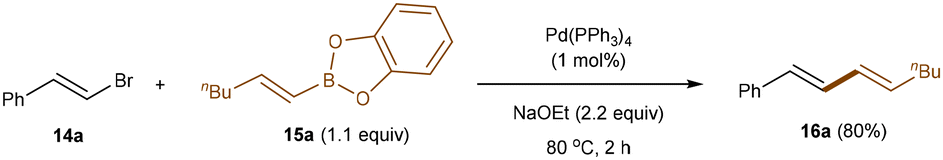DOI:
10.1039/D4QO01703J
(Review Article)
Org. Chem. Front., 2024,
11, 7249-7277
The reactivity of alkenyl boron reagents in catalytic reactions: recent advances and perspectives
Received
11th September 2024
, Accepted 29th October 2024
First published on 29th October 2024
Abstract
Although organic boron reagents have found broad applications in organic synthesis, the development of new methodologies using alkenyl boron reagents has lagged behind compared to the more commonly employed aryl-substituted variants. However, in the past five years, several innovative methods have been introduced that exploit the unique reactivity of alkenyl and boron moieties. These methods, utilizing readily available alkenyl boron substrates, enable the synthesis of highly functionalized alkenes or alkyl boron products under mild conditions. This review highlights the most recent advances, focusing on the novel reactivity of alkenyl boron reagents in polar or radical pathways within catalytic reactions, employing transition metal catalysis, organocatalysis, or photocatalysis.
1. Introduction
Organoboronic acids and their derivatives are privileged scaffolds in materials science and medicinal chemistry, thanks to their unique physicochemical and biological properties.1 In addition to their accessibility, low toxicity, and versatility, organoboron reagents have become indispensable tools for synthetic chemists. They can be selectively transformed into a variety of valuable intermediates and products,2 or employed as Lewis acid catalysts to design novel reactions.3 These features have led to their extensive use in organic synthesis,2 and their significance has been underscored by the awarding of three Nobel Prizes (in 1976, 1979, and 2010).4
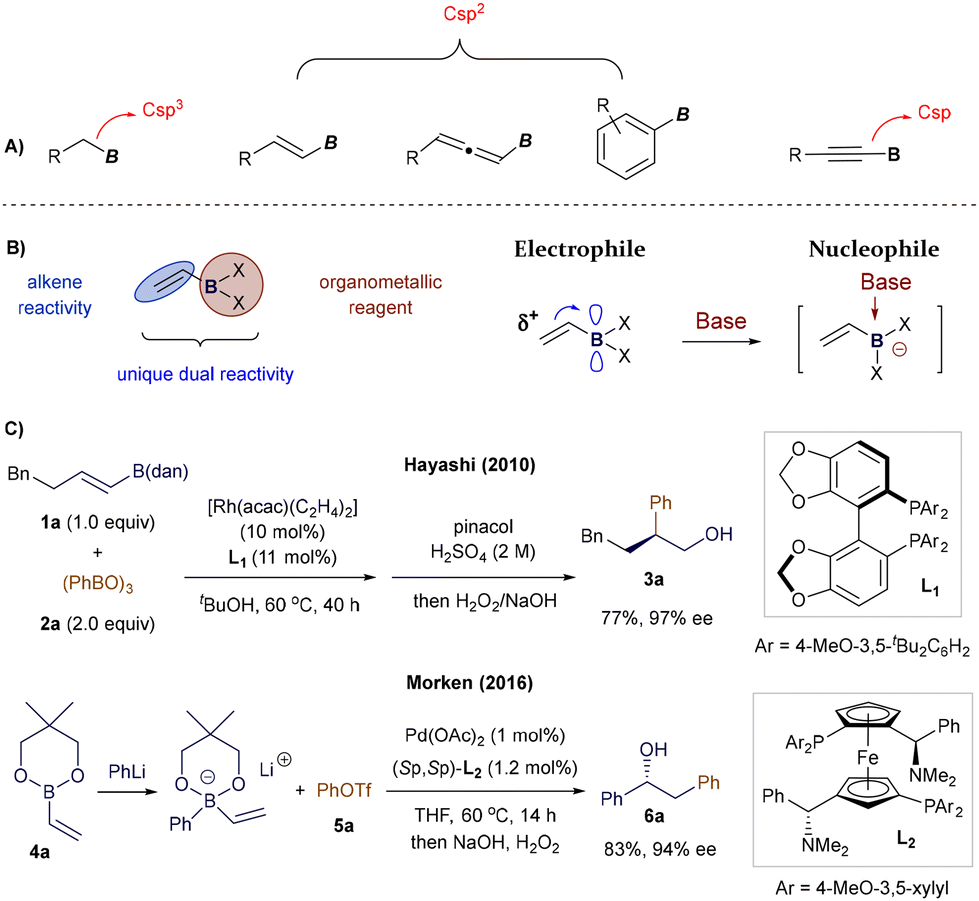
Among the various classes of organoboron compounds featuring Csp3 (alkyl), Csp2 (aryl, alkenyl, allenyl), or Csp (alkynyl) substituents, alkenyl boron reagents have recently garnered significant attention due to their unique reactivity, electronic properties, and structural characteristics (Fig. 1A). Unlike alkyl or aryl boron reagents, whose utility often centers around the manipulation of the boron moiety, Csp2- and Csp-substituted alkenyl boron reagents offer broader reactivity, stemming from the presence of additional unsaturated C–C double or triple bonds.2,5,6 Additionally, in contrast to allenyl5 or alkynyl6 boron compounds, a wide range of practical methods has been developed for the synthesis of diverse alkenyl boron species,2k,7 yielding numerous derivatives with varying substitution patterns at the boron atom, thus enabling a variety of transformations.
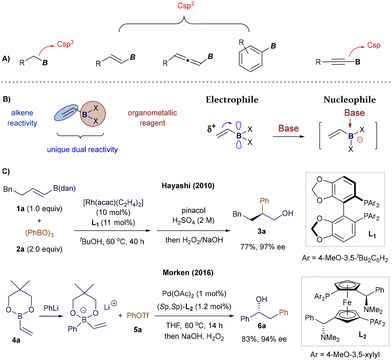 |
| | Fig. 1 Comparing the structure of different boron reagents and the typical reactivity of alkenyl boron reagents. | |
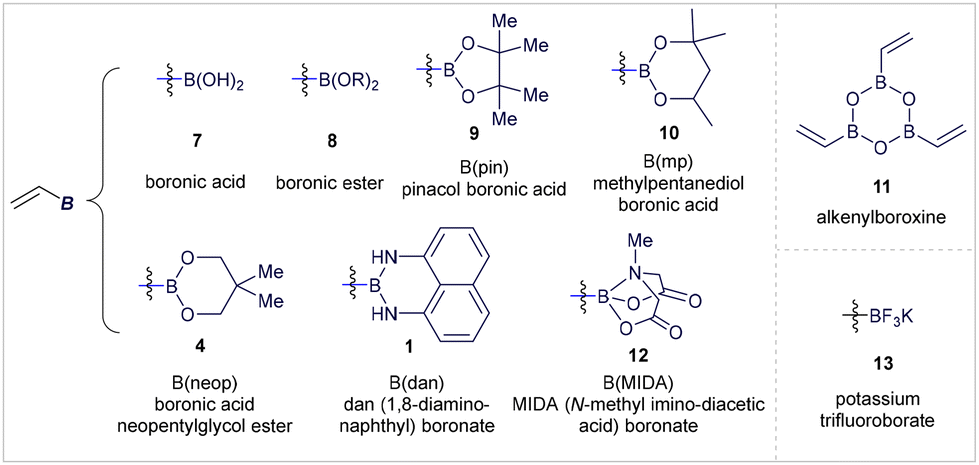
However, alkenyl boron reagents are more prone to protodeborylation, dimerization, and polymerization pathways in the presence of Brønsted acids, bases, transition metals, or oxidants, especially when compared to aryl boronic acid derivatives.2j,8 At first glance, the dual nature of alkenyl boron reagents—bearing both a boron center and an alkene double bond—can present selectivity challenges.2 While this increased reactivity may seem like a limitation, the synergistic interaction between the boron atom and the alkene moiety enables these compounds to exhibit unique chemical behaviours. The vacant p-orbital on the boron atom renders the adjacent double bond electron-deficient, while maintaining the boron center's Lewis acidity. As a result, alkenyl boron reagents possess ambiphilic reactivity, acting as electrophiles via their olefin portion and as nucleophiles when coordinated with a Lewis base (Fig. 1B). This dual reactivity is exemplified by key studies, such as Hayashi's Rh-catalyzed Michael addition to alkenyl boranes9 and Morken's Pd-catalyzed conjunctive cross-coupling via 1,2-migrations of alkenyl boronates (Fig. 1C).10 These characteristics make the chemistry of alkenyl boronates and boronic acids highly versatile, allowing for diverse modes of chemical activation and opening up exciting opportunities for designing novel synthetic protocols. The substitution at the boron center significantly impacts the reactivity and stability of the corresponding alkenyl boron compounds (Fig. 2).2d,11 Currently, a wide range of vinyl-boron reagents is available, including the commonly used boronic acids (7), esters (8), pinacol-type esters (9, 10, 4), as well as more stable derivatives such as 1,8-diaminonaphthalene (dan) boronates (1),12N-methyldiaminoacetic acid (MIDA) boronates (12),13 alkenylboroxines (11), and potassium trifluoroborates (13).14
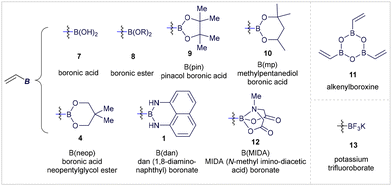 |
| | Fig. 2 Alkenyl boron reagents discussed within this review. | |
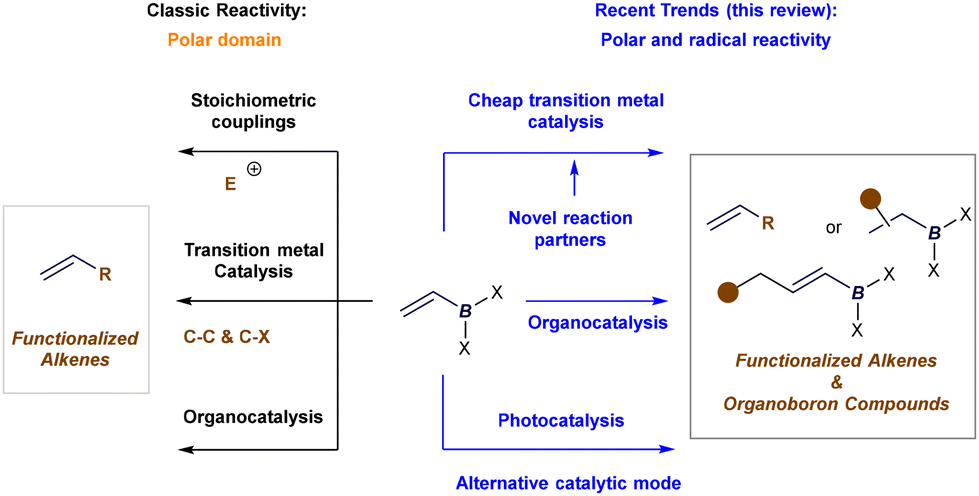
In synthetic chemistry, classic applications of alkenyl boron reagents have traditionally centered around their use in stoichiometric coupling reactions, such as the well-known Zweifel olefination.15 Additionally, their use in Pd- or Ni-catalyzed Suzuki–Miyaura cross-couplings—as organometallic precursors for vinyl groups—and in organocatalytic protocols has been well-documented (Fig. 3, left).2 However, most of these transformations focused on modifying the boron functionality, leaving the alkene moiety unaltered. Recent advances in catalysis—encompassing transition metal, organocatalysis, and more recently, photocatalysis—have introduced new methods for the catalytic activation of alkenyl boron reagents. These developments have expanded reactivity at both the boron terminus and the alkene or its adjacent positions, broadening the scope in terms of polar and radical reactivity, reaction partners, and reactive intermediates. As a result, key figures in boron chemistry have revisited the synthetic potential of alkenyl boron reagents. New protocols have emerged that enable the formation of densely functionalized olefins by exploiting reactivity at the boron site or the synthesis of complex organoboron compounds through selective manipulation of the double bond, or even its allylic position (Fig. 3, right).
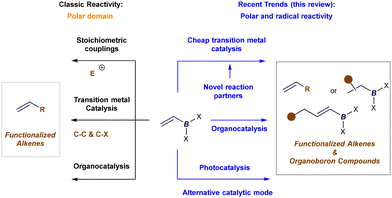 |
| | Fig. 3 Classic applications and scope of the review in the reactivity of alkenyl boron reagents in catalysis. | |
In this review, we highlight key catalytic protocols—primarily developed in the last five years—that have harnessed the unique reactivity of alkenyl boronates and boronic acids to advance catalytic methodologies. We will illustrate how these findings have already inspired, and will continue to inspire, the synthetic community to devise innovative solutions to long-standing challenges in synthesis. To enhance accessibility for readers, this review is structured around three major catalytic approaches—transition metal catalysis, organocatalysis, and photocatalysis—and further organized by reaction type. As discussed later, these three activation models enable efficient and versatile transformations through the selective activation of either alkenyl boron substrates or their reaction partners, yielding highly functionalized alkenes or alkyl boron products with excellent selectivity control. While several outstanding reviews on the synthetic applications of alkenyl boron reagents were published around 2020, they primarily focused on specific aspects of reactivity and do not reflect the most recent advancements in the field.2k,l,o–r
2. Cheap transition metal catalysis
Transition metal catalysis, recognized by several Nobel Prizes for enabling important and practical transformations,4 has been instrumental in developing sustainable synthetic methods with exceptional control of selectivity in modern organic chemistry. As discussed below, the use of boron reagents in cost-effective transition metal-catalysis is crucial for streamlining the formation of C–C and C–heteroatom bonds. Their ability to undergo transmetallation in the presence of a metal center, coupled with their relative stability and low toxicity, makes boron derivatives a preferred choice for cross-coupling reactions. However, the reactivity of alkenyl boronates and boronic acids in transition metal-catalysis extends beyond their common role as alkenylation agents in Suzuki–Miyaura-type cross-couplings, where they act as nucleophilic partners (Scheme 1).16 They can also be utilized in selective carbo- and hetero-functionalization of the olefin moiety, where alkenyl-boron reagents function as electrophilic partners, while preserving the boron functionality.
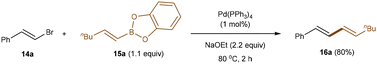 |
| | Scheme 1 Pioneering Suzuki–Miyaura cross-coupling by using alkenyl boron reagent. | |
2.1 Alkenyl boron reagents as the nucleophilic partner – transmetallation through the B-atom
In early contributions, alkenyl boron reagents were primarily employed as organometallic partners in Pd- and Ni-catalyzed Suzuki–Miyaura-type cross-couplings, often in combination with organohalides for constructing C–C bonds.2a,d,16 Additionally, their use in rhodium-catalyzed Hayashi-Miyaura additions to electron-deficient olefins has also been documented.2b In the context of carbon-heteroatom bond-forming couplings, inspired by Lam's work,17 Batey and co-workers developed a set of base-free Cu-catalyzed conditions that facilitate the oxidative coupling of potassium alkenyl trifluoroborate salts with amides, phenols, and carboxylates to prepare enamides, enols, and enol esters, respectively.18 Subsequent developments in this field have primarily focused on the use of alkenyl boron compounds for copper-catalyzed trifluoromethylation,19 allylic substitution,20 and addition reactions to CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 as well as electron-deficient alkenes22 and isatins.23 Furthermore, while investigating transition metal-catalyzed additions of boronic acids to unsaturated electrophiles, Cheng and co-workers found that Ni(II) and Co(II) salts facilitate the formation of nucleophilic organometallic intermediates from alkenyl boronic acids.24 This observation was leveraged to implement the addition of alkenyl boronic acids to α,β-unsaturated esters and methyl oct-2-ynoate. Building on this seminal contribution, the groups of Zhang25,26 and Zhao27 have disclosed the use of both Ni(II) and Co(II) catalysis to explore the asymmetric addition of alkenyl boronic acids to cyclic N-sulfimines and ketones, respectively.
21 as well as electron-deficient alkenes22 and isatins.23 Furthermore, while investigating transition metal-catalyzed additions of boronic acids to unsaturated electrophiles, Cheng and co-workers found that Ni(II) and Co(II) salts facilitate the formation of nucleophilic organometallic intermediates from alkenyl boronic acids.24 This observation was leveraged to implement the addition of alkenyl boronic acids to α,β-unsaturated esters and methyl oct-2-ynoate. Building on this seminal contribution, the groups of Zhang25,26 and Zhao27 have disclosed the use of both Ni(II) and Co(II) catalysis to explore the asymmetric addition of alkenyl boronic acids to cyclic N-sulfimines and ketones, respectively.
In this section, we will detail several recent and exciting examples that focus on the use of inexpensive metals, such as Co, Mn, and Ni, in catalyzed addition reactions to unsaturated bonds, cross-couplings with challenging electrophiles via radical or polar pathways, oxidative Heck reactions, and alkenylamination reactions.28 Mechanistically, we will describe how transition metal-catalyzed reactions of alkenyl boron derivatives proceed via transmetallation of the alkenyl fragment from the boron center to the metal catalyst, leading to the formation of densely functionalized olefinic products. In all these processes, the alkenyl-boron partner serves as the nucleophilic species.
Addition to unsaturated bonds.
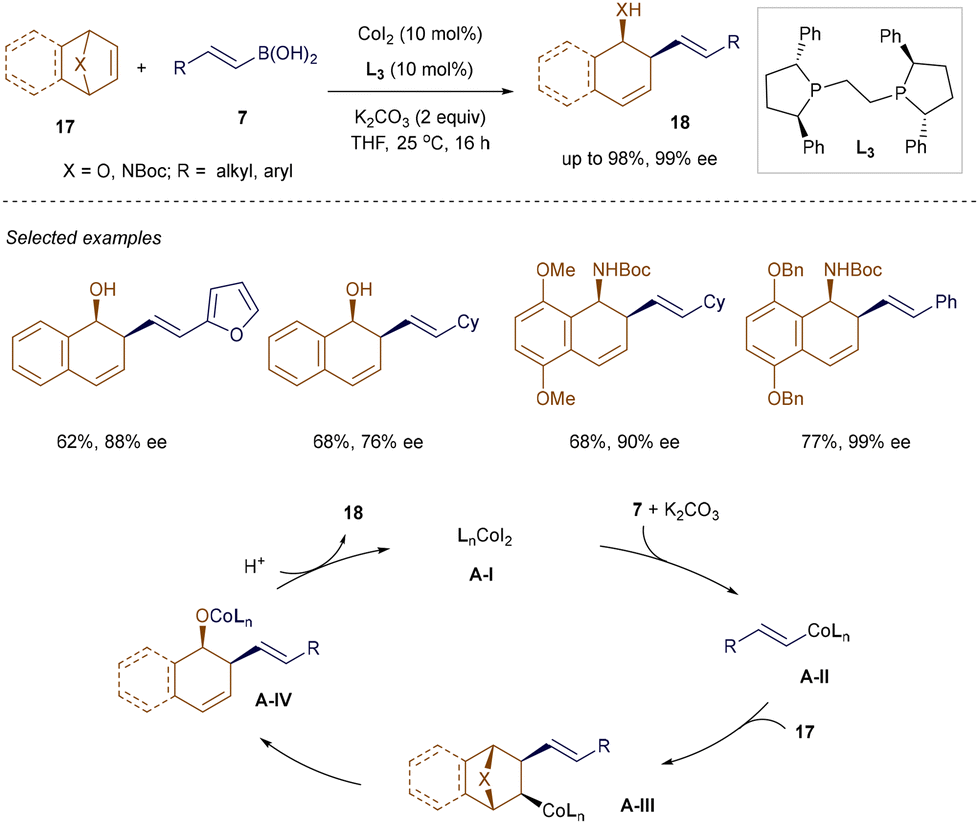
Catalytic asymmetric ring-opening reactions of heterobicyclic alkenes have emerged as a valuable strategy for accessing enantioenriched multisubstituted cyclohexenes,29 which are commonly found in various natural products and biologically active molecules.30 Although Hou and co-workers pioneered the asymmetric ring-opening of oxa- and aza-bicyclic alkenes using alkenylboronic acids in Pd-catalyzed reactions, the substrate scope was limited, and the enantioselectivity was moderate at very low temperatures.31 This limitation has been addressed by a recent method developed by Zhu, Chong, and Meng et al., which employs Co-catalysis.32 As illustrated in Scheme 2, the combination of the commercially available (R,R)-Ph-BPE L3 and CoI2 facilitates highly enantioselective ring-opening reactions of both β-alkyl and aryl-substituted alkenyl boronic acids 7 with oxa- or aza-bicyclic alkenes 17, yielding the corresponding products 18 with up to 98% yield and 99% enantiomeric excess (ee) at room temperature. The method demonstrates compatibility with various functional groups, including esters, chloro, bromo, and heterocycles. As proposed by the authors, the chiral phosphine-Co complex A-I undergoes transmetallation with alkenyl boronic acid 7 in the presence of K2CO3, followed by carbometallation to produce species A-III. β-Elimination and subsequent protonation then yields product 18 while regenerating Co complex A-I.
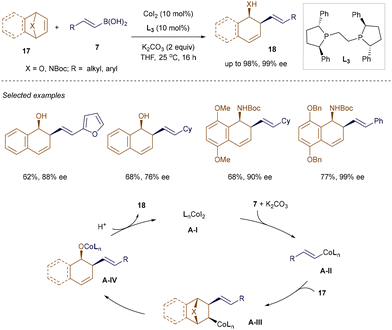 |
| | Scheme 2 Co(II)-catalyzed ring-opening reactions. | |
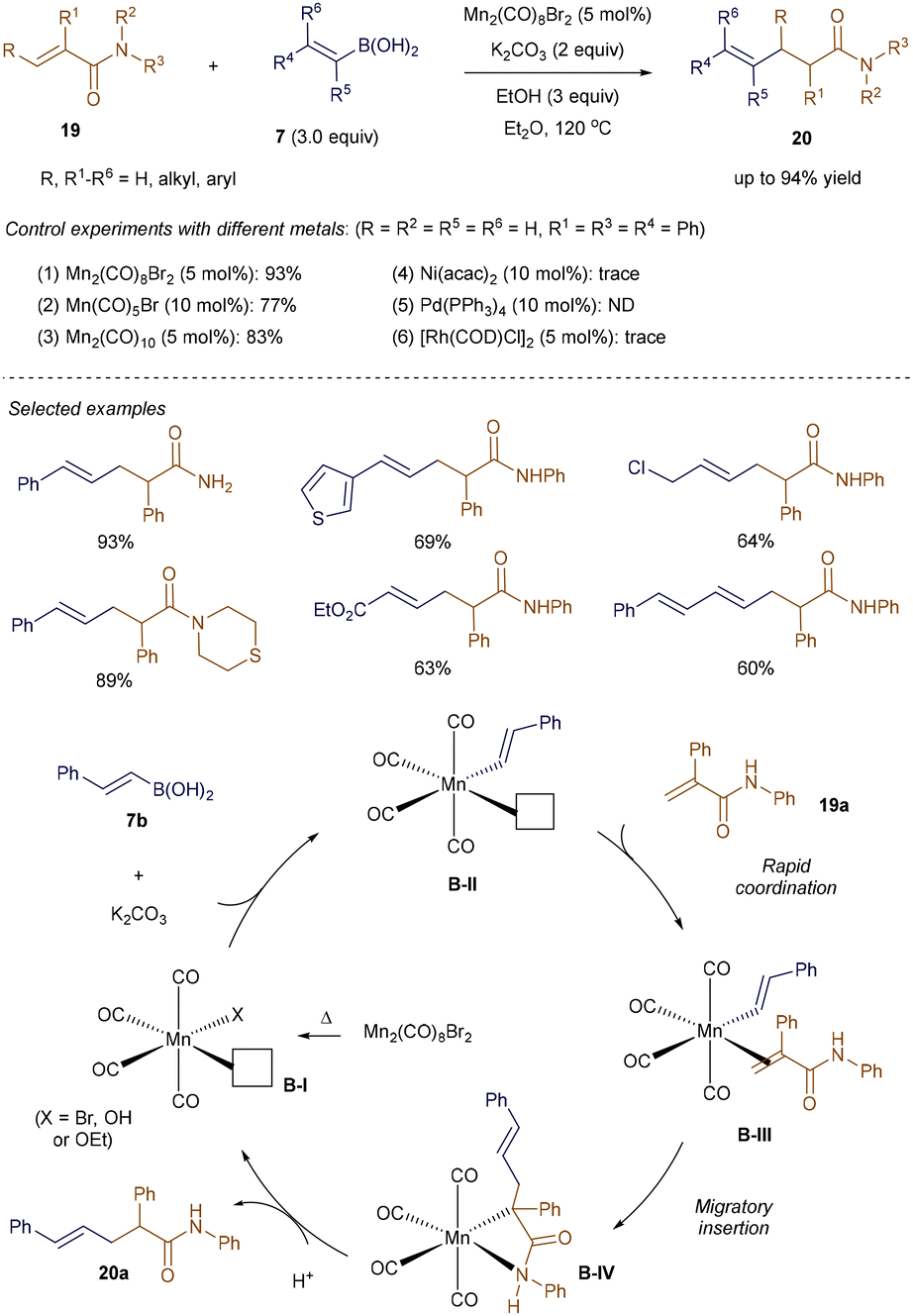
The high availability of manganese (the third most abundant transition metal in the Earth's crust), along with its low toxicity, has recently sparked renewed interest in Mn(I)-catalysis within the synthetic community.33 In line with this trend, Xie and colleagues leveraged the ability of vinyl-boronic acids to undergo transmetallation with Mn(I)-complexes,34 successfully applying this approach to Mn(I)-catalyzed addition reactions with α,β-unsaturated amides 19 (Scheme 3).35 Utilizing alkenyl boronic acids 7 and 5 mol% of Mn2(CO)8Br2, this method grants access to a diverse range of β-alkenylated amides 20 with remarkable efficiency, regio- and chemoselectivity, and high tolerance for various functional groups. The synthetic potential of this strategy is underscored by the fact that alternative metal sources—such as Ni(acac)2, Pd(PPh3)4, and [Rh(COD)Cl]2—failed to deliver the desired hydrovinylation product.
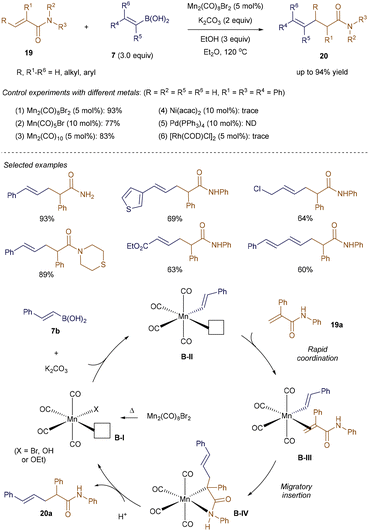 |
| | Scheme 3 Mn(I)-catalyzed addition to α,β-unsaturated amides. | |
As proposed by the authors (Scheme 3), the mechanism begins with the homolysis of the dimeric manganese pre-catalyst Mn2(CO)8Br2 at 120 °C, generating the active 16-electron Mn-species B-I, which features a vacant coordination site. Base-assisted transmetallation of alkenyl boronic acid 7b to B-I forms the nucleophilic intermediate B-II, which then coordinates with the incoming α, β-unsaturated amides 19a to yield complex B-III. Subsequent migratory insertion of the C–Mn bond across the double bond of the Michael acceptor generates B-IV, which upon protonation, produces the addition product 20a, thereby regenerating the active catalyst B-I.35
In the previous section, we discussed the ability of alkenyl boronic reagents to add to electron-deficient olefinic systems mediated by transition metals. Recently, cost-effective transition metal-catalyzed additions of alkenyl boron compounds to more challenging electron-rich double bonds have been reported, facilitating the realization of hydroalkenylation processes. In these reactions, fine-tuning of the conditions is often necessary to avoid over-addition of the vinyl precursor to the alkene products. Hydroalkenylation of unsaturated hydrocarbons, such as unbiased alkenes and cyclopropenes, has been successfully achieved using Mn, Ni, and Co catalysts, which will be reviewed in this section.
Hydroalkenylation.
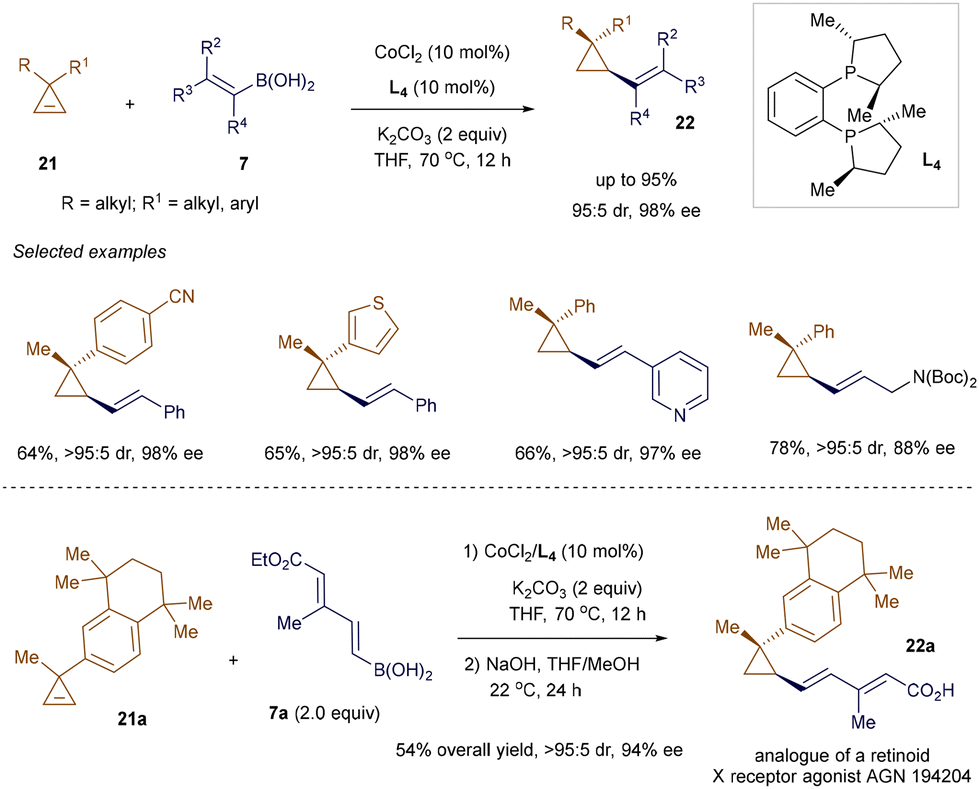
Alkenyl cyclopropanes have emerged as a key structural motif in medicinal chemistry and pharmaceuticals due to their presence in several biologically active natural products and drug candidates, as well as their distinct ‘strained-ring’ reactivity in chemical synthesis.36 Methods to produce these compounds in a stereoselective manner are highly sought after but remain somewhat underdeveloped. Addressing this gap, Meng's group employed the conjugate addition of alkenyl boronic acids 7 to 3,3-disubstituted cyclopropenes 21, under cobalt catalysis, to obtain enantioenriched alkenyl cyclopropanes 22 (Scheme 4).37 A Co(II)/chiral phosphine L4 complex was utilized to drive this enantioselective desymmetrization reaction,38 delivering cyclopropanes 22 with adjacent tertiary and quaternary carbon centers with both high efficiency and excellent enantioselectivity. The synthetic utility of this method was highlighted through the two-step synthesis of an analogue 22a of the retinoid X receptor agonist AGN 194204.
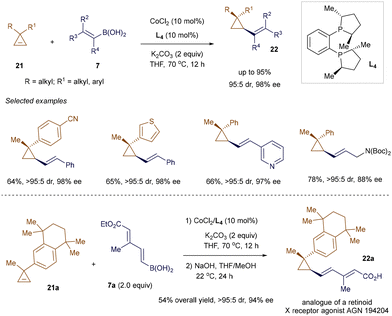 |
| | Scheme 4 Co(II)-catalyzed addition to cyclopropenes. | |
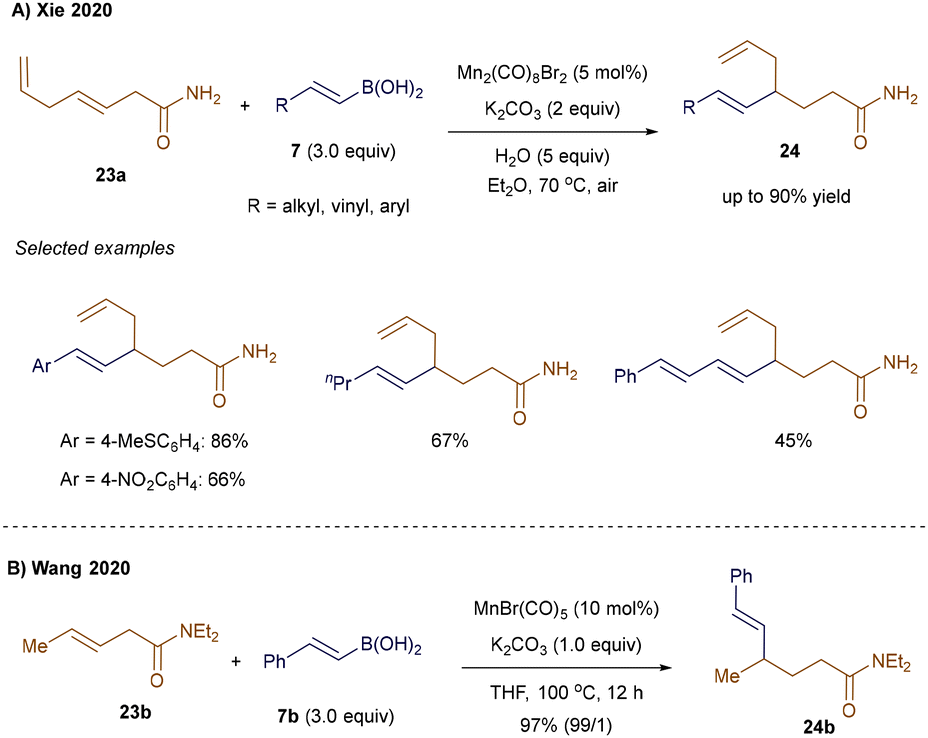
Compared to the previously reported Mn(I)-catalyzed Michael addition of alkenylboronic acids to α,β-unsaturated amides (see Scheme 3),35 the expansion of this approach to unbiased internal olefins presents a greater challenge. This difficulty arises from the rapid ligand exchange rate of the alkene, which competes with the slower migratory insertion of vinylmanganese species across unactivated alkenes.39 Xie and colleagues addressed this issue by employing a weakly coordinating primary amide directing group, which facilitates the carbometallation step and drives the regioselective Mn(I)-catalyzed hydroalkenylation of internal alkenes, even in the presence of terminal olefins (Scheme 5A).39 This strategy was demonstrated with substrate 23a, which contains both a terminal and an internal double bond, yielding the selectively 4,5-hydrovinylation product 24 in 45–90% yield. A similar transformation was reported by Wang and co-workers using tertiary amides as directing groups. This approach produced γ-vinylated amides 24b—derived from substrate 23b and boronic acid 7b—with excellent regio- and E/Z-selectivity control (Scheme 5B).40
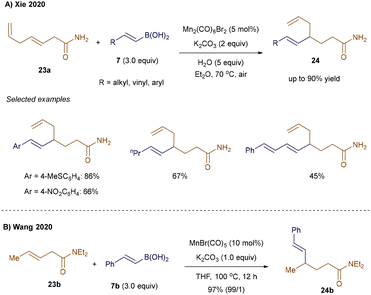 |
| | Scheme 5 Mn(I)-catalyzed hydroalkenylation of unbiased internal alkenes. | |
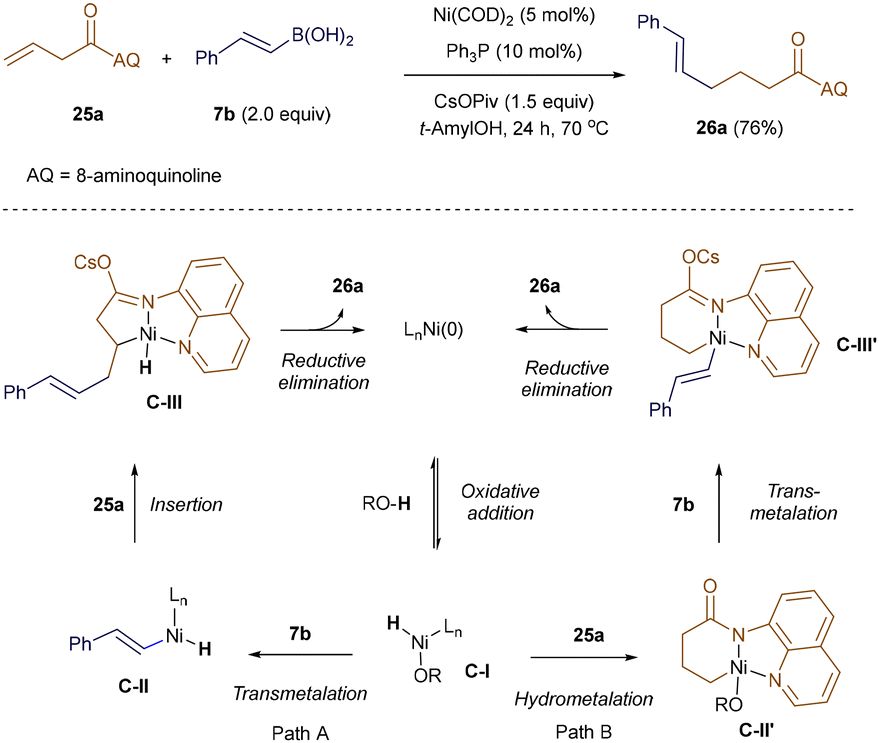
In 2018, Zhao and Zhou extended this reactivity to Ni-catalysis. They employed an 8-aminoquinoline (AQ) directing group, which enabled the linear-selective hydroalkenylation of unactivated terminal alkenes with alkenyl boronic acid 7b under redox-neutral conditions (Scheme 6).41 The AQ template was essential for overcoming the low reactivity of aliphatic alkenes. Drawing on previous work from the Zhou group,42 the authors proposed two potential catalytic cycles. Both cycles begin with the reversible formation of a Ni–H intermediate, C-I, with the alcohol additive serving as the proton source. In pathway A, the vinyl-Ni–H species C-II is generated through transmetallation with alkenyl boronic acid 7b. C-II then undergoes directed alkene insertion, forming a five-membered nickelacycle C-III, which upon reductive elimination yields the linear product 26a. Alternatively, in pathway B, sequential coordination and hydrometallation of intermediate I across the double bond of 25a produces a six-membered nickelacycle, C-II′. Transmetallation of C-II′ with boronic acid 7b forms C-III′, which then undergoes reductive elimination to generate product 26a, while regenerating the active Ni(0) catalyst.41
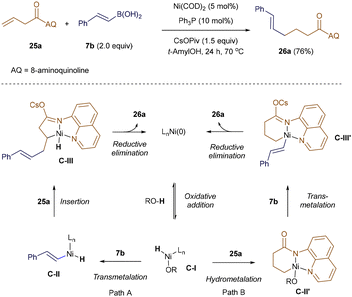 |
| | Scheme 6 Ni-catalyzed hydroalkenylation. | |
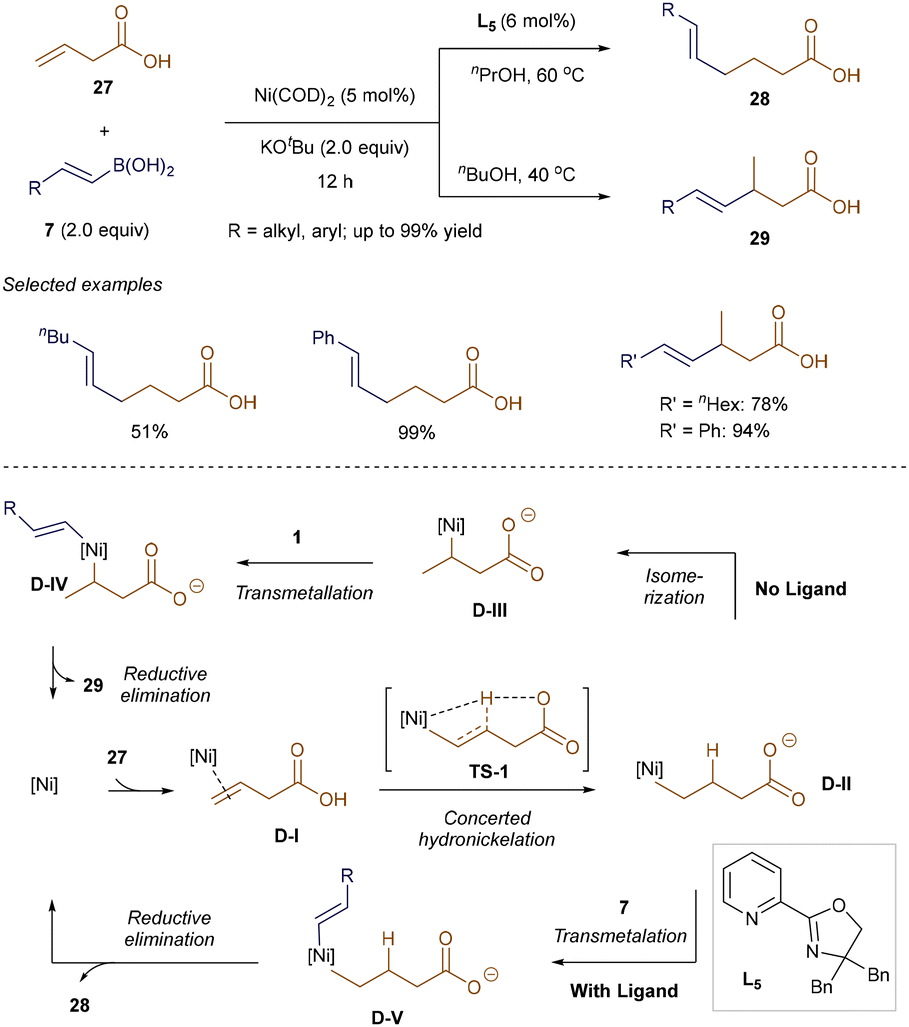
Liu and Engle revealed that the regioselectivity of nickel-catalyzed hydroalkenylation of β-alkenyl carboxylic acid 27 can be effectively tuned by ligands.43 As shown in Scheme 7, both regioisomers 28 and 29 were obtained with moderate to excellent yields, depending on the presence of Pyrox ligand L5. DFT calculations suggest that this reaction may proceed differently from Zhao and Zhou's Ni-catalyzed hydroalkenylation. Liu and Engle proposed that the key alkylnickel(II) intermediate D-II forms through concerted hydronickelation TS-1. Without a ligand, D-II isomerizes into a more stable five-membered heterocyclic intermediate D-IIIvia β-hydride elimination and alkene re-insertion, leading to Markovnikov products 29. In the presence of ligand L5, however, D-II directly undergoes transmetallation with alkenyl boronic acids 7 to form D-V, where steric hindrance from L5 prevents isomerization. In both pathways, transmetallation determines the rate and selectivity of the reaction.43
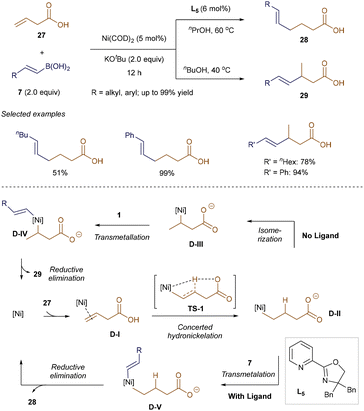 |
| | Scheme 7 Regiodivergent hydroalkenylation. | |
Asymmetric variants of Ni-catalyzed hydrovinylation processes using alkenyl boron nucleophiles have been developed, as demonstrated by Xiao and Zhou.44 In their work, a Ni(0)-precatalyst combined with spiro-aminophosphine ligand L6 enabled the branched hydroalkenylation of styrenes (Scheme 8A). Mei and co-workers reported similar asymmetric reactivity (Scheme 8B), using bisoxazoline L7 to achieve enantioselective branched alkenylated products 31.45 This method's scope was extended to include β-aryl-substituted alkenyl boronic acids. Notably, in 2022, Wang and colleagues accomplished the hydroalkenylation of internal styrenes, where a simple 1,3-diketone/Ni(0) system proved crucial for achieving good selectivity.46
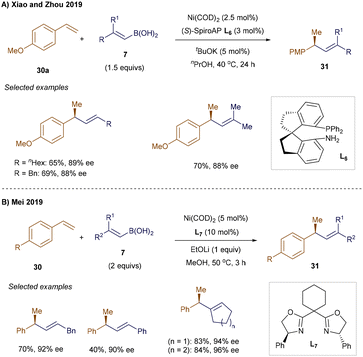 |
| | Scheme 8 Enantioselective hydroalkenylation of styrenes. | |
Cross-couplings.
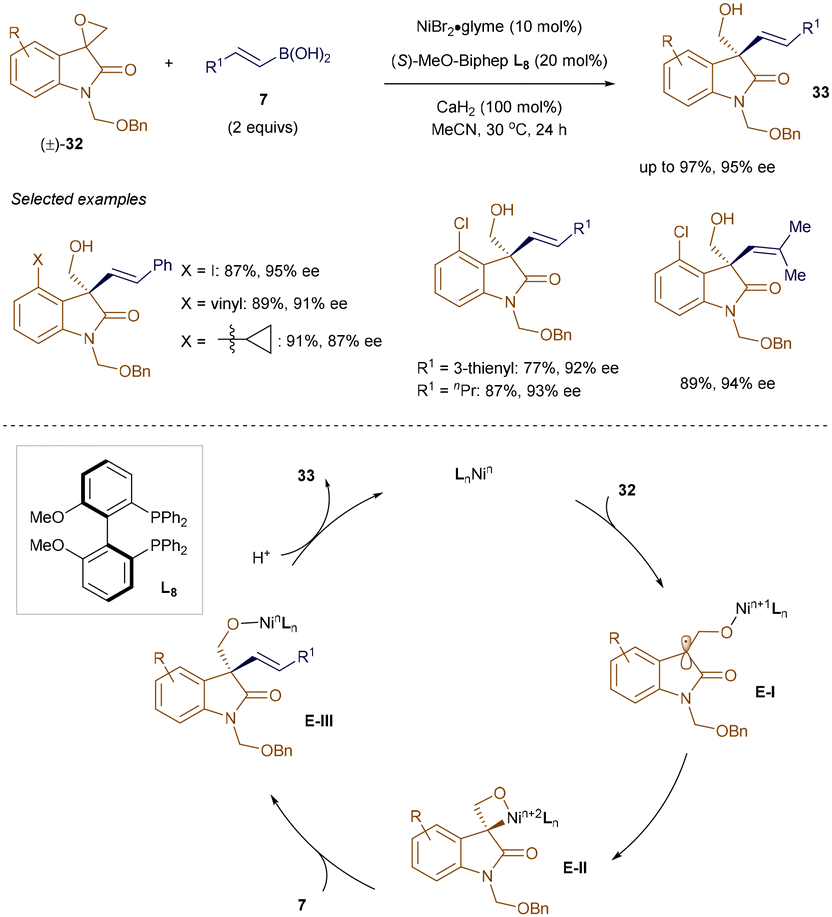
As discussed later in this section, both radical and polar pathways have been explored for challenging coupling transformations. Epoxides, due to their strained ring structure, are highly reactive intermediates and easily synthesized from alkenes and carbonyl compounds. Although transition metal-catalyzed47 and metal-free48 regioselective ring-opening of oxiranes with organoboron reagents is well documented, catalytic asymmetric variants have remained elusive. To address this, Zhang and colleagues developed a Ni-catalyzed enantioselective cross-coupling of racemic epoxides with alkenyl boronic acids.49 As depicted in Scheme 9, a NiBr2/L8 system enabled the coupling of racemic spiroepoxyoxindoles 32 with alkenyl boronic acids 7, yielding enantioenriched homoallylic alcohols 33 with all-carbon quaternary stereocenters. Control experiments suggest the reaction initiates through single-electron transfer (SET) from the Ni-catalyst to epoxide 32, forming a stabilized tertiary radical E-I. Radical recombination leads to a stereodefined Ni-oxetane adduct E-II, which couples with 7 to produce enantioenriched homoallylic alcohols 33. Additionally, Zhang's group achieved nickel-catalyzed ring-opening/cross-coupling of vinylaziridines with β-substituted alkenyl boronic acids, selectively producing skipped aminodienes.50
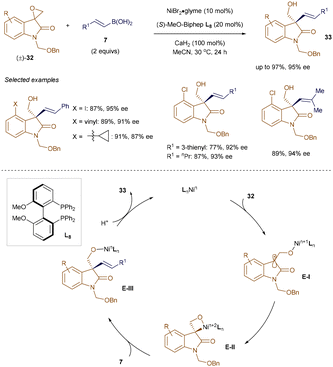 |
| | Scheme 9 Enantioconvergent coupling of spiroepoxyoxindoles with alkenyl boronic acids. | |
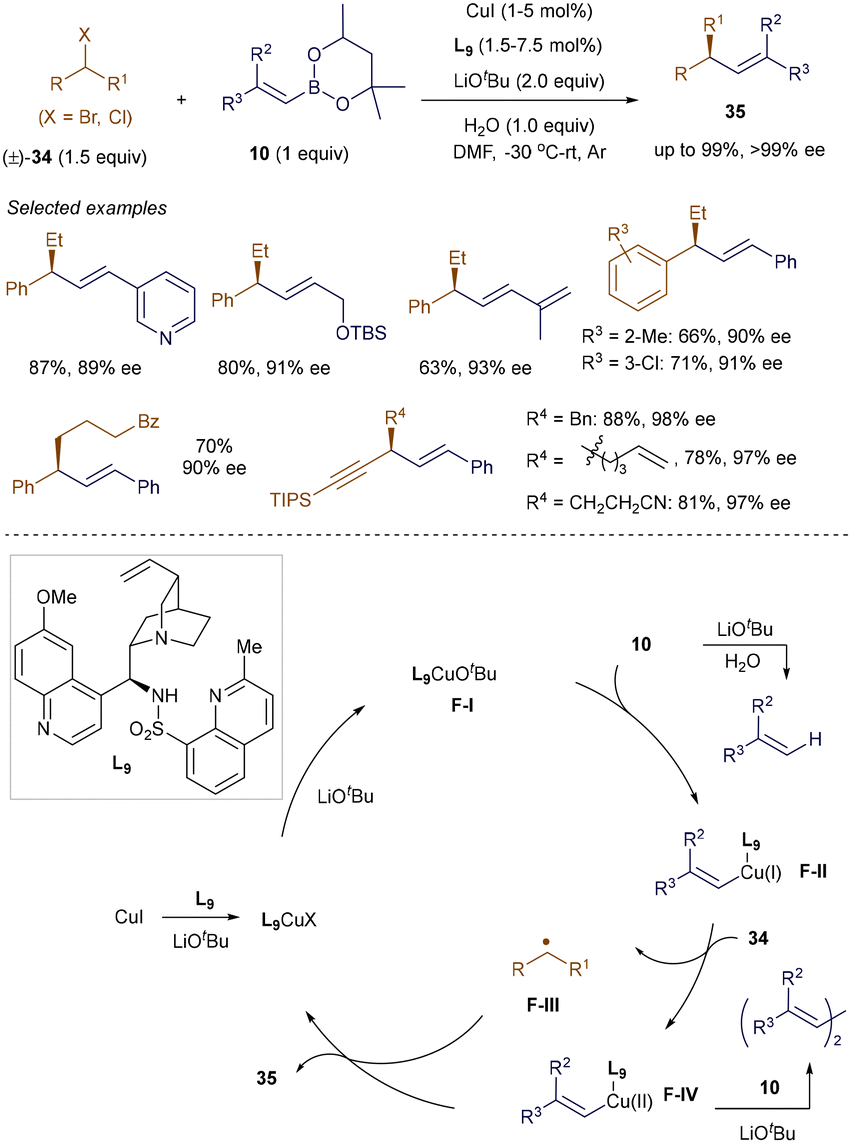
While enantioconvergent cross-coupling of racemic alkyl halides with alkenyl boronate esters is a promising strategy for constructing chiral Csp3–Csp2 bonds, it remains underdeveloped due to challenges in achieving both high reactivity and enantioselectivity.51 In 2022, Liu and co-workers successfully developed a chiral copper catalyst for the enantioconvergent cross-coupling of benzyl and propargyl halides with alkenyl boronate esters (Scheme 10).52 Control experiments revealed that a key to this success lies in the rational design of a hemilabile N,N,N-ligand L9 with steric hindrance at the ortho position of one coordinating quinoline ring. This novel ligand not only facilitates the radical cross-coupling process in its tridentate form, preventing homocoupling side reactions, but also provides high enantioselectivity by stabilizing reactive secondary alkyl radicals in its bidentate form. The system tolerated a wide variety of (hetero)benzyl and propargyl bromides and chlorides 34, as well as mono- and disubstituted alkenyl boronate esters 10 with diverse functional groups, achieving enantioenriched alkene products 35 with up to 98% yield and over 99% ee, even at 1 mol% catalyst loading.
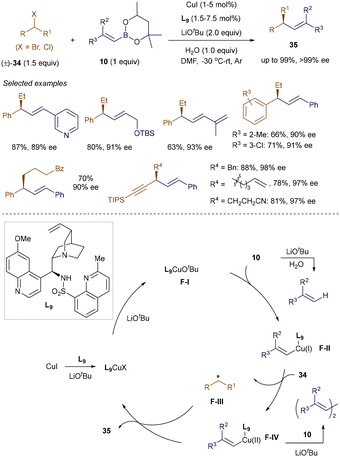 |
| | Scheme 10 Enantioconvergent coupling of secondary alkyl halides. | |
The proposed mechanism involves the formation of the active catalyst F-I from CuI, LiOtBu, and ligand L9 in its tridentate form. Transmetallation of F-I with alkenyl boronate ester 10 generates an alkenyl-Cu(I) species F-II and tBuO-B(mp). F-II then reacts with alkyl halides 34, generating a secondary alkyl radical F-III and an alkenyl-Cu(II) complex F-IVvia a single-electron transfer process. Ligand L9 efficiently suppresses the undesired homocoupling of F-IV. The enantioconvergent coupling of F-III and F-IV produces the desired product 35 and regenerates the active catalyst F-I. Notably, conducting the reaction at low temperatures substantially reduces the protodeboration of 10, aiding enantiocontrol.52
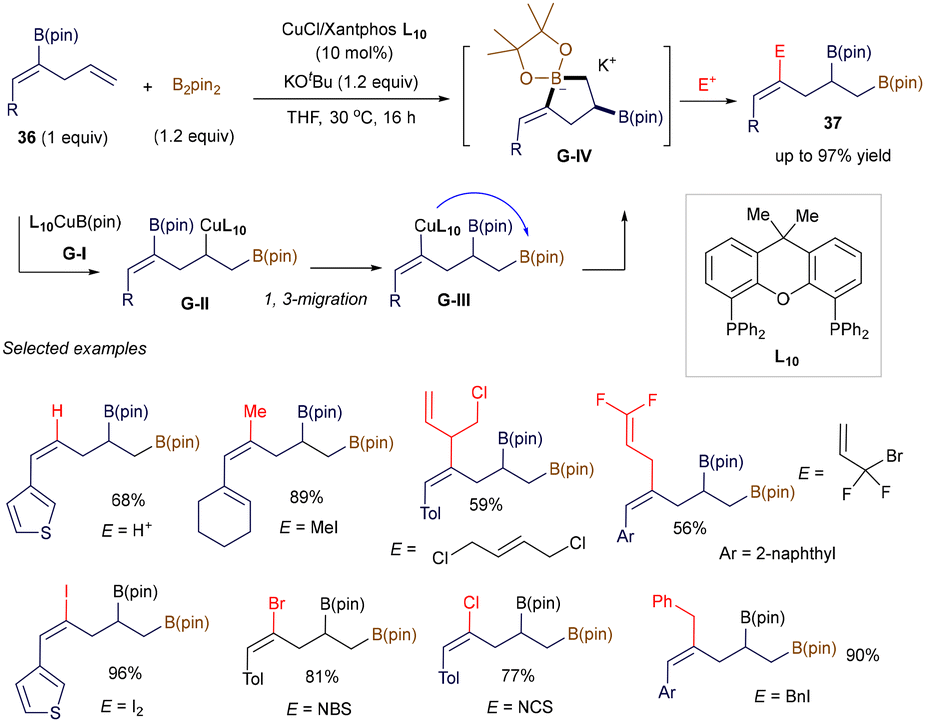
In addition to these radical-involved cross-coupling examples, three notable studies have recently demonstrated the use of alkenyl boronates as electrophiles via a polar pathway, significantly expanding the synthetic utility of these reagents. For instance, Carbó, Fernández, and co-workers revealed that the regioselective borylcupration of borylated skipped (Z)-dienes 36 generates the key electrophilic five-membered boracycle G-IV through a novel intramolecular, stereospecific B/Cu 1,3-rearrangement, where the B(pin) group migrates from a Csp2 to a Csp3 center (Scheme 11).53 By trapping G-IVin situ with various electrophiles—such as protons, alkyl halides, I2, NBS, or NCS—a wide range of functionalized homoallyl diborated products 37 can be easily prepared. DFT calculations suggest that, although alkoxides from bases like KOtBu or KOMe can form Lewis base adducts with intermediate G-I, the use of bulky tert-butoxide prevents stable adduct formation with L10CuBpin, thereby facilitating the initial borylcupration step.
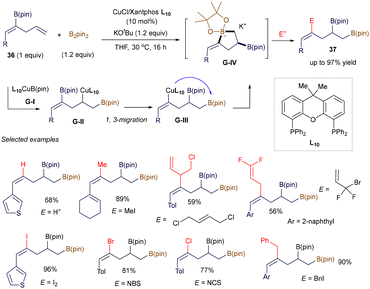 |
| | Scheme 11 Boron-copper 1,3-rearrangment. | |
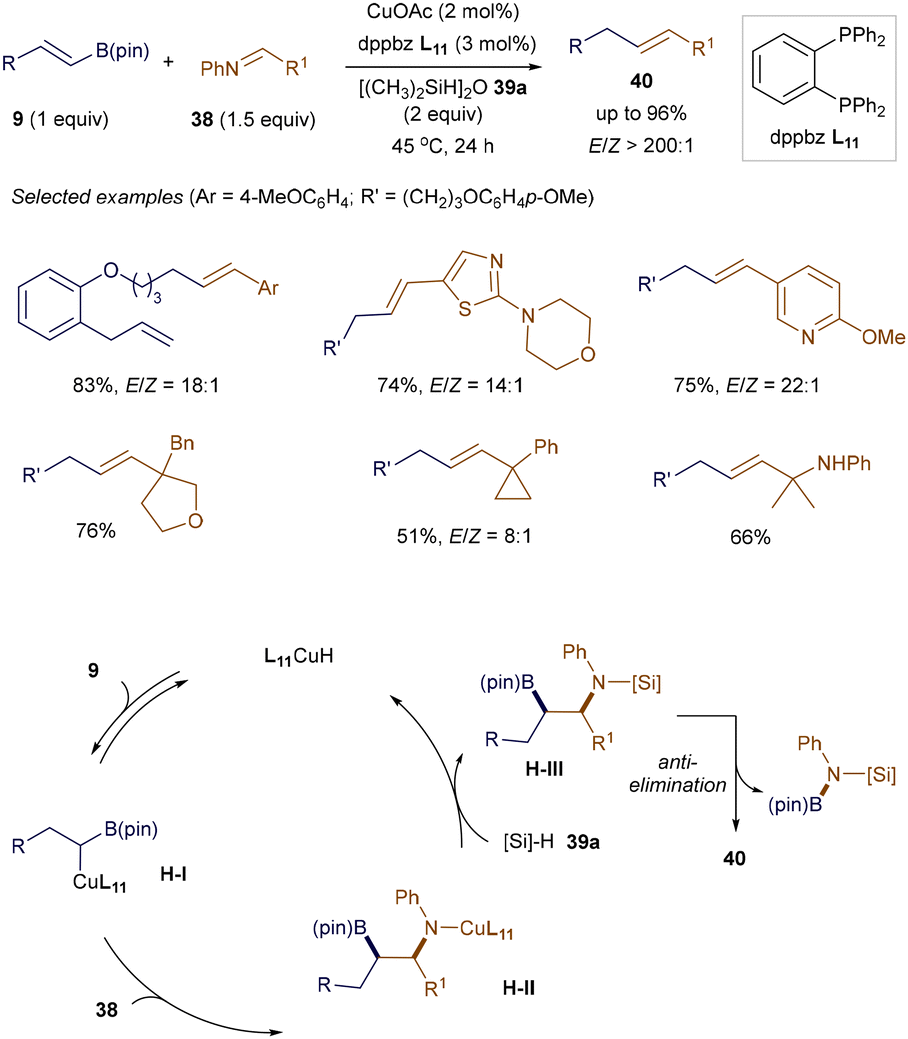
Alkenes are fundamental building blocks in organic synthesis, and stoichiometric Wittig-type olefination of carbonyls or imines is among the most widely used methods for constructing functionalized alkenes. However, catalytic versions of this strategy remain underdeveloped. Recently, Lalic and co-workers reported an excellent stereoselective copper-catalyzed olefination of aryl and sterically hindered alkyl imines with various functional groups (Scheme 12).54 The success of this method lies in the efficient generation of a copper boryl heterobimetallic complex H-I, formed through the regioselective addition of L9CuH to alkenyl boronates 9, acting as an ylide equivalent. Subsequent addition to imine 38, followed by interaction with hydrosilane 39a, produces intermediate H-III. This 1,2-boryl N-silylamine H-III then eliminates, furnishing the E-alkene products 40.
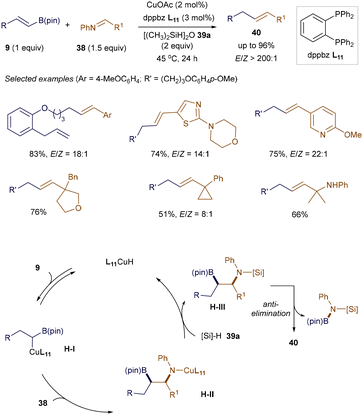 |
| | Scheme 12 Copper-catalyzed olefination of imines. | |
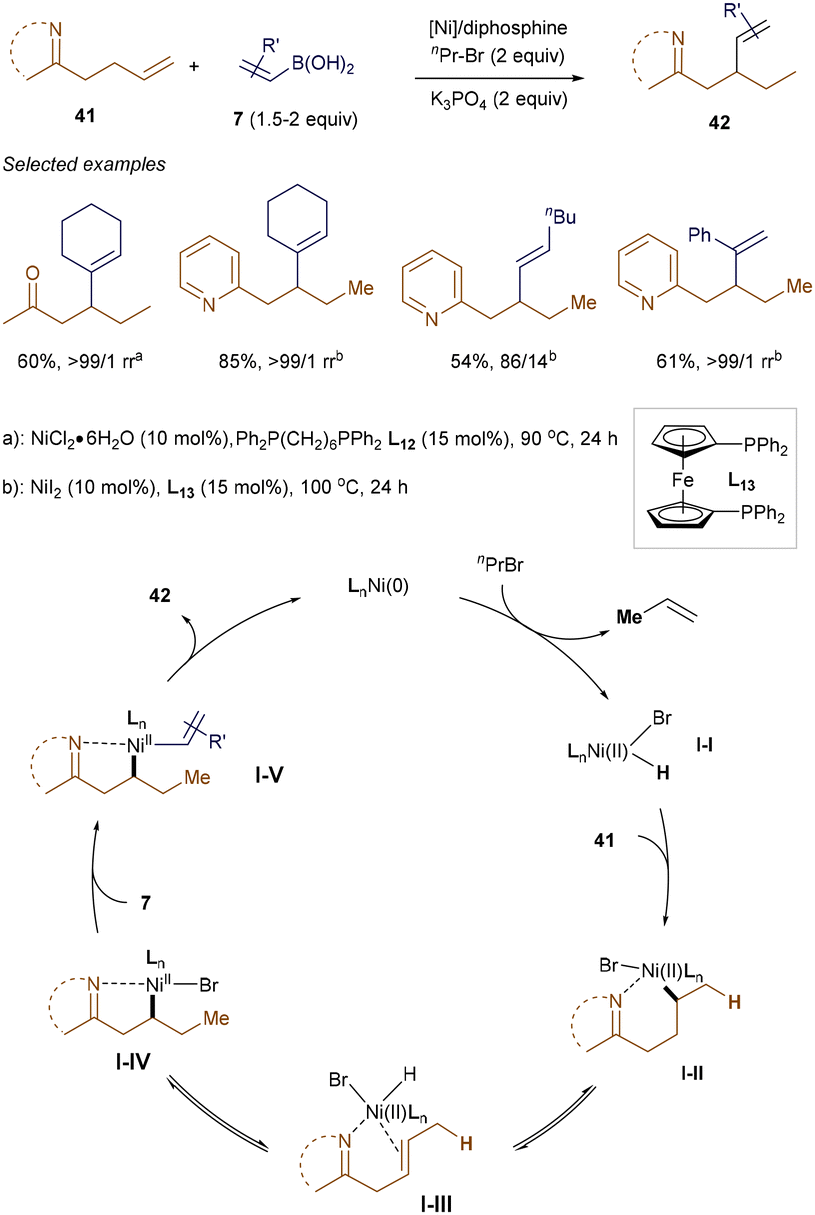
NiH-involved migratory hydrofunctionalization of alkenes has emerged as an efficient method for the selective functionalization of Csp3–H bonds.55 Recently, Cao and Wang developed a nickel-catalyzed, chelate-assisted strategy for the regioselective migratory hydrovinylation of alkenes using diphosphine ligands.56 As shown in Scheme 13, with imine or pyridine as directing groups, the NiH-promoted β-alkenylation proceeded smoothly, yielding vinylated products with excellent selectivity and good yields. Mechanistic studies suggest that the reaction involves the formation of a LnNi(II)Br–H intermediate I-Ivia the oxidative addition of LnNi(0) to nPrBr, followed by β-H elimination. After the migratory insertion of alkenes into I-I, assisted by nitrogen coordination, a stable nickellacycle I-IV is formed through a β-H elimination and reinsertion process. Transmetallation with alkenyl boronic acids 7, followed by reductive elimination, produces the desired vinylated products 42.
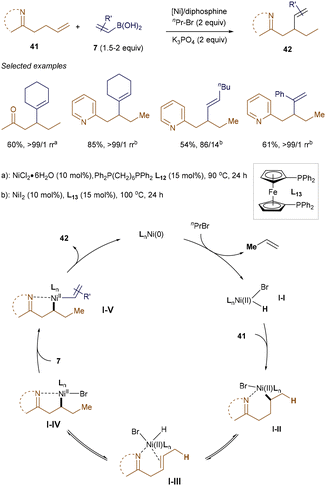 |
| | Scheme 13 Ni(II)H-catalyzed β-vinylation. | |
Oxidative Heck.
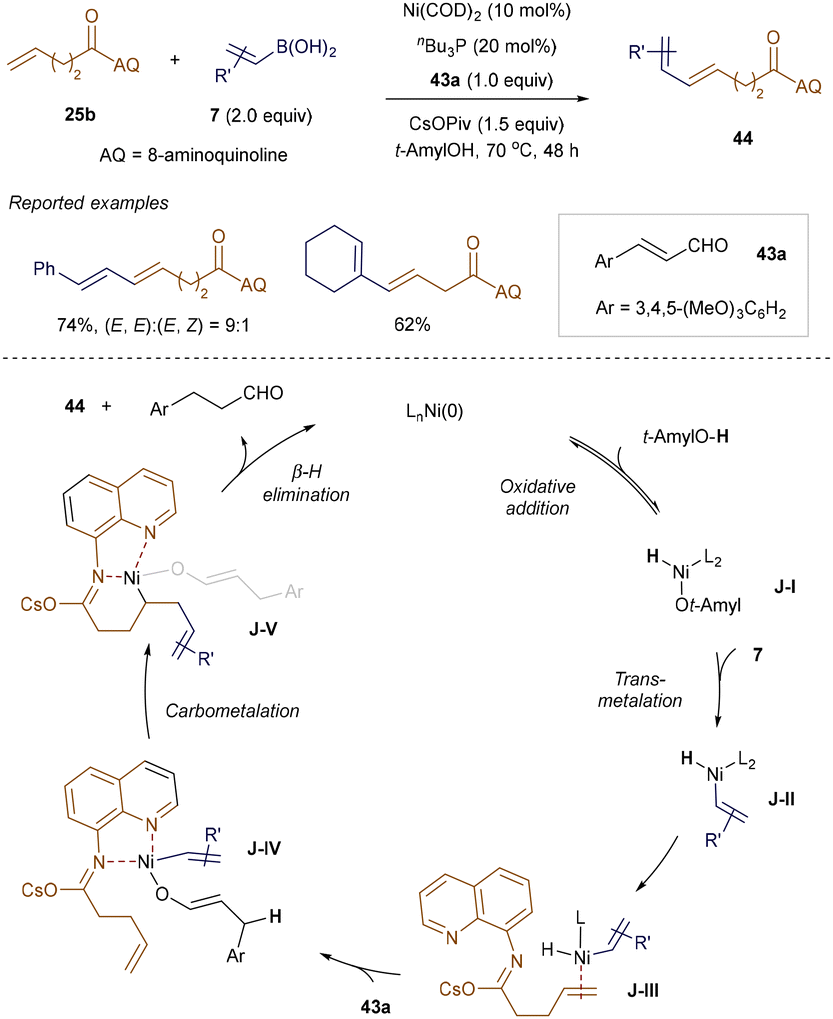
A related strategy to the hydrovinylation methods is the alkenylation of olefinic functionalities while preserving the integrity of the double bond. This was achieved in a collaboration between Xue, Engle, and Zhao, who reported a Ni(0)-catalyzed oxidative Heck reaction of unactivated alkenes with various boronic acids, using cinnamaldehyde 37a as the terminal oxidant (Scheme 14).57 The AQ template played a crucial role in enhancing reactivity at the unbiased alkene site. Both β-aryl and alkyl-substituted alkenyl boronic acids 7 were competent coupling partners, yielding conjugated dienoic acid derivatives 44. Mechanistic experiments and DFT calculations suggest that the reaction proceeds through oxidative addition of the Ni(0) catalyst into the O–H bond of the alcohol, followed by transmetallation with alkenyl boronic acid 7. Direct intramolecular carbometalation of alkene-coordinated adduct J-III was found to be highly endergonic, whereas the intermolecular 1,4-addition of the Ni–H bond of J-III to cinnamaldehyde 37a, forming J-IV, was more favorable. Coordination with the enolate ligand accelerates the carbometalation of J-IV, producing linear intermediate J-V. Finally, β-hydride elimination from J-V generates the product 44 and regenerates the Ni(0) catalyst.
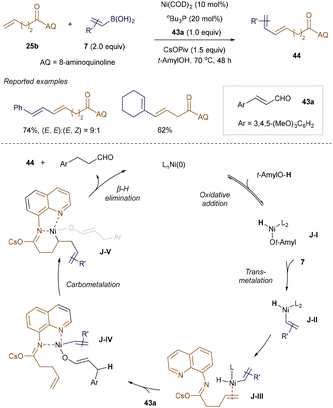 |
| | Scheme 14 Ni-catalyzed oxidative Heck reaction. | |
Alkenylamination.
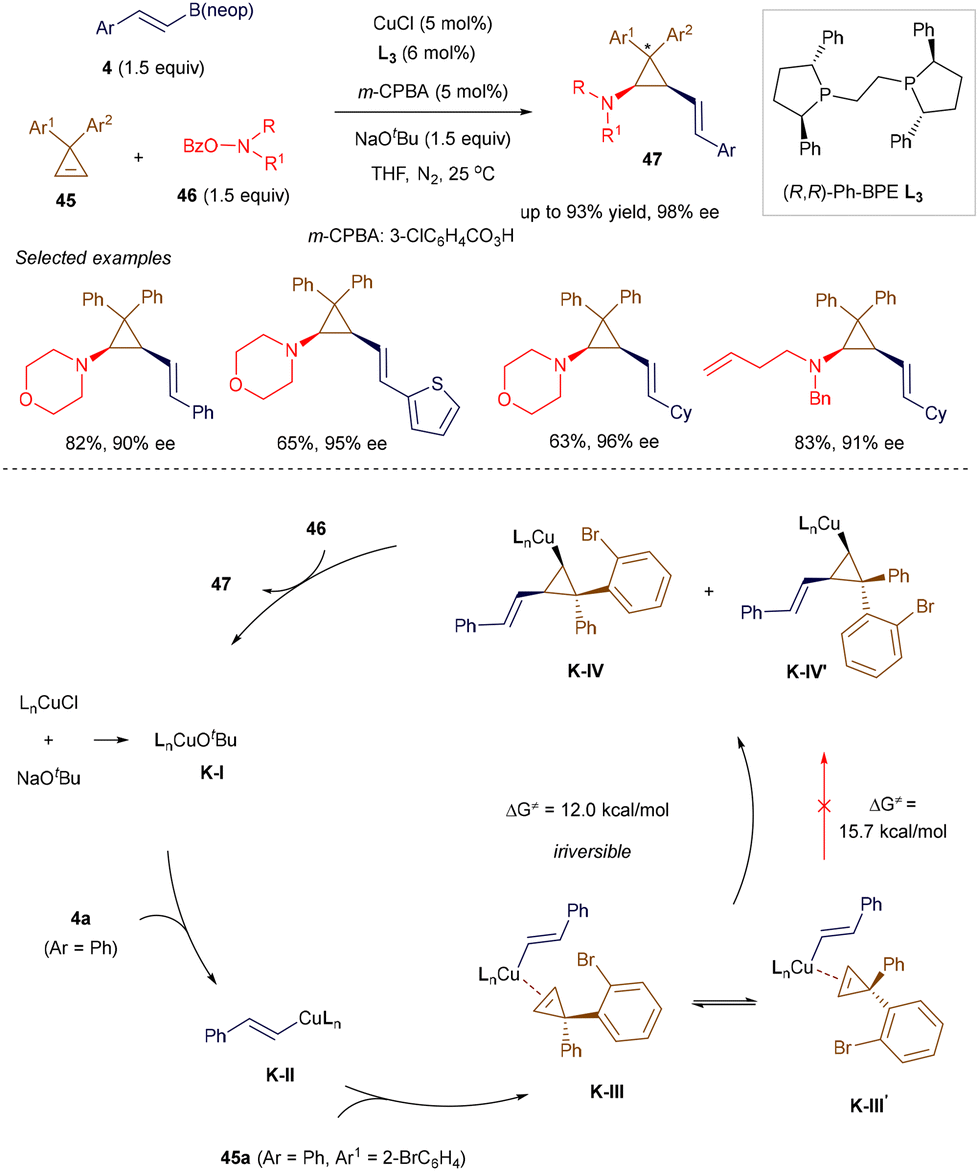
In 2020, Jia, Zhao, and co-workers introduced an elegant strategy for the highly stereoselective assembly of polysubstituted chiral cyclopropylamines.58 Cyclopropylamine functionalities are found in various drugs and drug candidates, and they also serve as useful synthons for constructing complex carbo- and heterocycles. The authors utilized commercially available ligand L3 in combination with copper(I) chloride to promote a three-component cyclopropene alkenylamination reaction, involving hydroxyamine esters 46 and alkenyl boron reagent 5, under mild conditions (Scheme 15). Similar to their earlier work,58a the addition of a catalytic amount of m-CPBA was crucial for successful alkenylamination. Interestingly, for cyclopropenes 45a bearing two different aryl groups on the C3 position, the catalytic system was able to distinguish subtle differences in ortho-substituents, producing the product as a single diastereomer. Although the scope of the alkenyl boron reagent was limited to simple β-aryl compounds, this method enables the synthesis of polysubstituted cis-1,2-alkenylcyclopropylamines 47 with up to three stereogenic carbon centers. The proposed mechanism (Scheme 15) begins with the formation of alkenyl Cu(I) K-IIvia transmetallation. The subsequent enantioselective migratory insertion step produces cyclopropyl Cu(I) K-IV through intermediate K-III. At this stage, due to steric interactions between the incoming alkenyl group and the larger 2-BrC6H4 substituent, K-III exists as a higher-energy intermediate compared to K-III′. However, K-III undergoes faster and irreversible migratory insertion, leading to the kinetically favorable isomer K-IV (over K-IV′). Trapping with aminating reagent 46 results in the formation of the observed diastereomer and regenerates the Cu(I) catalyst.
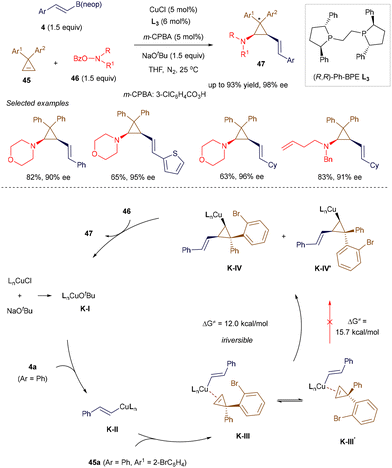 |
| | Scheme 15 Cu(I)-catalyzed alkenylamination. | |
2.2 Alkenyl boron reagents as the electrophilic partner – functionalization of the olefinic moiety
As shown in the previous transition metal-catalyzed examples, various alkenyl boron reagents have been used to incorporate carbon–carbon double bonds into products. Additionally, methods that directly functionalize the double bonds of alkenyl boron compounds have also been developed. A key feature of this strategy is that the boron moiety in the product is retained, opening new pathways for synthesizing useful alkyl boron compounds. The reactivity of the double bond in alkenyl boronates, compared to that of simple alkenes, can be unexpected and is influenced by the substituents on the boron atom, requiring careful consideration. From a bond-formation perspective, two main ionic pathways have been successfully utilized. The first pathway involves direct functionalization of the olefinic portion of alkenyl boronates through hydrogenation,59 Michael additions,9,60 hydroborylation,61 hydroamination,62 hydrosilylation,63 diborylation,64 Cu-mediated 1,2-difunctionalization,65 CuH-mediated hydroalkylation,66 and carbozincation.67 The second pathway focuses on conjunctive cross-coupling via 1,2-migrations of boronate complexes, largely pioneered by Morken and co-workers.2l,10,68 In this section, recent developments in nickel-, cobalt-, and copper-catalyzed hydroalkylation, 1,2-difunctionalization, and [2 + 2] reactions of alkenyl boronates are highlighted. In these cases, transition metals not only act as reductants to generate radicals but also mediate selective bond formation. Notably, the open-shell reactivity of alkenyl boron reagents has been applied in novel transformations under light-triggered reaction conditions (section 4).
Hydroalkylation.
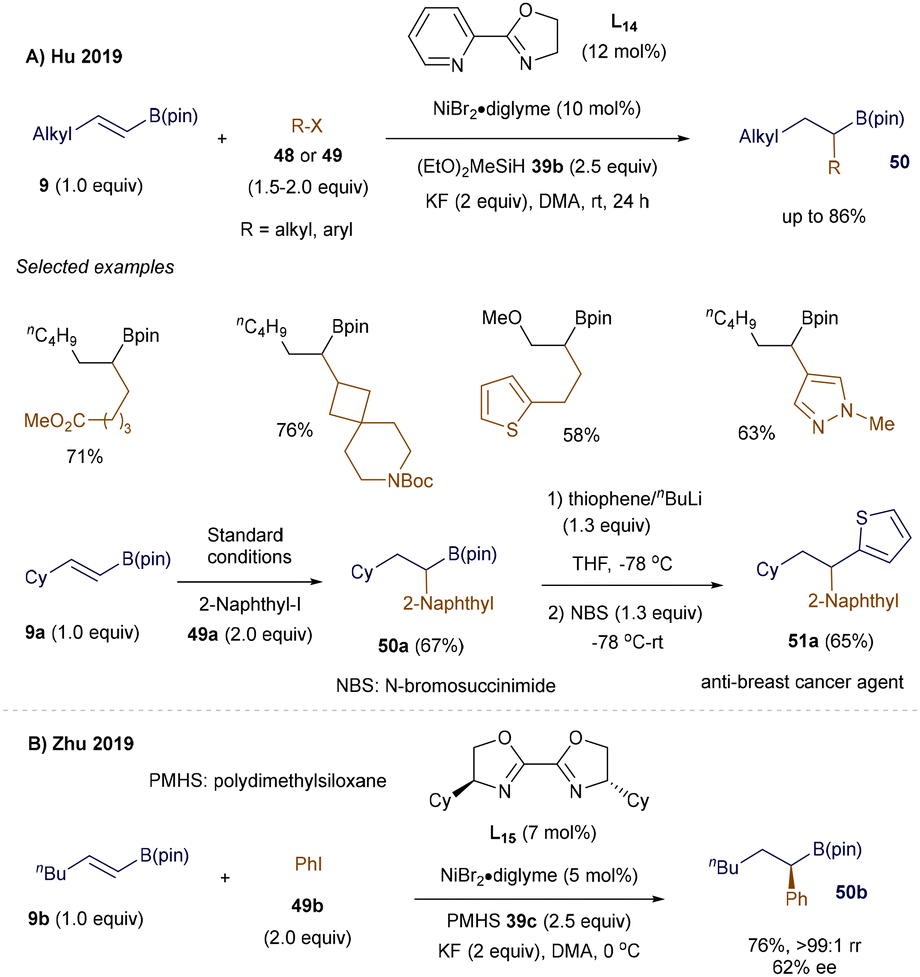
To broaden the scope of hydroalkylation of alkenyl boronates via CuH catalysis, 66 organic chemists have long been intrigued by the potential to form new carbon–carbon bonds through the open-shell reactivity of alkenyl boron reagents. In 2014, Baran's group reported an FeH-catalyzed hydroalkylation of alkenyl boron reagents.69 Around the same time, Hu70 and Zhu71 simultaneously developed NiH-catalyzed regioselective hydroalkylation and hydroarylation of β-alkyl alkenyl boronic pinacol esters 9. Directed by the B(pin) group, the strategy involves regioselective addition of the NiH intermediate to the double bond of 9, resulting in the formation of a stable α-boryl organonickel intermediate (Schemes 16 and 17). Subsequent cross-coupling with alkyl or aryl halides 48 or 49 leads to a wide range of secondary alkyl B(pin) derivatives 50, which serve as valuable synthetic intermediates in medicinal and materials chemistry.
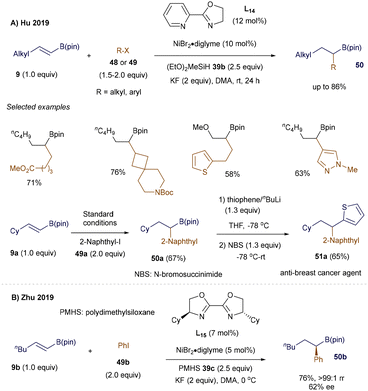 |
| | Scheme 16 NiH-catalyzed hydrocarbonation. | |
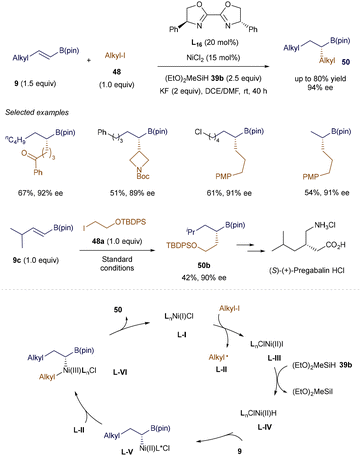 |
| | Scheme 17 NiH-catalyzed enantioselective hydroalkylation. | |
As shown in Scheme 16, under Hu's optimized conditions, both hydroalkylation and hydroarylation reactions proceeded smoothly with simple starting materials, demonstrating tolerance to a variety of functional groups (Scheme 16A). Additionally, this method was successfully applied to the synthesis of an anti-breast cancer agent 51a.70 Meanwhile, Zhu's group achieved similar success in the NiH-catalyzed remote hydroarylation of boron-containing alkenes, utilizing nearly identical reaction conditions. They also tested an asymmetric version of the reaction, though with moderate enantioselectivity (Scheme 16B).71
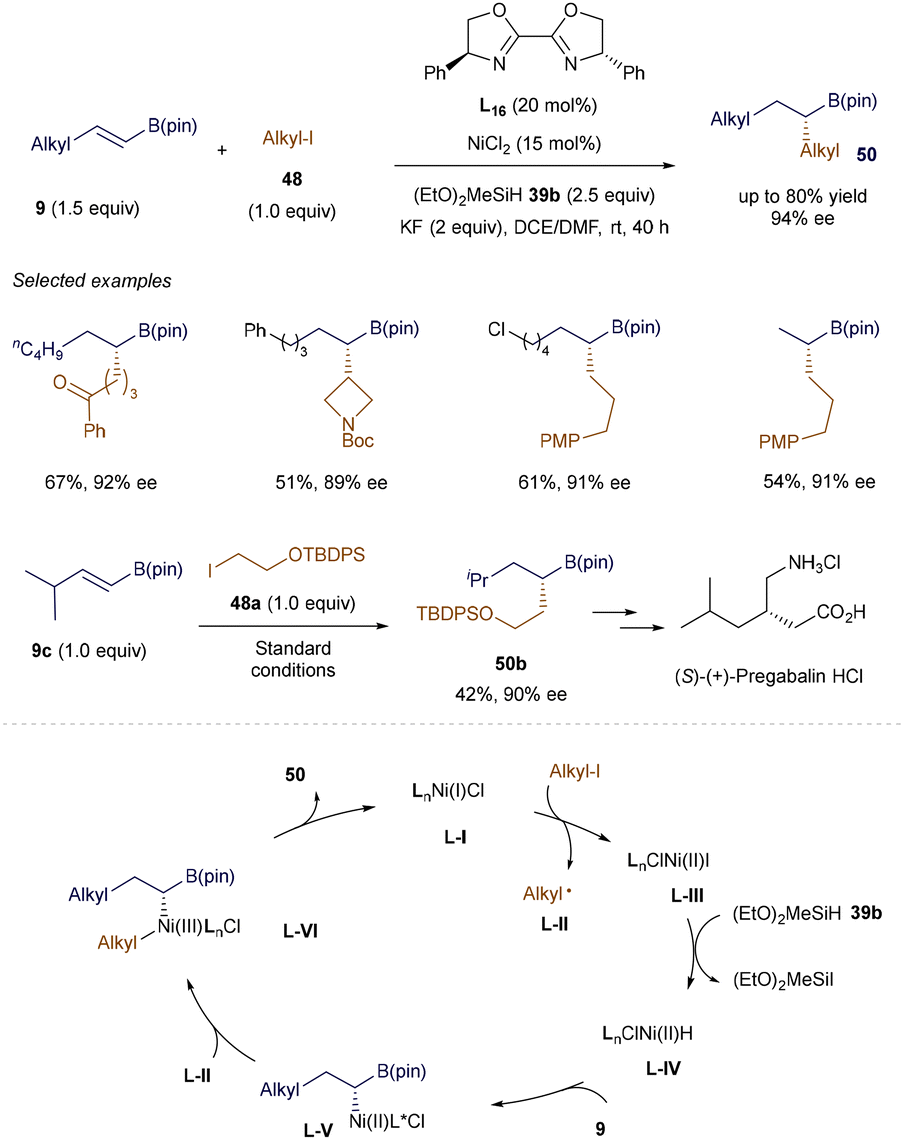
Hu's group further developed the asymmetric version of the NiH-catalyzed hydroalkylation reaction (Scheme 17).72 This strategy allows the stereoselective union of two alkyl fragments, including secondary alkyl groups, and exhibits a broad substrate scope with excellent tolerance for various functional groups. This enables the efficient synthesis of chiral alkyl B(pin) derivatives 50 under mild conditions. Moreover, the protocol has been applied to the functionalization of natural products and drug derivatives, and was successfully used in the facile synthesis of 50b, a key intermediate in the production of (S)-(+)-Pregabalin·HCl. A plausible mechanism was proposed based on a series of control experiments. It begins with the formation of a chiral LnNi(I)Cl species L-I, which reduces alkyl iodide to form an alkyl radical L-II and LnClNi(II) L-III. The reaction of L-III with (EtO)2MeSiH generates a Ni-H species L-IV. Subsequent regioselective insertion into alkenyl B(pin) 9 produces a Ni-alkyl intermediate L-V, which can recombine with the alkyl radical L-II to form a Ni(III) complex L-VI. The final reductive elimination yields the desired product 50 and closes the catalytic cycle.72
1,2-Difunctionalization.
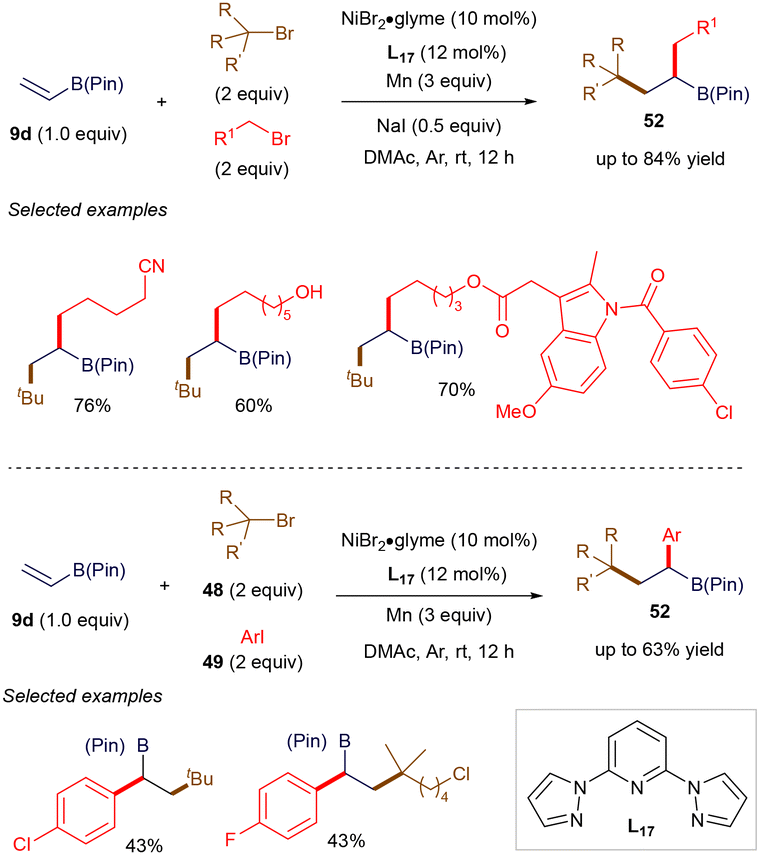
The open-shell reactivity of vinyl boronates plays a role in certain 1,2-carbofunctionalization reactions of vinyl boronate esters, catalyzed by inexpensive transition metals. For instance, in 2020, Lu, Fu, and co-workers reported a nickel-catalyzed three-component dialkylation and alkylarylation of vinyl boronate esters (Scheme 18).73 The use of nickel catalysis facilitates these transformations with high coupling efficiency and excellent functional group tolerance. The synthetic utility of this method has been showcased through late-stage modifications of bioactive derivatives. Mechanistically, the nickel catalyst can distinguish between the reactivities of tertiary alkyl bromides and either primary alkyl bromides or aryl iodides, yielding products with outstanding chemo- and regio-selectivities. Notably, as illustrated later in Scheme 33A, Martin reported a similar alkylarylation involving the synergistic use of nickel and photoredox catalysis. Following a similar mechanistic pathway to Martin's, the reaction likely proceeds through the generation of tertiary alkyl radicals via the reduction by a Ni(I) catalyst. This is followed by a 1,4-radical addition to vinyl boronate ester 9d and cross-coupling with primary alkyl bromides or aryl iodides, mediated by Ni(I), ultimately producing the observed product 52.
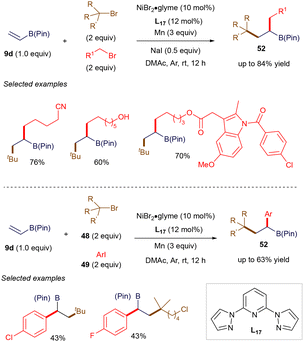 |
| | Scheme 18 Ni(I)-catalyzed selective 1,2-alkylation and 1,2-alkylarylation. | |
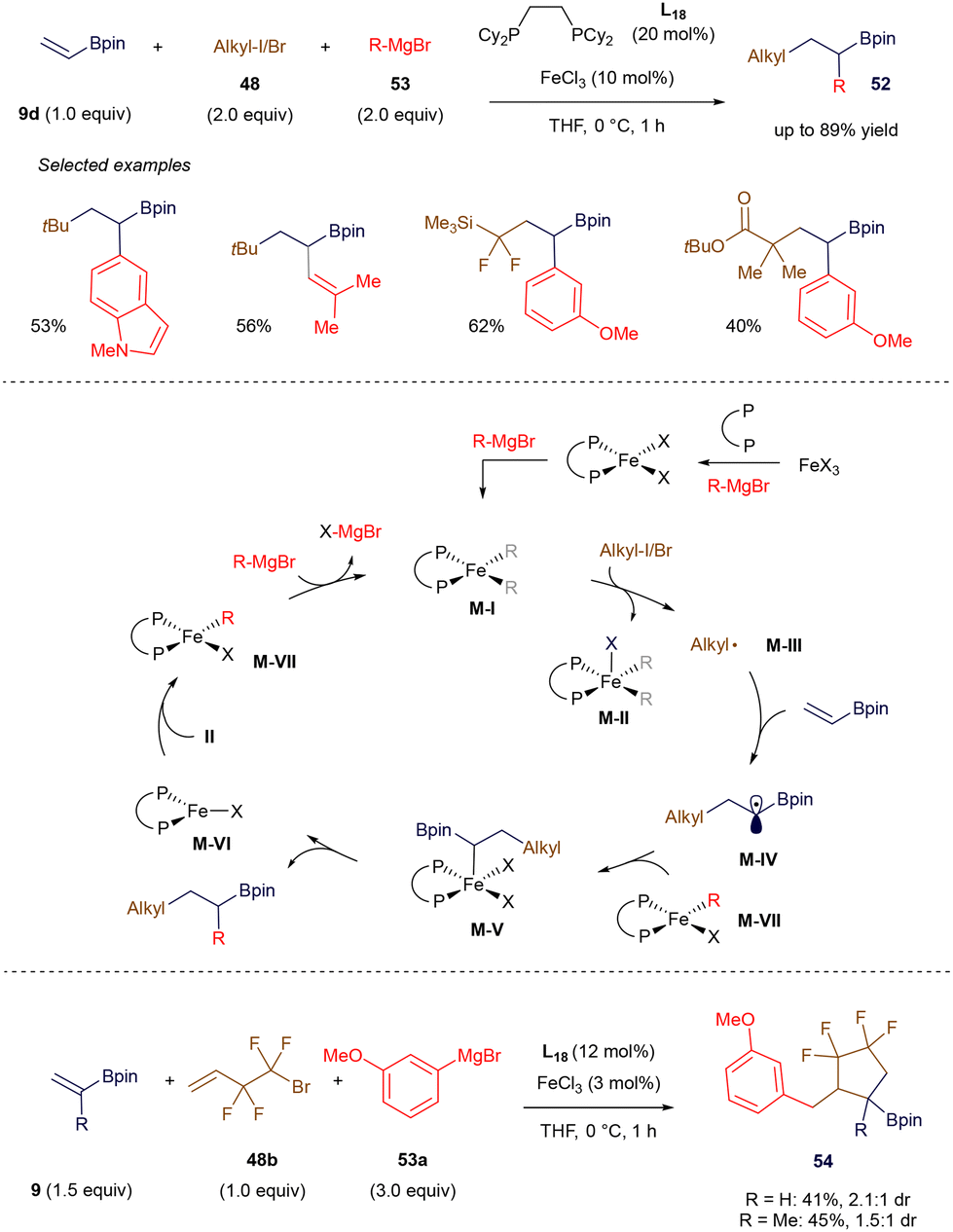
In 2021, Gutierrez's group developed an elegant Fe(II)-catalyzed three-component process involving alkyl halides, Grignard reagents, and vinyl boronate esters (Scheme 19).74 Following a thorough investigation utilizing Mössbauer spectroscopy, electron paramagnetic resonance (EPR), X-ray crystallography, and DFT calculations, the authors proposed that the in situ-formed bis-arylated Fe(II) species, M-I, is responsible for abstracting halogen from the alkyl halide, leading to the formation of the corresponding alkyl radicals M-III and M-II. Radical M-III undergoes a Giese-type addition to the π-system of alkenyl boronate 9d, generating the α-boryl radical M-IV (more examples of such radical additions are discussed in section 3). This radical is subsequently trapped by the monoarylated Fe(II) species M-VII, which facilitates C–C bond formation. Comproportionation between Fe(I) and Fe(III) species M-VI and M-II regenerates the Fe(II) species M-VII, allowing the catalytic cycle to continue in the presence of additional Grignard reagent. The authors further demonstrated the versatility of this method by extending it to alkyl halides with pendant olefins as radical precursors, enabling the synthesis of tetrafluoroethylene-containing cyclopentane derivatives 54.
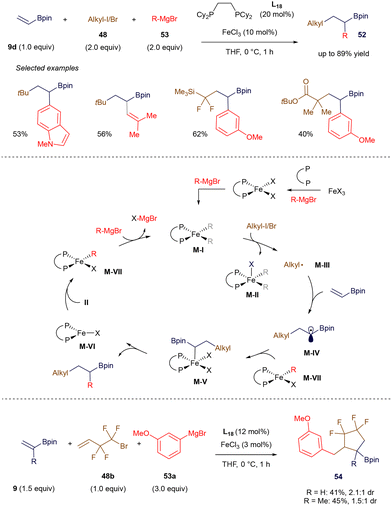 |
| | Scheme 19 Iron-catalyzed 1,2-biscarbofunctionalization. | |
α-Haloboronic esters are valuable building blocks in organic synthesis. Their synthesis via atom transfer radical addition (ATRA) presents an appealing but challenging strategy due to the relative stability of the α-boryl radical, which makes efficient halogen transfer difficult except with highly electron-deficient radical precursors. To address this challenge, Hull and co-workers developed a Cu-mediated approach.75 As illustrated in Scheme 20, using 4 mol% of [Cu(dtbbpy)2](OTf)2 as a catalyst efficiently promotes the ATRA reaction of various primary, secondary, and tertiary alkyl halides 48 with both unsubstituted and α-substituted vinyl pinacol boronic esters 9, producing α-haloboronic esters 55 with yields of up to 95%. The proposed mechanism involves the reduction of 48 by LnCu(I), generating radical N-I and LnCu(II)X. After radical N-I adds to 9, the resulting α-boryl radical N-II undergoes halogen atom transfer (XAT) with LnCu(II)X, forming the desired product 55 and regenerating LnCu(I).
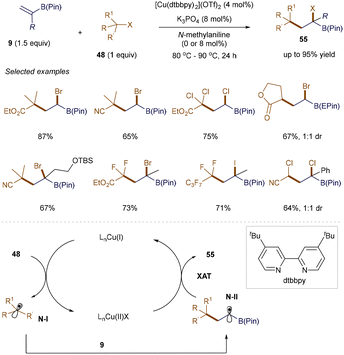 |
| | Scheme 20 ATRA reaction. | |
[2 + 2] cycloaddition.
In 2019, Rajanbabu and co-workers introduced an excellent and practical method for the enantioselective synthesis of cyclobutyl boronates 57via a Co(I)-catalyzed [2 + 2] cycloaddition between simple alkynes 56 and alkenyl boronate 9d (Scheme 21).76 This method demonstrated high efficiency and selectivity under mild conditions, accommodating both symmetric and asymmetric alkynes with ease. Mechanistic studies suggest that the reaction begins with Co(I)-catalyzed selective oxidative cyclization to generate intermediate O-II, followed by reductive elimination to furnish the desired cyclobutyl boronate product 57. Notably, a radical-mediated [2 + 2] cycloaddition strategy involving alkenyl boronates has also been developed for the stereoselective synthesis of cyclobutylboronates, with further examples discussed in section 4.3.
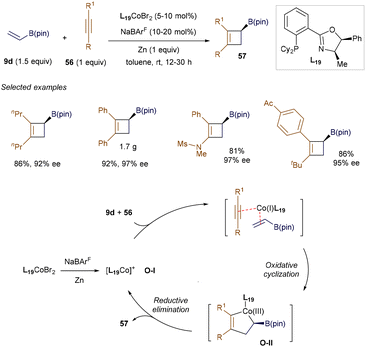 |
| | Scheme 21 Co(I)-catalyzed [2 + 2] cycloaddition. | |
3. Organocatalysis
Although the use of simple chiral organic molecules to promote asymmetric transformations has been documented since the 1970s,77 it wasn't until 2000 that the field experienced significant growth.78 During this time, small chiral organic molecules with minimal functional groups were employed to mimic biocatalysts, adopting diverse catalytic modes.79 Compared to transition-metal catalysis, organocatalysis offers several advantages, such as low toxicity, insensitivity to moisture and air, and ready availability from natural chiral sources. As a result, the field was awarded the Nobel Prize in 2021. However, organocatalytic transformations using alkenyl boron reagents have been far less explored than those using transition-metal catalysis. To date, several organocatalytic models relying on chiral diols2c,f and secondary amines80 have been developed. As summarized below, alkenyl boron reagents can be directly activated by organocatalysts or used to trap reactive organic intermediates formed in situ via Lewis acid or Lewis base catalysis. These new protocols have been successfully applied for the facile synthesis of functionalized alkenes or alkyl boronates, including enantioselective transformations. For clarity, the discussion in this section will be organized based on the types of organocatalysts used.
3.1 Chiral diol-catalyzed insertion reactions
One exceptionally good example that uses chiral small molecules to activate boronic reagents for asymmetric organocatalysis was reported in 2006. Because of the dynamic covalent bonding ability of boron,81 Schaus and co-workers noticed that a catalytic amount of chiral BINOL derivative can act as an exchangeable chiral ligand with neutral organoboronates, promoting the first enantioselective organocatalytic allylation of ketones via direct activation of allylboronates.82 Inspired from this seminal contribution, Schaus further sought suitable organocatalytic conditions to achieve the asymmetric vinylation of in situ formed iminium salts (Petasis reaction) with alkenylboronates.83 After this, the group further expanded the scope of electrophiles to acyl imines,84o-quinone methides,85 and 1-ethoxycarbonyl-1,2-dihydroquinolines,86 respectively. Furthermore, the groups of Chong,87 Takemoto,88 Sugiura,89 May,90 and Liu91 also made a significant contribution to this area. Considering that these findings have been well summarized by Matteson2c and May,2f the related chemistry will not be detailed in this section.
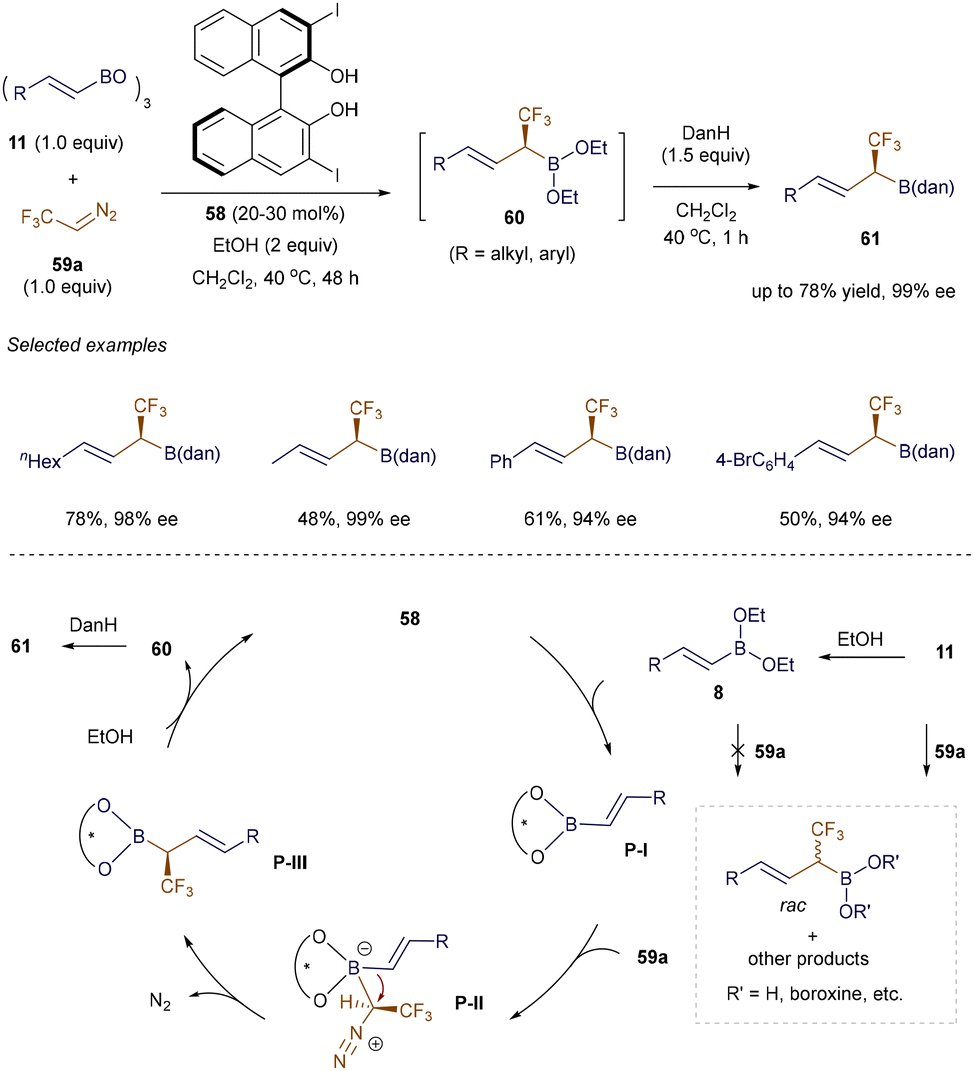
In 2020, Szabó et al. introduced a practical method for synthesizing chiral α-CF3 or α-SiMe3 allylic boron molecules, which are valuable in drug design and natural product synthesis (Scheme 22).92 Using a BINOL catalyst 58 and ethanol, the catalytic asymmetric homologation of both β-alkyl and aryl-substituted alkenyl boroxine 11 with CF3-diazomethane 59a proceeded smoothly under mild conditions. Notably, the reaction was scalable up to 2 mmol. The proposed mechanism begins with the formation of the unreactive ethyl alkenyl boronic ester 8. A transesterification step with catalyst 58 then produces the chiral alkenyl boronate P-I, which significantly increases the Lewis acidity of the boron atom, enabling the formation of the boron ate complex P-II and inducing stereoselectivity. This is followed by a stereoselective 1,2-migration of the alkenyl group, resulting in P-III. Ethanolysis of P-III yields the air-sensitive intermediate 60, which is ultimately protected with diaminonaphthalene (DanH), forming the final product 61.
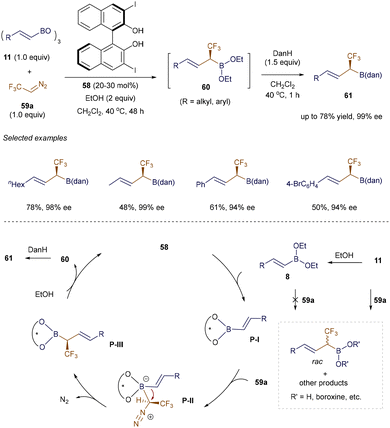 |
| | Scheme 22 Organocatalytic synthesis of α-CF3 allylboron compounds. | |
Furthermore, by reacting the chiral intermediate 60 with aldehydes 43 or under oxidative conditions (NaOH/H2O2), a variety of α-CF3-substituted alcohols 62 or 63 were obtained in good to high yields with good to excellent enantioselectivities (Scheme 23, eqn (1) and (2)). Additionally, the method is compatible with Me3Si-diazomethane 59b, opening a new pathway for the preparation of enantioenriched α-SiMe3 allylboronates 64 (eqn (3)). To further demonstrate the utility of these methods, the authors presented a one-pot strategy to convert the α-CF3 allyl boronic derivatives 61 into α-CF3 allylic boroxines 66. These boroxines 66 serve as highly reactive reagents for allylation reactions with aldehydes, ketones, and imines, as well as for dearomative allylation of indoles, yielding product 67 with excellent stereoselective control (eqn (4)).92
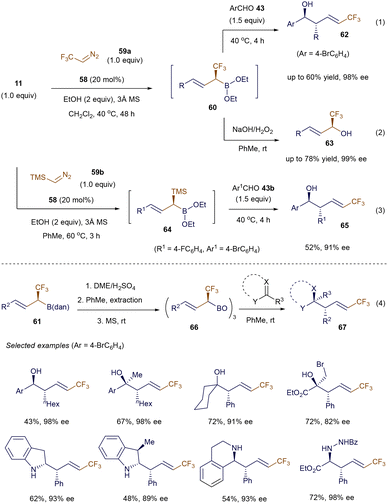 |
| | Scheme 23 Diverse reactivities of allylic boron and silicon reagents. | |
3.2 Phosphoric acid-catalyzed cycloadditions
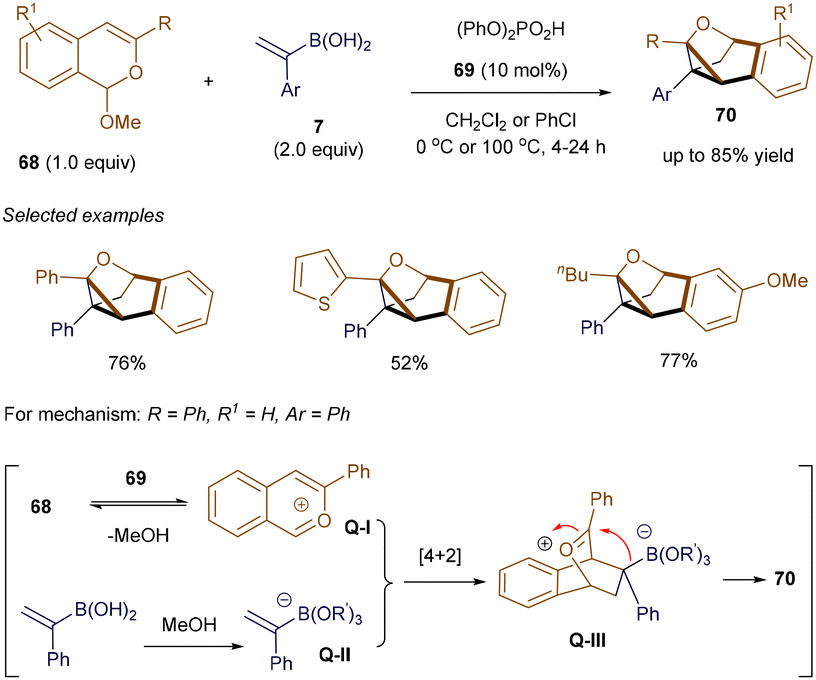
Considering the presence of double bonds in alkenyl boronic acids, the development of new catalytic examples that exploit this alkene moiety as a C2 synthon in organocatalysis has been explored. In 2015, Sun's group was among the first to utilize this reagent, employing an in situ generated 10π-electron isobenzopyrylium ion as a key intermediate to facilitate cycloaddition reactions.93 Soon after, Huang and Sun further demonstrated that using (1-arylvinyl)boronic acid as the nucleophilic partner to trap the isobenzopyrylium ion enabled the formation of cyclopropane-containing tricyclic products.94 As shown in Scheme 24, condition optimization revealed that (PhO)2PO2H 69 with appropriate acidity serves as an effective catalyst for this unusual process. A variety of highly strained products 70 were easily isolated in yields ranging from 41% to 85%, even on a gram scale. According to the proposed mechanism, the isobenzopyrylium ion Q-I forms through the interaction of catalyst 69 with substrate 68, accompanied by the release of MeOH. Meanwhile, boronic acid 7 is activated by MeOH, forming borate Q-II. A subsequent [4 + 2] cycloaddition between these two species generates the bicyclic oxonium intermediate Q-III. Unlike previous examples, where the C–O bond of the oxonium bridge in Q-III cleaves, in this case, the nucleophilic borate preferentially attacks the electrophilic carbon of the oxonium bridge, resulting in the formation of the strained C–C bond and the observed product structure.
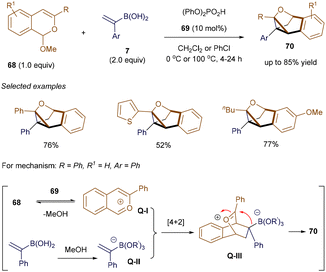 |
| | Scheme 24 Synthesis of cyclopropane-containing tricyclic skeleton. | |
3.3 Lewis base-catalysed allylic amination
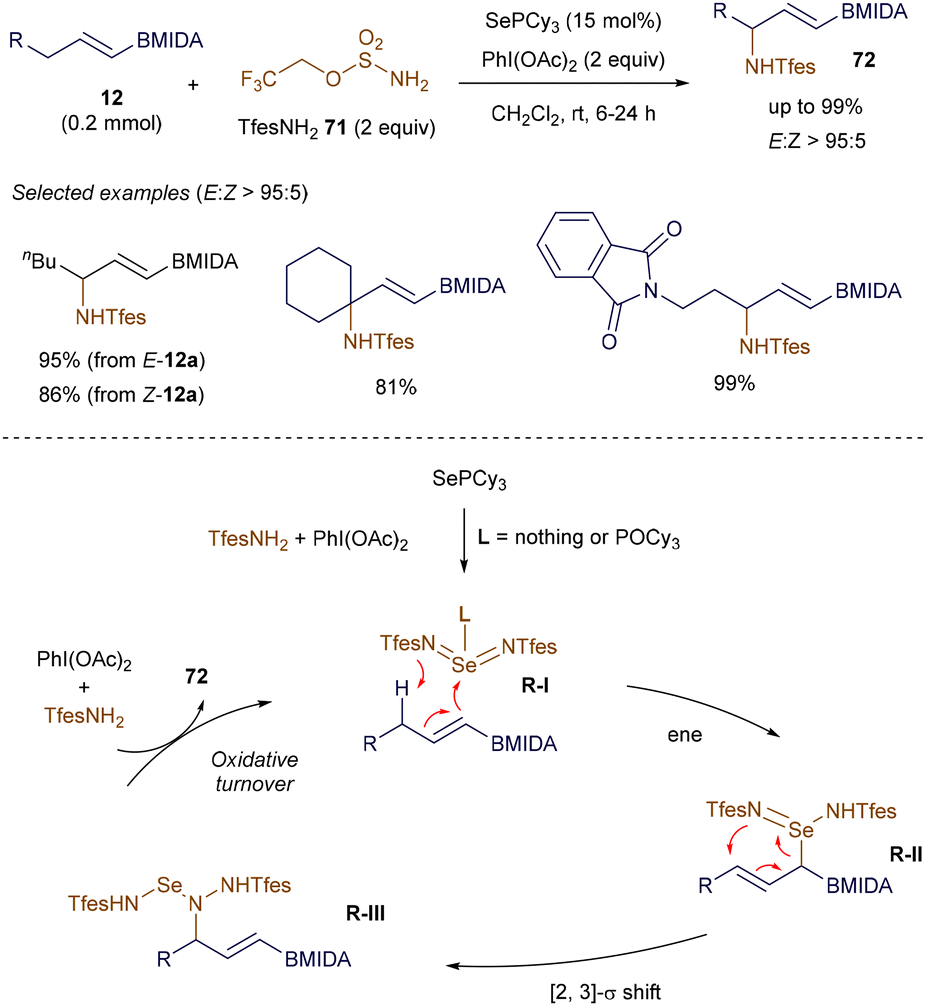
In addition to the stereospecific 1,2-migrations of boronate complexes induced by π-acidic transition metal complexes developed by Morken, an alternative organocatalytic 1,2-migration of alkenyl boronates was reported by Denmark in 2018. This method showcases the effectiveness of Lewis base catalysis in developing novel transformations using alkenyl boron reagents.95 In 2022, Michael introduced a highly selective and unusual allylic C–H amination of alkenyl boronates, also employing Lewis base catalysis (Scheme 25). This novel transformation further illustrates the expanding utility of alkenyl boronates in synthetic chemistry.96
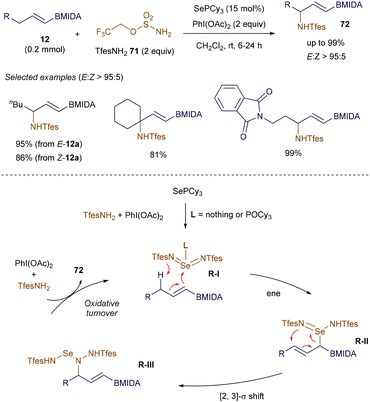 |
| | Scheme 25 Lewis base-catalyzed allylic amination. | |
The allylic amination of various alkenyl B(MIDA) 12 proceeds smoothly under the catalysis of SePCy3, forming a new C–N bond while avoiding potential issues such as transmetallation or alkene additions. Notably, the configuration of the double bond in substrates 12 has little impact on the outcome, and the reaction can be performed under very mild conditions. Preliminary mechanistic studies suggest that the reaction begins with the formation of a highly reactive selenium bis(imide) intermediate R-I through the interaction of the SePCy3 catalyst with the in situ generated iminoiodinane. This intermediate then undergoes an ene reaction, followed by a [2,3]-σ rearrangement, to afford the aminated product 72 with excellent selectivity.
4. Photocatalysis
The recent resurgence of photocatalytic methods has driven the development of numerous protocols for generating highly reactive intermediates, such as free radicals and triplet states, under very mild conditions—typically through light irradiation in the presence of catalytic amounts of a photocatalyst at room temperature.97 Upon photoexcitation, the photocatalyst gains enhanced redox properties, enabling it to facilitate either single-electron transfer (SET) or energy transfer (EnT) processes with the target substrate.98 One well-established application of this open-shell reactivity involves radical additions to alkenyl boron reagents, a strategy that has been explored for over 60 years.99 Due to the low polarization effect of the boron-based substituent, radical addition can occur either at the α or β position of the vinyl organoboron reagent, depending on the nature of the radicals and the substituents on the alkenyl or boron moiety. For example, as previously mentioned, the groups of Gutierrez74 and MacMillan80 have reported selective functionalization of alkenyl boron reagents at the β position of alkenyl boronate esters or the α position of trifluoroborate salts under thermal radical conditions, respectively. The group of Studer pioneered the investigation of open-shell reactivity in vinylboronate complexes through a thermal process.100 In 2017, Aggarwal's group reported the first light-triggered catalytic procedure for the dicarbofunctionalization of vinyl boronate complexes.101 This work has further fuelled the development of analogous photochemical methods, driven by the resurgence of photocatalysis over the past decade.
In this section, based on the types of boryl radicals formed, the discussion will be divided into three categories: radical additions at the α or β position of vinyl boronates, and the simultaneous formation of biradicals at both the α and β positions. Both single-electron transfer (SET) and energy transfer (EnT) activation strategies have been employed in these processes, aiming to broaden the reactivity scope of alkenyl boron reagents.
4.1 Radical addition at α position of vinyl boron reagents: synthesis of alkenes
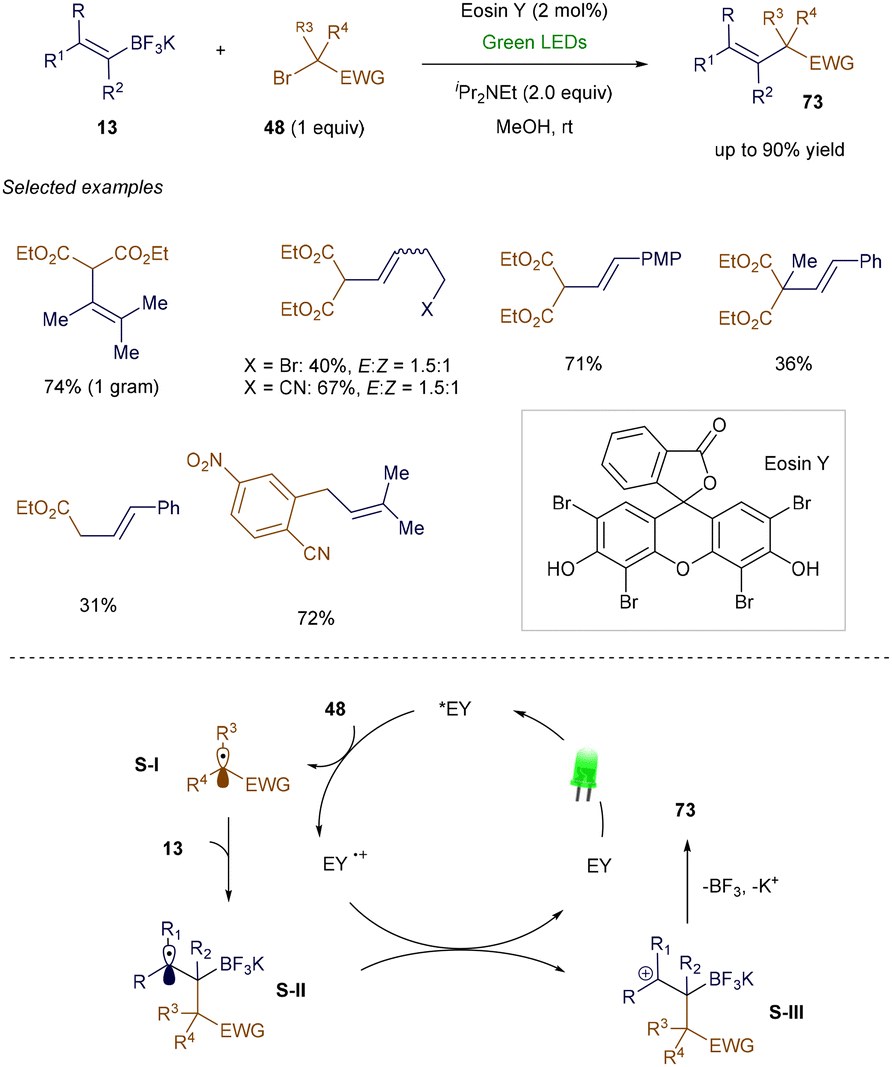
In 2013, Koike and Akita's group introduced the first photochemical strategy for synthesizing trifluoromethylated alkenes from vinyl boronates and Togni's reagent.102 Building on this work, Leonori's group later developed a general procedure for the vinylation of various electron-poor alkyl bromide derivatives (Scheme 26).103 In this reaction, Eosin Y, upon absorbing light, transfers an electron to electron-poor bromides 48, generating an electrophilic radical. This radical adds to the vinyl potassium trifluoroborate 13, forming a secondary carbon-centered radical. This radical then undergoes single-electron oxidation, resulting in a β-borato cation, which ultimately forms the desired product 73via elimination.
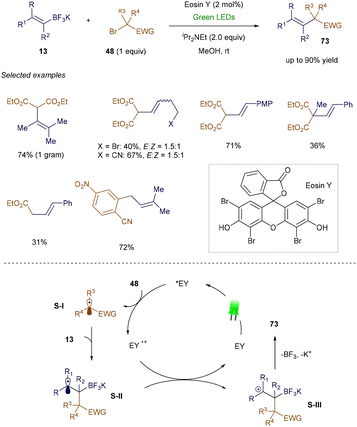 |
| | Scheme 26 Synthesis of functionalized alkenes from potassium vinyltrifluoroborates. | |
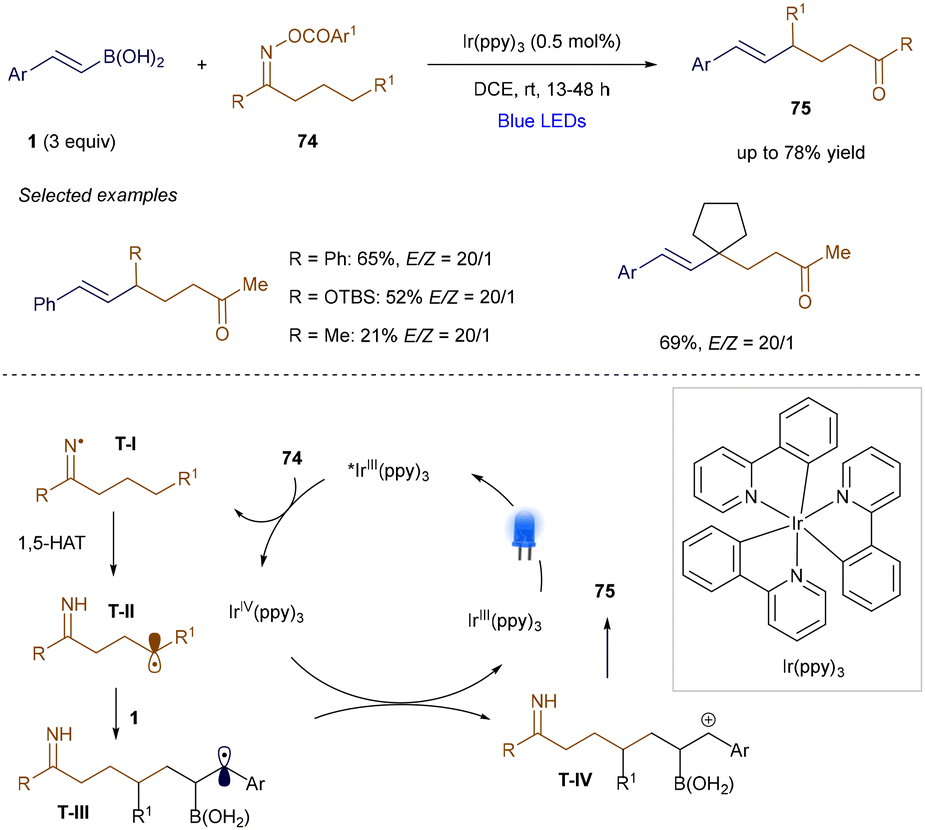
In addition to electrophilic radicals, nucleophilic radicals can also participate in α-position additions to vinyl borons, as demonstrated by Yu's group in 2018 (Scheme 27).104 In this case, O-acyl oximes undergo photoinduced single-electron reduction, generating nitrogen-centered radical T-I. This radical undergoes a 1,5-hydrogen atom transfer, producing the corresponding carbon-centered radical T-II. Despite having a different polarity compared to the electron-poor radicals discussed in Scheme 25, T-II adds at the α-position of a styrenyl boronic acid derivative, forming benzylic radical T-III. After single-electron oxidation, this radical undergoes elimination, yielding the alkene product 75. This method is versatile, enabling the generation of primary, secondary, and tertiary alkyl radicals. However, its main limitation is that it applies primarily to styrenyl boronic derivatives, where the benzyl radical formed after addition is stabilized.
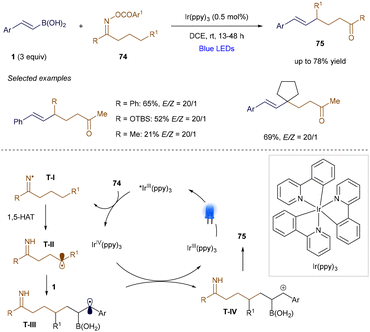 |
| | Scheme 27 Remote alkenylation. | |
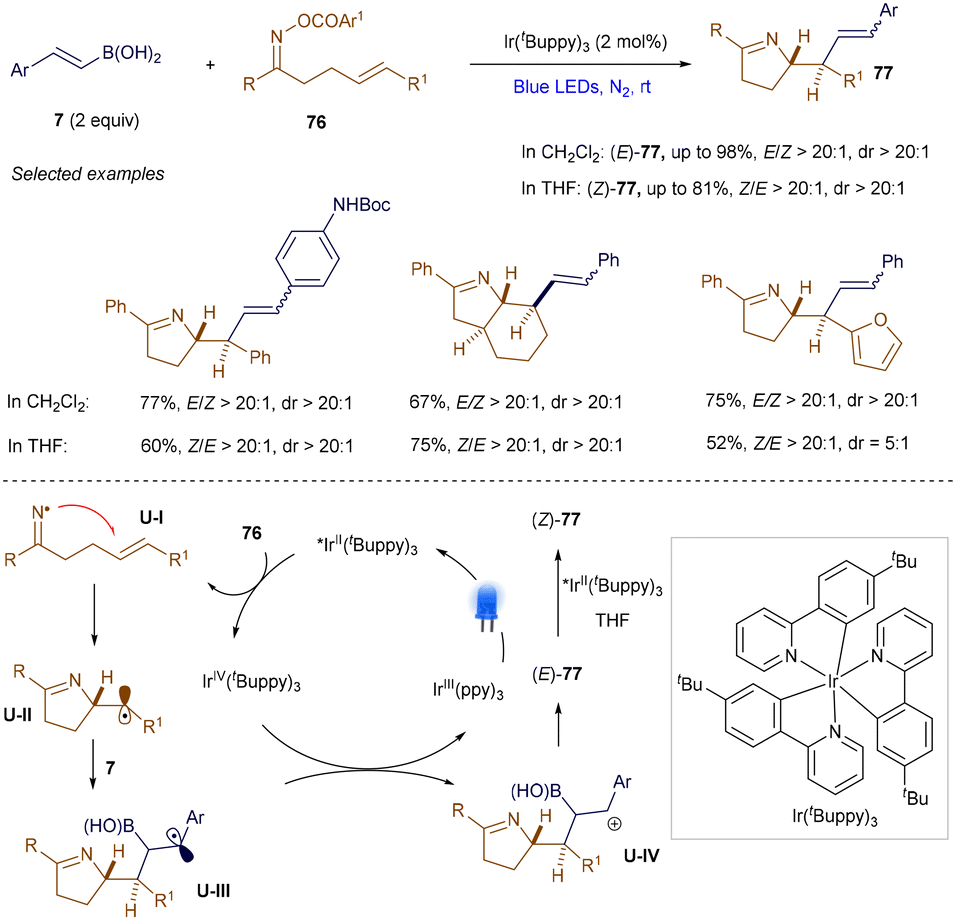
Using a similar strategy, Yu and co-workers also achieved the iminoalkenylation of alkenes via photoredox catalysis (Scheme 28).105 Notably, depending on the choice of solvent, both (E)- and (Z)-configurations of the alkene products 77 can be selectively obtained. Detailed mechanistic studies suggest that the formation of (Z)-77 results from a photocatalytic, contra-thermodynamic E to Z isomerization, facilitated by an energy transfer (EnT) pathway.
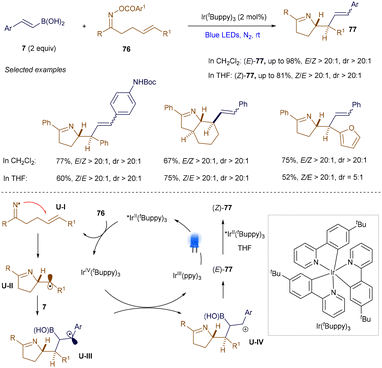 |
| | Scheme 28 Iminoalkenylation of alkenes. | |
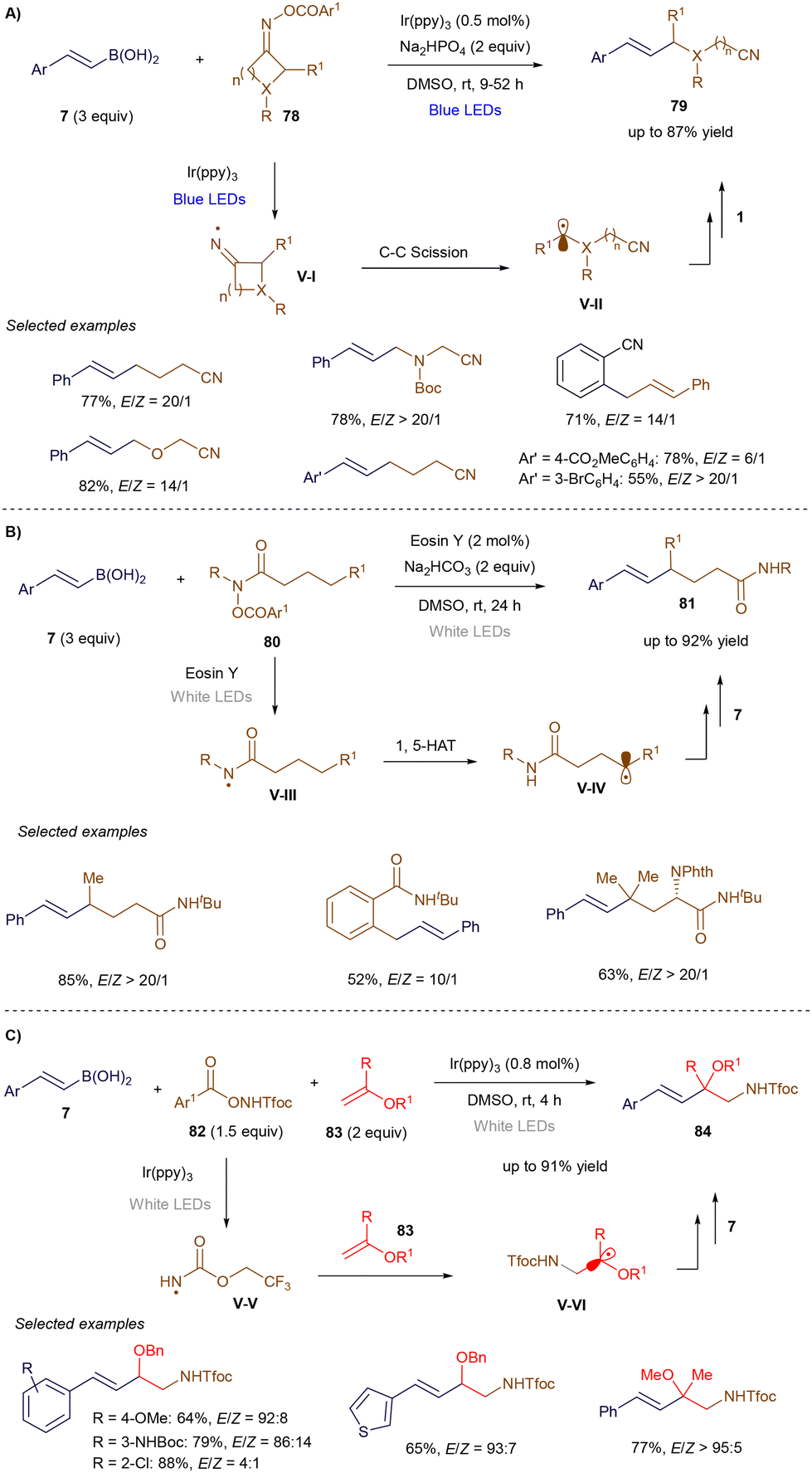
In the same work, Yu's group extended this process to the use of hydroxamic acid derivatives 78 (Scheme 29A). Following the photoinduced generation of a nitrogen-centered radical V-Ivia single-electron reduction, the radical undergoes carbon–carbon bond cleavage, yielding a carbon-centered radical V-II. This radical then adds at the α-position of styrenyl boronic acid 7, and the resulting benzylic radical, after undergoing single-electron oxidation, leads to alkene product formation through elimination.104 Later that year, the group demonstrated that amide derivatives 80 (Scheme 29B) could promote a 1,5-hydrogen atom shift to generate a carbon-centered radical V-IV. This radical reacts with styrenyl boronic acid 7 to ultimately produce alkene 81.106 Finally, Yu and co-workers expanded this methodology to a three-component reaction (Scheme 29C), where hydroxylamine derivatives 82 were combined with a photoredox catalyst to generate nitrogen-centered radicals V-V. These radicals were trapped in an intermolecular reaction with electron-rich enol ethers, forming nucleophilic α-oxo radicals V-VI, which added to styrenyl boronic acids 7. The resulting radicals, similar to previous steps, underwent further reactions to yield alkene products 84.107
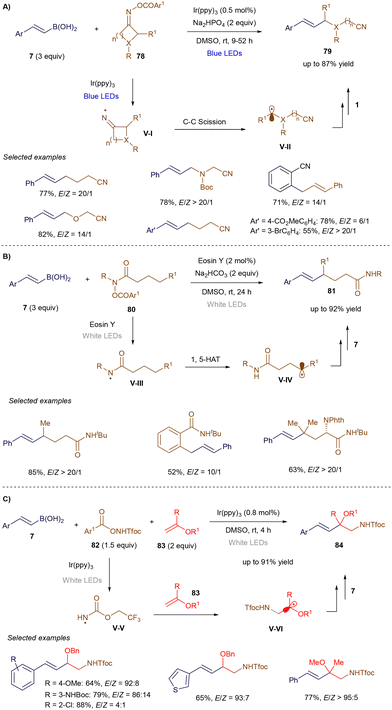 |
| | Scheme 29 Remote vinylation reactions. | |
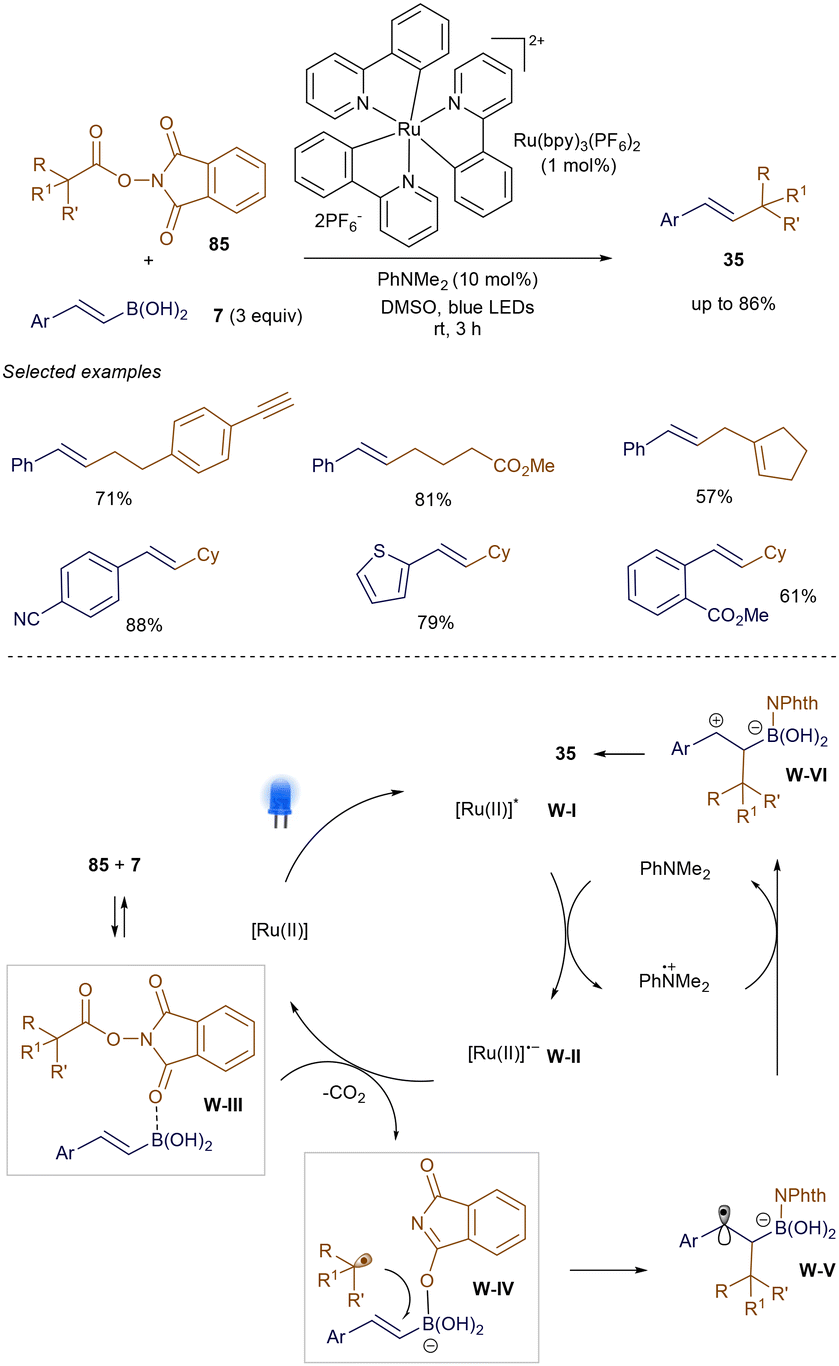
Unlike Baran's earlier work using NiH catalysis,69 Watson and co-workers recently reported that photoredox catalysis can effectively promote the decarboxylative cross-coupling of NHPI ester 85 with various styrene boronic acids 7 at room temperature.108 As shown in Scheme 30, under blue LED irradiation, the decarboxylative coupling proceeds smoothly, catalyzed by Ru(bpy)3(PF6)2 and PhNMe2, producing product 35 with versatile functional groups. Control experiments revealed that 85 can be activated through coordination with 7, facilitating its reduction by intermediate W-II. After decarboxylation of W-III, a nucleophilic rebound of the phthalimide anion generates the alkyl radical W-IV and boronate intermediate W-V. Subsequent radical addition, followed by radical-polar crossover via oxidation, forms W-VI, with final elimination leading to the cross-coupled product 35.
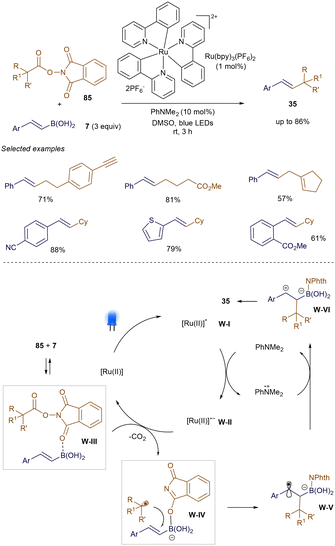 |
| | Scheme 30 Light-promoted decarboxylative cross-coupling. | |
4.2 Radical addition at β position of vinyl boron reagents: synthesis of alkyl boronates
Radical addition.
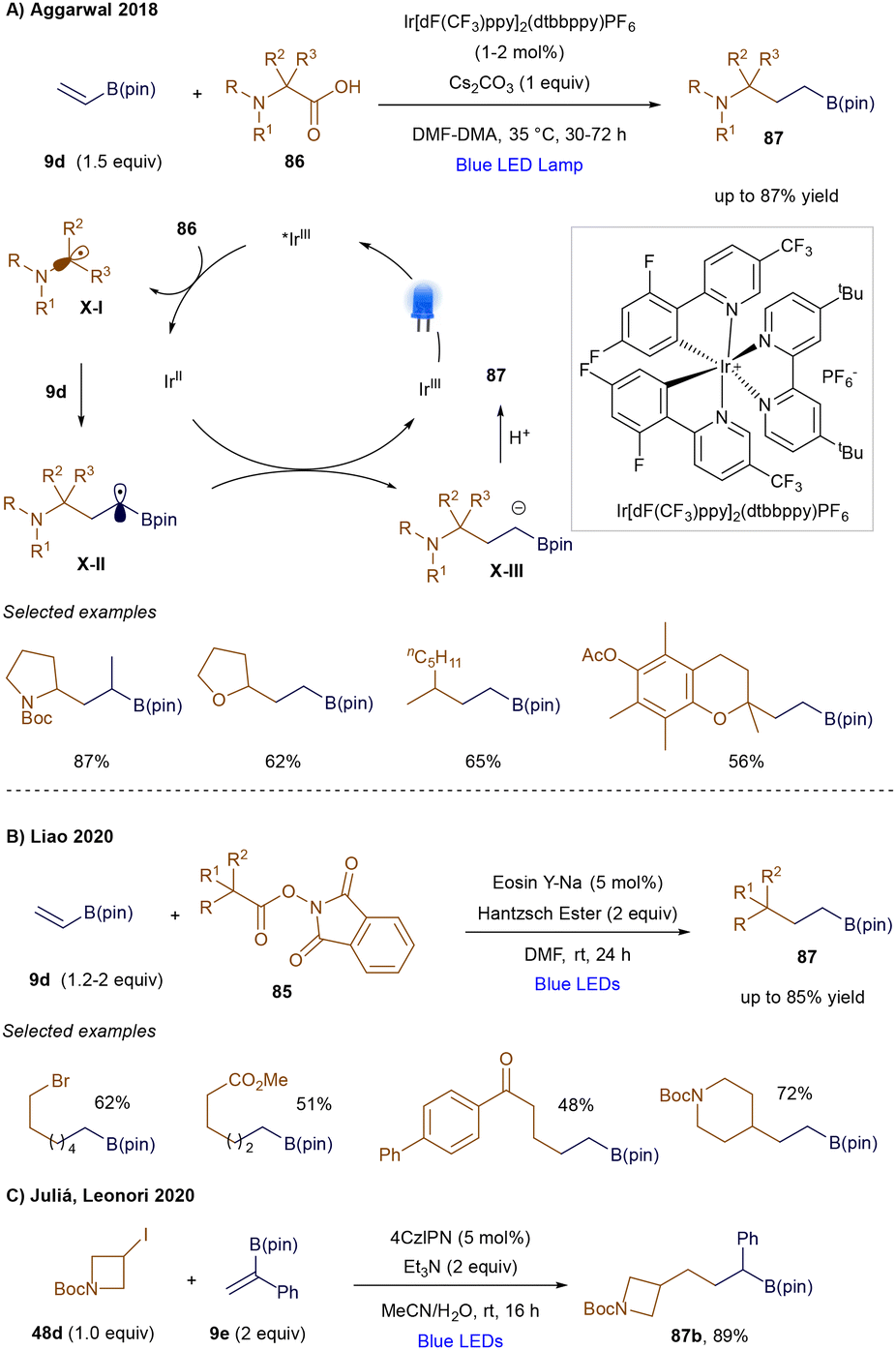
Despite their power, these photochemical methodologies lead to the loss of the boronic ester/acid group due to the formation of a β-borato cation followed by elimination. This group is typically useful for further synthetic modifications. In contrast, a radical addition at the β-position would prevent the formation of a vicinal carbenium ion, as the unpaired electron would now be located on the α-carbon. While this type of reactivity is well-established in thermally-promoted radical chemistry80 and non-catalytic photochemistry,109 the photocatalytic variant was first developed by Aggarwal's group in 2018.110 In this pioneering example, carboxylic acids 86 are converted into the corresponding radicals X-I through photo-induced single-electron oxidation and subsequent decarboxylation (Scheme 31A). The radical then undergoes conjugate addition to the vinyl boronic ester 9d, generating an α-boryl radical X-II, which is stabilized by delocalizing the unpaired electron into the adjacent boron's empty p-orbital. Single-electron reduction of this intermediate yields an α-boryl anion X-III, which, after protonation, provides the desired γ-amino boronic ester product. Subsequently, Liao's group reported that alkyl N-(acyloxy)phthalimides (NHPI, 85) could also engage in this type of reactivity (Scheme 31B), with Hantzsch ester serving as the terminal reductant.111 Moreover, Juliá and Leonori demonstrated a successful example of 1,4-radical addition using an aminoalkyl radical as a halogen atom transfer agent to activate secondary alkyl iodides, forming the key secondary alkyl radical. This radical then added to α-phenyl vinyl boronate ester 87b, yielding the product in 89% yield (Scheme 31C).112
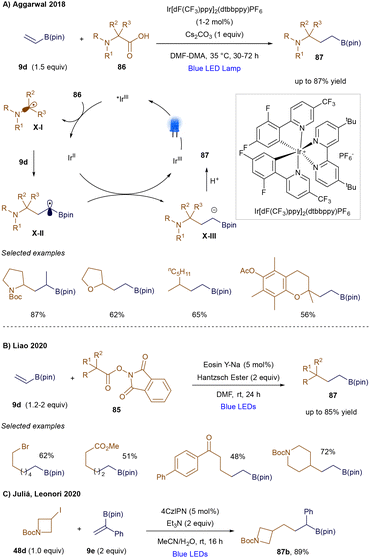 |
| | Scheme 31 1,4-Radical additions of vinyl boronic esters. | |
Cyclopropanation.
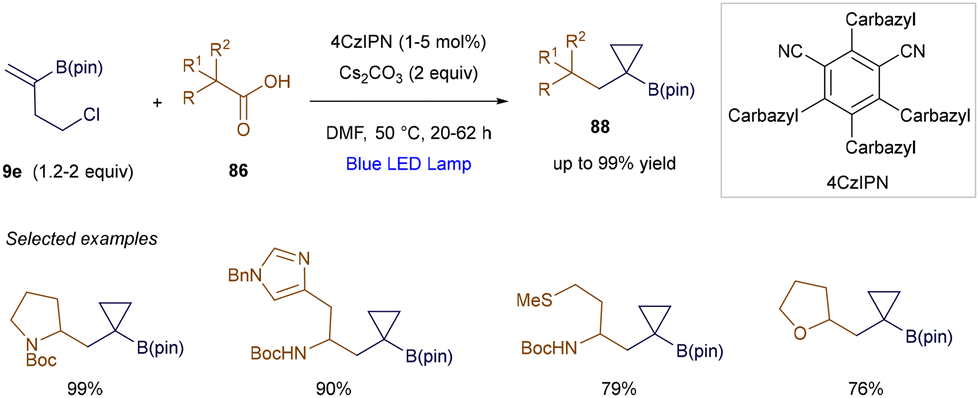
Aggarwal's group further expanded this reactivity by developing a photocatalytic 1,2-difunctionalization of vinyl boronic esters (Scheme 32).113 The key to this method is leveraging the intermediacy of α-boryl anions, which enabled the authors to design a photochemical cyclization sequence. In this sequence, the previously generated carbon-centered anion participates in an intramolecular SN2 step. This radical-polar crossover reactivity allowed for the efficient preparation of synthetically valuable cyclopropyl boronates 88, with yields reaching up to 99%.
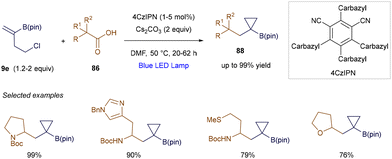 |
| | Scheme 32 Cyclopropanation of vinyl boronic esters. | |
1,2-Carboarylation.
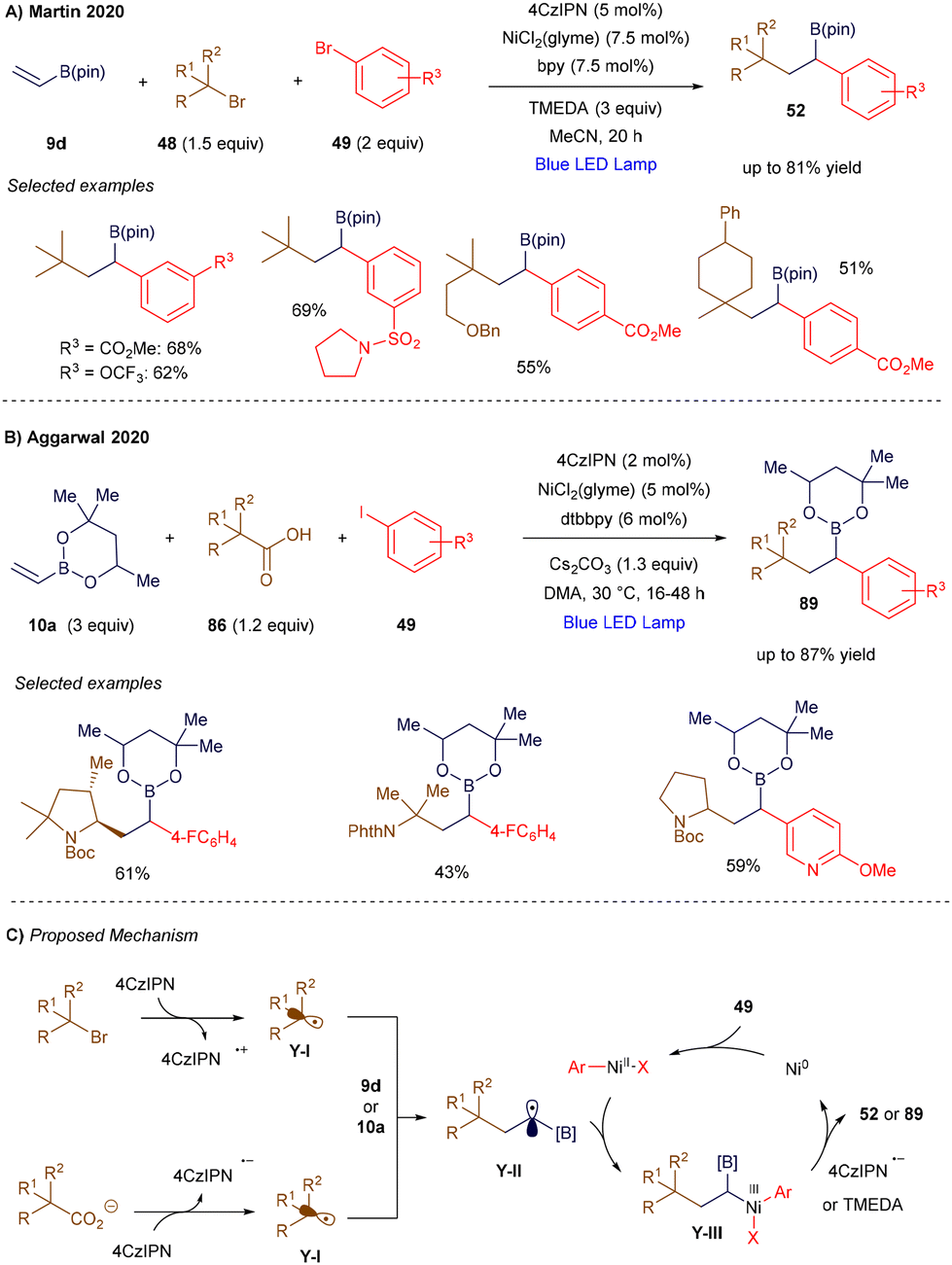
The versatility of vinyl borons also enabled the development of a synergistic system involving a nickel catalyst and an organic photocatalyst to promote their difunctionalization. In 2020, Martin's group (Scheme 33A)114 and Aggarwal's group (Scheme 33B)115 independently reported two nickel-catalyzed metallaphotoredox strategies for the difunctionalization of vinyl organoborons, each using different radical precursors. Martin's group employed alkyl bromides, which are prone to reduction, while Aggarwal's group used carboxylic acids, which are susceptible to oxidation. In both methodologies, a photochemically generated radical (Y-I, Scheme 33C) is trapped by the vinyl boron reagent, resulting in an α-boryl radical Y-II. Concurrently, the nickel catalyst oxidatively inserts into an aryl bromide, forming a Ni(II) intermediate that intercepts the α-boryl radical, generating a nickel(III) species Y-III. This species undergoes reductive elimination to yield the cross-coupled product. After the reaction, the resulting nickel(I) species is regenerated to the active nickel(0) state by the reduced form of the photoredox catalyst.
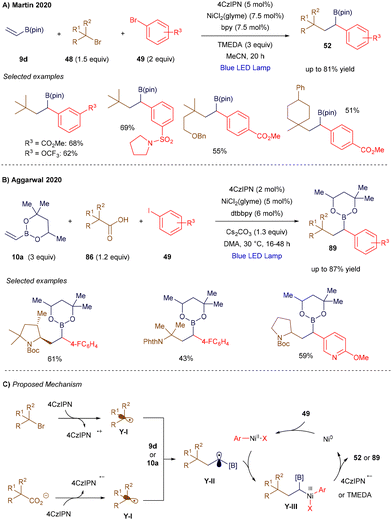 |
| | Scheme 33 Light-triggered 1,2-carboarylation of vinyl boronic derivatives. | |
In Martin's work, the net reductive transformation required a stoichiometric amount of tetramethylethylenediamine (TMEDA) as a reductant. More recently, Noël116 and Chu117 have expanded this approach, applying it to halogen atom transfer processes and developing an asymmetric variant, respectively.
All the previously discussed photochemical radical additions at the β-position of vinyl boronic esters involve nucleophilic radical precursors. This is because the boron functionality makes the π-system slightly electron-deficient. However, nucleophiles can interact with the empty p-orbital of the boron atom, forming boron-ate complexes. These complexes, in turn, render the π-system electron-rich, which explains why these intermediates preferentially react with electrophilic radicals.
4.3 Triplet energy-transfer catalysis
[2 + 2] cycloadditions.
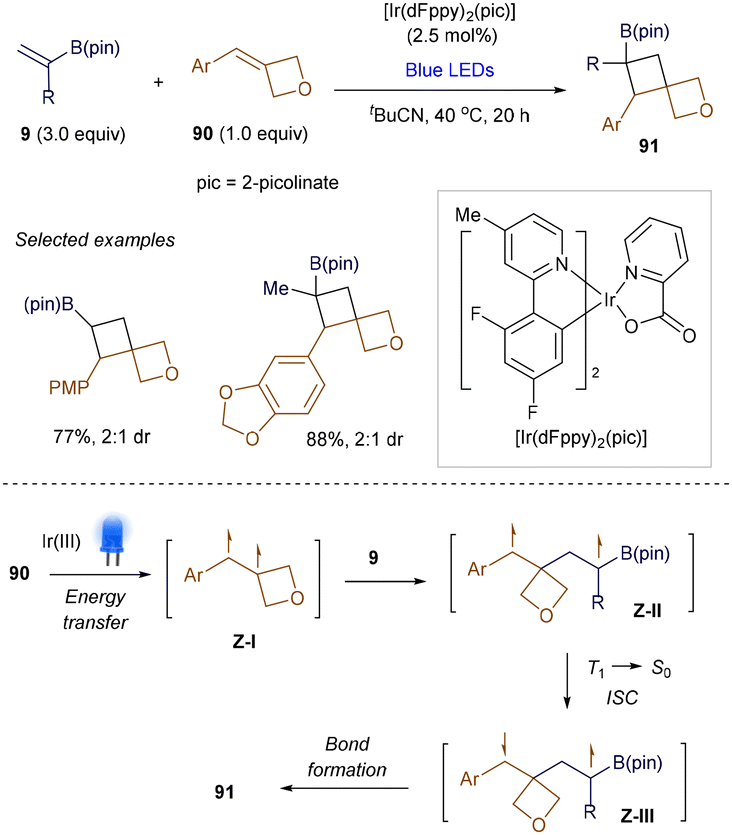
In addition to established examples of synthesizing cyclobutyl boronate via thermal [2 + 2] cycloaddition strategies, recent studies have revealed a new approach utilizing visible light-promoted, radical-mediated strategies. Although Hollis tested UV light-mediated [2 + 2] photocycloadditions of alkenyl boronate esters with 2-cyclopentenone as early as 1993,118 a synthetically useful, visible light-promoted catalytic version was reported by Knowles’ group in 2021.119 Two proposed pathways, discussed below, suggest the photosensitization of either styrenes or β-aryl vinyl boronates to generate key 1,2-triplet biradical intermediates. Knowles demonstrated that the intermolecular, cross-selective [2 + 2] cycloaddition between exocyclic arylidene oxetanes 90 and vinyl boronates 9 proceeds efficiently under Ir(III) photosensitizer catalysis (Scheme 34).119
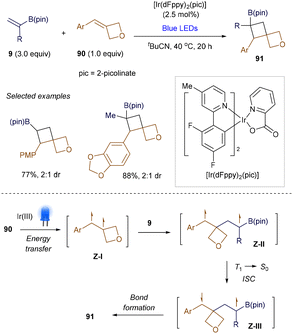 |
| | Scheme 34 Catalytic [2 + 2] photocycloaddition. | |
To ensure the desired [2 + 2] reaction occurs in an intermolecular fashion, the triplet energies of the substrates must differ. Preliminary mechanistic studies revealed that visible light excitation of the Ir(III) catalyst generates an excited-state complex, Ir(III)* (ET = 61.1 kcal mol−1). This species can selectively sensitize oxetane 90 (Ar = Ph, ET = 57.4 kcal mol−1) via energy transfer, producing the 1,2-triplet styrene intermediate Z-I. The subsequent [2 + 2] cycloaddition proceeds through 1,4-triplet and 1,4-singlet intermediates (Z-II and Z-III), ultimately yielding the desired products 91. Shortly after, intramolecular versions of this reaction were also successfully demonstrated by the groups of Yoon120 and Collins,121 offering a novel approach to constructing fused cyclobutanes.
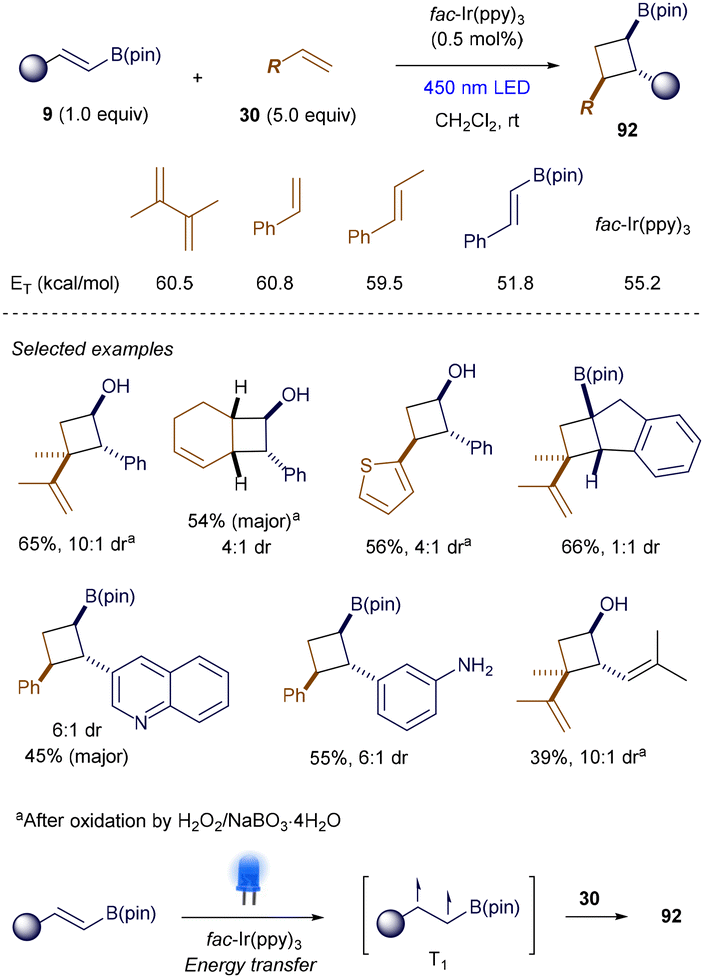
In 2022, Swierk and Brown reported that intermolecular [2 + 2] photocycloaddition reactions could also occur through energy transfer involving alkenyl boronates.122 The key to their success was the use of alkenyl boronates 9 with aryl or vinyl groups at the β-position, which significantly lowered the triplet energies compared to simple styrenes or 1,3-dienes (Scheme 35). Under the catalysis of 0.5 mol% fac-Ir(ppy)3, the cross [2 + 2] cycloaddition proceeded smoothly, producing polysubstituted cyclobutylboronates 92 with moderate to good yields and diastereoselectivity. The protocol exhibited good functional group tolerance and was scalable to 5 mmol. Mechanistic studies suggested that triplet energy transfer occurs preferentially from the photoexcited triplet state of fac-Ir(ppy)3 to alkenyl boronates 9, generating the crucial T1 state that initiates the cycloaddition.
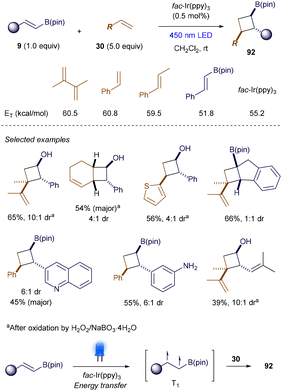 |
| | Scheme 35 Catalytic [2 + 2] photocycloadditions. | |
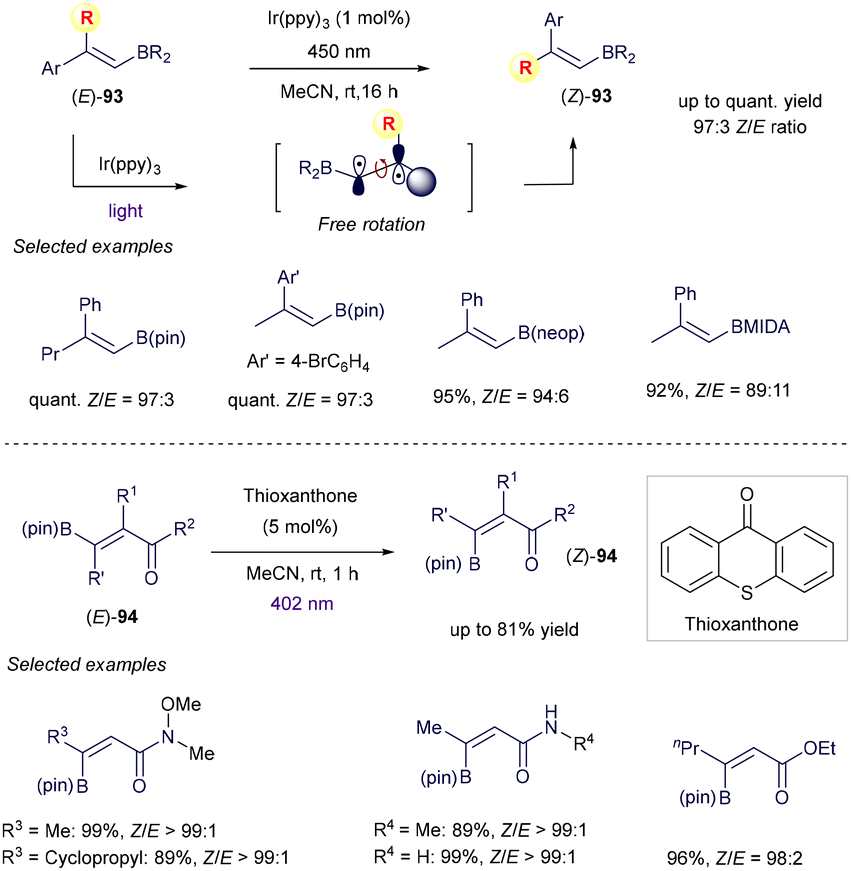
Finally, the use of vinyl boron reagents in the field of synthetic photochemistry is not limited to the above addition of radicals on the π-system, but also to photoinduced energy transfer manifold, as demonstrated by excellent examples from Gilmour and co-workers (Scheme 36).123–125 In this case, the authors proved how photosensitized alkene isomerization, usually restricted to styrenyl chromophores because of the different absorption properties of the E and Z isomers, could be expanded to contra-thermodynamic vinyl boron reagents as well. Specifically, the authors discovered how the presence of the boron-based group within the E isomers extends the π-system of the whole molecule and therefore permits the energy transfer from the excited state of the simple photocatalyst. After this event and subsequent isomerization, the C–B bond in the Z isomer is twisted by 90° and in a dative interaction with an aryl or carbonyl group. Because of this, the Z isomer is blue shifted with respect to the E analogue and full E to Z isomerization is possible. As shown in Scheme 36, the isomerization of cis-α-substituted styrenyl boron reagents 93 or 94 takes place smoothly with high fidelity under visible light irradiation,124 providing a new way for the stereoselective synthesis of sterically more hindered alkenes.
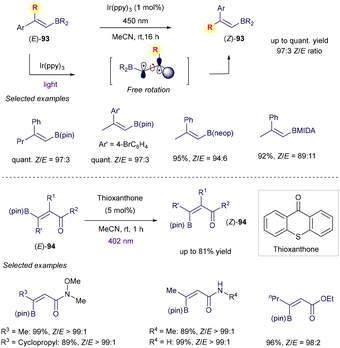 |
| | Scheme 36 Photochemical isomerization. | |
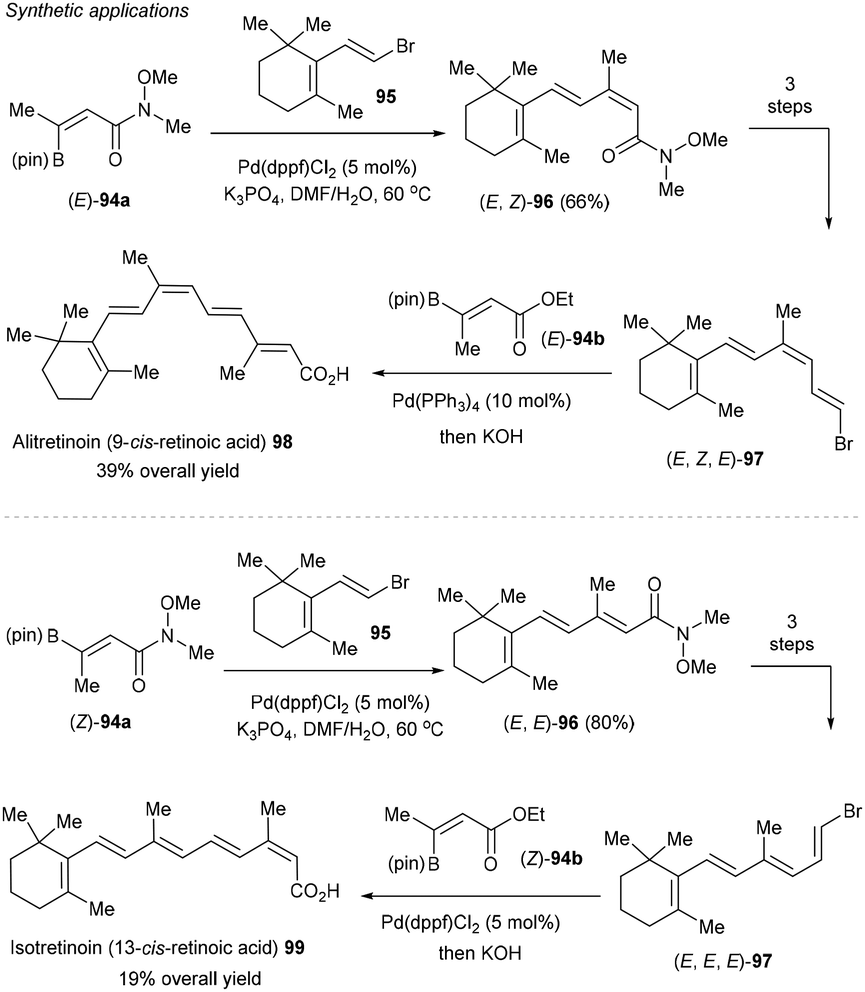
Furthermore, the method can be applied for the facile synthesis of polyene pharmaceuticals alitretinoin (9-cis-retinoic acid) 98 and isotretinoin (13-cis-retinoic acid) 99 with full control of the geometry of the alkenes, highlighting the usefulness of the methodology (Scheme 37).124 For example, starting from easily accessible (Z)- and (E)-94a, stereospecific Suzuki–Miyaura cross-coupling using vinyl bromide 95 gave rise to geometrical defined (E,E)- and (E,Z)-96 in high yields. Subsequent reduction, boron-Wittig reaction, followed by bromodeboronation furnished stereodefined intermediate (E,E,E)- and (E,Z,E)-97. A second Suzuki–Miyaura cross-coupling with (E)-94b worked smoothly to deliver 13-cis-retinoic acid 98 after hydrolysis. The analogous sequence with (Z)-94a enabled the synthesis of 9-cis-retinoic acid 99 (alitretinoin).
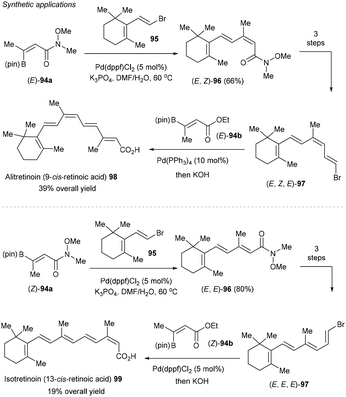 |
| | Scheme 37 Synthetic applications. | |
5. Conclusions
This review highlights emerging trends in catalytic synthetic methods using alkenyl boron reagents. The versatility provided by the boron atom within the olefinic system allows these reagents to function as either electrophiles or nucleophiles, depending on reaction conditions. This flexibility has been harnessed to develop metal-catalyzed processes, as detailed in section 2, where alkenyl boron compounds serve as either nucleophilic transmetallation reagents or electrophiles in various innovative transformations. Beyond metal catalysis, this versatility extends to organocatalytic protocols (section 3). Moreover, these reagents have proven useful in photocatalytic processes (section 4), demonstrating their broad reactivity, including polar and radical chemistries. These successful strategies offer new protocols for the efficient and stereoselective synthesis of valuable alkenes and alkyl boronates. The practical applications of these methods are exemplified by their use in synthesizing natural products and bioactive molecules.
Despite the progress made, many challenges and opportunities remain, offering ample room for further investigation. (1) The substrate scope is still limited. While many examples focus on aryl boron reagents, less attention is given to alkenyl boron reagents, even though the transition from aryl- to alkenyl-substituted boron reagents is non-trivial. (2) Compared to the polar reactivity of alkenyl boron reagents, their intriguing open-shell reactivity has yet to be fully explored. (3) Although several elegant examples have been reported, their asymmetric variants lag behind due to the challenge of controlling enantioselectivity, alongside chemo-, regio-, and diastereoselectivity. (4) Lastly, there is still a high demand within the synthetic community for practical methods that employ these protocols for the synthesis of highly valued molecules. We believe that the development of novel catalytic systems is crucial to addressing these challenges. For example, beyond the established metal, organo-, and photocatalytic systems, electrosynthesis126 could further drive this chemistry forward. Additionally, DFT studies could offer deeper insights and guidance in this area. We anticipate that these findings will significantly impact this field in the near future.
Data availability
All relevant data are within the paper.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from NSFC (22201062, 22201064, 22471060, 22401077) are gratefully acknowledged. This project is also funded by China Postdoctoral Science Foundation (2024T170222, 2023M741000) and Postdoctoral Research Grant of Henan Province (HN2024001), Natural Science Foundation of Henan Province (242300420532). It also supported by the 2023 Zhongyuan Talent Program (Talent Cultivation Series) – Zhongyuan Young Postdoctoral Innovative Talents Program and 2024 Henan Provincial Science and Technology Research and Development Plan Joint Fund (Advantageous Discipline Cultivation Category). We sincerely thank Prof. Giacomo E. M. Crisenza (University of Manchester) for very helpful discussions.
References
-
(a)
Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, ed. D. G. Hall, Wiley–VCH, Weinheim, Germany, 2nd edn, 2011 Search PubMed;
(b) C. D. Entwistle and T. B. Marder, Boron Chemistry Lights the Way: Optical Properties of Molecular and Polymeric Systems, Angew. Chem., Int. Ed., 2012, 41, 2927–2931 CrossRef;
(c) W. L. A. Brooks and B. S. Sumerlin, Synthesis and Applications of Boronic Acid-Containing Polymers: From Materials to Medicine, Chem. Rev., 2016, 116, 1375–1397 CrossRef CAS PubMed;
(d) B. Akgun and D. G. Hall, Boronic Acids as Bioorthogonal Probes for Site-Selective Labeling of Proteins, Angew. Chem., Int. Ed., 2018, 57, 13028–13044 CrossRef CAS;
(e) J. P. M. António, R. Russo, C. P. Carvalho, P. M. S. D. Cal and P. M. P. Gois, Boronic Acids as Building Blocks for the Construction of Therapeutically Useful Bioconjugates, Chem. Soc. Rev., 2019, 48, 3513–3536 RSC;
(f) S. K. Mellerup and S. Wang, Boron-Based Stimuli Responsive Materials, Chem. Soc. Rev., 2019, 48, 3537–3549 RSC;
(g) R. J. Grams, W. L. Santos, I. R. Scorei, A. Abad-García, C. A. Rosenblum, A. Bita, H. Cerecetto, C. Viñas and M. A. Soriano-Ursúa, The Rise of Boron-Containing Compounds: Advancements in Synthesis, Medicinal Chemistry, and Emerging Pharmacology, Chem. Rev., 2024, 124, 2441–2511 CrossRef PubMed.
-
(a) N. Miyaura and A. Suzuki, Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds, Chem. Rev., 1995, 95, 2457–2483 CrossRef;
(b) T. Hayashi and K. Yamasaki, Rhodium-Catalyzed Asymmetric 1, 4-Addition and Its Related Asymmetric Reactions, Chem. Rev., 2003, 103, 2829–2844 CrossRef PubMed;
(c) D. S. Matteson, Boronic Esters in Asymmetric Synthesis, J. Org. Chem., 2013, 78, 10009–10023 CrossRef PubMed;
(d) A. J. J. Lennox and G. C. Lloyd-Jones, Selection of Boron Reagents for Suzuki–Miyaura Coupling, Chem. Soc. Rev., 2014, 43, 412–443 RSC;
(e) S. Roscales and A. G. Csákÿ, Transition-Metal-Free C–C Bond Forming Reactions of Aryl, Alkenyl and Alkynylboronic Acids and Their Derivatives, Chem. Soc. Rev., 2014, 43, 8215–8225 RSC;
(f) T. N. Nguyen and J. A. May, Enantioselective Organocatalytic Conjugate Addition of Organoboron Nucleophiles, Tetrahedron Lett., 2017, 58, 1535–1544 CrossRef CAS;
(g) D. Chen and M.-H. Xu, Transition Metal-Catalyzed Asymmetric Addition of Organoboron Reagents to Imines, Chin. J. Org. Chem., 2017, 37, 1589–1612 CrossRef CAS;
(h) B. S. L. Collins, C. M. Wilson, E. L. Myers and V. K. Aggarwal, Asymmetric Synthesis of Secondary and Tertiary Boronic Esters, Angew. Chem., Int. Ed., 2017, 56, 11700–11733 CrossRef CAS;
(i) J. W. B. Fyfe and A. J. B. Watson, Recent Developments in Organoboron Chemistry: Old Dogs, New Tricks, Chem, 2017, 3, 31–55 CrossRef CAS;
(j) M. Quan, L. Wu, G. Yang and W. Zhang, Pd(II), Ni(II) and Co(II)-Catalyzed Enantioselective Additions of Organoboron Reagents to Ketimines, Chem. Commun., 2018, 54, 10394–10404 RSC;
(k) J. Carreras, A. Caballero and P. J. Pérez, Alkenyl Boronates: Synthesis and Applications, Chem. – Asian J., 2019, 14, 329–344 CrossRef CAS;
(l) S. Namirembe and J. P. Morken, Reactions of Organoboron Compounds Enabled by Catalyst-Promoted Metalate Shifts, Chem. Soc. Rev., 2019, 48, 3464–3474 RSC;
(m) X. Yang, S. J. Kalita, S. Maheshuni and Y.-Y. Huang, Recent Advances on Transition-Metal-Catalyzed Asymmetric Tandem Reactions with Organoboron Reagents, Coord. Chem. Rev., 2019, 392, 35–48 CrossRef CAS;
(n) Z. Kuang, K. Yang, Y. Zhou and Q. Song, Base-Promoted Domino-Borylation-Protodeboronation Strategy, Chem. Commun., 2020, 56, 6469–6479 RSC;
(o) G. J. Lovinger and J. P. Morken, Recent Advances in Radical Addition to Alkenylboron Compounds, Eur. J. Org. Chem., 2020, 2362–2368 CrossRef CAS;
(p) H. Wang, C. Jing, A. Noble and V. K. Aggarwal, Stereospecific 1, 2-Migrations of Boronate Complexes Induced by Electrophiles, Angew. Chem., Int. Ed., 2020, 59, 16859–16872 CrossRef CAS;
(q) D.-X. Zhu and M.-H. Xu, Transition Metal-Catalyzed Asymmetric Addition of Organoboron Reagents to Aldehydes and Ketones, Chin. J. Org. Chem., 2020, 40, 255–275 CrossRef;
(r) O. O. Grygorenko, V. S. Moskvina, O. V. Hryshchuk and A. V. Tymtsunik, Cycloadditions of Alkenylboronic Derivatives, Synthesis, 2020, 2761–2780 CrossRef;
(s) X. Wang, Y. Wang, W. Huang, C. Xia and L. Wu, Direct Synthesis of Multi(boronate) Esters from Alkenes and Alkynes via Hydroboration and Boration Reactions, ACS Catal., 2021, 11, 1–18 CrossRef;
(t) C. Haldar, M. E. Hoque, J. Chaturvedi, M. M. M. Hassan and B. Chattopadhyay, Chem. Commun., 2021, 57, 13059–13074 RSC;
(u) X. Ma, Z. Kuang and Q. Song, Recent Advances in the Construction of Fluorinated Organoboron Compounds, JACS Au, 2022, 2, 261–279 CrossRef PubMed;
(v) R. Bisht, C. Haldar, M. M. M. Hassan, M. E. Hoque, J. Chaturvedi and B. Chattopadhyay, Chem. Soc. Rev., 2022, 51, 5042–5100 RSC;
(w) X. Li, G. Zhang and Q. Song, Recent Advances in the Construction of Tetracoordinate Boron Compounds, Chem. Commun., 2023, 59, 3812–3820 RSC;
(x) C. Yin, S. Tang, J. Mei, X. Hu and H. Zhang, Electrochemical Synthesis and Transformation of Organoboron Compounds, Org. Chem. Front., 2023, 10, 3361–3377 RSC;
(y) M. M. M. Hassan, S. Guria, S. Dey, J. Das and B. Chattopadhyay, Transition Metal-Catalyzed Remote C−H Borylation: An Emerging Synthetic Tool, Sci. Adv., 2023, 9, eadg3311 CrossRef;
(z) X. Li, J. Chen and Q. Song, Recent Progress in the Catalytic Enantioselective Reactions of 1,1-Diborylalkanes, Chem. Commun., 2024, 60, 2462–2471 RSC;
(a
a) W. Xu and T. Xu, Dual Nickel- and Photoredox-Catalyzed Asymmetric Reductive Cross-Couplings: Just a Change of the Reduction System?, Acc. Chem. Res., 2024, 57, 1997–2011 CrossRef;
(a
b) S. Guria, M. M. M. Hassan and B. Chattopadhyay, C–H Borylation: a Tool for Molecular Diversification, Org. Chem. Front., 2024, 11, 929–953 RSC.
-
(a) D. G. Hall, Boronic Acid Catalysis, Chem. Soc. Rev., 2019, 48, 3475–3496 RSC;
(b) J. Lam, K. M. Szkop, E. Mosaferi and D. W. Stephan, FLP Catalysis: Main Group Hydrogenations of Organic Unsaturated Substrates, Chem. Soc. Rev., 2019, 48, 3592–3612 RSC;
(c) Y. Su and R. Kinjo, Small Molecule Activation by Boron-Containing Heterocycles, Chem. Soc. Rev., 2019, 48, 3613–3659 RSC.
-
https://www.nobelprize.org/prizes/lists/all-nobel-prizes-in-chemistry/
.
-
(a) S. Manna, K. K. Das, D. Aich and S. Panda, Synthesis and Reactivity of Allenylboron Compounds, Adv. Synth. Catal., 2021, 363, 2444–2463 CrossRef. For a recent example, please see:
(b) Q. Yan, C.-K. Ruan, Y.-Q. Deng, Y.-C. Pu, W.-D. Chu, C.-Y. He and Q.-Z. Liu, Copper-Catalyzed Asymmetric Propargylation of Imines Enabled by a Biphenol-Based Phosphoramidite Ligand, Org. Chem. Front., 2023, 10, 4935–4940 RSC.
- S. Nandy, S. Paul, K. K. Das, P. Kumar, D. Ghorai and S. Panda, Synthesis and Reactivity of Alkynyl Boron Compounds, Org. Biomol. Chem., 2021, 19, 7276–7297 RSC.
- For selected recent examples about the synthesis of alkenyl boron compounds, please see:
(a) F. Meng, K. P. McGrath and A. H. Hoveyda, Multifunctional Organoboron Compounds for Scalable Natural Product Synthesis, Nature, 2014, 513, 367–374 CrossRef;
(b) Q. Feng, K. Yang and Q. Song, Highly Selective Copper-Catalyzed Trifunctionalization of Alkynyl Carboxylic Acids: an Efficient Route to Bis-deuterated β-borylated α,β-Styrene, Chem. Commun., 2015, 51, 15394–15397 RSC;
(c) W. B. Reid, J. J. Spillane, S. B. Krause and D. A. Watson, Direct Synthesis of Alkenyl Boronic Esters from Unfunctionalized Alkenes: A Boryl-Heck Reaction, J. Am. Chem. Soc., 2016, 138, 5539–5542 CrossRef PubMed;
(d) K. Yeung, R. E. Ruscoe, J. Rae, A. P. Pulis and D. J. Procter, Enantioselective Generation of Adjacent Stereocenters in a Copper-Catalyzed Three-Component Coupling of Imines, Allenes, and Diboranes, Angew. Chem., Int. Ed., 2016, 55, 11912–11916 CrossRef PubMed;
(e) Q. Xuan, Y. Wei and Q. Song, Spiro(phosphoamidite) Ligand (SIPHOS)/Cu(OTf)2-C atalyzed Highly Regio- and Stereo-Selective Hydroborations of Internal Alkynes with Diborane in Water, Chin. Chem. Lett., 2017, 28, 1163–1166 CrossRef;
(f) J. Liu, M. Nie, Q. Zhou, S. Gao, W. Jiang, L. M. Chung, W. Tang and K. Ding, Enantioselective Palladium-Catalyzed Diboration of 1,1-Disubstituted Allenes, Chem. Sci., 2017, 8, 5161–5165 RSC;
(g) A. P. Pulis, K. Yeung and D. J. Procter, Enantioselective Copper Catalysed, Direct Functionalisation of Allenes via Allyl Copper Intermediates, Chem. Sci., 2017, 8, 5240–5247 RSC;
(h) J. J. Molloy, J. B. Metternich, C. G. Daniliuc, A. J. B. Watson and R. Gilmour, Contra-Thermodynamic, Photocatalytic E/Z Isomerization of Styrenyl Boron Species: Vectors to Facilitate Exploration of Two Dimensional Chemical Space, Angew. Chem., Int. Ed., 2018, 57, 3168–3172 CrossRef CAS;
(i) Z. Kuang, G. Gao and Q. Song, Base-Catalyzed Diborylation of Alkynes: Synthesis and Applications of cis-1,2-Bis(boryl)alkenes, Sci. China: Chem., 2019, 62, 62–66 CrossRef CAS;
(j) M. D. Aparece, C. Gao, G. J. Lovinger and J. P. Morken, Vinylidenation of Organoboronic Esters Enabled by a Pd-Catalyzed Metallate Shift, Angew. Chem., Int. Ed., 2019, 58, 592–595 CrossRef CAS PubMed;
(k) K. Yeung, F. J. T. Talbot, G. P. Howell, A. P. Pulis and D. J. Procter, Copper-Catalyzed Borylative Multicomponent Synthesis of Quaternary α-Amino Esters, ACS Catal., 2019, 9, 1655–1661 CrossRef CAS;
(l) S. Namirembe, C. Gao, R. P. Wexler and J. P. Morken, Stereoselective Synthesis of Trisubstituted Alkenylboron Reagents by Boron-Wittig Reaction of Ketones, Org. Lett., 2019, 21, 4392–4394 CrossRef CAS PubMed;
(m) S. Li, J. Li, T. Xia and W. Zhao, Stereoselective Synthesis of Vinylboronates by Rh-Catalyzed Borylation of Stereoisomeric Mixtures, Chin. J. Chem., 2019, 37, 462–468 CrossRef CAS;
(n) L. E. Longobardi and A. Fürstner,
trans-Hydroboration of Propargyl Alcohol Derivatives and Related Substrates, Chem. – Eur. J., 2019, 25, 10063–10068 CrossRef CAS PubMed;
(o) Z. Kuang, H. Chen, J. Qiu, Z. Ou, Y. Lan and Q. Song, Cu-Catalyzed Regio- and Stereodivergent Chemoselective sp2/sp3 1,3- and 1,4-Diborylations of CF3-Containing 1,3-Enynes, Chem, 2020, 6, 2347–2363 CrossRef CAS;
(p) S.-H. Yu, T.-J. Gong and Y. Fu, Three-Component Borylallenylation of Alkynes: Access to Densely Boryl-Substituted Ene-allenes, Org. Lett., 2020, 22, 2941–2945 CrossRef CAS PubMed;
(q) G.-M. Ho, L. Segura and I. Marek, Ru-Catalyzed Isomerization of ω-Alkenylboronates towards Stereoselective Synthesis of Vinylboronates with Subsequent in situ Functionalization, Chem. Sci., 2020, 11, 5944–5949 RSC;
(r) L. Tao, X. Guo, J. Li, R. Li, Z. Lin and W. J. Zhao, Rhodium-Catalyzed Deoxygenation and Borylation of Ketones: A Combined Experimental and Theoretical Investigation, J. Am. Chem. Soc., 2020, 142, 18118–18127 CrossRef PubMed;
(s) S. Gao, M. Duan, Q. Shao, K. N. Houk and M. J. Chen, Development of α,α-Disubstituted Crotylboronate Reagents and Stereoselective Crotylation via Brønsted or Lewis Acid Catalysis, J. Am. Chem. Soc., 2020, 142, 18355–18368 CrossRef;
(t) C. Wu and S. Ge, Ligand-controlled cobalt-catalyzed regiodivergent hydroboration of aryl,alkyl-disubstituted internal allenes, Chem. Sci., 2020, 11, 2783–2789 RSC;
(u) Y. Ping, R. Wang, Q. Wang, T. Chang, J. Huo, M. Lei and J. Wang, Synthesis of Alkenylboronates from N-Tosylhydrazones through Palladium-Catalyzed Carbene Migratory Insertion, J. Am. Chem. Soc., 2021, 143, 9769–9780 CrossRef;
(v) S.-H. Kim-Lee, P. Mauleón, R. G. Arrayás and J. C. Carretero, Dynamic Multiligand Catalysis: A Polar to Radical Crossover Strategy Expands Alkyne Carboboration to Unactivated Secondary Alkyl Halides, Chem, 2021, 7, 2212–2216 CrossRef;
(w) A. M. Y. Suliman, E. M. A. Ahmed, T.-J. Gong and Y. Fu, Cu/Pd-Catalyzed cis-Borylfluoroallylation of Alkynes for the Synthesis of Boryl-Substituted Monofluoroalkenes, Org. Lett., 2021, 23, 3259–3263 CrossRef;
(x) Z. Wang, J. Wu, W. Lamine, B. Li, J.-M. Sotiropoulos, A. Chrostowska, K. Miqueu and S.-Y. Liu, C−Boron Enolates Enable Palladium Catalyzed Carboboration of Internal 1,3-Enynes, Angew. Chem., Int. Ed., 2021, 60, 21231–21236 CrossRef;
(y) C. You, C. G. Daniliuc, K. Bergander, S. Yamaguchi and A. Studer, Regio- and Stereoselective 1,2-Carboboration of Ynamides with Aryldichloroboranes, Angew. Chem., Int. Ed., 2021, 60, 21697–21701 CrossRef;
(z) W. Li, S. Chen, J. Xie, Z. Fan, K. Yang and Q. Song, Synthesis of Axially Chiral Alkenylboronates through Combined Copper- and Palladium-Catalysed Atroposelective Arylboration of Alkynes, Nat. Synth., 2023, 2, 140–151 CrossRef;
(a
a) K. Liu, S. Jin and Q. Song, Base-Promoted Synthesis of Tetrasubstituted Alkenylboronates from Propargyl Amines and B2pin2 via dual 1,4-Metallate Shift and B–N Elimination, Org. Chem. Front., 2023, 10, 905–910 RSC;
(a
b) M. Chen, C. D. Knox, M. C. Madhusudhanan, T. H. Tugwell, C. Liu, P. Liu and G. Dong, Stereospecific Alkenylidene Homologation of Organoboronates by SNV Reaction, Nature, 2024, 631, 328–334 CrossRef PubMed;
(a
c) M. Chen, T. H. Tugwell, P. Liu and G. Dong, Synthesis of Alkenyl Boronates through Stereoselective Vinylene Homologation of Organoboronates, Nat. Synth., 2024, 3, 337–346 CrossRef;
(a
d) H.-W. Jiang, W.-L. Yu, D. Wang and P.-F. Xu, Photocatalyzed H2-Acceptorless Dehydrogenative Borylation by Using Amine Borane, ACS Catal., 2024, 14, 8666–8675 CrossRef;
(a
e) J. Wen, Y. Huang, Y. Zhang, H. Grϋtzmacher and P. Hu, Cobalt Catalyzed Practical Hydroboration of Terminal Alkynes with Time-Dependent Stereoselectivity, Nat. Commun., 2024, 15, 2208 CrossRef;
(a
f) Y. Jung, S. Y. Yoo, Y. Jin, J. You, S. Han, J. Yu, Y. Park and S. H. Cho, Iridium-Catalyzed Chemo-, Diastereo-, and Enantioselective Allyl-Allyl Coupling: Accessing All Four Stereoisomers of (E,)-1-Boryl-Substituted 1,5-Dienes by Chirality Pairing, Angew. Chem., Int. Ed., 2023, 62, e202218794 CrossRef PubMed;
(a
g) G.-L. Xu, C.-Y. Liu, X. Pang, X.-Y. Liu and X.-Z. Shu, Nickel-Catalyzed Cross-Electrophile Vinyl–Vinyl Coupling: An Approach to Structurally Versatile Dienylboronates, CCS Chem., 2022, 4, 864–871 CrossRef;
(a
h) Y. Ozawa, Y. Shiratori, H. Koriyama, K. Endo, H. Iwamoto and H. Ito, Synthesis of (Z)-Alkenyl Boronates via a Copper(I)-Catalyzed Linear-Selective Alkylboration of Terminal Allenes, Org. Chem. Front., 2023, 10, 4786–4793 RSC.
- D. V. Partyka, Transmetalation of Unsaturated Carbon Nucleophiles from Boron-Containing Species to the Mid to Late d-Block Metals of Relevance to Catalytic C−X Coupling Reactions (X = C, F, N, O, Pb, S, Se, Te), Chem. Rev., 2011, 111, 1529–1595 CrossRef.
- K. Sasaki and T. Hayashi, Rhodium-Catalyzed Asymmetric Conjugate Addition of Arylboroxines to Borylalkenes: Asymmetric Synthesis of β-Arylalkylboranes, Angew. Chem., Int. Ed., 2010, 49, 8145–8147 CrossRef.
- L. Zhang, G. J. Lovinger, E. K. Edelstein, A. A. Szymaniak, M. P. Chierchia and J. P. Morken, Catalytic Conjunctive Cross-Coupling Enabled by Metal-Induced Metallate Rearrangement, Science, 2016, 351, 70–74 CrossRef.
- For selected examples in a non-catalytic way, please see:
(a) C.-V. T. Vo, T. A. Mitchell and J. W. Bode, Expanded Substrate Scope and Improved Reactivity of Ether-Forming Cross-Coupling Reactions of Organotrifluoroborates and Acetals, J. Am. Chem. Soc., 2011, 133, 14082–14089 CrossRef PubMed;
(b) J. Kim and M. Movassaghi, Concise Total Synthesis and Stereochemical Revision of (+)-Naseseazines A and B: Regioselective Arylative Dimerization of Diketopiperazine Alkaloids, J. Am. Chem. Soc., 2011, 133, 14940–14943 CrossRef PubMed;
(c) M. Wan, Z. Meng, H. Lou and L. Liu, Practical and Highly Selective C-H Functionalization of Structurally Diverse Ethers, Angew. Chem., Int. Ed., 2014, 53, 13845–13849 CrossRef;
(d) Z. Xie, L. Liu, W. Chen, H. Zheng, Q. Xu, H. Yuan and H. Lou, Practical Metal-Free C(sp3)−H Functionalization: Construction of Structurally Diverse α-Substituted N-Benzyl and N-Allyl Carbamates, Angew. Chem., Int. Ed., 2014, 53, 3904–3908 CrossRef PubMed;
(e) W. Chen, Z. Xie, H. Zheng, H. Lou and L. Liu, Structurally Diverse α-Substituted Benzopyran Synthesis through a Practical Metal-Free C(sp3)–H Functionalization, Org. Lett., 2014, 16, 5988–5991 CrossRef PubMed;
(f) P. B. Brady and E. M. Carreira, Addition of Trifluoroborates to Oxetanyl N,O-Acetals: Entry into Spiro and Fused Saturated Heterocycles, Org. Lett., 2015, 17, 3350–3353 CrossRef PubMed;
(g) C. Li, Y. Zhang, Q. Sun, T. Gu, H. Peng and W. Tang, Transition-Metal-Free Stereospecific Cross-Coupling with Alkenylboronic Acids as Nucleophiles, J. Am. Chem. Soc., 2016, 138, 10774–10777 CrossRef;
(h) V. Ortega, E. del Castillo and A. G. Csákÿ, Transition-Metal-Free Stereocomplementary Cross-Coupling of Diols with Boronic Acids as Nucleophiles, Org. Lett., 2017, 19, 6236–6239 CrossRef;
(i) Z. Liu, L. Chen, J. Li, K. Liu, J. Zhao, M. Xu, L. Feng, R.-Z. Wan, W. Li and L. Liu, Oxidative C–H Functionalization of N-Carbamoyl 1,2-Dihydroquinolines, Org. Biomol. Chem., 2017, 15, 7600–7606 RSC;
(j) R. J. Armstrong, M. Nandakumar, R. M. P. Dias, A. Noble and E. L. Myers, Enantiodivergent Synthesis of Allenes by Point-to-Axial Chirality Transfer, Angew. Chem., Int. Ed., 2018, 57, 8203–8208 CrossRef CAS PubMed;
(k) S. Liu, X. Zeng, G. B. Hammond and B. Xu, Mild Base Promoted Nucleophilic Substitution of Unactivated sp3-Carbon Electrophiles with Alkenylboronic Acids, Adv. Synth. Catal., 2018, 360, 3667–3671 CrossRef CAS;
(l) S. Roscales, V. Ortega and A. G. Csákÿ, Selective Functionalization of Achmatowicz Rearrangement Products by Reactions with Potassium Organotrifluoroborates under Transition-Metal-Free Conditions, J. Org. Chem., 2018, 83, 11425–11436 CrossRef CAS PubMed;
(m) T. Fan, A. Wang, J.-Q. Li, J.-L. Ye, X. Zheng and P.-Q. Huang, Versatile One-Pot Synthesis of Polysubstituted Cyclopent-2-enimines from α, β-Unsaturated Amides: Imino-Nazarov Reaction, Angew. Chem., Int. Ed., 2018, 57, 10352–10356 CrossRef CAS;
(n) T. N. Nguyen, K. Setthakarn and J. A. May, Oxyallyl Cation Capture via Electrophilic Deborylation of Organoboronates:
Access to α, α′-Substituted Cyclic Ketones, Org. Lett., 2019, 21, 7837–7840 CrossRef CAS PubMed;
(o) M. Nandakumar, B. Rubial, A. Noble, E. L. Myers and V. K. Aggarwal, Ring-Opening Lithiation–Borylation of 2-Trifluoromethyl Oxirane: A Route to Versatile Tertiary Trifluoromethyl Boronic Esters, Angew. Chem., Int. Ed., 2020, 59, 1187–1191 CrossRef;
(p) N. Eghbarieh, N. Hanania, A. Zamir, M. Nassir, T. Stein and A. Masarwa, Stereoselective Diels–Alder Reactions of gem-Diborylalkenes: Toward the Synthesis of gem-Diboron-Based Polymers, J. Am. Chem. Soc., 2021, 143, 6211–6220 CrossRef;
(q) A. Music, C. M. Nuber, Y. Lemke, P. Spieß and D. Didier, Electro-alkynylation: Intramolecular Rearrangement of Trialkynylorganoborates for Chemoselective C(sp2)–C(sp) Bond Formation, Org. Lett., 2021, 23, 4179–4184 CrossRef.
- H. Noguchi, K. Hojo and M. Suginome, Boron-Masking Strategy for the Selective Synthesis of Oligoarenes via Iterative Suzuki-Miyaura Coupling, J. Am. Chem. Soc., 2007, 129, 758–759 CrossRef.
- E. P. Gillis and M. D. Burke, Iterative Cross-Couplng with MIDA Boronates: Towards a General Platform for Small Molecule Synthesis, Aldrichimica Acta, 2009, 42, 17–27 Search PubMed.
- G. A. Molander and N. Ellis, Organotrifluoroborates: Protected Boronic Acids That Expand the Versatility of the Suzuki Coupling Reaction, Acc. Chem. Res., 2007, 40, 275–286 CrossRef PubMed.
- R. J. Armstrong and V. K. Aggarwal, 50 Years of Zweifel Olefination: A Transition-Metal-Free Coupling, Synthesis, 2017, 3323–3336 Search PubMed.
-
(a) M. Norio, K. Yamada and A. Suzuki, A New Stereospecific Cross-Coupling by the Palladium-Catalyzed Reaction of 1-Alkenylboranes with 1-Alkenyl or 1-Alkynyl Halides, Tetrahedron Lett., 1979, 20, 3437–3440 CrossRef. For other recent excellent work, please see:
(b) X.-Y. Chen, X.-X. Nie, Y. Wu and P. Wang,
para-Selective Arylation and Alkenylation of Monosubstituted Arenes using Thianthrene S-oxide as a Transient Mediator, Chem. Commun., 2020, 56, 5058–5061 RSC;
(c) A. Chen, Y. Han, R. Wu, B. Yang, L. Zhu and F. Zhu, Palladium-Catalyzed Suzuki-Miyaura Cross-Couplings of Stable Glycal Boronates for Robust Synthesis of C-1 Glycals, Nat. Commun., 2024, 15, 5228 CrossRef.
- P. Y. S. Lam, G. Vincent, D. Bonne and C. G. Clark, Copper-Promoted/Catalyzed C-N and C-O Bond Cross-Coupling with Vinylboronic Acid and its Utilities, Tetrahedron Lett., 2003, 44, 4927–4931 CrossRef.
-
(a) T. D. Quach and R. A. Batey, Copper(II)-Catalyzed Ether Synthesis from Aliphatic Alcohols and Potassium Organotrifluoroborate Salts, Org. Lett., 2003, 5, 1381–1384 CrossRef PubMed;
(b) Y. Bolshan and R. A. Batey, Enamide Synthesis by Copper-Catalyzed Cross-Coupling of Amides and Potassium Alkenyltrifluoroborate Salts, Angew. Chem., Int. Ed., 2008, 47, 2109–2112 CrossRef PubMed;
(c) F. Huang, T. D. Quach and R. A. Batey, Copper-Catalyzed Nondecarboxylative Cross Coupling of Alkenyltrifluoroborate Salts with Carboxylic Acids or Carboxylates: Synthesis of Enol Esters, Org. Lett., 2013, 15, 3150–3153 CrossRef PubMed.
-
(a) L. Chu and F.-L. Qing, Copper-Mediated Trifluoromethylation of Boronic Acids, Org. Lett., 2010, 12, 5060–5063 CrossRef PubMed;
(b) M. Presset, D. Oehlrich, F. Rombounts and G. A. Molander, Copper-Mediated Radical Trifluoromethylation of Unsaturated Potassium Organotrifluoroborates, J. Org. Chem., 2013, 78, 12837–12843 CrossRef PubMed;
(c) J. Xu, D.-F. Luo, B. Xiao, Z.-J. Liu, T.-J. Gong, Y. Fu and L. Liu, Copper-Catalyzed Trifluoromethylation of Aryl Boronic Acids Using a CF3+ Reagent, Chem. Commun., 2011, 47, 4300–4302 RSC;
(d) T. Liu and Q. Shen, Copper-Catalyzed Trifluoromethylation of Aryl and Vinyl Boronic Acids with An Electrophilic Trifluoromethylating Reagent, Org. Lett., 2011, 13, 2342–2345 CrossRef CAS PubMed;
(e) J. Liu, Y. Xiao, J. Hao and Q. Shen, Copper-Catalyzed Trifluoromethylation of (Hetero)aryl Boronic Acid Pinacol Esters with YlideFluor, Org. Lett., 2023, 25, 1204–1208 CrossRef CAS PubMed;
(f) C.-P. Zhang, J. Cai, C.-B. Zhou, X.-P. Wang, X. Zheng, Y.-C. Gu and J.-C. Xiao, Copper-Mediated Trifluoromethylation of Arylboronic Acids by Trifluoromethyl Sulfonium Salts, Chem. Commun., 2011, 47, 9516–9518 RSC. For the reaction by using Ir(II) catalysis, please see:
(g) A. T. Parsons, T. D. Senecal and S. L. Buchwald, Iron(II)-Catalyzed Trifluoromethylation of Potassium Vinyltrifluoroborates, Angew. Chem., Int. Ed., 2012, 51, 2947–2950 CrossRef CAS.
-
(a) A. M. Whittaker, R. P. Rucker and G. Lalic, Catalytic SN2′-Selective Substitution of Allylic Chlorides With Arylboronic Esters, Org. Lett., 2010, 12, 3216–2318 CrossRef CAS;
(b) C.-T. Yang, Z.-Q. Zhang, Y.-C. Liu and L. Liu, Copper-Catalyzed Cross-Coupling Reaction of Organoboron Compounds with Primary Alkyl Halides and Pseudohalides, Angew. Chem., Int. Ed., 2011, 50, 3904–3907 CrossRef CAS PubMed;
(c) R. Shintani, K. Takatsu, M. Takeda and T. Hayashi, Copper-Catalyzed Asymmetric Allylic Substitution of Allyl Phosphates with Aryl- and Alkenylboronates, Angew. Chem., Int. Ed., 2011, 50, 8656–8659 CrossRef CAS PubMed;
(d) F. Gao, J. L. Carr and A. H. Hoveyda, Copper-Catalyzed Enantioselective Allylic Substitution with Readily Accessible Carbonyl- and Acetal-Containing Vinylboron Reagents, Angew. Chem., Int. Ed., 2012, 51, 6613–6617 CrossRef CAS PubMed;
(e) M. Yang, N. Yokokawa, H. Ohmiya and M. Sawamura, Org. Lett., 2012, 14, 816–819 CrossRef CAS PubMed. For an excellent example with Ir catalysis, please see:
(f) J. Y. Hamilton, D. Sarlah and E. M. Carreira, J. Am. Chem. Soc., 2013, 135, 994–997 CrossRef CAS.
-
(a) J. Takaya, S. Tadami, K. Ukai and N. Iwasawa, Copper(I)-Catalyzed Carboxylation of Aryl- and Alkenylboronic Esters, Org. Lett., 2008, 10, 2697–2700 CrossRef CAS PubMed;
(b) T. Ohishi, M. Nishiura and Z. Hou, Carboxylation of Organoboronic Esters Catalyzed by N-Heterocyclic Carbene Copper(I) Complexes, Angew. Chem., Int. Ed., 2008, 47, 5792–5795 CrossRef CAS.
-
(a) K. Takatsu, R. Shintani and T. Hayashi, Copper-Catalyzed 1,4-Addition of Organoboronates to Alkylidene Cyanoacetates: Mechanistic Insight and Application to Asymmetric Catalysis, Angew. Chem., Int. Ed., 2011, 50, 5548–5552 CrossRef CAS;
(b) Q. Chong, Z. Yue, S. Zhang, C. Ji, F. Cheng, H. Zhang, X. Hong and F. Meng,
N-heterocyclic Carbene−Cu-Catalyzed Enantioselective Conjugate Additions with Alkenylboronic Esters as Nucleophiles, ACS Catal., 2017, 7, 5693–5698 CrossRef CAS.
- R. Shintani, K. Takatsu and T. Hayashi, Copper-Catalyzed Asymmetric Addition of Arylboronates to Isatins: A Catalytic Cycle Involving Alkoxocopper Intermediates, Chem. Commun., 2010, 46, 6822–6824 RSC.
-
(a) P.-S. Lin, M. Jeganmohan and C.-H. Cheng, Nickel-Catalyzed Mizoroki–Heck- versus Michael-Type Addition of Organoboronic Acids to α, β-Unsaturated Alkenes through Fine-Tuning of Ligands, Chem. – Asian J., 2007, 2, 1409–1416 CrossRef;
(b) M.-H. Chen, S. Mannathan, P.-S. Lin and C.-H. Cheng, Cobalt(II)-Catalyzed 1, 4-Addition of Organoboronic Acids to Activated Alkenes: An Application to Highly cis-Stereoselective Synthesis of Aminoindane Carboxylic Acid Derivatives, Chem. – Eur. J., 2012, 18, 14918–14922 CrossRef.
- X. Wang, M. Quan, F. Xie, G. Yang and W. Zhang, Ni(II)/mono-RuPHOX-Catalyzed Asymmetric Addition of Alkenylboronic Acids to Cyclic Aldimines, Tetrahedron Lett., 2018, 59, 1573–1575 CrossRef.
- H. Wei, Y. Luo, J. Ren, Q. Yuan and W. Zhang, Ni(II)-Catalyzed Asymmetric Alkenylation and Arylation of Aryl Ketones with Organoborons via 1,5-Metalate Shift, Nat. Commun., 2024, 15, 8775 CrossRef PubMed.
- Y. Huang, R.-Z. Huang and Y. Zhao, Cobalt-Catalyzed Enantioselective Vinylation of Activated Ketones and Imines, J. Am. Chem. Soc., 2016, 138, 6571–6576 CrossRef PubMed.
- For two excellent examples by using gold catalysis, please see:
(a) S. Liao, A. Porta, X. Cheng, X. Ma, G. Zanoni and L. Zhang, Bifunctional Ligand Enables Efficient Gold-Catalyzed Hydroalkenylation of Propargylic Alcohol, Angew. Chem., Int. Ed., 2018, 57, 8250–8254 CrossRef;
(b) X. Li, M. D. Wodrich and J. Waser, Accessing Elusive σ-Type Cyclopropenium Cation Equivalents through Redox Gold Catalysis, Nat. Chem., 2024, 16, 901–921 CrossRef CAS PubMed.
- S. V. Kumar, A. Yen, M. Lautens and P. J. Guiry, Catalytic Asymmetric Transformations of Oxa- and Azabicyclic Alkenes, Chem. Soc. Rev., 2021, 50, 3013–3093 RSC.
-
(a) M. J. Fleming, H. A. McManus, A. Rudolph, W. H. Chan, J. Ruiz, C. Dockendorff and M. Lautens, Concise Enantioselective Total Syntheses of (+)-Homochelidonine, (+)-Chelamidine, (+)-Chelidonine, (+)-Chelamine and (+)-Norchelidonine by a PdII-Catalyzed Ring-Opening Strategy, Chem. – Eur. J., 2008, 14, 2112–2124 CrossRef CAS;
(b) M. Ito, F. Konno, T. Kumamoto, N. Suzuki, M. Kawahata, K. Yamaguchi and T. Ishikawa, Enantioselective Synthesis of Chelidonine, a B/C-cis,-11-hydroxyhexahydrobenzo[c]phenanthridine Alkaloid, Tetrahedron, 2011, 67, 8041–8049 CrossRef CAS.
- X. J. Huang, D.-L. Mo, C.-H. Ding and X.-L. Hou, Chiral P-Containing Palladacycle-Catalyzed Asymmetric Ring-Opening Reactions of Oxabicyclic Alkenes with Alkenyl Boronic Acids, Synlett, 2011, 943–946 CAS.
- D. Zhu, Y. Zhao, Q. Chong and F. Meng, Cobalt-Catalyzed Enantioselective Ring-Opening Reactions of Oxa- and Aza-bicyclic Alkenes with Alkenylboronic Acids, Chin. J. Chem., 2022, 40, 190–194 CrossRef.
-
(a) C. Wang, Manganese-Mediated C–C Bond Formation via C–H Activation: From Stoichiometry to Catalysis, Synlett, 2013, 1606–1613 CrossRef CAS;
(b) W. Liu and J. T. Groves, Manganese Catalyzed C–H Halogenation, Acc. Chem. Res., 2015, 48, 1727–1735 CrossRef CAS;
(c) J. R. Carney, B. R. Dillon and S. P. Thomas, Recent Advances of Manganese Catalysis for Organic Synthesis, Eur. J. Org. Chem., 2016, 3912–3929 CrossRef CAS;
(d) Y.-Y. Hu, B.-W. Zhou and C. Wang, Inert C–H Bond Transformations Enabled by Organometallic Manganese Catalysis, Acc. Chem. Res., 2018, 51, 816–827 CrossRef CAS PubMed;
(e) Z. Yan, C. Zhu and J. Xie, Manganese(I)-Catalyzed Selective Functionalization of Alkynes, Synlett, 2019, 124–128 CAS;
(f) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, 3d Transition Metals for C–H Activation, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS;
(g) K. Feng, R. E. Quevedo, J. T. Kohrt, M. S. Oderinde, U. Reilly and M. C. White, Late-Stage Oxidative C(sp3)–H Methylation, Nature, 2020, 580, 621–627 CrossRef CAS PubMed;
(h) J. P. Zhao, T. Nanjo, E. C. de Lucca Jr. and M. C. White, Chemoselective Methylene Oxidation in Aromatic Molecules, Nat. Chem., 2019, 11, 213–221 CrossRef PubMed.
-
(a) Z. F. Yan, X.-A. Yuan, Y. Zhao, C. J. Zhu and J. Xie, Selective Hydroarylation of 1, 3-Diynes Using a Dimeric Manganese Catalyst: Modular Synthesis of Z-Enynes, Angew. Chem., Int. Ed., 2018, 57, 12906–12910 CrossRef PubMed;
(b) Y. Pang, G. Liu, C. Huang, X.-A. Yuan, W. Li and J. Xie, A Highly Efficient Dimeric Manganese-Catalyzed Selective Hydroarylation of Internal Alkynes, Angew. Chem., Int. Ed., 2020, 59, 12789–12794 CrossRef PubMed.
- D. P. Wang, J. Dong, W. Fan, X.-A. Yuan, J. Han and J. Xie, Dimeric Manganese-Catalyzed Hydroarylation and Hydroalkenylation of Unsaturated Amide, Angew. Chem., Int. Ed., 2020, 59, 8430–8434 CrossRef.
-
(a) M. Rubin, M. Rubina and V. Gevorgyan, Transition Metal Chemistry of Cyclopropenes and Cyclopropanes, Chem. Rev., 2007, 107, 3117–3179 CrossRef PubMed;
(b) T. Hudlicky and J. Reed, Defying Ring Strain: New Approaches to Cyclopropanes, Angew. Chem., Int. Ed., 2010, 49, 486–488 CrossRef;
(c) L. Jiao and Z.-X. Yu, Vinylcyclopropane Derivatives in Transition-Metal-Catalyzed Cycloadditions for the Synthesis of Carbocyclic Compounds, J. Org. Chem., 2013, 78, 6842–6848 CrossRef PubMed.
- H. Zhang, W. Huang, T. Wang and F. Meng, Cobalt-Catalyzed Diastereo- and Enantioselective Hydroalkenylation of Cyclopropenes with Alkenylboronic Acids, Angew. Chem., Int. Ed., 2019, 58, 11049–11053 CrossRef CAS PubMed.
- X.-P. Zeng, Z.-Y. Cao, Y.-H. Wang, F. Zhou and J. Zhou, Catalytic Enantioselective Desymmetrization Reactions to All-Carbon Quaternary Stereocenters, Chem. Rev., 2016, 116, 7330–7396 CrossRef CAS.
- D. P. Wang, Y. He, H. Dai, C. Huang, X.-A. Yuan and J. Xie, Manganese-Catalyzed Hydrocarbofunctionalization of Internal Alkenes, Chin. J. Chem., 2020, 38, 1497–1502 CrossRef CAS.
- T. Liu, Y. Yang and C. Y. Wang, Manganese-Catalyzed Hydroarylation of Unactivated Alkenes, Angew. Chem., Int. Ed., 2020, 59, 14256–14260 CrossRef CAS.
- H. Lv, L.-J. Xiao, D. Zhao and Q.-L. Zhou, Nickel(0)-Catalyzed Linear-Selective Hydroarylation of Unactivated Alkenes and Styrenes with Aryl Boronic Acids, Chem. Sci., 2018, 9, 6839–6843 RSC.
- L.-J. Xiao, L. Chen, W.-M. Feng, M.-L. Li, J.-H. Xie and Q.-L. Zhou, Nickel(0)-Catalyzed Hydroarylation of Styrenes and 1, 3-Dienes with Organoboron Compounds, Angew. Chem., Int. Ed., 2018, 57, 461–464 CrossRef CAS PubMed.
- Z.-Q. Li, Y. Fu, R. Deng, V. T. Tran, Y. Gao, P. Liu and K. M. Engle, Ligand-Controlled Regiodivergence in Nickel-Catalyzed Hydroarylation and Hydroalkenylation of Alkenyl Carboxylic Acids, Angew. Chem., Int. Ed., 2020, 59, 23306–23312 CrossRef CAS.
- X.-Y. Lv, C. Fan, L.-J. Xiao, J.-H. Xie and Q.-L. Zhou, Ligand-Enabled Ni-Catalyzed Enantioselective Hydroarylation of Styrenes and 1,3-Dienes with Arylboronic Acids, CCS Chem., 2019, 1, 328–334 CrossRef CAS.
- Y.-G. Chen, B. Shuai, X.-T. Xu, Y.-Q. Li, Q.-L. Yang, H. Qiu, K. Zhang, P. Fang and T.-S. Mei, Nickel-catalyzed Enantioselective Hydroarylation and Hydroalkenylation of Styrenes, J. Am. Chem. Soc., 2019, 141, 3395–3399 CrossRef CAS PubMed.
- D.-M. Wang, L.-Q. She, Y. Wu, C. Zhu and P. Wang, Ligand-Enabled Ni-Catalyzed Hydroarylation and Hydroalkenylation of Internal Alkenes with Organoborons, Nat. Commun., 2022, 13, 6878 CrossRef CAS PubMed.
- Pd catalysis:
(a) N. Miyaura, Y. Tanabe, H. Suginome and A. Suzuki, Cross-Coupling Reactions of 1-alkenylboranes with 3,4-Epoxy-1-butene Catalyzed by Palladium or Nickel Complexes, J. Organomet. Chem., 1982, 233, C13–C16 CrossRef CAS;
(b) J. Kjellgren, J. Aydin, O. A. Wallner, I. V. Saltanova and K. J. Szabó, Palladium Pincer Complex Catalyzed Cross-Coupling of Vinyl Epoxides and Aziridines with Organoboronic Acids, Chem. – Eur. J., 2005, 11, 5260–5268 CrossRef CAS PubMed;
(c) S. Bonnet, J. H. van Lenthe, M. A. Siegler, A. L. Spek, G. van Koten and R. J. M. K. Gebbink, Bimetallic η6, η1 NCN-Pincer Ruthenium Palladium Complexes with η6-RuCp Coordination: Synthesis,
X-ray Structures, and Catalytic Properties, Organometallics, 2009, 28, 2325–2333 CrossRef CAS;
(d) D. K. Nielsen and A. G. Doyle, Nickel-Catalyzed Cross-Coupling of Styrenyl Epoxides with Boronic Acids, Angew. Chem., Int. Ed., 2011, 50, 6056–6059 CrossRef CAS;
(e) G. Hamasaka, T. Muto and Y. Uozumi, Molecular-Architecture-Based Administration of Catalysis in Water: Self-Assembly of an Amphiphilic Palladium Pincer Complex, Angew. Chem., Int. Ed., 2011, 50, 4876–4878 CrossRef CAS;
(f) X.-Y. Lu, C.-T. Yang, J.-H. Liu, Z.-Q. Zhang, X. Lu, X. Lou, B. Xiao and Y. Fu, Cu-Catalyzed Cross-Coupling Reactions of Epoxides with Organoboron Compounds, Chem. Commun., 2015, 51, 2388–2391 RSC;
(g) T. N. Nguygen and J. A. May, Tertiary and Quaternary Carbon Formation via Gallium-Catalyzed Nucleophilic Addition of Organoboronates to Cyclopropane, Org. Lett., 2018, 20, 112–115 CrossRef;
(h) T. N. Nguyen and J. A. May, Branched Amine Synthesis via Aziridine or Azetidine Opening with Organotrifluoroborates by Cooperative Brønsted/Lewis Acid Catalysis: An Acid-Dependent Divergent Mechanism, Org. Lett., 2018, 20, 3618–3621 CrossRef;
(i) X.-Y. Lu, L.-Y. Yan, J.-S. Li, J.-M. Li, H.-P. Zhou, R.-C. Jiang, C.-C. Liu, R. Lu and R. Hu, Base-free Ni-Catalyzed Suzuki-type Cross-Coupling Reactions of Epoxides with Boronic Acids, Chem. Commun., 2020, 56, 109–112 RSC.
-
(a) S. Roscales and A. G. Csákÿ, Metal-Free Ring-Opening of Epoxides with Potassium Trifluoroborates, Chem. Commun., 2014, 50, 454–456 RSC;
(b) M. Nandakumar, B. Rubial, A. Noble, E. L. Myers and V. K. Aggarwal, Ring-Opening Lithiation–Borylation of 2-Trifluoromethyl Oxirane: A Route to Versatile Tertiary Trifluoromethyl Boronic Esters, Angew. Chem., Int. Ed., 2020, 59, 1187–1191 CrossRef PubMed;
(c) V. Ortega and A. G. Csákÿ, Ring-Opening of Donor–Acceptor Cyclopropanes by Boronic Acids and Potassium Organotrifluoroborates under Transition-Metal-Free Conditions, J. Org. Chem., 2016, 81, 3917–3923 CrossRef PubMed.
- L. Wu, G. Yang and W. Zhang, Ni-Catalyzed Enantioconvergent Coupling of Epoxides with Alkenylboronic Acids: Construction of Oxindoles Bearing Quaternary Carbons, CCS Chem., 2019, 1, 623–631 CAS.
- L. Zhang, Q. Du, Y. Luo, Y. Wang, F. Gao, Y. Ye and W. Zhang, Synthesis of Skipped Aminodienes by a Ni-Catalyzed Ring-Opening/Cross-Coupling Reaction of Vinylaziridines with Multifunctional Organoboronic Acids, Chin. J. Chem., 2023, 41, 3003–3011 CrossRef CAS.
- For selected reviews on enantioconvergent cross-coupling of racemic alkyl (pseudo)halides, see:
(a) A. H. Cherney, N. T. Kadunce and S. E. Reisman, Enantioselective and Enantiospecific Transition-Metal-Catalyzed Cross-Coupling Reactions of Organometallic Reagents To Construct C−C Bonds, Chem. Rev., 2015, 115, 9587–9652 CrossRef CAS PubMed;
(b) J. Choi and G. C. Fu, Transition Metal-Catalyzed Alkyl-Alkyl Bond Formation: Another Dimension in Cross-Coupling Chemistry, Science, 2017, 356, eaaf7230 CrossRef PubMed;
(c) A. Lipp, S. O. Badir and G. A. Molander, Stereoinduction in Metallaphotoredox Catalysis, Angew. Chem., Int. Ed., 2021, 60, 1714–1726 CrossRef CAS.
- P.-F. Wang, J. Yu, K.-X. Guo, S.-P. Jiang, J.-J. Chen, Q.-S. Gu, J.-R. Liu, X. Hong, Z.-L. Li and X.-Y. Liu, Design of Hemilabile N, N, N-Ligands in Copper-Catalyzed Enantioconvergent Radical Cross-Coupling of Benzyl/Propargyl Halides with Alkenylboronate Esters, J. Am. Chem. Soc., 2022, 144, 6442–6452 CrossRef CAS.
- P. Dominguez-Molano, R. Weeks, R. J. Maza, J. J. Carbó and E. Fernández, Boron-Copper 1,3-Rearrangement: The New Concept Behind the Boryl Migration from C(sp2) in Alkenyl Boranes to C(sp3), Angew. Chem., Int. Ed., 2023, 62, e202304791 CrossRef CAS.
- J. E. Baumann, C. P. Chung and G. Lalic, Stereoselective Copper-Catalyzed Olefination of Imines, Angew. Chem., Int. Ed., 2024, 63, e202316521 CrossRef CAS.
-
(a) A. Vasseur, J. Bruffaerts and I. Marek, Remote Functionalization through Alkene Isomerization, Nat. Chem., 2016, 11, 209–219 CrossRef;
(b) H. Sommer, F. JuliáHernández, R. Martin and I. Marek, Walking Metals for Remote Functionalization, ACS Cent. Sci., 2018, 4, 153–165 CrossRef CAS;
(c) D. JanssenMüller, B. Sahoo, S.-Z. Sun and R. Martin, Tackling Remote sp3 C−H Functionalization via Ni-Catalyzed “chain-walking” Reactions, Isr. J. Chem., 2020, 60, 195–206 CrossRef;
(d) S. Ghosh, S. Patel and I. Chatterjee, Chain-Walking Reactions of Transition Metals for Remote C–H Bond Functionalization of Olefinic Substrates, Chem. Commun., 2021, 57, 11110–11130 RSC;
(e) Y. Wang, Y. He and S. Zhu, NiH-Catalyzed Functionalization of Remote and Proximal Olefins: New Reactions and Innovative Strategies, Acc. Chem. Res., 2022, 55, 3519–3536 CrossRef CAS;
(f) B. C. Lee, C.-F. Liu, L. Q. H. Lin, H. Z. Yap, N. Song, C. H. M. Ko, P. H. Chan and M. J. Koh,
N-Heterocyclic Carbenes as Privileged Ligands for Nickel-Catalysed Alkene Functionalization, Chem. Soc. Rev., 2023, 52, 2946–2991 RSC;
(g) C. Romano and R. Martin, Ni-Catalysed Remote C(sp3)–H Functionalization using Chain-Walking Strategies, Nat. Rev. Chem., 2024, 8, 833–850 Search PubMed.
- H.-D. He, R. Chitrakar, Z.-W. Cao, D.-M. Wang, L.-Q. She, P.-G. Zhao, Y. Wu, Y.-Q. Xu, Z.-Y. Cao and P. Wang, Diphosphine Ligand-Enabled Nickel-Catalyzed Chelate-Assisted Inner-Selective Migratory Hydroarylation of Alkenes, Angew. Chem., Int. Ed., 2024, 63, e202313336 CrossRef.
- H. Lv, H. Kang, B. Zhou, X. Xue, K. Engle and D. Zhao, Nickel-Catalyzed Intermolecular Oxidative Heck Arylation Driven by Transfer Hydrogenation, Nat. Commun., 2019, 10, 5025 CrossRef.
-
(a) Y. Zhang, Y. Li, W. Zhou, M. Zhang, Q. Zhang, R. Jia and J. Zhao, Assembly of Polysubstituted Chiral Cyclopropylamines via Highly Enantioselective Cu-Catalyzed Three-Component Cyclopropene Alkenylamination, Chem. Commun., 2020, 56, 12250–12253 RSC;
(b) Z. Li, M. Zhang, Y. Zhang, S. Liu, J. Zhao and Q. Zhang, Multicomponent Cyclopropane Synthesis Enabled by Cu-Catalyzed Cyclopropene Carbometalation with Organoboron Reagent: Enantioselective Modular Access to Polysubstituted 2-Arylcyclopropylamines, Org. Lett., 2019, 21, 5432–5437 CrossRef PubMed.
-
(a) M. Ueda, A. Saitoh and N. Miyaura, Asymmetric Hydrogenation of 1-Phenylethenylboronic Acid and Esters for the Synthesis of Chiral Organoboron Compounds, J. Organomet. Chem., 2002, 642, 145–147 CrossRef;
(b) J. B. Morgan and J. P. Morken, Catalytic Enantioselective Hydrogenation of Vinyl Bis(boronates), J. Am. Chem. Soc., 2004, 126, 15338–15339 CrossRef PubMed;
(c) A. Paptchikhine, P. Cheruku, M. Engman and P. G. Andersson, Iridium-Catalyzed Enantioselective Hydrogenation of Vinyl Boronates, Chem. Commun., 2009, 5996–5998 RSC;
(d) A. Ganić and A. Pfaltz, Iridium-Catalyzed Enantioselective Hydrogenation of Alkenylboronic Esters, Chem. – Eur. J., 2012, 18, 6724–6728 CrossRef PubMed;
(e) D. Fan, J. Zhang, Y. Hu, Z. Zhang, I. D. Gridnew and W. Zhang, Asymmetric Hydrogenation of α-Boryl Enamides Enabled by Nonbonding Interactions, ACS Catal., 2020, 10, 3232–3240 CrossRef CAS.
-
(a) J. C. H. Lee and D. G. Hall, Chiral Boronate Derivatives via Catalytic Enantioselective Conjugate Addition of Grignard Reagents on 3-Boronyl Unsaturated Esters and Thioesters, J. Am. Chem. Soc., 2010, 132, 5544–5545 CrossRef CAS;
(b) J. C. H. Lee, R. McDonald and D. G. Hall, Enantioselective Preparation and Chemoselective Cross-Coupling of 1, 1-Diboron Compounds, Nat. Chem., 2011, 3, 894–899 CrossRef CAS PubMed;
(c) H. J. Edwards, J. D. Hargrave, S. D. Penrose and C. G. Frost, Synthetic Applications of Rhodium Catalysed Conjugate Addition, Chem. Soc. Rev., 2010, 39, 2093–2105 RSC;
(d) M. Jean, B. Casanova, S. Gnoatto and P. van de Weghe, Strategy of Total Synthesis Based on the use of Rh-Catalyzed Stereoselective 1, 4-Addition, Org. Biomol. Chem., 2015, 13, 9168–9175 RSC.
-
(a) Y. Lee, H. Jang and A. H. Hoveyda, Vicinal Diboronates in High Enantiomeric Purity through Tandem Site-Selective NHC-Cu-Catalyzed Boron-Copper Additions to Terminal Alkynes, J. Am. Chem. Soc., 2009, 131, 18234–18235 CrossRef CAS PubMed;
(b) X. Feng, H. Jeon and J. Yun, Regio- and Enantioselective Copper(I)-Catalyzed Hydroboration of Borylalkenes: Asymmetric Synthesis of 1,1-Diborylalkanes, Angew. Chem., Int. Ed., 2013, 52, 3989–3992 CrossRef CAS PubMed;
(c) L. Dang, H. Zhao, Z. Lin and T. Marder, DFT Studies of Alkene Insertions into Cu−B Bonds in Copper(I) Boryl Complexes, Organometallics, 2007, 26, 2824–2832 CrossRef CAS;
(d) M. Zhang, Z. Liu and W. Zhao, Rhodium-Catalyzed Remote Borylation of Alkynes and Vinylboronates, Angew. Chem., Int. Ed., 2023, 62, e202215455 CrossRef CAS.
-
(a) D. Nishikawa, K. Hirano and M. Miura, Asymmetric Synthesis of α-Aminoboronic Acid Derivatives by Copper-Catalyzed Enantioselective Hydroamination, J. Am. Chem. Soc., 2015, 137, 15620–15623 CrossRef CAS;
(b) Y. Zhao, D. Qiao, M. Duan, Y. Wang and S. Zhu, Enantioselective Synthesis of α-Aminoboronates by NiH-Catalysed Asymmetric Hydroamidation of Alkenyl Boronates, Nat. Commun., 2022, 13, 5630 CrossRef.
-
(a) A. A. Szymaniak, C. Zhang, J. R. Coombs and J. P. Morken, Enantioselective Synthesis of Nonracemic Geminal Silylboronates by Pt-Catalyzed Hydrosilylation, ACS Catal., 2018, 8, 2897–2901 CrossRef CAS PubMed;
(b) Z. Kong, W. Hu and J. P. Morken, 1,2-Diborylsilanes: Catalytic Enantioselective Synthesis and Site-Selective Cross-Coupling, ACS Catal., 2023, 13, 11522–11527 CrossRef.
-
(a) J. R. Coombs, L. Zhang and J. P. Morken, Enantiomerically Enriched Tris(boronates): Readily Accessible Conjunctive Reagents for Asymmetric Synthesis, J. Am. Chem. Soc., 2014, 136, 16140–16143 CrossRef PubMed;
(b) H. Fang, N. Manoj, M. Popescu, A. Noble, R. Paton and V. K. Aggarwal, Platinum-Catalyzed Regio- and Enantioselective Diboration of Unactivated Alkenes with (pin)B−B(dan), Angew. Chem., 2024, 64, e202413504 Search PubMed.
-
(a) D. Nishikawa, K. Hirano and M. Miura, Copper-Catalyzed Regio- and Stereoselective Aminoboration of Alkenylboronates, Org. Lett., 2016, 18, 4856–4859 CrossRef PubMed;
(b) S. Radomkit, Z. Liu, A. Closs, M. S. Mikus and A. H. Hoveyda, Practical, Efficient, and Broadly Applicable Synthesis of Readily Differentiable Vicinal Diboronate Compounds by Catalytic Three-Component Reactions, Tetrahedron, 2017, 73, 5011–5017 CrossRef;
(c) I. Kageyuki, I. Osaka, K. Takaki and H. Yoshida, Copper-Catalyzed B(dan)-Installing Carboboration of Alkenes, Org. Lett., 2017, 19, 830–833 CrossRef PubMed;
(d) J. Carreras, A. Caballero and P. J. Pérez, Enantio- and Diastereoselective Cyclopropanation of 1-Alkenylboronates: Synthesis of 1-Boryl-2, 3-Disubstituted Cyclopropanes, Angew. Chem., Int. Ed., 2018, 57, 2334–2338 CrossRef PubMed. Arnold showed that the engineering of two variants of Rhodothermus marinus nitric oxide dioxygenase (RmaNOD) could catalyze the stereoselective and diastereodivergent cyclopropanation of unsubstituted alkenylboronate with ethyl diazoacetate, for details, please see:
(e) B. J. Wittmann, A. M. Knight, J. L. Hofstra, S. E. Reisman, S. B. J. Kan and F. H. Arnold, Diversity-Oriented Enzymatic Synthesis of Cyclopropane Building Blocks, ACS Catal., 2020, 10, 7112–7116 CrossRef CAS PubMed.
-
(a) W. J. Jang, J. Woo and J. Yun, Asymmetric Conjugate Addition of Chiral Secondary Borylalkyl Copper Species, Angew. Chem., Int. Ed., 2021, 60, 4614–4618 CrossRef;
(b) J. L. Knippel, A. Z. Ni, A. W. Schuppe and S. L. Buckwald, A General Strategy for the Asymmetric Preparation of α-Stereogenic Allyl Silanes, Germanes, and Boronate Esters via Dual Copper Hydride- and Palladium-Catalysis, Angew. Chem., Int. Ed., 2021, 61, e202212630 CrossRef;
(c) J. He, C. B. Seo, W. S. Yoon and J. Yun, Asymmetric Synthesis of β-Aminoboronates via Copper-Catalyzed Reductive Coupling of Vinyl Boronates with Imines, Org. Lett., 2023, 25, 5492–5497 CrossRef CAS PubMed;
(d) X. Gao, X. Ren, W. Deng and X. Zhang, Copper-Catalyzed Highly Stereoselective Hydrodifluoroallylation of Cyclopropenes and Alkenyl Boronates with 3, 3-Difluoroallyl Sulfonium Salts, Chin. J. Chem., 2023, 41, 3521–3527 CrossRef CAS.
- C. Zhang, W. Hu, G. J. Lovinger, J. Jin, J. Chen and J. P. Morken, Enantiomerically Enriched α-Borylzinc Reagents by Nickel-Catalyzed Carbozincation of Vinylboronic Esters, J. Am. Chem. Soc., 2021, 143, 14189–14195 CrossRef CAS.
-
(a) E. K. Edelstein, S. Namirembe and J. P. Morken, Enantioselective Conjunctive Cross-Coupling of Bis(alkenyl)borates: A General Synthesis of Chiral Allylboron Reagents, J. Am. Chem. Soc., 2017, 139, 5027–5030 CrossRef CAS;
(b) J. A. Myhill, C. A. Wilhelmsen, L. Zhang and J. P. Morken, Diastereoselective and Enantioselective Conjunctive Cross-Coupling Enabled by Boron Ligand Design, J. Am. Chem. Soc., 2018, 140, 15181–15185 CrossRef CAS;
(c) J. A. Myhill, L. Zhang, G. J. Lovinger and J. P. Morken, Enantioselective Construction of Tertiary Boronic Esters by Conjunctive Cross-Coupling, Angew. Chem., Int. Ed., 2018, 57, 12799–12803 CrossRef CAS;
(d) Y. Meng, Z. Kong and J. P. Morken, Catalytic Enantioselective Synthesis of anti-Vicinal Silylboronates by Conjunctive Cross-Coupling, Angew. Chem., Int. Ed., 2020, 59, 8456–8459 CrossRef CAS PubMed;
(e) C. Law, E. Kativhu, J. Wang and J. P. Morken, Angew. Chem., Int. Ed., 2020, 59, 10311–10315 CrossRef CAS PubMed;
(f) G. J. Lovinger and J. P. Morken, Ni-Catalyzed Enantioselective Conjunctive Coupling with C(sp3) Electrophiles: A Radical-Ionic Mechanistic Dichotomy, J. Am. Chem. Soc., 2017, 139, 17293–17296 CrossRef;
(g) C. A. Wilhelmsen, X. Zhang, J. A. Myhill and J. P. Morken, Enantioselective Synthesis of Tertiary B-Boryl Amides by Conjunctive Cross-Coupling of Alkenyl Boronates and Carbamoyl Chloride, Angew. Chem., Int. Ed., 2022, 61, e202116784 CrossRef;
(h) X. Zhang, C. Gao and J. P. Morken, Enantioselective Construction of Carbocyclic and Heterocyclic Tertiary Boronic Esters by Conjunctive Cross-Coupling Reaction, J. Am. Chem. Soc., 2023, 145, 16344–16349 CrossRef PubMed.
- J. C. Lo, J. Gui, Y. Yabe, C.-M. Pan and P. S. Baran, Functionalized Olefin Cross-Coupling to Construct Carbon–Carbon Bonds, Nature, 2014, 516, 343–348 CrossRef PubMed.
- S. Bera and X. Hu, Nickel-Catalyzed Regioselective Hydroalkylation and Hydroarylation of Alkenyl Boronic Esters, Angew. Chem., Int. Ed., 2019, 58, 13854–13859 CrossRef.
- Y. Zhang, B. Han and S. Zhu, Rapid Access to Highly Functionalized Alkyl Boronates by NiH-Catalyzed Remote Hydroarylation of Boron-Containing Alkenes, Angew. Chem., Int. Ed., 2019, 58, 13860–13864 CrossRef PubMed.
- S. Bera, R. Mao and X. Hu, Enantioselective C(sp3)-C(sp3) Cross-Coupling of Non-Activated Alkyl Electrophiles via Nickel Hydride Catalysis, Nat. Chem., 2021, 13, 270–277 CrossRef PubMed.
- X.-X. Wang, X. Lu, S.-J. He and Y. Fu, Nickel-Catalyzed Three-Component Olefin Reductive Dicarbofunctionalization to Access Alkylborates, Chem. Sci., 2020, 11, 7950–7956 RSC.
- L. Liu, M. C. Aguilera, W. Lee, C. R. Youshaw, M. L. Neidig and O. Gutierrez, General Method for Iron-Catalyzed Multicomponent Radical
Cascades-Cross-Couplings, Science, 2021, 374, 432–439 CrossRef CAS PubMed.
- T. D. Ho, B. J. Lee, C. Tan, J. A. Utley, N. Q. Ngo and K. L. Hull, Efficient Synthesis of α-Haloboronic Esters via Cu-Catalyzed Atom Transfer Radical Addition, J. Am. Chem. Soc., 2023, 145, 27230–27235 CrossRef.
-
(a) M. M. Parsutkar, V. V. Pagar and T. V. Rajanbabu, J. Am. Chem. Soc., 2019, 141, 15367–15377 CrossRef PubMed. Ogoshi has pioneered the Co(I)-catalyzed [2 + 2] cycloaddition of vinylboronate with 1,3-enyne, albeit with low efficiency, for details, please see:
(b) A. Nishimura, E. Tamai, M. Ohashi and S. Ogoshi, Chem. – Eur. J., 2014, 20, 6613–6617 CrossRef PubMed. For another recent work, please see:
(c) K. Prysiazhniuk, O. Polishchuk, S. Shulha, K. Gudzikevych, O. P. Datsenko, V. Kubyshkin and P. K. Mykhailiuk, Borylated Cyclobutanes via Thermal [2 + 2]-Cycloaddition, Chem. Sci., 2024, 15, 3249–3254 RSC.
- Z. G. Hajos and D. R. Parrish, Asymmetric Synthesis of Bicyclic Intermediates of Natural Product Chemistry, J. Org. Chem., 1974, 39, 1615–1621 CrossRef.
-
(a) B. List, R. A. Lerner and C. F. Barbas III, Proline-Catalyzed Direct Asymmetric Aldol Reactions, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef;
(b) K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, New Strategies for Organic Catalysis: The First Highly Enantioselective Organocatalytic Diels−Alder Reaction, J. Am. Chem. Soc., 2000, 122, 4243–4244 CrossRef.
-
(a) M. S. Taylor and E. N. Jacobsen, Asymmetric Catalysis by Chiral Hydrogen-Bond Donors, Angew. Chem., Int. Ed., 2006, 45, 1520–1543 CrossRef PubMed;
(b) E. A. Colby Davie, S. M. Mennen, Y. Xu and S. J. Miller, Asymmetric Catalysis Mediated by Synthetic Peptides, Chem. Rev., 2007, 107, 5759–5812 CrossRef CAS PubMed;
(c) T. Akiyama, Stronger Brønsted Acids, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS PubMed;
(d) D. Enders, O. Niemeier and A. Henseler, Organocatalysis by N-Heterocyclic Carbenes, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed;
(e) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Asymmetric Enamine Catalysis, Chem. Rev., 2007, 107, 5471–5569 CrossRef;
(f) K. L. Jensen, G. Dickmeiss, H. Jiang, Ł. Albrecht and K. A. Jørgensen, The Diarylprolinol Silyl Ether System: A General Organocatalyst, Acc. Chem. Res., 2012, 45, 248–264 CrossRef;
(g) S. Shirakawa and K. Maruoka, Recent Developments in Asymmetric Phase-Transfer Reactions, Angew. Chem., Int. Ed., 2013, 52, 4312–4348 CrossRef PubMed;
(h) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brønsted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates, Chem. Rev., 2014, 114, 9047–9153 CrossRef;
(i) S.-H. Xiang and B. Tan, Advances in Asymmetric Organocatalysis over the Last 10 Years, Nat. Commun., 2020, 11, 3786 CrossRef PubMed.
-
(a) S. Lee and D. W. C. MacMillan, Organocatalytic Vinyl and Friedel-Crafts Alkylations with Trifluoroborate Salts, J. Am. Chem. Soc., 2007, 129, 15438–15439 CrossRef;
(b) H. Kim and D. W. C. MacMillan, Enantioselective Organo-SOMO Catalysis: the α-Vinylation of Aldehydes, J. Am. Chem. Soc., 2008, 130, 398–399 CrossRef PubMed.
- R. Nishiyabu, Y. Kubo, T. D. James and J. S. Fossey, Boronic Acid Building Blocks: Tools for Self Assembly, Chem. Commun., 2011, 47, 1124–1150 RSC.
- S. Lou, P. N. Moquist and S. E. Schaus, Asymmetric Allylboration of Ketones Catalyzed by Chiral Diols, J. Am. Chem. Soc., 2006, 128, 12660–12661 CrossRef CAS.
- S. E. Schaus and S. Lou, Asymmetric Petasis Reactions Catalyzed by Chiral Biphenols, J. Am. Chem. Soc., 2008, 130, 6922–6923 CrossRef PubMed.
- J. A. Bishop, S. Lou and S. E. Schaus, Enantioselective Addition of Boronates to Acyl Imines Catalyzed by Chiral Biphenols, Angew. Chem., Int. Ed., 2009, 48, 4337–4340 CrossRef.
- T. Kodama, P. N. Moquist and S. E. Schaus, Enantioselective Boronate Additions to N-Acyl Quinoliniums Catalyzed by Tartaric Acid, Org. Lett., 2011, 13, 6316–6319 CrossRef.
-
(a) K. S. Barbato, Y. Luan, D. Ramella, J. S. Panek and S. Schaus, Enantioselective Multicomponent Condensation Reactions of Phenols, Aldehydes, and Boronates Catalyzed by Chiral Biphenols, Org. Lett., 2015, 17, 5812–5815 CrossRef;
(b) Y. Luan and S. E. Schaus, Enantioselective Addition of Boronates to o-Quinone Methides Catalyzed by Chiral Biphenols, J. Am. Chem. Soc., 2012, 134, 19965–19968 CrossRef PubMed.
- T. R. Wu and J. M. Chong, Asymmetric Allylboration of Acyl Imines Catalyzed by Chiral Diols, J. Am. Chem. Soc., 2007, 129, 4908–4909 CrossRef.
- T. Inokuma, K. Takasu, T. Sakaeda and Y. Takemoto, Hydroxyl Group-Directed Organocatalytic Asymmetric Michael Addition of α, β-Unsaturated Ketones with Alkenylboronic Acids, Org. Lett., 2009, 11, 2425–2433 CrossRef.
-
(a) M. Sugiura, R. Kinoshita and M. Nakajima,
O-Monoacyltartaric Acid Catalyzed Enantioselective Conjugate Addition of a Boronic Acid to Dienones: Application to the Synthesis of Optically Active Cyclopentenones, Org. Lett., 2014, 16, 5172–5175 CrossRef PubMed;
(b) M. Sugiura, M. Tokudomi and M. Nakajima, Enantioselective Conjugate Addition of Boronic Acids to Enones Catalyzed by O-Monoacyltartaric Acids, Chem. Commun., 2010, 46, 7799–7800 RSC.
-
(a) B. J. Lundy, S. Jansone-Popova and J. A. May, Enantioselective Conjugate Addition of Alkenylboronic Acids to Indole-Appended Enones, Org. Lett., 2011, 13, 4958–4961 CrossRef CAS PubMed;
(b) P. Q. Le, T. S. Nguyen and J. A. May, A General Method for the Enantioselective Synthesis of α-Chiral Heterocycles, Org. Lett., 2012, 14, 6104–6107 CrossRef CAS PubMed.
- X. Liu, Z. Meng, C. Li, H. Lou and L. Liu, Organocatalytic Enantioselective Oxidative C-H Alkenylation and Arylation of N-Carbamoyl Tetrahydropyridines and Tetrahydro-β-carbolines, Angew. Chem., Int. Ed., 2015, 54, 6012–6015 CrossRef PubMed.
- S. J. T. Jonker, R. Jayarajan, T. Kireilis, M. Deliaval, L. Eriksson and K. J. Szabó, Organocatalytic Synthesis of α-Trifluoromethyl Allylboronic Acids by Enantioselective 1, 2-Borotropic Migration, J. Am. Chem. Soc., 2020, 142, 21254–21259 CrossRef PubMed.
- H. Qian, W. Zhao, Z. Wang and J. Sun, Synthesis of Adjacent Quaternary Stereocenters by Catalytic Asymmetric Allylboration, J. Am. Chem. Soc., 2015, 137, 560–563 CrossRef.
- S. Liu, H. Qian, T. Zhang, H. Xie, Z. Han, W. Guo, H. Huang and J. Sun, Mild Intermolecular Synthesis of a Cyclopropane-Containing Tricyclic Skeleton: Unusual Reactivity of Isobenzopyryliums, Angew. Chem., Int. Ed., 2021, 60, 21272–21276 CrossRef PubMed.
- Z. Tao, K. A. Robb, J. L. Panger and S. E. Denmark, Enantioselective, Lewis Base-Catalyzed Carbosulfenylation of Alkenylboronates by 1, 2-Boronate Migration, J. Am. Chem. Soc., 2018, 140, 15621–15625 CrossRef PubMed.
- T. P. Maloney, J. L. Berman and F. E. Michael, Metal-Free Allylic C−H Amination of Vinylsilanes and Vinylboronates using Silicon or Boron as a Regioselectivity Switch, Angew. Chem., 2022, 134, e202210109 CrossRef.
-
(a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis, Chem. Rev., 2016, 81, 6898–6926 Search PubMed;
(b) N. A. Romero and D. A. Nicewicz, Organic Photoredox Catalysis, Chem. Rev., 2016, 116, 10075–10166 CrossRef PubMed;
(c) R. C. McAtee, E. J. McClain and C. R. J. Stephenson, Illuminating Photoredox Catalysis, Trends Chem., 2019, 1, 111–125 CrossRef PubMed;
(d) G. E. M. Crisenza and P. Melchiorre, Chemistry Glows Green with Photoredox Catalysis, Nat. Commun., 2020, 11, 803 CrossRef;
(e) N. L. Reed and T. P. Yoon, Oxidase Reactions in Photoredox Catalysis, Chem. Soc. Rev., 2021, 50, 2954–2967 RSC.
- D. M. Arias-Rotondo and J. K. McCusker, The Photophysics of Photoredox Catalysis: A Roadmap for Catalyst Design, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC.
-
(a) D. S. Matteson, Radical Catalyzed Additions to Dibutyl Ethyleneboronate, J. Am. Chem. Soc., 1959, 81, 5004–5005 CrossRef;
(b) D. S. Matteson, Radical-catalyzed Additions to α, β-Unsaturated Boronic Esters, J. Am. Chem. Soc., 1960, 82, 4228–4233 CrossRef.
- M. Kischkewitz, K. Okamoto, C. Mück-Lichtenfeld and A. Studer, Radical-Polar Crossover Reactions of Vinylboron Ate Complexes, Science, 2017, 355, 936–938 CrossRef.
- M. Silvi, C. Sandford and V. K. Aggarwal, Merging Photoredox with 1, 2-Metallate Rearrangements: The Photochemical Alkylation of Vinyl Boronate Complexes, J. Am. Chem. Soc., 2017, 139, 5736–5739 CrossRef.
- Y. Yasu, T. Koike and M. Akita, Visible-Light-Induced Synthesis of a Variety of Trifluoromethylated Alkenes from Potassium Vinyltrifluoroborates by Photoredox Catalysis, Chem. Commun., 2013, 49, 2037–2039 RSC.
- D. F. Reina, A. Ruffoni, Y. S. S. Al-Faiyz, J. J. Douglas, N. S. Sheikh and D. Leonori, Visible-Light-Mediated Reactions of Electrophilic Radicals with Vinyl and Allyl Trifluoroborates, ACS Catal., 2017, 7, 4126–4130 CrossRef.
- X. Shen, J.-J. Zhao and S. Yu, Photoredox-Catalyzed Intermolecular Remote C–H and C–C Vinylation via Iminyl Radicals, Org. Lett., 2018, 20, 5523–5527 CrossRef.
- X. Shen, C. Huang, X.-A. Yuan and S. Yu, Diastereoselective and Stereodivergent Synthesis of 2-Cinnamylpyrrolines Enabled by Photoredox-Catalyzed Iminoalkenylation of Alkenes, Angew. Chem., Int. Ed., 2021, 60, 9672–9679 CrossRef PubMed.
- H. Chen, L. Guo and S. Yu, Primary, Secondary, and Tertiary γ-C(sp3)–H Vinylation of Amides via Organic Photoredox-Catalyzed Hydrogen Atom Transfer, Org. Lett., 2018, 20, 6255–6259 CrossRef.
- X.-D. An, H. Zhang, Q. Xu, L. Yu and S. Yu, Stereodivergent Synthesis of α-Aminomethyl Cinnamyl Ethers via Photoredox-Catalyzed Radical Relay Reaction, Chin. J. Chem., 2018, 36, 1147–1150 CrossRef.
- J. Brals, T. M. McGuire and A. J. B. Watson, A Chemoselective Polarity-Mismatched Photocatalytic C(sp3) C(sp2) Cross-Coupling Enabled by Synergistic Boron Activation, Angew. Chem., Int. Ed., 2023, 62, e202310462 CrossRef CAS PubMed.
- M. R. Heinrich, L. A. Sharp and S. Z. Zard, A Convergent Approach to γ-Carbonyl Vinyl Boronates, Chem. Commun., 2005, 3077–3079 RSC.
- A. Noble, R. S. Mega, D. Pflästerer, E. L. Myers and V. K. Aggarwal, Visible-Light-Mediated Decarboxylative Radical Additions to Vinyl Boronic Esters: Rapid Access to γ-Amino Boronic Esters, Angew. Chem., Int. Ed., 2018, 57, 2155–2159 CrossRef PubMed.
- X. Shu, R. Xu, Q. Ma and S. Liao, Accessing Alkyl Boronic Esters via Visible Light-Mediated Decarboxylative Addition Reactions of Redox-Active Esters, Org. Chem. Front., 2020, 7, 2003–2007 RSC.
- T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Aminoalkyl Radicals as Halogen-Atom Transfer Agents for Activation of Alkyl and Aryl Halides, Science, 2020, 367, 1021–1026 CrossRef PubMed.
- C. Shu, R. S. Mega, B. J. Andreassen, A. Noble and V. K. Aggarwal, Synthesis of Functionalized Cyclopropanes from Carboxylic Acids by a Radical Addition–Polar
Cyclization Cascade, Angew. Chem., Int. Ed., 2018, 57, 15430–15434 CrossRef PubMed.
- S.-Z. Sun, Y. Duan, R. S. Mega, R. J. Somerville and R. Martin, Site-Selective 1,2-Dicarbofunctionalization of Vinyl Boronates through Dual Catalysis, Angew. Chem., Int. Ed., 2020, 59, 4370–4374 CrossRef PubMed.
- R. S. Mega, V. K. Duong, A. Noble and V. K. Aggarwal, Decarboxylative Conjunctive Cross-coupling of Vinyl Boronic Esters using Metallaphotoredox Catalysis, Angew. Chem., Int. Ed., 2020, 59, 4375–4379 CrossRef.
- A. Luridiana, D. Mazzarella, L. Capaldo, J. A. Rincón, P. García-Losada, C. Mateos, M. O. Frederick, M. Nuño, W. J. Buma and T. Noël, The Merger of Benzophenone HAT Photocatalysis and Silyl Radical-Induced XAT Enables Both Nickel-Catalyzed Cross-Electrophile Coupling and 1,2-Dicarbofunctionalization of Olefins, ACS Catal., 2022, 12, 11216–11225 CrossRef CAS PubMed.
- X. Li, Y. Hu, Z. Huang, S. Zhu, F.-L. Qing and L. Chu, Metallaphotoredox-Catalyzed Three-Component Asymmetric Cross-Electrophile Coupling for Chiral Boronate Synthesis, ACS Catal., 2024, 14, 15790–15798 CrossRef CAS.
-
(a) W. G. Hollis, W. C. Lappenbusch, K. A. Everberg and C. M. Woleben, the use of Alkenylboronate Esters in [2+2] Enone-Olefin Photocycloadditions, Tetrahedron Lett., 1993, 34, 7517–7520 CrossRef CAS;
(b) S. C. Coote and T. Bach, Enantioselective Intermolecular [2+2] Photocycloadditions of Isoquinolone Mediated by a Chiral Hydrogen-Bonding Template, J. Am. Chem. Soc., 2013, 135, 14948–14951 CrossRef CAS PubMed;
(c) O. P. Demchuk, O. V. Hryshchuk, B. V. Vashchenko, A. V. Kozytskiy, A. V. Tymtsunik, I. V. Komarov and O. O. Gryorenko, Photochemical [2 + 2] Cycloaddition of Alkenyl Boronic Derivatives: An Entry into 3-Azabicyclo[3.2.0]heptane Scaffold, J. Org. Chem., 2020, 85, 5927–5940 CrossRef CAS PubMed.
- P. R. D. Murray, W. M. M. Bussink, G. H. M. Davies, F. W. van der Mei, A. H. Antropow, J. T. Edwards, L. A. D'Agostino, J. M. Ellis, L. G. Hamann, F. Romanov-Michailidis and R. R. Knowles, Intermolecular Crossed [2 + 2] Cycloaddition Promoted by Visible-Light Triplet Photosensitization: Expedient Access to Polysubstituted 2-Oxaspiro[3.3]heptanes, J. Am. Chem. Soc., 2021, 143, 4055–4063 CrossRef CAS PubMed.
- S. O. Scholz, J. B. Kidd, L. Capaldo, N. E. Flikweert, R. M. Littlefield and T. P. Yoon, Construction of Complex Cyclobutane Building Blocks by Photosensitized [2 + 2] Cycloaddition of Vinyl Boronate Esters, Org. Lett., 2021, 23, 3496–3501 CrossRef CAS PubMed.
- G. Roland, K. N. Hurdal, A. Bouchouicha, N. Dowe, R. L. Davis and S. K. Collins, [2 + 2] Photocycloadditions to Form Cyclobutanes and Bicyclo[2.1.1]hexanes Employing Copper-Based Photocatalysis, ACS Catal., 2024, 14, 11490–11497 CrossRef CAS.
- Y. Liu, D. Ni, B. G. Stevenson, V. Tripathy, S. E. Braley, K. Raghavachari, J. R. Swierk and M. K. Brown, Photosensitized [2+2]-Cycloadditions of Alkenylboronates and Alkenes, Angew. Chem., Int. Ed., 2022, 61, e202200725 CrossRef CAS PubMed.
- J. J. Molloy, J. B. Metternich, C. G. Daniliuc, A. J. B. Watson and R. Gilmour, Contra-Thermodynamic, Photocatalytic E→Z Isomerization of Styrenyl Boron Species: Vectors to Facilitate Exploration of Two-Dimensional Chemical Space, Angew. Chem., Int. Ed., 2018, 57, 3168–3172 CrossRef CAS PubMed.
- J. J. Molloy, M. Schäfer, M. Wienhold, T. Morack, C. G. Daniluc and R. Gilmour, Boron-enabled Geometric Isomerization of Alkenes via Selective Energy-Transfer Catalysis, Science, 2020, 369, 302–306 CrossRef CAS PubMed.
- J. J. Molloy, T. Morack and R. Gilmour, Positional and Geometrical Isomerisation of Alkenes: The Pinnacle of Atom Economy, Angew. Chem., Int. Ed., 2019, 58, 13654–13664 CrossRef CAS PubMed.
- For
selected examples, please see:
(a) P. Xiong, H. Long, J. Song, Y. Wang, J.-F. Li and H.-C. Xu, Electrochemically Enabled Carbohydroxylation of Alkenes with H2O and Organotrifluoroborates, J. Am. Chem. Soc., 2018, 140, 16387–16391 CrossRef CAS PubMed;
(b) H. Yan, Z.-W. Hou and H.-C. Xu, Photoelectrochemical C−H Alkylation of Heteroarenes with Organotrifluoroborates, Angew. Chem., Int. Ed., 2019, 58, 4592–4595 CrossRef.
|
| This journal is © the Partner Organisations 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  c,
Shengnan
Jin
c,
Daniele
Mazzarella
*d and
Zhong-Yan
Cao
c,
Shengnan
Jin
c,
Daniele
Mazzarella
*d and
Zhong-Yan
Cao