DOI:
10.1039/D4QO00301B
(Review Article)
Org. Chem. Front., 2024,
11, 2351-2374
Catalytic intermolecular hydrofunctionalizations of ynamides
Received
15th February 2024
, Accepted 13th March 2024
First published on 14th March 2024
Abstract
Ynamides are electron-rich alkynes with a carbon–carbon triple bond directly attached to the nitrogen atom bearing an electron-withdrawing group. This unique structure renders diverse reactivities to ynamides in synthetic chemistry. Among them, the catalytic intermolecular hydrofunctionalization of ynamides is of critical importance, which enables a rapid and atom-economical synthesis of multi-substituted enamides with high and tunable chemo-, regio-, and stereoselectivity. This review provides a comprehensive summary of the hydrofunctionalization of ynamides and is categorized by the bond formation types including C–C, C–X, C–O, C–N, C–S, C–P, C–Si, and C–Ge bonds. Based on their position in making new bonds, the reactions can be classified into four types: the syn-α-adduct, syn-β-adduct, anti-α-adduct and anti-β-adduct. This review will facilitate a better understanding of the factors that contribute to the varied selectivities and arouse more research interest in this fascinating topic.
 Ying-Ying Zhao | Ying-Ying Zhao was born in Hebei Province, China, in 1990. She received her B.S. degree from Jiangsu University in 2013 and PhD degree from the Dalian Institute of Chemical Physics (DICP) in 2018, under the guidance of Prof. Boshun Wan. Then she began her career at Liaoning Normal University as a lecturer. In 2023, she moved to Jiangsu Normal University, where she works on developing new reaction patterns of ynamides. |
 Yu-Jing Jia | Yu-Jing Jia was born in Shijiazhuang, Hebei Province, China. She received her B.S. degree from Handan University in 2013. Then she worked as an engineer in Qingke New Material Technology (Hebei) Company Limited. Currently, she is pursuing her M.S. degree under the guidance of Prof. Yan-Cheng Hu. |
 Yan-Cheng Hu | Yan-Cheng Hu was born in Jiangsu Province, China, in 1989. He obtained his B.S. degree from Nanjing Normal University in 2010 and PhD degree from the Dalian Institute of Chemical Physics (DICP) in 2015 under the supervision of Prof. Boshun Wan. He did his postdoctoral research with Prof. Tao Zhang at the same institute and was promoted to associate professor in 2019. In 2022, he moved to the China University of Mining and Technology as a professor. His research focuses on transition-metal catalyzed transformations of alkynes and alkenes. |
1. Introduction
Ynamides are a subclass of alkynes that are directly attached to the nitrogen atom bearing an electron-withdrawing group (EWG = sulfonyl, benzoyl etc.). The electronic bias imposed by the nitrogen atom makes the carbon–carbon triple bond highly reactive, but it can be finely tuned by the electron-withdrawing group.1–5 Because of their unique structural characteristics and potential coordination ability, ynamides have now emerged as versatile building blocks in organic synthesis and a variety of intriguing reactivities have been disclosed.6–10 Some special topics such as intra-11–16 and inter-molecular annulations,17–22 radical reactions,23,24 and asymmetric transformations25,26 have been carefully reviewed in the last few years.
Of particular note is that very recently, Sahoo et al. published an elegant review on the difunctionalization of ynamides.27 In addition to this useful strategy, another interesting reaction pattern is the catalytic hydrofunctionalization of ynamides, which can provide easy and atom-economical access to trisubstituted enamides with high and tunable chemo-, regio-, and stereoselectivity. These pioneering works before 2010 have been discussed in two excellent reviews by Hsung et al.2 and Evano et al.3 With deeper insights into the fundamental principles, ynamide hydrofunctionalizations have witnessed an ever-increasing number of publications over the past decade. Nevertheless, there is a lack of a comprehensive summary of these achievements.
In light of this, we wish to give an exhaustive overview of the hydrofunctionalization of ynamides involving the formation of C–C, C–X (X = halides), C–O, C–N, C–S, C–P, C–Si, and C–Ge bonds. This review is restricted to intermolecular one-step hydrofunctionalizations, while those multi-step reactions for accessing hydrofunctionalized products are not covered here. By highlighting product diversity, controllable regioselectivity, and the mechanistic rationale, we believe that this review will allow the readers to better understand the factors that contribute to the varied selectivities and prompt further investigation into this fascinating research topic.
2. C–C bond formation
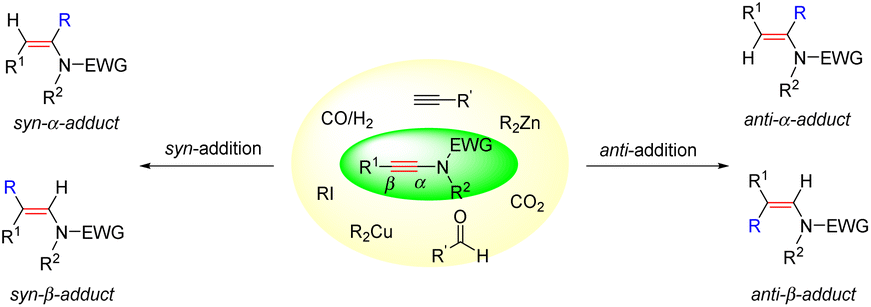
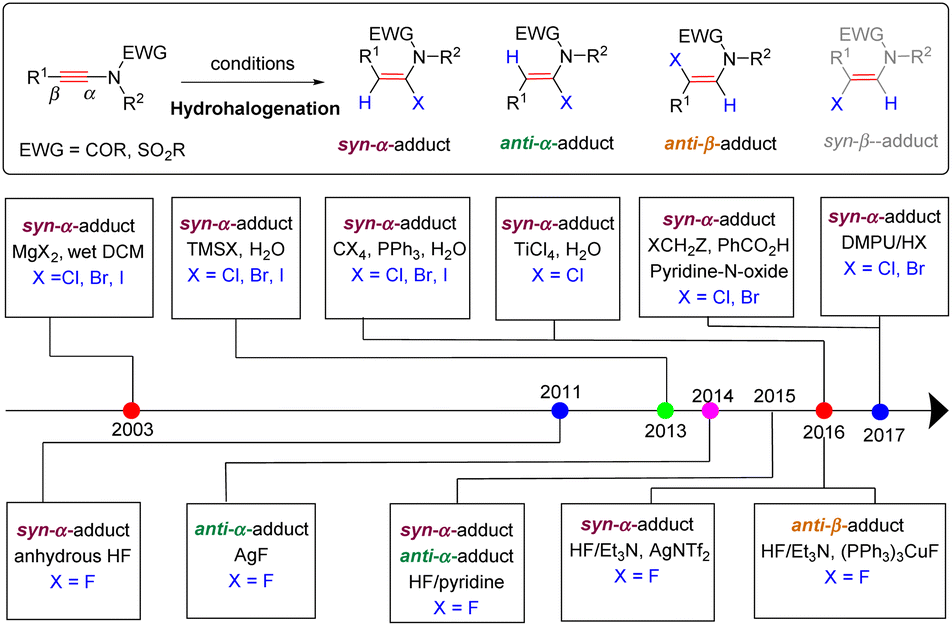
Catalytic hydrocarbonation of ynamides could construct C–C and C–H bonds at the same time with regio- and stereoselectivity. Ynamide, as a kind of unsymmetrical alkyne, has two possible reaction sites: α- and β-sites. Regioselectivity discussed herein refers to the position in making the C–C bond (α- vs. β-adduct). Stereoselectivity comes from the product enamides, which have two possible stereoisomers. According to the configuration of products, we classify the addition mode into syn- and anti-addition. As a consequence, the hydrofunctionalization of ynamides would generate four potential isomers: anti-α-, syn-α-, anti-β- and syn-β-adducts (Scheme 1).
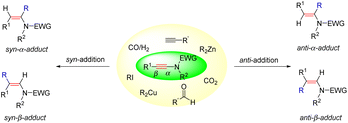 |
| | Scheme 1 Potential isomers for the hydrofunctionalization of ynamides. | |
2.1 Coupling with organocopper reagents
In 2005, Marek and co-workers reported an efficient hydrofunctionalization of ynamide 1 with organocopper reagents to form syn-β-enamide 2 with high regio- and stereoselectivity (Scheme 2).28 The chelate action of electron-withdrawing carbamoyl or sulfonyl groups with organometallic species plays a crucial role in tuning the regioselectivity (Int-1). The simultaneous coordination to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond and the electron-withdrawing group triggers a syn-attack of organocopper at the β-site of ynamide. The resulting vinylic metal species Int-2 could be trapped by H+ to afford syn-β-adduct 2.
C bond and the electron-withdrawing group triggers a syn-attack of organocopper at the β-site of ynamide. The resulting vinylic metal species Int-2 could be trapped by H+ to afford syn-β-adduct 2.
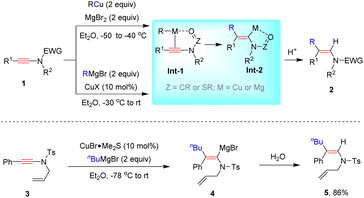 |
| | Scheme 2 Copper-catalyzed hydroalkylation of ynamides with organocopper reagents. | |
2.2 Coupling with organozinc reagents
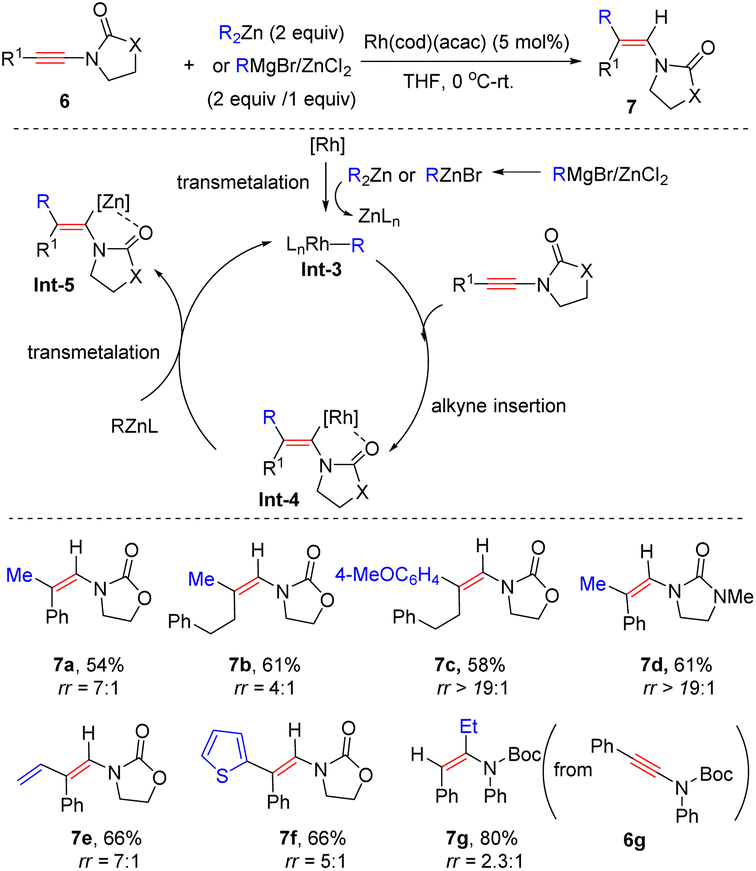
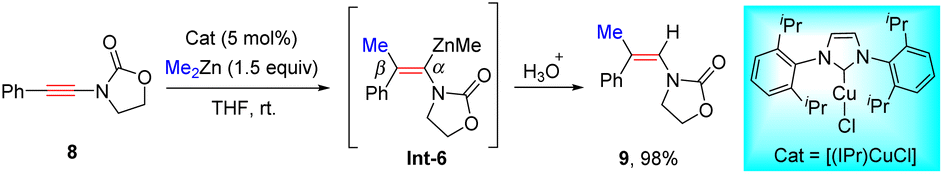
In 2009, Lam's group showcased that when using [Rh(cod)(acac)] as a catalyst, oxazolidinone-derived ynamides 6 reacted with organozincs smoothly to furnish syn-β-enamides 7 as the main products (Scheme 3).29 The process is initiated by ligand exchange to form the active R–Rh species Int-3. Subsequent alkyne insertion delivers vinylic rhodium Int-4, which can be stabilized by coordination with the proximal carbonyl group. A final transmetalation closes the catalytic cycle and gives vinylic zinc Int-5, followed by protonolysis to furnish the product. A series of aryl, heteroaryl, and alkenylzinc species were amenable to the transformation. It is noted that in the case of Boc-derived ynamide 6g, α-adduct 7g turned out to be predominant likely owing to the poor coordination ability of the Boc group. Later, Hou and co-workers found that a similar chelation-directed hydromethylation of ynamide with dimethylzinc took place efficiently in the presence of an (IPr)CuCl catalyst (Scheme 4).30
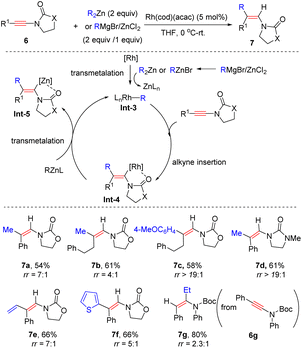 |
| | Scheme 3 Rh-catalyzed carbozincation of ynamides with in situ generated diorganozincs. | |
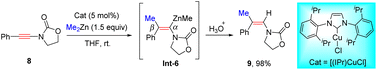 |
| | Scheme 4 Cu-catalyzed carbozincation of ynamides with dialkylzinc reagents. | |
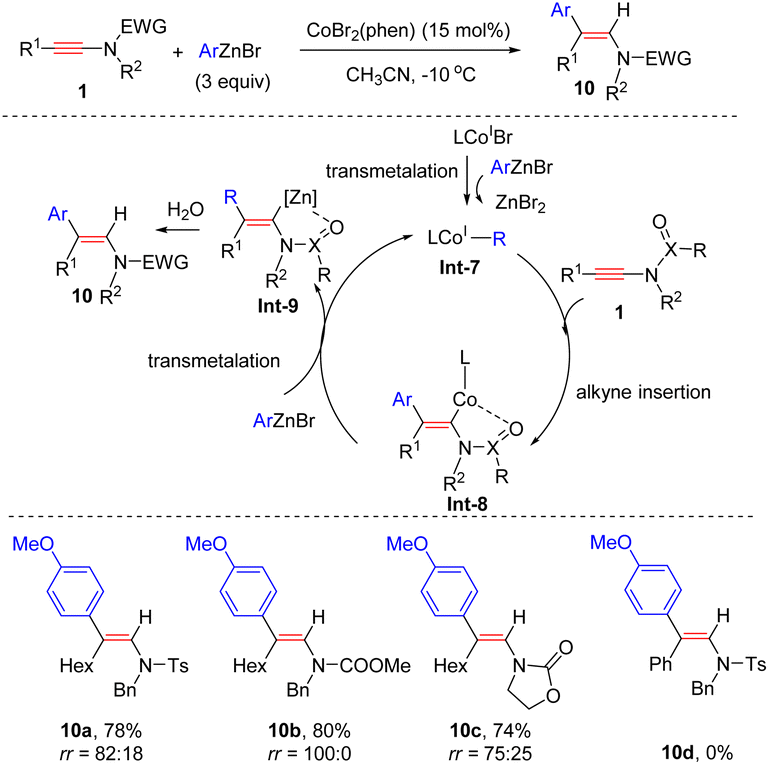
In 2017, Gillaizeau and Gosmini et al. documented a Co-catalyzed syn-hydroarylation of ynamides with arylzincs (Scheme 5).31 This catalytic system well complements previously developed rhodium- and copper-catalyzed methodologies. A wide variety of ynamides bearing an alkyl group at the terminus and a sulfonyl group on the nitrogen atom were compatible with the process. Analogously, the coordination between the carbonyl/sulfonyl group and the cobalt facilitates syn-β-addition.
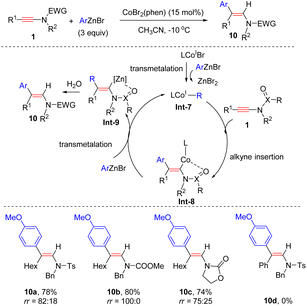 |
| | Scheme 5 Cobalt-catalyzed hydroarylation of ynamides with arylzincs. | |
2.3 Coupling with organoboron reagents
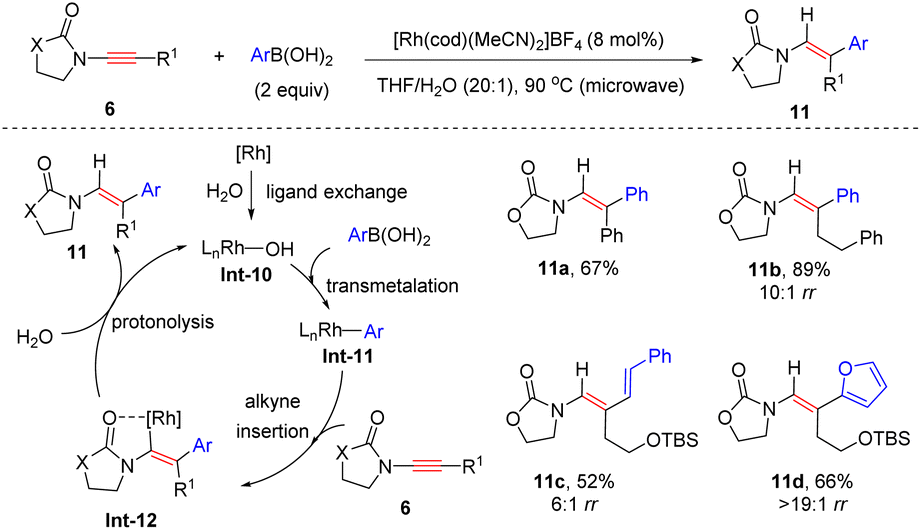
Lam and his colleagues, in 2010, showed that when arylboronic acids were employed as substrates, syn-hydroarylation of oxazolidinone-derived ynamides also occurred efficiently under rhodium catalysis (Scheme 6).32 The mechanism commences with the generation of rhodium(I) hydroxide species Int-10, which undergoes transmetalation with arylboronic acids to yield organorhodium Int-11. Then, chelation-directed syn-β-addition to ynamide and protonolysis with water release enamides 11 with the regeneration of rhodium(I) hydroxide.
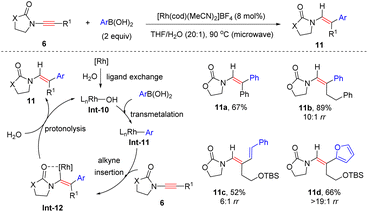 |
| | Scheme 6 Rhodium-catalyzed carbometalation of ynamides with arylboronic acids. | |
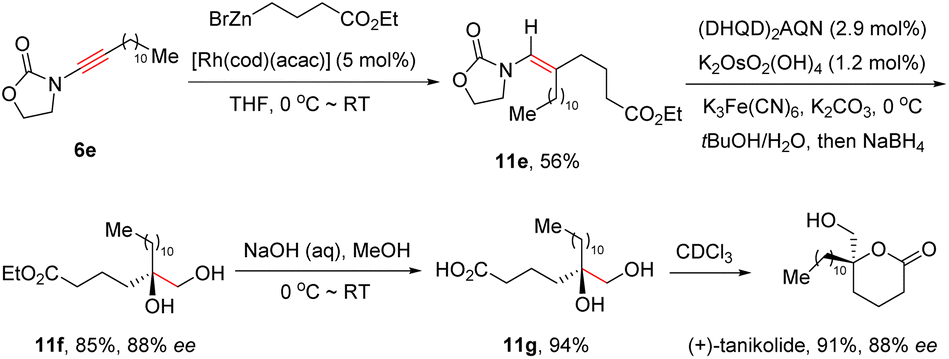
Remarkably, this elegant protocol was successfully applied in the total synthesis of the marine natural product (+)-tanikolide (Scheme 7).33 With the assistance of the [Rh(cod)(acac)] catalyst, oxazolidinone-derived ynamide 6e reacted with commercially available 4-ethoxy-4-oxobutylzinc bromide readily to afford enamide 11e in 56% yield. Subsequently, in the presence of (DHQD)2AQN (hydroquinidine anthraquinone-1,4-diyl diether) and K2OsO2(OH)4, Sharpless asymmetric dihydroxylation took place smoothly, followed by reduction with NaBH4 to furnish diol 11f in 85% yield and 88% ee. Finally, basic hydrolysis and lactonization resulted in the formation of the natural product (+)-tanikolide.
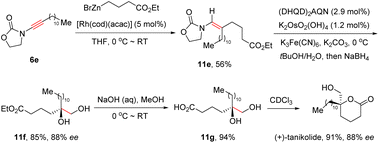 |
| | Scheme 7 Application in the total synthesis of the marine natural product (+)-tanikolide. | |
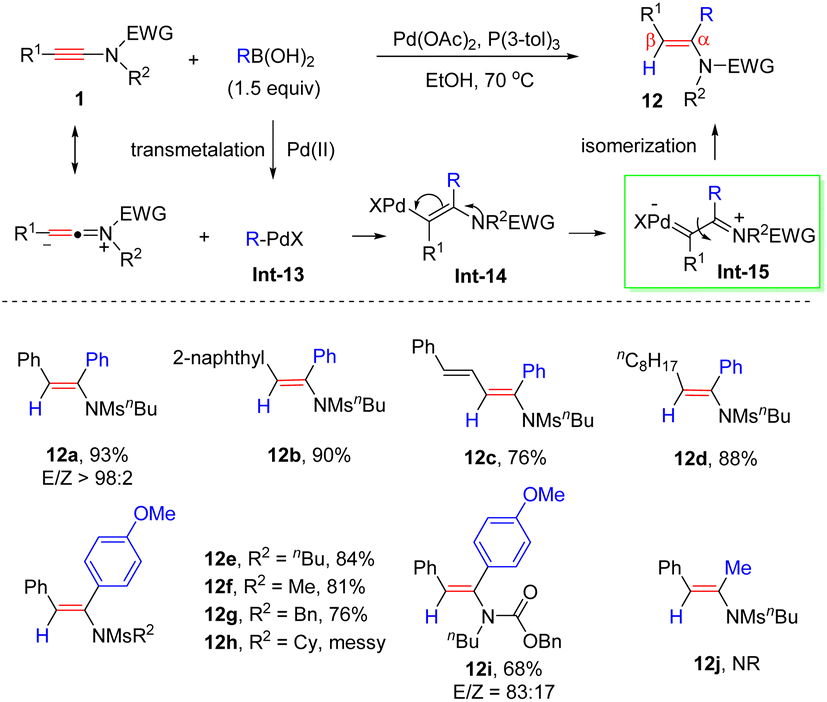
In 2014, an unprecedented anti-α-addition of boronic acids to ynamides under palladium catalysis was documented by Zhu et al. (Scheme 8).34 The properties of the substituents in the boronic acids had little impact on the reactivity. Besides, this approach tolerated a wide variety of aryl and alkyl substituents of ynamides, giving rise to anti-α-enamides 12 in good yields and regioselectivities. Mechanistically, the transmetallation of the Pd(II)-catalyst with boronic acids forms the R–PdX intermediate Int-13. The highly polarized CC bond, induced by the nitrogen atom, is preferably attacked by R–PdX at the α-site to yield the syn-type vinylic palladium intermediate Int-14. Subsequently, the palladium carbene species Int-15 undergoes E–Z isomerization to give the anti-α-adduct 12.
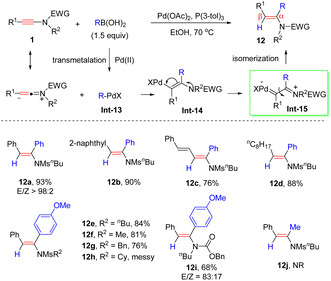 |
| | Scheme 8 Pd-catalyzed anti-addition of boronic acids to ynamides. | |
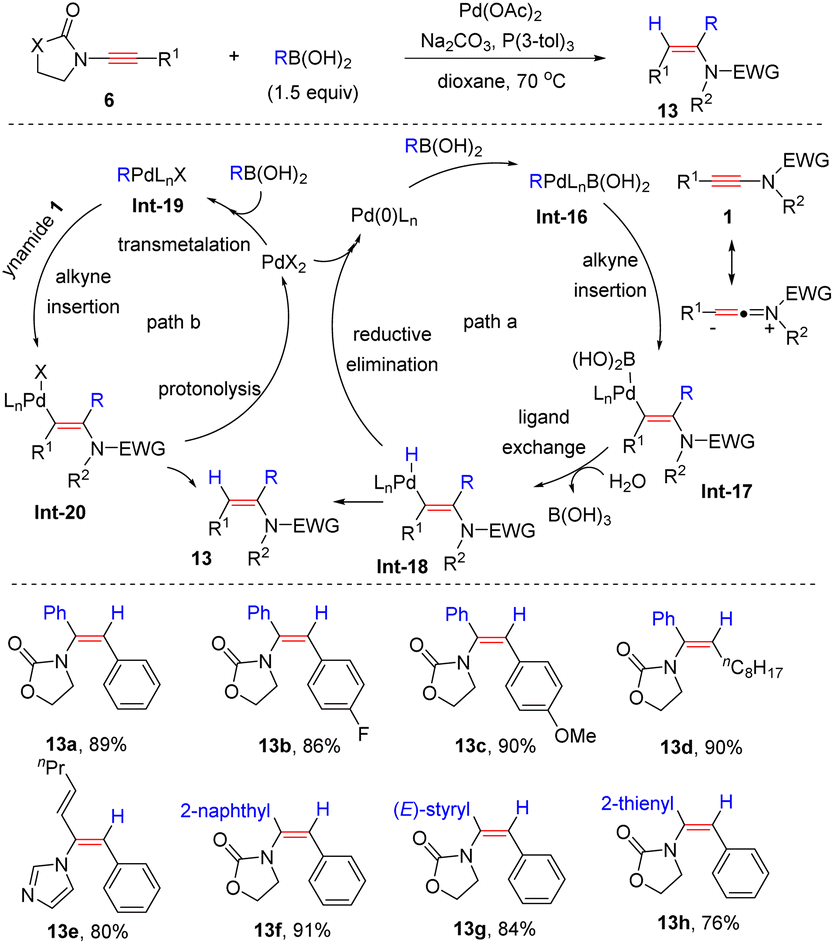
In comparison, they found that when oxazolidinone-substituted ynamides were used as starting materials, the selectivity switched to syn-α-addition (Scheme 9).35 Various boronic acids with electron-attracting and electron-repulsing groups were all suitable partners. Then two reasonable Pd(0) and Pd(II) catalytic cycles were proposed. An oxidative addition of the Pd(0) catalyst with boronic acids leads to the R–Pd–B complex Int-16 and subsequent syn-addition to ynamides affords the vinylic palladium intermediate Int-17. Eventually, ligand exchange of the Pd–B bond and reductive elimination give rise to the target products 13 (path a). Another pathway includes the transmetalation of Pd(II) with boronic acids, syn-addition to ynamides and protonolysis of the Pd–C bond (path b).
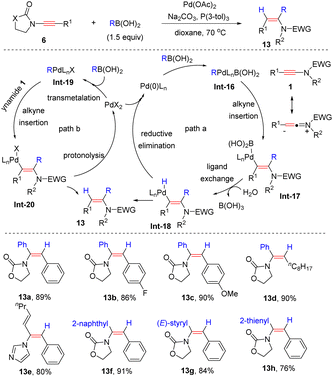 |
| | Scheme 9 Palladium-catalyzed syn-addition of boronic acids to ynamides. | |
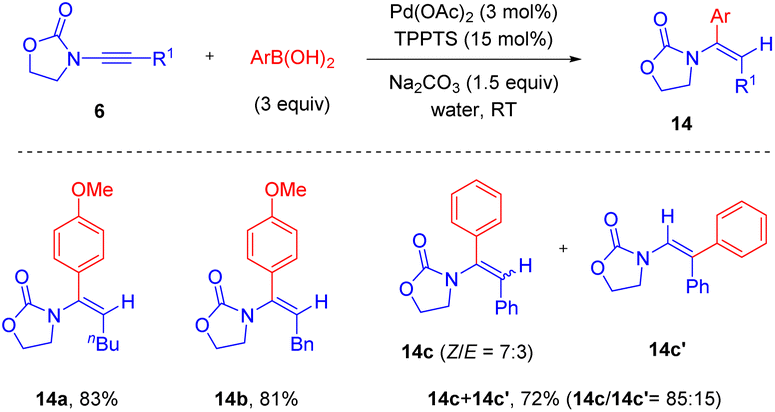
In 2020, Michelet and Arcadi et al. developed a mild alternative for the synthesis of α-aryl enamides 14via Pd-catalyzed hydroarylation of ynamides with boronic acids (Scheme 10).36 By employing the commercially available triphenylphosphine-3,3′,3′′-trisulfonic acid trisodium salt [TPPTS, P(m-C6H4SO3Na)3] as a ligand, this hydroarylation could take place smoothly in water. The oxazolidinone motif on ynamides played a crucial role in modulating the regioselectivity. Alkyl groups on the alkyne terminus gave syn-α-adducts with complete regiocontrol, while the phenyl-derived substrate resulted in a mixture of regioisomers but α-phenyl enamide was still the major product with moderate stereoselectivity.
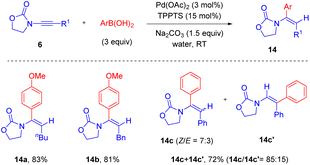 |
| | Scheme 10 Pd-catalyzed hydroarylation of oxazolidinone-substituted ynamides with arylboronic acids in water. | |
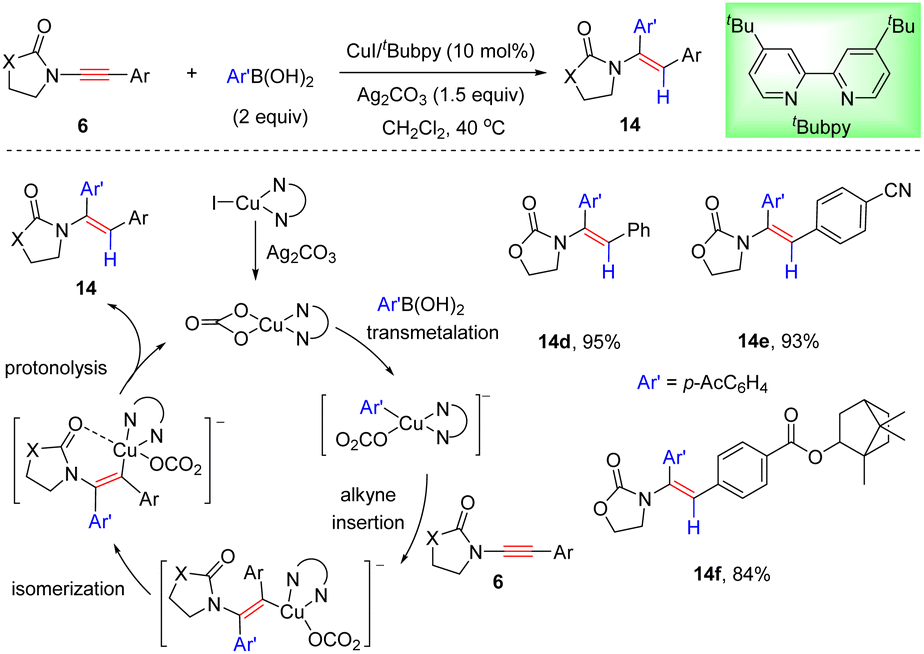
More recently, Sahoo and co-workers presented that the cheap metal catalyst system CuI/tBubpy could also promote the coupling of oxazolidinone-substituted ynamides with arylboronic acids and unexpected anti-α-adducts were obtained (Scheme 11).37 This protocol features mild reaction conditions, a broad substrate scope of ynamides, and high regio- and stereoselectivities. With the aid of Ag2CO3, the Cu(I) species is first oxidized to the active Cu(II) complex bearing the bidentate carbonate ligand. Subsequent transmetalation with arylboronic acid and further attack at the more electron-deficient α-site of ynamide deliver a syn α-carbocupration intermediate. Then chelation-directed isomerization occurs, leading to the formation of a thermodynamically stable anti isomer. An eventual protodecupration gives rise to the target anti-α-adduct with the regeneration of the active catalyst.
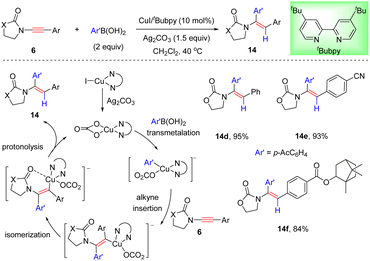 |
| | Scheme 11 Cu-catalyzed hydroarylation of oxazolidinone-substituted ynamides with arylboronic acids. | |
2.4 Coupling with aryl halides
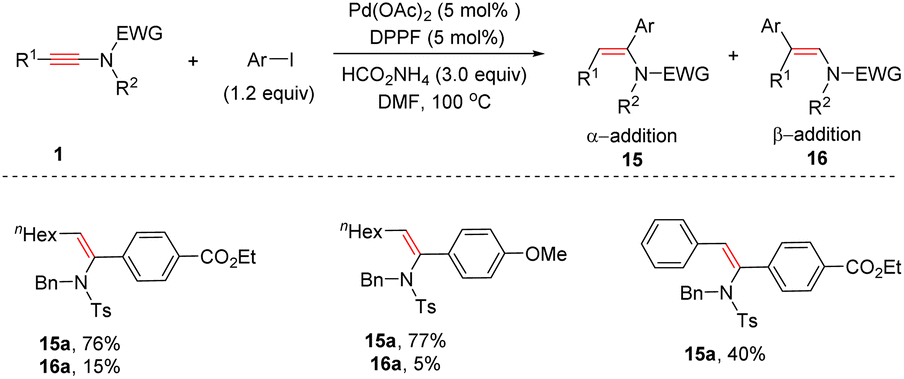
In 2017, Yoshimura and Wakamatsu et al. unveiled that aryl iodides could serve as effective coupling partners with ynamides in the presence of Pd(OAc)2 and 1,1′-bis(diphenylphosphino)ferrocene (DPPF), giving syn-α-enamides 15 as the major products (Scheme 12).38 Both electron-donating and electron-withdrawing groups on aryl iodides were tolerated, whereas the steric hindrance of the substituents on the phenyl ring exerted a crucial impact on the result. Both alkyl and aryl substituents on the alkyne terminus were amenable to the transformation.
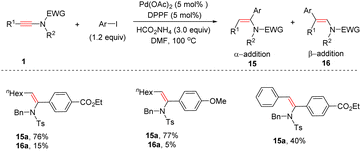 |
| | Scheme 12 Pd-catalyzed hydroarylation of ynamides with aryl iodides. | |
2.5 Coupling with terminal alkynes
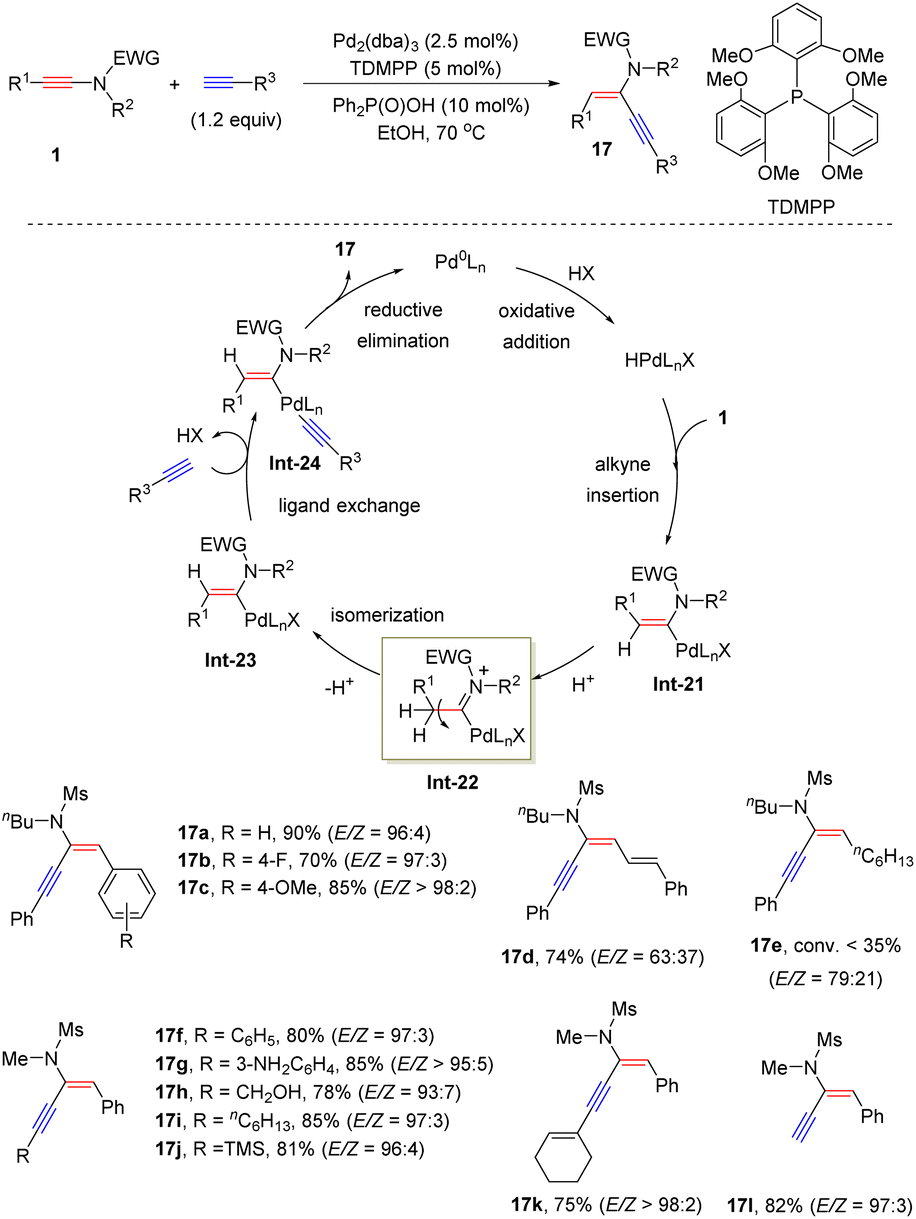
In 2014, Zhu and his colleagues established an interesting Pd-catalyzed hydroalkynylation of sulfonyl-derived ynamides with terminal alkynes (Scheme 13).39 This method enables a convenient synthesis of structurally useful anti-α-ynenamides 17. Aryl-substituted ynamides worked well under the standard conditions, while alkyl and alkenyl substituted ynamides led to a low stereoselectivity (17d and 17e). The oxidative addition of Pd(0) with an acid generates a Pd–H species, followed by migratory insertion of ynamides to produce the syn-α-vinylic palladium intermediate Int-21. Then E–Z isomerization under acidic conditions gives the anti-α-vinylic palladium intermediate Int-23. Subsequent ligand exchange with terminal alkynes and reductive elimination deliver anti-α-ynenamides 17.
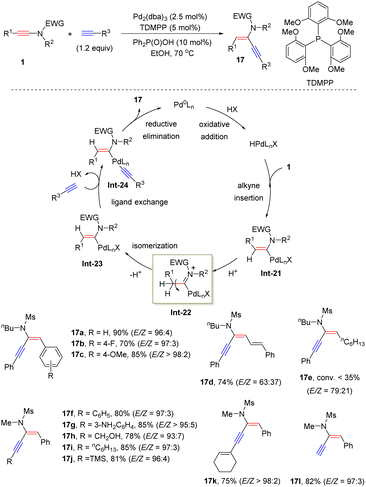 |
| | Scheme 13 Pd-catalyzed anti-addition of terminal alkynes to ynamides. | |
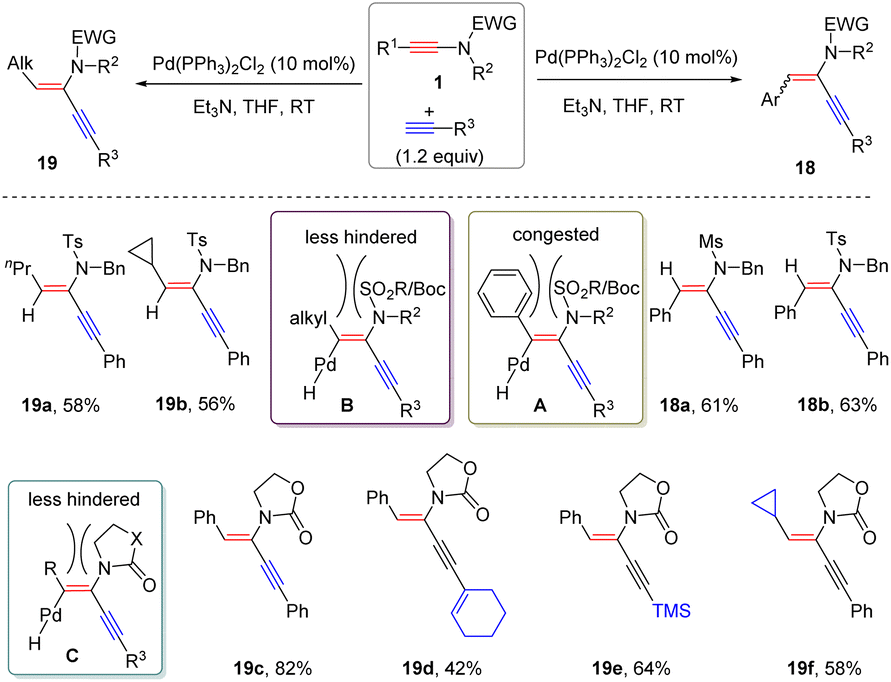
Later, Reddy and co-workers demonstrated that palladium-catalyzed α-hydroalkynylation of ynamides could also take place under alkaline conditions (Scheme 14).40 The substituents on the ynamides played a crucial role in controlling the stereoselectivity. For alkyl-derived sulfonyl ynamides, syn-α-adducts 19 were obtained exclusively, while the reaction of aryl-based substrates dominantly gave anti-α-adducts 18. This phenomenon is likely ascribed to the stronger steric hindrance between aryl and N-substituents. It is worthwhile to mention that in the case of oxazolidinone-derived ynamides, regardless of the nature of the substituents at the terminus, only syn-adducts were afforded due to the less steric hindrance.
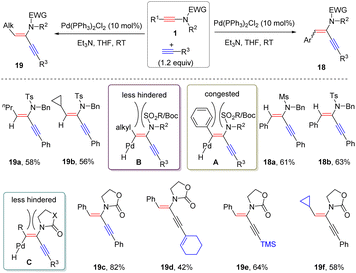 |
| | Scheme 14 Pd-catalyzed α-hydroalkynylation of ynamides with terminal alkynes. | |
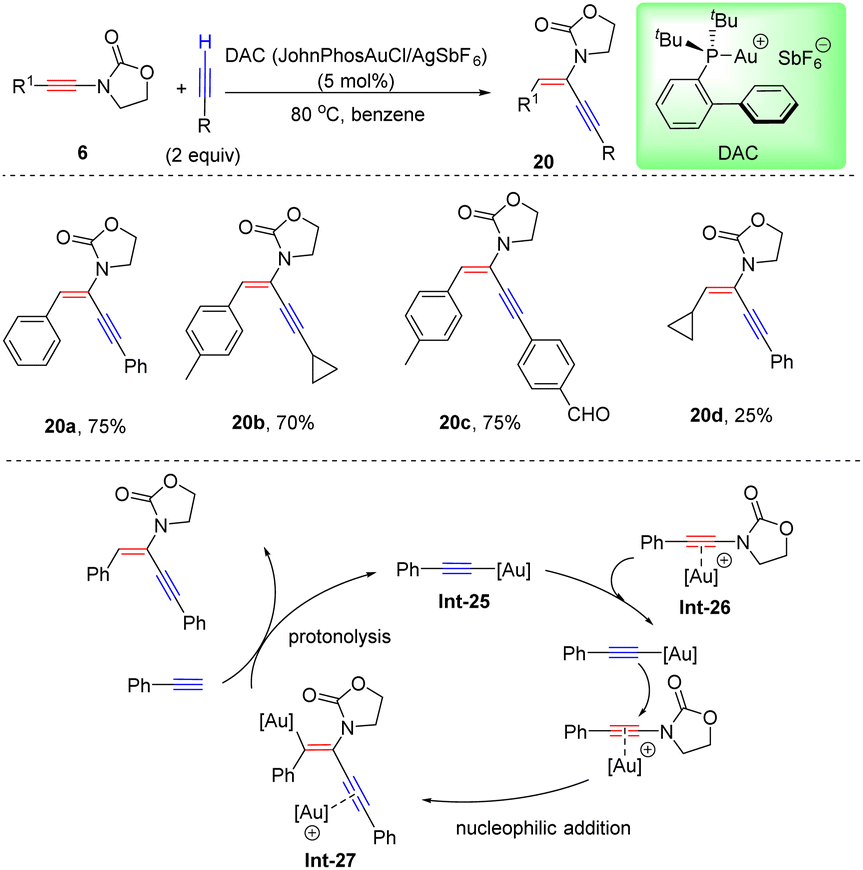
Recently, Haberhauer and co-workers found that the stereoselectivity for hydroalkynylation of oxazolidinone-derived ynamides could be altered to anti-adducts when a bulky cationic gold catalyst JohnphosAuSbF6 (DAC) was employed (Scheme 15).41 A gold acetylide complex, pre-synthesized with phenylacetylene and DAC, was demonstrated to be a better catalyst for hydroalkynylation of oxazolidinone-derived ynamides. Based on this fact, it is believed that the terminal alkyne first reacts with the cationic gold complex to deliver the active catalyst gold acetylide intermediate Int-25. Meanwhile, ynamides can also be activated by the [Au]+ fragment to form the π-complex intermediate Int-26, which is easily attacked by σ-activated gold acetylide from the back side to afford the vinyl gold species Int-27. A final protodeauration gives gold acetylide to close the cycle and results in product 20. Isotope labeling experiments and quantum chemical calculations both support the proposed dual-gold-catalyzed procedure.
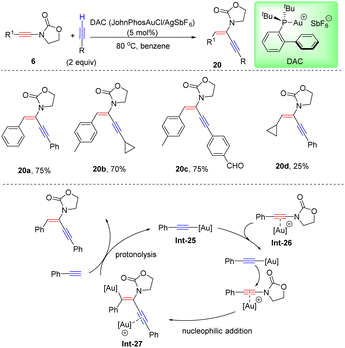 |
| | Scheme 15 Au-catalyzed hydroalkynylation of oxazolidinone-derived ynamides with terminal alkynes. | |
2.6 Coupling with electron-rich aromatics
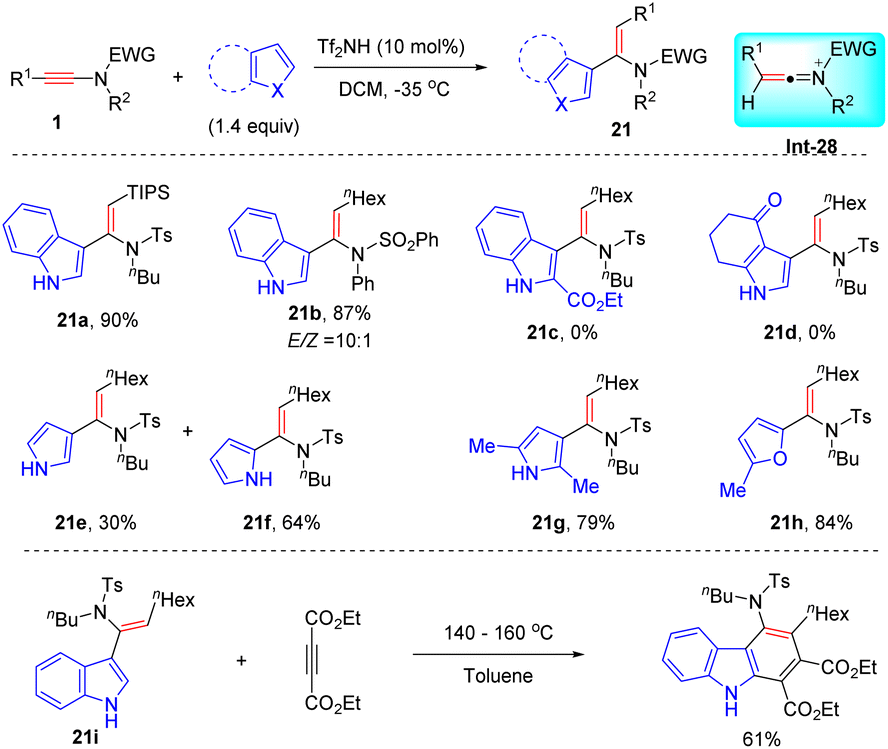
In 2005, Zhang developed a Brønsted acid-catalyzed syn-hydroarylation of ynamides for the synthesis of vinylindoles with high regio- and stereoselectivity (Scheme 16).42 The ynamide is first activated by Tf2NH to generate the keteniminium ion Int-28, which further undergoes a Friedel–Crafts step to afford the target vinylindole products. A variety of alkyl-derived ynamides and indoles were suitable for the process, while indoles with an electron-withdrawing group completely hampered the Friedel–Crafts step. Notably, the resulting adducts could easily undergo the Diels–Alder reaction with electron-deficient dimethyl acetylenedicarboxylate (DMAD) to furnish amino-substituted carbazoles.
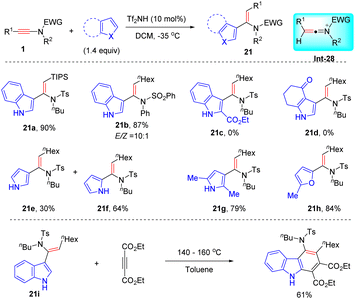 |
| | Scheme 16 Brønsted acid-catalyzed syn-hydroarylation of ynamides with indoles. | |
The same group depicted that other electron-rich aromatic heterocycles were also amenable to the process.43 For instance, the hydroarylation of a hexyl-derived ynamide with a simple pyrrole also took place readily, but afforded a mixture of C2- and C3-vinylated pyrroles (21e and 21f). When 2-methyl pyrrole and furan were engaged as substrates, the transformation could be executed with high efficiencies and regioselectivities (21g and 21h).
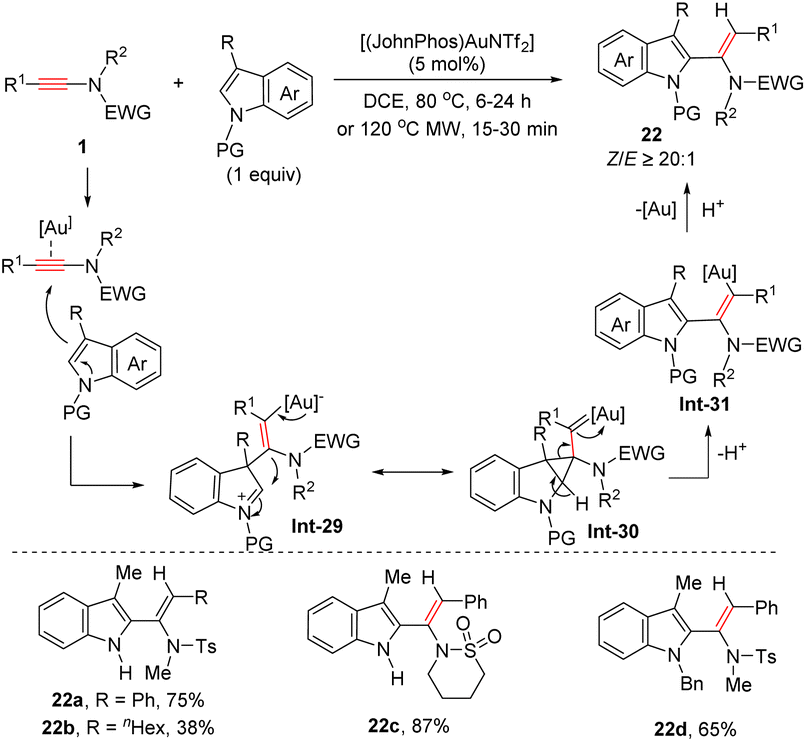
In 2016, Rossi and Pirovano et al. uncovered an elegant cationic gold-catalyzed syn-hydroarylation of ynamides with indoles, which provides a rapid access to various 2-vinylindoles with high regio- and stereoselectivities (Scheme 17).44 Mechanistically, the ynamide is initially activated by a cationic gold catalyst via coordination with the carbon–carbon triple bond, followed by the nucleophilic attack of the C3 site of indoles to generate the indolenium intermediate Int-29. Subsequently, a vinyl gold species attacks the electrophilic C2 site of indolenium to afford the cyclopropyl gold carbenoid Int-30. Then, the deprotonation and ring-opening of cyclopropane lead to a migration of the vinyl motif from the C3 to C2 site. An eventual protodeauration produces the desired 2-vinylindoles with the regeneration of the gold catalyst.
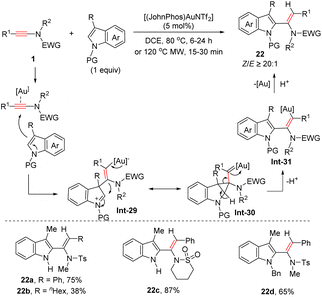 |
| | Scheme 17 Au-catalyzed syn-addition of indoles to ynamides. | |
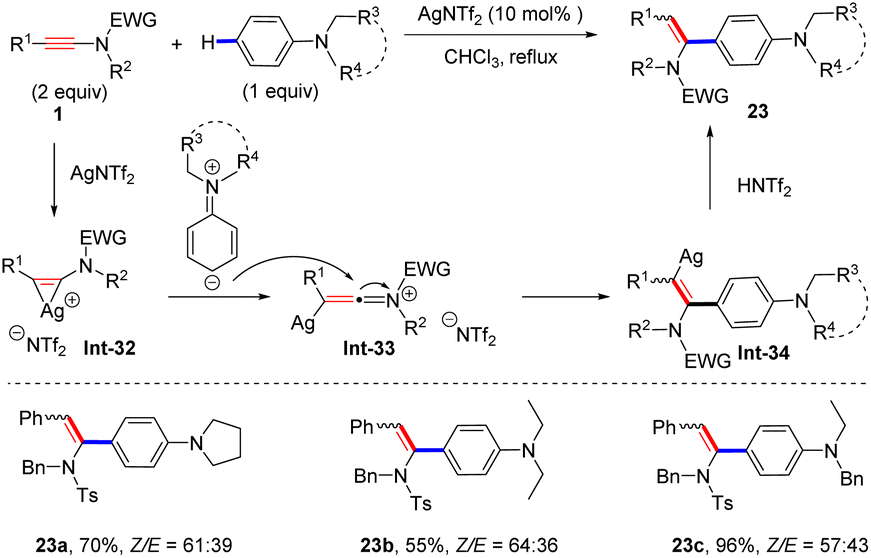
Recently, Wei and Si found that when N,N-dialkylanilines were employed as coupling partners, AgNTf2 was demonstrated to be a better catalyst for the hydroarylation of ynamides and syn-adducts were obtained as the main products (Scheme 18).45 In the presence of a cationic silver catalyst, the ynamide is first converted into the three-membered silver complex Int-32, which further undergoes rearrangement to form the silver keteniminium species Int-33. Subsequently, nucleophilic attack of the para-C atom of the aniline at the α-site of keteniminium yields the α-aryl vinyl silver intermediate. An eventual protonation leads to the formation of the expected product 23.
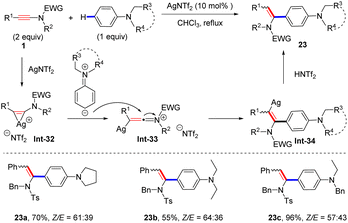 |
| | Scheme 18 Ag-catalyzed syn-addition of ynamides with N,N-dialkylanilines. | |
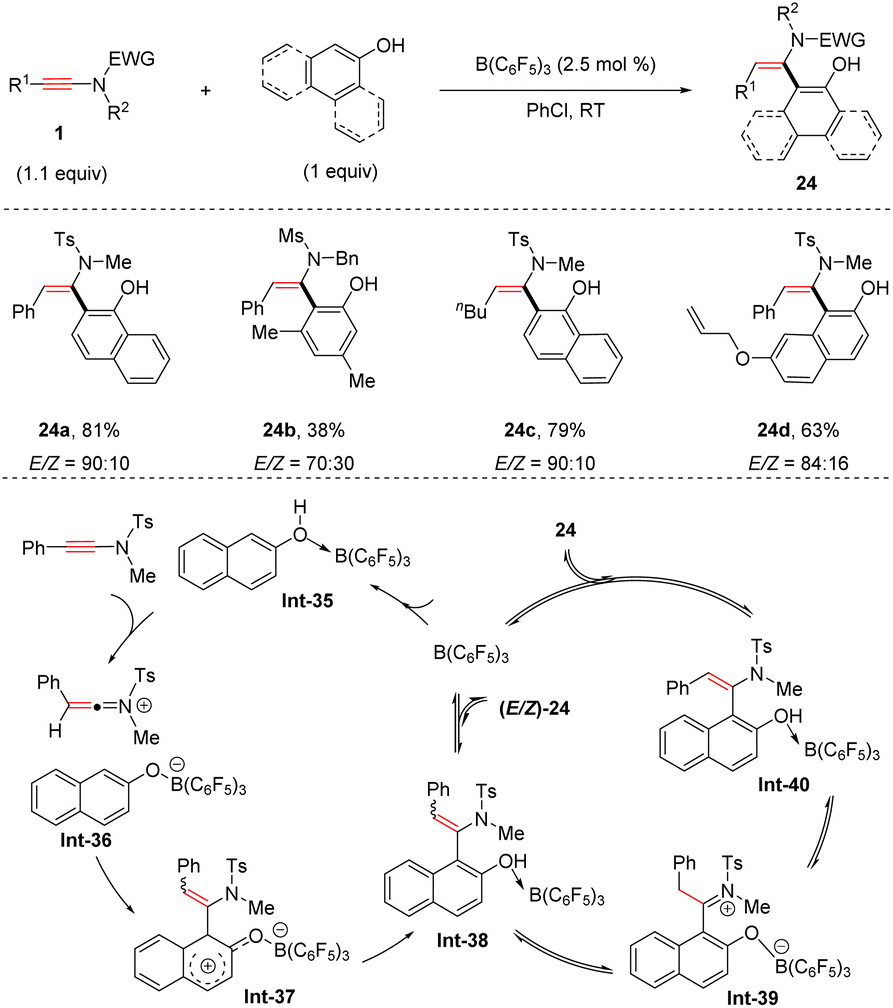
More recently, the group of Feng developed an intriguing anti-hydroarylation of ynamides with phenols with a catalytic amount of B(C6F5)3 (Scheme 19).46 The protocol features mild reaction conditions, a broad substrate scope and moderate to good anti-selectivity. Phenols are initially activated by B(C6F5)3via coordination with the oxygen atom. Afterwards, nucleophilic attack of the borate-naphthol anion on the keteniminium cation gives rise to the dearomatized intermediate Int-37. Subsequent aromatization and dissociation of B(C6F5)3 result in a mixture of Z- and E-enamides. It is worthwhile to mention that (Z)-enamides could be transformed into (E)-isomers with the assistance of B(C6F5)3via an iminium ion intermediate Int-39.
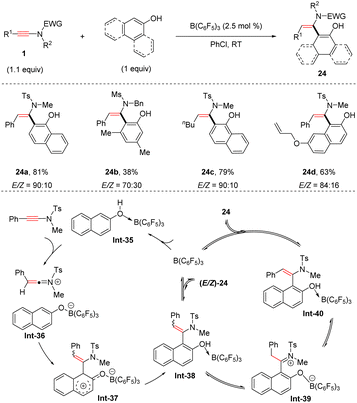 |
| | Scheme 19 B(C6F5)3-catalyzed anti-hydroarylation of ynamides with phenols. | |
2.7 Coupling with CO2
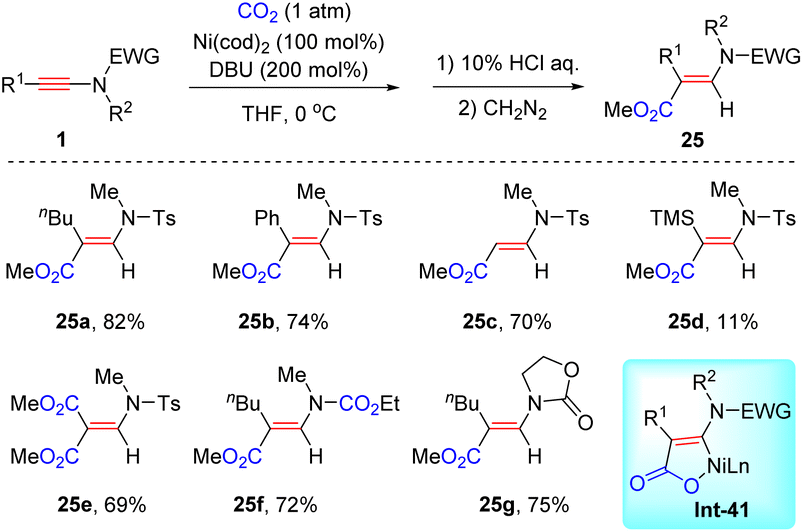
In 2016, Saito et al. pioneered the first example of the coupling between ynamides and CO2 (Scheme 20).47 In the presence of a stoichiometric amount of Ni(cod)2 and DBU, syn-β-hydrocarboxylation of ynamides took place smoothly, producing a variety of amino unsaturated esters 25. The features include mild reaction conditions, wide functional group tolerance and exclusive β-regioselectivity. The authors proposed that the key intermediate nickelactone Int-41 contributes to the high syn-β-selectivity.
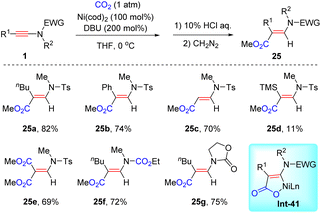 |
| | Scheme 20 Nickel-catalyzed syn-β-hydrocarboxylation of various ynamides with CO2. | |
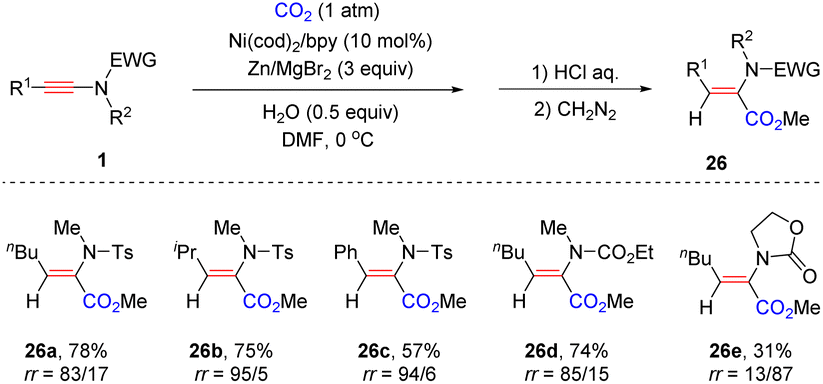
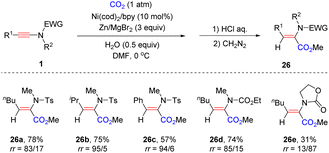
Later, the same group presented that on employing a catalytic amount of Ni(cod)2/bpy and Zn/MgBr2 as reducing agents, an unexpected syn-α-hydrocarboxylation of the ynamide with CO2 was achieved (Scheme 21).48 A wide range of alkyl and aryl substituents on the ynamides all worked well in the reaction. Besides, a carbamate-derived ynamide was compatible with the transformation as well (26d). In contrast, an oxazolidinone derived ynamide reversed the regioselectivity and β-carboxyl enamide was obtained as the main product (26e). Initially, an active nickel hydride complex is formed with H2O under reductive conditions. The subsequent syn-addition of Ni–H with ynamides generates α-nickel enamides, followed by the insertion of CO2 to yield α-carboxyl enamides. The steric hindrance of oxazolidinone reverses the regioselectivity for the addition between Ni–H and ynamide.
 |
| | Scheme 21 Nickel-catalyzed syn-α-hydrocarboxylation of ynamides with CO2 and H2O. | |
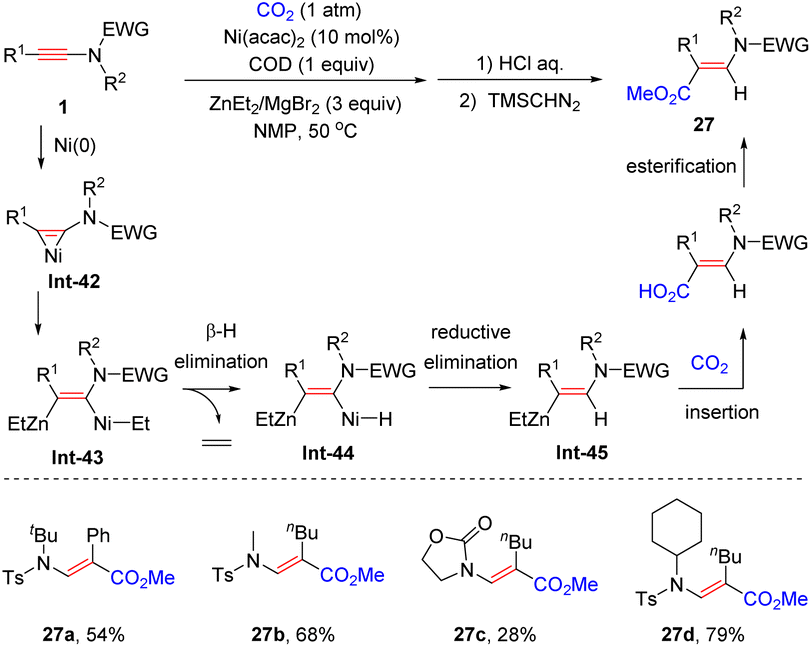
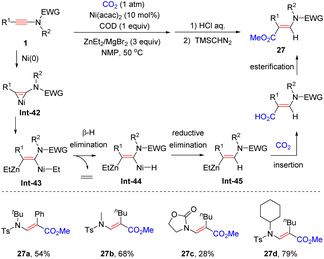
In 2019, the same group showcased that when diethylzinc was employed as the reductant, Ni(II)-catalyzed hydrocarboxylation of ynamides with CO2 led to a series of β-carboxyl enamides with complete regiocontrol (Scheme 22).49 The ynamide is initially activated by a Ni(0) species, in situ generated via the reduction of Ni(II) with ZnEt2, to form the nickelacyclopropene intermediate Int-42, which then reacts with ZnEt2 to generate the β-zinc alkenyl-nickel intermediate Int-43. Subsequent β-hydrogen elimination and reductive elimination yield the vinylzinc species Int-45. Finally, the insertion of CO2 and esterification result in the formation of β-carboxyl enamides.
 |
| | Scheme 22 Ni(acac)2-catalyzed hydrocarboxylation of ynamides with CO2. | |
2.8 Coupling with other reagents
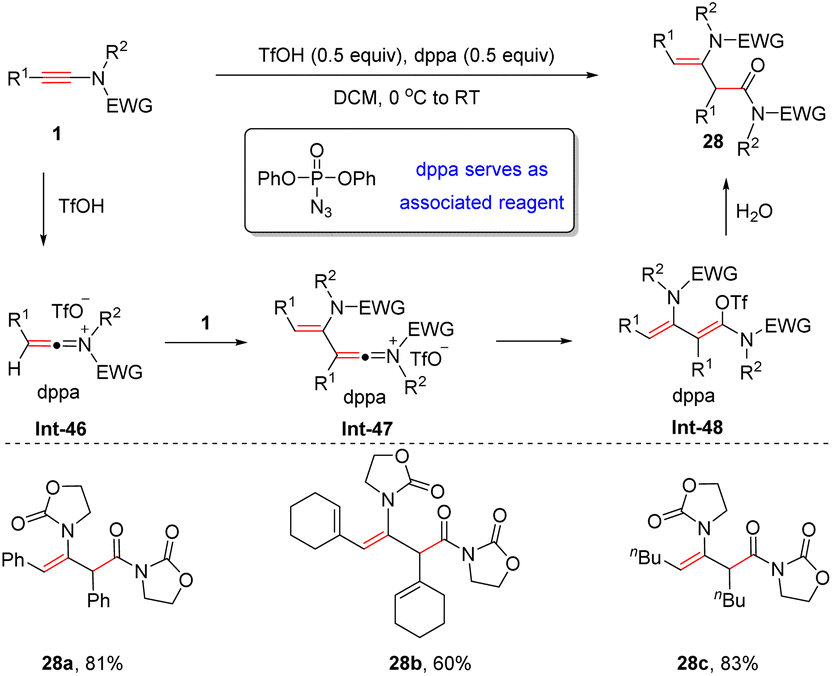
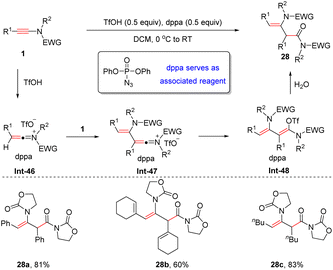
In 2016, Maulide and González et al. discovered that in the presence of TfOH and diphenylphosphoryl azide (dppa), ynamides can undergo hydrative dimerization to form syn-α-enamides (Scheme 23).50 The process commences with the formation of the keteniminium ion Int-46, which is captured by another ynamide to furnish enamide Int-47. A final hydrolysis furnishes the desired adduct. It should be noted that in the absence of dppa, only simple hydrolysis of the ynamide takes place and thus, dppa is crucial for this dimerization. DFT calculations indicate that dppa serves as an associated reagent, which can interact with the styrene-like transition state and keteniminium intermediate via π–π stacking. This interaction can lower the activation energy for the protonation and the energy of the transient species.
 |
| | Scheme 23 TfOH/dppa-catalyzed hydrative dimerization of ynamides. | |
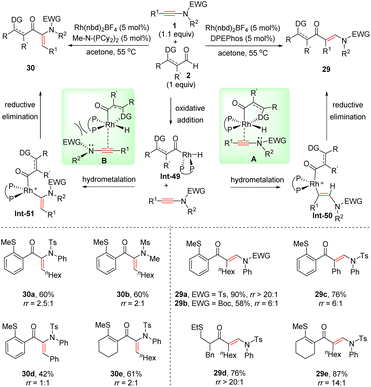
In 2017, Willis and co-workers established a fascinating regiodivergent syn-hydroacylation of ynamides with thioether-substituted aldehydes by rhodium catalysis (Scheme 24).51 In the presence of the electron-rich ligand Me-N-(PCy2)2 with a small bite angle, the Rh(nbd)2BF4-catalyzed coupling provided syn-α-adducts 30 as the major products, whereas the selectivity was diverted to syn-β-adducts 29 when using bis[(2-diphenylphosphino)phenyl] ether (DPEphos) as a ligand. The mechanism includes the oxidative addition of aldehydes with the Rh(I) catalyst, ynamide insertion into Rh–H to produce hydrometallated enamides(Int-50 or Int-51), and reductive elimination. Due to its π-acidity, DPEphos leads to an electron-poor rhodium centre, which is compensated by stronger coordination with ynamides (A). Because of this stronger interaction, as well as the steric repulsion between the EWG group and the ligand, the formation of β-hydrometallated enamides is more favourable. In contrast, strongly σ-donating ligand Me-N-(PCy2)2 increases the electron density on the rhodium centre, resulting in a more weak coordination with ynamides (B). Accordingly, in this case, α-hydrometallation of ynamides could also occur, eventually leading to syn-α-adducts 30.
 |
| | Scheme 24 Rhodium-catalyzed regiodivergent hydroacylation of ynamides with thioether-substituted aldehydes. | |
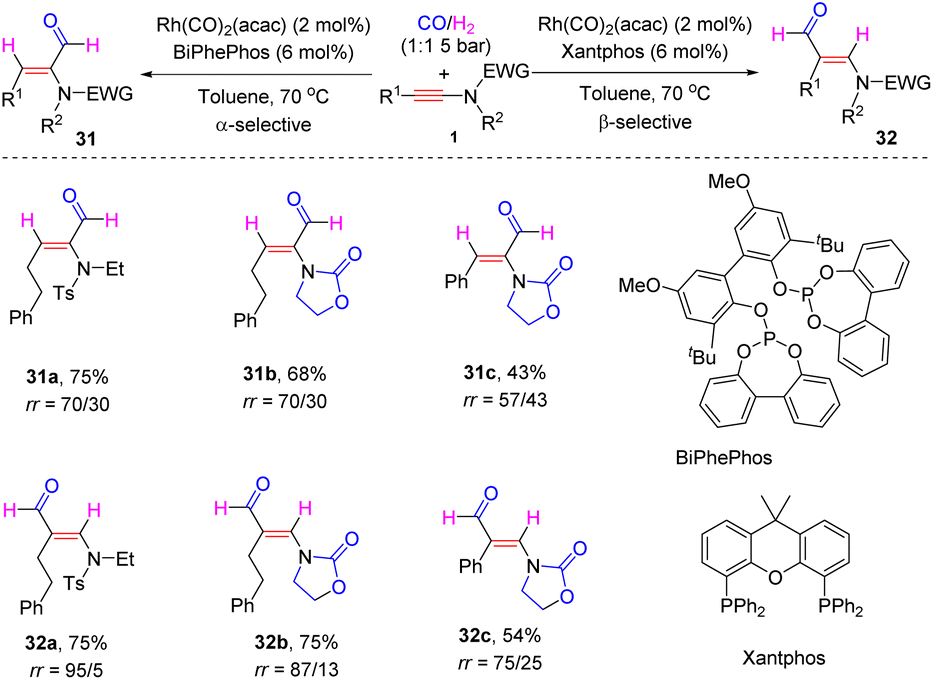
In 2019, Girard and Donnard et al. documented a rhodium-catalyzed ligand-controlled regiodivergent hydroformylation of ynamides (Scheme 25).52 When using the phosphite ligand BiPhePhos, syn-α-adducts 31 were produced with decent regioselectivities, whereas the employment of a Xantphos ligand altered the selectivity to syn-β-hydroformylation. The regioselectivity is controlled by the hydrometallation step via ynamide insertion into Rh–H. Compared with the phosphite ligand BiPhePhos, the phosphorus in the Xantphos ligand is more electron-rich with high σ-donating ability, which leads to weak coordination of the rhodium centre with ynamides. Thus, the polarity and steric hindrance of ynamides dominate the regioselectivity. The rhodium is preferentially incorporated at the electron rich and less hindered β-position. Subsequently, CO insertion, oxidative addition of H2 and reductive elimination deliver β-hydroformylated products 32. Conversely, the less σ-donating phosphite ligand BiPhePhos results in a decreased electron-density of the rhodium centre, but which can be compensated by the coordination of the EWG group in ynamides with rhodium. This chelation renders hydrometallation to preferably take place at the α-site of ynamides and α-hydroformyl enamides are finally obtained.
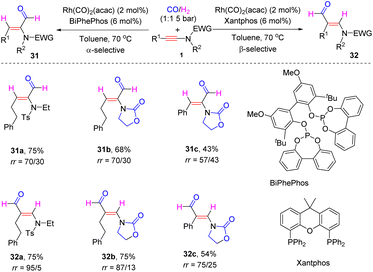 |
| | Scheme 25 Rhodium-catalyzed regiodivergent hydroformylation of ynamides with CO/H2. | |
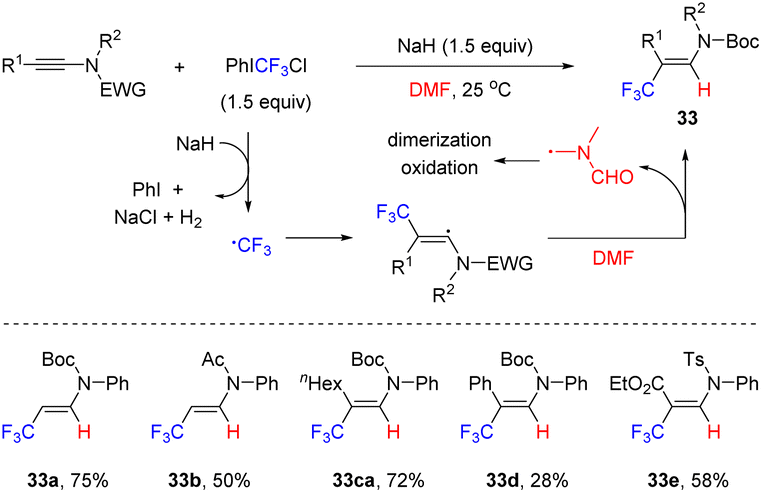
In 2022, Wang and co-workers reported an interesting radical hydrotrifluoromethylation of ynamides, which enabled a rapid access to β-CF3 enamides (Scheme 26).53 Using PhICF3Cl as the trifluoromethyl source and N,N-dimethylformamide (DMF) as the hydrogen donor, the transformation took place smoothly at room temperature in the presence of NaH. A wide variety of ynamides, regardless of their electronic properties, were all compatible with this approach. It is suggested that PhICF3Cl is first reduced by NaH to generate the trifluoromethyl radical by releasing iodobenzene, sodium chloride and hydrogen. Subsequently, the trifluoromethyl radical is captured by the electron-rich β-site of ynamide to obtain the β-CF3 alkenyl radical. Finally, the hydrogen atom of the N-methyl moiety in DMF is abstracted by the electrophilic trifluoromethyl radical to produce β-CF3 enamide. The formed DMF-based radical may undergo oxidation, dimerization or hydrolysis to afford complex mixtures.
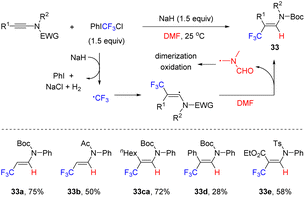 |
| | Scheme 26 NaH-promoted radical hydrotrifluoromethylation of ynamides with PhICF3Cl. | |
3. C–X bond formation
Haloenamides, owing to the weakly bonded halogen atom, are versatile building blocks in transition-metal-catalyzed cross-couplings for constructing new C–C and C-heteroatom bonds. Therefore, tremendous efforts have been devoted to the synthesis of such important frameworks over the past decades.54 The hydrohalogenation of ynamides, featuring high atom- and step-economy, is arguably the simplest and most straightforward strategy. On the basis of the addition mode to deliver regio- and stereoisomers, the adducts can be classified into three types: the syn-α-adduct, anti-α-adduct and anti-β-adduct, while the syn-β-adduct has not been reported yet. Thus far, a series of haloenamides (halogen atoms can be fluorine, chlorine, bromine, and iodine) has been synthesized by this strategy (Scheme 27).
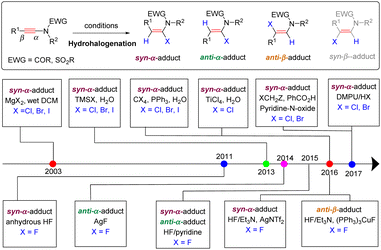 |
| | Scheme 27 An overview diagram for hydrohalogenation of ynamides. | |
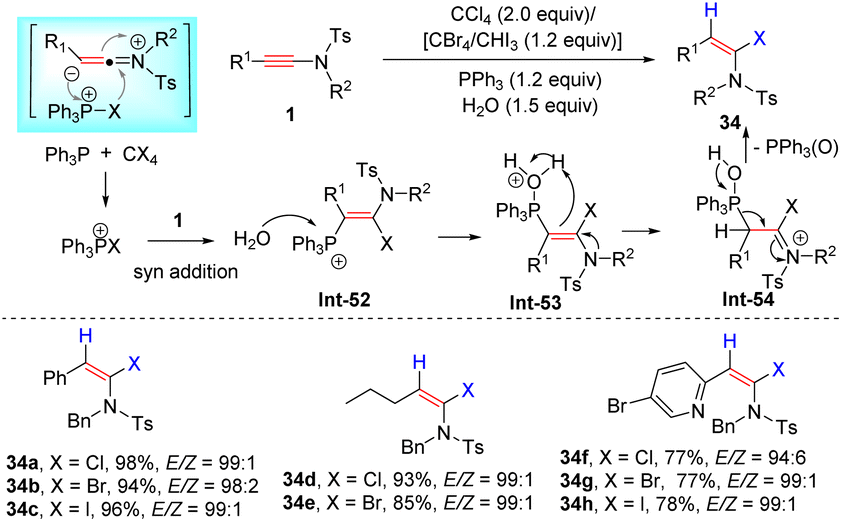
In 2003, Mulder and Hsung et al. uncovered the first syn-hydrohalogenation of ynamides to form α-iodo, -bromo, and -chloroenamides with high stereoselectivities (Scheme 27).55 The halogen source (HX) was in situ generated from a magnesium salt MgX2 and H2O in the reaction. Independently, using TMSX and H2O as the source, Iwasawa's group also accomplished this transformation (Scheme 27).56 In 2016, Matsuo and co-workers demonstrated that titanium tetrachloride was a suitable chlorine source, leading to syn-α-chloroenamides in good yields (Scheme 27).57 Alternatively, the halophosphonium salt, generated from PPh3 and CCl4/CBr4/CHI3, could undergo syn-α-addition with ynamides as well, followed by reaction with water and elimination of Ph3P(O) to give α-haloenamides in excellent regio- and stereoselectivities (Scheme 28).58
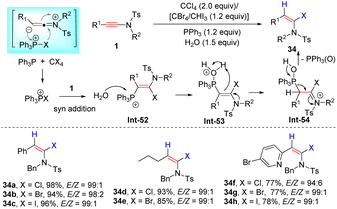 |
| | Scheme 28 The syn-α-addition of ynamides with the halogen source (HX generated from PPh3 and CCl4/CBr4/CHI3). | |
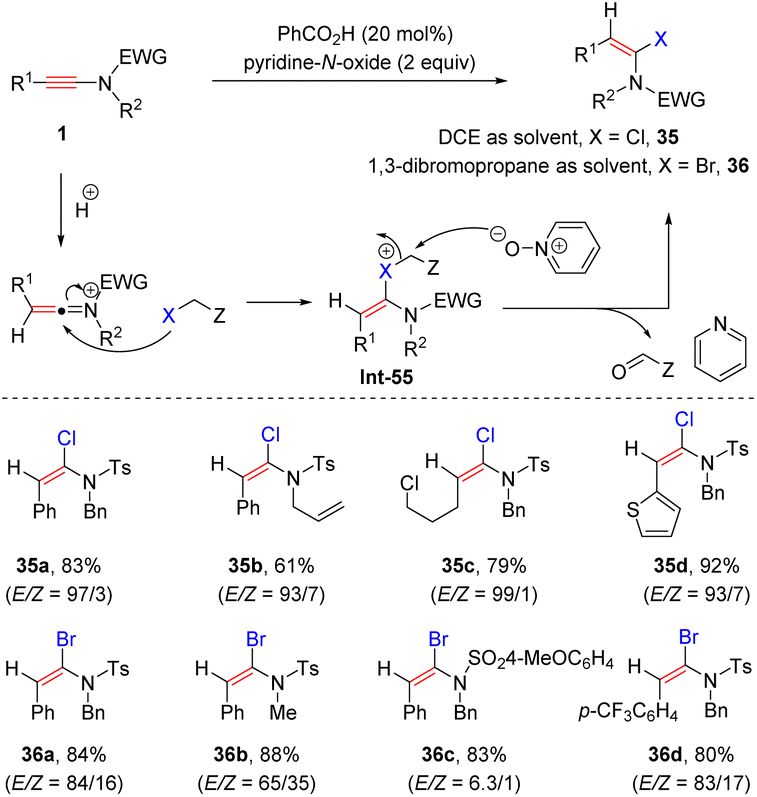
In 2017, Shin and co-workers discovered an acid-catalyzed pyridine-N-oxide promoted syn-α-hydrohalogenation of ynamides (Scheme 29).59 When the reaction was performed in 1,2-dichloroethane (DCE), syn-α-chloroenamides were afforded with high efficiency. The use of 1,3-dibromopropane as the solvent gave rise to syn-α-bromoenamides in decent stereoselectivities. The process commences with the formation of an active keteniminium ion, which is captured by the halogen atom of the solvent to generate the adduct Int-55. Subsequent attack by pyridine-N-oxide leads to the target product 35 with the release of pyridine and carbonyl compounds.
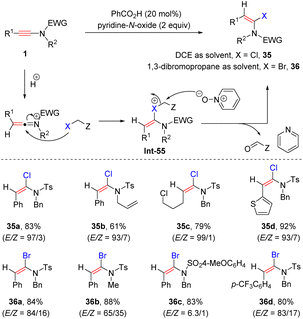 |
| | Scheme 29 Catalytic syn-α-addition of ynamides with 1,2-dichloroethane or 1,3-dibromopropane. | |
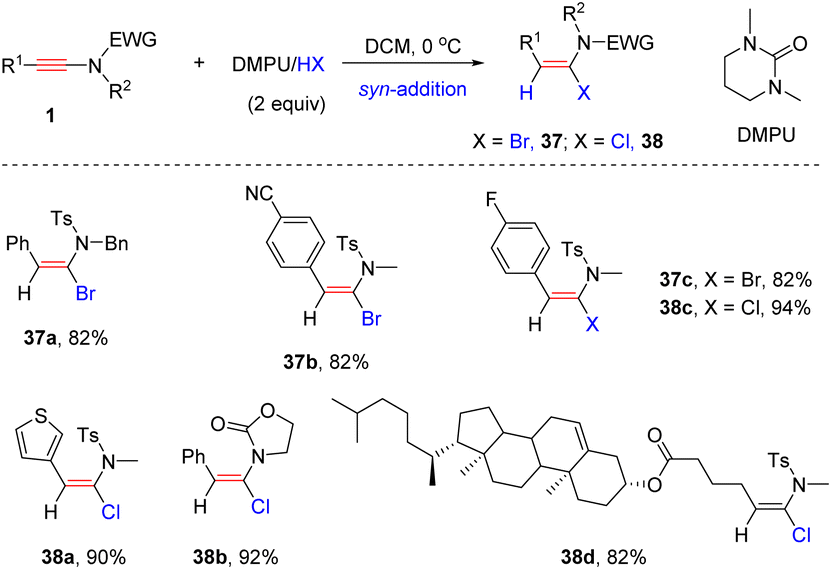
The keteniminium ion, generated by the protonation of the ynamide, is the key intermediate in the hydrohalogenation of the ynamide, and is easily captured by H2O to furnish the side product amide. To prevent this competitive hydrolysis pathway, Xu and Hammond et al. adopted a neutral aprotic medium (formed by mixing DMPU and HX) to accomplish the syn-α-hydrohalogenation of the ynamide (Scheme 30).60 The steric repulsion between the R1 group and the nucleophile (bromide/chloride anion) can account for the high stereoselectivity of the syn-addition.
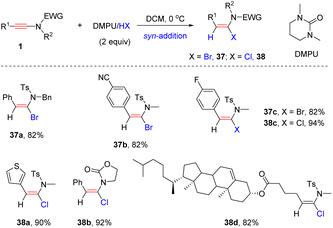 |
| | Scheme 30 The syn-α-hydrohalogenation of ynamide with a neutral aprotic medium (formed by mixing DMPU and HX). | |
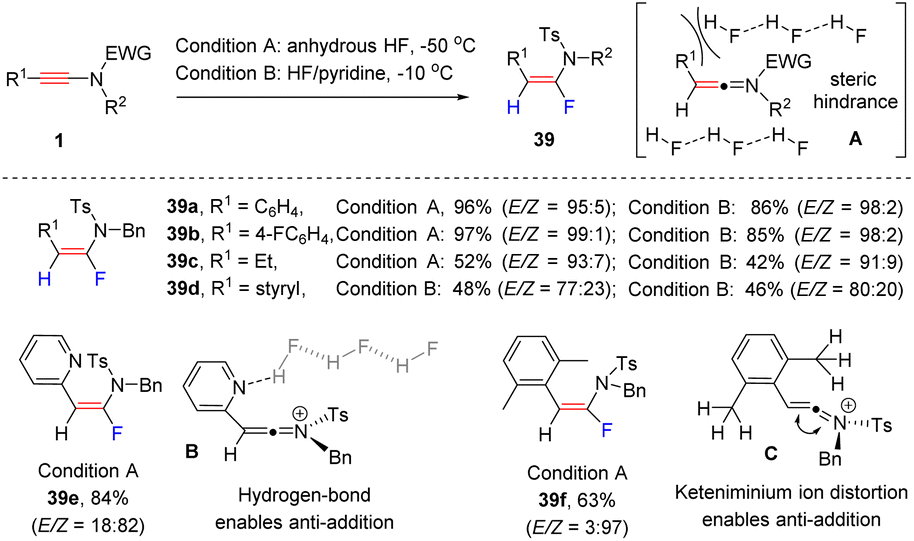
In 2012, Thibaudeau and Evano et al. explored the α-hydrofluorination of ynamides with HF (Scheme 31).61 Considering the safety issues in handling anhydrous HF, they also investigated a more user-friendly procedure (Scheme 29).62 When using HF/pyridine reagents, the reaction delivered syn-α-fluoroenamide as a major isomer. A broad range of aryl and alkyl substituents were compatible with the process. Surprisingly, the employment of a 2-pyridyl substituted ynamide resulted in anti-α-hydrofluorination (39e). The authors claimed that the hydrogen-bonding interaction between the pyridine ring and HF promoted the anti-addition of HF (B). For a xylyl-derived ynamide, the hyperconjugation distorted the keteniminium ion species (C), thus also enabling the anti-addition of HF (39f).
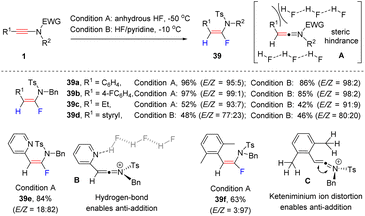 |
| | Scheme 31 The syn-α-hydrofluorination of ynamide with HF or HF/pyridine reagents. | |
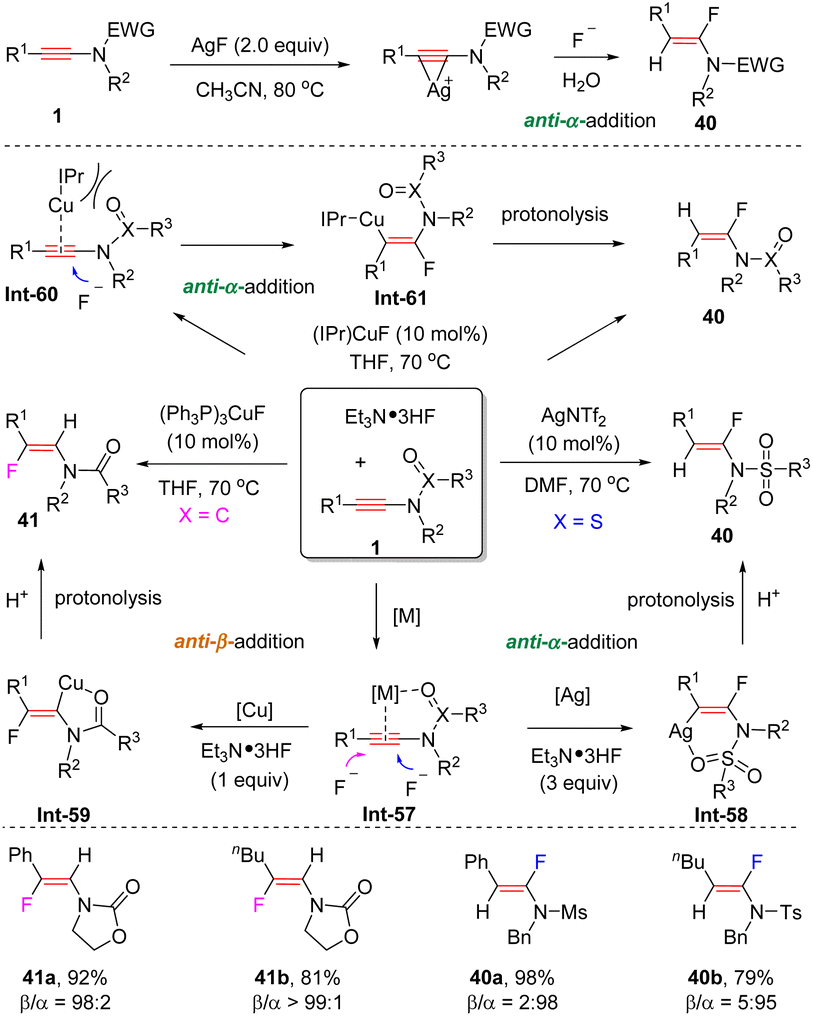
In 2014, Zhu and Zheng et al. unveiled a silver-promoted hydrofluorination of N-sulfonyl ynamides to deliver anti-α-adducts (Scheme 32).63 Ynamides initially coordinate with the sliver center via a three-membered ring, followed by the attack of a fluoride anion from the backside to afford the expected product. Later, it was found that when Et3N·HF was used as a partner, this reaction could be executed with a catalytic amount of AgNTf2 (Scheme 30).64 In comparison, by employing (PPh3)3CuF as a catalyst, the selectivity for hydrofluorination of oxazolidinone-derived ynamides was altered to anti-β-addition. The coordination between the carbonyl group of the ynamide and the copper center can account for this reversed regioselectivity. More importantly, the chemoselectivity could be switched by varying the ligand. With the catalyst (IPr)CuF, the reaction mainly produced anti-α-fluoroenamides 40.65 Because of the presence of the sterically hindered IPr ligand, the coordination between copper and the carbonyl oxygen is hampered. Thus, the nucleophilic fluorination preferentially takes place at the more electrophilic α-carbon.
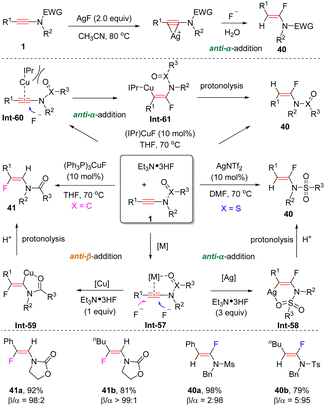 |
| | Scheme 32 The anti-hydrofluorination of ynamides. | |
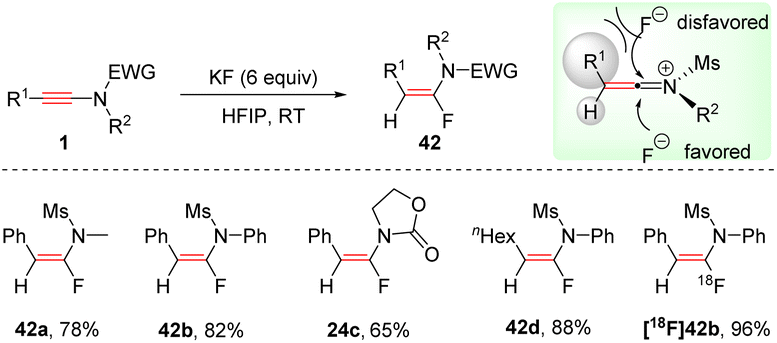
In 2018, Xu and Hammond et al. developed a mild alternative for the synthesis of α-fluoroenamides via a syn-hydrofluorination of ynamides with KF promoted by HFIP (hexafluoro-2-propanol) (Scheme 33).66 HFIP acts as a hydrogen-bond donor to interact with KF, which could enhance the nucleophilicity of the fluoride and increase the solubility of KF. The ynamide is activated by HFIP to afford a keteniminium ion. Then fluoride anion favourably attacks the more electrophilic α-carbon from the less hindered side. This protocol features mild reaction conditions, excellent regioselectivity, and a broad substrate scope. The steric and electronic effects of the substituents on the phenyl ring had a negligible impact on the outcome. Alkyl and natural product-derived ynamides also worked well. When available K18F was used as a substrate, and the corresponding radio fluoroenamides were readily obtained.
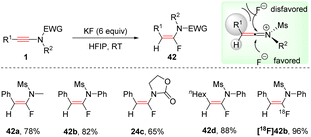 |
| | Scheme 33 The syn-hydrofluorination of ynamides with KF promoted by HFIP. | |
4. C–O bond formation
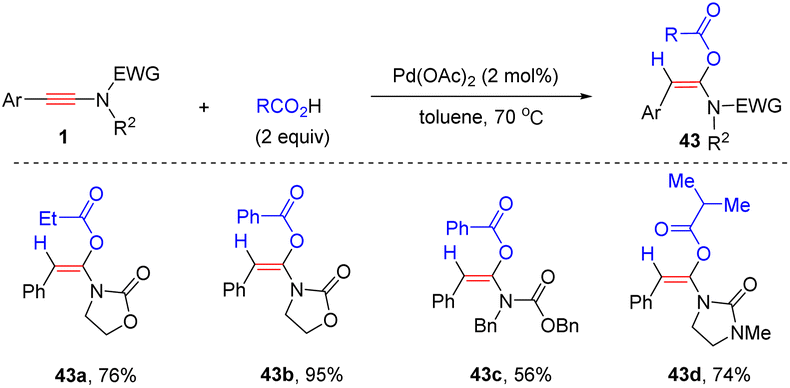
In 2012, Lam's group documented a palladium-catalyzed regio- and stereoselective syn-α-hydroacyloxylation of oxazolidinone-derived ynamides with carboxylic acids (Scheme 34).67 Only aryl substituents at the terminus of ynamides were amenable for this conversion. A wide variety of α-acyloxyenamides were prepared via this strategy, but the electron-donating group OMe on the phenyl ring of R1 gave slightly decreased yields. Alkyl and aryl carboxylic acids were suitable for the transformation.
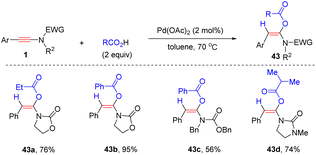 |
| | Scheme 34 Pd-catalyzed syn-α-hydroacyloxylation of oxazolidinone-derived ynamides with carboxylic acids. | |
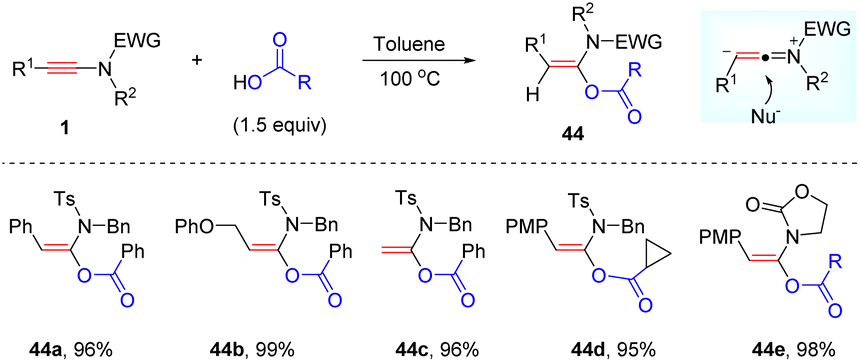
Interestingly, Bi and Hu et al. presented that the addition between ynamides and carboxylic acids could also take place efficiently without any catalyst at elevated temperatures, leading to the desired syn-α-adducts in excellent yields and selectivities (Scheme 35).68 Both N-sulfonyl ynamides and oxazolidinone-derived ynamides were amenable for the process. Besides, a wide array of carboxylic acids were tolerated as well. This protocol features metal-free conditions, high efficiency, broad functional group tolerance and simple operation.
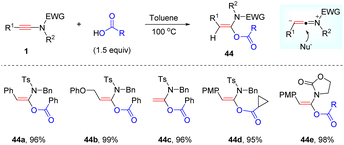 |
| | Scheme 35 Metal-free syn-α-hydroacyloxylation of ynamides with carboxylic acids. | |
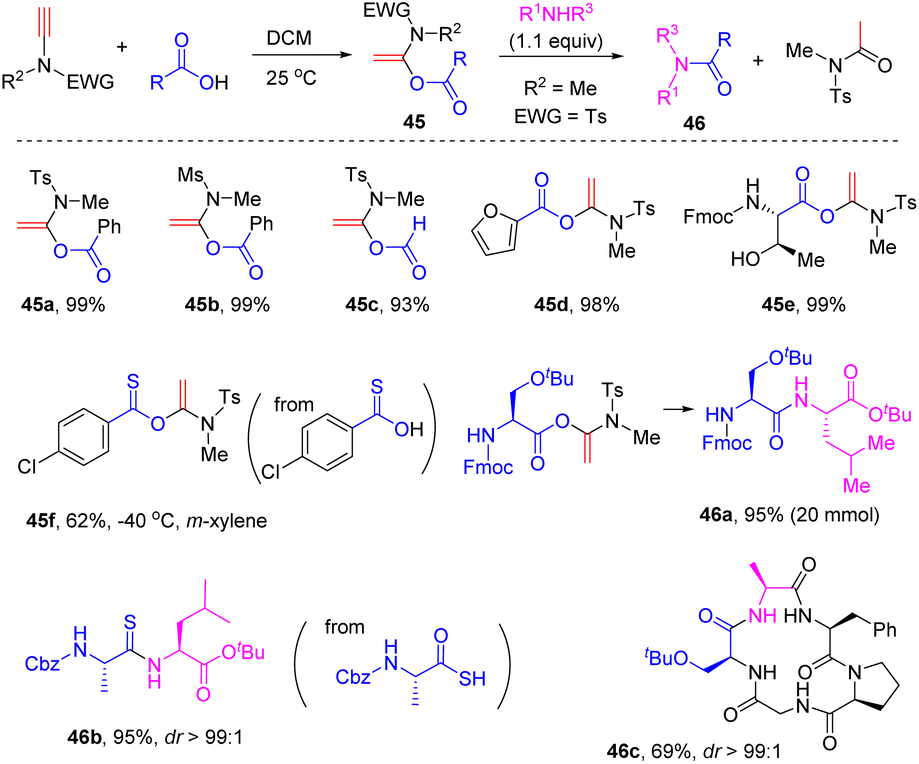
In 2016, Zhao and his colleagues found that the nature of the solvent exerted an impact on the hydroacyloxylation of ynamides (Scheme 36).69 When the reaction was conducted in dichloromethane, α-acyloxyenamides could be obtained with high efficiency just at room temperature. Remarkably, the resulting α-acyloxyenamides could undergo coupling with amines to form amides. This strategy was successfully applied in the construction of diverse peptides with the total preservation of the chiral integrity. It is worth mentioning that it is not necessary to isolate the α-acyloxyenamide intermediates in the synthesis of peptides and thus terminal ynamides can be regarded as an excellent peptide coupling reagent. By employing this elegant strategy, Zhao's group has accomplished the efficient synthesis of polypeptides,70–72 thionoesters,73 peptide thioesters,74 thioamides,75 macrolactones,76 and macrocyclic peptides.77
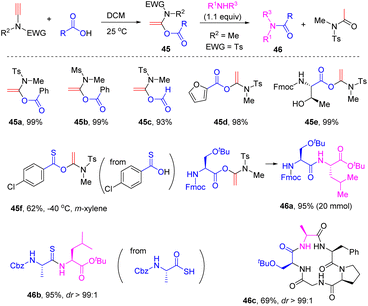 |
| | Scheme 36 The syn-α-hydroacyloxylation of terminal ynamides with carboxylic acids and applications in the construction of diverse peptides. | |
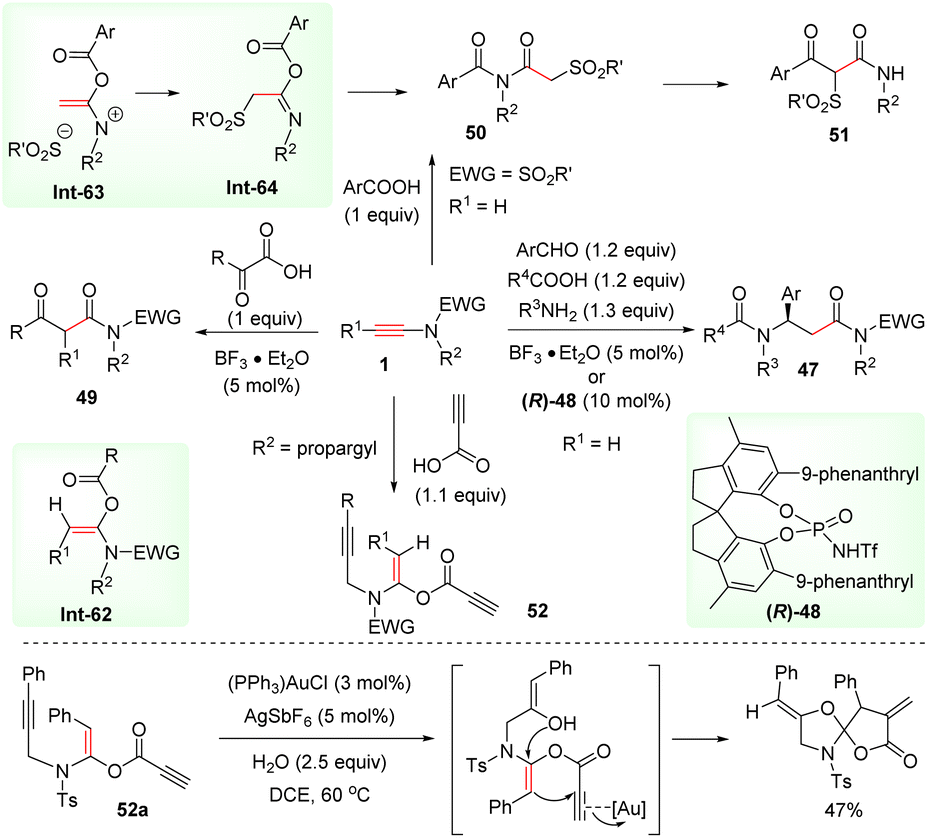
Meanwhile, by adopting the ynamide hydroacyloxylation strategy, Cui and co-workers realized the rapid assembly of β-amino amides 47via BF3·Et2O-catalyzed four-component coupling of terminal ynamides, carboxylic acids, aldehydes, and amines (Scheme 37).78,79 This process includes syn-α-hydroacyloxylation of the ynamide, enamide addition to the imine intermediate, and the Mumm rearrangement. More recently, Tan's group accomplished an asymmetric version by employing the chiral strong Brønsted acid N-triflylphosphoramide (R)-48 as the catalyst.80 Cui et al. disclosed that α-keto acids could also react with ynamides to produce β-keto amides 49via releasing carbon monoxide.81 In addition, aromatic carboxylic acids could be coupled with ynamides to form α-acyloxyenamides Int-63, followed by sulfonyl migration and the Mumm rearrangement to yield the imides 50.82 A further treatment with a strong base could give rise to β-keto amides 51via acyl migration. The group of Sahoo prepared useful building block enediynes 52via the syn-α-addition of propiolic acids with ynamides.83 With the aid of a gold catalyst, enediynes 52 and water underwent cascade addition and intramolecular cyclization to afford spiro-heterocycles.
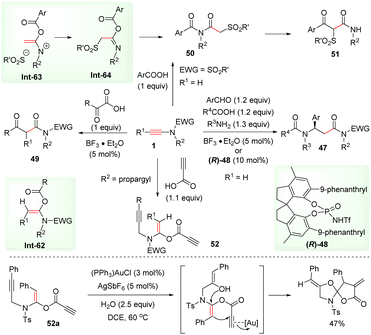 |
| | Scheme 37 The ynamide hydroacyloxylation strategy in a series of elegant synthetic transformations. | |
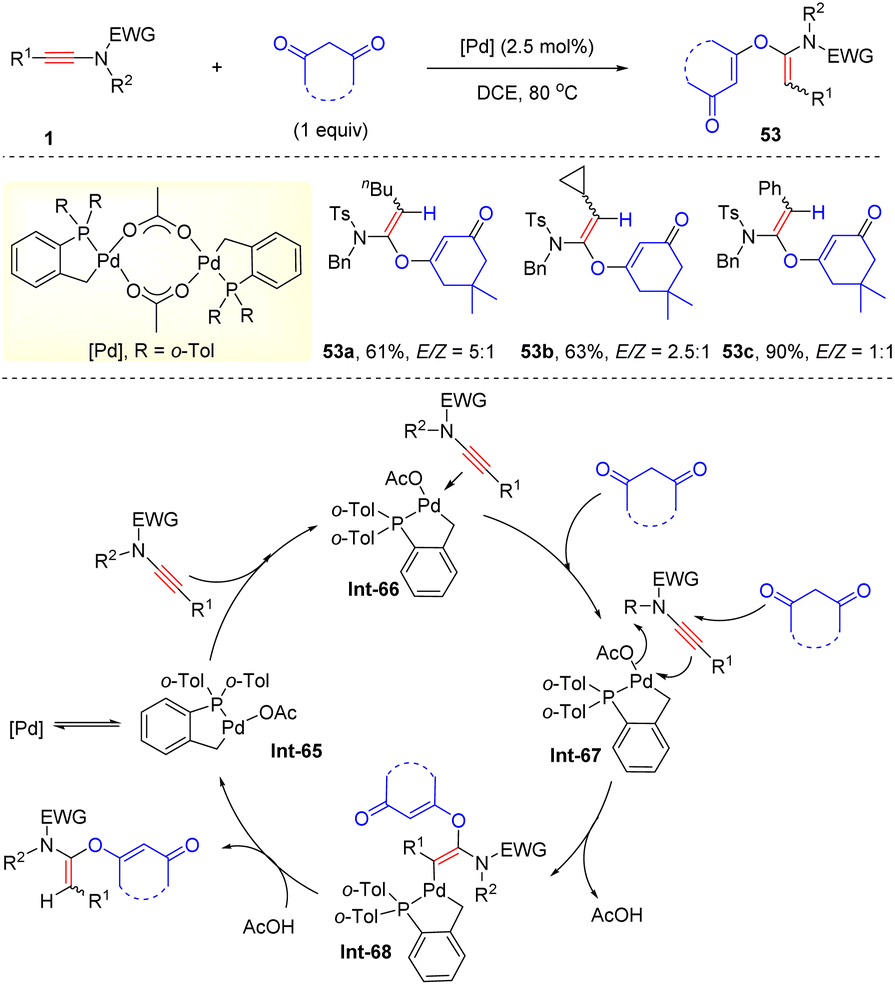
In 2014, Clavier and co-workers described that with the bulky phosphapalladacycle complex as the catalyst, ynamides could undergo hydroalkoxylation with 1,3-diones, delivering α-alkoxy enamides 53 with a moderate stereoselectivity (Scheme 38).84 Mechanistically, the ynamide is initially activated by the palladium catalyst, followed by the nucleophilic attack of 1,3-dione at the more electrophilic α-position of the carbon–carbon triple bond and protonolysis to afford the desired products.
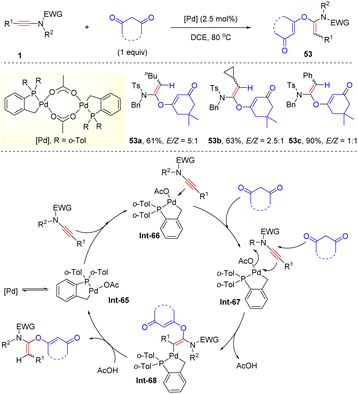 |
| | Scheme 38 Pd-catalyzed hydroalkoxylation of ynamides with 1,3-diones. | |
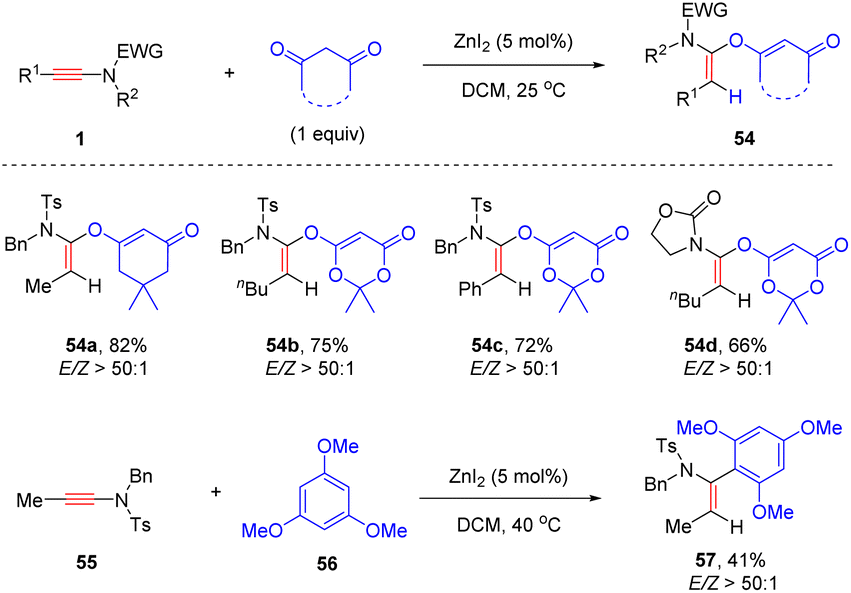
Later, the same group showed that when the Lewis acid ZnI2 was employed as the catalyst, hydroalkoxylation of internal ynamides with 1,3-diones could occur readily at room temperature and α-alkoxy enamides were afforded with high yields and selectivities (Scheme 39).85 Aryl and alkyl-substituted ynamides were well tolerated in the process. Oxacyclic 1,3-dione was a suitable coupling partner as well. Besides, this protocol was successfully extended to the selective addition of electron-rich trimethoxybenzene with ynamides.
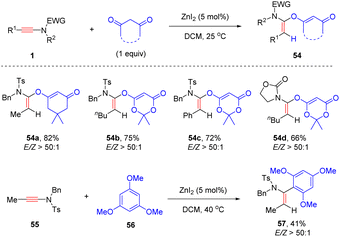 |
| | Scheme 39 ZnI2-catalyzed hydroalkoxylation of ynamides with 1,3-diones. | |
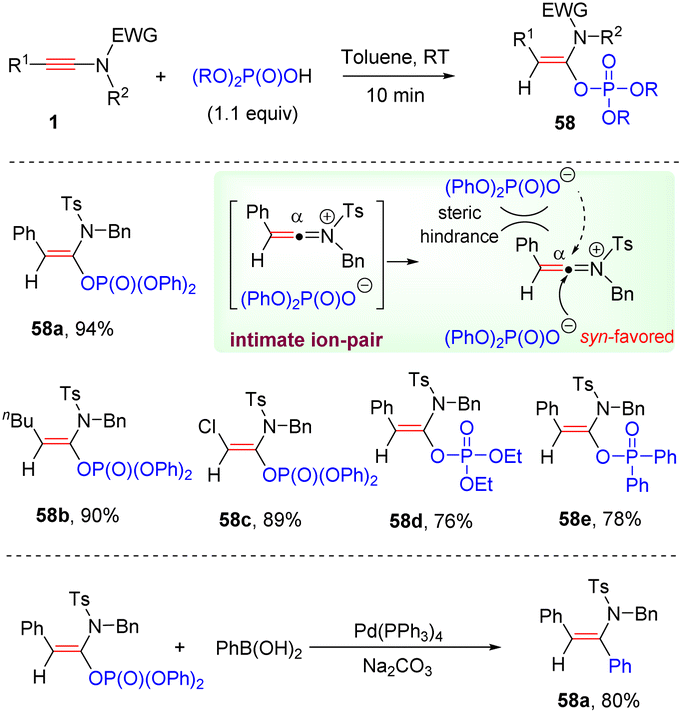
In 2021, Zhao and her colleagues documented a metal-free syn-hydrophosphoryloxylation of ynamides with phosphoric acids, resulting in the formation of a C–O bond at the α-site of ynamides (Scheme 40).86 This methodology features exclusive regio- and stereoselectivity, broad substrate scope, mild reaction conditions, and very short reaction time (10 min). The resulting enol phosphates could serve as effective building blocks in the Suzuki-type cross-coupling. The process is believed to proceed through the formation of an intimate ion-pair between the keteniminium ion and the phosphate anion. Subsequent nucleophilic addition favors syn-attack from the small H side to yield the target products.
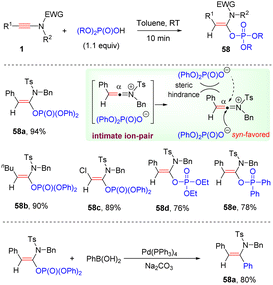 |
| | Scheme 40 Metal-free syn-hydrophosphoryloxylation of ynamides with phosphoric acids. | |
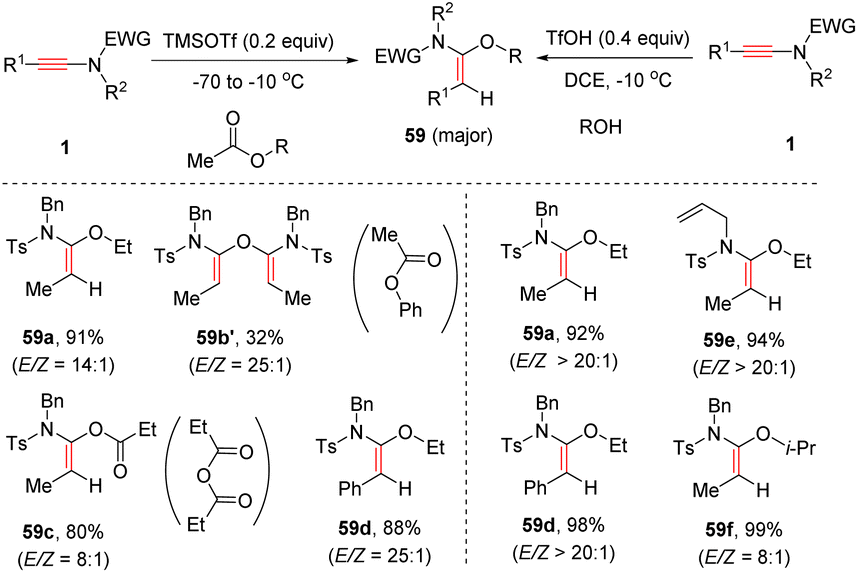
Chang and Wang et al. developed two acid systems to promote syn-α-hydroalkoxylation of ynamides (Scheme 41). In 2021, with the aid of TMSOTf, N-sulfonyl ynamides could couple with esters to furnish diverse α-alkoxy enamides with a decent stereoselectivity.87 Besides esters, anhydrides were suitable partners as well, albeit giving a decreased yield and selectivity. However, when phenyl acetate was used as a substrate, no desired product was observed but enamide 59b′ was afforded as a major product. Later, it was found that in the presence of TfOH, the addition between ynamides and simple alcohols proceeded efficiently, exclusively leading to the formation of (E)-α-alkoxy enamides 59.88
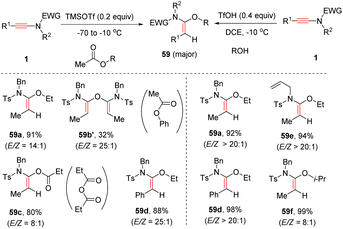 |
| | Scheme 41 Catalytic syn-α-hydroalkoxylation of ynamides. | |
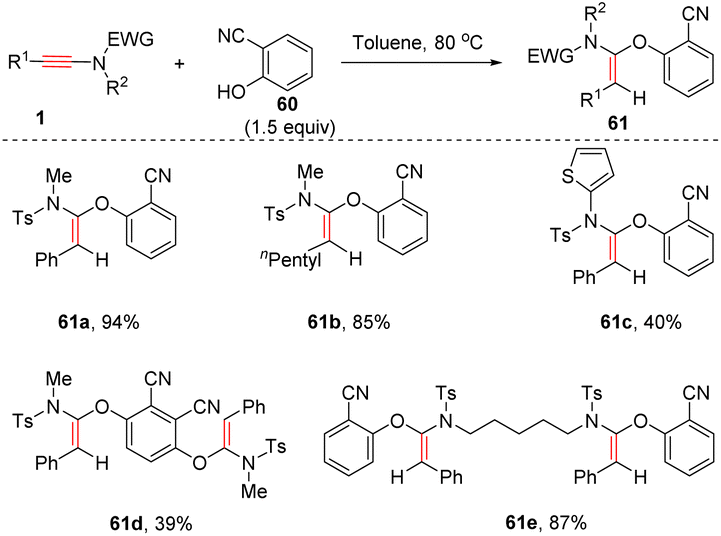
Very recently, Zeng, Yi and Zhou et al. developed a stereospecific syn-α-addition of phenols with ynamides (Scheme 42).89 The acidity of phenols proved to be crucial for this transformation. Electron-deficient 2-cyanophenol and their derivatives underwent this transformation smoothly to yield (E)-α-phenoxy enamides 61 as single products. The substituents on ynamides had a minimal effect on the outcome. Besides, pentafluorophenol was a suitable substrate as well.
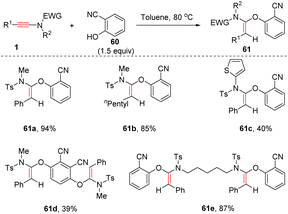 |
| | Scheme 42 The syn-α-hydrophenoxylation of ynamides with acidic phenols. | |
5. C–N bond formation
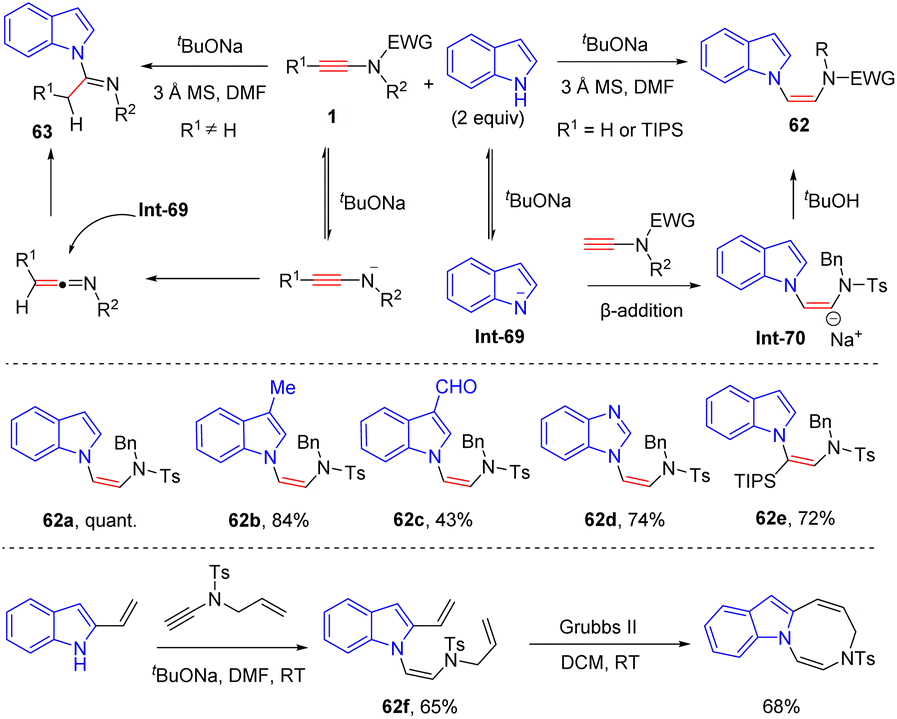
In 2014, Dodd and co-workers unveiled an interesting metal-free N-addition of indoles to the β-position of ynamides in the presence of tBuONa (Scheme 43).90 Terminal N-tosyl ynamides were tolerated in this procedure, providing vinyldiamines 62 in good yields and selectivities. In contrast, in the case of N-carboxyl ynamides, carboxyl was a good leaving group under these basic conditions and the resulting ketenimine was then trapped by the nitrogen atom of indole to form amidines. This mechanism is supported by density functional theory (DFT) calculations. It is to be noted that the adduct of N-allyl ynamide and 2-vinyl indole could further undergo intramolecular alkene metathesis to construct a seven-membered heterocycle.
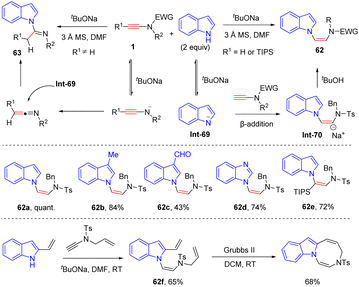 |
| | Scheme 43 Metal-free N-addition of indoles to the β-position of ynamides in the presence of tBuONa. | |
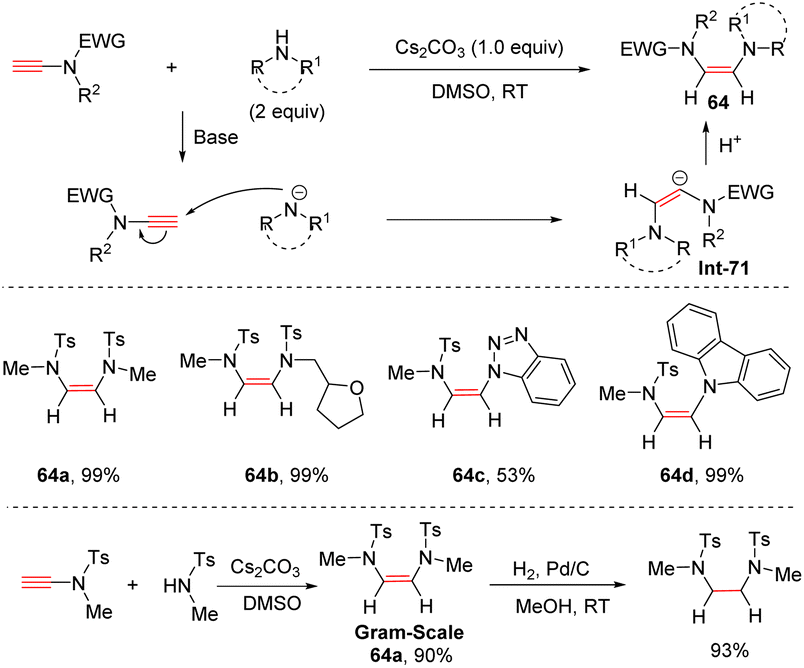
Later, following a similar strategy, using Cs2CO3 as a base, Zhao's group realized anti-hydroamination of ynamides with simple secondary amines, resulting in the formation of a C–N bond at the β-site with high regio- and stereoselectivities (Scheme 44).91 Aza-heterocycles including benzotriazole, indole and carbazole were all tolerated in this transformation. Over Pd/C catalyst, the resulting adducts could be easily hydrogenated to saturated diamines.
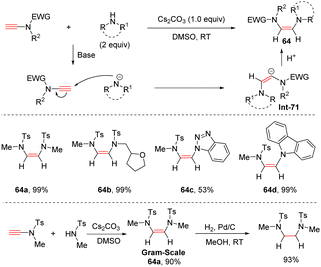 |
| | Scheme 44 The anti-hydroamination of ynamides with simple secondary amines. | |
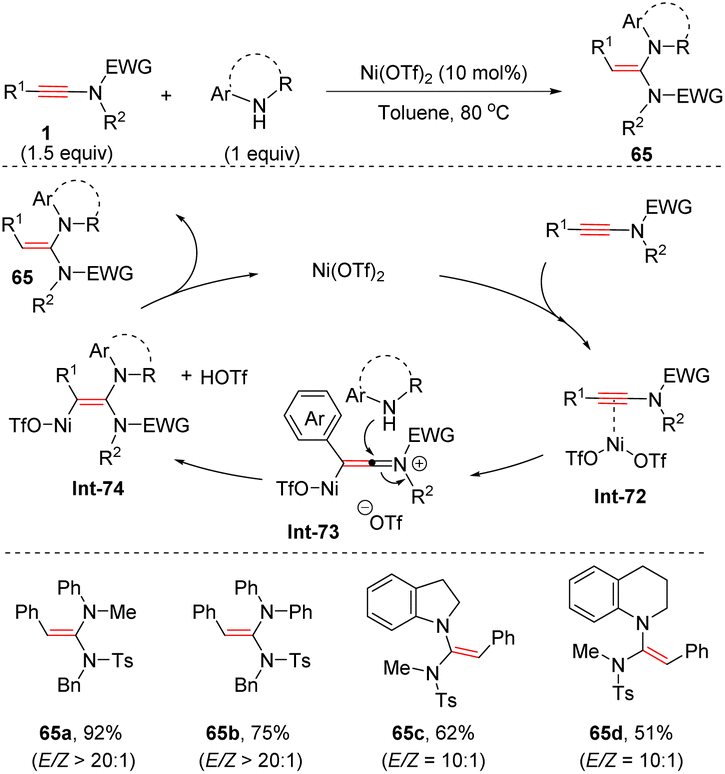
In contrast, Wei and Si et al. found that with the assistance of Ni(OTf)2, the coupling of aryl-derived ynamides with secondary amines showed a distinct regioselectivity from base-mediated addition, providing anti-α-adducts in appreciable yields and selectivities (Scheme 45).92 The ynamide is initially activated by Ni(OTf)2 to form the vinyl Ni(II) intermediate Int-73, followed by the nucleophilic attack of the amine at the more electrophilic α-site from the back side. A final protonolysis afforded the desired products with the regeneration of Ni(OTf)2 to close the catalytic cycle. A series of anilines and bicyclic secondary amines were applicable to the transformation.
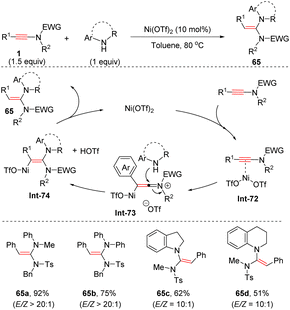 |
| | Scheme 45 Ni-catalyzed anti-α-hydroamination of ynamides with secondary amines. | |
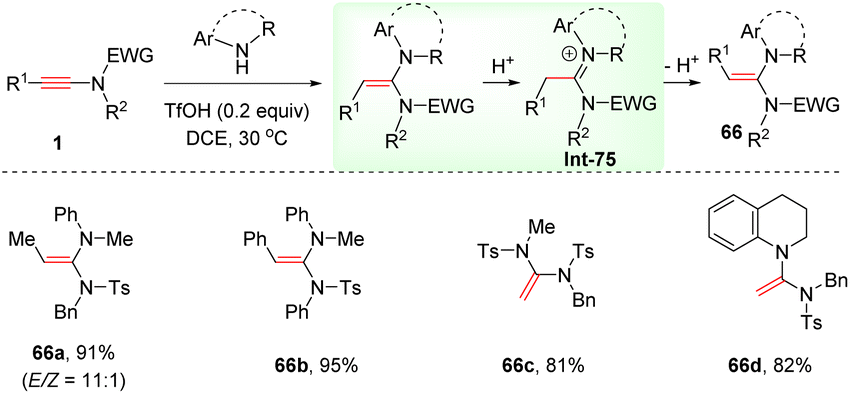
Interestingly, this anti-α-hydroamination of ynamides with secondary amines could also take place in the presence of the strong Brønsted acid TfOH (Scheme 46).93 Both alkyl and aryl substituents on the alkyne terminus were amenable for the conversion, delivering (E)-ethene-1,1-diamines 66 in moderate to good stereoselectivities. It is suggested that an initial syn-α-addition delivers (Z)-ethene-1,1-diamines, which are prone to undergo isomerization to produce thermodynamically stable (E)-ethene-1,1-diamines via the iminium ion intermediate Int-75.
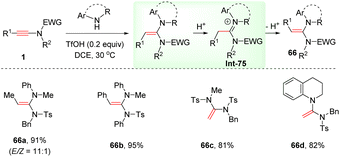 |
| | Scheme 46 TfOH-mediated anti-α-hydroamination of ynamides with secondary amines. | |
In 2016, Chen and co-workers developed a gold-catalyzed hydrosulfoximination of ynamides with NH-sulfoximines, producing a mixture of (E)- and (Z)-α-sulfoximidoyl enamides (Scheme 47).94 With the aid of [Ru(p-cymene)Cl2]2, this isomer mixture could be oxidized by H2O2 to furnish a urea-type sulfoximine via cleavage of the carbon–carbon double bond. The process includes the formation of the reactive keteniminium gold species Int-76, nucleophilic attack of sulfoximine and protodeauration.
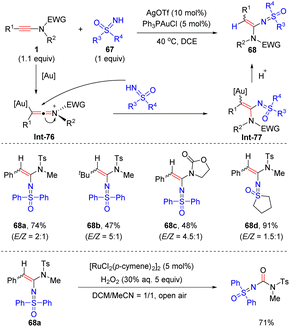 |
| | Scheme 47 Au-catalyzed hydrosulfoximination of ynamides with NH-sulfoximines. | |
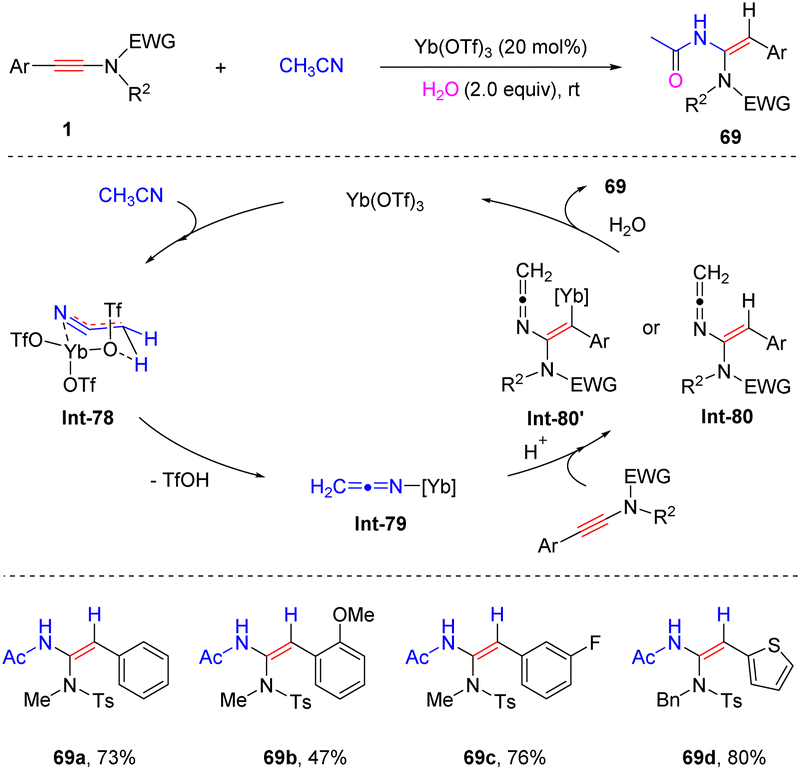
In 2017, Sahoo and his colleagues documented a Yb(OTf)3-catalyzed hydroamidation of aryl-ynamides with CH3CN in the presence of H2O (Scheme 48).95 The electron-withdrawing group on the phenyl and 3-thienyl-derived ynamides were compatible with the procedure, while the substrate with an electron-donating OMe group gave an inferior result. Initially, acetonitrile is activated by Yb(OTf)3 to form the keteniminium-metal intermediate Int-78via a six-membered ring transition state. Subsequently, syn-addition to the ynamide and protonolysis lead to α-hydroamidation products with high stereoselectivities.
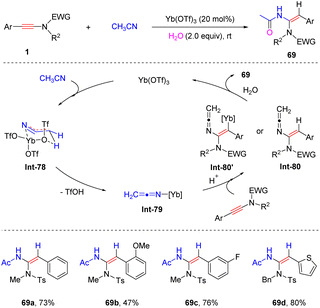 |
| | Scheme 48 Yb(OTf)3-catalyzed hydroamidation of aryl-ynamides with CH3CN. | |
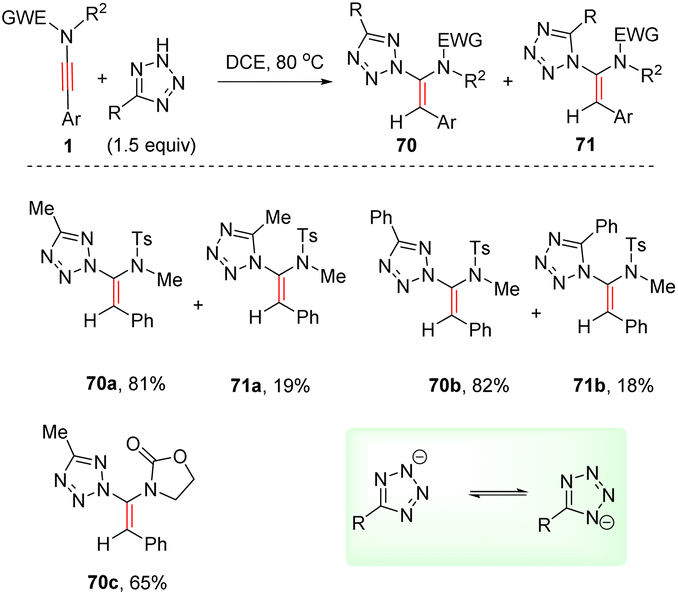
In 2019, Cao and Meng et al. presented a metal-free syn-hydroheteroarylation of ynamides with 2H-tetrazoles to construct the C–N bond at the α-site of ynamides (Scheme 49).96 Owing to the isomerization of 2H-tetrazole anion, both N1 and N2 could serve as nucleophiles in most cases and two α-adducts 70 and 71 were afforded. Only aryl substituents at the terminus were applicable to the transformation. The steric hindrance between terminal substituent on the alkyne and tetrazole anion accounts for the high regio and stereoselectivity.
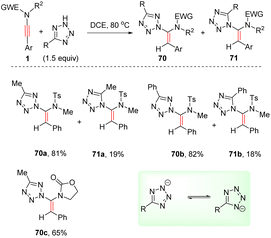 |
| | Scheme 49 Metal-free syn-hydroheteroarylation of ynamides with 2H-tetrazoles. | |
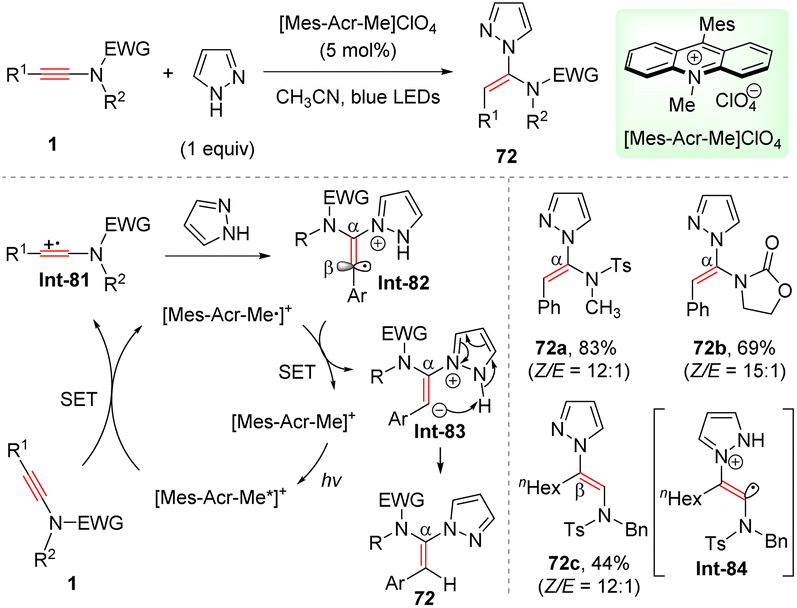
Recently, Deng and co-workers established a mild syn-α-hydroamination of ynamides with azoles enabled by visible light catalysis (Scheme 50).97 Visible light activation of photocatalyst [Mes-Acr-Me]ClO4 (9-mesityl-10-methylacridinium perchlorate) produces its excited state [Mes-Acr-Me*]+. Subsequent reductive quenching by ynamides leads to ynamide cation radical Int-81, which is trapped by pyrazole to afford α-amino vinyl radical Int-82. Then, single electron transfer (SET) from reduced photocatalyst [Mes-Acr-Me˙]+ to vinyl radical delivers α-amino vinyl anion Int-83 and regenerates photocatalyst. A final intramolecular proton transfer furnishes the target product. For aryl-derived ynamides, ynamide cation radical is favourably trapped at α-site, as the resulting vinyl radical could be stabilized by the aromatic resonance. In comparison, in the case of alkyl-substituted ynamide, sulfonamide unit would stabilize the vinyl radical Int-84, thus preferentially leading to β-adducts. Besides, 1H- and 2H-tetrazole could participate in this useful transformation as well.
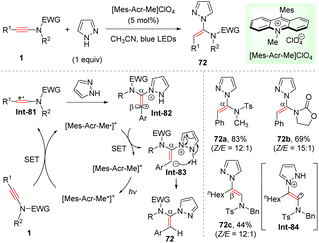 |
| | Scheme 50 Photocatalytic syn-α-hydroamination of ynamides with azoles. | |
6. C–P bond formation
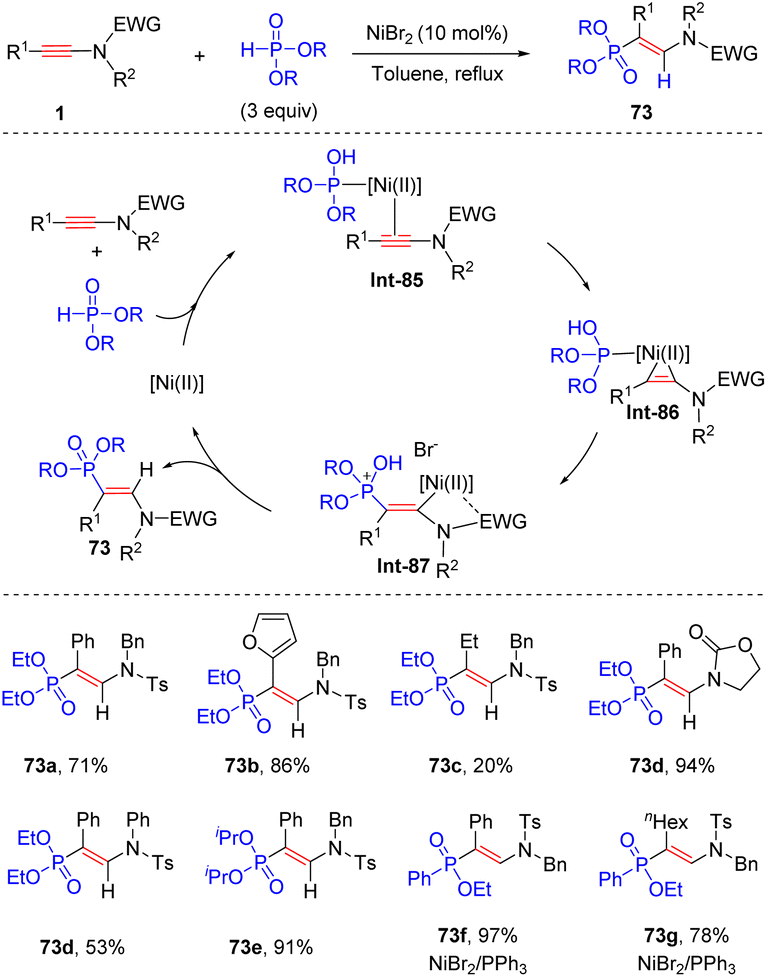
In 2011, Rabasso and Evano et al. disclosed an intriguing nickel-catalyzed syn-β-hydrophosphinylation of ynamides with dialkyl phosphonate, delivering vinylphosphonates with high regio- and stereoselectivity (Scheme 51).98 An array of aryl-derived ynamides worked well in the process, while alkyl substituents at the alkyne terminus gave modest yields. The alkyl groups in phosphonate had little impact on the outcome. P-Tautomerization of phosphonate forms hydrogen phosphite and its lone pair electrons can bind with NiBr2, followed by coordination with ynamide to deliver a three-membered vinylnickel specie Int-86. The coordination between electron-withdrawing group on the nitrogen atom and nickel facilitates a β-selective addition of hydrogen phosphite. Finally, P-isomerization and protonolysis lead to the expected product. It is noteworthy that a combination of NiBr2 and PPh3 turned out to be a better catalytic system for the addition of ynamides with phenylphosphinates.99
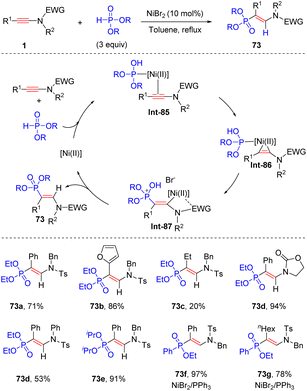 |
| | Scheme 51 Ni-catalyzed syn-β-hydrophosphinylation of ynamides with dialkyl phosphonate. | |
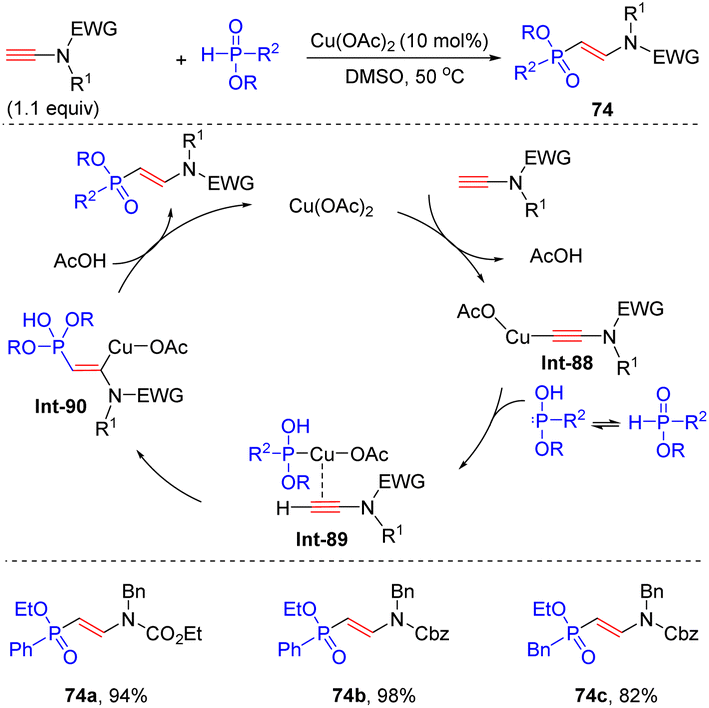
In 2017, Yamagishi's group reported that aided by Cu(OAc)2, phosphinates could react with terminal N-ester ynamides to prepare vinylphosphinates in high yields via β-hydrophosphinylation (Scheme 52).100 In this case, copper ynamide complex Int-88 is first formed via the reaction of copper catalyst with terminal ynamide. P-Tautomerization of phosphinate produces hydrogen phosphite, which binds with copper catalyst and further activates ynamides via π-coordination. Subsequent syn-β-addition and protonolysis give rise to the target products. A wide variety of H-phosphinates were all well applicable to the transformation. It is worthwhile to mention that P-chiral H-phosphinate was a suitable substrate as well and no erosion of enantioselectivity was observed in the process.
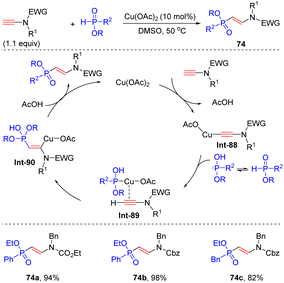 |
| | Scheme 52 Cu-catalyzed syn-β-hydrophosphinylation of ynamides with H-phosphinates. | |
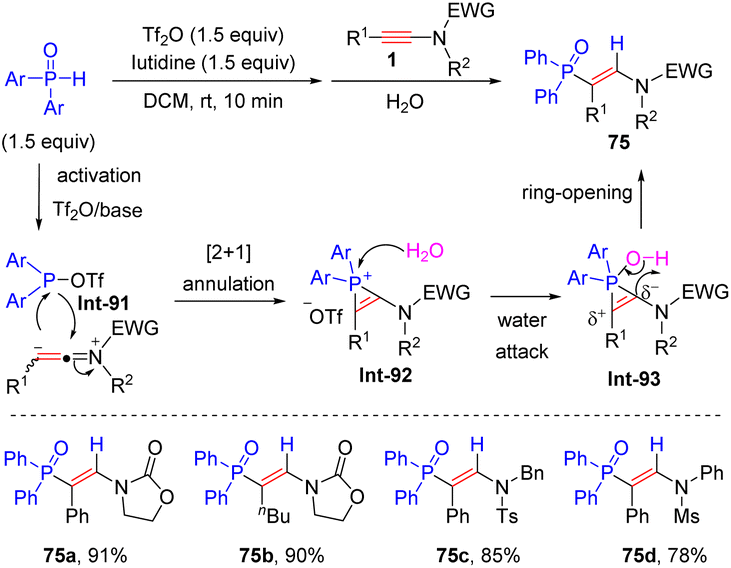
In 2018, Kang and his colleagues developed a metal-free strategy to realize regio- and stereoselective hydrophosphorylation of ynamides with diarylphosphine oxides (Scheme 53).101 This methodology tolerated a wide array of alkyl and aryl groups in R1 and R2 of ynamides. In most cases, the electronic factors of substituents on diarylphosphine oxides had a negligible impact on the results, whereas sterically hindered ortho-substituted diarylphosphine oxide was inapplicable. An electrophilic sulfonyloxy phosphane Int-91 is first generated from the reaction of diarylphosphine oxides and Tf2O in the presence of organic base lutidine. Subsequent [2 + 1] annulation with ynamide furnishes phosphirenium cation Int-92. Finally, water-assisted ring-opening of the three-membered ring delivers syn-β-adducts.
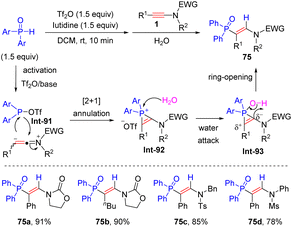 |
| | Scheme 53 Metal-free hydrophosphorylation of ynamides with diarylphosphine oxides. | |
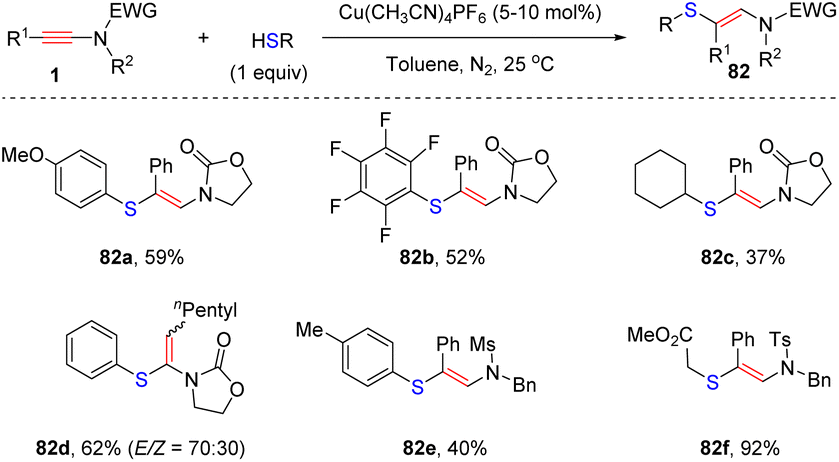
7. C–S bond formation
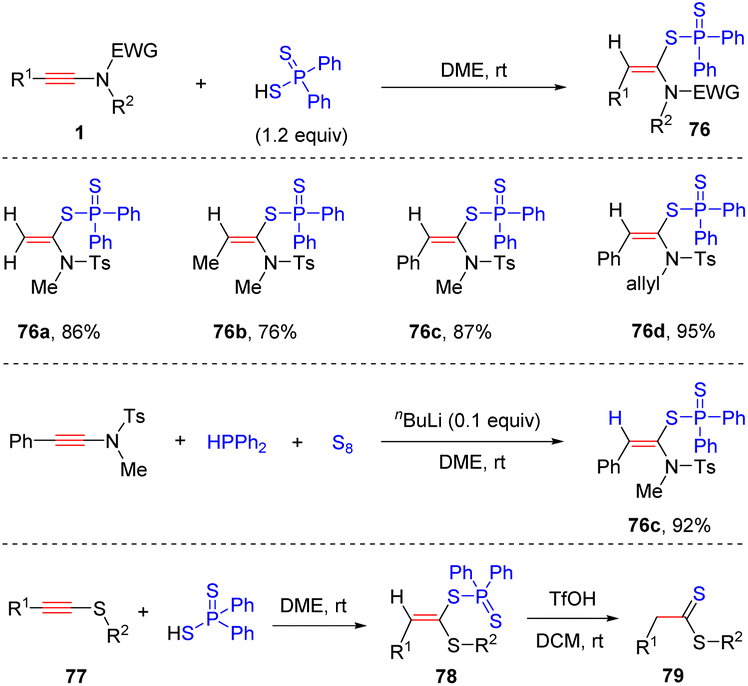
In 2008, Oshima and Yorimitsu et al. reported an intriguing syn-hydrothiolation of ynamides with diphenyldithiophosphinic acids, delivering α-adducts ketene N,S-acetal derivatives with high regio- and stereoselectivity (Scheme 54).102 A variety of aryl and alkyl groups on the terminus of ynamides were tolerated. When using diphenylphosphine and sulfur as substrates, the desired products 76c could also be obtained with a catalytic amount of nBuLi.103 Subsequently, they successfully expanded the scope to thioalkynes 77 and the resulting ketene S,S-acetals could be converted to dithioate in the presence of trifluoroacetic acid.
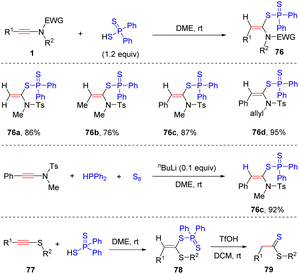 |
| | Scheme 54 Metal-free syn-hydrothiolation of ynamides with diphenyldithiophosphinic acids. | |
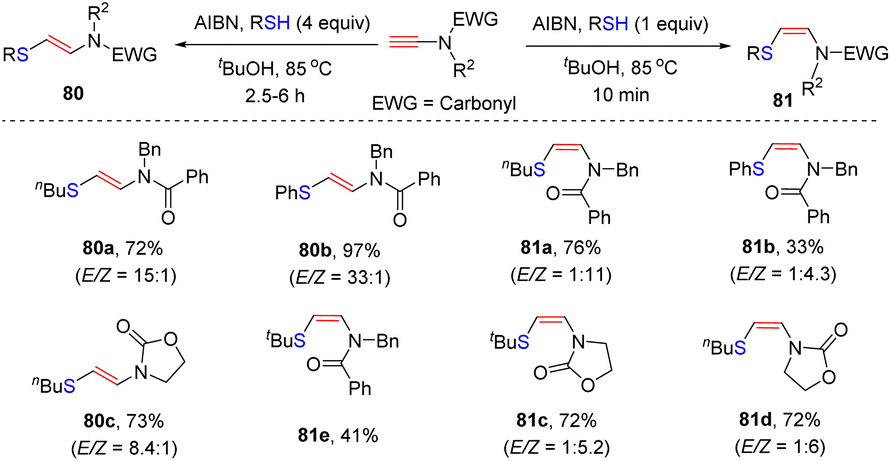
In 2010, Castle and co-workers disclosed an elegant stereodivergent addition of thiols with terminal N-carbonyl ynamides at the β-site in the presence of α,α′-azoisobutyronitrile (AIBN) via a radical pathway (Scheme 55).104 AIBN serves as a radical initiator in this transformation. The amount of thiols plays a crucial role for the stereoselectivity. When using 1.0 equivalent of thiols, kinetic products (Z)-β-thiylenamides were mainly afforded within 10 min. In comparison, the use of excessive thiols preferably led to the formation of thermodynamically stable (E)-β-thiylenamides. There exists an equilibrium between the Z- and E-vinyl radical intermediates. The excess thiol could rapidly quench the E-vinyl radical via hydrogen abstraction, thus shifting the equilibrium to E-configuration. However, in the case of bulky tert-butylthiol, both conditions gave rise to the Z-adduct 81e as a major product.
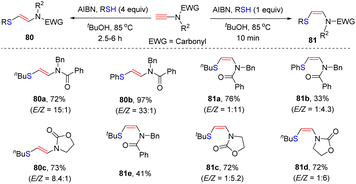 |
| | Scheme 55 Radical-triggered anti-β-hydrothiolation of terminal N-carbonyl ynamides with thiols. | |
Recently, Taillefer and Monnier et al. exploited an efficient syn-β-hydrothiolation of internal ynamides with thiols enabled by copper catalysis (Scheme 56).105 The aryl substituents on the ynamide terminus were well compatible with the transformation, exclusively providing (E)-β-thiylenamides in moderate to good yields. In comparison, subjecting alkyl-substituted ynamides to the standard conditions afforded unexpected α-thiylenamides with a moderate ratio of E- and Z-configuration. This approach tolerated a series of aromatic and aliphatic thiols, and was successfully extended to allenamides and enamides.
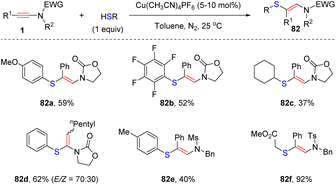 |
| | Scheme 56 Cu-catalyzed syn-β-hydrothiolation of internal ynamides with thiols. | |
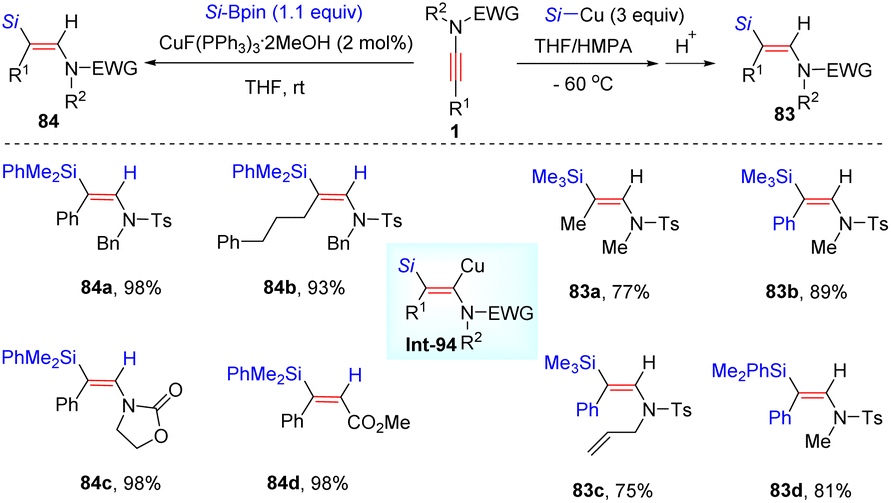
8. C–Si bond formation
In 2008, Oshima and Yorimitsu et al. reported the addition between ynamides and silylcopper for the preparation of (E)-β-silylenamides in appreciable yields (Scheme 57).106 The procedure includes syn-silylcupration of ynamides and subsequent protonolysis. When using other electrophiles instead of proton, for example allyl bromide, the corresponding coupling products could be obtained. In 2016, using silylboronates as partners, a copper-catalyzed syn-β-hydrosilylation of internal ynamides was achieved by Evano and Riant et al. (Scheme 57).107 The process features mild reaction conditions, easy operation, excellent regioselectivity, and broad substrate scope.
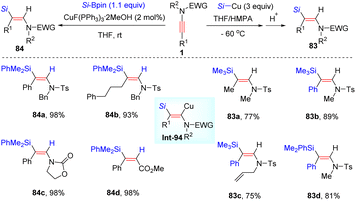 |
| | Scheme 57 Copper-catalyzed syn-β-hydrosilylation of internal ynamides. | |
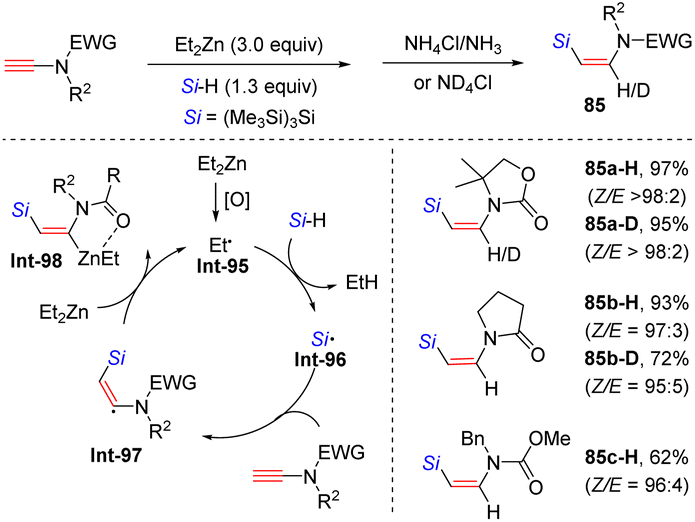
In 2014, Perez-Luna and co-workers presented a diethylzinc-triggered hydrosilylation of terminal ynamides with hydrosilanes, affording (Z)-β-silylenamides (e.g. anti-β-adducts of ynamides) in high regio- and stereoselectivity (Scheme 58).108 The ethyl radical Int-95, in situ generated from diethylzinc, first reacts with hydrosilane to give the silyl radical Int-96. Subsequent attack at the β-site of the ynamide delivers the β-silylvinyl radical Int-97. This species is further quenched by diethylzinc to yield the β-silylvinylzinc intermediate Int-98, which can be stabilized by the coordination between zinc and the carbonyl group on the nitrogen atom. A final protonolysis yields the expected product. It was noted that the protonolysis with ND4Cl could furnish deuterated products. Moreover, the mechanism is certified by a cascade copper-mediated electrophilic trapping of the vinyl zinc intermediate with allyl halides.
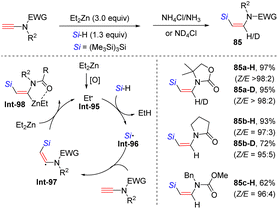 |
| | Scheme 58 Diethylzinc-triggered hydrosilylation of terminal ynamides with hydrosilanes. | |
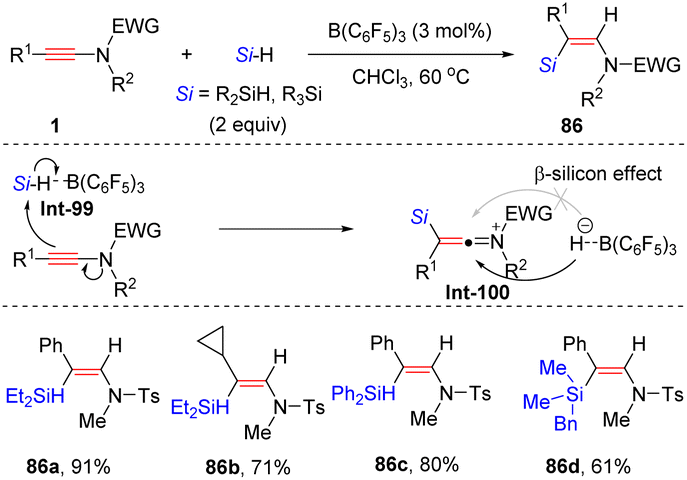
By leveraging frustrated Lewis pair catalysis, Chang and his colleagues developed a complementary anti-hydrosilylation of internal ynamides with the aid of B(C6F5)3, providing (Z)-β-silylenamides with high regio- and stereoselectivity (Scheme 59).109 Hydrosilane is initially activated by borane B(C6F5)3, followed by the nucleophilic attack of the β-site of the ynamide to form the silyl keteniminium ion. The α-carbon of keteniminium is then attacked by the borohydride from the opposite site of the silyl group to afford the target product with the regeneration of B(C6F5)3 for the next catalytic cycle. The DFT calculations revealed that the stereoselectivity of the hydride abstraction is triggered by the β-silicon effect.
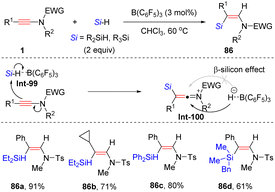 |
| | Scheme 59 B(C6F5)3-catalyzed anti-β-hydrosilylation of internal ynamides. | |
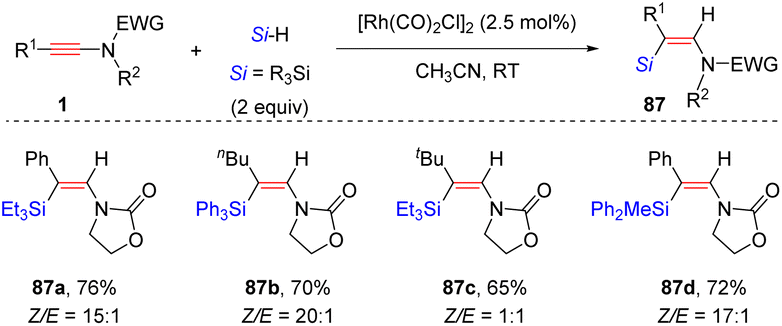
Later, Song and co-workers discovered that besides B(C6F5)3 catalysis, the rhodium complex [Rh(CO)2Cl]2 could also promote highly selective anti-β-hydrosilylation of ynamides with silanes (Scheme 60).110 In most cases, diverse alkyl and aryl substituents on ynamides were suitable for the process. For bulky the tBu-derived ynamide, a mixture of E- and Z-silylenamides was obtained. An array of tertiary silanes were tolerated, whereas the secondary silane (diphenylsilane) and primary silane (phenylsilane) were ineffective. The high regio- and stereoselectivity originates from the strong coordination between the carbonyl group on the nitrogen atom and the rhodium center, followed by hydride attack at the α-site of the keteniminium intermediate.
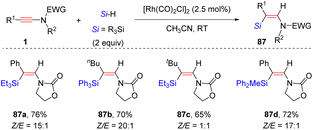 |
| | Scheme 60 Rh-catalyzed anti-β-hydrosilylation of ynamides with silanes. | |
9. C–Ge bond formation
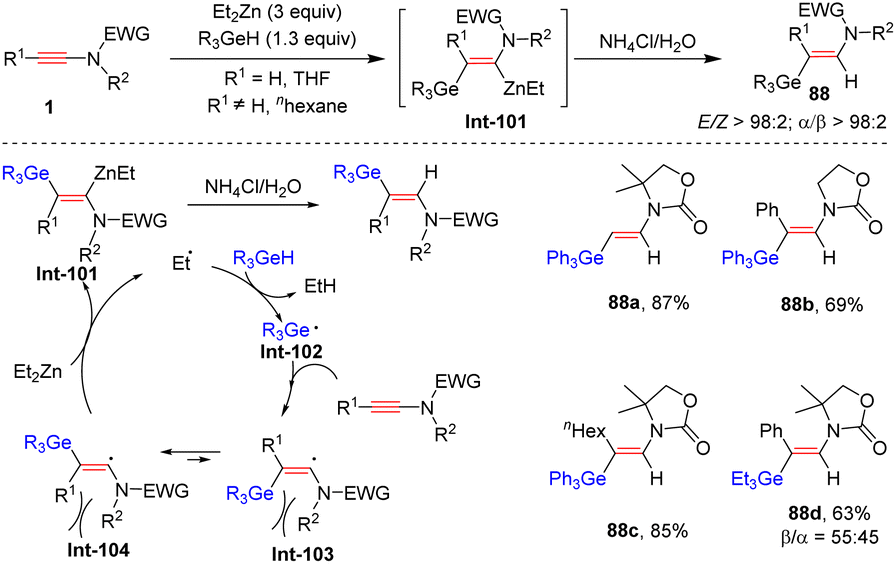
Prompted by ZnEt2-promoted hydrosilylation of ynamides, Perez-Luna and co-workers, in 2018, disclosed that with the assistance of ZnEt2, ynamides and germanes could undergo syn-β-hydrogermylation with high efficiency (Scheme 61).111 The ethyl radical, generated from ZnEt2, reacts with R3GeH to give the germanium-centered radical Int-102. Subsequent addition to the β-position of the ynamide delivers the (Z)-β-germanylvinyl radical Int-103, which rapidly shifts to the E-isomer Int-104 because of steric hindrance. Then, zinc transfer with ZnEt2 furnishes the (E)-β-germanylvinylzinc intermediate Int-101, which can be stabilized by the coordination between zinc and the carbonyl group on the nitrogen atom. An eventual protonolysis results in the expected product. Only oxazolidinone-derived ynamides were suitable substrates. Notably, the formed (E)-β-germanylvinylzinc intermediate can also participate in diverse cascade copper- and palladium-mediated electrophilic coupling to construct new C–C or C–heteroatom bonds with the retention of the E-configuration.
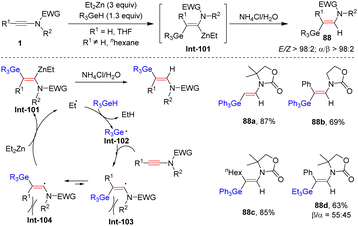 |
| | Scheme 61 ZnEt2-promoted syn-β-hydrogermylation of ynamides with germanes. | |
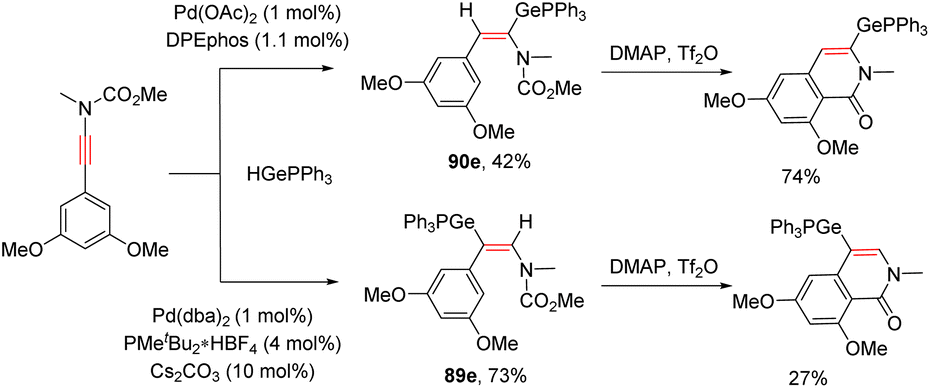
In 2020, Houk and Bizet et al. developed an elegant ligand-controlled regiodivergent syn-hydrogermylation of ynamides with germanes under palladium catalysis (Scheme 62).112 An initial oxidative addition of Pd(0) with the germanium hydride delivers the Ge–Pd–H complex Int-105, which activates the ynamide via coordination with the carbon–carbon triple bond. The ligand plays a crucial role for the subsequent hydropalladation. With the DPEphos ligand, the reaction predominantly gave α-palladium enamides Int-108, whereas the use of PMetBu2 altered the regioselectivity to the β-site owing to the steric hindrance between the bulkier ligand and the substituents on the nitrogen atom. Finally, reductive elimination delivers the corresponding α,E- and β,E-germanylenamides, respectively. DFT calculations indicated that the regioselectivity is derived from the steric requirements of the phosphine ligands in the hydropalladation step. N-Ester ynamides could also participate in this ligand-controlled regiodivergent transformation and the resulting adducts were readily converted into their corresponding germylated isoquinolinones in the presence of DMAP and triflic anhydride (Scheme 63).
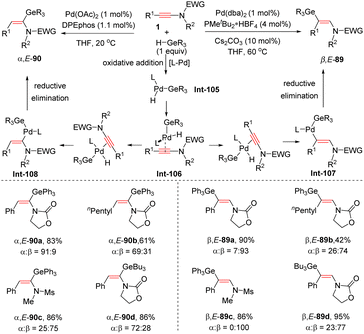 |
| | Scheme 62 Pd-catalyzed regiodivergent syn-hydrogermylation of ynamides with germanes. | |
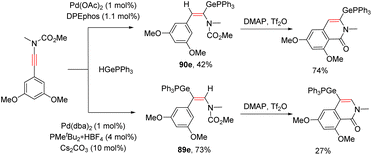 |
| | Scheme 63 Synthesis of germylated isoquinolinones. | |
10. Conclusion
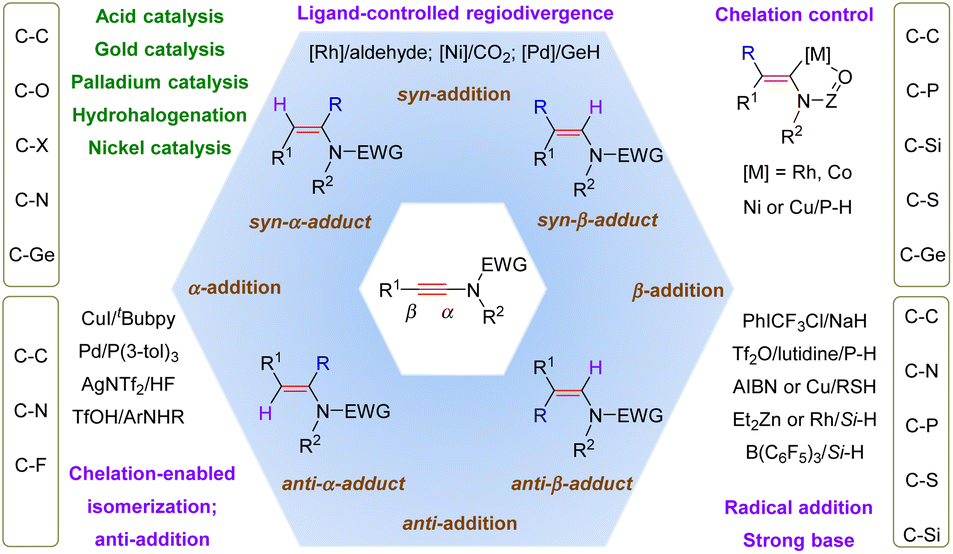
This review carefully summarizes the advances achieved in the regioselective hydrofunctionalization of ynamides (Scheme 64). These transformations enable the expedient synthesis of multi-substituted enamides that are otherwise difficult to access. When nucleophilic reagents are employed as partners, most additions favorably take place at the more electrophilic α-site of ynamides from the opposite side of the terminal group, thus finally leading to syn-α-adducts. In comparison, with the aid of transition metals, β-addition turns out to be predominant since the coordination of transition metals and the electron-withdrawing group on the nitrogen atom can stabilize β-functionalized α-metal enamides (chelation control), followed by protonolysis to give syn-β-adducts. However, the use of a bulky ligand can alter the regioselectivity due to its steric hindrance with the substituents on the nitrogen atom (ligand-controlled regiodivergence). Another exception is that the mechanism starts with hydrometalation and closes with reductive elimination, and in this circumstance, α-adducts are afforded and the stereoselectivity remains variable due to the potential E/Z isomerization of vinyl metals. For radical-enabled hydrofunctionalization, a less hindered β-position is more likely to be attacked by the radical species and E/Z isomerization can also be involved, thus producing β-adducts with tunable stereoselectivity.
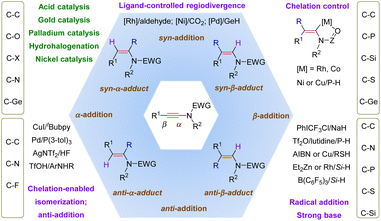 |
| | Scheme 64 Summary of the reaction patterns of the hydrofunctionalization of ynamides. | |
While impressive progress has been made, there is ample room for further development in this fascinating field. In most hydrofunctionalizations, the mechanism of stereoselectivity control still remains unclear. Therefore, theoretical studies will be needed to address this issue, which can offer guidance for the rational design of new hydrofunctionalizations. Additional efforts will be directed towards their applications in the synthesis of natural products and functional materials. Furthermore, some modern technologies are less explored in ynamide chemistry. We anticipate that photo-redox catalysis and electro-catalysis will be two important tools in developing green and sustainable hydrofunctionalizations of ynamides, which holds great promise for more exciting breakthroughs. Overall, this comprehensive overview may serve as a handy source for chemists interested in ynamide chemistry and provide valuable insights that can pave the way for future discoveries.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by the Xuzhou Basic Research Project (KC23018), the Fundamental Research Funds for the Central Universities (no. 2023-00104), and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
References
- C. A. Zificsak, J. A. Mulder, R. P. Hsung, C. Rameshkumar and L.-L. Wei, Recent advances in the chemistry of ynamines and ynamides, Tetrahedron, 2001, 57, 7575–7606 CrossRef
 .
.
- K. A. DeKorver, H. Y. Li, A. G. Lohse, R. Hayashi, Z. J. Lu, Y. Zhang and R. P. Hsung, Ynamides: A Modern Functional Group for the New Millennium, Chem. Rev., 2010, 110, 5064–5106 CrossRef CAS PubMed .
- G. Evano, A. Coste and K. Jouvin, Ynamides: Versatile Tools in Organic Synthesis, Angew. Chem., Int. Ed., 2010, 49, 2840–2859 CrossRef CAS PubMed .
- G. Evano, C. Theunissen and M. Lecomte, Ynamides: Powerful and Versatile Reagents for Chemical Synthesis, Aldrichimica Acta, 2015, 48, 59–70 CAS .
- G. Evano, B. Michelet and C. Zhang, The anionic chemistry of ynamides: A review, C. R. Chim., 2017, 20, 648–664 CrossRef CAS .
- B. Zhou, T.-D. Tan, X.-Q. Zhu, M. Shang and L.-W. Ye, Reversal of Regioselectivity in Ynamide Chemistry, ACS Catal., 2019, 9, 6393–6406 CrossRef CAS .
- Y.-B. Chen, P.-C. Qian and L.-W. Ye, Brønsted acid-mediated reactions of ynamides, Chem. Soc. Rev., 2020, 49, 8897–8909 RSC .
- Z.-S. Wang, Y.-B. Chen, H.-W. Zhang, Z. Sun, C. Zhu and L.-W. Ye, Ynamide Smiles Rearrangement Triggered by Visible-Light-Mediated Regioselective Ketyl–Ynamide Coupling: Rapid Access to Functionalized Indoles and Isoquinolines, J. Am. Chem. Soc., 2020, 142, 3636–3644 CrossRef CAS PubMed .
- L. Zeng, Y. Lin, J. Li, H. Sajiki, H. Xie and S. Cui, Skeletal reorganization divergence of N-sulfonyl ynamides, Nat. Commun., 2020, 11, 5639 CrossRef CAS PubMed .
- M. R. Mutra and J. J. Wang, Photoinduced ynamide structural reshuffling and functionalization, Nat. Commun., 2022, 13, 2345 CrossRef CAS PubMed .
- X. N. Wang, H. S. Yeom, L. C. Fang, S. H. He, Z. X. Ma, B. L. Kedrowski and R. P. Hsung, Ynamides in Ring Forming Transformations, Acc. Chem. Res., 2014, 47, 560–578 CrossRef CAS PubMed .
- F. Pan, C. Shu and L.-W. Ye, Recent progress towards gold-catalyzed synthesis of N-containing tricyclic compounds based on ynamides, Org. Biomol. Chem., 2016, 14, 9456–9465 RSC .
- B. Prabagar, N. Ghosh and A. K. Sahoo, Cyclization and Cycloisomerization of π-Tethered Ynamides: An Expedient Synthetic Method to Construct Carbo- and Heterocycles, Synlett, 2017, 2539–2555 CAS .
- R. H. Dodd and K. Cariou, Ketenimines Generated from Ynamides: Versatile Building Blocks for Nitrogen-Containing Scaffolds, Chem. – Eur. J., 2018, 24, 2297–2304 CrossRef CAS PubMed .
- C.-Y. Shi, L. Li, W. Kang, Y.-X. Zheng and L.-W. Ye, Claisen rearrangement triggered by transition metal-catalyzed alkyne alkoxylation, Coord. Chem. Rev., 2021, 446, 214131 CrossRef CAS .
- S. Ghosh, Y. Zheng, D. Maccarone, F.-A. Korsaye, I. Ciofini and L. Miesch, Triton B-promoted regioselective intramolecular addition of enolates to tethered ynamides for the construction of 8-membered O-heterocycles, Org. Chem. Front., 2022, 9, 4057–4062 RSC .
- Y.-C. Hu, Y. Zhao, B. Wan and Q.-A. Chen, Reactivity of ynamides in catalytic intermolecular annulations, Chem. Soc. Rev., 2021, 50, 2582–2625 RSC .
- F.-L. Hong and L.-W. Ye, Transition Metal-Catalyzed Tandem Reactions of Ynamides for Divergent N-Heterocycle Synthesis, Acc. Chem. Res., 2020, 53, 2003–2019 CrossRef CAS PubMed .
- H.-J. Xu, C.-T. Li, C.-M. Chen, J. Chen, X.-Q. Zhu, B. Zhou and L.-W. Ye, Copper-catalyzed formal
[4 + 1] annulation of N-propargyl ynamides with diketones, Org. Chem. Front., 2023, 10, 203–208 RSC .
- N. V. Shcherbakov, G. D. Titov, E. I. Chikunova, I. P. Filippov, N. V. Rostovskii, V. Y. Kukushkin and A. Y. Dubovtsev, Modular approach to non-aromatic and aromatic pyrroles through gold-catalyzed [3 + 2] cycloaddition of 2H-azirines and ynamides, Org. Chem. Front., 2022, 9, 5133–5140 RSC .
- L. Ding, Z. Zhu, X. Zhou, G. Zhu, J.-H. Wang, S.-T. Zhu, B. Hu, X.-N. Wang and J. Chang, Metal-free [2 + 2] and [4 + 2] cycloadditions of N-aryl-substituted ynamides to construct functionalized aminocyclobutenes and 4-aminoquinolines, Org. Chem. Front., 2023, 10, 4043–4054 RSC .
- H. Xu, Y. Liu, L. Ding, X.-N. Wang and J. Chang, Lewis acid-promoted cyclizations of o-alkyloxyphenyl-substituted ynamides to construct 2-amidobenzofurans, Org. Chem. Front., 2022, 9, 5637–5642 RSC .
- C. Mahe and K. Cariou, Ynamides in Free Radical Reactions, Adv. Synth. Catal., 2020, 362, 4820–4832 CrossRef CAS .
- T.-D. Tan, Z.-S. Wang, P.-C. Qian and L.-W. Ye, Radical Reactions of Ynamides, Small Methods, 2021, 5, 2000673 CrossRef CAS PubMed .
- J. Luo, G.-S. Chen, S.-J. Chen, J.-S. Yu, Z.-D. Li and Y.-L. Liu, Exploiting Remarkable Reactivities of Ynamides: Opportunities in Designing Catalytic Enantioselective Reactions, ACS Catal., 2020, 10, 13978–13992 CrossRef CAS .
- C. C. Lynch, A. Sripada and C. Wolf, Asymmetric synthesis with ynamides: unique reaction control, chemical diversity and applications, Chem. Soc. Rev., 2020, 49, 8543–8583 RSC .
- S. Dutta, R. K. Mallick and A. K. Sahoo, Regioselective Difunctionalization and Annulation of Ynamide, Angew. Chem., Int. Ed., 2023, 62, e202300816 CrossRef CAS PubMed .
- H. Chechik-Lankin, S. Livshin and I. Marek, Regiocontrolled carbometallation reactions of ynamides, Synlett, 2005, 2098–2100 CAS .
- B. Gourdet and H. W. Lam, Stereoselective synthesis of multisubstituted enamides via rhodium-catalyzed carbozincation of ynamides, J. Am. Chem. Soc., 2009, 131, 3802–3803 CrossRef CAS PubMed .
- M. Takimoto, S. S. Gholap and Z. Hou, Cu-Catalyzed Alkylative Carboxylation of Ynamides with Dialkylzinc Reagents and Carbon Dioxide, Chem. – Eur. J., 2015, 21, 15218–15223 CrossRef CAS PubMed .
- R. Sallio, M. Corpet, L. Habert, M. Durandetti, C. Gosmini and I. Gillaizeau, Cobalt-Catalyzed Carbozincation of Ynamides, J. Org. Chem., 2017, 82, 1254–1259 CrossRef CAS PubMed .
- B. Gourdet, D. L. Smith and H. W. Lam, Rhodium-catalyzed carbometalation of ynamides with organoboron reagents, Tetrahedron, 2010, 66, 6026–6031 CrossRef CAS .
- B. Gourdet and H. W. Lam, Catalytic asymmetric dihydroxylation of enamides and application to the total synthesis of (+)-tanikolide, Angew. Chem., Int. Ed., 2010, 49, 8733–8737 CrossRef CAS PubMed .
- Y. Yang, L. Wang, J. Zhang, Y. Jin and G. Zhu, An unprecedented Pd-catalyzed trans-addition of boronic acids to ynamides, Chem. Commun., 2014, 50, 2347–2349 RSC .
- Y. Yang, L. Wang, F. Zhang and G. Zhu, Preparation of (Z)-α,β-disubstituted enamides via palladium-catalyzed addition of boronic acids to ynamides, J. Org. Chem., 2014, 79, 9319–9324 CrossRef CAS PubMed .
- A. Di Nicola, V. Marsicano, A. Arcadi and V. Michelet, Room temperature palladium-catalyzed hydroarylation of ynamides in water, Tetrahedron Lett., 2020, 61, 151725 CrossRef CAS .
- A. Maity and A. K. Sahoo, Copper-Catalyzed Regio- and Stereoselective Hydroarylation of Ynamide, J. Org. Chem., 2024, 89, 852–863 CrossRef CAS PubMed .
- H. Wakamatsu, R. Yanagisawa, S. Kimura, N. Osawa, Y. Natori and Y. Yoshimura, Palladium-Catalyzed Regioselective Hydroarylation of Ynamides with Aryl Iodides: Easy Synthesis of Various Substituted Enamides Containing Stilbene Derivatives, Synlett, 2017, 2135–2138 CrossRef CAS .
- G. Liu, W. Kong, J. W. Che and G. G. Zhu, Palladium-Catalyzed Cross Addition of Terminal Alkynes to Aryl Ynamides: An Unusual trans-Hydroalkynylation Reaction, Adv. Synth. Catal., 2014, 356, 3314–3318 CrossRef CAS .
- V. Dwivedi, M. H. Babu, R. Kant and M. S. Reddy, N-Substitution dependent stereoselectivity switch in palladium catalyzed hydroalkynylation of ynamides: a regio and stereoselective synthesis of ynenamides, Chem. Commun., 2015, 51, 14996–14999 RSC .
- H. Siera, M. Kreuzahler, C. Wölper and G. Haberhauer, Regio- and Stereoselective Synthesis of Ynenamides through Gold(I)-Catalyzed Hydroalkynylation of Ynamides, Eur. J. Org. Chem., 2022, e202200591 CrossRef CAS .
- Y. Zhang, Syntheses of vinylindoles via a Bronsted acid catalyzed highly regio- and stereoselective cis-hydroarylation of ynamides, Tetrahedron Lett., 2005, 46, 6483–6486 CrossRef CAS .
- Y. S. Zhang, Synthesis of vinylpyrroles, vinylfurans and vinylindoles via a Bronsted acid catalyzed highly regio- and stereoselective cis-hydroarylation of ynamides, Tetrahedron, 2006, 62, 3917–3927 CrossRef CAS .
- V. Pirovano, M. Negrato, G. Abbiati, M. Dell'Acqua and E. Rossi, Gold-Catalyzed cis-Hydroarylation of Ynamides with Indoles: Regio- and Stereoselective Synthesis of a Class of 2-Vinylindoles, Org. Lett., 2016, 18, 4798–4801 CrossRef CAS PubMed .
- X. D. Nie, Z. Y. Mao, J. M. Guo, C. M. Si, B. G. Wei and G. Q. Lin, AgNTf2-Catalyzed Regioselective C-H Alkenylation of N,N-Dialkylanilines with Ynamides, J. Org. Chem., 2022, 87, 2380–2392 CrossRef CAS PubMed .
- Y. Xiao, L. Tang, T. T. Xu and J. J. Feng, Boron Lewis Acid Catalyzed Intermolecular trans-Hydroarylation of Ynamides with Hydroxyarenes, Org. Lett., 2022, 24, 2619–2624 CrossRef CAS PubMed .
- N. Saito, I. Abdullah, K. Hayashi, K. Hamada, M. Koyama and Y. Sato, Enantioselective synthesis of β-amino acid derivatives via nickel-promoted regioselective carboxylation of ynamides and rhodium-catalyzed asymmetric hydrogenation, Org. Biomol. Chem., 2016, 14, 10080–10089 RSC .
- R. Doi, I. Abdullah, T. Taniguchi, N. Saito and Y. Sato, Nickel-catalyzed hydrocarboxylation of ynamides with CO2 and H2O: observation of unexpected regioselectivity, Chem. Commun., 2017, 53, 7720–7723 RSC .
- R. Doi, T. Okano, I. Abdullah and Y. Sato, Nickel-Catalyzed -Carboxylation of Ynamides with Carbon Dioxide, Synlett, 2019, 1048–1052 CAS .
- V. Tona, S. A. Ruider, M. Berger, S. Shaaban, M. Padmanaban, L. G. Xie, L. Gonzalez and N. Maulide, Divergent ynamide reactivity in the presence of azides - an experimental and computational study, Chem. Sci., 2016, 7, 6032–6040 RSC .
- R. N. Straker, M. K. Majhail and M. C. Willis, Exploiting rhodium-catalysed ynamide hydroacylation as a platform for divergent heterocycle synthesis, Chem. Sci., 2017, 8, 7963–7968 RSC .
- P. Wagner, M. Donnard and N. Girard, Ligand-Controlled Regiodivergent Hydroformylation of Ynamides: A Stereospecific and Regioselective Access to 2- and 3-Aminoacroleins, Org. Lett., 2019, 21, 8861–8866 CrossRef CAS PubMed .
- W. Huang, R. Zhang, R. Zhang, J. Yu and M. Wang, Radical hydrotrifluoromethylation of ynamides: a route toward β-CF3 enamides, Org. Chem. Front., 2022, 9, 2169–2175 RSC .
- L. Feray, M. P. Bertrand and A. Galibert-Guijarro, α-Haloenamides: Synthesis and Subsequent Transformations, Synthesis, 2023, 27–44 CrossRef CAS .
- J. A. Mulder, K. C. Kurtz, R. P. Hsung, H. Coverdale, M. O. Frederick, L. Shen and C. A. Zificsak, Highly stereoselective synthesis of novel α-haloenamides
via a mild and efficient hydrohalogenation of ynamides, Org. Lett., 2003, 5, 1547–1550 CrossRef CAS PubMed .
- A. H. Sato, K. Ohashi and T. Iwasawa, Regio- and stereospecific synthesis of (E)-α-iodoenamide moieties from ynamides through iodotrimethylsilane-mediated hydroiodation, Tetrahedron Lett., 2013, 54, 1309–1311 CrossRef CAS .
- Y. Yabuuchi, T. Kuzuguchi, T. Yoshimura and J. I. Matsuo, Synthesis of α-Halo-γ-hydroxyenamides by Titanium Tetrahalide Mediated Addition of Aldehydes or Ketones to Ynamides, Org. Lett., 2016, 18, 4951–4953 CrossRef CAS PubMed .
- B. Prabagar, S. Nayak, R. K. Mallick, R. Prasad and A. K. Sahoo, Triphenylphosphine promoted regio and stereoselective α-halogenation of ynamides, Org. Chem. Front., 2016, 3, 110–115 RSC .
- S. W. Kim, T. W. Um and S. Shin, Bronsted acid-catalyzed α-halogenation of ynamides from halogenated solvents and pyridine-N-oxides, Chem. Commun., 2017, 53, 2733–2736 RSC .
- X. Zeng, Z. Lu, S. Liu, G. B. Hammond and B. Xu, Metal-free, Regio-, and Stereo-Controlled Hydrochlorination and Hydrobromination of Ynones and Ynamides, J. Org. Chem., 2017, 82, 13179–13187 CrossRef CAS PubMed .
- G. Compain, K. Jouvin, A. Martin-Mingot, G. Evano, J. Marrot and S. Thibaudeau, Stereoselective hydrofluorination of ynamides: a straightforward synthesis of novel α-fluoroenamides, Chem. Commun., 2012, 48, 5196–5198 RSC .
- B. Métayer, G. Compain, K. Jouvin, A. Martin-Mingot, C. Bachmann, J. Marrot, G. Evano and S. Thibaudeau, Chemo- and Stereoselective Synthesis of Fluorinated Enamides from Ynamides in HF/Pyridine: Second-Generation Approach to Potent Ureas Bioisosteres, J. Org. Chem., 2015, 80, 3397–3410 CrossRef PubMed .
- J. W. Che, Y. L. Li, F. Zhang, R. W. Zheng, Y. H. Bai and G. G. Zhu, Silver-promoted trans-hydrofluorination of ynamides: a regio- and stereoselective approach to (Z)-α-fluoroenamides, Tetrahedron Lett., 2014, 55, 6240–6242 CrossRef CAS .
- G. K. He, S. N. Qiu, H. Huang, G. H. Zhu, D. M. Zhang, R. Zhang and H. J. Zhu, Cu(I)- or Ag(I)-Catalyzed Regio- and Stereocontrolled trans-Hydrofluorination of Ynamides, Org. Lett., 2016, 18, 1856–1859 CrossRef CAS PubMed .
- G. H. Zhu, S. N. Qiu, Y. Xi, Y. Ding, D. M. Zhang, R. Zhang, G. K. He and H. J. Zhu, (IPr)CuF-catalyzed α-site regiocontrolled trans-hydrofluorination of ynamides, Org. Biomol. Chem., 2016, 14, 7746–7753 RSC .
- X. J. Zeng, J. L. Li, C. K. Ng, G. B. Hammond and B. Xu, (Radio)fluoroclick Reaction Enabled by a Hydrogen-Bonding Cluster, Angew. Chem., Int. Ed., 2018, 57, 2924–2928 CrossRef CAS PubMed .
- D. L. Smith, W. R. F. Goundry and H. W. Lam, Palladium-catalyzed hydroacyloxylation of ynamides, Chem. Commun., 2012, 48, 1505–1507 RSC .
- S. J. Xu, J. Q. Liu, D. H. Hu and X. H. Bi, Metal-free hydroacyloxylation and hydration reactions of ynamides: synthesis of α-acyloxyenamides and N-acylsulfonamides, Green Chem., 2015, 17, 184–187 RSC .
- L. Hu, S. Xu, Z. Zhao, Y. Yang, Z. Peng, M. Yang, C. Wang and J. Zhao, Ynamides as Racemization-Free Coupling Reagents for Amide and Peptide Synthesis, J. Am. Chem. Soc., 2016, 138, 13135–13138 CrossRef CAS PubMed .
- T. Liu, X. Zhang, Z. J. Peng and J. F. Zhao, Water-removable ynamide coupling reagent for racemization-free syntheses of peptides, amides, and esters, Green Chem., 2021, 23, 9916–9921 RSC .
- J. Yang, H. Huang and J. Zhao, Active ester-based peptide bond formation and its application in peptide synthesis, Org. Chem. Front., 2023, 10, 1817–1846 RSC .
- T. Liu, Z. Peng, M. Lai, L. Hu and J. Zhao, Inverse Peptide Synthesis Using Transient Protected Amino Acids, J. Am. Chem. Soc., 2024, 146, 4270–4280 CrossRef CAS PubMed .
- C. Yao, J. Yang, X. Lu, S. Zhang and J. Zhao, Ynamide-Mediated Thionoester and Dithioester Syntheses, Org. Lett., 2020, 22, 6628–6631 CrossRef CAS PubMed .
- X. Wang, Y. Zhao, J. Yang, Y. Li, Y. Luo, M. Xu and J. Zhao, Ynamide-Mediated Thioester Synthesis, J. Org. Chem., 2021, 86, 18265–18277 CrossRef CAS PubMed .
- C. Wang, C. Han, J. Yang, Z. Zhang, Y. Zhao and J. Zhao, Ynamide-Mediated Thioamide and Primary Thioamide Syntheses, J. Org. Chem., 2022, 87, 5617–5629 CrossRef CAS PubMed .
- M. Yang, X. Wang and J. Zhao, Ynamide-Mediated Macrolactonization, ACS Catal., 2020, 10, 5230–5235 CrossRef CAS .
- S. Xu, D. Jiang, Z. Peng, L. Hu, T. Liu, L. Zhao and J. Zhao, Ynamide-Mediated Peptide Bond Formation: Mechanistic Study and Synthetic Applications, Angew. Chem., Int. Ed., 2022, 61, e202212247 CrossRef CAS PubMed .
- B. Huang, L. Zeng, Y. Shen and S. Cui, One-Pot Multicomponent Synthesis of β-Amino Amides, Angew. Chem., Int. Ed., 2017, 56, 4565–4568 CrossRef CAS PubMed .
- L. Zeng, S. Xu, S. Cui and F. Zhang, Three-component synthesis of β-aminoxy amides, Org. Chem. Front., 2022, 9, 3757–3762 RSC .
- J. Wei, J. Zhang, J. K. Cheng, S. H. Xiang and B. Tan, Modular enantioselective access to β-amino amides by Bronsted acid-catalysed multicomponent reactions, Nat. Chem., 2023, 15, 647–657 CrossRef CAS PubMed .
- R. J. Chen, L. W. Zeng, B. Huang, Y. Y. Shen and S. L. Cui, Decarbonylative Coupling of α-Keto Acids and Ynamides for Synthesis of β-Keto Imides, Org. Lett., 2018, 20, 3377–3380 CrossRef CAS PubMed .
- Y. Y. Shen, Q. Li, G. Xu and S. L. Cui, Coupling of Carboxylic Acids with Ynamides and Subsequent Rearrangement for the Synthesis of Imides/Amides, Org. Lett., 2018, 20, 5194–5197 CrossRef CAS PubMed .
- R. Prasad, S. Kanikarapu, S. Dutta, S. Vangara and A. K. Sahoo, Metal-free stereoselective addition of propiolic acids to ynamides: a concise synthetic route to highly substituted enediyne/dienyne-(E)-N,O-acetals, New J. Chem., 2022, 46, 13981–13986 RSC .
- L. V. Graux, H. Clavier and G. Buono, Palladium-Catalyzed Addition of 1,3-Diones to Ynamides: An Entry to Alkoxy-Substituted Enamides, ChemCatChem, 2014, 6, 2544–2548 CrossRef CAS .
- R. Plamont, L. V. Graux and H. Clavier, Highly Selective Syn Addition of 1,3-Diones to Internal Ynamides Catalyzed by Zinc Iodide, Eur. J. Org. Chem., 2018, 1372–1376 CrossRef CAS .
- D. L. An, W. N. Zhang, B. Pan and Y. Y. Zhao, Metal-Free Hydrophosphoryloxylation of Ynamides: Rapid Access to Enol Phosphates, Eur. J. Org. Chem., 2021, 314–317 CrossRef CAS .
- E. R. Gao, C. Peng, J. Y. Zhang, X. N. Wang and J. B. Chang, Metal-free hydroalkoxylation of ynesulfonamides with esters, Org. Biomol. Chem., 2021, 19, 2182–2185 RSC .
- H. H. Wang, M. J. Hu, X. N. Wang and J. B. Chang, Metal-free hydroalkoxylation of ynesulfonamides with alcohols, Org. Biomol. Chem., 2022, 20, 3408–3412 RSC .
- T. Wang, Y. Y. Wan, M. Y. Xu, Y. Wang, X. J. Hong, H. Gao, Z. Zhou, W. Yi and Z. Y. Zeng, Regio- and stereospecific
cis-hydrophenoxylation of ynamides with acidic phenols, Org. Biomol. Chem., 2023, 21, 3073–3078 RSC .
- A. Hentz, P. Retailleau, V. Gandon, K. Cariou and R. H. Dodd, Transition-Metal-Free Tunable Chemoselective N Functionalization of Indoles with Ynamides, Angew. Chem., Int. Ed., 2014, 53, 8333–8337 CrossRef CAS PubMed .
- Z. Y. Peng, Z. M. Zhang, Y. L. Tu, X. Z. Zeng and J. F. Zhao, Regio- and Stereo-Selective Intermolecular Hydroamidation of Ynamides: An Approach to (Z)-Ethene-1,2-Diamides, Org. Lett., 2018, 20, 5688–5691 CrossRef CAS PubMed .
- X. D. Nie, X. L. Han, J. T. Sun, C. M. Si, B. G. Wei and G. Q. Lin, Nickel-Catalyzed Regioselective Hydroamination of Ynamides with Secondary Amines, J. Org. Chem., 2021, 86, 3433–3443 CrossRef CAS PubMed .
- Y. R. Wang, Z. Zhao, S. K. Lv, L. X. Ding, X. N. Wang and J. B. Chang, Nonmetal-catalyzed hydroamination of ynamides with amines, Org. Chem. Front., 2021, 8, 6244–6251 RSC .
- Z. Y. Chen, J. P. Huang and Z. J. Wang, Transition-Metal-Catalyzed Hydrosulfoximination and Oxidation Reaction for the Synthesis of Sulfoximine Derivatives, J. Org. Chem., 2016, 81, 9308–9314 CrossRef CAS PubMed .
- R. K. Mallick, B. Prabagar and A. K. Sahoo, Regioselective Synthesis of 2,4,5-Trisubstituted Oxazoles and Ketene Aminals via Hydroamidation and lodo-Imidation of Ynamides, J. Org. Chem., 2017, 82, 10583–10594 CrossRef CAS PubMed .
- J. K. Zhu, Q. B. Wang, X. Meng, C. Y. Zhao, X. J. Sun, L. J. Tian and Z. P. Cao, Direct Hydroheteroarylation of Ynamides with 2H-Tetrazoles: Regio- and Stereoselective Synthesis of (Z)-α-Tetrazole Enamides, Eur. J. Org. Chem., 2019, 4066–4070 CrossRef CAS .
- B. Wang, G. E. McCabe, M. J. Parrish, J. Singh, M. Zeller and Y. M. Deng, Organic Photoredox Catalyzed Direct Hydroamination of Ynamides with Azoles, Adv. Synth. Catal., 2022, 364, 1179–1184 CrossRef CAS .
- A. Fadel, F. Legrand, G. Evano and N. Rabasso, Highly Regio- and Stereoselective Nickel-Catalyzed Addition of Dialkyl Phosphites to Ynamides: an Efficient Synthesis of β-Aminovinylphosphonates, Adv. Synth. Catal., 2011, 353, 263–267 CrossRef CAS .
- A. Kinbara, H. Kasai, K. Sato, Y. Watanabe and T. Yamagishi, Nickel-catalyzed regio- and stereoselective hydrophosphinylation of internal ynamides with H-phosphinates, Tetrahedron, 2018, 74, 7068–7073 CrossRef CAS .
- A. Kinbara, M. Sato, K. Yumita and T. Yamagishi, Copper-catalyzed hydrophosphinylation of terminal ynamides with H-phosphinates, Tetrahedron, 2017, 73, 1705–1710 CrossRef CAS .
- H. Huang, H. J. Zhu and J. Y. Kang, Regio- and Stereoselective Hydrophosphorylation of Ynamides for the Synthesis of β-Aminovinylphosphine Oxides, Org. Lett., 2018, 20, 2778–2781 CrossRef CAS PubMed .
- S. Kanemura, A. Kondoh, H. Yasui, H. Yorimitsu and K. Oshima, Regio- and stereoselective additions of diphenyldithiophosphinic acid to N,-(1-alkynyl)amides and 1-alkynyl sulfides, Bull. Chem. Soc. Jpn., 2008, 81, 506–514 CrossRef CAS .
- H. Yasui, H. Yorimitsu and K. Oshima, Regio- and stereoselective hydrothiolation reactions of ynamides with diphenyldithiophosphinic acid:: straightforward synthesis of ketene N,S-acetal derivatives, Chem. Lett., 2008, 37, 40–41 CrossRef CAS .
- B. Banerjee, D. N. Litvinov, J. Kang, J. D. Bettale and S. L. Castle, Stereoselective Additions of Thiyl Radicals to Terminal Ynamides, Org. Lett., 2010, 12, 2650–2652 CrossRef CAS PubMed .
- L. Pagès, M. Bouquin, F. Jaroschik, F. Monnier and M. Taillefer, Copper-Catalyzed Regio- and Stereoselective Hydrothiolation of Allenamides, Enamides, and Ynamides, J. Org. Chem., 2023, 88, 1168–1176 CrossRef PubMed .
- H. Yasui, H. Yorimitsu and K. Oshima, Silylcupration and copper-catalyzed carbomagnesiation of ynamides:
Application to aza-Claisen rearrangement, Bull. Chem. Soc. Jpn., 2008, 81, 373–379 CrossRef CAS .
- S. Vercruysse, K. Jouvin, O. Riant and G. Evano, Copper-Catalyzed Silylcupration of Activated Alkynes, Synthesis, 2016, 3373–3381 CAS .
- E. Romain, C. Fopp, F. Chemla, F. Ferreira, O. Jackowski, M. Oestreich and A. Perez-Luna, Trans-Selective Radical Silylzincation of Ynamides, Angew. Chem., Int. Ed., 2014, 53, 11333–11337 CrossRef CAS PubMed .
- Y. Kim, R. B. Dateer and S. Chang, Borane-Catalyzed Selective Hydrosilylation of Internal Ynamides Leading to β-Silyl (Z)-Enamides, Org. Lett., 2017, 19, 190–193 CrossRef CAS PubMed .
- N. Zheng, W. Z. Song, T. X. Zhang, M. Li, Y. B. Zheng and L. Y. Chen, Rhodium-Catalyzed Highly Regioselective and Stereoselective Intermolecular Hydrosilylation of Internal Ynamides under Mild Conditions, J. Org. Chem., 2018, 83, 6210–6216 CrossRef CAS PubMed .
- K. de la Vega-Hernández, E. Romain, A. Coffinet, K. Bijouard, G. Gontard, F. Chemla, F. Ferreira, O. Jackowski and A. Perez-Luna, Radical Germylzincation of α-Heteroatom-Substituted Alkynes, J. Am. Chem. Soc., 2018, 140, 17632–17642 CrossRef PubMed .
- V. Debrauwer, A. Turlik, L. Rummler, A. Prescimone, N. Blanchard, K. N. Houk and V. Bizet, Ligand-Controlled Regiodivergent Palladium-Catalyzed Hydrogermylation of Ynamides, J. Am. Chem. Soc., 2020, 142, 11153–11164 CrossRef CAS PubMed .
|
| This journal is © the Partner Organisations 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *b
*b