
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHollow carbon-based materials for electrocatalytic and thermocatalytic CO2 conversion
Kaining
Li
a,
Yasutaka
Kuwahara
 *ab and
Hiromi
Yamashita
*ab
*ab and
Hiromi
Yamashita
*ab
aDivision of Materials and Manufacturing Science, Graduate School of Engineering, Osaka University, 2-1 Yamada-oka, Osaka 565-0871, Japan. E-mail: kuwahara@mat.eng.osaka-u.ac.jp; yamashita@mat.eng.osaka-u.ac.jp
bInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (OTRI), Osaka University, 2-1 Yamada-oka, Suita, Osaka 565-0871, Japan
First published on 7th December 2023
Abstract
Electrocatalytic and thermocatalytic CO2 conversions provide promising routes to realize global carbon neutrality, and the development of corresponding advanced catalysts is important but challenging. Hollow-structured carbon (HSC) materials with striking features, including unique cavity structure, good permeability, large surface area, and readily functionalizable surface, are flexible platforms for designing high-performance catalysts. In this review, the topics range from the accurate design of HSC materials to specific electrocatalytic and thermocatalytic CO2 conversion applications, aiming to address the drawbacks of conventional catalysts, such as sluggish reaction kinetics, inadequate selectivity, and poor stability. Firstly, the synthetic methods of HSC, including the hard template route, soft template approach, and self-template strategy are summarized, with an evaluation of their characteristics and applicability. Subsequently, the functionalization strategies (nonmetal doping, metal single-atom anchoring, and metal nanoparticle modification) for HSC are comprehensively discussed. Lastly, the recent achievements of intriguing HSC-based materials in electrocatalytic and thermocatalytic CO2 conversion applications are presented, with a particular focus on revealing the relationship between catalyst structure and activity. We anticipate that the review can provide some ideas for designing highly active and durable catalytic systems for CO2 valorization and beyond.
 Kaining Li | Kaining Li received his MS degree in Environmental Chemistry from South-Central University for Nationalities in 2021. He is pursuing a PhD degree from the Graduate School of Engineering, Osaka University, under the supervision of Prof. Hiromi Yamashita. His current research interests are novel nanomaterials and their applications in catalytic CO2 conversion. |
 Yasutaka Kuwahara | Yasutaka Kuwahara received his PhD degree in engineering from Osaka University in 2011. He was a researcher at National Institute of Advanced Industrial Science and Technology (AIST), Japan, in 2012–2014. He was appointed as an assistant professor at Osaka University in 2014, and has been an associate professor since 2021. His current research interests include the design of nanostructured catalysts with multi-functionalities using porous materials and their applications to green chemical reactions and the valorization of CO2. |
 Hiromi Yamashita | Hiromi Yamashita has been a professor at Osaka University since 2004. He received a PhD degree from Kyoto University in 1987. He was an assistant professor at Tohoku University and an associate professor at Osaka Prefecture University. He has been the editor of Applied Catalysis B: Environmental since 2006, the president of the Asia-Pacific Association of Catalysis Societies (2019–2023), and the president of the Catalysis Society of Japan (2019–2020). His research interests include the design of single-site photocatalysts and nanostructured catalysts. |
1. Introduction
The rapid advancement of industrialization has led to high fossil-fuel consumption, causing an ever-increasing atmospheric CO2 concentration. Excessive CO2 emissions are the main contributors to global warming and the resulting abnormal climatic variation, glacial ablation, ecosystem destruction, etc.,1,2 which seriously threaten the shared future of mankind. In the past decades, many efforts have been devoted to controlling the CO2 concentration worldwide (e.g., adoptions of the Kyoto Protocol and the Paris Agreement). The objective of the Paris Agreement is to keep the global average temperature below 2 °C above the pre-industrial mean, with a temperature increase of 1.5 °C limit.3,4 Nevertheless, the current trend of anthropogenic CO2 growth makes achieving this goal an urgent task for a sustainable community and economic society.5Catalytic CO2 reduction conversions (e.g., photocatalysis, thermocatalysis, and electrocatalysis) to produce high-value-added fuels and feedstocks, offer a promising means of realizing carbon neutrality and potentially help to mitigate the global warming issue.6–9 In comparison to photocatalysis, thermal catalytic and electrocatalytic CO2 conversions are easier to scale up, due to the appropriate reaction kinetics and lack of dependence on the availability and cost of high-intensity light sources. Herein, our review mainly focuses on these two reactions.
Electrochemical CO2 reduction reaction (eCO2RR), powered by electrical energy, refers to the process of using electrochemical cells to convert CO2 to valuable compounds, such as carbon monoxide (CO), methane (CH4), methanol (CH3OH), ethylene (C2H4), etc. A typical electrocatalytic CO2 reduction reaction involves four main steps:10,11 (1) diffusion of CO2 to the electrolyte/cathode interface; (2) CO2 molecules are adsorbed on the cathode surface; (3) when an external bias is applied, the oxygen evolution takes place at the anode due to the water oxidation, and the generated electrons are then transferred to the cathode through an external circuit, where they can participate in the CO2 reduction processes; (4) desorption of final products from the cathode. These steps jointly determine the overall efficiency of eCO2RR. Recently, eCO2RR has become the research hotspot in CO2 utilization owing to its multifarious advantages, including mild operation conditions (room temperature and atmospheric pressure), flexible reactor platforms, and relatively high conversion efficiency.12–16 Integrating renewable energy with eCO2RR, simultaneously achieving a net CO2 reduction and chemical raw material supply is possible. Despite eCO2RR possesses numerous advantages, its pragmatic commercialization is still confronting great challenges. Firstly, as a chemically inert and thermodynamically stable molecule, CO2 is difficult to be activated because of the stable C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond (806 kJ mol−1);17,18 thus, a high potential is inevitably needed to drive the eCO2RR. Secondly, most eCO2RR can yield multiple products, which makes the direct utilization of products difficult, requiring an extra target product separation process. Third, noble metal-based catalysts (Au, Ag) usually deliver high electroactivity, but are less desirable considering their cost-efficiency and scarcity. For these reasons, efficient electrocatalysts with the characteristics of highly exposed active sites, good conductivity, abundant pore structure for target molecule transfer, and high earth-abundance are urgently needed.19,20
O bond (806 kJ mol−1);17,18 thus, a high potential is inevitably needed to drive the eCO2RR. Secondly, most eCO2RR can yield multiple products, which makes the direct utilization of products difficult, requiring an extra target product separation process. Third, noble metal-based catalysts (Au, Ag) usually deliver high electroactivity, but are less desirable considering their cost-efficiency and scarcity. For these reasons, efficient electrocatalysts with the characteristics of highly exposed active sites, good conductivity, abundant pore structure for target molecule transfer, and high earth-abundance are urgently needed.19,20
Thermocatalytic CO2 conversion, mainly including CO2 hydrogenation and CO2 dry reforming of methane (DRM), is a feasible process to alleviate the large CO2 emissions from the viewpoint of cost-efficiency.21 Among them, DRM is a highly endothermic reaction with the equation CH4 + CO2 → 2CO + 2H2, ΔH° = 247 kJ mol−1.22 The large energy consumption and the deactivation of catalysts caused by active metal sintering or coke deposition during the DRM process seriously hindered its applicability. Compared to DRM, thermal hydrogenation of CO2 shows a lower thermodynamic limitation, which involves the reaction of CO2 and H2 and can afford high levels of variety and tailorability for the production of desirable hydrocarbons and oxygenates.23,24 Conventionally, the catalytic performance and product selectivity highly depend on the catalyst. Accordingly, the development of CO2 hydrogenation catalysts with high CO2 adsorption and activation ability, robust stability, and good selectivity, is of great practical relevance.
In nature, cells with hollow structures are the fundamental unit of an organism, and this unique structure endows them with distinctive properties that allow for the adjustment of adsorption, separation, and exchange for substrates required to maintain vital life activities.25,26 Inspired by nature, many encouraging hollow-structure nanoreactors have been developed for energy storage,27–31 biomass conversion,32,33 and environmental catalysis applications.34,35 In recent years, hollow-structured carbon (HSC) and its derived nanomaterials have attracted considerable attention in the fields of thermocatalysis and electrocatalysis for CO2 conversion due to their advantageous features. Firstly, guest molecules and catalytically active species can be encapsulated in the cavity; by limiting the moving space of the inclusions, the leaching and aggregation of catalytically active species can be suppressed during the reaction, leading to the excellent durability and reusability of the catalyst.36,37 Secondly, adjustable pore structure and size of the carbon shell can regulate the mass transport and reaction characteristics, and further alter the catalytic selectivity.38 Thirdly, the carbon shell provides a platform to anchor single atoms,39,40 load metal particles,41,42 and couple with multi-functional polymers,43 which shows vast potential in variable catalysis applications. The rational design of nano-cavity or channel of HSC material can confine the metal nanoparticles (NPs) or cluster in a limited volume space, offering nano-confinement effects to improve catalytic CO2 reduction performance in terms of activity and selectivity. The positive influences of nanoconfinement mainly include (1) increasing reactant molecule concentration in the confined space and enriching the surface overage of adsorbed species on the active sites,44,45 (2) regulating the local pH value to hinder the competitive hydrogen evolution reaction during the eCO2RR process,46 and (3) tailoring the intrinsic electronic structures and optimizing the intermediate adsorption energy of the catalytic sites.47 For instance, Zheng et al. reported that the confinement effect of N-doped carbon nanotubes (NCNT) enabled the attenuation of the binding strengths between Ni NPs and *CO intermediates; therefore, the NCNT-confined Ni NPs sample (Ni@NCNT) showed enhanced CO selectivity in eCO2RR in comparison with the Ni NPs-supported sample (Ni-NCNT).48 Pan et al. proposed that, in the hollow mesoporous carbon spheres (HMCS) confining Cu clusters electrocatalyst system, the nano-confinement effect of HMCS contributed to the facilitation of C–C bond coupling to produce C2 products in eCO2RR.37 In our previous works, a series of novel hollow carbon nanocatalysts encapsulating alloys have been developed, which exhibit excellent thermocatalytic activity and stability for CO2 hydrogenation to produce formate owing to the nanoconfinement effects provided by the hollow carbon sphere.49–51 Apart from CO2 thermal conversion, our recent research demonstrated that HSC with superior conductivity and abundant pore structure can also serve as an effective support for syngas electrosynthesis from CO2.52 In our opinion, HSC-based catalysts are promising materials for CO2 conversion, and they will continue to be hotspots in the thermocatalytic and electrocatalytic fields.
In the past few years, several valuable review articles related to the advancement and current status of hollow nanoreactors have been published. For instance, the review from Das et al. captured the advancements in catalytic CO2 conversion, with a focus on novel core–shell catalyst development, activity, and selectivity improvement.10 Kuang et al. reviewed the recent advances of metal@hollow carbon sphere catalysts in the fields of sustainable biomass and CO2 conversion.25 Li and co-workers reviewed the recent achievements of hollow carbon nanocages in the fields of energy storage and electrocatalysis.53 Yu et al. reviewed the theoretical development of the hollow nanoreactor-based system, emphasizing the opportunities for the study of molecular kinetic behaviors in the hollow nanoreactors for achieving controllable catalysis.54
However, most of the reviews of HSC materials mainly focus on the energy storage field and biomass conversion, with a small amount concentrating on thermo- and electrocatalysis towards CO2 conversion. Besides, a comprehensible summary of the structure–performance relationship of hollow carbon-based materials for CO2 conversion remains to be refined. More importantly, the review content related to the popularly studied single-atom anchored on hollow carbon-based materials is relatively scarce. Considering the rapid development of studies on HSC-based catalysts in thermocatalytic and electrocatalytic CO2 conversion, a timely cutting-edge review on this topic is of imperative need.
This review aims to offer a critical overview of the latest progress of HSC-based materials in thermocatalytic and electro-catalytic CO2 conversion fields, with a discussion on the linkage of catalyst structure and activity (Fig. 1). The synthesis methods and functionalization strategies of HSC are also introduced. In the last section, an outlook for future research direction is proposed, targeting the current challenges.
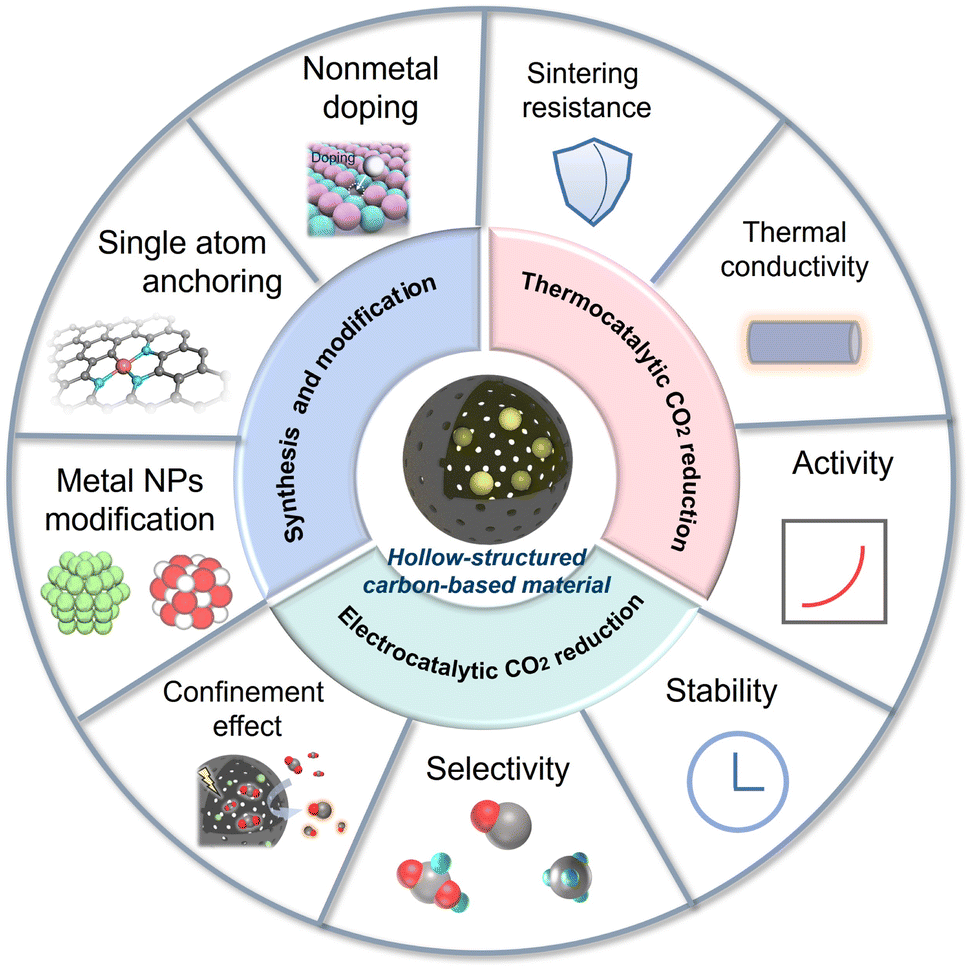 | ||
| Fig. 1 Overview of the topics covered in this review. This review presents the syntheses, functionalization strategies of hollow-structured carbon (HSC) based materials, as well as their fascinating applications in electrocatalytic and thermocatalytic CO2 conversion. | ||
2. Synthesis and functionalization of hollow-structured carbon materials
Generally, the HSC materials can be classified as hollow carbon spheres, hollow carbon nanotubes, hollow carbon polyhedrons, etc. motivated by the potentially utilitarian value for catalysis and energy storage, many efforts have been made toward the controllable synthesis and accurate design of hollow carbon with well-defined structures.55–57 In this section, the synthetic methods and functionalization strategies are discussed.2.1 Synthesis methods of HSC
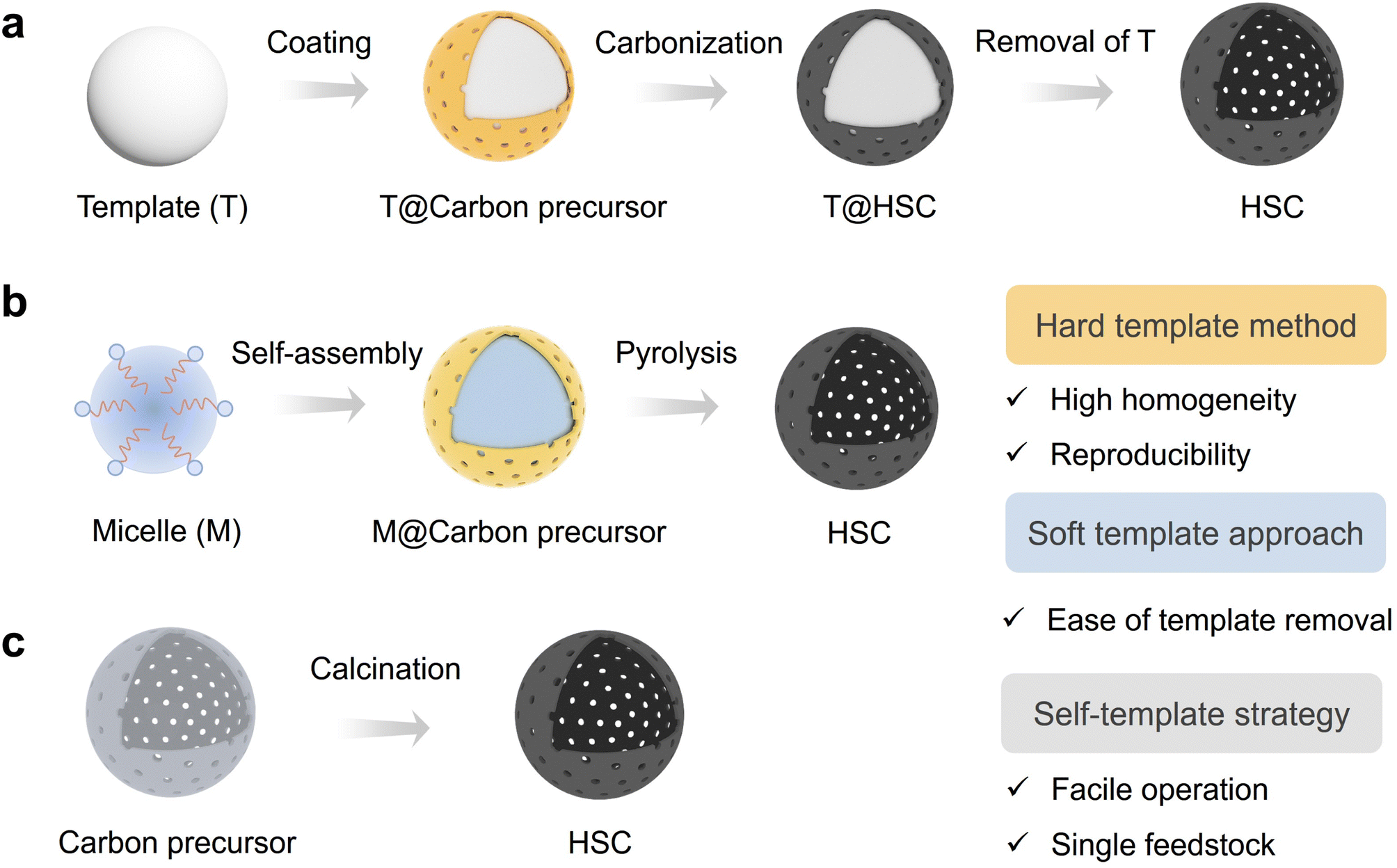 | ||
| Fig. 2 Synthetic methods for hollow structured carbon (HSC). (a) Hard template method, (b) soft template approach, and (c) self-template strategy. | ||
| Template | Carbon precursor | Template removal reagent | Hollow carbon type | Application | Ref. |
|---|---|---|---|---|---|
| SiO2 | Resorcinol and formaldehyde | HF | Mesoporous hollow carbon sphere | Supercapacitor | 59 |
| PMMA | Resorcinol and formaldehyde | Calcination | Hollow carbon sphere | Battery | 64 |
| PMMA | Dopamine | Calcination | Hollow carbon sphere | Electrocatalysis | 65 |
| SiO2 | Aniline | HF | Hollow carbon sphere | Supercapacitor | 66 |
| CaCO3 | Dopamine | HCl | Mesoporous hollow carbon sphere | Battery | 67 |
| SiO2 | Resorcinol and formaldehyde | NaOH | Mesoporous hollow carbon sphere | Microwave absorption | 68 |
| SiO2 | Dopamine | NaOH | Mesoporous hollow carbon sphere | Electrocatalysis | 69 |
| SiO2 | Resorcinol and formaldehyde | NaOH | Hollow carbon sphere | Thermal catalysis | 51 |
| CuO2 | 3-Aminophenol and formaldehyde | HCl, H2O2 | Hollow carbon sphere | Supercapacitor | 70 |
| Bi2S3 | L-Cysteine and resorcinol | Calcination | Nanotube | Electrocatalysis | 71 |
| SiO2 | Resorcinol and formaldehyde | HF | Hollow carbon sphere | Supercapacitor | 72 |
| Urchin-like nickel | Urea | HCl | Urchin-like hollow carbon | Electrocatalysis | 60 |
| SiO2 | Pyrrole | HF | Hollow carbon sphere | Battery | 73 |
| SiO2 | Resorcinol and formaldehyde | HF | Hollow carbon sphere | Battery | 74 |
| SiO2 | Resorcinol, formaldehyde and melamine | KOH | Hollow carbon sphere | Electrocatalysis | 75 |
| SiO2 | Resorcinol and formaldehyde | HF | Hollow carbon sphere | Oil emulsification | 76 |
| SiO2 | Dopamine | NaOH | Hollow carbon sphere | Microwave absorption | 77 |
| SiO2 | Resorcinol and formaldehyde | NaOH | Hollow carbon sphere | Electrocatalysis | 52 |
SiO2 spheres are often used as hard templates to fabricate the HSC material due to the advantages of high stability, good uniformity, and ease of preparation. According to the study of Zhang et al., mesoporous carbon hollow spheres (MCHS) were prepared using SiO2 core particle and resorcinol–formaldehyde (RF) resin as a hard template and a carbon precursor, respectively (Fig. 3a).59 As displayed in Fig. 3b and c, hollow cavity spaces were clearly seen in the spherical structure of MCHS. Notably, the pore size of carbon spheres can be manipulated by adjusting the synthesis conditions, such as the ratios of SiO2 precursors (TEOS/TPOS) or solvents (ethanol/water). Taking the TEOS/TPOS ratio as an example, as the proportion of TPOS increases, the pore size of MCHS enlarges. In contrast to TEOS, the polymerization and condensation speeds of TPOS are slower. Consequently, larger silica aggregates are easily formed in the case of high-fraction TPOS, which contributes to the large mesopore formation in the following interaction process with RF oligomers. Therefore, the optimized preparation parameter is of great significance to obtain the desired HSC material. In addition to SiO2, a metallic hard template is also available for the HSC preparation. Recent work from Li et al., for instance, provided a strategy to prepare a N-doped urchin-like hollow carbon loaded with single-Ni atoms using solid urchin-like Ni and urea (providing C, N sources) as raw materials (Fig. 3d and e), where the Ni not only served as a hard template to assist the creation of hollow structure, but also offered the Ni source for the formation of Ni single sites.60 Actually, the aforementioned method of integrating the metallic hard template with appropriate carbon sources may offer a new perspective for developing advanced nano-structured single-atom carbon-based catalysts.
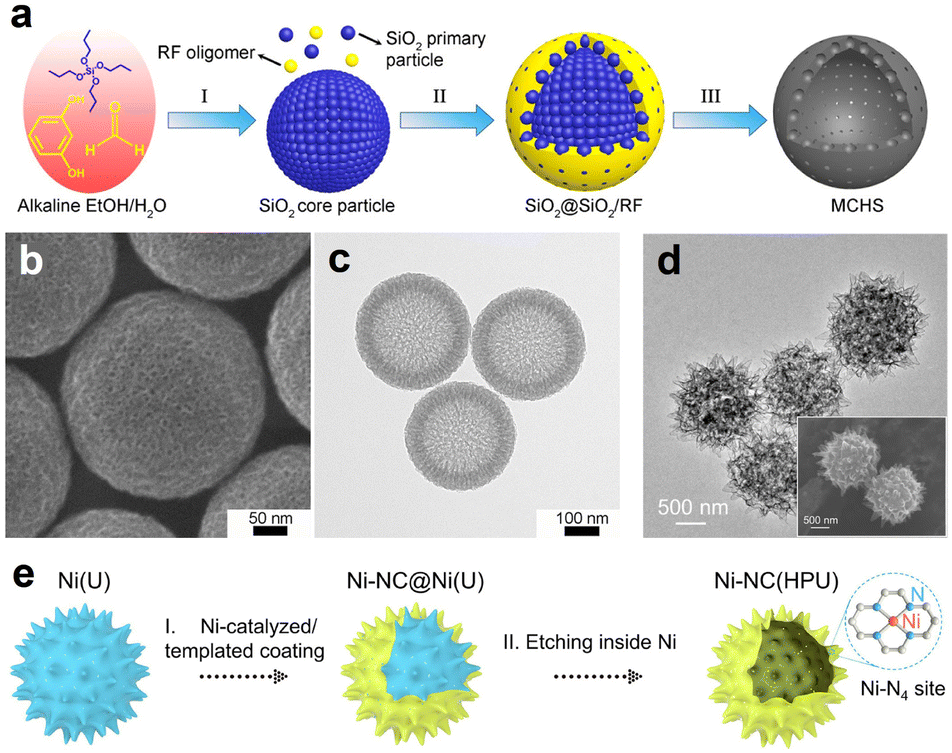 | ||
| Fig. 3 (a) Schematic illustration of the synthesis procedure for mesoporous carbon hollow spheres (MCHS). (b) SEM and (c) TEM images of MCHS. Reproduced with permission from ref. 59. Copyright 2016, American Chemical Society. (d) TEM image, inset of (d) SEM image, and (e) preparation for single-Ni atoms anchored on hollow porous urchin-like N-doped carbon (Ni-NC(HPU)). Reproduced with permission from ref. 60. Copyright 2022, Wiley-VCH. | ||
The soft templating approach for preparing HSC has gained considerable interest (Table 2), mainly due to the facile template fabrication and removal processes. In 2014, Wang et al. proposed a strategy to synthesize HSC using the mixed micelle converted from the P123 (EO20–PO70–EO20) and sodium oleate as a soft template;79 following this method, 2,4-dihydroxybenzoic acid and formaldehyde were served as carbon sources. Their results demonstrated that the diameter and shell thickness of HSC are highly related to the synthesis parameters, such as reaction temperature and the usage of polymer precursors.
| Preparation method | Carbon precursor | Template | Morphology | Application | Ref. |
|---|---|---|---|---|---|
| Soft template | Glucose | Latex | Hollow carbon sphere | Battery | 78 |
| 2,4-Dihydroxybenzoic acid and formaldehyde | P123 and sodium oleate | Hollow carbon sphere | Hydrogenolysis of 5-hydroxymethylfurfural | 79 | |
| Ribose | Oleic acid emulsion | Flasklike hollow carbon | Supercapacitor | 80 | |
| Glucose | Oleic acid emulsion | Hollow carbon sphere | Biochemistry and supercapacitor | 81 | |
| Aniline and pyrrole | F127 | Hollow carbon sphere | Sodium storage devices | 82 | |
| 2,4-Dihydroxybenzoic acid and hexamethylenetetramine | P123 and sodium oleate | Single-hole hollow carbon sphere | Liquid-phase adsorption | 63 | |
| Self-template | Melamine and formaldehyde resin | — | Hollow carbon microsphere | Supercapacitor | 83 |
| Poly(amic acid) | — | Hollow carbon microsphere | Supercapacitor | 84 | |
| Monocrystalline zeolitic imidazolate framework (ZIF-8) nanobubble | — | Hollow carbon nanobubble | Battery | 85 | |
| Deep eutectic solvent (urea, 2,5-dihydroxy-1,4-benzoquinone and ZnCl2) | — | Hollow carbon nanorod | Supercapacitor | 86 | |
| Poly(hexachlorocyclotriphosphazene-tannic acid-4,4′-sulfonyldiphenol) hollow nanosphere | — | Hollow carbon sphere | Electrocatalysis | 87 | |
| Polydopamine vesicle | — | Hollow carbon cage | Cathode for Li–S batteries | 88 | |
| Sulfur bridge covalent triazine framework sphere | — | S, N-doped hollow carbon sphere | Electrocatalysis | 89 | |
| Tetrahydrofuran-treated resorcinol-formaldehyde (RF) polymer sphere | — | Concave hollow carbon sphere | Anode material for Na/K-ion battery | 90 |
The preparation of HSC with a single opening on the shell is an attractive method for increasing the diffusivity and accessibility of the guest species to the interior surface, while preserving the porous structure. Therefore, based on the traditional soft templating technique, Yu and co-workers developed a single-hole hollow carbon sphere through a poly(ethylene glycol) (PEG)-assisted emulsion-templating method, where PEG molecule functioned as a reverse demulsifier to manipulate the structure of HSC (Fig. 4a).63 As revealed in Fig. 4b–d, hollow structured carbon spheres with a single hole can be observed, and the sizes of the hole and sphere are determined to be ∼38 nm and ∼138 nm, respectively. Additionally, by adjusting the molecular weight of PEG molecules, a closed-shell hollow carbon sphere, or bowl-like carbon can also be obtained. This work provides a new inspiration for the subtle preparation of HSC materials.
 | ||
| Fig. 4 (a) Schematic diagram of the synthesis for single-hole hollow carbon spheres (HCH). (b) SEM and (c) TEM images, and (d) diameter distribution of HCH. Reproduced with permission from ref. 63. Copyright 2022, American Chemical Society. | ||
Zhang et al. reported the fabrication of hollow carbon nanobubble with a shell thickness of only ∼10 nm by simply calcining hollow ZIF-8 in a N2 atmosphere (Fig. 5a).85 It was proposed that, the distinctive hollow structured carbon featuring with ultra-thin shell not only serves to shorten the distance of ion diffusion distance but also provides a more accessible surface area for ions. Consequently, hollow carbon nanobubbles showed enhanced ion storage performance, as compared to the solid carbon nanoparticles. This synthesis concept is expected to stimulate the exploitation of more multifunctional hollow carbon materials derived from MOFs. In a separate study, Zheng et al. synthesized N, S co-doped hollow mesoporous carbon spheres (N/S-HMCS) using sulfur-bridged covalent triazine frameworks (S-CTF) sphere as a precursor (Fig. 5b).89 The key point of this synthesis method is to control the pyrolysis temperature to regulate the polymerization degree of the inner and outer layers of S-CTF, which triggers the formation of the inner cavity. During the pyrolysis process, the volatile substance generated from the thermal decomposition of the central oligomer with a lower polymerization degree gradually migrated to the outside to reduce the surface energy and maintain the structural stability. With the temperature increase from 500 °C to 900 °C, the internal cavity size gradually became larger and the specific surface area increased from 12 m2 g−1 (N/S-HMCS500) to 331 m2 g−1 (N/S-HMCS900). This study proposes a promising route toward the controllable synthesis of hollow carbon material with tailored heteroatom doping.
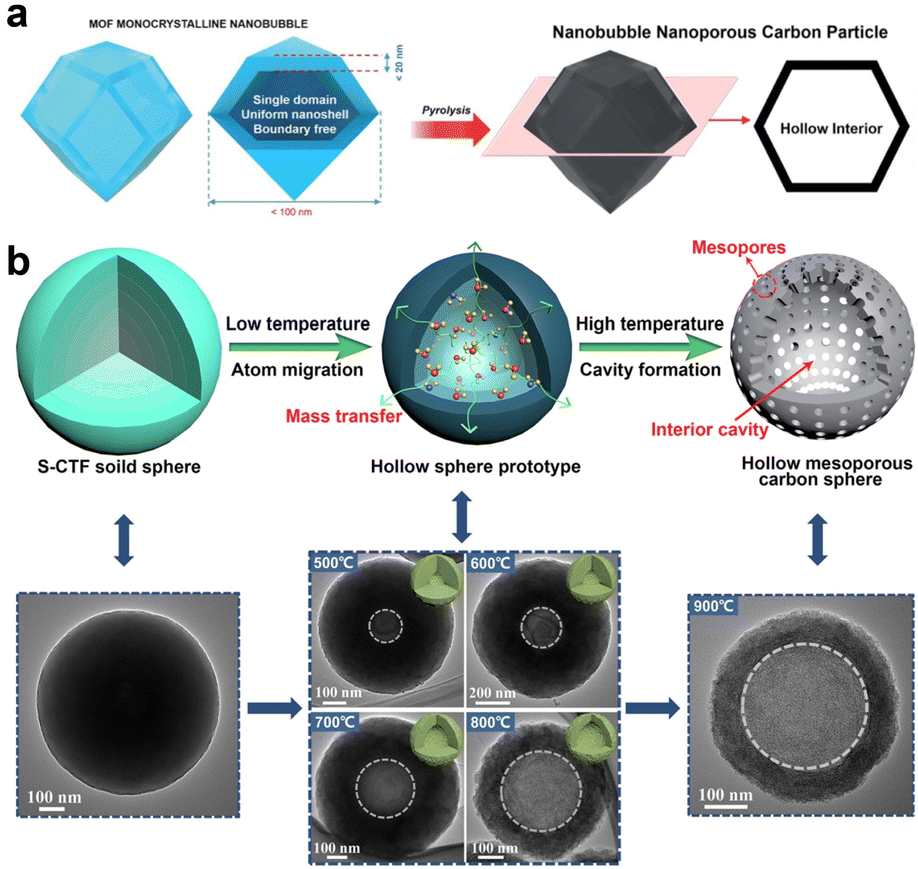 | ||
| Fig. 5 (a) Preparation of nanobubble carbon via the pyrolysis of MOF nanobubbles. Reproduced with permission from ref. 85. Copyright 2017, Royal Society of Chemistry. (b) The fabrication process of N, S co-doped hollow mesoporous carbon sphere (N/S-HMCS) and the corresponding TEM images. S-CTF represents the sulfur-bridged covalent triazine frameworks. Reproduced with permission from ref. 89. Copyright 2021, Elsevier. | ||
In general, the selection of an appropriate synthesis method for HSC should meet the demand of the intended application scenario. The hard template method is more recommended for applications requiring high product homogeneity and experimental reproducibility. Nevertheless, this approach still has some limitations, including the complex and time-consuming procedures and the use of corrosive reagents for template removal. In contrast, the soft template method offers a relatively easy template removal process; however, challenges arise concerning the maintenance of structure rigidity throughout subsequent applications. Although the template method shows some advantages in the aspect of controlling the particle size uniformity and dispersion of HSC, the self-templated method is still regarded as a promising route for achieving scalable production due to its facile operation and utilization of only a single feedstock.
2.2 Functionalization strategies
In addition to N, F doping is identified as an efficient way to tailor the electronic structure of carbon catalysts and enhance eCO2RR activity by forming a local positive charge area, and the local positive charge area can elevate COOH* (an important intermediate) adsorption and suppress the side-competing hydrogen evolution reaction (HER).100,101 To further increase the surface area, Ni et al. developed a F-doped cagelike carbon electrocatalyst, which exhibits excellent CO product selectivity at high overpotential.102 The results demonstrated that hollow morphology with abundant mesopores and micropores can facilitate the capture and diffusion of CO2 molecules. Additionally, a strong electric field formed at the edge site of the opening pores on the F-doped carbon shell can increase the partial concentration of electrolyte cations (K+), thus lowering the eCO2RR thermodynamic energy barrier and boosting the activation of CO2.
Besides single-element doping, multiple-element doping has recently been proven to be effective in synergistically improving the eCO2RR performance of HSC materials. Li et al. developed a N, S dual co-doped hollow carbon sphere, which showed a high CO faradaic efficiency (FECO) of 93% at a low potential of −0.6 V (vs. RHE), surpassing that of single-N-doped counterpart.103 The addition of S doping provided more active sites, and decreased the Gibbs free energy for the formation of COOH* intermediate, thus promoting the eCO2RR activity. Furthermore, N, B co-doped hollow carbon spheres reported by Cheng et al. also exhibited a high FECO of 95.1% at a low overpotential of 310 mV.104 It was suggested that doped B sites enhance CO2 adsorption, while N sites promote the hydrogenation of *CO2 to form COOH*.
Although encouraging progress has been made in developing non-metal-doped carbon materials, some issues still need to be solved. Taking N-doping carbon as an example. The identification of actual active sites in N-doped carbon catalyst is still controversial;105 because, during the doping process, different kinds of nitrogen configurations, including pyridinic-N, pyrrolic-N, and graphitic-N, will be introduced to the carbon framework, resulting in structural inhomogeneity.106 It is difficult to precisely control the N configuration. Furthermore, interactions between different types of nitrogen potentially exist, which may affect the CO2 catalytic reaction process.96 The combination of advanced in situ characterization techniques and DFT simulation is needed to further clarify the structure–activity relationship and understand the relevant reaction mechanism.
Normally, the isolated metal atoms with high surface energy are unstable and easy to migrate and coalesce.113 Integrating single-atom active sites with the aforementioned heteroatoms-doping HSC material prevents the aggregation of single atoms and endows HSC considerable catalytic activity simultaneously. For example, Pan et al. developed a highly dispersed Co–N5 site anchored on a hollow carbon sphere (Fig. 6a and b), and it showed high-efficiency CO2 electroreduction reactivity with FECO reaching 99.2% and 99.4% at −0.73 and −0.79 V vs. RHE, respectively (Fig. 6c).114 In contrast, cobalt phthalocyanine (CoPc, Co–N coordination number is 4) showed relatively low FECO, implying the positive roles of Co–N5 sites. As verified by the DFT calculation results (Fig. 6d), the excellent eCO2RR activity is associated with the existence of single-atom Co–N5 sites and the resulting enhanced CO2 activation, rapid formation of COOH*, and improved CO desorption. Similarly, by loading single-atom Ni–Nx sites on the well-regulated carbon structure of HSC-based catalysts, desired electrochemical eCO2RR performance can be achieved.115–117 Furthermore, atomically dispersed Ru(III) supported on a N-doped carbon sphere also showed satisfactory activity and stability in continuous CO2 hydrogenation test, owing to the strong binding strength between Ru(III) species and pyridinic N/pyrrolic N site.118
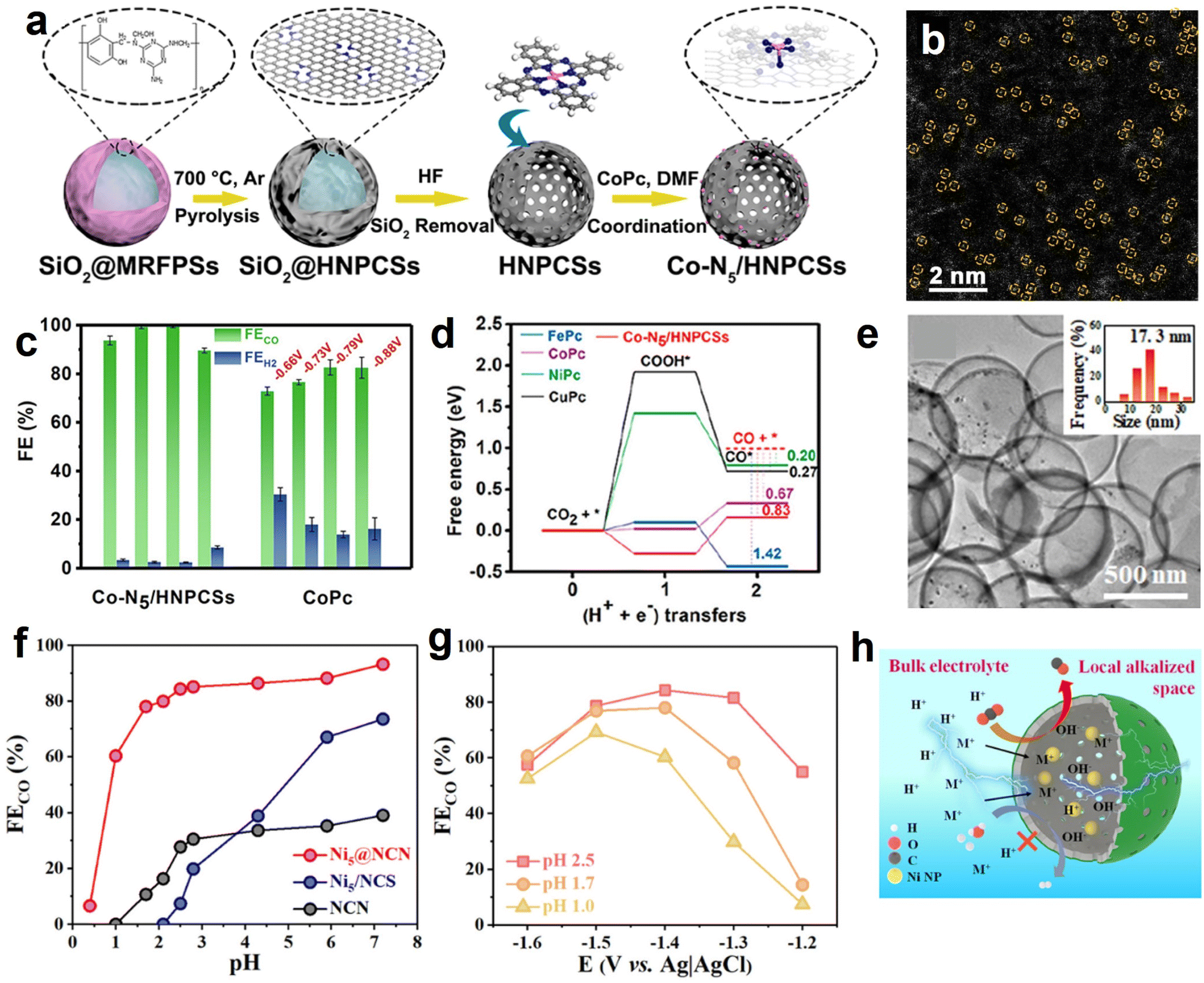 | ||
| Fig. 6 (a) Synthesis procedures and (b) magnified HAADF-STEM image of hollow N-doped porous carbon spheres with single-atom Co–N5 sites (Co-N5/HNPCSs). (c) Faradaic efficiencies for CO and H2 production at the given potentials over Co-N5/HNPCSs and CoPc. (d) Calculated eCO2RR free energy profiles of Co-N5/HNPCSs catalyst and MPc (M = Co, Fe, Ni, Cu). Reproduced with permission from ref. 114. Copyright 2018, American Chemical Society. (e) TEM image of Ni NPs@N-doped carbon nanocages (Ni@NCN). The inset shows the metal NPs size distribution. (f) FECO and (g) current density of the best-performing Ni@NCN sample in acidic electrolyte condition. (h) Schematic diagram of the pH environment and ions transportation around the Ni@NCN catalyst. Reproduced with permission from ref. 46. Copyright 2022, American Chemical Society. | ||
As mentioned above, single-atom modification plays a significant role in improving the eCO2RR performance of HSC-based materials. However, some important aspects still need to be further investigated: (1) the coordination environment has great impact on the electronic structure of SACs, so unveiling the relationship between the coordination number or connecting heteroatom type of SACs and eCO2RR activity is important; (2) at present, most of the SACs syntheses are still at the laboratory stage; a universal approach for large-scale SACs production is required to meet the demands of practical applications;119 (3) some studies suggest that single-atom site configuration only shows high reactivity in the primary reaction process and is inadequate for the subsequent catalytic reaction steps involving multiple electron transfer and intermediate conversion, accordingly, developing novel catalysts with dual atomic sites may overcome this limitation.120,121
In view of the fact that HCS coupling with metal NPs can build a high-efficiency nanoreactor, Liu et al. designed a hollow N-doped carbon nanocage catalyst with Ni nanoparticles being encapsulated within the cavity (Ni@NCN) (Fig. 6e).46 Benefiting from nanoconfinement, the optimized sample (Ni5@NCS) showed excellent eCO2RR activity, achieving a high FECO of 93.2% in neutral media. Furthermore, Ni5@NCS likewise exhibited decent performance in acid conditions, markedly exceeding the control samples of non-hollow-structured Ni/N-doped carbon sphere (Ni-NCS) and Ni-free N-doped carbon nanocage (NCN) (Fig. 6f). It is noteworthy that the FECO of Ni5@NCS can achieve 84.3% even in the electrolyte of pH 2.5 at −1.4 V vs. Ag/AgCl (Fig. 6g). The superior activity is ascribed to the local pH-regulator effects of hollow carbon nanoreactor by concentrating the OH− ions within the cavity (Fig. 6h). Specifically, the consumption rate of the H+ in the hollow carbon nanocage is faster than the replenishment rate of H+ because of the proton transport limitation, leading to a local alkaline environment and suppressing the competing hydrogen evolution reaction (HER). Recently, the effectiveness of metal NPs and HSC composite in electrochemical CO2 reduction for CO production has also been demonstrated on the Ni NPs@N-doped nanotube systems.125,126 Previous studies have shown that alloy NPs show superior performance over monometallic NPs in CO2 activation and formate formation.36,127 Motivated by this, our group developed a PdCu alloy NPs confined within hollow carbon sphere catalyst (PdCu-N@HCS) (Fig. 7a and b), which showed an excellent thermal-catalytic CO2 hydrogenation activity, delivering the highest turnover number hydrogenation activity, delivering the highest turnover number (TON) of 1432 at 100 °C for 24 h compared to single-metal-component samples (Fig. 7c);49 DFT calculation result demonstrated that the alloying of Pd with Cu effectively reduces the energy barrier of the H2 dissociation, and is capable of facilitating the HCO3− transformation.
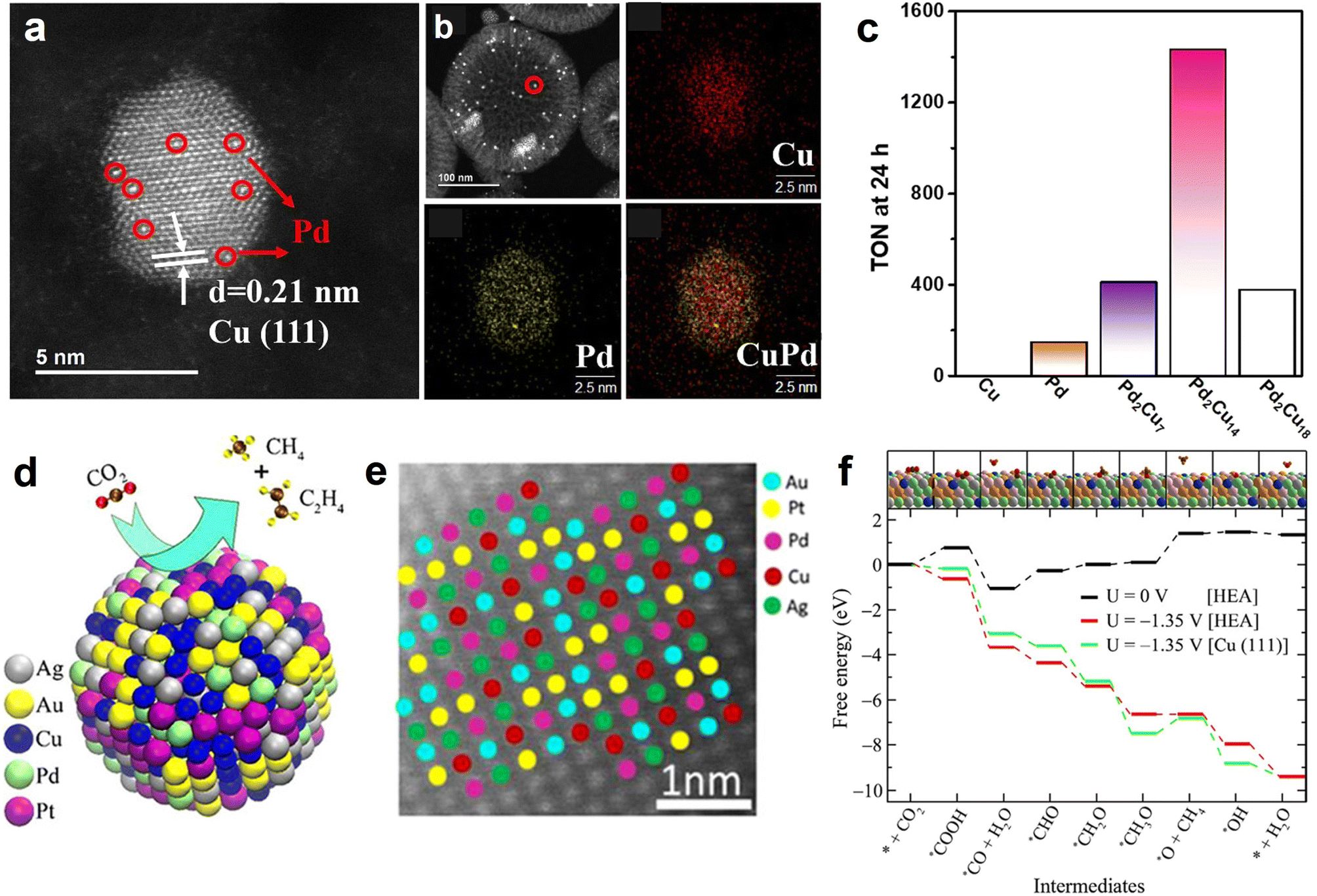 | ||
| Fig. 7 (a) Magnified HAADF-STEM image of CuPd alloy NPs and (b) STEM image and the corresponding EDS elemental maps of Cu, Pd and CuPd of PdCu-N@MHCS sample. (c) Effect of the Pd/Cu molar ratio on PdxCuy-N@MHCS for the hydrogenation of CO2 to formate. Reproduced with permission from ref. 49. Copyright 2021, American Chemical Society. (d) Illustration of electrochemical CO2 conversion and (e) HR-STEM image of HEA alloy NPs (AuAgPtPdCu). (f) Comparision of eCO2RR free energy on AuAgPtPdCu HEA and Cu (111). The Pt, Pd, Ag, Au, Cu, C, O, and H atoms are shown in gray, green, pink, yellow, blue, brown, red, and orange, respectively. Reproduced with permission from ref. 128. Copyright 2020, American Chemical Society. | ||
In conclusion, metal NPs coupled with HSC showed satisfactory catalytic activity in some electrocatalysis and thermal catalysis applications. However, the activity improvement may still profit from the consideration of the following aspects. Incorporating HSC with novel high-entropy alloys (HEAs), a new class of multi-component alloys that possess numerous advantages (e.g., flexible component adjustability, high oxidation resistance and excellent corrosion resistance), is expected to design high-efficiency catalysts. Recently, Nellaiappan et al., delicately fabricated a HEA catalyst (AuAgPtPdCu) (Fig. 7d and e), which can be used for efficient electrochemical CO2 conversion, with the FE of 67.5% for gaseous hydrocarbons (CH4, C2H4) at −0.3 V vs. RHE;128 the free energy diagram of eCO2RR results showed that, at a given potential of −1.35 V, the reaction profiles of HEA and Cu (111) are downhill in general. Notably, at the *OCH3 to *O transition step, the energy barrier on Cu (111) surface is obviously larger than that of HEA (Fig. 7f), suggesting that the thermodynamic favorability of eCO2RR on the HEA system is higher than on pristine Cu. This work demonstrates that HEA can effectively alter the reaction energy barrier of the intermediates, and accelerate the CO2 multi-electron reduction process. In another study, Pedersen et al. corroborated the feasibility that HEA material with optimized components and content can be employed as an efficient eCO2RR catalyst, by using DFT calculation and machine learning.129 In addition, the introduction of aminopolymers into metal NPs@HSC catalyst system is also worth studying, because aminopolymers contain plenty of amine groups with basicity that can promote CO2 capture ability.130 Another merit of aminopolymer is the anchoring of metal ions prior to the metal nucleation, thus facilitating the dispersion of metal NPs.131 Our previous works have proved the positive effects of aminopolymer in the nanoconfined reactor for CO2 electroreduction and thermo-catalytic conversion.52,132 The above-mentioned directions may provide more possibilities for designing high-performance metal NPs@HSC-based catalysts.
3. Electrocatalytic CO2 conversion of hollow carbon-based materials
Electrochemical CO2 conversion to produce fuels and chemicals is considered as an ideal option for responding to the increasing energy supply requirement and environmental concerns.135,136 Typically, a CO2 electroreduction cell comprises two electrodes: cathode (for eCO2RR) and anode (for water oxidation). By applying an external bias, the eCO2RR occurs at the three-phase interface of the cathode, electrolyte, and the adsorbed CO2, with value-added products (CO, HCOO−, C2H4, etc.) being obtained. The commonly used reactors for eCO2RR can be categorized into three types: (1) H-type cell (Fig. 8a), (2) liquid-flow cell (Fig. 8b), and (3) membrane electrode assembly (MEA) cell (Fig. 8c). The H-type cell involves the use of aqueous electrolytes, wherein the solubility of CO2 is relatively low (approximately 34 mM). Therefore, CO2 electrolysis on the H-type cell is susceptible to the influence of mass transport limitations, leading to low current densities (<100 mA cm−2).110,133 In the liquid-flow cell, an ion exchange membrane is used to separate the cathode and anode to prevent the CO2 reduction products in the cathode from transferring to the anode and undergoing re-oxidation. Additionally, the anolyte and catholyte are continuously cycled. Different from the H-type cell, eCO2RR occurs at the gas diffusion electrode (GDE), overcoming the solubility and mass transport limitations of CO2 in aqueous electrolytes and enabling a high current density to be achieved (>100 mA cm−2).134 In contrast to H-cell and liquid-flow cells, the anode and cathode in MEA cell are directly compressed with the ion exchange membrane, forming a sandwich structure (Fig. 8c). This zero-gap structure significantly reduces the distance between the anode and cathode, thus minimizing ohmic resistance and markedly enhancing energy efficiency and stability.137 Generally, the H-type cell is suitable for the fundamental study of eCO2RR as well as the screening of efficient catalysts, due to the ease of operation and the rapid testing. Among the above-mentioned eCO2RR cells, the MEA cell holds the most promising industrial potential for eCO2RR, as it shows better energy utilization efficiency and stability. The development of more high-performance membranes will benefit the further improvement of the efficiency for MEA cells.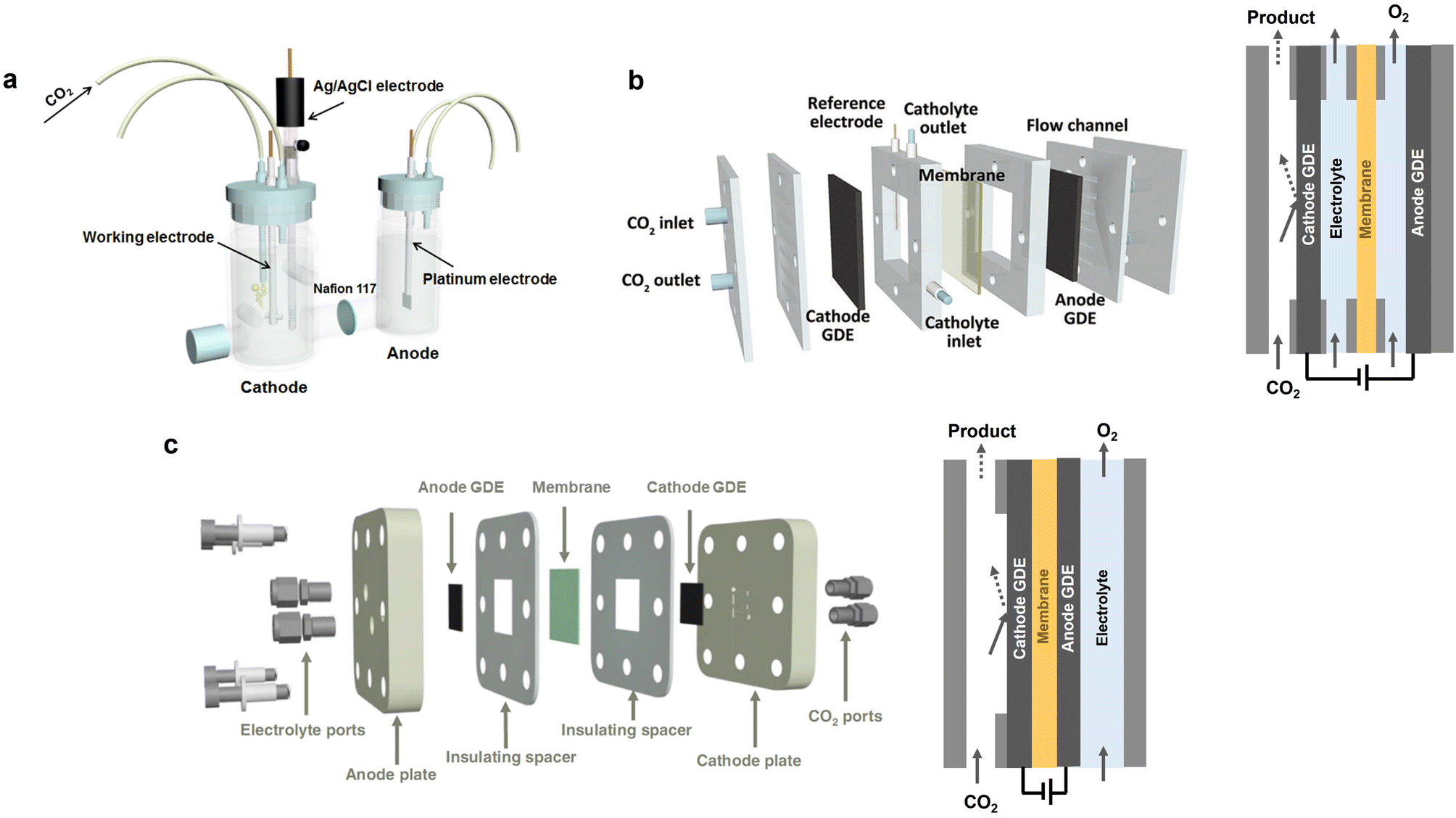 | ||
| Fig. 8 Schematic illustrations of the eCO2RR reactors. (a) H-cell. Reproduced with permission from ref. 133. Copyright 2018, Elsevier. (b) Liquid-flow cell. Reproduced with permission from ref. 134. Copyright 2021, Wiley-VCH. (c) MEA cell. Reproduced with permission from ref. 110. Copyright 2023, Springer Nature. | ||
The choice of electrolytes is important to increase the activity and selectivity of electrochemical CO2 reduction.138,139 Taking the widely used alkali metal bicarbonate electrolytes as examples, the size of alkali metal cation is able to affect the hydrolysis constants of hydrated cations, thereby altering the local pH value and CO2 concentration around the electrode; eventually, it will cause a change of the selectivity toward C1 products on Ag and C2 products on Cu.140 Additionally, the selectivity of eCO2RR is also associated with the concentration of cations. For instance, Marcandalli et al. reported that at low potentials (>−0.4 V vs. SHE), a high concentration of Na+ cations is beneficial for enhancing the FECO, contrasting with the situation at more negative potentials (<−0.4 V vs. SHE), where a low concentration of Na+ cations is more favorable for suppressing hydrogen evolution and improving the FECO.141 In general, the electrolyte type can serve as a crucial design parameter, enabling the tailoring of the electrocatalyst–electrolyte interfacial environment to obtain desired products in the reduction of CO2.
eCO2RR involving multiple proton-coupled electron transfer processes usually requires overcoming high thermodynamic and kinetic barriers.142,143 The development of reliable electrocatalysts is of great significance to regulate the charge transfer and reaction intermediate adsorption/activation, overcoming the aforementioned obstacles, and achieving high catalytic activity and selectivity. As a result of recent research efforts, a series of high-performance HSC-based electrocatalysts have emerged (Table 3), which have creatively exploited the structural advantages of HSC, broadening the understanding of the mechanism behind eCO2RR. In this section, the discussion mainly focuses on product selectivity with the aim of providing insights into the structure–activity relationships of HSC-based materials.
| Catalyst | Faradaic efficiency (%) | Partial current density (mA cm−2) | Potential (V vs. RHE) | Electrolyte | Reactor type | Ref. |
|---|---|---|---|---|---|---|
| Co-N5/HNPCSs | CO, 99.4 | ∼4.5 | −0.73 | 0.2 M NaHCO3 | H-type cell | 114 |
| Ni/N-doped carbon sphere | CO, ∼80 | ∼4 | −0.78 | 0.5 M KHCO3 | H-type cell | 116 |
| F-doped cagelike carbon | CO, 88.3 | ∼32 | −1.0 | 0.5 M KHCO3 | H-type cell | 102 |
| Ni SAs/N-doped carbon sphere | CO, 95.1 | ∼7.6 | −0.8 | 0.5 M KHCO3 | H-type cell | 155 |
| Ni-N4/carbon sphere | CO, 95% | 10.5 | −1.0 | 0.5 M KHCO3 | H-type cell | 115 |
| Ni/N-HCS | CO, 97% | 10 | −0.8 | 0.5 M KHCO3 | H-type cell | 117 |
| N, B co-doped HCS | CO, 95.1 | ∼1 | −0.42 | 1.0 M KHCO3 | H-type cell | 104 |
| Ni/N doped hollow carbon plates | CO, ∼100 | 18.2 | −1.0 | 0.5 M KHCO3 | H-type cell | 156 |
| Ni-N3/NHCSs | CO, 98.57 | ∼14.2 | −0.87 | 0.5 M KHCO3 | H-type cell | 146 |
| Ni NPs/N doped carbon cage | CO, 93.2 | 17.1 | −0.8 | 0.5 M KHCO3 | H-type cell | 46 |
| Ni-N doped HCS-Micro/Meso | CO, 98.3 | 16.2 | −1.0 | 0.5 M KHCO3 | H-type cell | 38 |
| Ni SAs/N-doped urchin-like carbon | CO, 91 | 24.7 | −0.8 | 0.5 M KHCO3 | H-type cell | 60 |
| Hollow carbon with CoN4O and ZnN4 dual atomic sites | CO, 92.6 | 15.57 | −1.0 | 0.5 M KHCO3 | H-type cell | 157 |
| Ni SAs/N-doped carbon sphere | CO, 94.91 | 15.35 | −0.8 | 0.5 M NaHCO3 | H-type cell | 147 |
| CO, 98.41 | 100 | — | 1 M KHCO3 | Flow cell | ||
| SnO2/carbon sphere | Formate, 54.2 | Formate, 3.7 | −0.9 | 0.1 M KHCO3 | H-type cell | 158 |
| CO, 21.8 | CO, 1.4 | |||||
| Bi NRs@NCNT | Formate, 90.9 | 5.9 | −0.9 | 0.1 M KHCO3 | H-type cell | 71 |
| Cu/HCS | Formate, 82.4 | 26 | −0.81 | 0.5 M KHCO3 | H-type cell | 41 |
| SnS2−x/NHCS | Formate, >80 | ∼30 | −1.2 | 0.5 M KHCO3 | H-type cell | 159 |
| Cu cluster/HMCS | C2+ products (C2H5OH and C2H4), 88.7 | ∼240 | −1.0 | 1.0 M KOH | Flow cell | 37 |
| Cu2O/NCNT | C2+ products (C2H5OH and C2H4), 77.61 | 16.79 | −1.1 | 0.1 M KHCO3 | H-type cell | 160 |
3.1 Selective production of CO or syngas (CO and H2)
Atomically dispersed metal–nitrogen sites (M–Nx) modified carbon materials are widely studied in electrocatalysis, especially electrocatalytic CO2 reduction, due to the well-tuned electronic characteristic and appropriate binding strength for the reaction intermediate. The merits of abundantly accessible sites, unique cavity space, and good permeability enable HSC materials to be appealing supports for anchoring the atomically dispersed M–Nx sites.Notably, a majority of the recent studies focus on HSC-based catalysts with Ni–Nx sites and their electrochemical CO2-to-CO conversion applications (Table 3). Herein, we use this type of material as the main line to introduce their CO2-to-CO applications. To begin with, Xiong et al. developed a single-atom hollow carbon sphere catalyst containing Ni–N4 atomic sites for eCO2RR to CO.115 The best-performing sample (Ni-HMCS-3-800) delivered an extremely high turnover frequency value of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 608 h−1, and the FECO above 95% at a wide potential range (−0.7 to −1.1 V vs. RHE) (Fig. 9a and b). It was demonstrated that the remarkable activity originated from the improved CO2 adsorption and activation capacity, which are associated with the optimization of the geometrical structures of carbon support, including the shell thickness and compactness. Besides, this work also highlighted the importance of pore structure in eCO2RR; the appropriate mesopore size distribution (∼7–10 nm) endows Ni-HMCS-3-800 with enhanced diffusion and overflow of the reactants. As a result, Ni-HMCS-3-800 showed higher catalytic activity in comparison to the Ni-HMCS-3-700 sample with a smaller mesopore size (∼3.1 nm).
608 h−1, and the FECO above 95% at a wide potential range (−0.7 to −1.1 V vs. RHE) (Fig. 9a and b). It was demonstrated that the remarkable activity originated from the improved CO2 adsorption and activation capacity, which are associated with the optimization of the geometrical structures of carbon support, including the shell thickness and compactness. Besides, this work also highlighted the importance of pore structure in eCO2RR; the appropriate mesopore size distribution (∼7–10 nm) endows Ni-HMCS-3-800 with enhanced diffusion and overflow of the reactants. As a result, Ni-HMCS-3-800 showed higher catalytic activity in comparison to the Ni-HMCS-3-700 sample with a smaller mesopore size (∼3.1 nm).
 | ||
| Fig. 9 (a) Turnover frequency (TOF) and (b) faradaic efficiencies of CO and H2 of as-synthesized samples at given potentials. Reproduced with permission from ref. 115. Copyright 2020, Wiley-VCH. (c) Ni K-edge fitting curve of Ni-NC/NHCSs-600 in R space. The upper and lower traces represent the magnitude and imaginary part, respectively. The inset shows the model of Ni-N3 moiety. Reproduced with permission from ref. 146. Copyright 2022, Elsevier. (d) COMSOL multiphysics finite element simulation results of CO2 diffusion time in 0.5 M NaHCO3 aqueous solution for solid sphere structure (model I, Ni-NC-based electrode) and hollow nano-reactor structure (model II, I-Ni SA/NHCRs-based electrode). (e) Bottom and (f) average concentration of CO2 for model I and model II via probe monitoring. (g) Calculated eCO2RR free energy profiles of Ni-C3N1 and Ni-C2N2 moiety. Reproduced with permission from ref. 147. Copyright 2023, Wiley-VCH. | ||
Recently, the effects of the coordination environment of the central metal Ni–Nx on eCO2RR activity have received considerable attention.144 As documented in the literature, the energy barrier for *COOH intermediate formation on the Ni–N3 site is lower than that of the Ni–N4 site, suggesting coordinatively unsaturated Ni–N3 may be favorable for eCO2RR.145 Accordingly, Gong et al. synthesized Ni–N3 units embedded in nitrogen-doped hollow carbon spheres (Ni-NC/NHCSs) by a hard template method using NiPc (Ni source) and dopamine (carbon and nitrogen sources) as precursors (Fig. 9c).146 The optimized catalyst (Ni-NC/NHCSs-600) exhibited a maximal FECO of 98.57% at −0.87 V vs. RHE, which is significantly larger than the value of its counterpart NiPc@NHCSs (FECO = 19.61%). Additionally, in sharp contrast to NiPc@NHCSs (stability < 2000 s), Ni-NC/NHCSs-600 showed a long-term stability of 14 h with a negligible current attenuation. In a separate study, the researchers of the same group rationally developed a novel catalyst consisting of Ni-C3N1 moiety confined in hollow carbon reactors (I-Ni SA/NHCRs), which was reported to achieve a high FECO of 94.91% at −0.80 V in H-cell, and showing higher CO partial current density (−15.35 mA cm−2) than that with outer Ni-C2N2 moiety (O–Ni SA/NHCRs, −12.06 mA cm−2), or without hollow structure (Ni-NC, −5.39 mA cm−2).147 More importantly, a high FECO of up to 98.41% was achieved for I-Ni SA/NHCRs at 100 mA cm−2 in a flow cell with 1 M KHCO3 electrolyte, implying a huge potential for practical application. It was proposed, based on COMSOL multiphysics finite element simulations, that the diffusion time of CO2 molecules in hollow nano-reactor structure (model II, I-Ni SA/NHCRs-based electrode) (CO2 diffusion time, ∼1.25 s) is much faster than that of solid sphere structure (model I, Ni-NC-based electrode) (CO2 diffusion time, >2.0 s) (Fig. 9d), as a result of the intrinsically structural virtue; in the meantime, the bottom and average concentration of CO2 in the above two models was dynamically monitored via the simulation probe (Fig. 9e and f), and the results fully confirmed the facilitated CO2 diffusion rate in hollow structural carbon. Obviously, it is conducive to the accessibility of isolated Ni active sites in I-Ni SA/NHCRs to CO2 molecules. The comparison of calculated free-energy diagrams for eCO2RR of Ni-C3N1 and Ni-C2N2 configurations is shown in Fig. 9g, and the energy barrier for the protonation of *CO2 to *COOH over Ni-C3N1 moiety (0.60 eV) is lower than the value of Ni-C2N2 structure (0.94 eV), implying that the formation of the key *COOH intermediate is more thermodynamically favorable on Ni-C3N1 configuration. These works provide inspiration for designing efficient HSC materials with tailored coordination of single atoms.
Despite most of the current studies being dedicated to improving CO selectivity and suppressing the competitive hydrogen evolution reaction from CO2 reduction, the production of the mixture of CO and H2 (syngas) is of great applied relevance. The syngas with specific ratios can serve as the feedstock for the synthetic chemical industry (e.g., Fischer–Tropsch synthesis), especially in the range of 0.25–3.33 CO/H2.148 HSC-based materials are reported to function as feasible catalysts for controllable syngas production. For example, Song et al., reported a nitrogen and cobalt co-doped hollow carbon catalyst (Co–HNC), with the Co–C2N2 moieties acting as catalytic sites for CO2 reduction and the N functional groups (i.e., pyridinic and graphitic N) serving as HER sites.149 The obtained Co-HNC sample showed a faradaic efficiency of nearly 100% and a production rate of ∼425 mmol g−1 h−1 for syngas evolution at −1.0 V vs. RHE. Notably, the syngas ratio is close to the desired 1/2 (CO/H2) that is appropriate for downstream methanol production.150 The hollow structure and sponge-like thin shell were claimed to be helpful for accelerating the mass transport and maximizing the catalytically active area, rendering the catalyst an excellent electrocatalytic performance. Additionally, in our previous work, a tunable syngas production (CO/H2 ratio, 1.09 to 2.54) could be achieved from eCO2RR over a wide electrochemical window from −0.7 to −1.1 V vs. RHE, using an aminopolymer-modified hollow carbon sphere incorporating Ag NPs catalyst (Ag-P@HCS).52 Detailed characterizations suggested that the introduction of aminopolymers led to the finely increased dispersion of Ag NPs and improved CO2 affinity of Ag-P@HCS, thus optimizing its eCO2RR performance. The work emphasized the positive effects of aminopolymers in HSC-based eCO2RR systems.
3.2 Selective production of formate
As an important liquid-phase product of eCO2RR, formate has extensive applications in the pharmaceutical industry, animal feed, and hydrogen energy transportation.151 Nowadays, indium (In), copper (Cu), and tin (Sn)-containing catalysts are widely studied in eCO2RR for CO2-to-formate production,152–154 owing to the favorable *OCHO formation and suitable binding intensity with *HCOOH; but some limitations towards pragmatic application still exist, such as unsatisfied selectivity, moderate stability, and large overpotential. In fact, except for the shape and constituent control of the metal/oxides NPs, the electrocatalytic performance is also determined by the electronic and morphologic characteristics of carbon substrates to some extent. Encouragingly, some interesting works have been reported on constructing metal-carbon hollow nano-structured hybrids with space-confinement effects and abundant exposed active sites to address the problems and achieve desirable electro-reactivity. As exemplified by the study conducted by Zhang et al., the pipet-like bismuth (Bi) nanorods semifilled in N-doped carbon nanotubes (Bi-NRs@NCNTs) catalyst (Fig. 10a) delivered a highly efficient CO2-to-formate electro-conversion, showing a high selectivity for formate production with the peak faradaic efficiency value of 90.9% at −0.9 V vs. RHE (Fig. 10b).71 Different from Bi granules, no obvious current density attenuation was observed on Bi-NRs@NCNTs throughout the 24 h electrolysis (Fig. 10c), implying its structural superiority. Specifically, the spatial restriction function of NCNTs efficiently prevents the self-aggregation and oxidation of metallic Bi nanorods, ensuring good durability during long-term testing. Meanwhile, the results demonstrated that the synergy of nanocapillary and nanoconfinement effects induced by NCNTs contributed to the accelerated mass transfer, improved CO2 adsorption, and concentrated CO2 around the active sites. This work not only highlights the functions of HSC material in CO2 valorization, but also provides a promising design philosophy on HSC-based material for energy conversion and beyond.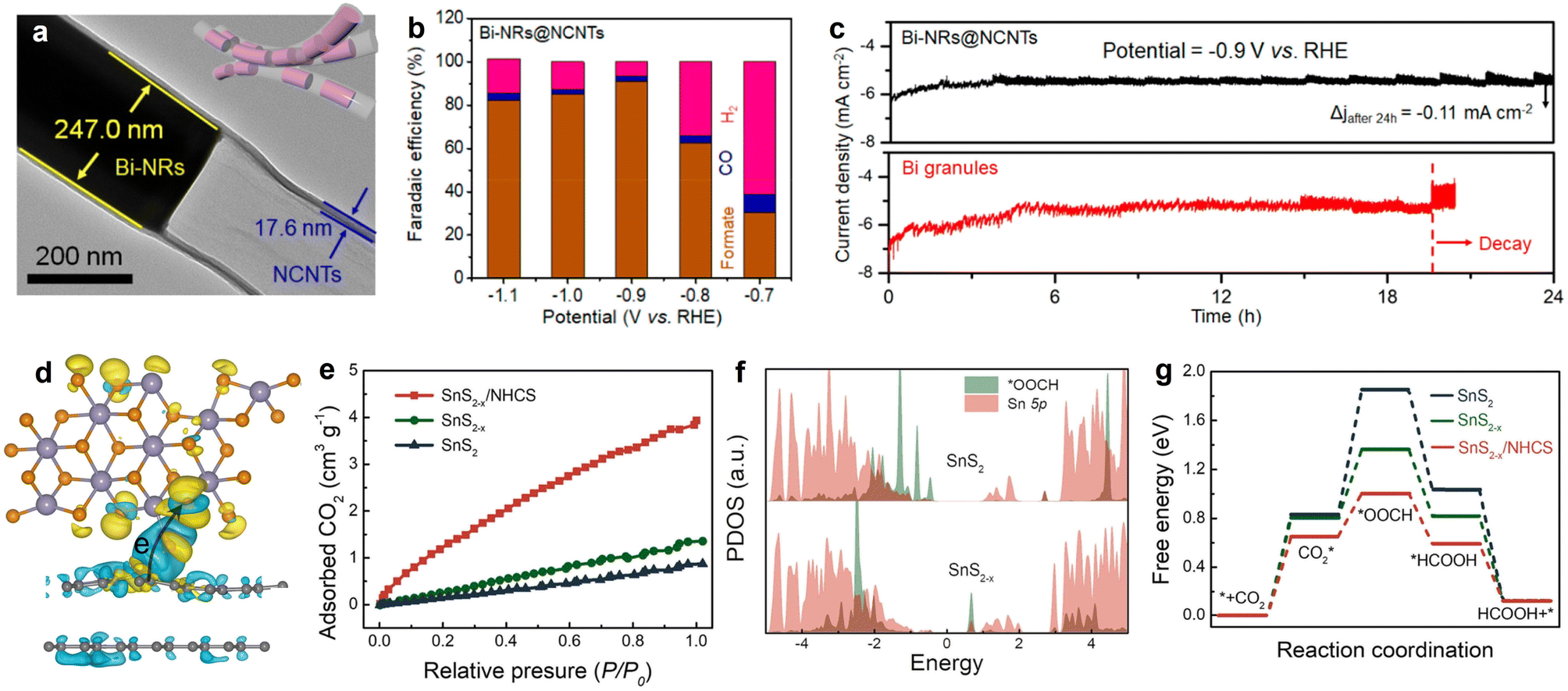 | ||
| Fig. 10 (a) HRTEM image and morphology illustration (inset) of Bi-NRs@NCNTs. (b) Faradaic efficiencies of the eCO2RR products of Bi-NRs@NCNTs. (c) Stability comparison of Bi-NRs@NCNTs and Bi granules. Reproduced with permission from ref. 71. Copyright 2021, American Chemical Society. (d) Charge density difference diagram SnS2−x/N-doped carbon. (e) CO2 adsorption isotherms. (f) Partial density of states (PDOS) of *OOCH and Sn 5p orbital for SnS2 and SnS2−x. (g) Calculated CO2RR free energy profiles. Reproduced with permission from ref. 159. Copyright 2022, Elsevier. | ||
In another example, Du et al. developed a hollow nano-structured hybrid electrocatalyst consisting of Cu, Cu2O, and nitrogen-doped carbon sphere (HCS/Cu) for highly selective formate production from eCO2RR.41 The best-performing sample HCS/Cu-0.12 showed 82.4% FEformate with a current density of 26 mA cm−2 at −0.81 V vs. RHE, in which the optimized Cu/C heterogenous interfaces could be responsible for the enhanced electron transport and the resulting excellent activity.
Very recently, nitrogen-doped hollow carbon spheres (NHCS) were employed to couple with tin disulfide containing sulfur vacancies to construct a catalyst (SnS2−x/NHCS) with improved CO2 activation ability and excellent conductivity.159 DFT simulation analysis confirmed that a migration channel of electrons was built with the direction from NHCS to SnS2−x (Fig. 10d). Benefited from the synergy of S defects and the integration with NHCS, SnS2−x/NHCS exhibited the highest uptake of CO2 (Fig. 10e). Moreover, simulations of projected density of states (PDOS) for *OOCH and Sn 5p on SnS2−x show a larger overlapping area than on its SnS2 counterpart, indicating the stronger interaction between *OOCH and SnS2−x (Fig. 10f). In addition, significantly decreased energy barriers for *CO2 formation and *OOCH generation can be observed on SnS2−x/NHCS compared with those of SnS2 and SnS2−x, signifying the promoted CO2 activation and conversion capacities (Fig. 10g). As a consequence, the partial current density and faradaic efficiency of SnS2−x/NHCS for formate production increased to 3.4 and 1.2 times, respectively, in comparison to the value of pristine SnS2. In general, the work disclosed the fundamental correlation between catalyst structure and performance, which may trigger more consideration of the material design by coupling defect engineering and HSC material with matched energy-level structure.
3.3 Selective production of multicarbon products
Of the prevalent eCO2RR HSC-based electrocatalysts, most mainly produce C1 products (CO or HCOO−), as presented in Table 3. The production of energy-dense and valuable multi-carbon (C2+) oxygenates and hydrocarbons, such as ethylene (C2H4) and ethanol (C2H5OH), is of great value for industrial manufacture, but the relevant reports are relatively rare; the primary obstacle lies in the difficulty of coupling C–C bonds.161Cu-based catalysts are of particular interest in the field of eCO2RR and are capable of converting CO2 to a series of C1/C2 products. However, suffering from product complexity and unsatisfactory activity, more efficient Cu-based catalysts are required. Considering the structural features of HSC materials for the carbonaceous intermediates, Pan et al. designed hollow mesoporous carbon spheres confining Cu cluster catalyst (Cu/HMCS) for improving the C2 selectivity.37In situ Fourier transform infrared spectroscopy (FTIR) analysis revealed that, under the aid of nanoconfinement provided by HMCS, with suitable Cu cluster loading, the conversion of *CO to *CHO could be facilitated; additionally, the nanocavity protection effects of HMCS allowed to boost the C–C bond coupling to generate C2 products. As a result, the sample (Cu/HMCS-20%) with optimized conditions exhibits a C2 (C2H4 and C2H5OH) faradaic efficiency of 88.7% at −1.0 V vs. RHE. This work provides insights for designing robust catalysts with high C2 selectivity.
In addition to hollow carbon spheres, carbon nanotubes (CNTs) are also commonly used in electrocatalysis; they can not only offer excellent conductivity, but also prevent the agglomeration of catalytic components.162 Liu et al. investigated the surface modification of N-doped CNTs on Cu2O NPs for enhanced CO2-to-C2 electro-conversion.160 The Cu2O NPs-NCNT sample showed a maximal C2 (C2H4 and C2H5OH) faradaic efficiency of 77.61% at −1.1 V vs. RHE, suppressing the value of the NCNT-free sample (Cu2O NPs, FEC2 38.15%); noteworthily, the C2 partial current density of Cu2O NPs-NCNT (16.79 mA cm−2) is 7.30 folds higher than that of Cu2O NPs (2.30 mA cm−2) at the same applied potential. It is proposed that NCNT is helpful for catalyzing the conversion of CO2 to CO, which subsequently enriches the CO concentration at the heterogeneous interface of NCNT and Cu2O NPs and is conducive to C–C coupling to generate C2 products.
Recently, some strategies (e.g., alloying and surface doping) have been demonstrated to enhance the FE towards C2+ production of Cu materials. For instance, Chen et al. reported Cu–Ag tandem alloy catalysts, and the results illustrated that the CO-enriched environment introduced by Ag facilitated the subsequent C–C coupling processes occurring at the Cu sites, thereby enhancing the formation of C2+.163 Additionally, Wu et al. found that employing molecular doping strategies, including the introduction of aromatic heterocycles (e.g., thiadiazole and triazole derivatives) to CuAg alloy, can orient the reaction route towards C2+ production, achieving a high FE of C2+ (≈80%); detailed analyses demonstrated that the functional groups with electron-withdrawing feature are capable of modulating the electronic structure of Cu atoms and thereby enhancing the catalytic reaction rate.164 In general, incorporating modern strategies such as alloying and molecular doping into the design of copper-containing HSC-based catalysts, alongside the optimization of component interactions, holds the potential to further enhance the efficiency of catalyzing C2+ production and accelerate the pace of approaching practical application.
4. Thermocatalytic CO2 conversion of hollow carbon-based materials
Thermocatalytic CO2 conversion to produce commodity chemicals is a promising way to alleviate the dependence on fossil resources; it can be classified as hydrogenation and reforming routes loosely. For the reforming reaction, CO2 serves as an oxidant for catalytically reforming methane to generate syngas (CO and H2) under high temperature condition (600–1000 °C).10 Nevertheless, the extremely high temperature of the reforming reaction may lead to the decomposition of the carbon material; and there are few researches of HSC-related material reported in this field. As for CO2 hydrogenation, it refers to the conversion of CO2 with H2 to formic acid, methane and olefins, etc., under a relatively mild ambient condition. CO2 hydrogenation is in line with the concept of green chemistry, and holds important industrial application prospects, therefore, many efficient HSC-based materials were designed for this reaction (Table 4). In this section, the recent applications of HSC-based materials in relatively popular CO2 hydrogenation applications will be summarized and discussed.| Catalyst | Target product | Reaction condition | Catalytic performance | Ref. |
|---|---|---|---|---|
| NiCe/CNT | CH4 | 0.1 g catalyst, 350 °C, atmospheric pressure, H2/CO2 = 4:1 (flow rate), continuous fixed-bed, gas hourly space velocity (GHSV) = 30 L gcat−1 h−1 |
CO2 conversion rate: 83.8% | 165 |
| Selectivity: 99.8% | ||||
| Ni NPs/N-CNT | CH4 | 0.2 g catalyst, 360 °C, atmospheric pressure, H2/CO2 = 6:1, continuous fixed-bed, GHSV = 120 L gcat−1 h−1 |
CO2 conversion rate: 92% | 45 |
| Selectivity: 98% | ||||
| Ni NPs@carbon hollow sphere | CH4 | 0.1 g catalyst, 325 °C, atmospheric pressure, H2/CO2 = 4:1, fixed-bed flow reactor, GHSV = 33 L gcat−1 h−1 |
CO2 conversion rate: ∼100% | 166 |
| Selectivity: 99.9% | ||||
| Ni NPs/NCNT | CH4 | 0.04 g catalyst, 340 °C, atmospheric pressure, H2/CO2/Ar = 4:1:0.6, fixed-bed flow reactor, GHSV = 50 L gcat−1 h−1 |
CO2 conversion rate: ∼50% | 42 |
| Selectivity: 95% | ||||
| Ni NPs/NCNT | CH4 | 0.1 g catalyst, 400 °C, atmospheric pressure, H2/CO2/He = 4:1:5, weight hourly space velocity (WHSV) = 60 L gcat−1 h−1 |
CO2 conversion rate: 81.2% | 162 |
| Selectivity: 99.2% | ||||
| PdAg-aminopolymer@MHCS | Formate | 100 °C, 2 MPa (1 MPa H2 and 1 MPa CO2), 15 mL 1.0 M NaHCO3 aqueous reaction solution, closed reactor, 24 h | Turnover number (TON): 2680 | 51 |
| Turnover frequency (TOF): 112 h−1 | ||||
| PdAg@NHCS | Formate | 100 °C, 2 MPa (1 MPa H2 and 1 MPa CO2), 15 mL 1.0 M NaHCO3 aqueous solution, closed reactor, 24 h | TON: 2750 | 36 |
| TOF: 115 h−1 | ||||
| PdCu@MHCS | Formate | 100 °C, 2 MPa (1 MPa H2 and 1 MPa CO2), 15 mL 1.0 M NaHCO3 aqueous solution, closed reactor, 24 h | TON: 1432 | 49 |
| TOF: 60 h−1 | ||||
| Ru3+-N,P-containing porous organic polymers@MHCS | Formate | 120 °C, 4 MPa (2 MPa H2 and 2 MPa CO2), 7.8 mL 1.0 M NaHCO3 aqueous solution, closed reactor, 24 h | TON: 1228 | 50 |
| TOF: 51 h−1 | ||||
| Ru/N-MHCS | Formate | 120 °C, 8 MPa (4 MPa H2 and 4 MPa CO2), 40 mL 3 M aqueous Et3N solution, closed reactor, 2 h | TON: 7550 | 118 |
| TOF: 3775 h−1 | ||||
| Fe@CNTs | C2+ products | 0.4 g catalyst, 370 °C, atmospheric pressure, H2/CO2 = 3:1 (flow rate), fixed-bed reactor |
CO selectivity: 45.1% | 167 |
| CH4 selectivity: 29.3% | ||||
| C2–C4 selectivity: 24.3% | ||||
| C5+ selectivity: 1.3% | ||||
| FeZn-NC (Fe@C matrix) | C2+ products | 0.5 g catalyst, 320 °C, 3 MPa, H2/CO2 = 3:1 (molar ratio), fixed-bed reactor, space velocity = 7200 mL g−1 h−1, 5 h |
CO selectivity: 19.9 C mol% | 168 |
| CH4 selectivity: 16.6 C mol% | ||||
| Long-chain hydrocarbons (C5+) selectivity: 27.4 C mol% | ||||
| Ligh olefins (C2–C4) selectivity: 30.0 C mol% |
||||
| FeZnK-NC (Fe inserted in carbon shell) | CO selectivity: 21.2 C mol% | |||
| CH4 selectivity: 19.1 C mol% | ||||
| C5+ selectivity: 22.1 C mol% | ||||
| C2–C4 selectivity: 32.0 C mol% |
||||
| FeK/SWNT | C2+ products | 0.2 g catalyst, 340 °C, 2 MPa, H2/CO2/N2 = 72:24:4 (volume ratio), fixed-bed reactor, GHSV = 9000 mL gcat−1 h−1, 24 h |
Olefins selectivity = 62.3% (C5+ selectivity = 39.8%, C2–C4 selectivity = 22.5%) |
169 |
| FTY (iron-time yield) for heavy olefins = 26.5 μmolCO2 gFe−1 s−1 | ||||
| FeK/MWNT | Olefins selectivity = 52.5% (C5+ selectivity = 21.8%, C2–C4 selectivity = 30.7%) |
|||
| FTY (iron-time yield) for heavy olefins = 10.2 μmolCO2 gFe−1 s−1 | ||||
| Spark plasma sintered Fe/CNTs | C2+ products | 0.5 g catalyst, 350 °C, 8.5 MPa, H2/CO2 = 2:1, fixed-bed reactor, GHSV = 14.4 L gcat−1 h−1 |
CO selectivity = 18 mol% | 170 |
| CH4 selectivity = 35 mol% | ||||
| C2–4 selectivity = 35 mol% | ||||
| C5+ selectivity = 12 mol% | ||||
| Iron-time yield = 12.2 molCO2 gFe−1 s−1 |
4.1 CO2 hydrogenation to formate
Formate synthesis from CO2 hydrogenation is regarded as one of the most sustainable and economically feasible for utilization of CO2. However, designing efficient catalysts with high stability for formate production from CO2 hydrogenation is challenging because of the difficulty of CO2 activation and harsh reaction conditions. In this regard, our group has developed a series of hollow carbon sphere-based materials via rationally engineering the catalyst component/structure, and achieved desirable performance. Compared to the conventional supported material, encapsulation effects of the cavity in hollow carbon spheres efficiently prevent the leaching of some functionalized modifiers (e.g., aminopolymer) and the active metals, resulting in improved robustness and formate conversion efficiency. To begin with, a nanostructured catalyst consisting of sub-3-nm-sized PdAg NPs with high dispersity and aminopolymers encapsulated in mesoporous hollow carbon spheres (PdAg-P@MHCS) was synthesized (Fig. 11a–e).51 MHCS provided immobilization spaces for aminopolymers; and the amine groups of polymers promoted the metal ions adsorption, enabling the selective formation of PdAg NPs within P@MHCS. As a result, the turnover number (TON) of optimized PdAg-P@MHCS for formate production reached 2680 at 100 °C for 24 h. With the combination of experimental characterization and kinetics analysis, the possible reaction mechanism was proposed, as illustrated in Fig. 11f. Firstly, H2 dissociation occurs and further forms Pd-hydride species, which are active in the hydrogenation process (step 1). Then, the bicarbonate (HCO3−) adsorbs onto Ag atoms that are in an electron-deficient state after alloying with Pd (step 2); meanwhile, CO2 dissolution in the alkaline reaction solution continuously provides HCO3− (step 3). The dissociative formate is formed under the attack of active H to HCO3− adsorbed on Ag (step 4). This work demonstrated a new design strategy for developing highly efficient CO2 hydrogenation catalysts for formate production. | ||
| Fig. 11 (a) HAADF-STEM image of PdAg-P@MHCS. (b) Magnified image of the red frame zone in (a). (c) Elemental line scan across the PdAg NPs in (b). (d and e) Elemental maps of (d) Ag and (e) Pd. (f) Hydrogenated CO2-to-formate conversion pathway over PdAg-P@MHCS catalyst. Reproduced with permission from ref. 51. Copyright 2020, Royal Society of Chemistry. (g) Synthesis of Ru3+-POPs@MHCS catalyst. (h) Reaction time profile of hydrogenated CO2-to-formate conversion over the as-prepared samples. (i–k) Double logarithm plots of TON and the partial pressure of (i) CO2 and (j) H2 in the hydrogenation process, and (k) Arrhenius plots over optimized Ru3+-0.5POPs@MHCS and Ru3+-0.5POPs (MHCS-free) samples. Reproduced with permission from ref. 50. Copyright 2021, Tsinghua University Press and Springer-Verlag GmbH Germany, part of Springer Nature. | ||
To alleviate the use of hazardous reagents (i.e., carbon tetrachloride) and the complex operation procedures in the aforementioned work, we further exploited a novel and facile synthesis method for monodispersed N-doped microporous hollow carbon spheres encapsulating PdAg alloy catalyst via an ethylenediamine-assisted pyrolysis.36 And it showed competitive CO2 hydrogenation activity with the TON value of 2750 at 100 °C for 24 h.
In addition to alloy, the MHCS was also employed to confine a composite of mononuclear Ru3+ and N, P-containing porous organic polymers (Ru3+-POPs) in another study of ours (Fig. 11g).50 The Ru3+-POPs@MHCS catalyst with optimized content of Ru3+-POPs exhibited good activity for CO2 hydrogenation to formate (TON = 1228 at 100 °C for 24 h), surpassing that of Ru3+-POPs (without MHCS) sample (Fig. 11h). The superior performance originated from the promoted adsorption abilities of CO2 and H2 induced by MHCS, as evidenced by the kinetics analysis (Fig. 11i and j). Furthermore, the Arrhenius curve displayed that the activation energy of hydrogenated CO2-to-formate conversion decreased with Ru3+-POPs being encapsulated in MHCS (Fig. 11k). Based on the above discussion and reasoning, we demonstrate here that hollow carbon spheres benefit the overall performance of formate production from CO2 hydrogenation. The optimization of nanoconfined space is still worthy of further study, and our relevant researches are in progress.
4.2 CO2 methanation
Methane synthesis from CO2 hydrogenation (CO2 + 4H2 → CH4 + 2H2O, ΔH° = −165 kJ mol−1), known as the Sabatier reaction, is an exothermic process that is more thermodynamically favorable than many other hydrogenation routes.21,171 However, it still suffers from significant kinetic limitations due to the intrinsical inertness of linear CO2 and the involvement of an eight-electron transfer process.165,172,173 Several metal-containing catalysts have been demonstrated as effective candidates for this reaction, typically based on Ru, Rh, Pd, and Ni NPs on supported materials, with Ni-based catalysts being the most popular one because of the low cost and rich reserves.174–176 Nevertheless, the high temperature in the catalytic reaction may give rise to issues of stability and sintering in Ni-based catalysts. The incorporation of hollow structured carbon enables the encapsulation or embedding of metal sites inside its structure, which can effectively alleviate the problems of coke formation and metal sintering, and ultimately improve the activity, stability, and selectivity for methane synthesis from CO2 hydrogenation.42,177 For instance, Lin et al., presented a MOF-derived hollow carbon nanostructured catalyst encapsulating dispersed Ni nanoparticles (Ni@C) which affords better CO2 methanation performance than Ni/C (control sample, Ni NPs on graphite carbon) owing to the unique carbon-confined configuration.166 Additionally, no obvious deactivation of Ni@C catalyst was observed in the long-term stability test at 250 °C for 24 h; in contrast, Ni/C without carbon shell protection displayed a 50% decline in CH4 yield by 24 h.Generally, the excessive local heat generated during CO2 methanation may result in the formation of “hot spots” that affect the stability duration and methane selectivity. Introducing an appropriate carbon material with good thermal conductivity can benefit the uniform dispersion of the reaction heat and ensure the methanation process. For example, Wang et al. reported that Ni NPs decorated N-doped carbon nanotubes (N-CNT) catalyst (Ni/N-CNT) (Fig. 12a and b) exhibited high methane selectivity of 98% and CO2 conversion rate of 92%, even at a high gas hourly space velocity (GHSV) of 120000 mL g−1 h−1 with a H2-to-CO2 v/v ratio of 6 (Fig. 12c).45 The existence of N-CNT with unique characteristics contributed to the superior performance because (1) the excellent thermal conductivity limited the generation of local “hot spots”, (2) the nanotube configuration optimized the diffusion pathway of the reactants, and (3) the alkalized surface of N-CNT is favorable for concentrating CO2 in the proximity of catalytically active Ni sites. Recently, another study disclosed the importance of N-doping catalyst towards CO2 methanation, and the results demonstrated that N-doping enabled the formation of highly dispersive Ni sites that were responsible for the enhanced performance.162 Inspired by these intriguing studies, in our opinion, it is deemed to be of great significance to conduct more comprehensive investigations of the metal–support interaction, namely, the correlation between the intrinsic characteristics of hollow carbon internal/external surfaces and the active metals.
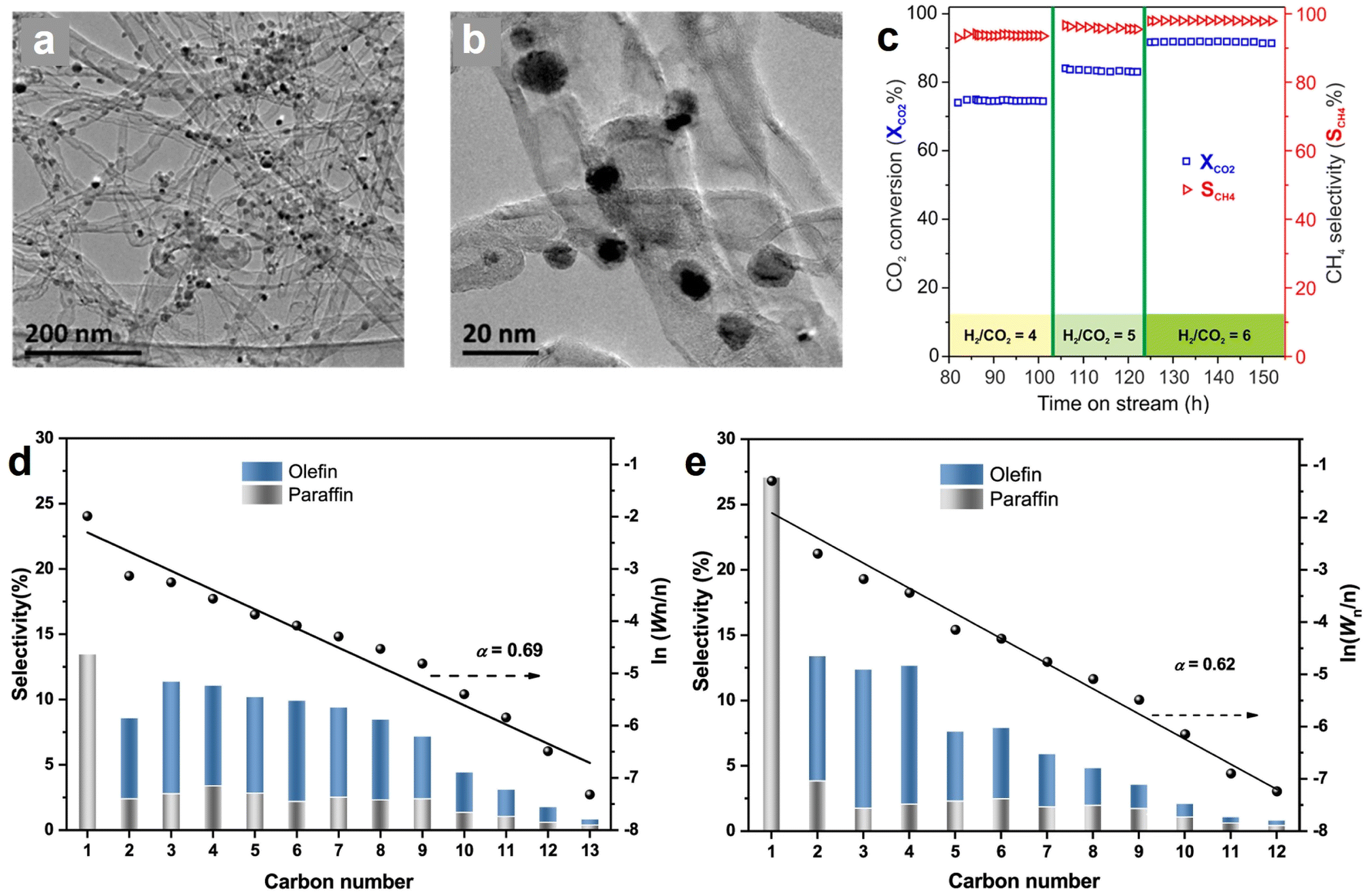 | ||
| Fig. 12 (a and b) TEM images of Ni/N-CNT catalyst in different magnifications. (c) Influence of H2/CO2 ratio on the CO2 methanation activity and selectivity Ni/N-CNT catalyst at a GHSV of 120000 mL g−1 h−1. Reproduced with permission from ref. 45. Copyright 2018, American Chemical Society. Hydrocarbon production distributions over the (d) FeK/SWNTs and (e) FeK/MWNTs catalysts after CO2 hydrogenation test for 24 h. Reaction conditions: 0.2 g of catalyst, T = 613 K, P = 2.0 MPa, H2/CO2 = 3, GHSV = 9000 mL gcat−1 h−1. Anderson–Schulz–Flory (ASF) plots and α-values (chain propagation probability) are also presented; Wn represents the weight fraction of a hydrocarbon with n carbon atoms. Reproduced with permission from ref. 169. Copyright 2021, American Chemical Society. | ||
4.3 CO2 hydrogenation to multicarbon products
Among the potential products from CO2 hydrogenation, C2+ hydrocarbon products such as olefins and paraffins are of particular interest due to their large energy density and industrial relevance.178,179 CO2 hydrogenation to hydrocarbons commonly involves reverse water–gas shift (RWGS) reaction and subsequent Fischer–Tropsch synthesis (FTS); the tandem of these two processes is thermodynamically feasible, but requires an effective catalyst to drive.180 The carbon-supported Fe-based catalysts receive considerable attention because they show good activity in both RWGS and FTS.168,181 For the potential hollow carbon support candidates, the CNTs with tubular morphology is the most popular one, owing to their good adhesion to metal NPs, high mechanical strength, good thermal conductivity, and ease of surface functionalization.182,183In 2013, Fe NPs@CNT catalyst was proved to be active for CO2 hydrogenation to produce hydrocarbons by O'Byrne et al., but the C2+ products selectivity and iron time yield (FTY) were relatively low.167 Encouragingly, an iron–potassium catalyst supported on single-wall carbon nanotubes (FeK/SWNT) reported by Wang et al. delivered an extremely competitive CO2 hydrogenation performance, with the olefins selectivity achieving 62.3%, where the heavy olefins (C5+) selectivity and light olefins (C2–C4) were 39.8% and 22.5%, respectively.169Fig. 12d and e show the comparison of CO2 hydrogenation results after 24 h measurement over FeK/SWNT and FeK/MWNT (control sample with multi-walled carbon nanotubes). According to the distribution of the hydrocarbons, both of the samples show higher selectivity for olefins instead of paraffins. Notably, FeK/SWNTs favor the formation of heavy olefins, whereas the production of light olefins is preferred on FeK/MWNT (see Table 4 Row 14 and 15 for more details), as reflected by the larger α-values (chain propagation probability) of FeK/SWNTs (0.69) as compared to that of FeK/MWNT (0.62). Detailed characterizations demonstrated that the excellent catalytic activity of FeK/SWNTs catalyst was associated with the presence of SWNTs with large curvature, which manipulated the electron distribution184 and accelerated the dissociation of CO on Hägg carbide, eventually boosting CO2 hydrogenation to heavy olefins. In extension to the conventional CNTs, the dense CNT materials are suitable for specific catalytic applications. Chernyak et al. prepared a new type of densely packed CNTs with embedding Fe NPs catalyst (Fe/CNT) via a spark plasma sintering method.170 Under supercritical conditions (350 °C, 8.5 MPa), the catalyst exhibited high specific activities at low H2/CO2 ratios (1 or 2), with the iron-time yields of 5.4–12.2 molCO2 gFe−1 s−1 and C2+ products selectivity of 40–50 mol%, in CO2 hydrogenation without pretreatment. This work highlighted the importance of CNT support with increased density for stabilizing the Fe NPs, which contributed to the strong metal–support interaction and CO intermediate activation.
Apart from the catalyst modification, the rational design of thermocatalytic reactors for CO2 reduction also contributes to enhancing catalytic activity and selectivity. It is recommended to establish an appropriate dynamic mathematical model to comprehend heat conduction and mass transfer during the reaction process. Furthermore, computational fluid dynamics analysis can be employed to investigate parameters such as fluid flow and pressure drop. The integration of dynamic mathematical models, computational simulation tools, and experimental validation can guide the development of efficient thermocatalytic CO2 reduction systems.
5. Conclusion and outlook
Herein, the recent progress of HSC-based materials in the fields of electrocatalytic and thermocatalytic CO2 utilization has been reviewed. The synthetic methods of HSC were first introduced. Then, a comprehensive overview of functionalization strategies for HSC was provided, including nonmetal doping, metal single atom anchoring, and metal nanoparticle modification; and they were found to offer numerous benefits, such as optimization of electronic structure, superior CO2 adsorption affinity, and improved reaction intermediate activation, allowing for the efficient enhancement of overall catalytic CO2 conversion behavior. Finally, the discussion of HSC-based materials towards electrocatalytic and thermocatalytic CO2 conversion applications was presented, based on the CO2 reduction product category. While encouraging progress has been witnessed in these fields, some key issues still exist that merit further study.(1) The synthesis of HSC materials with good rigidity, homogeneous morphology, and favorable reproducibility is mostly dependent on the utilization of the hard template method. However, this approach involves the use of highly corrosive agents to remove the template, potentially leading to adverse environmental consequences. Hence, the development of a green and environmentally friendly synthesis for high-quality HSC is urgently needed.
(2) The regulation of the pore structure of HSC is an important issue, as the porosity can affect the mass transport and the number of active sites, both of which have large influences on the activity and product selectivity. As exemplified by the work from Cao et al., in Ni–N doped hollow carbon sphere catalyst system used for eCO2RR, the mesopores on carbon shell were found to be favorable for the CO2 diffusion; while the micropores with confinement effect were conducive to the formation of Ni–N active sites during the synthesis process. By balancing the content of micropore and mesopore, the CO2-to-CO reaction rate can be efficiently improved.38 In thermocatalytic CO2 hydrogenation, the carbon-supported Fe–Co–K oxide catalyst using carbon support containing both micropores and mesopores shows higher olefin selectivity, as compared to that of the analogue with only micropores; the results suggest that the mesopores with sufficient accommodation space enable control over the intimate contact of K and FexOy species, making it possible to realize the boosted olefin production.185
(3) Most of the works are rather ambiguous in describing the contribution of confinement effects of HSC to the enhanced catalytic performance, which should be clarified. Here are some good examples. In CO2 electroreduction, the confinement effects of hollow carbon spheres contribute to the pH modulation of the internal cavity, which inhibits the competitive hydrogen evolution reaction by controlling the rate of H+ replenishment and creating a localized alkaline environment in the proximity of the catalytically active sites.46 In thermocatalytic CO2 reduction, in addition to improving the dispersion and stability of metal nanoparticles, HSC contributes to concentrating CO2 around the catalytically active sites and thus accelerating the reaction rate.45 In our opinion, more research efforts should be placed on emphasizing and clarifying the relationship between the confinement structure, active site, and substrate molecule, which allows to better utilize the positive effects and develop more advanced catalysts.
(4) Currently, the research on single-atom-based HSC materials is mainly focused on CO2 electroreduction, and only few cases in thermocatalytic CO2 conversion because the stabilization of single atoms with high surface free energy is a major challenge under the harsh conditions of thermocatalysis. Notably, in some specific situations, single atoms exhibit surprising stability, even better than metal nanoparticles. For example, the transformation of metal nanoparticles (Ru, Rh, and Pd) can be achieved by phosphine (PH3)-assisted pyrolysis to form single-atom materials, in which single-atom species are firmly fixed on thermodynamically stable sites, which help inhibit the agglomeration and leakage of single-atom species.186 Additionally, there are some studies reported the thermal atomic capture method allows the conversion of noble metal Pd, Pt, and Au nanoparticles to thermally stable single atoms.187,188 As a result, the stabilized single atoms showed better catalytic performance than nanoparticles; these works may offer some inspiration for the development of stable single-atom-based HSC materials for thermocatalytic CO2 conversion.
(5) Furthermore, the types of single-atom-related HSC catalysts are limited, with the majority of them focusing only on Ni or Co single atoms in eCO2RR for CO production (Table 3), and it is important to further exploit high-performance catalysts. In this regard, the following aspects are worthy of further consideration. To begin with, novel active metal single atoms are suggested to be introduced into the matrix of HSC material. For instance, the fluxional property of gallium (Ga) has been proven to be utilized to efficiently enhance eCO2RR behavior;189 the analogous unique characteristics of more single atoms need to be explored. Secondly, studies on the coordination environment of the central metal atom should be conducted, because the coordinated non-metal atoms can affect the electronic structure of the central metal atom, resulting in the change of the adsorption/activation ability towards reactant molecules. Thirdly, rationally designing dual single-atom sites with different functions is helpful in realizing some complex tandem reactions and in achieving the controllable production of specific products.
(6) HSC materials possess unique cavities and abundant pore structures, which provide a flexible platform for designing multifunctional catalysts. Integrating with other novel materials, the hybrid catalyst suitable for various catalytic application scenarios can be expected. As an example, the novel high-entropy alloys are active for both eCO2RR128 and thermocatalytic CO2 hydrogenation.190 Combining similar materials with HSC carbon materials to develop novel catalysts is also a research direction of interest.
(7) During the processes of electro-/thermocatalytic CO2 conversion, the intrinsic structure of the HSC materials may be changed under harsh conditions, such as large applied potential or high temperature. In this regard, ex situ characterizations may not be able to accurately characterize the catalysts; therefore, in situ measurements, such as in situ XAFS, Raman, XRD, and DRIFTS, should be employed to dynamically assess the structural change, which enables the guidance of a reasonable HSC material design. Theoretical simulation is a powerful tool to uncover catalytic mechanisms. Except for the commonly used DFT theoretical simulation, finite element modeling191,192 and molecular dynamics193,194 simulations are recommended to understand the mass transport behavior during the catalytic CO2 reduction reaction.
(8) The continuous exploration of the unique properties of HSC materials has pushed their application boundaries. For instance, a recent study revealed that HSC played an important role in enhancing photocatalytic CO2 reduction performance in a ternary HSC/CdS@ZnIn2S4 photocatalyst by offering multiple benefits, including improved light harvesting, increased CO2 adsorption, protection of CdS against photocorrosion, etc.195 In another work, HSC was demonstrated to be effective in boosting the separation of photoexcited electron–hole pairs and providing more catalytically active sites in the g-C3N4/Bi2O3@HSC photocatalyst system; as a result, enhanced photocatalytic antibiotic degradation can be achieved.196 In fact, the carbon shell of HSC material can absorb and convert light to heat under irradiation, resulting in the formation of a local hotspot, which is helpful for boosting the reaction kinetics.197 As there is a lack of relevant reports, further study on the photo-thermal features of HSC and the development of HSC-based photothermal catalytic reactors will be of great significance.
The unprecedented development of HSC-based materials in recent years provides infinite possibilities for sustainable CO2 utilization. We expect that this timely review will bring fellow researchers some inspiration to design more advanced HSC-based materials.
Author contributions
All of the authors contributed to the manuscript preparation. K. L. and Y. K. conceived the outline of the manuscript. K. L. wrote the original draft of the manuscript. Y. K. and H. Y. discussed and helped revise the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. K. and H. Y. gratefully acknowledge the Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS) (grant no. 22H00275). Y. K. acknowledges the cooperative research program of “Network Joint Research Center for Materials and Devices” (no. 20231065). K. L. gratefully acknowledges the financial support from the China Scholarship Council (No. 202108420065).References
- W. Gao, S. Liang, R. Wang, Q. Jiang, Y. Zhang, Q. Zheng, B. Xie, C. Y. Toe, X. Zhu, J. Wang, L. Huang, Y. Gao, Z. Wang, C. Jo, Q. Wang, L. Wang, Y. Liu, B. Louis, J. Scott, A. C. Roger, R. Amal, H. He and S. E. Park, Chem. Soc. Rev., 2020, 49, 8584–8686 RSC.
- D. I. A. McKay, A. Staal, J. F. Abrams, R. Winkelmann, B. Sakschewski, S. Loriani, I. Fetzer, S. E. Cornell, J. Rockström and T. M. Lenton, Science, 2022, 377, eabn7950 CrossRef CAS.
- J. Rogelj, M. Den Elzen, N. Höhne, T. Fransen, H. Fekete, H. Winkler, R. Schaeffer, F. Sha, K. Riahi and M. Meinshausen, Nature, 2016, 534, 631–639 CrossRef CAS PubMed.
- M. G. Lawrence and S. Schäfer, Science, 2019, 364, 829–830 CrossRef CAS PubMed.
- M. Meinshausen, J. Lewis, C. McGlade, J. Gütschow, Z. Nicholls, R. Burdon, L. Cozzi and B. Hackmann, Nature, 2022, 604, 304–309 CrossRef CAS PubMed.
- M. B. Ross, P. De Luna, Y. Li, C. T. Dinh, D. Kim, P. Yang and E. H. Sargent, Nat. Catal., 2019, 2, 648–658 CrossRef CAS.
- Y. Dong, P. Duchesne, A. Mohan, K. K. Ghuman, P. Kant, L. Hurtado, U. Ulmer, J. Y. Y. Loh, A. A. Tountas, L. Wang, F. M. Ali, M. Xia, R. Dittmeyer and G. A. Ozin, Chem. Soc. Rev., 2020, 49, 5648–5663 RSC.
- H. Ou, G. Li, W. Ren, B. Pan, G. Luo, Z. Hu, D. Wang and Y. Li, J. Am. Chem. Soc., 2022, 144, 22075–22082 CrossRef CAS PubMed.
- S. Wang, L. Wang, D. Wang and Y. Li, Energy Environ. Sci., 2023, 16, 2759–2803 RSC.
- S. Das, J. Pérez-Ramírez, J. Gong, N. Dewangan, K. Hidajat, B. C. Gates and S. Kawi, Chem. Soc. Rev., 2020, 49, 2937–3004 RSC.
- S. Garg, M. Li, A. Z. Weber, L. Ge, L. Li, V. Rudolph, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2020, 8, 1511–1544 RSC.
- C. J. Chang, Y. A. Lai, Y. C. Chu, C. K. Peng, H. Y. Tan, C. W. Pao, Y. G. Lin, S. F. Hung, H. C. Chen and H. M. Chen, J. Am. Chem. Soc., 2023, 145, 6953–6965 CrossRef CAS PubMed.
- X. Wang, Y. Jiang, K. Mao, W. Gong, D. Duan, J. Ma, Y. Zhong, J. Li, H. Liu, R. Long and Y. Xiong, J. Am. Chem. Soc., 2022, 144, 22759–22766 CrossRef CAS.
- T. Wang, J. Wang, C. Lu, K. Jiang, S. Yang, Z. Ren, J. Zhang, X. Liu, L. Chen, X. Zhuang and J. Fu, Adv. Mater., 2023, 2205553, 1–8 Search PubMed.
- Y. Hao, F. Hu, S. Zhu, Y. Sun, H. Wang, L. Wang, Y. Wang, J. Xue, Y. F. Liao, M. Shao and S. Peng, Angew. Chem., Int. Ed., 2023, 62, e202304179 CrossRef CAS PubMed.
- X. Tan, S. Jia, X. Song, X. Ma, J. Feng, L. Zhang, L. Wu, J. Du, A. Chen, Q. Zhu, X. Sun and B. Han, Chem. Sci., 2023, 14, 8214–8221 RSC.
- H. Jiang, L. Wang, H. Kaneko, R. Gu, G. Su, L. Li, J. Zhang, H. Song, F. Zhu, A. Yamaguchi, J. Xu, F. Liu, M. Miyauchi, W. Ding and M. Zhong, Nat. Catal., 2023, 6, 519–530 CrossRef CAS.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS.
- C. S. Diercks, Y. Liu, K. E. Cordova and O. M. Yaghi, Nat. Mater., 2018, 17, 301–307 CrossRef CAS PubMed.
- Y. Chen, Y. Yao, Y. Xia, K. Mao, G. Tang, Q. Wu, L. Yang, X. Wang, X. Sun and Z. Hu, Nano Res., 2020, 13, 2777–2783 CrossRef CAS.
- M. Kosari, A. M. H. Lim, Y. Shao, B. Li, K. M. Kwok, A. M. Seayad, A. Borgna and H. C. Zeng, J. Mater. Chem. A, 2023, 11, 1593–1633 RSC.
- J. Tian, B. Ma, S. Bu, Q. Yuan and C. Zhao, Chem. Commun., 2018, 54, 13993–13996 RSC.
- R.-P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell, Q. Li, M. Fan and Y.-G. Yao, Nat. Commun., 2019, 10, 5698 CrossRef CAS.
- E. C. Ra, K. Y. Kim, E. H. Kim, H. Lee, K. An and J. S. Lee, ACS Catal., 2020, 10, 11318–11345 CrossRef CAS.
- Y. Kuang and H. Li, J. Mater. Chem. A, 2022, 10, 7557–7603 RSC.
- Z. Yu, X. Lu, L. Sun, J. Xiong, L. Ye, X. Li, R. Zhang and N. Ji, ACS Sustain. Chem. Eng., 2021, 9, 2990–3010 CrossRef CAS.
- Q. Wu, L. Yang, X. Wang and Z. Hu, Adv. Mater., 2020, 32, 1–23 Search PubMed.
- G. H. Kim, W. H. Choi, J. W. Choi, K. H. Kim, D. G. Park, M. G. Park, M. G. Kim, H. Jang, U. H. Kim and J. K. Kang, ACS Nano, 2022, 16, 6552–6564 CrossRef CAS PubMed.
- J. Wang, Y. Cui and D. Wang, Adv. Mater., 2019, 31, 1–24 CAS.
- W. Hu, M. Zheng, B. Xu, Y. Wei, W. Zhu, Q. Li and H. Pang, J. Mater. Chem. A, 2021, 9, 3880–3917 RSC.
- Y. Boyjoo, H. Shi, Q. Tian, S. Liu, J. Liang, Z. S. Wu, M. Jaroniec and J. Liu, Energy Environ. Sci., 2021, 14, 540–575 RSC.
- H. Huang, R. Zong and H. Li, ACS Sustain. Chem. Eng., 2020, 8, 15998–16009 CrossRef CAS.
- S. S. R. Gupta, A. Vinu and M. L. Kantam, Chempluschem, 2021, 86, 259–269 CrossRef CAS PubMed.
- C. Cheng, D. Chen, N. Li, Q. Xu, H. Li, J. He and J. Lu, J. Hazard. Mater., 2020, 391, 122205 CrossRef CAS PubMed.
- Z. Xu, Y. Zhang, F. Wang, Z. Li, W. Gu, Y. Zhang and H. Xie, Chem. Eng. J., 2023, 452, 139229 CrossRef CAS.
- G. Yang, Y. Kuwahara, K. Mori, C. Louis and H. Yamashita, Appl. Catal., B, 2021, 283, 119628 CrossRef CAS.
- Y. Pan, H. Li, J. Xiong, Y. Yu, H. Du, S. S. S. Li, Z. Wu, S. S. S. Li, J. Lai and L. Wang, Appl. Catal., B, 2022, 306, 121111 CrossRef CAS.
- Z. Cao, P. Su, X. Wang, X. Liu, Y. Ma, C. Li, S. Ping Jiang and J. Liu, Fuel, 2022, 321, 124043 CrossRef CAS.
- T. Sun, S. Zhao, W. Chen, D. Zhai, J. Dong, Y. Wang, S. Zhang, A. Han, L. Gu, R. Yu, X. Wen, H. Ren, L. Xu, C. Chen, Q. Peng, D. Wang and Y. Li, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 12692–12697 CrossRef CAS PubMed.
- Y. Chen, S. Ji, S. Zhao, W. Chen, J. Dong, W.-C. Cheong, R. Shen, X. Wen, L. Zheng, A. I. Rykov, S. Cai, H. Tang, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 5422 CrossRef CAS PubMed.
- J. Du, Y. Xin, M. Dong, J. Yang, Q. Xu, H. Liu and B. Han, Small, 2021, 17, 1–7 Search PubMed.
- J. Gödde, M. Merko, W. Xia and M. Muhler, J. Energy Chem., 2021, 54, 323–331 CrossRef.
- F. Li, S. F. Zhao, L. Chen, A. Khan, D. R. MacFarlane and J. Zhang, Energy Environ. Sci., 2016, 9, 216–223 RSC.
- J. Lu, L. Yang, Y. Zhang, C. Wang, C. Zhang and X. S. Zhao, ACS Appl. Nano Mater., 2023, 6, 20746–20756 CrossRef CAS.
- W. Wang, C. Duong-Viet, H. Ba, W. Baaziz, G. Tuci, S. Caporali, L. Nguyen-Dinh, O. Ersen, G. Giambastiani and C. Pham-Huu, ACS Appl. Energy Mater., 2019, 2, 1111–1120 CrossRef CAS.
- Z. Liu, T. Yan, H. Shi, H. Pan, Y. Cheng and P. Kang, ACS Appl. Mater. Interfaces, 2022, 14, 7900–7908 CrossRef CAS PubMed.
- Y. Fu, T. Wang, W. Zheng, C. Lei, B. Yang, J. Chen, Z. Li, L. Lei, C. Yuan and Y. Hou, ACS Appl. Mater. Interfaces, 2020, 12, 16178–16185 CrossRef CAS PubMed.
- W. Zheng, C. Guo, J. Yang, F. He, B. Yang, Z. Li, L. Lei, J. Xiao, G. Wu and Y. Hou, Carbon, 2019, 150, 52–59 CrossRef CAS.
- G. Yang, Y. Kuwahara, K. Mori, C. Louis and H. Yamashita, J. Phys. Chem. C, 2021, 125, 3961–3971 CrossRef CAS.
- G. Yang, Y. Kuwahara, K. Mori, C. Louis and H. Yamashita, Nano Res., 2021, 16, 1–9 Search PubMed.
- G. Yang, Y. Kuwahara, S. Masuda, K. Mori, C. Louis and H. Yamashita, J. Mater. Chem. A, 2020, 8, 4437–4446 RSC.
- K. Li, Y. Kuwahara and H. Yamashita, Appl. Catal., B, 2023, 331, 122713 CrossRef CAS.
- Z. Li, B. Li, C. Yu, H. Wang and Q. Li, Adv. Sci., 2023, 2206605, 1–53 Search PubMed.
- Z. Yu, N. Ji, X. Li, R. Zhang, Y. Qiao, J. Xiong, J. Liu and X. Lu, Angew. Chem., Int. Ed., 2023, 62, e202213612 CrossRef CAS PubMed.
- T. Liu, L. Zhang, B. Cheng and J. Yu, Adv. Energy Mater., 2019, 9, 1–55 Search PubMed.
- P. Kuang, Y. Wang, B. Zhu, F. Xia, C. W. Tung, J. Wu, H. M. Chen and J. Yu, Adv. Mater., 2021, 33, 1–9 Search PubMed.
- A. Fu, C. Wang, F. Pei, J. Cui, X. Fang and N. Zheng, Small, 2019, 15, 1–21 Search PubMed.
- J. Liu, N. P. Wickramaratne, S. Z. Qiao and M. Jaroniec, Nat. Mater., 2015, 14, 763–774 CrossRef CAS PubMed.
- H. Zhang, O. Noonan, X. Huang, Y. Yang, C. Xu, L. Zhou and C. Yu, ACS Nano, 2016, 10, 4579–4586 CrossRef CAS PubMed.
- Y. Li, X. F. Lu, S. Xi, D. Luan, X. Wang and X. W. (David) Lou, Angew. Chem., Int. Ed., 2022, 61, e202201491 CrossRef CAS PubMed.
- S. Li, A. Pasc, V. Fierro and A. Celzard, J. Mater. Chem. A, 2016, 4, 12686–12713 RSC.
- H. Tian, J. Liang and J. Liu, Adv. Mater., 2019, 31, 1–30 Search PubMed.
- Q. Yu, D. C. Li, Z. Tian, C. Zhu, C. Jiao, Q. Zhang, Y. Chen, Y. Zhu, H. Jiang, J. Liu and G. H. Wang, Chem. Mater., 2022, 34, 3715–3723 CrossRef CAS.
- J. Ye, J. Zang, Z. Tian, M. Zheng and Q. Dong, J. Mater. Chem. A, 2016, 4, 13223–13227 RSC.
- S. Cai, Z. Meng, H. Tang, Y. Wang and P. Tsiakaras, Appl. Catal., B, 2017, 217, 477–484 CrossRef CAS.
- M. Karuppannan, Y. Kim, Y. E. Sung and O. J. Kwon, J. Mater. Chem. A, 2018, 6, 7522–7531 RSC.
- D. Ni, W. Sun, Z. Wang, Y. Bai, H. Lei, X. Lai and K. Sun, Adv. Energy Mater., 2019, 9, 1–10 Search PubMed.
- J. Chen, X. Liang, W. Liu, W. Gu, B. Zhang and G. Ji, Dalton Trans., 2019, 48, 10145–10150 RSC.
- W. Xiong, H. Li, H. You, M. Cao and R. Cao, Natl. Sci. Rev., 2020, 7, 609–619 CrossRef CAS PubMed.
- D. Zhang, S. Shen, X. Xiao, D. Mao and B. Yan, RSC Adv., 2020, 10, 26546–26552 RSC.
- W. Zhang, S. Yang, M. Jiang, Y. Hu, C. Hu, X. Zhang and Z. Jin, Nano Lett., 2021, 21, 2650–2657 CrossRef CAS PubMed.
- J. Du, A. Chen, X. Gao, Y. Zhang and H. Lv, ACS Appl. Mater. Interfaces, 2022, 14, 11750–11757 CrossRef CAS PubMed.
- K. Min, K. Kim, H. An, Y. Go, Y. Lee, D. Lim and S. H. Baeck, J. Power Sources, 2022, 543, 231849 CrossRef CAS.
- W. Ye, X. Li, B. Zhang, W. Liu, Y. Cheng, X. Fan, H. Zhang, Y. Liu, Q. Dong and M. S. Wang, Adv. Mater., 2023, 35, 1–11 Search PubMed.
- K. Eid, A. A. Abdelhafiz, S. Abdel-Azeim, R. S. Varma and M. F. Shibl, Green Chem., 2023, 25, 6748–6758 RSC.
- L. Dong, Y. Li, J. Li, Y. Guan, X. Chen, D. Zhang and Z. Wang, J. Hazard. Mater., 2023, 451, 131112 CrossRef CAS PubMed.
- B. Li, Z. Ma, X. Zhang, J. Xu, Y. Chen, X. Zhang and C. Zhu, Small, 2023, 19, 1–12 Search PubMed.
- K. Tang, L. Fu, R. J. White, L. Yu, M. M. Titirici, M. Antonietti and J. Maier, Adv. Energy Mater., 2012, 2, 873–877 CrossRef CAS.
- G. H. Wang, J. Hilgert, F. H. Richter, F. Wang, H. J. Bongard, B. Spliethoff, C. Weidenthaler and F. Schüth, Nat. Mater., 2014, 13, 293–300 CrossRef CAS PubMed.
- C. Chen, H. Wang, C. Han, J. Deng, J. Wang, M. Li, M. Tang, H. Jin and Y. Wang, J. Am. Chem. Soc., 2017, 139, 2657–2663 CrossRef CAS PubMed.
- X. Liu, P. Song, J. Hou, B. Wang, F. Xu and X. Zhang, ACS Sustain. Chem. Eng., 2018, 6, 2797–2805 CrossRef CAS.
- L. Liu, X. Sun, Y. Dong, D. Wang, Z. Wang, Z. Jiang, A. Li, X. Chen and H. Song, J. Power Sources, 2021, 506, 230170 CrossRef CAS.
- F. Ma, H. Zhao, L. Sun, Q. Li, L. Huo, T. Xia, S. Gao, G. Pang, Z. Shi and S. Feng, J. Mater. Chem., 2012, 22, 13464–13468 RSC.
- H. Sun, Y. Zhu, B. Yang, Y. Wang, Y. Wu and J. Du, J. Mater. Chem. A, 2016, 4, 12088–12097 RSC.
- W. Zhang, X. Jiang, Y. Zhao, A. Carné-Sánchez, V. Malgras, J. Kim, J. H. Kim, S. Wang, J. Liu, J. Sen Jiang, Y. Yamauchi and M. Hu, Chem. Sci., 2017, 8, 3538–3546 RSC.
- D. Xue, D. Zhu, H. Duan, Z. Wang, Y. Lv, W. Xiong, L. Li, M. Liu and L. Gan, Chem. Commun., 2019, 55, 11219–11222 RSC.
- X. Wei, D. Zheng, M. Zhao, H. Chen, X. Fan, B. Gao, L. Gu, Y. Guo, J. Qin, J. Wei, Y. Zhao and G. Zhang, Angew. Chem., Int. Ed., 2020, 59, 14639–14646 CrossRef CAS PubMed.
- S. Zeng, G. M. Arumugam, X. Liu, Y. Yang, X. Li, H. Zhong, F. Guo and Y. Mai, Small, 2020, 16, 1–8 Search PubMed.
- Y. Zheng, S. Chen, K. A. I. Zhang, J. Guan, X. Yu, W. Peng, H. Song, J. Zhu, J. Xu, X. Fan, C. Zhang and T. Liu, J. Colloid Interface Sci., 2022, 608, 3168–3177 CrossRef CAS PubMed.
- Y. Chen, X. Shi, B. Lu and J. Zhou, Adv. Energy Mater., 2022, 12, 1–8 CAS.
- B. Pan, X. Zhu, Y. Wu, T. Liu, X. Bi, K. Feng, N. Han, J. Zhong, J. Lu, Y. Y. Li and Y. Y. Li, Adv. Sci., 2020, 7, 2–7 Search PubMed.
- R. Li, F. Liu, Y. Zhang, M. Guo and D. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 44578–44587 CrossRef CAS PubMed.
- H. Chen, Y. Zhang, T. Yang, Y. Shang, Q. Zhu, S. Cao, X. Lin, S. Liu, S. Wei, B. Wei, Z. Wang and X. Lu, Dalton Trans., 2022, 51, 15883–15888 RSC.
- W. Peng, H. Tan, X. Liu, F. Hou and J. Liang, Energy Fuels, 2023, 37, 17863–17874 CrossRef CAS.
- P. Song, H. Wang, L. Kang, B. Ran, H. Song and R. Wang, Chem. Commun., 2019, 55, 687–690 RSC.
- Y. Shang, Y. Ding, P. Zhang, M. Wang, Y. Jia, Y. Xu, Y. Li, K. Fan and L. Sun, Chin. J. Catal., 2022, 43, 2405–2413 CrossRef CAS.
- W. Peng, J. Liu, X. Liu, L. Wang, L. Yin, H. Tan, F. Hou and J. Liang, Nat. Commun., 2023, 14, 4430 CrossRef CAS PubMed.
- J. Wu, R. M. Yadav, M. Liu, P. P. Sharma, C. S. Tiwary, L. Ma, X. Zou, X. Zhou, B. I. Yakobson, J. Lou and P. M. Ajayan, ACS Nano, 2015, 9, 5364–5371 CrossRef CAS PubMed.
- H. Wang, J. Jia, P. Song, Q. Wang, D. Li, S. Min, C. Qian, L. Wang, Y. F. Li, C. Ma, T. Wu, J. Yuan, M. Antonietti and G. A. Ozin, Angew. Chem., Int. Ed., 2017, 56, 7847–7852 CrossRef CAS PubMed.
- J. Xie, X. Zhao, M. Wu, Q. Li, Y. Wang and J. Yao, Angew. Chem., Int. Ed., 2018, 57, 9640–9644 CrossRef CAS PubMed.
- H. Chen, Z. Yang, C. L. Do-Thanh and S. Dai, ChemSusChem, 2020, 13, 6182–6200 CrossRef CAS PubMed.
- W. Ni, Y. Xue, X. Zang, C. Li, H. Wang, Z. Yang and Y. M. Yan, ACS Nano, 2020, 14, 2014–2023 CrossRef CAS PubMed.
- G. Li, Y. Qin, Y. Wu, L. Pei, Q. Hu, H. Yang, Q. Zhang, J. Liu and C. He, Chin. J. Catal., 2020, 41, 830–838 CrossRef CAS.
- C. Cheng, J. Shao, P. Wei, Y. Song, H. Li, D. Gao and G. Wang, ChemNanoMat, 2021, 7, 635–640 CrossRef CAS.
- H. Cui, Y. Guo, L. Guo, L. Wang, Z. Zhou and Z. Peng, J. Mater. Chem. A, 2018, 6, 18782–18793 RSC.
- X. Duan, J. Xu, Z. Wei, J. Ma, S. Guo, S. Wang, H. Liu and S. Dou, Adv. Mater., 2017, 29, 1–20 Search PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- Z. Zhang and D. Wang, J. Mater. Chem. A, 2021, 10, 5863–5877 RSC.
- X. He, Y. Deng, Y. Zhang, Q. He, D. Xiao, M. Peng, Y. Zhao, H. Zhang, R. Luo, T. Gan, H. Ji and D. Ma, Cell Rep. Phys. Sci., 2020, 1, 100004 CrossRef.
- J. W. Sun, X. Wu, P. F. Liu, J. Chen, Y. Liu, Z. X. Lou, J. Y. Zhao, H. Y. Yuan, A. Chen, X. L. Wang, M. Zhu, S. Dai and H. G. Yang, Nat. Commun., 2023, 14, 1599 CrossRef CAS PubMed.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS PubMed.
- X. Shi, L. N. Y. Cao, M. Chen and Y. Huang, Chin. Chem. Lett., 2022, 33, 5023–5029 CrossRef CAS.
- B. Wang, H. Cai and S. Shen, Small Methods, 2019, 3, 1–14 Search PubMed.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W. C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed.
- W. Xiong, H. Li, H. Wang, J. Yi, H. You, S. Zhang, Y. Hou, M. Cao, T. Zhang and R. Cao, Small, 2020, 16, 1–11 Search PubMed.
- S. Ma, P. Su, W. Huang, S. P. Jiang, S. Bai and J. Liu, ChemCatChem, 2019, 11, 6092–6098 CrossRef CAS.
- P. Yao, J. Zhang, Y. Qiu, Q. Zheng, H. Zhang, J. Yan and X. Li, ACS Sustain. Chem. Eng., 2021, 9, 5437–5444 CrossRef CAS.
- S. Ahn, K. Park, K. R. Lee, A. Haider, C. Van Nguyen, H. Jin, S. J. Yoo, S. Yoon and K. D. Jung, Chem. Eng. J., 2022, 442, 136185 CrossRef CAS.
- X. Hai, S. Xi, S. Mitchell, K. Harrath, H. Xu, D. F. Akl, D. Kong, J. Li, Z. Li, T. Sun, H. Yang, Y. Cui, C. Su, X. Zhao, J. Li, J. Pérez-Ramírez and J. Lu, Nat. Nanotechnol., 2022, 17, 174–181 CrossRef CAS PubMed.
- L. Wang, X. Gao, S. Wang, C. Chen, J. Song, X. Ma, T. Yao, H. Zhou and Y. Wu, J. Am. Chem. Soc., 2023, 145, 13462–13468 CrossRef CAS PubMed.
- M. Jafarzadeh and K. Daasbjerg, ACS Appl. Energy Mater., 2023, 6, 6851–6882 CrossRef CAS.
- S. Liu, H. Tao, L. Zeng, Q. Liu, Z. Xu, Q. Liu and J. L. Luo, J. Am. Chem. Soc., 2017, 139, 2160–2163 CrossRef CAS PubMed.
- H. Zhang, C. He, S. Han, Z. Du, L. Wang, Q. Yun, W. Cao, B. Zhang, Y. H. Tian and Q. Lu, Chin. Chem. Lett., 2022, 33, 3641–3649 CrossRef CAS.
- D. Wei, Y. Wang, C. Dong, Z. Zhang, X. Wang, Y. Huang, Y. Shi, X. Zhao, J. Wang, R. Long, Y. Xiong, F. Dong, M. Li and S. Shen, Angew. Chem., Int. Ed., 2023, 62, e202217369 CrossRef CAS PubMed.
- Z. Zhu, Z. Li, X. Wei, J. Wang, S. Xiao, R. Li, R. Wu and J. S. Chen, Carbon, 2021, 185, 9–16 CrossRef CAS.
- J. Meng, Z. Miao, J. Zhang, Z. Wang, R. Zhang, L. Xu, L. Diao, J. Zhou and S. Zhuo, J. Alloys Compd., 2023, 939, 168798 CrossRef CAS.
- K. Mori, T. Sano, H. Kobayashi and H. Yamashita, J. Am. Chem. Soc., 2018, 140, 8902–8909 CrossRef CAS PubMed.
- S. Nellaiappan, N. K. Katiyar, R. Kumar, A. Parui, K. D. Malviya, K. G. Pradeep, A. K. Singh, S. Sharma, C. S. Tiwary and K. Biswas, ACS Catal., 2020, 10, 3658–3663 CrossRef CAS.
- J. K. Pedersen, T. A. A. Batchelor, A. Bagger and J. Rossmeisl, ACS Catal., 2020, 10, 2169–2176 CrossRef CAS.
- H. J. Moon, J. M. Carrillo, J. Leisen, B. G. Sumpter, N. C. Osti, M. Tyagi and C. W. Jones, J. Am. Chem. Soc., 2022, 144, 11664–11675 CrossRef CAS PubMed.
- Z. A. Qiao, P. Zhang, S. H. Chai, M. Chi, G. M. Veith, N. C. Gallego, M. Kidder and S. Dai, J. Am. Chem. Soc., 2014, 136, 11260–11263 CrossRef CAS PubMed.
- Y. Kuwahara, H. Kango and H. Yamashita, ACS Catal., 2019, 9, 1993–2006 CrossRef CAS.
- W. Zhang, Y. Hu, L. Ma, G. Zhu, P. Zhao, X. Xue, R. Chen, S. Yang, J. Ma, J. Liu and Z. Jin, Nano Energy, 2018, 53, 808–816 CrossRef CAS.
- J. Chen and L. Wang, Adv. Mater., 2022, 34, 1–30 Search PubMed.
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C. T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, 1–24 CrossRef PubMed.
- N. J. Harmon and H. Wang, Angew. Chem., Int. Ed., 2022, 61, e202213782 CrossRef CAS PubMed.
- D. Gao, P. Wei, H. Li, L. Lin, G. Wang and X. Bao, Acta Phys.-Chim. Sin., 2021, 37, 2009021 Search PubMed.
- Y. J. Sa, C. W. Lee, S. Y. Lee, J. Na, U. Lee and Y. J. Hwang, Chem. Soc. Rev., 2020, 49, 6632–6665 RSC.
- M. Moura de Salles Pupo and R. Kortlever, ChemPhysChem, 2019, 20, 2926–2935 CrossRef CAS PubMed.
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager and A. T. Bell, J. Am. Chem. Soc., 2016, 138, 13006–13012 CrossRef CAS PubMed.
- G. Marcandalli, A. Goyal and M. T. M. Koper, ACS Catal., 2021, 11, 4936–4945 CrossRef CAS PubMed.
- X. Wang, X. Sang, C. Dong, S. Yao, L. Shuai, J. Lu, B. Yang, Z. Li, L. Lei, M. Qiu, L. Dai, Y. Hou, Q. Zhao, J. M. P. Martirez and E. A. Carter, Angew. Chem., Int. Ed., 2021, 60, 11959–11965 CrossRef CAS PubMed.
- Q. Zhao, J. M. P. Martirez and E. A. Carter, J. Am. Chem. Soc., 2021, 143, 6152–6164 CrossRef CAS PubMed.
- N. Wang, H. Li, H. Wang, H. Yang, Z. Ren and R. Xu, Small, 2023, 19, 2301469 CrossRef CAS PubMed.
- C. Yan, H. Li, Y. Ye, H. Wu, F. Cai, R. Si, J. Xiao, S. Miao, S. Xie, F. Yang, Y. Li, G. Wang and X. Bao, Energy Environ. Sci., 2018, 11, 1204–1210 RSC.
- S. Gong, W. Wang, R. Lu, M. Zhu, H. Wang, Y. Zhang, J. Xie, C. Wu, J. Liu, M. Li, S. Shao, G. Zhu and X. Lv, Appl. Catal., B, 2022, 318, 121813 CrossRef CAS.
- S. Gong, S. Yang, W. Wang, R. Lu, H. Wang, X. Han, G. Wang, J. Xie, D. Rao, C. Wu, J. Liu, S. Shao and X. Lv, Small, 2023, 2207808, 1–15 Search PubMed.
- M. B. Ross, Y. Li, P. De Luna, D. Kim, E. H. Sargent and P. Yang, Joule, 2019, 3, 257–264 CrossRef CAS.
- X. Song, H. Zhang, Y. Yang, B. Zhang, M. Zuo, X. Cao, J. Sun, C. Lin, X. Li and Z. Jiang, Adv. Sci., 2018, 5, 1–8 Search PubMed.
- W. Yang, J. H. Zhang, R. Si, L. M. Cao, D. C. Zhong and T. B. Lu, Inorg. Chem. Front., 2021, 8, 1695–1701 RSC.
- X. Zheng, P. De Luna, F. P. García de Arquer, B. Zhang, N. Becknell, M. B. Ross, Y. Li, M. N. Banis, Y. Li, M. Liu, O. Voznyy, C. T. Dinh, T. Zhuang, P. Stadler, Y. Cui, X. Du, P. Yang and E. H. Sargent, Joule, 2017, 1, 794–805 CrossRef CAS.
- I. M. Badawy, G. E. Khedr, A. Hafez, E. A. Ashour and N. K. Allam, Chem. Commun., 2023, 59, 7974–7977 RSC.
- B. Ren, G. Wen, R. Gao, D. Luo, Z. Zhang, W. Qiu, Q. Ma, X. Wang, Y. Cui, L. Ricardez-Sandoval, A. Yu and Z. Chen, Nat. Commun., 2022, 13, 1–11 Search PubMed.
- S. H. Li, S. Hu, H. Liu, J. Liu, X. Kang, S. Ge, Z. Zhang, Q. Yu and B. Liu, ACS Nano, 2023, 17, 9338–9346 CrossRef CAS PubMed.
- C. Z. Yuan, L. Y. Zhan, S. J. Liu, F. Chen, H. Lin, X. L. Wu and J. Chen, Inorg. Chem. Front., 2020, 7, 1719–1725 RSC.
- Y. Li, S. L. Zhang, W. Cheng, Y. Chen, D. Luan, S. Gao and X. W. Lou, Adv. Mater., 2022, 34, 1–8 Search PubMed.
- M. Liu, S. Liu, Q. Xu, Q. Miao, S. Yang, S. Hanson, G. Z. Chen, J. He, Z. Jiang and G. Zeng, Carbon Energy, 2023, 5, 1–12 Search PubMed.
- Yiliguma, Z. Wang, C. Yang, A. Guan, L. Shang, A. M. Al-Enizi, L. Zhang and G. Zheng, J. Mater. Chem. A, 2018, 6, 20121–20127 RSC.
- Y. Li, W. Niu, T. Chen, Y. Sun and M. Yu, Appl. Catal., B, 2023, 321, 122037 CrossRef CAS.
- J. Liu, K. Yu, Q. Zhu, Z. Qiao, H. Zhang and J. Jiang, ACS Appl. Mater. Interfaces, 2023, 15, 36135–36142 CrossRef CAS PubMed.
- P. P. Yang, X. L. Zhang, F. Y. Gao, Y. R. Zheng, Z. Z. Niu, X. Yu, R. Liu, Z. Z. Wu, S. Qin, L. P. Chi, Y. Duan, T. Ma, X. S. Zheng, J. F. Zhu, H. J. Wang, M. R. Gao and S. H. Yu, J. Am. Chem. Soc., 2020, 142, 6400–6408 CrossRef CAS PubMed.
- L. P. L. Gonçalves, M. Meledina, A. Meledin, D. Y. Petrovykh, J. P. S. Sousa, O. S. G. P. Soares, Y. V. Kolen'ko and M. F. R. Pereira, Carbon, 2022, 195, 35–43 CrossRef.
- C. Chen, Y. Li, S. Yu, S. Louisia, J. Jin, M. Li, M. B. Ross and P. Yang, Joule, 2020, 4, 1688–1699 CrossRef CAS.
- H. Wu, J. Li, K. Qi, Y. Zhang, E. Petit, W. Wang, V. Flaud, N. Onofrio, B. Rebiere, L. Huang, C. Salameh, L. Lajaunie, P. Miele and D. Voiry, Nat. Commun., 2021, 12, 1–11 CrossRef PubMed.
- W. Wang, W. Chu, N. Wang, W. Yang and C. Jiang, Int. J. Hydrogen Energy, 2016, 41, 967–975 CrossRef CAS.
- X. Lin, S. Wang, W. Tu, Z. Hu, Z. Ding, Y. Hou, R. Xu and W. Dai, Catal. Sci. Technol., 2019, 9, 731–738 RSC.
- J. P. O'Byrne, R. E. Owen, D. R. Minett, S. I. Pascu, P. K. Plucinski, M. D. Jones and D. Mattia, Catal. Sci. Technol., 2013, 3, 1202–1207 RSC.
- J. Liu, Y. Sun, X. Jiang, A. Zhang, C. Song and X. Guo, J. CO2 Util., 2018, 25, 120–127 CrossRef CAS.
- S. Wang, T. Wu, J. Lin, Y. Ji, S. Yan, Y. Pei, S. Xie, B. Zong and M. Qiao, ACS Catal., 2020, 10, 6389–6401 CrossRef CAS.
- S. A. Chernyak, A. S. Ivanov, D. N. Stolbov, S. V. Maksimov, K. I. Maslakov, P. A. Chernavskii, Y. A. Pokusaeva, A. E. Koklin, V. I. Bogdan and S. V. Savilov, Carbon, 2020, 168, 475–484 CrossRef CAS.
- H. Kim, K. Mori, T. Nakano and H. Yamashita, Adv. Funct. Mater., 2023, 33, 2303994 CrossRef CAS.
- X. Li, Y. Sun, J. Xu, Y. Shao, J. Wu, X. Xu, Y. Pan, H. Ju, J. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690–699 CrossRef CAS.
- X. Li, K. Li, D. Ding, J. Yan, C. Wang, S. A. C. Carabineiro, Y. Liu and K. Lv, Sep. Purif. Technol., 2023, 309, 123054 CrossRef CAS.
- X. Cui, S. Shyshkanov, T. N. Nguyen, A. Chidambaram, Z. Fei, K. C. Stylianou and P. J. Dyson, Angew. Chem., Int. Ed., 2020, 59, 16371–16375 CrossRef CAS PubMed.
- Y. Dai, M. Xu, Q. Wang, R. Huang, Y. Jin, B. Bian, C. Tumurbaatar, B. Ishtsog, T. Bold and Y. Yang, Appl. Catal., B, 2020, 277, 119271 CrossRef CAS.
- Y. Wang, H. Arandiyan, S. A. Bartlett, A. Trunschke, H. Sun, J. Scott, A. F. Lee, K. Wilson, T. Maschmeyer, R. Schlögl and R. Amal, Appl. Catal., B, 2020, 277, 119029 CrossRef CAS.
- J. Gao, Q. Jiang, Y. Liu, W. Liu, W. Chu and D. S. Su, Nanoscale, 2018, 10, 14207–14219 RSC.
- D. Wang, Z. Xie, M. D. Porosoff and J. G. Chen, Chem, 2021, 7, 2277–2311 CAS.
- M. Cui, Q. Qian, J. Zhang, Y. Wang, B. B. Asare Bediako, H. Liu and B. Han, Chem, 2021, 7, 726–737 CAS.
- D. R. Minett, J. P. O'Byrne, S. I. Pascu, P. K. Plucinski, R. E. Owen, M. D. Jones and D. Mattia, Catal. Sci. Technol., 2014, 4, 3351–3358 RSC.
- H. Yang, Y. Dang, X. Cui, X. Bu, J. Li, S. Li, Y. Sun and P. Gao, Appl. Catal., B, 2023, 321, 122050 CrossRef CAS.
- K. Jin, C. Wen, L. Chen, Q. Jiang, X. Zhuang, X. Xu, H. Wang, L. Ma, C. Wang and Q. Zhang, Fuel, 2023, 333, 126412 CrossRef CAS.
- H. Singh Malhi, Z. Zhang, Y. Shi, X. Gao, W. Liu, W. Tu and Y. F. Han, Fuel, 2023, 339, 127267 CrossRef CAS.
- H. Tan, W. Si, W. Peng, X. Chen, X. Liu, Y. You, L. Wang, F. Hou and J. Liang, Nano Lett., 2023, 23, 10571–10578 CrossRef CAS PubMed.
- T. Witoon, T. Numpilai, K. Nueangnoraj, C. K. Cheng, M. Chareonpanich and J. Limtrakul, Int. J. Hydrogen Energy, 2022, 47, 42185–42199 CrossRef CAS.
- P. Zhou, N. Li, Y. Chao, W. Zhang, F. Lv, K. Wang, W. Yang, P. Gao and S. Guo, Angew. Chem., Int. Ed., 2019, 58, 14184–14188 CrossRef CAS PubMed.
- S. Wei, A. Li, J. C. Liu, Z. Li, W. Chen, Y. Gong, Q. Zhang, W. C. Cheong, Y. Wang, L. Zheng, H. Xiao, C. Chen, D. Wang, Q. Peng, L. Gu, X. Han, J. Li and Y. Li, Nat. Nanotechnol., 2018, 13, 856–861 CrossRef CAS PubMed.
- Y. Chen, P. Wang, H. Hao, J. Hong, H. Li, S. Ji, A. Li, R. Gao, J. Dong, X. Han, M. Liang, D. Wang and Y. Li, J. Am. Chem. Soc., 2021, 143, 18643–18651 CrossRef CAS PubMed.
- Z. Zhang, J. Zhu, S. Chen, W. Sun and D. Wang, Angew. Chem., Int. Ed., 2023, 62, e202215136 CrossRef CAS PubMed.
- K. Mori, N. Hashimoto, N. Kamiuchi, H. Yoshida, H. Kobayashi and H. Yamashita, Nat. Commun., 2021, 12, 1–11 CrossRef PubMed.
- W. Liu, P. Zhai, A. Li, B. Wei, K. Si, Y. Wei, X. Wang, G. Zhu, Q. Chen, X. Gu, R. Zhang, W. Zhou and Y. Gong, Nat. Commun., 2022, 13, 1–12 Search PubMed.
- R. G. Mariano, M. Kang, O. J. Wahab, I. J. McPherson, J. A. Rabinowitz, P. R. Unwin and M. W. Kanan, Nat. Mater., 2021, 20, 1000–1006 CrossRef CAS PubMed.
- L. Fan, C. Y. Liu, P. Zhu, C. Xia, X. Zhang, Z. Y. Wu, Y. Lu, T. P. Senftle and H. Wang, Joule, 2022, 6, 205–220 CrossRef CAS.
- J. Wang, T. Cheng, A. Q. Fenwick, T. N. Baroud, A. Rosas-Hernández, J. H. Ko, Q. Gan, W. A. Goddard and R. H. Grubbs, J. Am. Chem. Soc., 2021, 143, 2857–2865 CrossRef CAS PubMed.
- X. Zhang, P. Wang, X. Lv, X. Niu, X. Lin, S. Zhong, D. Wang, H. Lin, J. Chen and S. Bai, ACS Catal., 2022, 12, 2569–2580 CrossRef CAS.
- B. Shao, Z. Liu, G. Zeng, Z. Wu, Y. Liu, M. Cheng, M. Chen, Y. Liu, W. Zhang and H. Feng, ACS Sustain. Chem. Eng., 2018, 6, 16424–16436 CrossRef CAS.
- H. C. Zhang, Z. X. Kang, J. J. Han, P. Wang, J. T. Fan and G. P. Sheng, Angew. Chem., Int. Ed., 2022, 61, e202200093 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |