–1671 CrossRef CAS
- R. Shen, C. Pu, X. Xu, Y. Xu, Z. Li and Z. Wu, Effect of Mischmetal Introduction on Hydrogen Storage Properties in Impure Hydrogen Gas of Ti-Fe-Mn-Co Alloys, Metals, 2020, 10, 1574 CrossRef CAS
- A. Seiler, F. Stucki and P. Charpié, How additives of Mn improve the hydrogenation characteristics of FeTi-and the role of its subsurface, Solid State Commun., 1982, 42, 337–341 CrossRef CAS
- P. Lv and Z. Liu, Effect of high zirconium content on hydrogenation properties and anti-poisoning ability of air-exposed TiFe alloy, J. Mater. Res. Technol., 2019, 8, 5972–5983 CrossRef CAS
- P. Jain, C. Gosselin and J. Huot, Effect of Zr, Ni and Zr7Ni10 alloy on hydrogen storage characteristics of TiFe alloy, Int. J. Hydrogen Energy, 2015, 40, 16921–16927 CrossRef CAS
- P. Modi and K.-F. Aguey-Zinsou, Titanium-iron-manganese (TiFe0.85Mn0.15) alloy for hydrogen storage: Reactivation upon oxidation, Int. J. Hydrogen Energy, 2019, 44, 16757–16764 CrossRef CAS
- S. Pati, S. Trimbake, M. Vashistha and P. Sharma, Tailoring the activation behaviour and oxide resistant properties of TiFe alloys by doping with Mn, Int. J. Hydrogen Energy, 2021, 46, 34830–34838 CrossRef CAS
- K. B. Park, W.-S. Ko, J. O. Fadonougbo, T.-W. Na, H.-T. Im, J.-Y. Park, J.-W. Kang, H.-S. Kang, C.-S. Park and H.-K. Park, Effect of Fe substitution by Mn and Cr on first hydrogenation kinetics of air-exposed TiFe-based hydrogen storage alloy, Mater. Charact., 2021, 178, 111246 CrossRef CAS
- K. B. Park, J. O. Fadonougbo, C.-S. Park, J.-H. Lee, T.-W. Na, H.-S. Kang, W.-S. Ko and H.-K. Park, Effect of Fe substitution on first hydrogenation kinetics of TiFe-based hydrogen storage alloys after air exposure, Int. J. Hydrogen Energy, 2021, 46, 30780–30789 CrossRef CAS
- S.-M. Lee and T.-P. Perng, Effect of the second phase on the initiation of hydrogenation of TiFe1−xMx (M = Cr, Mn) alloys, Int. J. Hydrogen Energy, 1994, 19, 259–263 CrossRef CAS
- H. Kim, M. Faisal, S.-I. Lee, J. Y. Jung, H.-J. Kim, J. Hong, Y.-S. Lee, J.-H. Shim, Y. W. Cho and D. H. Kim, Activation of Ti–Fe–Cr alloys containing identical AB2 fractions, J. Alloys Compd., 2021, 864, 158876 CrossRef CAS
- W. Zheng, W. Song, T. Wu, J. Wang, Y. He and X.-G. Lu, Experimental investigation and thermodynamic modeling of the ternary Ti–Fe–Mn system for hydrogen storage applications, J. Alloys Compd., 2022, 891, 161957 CrossRef CAS
- P. Lv and J. Huot, Hydrogenation improvement of TiFe by adding ZrMn2, Energy, 2017, 138, 375–382 CrossRef CAS
- Z. Dehouche, J. Goyette, T. Bose and R. Schulz, Moisture effect on hydrogen storage properties of nanostructured MgH2−V–Ti composite, Int. J. Hydrogen Energy, 2003, 28, 983–988 CrossRef CAS
- J.-R. Ares-Fernández and K.-F. Aguey-Zinsou, Superior MgH2 kinetics with MgO addition: A tribological effect, Catalysts, 2012, 2, 330–343 CrossRef
- K.-F. Aguey-Zinsou, J. A. Fernandez, T. Klassen and R. Bormann, Using MgO to improve the (de) hydriding properties of magnesium, Mater. Res. Bull., 2006, 41, 1118–1126 CrossRef CAS
- J. Zhang, H. Liu, P. Sun, C. Zhou, X. Guo and Z. Z. Fang, The role of oxide in hydrogen absorption and desorption kinetics of MgH2-based material, J. Alloys Compd., 2023, 934, 167757 CrossRef CAS
- L. Xie, J. Li, T. Zhang and L. Song, Air-stable MgH2–CeO2 composite with facilitated de/hydrogenation kinetics synthesized by high energy ball milling, Mater. Charact., 2017, 133, 94–101 CrossRef CAS
- L. Xie, J. Li, T. Zhang and H. Kou, De/hydrogenation kinetics against air exposure and microstructure evolution during hydrogen absorption/desorption of Mg-Ni-Ce alloys, Renewable Energy, 2017, 113, 1399–1407 CrossRef CAS
- Z. Ma, Y. Zhao, Z. Wu, Q. Tang, J. Ni, Y. Zhu, J. Zhang, Y. Liu, Y. Zhang and H.-W. Li, Air-stable magnesium nickel hydride with autocatalytic and self-protective effect for reversible hydrogen storage, Nano Res., 2022, 15, 2130–2137 CrossRef CAS
- Z. Ma, Q. Tang, J. Ni, Y. Zhu, Y. Zhang, H.-W. Li, J. Zhang, Y. Liu, Z. Ba and L. Li, Synergistic effect of TiH2 and air exposure on enhancing hydrogen storage performance of Mg2NiH4, Chem. Eng. J., 2022, 433, 134489 CrossRef CAS
- R. Shi, H. Gao, J. Zhang, H. Yan, Y. Zhao, Y. Zhu, J. Wang, Y. Liu, X. Hu and L. Li, Improved Hydrogen Storage Properties and Air Stability of Metal Hydrides by Constructing Heterophase Composites, Chem. Mater., 2023, 35, 3206–3217 CrossRef CAS
- X. Shan, J. H. Payer and J. S. Wainright, Improved durability of hydrogen storage alloys, J. Alloys Compd., 2007, 430, 262–268 CrossRef CAS
- P. Lv, C. Zhong, D. Huang, X. Zhou, Z. Liu and D. Huang, Effect of introducing manganese as additive on microstructure, hydrogen storage properties and rate limiting step of Ti–Cr alloy, Int. J. Hydrogen Energy, 2022, 47, 459–469 CrossRef CAS
- P. Lv, C. Zhong, D. Huang, X. Zhou, Z. Liu and R. Zhao, Superior anti-impurity gas poisoning ability and hydrogen storage properties of Ti–Cr alloy by introducing zirconium as additive, Int. J. Hydrogen Energy, 2022, 47, 18772–18785 CrossRef CAS
- T. Zhang, X. Yang, J. Li, R. Hu, X. Xue and H. Fu, On the poisoning effect of O2 and N2 for the Zr0.9Ti0.1V2 hydrogen storage alloy, J. Power Sources, 2012, 202, 217–224 CrossRef CAS
- Y. Li, H. Shang, Y. Zhang, P. Li, Y. Qi and D. Zhao, Investigations on gaseous hydrogen storage performances and reactivation ability of as-cast TiFe1−xNix (x = 0, 0.1, 0.2 and 0.4) alloys, Int. J. Hydrogen Energy, 2019, 44, 4240–4252 CrossRef CAS
- P. Modi, W. Liu and K.-F. Aguey-Zinsou, Effect of chromium addition on the reactivation of the titanium-iron-manganese (TiFe0.85Mn0.15) alloy, J. Alloys Compd., 2022, 891, 161943 CrossRef CAS
- P. Lv, Z. Liu, A. K. Patel, X. Zhou and J. Huot, Influence of ball milling, cold rolling and doping (Zr + 2Cr) on microstructure, first hydrogenation properties and anti-poisoning ability of TiFe alloy, Met. Mater. Int., 2021, 27, 1346–1357 CrossRef CAS
- M. Park, J.-H. Shim, Y.-S. Lee, Y. H. Im and Y. W. Cho, Mitigation of degradation in the dehydrogenation behavior of air-exposed MgH2 catalyzed with NbF5, J. Alloys Compd., 2013, 575, 393–398 CrossRef CAS
- J. Ni, Y. Zhu, J. Zhang, Z. Ma, Y. Liu, A. Wang and L. Li, Air exposure improving hydrogen desorption behavior of Mg–Ni-based hydrides, Int. J. Hydrogen Energy, 2023, 48, 22183–22191 CrossRef CAS
- F. Yang, X. Chen, Z. Wu, S. Wang, G. Wang, Z. Zhang and Y. Wang, Experimental studies on the poisoning properties of a low-plateau hydrogen storage alloy LaNi4.3Al0.7 against CO impurities, Int. J. Hydrogen Energy, 2017, 42, 16225–16234 CrossRef CAS
- T. Zhang, T. Wu, X. Xue, R. Hu, H. Kou and J. Li, Hydrogen storage performance of a pseudo-binary Zr-V-Ni Laves phase alloy against gaseous impurities, Renewable Energy, 2017, 103, 786–793 CrossRef CAS
- L. Hualing, W. Shumao, L. Jiang, L. Zhang, L. Xiaopeng and L. Zhinian, Preparation and research on poisoning resistant Zr-Co based hydrogen storage alloys, Rare Met., 2008, 27, 367–370 CrossRef
- Y. Zhang, J. Li, T. Zhang, H. Kou, R. Hu and X. Xue, Hydrogen storage properties of non-stoichiometric Zr0.9TixV2 melt-spun ribbons, Energy, 2016, 114, 1147–1154 CrossRef CAS
- S. Suwarno, M. Williams, J. K. Solberg and V. Yartys, Effect of nanoparticle (Pd, Pd/Pt, Ni) deposition on high temperature hydrogenation of Ti-V alloys in gaseous flow containing CO, Prog. Nat. Sci.: Mater. Int., 2017, 27, 93–98 CrossRef CAS
- X.-L. Wang and S. Suda, Stability and tolerance to impurities of the fluorinated surface of hydrogen-absorbing alloys, J. Alloys Compd., 1995, 227, 58–62 CrossRef CAS
- X.-L. Wang and S. Suda, Surface characteristics of fluorinated hydriding alloys, J. Alloys Compd., 1995, 231, 380–386 CrossRef CAS
- X.-L. Wang, K. Iwata and S. Suda, Hydrogen purification using fluorinated LaNi4.7Al0.3 alloy, J. Alloys Compd., 1995, 231, 860–864 CrossRef CAS
- H. Uchida, T. Inoue, T. Tabata, S. Seki, H. Uchida, F. Aono, T. Nakazawa, H. Kikuyama and R. Hirayama, Effect of HF pretreatment on H2 reactivity with LaNi5 and LaNi4.7Al0.3, J. Alloys Compd., 1997, 253, 547–549 CrossRef
- E. Ivanov, I. Konstanchuk, B. Bokhonov and V. Boldyrev, Hydrogen interaction with mechanically alloyed magnesium–salt composite materials, J. Alloys Compd., 2003, 359, 320–325 CrossRef CAS
- Y.-M. Sun and S. Suda, Studies on the fluorination method for improving surface properties and characteristics of AB5-types of hydrides, J. Alloys Compd., 2002, 330, 627–631 CrossRef
- X.-L. Wang and S. Suda, Surface Structure of Highly Reactive Hydriding Alloys Produced by Surface Treatment, Z. Phys. Chem., 1994, 183, 385–390 CrossRef CAS
- X.-L. Wang and S. Suda, Surface study of chemically treated LaNi4.7Al0.3 alloy, J. Alloys Compd., 1993, 194, 73–76 CrossRef CAS
- F.-J. Liu and S. Suda, A method for improving the long-term storability of hydriding alloys by air/water exposure, J. Alloys Compd., 1995, 231, 411–416 CrossRef CAS
- D. Rodriguez and G. Meyer, Improvement of the activation stage of MmNi4.7Al0.3 hydride-forming alloys by surface fluorination, J. Alloys Compd., 1999, 293, 374–378 CrossRef
- H. Park, W. Cho, B. Cho, S. R. Lee and K. Yun, Effect of fluorination on the lanthanum-doped AB2-type metal hydride electrodes, J. Power Sources, 2001, 92, 149–156 CrossRef CAS
- F.-J. Liu and S. Suda, Properties and characteristics of fluorinated hydriding alloys, J. Alloys Compd., 1995, 231, 742–750 CrossRef CAS
- F.-J. Liu and S. Suda, F-treatment effect on the hydriding properties of the La-substituted AB2 compound (Ti, Zr)(Mn, Cr, Ni)2, J. Alloys Compd., 1995, 231, 666–669 CrossRef CAS
- T. Zhang, M. Zhang, R. Hu, H. Kou, J. Li and X. Xue, Hydrogen absorption behavior of a Pd-coated Zr70Fe5.4V24.6 getter material against gaseous impurities, Vacuum, 2015, 122, 222–229 CrossRef CAS
- L. Zaluski, A. Zaluska, P. Tessier, J. Ström-Olsen and R. Schulz, Catalytic effect of Pd on hydrogen absorption in mechanically alloyed Mg2Ni, LaNi5 and FeTi, J. Alloys Compd., 1995, 217, 295–300 CrossRef CAS
- M. Williams, M. Lototsky, M. Davids, V. Linkov, V. Yartys and J. Solberg, Chemical surface modification for the improvement of the hydrogenation kinetics and poisoning resistance of TiFe, J. Alloys Compd., 2011, 509, S770–S774 CrossRef CAS
- K. Ashida, Y. Hatano, W. Nishida, K. Watanabe, A. Amano, K. Matsuda and S. Ikeno, Recovery of hydrogen isotopes by Pd-coated ZrNi from inert gas atmosphere containing impurities, J. Nucl. Sci. Technol., 2001, 38, 952–958 CrossRef CAS
- M. W. Davids, M. Lototskyy, A. Nechaev, Q. Naidoo, M. Williams and Y. Klochko, Surface modification of TiFe hydrogen storage alloy by metal-organic chemical vapour deposition of palladium, Int. J. Hydrogen Energy, 2011, 36, 9743–9750 CrossRef CAS
- H. Uchida, H.-G. Wulz and E. Fromm, Catalytic effect of nickel, iron and palladium on hydriding titanium and storage materials, J. Less-Common Met., 1991, 172, 1076–1083 CrossRef
- X. Shan, J. H. Payer and W. D. Jennings, Mechanism of increased performance and durability of Pd-treated metal hydriding alloys, Int. J. Hydrogen Energy, 2009, 34, 363–369 CrossRef CAS
- X. Guo, S. Wang, Z. Li, B. Yuan, J. Ye, H. Qiu, Y. Wu, X. Liu and L. Jiang, Study on the poisoning resistance of Pd-coated ZrCo alloy prepared by electroless plating method, Fusion Eng. Des., 2016, 113, 195–200 CrossRef CAS
- S. Yun and S. T. Oyama, Correlations in palladium membranes for hydrogen separation: A review, J. Membr. Sci., 2011, 375, 28–45 CrossRef CAS
- T. Zhang, Y. Zhang, M. Zhang, R. Hu, H. Kou, J. Li and X. Xue, Hydrogen absorption behavior of Zr-based getter materials with Pd-Ag coating against gaseous impurities, Int. J. Hydrogen Energy, 2016, 41, 14778–14787 CrossRef CAS
- Z. Botao, L. Lifei, Y. Yiming, H. Shilin, W. Dong and Z. Pingzhu, Enhanced hydrogen capacity and absorption rate of LaNi4.25Al0.75 alloy in impure hydrogen by a combined approach of fluorination and palladium deposition, Int. J. Hydrogen Energy, 2016, 41, 3465–3469 CrossRef
- E. S. Cho, A. M. Ruminski, S. Aloni, Y.-S. Liu, J. Guo and J. J. Urban, Graphene oxide/metal nanocrystal multilaminates as the atomic limit for safe and selective hydrogen storage, Nat. Commun., 2016, 7, 1–8 Search PubMed
- R. Shi, H. Yan, J. Zhang, H. Gao, Y. Zhu, Y. Liu, X. Hu, Y. Zhang and L. Li, Vacancy–Mediated Hydrogen Spillover Improving Hydrogen Storage Properties and Air Stability of Metal Hydrides, Small, 2021, 17, 2100852 CrossRef CAS PubMed
- M. Lototsky, M. Williams, V. Yartys, Y. V. Klochko and V. Linkov, Surface-modified advanced hydrogen storage alloys for hydrogen separation and purification, J. Alloys Compd., 2011, 509, S555–S561 CrossRef CAS
- M. Williams, M. Lototsky, V. Linkov, A. Nechaev, J. Solberg and V. Yartys, Nanostructured surface coatings for the improvement of AB5-type hydrogen storage intermetallics, Int. J. Energy Res., 2009, 33, 1171–1179 CrossRef CAS
- K. Modibane, M. Williams, M. Lototskyy, M. Davids, Y. Klochko and B. G. Pollet, Poisoning-tolerant metal hydride materials and their application for hydrogen separation from CO2/CO containing gas mixtures, Int. J. Hydrogen Energy, 2013, 38, 9800–9810 CrossRef CAS
- B. Zhao, L. Liu, Y. Ye, S. Hu, D. Wu and P. Zhang, Enhanced hydrogen capacity and absorption rate of LaNi4.25Al0.75 alloy in impure hydrogen by a combined approach of fluorination and palladium deposition, Int. J. Hydrogen Energy, 2016, 41, 3465–3469 CrossRef CAS
- S. Thangarasu, G. Palanisamy, Y. M. Im and T. H. Oh, An alternative platform of solid-state hydrides with polymers as composite/encapsulation for hydrogen storage applications: Effects in intermetallic and complex hydrides, Int. J. Hydrogen Energy, 2023, 48, 21429–21450 CrossRef CAS
- G. R. de Almeida Neto, F. H. Matheus, C. A. G. Beatrice, D. R. Leiva and L. A. Pessan, Fundamentals and recent advances in polymer composites with hydride-forming metals for hydrogen storage applications, Int. J. Hydrogen Energy, 2022, 47, 34139–34164 CrossRef
- S. Thangarasu and T. H. Oh, Impact of Polymers on Magnesium-Based Hydrogen Storage Systems, Polymers, 2022, 14, 2608 CrossRef CAS
- P. Selvam, B. Viswanathan and V. Srinivasan, The influence of atmospheric CO2 on the surface properties of Mg2NiH4 and a comparison with some hydrogen storage alloys, J. Less-Common Met., 1990, 158, L1–L7 CrossRef CAS
- S. Suwarno, Y. Gosselin, J. Solberg, J. Maehlen, M. Williams, B. Krogh, B. Børresen, E. Rytter, E. Ochoa-Fernández and V. Yartys, Selective hydrogen absorption from gaseous mixtures by BCC Ti-V alloys, Int. J. Hydrogen Energy, 2012, 37, 4127–4138 CrossRef CAS
- A. S. Pedersen, B. Vigeholm, J. Kjøller and B. Larsen, The effect of cycling in impure hydrogen on the hydrogen capacity of magnesium powder, Int. J. Hydrogen Energy, 1987, 12, 765–771 CrossRef CAS
- Y. Zhang, J. Li, T. Zhang, H. Kou and X. Xue, Hydrogen absorption properties of a non-stoichiometric Zr-based Laves alloy against gaseous impurities, Int. J. Hydrogen Energy, 2017, 42, 10109–10116 CrossRef CAS
- K. B. Park, J. O. Fadonougbo, J.-S. Bae, G. B. Kang, J. I. Choi, Y. D. Kim, T.-W. Na and H.-K. Park, The Evolution of Surface Oxides during TiFe0.9M0.1 (M = Ni, Mn) Activation: An In Situ XPS Investigation, Metals, 2022, 12, 2093 CrossRef CAS
- H. Zhu, J. Wu and Q. Wang, Reactivation behaviour of TiFe hydride, J. Alloys Compd., 1994, 215, 91–95 CrossRef CAS
- S. Bouaricha, J. Huot, D. Guay and R. Schulz, Reactivity during cycling of nanocrystalline Mg-based hydrogen storage compounds, Int. J. Hydrogen Energy, 2002, 27, 909–913 CrossRef CAS
- C.-C. Shen and H.-C. Li, Passivation and reactivation of Ti25V35Cr40 hydrides by cycling with impure hydrogen gas, Int. J. Hydrogen Energy, 2015, 40, 3277–3282 CrossRef CAS
- F. Meli, Z. Sheng, I. Vedel and L. Schlapbach, XPS analysis of the getter mechanism and getter activation process, Vacuum, 1990, 41, 1938–1940 CrossRef CAS
- S. Chia-Chieh and L. Hsueh-Chih, Passivation and reactivation of Ti25V35Cr40 hydrides by cycling with impure hydrogen gas, Int. J. Hydrogen Energy, 2015, 40, 3277–3282 CrossRef
- H. Emami, K. Edalati, J. Matsuda, E. Akiba and Z. Horita, Hydrogen storage performance of TiFe after processing by ball milling, Acta Mater., 2015, 88, 190–195 CrossRef CAS
- J. Manna, B. Tougas and J. Huot, Mechanical activation of air exposed TiFe+ 4 wt% Zr alloy for hydrogenation by cold rolling and ball milling, Int. J. Hydrogen Energy, 2018, 43, 20795–20800 CrossRef CAS
- S. Vincent and J. Huot, Effect of air contamination on ball milling and cold rolling of magnesium hydride, J. Alloys Compd., 2011, 509, L175–L179 CrossRef CAS
- F. Sun, M.-y. Yan, X.-p. Liu, J.-h. Ye, Z.-n. Li, S.-m. Wang and L.-j. Jiang, Effect of CO on hydrogen storage performance of 2LiNH2 + MgH2 system, Int. J. Hydrogen Energy, 2014, 39, 9288–9292 CrossRef CAS
Footnote |
| † These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2024 |