
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advancements in Ni/photoredox dual catalysis for Csp3–Csp3 cross-coupling reactions
Qi-Yun
Huang
a and
Min
Shi
 *ab
*ab
aKey Laboratory for Advanced Materials and Institute of Fine Chemicals, School of Chemistry & Molecular Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China
bState Key Laboratory of Organometallic Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Ling-Ling Lu, Shanghai, 200032, China. E-mail: mshi@mail.sioc.ac.cn
First published on 16th July 2024
Abstract
For the construction of Csp3–Csp3 bonds, cross-coupling reactions in Ni/photoredox dual catalysis have recently achieved significant progress compared to traditional ones. In the past five years, a variety of free radical precursors have been developed to participate in redox neutral and reductive cross-coupling reactions via a dual photoredox/Ni-catalytic process. In this minireview, we mainly focus on the recent advancements (from 2019 to 2023) in Ni/photoredox dual catalysis for Csp3–Csp3 cross-coupling reactions.
1. Introduction
The construction of carbon–carbon bonds is one of the core research topics in the field of organic chemistry.1 A transition-metal-catalyzed cross-coupling reaction is one of the most effective methods to forge carbon–carbon bonds,1d and typical transition metal catalyzed cross-coupling reactions include the Kumada, Negishi, and Suzuki cross-coupling reactions. These reactions could produce Csp2–Csp2 cross-coupling products very efficiently in the presence of a catalytic amount of transition metal under mild conditions. However, the formation of Csp3–Csp3 bonds is usually difficult to achieve under these transition metal catalytic conditions due to the following factors:2 (1) severe reaction conditions are required for the oxidative addition of more electron-rich Csp3–X bonds with the transition metal and reductive elimination of the alkyl group from the metal center, (2) the Csp3–M intermediate easily undergoes β-H elimination, affording alkene byproducts, and (3) alkyl halides are susceptible to base-assisted H–X elimination and halide exchange side reactions under cross-coupling conditions. In 2017, Fu and his co-workers summarized transition-metal-catalyzed alkyl–alkyl cross-coupling reactions,3 demonstrating the initial progress of unactivated alkyl electrophilic reagents for Csp3–Csp3 coupling, which also provided a key driving force for the development of Ni/photoredox dual catalysis.Fortunately, odd-electron processes are not uncommon for nickel complexes. Thus, nickel species have abundant and adjustable valence states (Ni0/NiI/NiII/NiIII/NiIV).4a The reactivity of the Ni species is enriched by the availability of the NiI and NiIII states as well as the possible involvement of radical processes.4b,c Moreover, under visible light irradiation, nickel is capable of undergoing single electron transfer (SET) smoothly, and the corresponding valence state will be raised or lowered by a single valence, making it more likely for free radicals to be generated mildly when exposed to light.4d,e,f
Traditional nickel-catalyzed coupling reactions involve three key steps:5a oxidative addition, transmetallation, and reductive elimination. Transmetallation is usually the decisive step in this type of reaction, with the exchange rate of different forms of carbon hybridization being Csp > Csp2 > Csp3, making the direct construction of Csp3–Csp3 bonds particularly difficult.5b Owing to their radical-generating properties and abilities to modulate the oxidation state of metal catalysts,5c photocatalysts greatly broaden the range of electrophilic and nucleophilic reagents for cross-coupling reactions and are compatible with more reactive groups. Photocatalytic single-electron transmetallation is more suitable for Csp3–Csp3 cross-coupling reactions than the conventional nickel-catalyzed transmetallation process using metal catalysis alone.
In recent years, the introduction of photocatalysts has expanded the range of coupling partners (such as Csp3–Cl bonds, Csp3–Br bonds, Csp3–C bonds, Csp3–O bonds, Csp3–H bonds, etc.) and has greatly changed organic synthetic chemistry (Scheme 1). In this review, we summarize recent advancements in Ni/photoredox dual catalysis for Csp3–Csp3 cross-coupling reactions over the last five years (2019–2023). We attempted to classify this context into three broad categories: neutral, reductive and oxidative cross-coupling according to the electrical properties of the coupling partners. Please note that some relevant information referred to can be found in the ESI section of the cited articles.
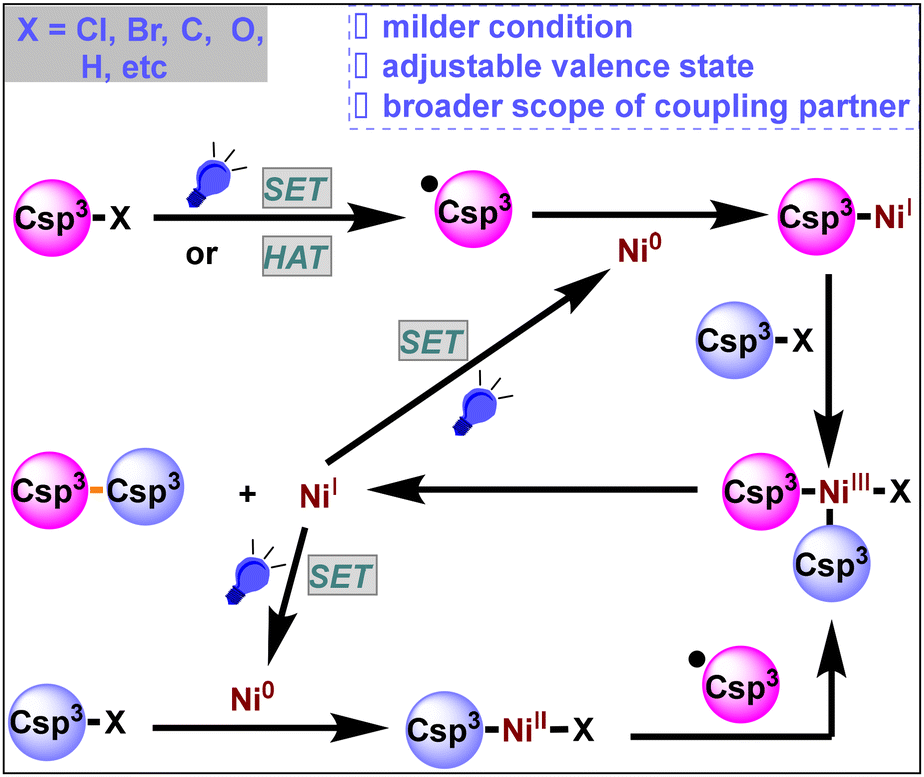 | ||
| Scheme 1 Ni/photoredox dual catalyzed cross-coupling reactions. | ||
2. Redox neutral cross-coupling
Redox neutral cross-coupling is a reaction strategy of great importance in organic synthetic chemistry.6 It is characterized by the design and selection of suitable reaction conditions so that the functional groups involved in the reaction do not undergo significant oxidation state changes during the course of the reaction, thereby improving the atom economy and selectivity of the reaction. In recent years, a number of redox neutral cross-coupling reactions under photoredox/nickel dual catalysis have been developed.In 2019, according to the fact that triplet benzaldehyde excited using ultraviolet light can interact with hydrogen-donating solvents to produce α-hydroxybenzyl and solvents such as tetrahydrofuran (THF) radicals as hydrogen atom transfer (HAT) catalysts possessing a strong ability to capture hydrogen, Hashmi and co-workers explored a platform for direct alkylation of Csp3–H bonds with alkyl halides via a HAT process (Scheme 2).7 The optimal reaction conditions are irradiation with UVA light, NiBr2·dme and L1 as the nickel catalyst, benzaldehyde as both the photosensitizer and the HAT catalyst, and K2HPO4 as the base. The substrate scope of bromide and ether coupling partners was investigated. However, the detailed mechanism requires more concrete study.
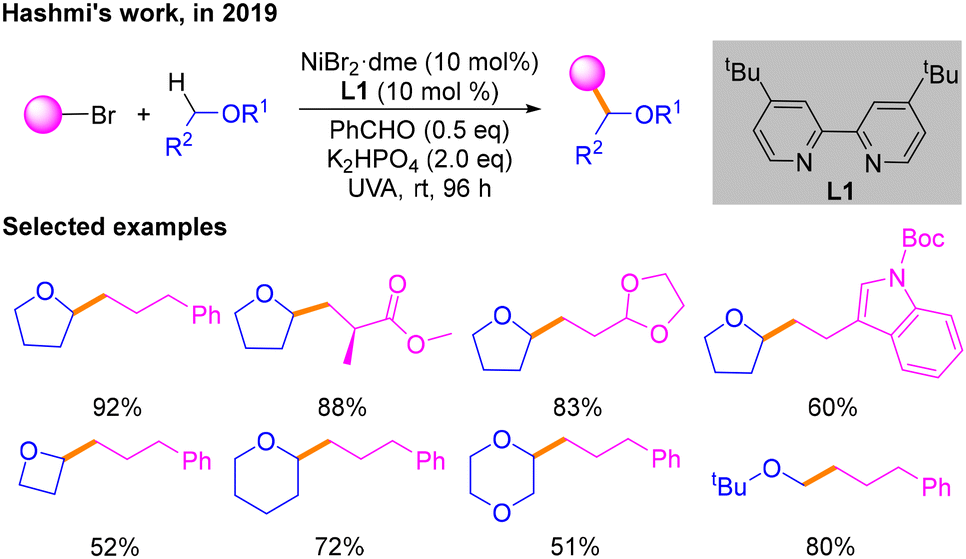 | ||
| Scheme 2 Csp3–Csp3 cross-coupling reaction of alkyl bromides with ethers (Hashmi's work). | ||
A few months later, Leonori and co-workers reported a divergent Csp3–Csp3 coupling reaction via iminium radical ring opening in conjunction with nickel catalysis and photocatalysis, enabling remote alkylation of nitriles (Scheme 3).8 The conditions for the highest yield are identified: Ir-1 as the photocatalyst (PC), Cs2CO3 as the base, NiCl2 with L1 as the nickel catalytic system and CH3CN as the solvent under blue LED irradiation. Single-electron oxidation and decarboxylation occur to provide radical 1 in the process of photocatalyst quenching in the excited state (PC*) to PC˙−, followed by single-electron β-scission to give alkyl radical 2. The nickel catalytic cycle begins with the oxidative addition of Ni0 species 3 with brominated hydrocarbon to produce NiII species 4, followed by the addition of 2 generated in the photocatalytic stage to give NiIII species 5, and finally reductive elimination to obtain a Csp3–Csp3 coupling product. The NiIBr 6 generated from the above process is reduced to 3, and PC˙− is oxidized to PC simultaneously via a SET process.
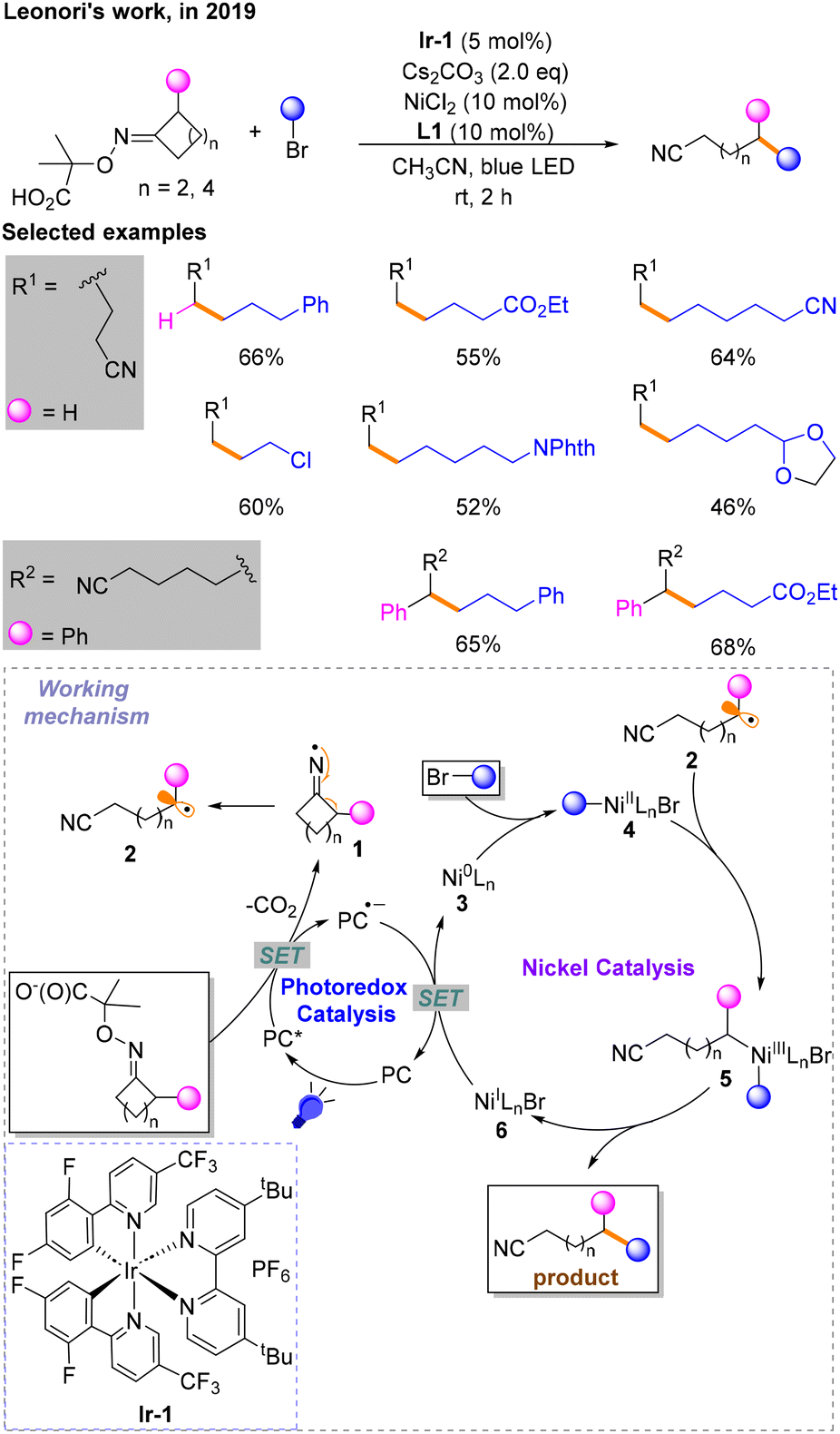 | ||
| Scheme 3 Coupling reaction of iminium radical ring-opening in conjunction with Ni-catalysis. | ||
Two months later, a method using 4-alkyl-1,4-dihydropyridines (DHPs) with oxa- and azabenzonorbornadienes to construct Csp3–Csp3 architectures was developed by Molander's group (Scheme 4).9 Notably, the regioselective and diastereoselective cis-1,2-dihydro-1-naphthyl alcohol backbone's structure can be achieved through this cross-coupling process under a nickel/photoredox dual catalytic system. The best conditions for the reaction are 4-CzIPN as the PC, NiCl2·dme with L2 or L3 as the nickel catalytic system, and acetone as the solvent under blue LED irradiation. A series of mechanistic experiments have proven the impossible activation of the substrate by by-products and the product-determining-step involving no benzyl residues. According to density functional theory (DFT) calculations, in this program, a π–σ isomerization process was believed to occur in NiIII species 7. The Curtin–Hammett principle supported the product produced with 1,2-regioselectivity.
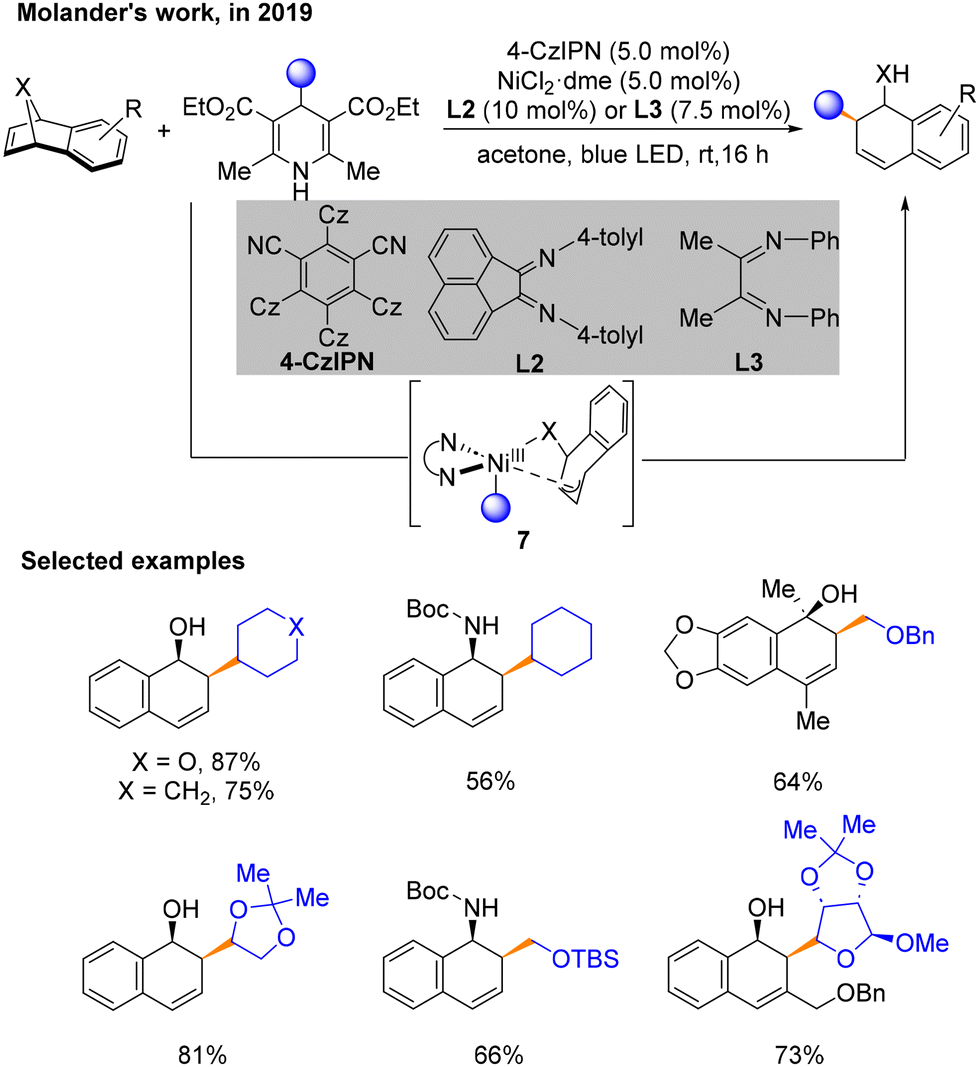 | ||
| Scheme 4 Cross-coupling of oxa- and azabenzonorbornadienes with DHP (Molander's work). | ||
In 2020, taking into account the previously mentioned study of other researchers, a protocol was introduced for a Csp3–Csp3 cross-coupling reaction of alkyl bromides with ethers using a Ni/photoredox dual catalyst via a halide-mediated HAT process by Paixão and König (Scheme 5).10 In this platform, they used 4-CzIPN as the photocatalyst to optimize the reaction conditions, and it is determined that the reaction should be carried out upon irradiation with a bule LED in the presence of Na2CO3 using Ni(acac)2 and L1 as the catalyst and 4-CzIPN as the photosensitizer in THF. The catalytic cycle begins with the oxidative addition of the Ni0 species 8 with a bromoalkane substrate to obtain the NiII species 9. Then, 9 is oxidized to afford NiIII10 by PC* to deliver PC˙−. Subsequently, 10 absorbs visible light, generating the NII species 11 and a halogen radical as a HAT agent. The generation of alkyl radicals is caused by the HAT of substrate ether and captured by 11 to provide 12. This HAT process is induced by the bromine radicals produced by the homolysis of the Ni–Br bond from 10 under the irradiation of a bule LED. The product comes from the reductive elimination of 12. Finally, NiI species 13 derived from 12 is reduced to Ni08 by PC˙− to deliver the PC, which makes the catalytic cycle complete.
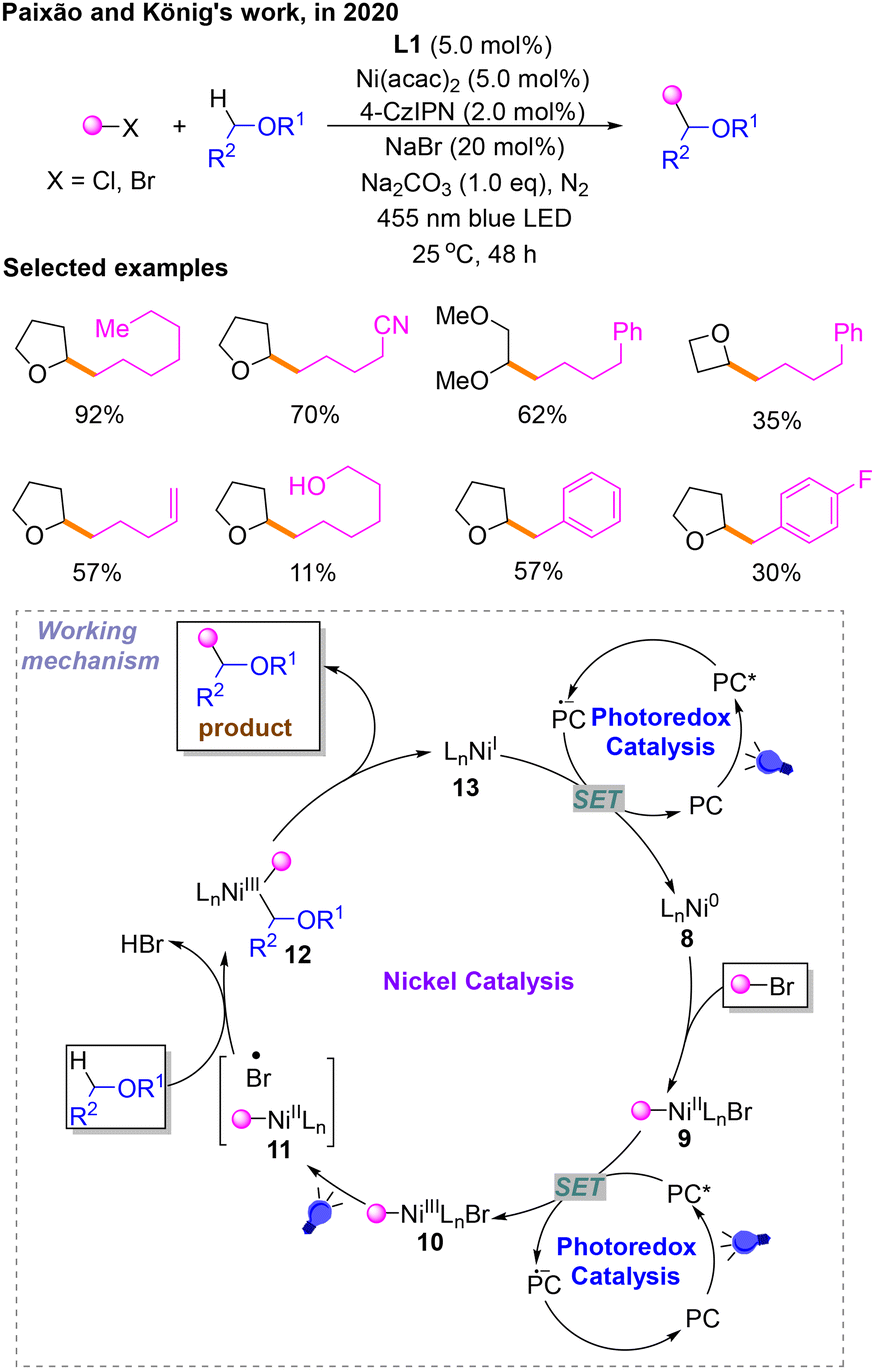 | ||
| Scheme 5 Csp3–Csp3 cross-coupling reaction of alkyl bromides with ethers (Paixão and König's work). | ||
After that, combining a light-induced system with nickel catalysis, redox-neutral and site-selectivity Csp3–H alkylation of an unactivated α-olefin with an alkyl bromide was reported by Martin's group in 2022 (Scheme 6).11 The reaction is able to obtain the maximum yield of primary alkyl bromides under blue LED irradiation, with NiBr2·dme and L4 as the nickel catalytic system, Ir-1 as the photocatalyst of light, K2CO3 as the base, and dimethylacetamide (DMA)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PhH (1:1) as the solvent. Surprisingly, the optimal conditions of secondary bromide hydrocarbons are not exactly the same in ligands and photocatalysts. For secondary bromides, L5 is the best ligand with 4-CzIPN as the photocatalyst.
:PhH (1:1) as the solvent. Surprisingly, the optimal conditions of secondary bromide hydrocarbons are not exactly the same in ligands and photocatalysts. For secondary bromides, L5 is the best ligand with 4-CzIPN as the photocatalyst.
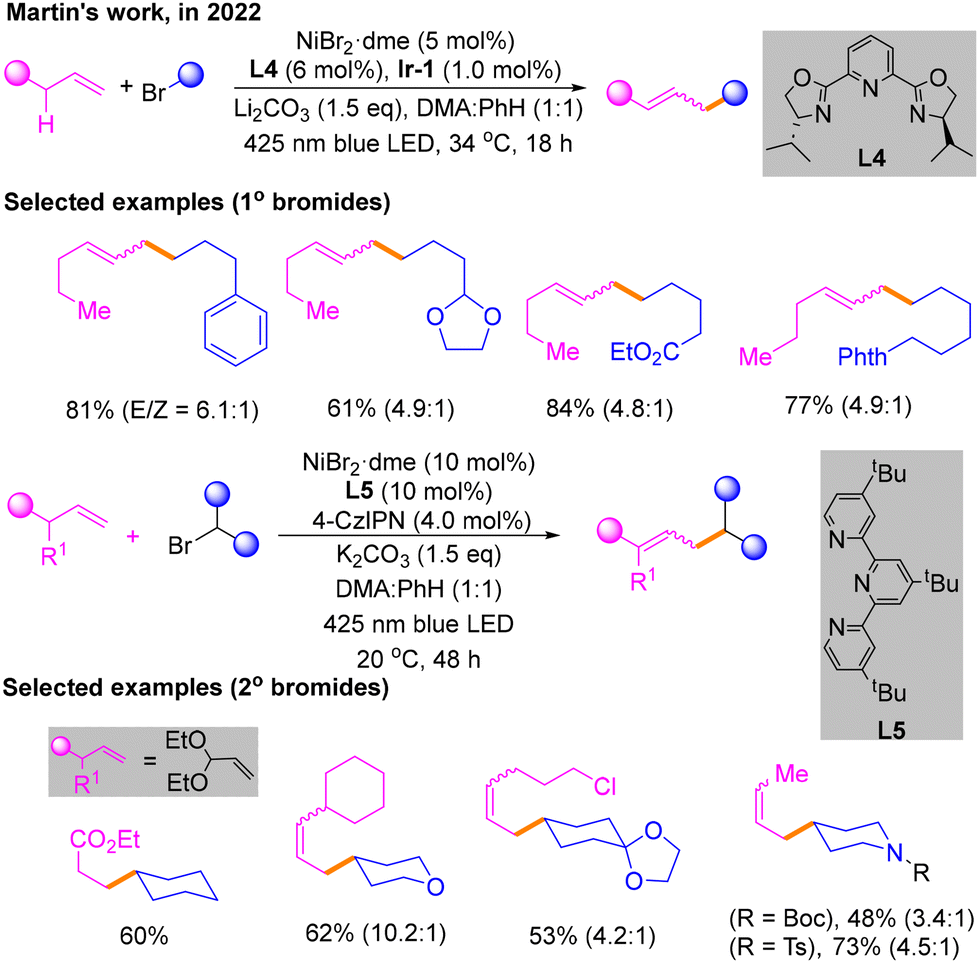 | ||
| Scheme 6 Csp3–H alkylation of an unactivated α-olefin. | ||
Soon after, MacMillan and co-workers developed a strategy for photo- and nickel-catalyzed Csp3–Csp3 cross-coupling reactions involving N-heterocyclic carbene (NHC)-mediated deoxygenation of alcohols and hypervalent iodine-mediated decarbonylation of carboxylic acids (Scheme 7).12 Methyl tertiary butyl ether (MTBE):dimethyl sulfoxide (DMSO) (1:1) is used as the solvent. Different from previously reported schemes, the two Csp3 radicals involved in the reaction are both generated during the photocatalytic SET process. In this event, the alcohol substrate reacts with NHC to afford the product 14, which can undergo SET with *IrIII. The resulting intermediate 15 undergoes the process of β-breakage to generate radical 16. Carboxylic acids are activated by iodine hypervalent to give 17. IrII then undergoes SET with 17 to generate another free radical 18 and IrIII. To maintain the catalytic cycle, blue light irradiation excites IrIII to *IrIII. The difficulty of this strategy is distinguishing between the two different free radicals 16 and 18 generated instantaneously. To solve this problem, based on the difference in the relative instability of the highly substituted metal-alkyl species and different strengths of nickel–carbon bonds, a radical sorting method was conceived. That is, 20 is more stable than 19, but their radical stabilities are the opposite with radical 18 being more stable than radical 16 (Scheme 8). After verifying the practicality of this idea, the author explored its generality with primary, secondary, and tertiary alkyl acids and alcohols under the optimal conditions.
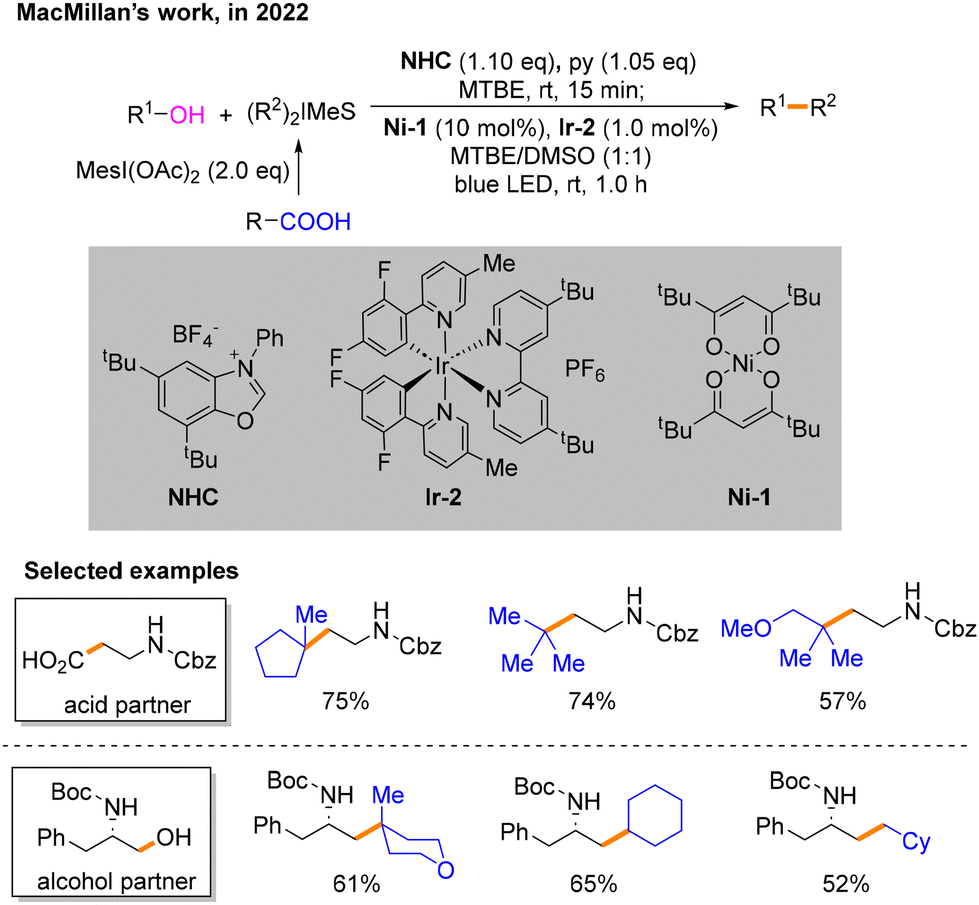 | ||
| Scheme 7 Cross-coupling reaction of alcohols and carboxylic acids. | ||
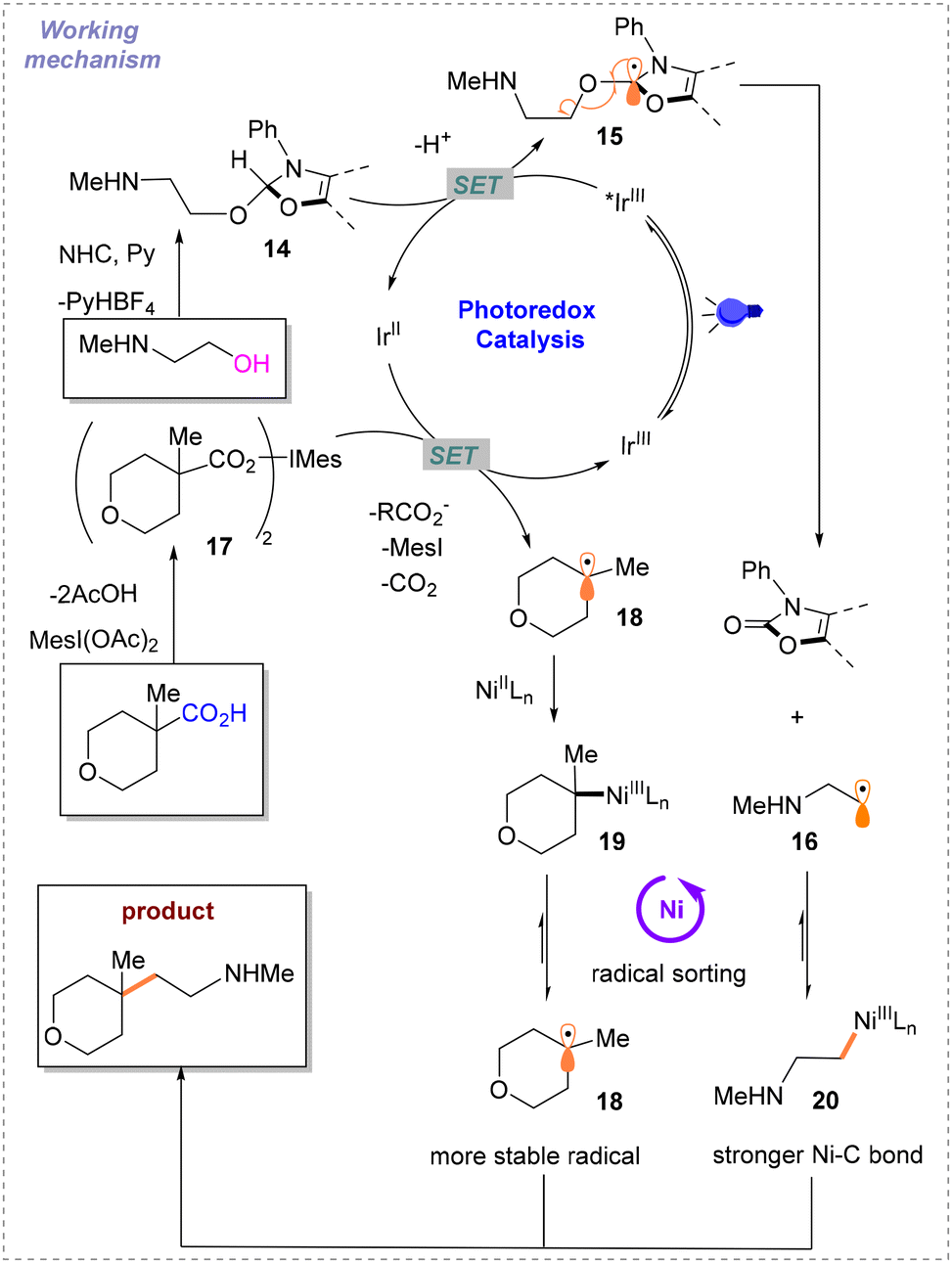 | ||
| Scheme 8 Proposed reaction design of cross-coupling reactions of carboxylic acids with alcohols. | ||
Two months later, a platform for cross-coupling mediated by Ni and a photocatalyst is developed by Martin's group to build Csp3–Csp3 bonds utilizing dihydroquinazolinones derived from ketones (Scheme 9).13 In this metal and photocatalyst dual catalytic system, the Ni and photo-induced catalytic cycle is similar to that discussed previously. Haloalkane substrates produce NiII species 21 in the Ni catalytic cycle. Alkyl radicals 22 are produced by the substrate dihydroquinazolinone in the photocatalytic phase. Then, 21 and 22 could undergo radical recombination to generate NiIII species 23. The product is also obtained by the reductive elimination of 23. However, a distinguishing feature of this reaction is that the dihydroquinazolinone serves as a radical source causing α C–C cleavage through aromatization, which is induced by single-electron oxidation of photocatalysis. The selected examples have also been shown in Scheme 9.
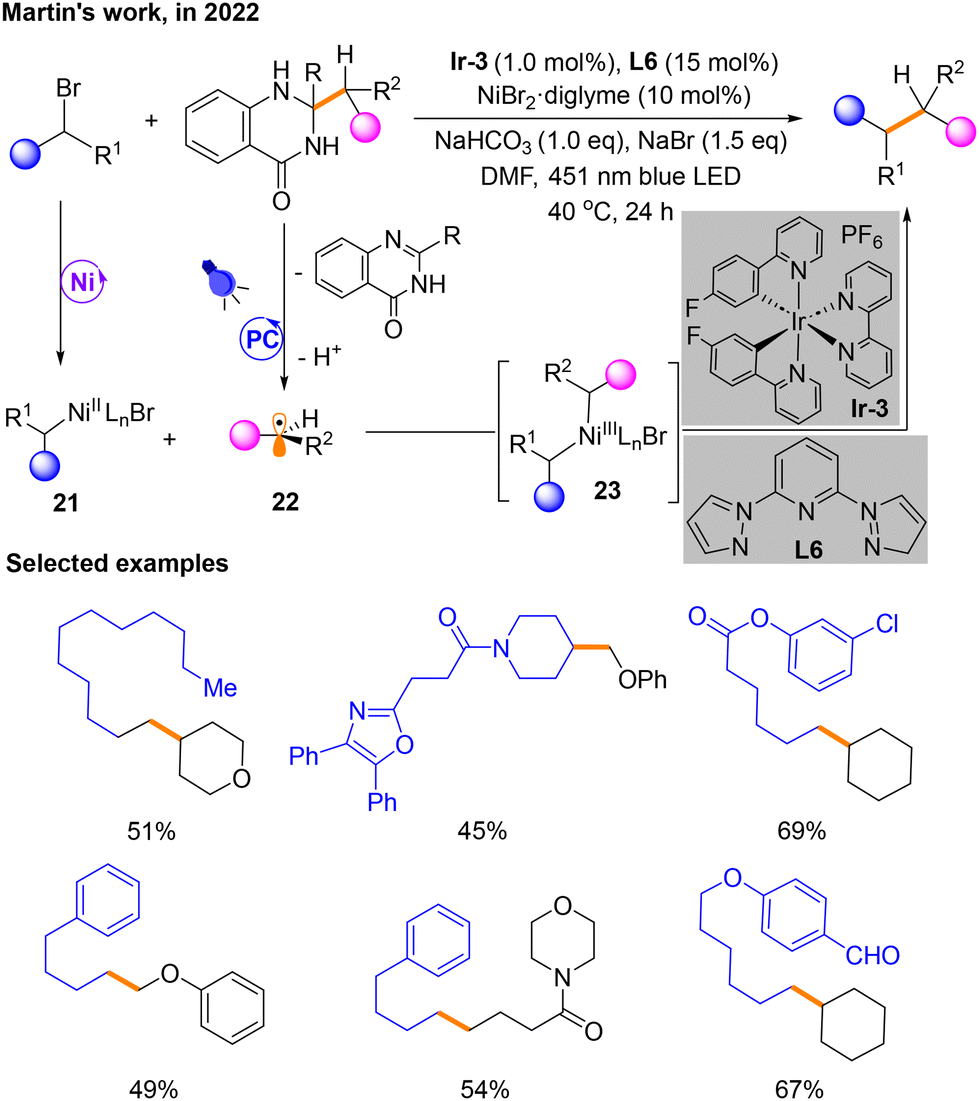 | ||
| Scheme 9 Csp3–Csp3 cross-coupling reaction of dihydroquinazolinones with bromoalkanes. | ||
Soon after, Breit and co-workers exploited a way for the construction of homoallylamines utilizing α-amino acids via photoredox Ni-catalytic cross-coupling (Scheme 10).14 This reaction enables the functionalization of allyl groups and diastereoselectively obtains branched allylation products involving α-amino radical intermediates. The first investigation was conducted using model substrates and determined the most optimal conditions: NiCl2·dme and L7 as the metal catalytic system, Cs2CO3 as the base, and [Ir(ppy)2(dtbbpy)]PF6 (Ir-4) as the PC. Subsequently, the arylallyl electrophile substrate was expanded using N-phenyl or N-p-methoxyphenyl (PMP) phenylalanine as the α-amino radical precursor. Mechanistic experiments illustrated that the free radicals involved in the catalytic cycle are generated by SET in the photocatalytic cycle and then decarboxylation. The diastereoselectivity of products was explained by DFT calculations. A critical step is the proton coupled electron transfer (PCET) process of the radical 24 and NiII species 25 to afford 26. Then, 26 in combination with the allyl group undergoes a nucleophilic attack on the imine to give the desired product.
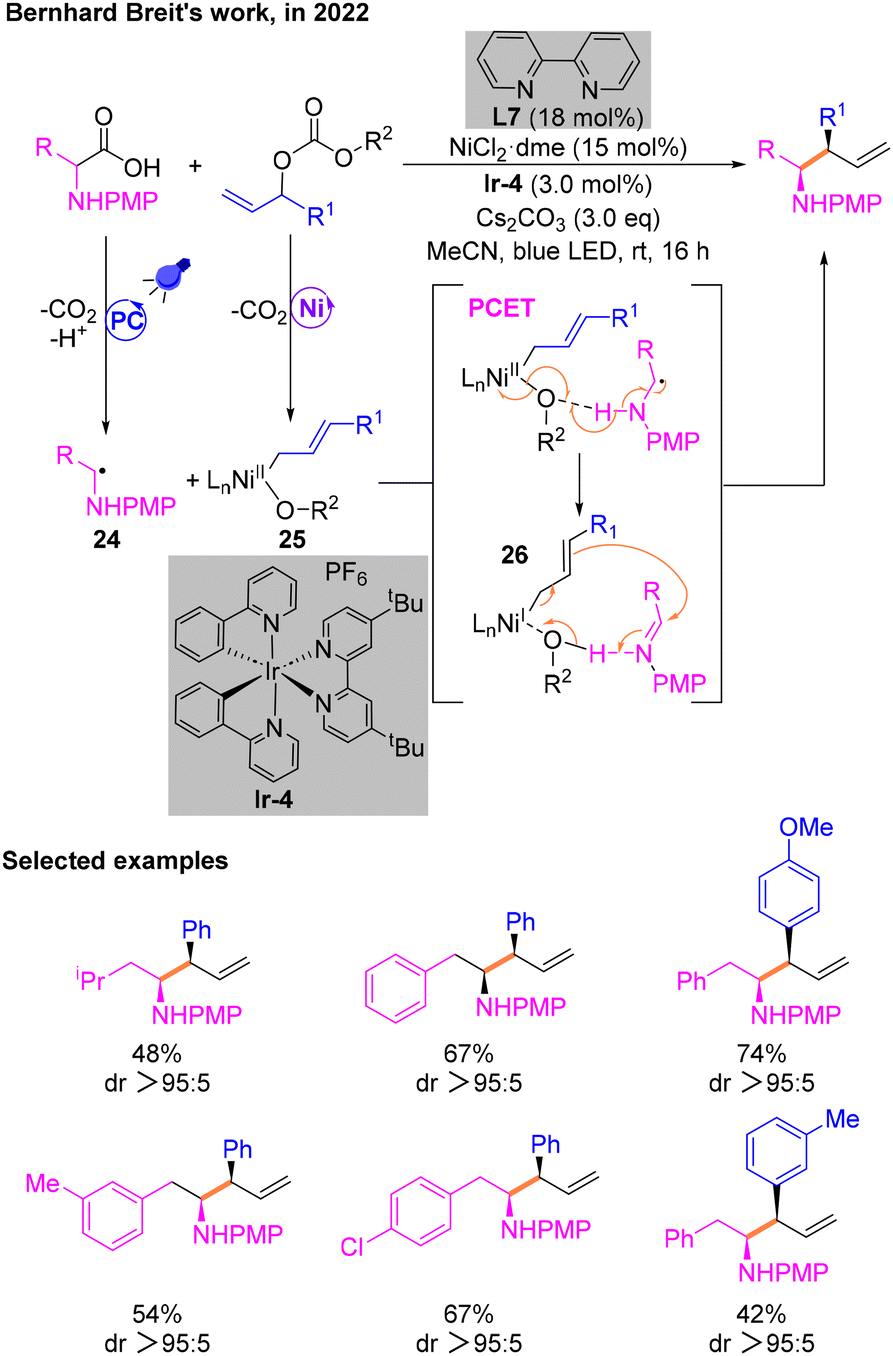 | ||
| Scheme 10 Synthesis of homoallylamines by a cross-coupling reaction. | ||
A few months later, Doyle's group utilizing benzaldehyde dialkyl acetals derived from alcohols developed a method for Csp3–Csp3 cross-coupling reactions with aziridines (Scheme 11).15 The product is obtained with NiBr2·dme and L1 as the nickel catalytic system using Ir-2 as the PC under blue light exposure in MeCN:PhH (1:1) mixed solvent. The function of the additive LiBr is to stabilize the anionic sulfonamide and facilitate the release of the product. Benzaldehyde dialkyl ketals are employed by HAT to produce radical species 27 under the stimulation of a photocatalyst and bromide ions. Mechanistic studies show that the activation of dialkyl acetals via HAT by Br− happens during the photocatalytic phase to give 27, and the oxidative addition of aziridines occurs in the Ni catalytic cycle to afford 28, explaining the differential activation of the two coupling partners. The product is released by the reductive elimination reaction from NiIII species 29.
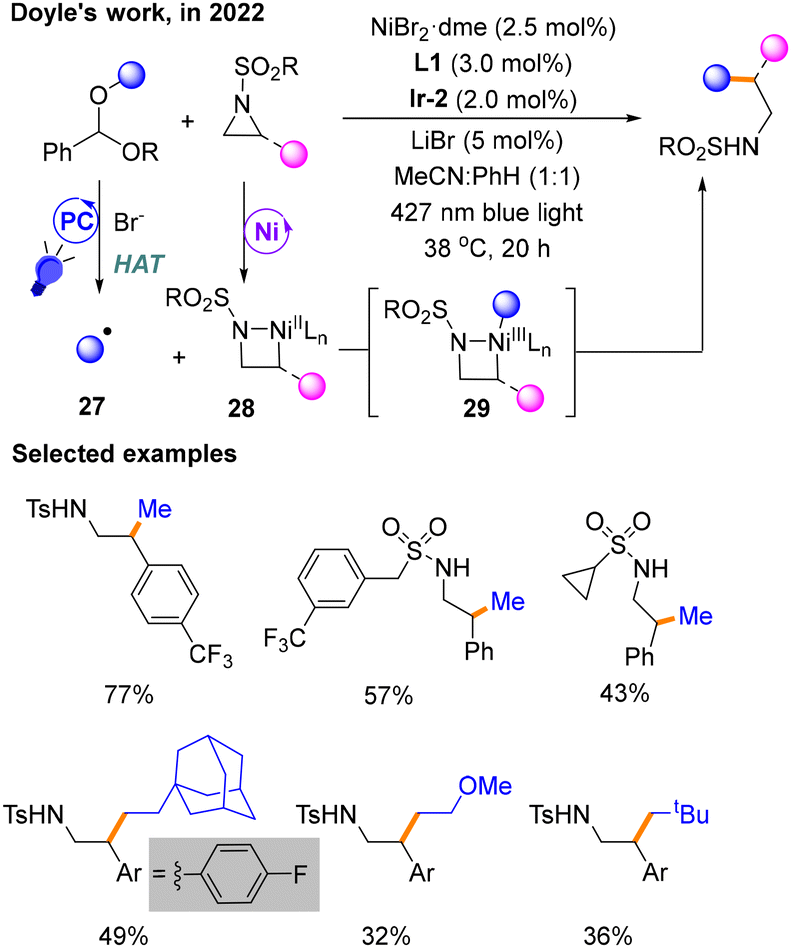 | ||
| Scheme 11 Csp3–Csp3 cross-coupling reaction of benzaldehyde dialkyl acetals with aziridines. | ||
Then, inspired by the Csp3-methyl group catalyzed by radical S-adenosylmethionine (SAM) methyltransferase in biological systems and some previous work, MacMillan and co-workers reported a Csp3–Csp3 coupling reaction through radical sorting and bimolecular homolytic substitution (SH2), in which direct methylation occurs on saturated heterocyclic Csp3–H bonds (Scheme 12).16 The most ideal conditions are irradiation under 365 nm light, Ni(acac)2·KTp* as the metal catalyst, Na3PO4 as the base and tetrabutylammonium decatungstate (TBADT) as the HAT reagent and photocatalyst in acetone. The universality of this strategy is also assessed. These two alkyl radicals are respectively produced by HAT and decarboxylation in the photocatalytic cycle. The excited triplet state of decatungstate *[W10O32]4− under light undergoes a HAT process at the hydridic α-amino C–H bond of the first substrate, resulting in reduced [W10O32]5− as well as alkyl radical 30. Another radical 31 is produced during the disproportionation cycle from the second substrate. Key to the success of this mechanism is the previously mentioned radical sorting effect mediated by nickel. Radicals 30 and 32 are more stable than 31 and 33, respectively. In addition, products are obtained from the SH2 process.
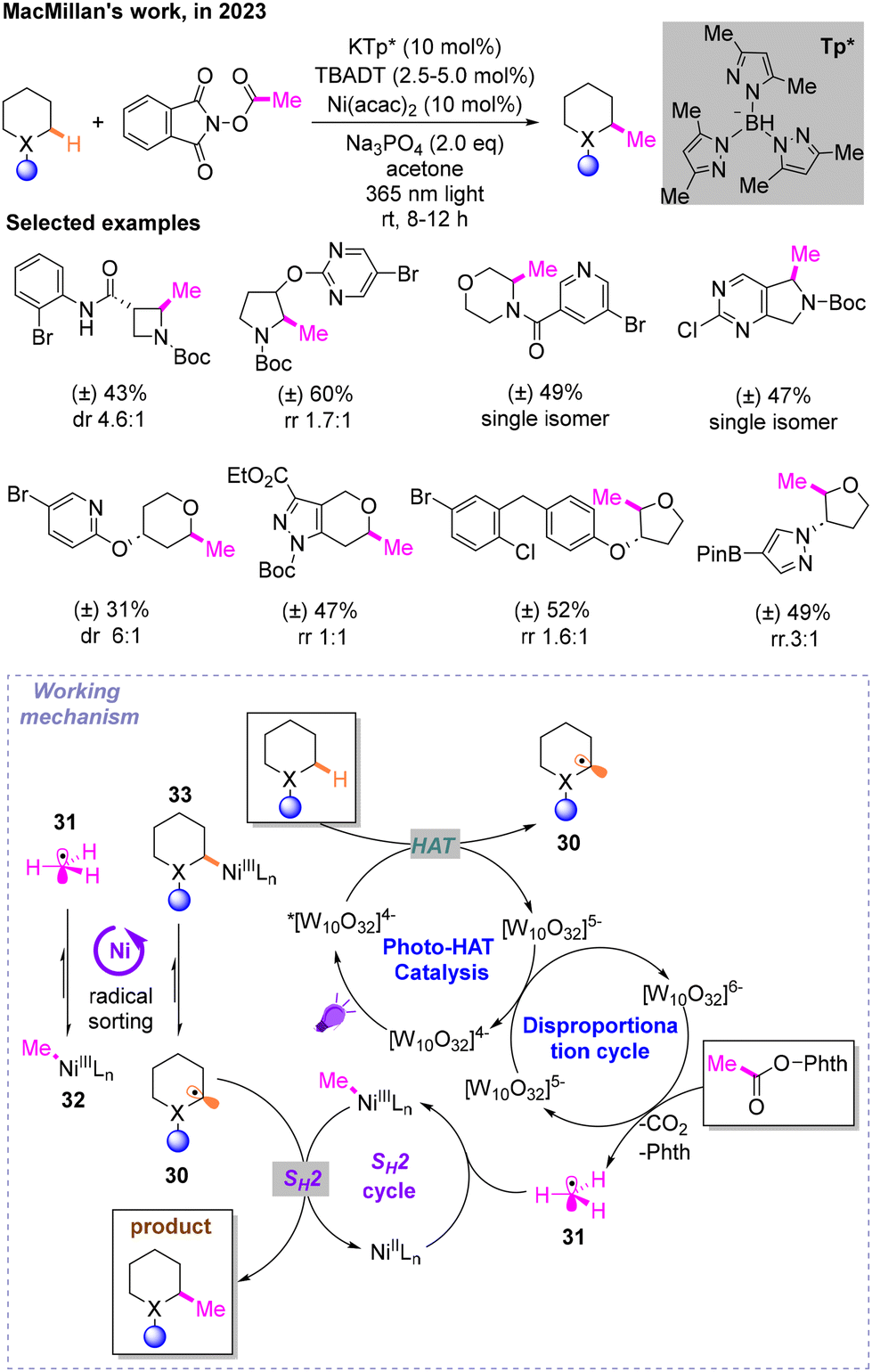 | ||
| Scheme 12 Methylation of saturated heterocyclic Csp3–H bonds. | ||
In 2023, Macmillan17 used NHC to activate alcohols and performed coupling reactions with brominated hydrocarbons to build Csp3–Csp3 bonds (Scheme 13). In the photocatalytic cycle, the alcohols are activated by NHC-mediated C–O homolysis to produce alkyl radicals, which is similar to their previous reports. Then, the resulting NiI species 34 during nickel and photo-induced catalysis undergoes oxidative addition with halogenated hydrocarbons to produce NiIII species 35, and finally the target product is obtained through reductive elimination of 35.
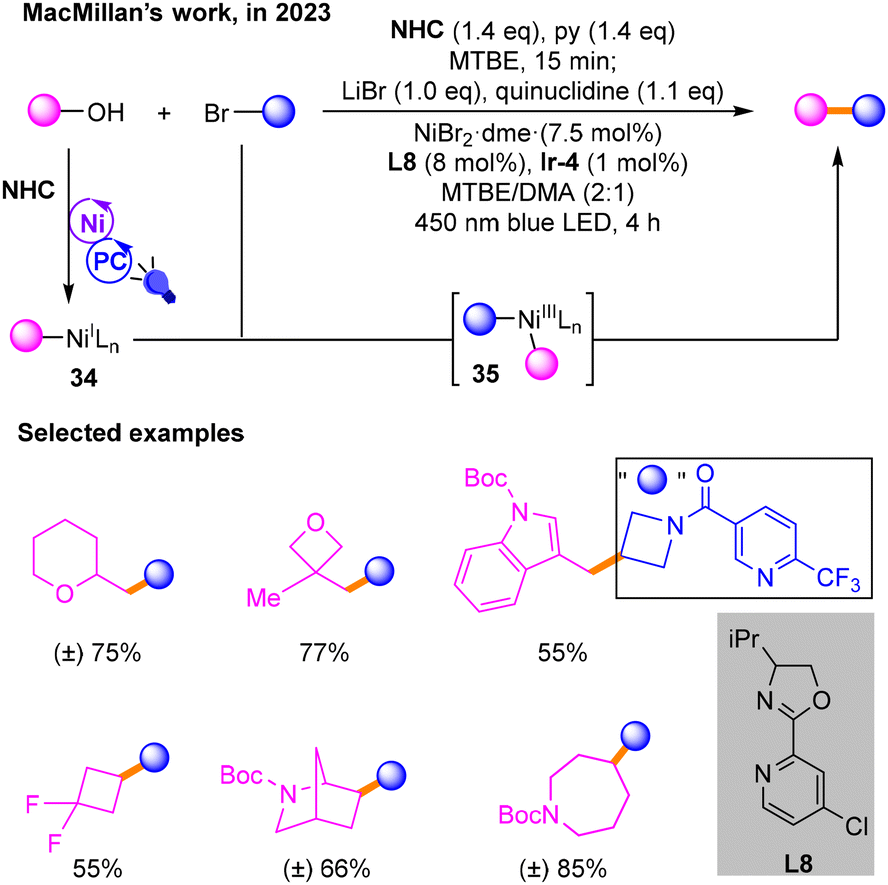 | ||
| Scheme 13 The cross-coupling reaction of activated alcohols with NHC and brominated hydrocarbons. | ||
Later, a light/nickel-catalyzed Csp3–Csp3 cross-coupling reaction of α-silylamines with unactivated alkyl halides, which could occur on the ipso and remote sites of alkyl halides selectively through ligand regulation, was for the first time described by Huang and Yuan in 2023 (Scheme 14).18 They found that photosensitizer Ir-4 is the most suitable for the ipso-site reaction, and 4-CzIPN is best for the remote-site reaction. After careful screening of the reaction conditions. α-N-Alkyl radicals participating in the nickel catalytic cycle are generated during the photocatalytic cycle. When L9 is used as the ligand, the olefin substrate will isomerize to form the ipso-site product (Path A). Moreover, the remote-site product can be output when the ligand is L10 (Path B). Remote- and ipso-site products are obtained from different NiIII species: 41 and 42. Mechanistic experiments have shown that β-hydrogen is necessary for the generation of remote-site products during the chain-walking process. DFT calculations proved that the steric hindrance of ligands is a factor affecting site selectivity.
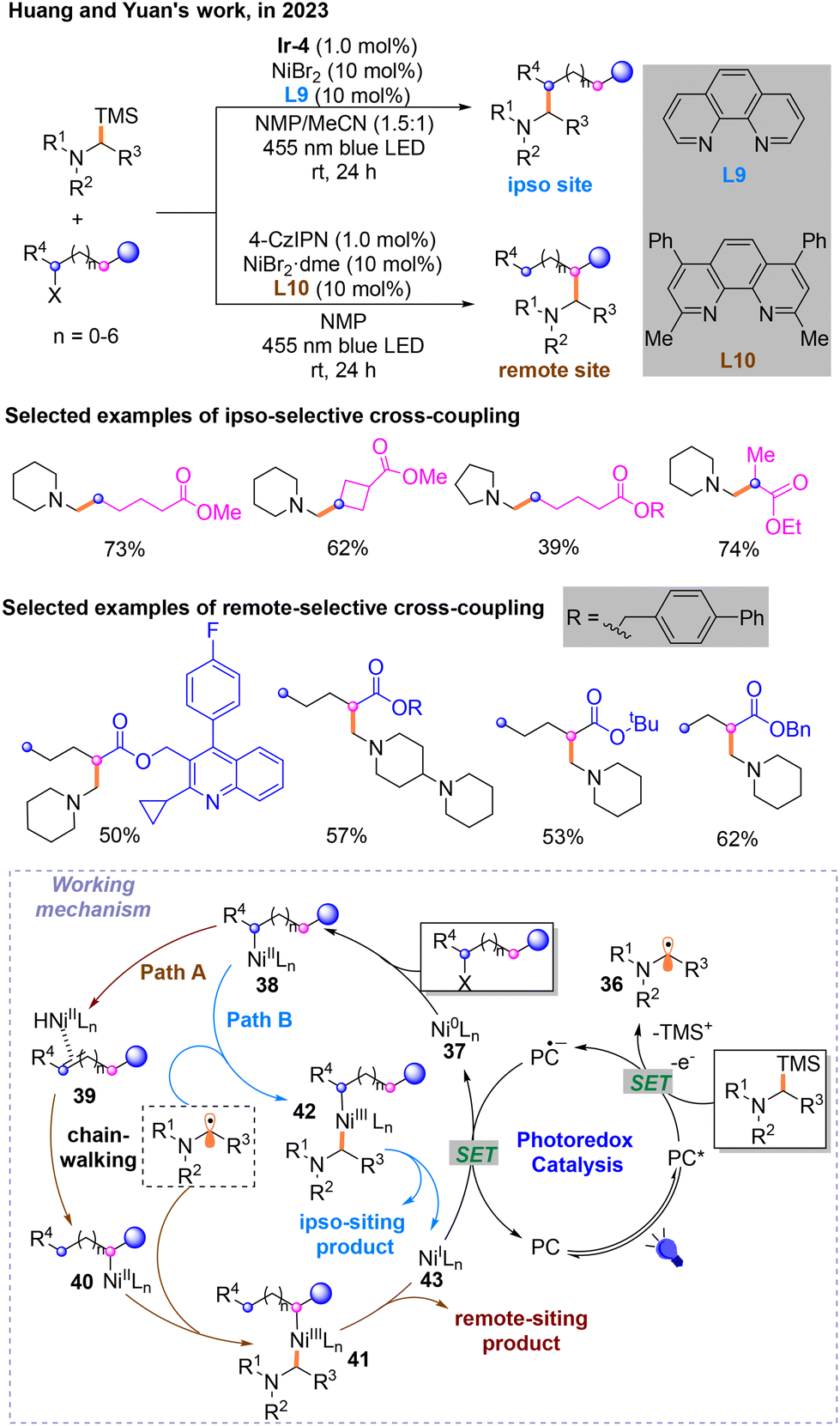 | ||
| Scheme 14 Csp3–Csp3 cross-coupling reaction of α-silylamines with alkyl bromides. | ||
After that, Liu and Fang reported a new Csp3–Csp3 cross-coupling system through decarboxylation19 mediated by a PC and the subsequent coupling of two radicals by a nickel catalyst (Scheme 15). This reaction was performed under the irradiation of a purple LED with dichloroethane (DCE) as the solvent, unlike most reactions that use a blue LED as the light source. Both radicals 44 and 45 are generated via an energy transfer (EnT) process during the photocatalytic stage, followed by a nickel-mediated cross-coupling reaction to afford NiIII species 46. The reductive elimination of 46 occurs to produce the target product.
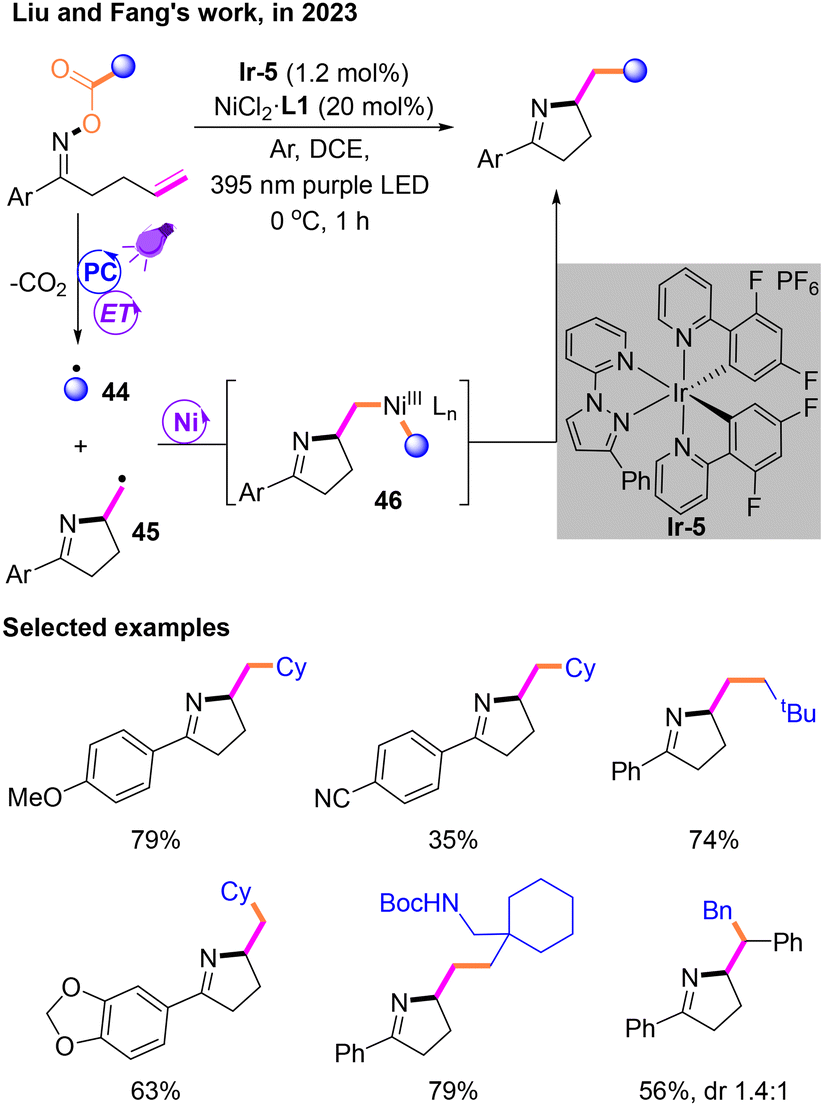 | ||
| Scheme 15 Csp3–Csp3 decarboxylated cross-coupling reaction via EnT. | ||
Subsequently, a photonickel-cocatalyzed Stille coupling reaction was developed by Zhu and Xue (Scheme 16).20 After determining the reaction conditions for Csp2–Csp3 coupling, the author evaluated the scope of this protocol, in which only a Csp3–Csp3 coupling product was reported. Several mechanistic studies and DFT calculations were carried out to show how this reaction proceeds. There are two pathways A and B for this reaction. The difference between them is whether conventional oxidative addition occurs first to give 48 followed by single-electron oxidative addition to give 49 (Path A), or whether the opposite occurs to give 50, followed by 49 (Path B). The products and NiI species 51 of both pathways are obtained by reductive elimination of NiIII species 52. Radical 53 could be captured by 51 to get 52, which would be reduced to 50.
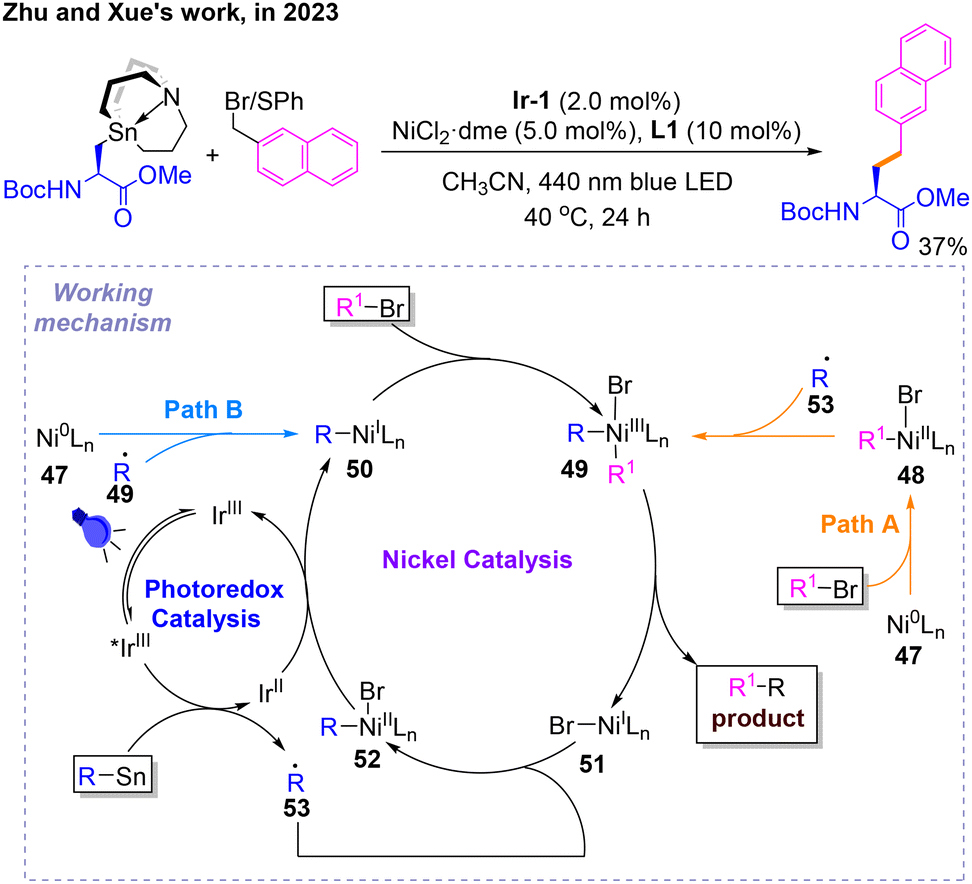 | ||
| Scheme 16 Photo- and nickel-catalyzed Stille coupling reaction. | ||
A month later, a Csp3–Csp3 cross-coupling reaction of α-bromoboronates and β-amino boronates through C–H bond activation is reported by Xu and co-workers to forge β-amino boronates (Scheme 17),21 whose synthesis is currently rare. It is worth noting that the additional use of ligands will decrease the yield, and thus this strategy does not involve supernumerary ligands. In addition, unlike most of the above methods, this strategy uses the irradiation of purple light to maintain the catalytic cycle. Mechanistically, α-bromoboronate undergoes a one-electron redox reaction in nickel catalysis to produce α-borate radical 54. The α-N-alkyl radical produced by the photocatalytic cycle via the HAT process is captured by the Ni catalytic cycle to afford NiII species 55 and then combined with 54. Finally, the product is obtained through reductive elimination of NiIII species 56.
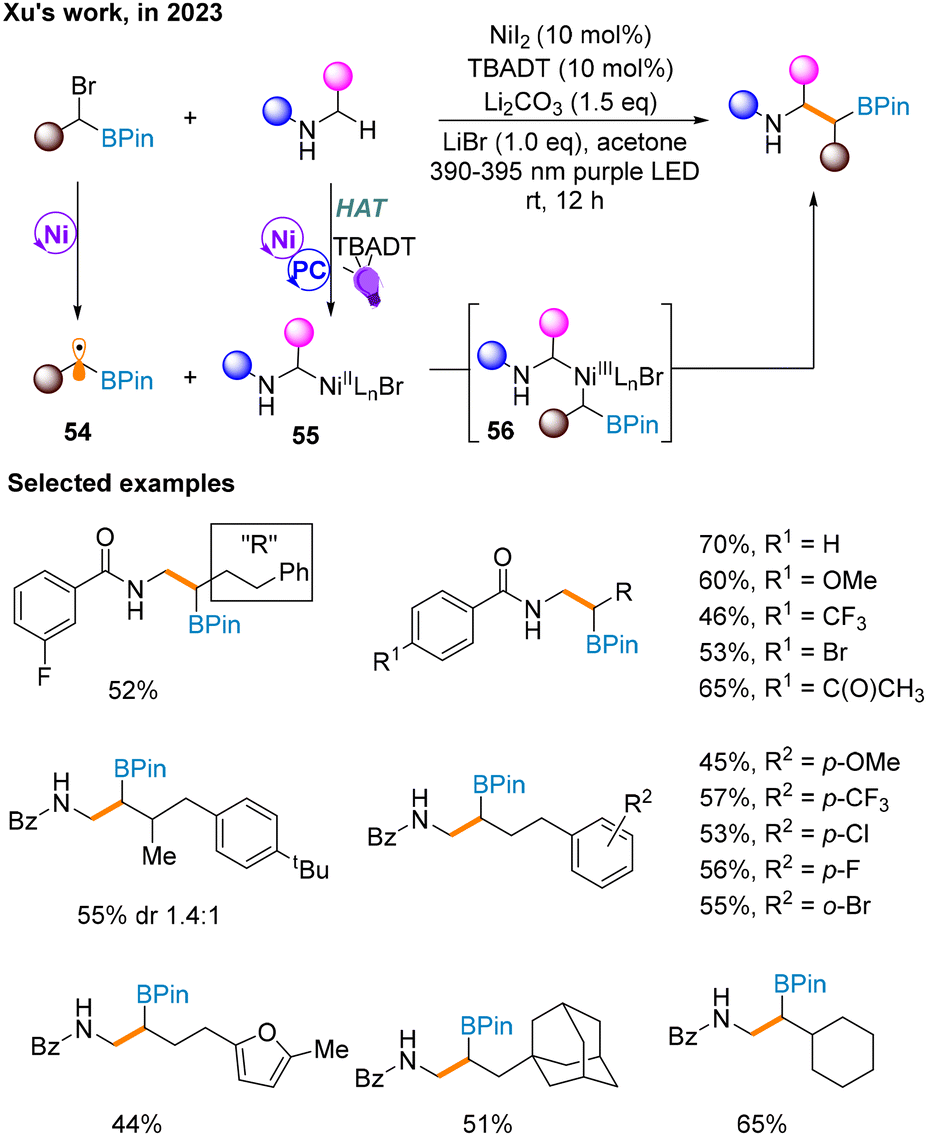 | ||
| Scheme 17 Synthesis of β-amino boronates via a Csp3–Csp3 cross-coupling reaction. | ||
3. Reductive cross-coupling
Reductive cross-coupling reactions are also an important class of reactions in organic chemistry.22 Reactants usually undergo electron transfer and recombination of chemical bonds in the presence of a reducing agent. In this reaction, a reducing agent is used to facilitate the formation of new carbon–carbon bonds between different organic fragments. In recent years, there have been a number of reductive coupling reactions under photoredox/nickel dual catalysis.In 2020, Martin and co-workers demonstrated the successful development of a dual photoredox/Ni-catalytic process that gives access to alkylation of Csp3via β-scission of a special class of aliphatic alcohol derivatives, N-phthalimide ethers (Scheme 18).23 Specifically, utilizing 4-CzIPN as the PC, K2CO3 as the base, Hantzsch ester (HE-1) as the electron donor (ED) and NiCl2·L5 as the metal catalyst under the irradiation of blue light, a series of Csp3–Csp3 coupling structures were produced. The range of Csp3 alkylation was studied, explaining the influence of ligand denticity. Some examples are selected and shown in Scheme 18. In this protocol, HE-1 is not only able to reduce N-phthalimide ethers (Ophth) to provide radicals 57, but it can also maintain the photocatalytic cycle. The photocatalytic cycle does not generate 57 involved in coupling, but 57 is generated by HE. Subsequently, the β-breakage of 57 occurs through the single electron activity of the ether with HE˙+, generating alkyl radical species 58 that participate in the nickel and photocatalytic cycle to afford the product. In addition, the authors suggest that there may be an electron-donor–acceptor (EDA) complex between HE and the substrate prior to β-scission.
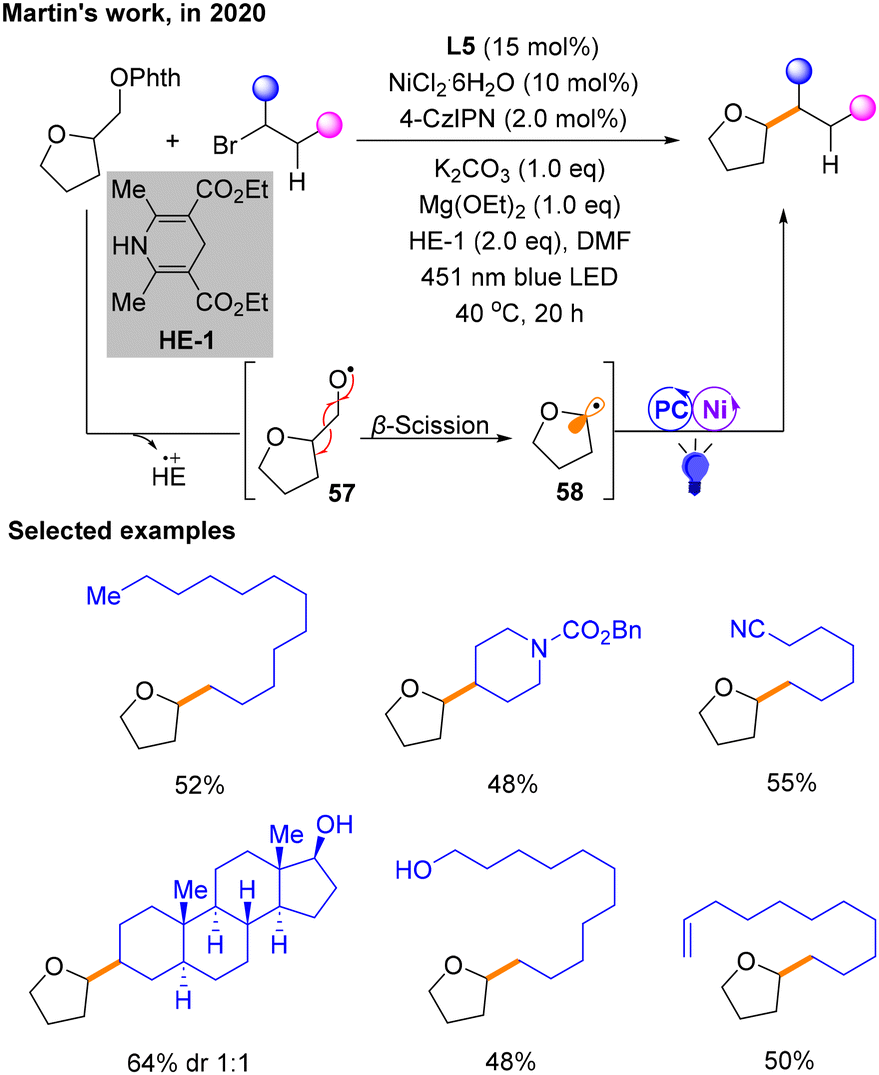 | ||
| Scheme 18 Cross-coupling reaction of sp3 alkylation via β-scission of N-phthalimide ethers. | ||
A year later, affected by the redox active characteristics of N-alkylpyridinium salts, a technology for constructing a Csp3–Csp3 skeleton via a photo-induced and nickel-catalyzed cross-electrophile coupling reaction with N-alkylpyridinium salts as radical precursors was disclosed by Koh in 2021 (Scheme 19).24 After optimization studies, the author explored the substrate range of deamination alkylation reactions with bromides and iodides, respectively, and found that the iodides could perform Csp3–Csp3 cross-coupling reactions effectively. Then, several mechanistic experiments were conducted, and it is believed that the proposed mechanism may involve EDA complexes formed between N-alkylpyridinium salts and HE-1. Then, photoexcitation and SET occur to generate dihydropyridine radicals 59 and HE˙+60, followed by deamination cleavage triggered by aromatization to produce triphenylpyridine 61 and alkane radical 62 involved in the catalytic cycle. The species 60 and 61 will reduce NiI66 to Ni063. 63 would then undergo an oxidative addition reaction with halogenated hydrocarbons to produce NiII64, which could capture the previously generated alkyl radicals 62 affording NiIII species 65. To complete the catalytic cycle, reductive elimination of 65 occurs to produce the desired products and 66.
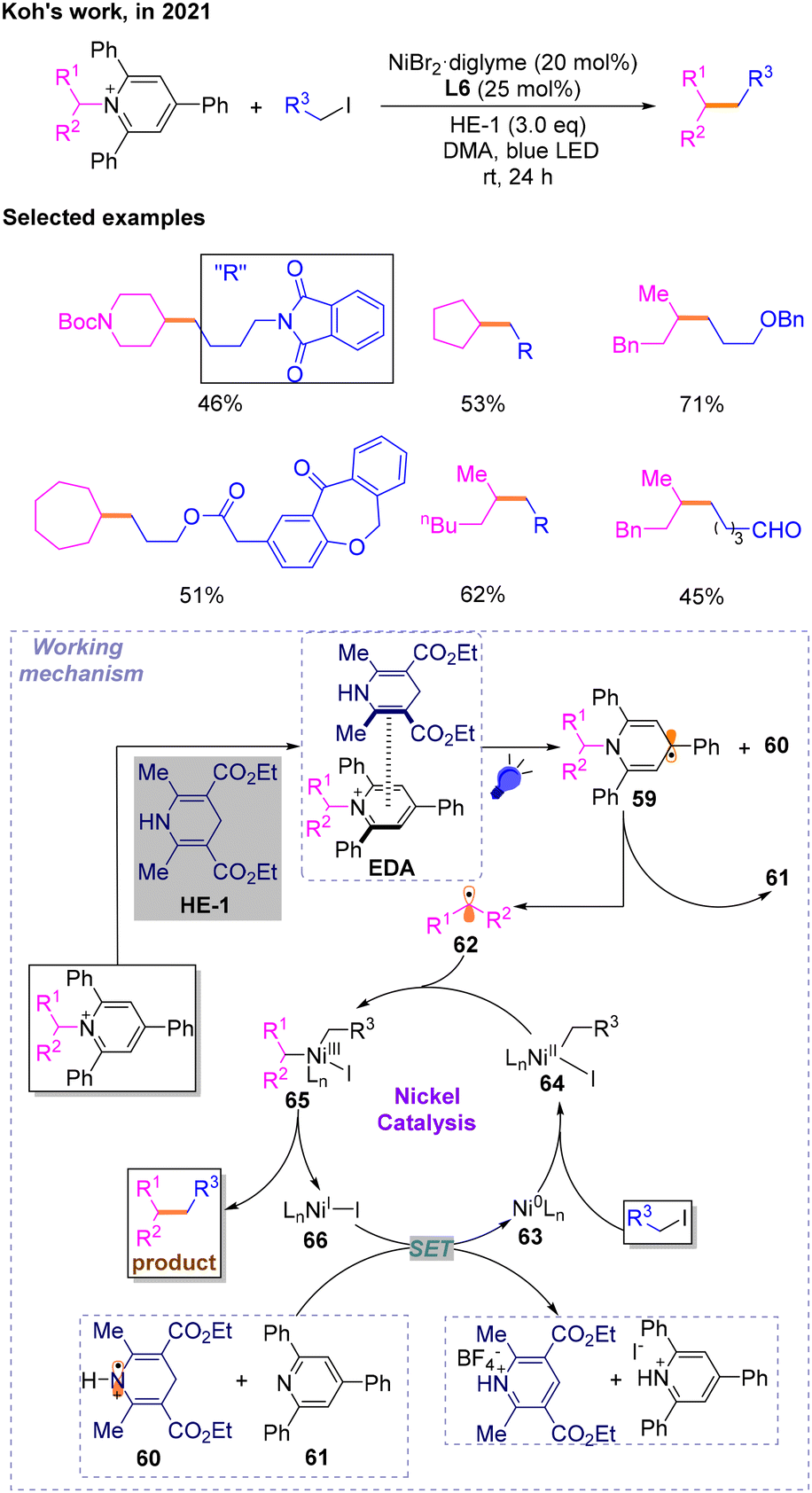 | ||
| Scheme 19 The coupling reaction involving N-alkylpyridinium salts as radical precursors. | ||
In 2022, Maji and co-workers proved that metallaphotoredox catalysis can be a prominent strategy to make the cyclopropane ring via an intramolecular Csp3–Csp3 cross-electrophile coupling reaction (Scheme 20) of 1,3-alkyl electrophiles.25 The reaction conditions were optimized using primary alkyl 1,3-dimesylate with an ester functionality as a model substrate, and the optimal reaction conditions were finally determined: Ir-6 as the PC, NiBr2 with L1 as the transition metal catalyst, nBu4NBr as the additive, triethanolamine (TEOA) as the ED, N,N-dimethylformamide (DMF) as the solvent and the reaction was performed at room temperature under the irradiation of a blue LED. In addition, secondary alkyl electrophiles could also react smoothly when one of the halogen atoms in the substrate is replaced with OMs. As the author speculated, PC* was quenched to PC˙− by the ED. The nickel catalytic cycle starts with NiI67 generated from a photocatalytic SET involving the ED, undergoing oxidative addition with halogenated hydrocarbons via a radical process to form NiIII68, which is subsequently reduced to NiII69 by PC˙− delivering the PC. PC˙− is regenerated under the blue light irradiation and ED to complete the photocatalytic cycle. Subsequently, 69 undergoes oxidative addition and reductive elimination to produce the target product. The new NiII species 70 produced in this step undergoes the same photocatalytic process and is reduced to NiI67 to enter the next catalytic cycle.
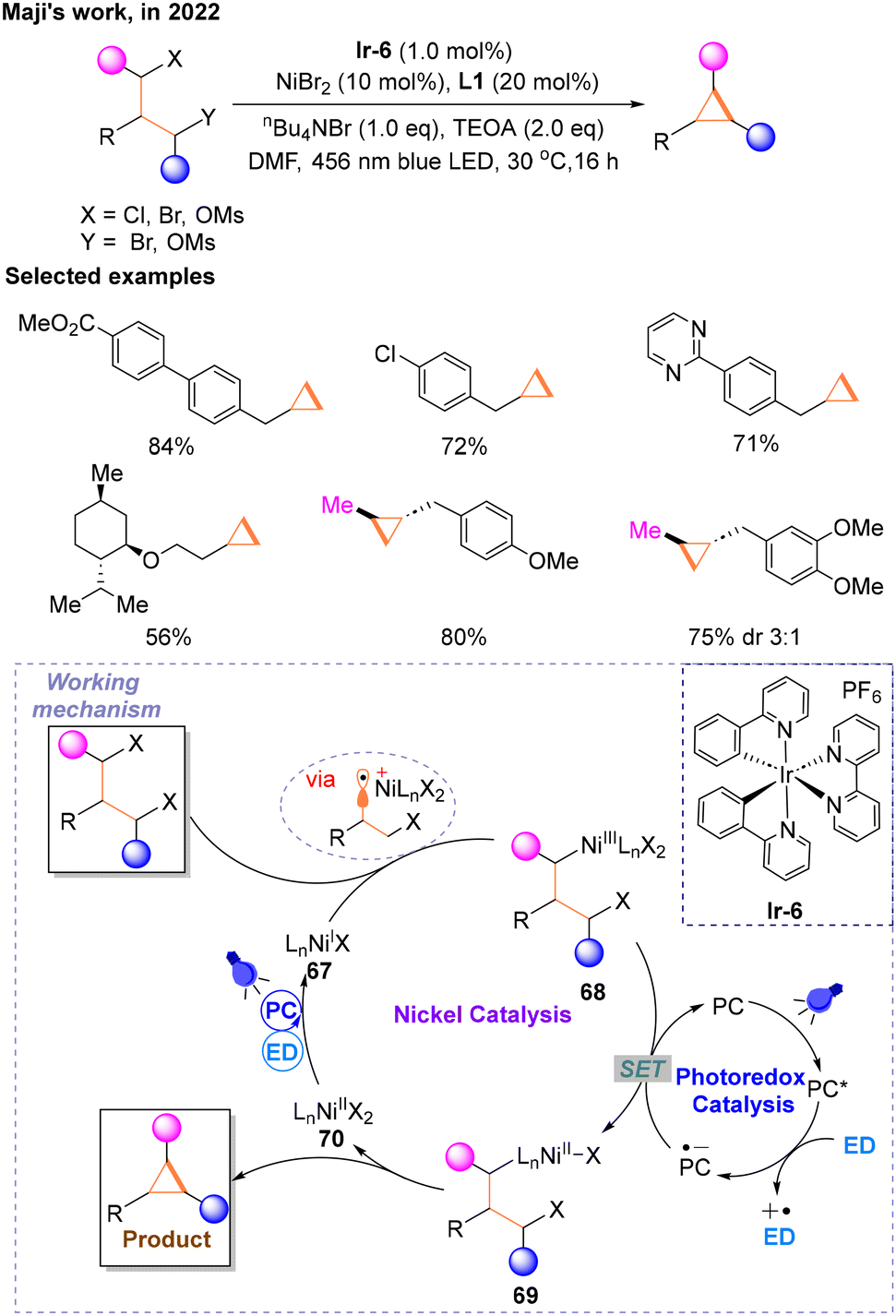 | ||
| Scheme 20 Synthesis of cyclopropanes via an intramolecular Csp3–Csp3 cross-coupling reaction. | ||
Later, Oestreich's research group achieved a reductive cross-coupling reaction between α-silylated alkyl bromides and an allylic sulfone using a HE as the reducing agent in 2022 (Scheme 21).26 It has been experimentally verified that the key to the successful conduction of the reaction is the ability of the methylsilane at the α-position of the alkyl bromide to stabilize the carbon radicals. After conditional screening, it is determined that the most qualified conditions are NiBr2·diglyme as the nickel precatalyst and terpyridine L11 as the ligand as well as HE-2 and Et3N under irradiation with a blue LED in DMA as the solvent. The reaction scope of this coupling reaction was subsequently evaluated under these conditions. Notably, this process does not require a photocatalyst but only blue light irradiation to maintain the catalytic cycle. Thus, further study is needed to gain mechanistic insights.
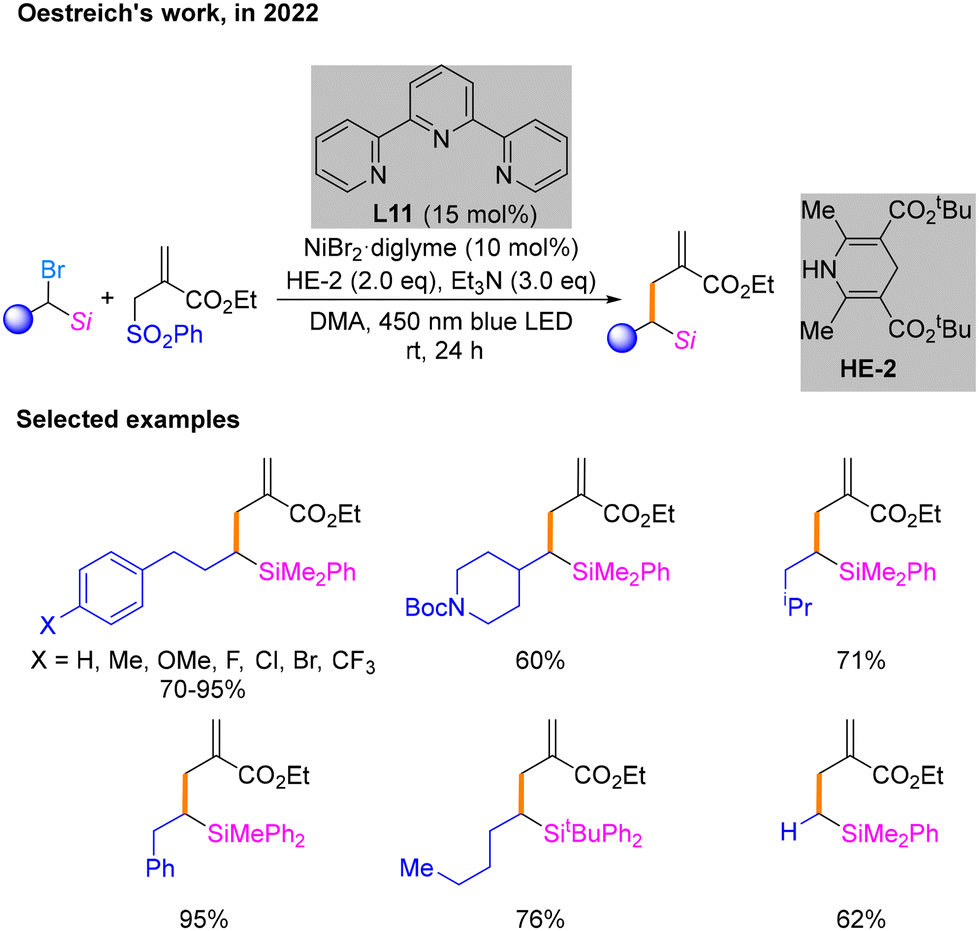 | ||
| Scheme 21 Cross-coupling reaction of α-silylated alkyl bromides and an allylic sulfone. | ||
In 2023, Xu and co-workers described Csp3–Csp3 cross-coupling with dual Ni/photocatalysis utilizing unactivated alkyl halides and racemic α-chloroboronates as substrates (Scheme 22).27 This strategy enables the highly selective synthesis of chiral secondary alkylborates, important intermediates in organic synthesis, making it possible to rapidly construct enantiomerically enriched complex molecules. This reaction could achieve the highest yield in the NiBr2·diglyme catalytic system using 4-CzIPN as the PC and DMA:PhCF3 (1:1) as the solvent in the presence of some additives. Then, the substrate scope of α-chloroborates and alkyl iodides for reductive cross-coupling was expanded. Based on radical-clock reactions and other mechanistic verification experiments, the author proposed that a Csp3 radical was formed in the photocatalytic stage from alkyl iodides under the irradiation of blue light. In addition, α-chloroboronates are activated in the nickel catalytic cycle to afford NiII species 71. Subsequently, free radical addition and reductive elimination with 72 occur successively to generate the final product through the NiIII species 73. HE-1 also acts as the ED to maintain the photocatalytic cycle.
 | ||
| Scheme 22 Cross-coupling of α-chloroboronates with alkyl iodides. | ||
4. Oxidative cross-coupling
Oxidative cross-coupling offers a new approach for constructing C–C bonds.28a In this type of reaction, transition-metal catalysts and oxidants are usually utilized to induce cross-coupling of two different nucleophilic organic molecules,28b,c resulting in the formation of new carbon–carbon or carbon–hetero bonds. In recent years, the construction of Csp3–Csp3 bonds by oxidative cross-coupling has been greatly developed, especially in the field of electrocatalysis.28d However, with regard to Ni/photoredox dual catalysis, there are limited cases of Csp3–Csp3 bond construction in oxidative cross-coupling.In 2022, a decarboxylative Csp3–Csp3 cross-coupling reaction29 of aliphatic acids was developed by MacMillan's group (Scheme 23). The two free radicals of this reaction are produced after the corresponding aliphatic carboxylic acid substrates are activated by MesI(OAc)2. Then radical 74 and NiIII species 75 are produced during the radical sorting process, and they finally produce the target product through SH2. When thioxanthone (TXO) is used as the photosensitizer, the reaction can occur under 365 nm violet light irradiation, and it is worth noting that when the photosensitizer is changed to 4-CzIPN, the product can also be obtained under the irradiation of a 450 nm blue LED. In addition, MesI(OAc)2 acts as the oxidant in this reaction.
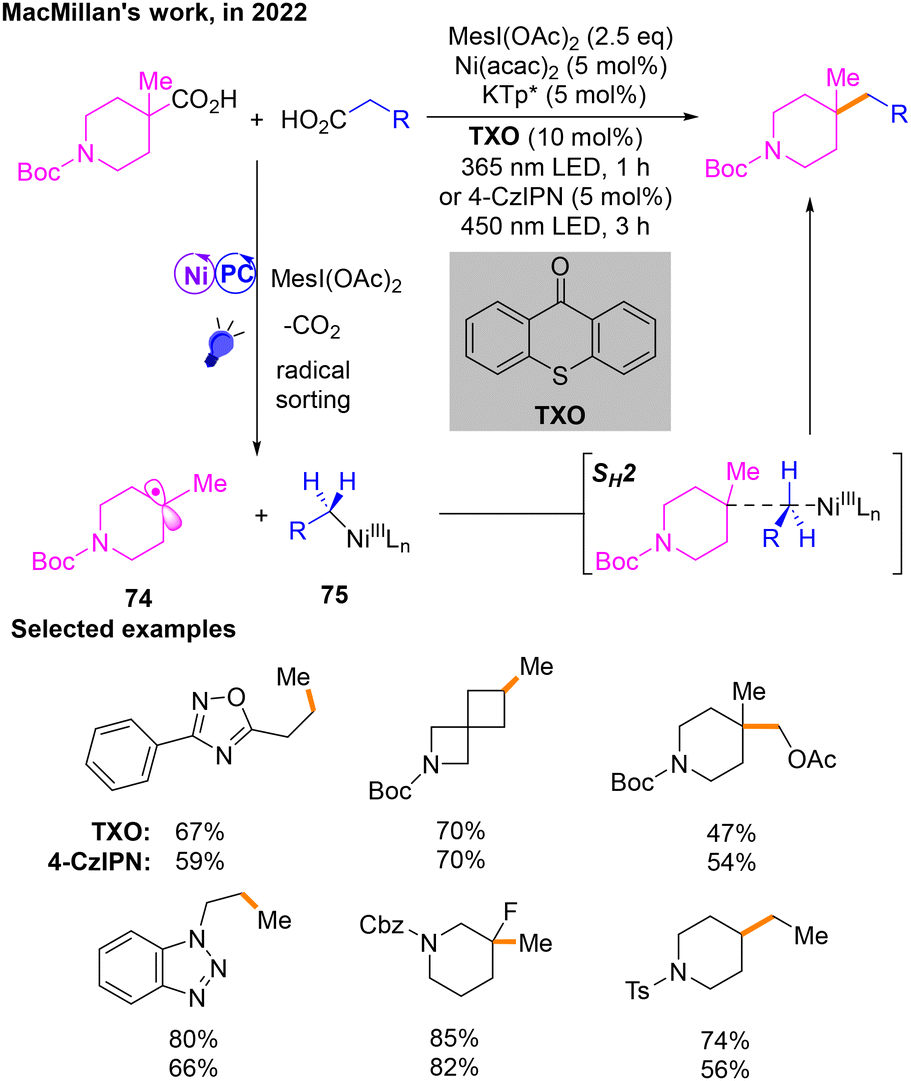 | ||
| Scheme 23 Decarboxylative cross-coupling of aliphatic acids. | ||
5. Conclusion and outlook
This minireview mainly covers the research on Ni/photoredox dual catalyzed Csp3–Csp3 cross-coupling reactions from 2019 to 2023. Under clean and mild light irradiation conditions and cheap nickel metal catalysis, various compounds containing C–C structures can be easily obtained, avoiding the difficulty of using traditional metal-catalyzed coupling reactions. To generate Csp3 radicals, a number of chemical bonds (Csp3–Cl bonds, Csp3–Br bonds, Csp3–C bonds, Csp3–O bonds, etc.) have flexibly participated in redox-neutral coupling reactions and reductive cross-coupling reactions in the past five years. In some specific transformations, coupling reactions have been developed that do not require additional photocatalysts or ligands. However, most coupling reactions are driven by blue LED light, and very few reactions are driven by purple light. In addition, the stereo- and regio-selectivities of these reactions still need further study.In the next stage, we anticipate that the development trend of the Csp3–Csp3 coupling reaction in Ni/photoredox dual catalysis may have the following six aspects: (a) recently, the asymmetric version of this field has experienced significant development.30 It is believed that more efforts will be devoted to exploring asymmetric Csp3–Csp3 coupling reactions by introducing chiral ligands into the catalytic system; (b) the mechanisms of some specific related reactions will be further clarified. Some new mechanisms will also emerge and become increasingly clear; (c) the formation method of C-centered sp3 hybridized radicals that can participate in Csp3–Csp3 cross-coupling reactions will be further expanded; (d) more classic conventional coupling reactions may be extended and also participate in the photo-nickel synergistic catalytic system to enable the formation of Csp3–Csp3 structures; (e) site-selective Csp3 alkylation via Csp3–Csp3 cross-coupling reactions of dual photoredox/Ni-catalysis is another important direction and requires future development. We believe that the challenges in this area will eventually be overcome and more efficient, green, and gentle methods will be developed. (f) More types of light sources (such as red and green light) may be used in Csp3–Csp3 cross-coupling reactions in the near future.
Author contributions
Shi, M. directed the perspective and revised the manuscript. Huang, Q. Y. carried out the literature collection, organization, wrote the manuscript, drew the schemes and reviewed them.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Key R&D Program of China (2023YFA1506700), the National Natural Science Foundation of China (21372250, 21121062, 21302203, 20732008, 21772037, 21772226, 21861132014, 91956115 and 22171078), a project supported by the Shanghai Municipal Science and Technology Major Project (Grant No. 2018SHZDZX03) and the Fundamental Research Funds for the Central Universities 222201717003.References
-
(a) S. Bhakta and T. Ghosh, Nickel-catalyzed hydroarylation reaction: a useful tool in organic synthesis, Org. Chem. Front., 2022, 9, 5074–5103 RSC
; (b) S. Zhu, H. Li, Y. Li, Z. Huang and L. Chu, Exploring visible light for carbon–nitrogen and carbon–oxygen bond formation via nickel catalysis, Org. Chem. Front., 2023, 10, 548–569 RSC
- W. Yuan, S. Zheng and Y. Hu, Recent Advances in C(sp3)–C(sp3) Cross-Coupling via Metalla-photoredox Strategies, Synthesis, 2021, 53, 1719–1733 CrossRef
- J. Choi and G. C. Fu, Transition metal–catalyzed alkyl-alkyl bond formation: Another dimension in cross-coupling chemistry, Science, 2017, 356, eaaf7230 CrossRef PubMed
-
(a) V. M. Chernyshev and V. P. Ananikov, Nickel and Palladium Catalysis: Stronger Demand than Ever, ACS Catal., 2022, 12, 1180–1200 CrossRef CAS
-
(a) C. G. Dong and Q. S. Hu, Ni(cod)2/PCy3-Catalyzed Cross-Coupling Reactions of Dihaloarenes with Arylboronic Acids, Synlett, 2012, 23, 2121–2125 CrossRef CAS
-
(a) L. J. Goossen, F. Collet and K. Goossen, Decarboxylative Coupling Reactions, Isr. J. Chem., 2010, 50, 617–629 CrossRef CAS
- L. Zhang, X. Si, Y. Yang, M. Zimmer, S. Witzel, K. Sekine, M. Rudolph and A. S. K. Hashmi, The Combination of Benzaldehyde and Nickel-Catalyzed Photoredox C(sp3)–H Alkylation/Arylation, Angew. Chem., Int. Ed., 2019, 58, 1823–1827 CrossRef CAS PubMed
- E. M. Dauncey, S. U. Dighe, J. J. Douglas and D. Leonori, A dual photoredox-nickel strategy for remote functionalization via iminyl radicals: radical ring-opening-arylation, -vinylation and -alkylation cascades, Chem. Sci., 2019, 10, 7728–7733 RSC
- Y. Luo, A. Gutierrez-Bonet, J. K. Matsui, M. E. Rotella, R. Dykstra, O. Gutierrez and G. A. Molander, Oxa- and Azabenzonorbornadienes
as Electrophilic Partners under Photoredox/Nickel Dual Catalysis, ACS Catal., 2019, 9, 8835–8842 CrossRef CAS PubMed
- M. S. Santos, A. G. Corrêa, M. W. Paixão and B. König, C(sp3)–C(sp3) Cross-Coupling of Alkyl Bromides and Ethers Mediated by Metal and Visible Light Photoredox Catalysis, Adv. Synth. Catal., 2020, 362, 2367–2372 CrossRef CAS
- M. B. Buendia, B. Higginson, S. Kegnæs, S. Kramer and R. Martin, Redox-Neutral Ni-Catalyzed sp3 C–H Alkylation of α-Olefins with Unactivated Alkyl Bromides, ACS Catal., 2022, 12, 3815–3820 CrossRef CAS
- H. A. Sakai and D. W. C. MacMillan, Nontraditional Fragment Couplings of Alcohols and Carboxylic Acids: C(sp3)–C(sp3) Cross-Coupling via Radical Sorting, J. Am. Chem. Soc., 2022, 144, 6185–6192 CrossRef CAS PubMed
- X. Y. Lv, R. Abrams and R. Martin, Dihydroquinazolinones as adaptative C(sp3) handles in arylations and alkylations via dual catalytic C–C bond-functionalization, Nat. Commun., 2022, 13, 2394 CrossRef CAS PubMed
- J. Zheng, C. Nopper, R. Bibi, A. Nikbakht, F. Bauer and B. Breit, Regio- and Diastereoselective Decarboxylative Allylation of N-Aryl α-Amino Acids by Dual Photoredox/Nickel Catalysis, ACS Catal., 2022, 12, 5949–5960 CrossRef CAS
- S. Dongbang and A. G. Doyle, Ni/Photoredox-Catalyzed C(sp3)–C(sp3) Coupling between Aziridines and Acetals as Alcohol-Derived Alkyl Radical Precursors, J. Am. Chem. Soc., 2022, 144, 20067–20077 CrossRef CAS PubMed
- E. Mao and D. W. C. MacMillan, Late-Stage C(sp3)–H Methylation of Drug Molecules, J. Am. Chem. Soc., 2023, 145, 2787–2793 CrossRef CAS PubMed
- W. L. Lyon and D. W. C. MacMillan, Expedient Access to Underexplored Chemical Space: Deoxygenative C(sp3)–C(sp3) Cross-Coupling, J. Am. Chem. Soc., 2023, 145, 7736–7742 CrossRef CAS PubMed
- W. Wang, X. Yan, F. Ye, S. Zheng, G. Huang and W. Yuan, Nickel/Photoredox Dual-Catalyzed Regiodivergent Aminoalkylation of Unactivated Alkyl Halides, J. Am. Chem. Soc., 2023, 145, 23385–23394 CrossRef CAS PubMed
- B. Yang, X. Y. Wang, X. T. Huang, Z. Y. Liu, X. Li, T. Huang, X. S. Li, L. Z. Wu, R. Fang and Q. Liu, Visible-Light-Mediated Two Transient C(sp3) Radical-Selective Cross-Coupling via Nickel Catalyst Continuous Capture: Synthesis of Pyrroline Derivatives, ACS Catal., 2023, 13, 15331–15339 CrossRef CAS
- Z. Zhou, J. Yang, B. Yang, Y. Han, L. Zhu, X. S. Xue and F. Zhu, Photoredox Nickel-Catalysed Stille Cross-Coupling Reactions, Angew. Chem., Int. Ed., 2023, 62, e202314832 CrossRef CAS PubMed
- Z. Hu, D. Wang and T. Xu, Direct Synthesis of β-Amino Boronates via Amide α-C–H Bond Activation and C(sp3)–C(sp3) Coupling under Dual Ni/Photoredox Catalysis, ACS Catal., 2024, 14, 547–553 CrossRef CAS
-
(a) W. Zhang, J. Chen, H. Wei and L. Wu, Development of Nickel-Catalyzed Cross-Coupling of Alcohol Derivatives to Construct Carbon-Carbon Bonds, Chin. J. Org. Chem., 2021, 41, 4208–4239 CrossRef
- F. Cong, X. Y. Lv, C. S. Day and R. Martin, Dual Catalytic Strategy for Forging sp2−sp3 and sp3−sp3 Architectures via β-Scission of Aliphatic Alcohol Derivatives, J. Am. Chem. Soc., 2020, 142, 20594–20599 CrossRef CAS PubMed
- T. Yang, Y. Wei and M. J. Koh, Photoinduced Nickel-Catalyzed Deaminative Cross-Electrophile Coupling for C(sp2)–C(sp3) and C(sp3)–C(sp3) Bond Formation, ACS Catal., 2021, 11, 6519–6525 CrossRef CAS
- S. K. Jana, M. Maiti, P. Dey and B. Maji, Photoredox/Nickel Dual Catalysis Enables the Synthesis of Alkyl Cyclopropanes via C(sp3)–C(sp3) Cross Electrophile Coupling of Unactivated Alkyl Electrophiles, Org. Lett., 2022, 24, 1298–1302 CrossRef CAS PubMed
- Y. Xu, M. L. Zhang and M. Oestreich, Photochemical, Nickel-Catalyzed C(sp3)–C(sp3) Reductive Cross-Coupling of α-Silylated Alkyl Electrophiles and Allylic Sulfones, ACS Catal., 2022, 12, 10546–10550 CrossRef CAS
- J. Zhou, D. Wang, W. Xu, Z. Hu and T. Xu, Enantioselective C(sp3)–C(sp3) Reductive Cross-Electrophile Coupling of Unactivated Alkyl Halides with α-Chloroboronates via Dual Nickel/Photoredox Catalysis, J. Am. Chem. Soc., 2023, 145, 2081–2087 CrossRef CAS PubMed
-
(a) Z. Huang, S. Tang and A. Lei, Sci. Bull., 2015, 60, 1391–1394 CrossRef CAS
- A. V. Tsymbal, L. D. Bizzini and D. W. C. MacMillan, Nickel Catalysis via SH2 Homolytic Substitution: The Double Decarboxylative Cross-Coupling of Aliphatic Acids, J. Am. Chem. Soc., 2022, 144, 21278–21286 CrossRef CAS PubMed
- J. Li, B. Cheng, X. Shu, Z. Xu, C. Li and H. Huo, Enantioselective alkylation of α-amino C(sp3)−H bonds via photoredox and nickel catalysis, Nat. Catal., 2024 DOI:10.1038/s41929-024-01192-7
| This journal is © the Partner Organisations 2024 |