
Recent advances in carbon atom addition for ring-expanding single-atom skeletal editing
Ting
Yuan
and
Lei
Shi
 *
*
School of Chemistry and Chemical Engineering, School of Science (Shenzhen), Harbin Institute of Technology, Harbin, 150001, China. E-mail: lshi@hit.edu.cn
First published on 29th October 2024
Abstract
Ring architecture not only influences molecular properties, spatial arrangement, and scaffold rigidity but also determines molecular function and regulation. Compared with the traditional construction of ring frameworks that often requires starting from scratch, the development of methods for directly editing the ring skeleton lags far behind, which is largely attributed to the inertness of chemical bonds that constitute the frameworks. This review focuses on recent progress (post 2021) in the development of ring-expanding single-atom skeletal editing via ring expansion or carbon-atom insertion, enabling a more efficient and accurate synthetic strategy for the synthesis of important scaffolds in drug discovery and beyond, as well as its application in late-stage molecular transformations and streamlined synthesis of bioactive molecules.
1. Introduction
The function and activities of small molecules are inherently dictated by their structural attributes such as ring system complexity, heteroatom content, fraction of sp3-hybridized carbons (Fsp3) and number of stereogenic centers.1 Among these features, ring systems form the structural core of diverse small molecules and determine their basic shape and conformational flexibility, as well as the spatial orientation of substituents.2 Indeed, the importance of rings has been well understood by modern medicinal chemists, since they play a key role in molecular properties such as lipophilicity, polarity, molecular reactivity, metabolic stability, and toxicity.2a Therefore, it is important that the core ring systems in molecules should have the correct size or composition patterns (fusion and connection) if the desirable functional molecules are to be manufactured as efficiently as possible. To provide compound libraries that are enriched in bioactivities and facilitate scaffold hopping3 that make structural changes to existing compounds, pseudo-natural products (pseudo-NPs) and natural products (NPs), which are composed of privileged ring substructures (Scheme 1A), have been widely employed to rapidly explore large areas of chemical space for starting points of molecular design in the search for active ingredients for drug discovery.4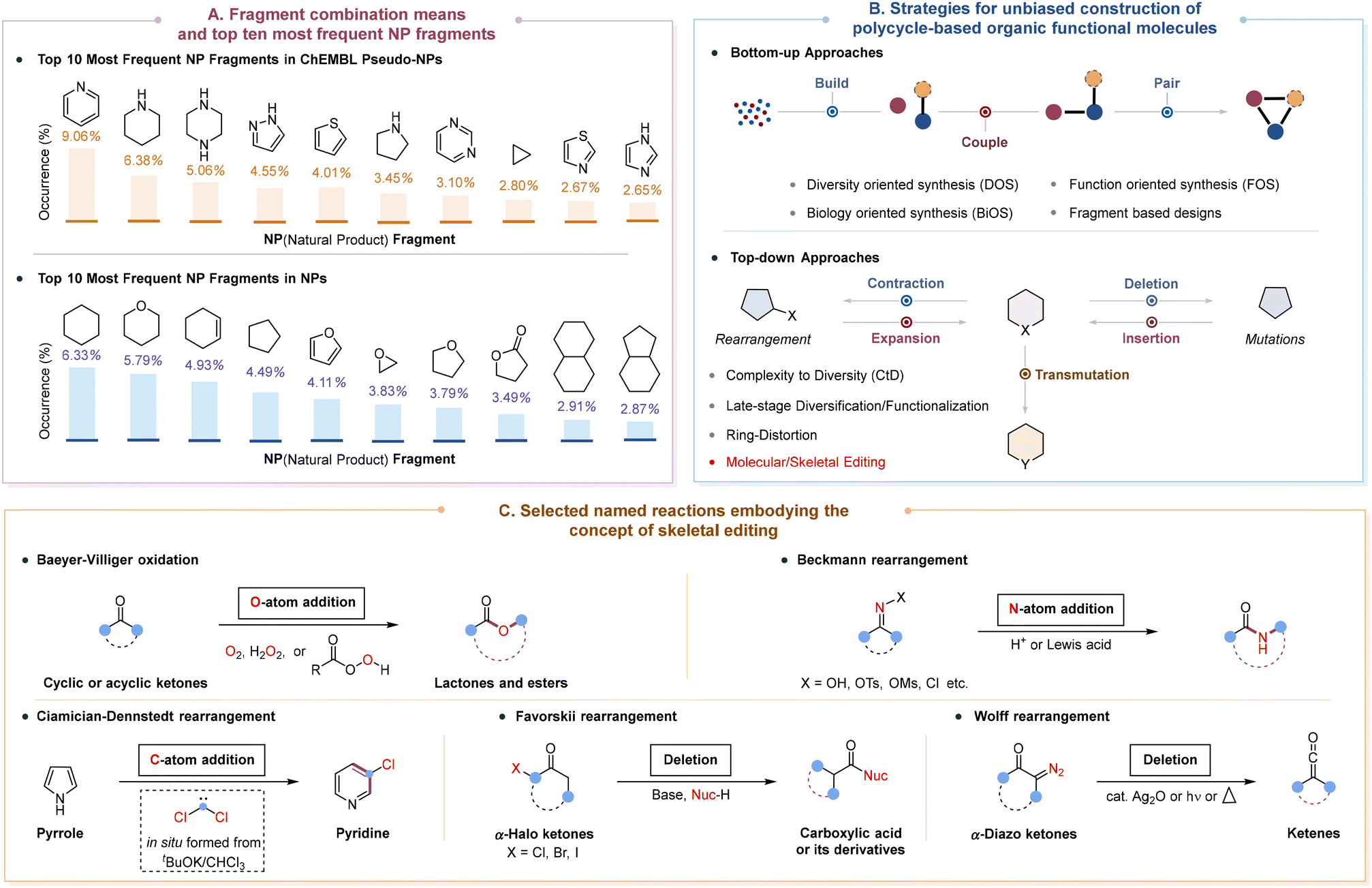 | ||
| Scheme 1 Background and concept. | ||
The past few decades have witnessed great achievements in efforts to create important functional molecules containing a diverse set of ring systems with various scaffold diversities and structural complexities in terms of bottom-up and top-down approaches (Scheme 1B).5 Generally, bottom-up strategies include diversity-oriented synthesis (DOS),6 function-oriented synthesis (FOS),7 biology-oriented synthesis (BiOS),8 and fragment based designs.9 Most of these efforts have a common feature to afford products with modular origins and chemically orthogonal handles via a build/couple/pair algorithm,10 which facilitates both systematic optimization and peripheral modification11 of the resulting product. On the other hand, top-down approaches are reminiscent of complexity-to-diversity (CtD)12 and late-stage functionalization,13 which usually take advantage of the molecular diversity and structural complexity inherent within attractive and abundant NPs and their derivatives to deliver markedly different core scaffolds that target NP-based underrepresented chemical space. Specifically, scaffold-hopping of NPs and the corresponding advanced synthetic intermediates could be achieved through attachment of an additional ring, ring cleavage, ring expansion/contraction, ring fusion, and/or ring rearrangement. However, the toolbox of traditional reactions utilized in this process is often limited by their compatibility and generality with existing functional groups, which in turn restricts access to the desired skeletal diversity and complexity in a screening collection of drug-like small molecules. Moreover, the limited options for inspection may result in an oversaturation of certain molecular structures within pharmaceuticals.14
Organic synthesis remains the most time-intensive step in drug-discovery campaigns, because of the requirement to rapidly increase the molecular diversity and complexity of compound libraries so as to maximize the coverage of the chemical space.14a The direct and specific modification of lead compounds at desired sites in any desired order allows the creation of new analogues for rapid structure–activity relationship exploration based on subtle or dramatic changes in the molecular structure, exhibiting an advantageous step and atom economy without resorting to de novo synthesis. In this regard, molecular editing15 has emerged as an attractive and straightforward strategy to selectively prune, insert, replace, or modify atoms or functional groups in a given molecular scaffold or pharmacophore, offering much easier and more flexible molecular design to enable the synthesis of innumerable chemical products that were previously out of reach. According to the characteristics of transformations, molecular editing can be generally categorized into peripheral editing and skeletal editing.16 Peripheral editing involves the direct and site-selective introduction of a variety of functional groups onto the periphery of the molecule without altering the molecular skeleton, with late-stage C–H functionalization serving as a representative example.17 In contrast, skeletal editing, which is in its infancy, enables the controlled and precise modification of aromatic or non-aromatic rings in the core of molecules, particularly by inserting, deleting, or switching a number of heavy atoms (Scheme 1B), typically including single-atom editing, multi-atom editing, ring insertion, atom transmutation, cut and sew reaction, etc.16,18 Notably, skeletal editing can facilitate the reorganization and refinement of underlying molecular skeletons during the late stages of synthesis and expedite the discovery of novel bioactive molecules by exploring an otherwise difficult-to-access chemical space. However, the implementation of skeletal editing reactions has been severely hindered by their non-intuitive retrosynthetic logic and has suffered from a lack of efficiency and generality. In particular, the current limitations of the available skeletal editing transformations are the employment of highly reactive reagents, inevitably generating undesirable by-products and thus requiring more intricate purification procedures, as well as the necessity for additional molecular fragments, shielding fragile functional groups during the transformative process.
According to the definition proposed by Sarpong and Levin in 2022,16a single-atom skeletal editing transformations can be categorized into three strategies in principle: rearrangements (expansion or contraction) and mutations (insertion or deletion) that enable the conversion of an n-membered ring directly into an n + 1 or n − 1 analogue,19 as well as atom transmutations that exchange one of the constituent atoms inside a ring system without altering the ring size.20 Historically, a limited number of named reactions to reshape molecular architectures, such as Baeyer–Villiger oxidation (1899),21 Beckmann rearrangement (1886),22 Ciamician–Dennstedt rearrangement (1881),23 Favorskii rearrangement (1894),24 and Wolff rearrangement (1902),25 have emerged as the original explorations for single-atom skeletal editing since the late nineteenth century (Scheme 1C), most of which were usually operated under harsh conditions or with narrow substrate scopes. In the past decades, a variety of well-known rearrangement reactions (e.g., Meerwein–Wagner, Semipinacol, Dowd–Beckwith, and Achmatowicz rearrangements) have already been extensively explored to access certain natural product frameworks and their analogues through single-atom manipulation for realizing skeletal reorganization of easily accessible molecules,26 wherein the atom being edited is well retained in the final core scaffold. Meanwhile, the direct interrogation of single-atom insertions and deletions of the constitutive elements of ring systems has still not reached the same level of precision and control over molecular structures, despite receiving increasing interest to make precise alterations to the connectivity of the molecular framework.
Selective ring-expanding single-atom skeletal editing, particularly involving the addition of a carbon atom, is becoming increasingly common as one of the most fundamental specific point changes to a molecular skeleton, leading to the formation of a new ring with one carbon atom more than in the original molecule without the requirement of substrate bias or high dilution. Importantly, this modification could enlarge an existing cyclic scaffold due to its ability to enable non-obvious retrosynthetic disconnections through controlled rearrangement and insertions at the single-atom precision. As a promising strategy to improve synthetic efficiency and structural flexibility, a systematic review of ring-expanding single-atom skeletal editing through the addition of a carbon atom could reveal critical success factors, such as stability factor and thermodynamic factor, as well as provide mechanistic insights and synthetic applications for their further design and application. Thus, we herein highlight the state-of-the-art protocols for ring-expanding single-atom skeletal editing via ring expansion or carbon-atom insertion with high site selectivity from January 2021 to September 2024, emphasizing the influence of the reaction conditions and the utilization of a wide range of structurally unusual species. It is instructive to classify these transformations by the type of skeleton being inserted together with a proper consideration for deciphering reaction mechanisms, and this review has been organized into two main sections: carbon atom addition into carbocyclic scaffolds and carbon atom addition into heterocyclic scaffolds. Important information including the reaction scheme of ring expansion or carbon-atom insertion, rational generation of possible reaction intermediates and their “mode-of-action” analysis, as well as late-stage functionalization and the resultant natural products, is depicted, showcasing their ever-growing importance and potential for diversifying the development of complexity-building reactions available to the synthetic community. While the selection presented is by no means a comprehensive coverage of this subject, our motivation to compose this review is to provide a useful overview and integrated perspective to imbue understanding of ring-expanding single-atom skeletal editing that can both enable the efficient and practical creation of most envisioned target structures and enrich the synthetic toolbox of chemical modification methods for drug discovery.
2. Carbon atom addition into carbocyclic scaffolds
The homologation of ketones with diazo compounds served as one of the most efficient strategies for extending the one-carbon chain of acyclic ketones or for expanding the ring structure of cyclic ketones.27 Feng and co-workers reported an efficient enantioselective catalytic homologation reaction of acyclic ketones or cyclic ketones with α-alkyl α-diazo esters to synthesize chiral β-keto esters with excellent regioselectivity and enantioselectivity by using chiral scandium(III) N,N′-dioxide as the Lewis acid catalyst in 2021. Density functional theory (DFT) calculations showed that the chiral N,N′-dioxide–Sc(OTf)3 complex served as the pivotal factor in activating substrates and precisely controlling the stereoselectivity of the addition/rearrangement process, and alkyl migration was the rate-determining step (Scheme 2A).28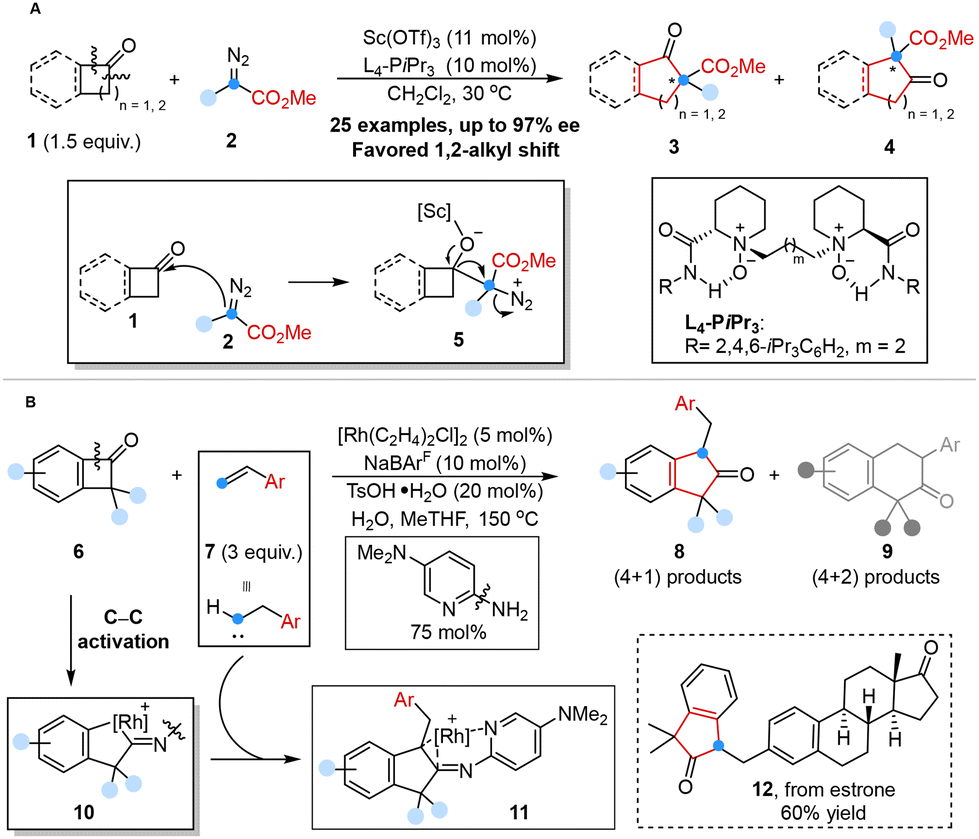 | ||
| Scheme 2 One-carbon homologation reaction with diazoalkanes reported by Feng (A); reaction of BCBs with olefins reported by Dong (B). | ||
Despite significant advances in novel Lewis acid catalysis described above, the regioselective insertion of a carbon atom into benzocyclobutenones remained a challenge,27d,29 including the potential explosiveness and toxicity of diazoalkanes and over-homologation of ring-expanding products.30
In 2022, Dong and co-workers proposed a unique one-carbon insertion strategy to obtain 1,1,3-trisubstituted 2-indanones from benzocyclobutenones (BCBs) via rhodium-catalyzed formal (4 + 1) cycloaddition using styrene-type alkenes as a less hazardous carbenoid equivalent. DFT studies revealed that the β-H elimination pathway was overall kinetically more favorable than the direct reductive elimination pathway, which contributed to the origin of the (4 + 1) selectivity. Notably, the employment of a cationic ‘ligandless’ rhodium catalyst served as the pivotal factor in obtaining high reactivity and achieving high selectivity of the (4 + 1) products (Scheme 2B).31
The well-known Büchner reaction,32 characterized by the elegant sequence of carbene addition, dearomative cyclopropanation, and subsequent transformation of norcaradienes into cycloheptatrienes via 6π electrocyclic opening, stood as an exceptionally valuable synthetic approach.33 However, it is only applicable to monocyclic arenes because of its inherent reactivity directing cyclopropanation to the 1,2-position, which could not readily undergo further ring expansion to obtain benzofused cycloheptatrienes.34,35
In 2022, Sarlah and co-workers optimized such a transformation and disclosed a two-step sequential strategy involving arenophile-based dearomative [4 + 2]-cycloaddition, Pd-catalyzed cyclopropanation and cycloreversion-initiated ring expansion, which successfully converted simple and non-activated polycyclic arenes to benzocycloheptenes (Scheme 3).36 This approach involved cyclopropanation and ring expansion at the 2,3-site positions of arenes, resulting in a Büchner-like reaction to achieve (aza)benzocycloheptatrienes that are common structural motifs in natural products and biologically active compounds.
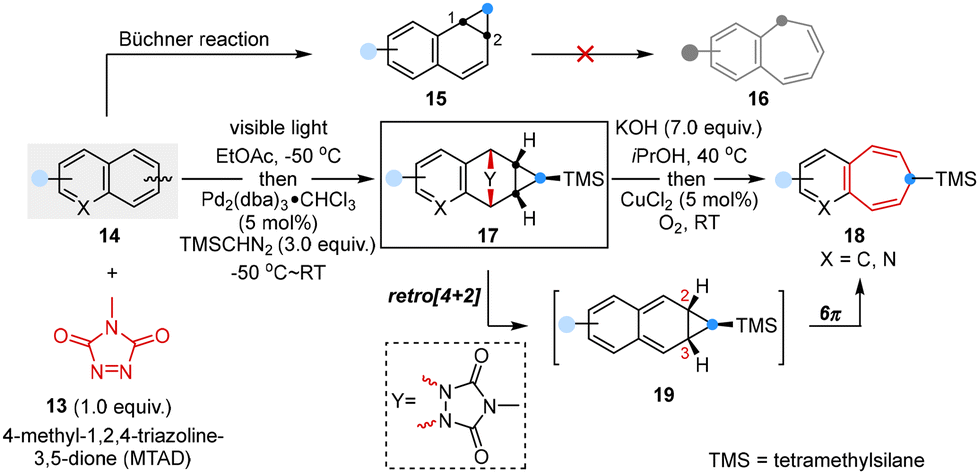 | ||
| Scheme 3 Ring expansion of arenophile-mediated ring expansion reported by Sarlah. | ||
Carbynyl cation equivalent reagents, including α-halo diazoalkanes37 and α-chlorodiazirines,38 have been commonly used for recent advancements in single-atom skeletal editing, specifically in the conversion of indoles to quinoline systems as we will discuss below. Conversely, the application of carbynyl radical equivalents in such reactions has been scarce, with minimal attention being given to the ring expansion of indenes to produce naphthalenes.
Inspired by this, Glorius and co-workers developed a photoredox-enabled ring expansion reaction in 2024, facilitating the insertion of functionalized carbon atoms into indenes, which is difficult to directly achieve by conventional methods. The employment of α-iodonium diazo compounds as masked carbyne equivalents and the simultaneous implementation of photoredox catalysis served as pivotal factors in realizing rapid construction of a library of 2-substituted naphthalenes. DFT studies have demonstrated a possible radical chain pathway along with cationic cyclization, which was likely to commence with the initial addition of a diazomethyl radical to indenes (Scheme 4).39
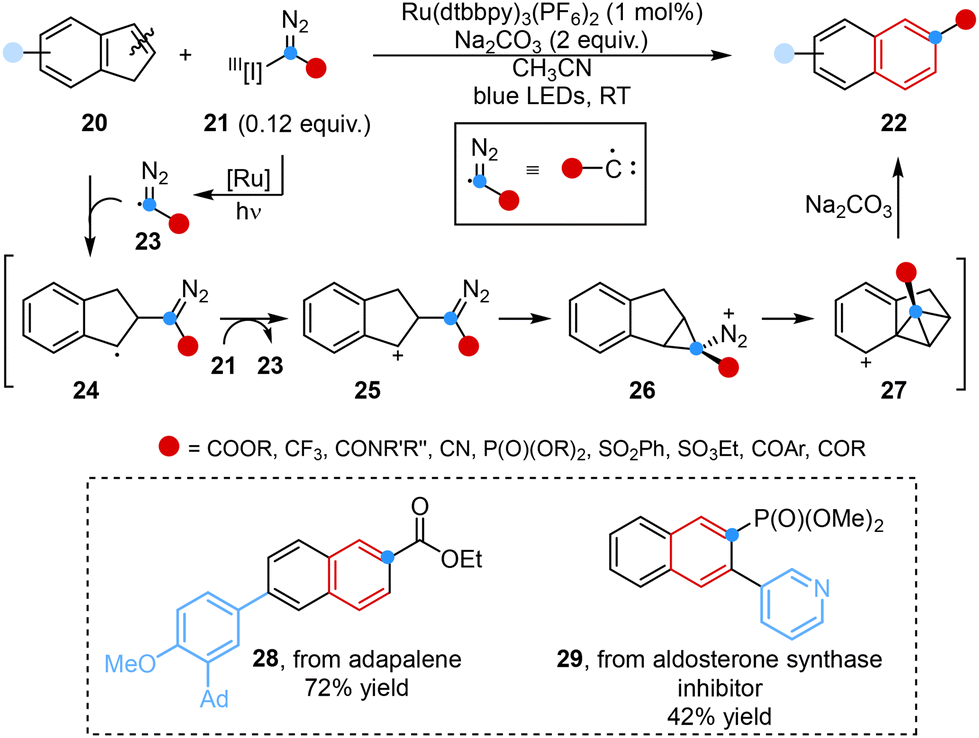 | ||
| Scheme 4 Reaction of indenes with α-iodonium diazo compounds reported by Glorius. | ||
3. Carbon atom addition into heterocyclic scaffolds
3.1 Carbon atom insertion into the C–C bond of pyrroles or indoles
Inspired by the mechanism of the Ciamician–Dennstedt rearrangement,23,40 Levin and co-workers achieved a notable breakthrough in 2021. They disclosed a method for the one-carbon ring expansion of indoles or pyrroles into quinolines or pyridines, employing chlorodiazirines as alternative, isolable and stable carbene precursors under mild thermolytic conditions (Scheme 5A).38a Chlorodiazirines could be prepared in a single step via Graham oxidation of amidinium salts.41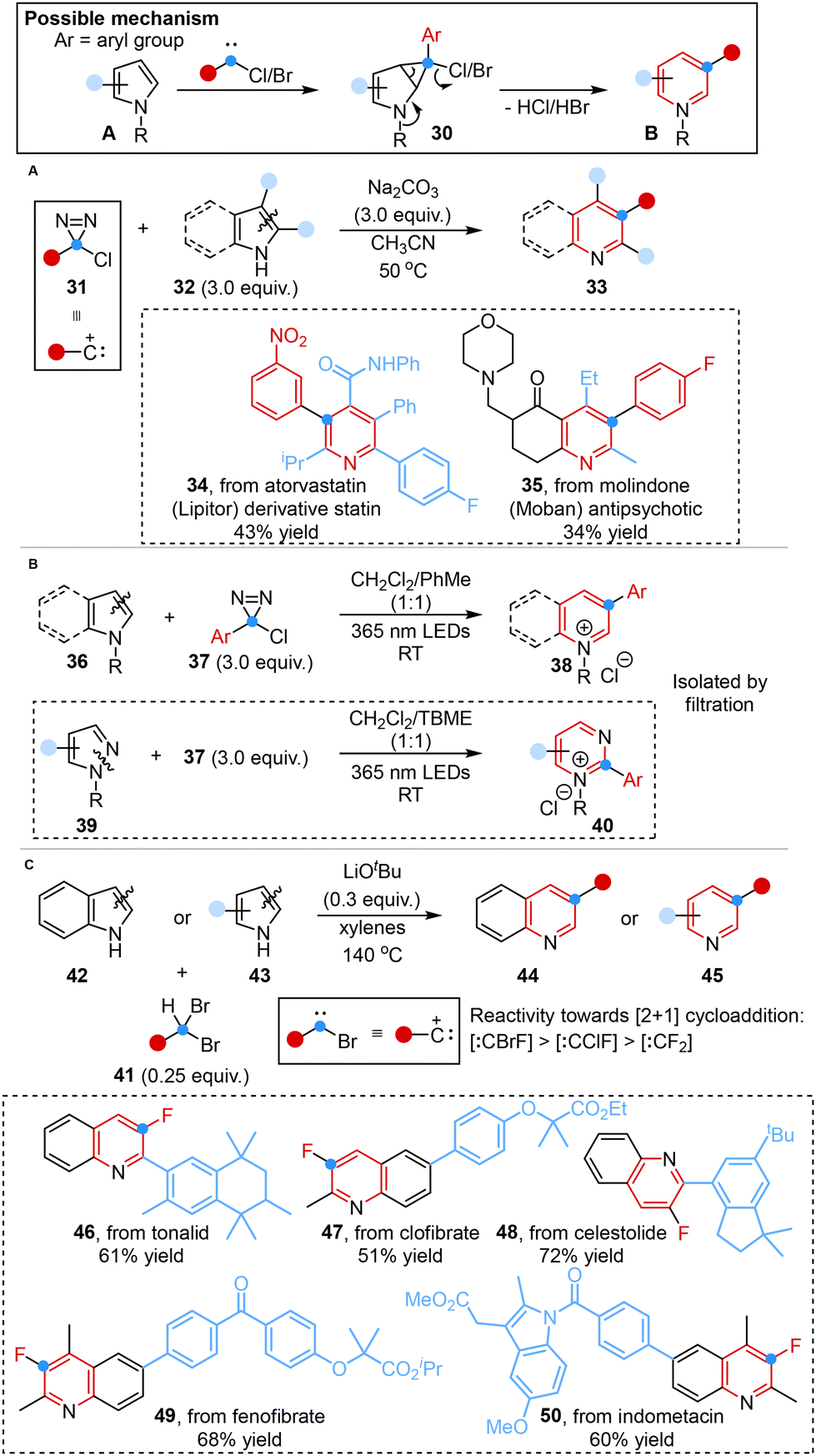 | ||
| Scheme 5 Carbon atom insertion into the C–C bond of heterocycles reported by Levin (A), Ball (B) and Xu (C). | ||
Despite its broad utility, the further application and practical implementation were limited by the fact that carbene reacts with chloride anion co-products or quinoline products42 and the low yields of 2-unsubstituted indoles. In light of this, Ball and co-workers developed an applicable method for achieving such ring expansion by using arylchlorodiazirines as photo-activated halocarbene precursors to afford pyridinium and quinolinium salts in high yields, which could be readily isolated by filtration, eliminating the necessity for a 2-substituent. Besides, pyrimidinium salts were also successfully obtained from N-substituted pyrazoles under standard conditions with tert-butyl methyl ether (TBME) as the solvent. The N-substituent of the substrates contributed to preventing product degradation, suppressing co-product inhibition and activating the azinium products for subsequent synthetic manipulations (Scheme 5B).42a
More recently, Xu and co-workers demonstrated a similar transformation to synthesize 3-fluorinated quinolines and pyridines under an air atmosphere in only 10 minutes using bench-stable 1,1-dibromoalkanes as versatile bromocarbene sources. This reaction featured the easy in situ formation of bromocarbene intermediates, facilitated by the presence of acidic C–H bonds and relatively weak C–Br bonds. Furthermore, the higher reactivity of bromofluorocarbene, in comparison with chlorofluorocarbene and difluorocarbene, stemmed from the fact that bromine had diminished ability to stabilize the carbene center (Scheme 5C).42b
These transformations involved the in situ generation of an electrophilic carbene intermediate that bore a suitable leaving group, overcoming the limitations of poor yield and low functional group tolerance in the Ciamician–Dennstedt rearrangement.
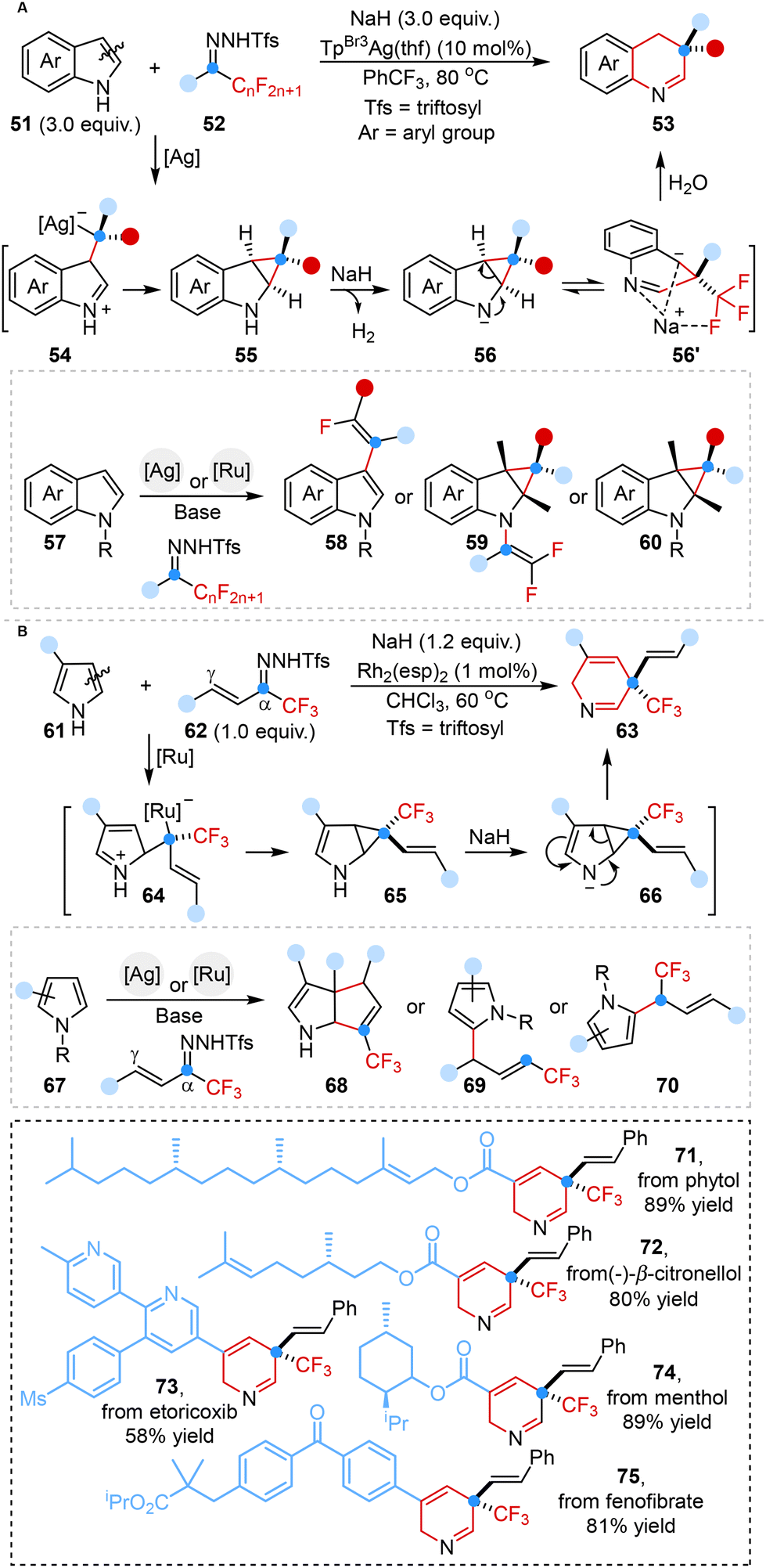
In 2024, Liu, Bi and co-workers disclosed a controllable molecular editing strategy ingeniously leveraging fluoroalkyl N-triftosylhydrazones as electrophilic fluoroalkyl carbene precursors, enabling both skeletal and peripheral editing to produce various fluorinated N-heterocyclic frameworks (Scheme 6A).43a Mechanistic investigations elucidated that the ring expansion process began with the attack of electrophilic metal carbene on the C3 site of indoles to generate intermediate 54, followed by the formation of the cyclopropane intermediate 55 under an [Ag]–NaH catalytic system via 2,3-cyclopropanation. Subsequently, the intermediate 55 was converted to intermediate 56 by using NaH as a hydrogen atom abstraction agent, followed by reversible ring opening and water-assisted protonation to ultimately deliver ring-expanding products.
 | ||
| Scheme 6 Carbene-initiated tunable skeletal and peripheral editing reported by Liu and Bi. | ||
The same group further advanced their innovative endeavor by devising a similar tunable synthetic strategy tailored for the synthesis of medicinally relevant CF3-containing N-heterocyclic analogues derived from pyrroles (Scheme 6B).43b This groundbreaking strategy harnessed trifluoromethyl vinyl N-triftosylhydrazones as versatile vinylcarbene precursors. Mechanistic studies revealed that the controllability of skeletal and peripheral editing depended on the electronic effects and/or non-covalent interactions of pyrrole substituents.
The implementation of tunable carbene-initiated single-atom skeletal editing methodologies significantly streamlined the synthetic routes for the expeditious construction of fluorine-containing and CF3-containing N-heterocyclic scaffolds.
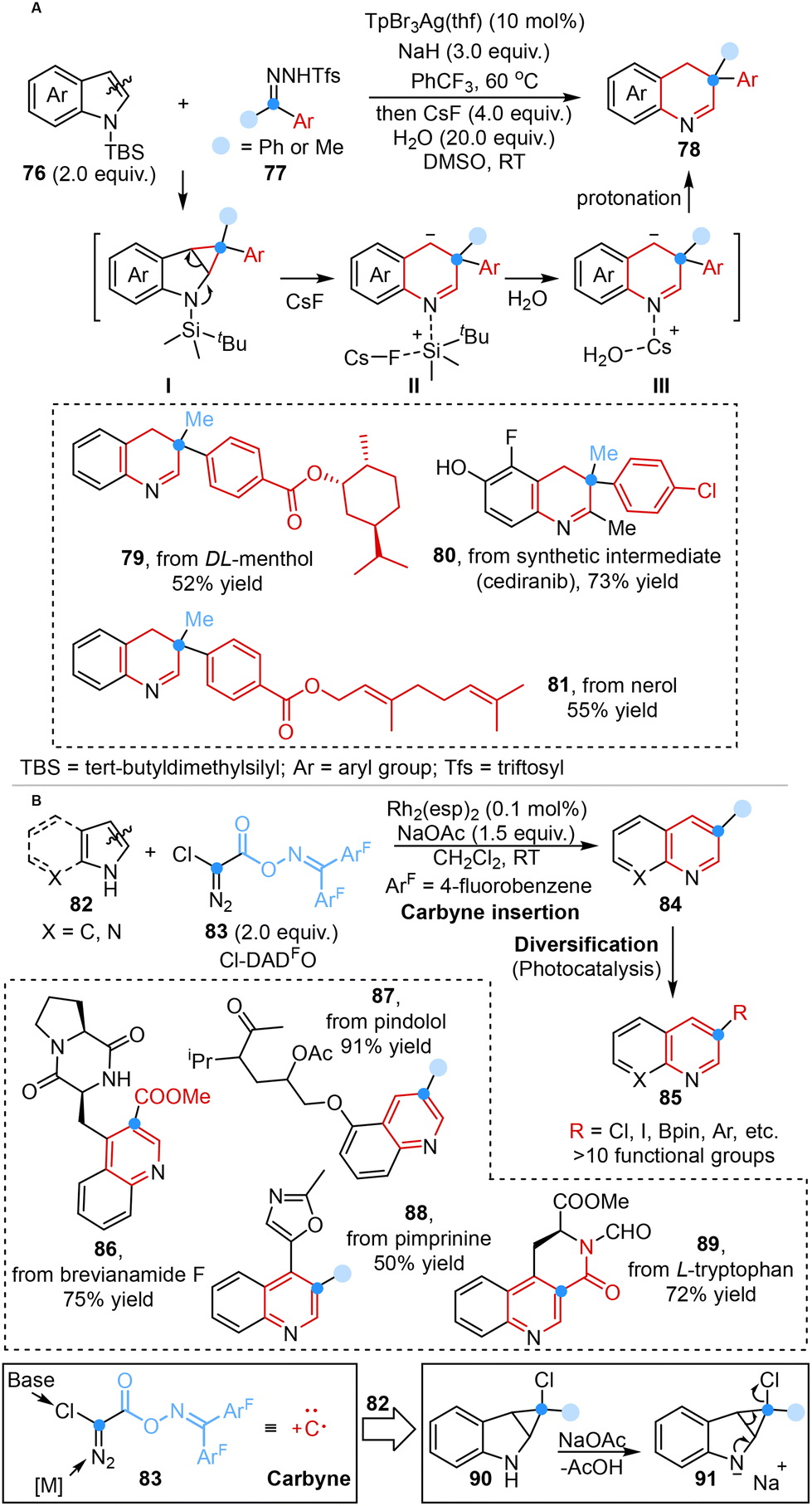
More recently, inspired by the methodologies mentioned above, Bi, Zhang, Nenajdenko and co-workers developed a one-pot, two-step silver-catalyzed ring expansion reaction for the rapid construction of dearomative quinoline frameworks employing N-triftosylhydrazones as masked donor–donor carbene precursors. This single carbon insertion strategy involved the formation of silver carbene, electrophilic attack, cyclopropanation, addition of CsF, atom-pair swap and protonation processes. Mechanistic studies and DFT simulations showcased the transient presence of a cyclopropane intermediate (I), which underwent removal of protecting groups and protonation processes in the presence of H2O to yield the desired product (Scheme 7A).44a
 | ||
| Scheme 7 Carbene-initiated ring expansion reaction reported by Bi, Zhang and Nenajdenko (A) and carbene-initiated ring expansion reaction reported by Glorius (B). | ||
In the same year, Glorius and co-workers disclosed the design and synthesis of the atomic carbon equivalent Cl-DADO, and its application in single-atom skeletal editing to form ring-expanding products via carbyne insertion. This innovative method showcased that the novel Cl-DADFO was capable of directly inserting a carbon atom into indole and pyrrole scaffolds and simultaneously enabling the formation of an additional bond at the carbon center, resulting in 3-functionalized ring-expansion products, which subsequently underwent versatile transformations under orthogonal reaction conditions to form diverse derivatives (Scheme 7B).44b
3.2 Carbon atom insertion into the N–X (X = N, O, and S) bond of heterocycles
The carbene-mediated approach devised by Levin also offered a feasible pathway for the structural transformations of pyrazoles or indazoles into pyrimidines or quinazolines by adding a carbon atom through N–N cleavage. Comprehensive mechanistic investigations, complemented by DFT studies, have collectively validated a mechanistic pathway encompassing the fragmentation of pyrazolium ylides and cyclization of the ring-opened diazahexatriene intermediate. This sequence of processes culminated in the formation of a new diazine core, thereby supporting the efficacy of the proposed carbene-mediated strategy (Scheme 8A).38b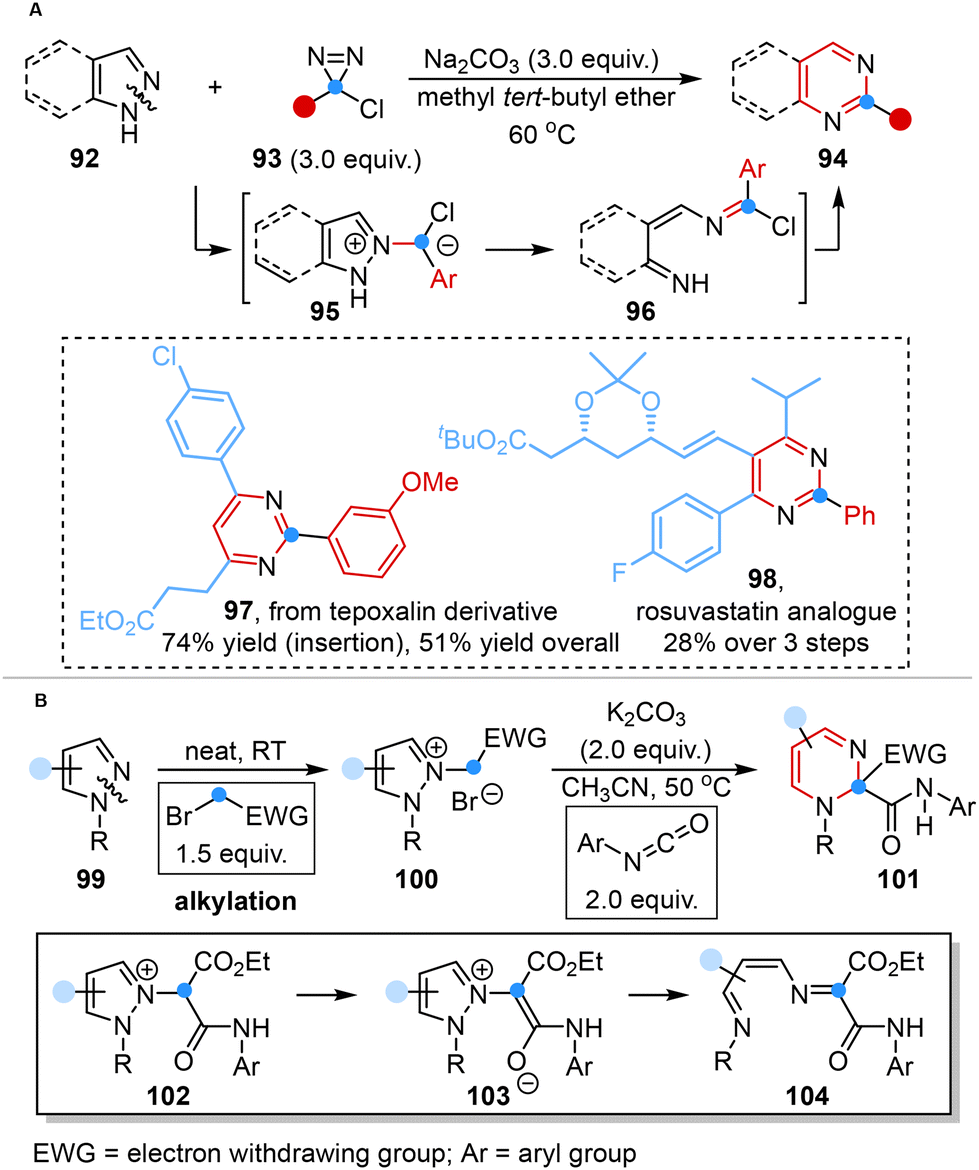 | ||
| Scheme 8 Carbon atom insertion into the N–N bond of heterocycles reported by Levin (A) and Derksen (B). | ||
In a parallel vein, Derksen and co-workers reported metal-free carbon atom insertion into the N–N bond of five-membered heterocyclic skeletons to obtain 1,2-dihydropyrimidines via the rearrangement of pyrazolium halide salts in the presence of isocyanates without harsh reaction conditions (high temperatures and strong bases) (Scheme 8B).45
In 2022, Lu, Wang and co-workers reported a one-carbon ring expansion reaction for the preparation of spiro[isoquinoline-4,2′-[1,3]oxazin]-3-ones from isoxazoles. This method featured the employment of tetrakis(acetonitrile) copper(I) hexafluorophosphate as a catalyst and 4-diazoisoquinolin-3-ones as copper carbene precursors (Scheme 9A).46a
 | ||
| Scheme 9 Carbon atom insertion into the N–X (X = N, O, and S) bond of heterocycles reported by Lu and Wang (A), Bi (B and C), and Nakamura (D). | ||
More recently, Bi and co-workers made a breakthrough in such analogous transformations that employed fluoroalkyl N-triftosylhydrazones as readily available and low-temperature accessible fluoroalkyl carbene precursors, enabling the efficient and versatile synthesis of a diverse array of ring-expanding products. DFT calculations elucidated that the cornerstone of success in this approach lay in the initial generation of an electrophilic Rh-carbene, followed by Rh-coordinated pyrazolium ylide formation and subsequent N–X (X = N, O, and S) bond cleavage, leading to the corresponding products (Scheme 9B).46b The same group also introduced an efficient silver carbene-mediated dearomative N–N cleavage method of indazoles (Scheme 9C).46c
In a parallel endeavor, Nakamura and co-workers developed a one-carbon ring expansion strategy for the precise and efficient synthesis of six-membered heterocycles via zinc carbenoid insertion into the N–X (X = N, O, and S) bond of five-membered heterocycles (Scheme 9D).46d DFT studies illuminated the mechanism wherein the zinc carbenoid was generated from a reliable and reproducible iodomethylzinc reagent (EtZnCH2I), triggering a cascade of reactions, encompassing subsequent N-alkylation, N–X bond cleavage and cyclization.
Apart from the aforementioned representative works, other transformations aimed at synthesizing quinazolinones through the insertion of a carbon atom into indazole skeletons were also worthy of discussion.
In 2023, Song and co-workers first disclosed a unique difluorocarbene-promoted one-carbon insertion of 2H-indazoles for the construction of quinazolin-4(3H)-ones. This innovative approach harnessed ethyl bromodifluoroacetate as a difluorocarbene source, marking its unprecedented utilization in the realm of single-atom skeletal manipulation within carbocyclic and heterocyclic frameworks (Scheme 10A).47a This reaction cascade encompassed several key steps: (1) difluorocarbene generation, (2) imine carbocation formation, (3) H2O attack, (4) intramolecular nucleophilic attack, and (5) base-assisted defluorination. Notably, an alternative mechanistic route was also elucidated, featuring the formation of an intermediate bearing an N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CFH moiety because of the instability of the NH–CF2H group and ultimately yielding the desired product as well (path b).
CFH moiety because of the instability of the NH–CF2H group and ultimately yielding the desired product as well (path b).
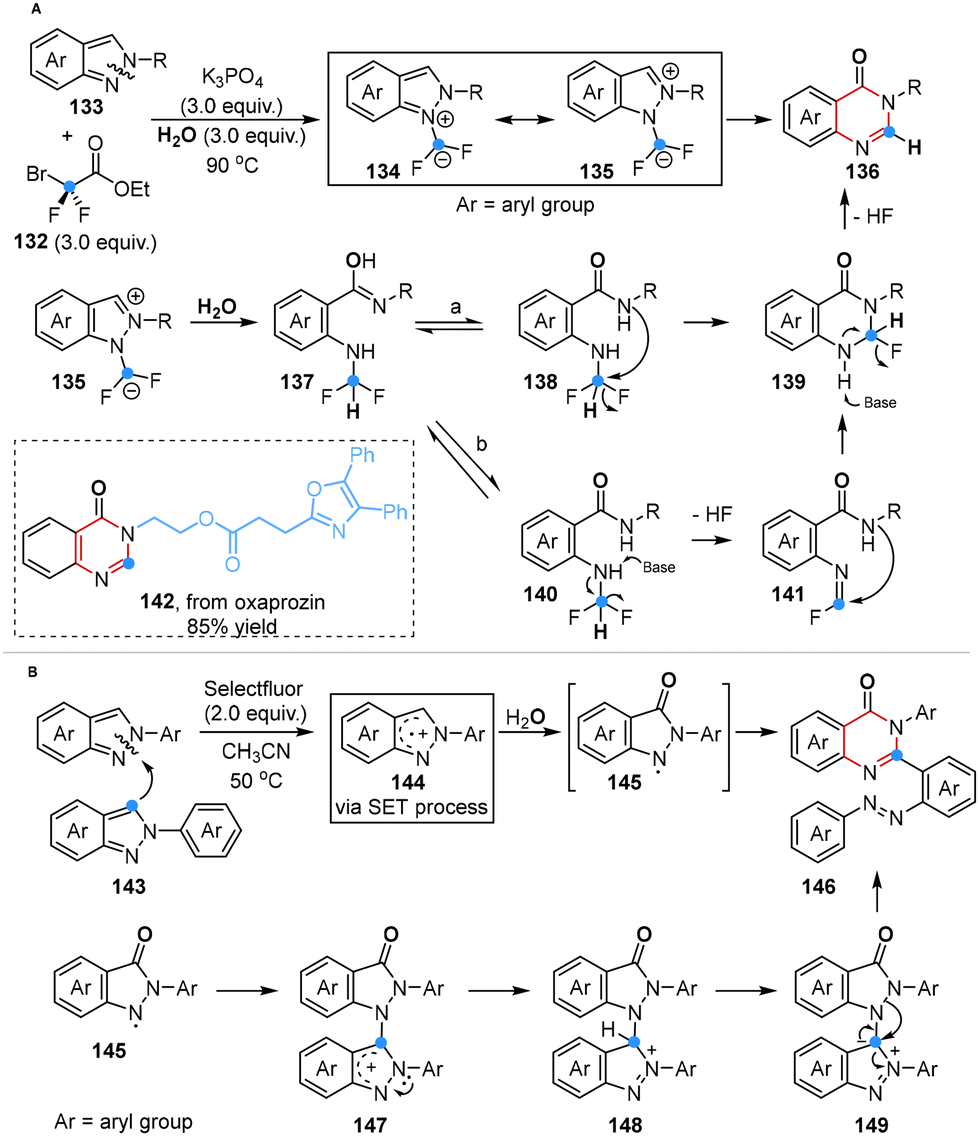 | ||
| Scheme 10 Ring expansion of indazoles to quinazolinones reported by Song (A) and Hajra (B). | ||
In addition, Hajra and co-workers reported, for the first time in 2023, a Selectfluor-mediated one-carbon insertion of 2H-indazoles through the utilization of two equivalents of indazoles, with one serving as the carbon source. Remarkably, the substrate underwent a single electron transfer (SET) process to yield a radical cation intermediate serving as a C-atom insertion reagent, which could be further oxidized to form an N-centered indazolone radical intermediate using H2O as an O atom source. Subsequently, these two intermediates rapidly reacted with each other, efficiently yielding azo-linked-2,3-disubstituted quinazolinones (Scheme 10B).47b
3.3 Carbon atom addition into the C–X (X = N and C) bond of heterocycles
In 2021, Yoo and co-workers introduced a regioselective one-carbon ring expansion reaction for the rapid synthesis of 4-substituted azepines by using a silver catalyst and diazoacetates. Mechanistic studies suggested that this reaction was successfully achieved through the regioselective 1,4-dearomative addition of diazoacetates with substrates and the generation of silver carbenoids (Scheme 11A).48a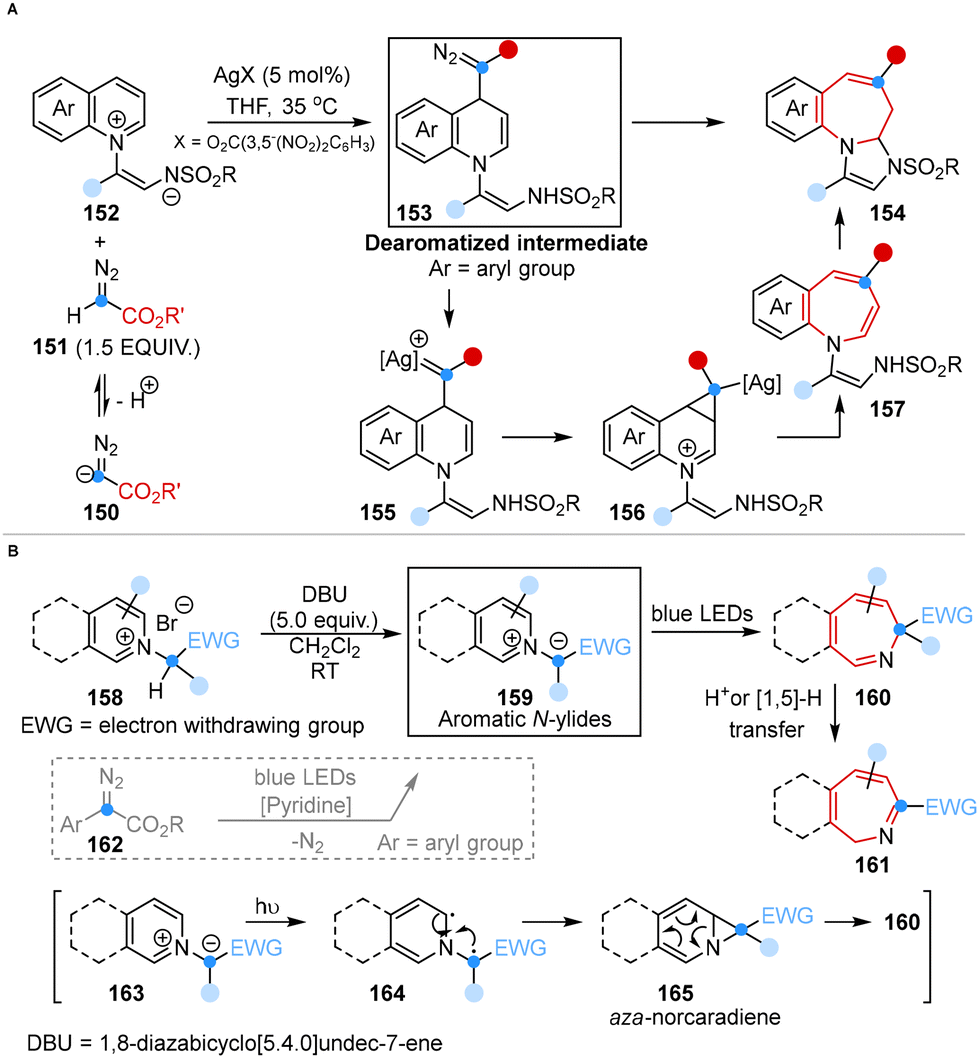 | ||
| Scheme 11 Ring expansion of aromatic N-heterocycles to azepines reported by Yoo (A) and Beeler (B). | ||
In the same year, Beeler and co-workers developed a photo-induced dearomative ring expansion of aromatic N-ylides to synthesize azepines, eliminating the need for an external diazo compound. This method featured the photo excitation of the ylides and diradical recombination 6π-electrocyclic ring opening. The application of N-ylides as substrates avoided the formation of carbene by-products, which are commonly generated under diazo conditions with pyridine (Scheme 11B).48b
These methods provided an easy-to-handle and convenient synthetic paradigm for the ring expansion of aromatic N-heterocycles, offering a robust route for the synthesis of fused azepines containing bioactive motifs.
In 2022, Singh and co-workers reported a novel one-pot, two-step Lewis acid-mediated one-carbon homologation reaction of isatins with para-quinone methides to provide 2-quinolinone-derived para-quinone methides in high yields and regioselectivity. DFT simulations illuminated the mechanistic pathway wherein isatins and para-quinone methides underwent P(NMe2)3-mediated [1,2]-phospha-Brook rearrangement to form a cyclopropane intermediate, followed by Lewis acid-mediated cyclopropane ring opening and subsequent ring expansion, resulting in the desired products via 1,2-aryl migration (Scheme 12).49
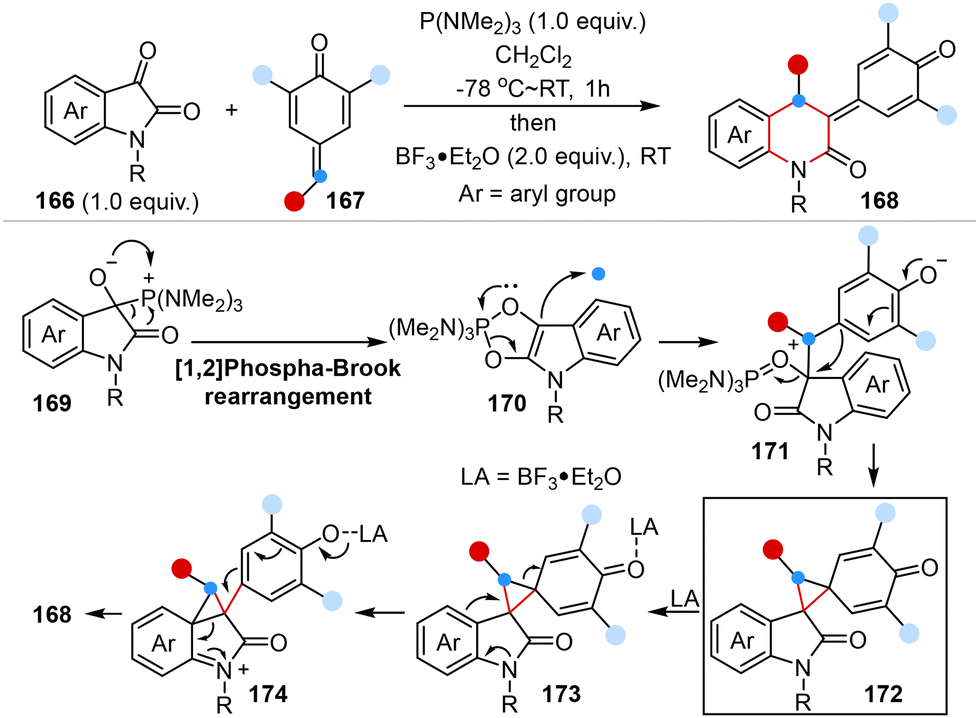 | ||
| Scheme 12 Lewis acid-mediated one-carbon homologation reaction reported by Singh. | ||
The introduction of an amine group into heteroaromatic systems represented a significant synthetic challenge and 3-aminated quinolin-2(1H)-ones are building blocks of pharmaceutically relevant compounds.50 In this regard, Roy and co-workers disclosed a light-induced regioselective C-3 amination reaction of 3-ylideneoxindoles to obtain 3-amino quinolin-2(1H)-ones using TMSN3 as the aminating agent in 2023. Mechanistic studies showed that an azide anion (N3−), which originated from the interaction of TMSN3, H2O and 4-dimethylaminopyridine (DMAP), reacted with a substrate to generate a triazoline intermediate via [3 + 2] cycloaddition. Subsequently, a SET process triggered the formation of the corresponding radical intermediate under light irradiation, followed by ring expansion, denitrogenation, protonation and enolization processes, leading to the desired products (Scheme 13).51
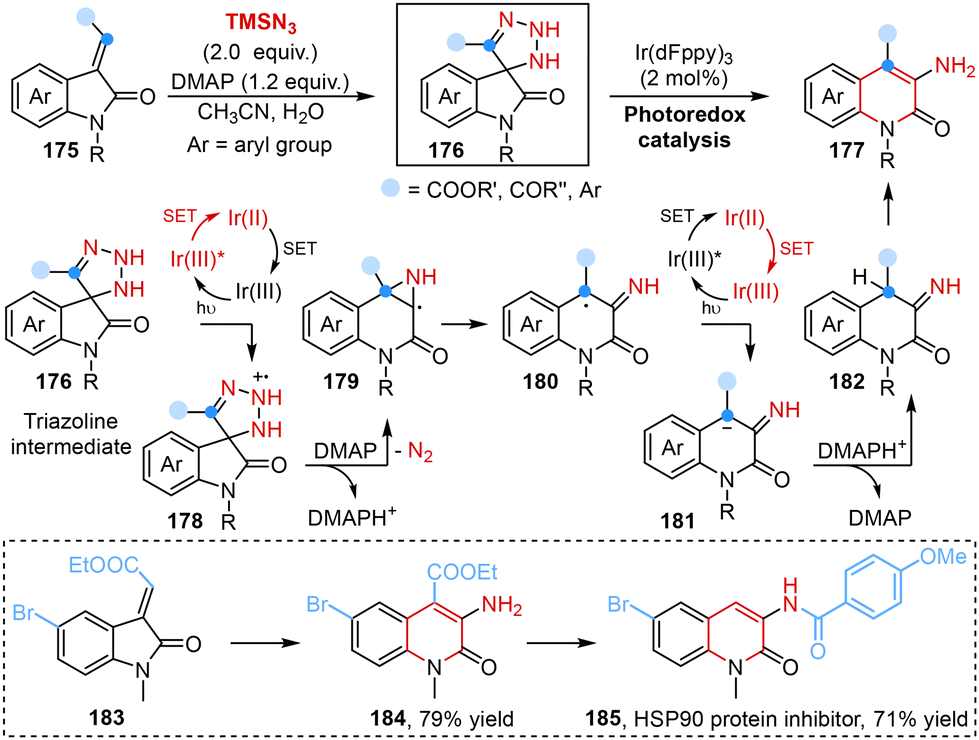 | ||
| Scheme 13 Light-induced ring expansion of 3-ylideneoxindoles to yield 3-amino quinolin-2(1H)-ones reported by Roy. | ||
More recently, Morandi and co-workers developed a rare regiodivergent ring expansion to obtain two quinolinone isomers from oxindoles under two distinct reaction conditions. Two possible mechanisms involving the formation of a highly energetic cationic isocyanate intermediate or a cyclopropyl alcoholate intermediate were proposed to elaborate this transformation (Scheme 14).52 Utilizing this unique regiodivergent strategy enabled late stage diversification of bioactive oxindoles and also provided an alternative and concise approach for the synthesis of a quinolinone drug (4-substitued) and its regioisomer (3-substitued) under lithium-bis-(trimethylsilyl)amide (LiHMDS)-mediated conditions and AgBF4 conditions, respectively.
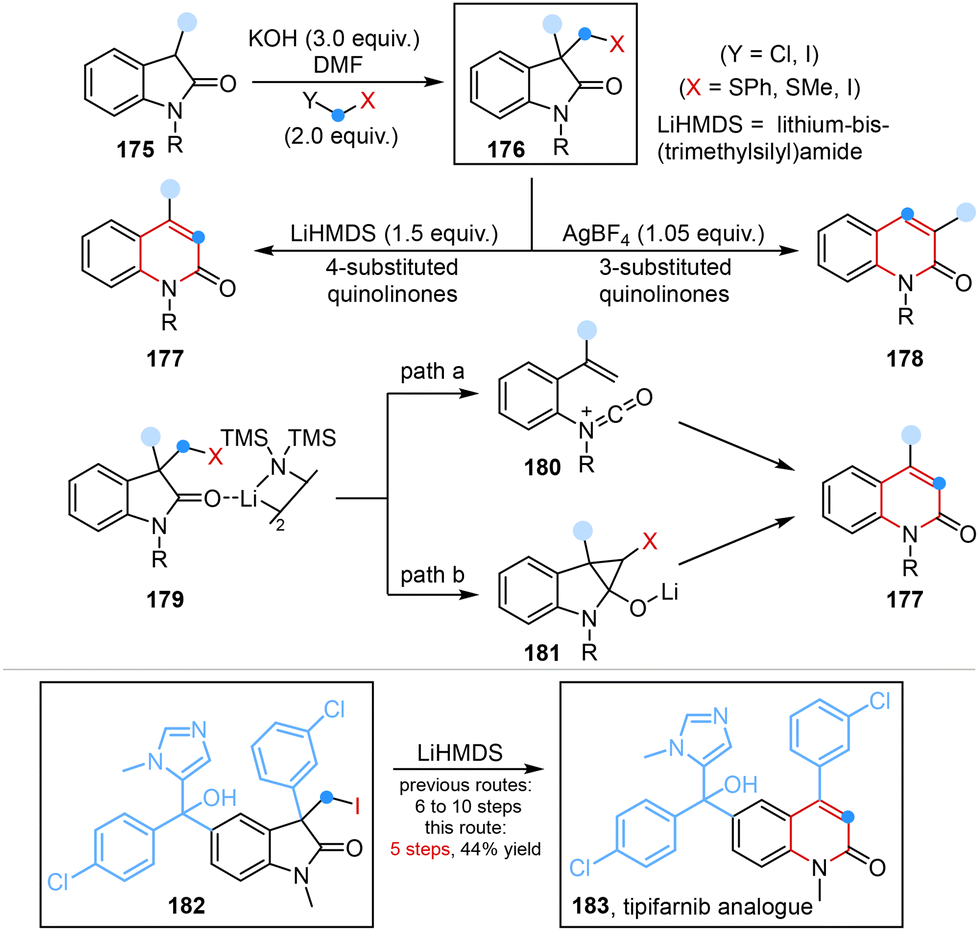 | ||
| Scheme 14 Regiodivergent ring expansion of oxindoles to access quinolinones reported by Morandi. | ||
In 2024, Suzuki and co-workers reported a carbon atom insertion reaction for the synthesis of 3,4-dihydroquinoxalin-2(1H)-ones in the presence of benzimidazoliums and 2-(methylsulfonyl)chromones under basic conditions. The possible mechanism showed that the deprotonation of benzimidazolium salt 185 could generate in situ a benzimidazolium derived N-heterocyclic carbene (186), which reacted with 2-(methylsulfonyl)chromone to form the key chromonylbenzimidazolium intermediate 187, subsequently leading to the generation of intermediate 188 in the presence of a hydroxide anion. Then, 188 underwent deprotonation of the hydroxyl group, insertion of chromone at the C2 position and ring opening processes to provide the desired product. Notably, no desired products were obtained by using other N-heterocyclic carbene precursor salts, indicating that the benzimidazole framework was important for this reaction (Scheme 15).53
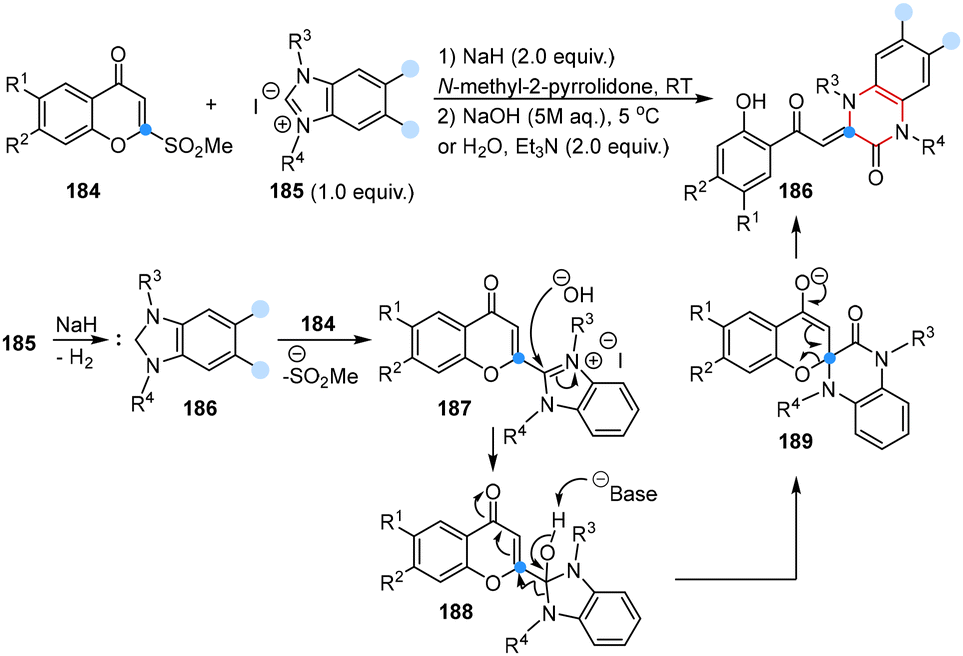 | ||
| Scheme 15 Carbon atom insertion into N-heterocyclic carbenes to obtain 3,4-dihydroquinoxalin-2(1H)-ones reported by Suzuki. | ||
In the same year, Ye, Sun and co-workers first reported an enantioselective copper-catalyzed one-carbon ring expansion reaction for the construction of chiral morpholin-2-ones, piperazin-2-ones, and piperidin-3-ones from N-heterocycle-tethered diynes in good to excellent yields with excellent enantioselectivities (Scheme 16).54 Mechanistic studies indicated that the key vinyl cation intermediates 194 were generated through intramolecular electrophilic cyclization between two alkyne moieties of substrates via Cu(I) catalysis. Subsequently, 194 underwent intramolecular trapping with the oxazolidone group, followed by 1,2-acyl migration to form ring-expanding intermediates 196, which ultimately provided the desired products through proton transfer and demetallation.
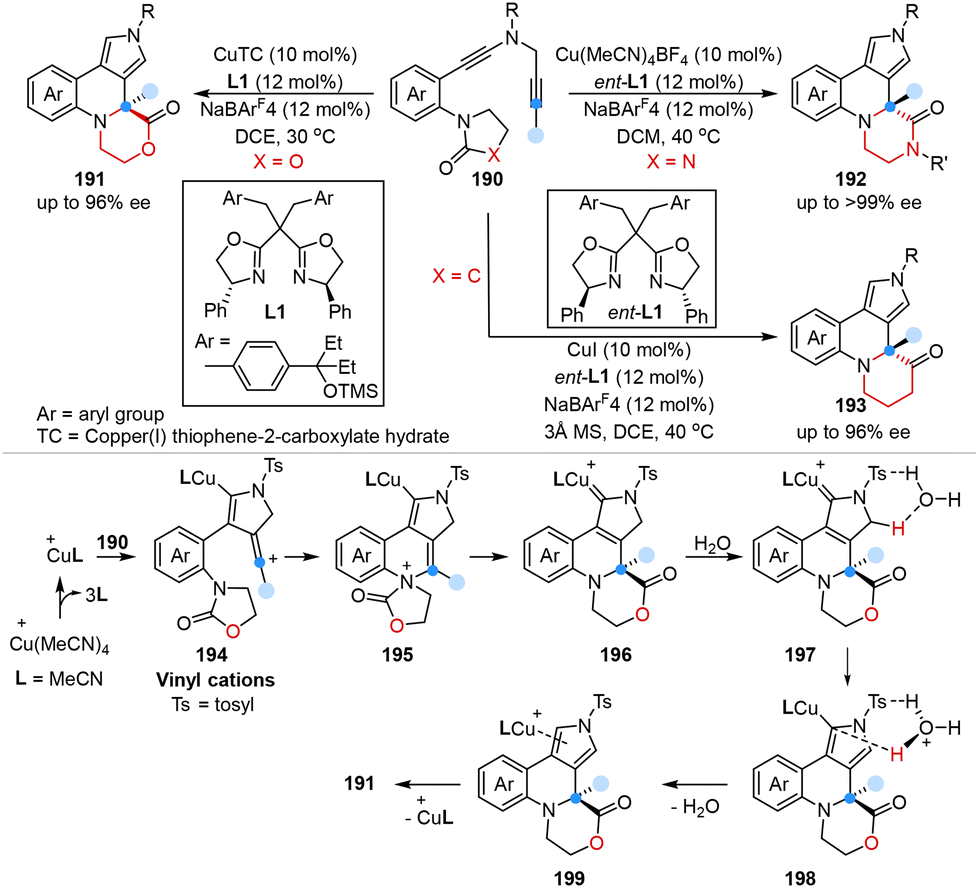 | ||
| Scheme 16 Ring expansion of N-heterocycle-tethered diynes via copper-catalyzed diyne cyclization reported by Ye and Sun. | ||
The one-carbon ring expansion reaction could be elegantly accomplished through the utilization of biocatalysts, showcasing the remarkable versatility and efficiency of biological systems in mediating such transformations.
In 2022, Arnold and co-workers reported a synthetic paradigm for the enantioselective synthesis of azetidines from aziridines using engineered ‘carbene transferase’ enzymes. The employment of this technology achieved unprecedented stereocontrol (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 er) in the [1,2]-Stevens rearrangement and overcame the inherent reactivity of aziridinium ylides, namely the cheletropic extrusion of olefins (Scheme 17A).55a
:1 er) in the [1,2]-Stevens rearrangement and overcame the inherent reactivity of aziridinium ylides, namely the cheletropic extrusion of olefins (Scheme 17A).55a
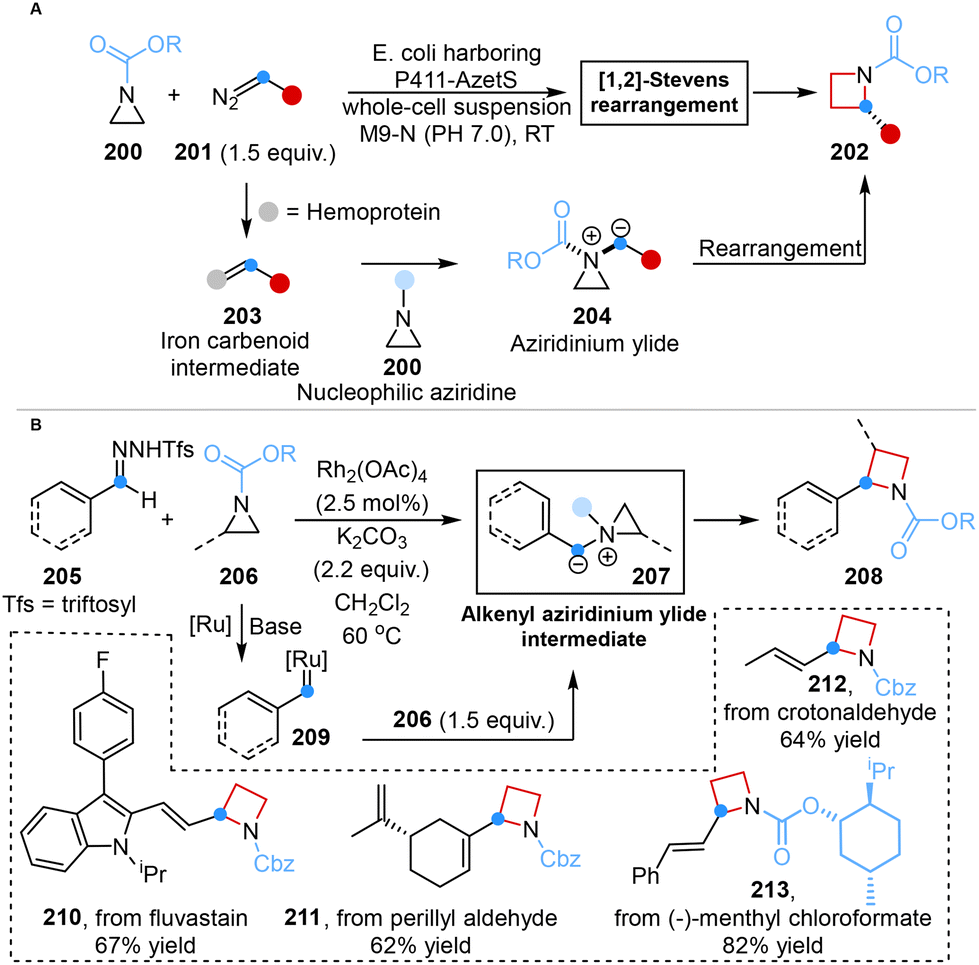 | ||
| Scheme 17 Ring expansion of aziridines to azetidines reported by Arnold (A) and Bi (B). | ||
In parallel, Bi and co-workers accomplished an analogous transformation in 2024, successfully synthesizing 2-vinyl azetidines via a Rh-catalyzed one-carbon ring expansion that involved a diradical mechanism. This achievement was characterized by the utilization of vinyl-N-triftosylhydrazones as carbene precursors, establishing the first synthetic paradigm within this specific realm. This versatile strategy involved the in situ generation of alkenyl aziridinium ylide intermediate, which served as the pivotal factor in successfully achieving carbon atom insertion into aziridine skeletons (Scheme 17B).55b
The utilization of the single-atom skeletal editing principle for adding a carbon atom into carbocyclic or heterocycle skeletons offered a versatile and effective strategy for the syntheses of natural products. Dai and co-workers presented a strategy involving one-carbon insertion and polarity inversion that enabled an efficient and concise total synthesis of complanadine A. Notably, the employment of the classical Ciamician–Dennstedt rearrangement served as the key step that transformed nucleophilic pyrrole into electrophilic pyridine along with the introduction of chloride at the C3-position for the next C–H arylation reaction (Scheme 18).56
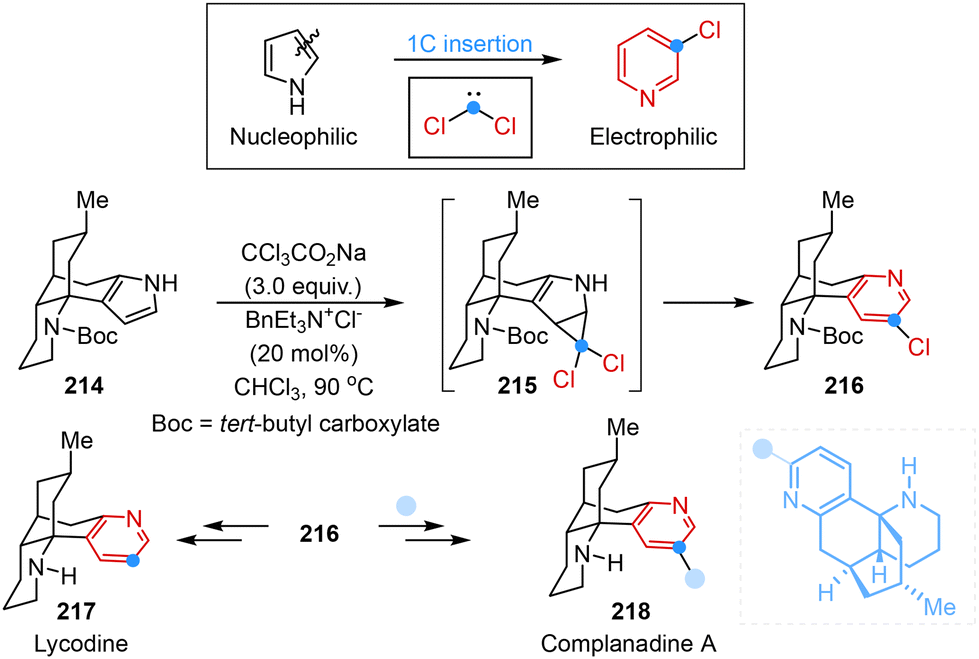 | ||
| Scheme 18 Single-atom skeletal editing logic in the total syntheses of complanadine A reported by Dai. | ||
More recently, Paton, Sarpong and co-workers demonstrated a contra-biosynthesis approach in which a carbon atom was inserted into benzenoid subfamily scaffolds to obtain troponoid congeners, followed by oxidative dearomatization and ring expansion to achieve a benzenoid-to-troponoid ring expansion, ultimately yielding harringtonolide from cephanolide A in two steps (Scheme 19).57 The Büchner–Curtius–Schlotterbeck reaction mechanism and extensive computational studies clearly demonstrated the designed synthetic transformation process, revealing that different Lewis acids with TMSCHN2 would change the relative energies of different transition structures, which had a significant effect on regioselectivity.
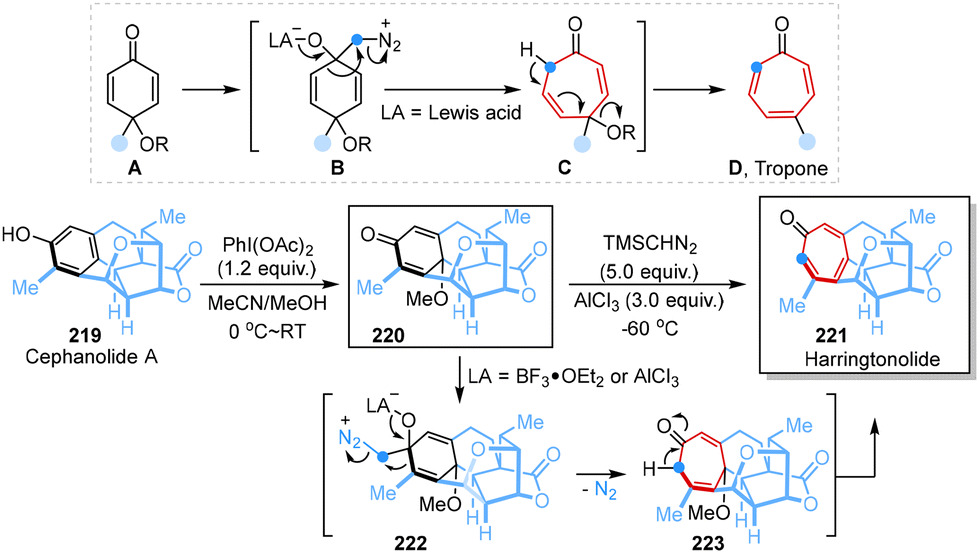 | ||
| Scheme 19 Single-atom skeletal editing logic in the syntheses of harringtonolide reported by Paton and Sarpong. | ||
4. Conclusions
In this review, we summarized the current state-of-the-art protocols with respect to ring-expanding skeletal editing for the synthesis of n + 1-membered core scaffolds by adding a single carbon atom into the ring systems, which represents an increasingly robust tool in molecular sciences and can be further utilized at more advanced stages of drug development and material discovery to subtly tune the molecular properties. Importantly, these methods allow for precise alteration and reorganization of the more readily available structural features of the corresponding precursors into ring-expanding products with structural uniqueness and complexity that were previously considered difficult to achieve. The utility of these synthetic transformations also highlights the importance and feasibility for the future continued discovery and development of general and selective methods for the ring-expanding single-atom skeletal editing of complex molecules.Despite the remarkable recent progress, selective ring-expanding single-atom skeletal editing is arguably still in its infancy and the current toolbox available is still limited. In other words, a number of challenges linked with generality, selectivity, efficiency, and atom economy remain to be addressed before its capability can be fully exploited in drug development campaigns and beyond. For example, the established methodologies by carbon atom insertion have mainly contributed to the prevalence of flat aromatic molecules, while direct insertion of carbon atoms into sp3-carbon-rich scaffolds is relatively limited, yet it is tremendously important for the accurate disposition of functionalities across three-dimensional chemical space. Additionally, enantioselective transformations in ring-expanding single-atom skeletal editing by employing a chiral catalyst or a ligand remain largely underexplored. Also, another key challenge is to overcome the relevant issues facing late-stage skeletal editing of complex and readily available molecules as well as the underdeveloped reaction manifolds, which will enable access to currently inaccessible substrate classes and unavailable reactivity patterns in a more reliable and predictable manner.
Besides these limitations, in-depth mechanistic investigations and detailed computational studies are essential for providing a more comprehensive understanding of the factors responsible for the reaction outcome via one mechanism or the other, which could accelerate the discovery of practical and scalable methods in the field of ring-expanding single-atom skeletal editing. Regardless, the successful progress made in the past four years has provided a new retrosynthetic paradigm for unique opportunities to rapidly build up molecular complexity and access valuable chemical space. We anticipate that the continued development and use of ring-expanding single-atom skeletal editing as a means to streamline efficiency will contribute considerably to offering solutions to the unmet synthetic needs for new chemistry to prepare most envisioned target structures by changing prevailing dogmas in retrosynthetic analysis, as well as facilitating chemical space exploration, drug discovery and other diverse applications.
Author contributions
Ting Yuan and Lei Shi wrote the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (Nos. 22271069 and 21871067), the Guangdong Basic and Applied Basic Research Foundation (Nos. 2023A1515012457 and 2021A1515010190) and the Shenzhen Science and Technology Program (No. GXWD20231130100539001).References
- (a) H. Lachance, S. Wetzel, K. Kumar and H. Waldmann, Charting, navigating, and populating natural product chemical space for drug discovery, J. Med. Chem., 2012, 55, 5989–6001 CrossRef CAS PubMed; (b) S. E. Motika and P. J. Hergenrother, Re-engineering natural products to engage new biological targets, Nat. Prod. Rep., 2020, 37, 1395–1403 RSC; (c) K. E. Kim, A. N. Kim, C. J. McCormick and B. M. Stoltz, Late-stage diversification: a motivating force in organic synthesis, J. Am. Chem. Soc., 2021, 143, 16890–16901 CrossRef CAS PubMed.
- (a) P. Ertl, S. Jelfs, J. Mühlbacher, A. Schuffenhauer and P. Selzer, Quest for the rings. In silico exploration of ring universe to identify novel bioactive heteroaromatic scaffolds, J. Med. Chem., 2006, 49, 4568–4573 CrossRef CAS PubMed; (b) R. D. Taylor, M. MacCoss and A. D. G. Lawson, Rings in drugs, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed; (c) Y. Chen, C. Rosenkranz, S. Hirte and J. Kirchmair, Ring systems in natural products: structural diversity, physicochemical properties, and coverage by synthetic compounds, Nat. Prod. Rep., 2022, 39, 1544–1556 RSC.
- (a) G. Schneider, W. Neidhart, T. Giller and G. Schmid, “Scaffold-Hopping” by topological pharmacophore search: a contribution to virtual screening, Angew. Chem., Int. Ed., 1999, 38, 2894–2896 CrossRef CAS; (b) Y. Hu, D. Stumpfe and J. Bajorath, Recent advances in scaffold hopping, J. Med. Chem., 2017, 60, 1238–1246 CrossRef CAS PubMed; (c) T. B. Callis, T. R. Garrett, A. P. Montgomery, J. J. Danon and M. Kassiou, Recent scaffold hopping applications in central nervous system drug discovery, J. Med. Chem., 2022, 65, 13483–13504 CrossRef CAS PubMed.
- (a) M. Grigalunas, S. Brakmann and H. Waldmann, Chemical evolution of natural product structure, J. Am. Chem. Soc., 2022, 144, 3314–3329 CrossRef CAS; (b) J.-M. Gally, A. Pahl, P. Czodrowski and H. Waldmann, Pseudonatural products occur frequently in biologically relevant compounds, J. Chem. Inf. Model., 2021, 61, 5458–5468 CrossRef PubMed.
- S. E. Motikaa and P. J. Hergenrother, Re-engineering natural products to engage new biological targets, Nat. Prod. Rep., 2020, 37, 1395–1403 RSC.
- M. D. Burke and S. L. Schreiber, A planning strategy for diversity-oriented synthesis, Angew. Chem., Int. Ed., 2004, 43, 46–58 CrossRef.
- P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Function-oriented synthesis, step economy, and drug design, Acc. Chem. Res., 2008, 41, 40–49 CrossRef CAS PubMed.
- S. Wetzel, R. S. Bon, K. Kumar and H. Waldmann, Biology-oriented synthesis, Angew. Chem., Int. Ed., 2011, 50, 10800–10826 CrossRef CAS PubMed.
- B. Over, S. Wetzel, C. Grütter, Y. Nakai, S. Renner, D. Rauh and H. Waldmann, Natural-product-derived fragments for fragment-based ligand discovery, Nat. Chem., 2013, 5, 21–28 CrossRef CAS PubMed.
- (a) T. E. Nielsen and S. L. Schreiber, Towards the optimal screening collection: a synthesis strategy, Angew. Chem., Int. Ed., 2008, 47, 48–56 CrossRef CAS PubMed; (b) M. Uguen, G. Davison, L. J. Sprenger, J. H. Hunter, M. P. Martin, S. Turberville, J. E. Watt, B. T. Golding, M. E. M. Noble, H. L. Stewart and M. J. Waring, Build–couple–transform: a paradigm for lead-like library synthesis with scaffold diversity, J. Med. Chem., 2022, 65, 11322–11339 CrossRef CAS PubMed.
- (a) K. Chen and P. S. Baran, Total synthesis of eudesmane terpenes by site-selective C–H oxidations, Nature, 2009, 459, 824–828 CrossRef CAS PubMed; (b) Y. Kanda, Y. Ishihara, N. C. Wilde and P. S. Baran, Two-phase total synthesis of taxanes: tactics and strategies, J. Org. Chem., 2020, 85, 10293–10320 CrossRef CAS PubMed.
- (a) K. C. Morrisona and P. J. Hergenrother, Natural products as starting points for the synthesis of complex and diverse compounds, Nat. Prod. Rep., 2014, 31, 6–14 RSC; (b) R. W. Huigens III, K. C. Morrison, R. W. Hicklin, T. A. Flood Jr., M. F. Richter and P. J. Hergenrother, A ring-distortion strategy to construct stereochemically complex and structurally diverse compounds from natural products, Nat. Chem., 2013, 5, 195–202 CrossRef.
- (a) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachalb and S. W. Krska, The medicinal chemist's toolbox for late stage functionalization of drug-like molecules, Chem. Soc. Rev., 2016, 45, 546–576 RSC; (b) J. Börgel and T. Ritter, Late-stage Functionalization, Chem, 2020, 6, 1877–1887 CrossRef; (c) J. D. Lasso, D. J. Castillo-Pazos and C.-J. Li, Green chemistry meets medicinal chemistry: a perspective on modern metal-free late-stage functionalization reactions, Chem. Soc. Rev., 2021, 50, 10955–10982 RSC.
- (a) D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Organic synthesis provides opportunities to transform drug discovery, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed; (b) J. Boström, D. G. Brown, R. J. Young and G. M. Keserü, Expanding the medicinal chemistry synthetic toolbox, Nat. Rev. Drug Discovery, 2018, 17, 709–727 CrossRef.
- (a) R. M. Wilson and S. J. Danishefsky, Small molecule natural products in the discovery of therapeutic agents: the synthesis connection, J. Org. Chem., 2006, 71, 8329–8351 CrossRef CAS; (b) A. M. Szpilman and E. M. Carreira, Probing the biology of natural products: molecular editing by diverted total synthesis, Angew. Chem., Int. Ed., 2010, 49, 9592–9628 CrossRef CAS; (c) K. R. Campos, P. J. Coleman, J. C. Alvarez, S. D. Dreher, R. M. Garbaccio, N. K. Terrett, R. D. Tillyer, M. D. Truppo and E. R. Parmee, The importance of synthetic chemistry in the pharmaceutical industry, Science, 2019, 363, eaat0805 CrossRef CAS.
- (a) J. Jurczyk, J. Woo, S. F. Kim, B. D. Dherange, R. Sarpong and M. D. Levin, Single-atom logic for heterocycle editing, Nat. Synth., 2022, 1, 352–364 CrossRef PubMed; (b) C. Ma, C. W. Lindsley, J. Chang and B. Yu, Rational molecular editing: a new paradigm in drug discovery, J. Med. Chem., 2024, 67, 11459–11466 CrossRef CAS.
- (a) L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Late-stage C–H functionalization offers new opportunities in drug discovery, Nat. Rev. Chem., 2021, 5, 522–545 CrossRef CAS PubMed; (b) N. J. Castellino, A. P. Montgomery, J. J. Danon and M. Kassiou, Late-stage functionalization for improving drug-like molecular properties, Chem. Rev., 2023, 123, 8127–8153 CrossRef CAS PubMed.
- For reviews, see: (a) Y. Xia and G. Dong, Temporary or removable directing groups enable activation of unstrained C–C bonds, Nat. Rev. Chem., 2020, 4, 600–614 CrossRef CAS PubMed; (b) C. Hui, Z. Wang, S. Wang and C. Xu, Molecular editing in natural product synthesis, Org. Chem. Front., 2022, 9, 1451–1457 RSC; (c) Y. Xue and G. Dong, Deconstructive synthesis of bridged and fused rings via transition-metal-catalyzed “cut-and-sew” reactions of benzocyclobutenones and cyclobutanones, Acc. Chem. Res., 2022, 55, 2341–2354 CrossRef CAS; (d) J. Huang, F. Liu, X. Wu, J.-Q. Chen and J. Wu, Recent advances in the reactions of silacyclobutanes and their applications, Org. Chem. Front., 2022, 9, 2840–2855 RSC; (e) Z. Liu, P. Sivaguru, Y. Ning, Y. Wu and X. Bi, Skeletal editing of (hetero)arenes using carbenes, Chem. – Eur. J., 2023, 29, e202301227 CrossRef CAS PubMed; (f) B. W. Joynson and L. T. Ball, Skeletal editing: interconversion of arenes and heteroarenes, Helv. Chim. Acta, 2023, 106, e202200182 CrossRef CAS; (g) Y. Nakano and D. W. Lupton, One carbon—four new bonds, Science, 2023, 379, 439–440 CrossRef CAS; (h) X. Li, J. Xu and Z.-G. Xu, Precision single-atom editing: new frontiers in nitrogen insertion and substitution for the generation of N-heterocycles, Org. Chem. Front., 2024, 11, 4041–4053 RSC. For works, see: (i) Y. He, Q. Liu, Z. Du, Y. Xu, L. Cao, X. Zhang and X. Fan, B(C6F5)3-Catalyzed α,β-difunctionalization and C–N bond cleavage of saturated amines with benzo[c]isoxazoles: access to quinoline derivatives, J. Org. Chem., 2022, 87, 14840–14845 CrossRef CAS PubMed; (j) F. Xie, Y. Chen, Y. Li, Z. Wang, J. Zhang and Y. Tang, Photo-induced two-carbon ring expansion of N-alkenyl lactams and N-alkenyl/phenyl benzoazetinones, Org. Chem. Front., 2023, 10, 928–935 RSC; (k) Q.-H. Shi, Y.-H. Wang, Z.-Y. Chen, X.-R. Wang, W.-H. Zhang, F.-L. Tian, L.-J. Peng, Y. Zhou and X.-L. Liu, Formal oxygen atom insertion as a skeletal-editing step: rapid access natural-product-inspired bispiro[oxindole-oxazinane] hybrids, Org. Chem. Front., 2023, 10, 3307–3312 RSC.
- (a) C. Hui, L. Brieger, C. Strohmann and A. P. Antonchick, Stereoselective synthesis of cyclobutanes by contraction of pyrrolidines, J. Am. Chem. Soc., 2021, 143, 18864–18870 CrossRef CAS PubMed; (b) S. H. Kennedy, B. D. Dherange, K. J. Berger and M. D. Levin, Skeletal editing through direct nitrogen deletion of secondary amines, Nature, 2021, 593, 223–227 CrossRef CAS PubMed; (c) G. L. Bartholomew, F. Carpaneto and R. Sarpong, Skeletal editing of pyrimidines to pyrazoles by formal carbon deletion, J. Am. Chem. Soc., 2022, 144, 22309–22315 CrossRef CAS PubMed; (d) J. Woo, A. H. Christian, S. A. Burgess, Y. Jiang, U. F. Mansoor and M. D. Levin, Scaffold hopping by net photochemical carbon deletion of azaarenes, Science, 2022, 376, 527–532 CrossRef CAS PubMed.
- (a) H. Zhong, D. T. Egger, V. C. M. Gasser, P. Finkelstein, L. Keim, M. Z. Seidel, N. Trapp and B. Morandi, Skeletal metalation of lactams through a carbonyl-to-nickel-exchange logic, Nat. Commun., 2023, 14, 5273 CrossRef CAS; (b) J. Woo, C. Stein, A. H. Christian and M. D. Levin, Carbon-to-nitrogen single-atom transmutation of azaarenes, Nature, 2023, 623, 77–82 CrossRef CAS; (c) T. J. Pearson, R. Shimazumi, J. L. Driscoll, B. D. Dherange, D.-I. Park and M. D. Levin, Aromatic nitrogen scanning by ipso-selective nitrene internalization, Science, 2023, 381, 1474–1479 CrossRef CAS; (d) H. Lu, Y. Zhang, X.-H. Wang, R. Zhang, P.-F. Xu and H. Wei, Carbon–nitrogen transmutation in polycyclic arenol skeletons to access N-heteroarenes, Nat. Commun., 2024, 15, 3772 CrossRef CAS PubMed; (e) Q. Cheng, D. Bhattacharya, M. Haring, H. Cao, C. Mück-Lichtenfeld and A. Studer, Skeletal editing of pyridines through atom-pair swap from CN to CC, Nat. Chem., 2024, 16, 741–748 CrossRef CAS PubMed; (f) L. Li, P. Sivaguru, D. Wei, M. Liu, Q. Zhu, S. Dong, E. Casali, N. Li, G. Zanoni and X. Bi, Silver-catalyzed direct conversion of epoxides into cyclopropanes using N-triftosylhydrazones, Nat. Commun., 2024, 15, 1951 CrossRef CAS PubMed; (g) G. L. Bartholomew, S. L. Kraus, L. J. Karas, F. Carpaneto, R. Bennett, M. S. Sigman, C. S. Yeung and R. Sarpong, 14N to 15N isotopic exchange of nitrogen heteroaromatics through skeletal editing, J. Am. Chem. Soc., 2024, 146, 2950–2958 CrossRef CAS.
- A. Baeyer and V. Villiger, Einwirkung des caro'schen reagens auf ketone, Ber. Dtsch. Chem. Ges., 1899, 32, 3625 CrossRef.
- E. Beckmann, Zur kenntniss der isonitrosoverbindungen, Ber. Dtsch. Chem. Ges., 1886, 19, 988–993 CrossRef.
- G. L. Ciamician and M. Dennstedt, Ueber die einwirkung des chloroforms auf die kaliumverbindung pyrrols, Ber. Dtsch. Chem. Ges., 1881, 14, 1153–1163 CrossRef.
- A. E. Favorskii, J. Russ. Phys.-Chem. Soc., 1894, 26, 559 Search PubMed.
- L. Wolff, Ueber diazoanhydride, Justus Liebigs Ann. Chem., 1902, 325, 129–195 CrossRef.
- (a) Z.-L. Song, C.-A. Fan and Y.-Q. Tu, Semipinacol rearrangement in natural product synthesis, Chem. Rev., 2011, 111, 7523–7556 CrossRef CAS; (b) K. Gao, J. Hu and H. Ding, Tetracyclic diterpenoid synthesis facilitated by ODI-cascade approaches to bicyclo[3.2.1]octane skeletons, Acc. Chem. Res., 2021, 54, 875–889 CrossRef CAS PubMed; (c) L. Liang, L.-D. Guo and R. Tong, Achmatowicz rearrangement-inspired development of green chemistry, organic methodology, and total synthesis of natural products, Acc. Chem. Res., 2022, 55, 2326–2340 CrossRef CAS PubMed.
- (a) T. Ye and M. A. McKervey, Organic synthesis with α-diazocarbonyl compounds, Chem. Rev., 1994, 94, 1091–1160 CrossRef CAS; (b) M. P. Doyle, M. A. Mckervey and T. Ye, Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides, Wiley, New York, 1998 Search PubMed; (c) Y. Zhang and J. Wang, Chem. Commun., 2009, 5350–5361 RSC; (d) N. R. Candeias, R. Paterna and P. M. P. Gois, Homologation reaction of ketones with diazo compounds, Chem. Rev., 2016, 116, 2937–2981 CrossRef CAS.
- F. Tan, M. Pu, J. He, J. Li, J. Yang, S. Dong, X. Liu, Y.-D. Wu and X. Feng, Catalytic asymmetric homologation of ketones with α-alkyl α-diazo esters, J. Am. Chem. Soc., 2021, 143, 2394–2402 CrossRef CAS PubMed.
- (a) E. Leemans, M. D′hooghe and N. De Kimpe, Ring expansion of cyclobutylmethylcarbenium ions to cyclopentane or cyclopentene derivatives and metal-promoted analogous rearrangements, Chem. Rev., 2011, 111, 3268–3333 CrossRef CAS PubMed; (b) D. C. Moebius, V. L. Rendina and J. S. Kingsbury, Catalysis of diazoalkane–carbonyl homologation. How new developments in hydrazone oxidation enable the carbon insertion strategy for synthesis, Top. Curr. Chem., 2014, 346, 111–162 CrossRef CAS PubMed.
- (a) F. Alonso, I. Micó, C. Nájera, J. M. Sansano, M. Yus, J. Ezquerra, B. Yruretagoyena and I. Gracia, Synthesis of 3- and 4-substituted cyclic α-amino acids structurally related to ACPD, Tetrahedron, 1995, 51, 10259–10280 CrossRef CAS; (b) V. L. Rendina, D. C. Moebius and J. S. Kingsbury, An enantioselective synthesis of 2-aryl cycloalkanones by Sc-catalyzed carbon insertion, Org. Lett., 2011, 13, 2004–2007 CrossRef CAS PubMed; (c) B. C. Kang, S. Y. Shim and D. H. Ryu, Highly stereoselective oxazaborolidinium ion catalyzed synthesis of (Z)-silyl enol ethers from alkyl aryl ketones and trimethylsilyldiazomethane, Org. Lett., 2014, 16, 2077–2079 CrossRef CAS PubMed.
- S. Ochi, Z. Zhang, Y. Xia and G. Dong, Rhodium-catalyzed (4 + 1) cycloaddition between benzocyclobutenones and styrene-type alkenes, Angew. Chem., Int. Ed., 2022, 134, e202202703 CrossRef.
- (a) E. Büchner and T. Curtius, Ueber die einwirkung von diazoessigäther auf aromatische kohlenwasserstoffe, Ber. Dtsch. Chem. Ges., 1885, 18, 2377–2379 CrossRef; (b) E. Büchner, Ueber pseudophenylessigsäure, Ber. Dtsch. Chem. Ges., 1896, 29, 106–109 CrossRef.
- (a) T. Ye and M. A. McKervey, Organic synthesis with α-diazo carbonyl compounds, Chem. Rev., 1994, 94, 1091–1160 CrossRef CAS; (b) S. E. Reisman, R. R. Nani and S. Levin, Buchner and beyond: arene cyclopropanation as applied to natural product total synthesis, Synlett, 2011, 2437–2442 CrossRef CAS.
- O. A. McNamara and A. R. Maguire, The norcaradiene–cycloheptatriene equilibrium, Tetrahedron, 2011, 67, 9–40 CrossRef CAS.
- (a) K. L. Smith, C. L. Padgett, W. D. Mackay and J. S. Johnson, Catalytic, asymmetric dearomative synthesis of complex cyclohexanes via a highly regio- and stereoselective arene cyclopropanation using α-cyanodiazoacetates, J. Am. Chem. Soc., 2020, 142, 6449–6455 CrossRef CAS PubMed; (b) R. J. Baker, J. Ching, T. R. Hou, I. Franzoni and M. Lautens, Dearomative cyclopropanation of naphthols via cyclopropene ring-opening, Angew. Chem., Int. Ed., 2022, 61, e202116171 CrossRef CAS PubMed.
- P. Piacentini, T. W. Bingham and D. Sarlah, Dearomative ring expansion of polycyclic arenes, Angew. Chem., Int. Ed., 2022, 61, e202208014 CrossRef CAS PubMed.
- M. Morten, M. Hennum and T. Bonge-Hansen, Synthesis of quinoline-3-carboxylates by a Rh(II)-catalyzed cyclopropanation–ring expansion reaction of indoles with halodiazoacetates, Beilstein J. Org. Chem., 2015, 11, 1944–1949 CrossRef CAS PubMed.
- (a) B. D. Dherange, P. Q. Kelly, J. P. Liles, M. S. Sigman and M. D. Levin, Carbon atom insertion into pyrroles and indoles promoted by chlorodiazirines, J. Am. Chem. Soc., 2021, 143, 11337–11344 CrossRef CAS PubMed; (b) E. E. Hyland, P. Q. Kelly, A. M. McKillop, B. D. Dherange and M. D. Levin, Unified access to pyrimidines and quinazolines enabled by N–N cleaving carbon atom insertion, J. Am. Chem. Soc., 2022, 144, 19258–19264 CrossRef CAS PubMed.
- F.-P. Wu, C. C. Chintawar, R. Lalisse, P. Mukherjee, S. Dutta, J. Tyler, C. G. Daniliuc, O. Gutierrez and F. Glorius, Ring expansion of indene by photoredox-enabled functionalized carbon-atom insertion, Nat. Catal., 2024, 7, 242–251 CrossRef CAS.
- Z. Wang, Ciamician–Dennstedt Reaction, in Comprehensive organic name reactions and reagents, American Cancer Society, John Wiley & Sons, 2010, pp. 646–648 Search PubMed.
- M. B. Jones and M. S. Platz, Solvent and substituent effects on the reaction of phenylchlorocarbene with pyridine, J. Org. Chem., 1991, 56, 1694–1695 CrossRef CAS.
- (a) B. W. Joynson, G. R. Cumming and L. T. Ball, Photochemically mediated ring expansion of indoles and pyrroles with chlorodiazirines: synthetic methodology and thermal hazard assessment, Angew. Chem., Int. Ed., 2023, 135, e202305081 CrossRef; (b) H. Guo, S. Qiu and P. Xu, One-carbon ring expansion of indoles and pyrroles: a straightforward access to 3-fluorinated quinolines and pyridines, Angew. Chem., Int. Ed., 2024, 63, e202317104 CrossRef CAS PubMed.
- (a) S. Liu, Y. Yang, Q. Song, Z. Liu, Y. Lu, Z. Wang, P. Sivaguru and X. Bi, Tunable molecular editing of indoles with fluoroalkyl carbenes, Nat. Chem., 2024, 16, 988–997 CrossRef CAS PubMed; (b) Y. Yang, Q. Song, P. Sivaguru, Z. Liu, D. Shi, T. Tian, G. de Ruiter and X. Bi, Controllable skeletal and peripheral editing of pyrroles with vinylcarbenes, Angew. Chem., Int. Ed., 2024, 63, e202401359 CrossRef CAS PubMed.
- (a) L. Li, H. Chen, X. Zhang, K. Murali, Q. Zhu, M. Liu, H. Zhang, V. Nenajdenko and X. Bi, Silver-catalyzed single-carbon insertion of indoles with acetophenone N-triftosylhydrazones, Org. Lett., 2024, 26, 7207–7211 CrossRef CAS PubMed; (b) F.-P. Wu, J. L. Tyler, C. G. Daniliuc and F. Glorius, Atomic carbon equivalent: design and application to diversity generating skeletal editing from indoles to 3-functionalized quinolines, ACS Catal., 2024, 14, 13343–13351 CrossRef CAS.
- R. Tran, D. K. Brownsey, L. O'Sullivan, C. M. J. Brandow, E. S. Chang, W. Zhou, K. V. Patel, E. Gorobets and D. J. Derksen, Leveraging pyrazolium ylide reactivity to access indolizineand 1,2-dihydropyrimidine derivatives, Chem. – Eur. J., 2024, 30, e202400421 CrossRef CAS PubMed.
- (a) M. Qi, M. Suleman, J. Fan, P. Lu and Y. Wang, Cu(I)-catalyzed synthesis of spiro[isoquinoline-4,2′-[1,3]oxazin]-3-ones via ring expansion reactions of isoxazoles with 4-diazoisoquinolin-3-ones, Tetrahedron, 2022, 128, 133092 CrossRef CAS; (b) L. Li, Y. Ning, H. Chen, Y. Ning, P. Sivaguru, P. Liao, Q. Zhu, Y. Ji, G. de Ruiter and X. Bi, Dearomative insertion of fluoroalkyl carbenes into azoles leading to fluoroalkyl heterocycles with a quaternary center, Angew. Chem., Int. Ed., 2024, 63, e202313807 CrossRef CAS PubMed; (c) L. Li, H. Chen, M. Liu, Q. Zhu, H. Zhang, G. de Ruiter and X. Bi, Silver-catalyzed dearomative skeletal editing of indazoles by donor carbene insertion, Chem. – Eur. J., 2024, 30, e202304227 CrossRef CAS PubMed; (d) M. Tsuda, T. Morita, Y. Morita, J. Takaya and H. Nakamura, Methylene insertion into nitrogen-heteroatom single bonds of 1, 2-azoles via a zinc carbenoid: an alternative tool for skeletal editing, Adv. Sci., 2024, 11, 2307563 CrossRef CAS PubMed.
- (a) Y. Zhou, F. Chen, Z. Li, J. Dong, J. Li, B. Zhang and Q. Song, Single-atom skeletal editing of 2H-indazoles enabled by difluorocarbene, Sci. China: Chem., 2023, 66, 1975–1981 CrossRef CAS; (b) S. Bhattacharjee and A. Hajra, Skeletal editing through molecular recombination of 2H-Indazoles to azo-linked-quinaz, Chem. – Eur. J., 2024, 30, e202303240 CrossRef CAS PubMed.
- (a) J. Kim and E. J. Yoo, Catalytic ring expansion of activated heteroarenes enabled by regioselective dearomatization, Org. Lett., 2021, 23, 4256–4260 CrossRef CAS PubMed; (b) M. J. Mailloux, G. S. Fleming, S. S. Kumta and A. B. Beeler, Unified synthesis of azepines by visible-light-mediated dearomative ring expansion of aromatic N-ylides, Org. Lett., 2021, 23, 525–529 CrossRef CAS PubMed.
- A. Ali, H. K. Harit, M. Devi, D. Ghosh and R. P. Singh, Ring expansion of isatins via 1,2-phospha-brook rearrangement: a route to the synthesis of 2-quinolinone-derived p-quinone methides, J. Org. Chem., 2022, 87, 16313–16327 CrossRef CAS PubMed.
- (a) J. Escribano, C. Rivero-Hernández, H. Rivera, D. Barros, J. Castro-Pichel, E. Pérez-Herrán, A. Mendoza-Losana, Í. Angulo-Barturen, S. Ferrer-Bazaga, E. Jiménez-Navarro and L. Ballell, 4-Substituted thioquinolines and thiazoloquinolines: potent, selective, and Tween-80 in vitro dependent families of antitubercular agents with moderate in vivo activity, ChemMedChem, 2011, 6, 2252–2263 CrossRef CAS PubMed; (b) D. Audisio, S. Messaoudi, L. Cegielkowski, J.-F. Peyrat, J.-D. Brion, D. Methy-Gonnot, C. Radanyi, J.-M. Renoir and M. Alami, Discovery and Biological Activity of 6BrCaQ as an Inhibitor of the Hsp90 Protein Folding Machinery, ChemMedChem, 2011, 6, 804–815 CrossRef CAS PubMed.
- S. Singh, G. Chakrabortty and S. R. Roy, Skeletal rearrangement through photocatalytic denitrogenation: access to C-3 aminoquinolin-2(1H)-ones, Chem. Sci., 2023, 14, 12541–12547 RSC.
- H. L. Schmitt, D. Martymianov, O. Green, T. Delcaillau, Y. S. P. Kim and B. Morandi, Regiodivergent ring-expansion of oxindoles to quinolinones, J. Am. Chem. Soc., 2024, 146, 4301–4308 CrossRef CAS PubMed.
- J. S. Lamb, F. Koyama, N. Suzuki and Y. Suzuki, Carbon atom insertion into N-heterocyclic carbenes to yield 3,4-dihydroquinoxalin-2(1H)-ones, Org. Chem. Front., 2024, 11, 277–283 RSC.
- F.-S. Li, X.-Y. Zou, T.-Q. Hu, Q. Sun, Z. Xu, B. Zhou and L.-W. Ye, Asymmetric one-carbon ring expansion of diverse N-heterocycles via copper-catalyzed diyne cyclization, Sci. Adv., 2024, 10, eadq7767 CrossRef PubMed.
- (a) D. C. Miller, R. G. Lal, L. A. Marchetti and F. H. Arnold, Biocatalytic one-carbon ring expansion of aziridines to azetidines via a highly enantioselective [1,2]-stevens Rearrangement, J. Am. Chem. Soc., 2022, 144, 4739–4745 CrossRef CAS PubMed; (b) Y. Ning, H. Chen, Y. Ning, J. Zhang and X. Bi, Rhodium-catalyzed one-carbon ring expansion of aziridines with vinyl-N-triftosylhydrazones for the synthesis of 2-vinyl azetidines, Angew. Chem., Int. Ed., 2024, 63, e202318072 CrossRef CAS PubMed.
- (a) D. Ma, B. S. Martin, K. S. Gallagher, T. Saito and M. Dai, One-carbon insertion and polarity inversion enabled a pyrrole strategy to the total syntheses of pyridine-containing lycopodium alkaloids: complanadine A and lycodine, J. Am. Chem. Soc., 2021, 143, 16383–16387 CrossRef CAS PubMed; (b) B. S. Martin, D. Ma, T. Saito, K. S. Gallagher and M. Dai, Concise total synthesis of complanadine a enabled by pyrrole-to-pyridine molecular editing, Synthesis, 2024, 107–117 CrossRef CAS.
- S. Wiesler, G. Sennari, M. V. Popescu, K. E. Gardner, K. Aida, R. S. Paton and R. Sarpong, Late-stage benzenoid-to-troponoid skeletal modification of the cephalotanes exemplified by the total synthesis of harringtonolide, Nat. Commun., 2024, 15, 4125 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2024 |