
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Adamantane-type clusters: compounds with a ubiquitous architecture but a wide variety of compositions and unexpected materials properties
Niklas
Rinn
a,
Irán
Rojas-León
 a,
Benjamin
Peerless
a,
Saravanan
Gowrisankar
de,
Ferdinand
Ziese
de,
Nils W.
Rosemann
b,
Wolf-Christian
Pilgrim
c,
Simone
Sanna
de,
Peter R.
Schreiner
de and
Stefanie
Dehnen
*a
a,
Benjamin
Peerless
a,
Saravanan
Gowrisankar
de,
Ferdinand
Ziese
de,
Nils W.
Rosemann
b,
Wolf-Christian
Pilgrim
c,
Simone
Sanna
de,
Peter R.
Schreiner
de and
Stefanie
Dehnen
*a
aInstitute of Nanotechnology, Karlsruhe Institute of Technology, Herrmann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
bLight Technology Institute, Karlsruhe Institute of Technology, Engesserstr. 13, 76131 Karlsruhe, Germany
cFachbereich Chemie and Wissenschaftliches Zentrum für Materialwissenschaften, Philipps University Marburg, Hans-Meerwein-Straße 4, 35043 Marburg, Germany
dDepartment of Chemistry, Justus Liebig University Giessen, Heinrich-Buff-Ring 17, 35392 Giessen, Germany
eCenter for Materials Research, Justus Liebig University, Giessen, Germany
First published on 2nd May 2024
Abstract
The research into adamantane-type compounds has gained momentum in recent years, yielding remarkable new applications for this class of materials. In particular, organic adamantane derivatives (AdR4) or inorganic adamantane-type compounds of the general formula [(RT)4E6] (R: organic substituent; T: group 14 atom C, Si, Ge, Sn; E: chalcogenide atom S, Se, Te, or CH2) were shown to exhibit strong nonlinear optical (NLO) properties, either second-harmonic generation (SHG) or an unprecedented type of highly-directed white-light generation (WLG) – depending on their respective crystalline or amorphous nature. The (missing) crystallinity, as well as the maximum wavelengths of the optical transitions, are controlled by the clusters' elemental composition and by the nature of the organic groups R. Very recently, it has been additionally shown that cluster cores with increased inhomogeneity, like the one in compounds [RSi{CH2Sn(E)R′}3], not only affect the chemical properties, such as increased robustness and reversible melting behaviour, but that such ‘cluster glasses’ form a conceptually new basis for their use in light conversion devices. These findings are likely only the tip of the iceberg, as beside elemental combinations including group 14 and group 16 elements, many more adamantane-type clusters (on the one hand) and related architectures representing extensions of adamantane-type clusters (on the other hand) are known, but have not yet been addressed in terms of their opto-electronic properties. In this review, we therefore present a survey of all known classes of adanmantane-type compounds and their respective synthetic access as well as their optical properties, if reported.
1. Introduction
Diamond, in its cubic modification, is the hardest solid on Earth, which is due to the unique structure and bonding with four strong bonds directing in a perfectly tetrahedral manner to four neighbors in a three-dimensional network of face-centered cubic (F![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) symmetry.1 It is therefore reasonable that the heavier congeners, Si, Ge, and α-Sn also adopt this structure. However, not only those, but also isoelectronic binary or multinary solids follow this structural concept, as the same overall electron count allows for a corresponding electronic structure of the material. The most well-known examples are 1
m) symmetry.1 It is therefore reasonable that the heavier congeners, Si, Ge, and α-Sn also adopt this structure. However, not only those, but also isoelectronic binary or multinary solids follow this structural concept, as the same overall electron count allows for a corresponding electronic structure of the material. The most well-known examples are 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 combinations of atoms of groups 13 and 15 or 12 and 16 such as GaAs or ZnS, but more complex compositions, like CuFeS2, can also be derived from the cubic diamond network by replacement of the atomic sites in a tetragonal superstructure. Moreover, there are also “filled” versions, like the Zintl phase NaTl with two intertwining diamond networks of covalently bonded Tl atoms and non-bonding Na+ cations, or crystobalite-type SiO2 with O atoms bridging between the Si atoms that are arranged in a diamond-type pattern. Naturally, the chemical and physical properties of the materials vary as a consequence of the different elemental combinations and corresponding changes in bond strengths and electronic structures. This is extensively taken advantage of in technical applications – starting with the electrical insulator and heat conductor diamond, via all kinds of semiconductor applications of the heavier homologues and the binary analogs, to more specific applications of the more complex compounds.
:1 combinations of atoms of groups 13 and 15 or 12 and 16 such as GaAs or ZnS, but more complex compositions, like CuFeS2, can also be derived from the cubic diamond network by replacement of the atomic sites in a tetragonal superstructure. Moreover, there are also “filled” versions, like the Zintl phase NaTl with two intertwining diamond networks of covalently bonded Tl atoms and non-bonding Na+ cations, or crystobalite-type SiO2 with O atoms bridging between the Si atoms that are arranged in a diamond-type pattern. Naturally, the chemical and physical properties of the materials vary as a consequence of the different elemental combinations and corresponding changes in bond strengths and electronic structures. This is extensively taken advantage of in technical applications – starting with the electrical insulator and heat conductor diamond, via all kinds of semiconductor applications of the heavier homologues and the binary analogs, to more specific applications of the more complex compounds.
However, the structure and bonding concept of diamond, which is overwhelmingly successful in solid state compounds, is not restricted to the three-dimensional extension. On the contrary, molecular fragments of these structures are even more diverse. The parent structural fragment of diamond is adamantane (derived from the greek adamas for diamond). The adamantane-type topology (or adamantane-type scaffold) is based on a core structure with ten atoms, four of which represent the bridgehead atoms, and six of which occupy the briding positions. It has a sum formula of C10H16 (or (CH)4(CH2)6) and was first proposed in 1924 by Decker, who investigated the compound under the name “decaterpene,” which would later be recognized as adamantane.2 However, due to its exceedingly low natural abundance (0.0004%),3 it took another decade until adamantane was identified in crude oil in Hodonin, Czechoslovakia in 1933. The adamantane-type scaffold, just like its parent solid state structures, is found in a multitude of compounds scattered throughout the periodic table. Innumerous admantane derivatives have been realized – either by replacing H with other atoms or molecules, or by isoelectronic replacement of some or all of the C atoms or CH2 units – like in the related solids with diamond-type structures. A very prominent derivative of the admantane molecule is urotropine, N4(CH2)6, a condensation product of ammonia and formaldehyde, in which the C–H bridgehead units are replaced with isoelectronic N atoms. There are also purely inorganic analogs. One of the first purely inorganic adamantane-type molecules, and maybe the most prominent example, is phosphorous pentaoxide that consists of binary molecules of P4O10, the structure of which was suggested in the late 19th century.4 Inorganic cluster cores of the type {Q4E6} are obtained when replacing the bridgehead C atoms (position Q) with atoms of another group 14 element and the CH2 groups (position E) with atoms of a group 16 element. Saturation of the bridgehead atoms requires a substituent to form either binary anions [Q4E10]4− (Q = Si, Ge, Sn) or hybrid clusters of the type [(RQ)4E6], with Q = group 14 element Si, Ge, or Sn and R = organic or organometallic group substituent.
While the first observation of these molecules was unintended and caused excitement for the beautiful structure, the development is now in the direction of the compounds' intriguing chemical and physical properties. With regard to the effects of the substitution of elements on these features, the same rules apply to molecules as to solids, which enables fine-tuning across a broad spectrum. To make use of these properties, however, it is necessary to know all about the synthetic approaches and their respective modifications, and develop them further. In this review, we therefore aim at giving a comprehensive overview of the various synthesis pathways of compounds with a molecular adamantane-type structure across the periodic table, and discuss methods for the functionalization of the organic adamantane. To keep in scope, we have decided to limit the organic synthesis to tetrasubstituted adamantanes. Based on this, we will further elaborate on the optical (nonlinear) properties and structural features of the different compounds in the solid state.
2. Variety of compositions and syntheses
2.1 Inorganic and hybrid compounds
Inorganic and hybrid compounds featuring adamantane-type architectures are formed with elements from nearly all groups across the periodic table. In this section, we will discuss their synthetic access and elaborate on prevalent methods for the formation of molecules with specific elemental combinations. This will be discussed for each combination of groups from the periodic table using the Q/E nomenclature introduced above, with Q representing the atom(s) featuring three bonds within the adamantane-type structure, and E representing the atoms or groups bridging between two of the former positions. Some methods are commonly used for all elements and will be briefly discussed first with one example given for each; for easier tracking, a letter will be assigned to those procedures, to be referred to later in the course of this article.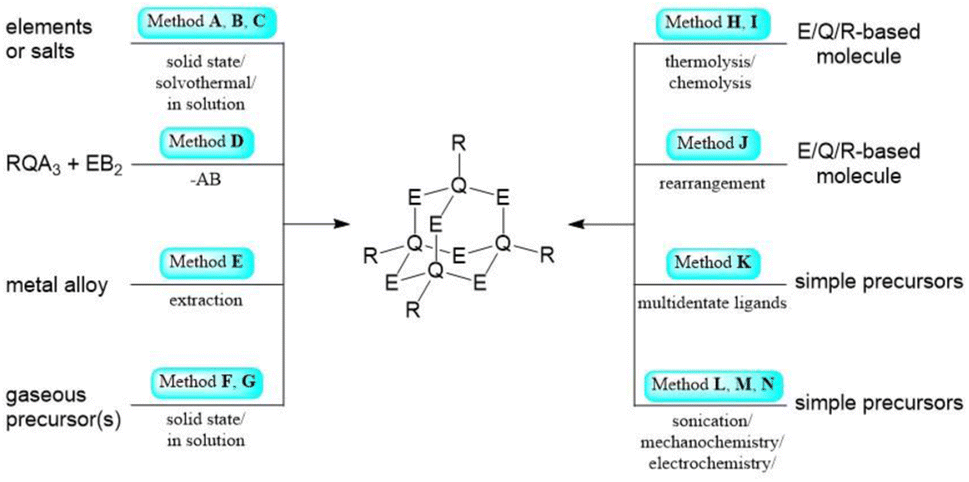 | ||
| Scheme 1 Simplified representation of the synthetic Methods A–N for the formation of adamantane-type clusters. | ||
Example:253
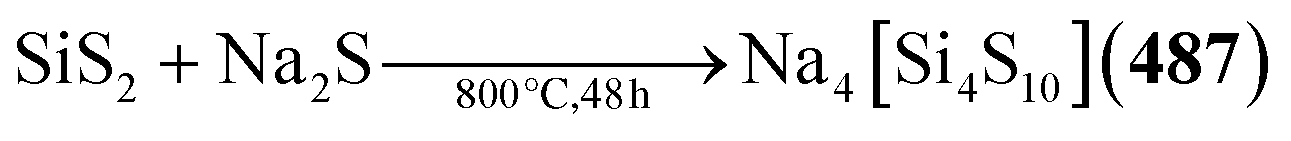
Example:65
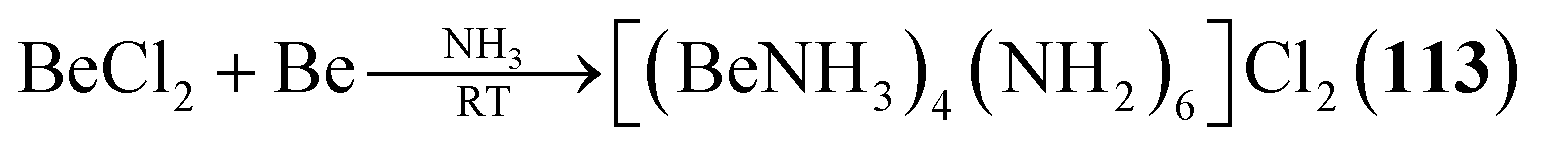
Example:253
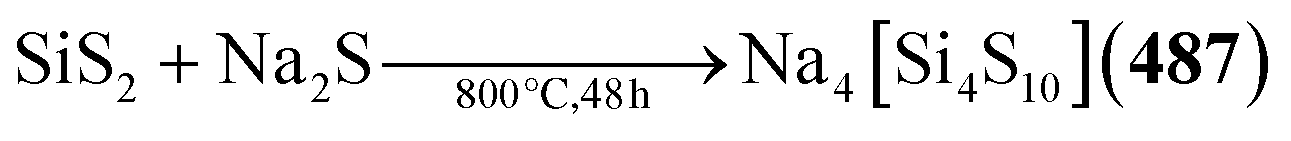
Example:304
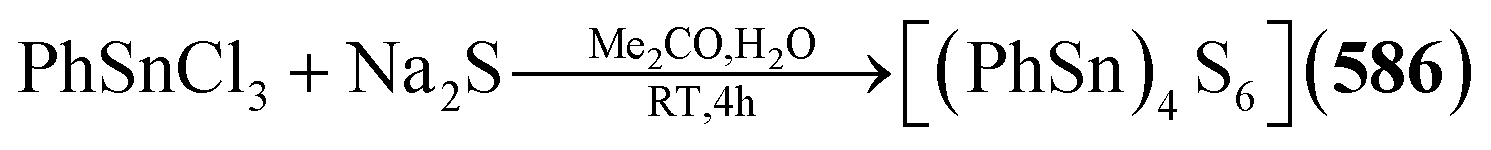
Example:282
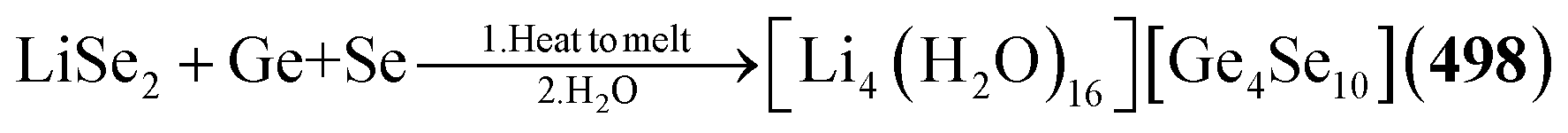
Example:304
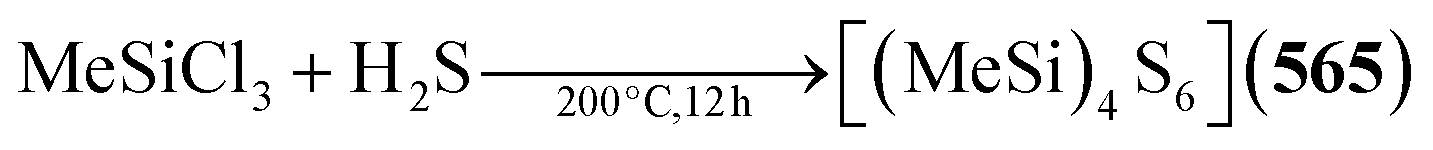
Example:78,79
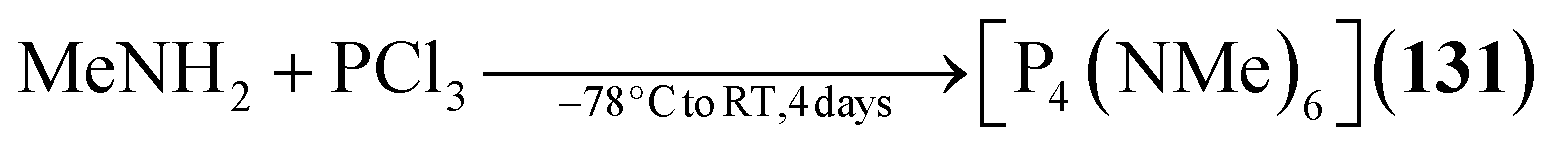
Example:62,63
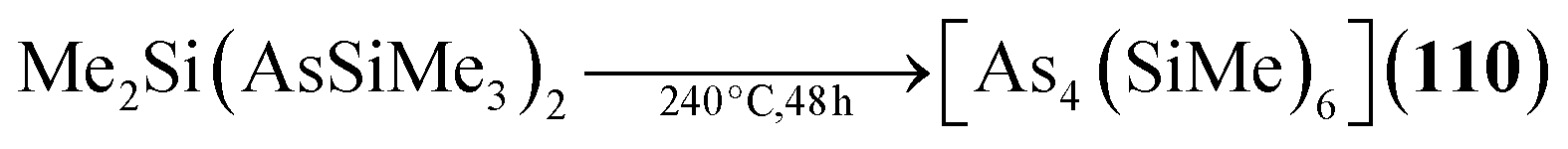
Example:116
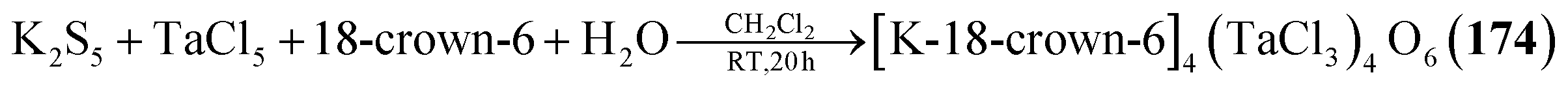
Example:28
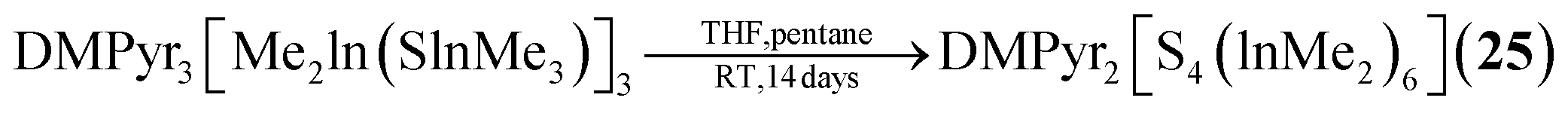
Example:30
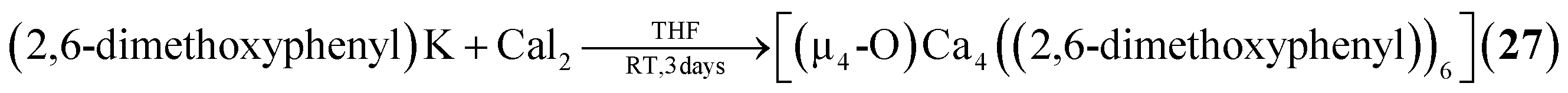
Example:29
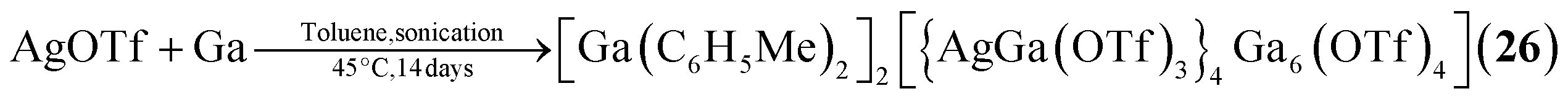
Example:86
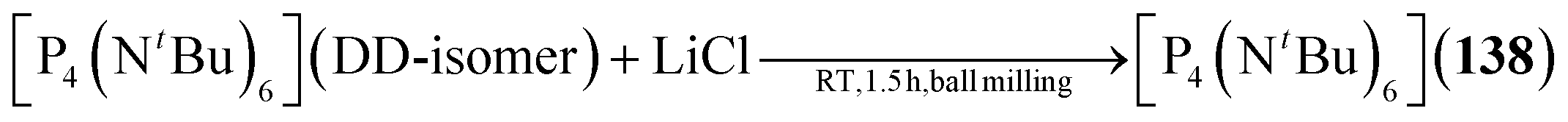
Example:194
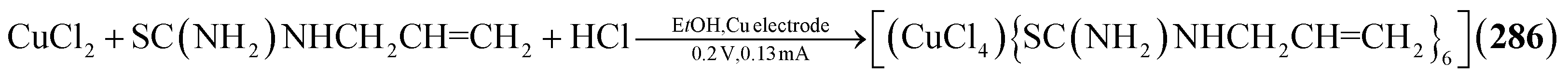
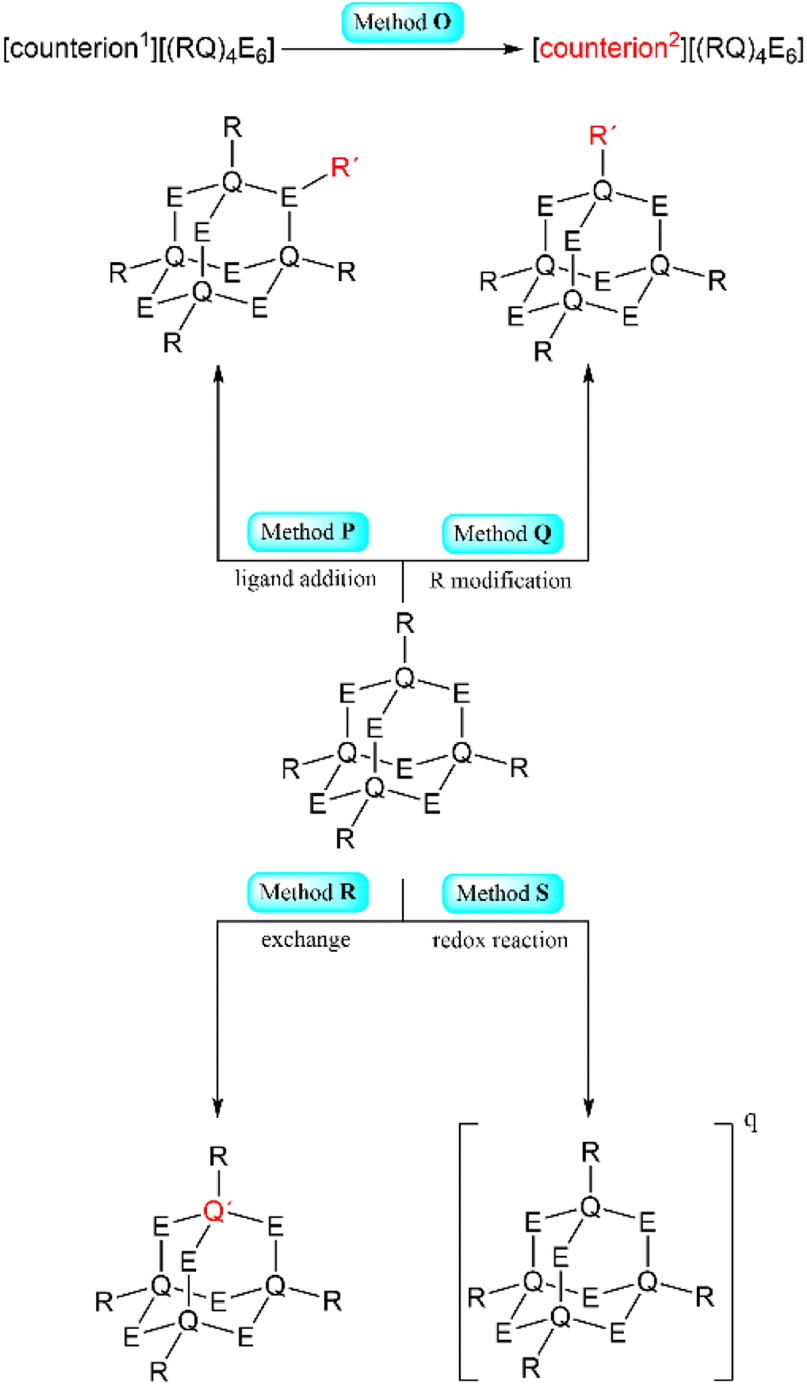 | ||
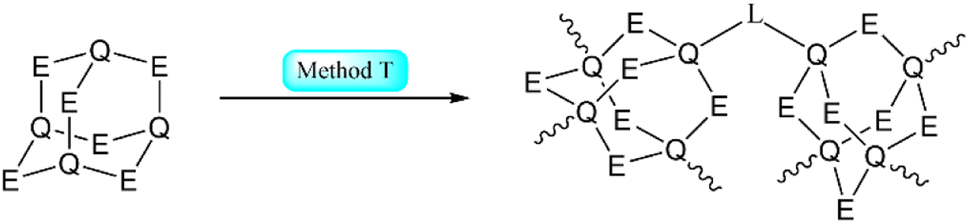
| Scheme 2 Simplified representation of the synthetic Methods O–S for the formation of adamantane-type clusters by modification of an adamantane-type cluster compound. | ||
Example:293
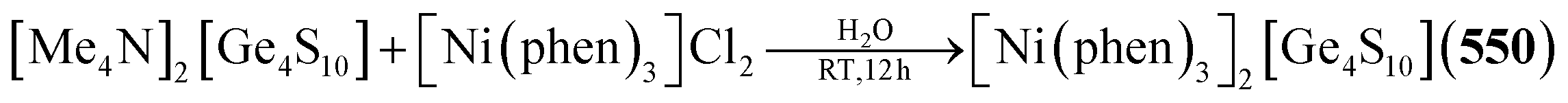
Example:345–347
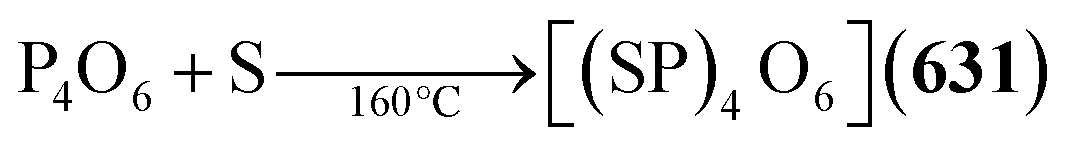
Example:512
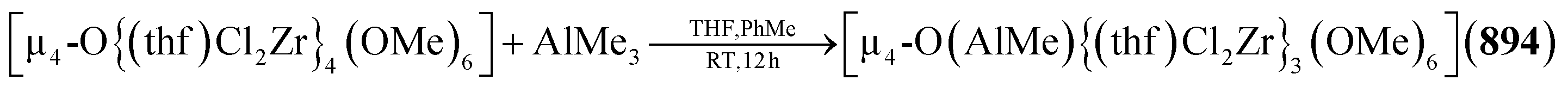
Example:131
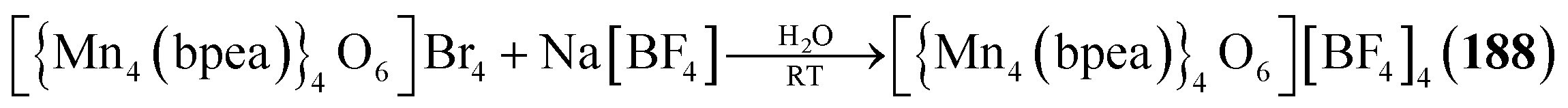
Example:131
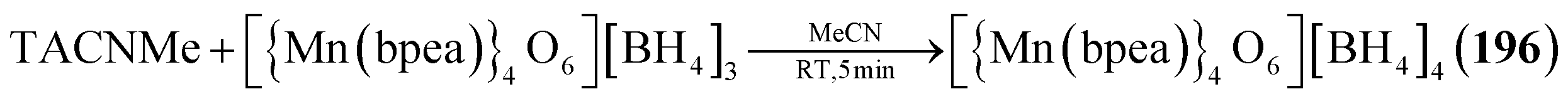
 | ||
| Scheme 3 Simplified representation of the synthetic Method T to generate a polymeric compound from adamantane-type clusters. | ||
Example:533

In the following, we will dicuss all different families of adamantane-type compounds in groups sorted by their elemental combination. This will be done in order of the group of the atoms in the E position, starting with hydride clusters and moving along to halide species. The only main groups that do not occur in the E position are groups 2 and 18.
Being rather uncommon, examples with transition metal atoms in the Q positions will be discussed last. In some of the final subsection, we will give an overview of clusters comprising elements from different groups in their scaffold, as well as extended and polymeric species.
All cluster examples, along with their simplified synthesis/reaction methods, are given in tables at the end of each section (Tables 1–23); for the sake of readability, the respective synthesis methods are not always referred to in the main text though. If the reaction temperature is not specified in the table, the reaction was carried out at ambient temperature. Similarly, reactions without specified durations occur instantaneously. Purification times and methods are not included for purifications that occur in additional, subsequent steps.
| Compound | Reagents/conditions | Method |
|---|---|---|
| a IDipp = 1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene, HMDS = 1,1,1,3,3,3-hexamethyldisilazide, TACNMe = 1,4,7-trimethyl-1,4,7-triazacyclononane, Bn = benzyl, Cp* = pentamethylcyclopentadienyl, DMAP = dimethylamine borane, BArF = [B[3,5-(CF3)2C6H3]. | ||
| [(MgIDipp)2(MgHMDS)2H6] (1) | IDipp, [Mg{N(SiMe3)2}2], PhSiH3/hexane, 60 °C, 3 h | C11 |
| [(CaTACNMe)4H6][B(C6H3-3,5-Me2)] (2) | H2 (1 bar), [(Me3TACNMe)Ca(CH2Ph)(thf)x][B(C6H3-3,5-Me2)]/THF, 70 °C, 6 h | G12 |
| [AsPh4]2[Re4(CO)12H6] (3) | Re2(CO)10, NaBH4, (C6H5)4AsCl/THF, EtOH | C13 |
| [Me3BnN]2[Re4(CO)12H6] (4) | Re2(CO)10, KOH, [Me3BnN]Cl/MeOH, 65 °C, prolonged heating | H14 |
| [(Cp*Zr)4H6] (5) | [(μ-H)(μ3-H)(Cp*ZrCl)]4, Na in Hg/Et2O, 1 month | J5 |
| [(ZnIDipp)2(ZnHMDS)2H6] (6) | Zn(HMDS)2, IDipp, DMAB/cyclohexane, RT, 30 min | C15 |
| [Ir4(IMe)7(CO)H10][BF4]2 (7) | [Ir(cod)(IMe)2][BF4], KOH, Na[BarF]/glycerol, H2O, 120 °C, 24 h | J16 |
| [Ir4(IMe)7(CO)H10][BArF]2 (8) | [Ir4(IMe)7(CO)H10][BF4]2 (7), Na[BarF]/dichlormethane, 2 h | O16 |
| [Ir4(IMe)8H10][BArF]2 (9) | [Ir(cod)(IMe)2] [BF4], KOH, NaBarF/glycerol, H2O, 120 °C, 24 h | J17 |
| [{Me2P(BH3)CHSiMe2OLi}4Li4(Et2O)2.75(thf)1.25] (10) | 1. Me3P(BH3), n-BuLi/THF, 2 h | J18 |
| 2. (Me2SiO)3/Et2O, 2 h | ||
We will illustrate examples of molecular structures of all cluster types that were obtained in single-crystalline form. For crystallographic details, we refer to the original literature.
An N-heterocyclic carbene can coordinate to [Mg(HMDS)2] (HMDS = 1,1,1,3,3,3-hexamethyldisilazide) and in turn be reacted with PhSiH3, resulting in the adamantane-type cluster [(MgIDipp)2(MgHMDS)2H6] (1, IDipp = 1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene, Fig. 1), where the magnesium atoms carry either an IDipp or N(SiMe3)2 ligand.11 A calcium congener [(CaTACNMe)4H6][B(C6H3-3,5-Me2)] (2, TACNMe = 1,4,7-trimethyl-1,4,7-triazacyclononane) is obtained from an in situ-formed complex [(TACNMe)Ca(CH2Ph)(thf)x][B(C6H3-3,5-Me2)] after treatment with H2 gas under elimination of toluene, with all Ca atoms carrying the same tridentate ligand.12
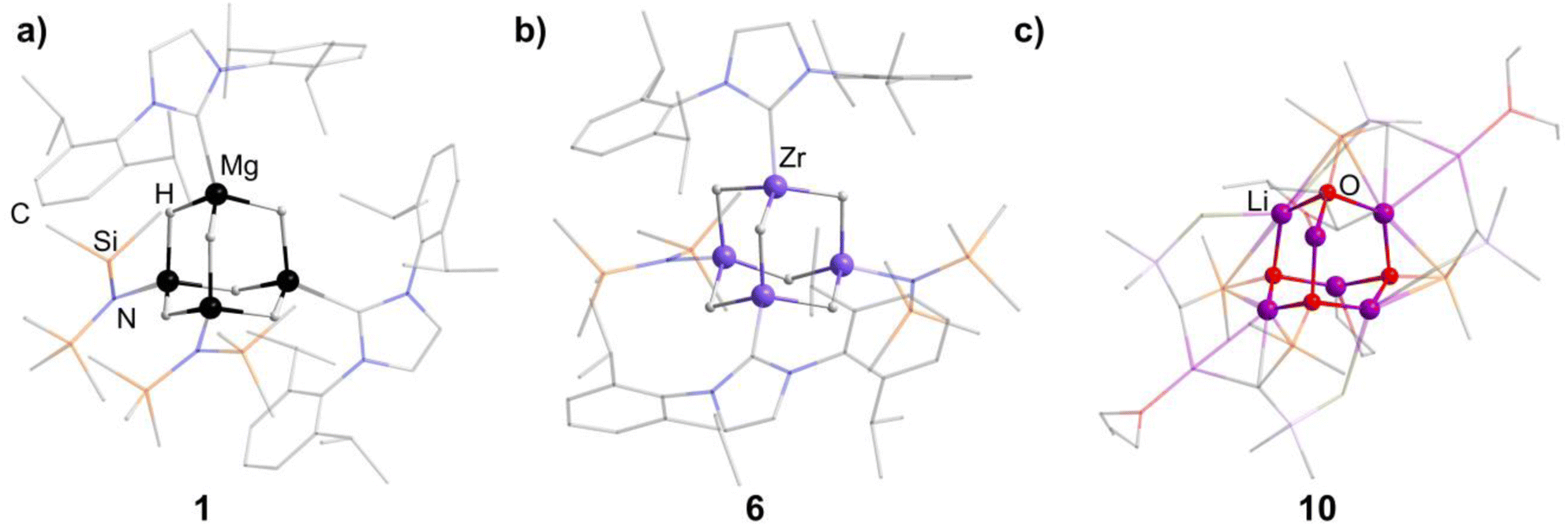 | ||
| Fig. 1 Examples of adamantane-type compounds with hydrogen or group 1 atoms in the E position: [(MgIDipp)2(MgHMDS)2H6] (1, left (a)), [(ZnIDipp)2(ZnHMDS)2H6] (6, middle (b)) and [{Me2P(BH3)CHSiMe2OLi}4Li4(Et2O)2.75(thf)1.25] (10, right (c)). Hydrogen atoms in the ligands are omitted for clarity. | ||
The first transition metal cluster anion in this group, [{(CO)3Re}4H6]2− (in 3 and 4) was formed from [Re2(CO)10], either by reaction with Na[BH4]13 or by prolonged heating under basic conditions in methanol as one of multiple products.14
The adamantane-type compound [(Cp*Zr)4H6] (5, Cp* = pentamethylcyclopentadienyl) was found as the final piece in a row of tetrahedral compounds with fewer hydrides by reduction of [(μ-H)(μ3-H)(Cp*ZrCl)]4 with Na amalgam.5 This led to a mixed-valence ZrII/ZrIII situation in the cluster core.
An analog to the aforementioned [(MgIDipp)2(MgHMDS)2H6] cluster was realized with zinc in [(ZnIDipp)2(ZnHMDS)2H6] (6, Fig. 1).15 The synthesis strategy runs in parallel as well, with Zn(HMDS)2 as the metal precursor and dimethylamine borane as a hydride source.
A number of iridium hydride clusters (7–9) could be obtained upon dehydrogenation reactions catalyzed by [Ir(Ime)2(cod)][BF4] (cod = 1,5-cyclooctadiene) of glycerol.16,17 This results in the formation of the hydride as well as CO ligands at the metal center in some cases.
A {Li4O6} adamantane-like core can be observed in the larger complex [{Me2P(BH3)CHSiMe2OLi}4Li4(Et2O)2.75(thf)1.25] (10, Fig. 1). It is formed as the tetramer of the in situ generated linear molecule Me2P(BH)CH(Li)Si(Me2)OLi coordinated by additional solvent molecules.18
Different approaches for the formation of the few known carbon/group 13 adamantane-type compounds have been showcased in the literature. The boron congeners [(RC)4(R′B)6] (11–13, Fig. 2) can be synthesized at higher temperatures by pyrolysis of BMe3 or (Cl2B)2CH2,19–21 or by a solid state reaction of HC(BEt2)3 and BEt3 in the presence of AlEt3.22 At room temperature, the rearrangement of (BEt)3(CMe)2 to [(CMe)4(BEt)6] (14) was observed, induced by elemental potassium and I2.23
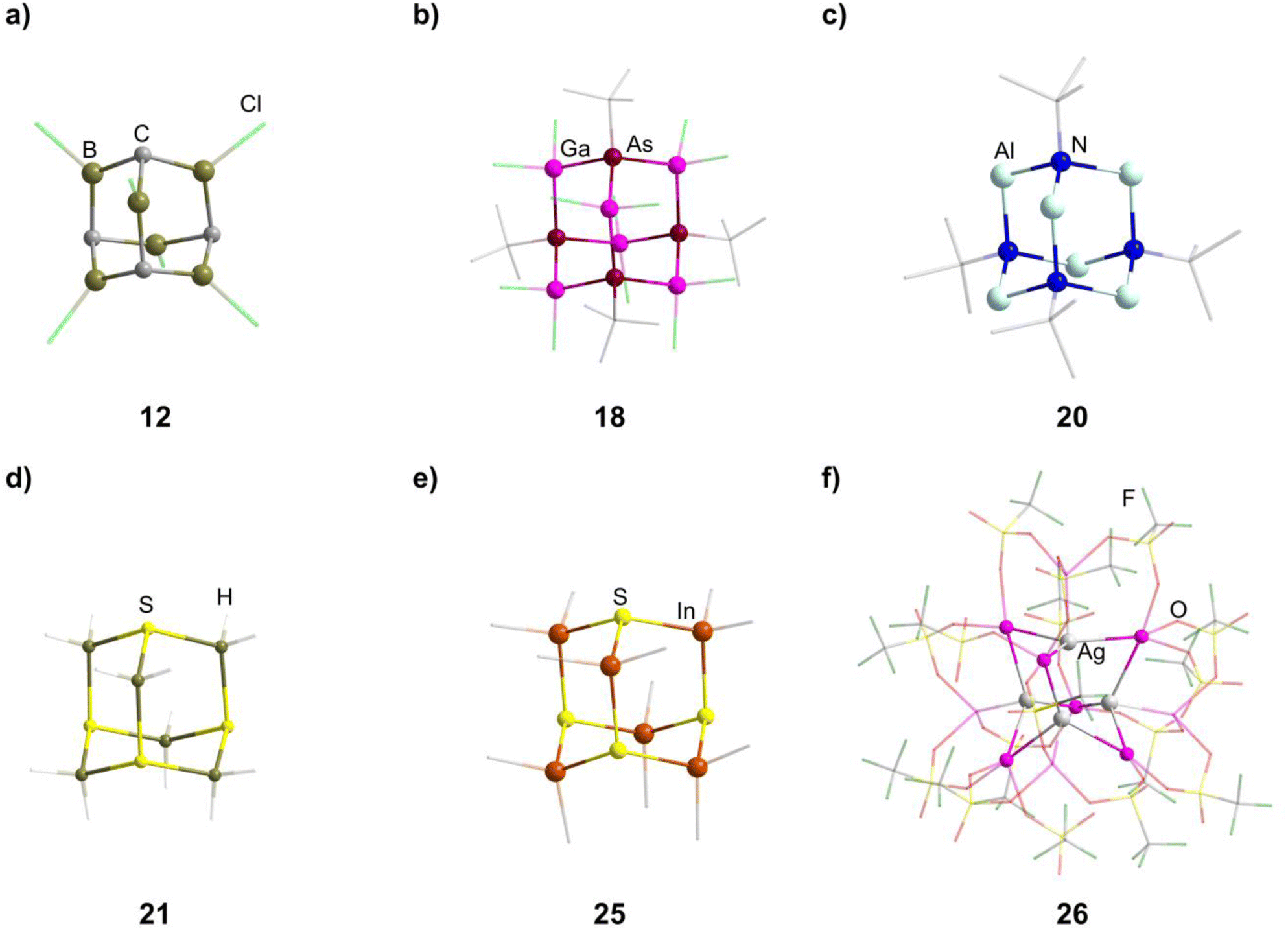 | ||
| Fig. 2 Examples of adamantane-type compounds with group 13 atoms in the E position: [(CH)4(BCl)6] (12, top left (a)), [Li(thf)4]2[(tBuAs)4(GaCl2)6] (18, top center (b)), Li2[(tBuN)4(AlH2)6] (20, top right (c)), Na2[S4(BH2)6] (21, bottom left (d)), DMPyr2[S4(InMe2)6] (25, bottom center (e)) and [Ga(C6H5Me)2]2[{AgGa(OTf)3}4Ga6(OTf)4] (26, bottom right (f)). Hydrogen atoms in the organic ligands and counterions, if present, are omitted for clarity. | ||
A unique synthetic approach, featuring R2GaH and alkenes HC≡CR′, leads to the formation of carbagallane adamantane-type structures [(R′C)4(RGa)6] (15–17).24 It involves the intermediate formation of dialkyl(alkenyl)gallium compounds, which react with additional R2GaH to form the clusters under elimination of GaR3.
Three dianionic group 15 congeners exist. An As/Ga compound [Li(thf)4]2[(tBuAs)4(GaCl2)6] (18, tBu = tertiary butyl, Fig. 2) is isolated by a simple condensation reaction of InCl3 and Li2AstBu at low temperatures,25 while the compounds Li2[(RN)4(AlH2)6] (19–20, R = Me, tBu, Fig. 2) are formed by condensation of Li[AlH4] and [RNH3]Cl.26
The sulfur containing Na2[S4(BH2)6] (21, Fig. 2) adamantane-type cluster is obtained by a stepwise condensation reaction of THF·BH3, and Na[BH4] with H2S under elimination of H2.27 In the reaction, an intermediate species (BH3)S(B2H5) is formed, which reacts with additional H2S to give the final product. The Se congener is formed via a different species with elemental Se and Na[BH4]. This leads to Na2[H3BSe–SeBH3] which, under the influence of elevated temperature and BH3, reforms Na2[Se4(BH2)6] (22). Both the sulfur and selenium homologs undergo a cation exchange to the Cs compounds (23–24) with CsBr. The only other example of a group 16-based adamantane in this category is DMPyr2[S4(Me2In)6] (25, Fig. 2), which is a decomposition side product of the six membered ring DMPyr3[Me2In(SInMe3)]3, which could not yet be synthesized in a pure form.28
The single example featuring a transition metal [Ga(C6H5Me)2]2[{AgGa(OTf)3}4Ga6(OTf)4] (26, OTf = O3SCF3, Fig. 2) comprises bridging triflate ligands between the gallium atoms, with the terminal gallium moieties connecting to three, and the atoms in the E position to four, ligands.29 It is formed by silver triflate reacting with elemental gallium after ultrasonic activation.
| Compound | Reagents/conditions | Method |
|---|---|---|
| a n Bu = normal butyl, tBu = tertiary butyl, diglyme = bis(2-methoxyethyl) ether, DMPyr = 1,1-dimethylpyrrolidinium, OTf = O3SCF3 | ||
| [(CH)4(BMe)6] (11) | BMe3/450 °C, 40 min | H19,20 |
| [(CH)4(BCl)6] (12) | (Cl2B)2CH2/450 °C to RT, 12 h | H21 |
| [(CH)4(BEt)6] (13) | HC(BEt2)3, BEt2, AlEt3/150 °C | A22 |
| [(CMe)4(BEt)6] (14) | (BEt)3(CMe)2, I2, K/THF | J23 |
| [(EtC)4(GaEt)6] (15) | Et2GaH, HC≡CEt/−196 °C to RT, 4 h | U24 |
| [(nBuC)4(GaEt)6] (16) | Et2GaH, HC≡CnBu/4 h | U24 |
| [(EtC)4(GaMe)6] (17) | Me2GaH, HC≡CEt/−196 °C to RT, 4 h | U24 |
| [Li(thf)4]2[(tBuAs)4(GaCl2)6] (18) | Li2AstBu, GaCl3/Et2O, −78 °C to RT, 3 days | C25 |
| Li2[(RN)4(AlH2)6] (19–20, R = Me, tBu) | Li[AlH4], [RNH3]Cl,/Et2O, 4 weeks | C26 |
| Na2[S4(BH2)6] (21) | THF·BH3, Na[BH4], H2S/0 °C | C27 |
| Na2[Se4(BH2)6] (22) | 1. Se, Na[BH4]/diglyme, 0 °C to 110 °C, 8 h | B27 |
| 2. THF·BH3/diglyme | ||
| Cs2[S4(BH2)6] (23) | Na2[S4(BH2)6] (21), CsBr/H2O | O27 |
| Cs2[Se4(BH2)6] (24) | Na2[Se4(BH2)6] (22), CsBr/H2O | O27 |
| DMPyr2[S4(InMe2)6] (25) | DMPyr3[Me2In(SInMe3)]3/THF, pentane, 14 days | J28 |
| [Ga(C6H5Me)2]2[{AgGa(OTf)3}4Ga6(OTf)4] (26) | AgOTf, Ga/toluene, 45 °C, 1.5 h (ultrasonic activation) | L29 |
2.1.3.1 Group 2/group 14 adamantane-type clusters. This unique group 2/14 adamantane-type, [(μ4-O)Ca4(2,6-dimethoxyphenyl)6] (27, Fig. 3), which is formed around a central oxygen atom, uses the tridentate dimethoxyphenyl group as a templating ligand.30 These ligands bridge the Ca sites both by a carbon atom in the E position, as well as by coordination with their oxygen atoms. The origin of the central μ4-O atom could not be determined and might stem either from H2O or O2 impurities during the inert gas protected reaction, or decomposition of the solvent/ligand.
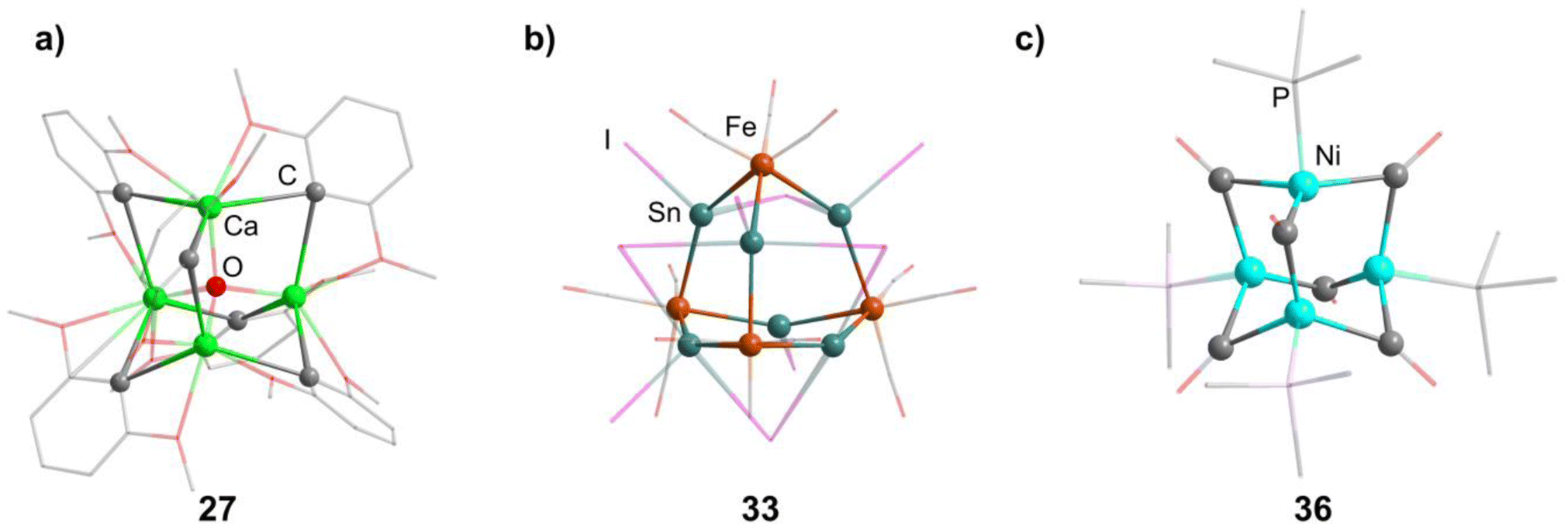 | ||
| Fig. 3 Examples of adamantane-type compounds with group 2 and transition metals in the Q position and group 14 atoms in the E position: [(μ4-O)Ca4(2,6-dimethoxyphenyl)6] (27, left (a)), [BMIm]6[S][{Fe(CO)3}4Sn6I10]2 (33, center (b)) and [(NiPMe3)4(CO)6] (36 right (c)). Hydrogen atoms and counterions, if present, are omitted for clarity. | ||
2.1.3.2 Group 8/14 adamantane-type clusters. In two studies, Fe clusters have been characterized. In the cluster family [(Fe)4(aryl)6(thf)x] (28–31), the carbon atoms of aryls are found in the E position. These clusters are prepared by reacting Fe(acac)3 (acac = acetylacetonate) with the aryl Grignard reagent (aryl)MgBr.31
In the other study, reactions of SnI4 and Fe(CO)5 in ionic liquids lead to Fe/Sn compounds. [BMIm]2[{Fe(CO)3}4Sn6I10] (32, BMIm = 1-butyl-3-methyl-imidazolium) or [BMIm]6[S][{Fe(CO)3}4Sn6I10]2 (33, Fig. 3) depending on the counterion in the ionic liquid.32 They each feature different Sn coordination sites. In 32, three Sn atoms carry two iodo ligands, one is connected to only one iodine and the final two carry one terminal iodine and one bridging μ-I connecting them to each other. The second cluster comprises three tin atoms carrying two iodine ligands, while the other three only connect to one terminal iodide each and are connected via a μ3-I bridge.
2.1.3.3 Group 8/14 adamantane-type clusters. Group 10 clusters with group fourteen elements in the E position are known for combinations with Ni and Pd.
The first family of such compounds with the general compositon [(NiPR3)4(CO)6] (34–37, Fig. 3) comprise CO bridged Ni tetrahedra with terminal phosphine ligands.33–35 They are generally prepared by reacting a Ni complex with the desired phosphine and CO gas, if the original complex does not contain such ligands already. These results could be transferred to palladium in the case of [(PdPnBu3)4(CO)6] (38).36
| Compound | Reagents/conditions | Method |
|---|---|---|
| a cod = 1,5-cyclooctadiene, OAc = acetate, acac = acetylacetonate, BMIm = 1-butyl-3-methyl-imidazolium, NTf2 = bistrifluoridomethansulfonimide. | ||
| [(μ4-O)Ca4(2,6-dimethoxyphenyl)6] (27) | (2,6-Dimethoxyphenyl)K, CaI2/THF, 3 days | K30 |
| [Fe4(Ph)6(THF)4] (28) | Fe(acac)3, PhMgBr/THF, −30 °C, 25 min | C31 |
| [Fe4(p-tolyl)6(THF)4] (29) | Fe(acac)3, p-tolylMgBr/THF, −30 °C, 25 min | C31 |
| [Fe4(p-tolyl)6(THF)3] (30) | Fe(acac)3, p-tolylMgBr/THF, −30 °C, 25 min | C31 |
| [Fe4(4-F-Ph)6(THF)4] (31) | Fe(acac)3, 4-F-PhMgBr/THF, −30 °C, 25 min | C31 |
| [BMIm]2[{Fe(CO)3}4Sn6I10] (32) | SnI4, Fe(CO)5, [BMIm][NTf2]/130 °C, 4 days | B32 |
| [BMIm]6[S][{Fe(CO)3}4Sn6I10]2 (33) | SnI4, NH4I, Fe(CO)5, [BMIm][OTf]/130 °C, 4 days | B32 |
| [{NiP(CH2CH2CN)3}4(CO)6] (34) | Tris-(2-cyanoethyl)phosphine, Ni(CO)4/MeOH, 70 °C, 24 h | B33 |
| [(NiPMe3)4}[BF4][(NiPMe3)4(CO)6] (35) | Ni(COMe)Cl(PMe3)2, PMe3, Tl[BF4]/CH2Cl2, RT, 30 min | C34 |
| [(NiPMe3)4(CO)6] (36) | Bis(cod)nickel, PMe3, CO/toluene, RT, 6 h | F35 |
| [(NiPnBu3)4(CO)6] (37) | Bis(cod)nickel, PnBu3, CO/toluene, RT, 6 h | F35 |
| [(PdPnBu3)4(CO)6] (38) | Pd4(CO)5(PBu3n)4, CH3COOH/EtOH, pentane, RT, 2 days or Pd(OAc)2, CH3COOH, CO, PBu3n/dioxane, Me2CO, 5 days | J/F36 |
2.1.3.4 Group 14/14 adamantane-type clusters. A family of tetrasilaadamantanes of the composition [(RSi)4(CH2)6] has been investigated, with the first examples being obtained in high temperature reactions of either SiMe4 to form [(SiMe)4(CH2)6] or SiCl4 and Me3SiCl in the presence of AlCl3 to yield [(SiCl)4(CH2)6] (39).37–40 In subsequent work, access to such compounds was made at considerably lower temperatures and in higher yields. [(SiMe)4(CH2)6] (40, Fig. 4) could be obtained from an AlBr3 induced rearrangement of (Me2SiCH2)3 at 100 °C, which could then in turn be reacted with Cl2 and I2 to form 39 or be treated with Li[AlH4] to form the hydrogen terminated [(SiH)4(CH2)6] (41).39,41,42Via both of these routes, tetrasilaadamantanes with mixed methyl and halide positions can be isolated as well.41–43 These clusters described so far are used as the basis for ligand exchange reactions at the Si sites (Method Q, leading to 42–55), often by exchanging the halides found in various positions.38,44–46 Asides from derivatization on the silicon atom, the CH2 moiety can also be a target for lithiation to give stepwise addition of longer C/Si chains (56–60).47 Lastly, it was also shown that the ligand of a single Si site can be abstracted to obtain a charged cluster cation [(SiMe)3Si(CH2)6][CHB11Cl11] (61) by reacting the carbocation [Ph3C]+ with [(SiMe)3SiH(CH2)6] (49).48
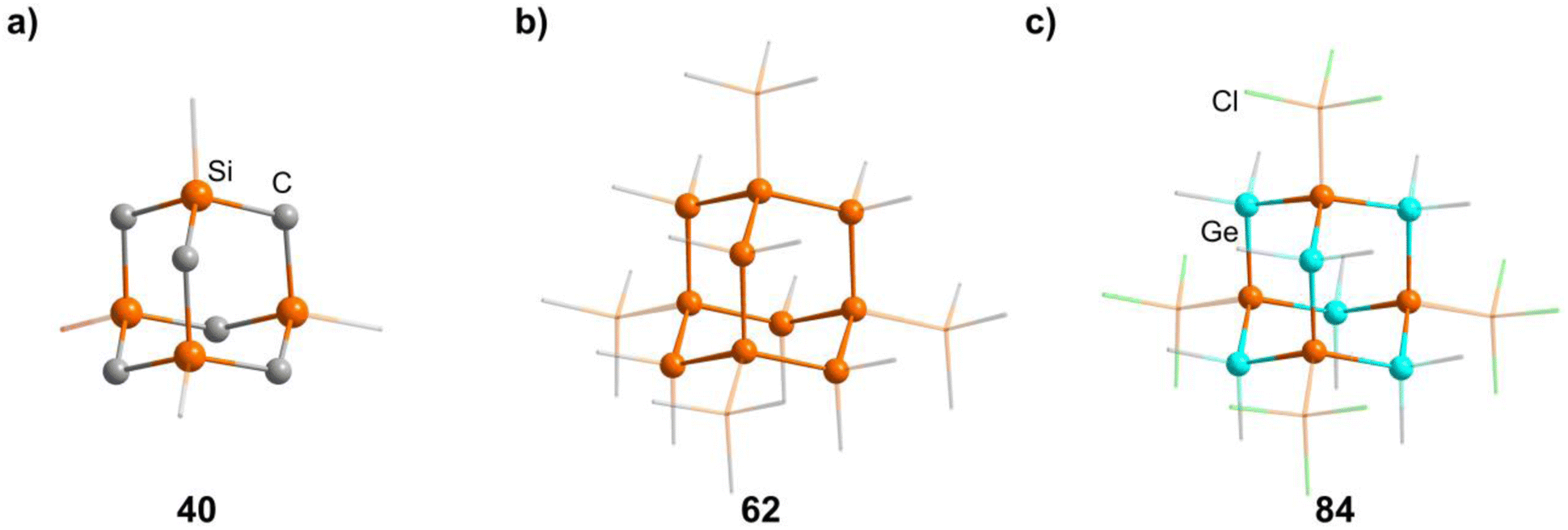 | ||
| Fig. 4 Examples of adamantane-type compounds with group 14 elements in the Q position and group 14 atoms in the E position: [(SiMe)4(CH2)6] (40, left (a)), [(SiSiMe3)4(SiMe2)6] (62, center (b)) and [(SiSiCl3)4(GeMe2)6] (84, right (c)). Hydrogen atoms are omitted for clarity. | ||
Realizing the first purely Si based adamantanes took a 9 step synthesis, the last one being a rearrangement of a tricyclic compound Si14Me24 to [(SiMe)4(SiMe2)6] (62, Fig. 4) reminiscent of a synthesis route to organic adamantanes by Schleyer (see section 2.2).49 In recent times, the topic has been reinvestigated, resulting in a streamlined gram scale synthesis method, and strategies for a site selective functionalization, which can lead to one or more methyl groups being substituted at the Q position (63–81).50
While the pure silaadamantanes were not obtainable from simple building blocks, compounds with mixed Ge/Si sites were isolated by a mixture of Me2GeCl2, Si2Cl6 and [nBu4N]Cl, leading to [(SiSiCl3)4(GeMe2)6−x(SiCl2)x] (82–84, x = 0–2, Fig. 4), with the amount of Ge incorporated rising with the use of higher amounts of [nBu4N]Cl.51 In follow up investigations, site selective methylation at the Q position of these compounds was realized using the Grignard reagent MeMgBr (85–87).52 It was also possible to obtain the corresponding stannasilaadamantanes [(SiSiCl3)4(SnMe2)6−x(SiCl2)x] ((88–89, x = 1–2) by substituting the Ge component for the higher homolog Me2SnCl2 in the reaction.
| Compound | Reagents/conditions | Method |
|---|---|---|
| a TMEDA = tetramethylethylenediamine, iPr = isopropyl. | ||
| [(SiCl)4(CH2)6] (39) | SiCl4, Me3SiCl, AlCl3/500 °C or [(SiMe)4(CH2)6], Cl2, I2/CCl4 | B/Q37,39,42 |
| [(SiMe)4(CH2)6] (40) | SiMe4/700 °C or (Me2SiCH2)3, AlBr3/100 °C | A38,40,41 |
| [(SiH)4(CH2)6] (41) | Li[AlH4], [(SiMe)4(CH2)6] (40) | Q39 |
| [(SiMe)3SiBr(CH2)6] (42) | (Me2SiCH2)3, AlBr3/100 °C | J41,43 |
| [(SiMe)2(SiBr)2(CH2)6] (43) | (Me2SiCH2)3, AlBr3/100 °C | J41 |
| [(SiMe)3SiCl(CH2)6] (44) | (Me2SiCH2)3, AlCl3/100 °C | J41 |
| [(SiMe)2(SiCl)2(CH2)6] (45) | (Me2SiCH2)3, AlCl3/100 °C | J41 |
| [SiMe(SiCl)3(CH2)6] (46) | [(SiMe)4(CH2)6] (40), Cl2, I2/CCl4 | Q42 |
| [(SiMe)3SiOH(CH2)6] (47) | [(SiMe)3SiCl(CH2)6] (44), [NBu4]Cl, KOH/2-methyl-2-butanol, H2O, 80 °C, 30 min | Q44 |
| [(SiMe)3SiOCH2CH2NMe2(CH2)6] (48) | [(SiMe)3SiCl(CH2)6] (44), HOCH2CH2NMe2, n-BuLi/hexane, 69 °C, 9 h | Q44 |
| [(SiMe)3SiH(CH2)6] (49) | [(SiMe)3SiBr(CH2)6] (42), Li[AlH4]/Et2O, 35 °C, 4 days | Q44 |
| [(SiMe)3SiNEt2(CH2)6] (50) | [(SiMe)3SiCl(CH2)6] (44), LiNEt2/hexane, 24 h | Q44 |
| [(SiMe)3SiPh(CH2)6] (51) | [(SiMe)3SiCl(CH2)6] (44), LiPh/Et2O | Q45 |
| [(SiMe)3SiOMe(CH2)6] (52) | [(SiMe)3SiBr(CH2)6] (42), NaOMe,/MeOH | Q45 |
| [(SiMe)3SiF(CH2)6] (53) | [(SiMe)3SiBr(CH2)6] (42), c-C6H11NH3F,/CHCl3 | Q45 |
| [(SiMe)3SiOTf(CH2)6] (54) | [(SiMe)4(CH2)6] (40), ICl, AgOTf/CH2Cl2, 1 day | Q46 |
| [(SiOTf)2(SiMe)2(CH2)6] (55) | [(SiMe)3SiOTf(CH2)6] (54), ICl, AgOTf/CH2Cl2, 24 h | Q46 |
| [(SiMe)4(CH2)5CHSiMe2Ph] (56) | [(SiMe)4(CH2)6] (40), ClSiMe2Ph, n-BuLi, KOCMe3/THF, 0 °C, 10 h | Q47 |
| [(SiMe)4(CH2)5CHSiMe2CH2SiMe2Ph] (57) | [(SiMe)4(CH2)5CHSiMe2Ph] (56), Br2, LiCH2SiMe2Ph/ | Q47 |
| [(SiMe)4(CH2)5CHSiMe2CH2SiMe2CH2SiMe3] (58) | [(SiMe)4(CH2)6] (40), Me3SiCH2SiMe2CH2SiMe2Br, n-BuLi, TMEDA/hexane, 40 °C, 5 h | Q47 |
| [(SiMe)3(CH2)5CH(SiMe2CH2SiMe2CH2)Si] (59) | [(SiMe)4(CH2)5CHSiMe2CH2SiMe2CH2SiMe3] (59), AlBr3/30 °C, 20 h | Q47 |
| [(SiBr)(SiMe)2(CH2)5CH(SiMe2CH2SiMe2CH2)Si] (60) | [(SiMe)4(CH2)5CHSiMe2CH2SiMe2CH2SiMe3] (59), AlBr3/30 °C, 20 h | Q47 |
| [(SiMe)3Si(CH2)6][CHB11Cl11] (61) | [Ph3C][CHB11Cl11], [(SiMe)3SiH(CH2)6] (49)/PhBr | Q48 |
| [(SiSiMe3)4(SiMe2)6] (62) | Si14Me24, [CPh3][B(C6F5)4]/Toluene, 48 h | J49,50 |
| [(SiSiMe3)4(SiMe2)5(SiMeCl)] (63) | Si14Me24, AlCl3, MeI, Me3SiCl/C6H6, 48 h | J50 |
| [(SiSiMe3)4(SiMe2)5(SiMeBr)] (64) | Si14Me24, AlBr3, MeI, Me3SiBr/C6H6, 17 days | J50 |
| [(SiSiMe2Cl)(SiSiMe3)3(SiMe2)6] (65) | 1. [(SiSiMe3)4(SiMe2)6] (62), KOCMe3, 18-crown-6/toluene, 16 h | Q50 |
| 2. Me2SiCl2/1 h | ||
| [(SiSiMe2Ph)(SiSiMe3)3(SiMe2)6] (66) | 1. [(SiSiMe3)4(SiMe2)6] (62), KOCMe3, 18-crown-6/toluene, 16 h | Q50 |
| 2. Me2PhSiCl/3 h | ||
| [(SiSiPh3)(SiSiMe3)3(SiMe2)6] (67) | 1. [(SiSiMe3)4(SiMe2)6] (62), KOCMe3, 18-crown-6/toluene, 16 h | Q50 |
| 2. Ph3SiCl/3 h | ||
| [(SiSnMe3)(SiSiMe3)3(SiMe2)6] (68) | 1. [(SiSiMe3)4(SiMe2)6] (62), KOCMe3, 18-crown-6/toluene, 16 h | Q50 |
| 2. Me3SnCl/3 h | ||
| [(SiGeMe3)(SiSiMe3)3(SiMe2)6] (69) | 1. [(SiSiMe3)4(SiMe2)6] (62), KOCMe3, 18-crown-6/toluene, 16 h | Q50 |
| 2. Me3GeCl/3 h | ||
| [(SiH)(SiSiMe3)3(SiMe2)6] (70) | 1. [(SiSiMe3)4(SiMe2)6] (62), KOCMe3, 18-crown-6/toluene, 16 h | Q50 |
| 2. HCl/3 h | ||
| [{SiP(NET2)2}(SiSiMe3)3(SiMe2)6] (71) | 1. [(SiSiMe3)4(SiMe2)6] (62), KOCMe3, 18-crown-6/toluene, 16 h | Q50 |
| 2. P(NET2)2Cl/3 h | ||
| [(SiCH2SMe)(SiSiMe3)3(SiMe2)6] (72) | 1. [(SiSiMe3)4(SiMe2)6] (62), KOCMe3, 18-crown-6/toluene, 16 h | Q50 |
| 2. ClCH2SMe/3 h | ||
| [(SiMe)(SiSiMe3)3(SiMe2)6] (73) | 1. [(SiSiMe3)4(SiMe2)6] (62), KOCMe3, 18-crown-6/toluene, 16 h | Q50 |
| 2. Methyl-p-toluenesulfonate/3 h | ||
| [(SiBr)(SiSiMe3)3(SiMe2)6] (74) | 1. [(SiSiMe3)4(SiMe2)6] (62), KOCMe3, 18-crown-6/toluene, 16 h | Q50 |
| 2. 1,2-Dibromoethane/3 h | ||
| [(SiCl)(SiSiMe3)3(SiMe2)6] (75) | 1. [(SiSiMe3)4(SiMe2)6] (62), KOCMe3, 18-crown-6/toluene, 16 h | Q50 |
| 2. PCl3/−78 °C, 3 h | ||
| [(SiCH2SMe)2(SiSiMe3)2(SiMe2)6] (76) | 1. [(SiSiMe3)4(SiMe2)6] (62), KOCMe3, 18-crown-6/toluene, 16 h | Q50 |
| 2. ClCH2SMe/3 h | ||
| [(SiCH2SMe)3(SiSiMe3)(SiMe2)6] (77) | 1. [(SiSiMe3)4(SiMe2)6] (62), KOCMe3, 18-crown-6/toluene, 16 h | Q50 |
| 2. ClCH2SMe/3 h | ||
| [(SiCH2SMe)4(SiMe2)6] (78) | 1. [(SiSiMe3)4(SiMe2)6] (62), KOCMe3, 18-crown-6/toluene, 16 h | Q50 |
| 2. ClCH2SMe/3 h | ||
| [(SiMe)(SiiPr)(SiSiMe3)2(SiMe2)6] (79) | 1. [(SiMe)(SiSiMe3)3(SiMe2)6] (62), KOCMe3, 18-crown-6/toluene, 16 h | Q50 |
| 2.Chlorotriisopropylsilane/3 h | ||
| [(SiMe)(SiiPr)(SiCH2SMe) (SiSiMe3)(SiMe2)6] (80) | 1. [(SiMe)(SiiPr)(SiSiMe3)2(SiMe2)6] (79), KOCMe3, 18-crown-6/toluene, 16 h | Q50 |
| 2.ClCH2SMe/3 h | ||
| [(SiMe)(SiiPr)(SiCH2SMe)(SiBr)(SiMe2)6] (81) | 1. [(SiMe)(SiiPr)(SiCH2SMe) (SiSiMe3)(SiMe2)6] (80), KOCMe3, 18-crown-6/toluene, 16 h | Q50 |
| 2. 1,2-Dibromoethane/3 h | ||
| [(SiSiCl3)4(GeMe2)4(SiCl2)2] (82) | Me2GeCl2, Si2Cl6, [Bu4N]Cl/CH2Cl2, 13 days | C51 |
| [(SiSiCl3)4(GeMe2)5(SiCl)] (83) | Me2GeCl2, Si2Cl6, [Bu4N]Cl/CH2Cl2, 19 days | C51 |
| [(SiSiCl3)4(GeMe2)6] (84) | Me2GeCl2, Si2Cl6, [Bu4N]Cl/CH2Cl2, 60 °C, 6 days | C51 |
| [(SiSiMe3)4(GeMe2)6] (85) | [(SiSiCl3)4(GeMe2)6] (85), MeMgBr/Et2O, 60 °C, 1 day | Q52 |
| [(SiSiMe3)4(GeMe2)4(GeMe2)2] (86) | [(SiSiCl3)4(GeMe2)4(GeMe2)2] (86), MeMgBr/Et2O, 60 °C, 1 day | Q52 |
| [(SiSiMe3)4(GeMe2)5(GeMe2)] (87) | [(SiSiCl3)4(GeMe2)5(GeMe2)] (87), MeMgBr/THF, Et2O, 1 day | Q52 |
| [(SiSiCl3)4(SnMe2)4(SiCl2)2] (88) | Me2SnCl2, Si2Cl6, [Bu4N]Cl/CH2Cl2, 3 days | Q52 |
| [(SiSiCl3)4(SnMe2)5(SiCl2)] (89) | [(SiSiCl3)4(SnMe2)4(SiCl2)2] (88), [Bu4N]Cl/CH2Cl2, 60 °C, 1 day | Q52 |
2.1.3.5 Group 15/14 adamantane-type clusters. [P4(SiR2)6] (90–95) adamantane-type clusters with different ligands R are formed by adding Cl2SiR2 to a solution of Na, K and P4.53–56 A route to mixtures of such compounds with a heterogeneous ligand sphere
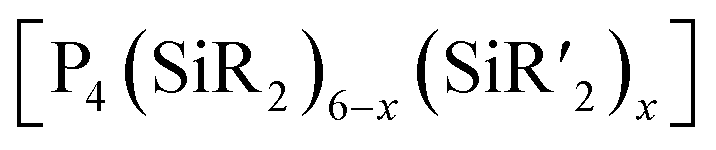 (90–92, 96–105) is by the thermolysis of
(90–92, 96–105) is by the thermolysis of 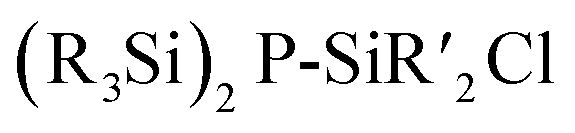 .57
.57
The germanium compound [P4(GeMe2)6] (106, Fig. 5) is obtained by a Hg catalyzed reaction of Cl2GeMe2,58,59 while the heaviest congeners [P4(SnR2)6] (107–109) were first suggested to be detected as a side product in the condensation reaction of PH3 and R2SnCl2.60 The first larger yield synthesis and crystallographic investigation of 107 (Fig. 5) was carried out after an unexpected rearrangement of P(SnMe3)3 catalyzed by [(ZnCl)2Fe(CO)4(THF)2] was observed.61
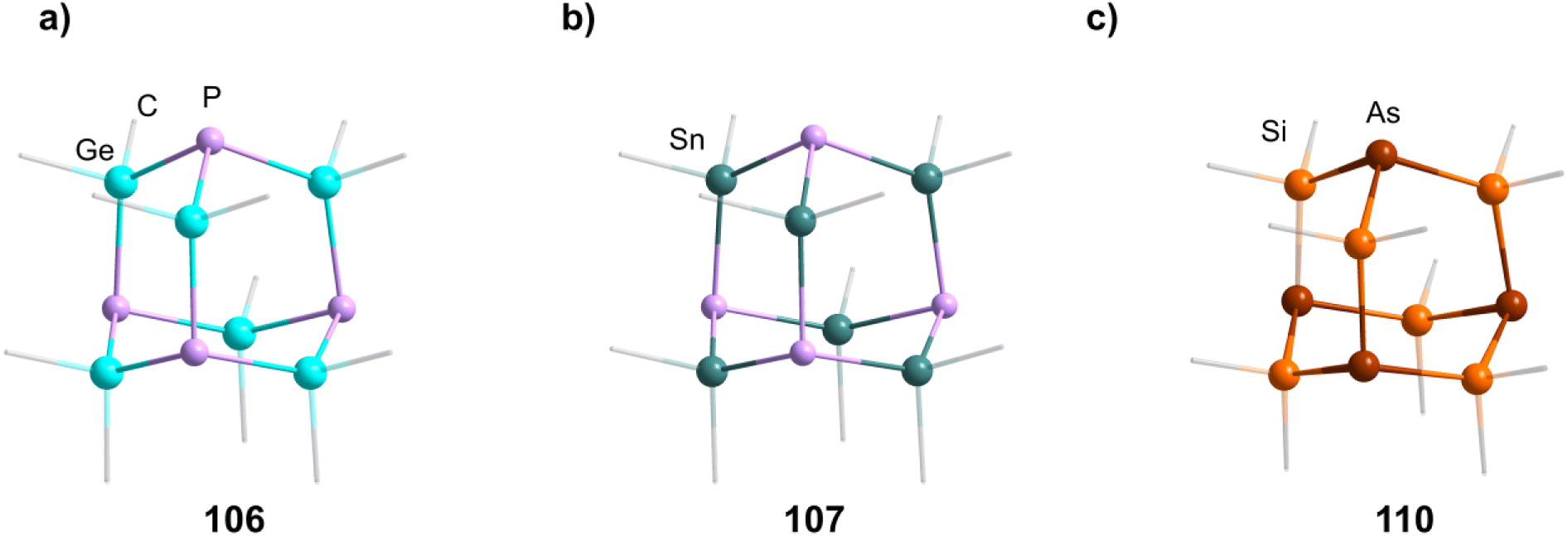 | ||
| Fig. 5 Examples of adamantane-type compounds with group 15 elements in the Q position and group 14 atoms in the E position: [P4(GeMe2)6] (106, left (a)), [P4(SnMe2)6] (107, center (b)) and [As4(SiMe2)6] (110, right (c)). Hydrogen atoms are omitted for clarity. | ||
The analogous [As4(SiMe2)6] (110, Fig. 5) is only found as a side product in the thermolysis of Me2Si(AsSiMe3)2.62,63
| Compound | Reagents/conditions | Method |
|---|---|---|
| a DME = 1,2-dimethoxyethane. | ||
| [P4(SiR2)6] (90–95, R2 = Me2, MeEt, Et2, MePh, (Me)(C2H3), MeH) | 1. P4, K, Na,/DME, −78 °C | D53–56 |
| 2. Cl2SiR2/DME, 24 h | ||
| [P4(SiMe2)6−x(SiEt2)x] (90, 96–100, 92, x = 0–6) | (Me3Si)2P–SiEt2Cl/300 °C | H57 |
| [P4(SiMe2)6−x(SiMeEt)x] (90, 101–105, 91, x = 0–6) | (Me3Si)2P–SiMeEtCl/300 °C | H57 |
| [P4(GeMe2)6] (106) | Me2Ge(PH2)2, Hg/100 °C, 24 h | H58,59 |
| [P4(SnR2)6] (107–109, R = Me, nBu, Ph) | R2SnCl2, PH3/or P(SnMe3)3, [(ZnCl)2Fe(CO)4(thf)2]/THF, 4 days | D/J60,61 |
| [As4(SiMe2)6] (110) | Me2Si(AsSiMe3)2/240 °C, 48 h | H62,63 |
2.1.4.1 Group 2/15 adamantane-type clusters. Two studies have investigated Be/N adamantane-type clusters. One publication found the anionic azide compounds [(BeX)4(N3)6] (111–112, Fig. 6) by reactions of Me3SiN3 with (Ph4P)2[Be2X6].64 The other investigated the formation of amido adamantanes [(BeNH3)4(NH2)6]2+ (in compounds 113–118) in liquid ammonia from elemental Be with varying counterions.65
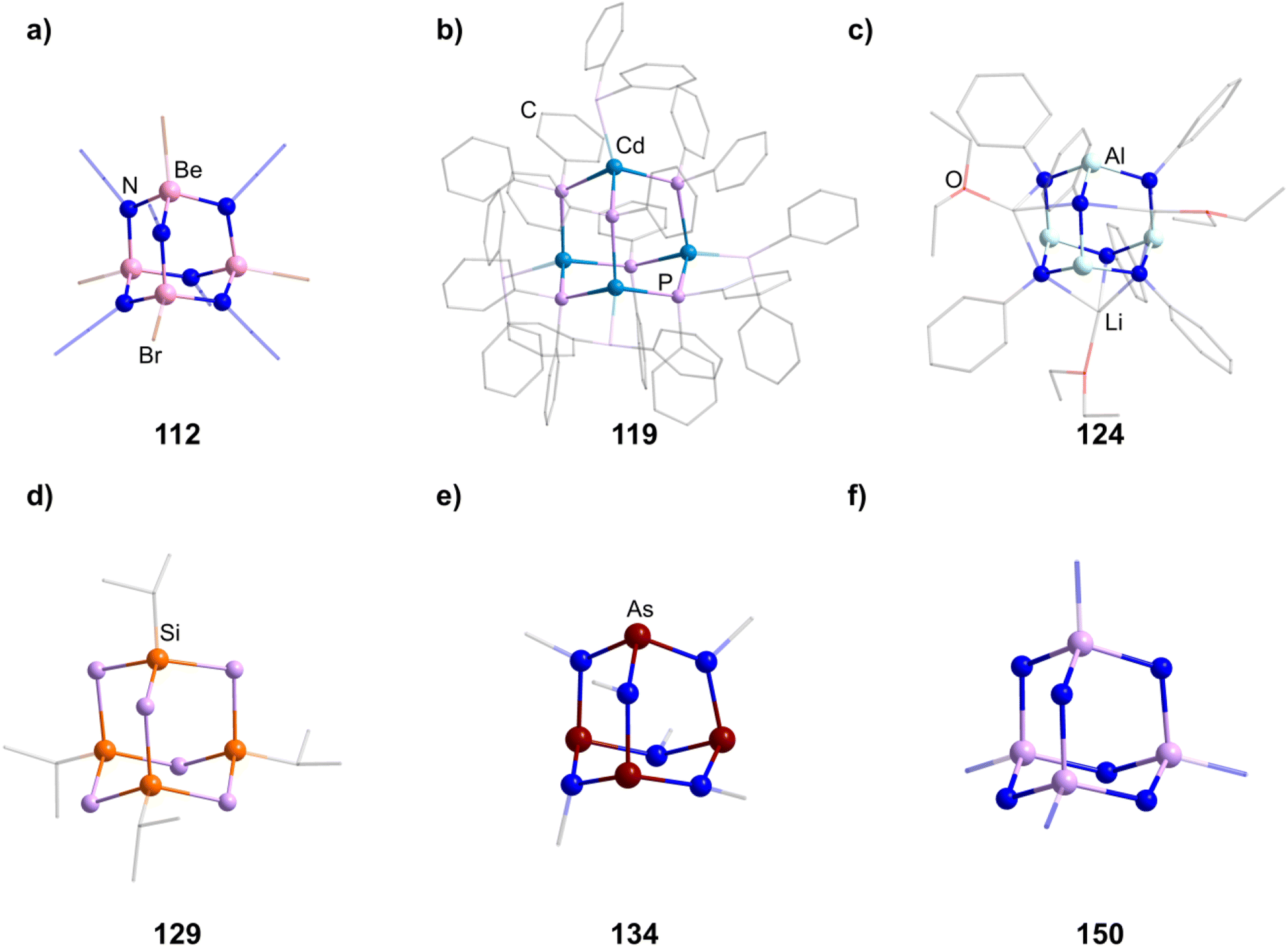 | ||
| Fig. 6 Examples of adamantane-type compounds with group 15 atoms in E position: (Ph4P)2[(Be4Br)4(N3)6] (112, top left (a)), [Li(thf)4]2[(CdPPh2)4(PPh2)6] (119, top center (b)), [Li(OEt2)3][(HAl)4(NPh)6{Li(OEt2)}3] (124, top right (c)), [(iPrSi)4(PH)6] (129, bottom left (d)), [As4(NMe)6] (134,bottom center (e)) and Na10[P4(NH)6N4](NH2)6(NH3)0.5 (150, bottom right (f)). Hydrogen atoms and counterions, if present, are omitted for clarity. | ||
2.1.4.2 Transition metal/15 adamantane-type clusters. There are only a few examples of group 15 containing adamantanes with transition metals. Two of them can be formed by the addition of Ph2PH to a metal salt in the presence of nBuLi to yield [Li(thf)4]2[(CdPPh2)4(PPh2)6] (119, Fig. 6) or [Li(thf)4]2[Cu4(PPh2)6] (120) depending on the element used.66,67 Two neutral iron clusters with a [Fe4]6+ core, comprising iron centers in a formal oxidation state of +1.5 were investigated.68,69 One could be obtained with a phosphide ligand, [Fe4(PiPr2)6] (121), and the other with a ketimide ligand, [Fe4(N
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CPh2)6] (122). Both are prepared in the same way as the Cd and Cu adamantanes by reacting the lithiated ligand with a metal salt.
CPh2)6] (122). Both are prepared in the same way as the Cd and Cu adamantanes by reacting the lithiated ligand with a metal salt.
2.1.4.3 Group 13/15 adamantane-type clusters. [(NMe2)2AlCl]2 can dimerize under elimination of NMe3 to form [(AlCl)4(NMe2)4(NMe)2] (123), with higher yields achieved in the presence of B2(NMe2)4.70,71 An anionic derivative [(HAl)4(NPh)6{Li(OEt2)}3]− (in 124, Fig. 6) featuring a Li capped adamantane is prepared by the combination of PhN(H)Li and AlH3.72
The only known Al/P adamantane compound, [(ArMe6Al)4(PH2)4(PH)2] (125), is isolated after the reaction of PH3 with [ArMe6AlH2]2.73 An example for Ga in the Q position [(PhGa)4(NHiBu)4(NiBu)2] (126) can be synthesized from [PhGa(NMe2)2]2 and H2NiBu.74
2.1.4.4 Group 14/15 adamantane-type clusters. Group 14/15 adamantanes have been investigated for E = P. [(PhSi)4(PPh)6] (127) and its germanium congener (128) are obtainable by a simple condensation reaction of PhQCl3 and K2PPh.75 The same principle can be used for the synthesis of [(iPrSi)4(PH)6] (129, Fig. 6).76 Another synthesis method, utilizing preformed fragments [Li(tmeda)]2[C6H4(PSiMe3)2-1,2] with SitBuCl3, leads to the formation of the asymmetrical [(C6H4{P(SitBuP)1,2})3(SitBu)] (130).77
2.1.4.5 Group 15/15 adamantane-type clusters. Compounds of the type [P4(NR)6] form the vast majority of clusters within this group combination. They are mostly obtained by condensation reactions of PCl3 with RNH2 (131–133),78–81 a synthesis strategy which also works when substituting PCl3 for AsCl3 to form the lesser investigated congeners [As4(NR)6] (134–136, Fig. 6).82,83 Notably, another method of achieving an adamantane-type topology is a reaction starting from a precursor featuring a P2N2 four membered ring, ClP(NiPr)2PNiPrSiMe3, which dimerizes when heated to form the so called double decker-type cluster [P4(NiPr)6], an isomer to the adamantane-type architecture consisting of two four membered rings bridged by two bridging atoms.84,85 This cluster will in turn rearrange to the adamantane compound (137); an isomerization that also plays a major role in the chemistry of group 14/16 adamantane-type structures and for one Mn/O cluster. The same rearrangement from the double decker was required to form [P4(NtBu)6] (138), albeit that ball milling was needed instead of higher temperatures to prompt the rearrangement.86
These compounds can be used as precursors in ligand addition reactions to the pnictogen. The first one investigated was the addition of MeI resulting in [P3(PMe)(NMe)6Me]I (139).78,79,87 Adding S or O atoms in the form of Me3NO or elemental sulfur leads to [(OP)4(NR)6] (140) or [(SP)4(NR)6] (141) respectively. The addition of sulfur can be carried out stepwise to achieve the desired degree of sulfurization (142–145).80,88–93 The addition of transition metal moieties was also realized by reactions with [Ni(CO)4] to 131 and 134, resulting in adamantanes with terminal Ni(CO)3 groups (146 and 147).94 The ligand sphere on the phosphorous atom can also be expanded stepwise by introducing a SiMe3 group in [(PNSiMe3)4(NMe)6] (148), which can subsequently be exchanged for PPh3 (149).95 Lastly, purely inorganic and anionic clusters were obtained by the rearrangement of P3N5 with addition of alkaline metal NH2 salts to yield cluster cores [(PN)4N6] (150–151, Fig. 6) with different degrees of protonation.96,97
| Compound | Reagents/conditions | Method |
|---|---|---|
| a ArMe6 = C6H3-2,6(C6H2-2,4,6-Me3)2. | ||
| (Ph4P)2[(BeCl)4(N3)6] (111) | Me3SiN3, (Ph4P)2[Be2Cl6]/CH2Cl2, 2 days | C64 |
| (Ph4P)2[(Be4Br)4(N3)6] (112) | Me3SiN3, (Ph4P)2[Be2Br6]/CH2Br2, 2 days | C64 |
| [(BeNH3)4(NH2)6]Cl2 (113) | BeCl2, Be/NH3, 2 days | B65 |
| [(BeNH3)4(NH2)6]Br2 (114) | BeBr2, Be/NH3, 2 days | B65 |
| [(BeNH3)4(NH2)6]I2 (115) | NH4I, Be/NH3, 29 days | B65 |
| [(BeNH3)4(NH2)6](CN)2 (116) | Me3SiCN, Be/NH3, 2 days | B65 |
| [(BeNH3)4(NH2)6](SCN)2 (117) | NH4SCN, Be/NH3, 4 days | B65 |
| [(BeNH3)4(NH2)6](N3)2 (118) | Me3SiN3, Be/NH3, 4 days | B65 |
| [Li(thf)4]2[(CdPPh2)4(PPh2)6] (119) | n BuLi, Ph2PH, [Cd{N(SiMe3)2}2]/THF, 80 °C to RT, 12 h | C66 |
| [Li(thf)4]2[Cu4(PPh2)6] (120) | n BuLi, Ph2PH, CuCN/toluene, −78 °C | C67 |
| [Fe4(PiPr2)6] (121) | [FeBr2(thf)2], iPr2PLi/DME, RT | C68 |
| [Fe4(NCPh2)6] (122) |
FeBr2, LiNCPh2, Zn/THF, −25 °C to RT, 18 h |
C69 |
| [(AlCl)4(NMe2)4(NMe)2] (123) | (NMe2)2AlCl, B2(NMe2)4/240 °C, 10 h | B70,71 |
| [Li(OEt2)3][(HAl)4(NPh)6{Li(OEt2)}3] (124) | PhN(H)Li, H3Al·N(Me)C5H8/Et2O | D72 |
| [(ArMe6Al)4(PH2)4(PH)2] (125) | (ArMe6AlH2)2, PH3/toluene, 80 psi, 24 h | G73 |
| [(PhGa)4(NHiBu)4(NiBu)2] (126) | [PhGa(NMe2)2]2, H2NiBu/2 h | C74 |
| [(PhSi)4(PPh)6] (127) | PhSiCl3, K2PPh/C6H6, Et2O, DME, 10 h | D75 |
| [(PhGe)4(PPh)6] (128) | PhGeCl3, K2PPh/C6H6, Et2O, DME, 10 h | D75 |
| [(iPrSi)4(PH)6] (129) | Li[Al(PH2)4], iPrSiCl3/1,2-DME, −30 °C, 3 h | C76 |
| [(C6H4{P(SitBuP)1,2})3(SitBu)] (130) | [Li(tmeda)]2[C6H4(PSiMe3)2-1,2], SitBuCl3/THF, −78 °C | J77 |
| [P4(NMe)6] (131) | MeNH2, PCl3/−78 °C to RT, 4 days | G78,79 |
| [P4(NEt)6] (132) | PCl3, EtNH2/−60 °C to 150 °C | G80,81 |
| [P4(NBn)6] (133) | PCl3, nBuLi, BnNH2 NEt3/THF, −60 °C to RT, 5 days | D81 |
| [As4(NMe)6] (134) | AsCl3, MeNH2/C6H6, 0 °C, 1 h | G82,83 |
| [As4(NiPr)6] (135) | AsCl3, iPrNH2/pentane, 1 h | D82 |
| [As4(NnBu)6] (136) | AsCl3, nBuNH2/C6H6, 60 °C, 30 min | D82 |
| [P4(NiPr)6] (137) | 1. ClP(NiPr)2PNiPrSiMe3/MeCN, 82 °C, 15 h | K84,85 |
| 2. 158 °C, 3 days | ||
| [P4(NtBu)6] (138) | [P4(NtBu)6] (double decker isomer), LiCl/ball milling, 90 min | M86 |
| [P3(PMe)(NMe)6Me]I (139) | [P4(NMe)6] (131), MeI/0 °C | P78,79,87 |
| [(SP)4(NEt)6] (140) | [P4(NEt)6] (132), S/toluene, 95 °C, 9 h | P80 |
| [(OP)4(NMe)6] (141) | [P4(NMe)6] (131), Me3NO/C6H6, 12 h | P90,91 |
| [Pn(SP)4−n(NMe)6] ((142–145, n = 1–4) | [P4(NMe)6] (131), S or [P4(NMe)6] (131), S/CS2, −20 °C, 12 h | P88–93 |
| [{(CO)3NiP}4(NMe)2] (146) | [P4(NMe)6] (131), [Ni(CO)4]/3 h | P94 |
| [{(CO)3NiAs}4(NMe)2] (147) | [As4(NMe)6] (134), [Ni(CO)4]/CHCl3, 3 h, 5 min | P94 |
| [(PNSiMe3)4(NMe)6] (148) | [P4(NMe)6] (131), Me3SiN3/toluene, 100 °C, 12 weeks | P95 |
| [(PNPPh3)4(NMe)6] (149) | [(PNSiMe3)4(NMe)6] (148), Ph3PBr2/MeCN, 55 °C, 3 days | P95 |
| Na10[P4(NH)6N4](NH2)6(NH3)0.5 (150) | P3N5, NaNH2/600 °C, 5 days | A96 |
| Rb8[(PNH)4N6](NH2)2 (151) | P3N5, RbNH2/400 C, 5 days | A97 |
Chalcogenide adamantane-type clusters of the general composition [(QR0–3)4E6]q are found in a large family of compounds of the groups 13 and 14 as well as a single example with Ru. They are obtainable by condensation reactions using a metal (pseudo)halide and a chalcogenide source such as alkaline metal chalcogenides, H2E or (SiMe3)2E.
An additional family of purely inorganic adamantane-type clusters [Q4E10] is found for the groups 13–15. They are mostly accessible from the elements and simple salts by Methods A–C or by extracting alloys in accordance with Method E.
2.1.5.1 Group 2/16 adamantane-type clusters. In group 2, a Be hydroxide cluster Na2[(BeOH)4(OH)6] (152) is reported to form from BeSO4 in basic aqueous solution.98 Two further oxygen centered species are obtainable with Ba. One, [(μ4-O)Ba4(μ-OC6H2(CH2NMe2)3-2,4,6)6] (153, Fig. 7), is formed with a tridentate ligand, which both delivers the oxygen in the E position and coordinates to the two closest barium atoms via nitrogen atoms.99 The other is obtained from a Ba dimer [Ba{N(SiMe3)2}2]2 assembling around (mes)2BOH to form [(μ4-O)Ba4{OB(mes)2}6] (147).100
 | ||
| Fig. 7 Examples of adamantane-type compounds with group 2–6 elements in Q position and group 16 atoms in the E position: [(μ4-O)Ba4(μ-OC6H2(CH2NMe2)3-2,4,6)6] (153, top left (a)), [(μ4-S)(TpMe2Y)4(SBn)6] (155, top center (b)), [(TiCp*)4O6] (158, top right (c)), [(μ4-O){Zr(acac)}4{Zr(OMe)(acac)}(DBcat)3(OMe)3] (171, bottom left (d)), [K-18-crown-6]4[(TaCl3)4O6] (174,bottom center (e)) and (enH2)[Cr4(OH)4(hpdta)2] (178, bottom right (f)). Hydrogen atoms and counterions, if present, are omitted for clarity. | ||
2.1.5.2 Group 3/16 adamantane-type clusters. [(μ4-S)(TpMe2Y)4(SBn)6] (154, Fig. 7) is a unique compound in two ways, as it is both the only group 3 and S centered compound in this review.101 It is created by adding the Y complex [TpMe2YBn2(thf)] with a tridentate ligand to elemental sulfur, which creates the cluster in a redox reaction.
2.1.5.3 Group 4/16 adamantane-type clusters. All but one literature known compound in this category feature a TiIV4O6 core. The first two examples were cationic in nature, [(TiL3)4O6]4+, with each Ti exhibiting three bonds to neutral ligands L. For [{Ti(TACN)}4O6]Br4 (156, TACN = 1,4,7-triazacyclononane), this was achieved by hydrolysis of TiO(acac)2 in the presence of TACN and NaBr,102 while the second example [{Ti(dmso)3}4O6]Cl4 (157) was generated in a solution of TiCl4, Na2S4 and PPh3 in DMSO under partial decomposition of the solvent to yield the required oxygen atoms.103
A larger family of neutral compounds contains derivatives of cyclopentadienyl at the Ti centers [(TiCpR)4O6] (158–164, 158 in Fig. 7), mainly obtained through hydrolysis of various Ti cyclopentadienyl complexes or through reactions with other O sources.104–108
More complex neutral clusters are isolated when the Ti4O6 is formally extended by additional M/O fragments. This could be observed for [Ti4(dmae)6(OH)(O)6Cu6(benzoate)9] (165, dmae = N,N-dimethylaminoethanolate) and its methyl derivative (166).109 They form from the respective hydrated Cu benzoates and Ti(dmae)4 in toluene and feature different coordination modes of the Cu/O fragments.
Two isostructural compounds [{Ti(thf)}4O6M2(TFA)8(thf)2] (167–168, M = Fe, Cd; TFA = trifluoroacetic acid) show a symmetric buildup, with the M centers being connected to opposing oxygen atoms in E position and via four TFA groups each to the neighboring Ti centers.110,111 They are obtained from [Fe3O(OAc)6(H2O)3]NO3 (OAc = acetate) or [(OAc)2Cd(H2O)2], and after addition of a Ti complex and TFA in THF.
A highly charged anion [Ti4O6(Hcit)3(cit)]9− (in 169, H4cit = citric acid) is crystallized from a reactive solution of citric acid and [Ti{iPrO)4] in a H2O/THF mix. The addition of [Co(NH3)6]Cl3 yields the cobaltate salt, which can be converted to the Na analog (170) by ion exchange chromatography.112
[(μ4-O){Zr(acac)}4{Zr(OMe)(acac)}(DBcat)3(OMe)3] (171, acac = acetylacetonate, H2DBcat = 3,5-di-tert-butylcatechol, Fig. 7), hydrolytically obtained from [Zr2(acac)4(DBcat)2], is a singular Zr example in this group in which half of the E positions are occupied by methoxy groups and half of them by DBcat groups, which also coordinate to one Zr center each.113
2.1.5.4 Group 5/16 adamantane-type clusters. There are only three unrelated examples of different group 5 oxides in this group.
The vanadium species [(VCp*)4O6] (172) stems from a rearrangement of the trimeric species [Cp*V(O)(μ-O)]3 after addition of PMe2Ph.114
The cluster compound [{HBO-3,5-(tBu)2NbCl}4O6] (173, HBO = 2-(2′-Hydroxyphenyl)benzoxazole) is the simple hydrolysis product of [HBO-3,5-(tBu)2NbCl4].115
Using a water containing sample of 18-crown-6 in a reaction of TaCl5 and K2S5 generates the heaviest congener [K-18-crown-6]4[(TaCl3)4O6] (174, Fig. 7) with an anionic cluster scaffold.116
2.1.5.5 Group 6/16 adamantane-type clusters. Two cationic hydroxo clusters of the type [(CrR)4(OH)6]q+ (in 175–177) can be obtained by hydrolysis of Cr precursor complexes.117,118 In the case of a combination of CrCl3 and the pentadentate ligand hpdta (H5hpdta = hydroxypropanediaminotetraacetic acid), a compound with the cationic cluster [Cr4(μ-OH)4(hpdta)2]2+ (in 178, Fig. 7) was isolated, in which two of the oxygen atoms in E position stem from the hpdta ligands.119
The only known Mo congener [{MoO(IPAP)}4O6] (179, HIPAP = N-(tert-butyl)-3-((3,5-di-tert-butyl-2-hydroxybenzylidene)amino)-propanamide) is formed as a side product during the reduction of the complex [Mo(O)2(IPAP)2] using PPh3 and could only be isolated in trace amounts.120
Two structurally related oxo clusters of tungsten, [(W(O)(tdmap)}4O6] (180, tdmap = OC(CH2NMe2)3) and [{(W(O)(S-Phoz)}4O6] (181, S-Phoz = 2-(4′,4′-dimethyloxazoline-2′-yl)thiophenolate), are known in the literature.121,122 The first from a reaction of [W(O)(OiPr)4] with Htdmap in the presence of water and the second by rearrangement of the complex [W(CO)(C2Me2)(S-Phoz)2] after oxidation using pyridine-N-oxide.
One sulfide containing adamantane-type cluster [(WPMe2Ph)4S6] (182) exists, which rearranges from the tetranuclear [W4(μ3-S)2(μ-S)4Cl2(PMe2Ph)6] after reduction with a Na/Hg amalgam in low yields.123
| Compound | Reagents/conditions | Method |
|---|---|---|
| a mes = 2,4,6-Me3-C6H2, TpMe2 = tri(3,5 dimethylpyrazolyl)borate), TACN = 1,4,7-triazacyclononane, DMSO = dimethyl sulfoxide, CpxPh = C5Me4Ph, OHF = 1,2,3,4,5,6,7,8-octahydrofluorenyl, dmae = N,N-dimethylaminoethanolate, TFA = trifluoacetic acid, H4cit = citric acid, H2DBcat = 3,5-di-tert-butylcatechol, HBO = 2-(2′-hydroxyphenyl)benzoxazole, tach = 1,3,5-triaminocyclohexane, en = ethylendiamine, H5hpdta = hydroxypropanediaminotetraacetic acid, HIPAP = N-(tert-butyl)-3-((3,5-di-tert-butyl-2-hydroxybenzylidene)amino)-propanamide, tdmap = OC(CH2NMe2)3, S-Phoz = 2-(4′,4′-dimethyloxazoline-2′-yl)thiophenolate. | ||
| Na2[(BeOH)4(OH)6] (152) | BeSO4, Ba(OH)2, NaOH/H2O, pH 13.2, 18 h | C98 |
| [(μ4-O)Ba4(μ-OC6H2(CH2NMe2)3–2,4,6)6] (153) | K[(OC6H2(CH2NMe2)3-2,4,6), BaI2/toluene | K99 |
| [(μ4-O)Ba4{OB(mes)2}6] (154) | (mes)2BOH, [Ba{N(SiMe3)2}2]2 | C100 |
| [(μ4-S)(TpMe2Y)4(SBn)6] (155) | S, [TpMe2YBn2(thf)]/THF, RT, 18 h | C101 |
| [{Ti(TACN)}4O6]Br4 (156) | TiO(acac)2, 9aneN3, NaBr/Me2CO, H2O, 50 °C, 30 min | I102 |
| [{Ti(dmso)3}4O6]Cl4 (157) | Na2S4, PPh4, TiCl4/DMSO, RT | C103 |
| [(TiCp*)4O6] (158) | Cp*TiCl3, NH4OH/toluene, RT, 3 days or Cp*Ti(OMe)3/H2O, RT | I104,105 |
| [(TiCpxPh)4O6] (159) | CpxPhTi(OME)3/Me2CO, H2O, 100 °C, 30 min | I106 |
| [{Ti(η5-C5Me4SiMe2NHNMe2)}4O6] (160) | [(η5-C5Me4)SiMe2(NNMe2)]Ti(NMe)2/H2O, toluene, RT, 5 h | I107 |
| [{Ti(OHF)}4O6] (161) | [(OHF)Ti(OMe)3]/Me2CO, H2O 56 °C | I106 |
| [{Ti(η5-C5Me4SiMe3)}4O6] (162) | (η5-C5Me4SiMe3)2Ti(O)/pentane, RT, 2 weeks | J108 |
| [{Ti(η5-C5Me4SiMe2Ph)}4O6] (163) | (η5-C5Me4SiMe2Ph)2Ti(O)/pentane, RT, 2 weeks | J108 |
| [{Ti(η5-C5Me4iPr)}4O] (164) | (η5-C5Me4iPr)2Ti(O), Na2O2/THF, RT, overnight | I108 |
| [Ti4(dmae)6(OH)(O)6Cu6(benzoate)9] (165) | Cu(benzoate)2·2H2O, Ti(dmae)4/toluene, RT, 2 h | C109 |
| [Ti4(dmae)6(OH)(O)6Cu6(2-methylbenzoate)9] (166) | Cu(2-methylbenzoate)2·2H2O, Ti(dmae)4/toluene, RT, 2 h | C109 |
| [{Ti(thf)}4O6Fe2(TFA)8(thf)2] (167) | [Fe3O(OAc)6(H2O)3]NO3, [(EtOEtO)4Ti], TFA/THF, RT, 1 h | J110 |
| [{Ti(thf)}4O6Cd2(TFA)8(thf)2] (168) | [(OAC)2Cd(H2O)2], [Ti{iPrO)4], TFA/THF, RT, 4 h | C111 |
| [Co(NH3)6]3[Ti4O6(Hcit)3(cit)] (169) | [Ti{iPrO)4], H4cit, [Co(NH3)6]Cl3/THF, H2O, 90 °C 1 h | I112 |
| Na9[Ti4O6(Hcit)3(cit)] (170) | [Co(NH3)6]3[Ti4O6(Hcit)3(cit)] (169)/ion exchange chromatography | O112 |
| [(μ4-O){Zr(acac)}4{Zr(OMe)(acac)}(DBcat)3(OMe)3] (171) | [Zr2(acac)4(DBcat)2]/CH2Cl2, MeOH, H2O, RT | I113 |
| [(VCp*)4O6] (172) | [Cp*V(O)(μ-O)]3, PMe2Ph/THF | J114 |
| [{HBO-3,5-(tBu)2NbCl}4O6] (173) | HBO-3,5-(tBu)2NbCl4, H2O/Toluene, THF, RT, 12 h | I115 |
| [K-18-crown-6]4[(TaCl3)4O6] (174) | K2S5, TaCl5, 18-crown-6, H2O/CH2Cl2, RT, 20 h | I116 |
| [(Cp*Cr)4(OH)6][Cp*Cr(CO)3] (175) | [(Cp*)2Cr2(CO)4]/H2O, toluene, 111 °C, 24 h | I117 |
| [{(Cp*)Cr}4(OH)6][BF4]2 (176) | [(Cp*Cr)4(OH)6][Cp*Cr(CO)3] (175), H[BF4] | J117 |
| [{Cr(tach)}4(OH)6](ClO4)n(CF3SO3)6–n (177) | [Cr(tach)(CF3SO3)3], NaOH,/H2O | I118 |
| (enH2)[Cr4(OH)4(hpdta)2] (178) | H5hpdta, en, CrCl3/H2O, 85 °C, 24 h | K119 |
| [{MoO(IPAP)}4O6] (179) | 1. HIPAP, [MoO2Br2(DMSO)2], NEt3, PMe3/MeOH, RT, 18 h | I120 |
| 2. PMe3/toluene, RT, 18 h | ||
| [{W(O)(tdmap)}4O6] (180) | [W(O)(OPri)4], Htdmap/toluene, H2O, iPrOH, reflux, 24 h | I121 |
| [{(W(O)(S-Phoz)}4O6] (181) | [W(CO)(C2Me2)(S-Phoz)2], pyridine-N-Oxide/CH2Cl2, RT, 24 h | J124 |
| [(WPMe2Ph)4S6] (182) | [W4(μ3-S)2(μ-S)4Cl2(PMe2Ph)6], Na(Hg)/THF, 8 h | J123 |
2.1.5.6 Group 7/16 adamantane-type clusters. All known adamantane compounds with an elemental combination of groups 7/16 are Mn clusters in the oxidation state IV, either with oxygen or thiolates in the E position. The oxides are mainly available via hydrolysis and can be derivatized by ligand or ion exchange.
{Mn(TACN)}4O6]4+ (in 183–185) is the first example of such an adamantane-type structure synthesized by addition of simple MnII salts to TACN in the presence of water and air to oxidize the metal centers.125–130
The related adamantane [{Mn(bpea)}4O6](ClO4)4 (186, bpea = N,N-bis(2-pyridylmethyl)ethylamine) also comprises of an N,N,N-tridentate ligand and cannot be obtained by air oxidation, but requires a comproportionation of two Mn compounds Mn(ClO4)2 and [nBu4N][MnO4] and bpea.131 Addition of [nBu4N]Br yields the bromide salt [{Mn(bpea)}4O6]Br 187, which can subsequently be treated with alkaline metal salts for anion exchange (188–192). Methylated bpea can also be used during the synthesis to form derivatives (193–194). The same study also investigated the single electron reduction of the compounds under retention of the adamantane-type scaffold, either by electrochemistry or via TACNMe as a reducing agent (195).
[Mn4O6(bpea)4](ClO4)4 can also be used as a basis for ligand exchange using other tridentate ligands (196–200).129 In the case of the charged N-substituted iminodicarboxylate ligands, used as their ammonium salts, only partial substitiution products in the form of [{Mn(R-ida)}2{Mn(bpea)}2O6] (201–206, R-ida = N-(R)iminodiacetate) could be isolated as stable compounds.
By a reaction of tame·3HOTf (tame = tert-amyl methyl ether), Mn(OTf)2 and Et3N in MeCN and under exposure to athmosperic O2, the mixed oxo/hydroxo species [{Mn(tame)}4O5(OH)](OTf)5 (207) was obtained, which could be completely deprotonated by additional Et3N, leading to 208.130 Protonation of [{Mn(TACN)}4O6]4+ to the corresponding [{Mn(tame)}4O5(OH)]5+ (in 209) by HClO4 was also proven to work.
The last literature-known oxide cluster [Mn4O4(tphpn)2](OTf)2(ClO4)3 (210, Htphpn = N,N,N′,N′-tetra-(2-methylpyridyl)-2-hydroxypropanediamine, Fig. 8) features a MnIII/MnIV mixed valency situation and a pentadentate ligand bridging two Mn moieties by coordination with its N sites as well as the O atom in the E position between the two metal centers.132 It is prepared by a reductively induced isomerization of the double decker type compound [{Mn2(μ-O)2(tphpn)}2].
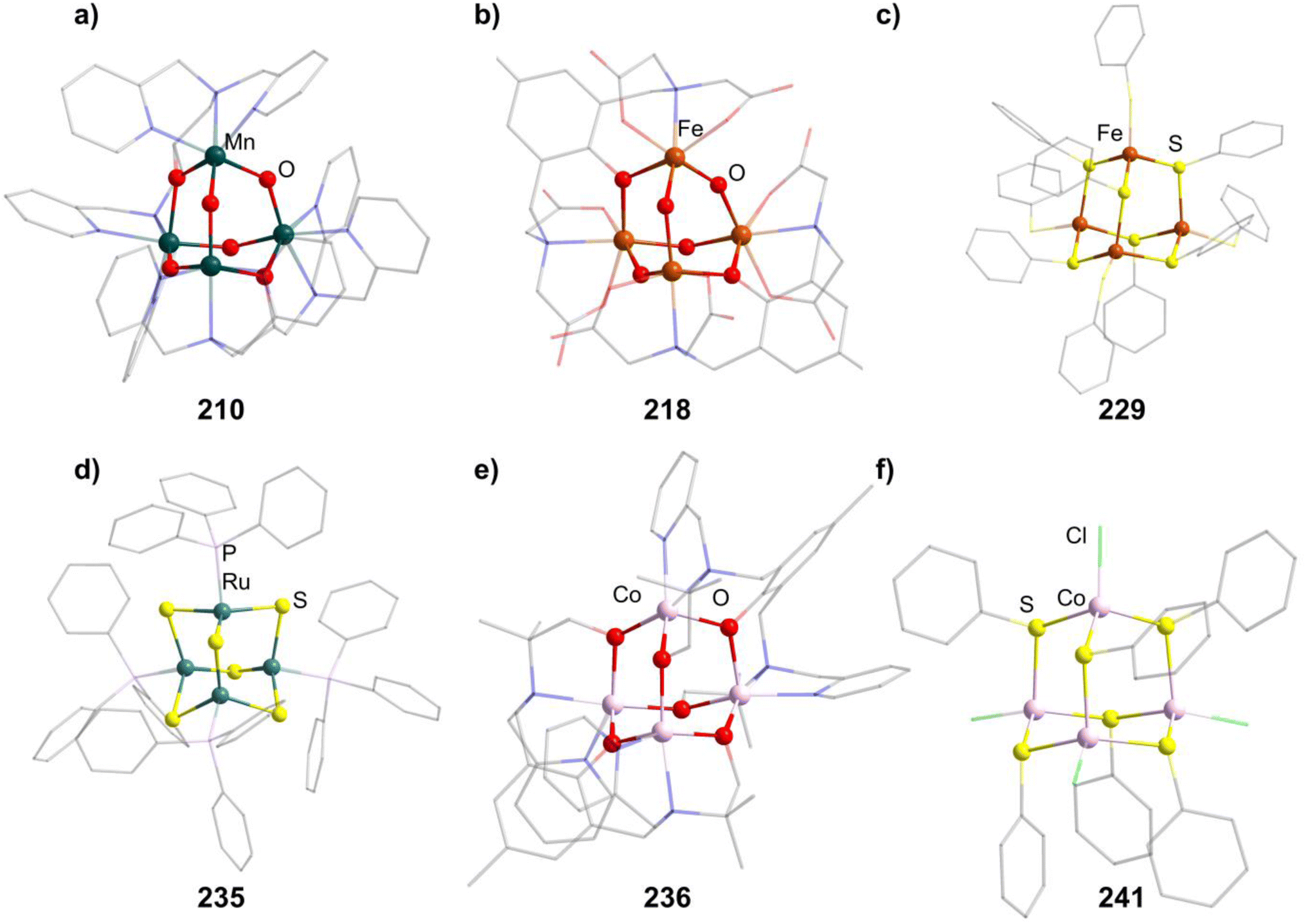 | ||
| Fig. 8 Examples of adamantane-type compounds with group 7–9 elements in the Q-position and group 16 atoms in the E-position: [Mn4O4(tphpn)2](CF3SO3)2(ClO4)3 (210, top left (a)), (HPy)3[{Fe2(HPhXCG)}2O(OH)3] (218, top center (b)), [Et4N]2[(FeSPh)4(SPh)6] (229, top right (c)), [(RuPPh3)4S6] (235, bottom left (d)), [Co4(HMPM)2](ClO4)2 (236,bottom center (e)) and [Et4N]2[{Co(Cl)}4(SPh)6] (241, bottom right (f)). Hydrogen atoms and counterions, if present, are omitted for clarity. | ||
Thiolate complexes with Mn exhibiting adamantane-type structure have also been studied. In the most simple case, dianionic [(MnSPh)4(SPh)6]2− clusters (in 211–212) are isolated after conversion of MnCl2 with NaSPh and an appropriate ammonium countercation.133 Unlike the oxygen species, they contain MnII sites in their inorganic core.
In those compounds, all Mn atoms also carry a thiolate ligand which can formally be substituted by halides by either using [Et4N]Br during the synthesis to form the brominated 213,134 or through a rearrangement by adding MnCl2 to [(Me4N)2{Mn2(SiPr)6}], leading to [Me4N]2((MnCl)4(SiPr)6] (214).135 The last method can also yield the corresponding selonlate [Me4N]2[(MnBr)4(SeiPr)6] (215) when using MnBr2 and [Me4N]2[Mn2(SeiPri)6] instead.
In [{Mn(BMAP)}3(MnCl)3]Cl (216, H2BMAP = 2-[bis(2-mercaptoethyl)aminomethyl-pyridine), the BMAP ligands coordinate to three of the Mn centers by their N atoms and also carry two thiols each, which make up the atoms in the E position.136 The last Mn atom is saturated by a chlorine atom. It forms when adding H2BMAP to MnCl2.
2.1.5.7 Group 8/16 adamantane-type clusters. All but one known compounds in this cluster family are iron compounds, which mainly form oxo/hydroxo compounds with polydentate ligands and FeIII centers, but also FeII thiolate complexes common for most transition metal groups.
A family of oxo/hydroxo clusters comprising heptadentate ligands of the type [{Fe2(L)}2O4−n(OH)n]q (in 217–221, n = 2, 3, Fig. 8) is obtainable from mostly basic conditions by providing the desired ligand and simple iron salts.137–141 The ligands in those systems bridge two Fe atoms by providing an O atom in the E position between them and coordinating via three Lewis basic sites to both of them. The charge of the resulting clusters depends on the charge of the ligand and the O/OH ratio. For [Fe4(N-Et-HPTB)2O4][BF4]2 (222), obtained from bubbling O2 through a solution of [Fe2(N-Et-HPTB)(dmf)4][BF4]3, all of the four E atoms not part of the organic ligand are oxo ligands.142
There is a distinctly different arrangement of bridging ligands found in the hydroxo cluster [{Fe(tBuOH)}4(dppoe)4(OH)6][PF6]2Cl4 (223, dppoe = 1,2-bis(diphenylphosphine oxide)ethane), in which the neutral ligands are not part of the adamantane architechture.143 It was unintentionally found to be the main product in a reaction of [(Cp)(dppe)FeCl] (dppe = 1,2-bis(diphenylphosphino)ethane) with the carborane [closo-1,12-C2B11H10(CN)2] while in contact to air, oxidizing both the dppe and iron atoms.
Clusters with the non bridging tridentate ligands TACN, [{Fe(TACN)}4O2(OH)4]X4 (224–225, X = I, CIO4), do also not comprise oxygen atoms from the ligand in their scaffold and were first obtained after the hydrolysis of [(TACN)2Fe2(acac)2(O)](ClO4)2 under addition of NaX,144 although examples of [{Fe(TACN)}4O4−n(OH)n]q (in 226–227, n = 2, 3) with different halide counterions could later be synthesized directly from [(TACN)FeCl3] with a sodium halide in basic solution.145,146
Thiolate clusters of the form [(FeSR)4(SR)6]2− (in 228–230, Fig. 8) and [(FeX)4(SR)6]2− (231–232, X = Cl, Br) both exist. The first type is generated by converting FeCl2 using thiosulfates147–149 and the second by adding the preformed thiol complex [Ph4P]2[Fe(SPh)4] to FeX2.150 [Et4N]2[(FeBr)4(SBn)6](233) can also be prepared by the first method.151
[{Fe(BMAP)}3(FeCl)3]Cl (234) is isostructural to the Mn congener 209 and prepared accordingly.136
[(RuPPh3)4S6] (235, Fig. 8) is a singular example, as it is a pure sulfide cluster and the only Ru compound.152 It can be formed in reactions of a sulfide source like (SiMe3)2S or NaSH with PPh3 and a RuII complex like RuCl2(DMSO)4 resulting in H2 or (SiMe3)2 as reduced side products.
2.1.5.8 Group 9/16 adamantane-type clusters. There are only a few adamantane-type structures comprising cobalt which are known in the literature.
One, [Co4(HMPM)2](ClO4)2 (236, H3HMPM = 2,6-bis[{{(1-hydroxy-2-methylpropan-2-yl)(pyridine-2-ylmethyl)}amino}methyl]-4-methylphenol, Fig. 8), is formed with two heptadentate ligands, which encompass the six oxygen atoms in the E position and coordinate terminally to the CoII moieties with two N atoms per metal center.153 It is formed by combining the deprotonated H3HMPM ligand and Co(ClO4)2.
All other Co adamantane-type clusters are thiolates with a CoIII core. Clusters of the form [{Co(SPh)}4(SR)6]2− (in 237–239) are obtained from the thiolates and common cobalt and ammonium salts.149,154 The terminal thiolates can be formally exchanged by halides, as seen in the compound [tBu4N]2[{Co(Cl)}4(SPh)6] (240) formed from [tBu4N][CoCl3(PPh3)] reacting with PhSSiMe3 and [Et4N]2[{Co(Cl)}4(SPh)6] (241, Fig. 8), which in turn forms in a solution of Na, PhSH, CoCl2 and [Et4N]Cl.155,156
A heterogenous substitution pattern is observed for [{Co(Cl)}2(CoPPh3)2(SPh)6] (242) and [{Co(Cl)}2(CoPOPh3)(CoPPh3)(SPh)6] (243), which could both be isolated as the products of the addition of PhSSiMe3 to the complex [CoCl2(PPh3)2], in the presence of O2 in the second case.155
| Compound | Reagents/conditions | Method |
|---|---|---|
a bpea = N,N-bis(2-pyridylmethyl)ethylamine,  = [3,5-(CF3)2C6H3]4B]−, dien = diethylenetriamine, medien = N′-methyldiethylenetriamine, R-ida = N-(R)iminodiacetate, CPe = cyclopentane, tame = tert-amyl methyl ether, Htphpn = N,N,N′,N′-tetra-(2-methylpyridyl)-2-hydroxypropanediamine, pz = pyrazolyl, H2BMAP = 2-[bis(2-mercaptoethyl)aminomethyl]pyridine, Py = pyridine, H5HMeXCG = N,N′-(2-hydroxy-5-methyl-1,3-xylylene)bis(N-(carboxymethyl)glycine), H5HPhXCG = N,N′-(2-hydroxy-5-phenyl-1,3-xylylene)bis(N-(carboxymethyl)glycine), dma = N,N-dimethylacetamid, Hbpbp = 2,6-bis((N,N′-bis-(2-picolyl)amino)methyl)-4-tert-butylphenol, {(TACN)CH2}2CHOH = 1,3-bis(1,4,7-triaza-1-cyclononyl)-2-hydroxypropane, N-Et-HPTB = N,N,N′,N′-tetrakis(2-(1-ethylbenzimidazolyl))-2-hydroxy-1,3-diaminopropane, dppoe = 1,2-bis(diphenylphosphine oxide)ethane, dppe = 1,2-bis(diphenylphosphino)ethane, H3HMPM = 2,6-bis[{{(1-hydroxy-2-methylpropan-2-yl)(pyridine-2-ylmethyl)}amino}methyl]-4-methylphenol. = [3,5-(CF3)2C6H3]4B]−, dien = diethylenetriamine, medien = N′-methyldiethylenetriamine, R-ida = N-(R)iminodiacetate, CPe = cyclopentane, tame = tert-amyl methyl ether, Htphpn = N,N,N′,N′-tetra-(2-methylpyridyl)-2-hydroxypropanediamine, pz = pyrazolyl, H2BMAP = 2-[bis(2-mercaptoethyl)aminomethyl]pyridine, Py = pyridine, H5HMeXCG = N,N′-(2-hydroxy-5-methyl-1,3-xylylene)bis(N-(carboxymethyl)glycine), H5HPhXCG = N,N′-(2-hydroxy-5-phenyl-1,3-xylylene)bis(N-(carboxymethyl)glycine), dma = N,N-dimethylacetamid, Hbpbp = 2,6-bis((N,N′-bis-(2-picolyl)amino)methyl)-4-tert-butylphenol, {(TACN)CH2}2CHOH = 1,3-bis(1,4,7-triaza-1-cyclononyl)-2-hydroxypropane, N-Et-HPTB = N,N,N′,N′-tetrakis(2-(1-ethylbenzimidazolyl))-2-hydroxy-1,3-diaminopropane, dppoe = 1,2-bis(diphenylphosphine oxide)ethane, dppe = 1,2-bis(diphenylphosphino)ethane, H3HMPM = 2,6-bis[{{(1-hydroxy-2-methylpropan-2-yl)(pyridine-2-ylmethyl)}amino}methyl]-4-methylphenol.
|
||
| [{Mn(TACN)}4O6]Br3.5OH0.5 (183) | TACN, MnCl2, NaBr, O2/H2O | S125,126 |
| [{Mn(TACN)}4O6](ClO4)4 (184) | Mn(NO3)2, Na2C2O4, TACN, NaClO4, NaOH, O2/MeOH, H2O, 60 °C, 3 h or TACN, [Mn4O6(bpea)4](ClO4)4 (186)/MeCN, RT, 1 h | S127–129 |
| [{Mn(TACN)}4O6](OTf)4 (185) | Mn(OTf)2, TACN, O2/MeCN | S130 |
| [{Mn(bpea)}4O6](ClO4)4 (186) | Mn(ClO4)2·6H2O, [nBu4N][MnO4],/MeCN, RT, 1 h | C131 |
| [{Mn(bpea)}4O6]Br4 (187) | [nBu4N][Br], [Mn4O6(bpea)4](ClO4)4 (186)/MeCN, RT, 24 h | O131 |
[{Mn(bpea)}4O6]X4 ((188–192, X = [BF4], OTf, [PF6], SCN,  ) ) |
[{Mn(bpea)}4O6)4]Br4 (187), NaX or KX/H2O, RT | O131 |
| [{Mn(4,4′-Me2bpea)}4O6](ClO4)4 (193) | Mn(4,4′-Me2bpma)2·6H2O, [n-Bu4N][MnO4],/MeCN, RT, 1 h | C131 |
| [{Mn(5,5′-Me2bpea)}4O6](ClO4)4 (184) | Mn(5,5′-Me2bpma)2·6H2O, [n-Bu4N][MnO4],/MeCN, RT, 1 h | C131 |
| [{Mn(bpea)}4O6](ClO4)3 (195) | [Mn4O6(bpea)4](ClO4)4, [nBu4N]ClO4/MeCN, THF, electrolysis (−0.1 V), 25 min | N131 |
[{Mn(bpea)}4O6](X)3 (196–200, X = [BF4], OTf, [PF6], SCN,  ) ) |
TACNMe, [Mn4O6(bpea)4](X)4 (188–192)/MeCN, 5 min | S131 |
| [{Mn(dien)}2{Mn(bpea)}2O6](ClO4)4 (201) | [Mn4O6(bpea)4](ClO4)4 (186), dien/MeCN, RT, 3 h | Q129 |
| [{Mn(Medien)}4](ClO4)4 (202) | [Mn4O6(bpea)4](ClO4)4 (186), medien/MeCN, RT, 45 min | Q129 |
| [{Mn(R-ida)}2{Mn(bpea)}2O6] (203–206, R = Me, Bn, tBu, CPe) | [Mn4O6(bpea)4](ClO4)4 (186), [tBu4N]2[R-ida]/MeCN, RT, 30 min | Q129 |
| [{Mn(tame)}4O5(OH)](OTf)5 (207) | tame·3HOTf, Mn(OTf)2·MeCN, Et3N, O2/MeCN, RT, 36 h | S130 |
| [{Mn(tame)}4O6](OTf)4 (208) | [{Mn(tame)}4O5(OH)](OTf)5[Mn4O6(bpea)4](ClO4)4 (207), NEt3/MeCN | Q130 |
| [{Mn(tame)}4O5(OH)](OTf)5 (209) | [{Mn(TACN)}4O6](OTf)4 (185), HClO4/MeCN | P130 |
| [Mn4O4(tphpn)2](CF3SO3)2(ClO4)3 (210) | [{Mn2(μ-O)2(tphpn)}2], [Mn((HB(3,5-Me2pz)3)2](ClO4)2/MeCN, RT, 10 min | S/K132 |
| [Et4N]2[(MnSPh)4(SPh)6] (211) | MnCl2·4H2O, PhSNa, Et4NCl·H2O/MeOH, RT, 40 min | C133 |
| [Me4N]2[(MnSPh)4(SPh)6] (212) | MnCl2·4H2O, PhSNa, Me4NCl/MeOH, RT, 40 min | C133 |
| [Et4N]2[(MnBr)4(SPh)6] (213) | MnBr2, NaSPh, [Et4N]Br/MeCN, RT, 2 h | C134 |
| [Me4N]2((MnCl)4(SiPr)6] (214) | [Me4N]2(Mn2(SiPr)6], MnCl2/MeCN, 35 °C, 5 h | J135 |
| [Me4N]2((MnBr)4(SeiPr)6] (215) | [Me4N]2(Mn2(SeiPr)6], MnBr2/MeCN, RT, 12 h | J135 |
| [{Mn(BMAP)}3(MnCl)3]Cl (216) | H2BMAP, MnCl2/MeOH, 60 °C, 5 min | K136 |
| [nBu4N]4[{Fe2(HXMeCG)}2O2(OH)2] (217) | FeCl3, [nBu4N]Cl, NaOH, H5HMeXCG/H2O | K137 |
| (HPy)3[{Fe2(HPhXCG)}2O(OH)3] (218) | Na3H2HPhXCG, Py, Fe(NO3)3/MeOH, RT, 1 month | K138 |
| (enH2)1.5[Fe4O(OH)3(hpdta)2] (219) | H5hpdta, Fe(NO3)3, en, dma/H2O, 3 days | K139 |
| [{Fe2(bpbp)}2O2(OH)2](ClO4)4 (220) | Hbpbp, Fe(ClO4)3/THF, H2O, RT, 2 days | I140 |
| [(Fe2{(TACN)CH2}2CHOH)O(OH)]2[PF6]4 (221) | {(TACN)CH2}2CHOH, FeCl3, NaOAc, NEt3, K[PF6]/iPrOH, 24–36 h | K141 |
| [Fe4(N-Et-HPTB)2O4][BF4]2 (222) | [Fe2(N-Et-HPTB)(dmf)4][BF4]3, O2/DMF | K142 |
| [{Fe(tBuOH)}4(dppoe)4(OH)6][PF6]2Cl4 (223) | [closo-1,12-C2B11H10(CN)2], [(Cp)(dppe)FeCl], [NH]4[PF6], tBuOH/THF, 66 °C, 18 h | J143 |
| [{Fe(TACN)}4O2(OH)4]X4 (224–225, X = I, CIO4) | NaX, [(TACN)2Fe2(acac)2(O)](ClO4)2/Me2CO, H2O, 2 weeks | I144 |
| [{Fe(TACN)}4O(OH)5](I)4I3 (226) | [(TACN)FeCl3], KI/H2O, Py, 72 h | D145 |
| [{Fe(TACN)}4O2(OH)]Br4 (227) | [(TACN)FeCl3], NaBr/H2O, 25 °C, pH = 10.28 | D146 |
| [R4N]2[(FeSPh)4(SPh)6] (228–229, R = Me, Et) | FeCl2, NaSPh, [R4N]Cl/MeOH | C147,148 |
| [Me4N]2[(FeSEt)4(SEt)6] (230) | FeCl2, NaSEt, [Me4N]Br/MeOH, 2 h | C149 |
| [Ph4P]2[(FeCl)4(SPh)6] (231) | FeCl2, [Ph4P]2[Fe(SPh)4]/MeCN, 30 min | C150 |
| [Ph4P]2[(FeBr)4(SPh)6] (232) | FeBr2, [Ph4P]2[Fe(SPh)4]/MeCN, 30 min | C150 |
| [Et4N]2[(FeBr)4(SBn)6] (233) | FeCl2, NaSBn, [Et4N]Br/MeCN | C151 |
| [{Fe(BMAP)}3(FeCl)3]Cl (234) | H2BMAP, FeCl2·4H2O/MeOH, 60 °C, 5 min | K136 |
| [(RuPPh3)4S6] (235) | RuCl2(DMSO)4, PPH3, (SiMe3)2S/THF, −50 °C | C152 |
| [Co4(HMPM)2](ClO4)2 (236) | Co(ClO4)2, H3HMPM, Et3N/MeOH, RT | K153 |
| [Me4N]2[{Co(SPh)}4(SPh)6] (237) | PhSH, Et3N, Co(NO3)2, [Me4N]Cl/EtOH | C154 |
| [Cy2NH2]2[{Co(SPh)}4(SPh)6] (238) | PhSH, Cy2NH, Co(NO3)2, [Me4N]Cl/EtOH | C154 |
| [Et4N]2[{Co(SEt)}4(SEt)6] (239) | NaSEt, CoCl2, [Et4N]Cl/MeCN | C149 |
| [tBu4N]2[{Co(Cl)}4(SPh)6] (240) | [tBu4N][CoCl3(PPh3)], PhSSiMe3/toluene, 3 h | C155 |
| [Et4N]2[{Co(Cl)}4(SPh)6] (241) | Na, PhSH, CoCl2, [Et4N]Cl/MeOH, RT | C156 |
| [{Co(Cl)}2(CoPPh3)2(SPh)6] (242) | [CoCl2(PPh3)2], PhSSiMe3/THF, 3 h | C155 |
| [{Co(Cl)}2(CoPPh3)(CoPOPh3)(SPh)6] (243) | [CoCl2(PPh3)2], PhSSiMe3, O2/THF, 3 h | C155 |
2.1.5.9 Group 11/16 adamantane-type clusters. The second largest family of compounds with group 16 elements in the E position is the 11/16 combination. Most of them exist for the elemental combination Cu and S, although some Ag examples and clusters with different chalcogenides are known.
Thiolate containing adamantane-type cluster anions of the general composition [Cu4(SR)6]2− (in 244–255) have been extensively studied, and can be obtained by reacting a copper salt with the desired thiolate or by using a monomeric precursor complex already containing the SR species in most cases.157–169 In some cases, this involves a reduction of the copper atoms from CuII to CuI.
Different synthetic approaches have also been showcased. An interesting alternative synthesis route features the inversion of Q and E positions during the transformation of the S/Cu adamantane-type structure [(NEt4]4[(SPh)4(CuBr)6] (847, see section 2.1.7) to the desired [Et4N]2[Cu4(SPh)6] (256) by addition of [Et4N]SPh in DMF.170
The polymer (CuSCH2CH2OH)n decomposes and dissolves in basic aqueous solutions to give the adamantane-type [(nBu)4N]2[Cu4(SCH2CH2OH)6] (257, Fig. 9).171
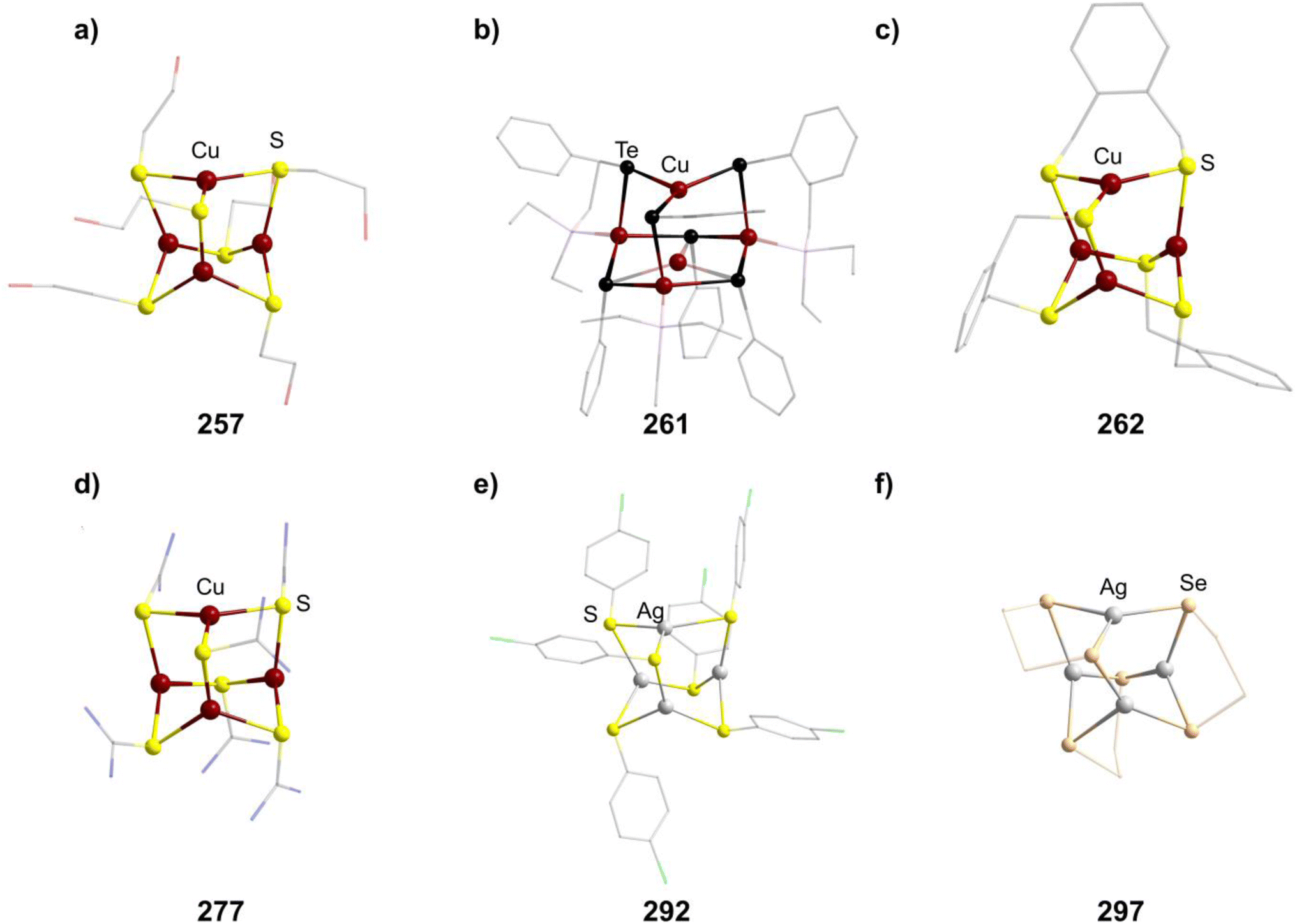 | ||
| Fig. 9 Examples of adamantane-type compounds with group 11 in the Q-position and group 16 atoms in the E-position: [(nBu)4N]2[Cu4(SCH2CH2OH)6] (257, top left (a)), [Et3PPh][μ3-Cu(CuPEt3)3Cu(TePh)6] (261, top center (b)), [Ph4P]2[Cu4{o-(SCH2)2C6H4}3] (262, top right (c)), [Cu4{SC(NH2)2}6](SO4)2 (277, bottom left (d)), [Et4N]2[Ag4(SC6H4-p-Cl)6] (292, bottom center (e)) and [nPr4N]2[Ag4(Se4)3] (297, bottom right (f)). Hydrogen atoms and counterions, if present, are omitted for clarity. | ||
An electrochemical synthesis route to the thiolate cluster [Cu(BIK)2]2[Cu4{S(o-tolyl)}6] (258, BIK = bis(2-methyl-imidazole-2-yl)ketone) is also feasible using a Cu anode in an electrolyte solution of BIK, the thiol HS(o-tolyl) and [nBu4N]ClO4 in MeCN.172
Analogous reactions can also generate the selenium congener [Me4N]2[Cu4(SePh)6] (259),173 while the only known Te congener [tBu3PH]2[Cu4(TePh)6] (260) has been obtained from a rearrangement of the cluster [(tBu3P)3(CuTePh)4].174
There is however another tellurium containing adamantane-type structure formally derived from this example. Unlike many other adamantanes discussed here containing a μ4-atom in the center, this one features a μ3-Cu atom. One six membered (CuPEt3)3Te3 ring of the adamantane-type scaffold in [Et3PPh][μ3-Cu(CuPEt3)3Cu(TePh)6] (261, Fig. 9) coordinates an additional Cu atom in its center opposite to a naked Cu atom in the Q position, leading to a more planar arrangement of the six membered ring.175 Isolation was possible if Te(Ph)SiMe3 was used as a tellurolate source in a solution with CuCl and PEt3.
A related family of adamantane-type ions [Cu4(SRS)3]2− (in 262–274, Fig. 9) comprises bridging bis-thiolates in the E position. This leads to two different copper sites: three copper moieties are coordinated by two sulfur atoms of the same bis-thiolate and one from another, while the last Cu atom is coordinated by three different ligands.
Their synthesis normally follows the same patterns as has been discussed for the monothiolates,176–181 although two examples can be found that form by rearrangement of other copper thiolate compounds.179,182,183
A purely inorganic S4 bridge in place of a bis-thiolate could also be observed in the compound [Ph4P]2[Cu4(S4)3] (275), prepared from a reaction of elemental sulfur, H2S and Cu(OAc)2, thus involving a reduction of the copper atoms.184
Utilizing neutral thiones in place of thiolates results in the formation of cationic adamantanes of the type [Cu4(SCR2)6]4+ (in 276–280, Fig. 9).185–189 This is achieved by addition of the thione to simple copper salts, mostly nitrates or sulfates, in common solvents. Depending on the concentrations and additives used, additional thione ligands can also coordinate to one or multiple Cu sites in the cluster, expanding their coordination number from three to four (281–283).185,190,191 When choosing CuI as a precursor, such an addition of iodide is observed on all copper atoms, resulting in neutral clusters [(CuI)4(SR2)6] (284–285).192,193 A Cl homolog [(CuCl)4{SC(NH2)NHCH2CHCH2}6] (286) is observed in an electrochemical reaction at copper electrodes in an electrolyte of CuCl2, HCl and SC(NH2)NHCH2CHCH2 in ethanol.194
Using linked phosphine sulfides or selenides (EPPh2)2N− (E = S, Se) results in cluster cations [Cu4{(EPPh2)2N}3]+ (in 287–290) with the same architecture as described for linked thiolates.195–198
[Cu4(O3N4)2](ClO4)2 (291, H3O3N4 = 1-Me-4-OH-3,4-bis(CH2N(CH2C5H4N)(CMe2CH2OH)–C6H2) represents the only example of a CuII as well as a Cu/O cluster compound. The two ligands deliver three oxygen atoms in the E position and additionally coordinate to two copper atoms each via four N moieties, resulting in a heptadentate coordination.199 Despite the differences, the reaction pathway is similar to the thiolate route as the ligand is deprotonated before reaction with a simple copper salt.
While fewer examples for silver exist, they can generally be seen as the simple heavier congeners of known Cu compounds. [Et4N]2[Ag4(SC6H4-p-Cl)6] (292, Fig. 9) results from transferring the chemistry of simple Cu thiolates to silver,166 while [Ph4P]2[Ag4{o-(SCH2)2C6H4}3] (293) and [nBu4N]2[Ag4(FcSe2)3] (294, Fc = ferrocenyl) can be isolated when using a bis-thiolate or bis-selenide respectively.200,201
Another silver thiolate could be found as the anion in an intercluster compound [Et4N][Br@Ag8(2-TBI)12(SO4)2][Ag4(2-TBI)6(SO4)3]2 (295, 2-TBI = 2-thiobenzimidadzol) together with an octomeric cluster, in which it is additionally coordinated by three sulfate ions. While the reactants are similar to those used in other reactions leading to thiolate adamantanes, solvothermal conditions and ultrasonic activation are used in this case.202
The only Te homolog in this compound family is found in [Ph4P]2[Ag4(C4H3STe)6] (296). The ligand of this cluster is made by addition of elemental Te to thiophene in the presence of nBuLi.203
An oligoselenide-containing cluster [nPr4N]2[Ag4(Se4)3] (297, Fig. 9) in analogy to the sulfide congener could also be obtained after using Na2Se5 as the selenide source.204
The nitrogen bridged phophine selenide [Ag4{(SePPh2)2N}3](OTf) (298) is another example of a silver compound that can be prepared according to the synthesis used for its copper homolog.205
Lastly, a second selenone [(AgPPh3)4(Mbis)3](OTf)4 (299, Mbis = 1,1′-methylenebis(3-methylimidazoline-2-selone)) unique to the chemistry with silver results from the addition of Mbis to [Ag(OTf)(PPh3)], which leads to an adamantane featuring PPh3 terminal ligands at the silver positions.206
| Compound | Reagents/conditions | Method |
|---|---|---|
| a Me2phen = 2,9-dimethyl-1,10-phenanthroline, BIK = bis(2-methyl-imidazole-2-yl)ketone, tBu2DED = 1,1-dicarbo-tert-butoxy-2,2-ethylenedithiolate, tpdt = 3,4-thiophenedithiolate, α-tpdt = 2,3-thiophenedithiolate, H4pymtH = 3,4,5,6-tetrahydropyrimidine-2-thione, H3O3N4 = 1-Me-4-OH-3,4-bis(CH2N(CH2C5H4N)(CMe2CH2OH)–C6H2, Fc = ferrocenyl, 2-TBI = 2-thiobenzimidadzol, Mbis = 1,1′-methylenebis(3-methylimidazoline-2-selone). | ||
| [Me4N]2[Cu4(SPh)6] (244) | Cu(NO3), PhSH, nBu3N, [Me4N]Cl/EtOH, 75 °C | C157–159 |
| [Ph4P]2[Cu4(SPh)6] (245) | [Ph4P]2[Cu(SPh)3], [Cu(MeCN)4]ClO4/MeCN, 82 °C, 5 min | J160,161 |
| [Li(diglyme)2]2[Cu4(SPh)6] (246) | CuN(SiMe3)2, LiN(SiMe3)2, HSPh/diglyme, 110 °C, 10 min | C162 |
| [Li(dme)3]2[Cu4(SPh)6] (247) | CuN(SiMe3)2, LiN(SiMe3)2, HSPh/DME, 84 °C, 10 min | C162 |
| [Li(15-crown-5)thf]2[Cu4(SPh)6] (248) | CuN(SiMe3)2, LiN(SiMe3)2, HSPh, 15-crown-5/THF, slight heat, 5 min | C162 |
| [Me4N]2[Cu4(SMe)6] (249) | [Me4N][CuCl2], NaSMe/EtOH, MeCN, 75 °C, 90 min | C159,163 |
| [nPr4N]2[Cu4(SMe)6] (250) | Cu2O, [nPr4N]Br, NaOMe/(CH2OH)2, MeOH, MeCN, 55 °C, 1 h | C164 |
| [Ph4P]2[Cu4(SEt)6] (251) | Cu2O, EtSH, [Ph4P]Br, NaOMe/(CH2OH)2, 55 °C | C165 |
| [Et4N]2[Cu4(SC6H4-p-Cl)6] (252) | Cu(NO3)2, HSC6H4-p-Cl, nBu3N, [Et4N]Cl/EtOH, MeOH, MeCN, 50 °C to 4 °C, 18 h | C166 |
| [Et4N]2[Cu4{S(o-tBuC6H4)}6] (253) | CuCl, HS(o-tBuC6H4, NaH, [Et4N]Cl/DMF | C167 |
| [Et4N]2[Cu4(SiPr)6] (254) | CuCl, HSiPr, NaH, [Et4N]Cl/THF, 24 h | C168 |
| [K(Me2phen)3]2[Cu4(SBn)6] (255) | CuCl, KSBn, Me2phen/THF | C169 |
| [Et4N]2[Cu4(SPh)6] (256) | [(NEt4]4[(SPh)4(CuBr)6] (847), HSPh, Et3N/DMF, 15 min | J170 |
| [(nBu)4N]2[Cu4(SCH2CH2OH)6] (257) | (CuSCH2CH2OH)n, [(nBu)4N]OH/H2O | I171 |
| [Cu(BIK)2]2[Cu4{S(o-tolyl)}6] (258) | BIK, HS(o-tolyl), Cu anode, [nBu4N]ClO4/MeCN, electrolysis | N172 |
| [Me4N]2[Cu4(SePh)6] (259) | CuCl, PhSeH, Et3N, [Me4N]Cl/DMF, MeOH | C173 |
| [tBu3PH]2[Cu4(TePh)6] (260) | [(tBu3P)3(CuTePh)4], Me3SiTePh, Me3GaOEt2/THF | J174 |
| [Et3PPh][μ3-Cu(CuPEt3)3Cu(TePh)6] (261) | PEt3, CuCl, Te(Ph)SiMe3/Pentane, RT, 18 h | C175 |
| [Ph4P]2[Cu4{o-(SCH2)2C6H4}3] (262) | Cu(NO3)2, o-(HSCH2)2C6H4, NEt3, [Ph4P]Br/EtOH, 5 h | C176 |
| [Ph4P]2[Cu4(SCH2CH2S)3] (263) | CuCl, HSCH2CH2SH, NEt3, [Ph4P]Br/MeCN, 5 h | C177,178 |
| [(Me3P)4Cu]2[Cu4(SCH2CH2S)3] (264) | [CuSCH2CH2SCu], PMe3/PhMe, 90 °C, 1.5 h | C179 |
| [Ph4P]2[Cu4{S(CH2)3S}3] (265) | HS(CH2)3SH, Cu2O, [Ph4P]Br, NaOMe/(CH2OH)2, MeOH, 55 °C, 1 h | C178 |
| [Me4N]2[Cu4{S(CH2)3S}3] (266) | HS(CH2)3SH, Cu2O, [Me4N]Cl, NaOMe/MeCN, MeOH, 50 °C, 1 h | C178 |
| [Et4N]2[Cu4{S(CH2)3S}3] (267) | HS(CH2)3SH, Cu2O, [Et4N]Br, NaOMe/MeCN, MeOH, 50 °C, 45 min | C178 |
| [Et4N]2[Cu4(SCH2CH2S)3] (268) | HSCH2CH2SH, Cu2O, [Et4N]OH/MeCN, MeOH, 50 °C | C178 |
| [Me3NCH2Ph]2[Cu4(SCH2CH2S)3] (269) | HSCH2CH2SH, Cu2O, [Me3NCH2Ph]Cl, NaOMe/glycerol, MeOH, 45 °C | C178 |
| [Me4N]2[Cu4(C8H6S8)3] (270) | [Cu(MeCN)4][PF6], C8H8S8, [Me4N]OH/THF, Me2CO, MeOH, 3 days | C180 |
| [Ph4P]2[Cu4(tpdt)3] (271) | CuCl2, 5,6-thieno[2,3-d]-1,3-dithiol-2-one, KOMe, [Ph4P]Br/MeOH, 1 h | C181 |
| [Ph4P]2[Cu4(α-tpdt)3] (272) | CuCl2, thieno[3,4-d]-1,3-dithiol-2-thione, KOMe, [Ph4P]Br/MeOH, 1 h | C181 |
| [(Me3P)4Cu][Cu4(SCH2CH2S)3(CuPPh3)] (273) | [(Me3P)4Cu]2[Cu4(SCH2CH2S)3]/THF | J179 |
| K[Ph4P][Cu4(tBu2DED)3] (274) | K4[Cu8(tBu2DED)6], [Ph4P]Cl, S/Me2CO, EtOH | J182,183 |
| [Ph4P]2[Cu4(S4)3] (275) | S, H2S, Cu(MeCO2)2, [Ph4P]Br, NH3/MeCN | C184 |
| [Cu4{SC(NH2)2}6](NO3)4 (276) | CuNO3, SC(NH2)2 HNO3/H2O | C185 |
| [Cu4{SC(NH2)2}6](SO4)2 (277) | CuSO4, SC(NH2)2, HOAc/H2O, 80° | C186 |
| [Cu4{SC(NH2)2}6](HSO4)2SO4 (278) | CuSO4, SC(NH2)2, H2SO4/H2O, 80 °C | C186,187 |
| [Cu4(H4pymtH)6](ClO4)4 (279) | [Cu(C2H4)ClO4], H4pymtH, C2H4/MeOH | C188 |
| [Cu4{SC(NH2)NHCH2CHCH2}6](OTf)4 (280) |
Cu(OTf)2, SC(NH2)NHCH2CHCH2/C6H6, 20 min |
C189 |
| [{CuSC(NH2)2}3Cu{SC(NH2)2}6](NO3)4 (281) | CuNO3, SC(NH2)2, HNO3/H2O | C185 |
| [{CuSC(NH2)2}Cu3{SC(NH2)2}6](SO4)2 (282) | CuSO4, SC(NH2)2, H2SO4/H2O | C190 |
| [{CuSC(NH2)2}(CuNO3)Cu2{SC(NH2)2}6](SO4)(NO3) (283) | Cu(NO3)2, SC(NH2)2/H2O, 80 °C to 5 °C, 5 days | C191 |
| [(CuI)4{SC(NH2)NHEt}6] (284) | CuI, SC(NH2)NHEt/EtOH, 50 °C, 3 h | C192 |
| [(CuI)4{SC(NH2)2}6] (285) | CuI, SC(NH2)2, KI/H2O, 80 °C | C193 |
| [(CuCl)4{SC(NH2)NHCH2CHCH2}6] (286) |
CuCl2, Cu electrode, SC(NH2)NHCH2CHCH2, HCl/EtOH, 0.2 V, 0.13 mA |
N194 |
| [Cu4{(SPPh2)2N}3][CuICl2] (287) | 1. NaN(SPPh2)2, CuCl2/H2O | J195,196 |
| 2. CCl4, CH2Cl2 | ||
| [Cu4{(SPPh2)2N}3][BF4] (288) | [Cu(MeCN)4][BF4], (SPPh2)2NH/CH2Cl2, 1 h | C197 |
| [Cu4{(SPPh2)2N}3]I3 (289) | Cu, (SPPh2)2NH·I2/Et2O, 2 days | C198 |
| [Cu4{(SePPh2)2N}3][BF4] (290) | [Cu(MeCN)4][BF4], (SePPh2)2NH/CH2Cl2, 1 h | C197 |
| [Cu4(O3N4)2](ClO4)2 (291) | Cu(ClO4)2, H3O3N4, Et3N/MeOH | C199 |
| [Et4N]2[Ag4(SC6H4-p-Cl)6] (292) | AgNO3, HSC6H4-p-Cl, nBu3N, [Et4N]Cl/EtOH, MeOH, MeCN, 50 °C to 4 °C, 18 h | C166 |
| [Ph4P]2[Ag4{o-(SCH2)2C6H4}3] (293) | AgNO3, Na2o-(SCH2)2C6H4, [Ph4P]Br/MeOH, 5 h | C200 |
| [nBu4N]2[Ag4(FcSe2)3] (294) | AgCl, Fc(SeSiMe3)2, [nBu4N]Br/THF | C201 |
| [Et4N][Br@Ag8(2-TBI)12(SO4)2][Ag4(2-TBI)6(SO4)3]2 (295) | Ag2SO4, 2-TBI, [Et4N]Br/MeCN, DMF, sonification, 120 °C, 2 days | B/L202 |
| [Ph4P]2[Ag4(C4H3STe)6] (296) | 1. Te, [Ph4P]Br, thiophene, nBuLi/THF | C203 |
| 2. AgNO3/DMF | ||
| [nPr4N]2[Ag4(Se4)3] (297) | AgNO3, Na2Se5, [nPr4N]Cl/DMF | C204 |
| [Ag4{(SePPh2)2N}3](OTf) (298) | Ag(OTf), K{(SePPh2)2N}/CH2Cl2, 30 min | C205 |
| [(AgPPh3)4(Mbis)3](OTf)4 (299) | [Ag(OTf)(PPh3)], Mbis/Me2CO, 1 h | C206 |
2.1.5.10 Group 12/16 adamantane-type clusters. This family of compounds has been studied systematically in regards to the influence of different ligands, elemental combinations and counter ions. Most of the studies on Zn compounds could be transferred to their cadmium and, unusually for period 6 elements, also to their Hg homologs. While the number of compounds investigated is very high, the types of compounds are not as diverse as for other combinations. With the exception of two clusters, all of them feature chalcogenolate groups in the E position. In the simplest case, this leads to anions of the type [(MER)4(ER)6]2− (M = Zn, Cd; E = S, Se, 300–319, Fig. 10).
 | ||
| Fig. 10 Examples of adamantane-type compounds with group 12 in the Q-position and group 16 atoms in the E-position: [Et3NH]2[(ZnSPh)4(SPh)6] (300, top left (a)), [Me4N]2[(CdBr)4(SPh)6] (412, top center (b)), [(HgPPh3)2(HgBr)2(TeoPy)6] (450, top right (c)), [Cd2(CdPPh3)2(SiPr)6][ClO4]2 (464, bottom left (d)), [2.2.2]-cryptH2[(ZnI)4(MeO)6] (465, bottom center (e)) and [Zn4(POPYH)3Cl] (466, bottom right (f)). Hydrogen atoms and counterions, if present, are omitted for clarity. | ||
While the first such compounds were obtained from electrolysis of metal anodes in basic thiol solutions,207,208 a simpler method involving reactions between chalcogenolate solutions and simple non-halide metal salts at mostly room temperature has subsequently been used.148,209–222
In solution, Cd clusters can exchange chalcogenolates, including partial substitution with tellurium, to form mixed compounds [Cd4(ER)n(E′R′)10−n]2− (in 320–368) by reacting them with 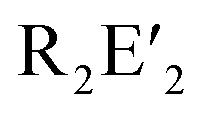 , or in equilibrium reactions with other similar clusters.222 The latter strategy also works to form the mixed metal compound [Me4N][CdnZn4−n(SPh)10] (369–371).
, or in equilibrium reactions with other similar clusters.222 The latter strategy also works to form the mixed metal compound [Me4N][CdnZn4−n(SPh)10] (369–371).
By utilizing a zwitter-ionic thiolate 4-(trimethylammonio)benzenethiolate (Tab), the cationic adamantane in [(MTab)4(Tab)6][PF6]8 (372–373, M = Zn, Cd) can be isolated by the above described method.223
The terminal chalcogenolates can be formally replaced by halides (374–433, Fig. 10). This can be done by ligand exchange reactions with PhICl2, Br2 or I2,214,224 or during cluster formation by using halide salts, which can also be used to stabilize Hg clusters including rare examples of Hg4Te6 scaffolds.225–230
As described for the pure chalcogenolate clusters, mixed metal adamantanes [Et4N]2[(MI)4(M′I)4−n(SnPr)6] (434–442, M = Zn, Cd, Hg, Fig. 10) can be obtained by exchange reactions between homometallic congeners.
Asymmetric substitution at the terminal position is possible as well. Depending on the ratio and chalcogenide used, anions of the type [(MX)n(SR)4−n(SR)6]2− (443–447, M = Zn, Cd; X = Cl, Br) can be isolated.210,231,232 Trying to obtain the HgI/SePh compound with a [(Ph3P)2N]+ countercation resulted in a charge reduced anion [Hg(HgI)3(SePh)6]− (in 448) with one Hg site not carrying any ligand.225
To reduce the negative charge of the cluster compounds, replacement of the terminal anionic ligands used previously with neutral lewis basic ligands like phosphines or arsines was necessary. With mercury, the neutral compounds [(HgPPh3)2(HgX)2(TeoPy)6] (449–451, X = Cl, Br, I; oPy = ortho-pyridyl) and [(HgPPh3)2(HgSePh)2(SePh)6] (452) with mixed terminal ligands were obtainable when using halide or acetate mercury salts.233,234 A complex precursor [M(L)2(ClO4)2] (M = Cd, Hg; L = PPh3, PEt3, AsPh3) in combination with M(ER)2 (E = S, Se) and free L leads to cationic clusters in [(ML)4(ER)6][ClO4]2 (453–464, Fig. 10).235–237 With certain L and R combinations, this can lead to clusters with a few terminally uncoordinated M sites, which do not, however, influence the charge.235
There are only two examples with oxygen in the E position. One, the methanolate cluster [2.2.2]-cryptH2[(ZnI)4(MeO)6] (465, [2.2.2]-crypt = 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane, Fig. 10) is obtained in a simple reaction of ZnI2 and [2.2.2]-crypt in MeOH in which the cryptand acts as a base.238
The other example, [Zn4(POPYH)3Cl] (466, POPYH4 = N,N′-bis(2-hydroxyphenyl)-pyridine-2,6-dicarboxamide, Fig. 10) is formed by the partially deprotonated multidentate ligand coordinating to ZnCl2, and comprises two different Zn sites.239 Three are coordinated by two oxygen and two nitrogen atoms of one ligand and one oxygen of another, while the last connects to three different ligands via their oxygen atoms and carries an additional terminal Cl ligand.
| Compound | Reagents/conditions | Method |
|---|---|---|
| a DAMS = trans-4-(4-dimethylamino-styryl)-N-methyl-pyridinium, bipy = bipyridine, nPr = normal propyl, secBu = secondary butyl, phen = 1,10-phenanthroline, oPy = ortho-pyridyl, Tab = 4-(trimethylammonio)benzenethiolate, [2.2.2]-crypt = 4,7,13,16,21,24-Hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane, POPYH4 = N,N′-bis(2-hydroxyphenyl)-pyridine-2,6-dicarboxamide. | ||
| [Et3NH]2[(ZnSPh)4(SPh)6] (300) | HSPh, Zn anode, Et3N, [Et4N]ClO4/MeCN, electrolysis or NaSPh, ZnCl2 [Et3NH]Cl/MeOH, 0 °C, 90 min | N,C207–209 |
| [Me4N]2[(ZnSPh)4(SPh)6] (301) | HSPh, Zn(NO3)2, Et3N, [nPr4N]Cl/MeOH, Me2CO, 3 days | C210 |
| (DAMS)2[(ZnSPh)4(SPh)6] (302) | HSPh, Zn(NO3)2, Et3N, (DAMS)I/MeOH | C211 |
| [Ru(2,2′-bipy)3][(ZnSPh)4(SPh)6] (303) | Cd(SPh)2, SC(NH2)2, [Ru(2,2′-bipy)3Cl2]/MeCN, H2O, 85 °C, 10 days | B212 |
| [Et4N]2[(ZnSBn)4(SBn)6] (304) | BnSH, NaOMe, Zn(NO3)2, [Et4N]2Cl/MeOH, 2 h | C213 |
| [Ph4P]2[(ZnSBn)4(SBn)6] (305) | BnSH, NaOMe, Zn(NO3)2, [Ph4P]2Cl/MeOH, 2 h | C213 |
| [Et3NH][Me4N][(ZnSC6H4-4-Cl)4(SC6H4-4-Cl)6] (306) | HSC6H4-4-Cl, Et3N, Zn(NO3)2 [Me4N]Cl/MeOH, 0 °C, 30 min | C209 |
| [Me4N]2[(ZnSC6H4-4-Cl)4(SC6H4-4-Cl)6] (307) | HSC6H4-4-Cl, NaOH, ZnCl2 [Me4N]Cl/MeOH, 0 °C, 2 h | C209 |
| [Et3NH][(ZnSC6H4-4-Cl)4(SC6H4-4-Cl)6] (308) | HSC6H4-4-Cl, Et3N, Zn(NO3)2 [Et3NH]Cl/MeOH, 0 °C, 30 min | C209 |
| [Me4N]2[(ZnSePh)4(SePh)6] (309) | Na, HSePh, Zn(NO3)2, Et3N, [nPr4N]Cl/H2O, MeOH, MeCN, 60 °C | C214,215 |
| [Et3NH][(CdSPh)4(SPh)6] (310) | HSPh, Cd anode, Et3N, [Et4N]ClO4/MeCN, electrolysis | N208 |
| [Me4N]2[(CdSPh)4(SPh)6] (311) | HSPh, Cd(NO3)2, Et3N, [Me4N]Cl/MeOH | C216 |
| [Et4N]2[(CdSPh)4(SPh)6] (312) | HSPh, CdCl2, Et3N/MeOH, H2O | C148,217 |
| (DAMS)2[(CdSPh)4(SPh)6] (313) | (DAMS)I, PhSH, Et3N, Cd(SCN)2,/MeOH, 10 min | C218 |
| [M(phen)3][(CdSPh)4(SPh)6] (314–316, M = Ru, Fe, Ni) | [Me4N][(CdSPh)4(SPh)6] (311), M(phen)3Cl2/MeCN, 30 min | O219 |
| [Et4N]2[(CdSCy)4(SCy)6] (317) | NaSCy, CdCl2, [Et3N]Cl/EtOH, MeCN | C220 |
| [Et3NH]2[(CdSC6H4-4-Me)4(SC6H4-4-Me)6] (318) | Cd[ClO4]2, SC6H4-4-Me, Et3N, [Me4N]Cl/MeOH, 1 h | C221 |
| [Me4N][(CdSePh)4(SePh)6] (319) | NaSePh, Cd(NO3)2, [Me4N]Cl/MeOH, H2O, MeCN, 80 °C | C215,222 |
| [Me4N][Cd4(SPh)10−n(SMe)n] (320–322, n = 8–10) | [Me4N][(CdSPh)4(SPh)6] (311), Me2S2/Me2CO | Q222 |
| [Me4N][Cd4(SPh)10−n(SnBu)n] (323–329, n = 4–10) | [Me4N][(CdSPh)4(SPh)6] (311), nBu2S2/Me2CO | Q222 |
| [Me4N][Cd4(SPh)10−n(SBn)n] (330–333, n = 7–10) | [Me4N][(CdSPh)4(SPh)6] (311), Bn2S2/Me2CO | Q222 |
| [Me4N][Cd4(SPh)10−n{S(2-C6H4Me)}n] (334–344, n = 0–10) | [Me4N][(CdSPh)4(SPh)6] (311), (2-C6H4Me)2S2/Me2CO | Q222 |
| [Me4N][Cd4(SePh)10−n(SnBu)n] (345–350, n = 5–10) | [Me4N][(CdSePh)4(SPh)6] (319), nBu2S2/Me2CO | R222 |
| [Me4N][Cd4(SPh)10−n(TePh)n] (351–353, n = 8–10) | [Me4N][(CdSPh)4(SPh)6] (311), Ph2Te2/Me2CO | R222 |
| [Me4N][Cd4(SePh)10−n(TePh)n] (354–357, n = 7–10) | [Me4N][(CdSePh)4(SePh)6] (319), Ph2Te2/Me2CO | R222 |
| [Me4N][Cd4(SPh)10−n(SePh)n] (358–368, n = 0–10) | [Me4N][(CdSPh)4(SPh)6] (311), [Me4N][(CdSePh)4(SePh)6]/ | R222 |
| [Me4N][CdxZn4−n(SPh)10] (369–371, n = 2–4) | [Me4N][(CdSPh)4(SPh)6] (311), [Me4N]2[(ZnSPh)4(SPh)6]/Me2CO | R222 |
| [(MTab)4(Tab)6][PF6]8 (372–373, M = Zn, Cd) | TabH[PF6], M(OAc)2/MeCN, DMF, MeOH, 70 °C, 1 h | C223 |
| [Me4N]2[(ZnCl)4(SPh)6] (374) | [Me4N]2[(ZnSPh)4(SPh)6] (301), PhICl2/MeCN, 10 min | Q214,224 |
| [Me4N]2[(ZnBr)4(SPh)6] (375) | [Me4N]2[(ZnSPh)4(SPh)6] (301), Br2/CCl4, Me2CO, 10 min | Q214,224 |
| [Me4N]2[(ZnI)4(SPh)6] (376) | [Me4N]2[(ZnSPh)4(SPh)6] (301), I2/Me2CO | Q214,224 |
| [nBu4N]2[(ZnI)4(SnPr)6] (377) | Zn(SnPr)2, ZnI2, [nBu4N]I/CH2Cl2 | C225 |
| [Et4N]2[(MX)4(SR)6] (378–406, R/M/X = iPr/Zn/Cl, Br, I; iPr/Cd/Cl, Br, I; Me/Zn/Br, I; nPr/Zn/I; nBu/Zn/I; nBu/Cd/I, Et/Zn/Cl, Br, I; Et/Cd/Cl, Br, I, Bn/Zn/Cl, Br, I; Bn/Cd/Cl, Br, I, secBu/Zn/Cl, Br, I; secBu/Cd/Cl, Br, I) | MX2, [Et4N]X, M(SR)2/CH2Cl2, 1 h | C226 |
| [Me4N]2[(ZnCl)4(SePh)6] (407) | [Me4N]2[(ZnSePh)4(SePh)6] (309), PhICl2/MeCN, 10 min | Q214 |
| [Me4N]2[(CdCl)4(SPh)6] (408) | [Me4N]2[(CdSPh)4(SPh)6] (309), PhICl2/MeCN, 10 min | Q214 |
| [R4N]2[(CdCl)4(SePh)6] (409–410, R = nPr, nBu) | CdCl2, (cat)Cl, Cd(SPh)2/CH2Cl2, 1 h | C227,228 |
| [nPr3PH]2[(CdCl)4(SeFc)6] (411) | n Pr3P, CdCl2, Me3SiSeFc/THF, 10 min | C229 |
| [Me4N]2[(CdBr)4(SPh)6] (412) | [Me4N]2[(CdSPh)4(SPh)6] (311), Br2/CCl4, Me2CO, 10 min | Q214 |
| [Me4N]2[(CdI)4(SPh)6] (413) | [Me4N]2[(CdSPh)4(SPh)6] (311), I2/Me2CO | Q214 |
| (DAMS)2[(CdI)4(SPh)6] (414) | (DAMS)I, PhSH, Et3N, Cd(NO3)2,/MeOH, 10 min | C218 |
| [Et4N]2[(CdI)4(SnPr)6] (415) | CdI2, [Et4N]I, Cd(SnPr)2/CH2Cl2 | C225 |
| [Me4N]2[(CdCl)4(SePh)6] (416) | [Me4N]2[(CdSePh)4(SePh)6] (319), PhICl2/MeCN, 10 min | Q214 |
| [Me4N]2[(CdBr)4(SePh)6] (417) | [Me4N]2[(CdSePh)4(SePh)6] (319), Br2/CCl4, Me2CO, 10 min | Q214 |
| [Me4N]2[(CdI)4(SePh)6] (418) | [Me4N]2[(CdSePh)4(SePh)6] (319), I2/Me2CO | Q214 |
| [Et4N]2[(HgX)4(SnPr)6] (419–421, X = Cl, Br, I) | HgX2, [Et4N]X, Hg(SnPr)2/CH2Cl2 | C225 |
| [Et4N]2[(HgX)4(SePh)6] (422–424, X = Cl, Br, I) | HgX2, [Et4N]X, Hg(SePh)2/CH2Cl2 | C225 |
| [Mg(CH2{P(O)Ph2}2)3][(HgX)4(SePh)6] (425–427, X = Cl, Br, I) | Hg(SePh)2, MgX2, CH2{P(O)Ph2}2/DMF, 1 h | C230 |
| [M(CH2{P(O)Ph2}2)3][(HgBr)4(SePh)6] (428–430, M = Fe, Co, Ni) | Hg(SePh)2, MBr2, CH2{P(O)Ph2}2/DMF, 1 h | C230 |
| [Et4N]2[(HgX)4(TePh)6] (431–433, X = Cl, Br, I) | HgX2, [Et4N]X, Hg(TePh)2/CH2Cl2, 30 min | C225 |
| [Et4N]2[(CdI)n(ZnI)4−n(SnPr)6] (434–436, n = 1–3) | [Et4N]2[(CdI)4(SnPr)6] (415), [nBu4N]2[(ZnI)4(SnPr)6] (377)/CH2Cl2 | R225 |
| [Et4N]2[(HgI)n(CdI)4−n(SnPr)6] (437–439, n = 1–3) | [Et4N]2[(HgI)4(SnPr)6] (421), [Et4N]2[(CdI)4(SnPr)6] (415)/CH2Cl2 | R225 |
| [Et4N]2[(HgI)n(ZnI)4−n(SnPr)6] (440–442, n = 1–3) | [Et4N]2[(HgI)4(SnPr)6] (421), [nBu4N]2[(ZnI)4(SnPr)6] (377)/CH2Cl2 | R225 |
| [Me4N]2[(ZnSPh)2(ZnX)2(SPh)6] (443–444, X = Cl, Br) | HSPh, Zn(NO3)2, Et3N, [Me4N]X/MeOH, Me2CO, 10 days | C210 |
| [Me4N]2[(ZnSPh)3(ZnCl)(SPh)6] (445) | HSPh, Zn(NO3)2, Et3N, [nPr4N]Cl/MeOH, Me2CO, 10 days | C210 |
| [Et3NH]2[(CdCl)3(p-tBu-C6H4SCd)(p-tBu-C6H4S)6] (446) | p-tBu-C6H4SH, Et3N, CdCl2/MeOH, 19.5 h | C231 |
| [Me4N]2[(CdSPh)3(CdCl)(SPh)6] (447) | HSPh, Cd(NO3)2, NEt3, [Me4N]Cl/MeOH, 1 h | C232 |
| [(Ph3P)2N]2[Hg(HgI)3(SePh)6] (448) | HgI2, [(Ph3P)2N]I, Hg(SePh)2/CH2Cl2 | C225 |
| [(HgPPh3)2(HgX)2(TeoPy)6] (449–451, X = Cl, Br, I) | o Py2Te2, Li[BH4], Hg(OAc)2, HgX2, PPh3/DMF, EtOH, THF, 2 h | C233 |
| [(HgPPh3)2(HgSePh)2(SePh)6] (452) | HgBr2, PPh3, HSePh, Et3N/MeCN, 3 days | C234 |
| [(CdPPh3)4(SPh)6][ClO4]2 (453) | [Cd(PPh3)2(ClO4)2], Cd(SPh)2, PPh3/CH2Cl2, 20 min | C235 |
| [(CdPPh3)4(SePh)6][ClO4]2 (454) | [Cd(PPh3)2(ClO4)2], Cd(SePh)2, PPh3/CH2Cl2, 20 min | C235 |
| [(HgPPh3)4(EPh)6][ClO4]2 (455–456, E = S, Se) | [Hg(PPh3)2(ClO4)2], Hg(EPh)2, PPh3/CHCl3, 10 min | C236,237 |
| [(HgPPh3)4(SMe)6][ClO4]2 (457) | [Hg(PPh3)2(ClO4)2], Hg(SMe)2, PPh3/CHCl3, 10 min | C236 |
| [(HgPPh3)4(SEt)6][ClO4]2 (458) | [Hg(PPh3)2(ClO4)2], Hg(SEt)2, PPh3/CH2Cl2, 10 min | C236 |
| [(HgAsPh3)4(SPh)6][ClO4]2 (459) | [Hg(AsPh3)2(ClO4)2], Hg(SPh)2, AsPh3/CHCl3, 10 min | C236 |
| [(HgPEt3)4(SPh)6][ClO4]2 (460) | [Hg(PEt3)2(ClO4)2], Hg(SPh)2, PEt3/Me2CO | C236 |
| [(HgPEt3)4(SePh)6][ClO4]2 (461) | [Hg(PEt3)2(ClO4)2], Hg(SePh)2, PEt3/CHCl3, 10 min | C236 |
| [Cd(CdPPh3)3(SnPr)6][ClO4]2 (462) | [Cd(PPh3)2(ClO4)2], Cd(SnPr)2, PPh3/CH2Cl2, 20 min | C235 |
| [Cd2(CdPPh3)2(SR)6][ClO4]2 (463–464, R = Cy, iPr) | [Cd(PPh3)2(ClO4)2], Cd(SR)2, PPh3/CH2Cl2, 20 min | C235 |
| [2.2.2]-cryptH2[(ZnI)4(MeO)6] (465) | [2.2.2]-crypt, ZnI2/MeOH, 1 day | C238 |
| [Zn4(POPYH)3Cl] (466) | POPYH4, Et3N, ZnCl2/MeCN, 70 °C, 3 h | K239 |
2.1.5.11 Group 13/16 adamantane-type clusters. Some group 13 examples with Al, Ga and In are known, although no examples with Te have been observed so far. The simplest examples of group 13/16 adamantane-type structures are [Q4E10]8− (467–469, Fig. 11) anionic clusters, which were the first to be realized for Ga/S, In/S and In/Se from the binary Q2E3 and K2E in water.240 The only other example of such clusters is [H2dap]4[Ga4Se10] (470, dap = 1,2-diaminopropane), also synthesized in aqueous solution, but directly from the elements and dap in solvothermal conditions.241
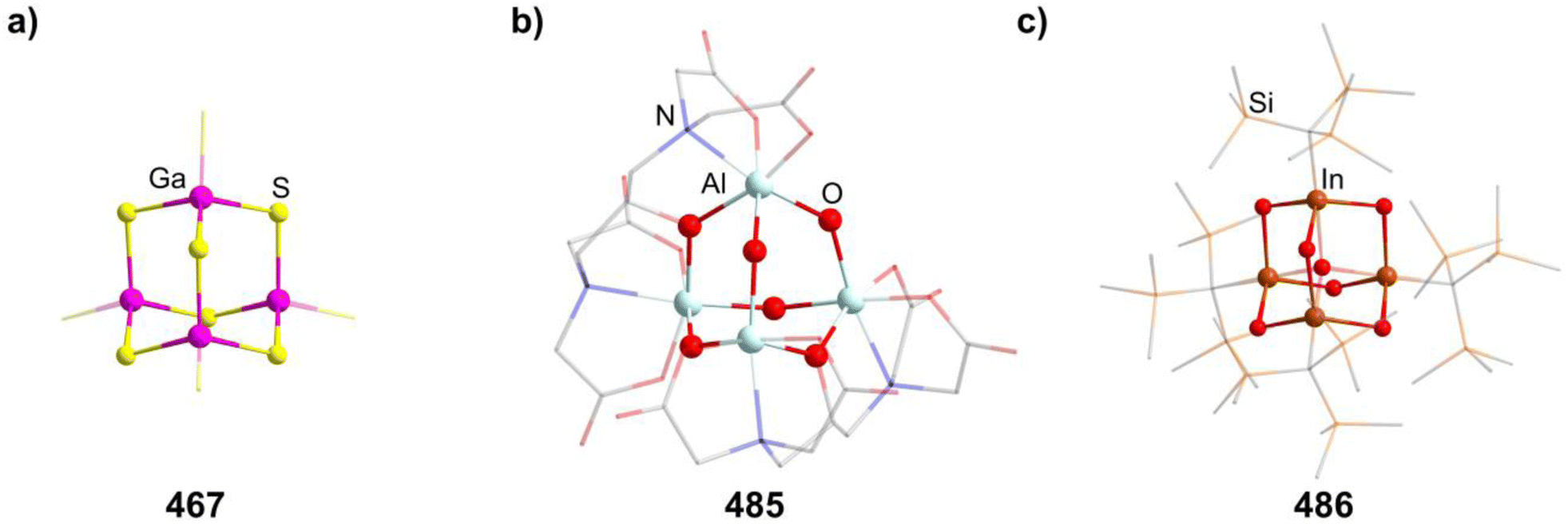 | ||
| Fig. 11 Examples of adamantane-type compounds with group 13 elements in the Q-position and group 16 atoms in the E-position: K8[Ga4S10] (467, left (a)), [enH2][Al4(OH)4(hpdta)2] (485, center (b)) and [(μ4-O){(Me3Si)3CIn}4(OH)6] (486, right (c)). Hydrogen atoms and counterions, if present, are omitted for clarity. | ||
Derivatization of the cluster archetype by protonation of the terminal sulfur atoms was presented for two compounds [(InSH)4S6]4− (in 471–472) with ammonium counterions, prepared by Method B.242,243 The addition of a larger fragment was reached in {[Ni(tepa)]2SO4}[Ni(tepa)(GaSH)4S6] (473, tepa = tetraethylenepentamine), which additionally comprises of a Ni complex coordinated by the cluster, obtained solvothermally from NiS, Ga and tepa.241
Formally substituting the terminal chalcogenides by neutral amine ligands yields neutral compounds [(QNR3)4S6]. A condensation of Me3N·AlH3 and (SiMe3)2S forms the Al congener [(AlNMe3)4S6] (474),244 while a Ga cluster [(GaNH3)4S6] (475) is isolated after the solvethermal reaction of Ga, S and [NMe4]Cl in hydrazine hydrate.243 A compound with a slightly larger ligand [(4-Me2N–C5H4NGa)4S6] (476) could be achieved in a two step synthesis via an intermediate [(4-Me2N–C5H4N)GaSH0.64Cl0.36] formed by (SiMe3)2S and the ligand decorated GaHCl2 species, which can then be converted to the target compound by an additional ligand.245
Another way to achieve neutral clusters is the partial functionalization of the chalcogenides in the E position observed in [(MI)4(SMe)4S2] (477–478, M = Al, Ga), prepared by solid state reactions from binary or elemental compounds.246,247
Hydroxo clusters of indium [(TACNIn)4(OH)6]6+ (479–480) were the first oxygen species reported, synthesized at room temperature by InCl3 and TACN in basic aqueous solution in the presence of different counterions.248
By utilizing a formally negative ligand, the charge reduced dication [(BuGa)4(OH)6][CHB11Br6Me5] (481) with a carborane counterion was obtained from hydrolysis of a low coordinate Ga complex.249
Another cluster type counterion is observed in [{(Me3Si)3Si}4Ga4O(OH)5][{(CO)3Fe}3{GaSi(SiMe3)3}2{GaFe(CO)4}] (482), which comprises a central Ga4O(OH)5 adamantane-type structure with mixed E sites decorated by hypersilyl groups (Si(SiMe3)3) leading to a monocationic cluster.250 It is formed by a rearrangement of [(Me3Si)3SiGaCl]4 in the presence of Na2Fe(CO)4·2 dioxane and NaOH.
Mixed oxo and hydroxo clusters [{(Me3Si)3CM}4O2(OH)4] (483–484, M = Al, Ga) can also be isolated as neutral compounds from the stepwise hydrolysis of a precursor complex [(Me3Si)3CMMe2], albeit in low yields.251
A pentadentate ligand was used to create a dianionic compound, [enH2][Al4(OH)4(hpdta)2] (485, en = ethane-1,2-diamine, Fig. 11), in which the hpdta ligands each use one oxygen moiety as a μ-bridging site in the E position while coordinating with the two N atoms and the other four oxygen positions to the Al atoms.139 The cluster was isolated after a simple condensation reaction between AlCl3 and the quintuply protonated ligand H5hpdta in ethane-1,2-diamine.
Lastly, a single oxo centered cluster [(μ4-O){(Me3Si)3CIn}4(OH)6] (486, Fig. 11) is synthesized by reacting the In complex Li[Me3SiInCl3] with Li[AlH4] to obtain a cyclic Li/In hydride compound [(Me3Si)(H)In(μ-H)Li(thf)2(μ-H)In(μ-H)(H)(SiMe3)], which will subsequently hydrolyze to the target compound.252
| Compound | Reagents/conditions | Method |
|---|---|---|
| a dap = 1,2-diaminopropane, tepa = tetraethylenepentamine | ||
| K8[Ga4S10] (467) | Ga2S3, K2S/H2O, 90 °C, 4 h | C240 |
| K8[In4S10] (468) | In2S3, K2S/H2O, 90 °C, 4 h | C240 |
| K8[In4Se10] (469) | In2Se3, K2S/H2O, 90 °C, 4 h | C240 |
| [H2dap]4[Ga4Se10] (470) | Ga, Se, dap, H2O/170 °C, 5 days | B241 |
| [(C3H7)2NH2]4[(InSH)4S6] (471) | In, S, dipropylamine/180 °C, 5 days | B242 |
| [NHMe3]4[(InSH)4S6] (472) | In, S, NMe3/EtOH, 140 °C, 5 days | B243 |
| {[Ni(tepa)]2SO4}[Ni(tepa)(GaSH)4S6] (473) | Ga, NiS, tepa/H2O, 180 °C, 7 days | B241 |
| [(AlNMe3)4S6] (474) | Me3N·AlH3, (SiMe3)2S/toluene, 110 °C, 5 days | D244 |
| [(GaNH3)4S6] (475) | Ga, S, [NMe4]Cl, urea/N2H4·H2O, 180 °C, 8 days | B243 |
| [(4-Me2N–C5H4NGa)4S6] (476) | 1. (4-Me2N–C5H4N)GaHCl2, (SiMe3)2S/MeCN, −25 °C to RT, 29 h | D245 |
| 2. 4-Me2N–C5H4N/MeCN, 82 °C, 8 h | ||
| [(AlI)4(SMe)4S2] (477) | 1. Ga, GaI3, AlI3/200 °C | A247 |
| 2. Me2S2/110 °C | ||
| [(GaI)4(SMe)4S2] (478) | Me2S2, Ga2I4/110 °C | A246 |
| [(TACNIn)4(OH)6](ClO4)6 (479) | InBr3, NaOH, NaClO4, TACN/H2O, 12 h | C248 |
| [(TACNIn)4(OH)6](S2O6)3 (480) | InBr3, NaOH, NaS2O6, TACN/H2O, 12 h | C248 |
| [(BuGa)4(OH)6][CHB11Br6Me5] (481) | [2,6-(2,6-Mes2C6H3)2C5H3GanBu][CHB11Br6Me5], H2O/C6D6, 16 h | I249 |
| [{(Me3Si)3Si}4Ga4O(OH)5][{(CO)3Fe}3{GaSi(SiMe3)3}2{GaFe(CO)4}] (482) | [(Me3Si)3SiGaCl]4, Na2Fe(CO)4·2 dioxane, NaOH/Et2O | J250 |
| [{(Me3Si)3CAl}4O2(OH)4] (483) | 1. AlMe2Cl, [(Me3Si)3CLi·2 thf]/THF, hexane, 15 h | I251 |
| 2. H2O/THF, −10 °C, 1 h | ||
| [{(Me3Si)3CGa}4O2(OH)4] (484) | 1. GaMe2Cl, [(Me3Si)3CLi·2 thf]/THF, hexane, 15 h | I251 |
| 2. H2O/THF, 24 h, 150 °C, 4 h | ||
| [enH2][Al4(OH)4(hpdta)2] (485) | H5hpdta, AlCl3, en/H2O | K139 |
| [(μ4-O){(Me3Si)3CIn}4(OH)6] (486) | 1. InCl3, (Me3Si)3CLi/THF, −40 °C | I252 |
| 2. LiAlH4/THF, −78 °C | ||
| 3. MeOH, H2O | ||
2.1.5.12 Group 14/16 adamantane-type clusters. The combination of group 14 and 16 elements entails the most compounds investigated until now. Most examples have been synthesized with the sulfides, selenides and, to a lesser degree, tellurides. Looking at the group 14 element, there are many examples for compounds with Si, Ge and Sn, but only a single one for a compound with Pb.
Two large groups of monomeric compounds can be defined: the first are purely inorganic cluster anions with a formal composition of [Q4E10]4− (487–563, Fig. 12) and their derivatives. They are the analogs to previously discussed group 13 compounds like [Ga4S10]8− but feature many more examples and a lower charge. They are mostly formed from the elements and/or simple binary precursors by the Methods A–C and E, resulting in regular adamantane-type anions with mostly (alkaline) metal or ammonium counterions.253–282 In a unique synthetic approach, it was also shown that those clusters can be made electrochemically using a Sn2Se3 cathode in a [Et4N]Br electrolyte solution in ethane-1,2-diamine to form [Et4N]4[Sn4Se10] (517).283
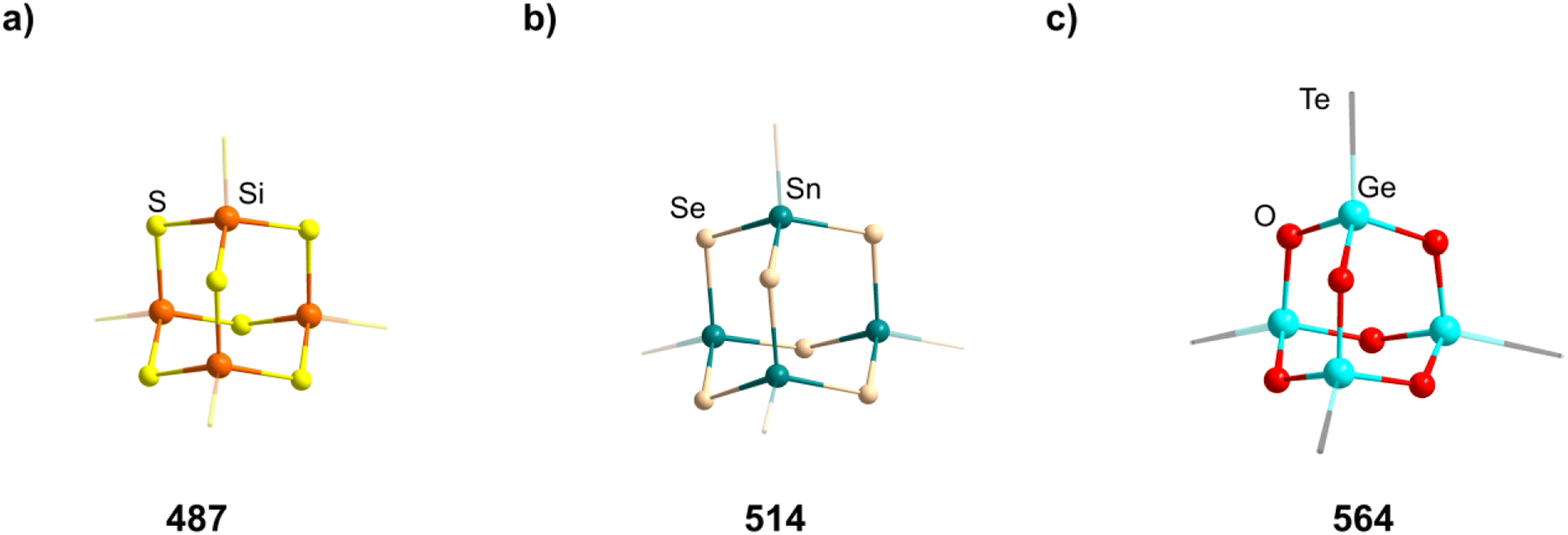 | ||
| Fig. 12 Examples of adamantane-type compounds with purely inorganic cluster anions with group 14 elements in the Q position and group 16 atoms in the E position: Na4[Si4S10] (487, left (a)), [18-crown-6-K]4[Sn4Se10] (514, center (b)) and [Mn(en)3]2[Ge4O6Te4] (564, right (c)). Counterions are omitted for clarity. | ||
The known [Ge4E10] cluster compounds are often used as reactants in ion exchange reactions (Method L) to introduce a desired functionality or structural template to the compound, such as larger ammonium cations forming lamellar structures (521–538),284–286 organic molecules (539–546)287–291 or transition metal complexes with interesting optical properties(547–551).289,292,293 The family of clusters with metal complex counterions could also be expanded by starting from elements and binary precursors in solvothermal reactions (Method B) to not only obtain more Ge clusters (552–556),294–297 but also Sn congeners as well as rare earth containing examples(557–562).298–300 In one case, the addition of antimony to such a reaction mix of GeO2 and elemental sulfur led to the formation of a compound with two distinct clusters, [(Me)2NH2]6[Ge2Sb2S7][Ge4S10] (563), one adamantane-type and another ternary molecule with noradamantane like topology.301
Unlike the other compounds in this section, the oxo cluster compound [Mn(en)3]2[Ge4O6Te4] (564, Fig. 12) deviates from the strict [Q4E10]4− cluster buildup and carries terminal Te groups at the Q position. It is obtained from a solvothermal reaction of Ge, Te, Mn(OAc)2 and [Me4N]I in ethane-1,2-diamine.302
| Compound | Reagents/conditions | Method |
|---|---|---|
| a Vio = viologen dication, TMPyP = 5,10,15,20-tetrakis(N-methyl-4-pyridyl)porphyrin, DMBPE = N,N′-dimethyl-1,2-bis(4-pyridinium)-ethylene, cyclam = 1,4,8,11-tetraazacyclotetradecane, trien = triethylentetramin, teta = triethylenetetramine. | ||
| Na4[Si4S10] (487) | SiS2, Na2S/800 °C, 48 h | A253 |
| Na4[Si4Se10] (488) | Na, Si, Se/800 °C | A254 |
| K4[Si4Te10] (489) | K, Si, Te/350 to 400 °C, 17 h | A255 |
| Na4[Ge4S10] (490) | GeS2, Na2S/H2O or GeS2, Na2S/800 °C, 48 h | A/C253,256–259 |
| K4[Ge4S10] (491) | GeS2, K2S/H2O | C256 |
| Rb4[Ge4S10] (492) | GeS2, Rb2S/H2O | C256 |
| Cs4[Ge4S10] (493) | GeS2, Cs2S/H2O or S, Ge, CsOH/H2O, 150 °C, 16 h | B/C 256,260,261 |
| Ba2[Ge4S10] (494) | GeS2, BaS/1250 °C | A253 |
| Tl4[Ge4S10] (495) | Tl2S, GeS2/500 °C, 10 days | A262 |
| [Me4N]4[Ge4S10] (496) | GeS2, [Me4N]HS, H2S/H2O, 150 °C, 4 days or GeS2, [Me4N]Cl, Na2CO3/H2O, 120 °C, 2 days | G/B263–266 |
| [EtNH3]3[MeNH3][Ge4S10] (497) | 1. GeO2, S, MeNH2/EtOH, 160 °C, 24 h | B281 |
| 2. EtNH2/EtOH, 160 °C, 24 h | ||
| [Li4(H2O)16][Ge4Se10] (498) | 1. LiSe2, Ge, Se/heat to melt | E282 |
| 2. H2O | ||
| [Li4(thf)12][Ge4Se10] (499) | 1. LiSe2, Ge, Se/heat to melt | E282 |
| 2. THF | ||
| Na4[Ge4Se10] (500) | Na, Ge, Se/800 °C | A267 |
| K4[Ge4Se10] (501) | K, Ge, Se/800 °C | A268 |
| Rb4[Ge4Se10] (502) | Rb2CO3, Ge, Se/MeOH, 190 °C, 24 h | B269 |
| Cs4[Ge4Se10] (503) | Cs2CO3, Ge, Se/MeOH, 190 °C to RT, 4 h | B270 |
| Tl4[Ge4Se10] (504) | Tl2Se, GeSe2/500 to 400 °C, 9 days | A271 |
| [Me4N]4[Ge4Se10] (505) | Ge, Se, Me4N]OH/H2O, 150 °C, 3 days | B272 |
| [(C3H7)3NH]4[Ge4Se10] (506) | Ge, Se, N(C3H7)3/H2O, 230 °C, 20 days | B273 |
| [Et4N]4[Ge4Te10] (507) | 1. K2Te, Ge, Te/heat to melt | E274 |
| 2. [Et4N]Br/en, 3 days | ||
| [R4N]4[Sn4E10], (508–513, R = Me, Et; E = S, Se, Te) | 1. K2E, E, Sn/heat to melt | E275 |
| 2. [R4N]Br/en, 100 °C, 12 h | ||
| [18-Crown-6-K]4[Sn4Se10] (514) | 1. K, Sn, Se/heat to melt | E276 |
| 2. 18-Crown-6/THF, en, 14 days | ||
| (K[2.2.2]-crypt)4[Sn4Se10] (515) | 1. K, Sn, Se/heat to melt | E277 |
| 2. [2.2.2]-crypt/en, NH3, -40 °C | ||
| [Me4N]4[Sn4Se10] (516) | Sn, Se, [Me4N]OH,/H2O, 150 °C, 16 days | B278 |
| [Et4N]4[Sn4Se10] (517) | [Et4N]Br, Sn2Se3 cathode, Ni anode/en, 300 μA, 5 V, 5 days | N283 |
| [(CHMeEt)2NH2]4[Sn4Se10] (518) | Sn, Se (CHMeEt)2NH/H2O, 160 °C, 25 days | B279 |
| [(C3H7)2NH2]4[Sn4Se10] (519) | Sn, Se, S, (C3H7)3N/H2O, 130 °C, 20 days | B280 |
| [18-Crown-6-K]4[Sn4Te10] (520) | 1. K2Te, Sn, Te/heat to melt | E276 |
| 2. 18-Crown-6/THF, en, 28 days | ||
| [CnH2n+1NMe3]4[Ge4S10] (521–524, n = 12, 14, 16, 18) | Na4[Ge4S10] (490), [CnH2n+1NMe3]Br/H2O, 18 h | O284 |
| [C8H17NMe3]4[Ge4Se10] (525) | K4[Ge4Se10] (501), [C8H17NMe3]Br/Me2CO, H2O, 3 days | O285 |
| [C9H19NMe3]4[Ge4Se10] (526) | K4[Ge4Se10] (501), [C9H19NMe3]Br/Me2CO, H2O, 45 °C, 1 day | O285 |
| [C8H17NMe2H]4[Ge4Se10] (527) | K4[Ge4Se10] (501), [C8H17NMe2H]Cl/Me2CO, H2O, 40 °C, 1 day | O285 |
| [CnH2n+1NMe3]4[Ge4Se10] (528–530, n = 10, 11, 12) | K4[Ge4Se10] (501), [CnH2n+1NMe3]Br/Me2CO, H2O, 80 °C, 1 day | O285 |
| [CnH2n+1NMe3]4[Ge4Se10] (531–573, n = 14, 16, 18) | K4[Ge4Se10] (501), [CnH2n+1NMe3]Br/Me2CO, H2O, 120 °C, 3 days | O285 |
| [(C4H9)3NH]4[Ge4Se10] (534) | K4[Ge4Se10] (501), (C4H9)3N, HCl/Me2CO, H2O, 50 °C, 3 days | O285 |
| [CnH2n+1NH3]4[Ge4Se10] (535–538, n = 12, 14, 16, 18) | Na4[Ge4Se10] (500), [CnHn+1NH3]Cl/EtOH, H2O, 60 °C 2 h | O286 |
| (H24,4′-bipy)2[Ge4S10]·4,4′-bipy (539) | [Me4N]4[Ge4S10] (496), Cu(NO3)2, 4,4′-bipy/140 °C, 3 days | O287 |
| [(CnH2n+1)2Vio]2[Ge4S10] (540–543, n = 0, 2, 3,4) | [Me4N]2[Ge4S10] (496), [(CnH2n+1)2Vio]/iPrOH, H2O, 3 days | O288 |
| [Me2Vio]2[Ge4S10] (544) | [Me4N]2[Ge4S10] (496), [MV]I2/H2O, MeOH, DMF | O289 |
| TMPyP[Ge4S10] (545) | [Me4N]2[Ge4S10] (496), TMPyP(PF6)4/MeOH, H2O, DMF, 80 °C, 7 days | O290 |
| [DMBPE]2[Ge4S10] (546) | [Me4N]2[Ge4S10] (496), [DMBPE]I2/H2O | O291 |
| [Ni(cyclam)]3[Ni(cyclam)(H2O)2][Ge4S10]2 (547) | [Me4N]2[Ge4S10] (496), [Ni(cyclam)](ClO4)2/MeCN, H2O, 3 days | O292 |
| [Mn(2,2′-bipy)2H2O][Ge4S10] (548) | [Me4N]2[Ge4S10] (496), [Mn(2,2′bipy)3](ClO4)2/MeCN, H2O, 3 days | O292 |
| [Fe(2,2′-bipy)3]2[Ge4S10] (549) | [Me4N]2[Ge4S10] (496), [Fe(2,2′bipy)3](ClO4)2/H2O, 1 day | O292 |
| [Ni(phen)3]2[Ge4S10] (550) | [Me4N]2[Ge4S10] (496), [Ni(phen)3]Cl2/MeOH, H2O, 12 h | O293 |
| MnTMPyP[Ge4S10] (551) | [Me4N]2[Ge4S10] (496), TMPyP(PF6)4, MnCl2/MeOH, H2O, DMF, 80 °C, 7 days | O290 |
| [Ni(trien)2]2[Ge4S10] (552) | GeO2, NiCl2, S/trien, 160 °C, 5 days | B294 |
| [M(dap)3]4[Ge4S10]Cl4 (553–554, M = Co, Ni)) | GeO2, Sb, S, MCl2/dap, 170 °C, 6 days | B295 |
| [Ni2(μ-teta)(teta)2][Ge4S10] (555) | GeO2, S, NiCl2, teta/H2O, 170 °C, 12 days | B296,297 |
| [Ni(teta)2]2[Ge4Se10] (556) | GeO2, Se, NiCl2, teta/H2O, 170 °C, 16 days | B296 |
| [Ho2(tepa)2(OH)2Cl2]2[Sn4Se10] (557) | SnCl4·H2O, Se, HoCl3/tepa,170 °C, 6 days | B298 |
| [Ni(teta)(en)][Ni(teta)(hda)][Sn4Se10] (558) | Sn, Se, Ni(OAc)2, hda/teta, 170 °C, 5 days | B299 |
| [Ln2(tepa)2(OH)2Cl2]2[Sn4Se10] (559–562, Ln = Y, Dy, Er, Tm) | SnCl4·5H2O, Se, LnCl3, Ag/tepa, 180 °C, 6 days | B300 |
| [(Me)2NH2]6[Ge2Sb2S7][Ge4S10] (563) | GeO2, Sb, S/DMF, 160 °C, 7 days | B301 |
| [Mn(en)3]2[Ge4O6Te4] (564) | Ge, Te, Mn(OAC)2, [Me4N]I/en, 150 °C, 80 h | B302 |
The other group contains predominantly neutral clusters with mostly organic ligands of the type [(RQ)4E6]. While at first reactions were carried out using gaseous H2E (E = S, Se) and a group 14 halide RQX3,303–305 most hybrid materials can be obtained through route D, using a solid or liquid chalcogenide source A2E (A = alkaline metal, SiMe3; E = S, Se) to prepare 565–612 (Fig. 13).306–322 As some of them are sensitive to water, the (SiMe3)2E precursors are often advantageous for their solubility in organic solvents. The clusters’ structure is heavily influenced by their organic component. In some cases, this leads to an equilibrium between compounds with an adamantane like cluster core architecture and compounds featuring the previously discussed double decker type (see section 2.1.4).306,307,323 Especially for tin compounds, back coordinating ligands shift the equilibrium away from the adamantane-type architecture, also resulting in defect heterocubane type arrangements, while some Ge congeners can be obtained in the adamantane topology.323
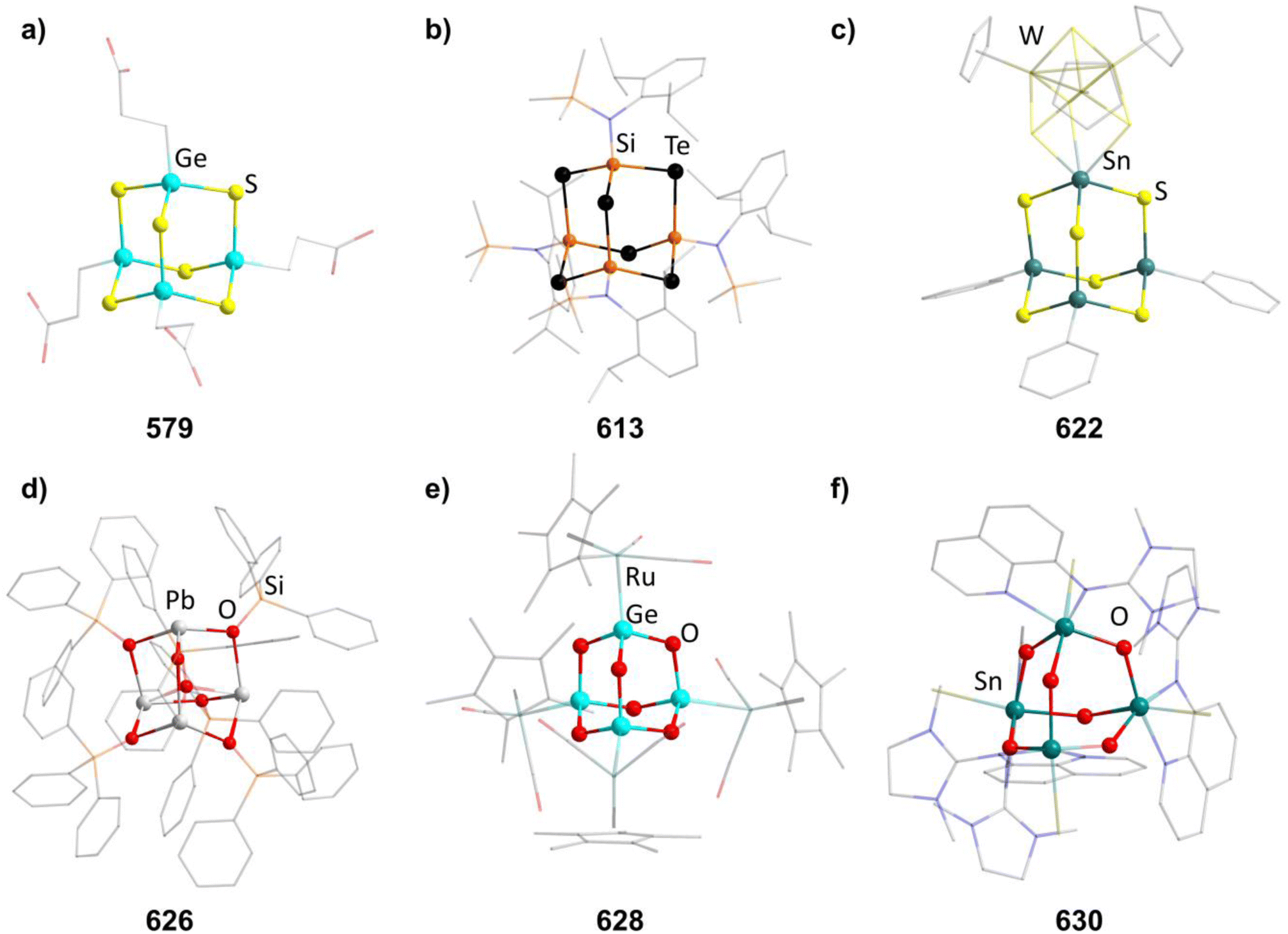 | ||
| Fig. 13 Examples of hybrid adamantane-type cluster compounds with group 14 elements in the Q position and group 16 atoms in the E position: [{HOOC(CH2)2Ge}4S6] (579, top left (a)), [(N(SiMe3)DippSi)4Te6] (613, top center (b)), [{(PhSn)3SnS6}{(WCp)3S4}] (622, top right (c)), [(μ4-O)Pb4(OSiPh3)6] (626, bottom left (d)), [({Cp*(CO)2Ru}2Ge)4O6] (628, bottom center (e)) and [(Sn(DMEGqu)Br)4O4(OH)2]Br2 (630, bottom right (f)). Hydrogen atoms are omitted for clarity. | ||
Reactive organic groups on the adamantanes can be used as a site to introduce new functionality. But to prevent the formation of defect heterocubane or double decker type cluster during the addition of Lewis basic ligands to an adamantane core, back-coordination must be prevented by using inflexible ligands.321
Tellurium containing adamantanes of the [(RQ)4E6] type have not been obtained yet by Method D. However, in one example, the silicon cluster [Si4{N(SiMe3)Dipp}4] (Dipp = 2,6-diisopropylphenyl) can be reacted with (nBu)3PTe to afford the desired [(N(SiMe3)DippSi)4Te6] (613, Fig. 13).324
In a unique oxidative addition of a SnII species N(2,6-iPr2C6H3)(SiMe3)SnCl with elemental sulfur or selenium, [{N(2,6-iPr2C6H3)(SiMe3)Sn}4E6] (614–615, E = S, Se) were isolated.325
Aside from purely organic ligands, organometallic fragments have also been used to stabilize adamantane-type clusters by the same RSnX3 and A2E method described above, either with {Cp(CO)xM} fragments (616–618)326–328 or ferrocenyl ligands (619–620).329,330
It was also possible to exchange one organic ligand in [(PhSn)4S6] with a M3S4 (M = Mo, W) fragment under retention of the adamantane framework by simple addition of [Cp(CO)3MCl] and (SiMe3)2S, resulting in [{(PhSn)3SnS6}{(MCp)3S4}] (621–622, Fig. 13).331
One case, leading to an anionic adamantane-type structure with a gold counterion, could be realized by the rearrangement of a defect heterocubane type cluster [{Me(O)CCH2CMe2Sn}3S4]Cl combined with a ligand extension to [Au(dppe)2][{Me(H2NN)CCH2CMe2Sn}4S6Cl] (623) in the presence of a gold complex.332
Compounds with oxygen in the E position are much rarer with only seven examples, one of which is the only known Pb containing adamantane [(μ4-O)Pb4(OSiPh3)6] (624, Fig. 13), featuring an endohedral μ4-O atom and silanolate μ-bridging groups.333,334624 was isolated after a reaction of plumbocene with Ph3SiOh in Et2O.
The stoichiometric hydrolysis of RSiCl3 with bulky R leads to the formation of adamantane type clusters [(RSi)4O6] (625–626, R = tBu, iPr), as the polymeric species are inhibited due to steric reasons.335
A reaction more closely related to the synthesis of the higher chalcogenide congeners is utilized for [{(Me3Si)3CSn}4O6] (627), which is made by combining (Me3Si)3CSnCl3 with Na2O.317
Two further examples obtained from hydrolysis are stabilized by transition metal fragments (628–629),336,337 with the last one being a cationic species [{Sn(DMEGqu)Br}4O4(OH)2]Br2 (630, DMEGqu = N-(1,3-dimethylimidazolidin2-ylidene)quinoline-8-amine) formed by SnBr4, DMEGqu and H2O and exhibiting a coordination number of 6 at the Sn center, unusual for adamantane-type structures.338
| Compound | Reagents/conditions | Method |
|---|---|---|
| a Thex = 1,1,2-trimethylpropyl, Np = naphthyl, Sty = para-styryl, Cy = cyclohexyl, Cp = cyclopentadienyl, Dipp = 2,6-diisopropylphenyl, DMEGqu = N-(1,3-dimethylimidazolidin2-ylidene)quinoline-8-amine. | ||
| [(MeSi)4S6] (565) | MeSiCl3, H2S/200 °C, 12 h | F304,305 |
| [(EtSi)4S6] (566) | EtSiCl3, H2S/150 °C, 3 h | F304 |
| [(ThexSi)4S6] (567) | 1. Li2S, ThexSiCl3/THF, 0 °C to RT, 14 days | D306,307 |
| 2. Decaline, 195 °C, 24 h | ||
| [(PhSi)4S6] (568) | PhSiCl3, Na2S/THF, 0 °C to RT, 24 h | D310 |
| [(RSi)4S6] (569–570, R = 1-Np, Sty) | Na2S, 1-NpSiCl3/THF, 0 °C, 18 h | D309 |
| [(MeSi)4Se6] (571) | MeSiCl3, H2Se/400 °C, 1 h | F304 |
| [(EtSi)4Se6] (572) | EtSiCl3, H2Se, Al/150 °C, 3 h | F304 |
| [(ThexSi)4Se6] (573) | 1. Li2S, ThexSiCl3/THF, 0 °C to RT, 5 days | D306,307 |
| 2. Decaline, 150 °C, 3 h | ||
| [(PhSi)4Se6] (574) | Na2Se, PhSiCl3/THF, 0 °C, 18 h | D308 |
| [(MeGe)4S6] (575) | MeGeBr3, H2S, NEt3/C6H6, 80 °C, 1 h | D311,312 |
| [(EtGe)4S6] (576) | EtGeCl3, (SiH3)2S, Al2Cl6/CS2, 75 °C, 7 days | D313 |
| [(CF3Ge)4S6] (577) | CF3GeCl3, (SiH3)2S, Al2Cl6/CS2, 80 °C, 10 days | D313 |
| [(ThexGe)4S6] (578) | 1. Li2S, ThexGeCl3/THF, 0 °C to RT, 24 h | D306,307 |
| 2. Decaline, 195 °C, 24 h | ||
| [{HOOC(CH2)2Ge}4S6] (579) | HOOC(CH2)2GeCl3, Na2S/Me2CO, H2O, 3 h | D323 |
| [{Me(O)CCH2CMe2Ge}4S6] (580) | MeOCCH2CMe2GeCl3, Na2S/Me2CO, H2O, 4 h | D323 |
| [{NC(CH2)2Ge}4S6] (581) | NC(CH2)2GeCl3, Na2S/Me2CO, H2O, 5 h | D314 |
| [(PhGe)4S6] (582) | PhGeCl3, Na2S/Me2CO, H2O, 1 h | D310 |
| [(CF3Ge)4Se6] (583) | CF3GeCl3, (SiH3)2Se, Al2Cl6/n-hexane, 110 °C, 4 days | D313 |
| [(ThexGe)4Se6] (584) | 1. Li2Se, ThexGeCl3/THF, 0 °C to RT, 24 h | D306,307 |
| 2.C6H6, 80 °C, 24 h | ||
| [{NC(CH2)2Ge}4Se6] (585) | NC(CH2)2GeCl3, Na2Se/THF, 30 h | D314 |
| [(MeSn)4S6] (586) | MeSnI3, H2S, HCl/H2O or MeSnCl3, Na2S/Me2CO, H2O, 3 h | G/D303,304,315,316 |
| [(PhSn)4S6] (587) | PhSnCl3, Na2S/Me2CO, H2O, 4 h | D304 |
| [(nBuSn)4S6] (588) | n BuSnCl3, Na2S/Me2CO, H2O, 3 h | D304,316,317 |
| [(nPrSn)4S6] (589) | (nPrSn)2O3, Na2S, HCl/H2O, 3 h | D316 |
| [(mesSn)4S6] (590) | mesSnCl3, Na2S/H2O, Me2CO, 0 °C, 12 h | D319 |
| [(1-NpSn)4S6] (591) | 1-NpSnCl3, Na2S/H2O, Me2CO, 0 °C, 18 h | D319 |
| [(4-MeC6H4Sn)4S6] (592) | 4-MeC6H4SnCl3, Na2S/H2O, Me2CO, 0 °C, 4 h | D319 |
| [(4-MeOC6H4Sn)4S6] (593) | 4-MeOC6H4SnCl3, Na2S/H2O, Me2CO, 0 °C, 2 h | D319 |
| [(4-FC6H4Sn)4S6] (594) | 4-FC6H4SnCl3, Na2S/H2O, Me2CO, 0 °C, 14 h | D319 |
| [(3-FC6H4Sn)4S6] (595) | 3-FC6H4SnCl3, (SiMe3)2S/10 °C, 1 h | D319 |
| [(C6F5Sn)4S6] (596) | C6F5SnCl3, (SiMe3)2S/C6H6, 10 °C, 15 min | D319 |
| [{(Me3Si)3CSn}4S6] (597) | [(Me3Si)3CSnBr3], Na2S/NH3, 24 h | D317 |
| [(StySn)4S6] (598) | PhSnCl3, Na2S/THF, 0 °C to RT, 24 h | D320 |
| [(CySn)4S6] (599) | CySnCl3, (SiMe3)2S/toluene, 24 h | D321 |
| [(BnSn)4S6] (600) | BnSnCl3, (SiMe3)2S/toluene, 5 min | D321 |
| [{EtO2C(C6H4)CH2CH2Sn}4S6] (601) | EtO2C(C6H4)CH2CH2SnCl3, (SiMe3)2S/toluene, 2 h | D321 |
| [(CpSn)4S6] (602) | 1. SnCl4, NaCp/toluene, 0 °C, 5 h | D321 |
| 2. (SiMe3)2S/toluene, 1 h | ||
| [(MeSn)4Se6] (603) | MeSnBr3, NaHSe, Na[BH4]/H2O, 1 h | D318 |
| [(nBuSn)4Se6] (604) | Na2Se, nBuSnCl3/NH3, −33 °C, 5 h | D317 |
| [{(Me3Si)3CSn}4Se6] (605) | [(Me3Si)3CSnBr3], Na2Se/NH3, 24 h | D317 |
| [(iPrSn)4Se6] (606) | iPrSnCl3, Na2S/H2O, Me2CO, 0 °C, 18 h | D322 |
| [(PhSn)4Se6] (607) | PhSnCl3, (SiMe3)2Se/toluene, 5 min | D321 |
| [(BnSn)4Se6] (608) | BnSnCl3, (SiMe3)2Se/toluene, 5 min | D321 |
| [EtO2C(C6H4)CH2CH2Sn)4Se6] (609) | EtO2C(C6H4)CH2CH2SnCl3, (SiMe3)2Se/toluene, 16 h | D321 |
| [(CpSn)4Se6] (610) | 1. SnCl4, NaCp/toluene, 0 °C, 5 h | D321 |
| 2. (SiMe3)2S/toluene, 5 min | ||
| [(CySn)4Se6] (611) | CySnCl3, (SiMe3)2Se/toluene, 1 h | D321 |
| [{Me(PhNHN)CCH2CMe2Ge}4S6] (612) | [(MeOCCH2CMe2Ge)4S6], H2NNHPh/CH2Cl2, 3 h | Q323 |
| [(N(SiMe3)DippSi)4Te6] (613) | [Si4{N(SiMe3)Dipp}4], (nBu)3PTe/toluene, 110 °C, 2 h | J324 |
| [{N(2,6-iPr2C6H3)(SiMe3)Sn}4E6] (614–615, E = S, Se) | N(2,6-iPr2C6H3)(SiMe3)SnCl, E/THF, 18 h | C325 |
| [({Cp(CO)2Fe}Sn)4Se6] (616) | [{Cp(CO)2Fe}2SnCl2], (SiMe3)2Se/THF | D326 |
| [({Cp(CO)3Mo}Sn)4Te6] (617) | [{Cp(CO)3Mo}SnCl3], (SiMe3)2Te/THF, −78 to −18 °C, 19 days | D327 |
| [({Cp(CO)Fe}2Sn)4S6] (618) | [{Cp(CO)2Fe}SnCl3], (Bu3Sn)2S/toluene, 12 h | D328 |
| [(FcSn)4S6] (619) | FcSnCl3, Na2S/THF, 0 °C, 31 h | D329 |
| [(FcSn)4Se6] (620) | FcSnCl3, K2Se/THF, 48 h | D330 |
| [{(PhSn)3SnS6}{(MCp)3S4}] (621–622, M = Mo, W) | [(PhSn)4S6] (587), [M(CO)3CpCl], (Me3Si)2S/THF, 15 h | R331 |
| [Au(dppe)2][{Me(H2NN)CCH2CMe2Sn}4S6Cl] (623) | 1. [{Me(O)CCH2CMe2Sn}3S4]Cl, AuCl, dppe, (Me3Si)2S,/CH2Cl2, 17 h | R332 |
| 2. PhNHNH2/CH2Cl2, 45 min | ||
| [(μ4-O)Pb4(OSiPh3)6] (624) | Ph3SiOH, [Pbcp2]/Et2O, 35 °C, 30, min | D333,334 |
| [(tBuSi)4O6] (625) | t BuSiCl3, H2O/Et2O, 24 h | I335 |
| [(iPrSi)4O6] (626) | iPrSiCl3, H2O/Et2O, −80 °C to RT, 2 days | I335 |
| [{(Me3Si)3CSn}4O6] (627) | [(Me3Si)3CSnBr3], Na2O/NH3, THF, −78 °C, 6 h | D317 |
| [({Cp*(CO)2Ru}2Ge)4O6] (628) | 1. [Cp*RuCO(GeCl2)]2, K/THF, 48 h | I336 |
| 2. O2 | ||
| [{(CO)5WGe}4O2(OH)4] (629) | 1. 2-Methoxybenzyl alcohol, Ge[N(SiMe3)2]2/Et2O, 30 min | I337 |
| 2. [W(CO)5(thf)]/THF, 12 h | ||
| 3. H2O in pentane | ||
| [(Sn(DMEGqu)Br)4O4(OH)2]Br2 (630) | SnBr4, 3,5-ditert-butyl-o-benzoquinone, DMEGqu/THF, H2O | I338 |
2.1.5.13 Group 15/16 adamantane-type clusters. The simplest adamantanes with the combination of 15/16 elements are P4O10, P4O6, P4S10, P4Se10 or As4O10. They are often used as precursors for further derivatives.
Simple derivatization reactions on [P4O6] can be carried out by adding terminal chalcogenide groups to the P moieties, oxidizing them from their +III to a +V state. A straightforward method is the thermal oxidation reaction in the presence of trace amount of water to form [P4O7] (631).339–342 Ligand exchange reactions using [P4S10] or [P4Se10] can be used with [P4O6] to obtain the series [(P4O6Sx] (632–635, x = 1–4) and [(P4O6Sex] (636–638, x = 1–3) with the four fold substituted selenium compound not being achieved due to the lower reactivity of the reagent, which would make temperatures above the decomposition point necessary.343,344 By employing this strategy and starting from 632, a mixed S/Se compound [(P4O6SSe] (639) is accessible as well.343 [(SP)4O6] (635) can also be obtained by reacting [P4O6] with elemental sulfur.345–347 Repeating the reactions with [P4O7] gives the corresponding mixed terminated adamantane-type structures [P4O7Sx] (640–642, x = 1–3) and [P4O7Se] (643), with impurities of [P4O8] (644) and [P4O8Sx] (645–646, x = 1–2) being found in the sulfur containing reaction mixture.343,348,349
P4O6 could also be used as a non-chelating tetradentate ligand to coordinate to Ni(CO)4 in a solventless reaction at room temperature.350–352 Depending on the ratio used, the complexes [P4O6{Ni(CO)3}x] (647–650, x = 1–4) or [(P4O6)xNi(CO)4−x] (651–652, x = 2–3) could be obtained if one reactant is given in excess. Using a stoichiometric ratio, the formation of coordination polymers has been reported. Reactions with the iron carbonyl [Fe(CO)5] can similarly be carried out, but proceed much slower and at higher temperatures (653–656).351,353
[P4S10] (657, Fig. 14) is most easily obtained from the elements using Method A, though many methods are available.354–356
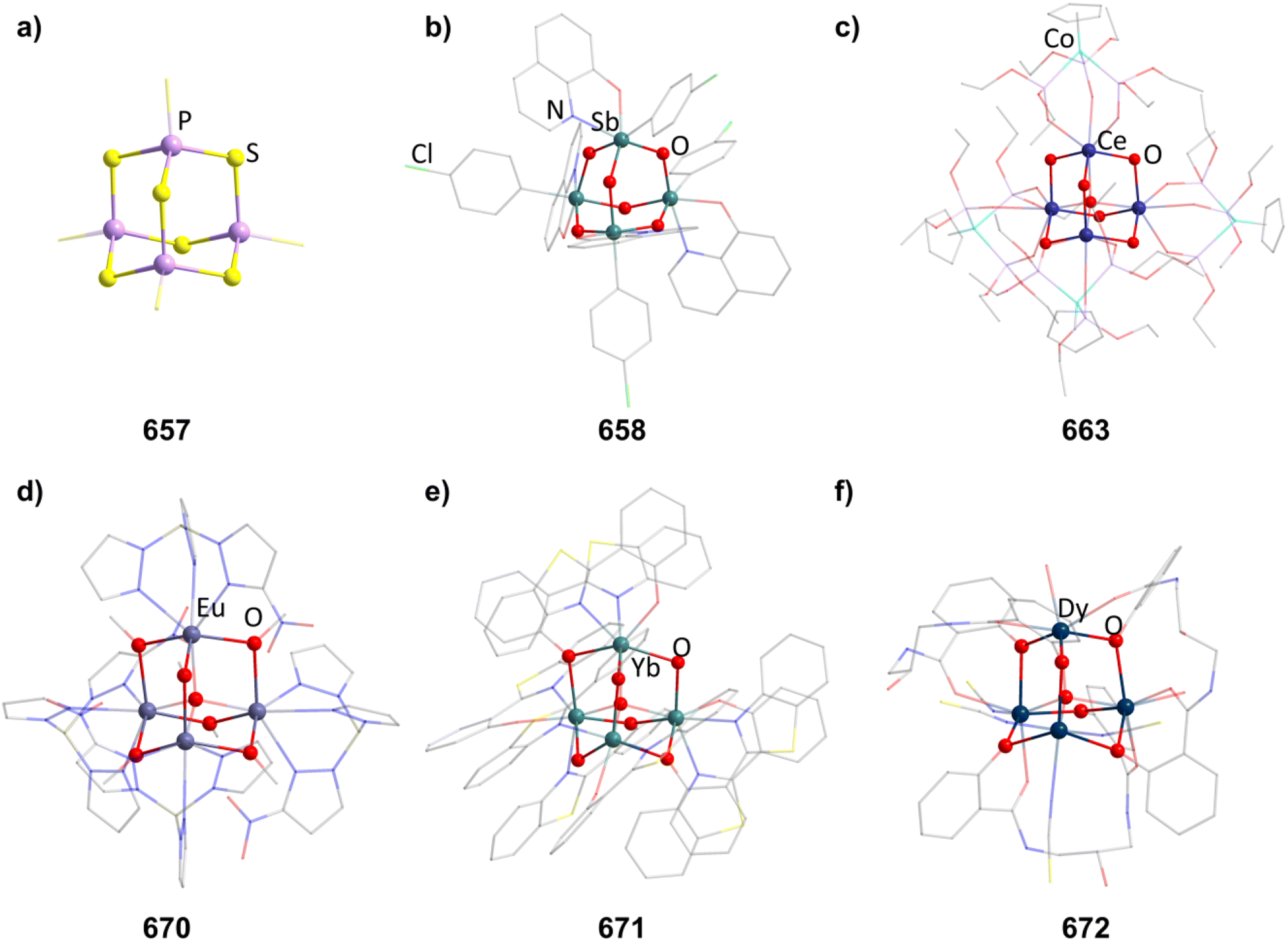 | ||
| Fig. 14 Examples of adamantane-type with group 15 and lanthanide elements in the Q position and group 16 atoms in the E position: [P4S10] (657, top left), [{(8-HQ)(p-Cl-C6H4)Sb}4O6] (658, top center (a)), [(μ4-O){Ce(LOEt)}4O4(OH)2] (663, top right (b)), [(μ4-O){Eu(3-NO2Tp)}4(μ2-OMe)6] (670, bottom left (c)), [(μ4-O){(SON)Yb}4(SON)4(OH)2] (671, bottom center (d)) and [(μ4-O)Dy4(HL)3(SCN)4(H2O)2] (672, bottom right (e)). Hydrogen atoms are omitted for clarity. | ||
Arylstibonic acids, RSbO3H2, can be used as precursors for adamantane-type structures with six coordinated Sb sites (658–661, Fig. 14) in combination with N,O-chelating ligands which trigger the rearrangement at elevated temperatures.357 A similar compound can also be achieved by treating the C,P-coordinated Sb complex (dpan)SbCl4 (dpan = 6-diphenylphosphinoacenaphth-5-yl) with a basic aqueous solution, yielding [{(dpan)(OH)Sb}4O6] (662).358
2.1.5.14 Lanthanide/group 16 adamantane-type clusters. Lanthanide atoms occupying positions within the adamantane-type scaffolds are only known in combination with oxygen in the E position for a number of oxygen centered compounds. In similarity to clusters with hydrogen (see section 2.1.1), related compounds derived from the adamantane-type architecture, in which some atoms in the E positions are formally replaced by two oxygen bridges are also known, but will not be further discussed here.359–365 In either case, the lanthanides prefer higher coordination numbers, often resulting in multiple or multidentate ligands.
[(μ4-O){Ce(LOEt)}4O4(OH)2] (663, Fig. 14) was the first example of such a compound, featuring the tripodal ligand LOEt = [Co(η5-C5H5){P(O)(OEt)2}3]−366 It was realized by the addition of [Et4N]OH to [LOEtCe(NO3)3], which led to a mix of oxo and hydroxy bridges. It is possible to treat this compound with H2O2, which will result in exchanging the oxo bridges with η2-O2 units in the Q position (664).
The series of clusters [(μ4-O){M(3-NO2Tp)}4(μ2-OMe)6] (665–670, M = Pr–Tb; 3-NO2Tp = 3-nitrotrispyrazolylborate, Fig. 14) also comprises a tripodal ligand on each metal center, but methoxy groups in the E position.367 The reaction path also involved the formation of the monomeric metal complex by addition of MCl3 to [Bu4N][3-NO2Tp] in the presence of methanol.
Another study resulted in a compound in which most oxygen atoms are part of a bridging ligand directly connected to the metal centers.368 [(μ4-O){(SON)Yb}4(SON)4(OH)2 (671, SON = (benzothiazole-2-yl)phenolate, Fig. 14) contains SON ligands with two different connecting modes: chelating a single Yb site or connecting two such atoms via one of its oxygens and two E positions.
Two clusters, [(μ4-O)M4(HL)3(SCN)4(H2O)2] (672–673, M = Dy, Eu, Fig. 14), were constructed by arranging the metal atoms stemming from M(SCN)3 around two polydentate ligands 2-hydroxy-N-[2-hydroxy-3-[(2hydroxybenzoyl)amino]propyl]benzamide (H3L), which comprise all oxygen atoms in the E position.369
| Compound | Reagents/conditions | Method |
|---|---|---|
| a 8-HQ = 8-hydroxyquinoline, H2naphpz = 2-[1H-pyrazol-5(3)-yl]naphthalene-1-ol, dpan = 6-diphenylphosphinoacenaphth-5-yl, LOEt = [Co(η5-C5H5){P(O)(OEt)2}3]−, 3-NO2Tp = 3-nitrotrispyrazolylborate, SON = (benzothiazole-2-yl)phenolate, HBT = 2-(2-hydroxyphenyl)benzothiazole, H3L = 2-hydroxy-N-[2-hydroxy-3-[(2hydroxybenzoyl)amino]propyl]benzamide. | ||
| [P4O7] (631) | P4O6, H2O/diglyme, 140 °C | P339–342 |
| [(P4O6Sx] (632–635, x = 1–4) | P4O6, [P4S10]/PhMe, 110 °C | P343 |
| [(SP)4O6] (635) | P4O6, S/160 °C | P345–347 |
| [(P4O6Sex] (636–638, x = 1–3) | P4O6, [P4Se10]/PhMe, 110 °C | P343,344 |
| [(P4O6SSe] (639) | [P4O6S] (632), [P4Se10]/PhMe, 110 °C | P343 |
| [P4O7Sx] (640–642, x = 1–3) | [P4O7] (631), [P4S10]/PhMe, 110 °C | P343,348 |
| [P4O7Se] (643) | [P4O7] (631), [P4Se10]/PhMe, 110 °C | P343,349 |
| [P4O8] (644) | [P4O7] (631), [P4S10]/PhMe, 110 °C | P343 |
| [P4O8Sx] (645–646, x = 1–2) | [P4O7] (631), [P4S10]/PhMe, 110 °C | P343 |
| [P4O6{Ni(CO)3}x] (647–650, x = 1–4) | P4O6, Ni(CO)4/10 min | P350–352 |
| [(P4O6)xNi(CO)4−x] (651–652, x = 2–3) | P4O6, Ni(CO)4/10 min | P350,351 |
| [P4O6{Fe(CO)4}x] (653–656, x = 1–4) | P4O6, [Fe(CO)5]/103 °C, 24 h | P351,353 |
| [P4S10] (657) | P, S,/100 °C | A354–356 |
| [{(8-HQ)(p-X-C6H4)Sb}4O6] (658–659, X = Cl, Br) | p-X-C6H4SbO3H2, 8-HQ/toluene, 110 °C, 6 h | C357 |
| [{(H2naphpz)(p-X-C6H4)Sb}4O6] (660–661, X = Cl, Br) | p-X-C6H4SbO3H2, H2naphpz/toluene, 110 °C, 6 h | C357 |
| [{(dpan)(OH)Sb}4O6] (662) | dpanSbCl4, NaOH/H2O, Et2O, 18 h | I358 |
| [(μ4-O){Ce(LOEt)}4O4(OH)2] (663) | [Et4N]OH, [CeLOEt(NO3)3]/MeCN, 1 h | C366 |
| [(μ4-O){Ce(LOEt)}4(O2)4(OH)2] (664) | [(μ4-O){Ce(LOEt)}4O4(OH)2] (663), H2O2/MeCN, 1 h | I366 |
| [(μ4-O){M(3-NO2Tp)}4(μ2-OMe)6] (665–666, M = Gd, Tb) | MCl3·H2O, [Bu4N][3-NO2Tp]/MeOH, 3 days | C367 |
| [(μ4-O){M(3-NO2Tp)}4(μ2-OMe)6] (667–670, M = Pr, Nd, Sm, Eu) | MCl3·H2O, [Bu4N][3-NO2Tp]/MeOH, 2–4 weeks | C367 |
| [(μ4-O){(SON)Yb}4(SON)4(OH)2] (671) | Yb[N(SiMe3)2], HBT/DME, H2O, 30 °C, 1 h | I/K368 |
| [(μ4-O)M4(HL)3(SCN)4(H2O)2] (672–673, M = Dy, Eu) | H3L, Et3N, M(SCN)3·6H2O/MeOH, MeCN, 100 °C, 2 days | K369 |
A study showed the formation of simple anionic [Be4Cl10]2− compounds (in 674–678) with various cations in solid state reactions of BeCl2 and chlorides.370
There is an oxygen centered example of a magnesium adamantane-type cluster [μ4-O{Mg(Et2O)}4Br6] (679, Fig. 15) prepared by directly reacting the Grignard reagent PhMgBr with O2 in ether.371,372
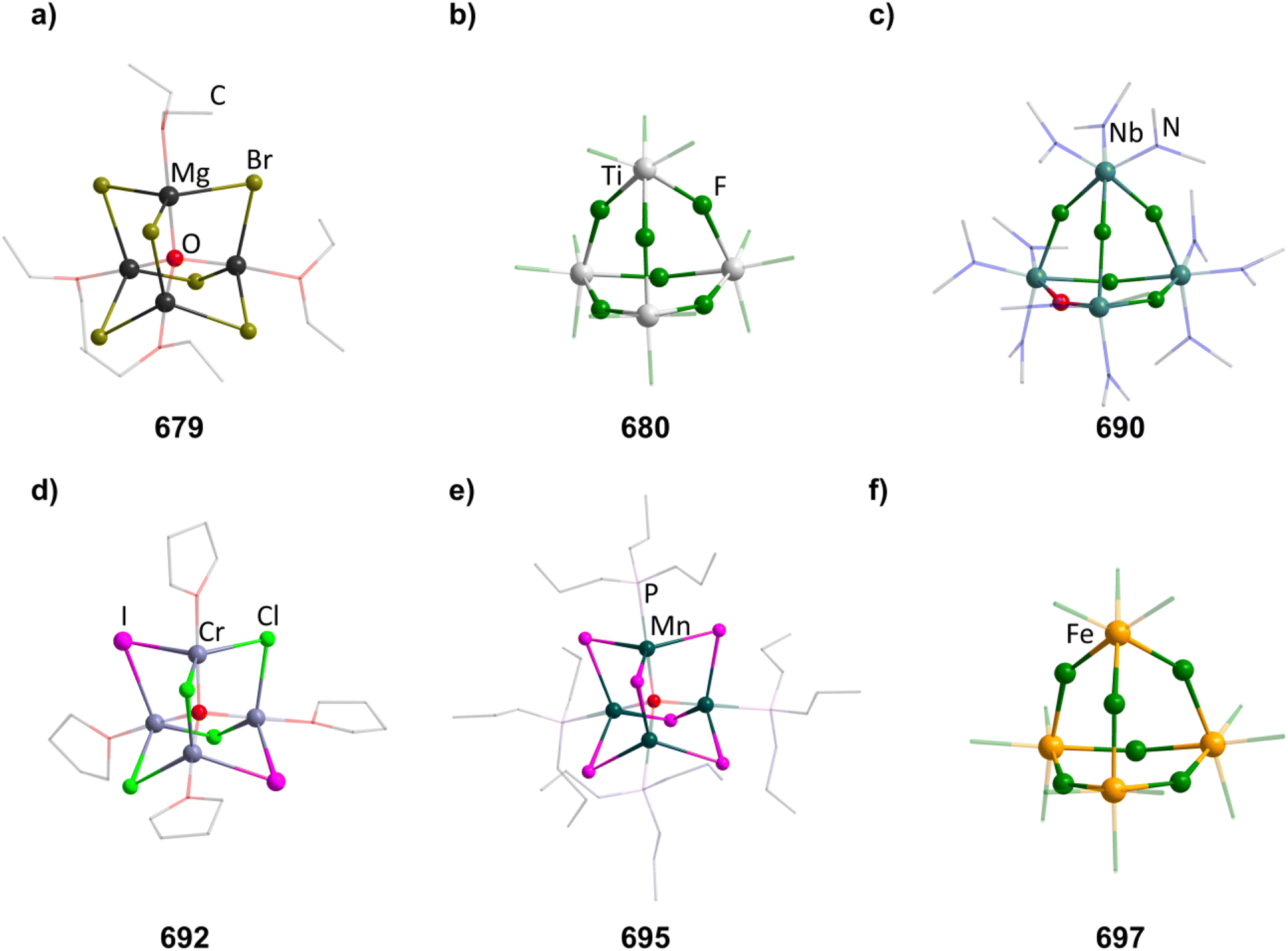 | ||
| Fig. 15 Examples of adamantane-type clusters with group 2 and 4–8 atoms in the Q position and group 17 atoms in the E position: [μ4-O{Mg(Et2O)}4Br6] (679, top left), [{Nb)NMe2)3}4{Nb)NMe2)2}F5O]Cl2 (680, top center (a)), [{Nb)NMe2)3}4{Nb)NMe2)2}F5O]Cl2 (690, top right (b)), [μ4-O{Cr(thf)}4Cl4I2] (692, bottom left (c)), [μ4-O{Mn(PnPr3)}4Cl6] (695, bottom center (d)) and [H8-HQ]6[(FeF3)4F6] (697, bottom right (e)). Hydrogen atoms and counterions, if present, are omitted for clarity. | ||
Titanium mostly forms adamantane-type clusters of the composition [(TiF3)4F6]2− (680–685, Fig. 15). All of them are formed from TiF4 in the presence of an appropriate counterion complex, such as crown ether coordinated alkaline metals, ammonium or phosphonium cations.373–375 These reactions can be carried out in conventional solvents like MeCN or in liquid HF.
In the presence of a macrocyclic arene during the formation of the adamantane, coordination to two Ti moieties under elimination of two fluorines at each position was observed, leading to [(TiCl3)2(Ti2{da6aH2(H2)})F6] (686, da6aH6 = p-methyl-dimethyldiazacalix[6]areneH6).376 Another formal, but this time complete, exchange of the terminal fluorine atoms by chlorine was observed for [C4mim]2[(TiCl3)4F6] (687, C4mim = 1-butyl-3-methylimidazolium) obtained after an ionothermal reaction of TiCl4 under decomposition of the [BF4] counterion of the ionic liquid.377
The cage compound [{Nb)NMe2)3}4F6]Cl2 (688) is obtainable by a synthesis using NbF5 and Me3SiNMe2 in chloroform and toluene.378 While the anion is exchanged by Br in CH2Br2 (689), dissolving the cluster in H2O exchanges one of the F atoms in the E position with an O atom and eliminates a ligand to form [{Nb(NMe2)3}4{Nb(NMe2)2}F5O]Cl (690, Fig. 15).
A Cr compound [μ4-O{Cr(thf)}4Cl6] (691) with a central oxygen and coordinated solvent molecule very similar to the Mg species 679 was obtained from CrCl2, nBuLi and LiOH·H2O in THF.379
Two derivatives with both Cl and I sites in the E position [μ4-O{Cr(solv)}4Cl4I2] (692–693, solv = THF, tetrahydropyran (thp), Fig. 15), could be found in small quantities while trying to synthesize the methylidine complexes [Cr3Cl3(μ-Cl)3(μ3-CH)(solv)6].380
A tungsten congener in oxidation state V+ features an anionic fluorine scaffold in [Cp2WCl2]2[(WF3)4F6] (694), resulting from the comproportionation reaction of WF6 and [Cp2WCl2].381
The Mn analogs [μ4-O{Mn(PR3)}4Cl6] (695–696, R3 = nPr3, PhMe2, Fig. 15) were prepared by bubbling O2 through an anhydrous solution of [MnI2(PR3)].382,383
Another fluorine cluster [H8-HQ]6[(FeF3)4F6] (8-HQ = 8-hydroxyquinoline, 697, Fig. 15) was isolated after a solvothermal reaction of FeF2, FeF3 and 8-HQ in the presence of HF.384
| Compound | Reagents/conditions | Method |
|---|---|---|
| a C4mim = 1-butyl-3-methylimidazolium, da6aH6 = p-methyl-dimethyldiazacalix[6]areneH6. | ||
| [H4N]2[Be4Cl10] (674) | BeCl2, NH4Cl/400–230 °C | A370 |
| Cs2[Be4Cl10] (675) | BeCl2, CsCl/400–230 °C | A370 |
| Rb2[Be4Cl10] (676) | BeCl2, RbCl/400–230 °C | A370 |
| K2[Be4Cl10] (677) | BeCl2, KCl/400–230 °C | A370 |
| Tl2[Be4Cl10] (678) | BeCl2, TlCl/400–230 °C | A370 |
| [μ4-O{Mg(Et2O)}4Br6] (679) | BrMgPh, O2/Et2O | G371,372 |
| [TiF2(15-crown-5)][(TiF3)4F6] (680) | TiF4, 15-crown-5/MeCN | C373 |
| [o-C6H4(PPh2H)2][(TiF3)4F6] (681) | TiF4, o-C6H4(PPh2)2/MeCN, CH2Cl2, 1 h | C374 |
| [o-C6H4(AsMe2H)2][(TiF3)4F6] (682) | TiF4, o-C6H4(AsMe2)2/MeCN, CH2Cl2, 1 h | C374 |
| [HiPrS(CH2)2SiPrH][(TiF3)4F6] (683) | TiF4, iPrS(CH2)2SiPr/MeCN, CH2Cl2, 1 h | C374 |
| [Me4N]2[(TiF3)4F6] (684) | TiF4, [Me4N]F/HF, −196 K to RT | F375 |
| [Ph4P]2[(TiF3)4F6] (685) | TiF4, [Ph4P]F/HF, −196 K to RT | F375 |
| [(TiCl3)2(Ti2{da6aH2(H2)})F6] (686) | TiF4, p-methyl-dimethyldiazacalix[6]areneH6/toluene, 110 °C | C376 |
| [C4mim]2[(TiCl3)4F6] (687) | TiCl4, [C4mim][BF4]/70 °C | B377 |
| [{Nb)NMe2)3}4F6]Cl2 (688) | NbF5, Me3SiNMe2/toluene, CHCl3 | C378 |
| [{Nb)NMe2)3}4F6]Br2 (689) | [{Nb(NMe2)3}4F6]Cl2 (688)/CH2Br2 | O378 |
| [{Nb)NMe2)3}4{Nb)NMe2)2}F5O]Cl2 (690) | [{Nb(NMe2)3}4F6]Cl2 (688)/H2O | I378 |
| [μ4-O{Cr(thf)}4Cl6] (691) | LiOH·H2O, nBuLi, CrCl2/THF, hexane | C379 |
| [μ4-O{Cr(thf)}4Cl4I2] (692) | [Cr3Cl3(μ-Cl)3(μ3-CH)(thf)6], benzaldehyde/THF | J380 |
| [μ4-O{Cr(thp)}4Cl4I2] (693) | CrCl2, CHI3/THP, −35 °C to RT, 19 h | C380 |
| [Cp2WCl2]2[(WF3)4F6] (694) | [Cp2WCl2], WF6/SO2 | C381 |
| [μ4-O{Mn(PnPr3)}4Cl6] (695) | [MnI2(PnPr3)], O2/n-pentane | F382 |
| [μ4-O{Mn(PnPhMe2)}4Cl6] (696) | [MnI2(PnPhMe2)], O2/n-pentane | F383 |
| [H8-HQ]6[(FeF3)4F6] (697) | FeF2, FeF3, 8-HQ, HF/H2O, EtOH, 120 °C, 72 h | B384 |
2.1.6.1 Group 11/17 adamantane-type clusters with a central μ4-O atom. Compounds with copper form by far the biggest group of this combination. The vast majority of compounds with Cl and Br in the E position comprise a central oxygen atom and will be discussed first.
The first compounds discovered were the neutral CuII complexes of the type [μ4-O{Cu(L)}4Cl6] (698–758, Fig. 16) with L being a neutral ligand. They were isolated after an addition of simple CuClx to L in the presence of ambient air, hydroxide or CuO.385–436 In some of those cases, the oxygen source could not be determined and is most likely a H2O or O2 impurity in the reaction, or stems from decomposition of the solvent. A deviating synthetic strategy uses oligomeric [LCuCl]x complexes already containing the desired ligand, which rearrange to the desired product.437–439 The clusters [μ4-O{Cu(solv)}4Cl6] (707 and 737, solv = MeCN, MeOH) can also be used in ligand exchange reactions to generate different compounds with more Lewis-basic ligands (748–749).394,440 A unique approach was taken in the formation of [μ4-O{Cu(Amt)}4Cl6] (758, Amt = 1,3-diamino-1,2,2-trimethylcyclopentane), which is formed after the ligand in [Cu(α-CgPAmtHMe)(Cl)][BF4] (CgP = 1,3,5,7-tetramethyl-2,4,6-trioxa-8-phosphatricyclo[3.3.1.1]-decane) decomposes after addition of KHMDS.441
 | ||
| Fig. 16 Examples of adamantane-type clusters with a central μ4-oxygen atom, Cu in the Q position and group 17 atoms in the E position: [μ4-O{Cu(Py)}4Cl6] (698, left (a)), [Me4N]4[μ4-O(CuCl)4Cl6] (766, center (b)) and [μ4-O{Cu(nicotine)}4Br6] (774, right (c)). Hydrogen atoms and counterions, if present, are omitted for clarity. | ||
Heterogenous substitution is possible as well in cases where multiple coordinating molecules are present (759–765).401,442–444
Anionic clusters can be generated when not all chloride atoms are substituted by a ligand during the reaction.416,436 When no coordinating ligand is present, tetraanions [μ4-O(CuCl)4Cl6]4− (in 766–772, Fig. 16) can be isolated readily with different counterions.445–451
While not as extensively studied, the Br homologs [μ4-O{Cu(L)}4Br6] (773–779, Fig. 16)394,432,452–455 were found to be achievable in a similar way by using the appropriate CuBrx salts.
The mixed cluster [μ4-O{Cu(L)}4Cl6−nBrn] (780–807) with n = 0–6 are available from using both CuBr2 and CuCl2 during the formation reaction, or by ligand exchange from [μ4-O{Cu(MeOH)}4Cl6−nBrn] (780–786).456,457
| Compound | Reagents/conditions | Method |
|---|---|---|
| a HMTA = hexamethylentetramine, HBDA = hexakis(trimethylsilyl)benzdiamidine, cpz = 2-chloro-10-(3-dimethylaminopropyl(phenothiazine), DENC = N,N-diethylnicotinamide, PziPr2H = 3,5-diisopropylpyrazole, DASO = diallyl sulfoxide, Amt = 1,3-diamino-1,2,2-trimethylcyclopentane, CgP = 1,3,5,7-tetramethyl-2,4,6-trioxa-8-phosphatricyclo[3.3.1.1]-decane, nmp = N-methyl-2-pyrrolidinone, teed = N,N,N′,N′-tetraethylethylenediamine, BPBACy = bis(1-propylbenzimidazol-2-yl)-trans-1,2-cyclohexane. | ||
| [μ4-O{Cu(Py)}4Cl6] (698) | CuCl2, NaOH/Py, 2 days | C385 |
| [μ4-O{Cu(2-methylpyridine)}4Cl6] (699) | CuCl2, 2-methylpyridine/MeOH, 65 °C, 24 h | C387 |
| [μ4-O{Cu(OPPh3)}4Cl6] (700) | CuCl2, CuO, OPPh3/MeNO2, 100 °C, 3 h | C386 |
| [μ4-O{Cu(3-quinuclidinone)}4Cl6] (701) | CuCl2·2H2O, 3-quinuclidinone, MeONa/MeOH, 65 °C, 15 min | C388 |
| [μ4-O{Cu(HMTA)}4Cl6] (702) | CuCl2·H2O, HMTA/Me2CO | C389 |
| [μ4-O{Cu(OSR2)}4Cl6] (703–704, R = Et, nPr) | CuCl2·2H2O, OSR2/Me2CO, 24 h | C390 |
| [μ4-O{Cu(N-Methylimidazole)}4Cl6] (705) | CuCl2·2H2O, N-methylimidazole/MeOH | C391,392 |
| [μ4-O{Cu(dmso)}4Cl6] (706) | Cu/CCl4, DMSO | C393,394 |
| [μ4-O{Cu(MeCN)}4Cl6] (707) | CuCl2·2H2O, HBDA/MeCN, 82 °C | C394,395 |
| [μ4-O{Cu(1,2-dimethylimidazole)}4Cl6] (708) | CuCl2·2H2O, 1,2-dimethylimidazole/EtOH, MeOH | C396 |
| [μ4-O{Cu(nictonie)}4Cl6] (709) | CuCl2·2H2O, nicotine/H2O, Me2CO | C397 |
| [μ4-O{Cu(3,4-dimethyl-5-phenylpyrazole)}4Cl6] (710) | CuCl2·2H2O, 3,4-dimethyl-5-phenylpyrazole/EtOH | C398 |
| [μ4-O{Cu(N,N-dimethylaminomethylferrocene)}4Cl6] (711) | CuCl, N,N-dimethylaminomethylferrocene, O2/CH2Cl2, 20 min | C399 |
| [μ4-O{Cu(7-azaindol)}4Cl6] (712) | CuCl2·2H2O, 7-azaindol/MeOH, 65 °C, 15 min | C400 |
| [μ4-O{Cu(Me2NH)}4Cl6] (713) | Cu/Me2NH·HCl, DMF, 50 °C, 30 min | C401 |
| [μ4-O{Cu(cpz)}4Cl6] (714) | CuCl2·2H2O, cpz/EtOH | C402 |
| [μ4-O{Cu(1-(4-picolylpyrrolidin-2-on)}4Cl6] (715) | CuCl2·2H2O, 1-(4-picolyl)pyrrolidin-2-on/MeOH | C403 |
| [μ4-O{Cu(morpholine)}4Cl6] (716) | CuCl, morpholine, Cl3CCOOMe/MeCN, H2O, 30 min | C404,405 |
| [μ4-O{Cu(Ph2SNH)}4Cl6] (717) | CuCl2·2H2O, Ph2SNH, air/MeCN, 1 day | C406 |
| [μ4-O{Cu(Imidazole)}4Cl6] (718) | CuCl2·2H2O, imidazole/MeOH | C407 |
| [μ4-O{Cu(thf)}4Cl6] (729) | CuCl2·2H2O/THF | C408 |
| [μ4-O{Cu(2-methyl-2-thiazoline)}4Cl6] (720) | CuCl2·2H2O, 2-methyl-2-thiazoline/MeOH | C409 |
| [μ4-O{Cu(2-ethylpyrazine)}4Cl6] (721) | CuCl, 2-ethylpyrazine, air/MeCN, 2 days | C410 |
| [μ4-O(Cu{1-(1-Isoquinolyl)benzotriazole})4Cl6] (722) | CuCl2·2H2O, 1-(1-isoquinolyl)benzotriazole/MeOH, CHCl3, 1 day | C411 |
| [μ4-O{Cu(3-mesitylpyrazole)}4Cl6] (723) | CuCl2·2H2O, 3-mesitylpyrazole, NaOH/MeOH, 18 h | C412 |
| [μ4-O{Cu(3-benzyl-benzimidazole)}4Cl6] (724) | CuSO4·5H2O, benzimidazole, benzlychloride/Py, 120 °C, 36 h | B413 |
| [μ4-O{Cu(2-ethyltetrazole)}4Cl6] (725) | CuCl2·2H2O, 2-ethyltetrazole/MeOH, 1 h | C414 |
| [μ4-O{Cu(1-Methylbenzotriazole)}4Cl6] (726) | CuCl2·2H2O, 1-methylbenzotriazole, CuO/MeOH, 65 °C, 1 h | C415 |
| [μ4-O{Cu(pyridine N-oxide)}4Cl6] (727) | CuCl2·2H2O, pyridine N-oxide/MeOH, 45 min | C416 |
| [μ4-O{Cu(2-Methylimidazole)}4Cl6] (728) | CuCl2·2H2O, 2-methylimidazole/MeOH, 45 min | C416 |
| [μ4-O(Cu{OP(NHtBu)3})4Cl6] (729) | CuCl2·2H2O, OP(NHtBu)3/hexane, 80 °C, 38 h | C417 |
| [μ4-O{Cu(3,5-dimethylpyrazole)}4Cl6] (730) | CuCl2·2H2O, acetylacetone, benzohydrazide/EtOH, 8 h | C418,419 |
| [μ4-O{Cu(1,4-dioxane)}4Cl6] (731) | CuCl2·2H2O, 1,4-dioxane, benzoylhydrazine/MeOH, CH2Cl2, 30 min | C420 |
| [μ4-O{Cu(1-ethyl-5-nitro-1,2,3-triazole)}4Cl6] (732) | CuCl2·2H2O, 1-ethyl-5-nitro-1,2,3-triazole/EtOH, 78 °C, 1 h | C421 |
| [μ4-O{Cu(3-hydroxyethylpyridine)}4Cl6] (733) | CuCl2·2H2O, 3-hydroxyethylpyridine/MeOH | C422 |
| [μ4-O{Cu(Quinuclidine)}4Cl6] (734) | CuCl, quinuclidine, air/MeCN, 82 °C, 30 min | C423 |
| [μ4-O(Cu{1-(pyridin-2-ylmethyl)-1H-benzimidazole})4Cl6] (735) | CuCl2·6H2O, 1-(pyridin-2-ylmethyl)-1H-benzimidazole, air/MeCN, H2O | C424 |
| [μ4-O{Cu(benzylamine)}4Cl6][Cu(benzylamine)2Cl2] (736) | CuCl2·2H2O, benzylamine/MeOH, 10 min | C394 |
| [μ4-O{Cu(MeOH)}4Cl6] (737) | CuCl2·2H2O, CuO/MeOH, 65 °C, 2 h | C394 |
| [μ4-O{Cu(PziPr2H)}4Cl6] (738) | CuCl2·2H2O, PziPr2H, sodium parafluorobenzoate/MeOH, 4 h | C425 |
| [μ4-O{Cu(DASO)}4Cl6] (739) | CuCl, DASO, air/allyl chloride, 3 h | C426 |
| [μ4-O{Cu(4-dimethylaminopyridine)}4Cl6] (740) | CuCl2·2H2O, 4-dimethylaminopyridine, 2,2,6,6-tetramethylpiperidinyl-1-oxyl, BnOH/MeOH, CH2Cl2, 10 min | C427 |
| [μ4-O{Cu(phenethylamine)}4Cl6]·[Cu(phenethylamine)2Cl2] (741) | CuCl2·2H2O, phenethylamine/MeOH, 10 min | C428 |
| [μ4-O{Cu(N,N-dimethylbenzylamine)}4Cl6] (742) | CuCl2·2H2O, N,N-dimethylbenzylamine/MeOH, 10 min | C428 |
| [μ4-O{Cu(cyclohexanemethylamine)}4Cl6]·1,5[Cu(cyclohexanemethylamine)2Cl2] (743) | CuCl2·2H2O, cyclohexanemethylamine/MeOH, 10 min | C428 |
| [μ4-O{Cu(pyrazole)}4Cl6] (744) | CuCl2·2H2O, pyrazole/MeOH, 65 °C, 2 h | C455 |
| [μ4-O{Cu(dimethyl acetamide)}4Cl6] (745) | CuCl2·2H2O, dimethyl acetamide/1,4-dioxane, 50 °C, 24 h | C429 |
| [μ4-O{Cu(1-vinylimidazole)}4Cl6] (746) | CuCl2·2H2O, 1-vinylimidazole/MeOH, H2O, 60 °C, 2 days | C430 |
| [μ4-O{Cu(metronidazole)}4Cl6] (747) | CuCl, metronidazole, air/MeOH | C431 |
| [μ4-O{Cu(NCNMe2)}4Cl6] (748) | CuCl2·2H2O/NCNMe2 | C432 |
| [μ4-O{Cu(4-(phenylethynyl)pyridine)}4Cl6] (749) | CuCl, 4-(phenylethynyl)pyridine, air/CH2Cl2, 24 h | C433 |
| [μ4-O{Cu(pyridine-3-carbaldehyde)}4Cl6] (750) | CuCl2·2H2O, pyridine-3-carbaldehyde/MeOH, CH2Cl2, 70 °C, 6 days | B434 |
| [μ4-O{Cu(2-ethylpyridine)}4Cl6] (751) | CuCl2·2H2O, 2-ethylpyridine, air/MeOH, 50 °C, 1 h | C435 |
| [μ4-O(Cu{N-(α-4-picolyl)piperidine})4Cl6] (752) | CuCl2·2H2O, N-(α-4-picolyl)piperidine/MeCN | C436 |
| [μ4-O{Cu(OPEt3)}4Cl6] (753) | [PEt3CuCl]4/CCl4, CH2Cl2, 4 days | C437 |
| [μ4-O{Cu(DENC)}4Cl6] (754) | [{(DENC)CuCl}4O2]/MeOH, CH2Cl2 | J438 |
| [μ4-O{Cu(benzimidazol)}4Cl6] (755) | [Cu2Cl3(benzimidazol)5]Cl/EtOH | J439 |
| [μ4-O{Cu(dmf)}4Cl6] (756) | [μ4-O{Cu(MeOH)}4Cl6] (737)/DMF | Q394 |
| [μ4-O{Cu(3-nonyl-8-fluoroimidazo[1,5-a]pyridine)}4Cl6] (757) | [μ4-O{Cu(MeCN)}4Cl6] (707), 3-nonyl-8-fluoroimidazo[1,5-a]pyridine/MeCN, 100 °C, 10 min | Q440 |
| [μ4-O{Cu(Amt)}4Cl6] (758) | [Cu(α-CgPAmtHMe)(Cl)][BF4], KHMDS/THF | J441 |
| [μ4-O{Cu(nmp)}3(CuOH2)Cl6] (759) | CuCl, O2/nmp, H2O | F442 |
| [μ4-O{Cu(Me2NH)}3{Cu(dmso)}Cl6] (760) | Cu/Me2NH·HCl, DMSO, 50 °C, 2 h | C401 |
| [μ4-O{Cu(Me2NH)}2{Cu(MeOH)}2Cl6] (761) | Cu/Me2NH·HCl, MeOH, 50 °C, 30 min | C401 |
| [μ4-O{Cu(thf)}3(CuOH2)Cl6]2[μ4-O{Cu(thf)}4Cl6]2 (762) | CuCl2·2H2O, tetra-μ-acetato-κ8O:Ot-dicopper(II) dehydrate/THF, 24 h | C443 |
| [μ4-O{Cu(urea)}3{Cu(thf)}Cl6] (763) | [μ4-O{Cu(MeOH)}4Cl6] (737), urea/THF, 2 h | Q444 |
| [4-phenylimidazolium][μ4-O{Cu(4-phenylimidazole)}3{CuCl}Cl6] (764) | CuCl2·2H2O, 4-phenylimidazole/MeOH, 45 min | C416 |
| [μ4-O(Cu{N-(α-4-picolyl)morpholine})2(Cu{N-(α-4-picolyl)morpholinium})(CuCl)Cl6] (765) | CuCl2·2H2O, N-(α-4-picolyl)morpholine/MeCN, H2O | C436 |
| [Me4N]4[μ4-O(CuCl)4Cl6] (766) | CuCl2, CuO, [Me4N]Cl/MeOH, 65 °C, 24 h | C445 |
| [teedH2]2[μ4-O(CuCl)4Cl6] (767) | CuCl2, teed/EtOH | C446 |
| [Et2NH2]4[μ4-O(CuCl)4Cl6] (768) | CuCl2, [Et2NH2]Cl/MeOH, 65 °C, 24 h | C447 |
| (C11H24C12N4O2)2[μ4-O(CuCl)4Cl6] (769) | CuCl2, N,N′-bis[2-(dimethylamino)-ethyl]propanediamide/CHCl3, H2O | C448 |
| [BMIm]4[μ4-O(CuCl)4Cl6] (770) | CuCl2·2H2O, [BMIm]Cl, O2, 2,3,6-trimethylphenol/nBuOH, 60 °C | C449 |
| [H2BPBACy]2[μ4-O(CuCl)4Cl6] (771) | CuCl2·2H2O, BPBACy/MeNO2, MeOH | C450 |
| [choline]4[μ4-O(CuCl)4Cl6] (772) | CuCl2·2H2O, air/choline chloride | C451 |
| [μ4-O{Cu(Py)}4Br6] (773) | CuBr2, CuO, Py/H2O, 100 °C | C452 |
| [μ4-O{Cu(nicotine)}4Br6] (774) | CuBr, 4-cyanopyridine, nicotine/DMF, 40 min | C458 |
| [μ4-O{Cu(2-bromo-1-methyl-imidazole)}4Br6] (775) | CuBr, 2-mercapto-1-methyl-imidazoline, air/MeCN, CHCl3 | C453 |
| [μ4-O{Cu(clotrimazole)}4Br6] (776) | CuBr2, clotrimazole/EtOH, 78 °C, 4 h | C454 |
| [μ4-O{Cu(benzylamine)}4Br6][Cu(benzylamine)2Br2] (777) | CuBr2, benzylamine/MeOH, 10 min | C394 |
| [μ4-O{Cu(3,5-dimethyl-4-bromo-pyrazole)}4Br6] (778) | CuBr2, acetylacetone, benzohydrazide/EtOH, 8 h | C455 |
| [μ4-O{Cu(NCNMe2)}4Br6] (779) | CuBr2/NCNMe2 | C432 |
| [μ4-O{Cu(MeOH)}4Cl6−nBrn] (780–786) | CuCl2·2H2O, CuBr2, CuO/MeOH, 60 °C, 4 h | C456 |
| [μ4-O{Cu(morpholine)}4Cl6−nBrn] (787–793, n = 0–6) | [μ4-O{Cu(MeOH)}4Cl6−nBrn] (780–786), morpholine/MeOH, 60 °C, 6 h | Q456 |
| [μ4-O{Cu(piperidine)}4Cl6−nBrn] (794–800, n = 0–6) | [μ4-O{Cu(MeOH)}4Cl6−nBrn] (780–786), piperidine/MeOH, 60 °C, 6 h | Q456 |
| [μ4-O{Cu(OPPh3)}4Cl6−nBrn] (801–807, n = 0–6) | [μ4-O{Cu(MeOH)}4Cl6−nBrn] (780–786), OPPh3/MeOH, 60 °C, 6 h | Q456,457 |
2.1.6.2 Group 11/17 adamantane-type clusters without central μ4-O atom. Unlinke the many oxygen centered chloride adamantane-type structures, there is only one example for an oxygen free species besides a binary copper complex, namely in [H2dpipa]3[Cu4Cl6][Cu2Cl6] (808, dpipa = N,N′-dimethylpiperazine), obtained from dissolving elemental Cu in HCl together with dpipa and treating it solvothermally in aqueous solution at 120 °C degree for 24 h.459
The analogous Br cluster [Cu4Br6]2− (in 809–816) is found in combination with different ammonium, phosphonium and a Mg complex counterions, always available through a reaction of CuBr with the corresponding complex bromide.460–464 One such cluster (806) was also found in a side reaction during the catalytical C–C bond formation between allyl bromide and a (C6F5)− ligand from a mixed Cu/Al complex.465 The congener of the only known Cl species discussed before [H2dpipa]3[Cu4Br6][Cu2Br6] (815) is synthesized in an analogous way by exchanging HCl with HBr.
Even larger complexes can be found in the compound [Ti12(μ3-O)14(OiPr)18][Cu4Br6] (816), in which a polyoxotitanium cluster formed alongside the adamantane when treating CuBr with [Ti(OiPr)4] under solvothermal conditions.466,467
There is only one example of a compound with a [Cu4Br6] inorganic core carrying terminal ligands: [{Cu(Hdabco)}4Br6](HCOO)2 (817, dabco = 1,4-diazabicyclo[2.2.2]octane). It is isolated from CuBr and dabco, and contains [{Cu(Hdabco)}4Br6]2+ cations forming loose networks by hydrogen bonding between the cluster units.468
Synthethic strategies for the preparation of [Cu4I6]2− (in 818–832, Fig. 17) are generally the same as for the bromide compounds. Simple species with ammonium, arsonium or phosphonium are isolated after reactions of CuI, or alternatively Cu and I2, with an appropriate complex salt (818–823).469–473 Another type of counterion often used are alkaline metal complexes with multidentate ligands such as crown ethers (824–827).474–476 They are accessible through iodine salts of Cu and the alkaline metal used, if a polyether of the appropriate size is present. [Cu4I6]2−, similar to its Br congener, is also present as a counterion with other complexes of interest. It is found either as the sole anion or together with [Cu2I4] in compounds with phosphine Mn complexes, depending on the phophine used (828–829).477 Reaction conditions apart from the nature of the ligand stay the same: MnI2 and CuI are reacted with R(PPh2O)2. Similarly, (BPPIP)[{(BPPIP)Cu2I3}2][Cu4I6] (830, BPPIP = Bis-triphenylphosphonio-isophosphindolide) comprises an additional phosphine coordinated linear Cu4I6 complex besides the adamantane.478 This formation of multiple Cu/I complexes in one compound is also observed for K[K(12-crown-4)]6[Cu4I6][Cu8I13] (831), prepared according to the strategy described for other ether complex species.479 This showcases the importance of the nature of the counterion for the structural motif of the cluster ion.
 | ||
| Fig. 17 Examples of adamantane-type clusters without a central μ4-oxygen atom, group 11–13 elements in the Q position and group 17 atoms in the E position: [(C7H16)4N]2[Cu4I6] (821, left (a)), [{Cp*NbClO}3][(Cp*Nb)3Cl2O3OH][(ZnCl)4Cl6] (833, center (b)) and [H3dien]2[Al4F18] (835, right (c)). Counterions are omitted for clarity. | ||
Lastly, the Cu/I-adamantane motif is observed as a counterion to a three dimensionally extended metal organic framework [Co(tib)2][Cu4I6] (832, tib = 1,3,5-tris(1-imidazolyl)benzene) after a reaction of CoO, CuI and tib according to Method B.480
2.1.6.3 Group 11/12 adamantane-type clusters. A very complex compound [{Cp*NbClO}3][(Cp*Nb)3Cl2O3OH][(ZnCl)4Cl6] (833, Fig. 17) featuring two cationic Nb clusters and [(ZnCl)4Cl6]2− was observed when reducing the bimetallic trigonal bipyramidal complex [(Cp*NbCl2)2ClO(OH)] with Zn in the presence of ZnO.481 It was also found as the counterion in [(Cp*TaCl)3O3(OH)2][(ZnCl)4Cl6] (834), obtained from a similar reaction of Zn, O2 and [(Cp*TaCl2)2Cl2O].482
2.1.6.4 Group 11/13 adamantane-type clusters. In group 13, [Al4F18]6− (in 835–837, Fig. 17) with varying organic countercations are obtained by solvothermal methods using microwave heating from Al(OH)3 and HF.483–485 In these compounds, each Al site carries three terminal fluorine ligands.
| Compound | Reagents/conditions | Method |
|---|---|---|
| a dpipa = N,N′-dimethylpiperazine, PoxIm = N-phenyl-N′-{bis(tertbutyl)phosphinoxide}-imidazolylidene, dabco = 1,4-diazabicyclo[2.2.2]octane, tib = 1,3,5-tris(1-imidazolyl)benzene, pyr = pyrrolidine, dppbO2 = 1,2-bis(diephenlyphospineoxide) benzol, tdpmO3 = tris(diephenlyphospineoxide) methan, BPPIP = bis-triphenylphosphonio-isophosphindolide, THP = tetrahydropyran, tren = tris(2-ethylamino)amine, gua = guanidine. | ||
| [H2dpipa]3[Cu4Cl6][Cu2Cl6] (808) | Cu, dpipa, HCl/H2O, 180 °C, 24 h | B459 |
| [iPr4N]2[Cu4Br6] (809) | CuBr, [iPr4N]Br,/EtOH | C460,461 |
| [nBuNPh3]2[Cu4Br6] (810) | CuBr, [nBuNPh3]Br/EtOH | C462 |
| [N(PPh3)2]2[Cu4Br6] (811) | CuBr, [N(PPh3)2]Br/EtOH, heat | C463 |
| [tBu3NMe]2[Cu4Br6] (812) | CuBr, [nBu3NMe]Br/iPrOH, 100 °C, 30 min | C461 |
| [Mg(thf)6][Cu4Br6] (813) | CuBr, MgBr2/THF, 50 °C, 18 h | C464 |
| [(Poxim)2AlBr][Cu4Br6] (814) | [Al(C6F5)3(toluene)0.5], CuOtBu, poxim, allyl bromide, C14H30/toluene, −30 °C to 80 °C, 7 h | U465 |
| (H2dpipa)3[Cu4Br6][Cu2Br6] (815) | Cu, HBr, dpipa/180 °C, 24 h | B459,467 |
| [Ti12(μ3-O)14(OiPr)18][Cu4Br6] (816) | CuBr, [Ti(OiPr)4]/iPrOH, 80 °C, 3 days | B466 |
| [{Cu(Hdabco)}4Br6](HCOO)2 (817) | CuBr, dabco/DMF, H2O, 85 °C, 72 h | C468 |
| [MePPh3]2[Cu4I6] (818) | CuI, [MePPh3]I CuI/MeNO2, EtOH, heat | C469,470 |
| [MeAsPh3]2[Cu4I6] (819) | CuI, [MePPh3]I/MeNO2, EtOH, heat | C469 |
| [Ph4P]2[Cu4I6] (820) | Cu, I2, [Ph4P]I/Me2CO, 56 °C | C471 |
| [(C7H16)4N]2[Cu4I6] (821) | Cu, I2, [(C7H16)4N]/hydroxyacetone, heat | C472 |
| [O{P(pyr)3}2][Cu4I6] (822) | CuI, KI, ClP(pyr)3/MeCN, 90 °C, 1 day | B473 |
| [KN{(CH2)2O(CH2)2OMe}3]2[Cu4I6] (823) | CuI, KI, N{(CH2)2O(CH2)2OMe}3 | C474 |
| [Li(benzo-15-crown-5)H2O]2(benzo-15-crown-5)[Cu4I6] (824) | CuI, LiI, benzo-15-crown-5, ascorbic acid/H2O, Me2CO, reflux, 4 h | C475 |
| [Cs(benzo-15-crown-5)]2[Cu4I6] (825) | CuI, CsI, benzo-15-crown-5, ascorbic acid/H2O, Me2CO, reflux, 2 h | C475 |
| [Na(18-crown-6)H2O]2(H2O)[Cu4I6] (826) | CuI, NaI, 18-crown-6, ascorbic acid/H2O, Me2CO, reflux, 6 h | C475 |
| [Rb(18-crown-6)]2(MeCN)[Cu4I6] (827) | RbI, Cu, [NH4]I, 18-crown-6/MeCN, 60 °C, 28 h | C476 |
| [Mn(tdpmO3)2][Cu4I6] (828) | CuI, MnI2, tdpmO3/MeCN, 30 min | C477 |
| [Mn(dppbO2)3]2[Cu4I6][Cu2I4] (829) | CuI, MnI2, dppbO2/MeCN, 1 h | C477 |
| (BPPIP)[{(BPPIP)Cu2I3}2][Cu4I6] (830) | (BPPIP)I, CuI/CH2Cl2, MeOH | C478 |
| K[K(12-crown-4)]6[Cu4I6][Cu8I13] (831) | CuI, KI, 12-crown-4/H2O, Me2CO | C479 |
| [Co(tib)2][Cu4I6] (832) | CoO, CuI, tib, KI, HI/MeOH, 145 °C, 7 days | B480 |
| [{Cp*NbClO}3][(Cp*Nb)3Cl2O3OH][(ZnCl)4Cl6] (833) | [(Cp*NbCl2)2ClO(OH], Zn, ZnO/CH2Cl2 | U481 |
| [(Cp*TaCl)3O3(OH)2][(ZnCl)4Cl6] (834) | [(Cp*TaCl2)2Cl2O] Zn, O2/CH2Cl2 | U482 |
| [H3dien]2[Al4F18] (835) | Al(OH)3, dien, HF/EtOH, 190°C microwave heating, 1 h |
B483 |
| [H3tren]2[Al4F18]·3.5H2O (836) | Al(OH)3, tren, HF/EtOH, 190°C microwave heating, 1 h |
B484 |
| (H3O)2[Hgua]16[Al4F18]3 H2O (837) | Al(OH)3, HguaCl, HF/EtOH, 190°C microwave heating, 1 h |
B485 |
The cyclic Q/Zn complexes [ZnI2{Q(SiMe3)3}]2 (Q = P, As) can be prompted to rearrange at elevated temperature when offered a proper cation to form the anionic adamantane-type structures [(QSiMe3)4(ZnI)6(thf)2] (838–839, Fig. 18).486,487
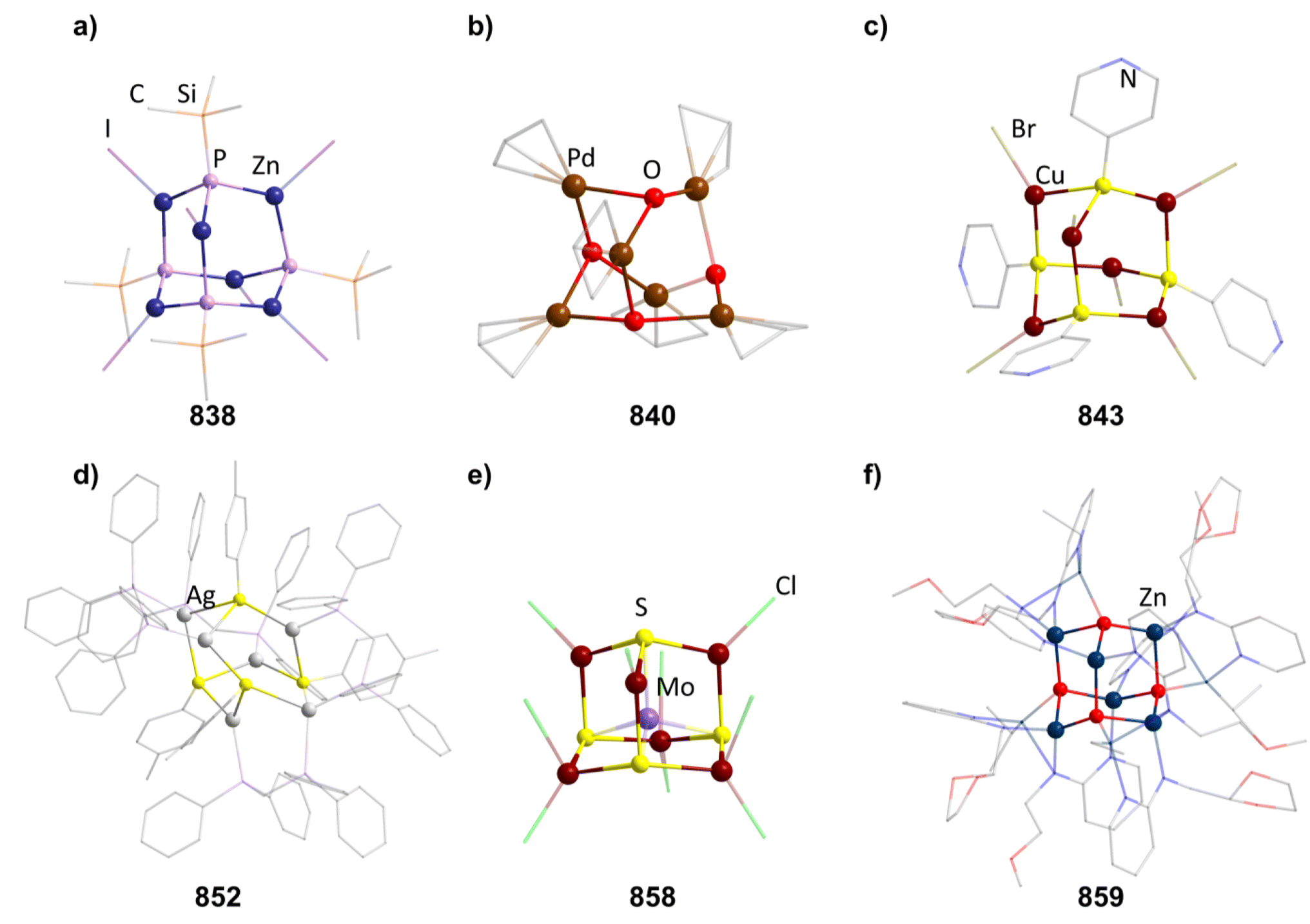 | ||
| Fig. 18 Examples of adamantane-type cluster with transition metal atoms in the E position: [nBu4N]2[(PSiMe3)4(ZnI)6(thf)2] (838, top left (a)), [(Pd{(η3-C3H5)}4(OH)6]CF3SO3 (840, top center (b)), [(4-SC5H4NH)4(CuBr)6] (843, top right (c)), [(SC6H4Me-p)4Ag6{(Ph2P)2Me}4][PF6]2 (852, bottom left (d)), [NMe4]5[(μ4-Mo)S4(CuCI)3(CuCI2)3] (858, bottom center(e)) and [O4(anpy)8Zn6(ZnEt)4] (859, bottom right (f)). Hydrogen atoms and counterions, if present, are omitted for clarity. | ||
Hydrolysis of a π-allyl Pd complex with an additional chelating and sterically hindered bispidine ligand leads to the formation of a cationic hydroxo cluster [(Pd{(η3-C3H5)}4(OH)6]2+ (in 840, Fig. 18).488
Simple addition reactions can be used to react a preformed Cu complex of a S2N2-tetradentate ligand with CuCl to form [Cu(bme*daco)}2(CuCl)4] (841, bme*daco = bis(N,N′-2-mercapto-2-methylpropyl)1,5-diazocyclooctane).489
In a redox reaction of CuIICl2 with KI and para-4-mercaptopyridine, a poylmeric CuI3I4 formed as the main product next to an adamantane-type cluster [(4-SC5H4NH)4(CuCl)6] (842) of neutral pyridine-4-thione and CuICl.490 The corresponding bromide compound (843; Fig. 18) could be obtained after cleaving the S–S bond in 4,4′-bipyridyldisulfide at higher temperatures and reacting with CuBr.491
Two isomers of [Cu6(phen)4(SPh)4Cl2] (844–845) with differing positions of the chlorine atoms in the cluster scaffold were found after a simple condensation of PhSH with CuOtBu under the addition of phenanthroline.492 The chlorine found in the compound is suspected to stem from decomposition of the solvent CH2Cl2.
[NEt3]X (X = Cl, Br) was found to break up polymeric [CuSPh]n to initiate a rearrangement to [(NEt4]4[(SPh)4(CuX)6] (846–847).170 This cluster could be prompted to reversibly invert its Q and E positions and form [Et4N]2[Cu4(SPh)6] (256) with an excess of [HNEt3][SPh], as described before.
Extreme levels of structural distortion are seen in compounds with cationic cluster molecules of the type [(ER)4M6{(Ph2P)2R}4]2+ (848–857, E = S, Se, M = Cu, Ag, Fig. 18), which are made by combination of dimeric complexes [M2{(Ph2P)2R}2(MeCN)2]2+ with phosphine ligand bridged metal centers rearranging around RE− units,493–495 or the reaction of polymeric [AgER]n with the phosphine ligand.496
In one case, an adamantane-type structure could be built around a central [MoS4] fragment by coordination of the tetrahedral [NMe4]2[MoS4] with CuCl to isolate crystals of [NMe4]5[(μ4-Mo)S4(CuCI)3(CuCI2)3] (858, Fig. 18).497
A [Zn10O4] oxo adamantane is found at the centre of [O4(anpy)8Zn6(ZnEt)4] (859, anpy = anilido-pyridinate, Fig. 18). It comprises four terminal ZnEt and six bridging Zn units, which are interconnected by eight bidentate organic ligands. It is obtained from the hydrolysis of ZnEt2 in the presence of the templating anilido-pyridinate.498
| Compound | Reagents/conditions | Method |
|---|---|---|
| a bme*daco = bis(N,N′-2-mercapto-2-methylpropyl)1,5-diazocyclooctane, bdpman = N,N′-bis(diphenylmethyl)-3,7-diazabicyclo[3.3.1]nonane. | ||
| [nBu4N]2[(PSiMe3)4(ZnI)6(thf)2] (838) | [ZnI2{P(SiMe3)3}]2, [nBu4N]2I/THF, 24 h | J486 |
| [nBu4P]2[(AsSiMe3)4(ZnI)6(thf)2] (839) | [ZnI2{As(SiMe3)3}]2, [nBu4P]2I/THF, 24 h | J487 |
| [(Pd{(η3-C3H5)}4(OH)6]CF3SO3 (840) | [(Bdpman)Pd(η3-C3H5)]CF3SO3/H2O, pentane | I488 |
| [{Cu(bme*daco)}2(CuCl)4] (841) | (bme*daco)Cu, CuCl/MeCN | K489 |
| [(4-SC5H4NH)4(CuCl)6] (842) | CuCl2 HS-4-C5H4N, KI/EtOH, 160 °C, 60 h | B490 |
| [(4-SC5H4NH)4(CuBr)6] (843) | CuBr, 4,4′-bipyridyldisulfide/EtOH, 120 °C, 3 days | B491 |
| [Cu6(phen)4(SPh)4Cl2] (844–845) | PhSH, phen, CuOtBu/THF, CH2Cl2, 18 h | C492 |
| [(NEt4]4[(SPh)4(CuX)6] (846–847, X = Cl, Br) | [CuSPh]n, [NEt3]X/DMF, 10 min | J170 |
| [(SePh)4Cu6{(Ph2P)2R}4][BF4]2 (848–849, RCH2, NH) |
HSePh, NEt3 [Cu2{(Ph2P)2R}2(MeCN)2][BF4]2/THF, Me2CO, 12 h | J493 |
| [(ER)4Ag6{(Ph2P)2Me}4][PF6]2 (850–853, ER = SPh, SC6H4Me-p, SePh, SeC6H4Cl-p) | NaER, [Ag2{(Ph2P)2Me}2(MeCN)2][PF6]2/CH2Cl2, 12 h | J494 |
| [(SC6H4(NH2)-m)4Ag6{(Ph2P)2NH}4][BF4]2 (854) | NaSC6H4(NH2)-m, [Ag2{(Ph2P)2MNH}2][BF4]2/MeCN, CH2Cl2 12 h | J495 |
| [(SC6H4Me-p)4Ag6{(Ph2P)2Me}4][PF6]2 (855) | [AgSC6H4Me-p]n, dppm, [NH4][PF6]/CH2Cl2, 4 h | J496 |
| [(SR)4Ag6{(Ph2P)2Me}4][ClO4]2 (856–857, RC6H4Me-p, 2-Np) |
[AgSR]n, dppm, LiClO4/CH2Cl2, 3 h | J496 |
| [NMe4]5[(μ4-Mo)S4(CuCI)3(CuCI2)3] (858) | [NMe4]2[MoS4], CuCl/MeCN, 1 h | C497 |
| [O4(anpy)8Zn6(ZnEt)4] (859) | ZnEt2, anpy,/H2O | I/K498 |
The earliest example of such a reported compound was the cage compound [S4(CH2)2(BH2)4] (860, Fig. 19), which is obtained by using THF-BH3 gas with the binary synthon and solvent CS2.499 Exchanging the borane for NaB3H8 leads to a slightly different reactivity, with only one intact CS2 unit in the product [S4(CH2)(BH2)5] (861).500
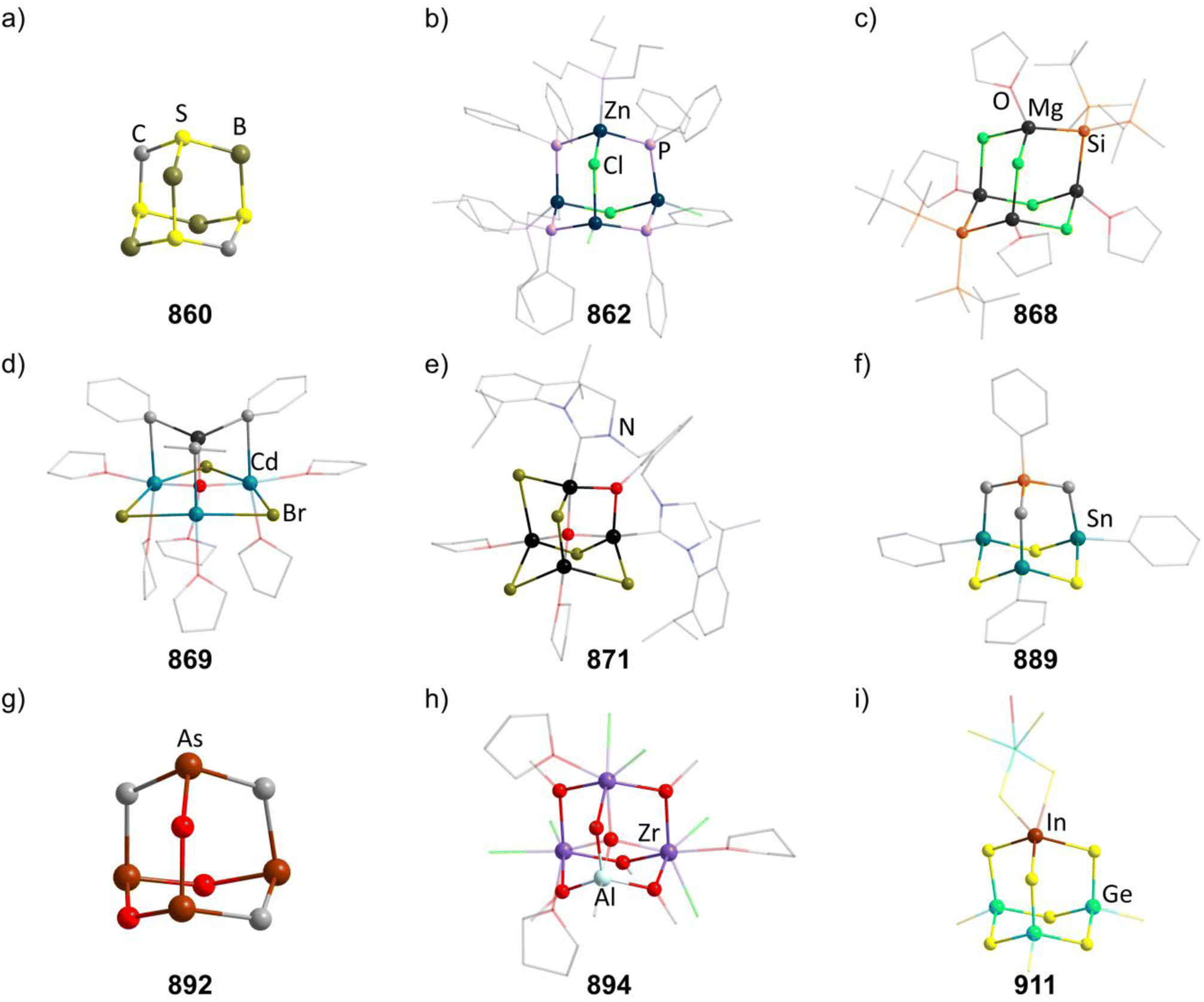 | ||
| Fig. 19 Examples of adamantane-type clusters with elements from different groups in equivalent positions.: [S4(CH2)2(BH2)4] (860, top left (a)), [(ZnCl)2(ZnPnPr3)2(PPh2)4Cl2] (862, top center (b)), [{(thf)Mg}4{Si(SiMe2tBu)2}2Cl4] (868, top right (c)), [μ4-O{(thf)2Ca}3MgPh3Br3] (869, middle left (d)), [{(1-C{NDippCH2CH2N})2(CH2)2PhO}Mg2(Mg(thf)}2Br4] (871, middle center (e)), [PhSi{CH2Sn(S)Ph}3] (889, middle right (f)), [As4(CH2)3O3] (892, bottom left (g)), [μ4-O(AlMe){(thf)Cl2Zr}3(OMe)6] (894, bottom center (h)) and [H3TAEA]2[InGe4S11(SH)2(OH)] (911, bottom right (i)). Hydrogen atoms and counterions, if present, are omitted for clarity. | ||
A mixture of Cl and P in the E position results from a stochiometric condensation reaction between four MCl2 (M = Zn, Cd) and four Ph2PSiMe3 molecules under elimination of ClSiMe3 in the presence of PnPr3 (nPr = normal propy). The resulting [(MCl)2(MPnPr3)2(PPh2)4Cl2] (862–863, Fig. 19) features two formally retained MCl2 fragments bridged by PPh2 units.501 The Zn compound was also synthesised with varying terminal phosphine ligands (864–866).502
A preformed complex dimer [(SiMe3)3PZnI)I]2 was observed to form the adamantane-type [nBu4N]2[(CdI)4{P(SiMe3)3}2I4] (867) after addition of [nBu4N]I, which also comprises mixed P and halide E sites, albeit with inverse ratios.486 This is formally achieved by a dimerization under elimination of two equivalents of (Me3Si)3PI.
A similar dimer with a four membered ring-structure [(thf)2Mg{Si(SiMe2tBu)2}]2 was rearranged under formal chlorination by tBuMgCl·2MgCl2 to form the adamantane-type dimer [{Mg(thf)}4{Si(SiMe2tBu)2}2Cl4] (868, Fig. 19).503
In the preparation of a calcium cuprate, using a CuPh precursor with residual MgBr2 from the Grignard reaction carried out in its synthesis can lead to a formal adduct of MgBr2 to the Ca complex, leading to [μ4-O{(thf)2Ca}3MgPh3Br3] (869, Fig. 19) with the central oxygen atom stemming from decomposition of THF.504 In this compound, three phenyl groups and three bromides occupy the E positions.
MgBr2 can also be used in a reaction with a tridentate carbene-ligand-stabilized adduct of lithium hexamethyldisilazide [{1-C(NDippCH2CH2N)}2(CH2)2PhOLi2N(SiMe3)2], leading to the substitution of the lithium azide with two MgBr fragments.505 As additional products, a symmetric and asymmetric adamantane-type cluster with endohedral μ4-O atoms were found. The symmetrical compound, [{(1-C{NDippCH2CH2N})2(CH2)2PhO}2Mg4Br4] (870), can be understood as a dimer of the carbene stabilized Mg complex, while the asymetrical example, [{(1-C{NDippCH2CH2N})2(CH2)2PhO}Mg2(Mg(thf)}2Br4] (871, Fig. 19), has lost one ligand and saturates the Mg moieties with THF.
In group 14/16 adamantane clusters, the group 16 elements in E position can be replaced by isoelectronic CR2 fragments. Corresponding compounds can be accessed from carbon-bridged fragments, which are connected by intermolecular or intramolecular condensation reactions with the desired group 16 precursor. For tin compounds, this was first shown for a series 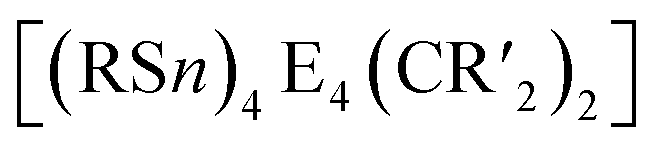 (872–876, R = Ph, CH2SiMe3, R′ = H, E = S; R = Ph, R′ = H, E = Se, Te; R = R′ = Me, E = Se), originating from an
(872–876, R = Ph, CH2SiMe3, R′ = H, E = S; R = Ph, R′ = H, E = Se, Te; R = R′ = Me, E = Se), originating from an 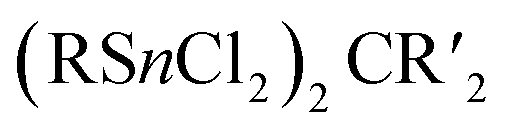 precursor reacted with Na2E or (tBu2SnE)2.506 Those compounds were also found to undergo exchange reactions, forming either a cluster with mixed organic ligands, [(PhSn)2(Me3SiCH2Sn)2S4(CH2)2] (877), by mixing 872 and 873 or clusters with mixed E sites, [(PhSn)4S4−xSex(CH2)2] (878–881) or [(PhSn)4Se4−xTex(CH2)2] (882–885), by mixing 872 with 874 or 874 with 875, respectively. Note that there are two possible isomers for the x = 2 case.
precursor reacted with Na2E or (tBu2SnE)2.506 Those compounds were also found to undergo exchange reactions, forming either a cluster with mixed organic ligands, [(PhSn)2(Me3SiCH2Sn)2S4(CH2)2] (877), by mixing 872 and 873 or clusters with mixed E sites, [(PhSn)4S4−xSex(CH2)2] (878–881) or [(PhSn)4Se4−xTex(CH2)2] (882–885), by mixing 872 with 874 or 874 with 875, respectively. Note that there are two possible isomers for the x = 2 case.
An analogous oxo-cluster, [{(Me3Si)2CH2Sn}4O4(CMe2)2] (886), was isolated after exposure of {(Me3Si)2CH2SnCl2}2CMe2 to a NaOH solution.507
When using a tetrameric precursor RSi(CH2SnPhX2)3 (X = Cl, I) in reactions with a chalcogenide source, the mixed-element clusters [MeSi{CH2Sn(E)Ph}3] (887–888, E = S, Se) and [PhSi{CH2Sn(E)Ph}3] (889–891, E = S, Se, Te; Fig. 19) were realized, with three instead of two E positions being occupied by CH2 and also mixed Si and Sn positions.508,509
Another reaction to mixed adamantane-type structures from preformed precursors is the synthesis of arsenicin A [As4(CH2)3O3] (892, Fig. 19) from the linear CH2(AsPhCH2AsPh3)2, which is isolated as a racemic mixture after treatment with HI to halogenate all the As positions and a subsequent hydrolysis with aqueous ammonia.510
A hydride cluster [(Cp*Ru)3H5] can coordinate the primary silane tBuSiH3 in a μ3-η2:η2:η2 mode under H2 elimination to form the compound [(Cp*Ru)3(tBuSi)H6] (893) with an adamantane-type scaffold.511 Hydrogen atoms can be abstracted to transform the multi center bonds into a simpler Ru–Si contact.
Another method to obtain such mixed adamantane-type structures is the substitution of one atom in an already synthesized cluster. One Zr atom in the previously described cluster [μ4-O{(thf)Cl2Zr}4(OMe)6] was treated with AlMe3 to incorporate a AlMe site in the Q position of the compound [μ4-O(AlMe){(thf)Cl2Zr}4(OMe)6] (894, Fig. 19).512
Multiple chalcogenolate clusters comprising transition metals of different groups, and in one study Ga, could be isolated. The earliest study achieves this for [Me4N][(MSPh)n(M′SPh)4−n(SPh)6] (895–899, M/M′ = Fe/Co, Fe/Zn, Fe/Cd, Co/Zn, Co/Cd) by exchange between the homometallic clusters, as has been described before in this review.148 Similar compounds [Me3NBn]2[(FeCl)3Cu(EiPr)6] (900–901, E = S, Se) and [nPr3N(CH2)6NnPr3][(FeBr)3Cu(SePh)6] (902) with Fe and Cu in the Q positions could also be obtained from a dimeric homometallic precursor complex [Me3NBn]2[(Fe2(EiPr)6] by addition of FeCl2 and CuCl or from a mixture of CuBr, Fe(OAc)2 and PhSeSiMe3, and under addition of [nPr3N(CH2)6NnPr3] counterions in the second case.513–515
As discussed for the Cu/Te cluster 254 before, there are examples for μ3-group 11 atoms located at the center of a M3Te3 six membered ring. In the following compounds, the Q position opposing this μ3 metal is occupied by an element from a different group. The first examples of this architecture are [(μ3-M)(CdPPh3)(MPPh3)3(TePh)3(μ3-TePh)3] (903–904, M = Cu, Ag), prepared from NaTePh, MCl and CdCl2 in the presence of PPh3.516 The Zn congener [(μ3-Cu)(ZnPiPr3)(CuPiPr3)3(TePh)3(μ3-TePh)3] (905) was later isolated by a more complex synthetic route starting from a tetranuclear cluster precursor [(PiPr3)3(CuTePh)4] which was reacted in a stepwise manner with ZnEt2, PiPr3 and PhTeSiMe3.517 The same motif could also be stabilized for compounds with the main group metal Ga, [μ3-Cu{Cu(PR3)3}3(GaMe)(EPh)6] (906–909, E = Se, R = Me, Et, Et2iPr; E = Te, R = Et).174 They are obtained after reacting the complexes [(PR3)5(CuEPh)6] with chalcogenidolates and a GaMe source.
A mixed W/Ti oxygen adamantane-type structure [(W(O)OiPr)2{Ti(OiPr)2}2(O)4(bdmap)2] (910, Hbdmap = 1,3-bis-(dimethylamino)-propan-2-ol) was obtained after a reaction of the complex [W(O)(OiPr)3(bdmap)] with Hbdmap and Ti(OiPr)4, followed by a hydrolysis in a H2O/iPrOH mixture.122
There are also examples of mixed adamantanes accessible directly from the elements and simple binary compounds if the correct additives and conditions are used. [H3TAEA]2[InGe4S11(SH)2(OH)] (911, TAEA = tris(2-aminoethyl)amine, Fig. 19) is obtained solvothermally from In(NO3) and GeO2.518 Its structure can be understood as a [Ge4S10]4− adamantane-type in which one GeS unit is substituted by an InS2GeOH(SH)2 fragment.
An example of two different transition metals in the E position is obtained when using a Ni complex instead of a copper complex in the reaction to give [Cu(bme*daco)}2(CuCl)4] (912), leading to the mixed derivative [Ni(bme*daco)}2(CuCl)4].489
| Compound | Reagents/conditions | Method |
|---|---|---|
| a Hbdmap = 1,3-bis-(dimethylamino)-propan-2-ol, nHep = normal heptane, TAEA = tris(2-aminoethyl)amine. | ||
| [S4(CH2)2(BH2)4] (860) | THF-BH3,/CS2, 50 °C, 3 h | G499 |
| [S4(CH2)(BH2)5] (861) | NaB3H8/CS2, 75 °C, 5 h | B500 |
| [(MCl)2(MPnPr3)2(PPh2)4Cl2] (862–863, M = Zn, Cd) | MCl2, PnPr3, Ph2PSiMe3/THF, 12 h | C501 |
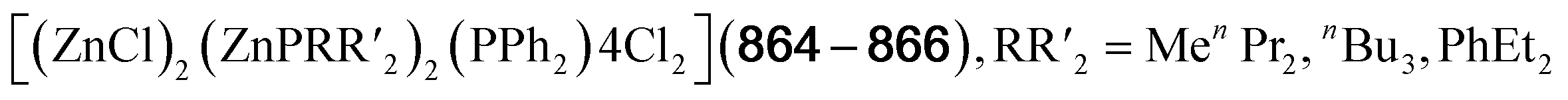
|
ZnCl2,  , Ph2PSiMe3/THF, 12 h , Ph2PSiMe3/THF, 12 h |
C502 |
| [nBu4N]2[(CdI)4{P(SiMe3)3}2I4] (867) | [CdI2{P(SiMe3)3}]2, [nBu4N]2I/THF, 24 h | J486 |
| [{(thf)Mg}4{Si(SiMe2tBu)2}2Cl4] (868) | [(thf)2Mg{Si(SiMe3tBu)2}]2, tBuMgCl·2 MgCl2/THF, C6H6, 0 °C | J503 |
| [μ4-O{(thf)2Ca}3MgPh3Br3] (869) | MgBr2, CuPh, Ca/THF, −78 °C to RT, 20 h | C504 |
| [{(1-C{NDippCH2CH2N})2(CH2)2PhO}2Mg4Br4] (870) | [{1-C(NDippCH2CH2N)}2(CH2)2PhOLi2N(SiMe3)2], MgBr2/THF, 12 h | K505 |
| [{(1-C{NDippCH2CH2N})2(CH2)2PhO}Mg2(Mg(thf)}2Br4] (871) | [{1-C(NDippCH2CH2N)}2(CH2)2PhOLi2N(SiMe3)2], MgBr2/THF, 12 h | K505 |
 (872–876, R = Ph, CH2SiMe3, R′ = H, E = S; R = ph, R′H, E = Se, Te; R = R′ = Me, E = Se) (872–876, R = Ph, CH2SiMe3, R′ = H, E = S; R = ph, R′H, E = Se, Te; R = R′ = Me, E = Se) |
 , Na2E/Me2CO, H2O, 0 °C to RT, 18 h , Na2E/Me2CO, H2O, 0 °C to RT, 18 h |
K506 |
| [(PhSn)2(Me3SiCH2Sn)2S4(CH2)2] (877) | [(PhSn)4S4(CH2)2] (872), [(Me3SiCH2Sn)4S4(CH2)2] (873)/CH2Cl2, 2 days | R506 |
| [(PhSn)4S4−xSex(CH2)2] (878–881) | [(PhSn)4S4(CH2)2] (872), [(PhSn)4Se4(CH2)2] (874)/CH2Cl2 | R506 |
| [(PhSn)4Se4−xTex(CH2)2] (882–885) | [(PhSn)4Se4(CH2)2] (874), [(PhSn)4Te4(CH2)2] (875)/CH2Cl2 | R506 |
| [{(Me3Si)2CH2Sn}4O4(CMe2)2] (886) | {(Me3Si)2CH2SnCl2}2CMe2, NaOH/H2O, PhMe, 80 °C, 18 h | K507 |
| [MeSi{CH2Sn(S)Ph}3] (887–888, E = S, Se) | PhSi(CH2SnPhI2)3, Na2S/Me2CO, MeOH, H2O | K508 |
| [PhSi{CH2Sn(S)Ph}3] (889–891, E = S, Se, Te) | PhSi(CH2SnPhCl2)3, (SiMe3)2E/toluene, 24 h | K509 |
| [As4(CH2)3O3] (892) | 1. CH2(AsPhCH2AsPh3)2, HI/CH2Cl2 | I510 |
| 2. NH3, H2O/THF | ||
| [(Cp*Ru)3(tBuSi)H6] (893) | [(Cp*Ru)3H5], tBuSiH3/hexane, 5 min | C511 |
| [μ4-O(AlMe){(thf)Cl2Zr}3(OMe)6] (894) | [μ4-O{(thf)Cl2Zr}4(OMe)6], AlMe3/THF, PhMe, 12 h | R512 |
| [Me4N][(MSPh)n(M′SPh)4−n(SPh)6] (895–899, M/M′ = Fe/Co, Fe/Zn, Fe/Cd, Co/Zn, Co/Cd) | [Me4N][(MSPh)4(SPh)6], [Me4N][(M′SPh)4 (SPh)6]/MeCN | C148 |
| [Me3NBn]2[(FeCl)3Cu(SiPr)6] (900) | [Me3NBn]2[(Fe2(SiPr)6], FeCl2, CuCl/THF, 2 days, 70 °C | K513 |
| [Me3NBn]2[(FeCl)3Cu(SeiPr)6] (901) | [Me3NBn]2[(Fe2(SeiPr)6], FeCl2, CuCl/MeCN, 1 day, 70 °C | J514 |
| [nPr3N(CH2)6NnPr3][(FeBr)3Cu(SePh)6] (902) | CuBr, Fe(OAc)2, PhSeSiMe3, [nPr3N(CH2)6NnPr3]Br2/MeCN | C515 |
| [(μ3-Cu)(CdPPh3)(CuPPh3)3(TePh)3(μ3-TePh)3] (903) | NaTePh, CuCl, CdCl2, PPh3/THF, 2 h | C516 |
| [(μ3-Ag)(CdPPh3)(AgPPh3)3(TePh)3(μ3-TePh)3] (904) | NaTePh, AgCl, CdCl2, PPh3/THF, 3 h | C516 |
| [(μ3-Cu)(ZnPiPr3)(CuPiPr3)3(TePh)3(μ3-TePh)3] (905) | [(PiPr3)3(CuTePh)4], ZnEt2, PiPr3, PhTeSiMe3/nHep, EtOH, 2 h, 0 °C to RT | C517 |
| [(μ3-Cu){Cu(PR3)3}3(GaMe)(SePh)3(μ3-SePh)3] ((906–907, R = Me, Et2iPr) | 1. CuOAc, PR3, Me3SiSePh/THF | J174 |
| 2. Me3SiSePh, [Me2GaSePh]n/THF | ||
| [(μ3-Cu){Cu(PEt3)3}3(GaMe)(SePh)3(μ3-SePh)3] (908) | [(PEt3)5(CuSePh)6], Me3SiSePh, [Me2GaSePh]n/THF | J174 |
| [(μ3-Cu){Cu(PEt3)3}3(GaMe)(TePh)3(μ3-TePh)3] (909) | [(PEt3)5(CuTePh)6], Me3SiTePh, Me3Ga·OEt2/THF | J174 |
| [(W(O)OiPr)2{Ti(OiPr)2}2(O)4(bdmap)2] (910) | [W(O)(OiPr)3(bdmap)], Hbdmap, Ti(OiPr)4/PhMe, H2O, HOiPr, 2 days, 110 °C to 0 °C | C/I122 |
| [H3TAEA]2[InGe4S11(SH)2(OH)] (911) | In(NO3), GeO2, TAEA/(CH2OH)2, nBuNH2, (CH2SH)2, 170 °C, 5 days | B518 |
| [{Ni(bme*daco)}2(CuCl)4] (912) | (bme*daco)Ni, CuCl/MeCN | K489 |
A formal addition of a Cu(PR3)2 unit to a [μ3-Cu(CuPR3)3Cu(EPh)6] (E = Se, Te) core, a structural motif observed in the previously discussed compound 261,175 leads to the neutral clusters [μ3-Cu(CuPR3)3{Cu(PR3)2}Cu(EPh)6] (913–915).519,520 The synthesis does not deviate much from the one for the anionic cluster. As in all cases, a Cu salt is reacted with PR3 and PhESiMe3, with the resulting compound depending only on the exact chalcogenide or PR3 used.
A related compound featuring silver atoms [Ag4{Ag(PEt3)2}2(TenBu)6] (916, Fig. 20) could be isolated using an analogous route.521 Here, both additional Ag(PEt3)2 units coordinate on the outside of the cluster, bridging two Te atoms each.
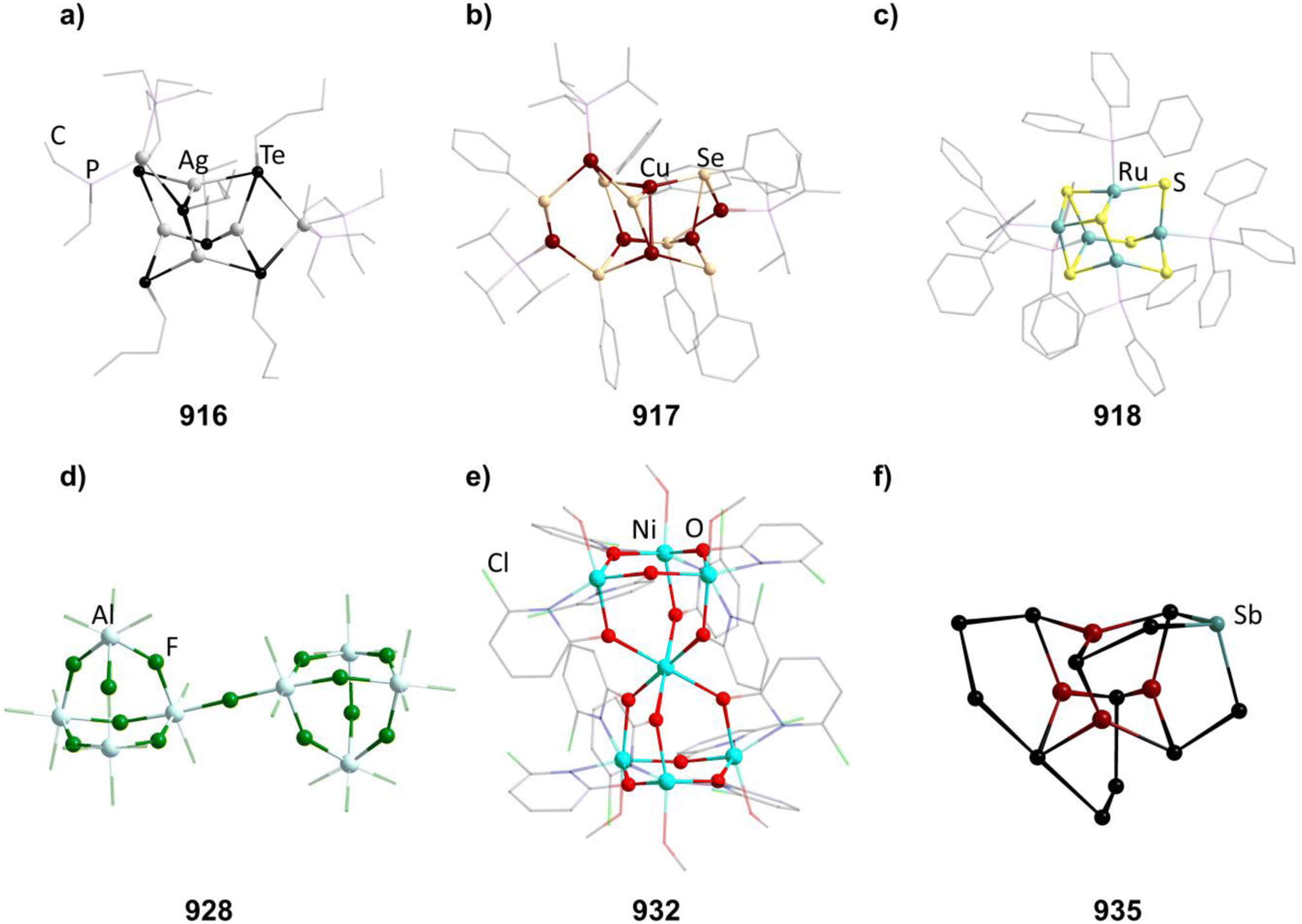 | ||
| Fig. 20 Examples of compounds with an extended adamantane-type structure: [Ag4{Ag(PEt3)2}2(TenBu)6] (916, top left (a)), [Cu4(CuPiPr3)3(SePh)7] (917, top center (b)), [μ3-(RuPPh3)(RuPPh3)4S6] (918, top right (c)), [H3tren]4[(Al4F17)2F]OH (928, bottom left (d)), [Ni{Ni(chp)2MeOH}6]Cl2 (932, bottom center (e)) and [Et4N]3[Cu4Sb(Te7)(Te2)2Te] (935, bottom right (f)). Hydrogen atoms and counterions, if present, are omitted for clarity. | ||
[Cu4(CuPiPr3)3(SePh)7] (917, Fig. 20) is an example of a larger expansion of a [Cu4(SePh)6] central adamantane. In this case, by addition of a μ-CuPiPr3 connecting two selenium atoms of the central scaffold and an additional μ3-(CuPiPr3)2SePh bridge between three other selenolates, an increase of the coordination number of all but one selenium atoms to 4 is achieved.520
Another structural motif of expanded adamantanes is achieved by formally capping one face of the octahedron formed by the six E atoms with an additional metal fragment. In contrast to some other molecules, we have discussed featuring μ3 Cu or Ag atoms in the center of a six membered ring of the adamantane-type scaffold, these metal moieties carry additional ligands and are located below the plane of the Q3E3-ring, which causes a greater deviation from planarity as opposed to a more planar arrangement when compared to an uncoordinated adamantane-type structure. This effect can be observed in [μ3-(RuPPh3)(RuPPh3)4S6] (918, Fig. 20) when compared to the non-coordinated 235 discussed beforehand.152 The extended compound is obtained by reacting S(SiMe3)2 with [Ru(PPh3)3Cl2] in hot THF, as opposed to using NaSH as a sulfur precursor, which leads to less oxidized metal centers.
This architecture has also been explored for two clusters [μ3-(FeCl)(VPEt3)(FePEt3)3S6] (919–920, M = V, Mo) capped by a FeCl unit.522,523 They are also obtained by using S(SiMe3)2 and a mixture of the metal complexes [M(thf)3Cl3] and [Fe(PEt3)2Cl2]. Sodium thiolates can be used to replace the chlorine atom at the added site by a SR group (921–925).
When NaS2 is used instead, the cluster will dimerize to [μ3-{(VPEt3)(FePEt3)3FeS6}2S] (926), comprising two adamantane-type cages connected by a μ-S bridge.524 A more distorted example of this dimer buildup is [μ3-(HgSPh)(AgPPh3)3Hg(SPh)6}2S] (927), in which the metal atoms on both sites of an Ag3S3 ring are Hg atoms.525 This is achieved by forming NaSPh in situ and reacting it with HgO and [Ag(PPh3)2]NO3.
This μ-bridged adamantane topology is also present in two further examples. One is [H3tren]4[(Al4F17)2F]OH (928, tren = tris(2-ethylamino)amine, Fig. 20), in which two [Al4F18] clusters are condensed by a μ-F.484 It is observed when adjusting the compound ratios in the synthesis of monomeric 836 (see section 2.1.6). The other is [{P4(NMe)6}2CuCl]2 (929) which was isolated as a side product when reacting an excess of [P4(NMe)6] with CuCl besides further polymeric products, that will be discussed in the next section.526
A different type of dimer could first be observed in the compounds [H2Ta(tdci)2]CI3 (tdci = 1,3,5-trideoxy-1,2,5-tris(dimethylamino)-cis-inositol, Hchp = 6-chloro-2-hydroxypyridine, 930) and [H11Ta7O12(tdci)6] (931), in which two adamantane-type clusters are condensed by one atom in the Q position.527 The first is an organometallic compound, in which a central Ta is trigonal prismatically coordinated by six oxygen atoms and the adamantane-type scaffolds are completed by hydrocarbons. This is achieved by coordinating tdci to TaCl5 in methanol. The second compound is obtained after hydrolysis of the first, and features two condensed Ta4O6 subunits decorated by tdci ligands on the three non-condensed Ta sites, which in turn resemble an organometallic adamantane-type structure. Thus, this compound could also be described as comprising 8 condensed adamantane-type scaffolds.
A further compound with the same dimer architechture is [Ni{Ni(chp)2MeOH}6]Cl2 (932, Hchp = 6-chloro-2-hydroxypyridine, Fig. 20), made at 130 °C under inert conditions by addition of Ni(OH)2 and Hchp.528 This compound is notable due to the fact that there are no monomeric group 10/16 adamantane-type structures at all.
Apart from examples with oxygen, there is an Al/F dimer [(C2H4NH3)3NH]2·(H3O)·[Al7F30] (933), formally made up of [Al4F18] clusters condensed by an Al site.529 It is obtained by solvothermal conversion from Al2O3 with HF.
By formally condensing two adamantanes at a face between a Q and two connected E atoms instead of just by one Q atom, a new structural motif is achievable. This was realized for [{(SiMe)3(CH2)4}2Si(CH)2] (934), which is formed by two [(SiMe)4(CH2)6] molecules condensed via one face.530,531 The presence of this compound was confirmed after heating SiMe4 at 700 °C.
A clear adamantane-type cluster Cu4Te6 core is also present in the cluster [Et4N]3[Cu4Sb(Te7)(Te2)2Te] (935, Fig. 20). However, the Te sites are mostly part of oligotellurides.532 One Cu atom coordinates to three sites of a linear Te7, all of which also coordinate to the three other Cu atoms which form the typical six membered ring opposed to the first copper together with a single Te and two Te2 units. Lastly these three Te fragments coordinate a Sb atom below the six membered ring. It was obtained by the extraction of the alloy KCuSbTe3, prepared from K2Te, Cu, Sb2Te3 and Te with ethane-1,2-diamine.
| Compound | Reagents/conditions | Method |
|---|---|---|
| a tdci = 1,3,5-trideoxy-1,2,5-tris(dimethylamino)-cis-inositol, Hchp = 6-chloro-2-hydroxypyridine. | ||
| [μ3-Cu(CuPEtPh2)3{Cu(PEtPh2)2}Cu(TePh)6] (913) | CuCl, PEtPh2, Te(Ph)SiMe3/THF:Et2O, RT | C519 |
| [μ3-Cu(CuPEt3)3{Cu(PEt3)2}Cu(SePh)6] (914) | CuOAc, Et3P, PhSeSiMe3/toluene, RT, 12 h | C520 |
| [μ3-Cu(CuPEt3)3{Cu(PEt3)2}Cu(TePh)6] (915) | CuOAc, Et3P, PhTeSiMe3/Et2O, 0 °C, 2 h | C520 |
| [Ag4{Ag(PEt3)2}2(TenBu)6] (916) | n BuTeSiMe3, AgCl–PEt3/pentane, −40 °C | C521 |
| [Cu4(CuPiPr3)3(SePh)7] (917) | CuOAc, iPr3P, PhSeSiMe3/THF, RT, 1 h | C520 |
| [μ3-(RuPPh3)(RuPPh3)4S6] (918) | [Ru(PPH3)3Cl2], S(SiMe3)2/MeCN, 85 °C, 6.5 h | C152 |
| [μ3-(FeCl)(MoPEt3)(FePEt3)3S6] (919) | [Mo(thf)3Cl3], S(SiMe3)2, [Fe(PEt3)2Cl2]/THF, 50 °C, 4 h | D522,523 |
| [μ3-(FeCl)(VPEt3)(FePEt3)3S6] (920) | [V(thf)3Cl3], S(SiMe3)2, [Fe(PEt3)2Cl2]/THF, RT | D522 |
| [μ3-(FeSPh)(VPEt3)(FePEt3)3S6] (921) | [μ3-(FeCl)(VPEt3)(FePEt3)3S6] (920), NaSPh/THF, MeCN, RT | Q523 |
| [μ3-(FeSPh)(MoPEt3)(FePEt3)3S6] (922) | [μ3-(FeCl)(MoPEt3)(FePEt3)3S6] (919), NaSPh/THF, MeCN, RT, 30 min | Q523 |
| [μ3-(FeSEt)(VPEt3)(FePEt3)3S6] (923) | [μ3-(FeCl)(VPEt3)(FePEt3)3S6] (920), NaSEt/THF, MeCN, RT | Q523 |
| [μ3-(FeSEt)(MoPEt3)(FePEt3)3S6] (924) | [μ3-(FeCl)(MoPEt3)(FePEt3)3S6] (919), NaSEt/THF, MeCN, RT, 30 min | Q523 |
| [μ3-(FeS-p-C6H4OMe)(MoPEt3)(FePEt3)3S6] (925) | [μ3-(FeCl)(MoPEt3)(FePEt3)3S6] (919), NaS-p-C6H4OMe/THF, MeCN, RT, 30 min | Q523 |
| [μ3-(VPEt3)(FePEt3)3FeS6}2S] (926) | [μ3-(FeCl)(VPEt3)(FePEt3)3S6] (920), Li2S/MeCN, RT, overnight | Q524 |
| [μ3-(HgSPh)(AgPPh3)3Hg(SPh)6}2S] (927) | Na, HgO, PhSH, [Ag(PPh3)2]NO3/MeOH, CHCl3, 3 h | C525 |
| [H3tren]4[(Al4F17)2F]OH (928) | Al(OH)3, tren, HF/EtOH, 190°C microwave heating, 1 h |
B484 |
| [{P4(NMe)6}2CuCl]2 (929) | [P4(NMe)6], CuCl/MeCN, 2 days | T526 |
| [H2Ta(tdci)2]CI3 (930) | TaCl5, tdci/MeOH | J527 |
| [H11Ta7O12(tdci)6] (931) | [H2Ta(tdci)2]CI3/H2O | I527 |
| [Ni{Ni(chp)2MeOH}6]Cl2 (932) | Ni(OH)2, Hchp/130 °C | C528 |
| {[C2H4NH3)3NH]}2·(H3O)·[Al7F30] (933) | Al2O3, HF, tris(2-aminoethyl)amine/EtOH, 200 °C, 96 h | B529 |
| [Si7C16H36] (934) | SiMe4/700 °C | A530,531 |
| [Et4N]3[Cu4Sb(Te7)(Te2)2Te] (935) | 1. K2Te, Cu, Sb2Te3, Te/heat to melt | E532 |
| 2. [Et4N]Br/en | ||
Linking of the previously discussed P/N adamantane-type structures [P4(NR)6] (936–938, R = Me, Et, Bn, Fig. 21) can be achieved by the addition CuI to form one dimensional chains of [{P4(NR)6}CuI]n (R = Me, Et) with μ-bridging CuI moieties or [{P4(NMe)6}(CuI)2(MeCN)2]n comprising linking CuI and MeCN four membered rings.81,533 Similarly, reactions of [P4(NMe)6] with CuCl lead to a three dimensional network [{P4(NMe)6}2(CuCl)3(MeCN)2] (939) or a ladder like one dimensional polymer [{P4(NMe)6}(CuCl)2]n (940) depending on the cluster to CuCl ratio.526 Ligands on [(PNSiMe3)4(NMe)6] can be exchanged for TiCl3 or p-nBuPhPCl2, which polymerize to form extended networks that could not yet be structurally characterized (941–942).95
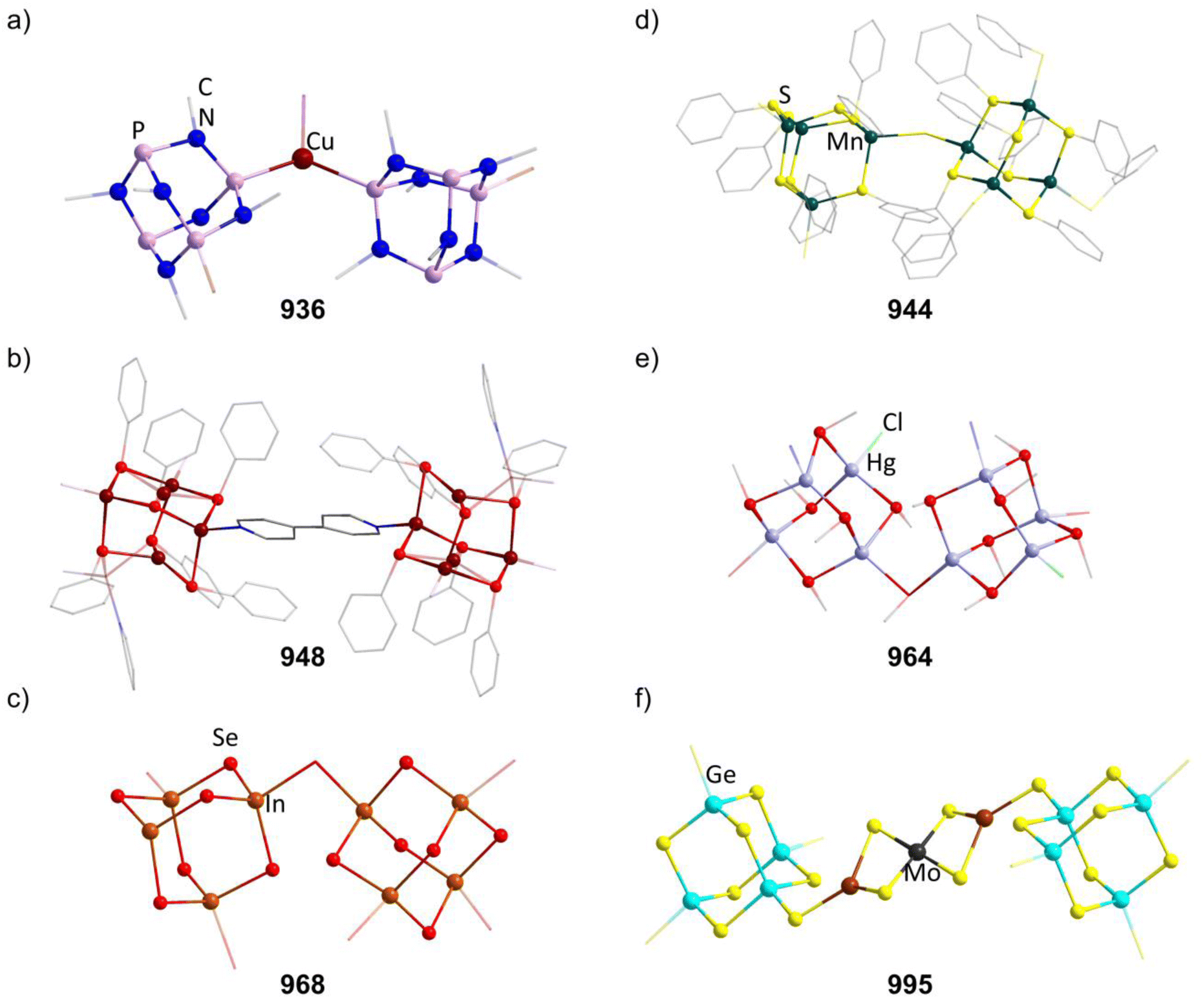 | ||
| Fig. 21 Examples of networks of type 2 supertetrahedra: [{P4(NMe)6}CuI]n (936, top left), [Mn4(SPh)8]n (944, top right (a)), [(μ3-Cu)Cu4(SePh)6(CuPPh3)3(4,4′-bipy)]n (948, middle left (b)), [Hg4(PhSe)7ClPy]n (964, middle right (c)), (C5H5NH2)24[In28Se54(H2O)4] (968, bottom left (d)) and {[Me4N]4[(Ge4S10)Cu4Mo2S8]}n (995, bottom right (f)). Hydrogen atoms and counterions, if present, are omitted for clarity. | ||
A previously discussed Cr/O adamantane-type cluster with hpdta ligands can be obtained as Ba linked chains in [Ba(OH2)5{Cr4(OH)4(hpdta)2}]n (943) by adding BaCl2 to the initial reaction mixture without ethane-1,2-diamine.119 This leads to two parallel cluster strands connected via interactions between Ba ions and the organic ligand.
The only known group 7 example is the thiolate network [Mn4(SPh)8]n (944, Fig. 21), in which all metal centers coordinate to the next cluster via a bridging thiolate, a composition often observed for transition metal chalcogenolates.534 It is isolated after a reaction of [Mn{N(SiMe3)2}2]2 with HSPh in THF at low temperatures.
A layered network of [{Cu4{SC(NH2)2}6}2{SC(NH2)2}3]n(SO4)4 (945) can be observed from dissolving Cu2SO4 in sulfuric acid in the presence of thiourea.187 Only three copper atoms per cluster carry a thiourea ligand forming the cluster sheets, while the last one only forms bonds within the adamantane-type scaffold.
Utilizing a multidentate thiolate ligand 4,5-dimercapto-1,3-dithiole-2-thionato (dmit) in a reaction with [Cu(MeCN)4][ClO4] an ammonium or pyridinium counterion form the dimeric anion in [{Cu4(dmit)3}2]n2− (in 946–947), which is made up of layers facilitated by further Cu–S and S–S interactions.
The structural motif of a μ3-Cu coordinating a six membered ring in an adamantane-type structure has been discussed several times before. Such a motif can also be found in a one dimensional zigzag chain polymer [(μ3-Cu)Cu4(SePh)6(CuPPh3)3(4,4′-bipy)]n (948, Fig. 21), in which such adamantanes are connected by 4,4′-bipy(CuPPh3)2 units to two Se atoms in the E position and another 4,4′-bipy terminally connected to an adamantane Cu moity.535 It forms from Cu(MeCOO), PhSeSiMe3, PPh3 and 4,4′-bipy.
Another dmit linked layered cluster exists in {[Et4N][(Ag4(dmit)3]dmf}n (949), in which a porous architecture filled with both cations and solvents is formed.
Linear chains of [Zn4(SPh)6] adamantane-type clusters are found in a series of compounds [(Zn4(SPh)8ROH]n (950–953, R = Me, Et, nPr, nBu) obtained from ZnCO3 and HSPh reacted in alcoholic solutions.536 Two opposing Zn atoms carry SPh ligands bridging to the next cluster to form the polymer, while the others carry a terminal alcohol or SPh ligand respectively which form hydrogen bonds to extend the structure to loose layers. There are a couple of related one dimensional adamantane-type clusters connected via organic ligands [(Zn4(SPh)8L]n (954–955, L = trans-1,2-bis(4-pyridyl)ethylene, 4,4′-bipy).537 The first one forms a zigzag chain, while the second exhibits a helical buildup. They both are obtained from solvothermal reactions in water with HSPh, Zn(MeCOO)2 and the appropriate ligand.
In the cadmium thiolate cluster network [(Cd4(SPh)8]n (956), all Cd moieties carry bridging thiolate ligands to form a three dimensional architechture with helical arrangement of the adamantane-type clusters similar to that in cristobalite.538 This is obtained by reacting Ca(NO3)2 with HSPh and NEt3 in ethanol. An alternative reaction strategy is the poylmerisation of 447 in THF/MeCN at elevated temperatures.232 The same structural motif, albeit in a different crystallographic space group, is found for the para-fluorinated species [(Cd4(SC6H4F-4)8]n (957).539
When exchanging the fluorine with Br or Me groups, the structure of the product varies significantly. The methylated species also forms a three-dimensional network, but arranges in cyclic groups of 4, 6 or 8 clusters, resulting in a porous zeolite like buildup. The bromide congener forms a layered structure [{Cd6(SC6H4Br-4)15}(CdSC6H4Br-4){Cd(dmf)3}]n (958) and incorporates solvent molecules in its buildup. This leads to two distinct clusters, each with three cadmium atoms linking to the next cluster via briding thiolate units, but also each with one terminally coordinated metal center, either by a thiolate or three dmf molecules. The chlorinated species has been synthesized as well, but could not be elucidated crystallographically due to a fast decomposition of the crystals.
956 can be partially decomposed to chains of [(Cd4(SPh)8PPh3]n (959) by the addition of PPh3.232
The higher homolog [(Cd4(SePh)8]n (960) is isostructural to its thiolate compound and prepared in the same manner by creating the selenolate and reacting with a Cd salt, CdCl2 in this case.540
Using two Cd sources, (PhSe)2Cd and CdX (Cl, Br), and a coordinating ligand, PR3, to stabilize intermediates leads to one dimensional chains of [Cd4(PhSe)7X(PR3)]n (961–962), where two Cd atoms in each cluster connect to the next via PhSe bridges and the others carry a PPh3 or X ligand.541,542
Four isostructural Hg species [Hg4(PhSe)7(X)solv]n (963–966, Fig. 21) with different (pseudo)halides (X) and solvent ligands were obtained by changing to the appropriate salt, solvent and stabilizing ligand.543–545 This chemistry could also be translated to a Te congener [Hg4(PhTe)7IPy]n (967), although in this case, (PhTe)2Hg was used alongside CdI2 instead of the mercury halide, which did not lead to the inclusion of Cd into the final compound.546
For the group 13/16 combination, some In/Se networks are known. (C5H5NH2)24[In28Se54(H2O)4] (968, Fig. 21), formed by the elements and piperidine in aqueous solution through Method B, features a three dimensional structure of corner condensed adamantane-type clusters with some indium sites coordinated by water molecules.547 These positions can be partially substituted by Bi atoms when adding Bi(NO3)3 to the reaction mixture, leading to a doped structure (969). Other linking modes are obtained for the three dimensional network [μ3-Se4]3.27[In49.88Se95.92](C5H12N)26.0·(C2H8N)42.4 (970), in which [In4Se10] clusters are linked by μ3 Se and [InSe4] fragments, a structure obtained from the elements in piperidine solvothermally.548 Adding 1,4-dioxane and 3,5-dimethylpyridine to the mixture changes the outcome to [In4Se10]·(C7H16N)1.8·(C2H8N)2.2 (971), comprising μ-Se3 linkers between the individual clusters.
In group 14/16 adamantane-type clusters, extended structures are produced mainly by adding transition metal complexes to ammonium or alkaline metal salts of [Q4E10] clusters (972–994).260,263,272,275,281,549–552 By utilizing two different transition metal complexes during the synthesis, a more complex Cu2MS6 (M = Mo, W) linker between Ge/S adamantane-type compounds was obtained to form the MOF-like {[Me4N]4[(Ge4S10)Cu4M2S8]}n (995–996, Fig. 21).553 The concept could also be used to introduce another group 14 element, here tin, to Ge/S adamantanes by adding SnCl2 to K+ or Cs+ salts of [Ge4S10]4− in ionic liquids, with the exact outcome dependent on the ionic liquid used (997–999).554,555 In one case, {[BMIm]2[Ge4Se9]}n (997), this approach did not lead to the incorporation of Sn into the structure.554
In another case, utilizing the functional ligand in 579, manganese complexes were used to form a coordination polymer forming a layered structure [Mn2{(OOCC2H4Ge)4S6}(MeOH)(dmf)2]n (1000) by using the transition metal as a linker between the acid moieties.556
Most other transition metal linked adamantanes are isolated after reactions of simple binary or elemental precursors in solution (1001–1005)549,557–560 or, in one case, the solid state,561 which results in clusters linked by disordered Cu0.44Ge0.56S4 sites (in 1006).
Two examples also showcase the possibility of creating manganese linked adamantane-type structures by Method E, the extraction of a solid created from a melt of simple precursors (1007–1008).275,282 This led to the only example of a tellurium adamantane in network structures (1007).
Pure group 14/16 structures can also be obtained, one of them containing the same polymeric chain [Ge4Se9]2− previously discussed as the surprising outcome of a reaction of a Ge/S adamantane with SnCl2. In this case, the compound {[Pr2NH2][PrEtNH2][Ge4S9]}n (1009) could be isolated from a solvothermal reaction of GeS2 and [Pr2NH2]Cl in the presence of NaHCO3.562
The other example {[Me4N]2[OSn5Se10]}n (1010) consists of a corner condensed oxygen centered [μ4-OSn4Se10] adamantane-type structure synthesized solvothermally from the elements and [Me4N]OH.278
Aside from ionic or ligand decorated networks, partial acidic decomposition of 497 led to a novel modification of GeS2, δ-GeS2 (1011), with corner condensed Ge4S10 adamantanes, which can be derived from two interpenetrating cristobalite-like structures of γ-GeS2.266
| Compound | Reagents/conditions | Method |
|---|---|---|
| a dmit = 4,5-dimercapto-1,3-dithiole-2-thionato, EDTA = ethylenediamine-tetraacetate, BMMIm = 1-butyl-2,3-dimethyl-imidazolium. | ||
| [{P4(NMe)6}CuI]n (936) | [P4(NMe)6] (131), CuI/MeCN, 2 days | T533 |
| [{P4(NEt)6}CuI]n (937) | [P4(NEt)6] (132), CuI/MeCN, 2 days | T81 |
| [{P4(NMe)6}(CuI)2(MeCN)2]n (938) | [P4(NBn)6] (133), CuI/MeCN, 3 days | T81 |
| [{P4(NMe)6}2(CuCl)3(MeCN)2]n (939) | [P4(NMe)6] (131), CuCl/MeCN, 90 min | T526 |
| [{P4(NMe)6}(CuCl)2]n (940) | [P4(NMe)6] (131), CuCl/MeCN, 2 days | T526 |
| [{PN(p-nBuPhP)0.5}4(NMe)6]n (941) | [(PNSiMe3)4(NMe)6] (148), p-nBuPhPCl2/THF, 90 °C, 5 days | T95 |
| [{PN(TiCl2)0.5}4(NMe)6]n (942) | [(PNSiMe3)4(NMe)6] (148), TiCl4/MeCN, 100 °C, 4 days | T95 |
| [Ba(H2O)5][Cr4(OH)4(hpdta)2] (943) | H5hpdta, CrCl3, BaCl2/H2O, 85 °C, 20 h | J119 |
| [Mn4(SPh)8]n (944) | [Mn{N(SiMe3)2}2]2, HSPh/THF, 0 °C, 2 h | C534 |
| [{Cu4{SC(NH2)2}6}2{SC(NH2)2}3]n(SO4)4 (945) | CuSO4, SC(NH2)2, H2SO4/H2O, 80 °C | C187 |
| [N-methylpyridinium]2[{Cu4(dmit)3}2]n (946) | [Cu(MeCN)4][ClO4], Na2dmit, [N-methylpyridinium]I/MeOH | C563 |
| [nBu4N]2[{Cu4(dmit)3}2]n (947) | [Cu(MeCN)4][ClO4], Na2dmit, [nBu4N]Br/MeOH | C563 |
| [(μ3-Cu)Cu4(SePh)6(CuPPh3)3(4,4′-bipy)]n (948) | Cu(MeCOO), PhSeSiMe3, PPh3, 4,4′-bipy/DME, 4 h | C535 |
| {[Et4N][(Ag4(dmit)3]dmf}n (949) | AgNO3, H2dmit, Na, [Et4N]Br, NH3/MeOH, DMF | C564 |
| [(Zn4(SPh)8MeOH]n (950) | HSPh, ZnCO3/MeOH, 55 °C, 5 days | C536 |
| [(Zn4(SPh)8EtOH]n (951) | HSPh, ZnCO3/EtOH, 78 °C, 2 h | C536 |
| [(Zn4(SPh)8nPrOH]n (952) | HSPh, ZnCO3/MeOH, nPrOH, 10 to 97 °C, 5 h | C536 |
| [(Zn4(SPh)8nBuOH]n (953) | HSPh, ZnCO3/MeOH, nBuOH, 10 to 117 °C, 5 h | C536 |
| [(Zn4(SPh)8(trans-1,2-bis(4-pyridyl)ethylene)]n (954) | HSPh, Zn(MeCOO)2, trans-1,2-bis(4-pyridyl)ethylene/H2O, 165 °C, 5 days | B537 |
| [(Zn4(SPh)8(4,4′-bipy)]n (955) | HSPh, Zn(MeCOO)2, 4,4′-bipy/H2O, 165 °C, 5 days | B537 |
| [(Cd4(SPh)8]n (956) | Cd(NO3)2, HSPh, NEt3/EtOH, DMF or [Me4N]2[(CdSPh)3(CdCl)(SPh)6] (447)/MeCN, H2O, 100 °C | C/T232,538 |
| [(Cd4(SC6H4F-4)8]n (957) | Cd(NO3)2, HSC6H4F-4, NEt3/EtOH, DMF | C539 |
| [{Cd6(SC6H4Br-4)15}(CdSC6H4Br-4){Cd(dmf)3}]n (958) | Cd(NO3)2, HSC6H4Br-4, NEt3/EtOH, DMF | C539 |
| [(Cd4(SPh)8PPh3]n (959) | [(Cd4(SPh)8]n (939), PPh3/THF, DMF | I232 |
| [(Cd4(SePh)8]n (960) | CdCl2, HSePh, NaOH/MeOH, H2O | C540 |
| [Cd4(PhSe)7X(PPh3)]n (961–962, X = Cl, Br) | (PhSe)2Cd, CdX2, PPh3/MeOH, 130 °C, 1 h | B541,542 |
| [Hg4(PhSe)7BrPy]n (963) | (PhSe)2Hg, HgBr2, 1,3-bis(4-nitrophenyl)triazene/THF, Py, 100 min | C543 |
| [Hg4(PhSe)7ClPy]n (964) | (PhSe)2Hg, HgCl2, PPh3/THF, Py, 5 h | C544 |
| [Hg4(PhSe)7Br(dmf)]n (965) | (PhSe)2Hg, HgI2, 4,4′-bipy/DMF, 5 h | C544 |
| [Hg4(PhSe)7(SCN)Py]n (966) | (PhSe)2Hg, Hg(SCN)2,/MeOH, THF, Py, 1 h | C545 |
| [Hg4(PhTe)7IPy]n (967) | (PhTe)2Hg, CdI2/THF, Py, 90 min | C546 |
| (C5H5NH2)24[In28Se54(H2O)4] (968) | In, Se, piperidine/H2O, 170 °C, 7 days | B547 |
| (C5H5NH2)24[In28−xBixSe54(H2O)4] (969) | In, Se, Bi(NO3)3, piperidine/H2O, 170 °C, 7 days | B547 |
| [μ3-Se4]3.27[In49.88Se95.92](C5H12N)26.0·(C2H8N)42.4 (970) | Se, In, piperidine/DMF, 170 °C, 5 days | B548 |
| [In4Se10](C7H16N)1.8 (C2H8N)2.2 (971) | Se, In, 1,4-dioxane, 3,5-dimethylpyridine/DMF, 170 °C, 7 days | B548 |
| {[Me4N]2[MnGe4S10]}n (972) | [Me4N]4[Ge4S10] (496), Mn(Me2CO)2/H2O, 24 h or GeS2, Mn(Me2CO)2, [Me4N]Cl, NaHCO3/H2O, 120 °C, 2 days | B/T263,549,550 |
| {[Me4N]2[FeGe4S10]}n (973) | [Me4N]4[Ge4S10] (496), Fe(Me2CO)2/H2O, 24 h or GeS2, FeCO3, [Me4N]Cl, [H4N]HCO3/H2O, 220 °C, 2 days | B/T260,549,557 |
| {[Me4N]2[CoGe4S10]}n (974) | [Me4N]4[Ge4S10] (496), Co(Me2CO)2/H2O, 24 h | T549 |
| {[Me4N]2[ZnGe4S10]}n (975) | [Me4N]4[Ge4S10] (496), Zn(Me2CO)2/H2O, 24 h | T549 |
| {[Me4N]2[Ag2Ge4S10]}n (976) | [Me4N]2[Ge4S10] (496), Na2S2O3, Ag2NO3/H2O, 16 h | T551 |
| {Cs2[FeGe4S10]}n (977) | Cs4[Ge4S10] (493), FeSO4/H2O | T260 |
| {[Me4N]2[Cu2Ge4S10]}n (978) | [Me4N]2[Ge4S10] (496), NaBr, CuCl/H2O, MeCN, 16 h | T551 |
| {[CnH2n+1NC5H5]2[Pt2Ge4S10]}n (979–984, n = 12, 14, 16, 18, 20, 22) | [Me4N]2[Ge4S10] (496), [CnH2n+1NC5H5]Br, K2[PtCl4]/formamide, 80 °C, 18 h | T552 |
| {[(CH3CH2)4N]3[AgGe4S10]}n (985) | [EtNH3]3[MeNH3][Ge4S10] (497), AgOAc, [(Et)4N]Br, methylurea/130 °C, 24 h | T281 |
| {[(Et)4N]3[CuGe4S10]}n (986) | [EtNH3]3[MeNH3][Ge4S10] (497), CuCl, [(Et)4N]Br, methylurea/130 °C, 24 h | T281 |
| {[Me4N]2[MnGe4Se10]}n (987) | [Me4N]4[Ge4Se10] (505), Mn(OAc)2/H2O | T272 |
| {[Me4N]2[FeGe4Se10]}n (988) | [Me4N]4[Ge4Se10] (505), FeSO4/H2O | T272 |
| {[C16H33NC5H5]2[Pt2Ge4Se10]}n (989) | [Me4N]2[Ge4Se10] (505), [C16H33NC5H5]Br, K2[PtCl4]/Formamide, 80 °C, 18 h | T552 |
| {[C16H33NC5H5]2[Pt2Sn4Se10]}n (990) | [Me4N]2[Sn4Se10] (516), [C16H33NC5H5]Br, K2[PtCl4]/Formamide, 80 °C, 18 h | T552 |
| {[Me4N]2[MSn4Se10]}n (991–993, M = Fe, Co, Mn) | [Me4N]2[Sn4Se10] (561), MCl2/H2O | T275 |
| {[Me4N]2[ZnSn4Se10]}n (994) | [Me4N]2[Sn4Se10] (561), ZnCl2, Na4EDTA/H2O | T275 |
| {[Me4N]4[(Ge4S10)Cu4M2S8]}n (995–996, M = Mo, W) | [Me4N]4[Ge4S10] (496), [Me4N]2[MS4], Cu(OAc)2/BuOH, H2O, DMF, 100 °C, 3 days | T553 |
| {[BMIm]2[Ge4Se9]}n (997) | K4[Ge4Se10] (501), SnCl2 2,6-dimethylmorpholine/[BMIm][BF4], 150 °C, 3 days | T554 |
| {[BMMIm]2[Ge4SnSe10]}n (998) | K4[Ge4Se10] (501), SnCl2 2,6-dimethylmorpholine/[BMMIm][BF4], 150 °C, 3 days | T554 |
| {(BMIm)2[SnII(GeIV4Se10)]}n (999) | Cs4[Ge4Se10] (503), SnCl2, 2,6-dimethylmorpholine, [Pt@Bi10][AlBr4]4/(BMIm)Cl, (BMIm)[BF4], 120 °C, 4 days | T555 |
| [Mn2{(OOCC2H4Ge)4S6}(MeOH)(dmf)2]n (1000) | [{HOOC(CH2)2Ge}4S6] (579), MnCl2/MeOH, DMF, 100 °C, 24 h | T556 |
| {[Me4N]2[Mn0.86Co0.14Ge4S10]}n (1001) | GeS2, [Me4N]HCO3, [Me4N]OH, Mn(Me2CO)2, Co(Me2CO)2, H2S/H2O, EtOH, 78 to 150 °C, 3 days | G549 |
| {[Me4N]2[CdGe4S10]}n (1002) | GeS2, CdSO4, [Me4N]Cl, [H4N]HCO3/H2O, 220 °C, 2 days | B557 |
| {(H2dabco)[MnGe4S10]}n (1003) | GeS2, MnCl2, dabco/H2O, CO2, 120 °C, 7 days | B558 |
| {(H2dabco)(H3O)[AgGe4S10]}n (1004) | GeS2, Ag(OAc), dabco/H2O, 130 °C, 2 days | B559 |
| {[Et4N]2[Cu2Ge4Se10]}n (1005) | GeS2, Cu(OAc), [Me4N]HCO3/H2O, 150 °C, 1 day | B560 |
| {[Me4N]6[(Cu0.44Ge0.56S2.23)4(Ge4S8)3]}n (1006) | GeS2, [Me4N]HCO3, Cu(OAc)2, [Me4N]Cl/150 °C, 24 h | A561 |
| {[Me4N]2[MnGe4Te10]}n (1007) | 1. K2Te, Te, Ge/heat to melt | E275 |
| 2. [Me4N]Br, MnCl2/en, 100 °C, 12 h | ||
| {[Li4(H2O)8][MnGe4Se10]}n (1008) | 1. LiSe2, Ge, Se/heat to melt | E282 |
| 2. MnCl2/MeOH, H2O, 24 h | ||
| {[Pr2NH2][PrEtNH2][Ge4S9]}n (1009) | GeS2, [Pr2NH2]Cl, NaHCO3/H2O, 125 °C, 24 h | B562 |
| {[Me4N]2[OSn5Se10]}n (1010) | Sn, Se, [Me4N]OH,/H2O, 150 °C, 16 days | B278 |
| δ-GeS2 (1011) | [Me4N]4[Ge4S10] (496), HCl/H2O, 50 °C, 24 h | I266 |
2.2 1,3,5,7-Tetrasubstituted adamantane derivatives
In 1941, the first synthesis of adamantane (1012), the smallest so-called diamondoid,565,566 was achieved by Prelog, yielding 1.5% from Meerwein's ester through a series of conventional enolate alkylation techniques, Wolff–Kishner reductions, and a final double decarboxylation step (Scheme 4).567 Subsequent refinements by Stetter increased the yield to 6.5%, but the method remained intricate, involving multiple stages for the removal of functional groups used in adamantane synthesis.568 In 1957, Paul von Ragué Schleyer introduced a groundbreaking Lewis acid-promoted rearrangement of tetrahydrodicyclopentadiene, offering an alternative pathway to adamantane synthesis. This isomerization method significantly enhanced the yield by approximately 40%.569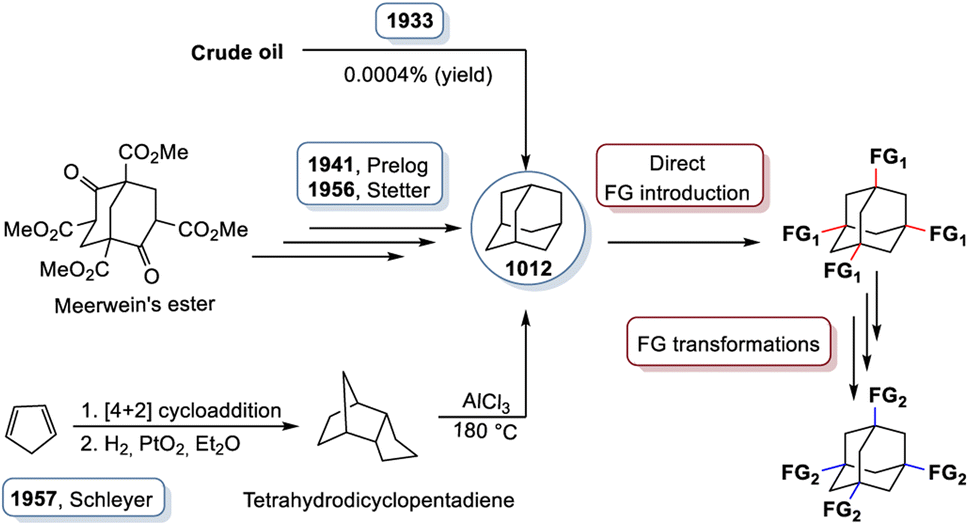 | ||
| Scheme 4 Synthesis and functionalization of tetra-substituted adamantane. | ||
These sections center on the synthesis of 1,3,5,7-tetra-substituted 1012 and explores its applications in advancing nonlinear optical properties.570,571 In these sections, we employ two distinct approaches. The first approach concentrates on directly incorporating functional groups onto the adamantane core. The second approach delves into functional group transformations, commencing from 1,3,5,7-tetra-substituted adamantane as the starting point.
In general, these conditions facilitate the abstraction of hydride ions while also serving as sources of nucleophilic species.
Direct bromination of 1012 leads to the formation of only 1-bromo adamantane.574,575 However, the presence of Friedel–Crafts-type catalysts like AlCl3 and AlBr3 allows for the gradual replacement of more tertiary C–H bonds with bromine. The successful synthesis of 1,3,5,7-tetrabromoadamantane (1013) has been achieved by utilizing AlCl3 and Br2 at 150 °C (Scheme 5).576,577 Note that the use of larger amounts of AlCl3 leads to the generation of not only 1013 but also small amounts of 1-chloro-3,5,7-tribromoadamantane in around 12% yield. In addition, synthesis of 1013 has been achieved in the presence of AlBr3 under sealed tube conditions at 150 °C. This approach avoids halogen exchange during the synthesis of 1013 by utilizing aluminum tribromide.578 The use of two equivalents of AlBr3 resulted in the clean formation of 1013 with 85% yield at room temperature.579 The established one-step method to synthesize 1,3,5,7-tetrachloroadamantane (1014) proceeds by refluxing adamantane in CCl4 in the presence of AlCl3 (Scheme 5).580
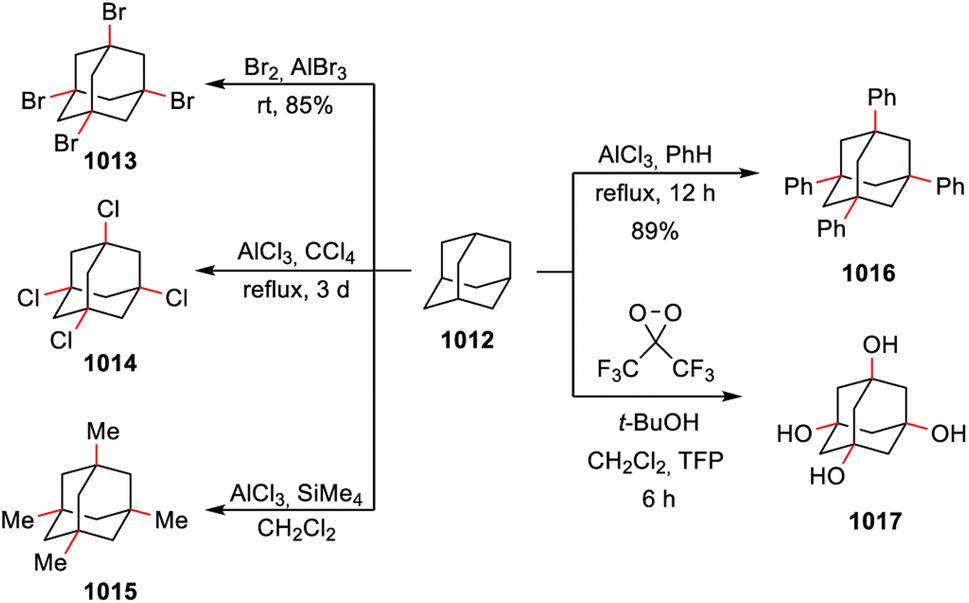 | ||
| Scheme 5 Direct tetra-functionalization of adamantane. | ||
Direct methylation of 1012 with tetramethylsilane as the methylation agent and a Friedel–Crafts catalyst has been explored and optimized for the synthesis of 1,3,5,7-tetramethyladamantane (1015, Scheme 5). With the introduction of four methyl groups in the presence of AlCl3, adamantane underwent fourfold methylation of all bridgehead carbons.581
As is well known, Friedel–Crafts alkylations can generate mixtures of substitution products, and the selective introduction of aryl groups at the 1,3,5,7-positions of 1012 requires precise control of reaction conditions.
In 1968, Stetter and Krause employed a two-step process to add phenyl groups to the adamantane molecule, resulting in the synthesis of 1,3,5,7-tetraphenyladamantane (1016, Scheme 5). Initially, they brominated adamantane using molecular bromine (Br2). Subsequently, in the presence of AlCl3 and benzene, phenyl groups were introduced via Friedel–Crafts alkylation.582
In 1972, Newman utilized the Friedel–Crafts catalyst along with tert-butyl bromide to synthesize 1016 from 1-bromoadamantane. This method allowed for selective Friedel–Crafts phenylation under controlled reaction conditions, resulting in the clean formation of 1016.583
Alternatively, 1016 was synthesized from adamantane under refluxing conditions, utilizing a catalytic amount of AlCl3. The reaction proceeded overnight giving a yield of 89%.584 Furthermore, 1012 can be directly converted into 1,3,5,7-tetrahydroxyadamantane (1017) under remarkably mild conditions, employing an excess of methyl(trifluoromethyl)dioxirane in solution (Scheme 5).585
Recently, we reported a new meta-selective adamantane tetraarylation using substituted benzenes. This Friedel–Crafts-type reaction yields a large amount of all-meta-tetrafluorophenyl adamantane derivatives (1018–1021) in the presence of tert-BuBr as the additive and AlCl3 as the catalyst (Scheme 6).586
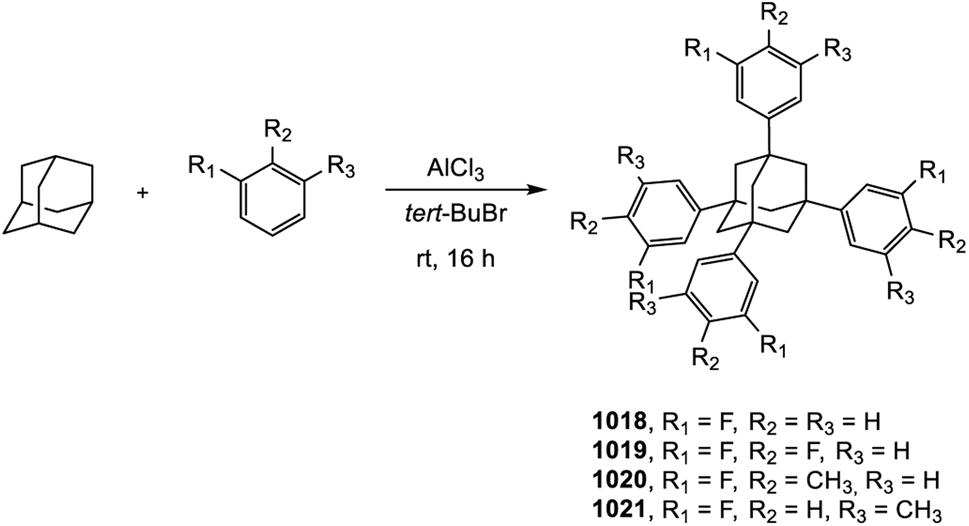 | ||
| Scheme 6 Direct meta-substituted tetra-functionalization of adamantane. | ||
The conversion of 1,3,5,7-tetracyanoadamantane (1022, Scheme 7) from 1013 was achieved through a nucleophilic radical substitution reaction. Interestingly, no reaction occurred in the dark. However, upon photolysis with sodium cyanide in DMSO in a quartz vessel using a Rayonet reactor, a mixture was obtained where 1022 was the dominant product.579 The synthesis of 1,3,5,7-tetraiodoadamantane (1023) did not proceed directly from 1012. Initially, a bromination reaction was conducted to substitute hydrogen atoms on the adamantane bridgeheads with bromine atoms. This process involves halogen exchange in the presence of methyliodide, aluminum powder, and bromine, carried out at 80–85 °C for 45 min, as illustrated in Scheme 7.606 An improved procedure for 1023 involves the use of methyliodide and AlBr3, resulting in a yield of 91% (Scheme 7).579
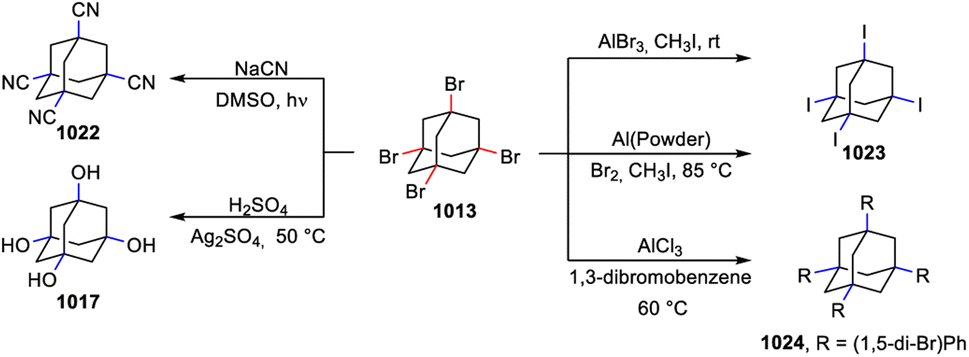 | ||
| Scheme 7 Functional Group transformations from 1,3,5,7-tetrabromoadamantane. | ||
The direct method to prepare 1,3,5,7-tetrahydroxyadamantane (1017) from 1012 utilizes a strong oxidation reagent such as dioxiranes, which poses a risk of explosion during their preparation (see Scheme 5). Target compound 1017 can be prepared conveniently from 1013 in the presence of concentrated H2SO4 and Ag2SO4.582 Exhaustive Soxhlet extraction improved the yield, increasing it to 98% compared to the 84% reported in the literature.607 Starting from 1013, the synthesis of 1,3,5,7-tetrakis(1,3-dibromophenyl)adamantane (1024) can be accomplished with 1,3-dibromobenzene and AlCl3 (Scheme 7).608
The nitration of 1012 with concentrated nitric acid in glacial acetic acid at elevated temperatures has been previously reported to yield 1-nitro-, 1,3-dinitro-, and 1,3,5-trinitroadamantanes, albeit in moderate to low yields.609 When adamantanes are subjected to nitration with nitrogen dioxide at elevated temperatures, the primary products are typically 1-nitro and 1,3-dinitro derivatives. Similarly, the photochemical reaction of N2O5 with 1012 primarily results in mononitration. Note that while the oxidation of tert-alkyl amines to their corresponding nitro compounds is a standard method, it has not been widely used in the past to prepare compounds containing more than two nitro groups. In a noteworthy synthesis, Sollot and Gilbert reported the hydrolysis of 1,3,5,7-tetraaminoadamantane·tetrahydrochloride (1025) to obtain the free tetraamine, which was subsequently oxidized using permanganate to yield the desired 1,3,5,7-tetranitrodamantane (1026) with a yield of 45% (Scheme 8).576Additionally, the powerful oxidizing agent dimethyldioxirane was employed to synthesize 1026, achieving an impressive yield of 91%.606
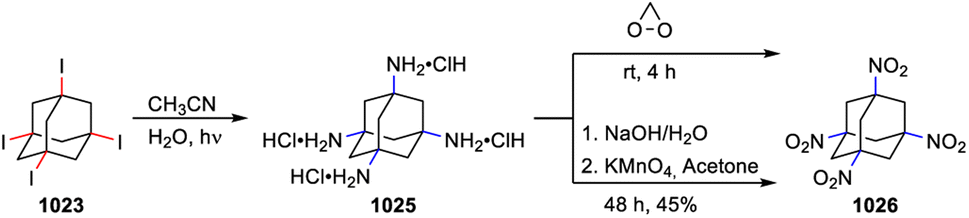 | ||
| Scheme 8 Synthesis of 1,3,5,7-tetranitroadamantane. | ||
The reduction of 1022 was accomplished using monochloroborane-methyl sulfide in refluxing THF. Subsequent reaction with dry methanolic HCl resulted in the formation of 1,3,5,7-tetrakis(aminomethyl)adamantane tetrahydrochloride with an impressive yield of 98%. To obtain the 1,3,5,7-tetrakis(aminomethyl)adamantane (1027), deprotonation of an aqueous solution with NaOH was performed (Scheme 9).579 Additionally, hydrolysis of 1022 led to 1,3,5,7-tetracarboxylic acid adamantane (1028). This method serves as an excellent alternative for preparing 1028, reducing the number of synthetic steps compared to those reported by others (Scheme 9).610
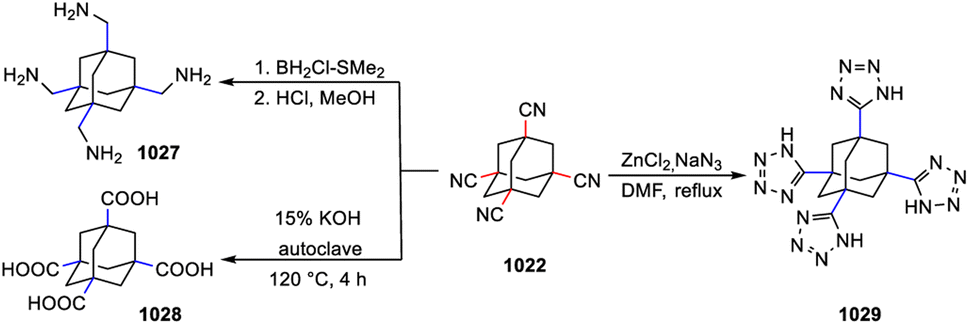 | ||
| Scheme 9 Functional Group transformations from 1,3,5,7-tetracynoadamantane. | ||
The use of ‘click chemistry,’ specifically tetrazole formation through the cycloaddition of azides to nitriles in the presence of ZnCl2, offers an especially cost-effective route to obtain tetrakis-tetrazole derivatives of adamantane. When applied to 1022, this process exhibited slightly slower kinetics compared to aromatic or unhindered aliphatic nitriles but ultimately yielded 1,3,5,7-tetrakis(tetrazol-5-yl)adamantane (1029, Scheme 9). The reaction can be conducted in DMF under reflux conditions for 48 h or at 175 °C in an autoclave within 6 h. The former conditions provide a purer product.611
The synthesis of 1,3,5,7-tetra(diphenylphosphate)adamantane (1030) with a yield of 62% can be achieved by reacting 1,3,5,7-tetrahydroxyadamantane (1017) with diphenyl chlorophosphate under controlled conditions (Scheme 10).612 Introduction of electron-rich arenes (substituted anisoles) to 1,3,5,7-tetrahydroxyadamantane through Friedel–Crafts alkylation results in symmetrical tetraaryladamantanes with yields ranging from 20–41% (1031–1034, Scheme 10). This alkylation process requires strong Brønsted acids, such as tosylic acid (TfOH), and elevated temperatures. The study reports that weaker acids or lower temperatures are ineffective, leading to low reactivity and consequently very low yields.607,613–615
 | ||
| Scheme 10 Functional group transformations from 1,3,5,7-tetrahydroxyadamantane. | ||
In this context, various functional groups were introduced onto the phenyl rings of 1016 through electrophilic substitution at the para-positions of the phenyl moieties, as outlined in Table 22. The direct functionalization of 1016 through electrophilic aromatic substitution can serve as a good starting point for synthetic modifications, enabling access to a wide array of functional groups (R) attached to the aromatic moieties of 1,3,5,7-tetraaryladamantanes (R = Br, I, SO2Cl, NO2, COCH3, and CHO).
|
|
||
|---|---|---|
| R = | Reagents/conditions | Ref. |
| Br (1035) | Br2/CHCl3, −78 °C, 12 h, 60% | 616 |
| Br (1035) | Fe, Br2/50 °C, 12 h, 94% | 617 |
| Br (1035) | Fe, Br2/70 °C, 7 h, 36% | 618 |
| I (1036) | PhI(CH3CO2)2, I2/CHCl3, 12 h, 80% | 619 |
| SO2Cl (1037) | HSO3Cl/1.6 h, 57% | 620 |
| NO2 (1038) | HNO3, Ac2O, AcOH/30 min, 35% | 621 and 622 |
| COCH3 (1039) | AlCl3, CHCOCl/16 h, 68% | 626 |
| CHO (1040) | TiCl4, CH3OCHCl2/CH2Cl2, −10 °C to rt, 12 h, 84% | 623 |
The bromination of 1016 in liquid bromine selectively occurs at the para-position, yielding 1,3,5,7-tetrakis(4-bromophenyl)adamantane (1035, Table 22) with 60% yield, without the need for additional catalysts.616 However, when bromination of 1016 is conducted in the presence of Fe, 1035 is obtained in a significantly improved yield of 94%,617 which further increases to 96% at elevated temperatures.618 The iodination of 1016, using PhI(OCOCF3)2 in a chloroform solution of iodine, leads to the formation of 1,3,5,7-tetrakis(4-iodophenyl)adamantane (1036, Table 22).619 The sulfonation of 1016 using chlorosulfuric acid efficiently produces 1,3,5,7-tetra(phenyl-4-sulfonyl chloride)adamantane (1037) with a yield of 57% (Table 22).620 Starting material 1016 was subjected to nitration in fuming nitric acid at −15 °C for 30 min, yielding 1038 in low yield (Table 22). The degree of nitration can be controlled by adjusting the reaction time.621,622 The Friedel–Crafts acetylation of 1016 results in the formation of 1039 with good yield (Table 22). Additionally, 1,3,5,7-tetrakis-4-formylphenyladamantane (1039)626 was synthesized using a modified patented procedure involving the titanium tetrachloride-promoted formylation of 1040 (Table 22).623
The synthesis of 1041 was achieved by reacting 1036 with NaOMe and Cu(I)Br in dry MeOH/DMF, yielding a 47% yield. Similarly, the reaction of 1,3,5,7-tetrakis(4-bromophenyl)adamantane (1035) with NaOMe and Cu(I)Br in dry MeOH/DMF results in the formation of 1041 with a yield of 52% (tableref>/tableref>).614
|
|
||
|---|---|---|
| R = | Reagents/conditions | Ref. |
| a MCPBA = m-chloroperbenzoic acid. | ||
| OMe (1041) | Cu(I)Br, NaOMe/MeOH, DMF, 100 °C, 12 h, 47–52% | 587, 622 |
| CN (1042) | CuCN/DMF, 160 °C, 16 h, 82% | 625 |
| PO(OEt)2 (1043) | 1. HPO(OEt)2, PdCl2(PPh3)2, Et3N/PhH, 80 °C | 627 |
| 2. HCl/reflux, 76% | ||
| Pyrrole (1044) | R–H, CuI, K2CO3, N,N-dimethylglycine/DMSO, 120 °C, 21–42% | 628 |
| Carbazole (1045) | ||
| Imidazole (1046) benzimidazole (1047) phenylimidazole (1048) | R–H, CuI, K2CO3, N,N-dimethylglycine/DMSO, 120 °C, 5 days, 35–43% | 628 |
| 2-CH3-imidazole (1049) | R–H, CuI, K2CO3, N,N-dimethylglycine/DMSO, 120 °C, 5 days, 41% | 629 |
| N(4-OMePh)2 (1050) | R–H, Pd(OAc)2, tBuOK, tBu3P/toluene, 140 °C, 48 h, 72% | 630 |
| Ph (1051) | R–B(OH)2, Pd(PPh3)4, NaOEt/EtOH, PhH, 80–100 °C, 24 h, 35–45% | 619 and 632 |
| Ethynyl (1052) | 1. Me3Si-ethynyl, Et3N, [PdCl2(PPh3)2], CuBr/80 °C, 72 h | 621 |
| 2. KF/CH3OH, 50 °C, 12 h, 74% | ||
| Ethynyl (1052) | Me3Si-ethynyl, Pd(PPh3)2Cl2, CuI, Et3N, KF/MeOH, 5 days, 81% | 621 and 631 |
| I(OAc)2 (1053) | MCPBA/CH2Cl2, AcOH, rt, 12 h, 97% | 633 |
| Stilbenyl (1054) | Styrene, Pd(OAc)2, K2CO3, nBu4NBr, DMA/105 °C, 24% | 634 |
The synthesis of 1,3,5,7-tetrakis(4-cyanophenyl)adamantane (1042) commenced with 1036, using the Rosenmund–von Braun reaction. Typically, in the literature, ethane-1,2-diamine is used to eliminate the nitrile–copper cyanide complexes and is followed by nitrile extraction to obtain the desired product.592,624 However, in this particular case, ethane-1,2-diamine proved to be inefficient, and the use of an excess of aqueous KCN was found to be more effective in the synthesis of 1042 (Table 23).625
1,3,5,7-Tetrakis(4-phosphonatophenyl)adamantane (1043), was synthesized through a two-step process but without isolating the intermediate. First, a palladium-catalyzed P–C coupling reaction between 1036 and diethylphosphite was carried out. Subsequently, the resulting phosphonic acid diethyl ester was subjected to acidic hydrolysis to obtain 1043 (Table 23).627
A copper(I)-catalyzed coupling reaction was employed to synthesize various derivatives of 1044–1049 (Table 23). This reaction involved the use of pyrrole, carbazole, imidazole, benzimidazole, phenylimidazole, and 2-CH3-imidazole as reactants. The reaction took place in the presence of N,N-dimethylglycine and DMSO at a temperature of 120 °C for a duration of 5 days.628,629
The synthesis of 4,4′,4′′,4′′′–(adamantane-1,3,5,7-tetrayl)tetrakis(N,N-bis(4-methoxyphenyl)aniline) (1050) was achieved by combining bis(4-methoxyphenyl)amine and 1036 in solution in the presence of Pd(OAc)2, t-Bu3P, and t-BuOK (Table 23).630 The reaction of 1036 with phenyl boronic acid under Suzuki coupling conditions yielded compound 1051 (Table 23). This compound is soluble in CHCl3, making it easy to purify and characterize.619 It can readily be converted to 1,3,5,7-tetrakis(4-trimethylsilyl-ethynylphenyl)adamantane by reacting it with Et3N, trimethylsilylacetylene, Pd(PPh3)2Cl2, and CuI in toluene. The crude product can then be deprotected to give 1,3,5,7-tetrakis(4-ethynylphenyl)adamantane (1052) in a yield of 74% (Table 23).621 This product was also prepared by a palladium/copper co-catalytic system for coupling 1036 with Me3Si-ethynyl in the presence of Et3N and DMSO.621,631
Oxidation of 1036 by conventional methods with peracetic acid (30% H2O2 and acetic anhydride), sodium perborate (NaBO3·nH2O) in acetic acid, or sodium periodate (NaIO4) unexpectedly gave 1,3,5,7-tetrakis[4-(diacetoxyiodo)phenyl]adamantane (1053) in low yield, accompanied by poorly soluble and unidentifiable polymeric products (Table 23). After further investigations, it was finally possible to synthesize 1053 in 97% yield by using MCPBA in CH2Cl2/AcOH (1:1) under dilute conditions.633 The synthesis of 1,3,5,7-tetrakis(4-stilbenylphenyl)adamantane (1054) is readily achieved by reacting compound 1036 with excess styrene under palladium-mediated Heck coupling conditions, resulting in an 86% yield. However, when starting with 1,3,5,7-tetrakis(4-bromophenyl)adamantane (1035) under analogous conditions, consistently lower yields were obtained.634
3. Optical properties: linear optical phenomena and photocurrent conversion
Compounds with an adamantane-type scaffold have most commonly been investigated for their luminescence properties over the years. Especially molecules with group 16 or group 17 elements in the E position have been the focus of such investigations, but other emissive examples have been reported as well.Compounds containing the highly distorted group 11 chalcogenide adamantane cations [(ER)4M6{(Ph2P)2R}4]2+ (848–853, 855–857) have been investigated systematically for their photoluminescence behavior.493,494,496 While the emission of the copper complexes [(SePh)4Cu6{(Ph2P)2R}4][BF4]2 (848–849, R = CH2, NH) in solution was of low intensity when compared to other related copper clusters, the long lifetime of the excited state suggests a spin-forbidden transition likely stemming from a ligand-to-metal charge transfer between the PhSe fragments and the Cu centers.493 Most silver congeners only feature a significant luminescence at low temperatures of about 77 K in the solid state, which consists of a single emission peak for [(ER)4Ag6{(Ph2P)2Me}4][PF6]2 (850–853, ER = SPh, SC6H4Me-p, SePh, SeC6H4Cl-p) at around 700 nm. With a rising electron richness of the ER fragments from 850 to 853, the signal shifts to higher energies (746 nm to 666 nm). This was attributed to the influence of the π-acceptor ability of the ER moiety, which affects the orbital splitting of the bonding and antibonding orbitals.494 In contrast, compounds 856–857 show a double emission at 77 K, while 855 is non-emissive.496 An explanation of the different behaviors of the silver homologs is still elusive.
The Cu-thiolate adamantane moieties in [(NEt4]4[(SPh)4(CuX)6] (846–847, X = Cl, Br) show a more symmetrical buildup and a strong photoluminescence with a single emission at around 560 nm.170
[Et4N]2[Cu4(SPh)6] (256), comprising an inverted adamantane-type core as compared to 846 and 847, shows a much weaker photoluminescence when being excited at 350 nm, but exhibits a dual emission at 436 nm and 573 nm, which is attributed to ligand-to-metal charge transfer or transitions between the metal centers, respectively, which has not been possible for the previously discussed compounds due to their larger Cu⋯Cu distances.170
As part of a study on differently sized Cd-selenolate supertetrahedra, the photoluminescence of [nPr4N]2[(CdCl)4(SePh)6] (409) was discussed.228 Significant emission is only detected at temperatures below ∼50 K and is attributed to forbidden transitions. Other adamantane-type thiolate clusters featuring ammonium counterions, like [Me4N]2[(CdSPh)4(SPh)6] (311) and [Et3NH]2[(CdSC6H4-4-Me)4(SC6H4-4-Me)6] (318), exhibit photoluminescence at room temperature, with low to moderate intensity.219,221
Group 12 chalcogenolate adamantane anions also have been subject to studies in combination with the chromophore cation DAMS. The clusters were found to affect the intramolecular charge transfer and reduce quenching.211
The cadmium cluster (DAMS)2[(CdSPh)4(SPh)6] (313) and its iodine derivative (DAMS)2[(CdI)4(SPh)6] (414) have been proven to show significant photoluminescence at room temperature.218 While (DAMS)I already exhibits an emission under similar conditions, 313 and especially 414 do so much more intensely, albeit slightly blue-shifted. Additionally, two-photon pumped lasing spectra revealed nonlinear optical properties for both compounds.
The first OLED constructed from such a compound comprises (DAMS)2[(ZnSPh)4(SPh)6] (302), which follows the trend of a more intense and slightly blue-shifted emission as compared to the pure chromophore.211 The device produces a narrow red emission at 630 nm with a full width at half heights of the measured peak of 80 nm.
Subsequently, combinations of adamantane anions and different chromophore cations were explored. In [Ru(phen)3][(CdSPh)4(SPh)6] (314), the fluorescence enhancing findings made for DAMS compounds could be repeated in a titration study, where an increase of the fluorescence intensity could be observed when adding cluster anions to a solution of the ruthenium complex.219 This effect reaches a plateau at a 1:1 ratio, which is in accordance with an anion–cation charge transfer indicated by both spectroscopic findings and theoretical studies.
The cluster-dye composite [Ru(2,2′-bipy)3][(ZnSPh)4(SPh)6] (303) was investigated for its photocurrent conversion behavior, which can be enhanced by substituting the adamantane type cluster with larger supertetrahedra.212
The earlier UV-vis measurements on group 14/16 adamantane anions without organic ligands revealed the intra molecular transitions to be responsible for the absorption behavior, with no or only negligible contributions of the counterions in case of ammonium cations.279,281,284,286 The optical properties of the adamantane-type cluster remain dominant even in the presence of some transition metal complexes, as long as no charge transfer between them is possible.295,296,298,300 Therefore, such charge transfer pathways have to be present to influence the band gap more significantly, as has been seen in [Ni(trien)2]2[Ge4S10] (552).288–291,294 In the case of [Me2Vio]2[Ge4S10] (544), fluorescence can be detected distinctly red-shifted from the fluorescence of [Me2Vio]I2 by the charge transfer between the cation and anion. The cluster compound also shows solid-state solvatochromicity, depending on the inclusion of water or MeOH into the crystal structure, and it is photoelectrically active.289 A similar behavior in regard to fluorescence and photocurrent was found for [DMBPE]2[Ge4S10] (546) and for the compounds [(CnH2n+1)2Vio]2[Ge4S10] (540–543, n = 0, 2, 3,4).288,291 By utilizing a porphyrin derivative as counterion, like in the fluorescing species TMPyP[Ge4S10] (545), even larger photocurrents could be measured.290 Lastly, there are cases, in which the anion plays nearly no role in the transitions at the band gap, such as in [Ni2(μ-teta)(teta)2][Ge4S10] (555), in which the photoluminescence does not deviate much from the one found for the amine hydrochloride.293,297,299 Similar to 544, [Ni(phen)3]2[Ge4S10] (550) additionally features the ability to reversibly change its color depending on the inclusion of water or MeOH in its crystal structure (Fig. 22).293
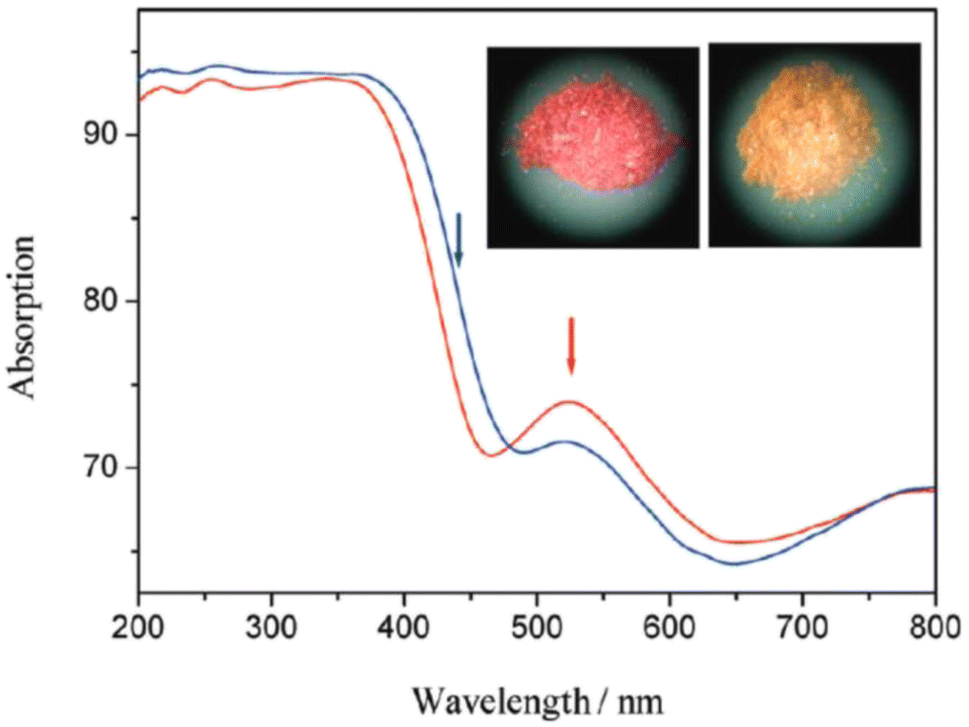 | ||
| Fig. 22 Solvatochromicity in [Ni(phen)3]2[Ge4S10] (550), depending on the inclusion of H2O (red curve) or MeOH (blue curve) into the crystal lattice. (reproduced from ref. 293 with permission from ACS) | ||
For [Ni(teta)(en)][Ni(teta)(hda)][Sn4Se10] (558) moderately weak photocurrent photoelectric conversion was observed.299
An extensive study of lanthanide complexes of the 3-NO2Tp ligand features adamantane type clusters in [(μ4-O){M(3-NO2Tp)}4(μ2-OMe)6] (665–670), with the photoluminescence behavior depending heavily on the lanthanide center.367
The Yb cluster 671 also shows photoluminescence when excited at 405 nm with an emission pattern in the near infrared region, as typical for Yb3+ complexes.368
The mixed-metal compounds [(μ3-M)(CdPPh3)(MPPh3)3(TePh)3(μ3-TePh)3] (903–904, M = Cu, Ag) show strong photoluminescence at around 480 nm when excited at 350 nm. This was assigned to transitions between the coinage metal and its ligands, rather then involving the Cd centers.516 The spectrum of 903 shows an additional shoulder attributed to intracopper ds/dp transitions owing to the small Cu⋯Cu distances.
Adamantane-type clusters with a group 17 element in E position are the second group that were heavily investigated for their optical properties, chiefly among them the copper halide anions [Cu4E6]2−. The first bromine congener investigated was [{Cu(Hdabco)}4Br6](HCOO)2 (817), although a cationic one due to the ligands at the Cu sites. It showed strong photoluminescence with a yellow emission at 556 nm.468 Thermochromic photoluminescence can be observed for (H2dpipa)3[Cu4Br6][Cu2Br6] (815), with different bromido cuprate anions in its structure, and its Cl homolog, which was extensively studied by DFT calculations.459,467 The luminescence of [iPr4N]2[Cu4Br6] (809) and [tBu3NMe]2[Cu4Br6] (812) was compared to other copper bromide compounds, and 809 was found to feature the most brilliant red-orange emission, which was utilized to manufacture a white-light emitting LED in conjunction with two other commercial phosphors.461
The combination of bromido cuprate anions, [Cu4Br6]2− among them, and a polyoxotitanium cluster in a series of compounds including [Ti12(μ3-O)14(OiPr)18][Cu4Br6] (816) showed a vast dependency of the absorption spectra on the anion and the resulting supersalt structure.466
Also [Cu4I6]2−-containing compounds have been investigated for their luminescence properties in several studies.471,472,474,476,477 [Mn(tdpmO3)2][Cu4I6] (828) and [Mn(dppbO2)3]2[Cu4I6][Cu2I4] (829) are part of a series of dual-emission compounds with both the cation and anion showing a distinct emission (Fig. 23).477 When grinding crystals of 829, a triboluminescence originating from the same centers as the photoluminescence is detectable.
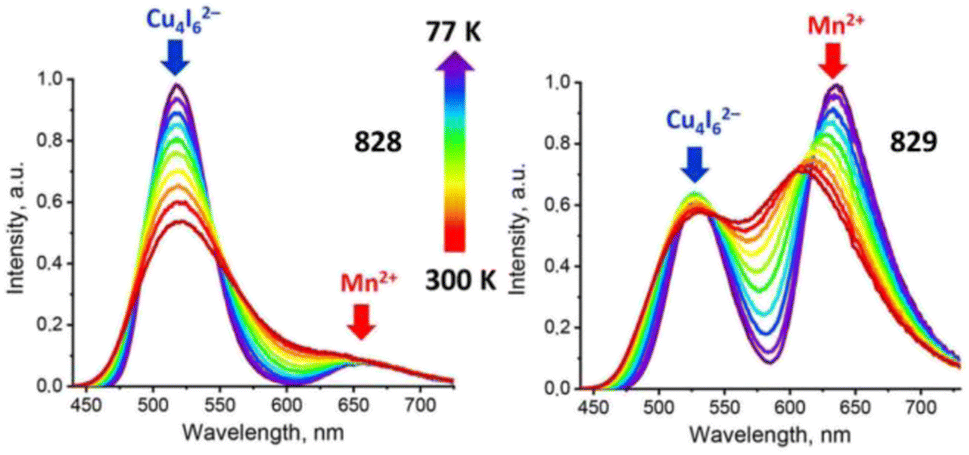 | ||
| Fig. 23 Photoluminescence at different temperatures of 828 (left, reproduced from ref. 477 with permission from ACS) and 829 (right, reproduced from ref. 477 with permission from ACS). | ||
The band gap in compounds with a [μ4-O(CuR)4Cl6] inorganic core is determined by ligand to metal charge transfer and therefore heavily influenced by the ligand used, but such compounds do not show luminescence as opposed to their anionic relatives.390,403,415
4. Optical properties: nonlinear white-light generation (WLG) and second-harmonic generation (SHG)
The huge compositional variety of adamantane based clusters offers the possibility to finely tune the optical properties for a multitude of applications. Obviously, the fundamental element of all optical properties is defined by the HOMO–LUMO gap, in the case of adamantane (1012), this gap is ∼6.49 eV.635 As a consequence, the absorption onset and corresponding photoluminescence is in the ultra-violet (UV) spectral range.635 An advantage of this large HOMO–LUMO gap is a very high photostability of adamantane, since it is virtually unaffected by light in the visible spectral range.636 At the same time, the large HOMO–LUMO gap makes it difficult to make use of pure adamantane in optoelectronic applications. However, by functionalization on both the Q and E site, the HOMO–LUMO gap can easily be tuned. Functionalization schemes include fluorination, addition of alkali-metals or introduction of very simple or complex organic ligands, only to name a few.637–640 Even with these rather simple functionalization schemes, it is already possible to tune the HOMO–LUMO gap and thus the optical properties of fully organic adamantanes to cover virtually the whole visible spectral range.An even larger space for tuning the optical properties opens up when stepping away from the fully organic adamantane by alternating the composition on the Q- and E-site too.
In the last decade, several hundreds of adamantane based clusters have been investigated theoretically.571 All the structures with adamantane like cores and the same ligand field show a variation in the HOMO–LUMO gaps in a range of about ΔE ≈ 2 eV. This is demonstrated in Fig. 24 for clusters with homogeneous phenyl ligands (tetraphenyl clusters) and different cluster cores. It should be noted that Fig. 24 is not exhaustive and only features a relatively small sample of possible modifications of the core which have been investigated recently.
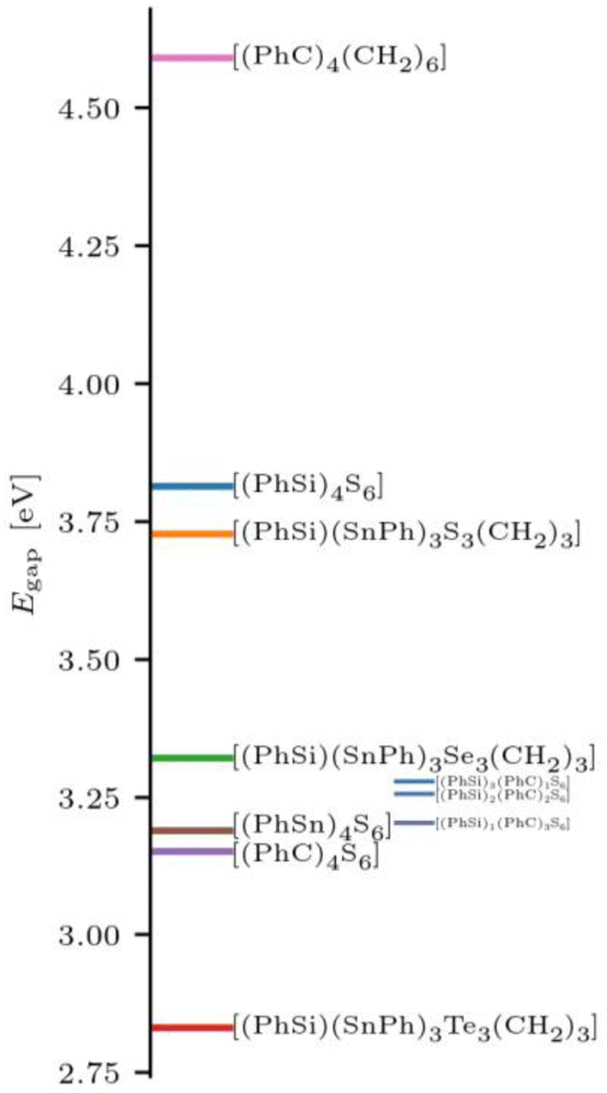 | ||
| Fig. 24 HOMO–LUMO energy gaps calculated within the density functional theory in the independent particle approximation for selected adamantane-based tetraphenyl clusters. | ||
The highest HOMO–LUMO gap is predicted (DFT calculations in the IPA) for the prototypical AdPh4 cluster (1016, Eg ≈ 4.65 eV). Modifications of the adamantane core (inorganic and/or heterogeneous composition) lower this value up to about 2 eV, with a minimum value calculated for the heterogeneous and inorganic [PhSi{CH2Sn(Te)Ph}3] (891) cluster. Interestingly, a fine tuning of the HOMO–LUMO gap value can be achieved by the gradual modification of the cluster core, as shown exemplarily by the stepwise transition from the inorganic [(PhSi)4S6] cluster (568) to the organic [(PhC)4S6] cluster.641 The first substitution largely modifies the HOMO–LUMO-gap, due to an abrupt modification of the bond lengths in the core. For the subsequent modifications, a quite gradual change is seen (as shown in Fig. 24). The fine tuning of the HOMO–LUMO gap is also expected for the stepwise transition between the other cores and has consequences for the manipulation of the optical response.
In addition to tuneability of the linear optical properties, it was found that a large number of cluster molecules of adamantane-type cluster compounds with composition [(RQ)4E6] presented in Table 13, with group 14 elements in the Q position and those of group 16 in the E-position, show strong nonlinear optical properties when they are irradiated by a simple near-infrared (NIR) low-power laser diode.310,320 This nonlinear response manifests itself in the emission of light covering virtually the whole visible spectrum from ∼400 nm to 800 nm (see Fig. 25 for emission spectra under various excitation energies with excitation region marked by dotted grey line). Because of the broad spectral-range, we will refer to the process as white-light emission, although due to the spectral intensity distribution the appearance to the human-eye is warm-white (see Fig. 25).
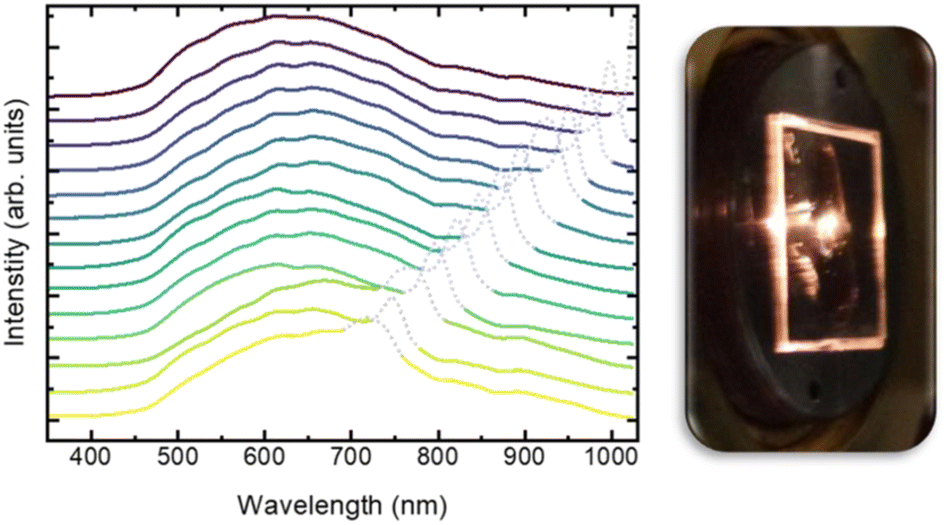 | ||
| Fig. 25 Broad white-light emission of [(StySn)4S6] (598), shown in differently colored lines for various excitation wavelength (indicated by the grey dotted part of the spectra) in the range of 700–1050 nm (left). Photograph of the emission of [(StySn)4S6] sandwiched between two glass slides and excited with 800 nm laser light (reproduced from ref. 320 with permission from AAAS). | ||
As it has been demonstrated that the nonlinear optical response of the clusters can be generally interpreted in terms of multiphoton processes,309,642,643 tuning the HOMO–LUMO gap makes it possible to tune the nonlinear response too, e.g., the frequency dependence of the main SHG and THG peaks. It is worth mentioning here that SHG can be observed on these materials even for compounds that crystallize in centrosymmetric space groups, which usually is an exclusion rule, as only crystals lacking inversion symmetry can produce bulk SHG. However, the SHG in such samples appears to be due to surface effects and/or defects in the crystal. With the SHG being very effective, the contribution of the surfaces of the (micro- or nanosized) crystals is sufficiently high to observe SHG in most cases.
The optical response of a wide class of adamantane-based clusters and cluster aggregates has been calculated from first principles in recent years.309,571,642–645 The calculations show that all adamantane-based clusters are characterized by a nonlinear optical response with the same structure, as long as they have the same ligand field. For example, a prominent peak above 2 eV followed by a dip and a second peak is common to all adamantane-based cores with phenyl ligands. This suggests that the nonlinear optical response is dominantly defined by the ligand structures and originates only to a minor extent in the core region. This is demonstrated exemplarily in Fig. 26, where we compare the SHG signal of AdPh4 (1016) with that calculated on the same footing for CPh4.646
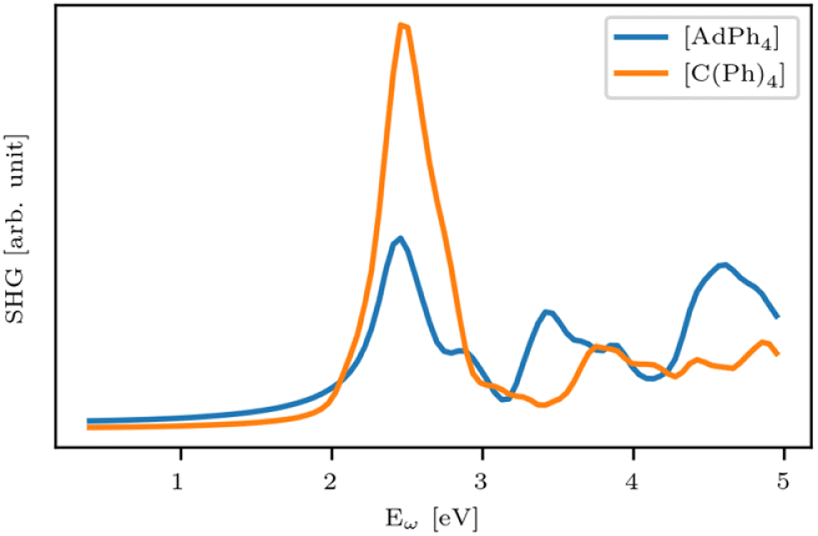 | ||
| Fig. 26 Second-harmonic generation (average of all tensor components, absolute values) calculated as a function of the incident laser wavelength for the isolated [AdPh4] cluster (1016) (blue line) and the [CPh4] cluster (orange line) within the independent particle approximation at the DFT equilibrium geometry.646 | ||
The structure of the second order nonlinear response is basically the same for the two clusters, although CPh4 has no adamantane core at all. This, however, does not mean that the cluster core does not affect the linear and nonlinear optical properties. As we will discuss in the following, the geometry and the chemistry of the core have an impact on the magnitude of the nonlinearities and, to a lesser extent, to their spectral position. Thus, modifications of the cluster core offer the possibility to manipulate the optical response towards desired energies and intensities. Yet, the main characteristics are preserved as long as the ligand structures are maintained.
Although the nonlinear optical response originates in and is dominated by the ligand field, as previously discussed, the optical nonlinearities are enhanced by disorder and structural asymmetry in the cluster core. This is particularly true for molecules featuring a heterogeneous core composition.642 This is clearly shown with clusters [PhSi{CH2Sn(E)Ph}3] (889–891, E = S, Se, Te) as a model system. The cluster structure is shown in Fig. 27, along with the distortion in the core, quantified by the ratio between the largest and shortest bond length. The core symmetry is greatly reduced from S to Te, while the optical susceptibilities are correspondingly enhanced, as displayed in Fig. 28. The AdPh4 cluster features a regular core and has the lowest SHG coefficients. This effect is quite remarkable, as a range of intensities spanning an entire magnitude of SHG responses can be achieved with these structures. Noticeably, the form of the second order optical response typical for tetraphenyl compounds (displaying a main peak above 2 eV, followed by a dip and a second peak) is maintained for all variations of the core.
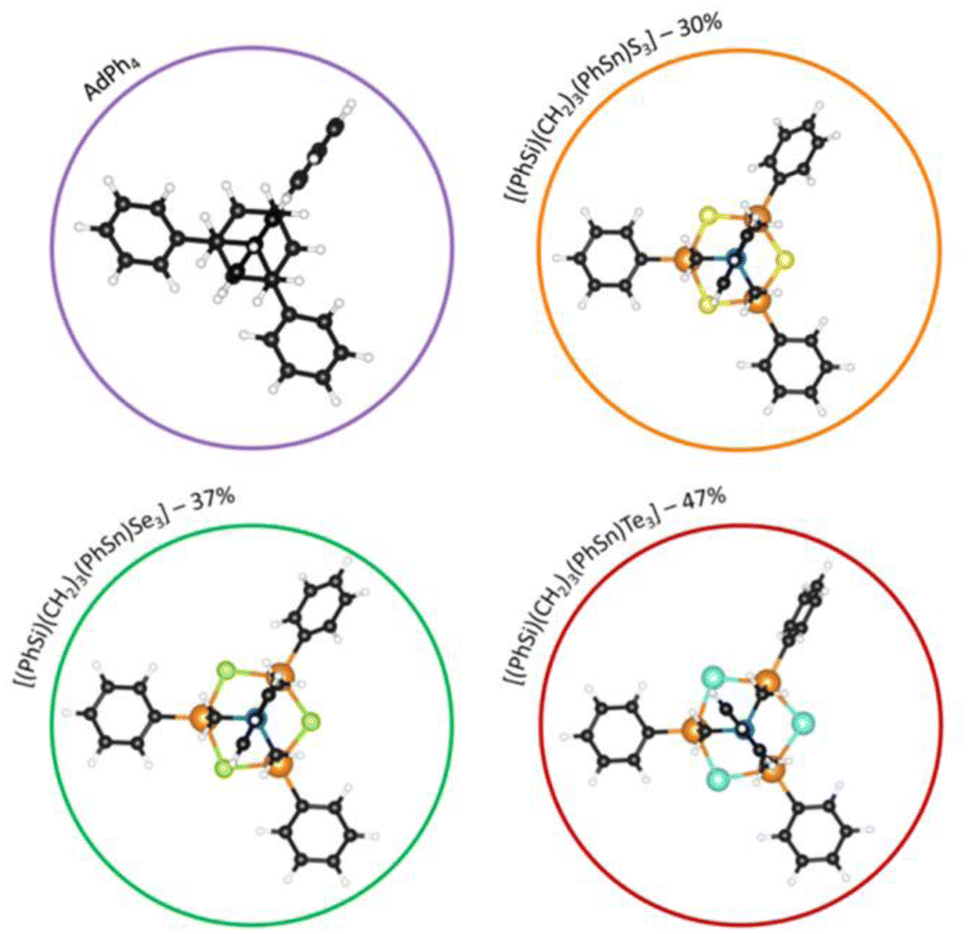 | ||
| Fig. 27 Atomic structure calculated within DFT in the independent particle approximation for AdPh4 (1016, top left) and different tetraphenyl clusters with modified cluster cores. The circle color corresponds to the respective line color of the second order optical response shown in Fig. 28.642,644 | ||
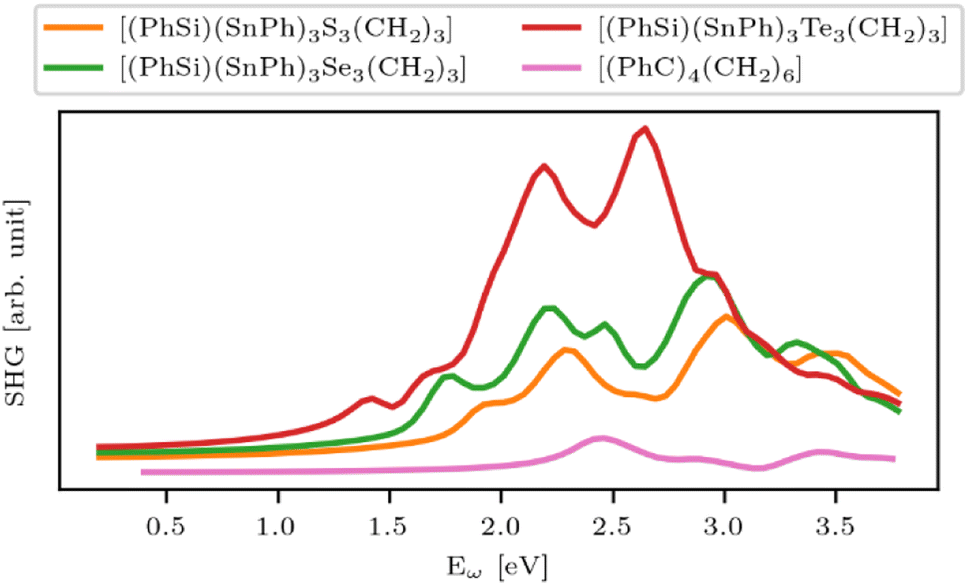 | ||
| Fig. 28 Second-harmonic generation per cluster (the average of all tensor components is shown) calculated within DFT in the independent particle approximation for different tetraphenyl clusters with modified cluster cores.642,644 | ||
Although a generally accepted theory of the observed white-light generation is still missing, it is known that higher order nonlinear effects like supercontinuum generation involve the whole catalogue of nonlinear-optical effects, which add up to produce emission with an extremely broad spectrum.646–648 These nonlinear effects include self- and cross-phase modulation, four-wave mixing, and many others, for which an exhaustive theory is still missing. Nevertheless, it is reasonable to assume that a more or less high nonlinear optical activity is a prerequisite for the mentioned effects leading to white-light emission. In this respect, the theoretical studies performed in the last years allowed for an (at least partial) understanding of the optical response of the white-light emitters and revealed several interesting aspects. As a general feature, all clusters showing WLE are characterized by strong optical nonlinearities of second and third order, at least as strong as that of the crystalline ferroelectrics.309,571,642–646,650,651 The optical nonlinearities feature high peaks and low dips at which the optical coefficients are almost quenched, as seen by the example in Fig. 28 for the prototypical AdPh4.
The optical response is thus strongly dependent on the incident photon energy, with most compounds showing a maximum of the SHG coefficients above 2 eV and a THG maximum just below 2 eV (see e.g., Fig. 29). In general, the white-light emission efficiency is expected to depend on the exciting wavelength.648 However, this does not seem to be the case for the adamantane-type cluster molecules.320,571 This might be related to the fact that in the adamantane-type cluster molecules, white-light emission is achieved by an excitation in a generally non-resonant region of the nonlinear optical spectrum (1.1–1.3 eV), however, where the onset of the optical nonlinearities is already pronounced. As this spectral region is followed by a steep gradient of the nonlinear optical susceptibilities, the white-light emission efficiency might be further increased, provided it correlates (as currently assumed) with the optical nonlinearities.
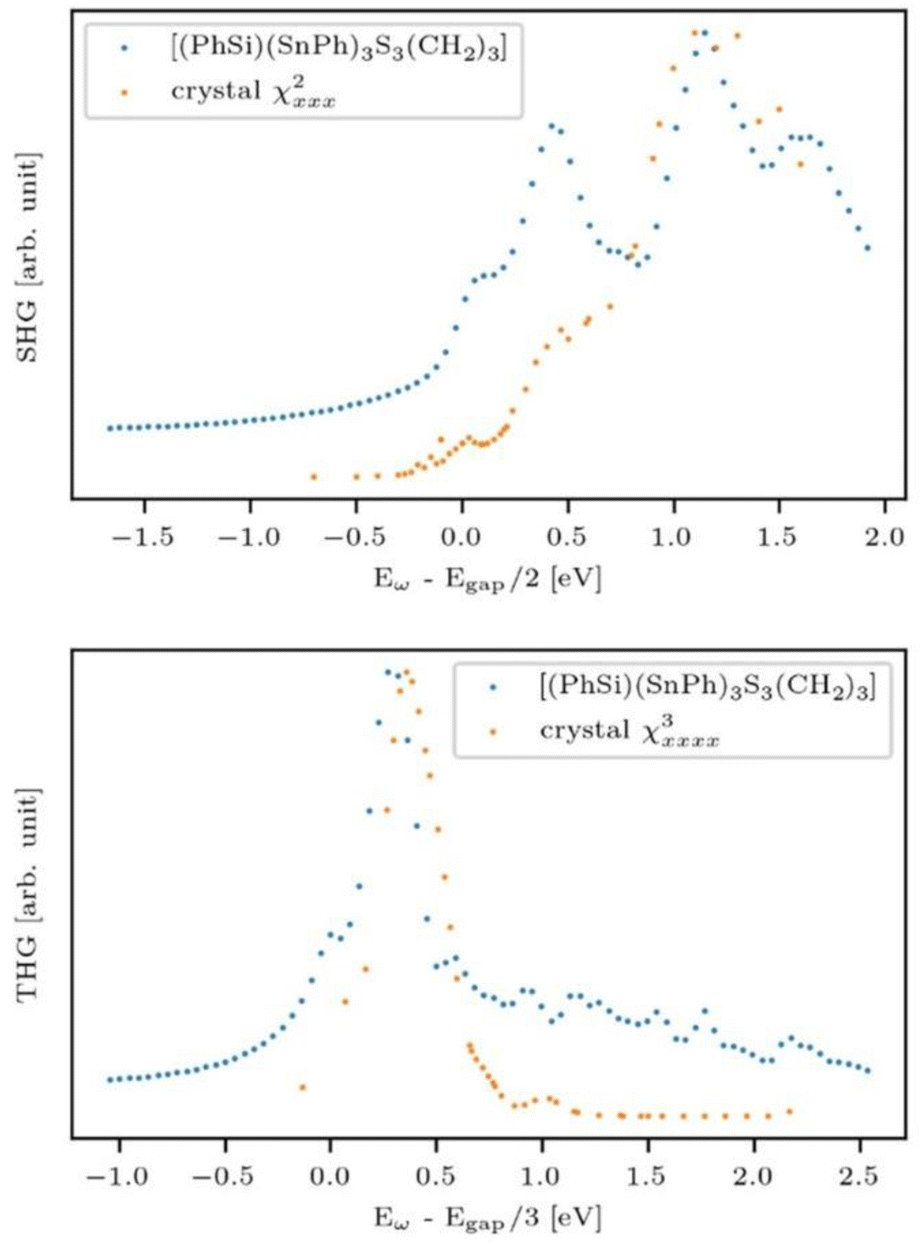 | ||
| Fig. 29 Second-harmonic generation (top, χ(2)xxx component) and third harmonic generation (bottom, χ(3)xxxx component) coefficients (absolute values) calculated as a function of the incident laser wavelength for the isolated [PhSi{CH2Sn(S)Ph}3] cluster (blue dots) and the corresponding crystal (orange dots) within the independent particle approximation at the DFT equilibrium geometry. Intensities are scaled by the respective maximum for each structure, and energies are shifted relative to the energy gap.642,644 | ||
5. Materials properties (crystalline/amorphous, glass formation): comprehension and prediction
A common trend identified in multiple structures with adamantane and adamantane-like cores is the inheritance of both the linear and the nonlinear optical response from the parent molecules to dimers and the crystal structures.642,644 This is admittedly shown in Fig. 29 for the [PhSi{CH2Sn(S)Ph}3] molecule. However, a similar trend has been demonstrated also for many other adamantane-based clusters such as [(PhSi)4S6] and [(NpSi)4S6].309,571Fig. 29 shows that all spectral features of the isolated molecules can be found in the optical response of the crystals, although the spectral weights are somewhat redistributed. This similarity offers the possibility to roughly estimate, e.g., the second and third order optical nonlinearity by the knowledge of the corresponding single molecule. This is a great advantage, in particular in theoretical studies, due to the computationally less extensive investigations of the single clusters as compared to molecular crystals.It has been discussed in many publications that an amorphous structure is a prerequisite for white-light generation.652 Although the connection between the habitus of the aggregate and its nonlinear optical answer is not completely understood, atomistic calculations show a clear relation between aggregate symmetry and intensity of the nonlinear optical response. We demonstrate this connection employing the prototypical adamantane-type cluster [AdPh4] as a model system.
Depending on the environment, the geometry and the symmetry of the cluster undergo slight modifications and so does the nonlinear optical response, quantified, in this example, by the average of all the components of the SHG tensor. [AdPh4] belongs to the space group P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 2n. This group lacks centrosymmetry, so that SHG generation is expected. Indeed, the isolated cluster in its calculated equilibrium geometry is characterized by a second-order optical response as shown by the blue line in Fig. 30. In aggregates such as [AdPh4] crystals, the material can be thought of as a periodic repetition of [AdPh4] dimers.644 The dimers are arranged in a manner that is still not centrosymmetric, however, the deviation from the centrosymmetry is lower than in the case of the isolated clusters. The SHG response per molecule, shown in Fig. 30 (orange line) features all the spectral signatures predicted for the isolated cluster, however, with a lower intensity.
2n. This group lacks centrosymmetry, so that SHG generation is expected. Indeed, the isolated cluster in its calculated equilibrium geometry is characterized by a second-order optical response as shown by the blue line in Fig. 30. In aggregates such as [AdPh4] crystals, the material can be thought of as a periodic repetition of [AdPh4] dimers.644 The dimers are arranged in a manner that is still not centrosymmetric, however, the deviation from the centrosymmetry is lower than in the case of the isolated clusters. The SHG response per molecule, shown in Fig. 30 (orange line) features all the spectral signatures predicted for the isolated cluster, however, with a lower intensity.
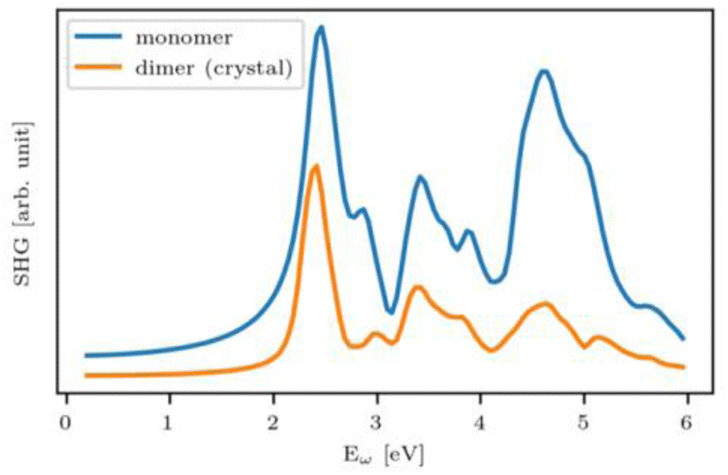 | ||
| Fig. 30 Second-harmonic generation per cluster (the average of the absolute value of all tensor components is shown) calculated within the independent particle approximation as a function of the incident laser wavelength for the isolated [AdPh4] cluster with the DFT equilibrium geometry (blue line) and with the geometry of a cluster dimer cut out from a [AdPh4] crystal (orange line).644 | ||
Artificially modifying the structure towards centrosymmetry further lowers the SHG coefficients. The same behavior is observed for other optically nonlinear molecular clusters. Differences in the symmetry of monomer and dimer structures can also greatly influence the magnitude of the optical response, yet maintaining its overall form. Especially the differences of the nonlinear optics of free-standing dimers compared to their crystal counterparts show that the habitus of the material offers a path to tuning the symmetry and thus the nonlinear optical properties. WLG is only observed in samples with amorphous morphology. The crystalline members of the class of adamantane-based cluster compounds show a different nonlinear optical response, generally SHG originating from the bulk (in the case of crystals lacking inversion symmetry) or from surfaces or interfaces and defects (in the case of centrosymmetric crystals).649 This is shown in Fig. 31 for the crystalline compound [(PhSi)4S6] (568) (a) and for the amorphous material [(PhSn)4S6] (587) (b) as examples. Both clusters are provided with the same organic phenyl ligands and differ just by the exchange of Si atoms by Sn atoms in the adamantane shaped cluster cores. The solid with the {SiS} cluster core (568) shows the typical powder diffractogram of a crystal (Fig. 31a). When irradiated by an optical laser line with wavelength 979 nm, it reacts with an intense second-harmonic at 489 nm. In contrast, the X-ray diffractogram obtained from the solid of the corresponding {SnS} cluster (587) yields a pattern typical for a completely disordered material like a glass, not comprising any information about structural long-range order between the molecules (Fig. 31b). However, the optical response to NIR irradiation at 979 nm is now found to be a broad white emission that spans the entire visible portion of the white-light spectrum.
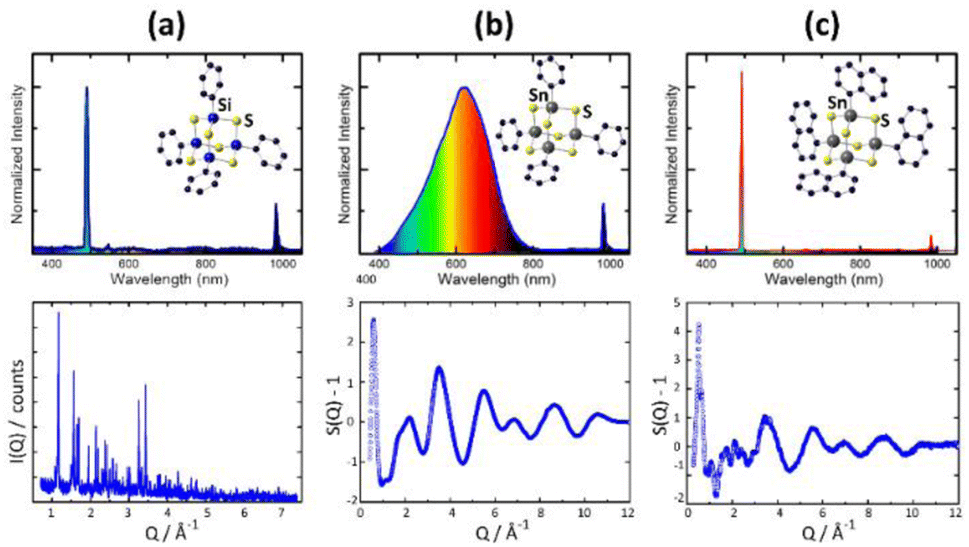 | ||
| Fig. 31 NLO responses from the clusters 568 (a) and 587 (b) (top). The driving excitation is visible at 979 nm (1.265 eV) in each spectrum. The 2nd-harmonics of (a) is seen at 489.5 nm (2.53 eV), while (b) depicts a broad white spectrum. The respective X-ray patterns are also shown below indicating that 568, the SHG-material, is clearly crystalline while the WLG material 587 shows a typical structure factor S(Q) of an amorphous solid. (c) shows the NLO response of cluster 591 indicating SHG, although the X-ray structure factor clearly designates an amorphous solid (reproduced from ref. 657 with permission from the authors under CC BY 4.0). | ||
Meanwhile, a large number of solids of this type could be synthesized showing this correlation between morphology and optical behavior.571 While all crystalline materials have been identified as materials for second-harmonic generation (SHG), it however turned out that among the amorphous materials, it is only the vast majority responding by white-light generators (WLG). A few apparently amorphous clusters whose diffractograms resemble those of glasses, with pronounced structural disorder at the molecular level, nevertheless exhibit SHG upon NIR irradiation. The optical response and X-ray diffractogram of one such example, the {SnS} cluster decorated with naphthyl ligands, [(NpSn)4S6] (591), are shown in Fig. 31c. Although the X-ray diffractogram of (591) does not show typical Bragg peaks but rather resembles the typical structure factor S(Q) for a glass, the optical response when irradiated with the NIR line at 979 nm is found to be SHG.
In trying to understand this behavior, two questions come to mind: What controls the solidification of these cluster molecules into either an amorphous or a crystalline state, and why is pronounced microscopic disorder a prerequisite for a nonlinear response in the form of WLG? In order to answer these questions, it is compulsory to first obtain precise knowledge of the interactions between the clusters, which however requires detailed knowledge of their electronic structure as a function of composition. Then, it must be further understood how the molecular and electronic structure is altered when the clusters aggregate into either a crystalline or amorphous condensed phase. This is a prerequisite for finding approaches from the observed differences that may lead to an understanding of the different optical behavior of these clusters in the two different solid-state morphologies. For this purpose, several optical studies were performed on different cluster systems.653 Furthermore, the electronic structures of many clusters from this group with different organic ligands and {QE} cluster cores were calculated, as well as interaction energies between the clusters for simple arrangements of two to four clusters as simple models to distinguish between different contributions to the interactions.309,644 Also, experimental investigations were accomplished to explore the mutual orientation of the clusters in the amorphous solid.654–657 It showed up thereby however, that the control of the morphology is complex and an assignment amorphous/WLG, crystalline/SHG is oversimplified. It was however found from EXAFS experiments658 that the molecular structure of adamantane-like cluster materials showing SHG (586 and 591) was close to that found from quantum chemical calculations,659 while those showing WLG (587 and 602) considerably deviated from these structures.
More detailed studies carried out on the WLG compound 587 and the SHG compound 591 using X-ray diffraction combined with molecular Reverse Monte Carlo (m-RMC) simulations further revealed that this observation results from significantly distorted cluster cores in the compounds showing WLG. The distortions originate from sulfur atoms moving out of their equilibrium positions, towards other sulfur atoms of neighboring molecules. This may result from strong interactions between the cores of the individual clusters, as was also suggested by quantum chemical calculations,309,660 where smaller cluster aggregates of two to four molecules were assumed as simple models for a disordered phase. Indeed, the mutual cluster orientations predicted there for the WLG compound 587 were also found in the experimental X-ray/RMC-studies657 supporting the theoretical findings. Similar experimental studies on the SHG compound 591 revealed no evidence for distorted cluster cores. The mutual molecular orientation of the clusters in the solid state also suggested stronger intermolecular interactions via the organic ligands, which also supports the quantum chemical calculations made earlier. In addition, electron microscopy could reveal sections of nanocrystallinity, which may additionally explain the findings by ordered parts of the material.
The observation of additional cluster core distortions in the material exhibiting strong WLG compound 587, further highlights the point made earlier that a clear relationship exists between structural and molecular symmetry and intensity of the nonlinear optical response.
6. Conclusion and outlook
The number of compounds based on adamantane-type core architechture is vast, and the elemental compositions of the cluster cores as well as the variety of the substituents is extremely diverse. While this naturally leads to different chemical reactivities and unique electronic structures, a number of them share luminescence phenomena. Only recently, strong second-harmonic generation as well as more uncommon nonlinear optical properties in the form of directed white-light emission have been observed, which seem to be a consequence of the unique adamantane type architecture – with its relatively dense electronic structure and its inherently missing inversion symmetry – as well as of the compounds' arrangement within the solid material.571 Assuming that these phenomena could in theory be observable for other adamantane-type cluster compounds, we have summarized all classes of inorganic and a selection of organic admantanes in this survey. Most of these compounds have not been fully investigated for their physical properties so far, but we suggest that doing so could be a great chance to gain understanding on the optical phenomena in this class of compounds and its potential use. The plethora of different compositions and structural peculiarities is an excellent basis for future investigations into this field, which could become a library of compounds that fulfils any desired properties in regards of wavelength, bandwidth, intensity, and directionality in combination of convenient synthetic access, robustness, and processability of the material.Abbreviations
| IDipp | 1,3-Bis(2,6-diisopropylphenyl)imidazole-2-ylidene |
| HMDS | 1,1,1,3,3,3-Hexamethyldisilazide |
| TACNMe | 1,4,7-Trimethyl-1,4,7-triazacyclononane |
| Bn | Benzyl |
| Cp* | Pentamethylcyclopentadienyl |
| DMAP | Dimethylamine borane |
| BArF | [B[3,5-(CF3)2C6H3] |
| n Bu | Normal butyl |
| t Bu | Tertiary butyl |
| Diglyme | Bis(2-methoxyethyl) ether |
| DMPyr | 1,1-Dimethylpyrrolidinium |
| OTf | O3SCF3 |
| cod | 1,5-Cyclooctadiene |
| OAc | Acetate |
| acac | Acetylacetonate |
| BMIm | 1-Butyl-3-methyl-imidazolium |
| NTf2 | Bistrifluoridomethansulfonimide |
| TMEDA | Tetramethylethylenediamine |
| iPr | Isopropyl |
| DME | 1,2-Dimethoxyethane |
| ArMe6 | C6H3-2,6(C6H2-2,4,6-Me3)2 |
| mes | 2,4,6-Me3-C6H2 |
| TpMe2 | Tri(3,5 dimethylpyrazolyl)borate) |
| TACN | 1,4,7-Triazacyclononane |
| DMSO | Dimethyl sulfoxide |
| CpxPh | C5Me4Ph |
| OHF | 1,2,3,4,5,6,7,8-Octahydrofluorenyl |
| dmae | N,N-Dimethylaminoethanolate |
| TFA | Trifluoacetic acid |
| H4cit | Citric acid |
| H2DBcat | 3,5-Di-tert-butylcatechol |
| HBO | 2-(2′-Hydroxyphenyl)benzoxazole |
| tach | 1,3,5-Triaminocyclohexane |
| en | Ethylendiamine |
| H5hpdta | Hydroxypropanediaminotetraacetic acid |
| HIPAP | N-(tert-Butyl)-3-((3,5-di-tert-butyl-2-hydroxybenzylidene)amino)-propanamide |
| tdmap | OC(CH2NMe2)3 |
| S-Phoz | 2-(4′,4′-Dimethyloxazoline-2′-yl)thiophenolate |
| bpea | N,N-Bis(2-pyridylmethyl)ethylamine |
| [3,5-(CF3)2C6H3]4B]− |
| dien | Diethylenetriamine |
| Medien | N′-Methyldiethylenetriamine |
| R-ida | N-(R)Iminodiacetate |
| C Pe | Cyclopentane |
| tame | tert-Amyl methyl ether |
| Htphpn | N,N,N′,N′-Tetra-(2-methylpyridyl)-2-hydroxypropanediamine |
| pz | Pyrazolyl |
| H2BMAP | 2-[bis(2-mercaptoethyl)aminomethyl]pyridine |
| Py | Pyridine |
| H5HMeXCG | N,N′-(2-Hydroxy-5-methyl-1,3-xylylene)bis(N-(carboxymethyl)glycine) |
| H5HPhXCG | N,N′-(2-Hydroxy-5-phenyl-1,3-xylylene)bis(N-(carboxymethyl)glycine) |
| Dma | N,N-Dimethylacetamid |
| Hbpbp | 2,6-Bis((N,N′-bis-(2-picolyl)amino)methyl)-4-tert-butylphenol |
| {(TACN)CH2}2CHOH | 1,3-Bis(1,4,7-triaza-1-cyclononyl)-2-hydroxypropane |
| N-Et-HPTB | N,N,N′,N′-Tetrakis(2-(1-ethylbenzimidazolyl))-2-hydroxy-1,3-diaminopropane |
| dppoe | 1,2-Bis(diphenylphosphine oxide)ethane |
| dppe | 1,2-Bis(diphenylphosphino)ethane |
| H3HMPM | 2,6-Bis[{{(1-hydroxy-2-methylpropan-2-yl)(pyridine-2-ylmethyl)}amino}methyl]-4-methylphenol |
| Me2phen | 2,9-Dimethyl-1,10-phenanthroline |
| BIK | Bis(2-methyl-imidazole-2-yl)ketone |
| t Bu2DED | 1,1-Dicarbo-tert-butoxy-2,2-ethylenedithiolate |
| tpdt | 3,4-Thiophenedithiolate |
| α-tpdt | 2,3-Thiophenedithiolate |
| H4pymtH | 3,4,5,6-Tetrahydropyrimidine-2-thione |
| H3O3N4 | 1-Me-4-OH-3,4-bis(CH2N(CH2C5H4N)(CMe2CH2OH)–C6H2 |
| Fc | Ferrocenyl |
| 2-TBI | 2-Thiobenzimidadzol |
| Mbis | 1,1′-Methylenebis(3-methylimidazoline-2-selone) |
| DAMS | Trans-4-(4-dimethylamino-styryl)-N-methyl-pyridinium |
| bipy | Bipyridine |
| n Pr | Normal propyl |
| secBu | Secondary butyl |
| phen | 1,10-Phenanthroline |
| o Py | ortho-Pyridyl |
| Tab | 4-(Trimethylammonio)benzenethiolate |
| [2.2.2]-crypt | 4,7,13,16,21,24-Hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane |
| POPYH4 | N,N′-Bis(2-hydroxyphenyl)-pyridine-2,6-dicarboxamide |
| dap | 1,2-Diaminopropane |
| tepa | Tetraethylenepentamine |
| Vio | Viologen dication |
| TMPyP | 5,10,15,20-Tetrakis(N-methyl-4-pyridyl)porphyrin |
| DMBPE | N,N′-Dimethyl-1,2-bis(4-pyridinium)-ethylene |
| Cyclam | 1,4,8,11-Tetraazacyclotetradecane |
| Trien | Triethylentetramin |
| Teta | Triethylenetetramine |
| Thex | 1,1,2-Trimethylpropyl |
| Np | Naphthyl |
| Sty | para-Styryl |
| Cy | Cyclohexyl |
| Cp | Cyclopentadienyl |
| Dipp | 2,6-Diisopropylphenyl |
| DMEGqu | N-(1,3-Dimethylimidazolidin2-ylidene)quinoline-8-amine |
| 8-HQ | 8-Hydroxyquinoline |
| H2naphpz | 2-[1H-Pyrazol-5(3)-yl]naphthalene-1-ol |
| dpan | 6-Diphenylphosphinoacenaphth-5-yl |
| LOEt | [Co(η5-C5H5){P(O)(OEt)2}3]− |
| 3-NO2Tp | 3-Nitrotrispyrazolylborate |
| SON | (Benzothiazole-2-yl)phenolate |
| HBT | 2-(2-Hydroxyphenyl)benzothiazole |
| H3L | 2-Hydroxy-N-[2-hydroxy-3-[(2hydroxybenzoyl)amino]propyl]benzamide |
| C4mim | 1-Butyl-3-methylimidazolium |
| da6aH6 | p-Methyl-dimethyldiazacalix[6]areneH6 |
| HMTA | Hexamethylentetramine |
| HBDA | Hexakis(trimethylsilyl)benzdiamidine |
| cpz | 2-Chloro-10-(3-dimethylaminopropyl(phenothiazine) |
| DENC | N,N-Diethylnicotinamide |
| PziPr2H | 3,5-Diisopropylpyrazole |
| DASO | Diallyl sulfoxide |
| Amt | 1,3-Diamino-1,2,2-trimethylcyclopentane |
| CgP | 1,3,5,7-Tetramethyl-2,4,6-trioxa-8-phosphatricyclo[3.3.1.1]-decane |
| nmp | N-Methyl-2-pyrrolidinone |
| teed | N,N,N′,N′-Tetraethylethylenediamine |
| BPBACy | Bis(1-propylbenzimidazol-2-yl)-trans-1,2-cyclohexane |
| dpipa | N,N′-Dimethylpiperazine |
| PoxIm | N-Phenyl-N′-{bis(tertbutyl)phosphinoxide}-imidazolylidene |
| Dabco | 1,4-Diazabicyclo[2.2.2]octane |
| tib | 1,3,5-Tris(1-imidazolyl)benzene |
| pyr | Pyrrolidine |
| dppbO2 | 1,2-bis(diephenlyphospineoxide) benzol |
| tdpmO3 | tris(diephenlyphospineoxide) methan |
| BPPIP | Bis-triphenylphosphonio-isophosphindolide |
| THP | Tetrahydropyran |
| tren | Tris(2-ethylamino)amine |
| gua | Guanidine |
| bme*daco | Bis(N,N′-2-mercapto-2-methylpropyl)1,5-diazocyclooctane |
| Bdpman | N,N′-Bis(diphenylmethyl)-3,7-diazabicyclo[3.3.1]nonane |
| Hbdmap | 1,3-Bis-(dimethylamino)-propan-2-ol |
| n Hep | Normal heptane |
| TAEA | Tris(2-aminoethyl)amine |
| tdci | 1,3,5-Trideoxy-1,2,5-tris(dimethylamino)-cis-inositol |
| Hchp | 6-Chloro-2-hydroxypyridine |
| dmit | 4,5-Dimercapto-1,3-dithiole-2-thionato |
| EDTA | Ethylenediamine-tetraacetate |
| BMMIm | 1-Butyl-2,3-dimethyl-imidazolium |
| MCPBA | m-Chloroperbenzoic acid |
Author contributions
All athors agreed on the concept of the article and co-wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Computations for this research were conducted on the Lichtenberg high-performance computer of the TU Darmstadt and at the Höchstleistungrechenzentrum Stuttgart (HLRS). The authors furthermore acknowledge the computational resources provided by the HPC Core Facility and the HRZ of the Justus-Liebig-Universität Gießen. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – 2082/1 – 390761711 and within the network of DFG Research Unit FOR 2824.Notes and references
- A. F. Holleman, E. Wiberg and N. Wiberg, Anorganische Chemie, De Gruyter, Berlin/Boston, 2016 Search PubMed.
- H. Decker, Z. Angew. Chem., 1924, 37, 795 Search PubMed.
- S. Landa and V. Macháček, Collect. Czech. Chem. Commun., 1933, 5, 1 CrossRef CAS.
- W. A. Tilden and R. E. Barnett, J. Chem. Soc. Trans., 1896, 69, 154 RSC.
- H.-H. Yang, C.-H. Chien, C.-C. Yang, F.-C. Liu, A. H. H. Chang, G.-H. Lee and S.-M. Peng, Dalton Trans., 2013, 42, 1168 RSC.
- O. Tardif, M. Nishiura and Z. Hou, Organometallics, 2003, 22, 1171 CrossRef CAS.
- J. Cheng, K. Saliu, G. Y. Kiel, M. J. Ferguson, R. McDonald and J. Takats, Angew. Chem., Int. Ed., 2008, 47, 4910 CrossRef CAS PubMed.
- T. Shima, Y. Luo, T. Stewart, R. Bau, G. J. McIntyre, S. A. Mason and Z. Hou, Nat. Chem., 2011, 3, 814 CrossRef CAS PubMed.
- T. Shima, S. Hu, G. Luo, X. Kang, Y. Luo and Z. Hou, Science, 2013, 340, 1549 CrossRef CAS PubMed.
- T. Höllerhage, P. Ghana, T. P. Spaniol, A. Carpentier, L. Maron, U. Englert and J. Okuda, Angew. Chem., Int. Ed., 2022, 61, e202115379 CrossRef PubMed.
- M. Arrowsmith, M. S. Hill, D. J. MacDougall and M. F. Mahon, Angew. Chem., Int. Ed., 2009, 48, 4013 CrossRef CAS PubMed.
- D. Schuhknecht, T. P. Spaniol, I. Douair, L. Maron and J. Okuda, Chem. Commun., 2019, 55, 14837 RSC.
- H. D. Kaesz, B. Fontal, R. Bau, S. W. Kirtley and M. R. Churchill, J. Am. Chem. Soc., 1969, 91, 1021 CrossRef CAS.
- G. Ciani, A. Sironi and V. G. Albano, J. Organomet. Chem., 1977, 136, 339 CrossRef CAS.
- A. J. Roberts, W. Clegg, A. R. Kennedy, M. R. Probert, S. D. Robertson and E. Hevia, Dalton Trans., 2015, 44, 8169 RSC.
- L. S. Sharninghausen, B. Q. Mercado, R. H. Crabtree, D. Balcells and J. Campos, Dalton Trans., 2015, 44, 18403 RSC.
- L. S. Sharninghausen, B. Q. Mercado, C. Hoffmann, X. Wang, J. Campos, R. H. Crabtree and D. Balcells, J. Organomet. Chem., 2017, 849–850, 17 CrossRef CAS.
- K. Izod, C. Wills, W. Clegg and R. W. Harrington, Organometallics, 2007, 26, 2861 CrossRef CAS.
- M. P. Brown, A. K. Holliday and G. M. Way, J. Chem. Soc., Dalton Trans., 1975, 148 RSC.
- I. Rayment and H. M. M. Shearer, J. Chem. Soc., Dalton Trans., 1977, 136 RSC.
- W. Haubold, W. Keller and G. Sawitzki, J. Organomet. Chem., 1989, 367, 19 CrossRef CAS.
- R. Köster, H.-J. Horstschäfer, P. Binger and P. K. Mattschei, Adv. Cycloaddit., 1975, 1975, 1339 Search PubMed.
- R. Köster, G. Seidel and B. Wrackmeyer, Angew. Chem., Int. Ed., 1985, 24, 326 CrossRef.
- W. Uhl, L. Cuypers, B. Neumüller and F. Weller, Organometallics, 2002, 21, 2365 CrossRef CAS.
- A. Dashti-Mommertz, B. Neumüller and Z. A. Aallg, Chem, 1999, 625, 954 CAS.
- C. Y. Tang, A. R. Cowley, A. J. Downs and S. Parsons, Inorg. Chem., 2007, 46, 5439 CrossRef CAS PubMed.
- H. Binder, H. Loos, K. Dermentzis, H. Borrmann and A. Simon, Chem. Ber., 1991, 124, 427 CrossRef CAS.
- J. Guschlbauer, T. Vollgraff, L. H. Finger, K. Harms and J. Sundermeyer, ChemistryOpen, 2021, 10, 83 CrossRef CAS PubMed.
- J. T. Boronski, M. P. Stevens, B. IJzendoorn, A. C. Whitwood and J. M. Slattery, Angew. Chem., Int. Ed., 2021, 60, 1567 CrossRef CAS PubMed.
- C. Ruspic and S. Harder, Organometallics, 2005, 24, 5506 CrossRef CAS.
- S. H. Carpenter, T. M. Baker, S. B. Muñoz, W. W. Brennessel and M. L. Neidig, Chem. Sci., 2018, 9, 7931 RSC.
- S. Wolf, F. Winter, R. Pöttgen, N. Middendorf, W. Klopper and C. Feldmann, Chem. Eur J., 2012, 18, 13600 CrossRef CAS PubMed.
- M. J. Bennett, F. A. Cotton and B. H. C. Winquist, J. Am. Chem. Soc., 1967, 89, 5366 CrossRef CAS.
- M. Bochmann, I. Hawkins, M. B. Hursthouse and R. L. Short, J. Organomet. Chem., 1987, 332, 361 CrossRef CAS.
- M. Bochmann, I. Hawkins, L. J. Yellowlees, M. B. Hursthouse and R. L. Short, Polyhedron, 1989, 8, 1351 CrossRef CAS.
- E. G. Mednikov, N. K. Eremenko, S. P. Gubin, Yu. L. Slovokhotov and Yu. T. Struchkov, J. Organomet. Chem., 1982, 239, 401 CrossRef CAS.
- A. L. Smith and H. A. Clark, J. Am. Chem. Soc., 1961, 83, 3345 CrossRef CAS.
- G. Fritz and J. Grobe, Z. Anorg. Allg. Chem., 1962, 315, 157 CrossRef CAS.
- G. Fritz, R. Haase and D. Kummer, Z. Anorg. Allg. Chem., 1969, 365, 1 CrossRef CAS.
- E. W. Krahé, R. Mattes, K.-F. Tebbe, H. G. V. Schnering and G. Fritz, Z. Anorg. Allg. Chem., 1972, 393, 74 CrossRef.
- C. L. Frye, J. M. Klosowski and D. R. Weyenberg, J. Am. Chem. Soc., 1970, 92, 6379 CrossRef CAS.
- G. Fritz and J. Honold, Z. Anorg. Allg. Chem., 1988, 556, 23 CrossRef CAS.
- S. N. Gurkova, A. I. Gusev, V. A. Sharapov, T. K. Gar and N. V. Alekseev, J. Struct. Chem., 1979, 20, 302 CrossRef.
- C. L. Frye and J. M. Klosowski, J. Am. Chem. Soc., 1972, 94, 7186 CrossRef CAS.
- G. D. Homer and L. H. Sommer, J. Am. Chem. Soc., 1973, 95, 7700 CrossRef CAS.
- K. K. Laali, G. F. Koser, S. D. Huang and M. Gangoda, J. Organomet. Chem., 2002, 658, 141 CrossRef CAS.
- G. Fritz, J. Neutzner and H. Volk, Z. Anorg. Allg. Chem., 1983, 497, 21 CrossRef CAS.
- R. J. Wehmschulte, K. K. Laali, G. L. Borosky and D. R. Powell, Organometallics, 2014, 33, 2146 CrossRef CAS.
- J. Fischer, J. Baumgartner and C. Marschner, Science, 2005, 310, 825 CrossRef CAS PubMed.
- T. C. Siu, M. Imex Aguirre Cardenas, J. Seo, K. Boctor, M. G. Shimono, I. T. Tran, V. Carta and T. A. Su, Angew. Chem., Int. Ed., 2022, 61, e202206877 CrossRef CAS PubMed.
- B. Köstler, M. Bolte, H. Lerner and M. Wagner, Chem. Eur J., 2021, 27, 14401 CrossRef PubMed.
- B. Köstler, J. Gilmer, M. Bolte, A. Virovets, H.-W. Lerner, P. Albert, F. Fantuzzi and M. Wagner, Chem. Commun., 2023, 59, 2295 RSC.
- G. Fritz, R. Uhlmann and W. Hölderich, Z. Anorg. Allg. Chem., 1978, 442, 86 CrossRef CAS.
- W. Hönle and H. G. von Schnering, Z. Anorg. Allg. Chem., 1978, 442, 91 CrossRef.
- G. Fritz and R. Biastoch, Z. Anorg. Allg. Chem., 1986, 535, 63 CrossRef CAS.
- G. Fritz and J. Reuter, Z. Anorg. Allg. Chem., 1989, 578, 27 CrossRef CAS.
- G. Fritz and J. Reuter, Z. Anorg. Allg. Chem., 1989, 575, 39 CrossRef CAS.
- A. R. Dahl and A. D. Norman, J. Am. Chem. Soc., 1970, 92, 5525 CrossRef CAS.
- A. R. Dahl, A. D. Norman, H. Shenav and R. Schaeffer, J. Am. Chem. Soc., 1975, 97, 6364 CrossRef CAS.
- H. Schumann, Angew. Chem., 1969, 81, 970 CrossRef.
- O. Fuhr and D. Fenske, Z. Anorg. Allg. Chem., 2004, 630, 244 CrossRef CAS.
- W. Hönle and G. von Schnering, Z. Naturforsch. B, 1980, 35, 789 CrossRef.
- G. Becker, G. Gutekunst and H. J. Wessely, Z. Anorg. Allg. Chem., 1980, 462, 113 CrossRef CAS.
- B. Neumüller and K. Dehnicke, Z. Anorg. Allg. Chem., 2004, 630, 347 CrossRef.
- M. Müller, A. J. Karttunen and M. R. Buchner, Chem. Sci., 2020, 11, 5415 RSC.
- M. A. Beswick, P. R. Raithby, C. A. Russell, A. Steiner, K. L. Verhorevoort, G. N. Ward and D. S. Wright, Angew. Chem., Int. Ed., 1996, 34, 2662 CrossRef.
- P. J. Harford, J. Haywood, M. R. Smith, B. N. Bhawal, P. R. Raithby, M. Uchiyama and A. E. H. Wheatley, Dalton Trans., 2012, 41, 6148 RSC.
- K. Kaniewska, Ł. Ponikiewski, N. Szynkiewicz, B. Cieślik, J. Pikies, J. Krzystek, A. Dragulescu-Andrasi, S. A. Stoian and R. Grubba, Dalton Trans., 2020, 49, 10091 RSC.
- A. W. Cook, J. D. Bocarsly, R. A. Lewis, A. J. Touchton, S. Morochnik and T. W. Hayton, Chem. Sci., 2020, 11, 4753 RSC.
- H. Nöth and P. Konrad, Chem. Ber., 1968, 101, 3423 CrossRef.
- U. Thewalt and I. Kawada, Chem. Ber., 1970, 103, 2754 CrossRef CAS.
- K. W. Henderson, A. R. Kennedy, A. E. McKeown and R. E. Mulvey, Angew. Chem., Int. Ed., 2000, 39, 3879 CrossRef CAS PubMed.
- C. E. Melton, J. W. Dube, P. J. Ragogna, J. C. Fettinger and P. P. Power, Organometallics, 2014, 33, 329 CrossRef CAS.
- B. Luo and W. L. Gladfelter, Inorg. Chem., 2002, 41, 6249 CrossRef CAS PubMed.
- H. Schumann and H. Benda, Angew. Chem., 1969, 81, 1049 CrossRef.
- M. Baudler, W. Oehlert and K.-F. Tebbe, Z. Anorg. Allg. Chem., 1991, 598, 9 CrossRef.
- P. B. Hitchcock, M. F. Lappert and P. Yin, J. Chem. Soc., Chem. Commun., 1992, 1598 RSC.
- R. R. Holmes and J. A. Forstner, J. Am. Chem. Soc., 1960, 82, 5509 CrossRef CAS.
- R. R. Holmes, J. Am. Chem. Soc., 1961, 83, 1334 CrossRef CAS.
- T. G. Hill, R. C. Haltiwanger, M. L. Thompson, S. A. Katz and A. D. Norman, Inorg. Chem., 1994, 33, 1770 CrossRef CAS.
- A. B. Spore, N. M. Rizzo, B. C. Noll and R. D. Sommer, Inorg. Chim. Acta, 2010, 364, 261 CrossRef CAS.
- H.-J. Vetter, H. Nöth and W. Jahn, Z. Anorg. Allg. Chem., 1964, 328, 144 CrossRef CAS.
- J. Weiss and W. Eisenhuth, Z. Anorg. Allg. Chem., 1967, 350, 9 CrossRef CAS.
- O. J. Scherer, K. Andres, C. Krüger, Y.-H. Tsay and G. Wolmerhäser, Angew. Chem., Int. Ed., 1980, 19, 571 CrossRef.
- A. Bashall, E. L. Doyle, F. García, G. T. Lawson, D. J. Linton, D. Moncrieff, M. McPartlin, A. D. Woods and D. S. Wright, Chem. Eur J., 2002, 8, 5723 CrossRef CAS PubMed.
- Y. X. Shi, K. Xu, J. K. Clegg, R. Ganguly, H. Hirao, T. Friščić and F. García, Angew. Chem., Int. Ed., 2016, 55, 12736 CrossRef CAS PubMed.
- G. W. Hunt and A. W. Cordes, Inorg. Chem., 1974, 13, 1688 CrossRef CAS.
- R. R. Holmes and J. A. Forstner, Inorg. Chem., 1963, 2, 377 CrossRef CAS.
- J. G. Riess and A. Wolff, J. Chem. Soc., Chem. Commun., 1972, 1050 RSC.
- F. Casabianca, A. A. Pinkerton and J. G. Riess, Inorg. Chem., 1977, 16, 864 CrossRef CAS.
- F. Casabianca, F. A. Cotton, J. G. Riess, C. E. Rice and B. R. Stults, Inorg. Chem., 1978, 17, 3232 CrossRef CAS.
- F. A. Cotton, J. G. Riess, C. E. Rice and B. R. Stults, Inorg. Chem., 1978, 17, 3521 CrossRef CAS.
- F. A. Cotton, J. G. Riess, C. E. Rice and B. R. Stults, Inorg. Chem., 1982, 21, 3123 CrossRef CAS.
- J. G. Riess and J. R. Van Wazer, J. Organomet. Chem., 1967, 8, 347 CrossRef CAS.
- J. Bedard, N. J. Roberts, M. Shayan, K. L. Bamford, U. Werner-Zwanziger, K. M. Marczenko and S. S. Chitnis, Angew. Chem., Int. Ed., 2022, 61, e202204851 CrossRef CAS PubMed.
- H. Jacobs, S. Pollok and F. Golinski, Z. Anorg. Allg. Chem., 1994, 620, 1213 CrossRef CAS.
- F. Golinski and H. Jacobs, Z. Anorg. Allg. Chem., 1995, 621, 29 CrossRef CAS.
- H. Schmidbaur, M. Schmidt, A. Schier, J. Riede, T. Tamm and P. Pyykkö, J. Am. Chem. Soc., 1998, 120, 2967 CrossRef CAS.
- K. F. Tesh and T. P. Hanusa, J. Chem. Soc., Chem. Commun., 1991, 879 RSC.
- E. Le Coz, J. Hammoud, T. Roisnel, M. Cordier, V. Dorcet, S. Kahlal, J. Carpentier, J. Saillard and Y. Sarazin, Chem. Eur J., 2021, 27, 11966 CrossRef CAS PubMed.
- F. Zhang, J. Zhang and X. Zhou, Inorg. Chem., 2017, 56, 2070 CrossRef CAS PubMed.
- K. Wieghardt, D. Ventur, Y. H. Tsai and C. Krüger, Inorg. Chim. Acta, 1985, 99, L25 CrossRef CAS.
- S. Rabe and U. Müller, Z. Naturforsch. B, 1997, 52, 1291 CrossRef CAS.
- L. M. Babcock, V. W. Day and W. G. Klemperer, J. Chem. Soc., Chem. Commun., 1987, 858 RSC.
- J. Schellenberg, Eur. Polym. J., 2004, 40, 2259 CrossRef CAS.
- M. Björgvinsson, S. Halldorsson, I. Arnason, J. Magull and D. Fenske, J. Organomet. Chem., 1997, 544, 207 CrossRef.
- S. C. Yoon, B.-J. Bae, I.-H. Suh and J. T. Park, Organometallics, 1999, 18, 2049 CrossRef CAS.
- T. E. Hanna, E. Lobkovsky and P. J. Chirik, Inorg. Chem., 2007, 46, 2359 CrossRef CAS PubMed.
- A. A. Tahir, M. Hamid, M. Mazhar, M. Zeller, A. D. Hunter, M. Nadeem and M. J. Akhtar, Dalton Trans., 2008, 1224 RSC.
- M. A. Ehsan, A. A. Tahir, M. Hamid, M. Mazhar, K. G. U. Wijayantha and M. Zeller, Inorg. Chim. Acta, 2011, 376, 189 CrossRef CAS.
- M. A. Ehsan, H. Khaledi, A. Pandikumar, N. M. Huang, Z. Arifin and M. Mazhar, J. Solid State Chem., 2015, 230, 155 CrossRef CAS.
- N. Engelberg, A. Bino and E. Y. Tshuva, Inorg. Chim. Acta, 2020, 503, 119429 CrossRef CAS.
- Y. Chi, J.-W. Lan, W.-L. Ching, S.-M. Peng and G.-H. Lee, J. Chem. Soc., Dalton Trans., 2000, 2923 RSC.
- F. Bottomley, C. P. Magill and B. Zhao, Organometallics, 1990, 9, 1700 CrossRef CAS.
- S. Pappuru, D. Chakraborty and V. Ramkumar, Dalton Trans., 2017, 46, 16640 RSC.
- A. Noll and U. Müller, Z. Anorg. Allg. Chem., 1999, 625, 1721 CrossRef.
- D. Wormsbächer, K. M. Nicholas and A. L. Rheingold, J. Chem. Soc., Chem. Commun., 1985, 721 RSC.
- J. Glerup, H. Weihe, P. A. Goodson and D. J. Hodgson, Inorg. Chim. Acta, 1993, 212, 281 CrossRef CAS.
- S. B. Mukkamala, R. Clérac, C. E. Anson and A. K. Powell, Polyhedron, 2006, 25, 530 CrossRef CAS.
- N. Zwettler, M. A. Ehweiner, J. A. Schachner, A. Dupé, F. Belaj and N. C. Mösch-Zanetti, Molecules, 2019, 24, 1814 CrossRef CAS PubMed.
- H. Choujaa, A. L. Johnson, G. Kociok-Köhn and K. C. Molloy, Dalton Trans., 2012, 41, 11393 RSC.
- H. Choujaa, A. L. Johnson, G. Kociok-Köhn and K. C. Molloy, Polyhedron, 2013, 63, 199 CrossRef CAS.
- S. Kuwata, Y. Mizobe and M. Hidai, J. Chem. Soc., Dalton Trans., 1997, 1753 RSC.
- L. M. Peschel, C. Vidovič, F. Belaj, D. Neshchadin and N. C. Mösch-Zanetti, Chem. Eur J., 2019, 25, 3893 CrossRef CAS PubMed.
- K. Wieghardt, U. Bossek and W. Gebert, Angew. Chem., Int. Ed., 1983, 22, 328 CrossRef.
- K. Wieghardt, U. Bossek, B. Nuber, J. Weiss, J. Bonvoisin, M. Corbella, S. E. Vitols and J. J. Girerd, J. Am. Chem. Soc., 1988, 110, 7398 CrossRef CAS.
- L. Zhang, S. Yan, C. Li, D. Liao, Z. Jiang, P. Cheng, G. Wang, L. Weng and X. Leng, J. Chem. Crystallogr., 2000, 30, 251 CrossRef CAS.
- T. H. Bennur, D. Srinivas, S. Sivasanker and V. G. Puranik, J. Mol. Catal. A: Chem., 2004, 219, 209 CrossRef CAS.
- C. E. Dubé, S. Mukhopadhyay, P. J. Bonitatebus, R. J. Staples and W. H. Armstrong, Inorg. Chem., 2005, 44, 5161 CrossRef PubMed.
- K. S. Hagen, T. D. Westmoreland, M. J. Scott and W. H. Armstrong, J. Am. Chem. Soc., 1989, 111, 1907 CrossRef CAS.
- C. E. Dubé, D. W. Wright, S. Pal, P. J. Bonitatebus and W. H. Armstrong, J. Am. Chem. Soc., 1998, 120, 3704 CrossRef.
- S. Mukhopadhyay, H. J. Mok, R. J. Staples and W. H. Armstrong, J. Am. Chem. Soc., 2004, 126, 9202 CrossRef CAS PubMed.
- T. Costa, J. R. Dorfman, K. S. Hagen and R. H. Holm, Inorg. Chem., 1983, 22, 4091 CrossRef CAS.
- H.-O. Stephan, K. Griesar, H. Wolfgang and G. Henkel, Z. Naturforsch. B, 1994, 49, 1620 CrossRef CAS.
- H.-O. Stephan and G. Henkel, Polyhedron, 1996, 15, 501 CrossRef CAS.
- M. Mikuriya and T. Kotera, Chem. Lett., 1998, 27, 971 CrossRef.
- B. P. Murch, P. D. Boyle and L. Que, J. Am. Chem. Soc., 1985, 107, 6728 CrossRef CAS.
- W. Schmitt, M. Murugesu, J. C. Goodwin, J. P. Hill, A. Mandel, R. Bhalla, C. E. Anson, S. L. Heath and A. K. Powell, Polyhedron, 2001, 20, 1687 CrossRef CAS.
- W. Schmitt, P. A. Jordan, R. K. Henderson, G. R. Moore, C. E. Anson and A. K. Powell, Coord. Chem. Rev., 2002, 228, 115 CrossRef CAS.
- M. Ghiladi, F. B. Larsen, C. J. McKenzie, I. Søtofte and J.-P. Tuchagues, Dalton Trans., 2005, 1687 RSC.
- J. L. Sessler, J. W. Sibert, A. K. Burrell, V. Lynch, J. T. Markert and C. L. Wooten, Inorg. Chem., 1993, 32, 4277 CrossRef CAS.
- A. Majumdar, U.-P. Apfel, Y. Jiang, P. Moënne-Loccoz and S. J. Lippard, Inorg. Chem., 2014, 53, 167 CrossRef CAS PubMed.
- A. Pietrzak, J. Guschlbauer and P. Kaszyński, Materials, 2021, 14, 6840 CrossRef CAS PubMed.
- S. Drueke, K. Wieghardt, B. Nuber, J. Weiss, E. L. Bominaar, A. Sawaryn, H. Winkler and A. X. Trautwein, Inorg. Chem., 1989, 28, 4477 CrossRef CAS.
- D. Zipse, K. A. Abboud and N. S. Dalal, J. Appl. Phys., 2003, 93, 7086 CrossRef CAS.
- M. K. Jin, Y. Kim, D.-Y. Jung, M. Heu, S. Yoon and B. J. Suh, Bull. Korean Chem. Soc., 2005, 26, 253 CrossRef CAS.
- K. S. Hagen, J. M. Berg and R. H. Holm, Inorg. Chim. Acta, 1980, 45, L17 CrossRef CAS.
- K. S. Hagen, D. W. Stephan and R. H. Holm, Inorg. Chem., 1982, 21, 3928 CrossRef CAS.
- K. S. Hagen and R. H. Holm, Inorg. Chem., 1984, 23, 418 CrossRef CAS.
- D. Coucouvanis, M. Kanatzidis, E. Simhon and N. C. Baenziger, J. Am. Chem. Soc., 1982, 104, 1874 CrossRef CAS.
- M. A. Whitener, J. K. Bashkin, K. S. Hagen, J. J. Girerd, E. Gamp, N. Edelstein and R. H. Holm, J. Am. Chem. Soc., 1986, 108, 5607 CrossRef CAS.
- A. L. Eckermann, M. Wunder, D. Fenske, T. B. Rauchfuss and S. R. Wilson, Inorg. Chem., 2002, 41, 2004 CrossRef CAS PubMed.
- P. Mondal, R. Singh, A. Hens, J. Cano, E. Colacio and K. K. Rajak, Polyhedron, 2013, 65, 60 CrossRef CAS.
- I. G. Dance, J. Am. Chem. Soc., 1979, 101, 6264 CrossRef CAS.
- D. Fenske, J. Meyer and K. Merzweiler, Z. Naturforsch. B, 1987, 42, 1207 CrossRef CAS.
- W. Tremel, B. Krebs, K. Greiwe, W. Simon, H.-O. Stephan and G. Henkel, Z. Naturforsch. B, 1992, 47, 1580 CrossRef CAS.
- I. G. Dance, J. Chem. Soc., Chem. Commun., 1976, 103 RSC.
- I. G. Dance and J. C. Calabrese, Inorg. Chim. Acta, 1976, 19, L41 CrossRef.
- I. G. Dance, G. A. Bowmaker, G. R. Clark and J. K. Seadon, Polyhedron, 1983, 2, 1031 CrossRef CAS.
- D. Coucouvanis, C. N. Murphy and S. K. Kanodia, Inorg. Chem., 1980, 19, 2993 CrossRef CAS.
- M. Baumgartner, W. Bensch, P. Hug and E. Dubler, Inorg. Chim. Acta, 1987, 136, 139 CrossRef CAS.
- A. Eichhöfer, H. Sommer, V. Andrushko, S. Indris and S. Malik, Eur. J. Inorg. Chem., 2013, 2013, 1531 CrossRef.
- G. Bowmaker and L. Tan, Aust. J. Chem., 1979, 32, 1443 CrossRef CAS.
- M. Baumgartner, H. Schmalle and C. Baerlocher, J. Solid State Chem., 1993, 107, 63 CrossRef CAS.
- M. Baumgartner, H. Schmalle and E. Dubler, Polyhedron, 1990, 9, 1155 CrossRef CAS.
- K. Fujisawa, S. Imai, S. Suzuki, Y. Moro-oka, Y. Miyashita, Y. Yamada and K. Okamoto, J. Inorg. Biochem., 2000, 82, 229 CrossRef CAS PubMed.
- S. Zeevi and E. Y. Tshuva, Eur. J. Inorg. Chem., 2007, 2007, 5369 CrossRef.
- A. Kohner-Kerten and E. Y. Tshuva, J. Organomet. Chem., 2008, 693, 2065 CrossRef CAS.
- C. Chen, Z. Weng and J. F. Hartwig, Organometallics, 2012, 31, 8031 CrossRef CAS PubMed.
- B. K. Maiti, K. Pal and S. Sarkar, Eur. J. Inorg. Chem., 2007, 2007, 5548 CrossRef.
- A. T. Royappa, R. J. Papoular, M. Gembicky, W. Shepard, A. D. Ross, A. G. Stemen, J. J. Bobbitt, D. T. Doan, S. H. Lapidus, D. H. Johnston and A. Filatov, Polyhedron, 2022, 222, 115873 CrossRef CAS.
- A. F. Stange, E. Waldhör, M. Moscherosch and W. Kaim, Z. Naturforsch. B, 1995, 50, 115 CrossRef CAS.
- X. Jin, K. Tang, Y. Long and Y. Tang, Acta Crystallogr., 1999, C55, 1799 CAS.
- O. Kluge and H. Krautscheid, Inorg. Chem., 2012, 51, 6655 CrossRef CAS PubMed.
- M. W. DeGroot, M. W. Cockburn, M. S. Workentin and J. F. Corrigan, Inorg. Chem., 2001, 40, 4678 CrossRef CAS PubMed.
- J. R. Nicholson, I. L. Abrahams, W. Clegg and C. D. Garner, Inorg. Chem., 1985, 24, 1092 CrossRef CAS.
- G. Henkel, B. Krebs, P. Betz, H. Fietz and K. Saatkamp, Angew. Chem., Int. Ed., 1988, 27, 1326 CrossRef.
- M. Baumgartner, H. Schmalle and E. Dubler, Inorg. Chim. Acta, 1993, 208, 135 CrossRef CAS.
- M. Kischel, G. Dornberg and H. Krautscheid, Inorg. Chem., 2014, 53, 1614 CrossRef CAS PubMed.
- J. Dai, M. Munakata, Y. Ohno, G. Bian and Y. Suenaga, Inorg. Chim. Acta, 1999, 285, 332 CrossRef CAS.
- D. Belo, M. J. Figueira, J. Mendonça, I. C. Santos, M. Almeida, R. T. Henriques, M. T. Duarte, C. Rovira and J. Veciana, Eur. J. Inorg. Chem., 2005, 2005, 3337 CrossRef.
- S. Kanodia and D. Coucouvanis, Inorg. Chem., 1982, 21, 469 CrossRef CAS.
- D. Coucouvanis, S. Kanodia, D. Swenson, S. J. Chen, T. Stuedemann, N. C. Baenziger, R. Pedelty and M. Chu, J. Am. Chem. Soc., 1993, 115, 11271 CrossRef CAS.
- A. Müller, N. H. Schladerbeck, E. Krickemeyer, H. Bögge, K. Schmitz, E. Bill and A. X. Trautwein, Z. Anorg. Allg. Chem., 1989, 570, 7 CrossRef.
- E. H. Griffith, G. W. Hunt and E. L. Amma, J. Chem. Soc., Chem. Commun., 1976, 432 RSC.
- M. Van Meerssche, R. Kamara, G. Germain and J. P. Declercq, Bull. Soc. Chim. Belg., 1982, 91, 553 CrossRef CAS.
- J. P. Declercq, R. Kamara, C. Moreaux, J. M. Dereppe, G. Germain and M. Van Meerssche, Acta Crystallogr., 1978, B34, 1036 CrossRef CAS.
- S. Kitagawa, Y. Nozaka, M. Munakata and S. Kawata, Inorg. Chim. Acta, 1992, 197, 169 CrossRef CAS.
- V. V. Olijnyk, Ya. E. Filinchuk and N. L. Pandiak, Z. Anorg. Allg. Chem., 2003, 629, 1904 CrossRef.
- R. C. Bott, G. A. Bowmaker, C. A. Davis, G. A. Hope and B. E. Jones, Inorg. Chem., 1998, 37, 651 CrossRef CAS.
- L. A. Solov’ev, A. D. Vasil’ev and N. N. Golovnev, Russ. J. Coord. Chem., 2002, 28, 587 CrossRef.
- C. Pakawatchai, Y. Thanyasirikul, T. Saepae, S. Pansook, H.-K. Fun and K. Chinnakali, Acta Crystallogr., 1998, C54, 1750 Search PubMed.
- G. A. Bowmaker, J. V. Hanna, C. Pakawatchai, B. W. Skelton, Y. Thanyasirikul and A. H. White, Inorg. Chem., 2009, 48, 350 CrossRef CAS PubMed.
- Ya. E. Filinchuk, V. V. Oliinik, T. Glovyak and M. G. Mys’kiv, Russ. J. Coord. Chem., 2001, 27, 126 CrossRef CAS.
- O. Siiman, C. P. Huber and M. L. Post, Inorg. Chim. Acta, 1977, 25, L11 CrossRef CAS.
- C. P. Huber, M. L. Post and O. Siiman, Acta Crystallogr., 1978, B34, 2629 CrossRef CAS.
- H. Liu, N. A. G. Bandeira, M. J. Calhorda, M. G. B. Drew, V. Félix, J. Novosad, F. F. D. Biani and P. Zanello, J. Organomet. Chem., 2004, 689, 2808 CrossRef CAS.
- M. C. Aragoni, M. Arca, M. B. Carrea, F. Demartin, F. A. Devillanova, A. Garau, M. B. Hursthouse, S. L. Huth, F. Isaia, V. Lippolis, H. R. Ogilvie and G. Verani, Eur. J. Inorg. Chem., 2006, 2006, 200 CrossRef.
- A. Banerjee, R. Singh, P. Mondal, E. Colacio and K. K. Rajak, Eur. J. Inorg. Chem., 2010, 2010, 790 CrossRef.
- G. Henkel, P. Betz and B. Krebs, Angew. Chem., Int. Ed., 1987, 26, 145 CrossRef.
- A. I. Wallbank and J. F. Corrigan, J. Cluster Sci., 2004, 15, 225 CrossRef CAS.
- P. Sun, X. Tang, W. Yang, X. Wang, R. Zhou, N. Chen and S.-F. Yuan, Inorg. Chem., 2022, 61, 9251 CrossRef CAS PubMed.
- J. Zhao, D. Adcock, W. T. Pennington and J. W. Kolis, Inorg. Chem., 1990, 29, 4358 CrossRef CAS.
- M. G. Kanatzidis and S.-P. Huang, Angew. Chem., Int. Ed., 1989, 28, 1513 CrossRef.
- S. Canales, O. Crespo, M. C. Gimeno, P. G. Jones, A. Laguna, A. Silvestru and C. Silvestru, Inorg. Chim. Acta, 2003, 347, 16 CrossRef CAS.
- M. T. Aroz, M. C. Gimeno, M. Kulcsar, A. Laguna and V. Lippolis, Eur. J. Inorg. Chem., 2011, 2011, 2884 CrossRef CAS.
- J. L. Hencher, M. Khan, F. F. Said and D. G. Tuck, Inorg. Nucl. Chem. Lett., 1981, 17, 287 CrossRef CAS.
- J. L. Hencher, M. A. Khan, F. F. Said and D. G. Tuck, Polyhedron, 1985, 4, 1263 CrossRef CAS.
- L. Pandolfo, R. Seraglia, A. Venzo, S. Gross and G. Kickelbick, Inorg. Chim. Acta, 2005, 358, 2739 CrossRef CAS.
- I. G. Dance, Inorg. Chem., 1981, 20, 2155 CrossRef CAS.
- J. Sun, X. Tao, Y. Chuai, F. Wang, D. Zou, J. Yang, Y. Ren, Z. Liu and M. Jiang, Inorg. Chem. Commun., 2006, 9, 942 CrossRef CAS.
- Y. Liu, Q. Lin, Q. Zhang, X. Bu and P. Feng, Chem. Eur J., 2014, 20, 8297 CrossRef CAS PubMed.
- M. Gelinsky and H. Vahrenkamp, Z. Anorg. Allg. Chem., 2002, 628, 1017 CrossRef CAS.
- P. A. W. Dean, J. J. Vittal and N. C. Payne, Inorg. Chem., 1987, 26, 1683 CrossRef CAS.
- J. J. Vittal, P. A. W. Dean and N. C. Payne, Can. J. Chem., 1992, 70, 792 CrossRef CAS.
- A. Choy, D. Craig, I. Dance and M. Scudder, J. Chem. Soc., Chem. Commun., 1982, 1246 RSC.
- K. S. Hagen and R. H. Holm, Inorg. Chem., 1983, 22, 3171 CrossRef CAS.
- X. Zhang, Y. Tian, F. Jin, J. Wu, Y. Xie, X. Tao and M. Jiang, Cryst. Growth Des., 2005, 5, 565 CrossRef CAS.
- J.-B. Jiang, G.-Q. Bian, Y.-P. Zhang, W. Luo, Q.-Y. Zhu and J. Dai, Dalton Trans., 2011, 40, 9551 RSC.
- Y. Matsunaga, K. Fujisawa, N. Ibi, M. Fujita, T. Ohashi, N. Amir, Y. Miyashita, K. Aika, Y. Izumi and K. Okamoto, J. Inorg. Biochem., 2006, 100, 239 CrossRef CAS PubMed.
- C. Xu, Z. Zhou and H. Han, Crystals, 2022, 12, 1236 CrossRef CAS.
- P. A. W. Dean and J. J. Vittal, Inorg. Chem., 1986, 25, 514 CrossRef CAS.
- X.-Y. Tang, R.-X. Yuan, J.-X. Chen, W. Zhao, A.-X. Zheng, M. Yu, H.-X. Li, Z.-G. Ren and J.-P. Lang, Dalton Trans., 2012, 41, 6162 RSC.
- P. A. W. Dean, N. C. Payne, J. Wranich and J. J. Vittal, Polyhedron, 1998, 17, 2411 CrossRef CAS.
- P. A. W. Dean and V. Manivannan, Inorg. Chem., 1990, 29, 2997 CrossRef CAS.
- P. A. W. Dean, J. J. Vittal and Y. Wu, Can. J. Chem., 1992, 70, 779 CrossRef CAS.
- K. S. Anjali, K. W. K. Thia, T. T. Low, M. Q. Chen, H. Huan and J. J. Vittal, Main Group Met. Chem., 2001, 24, 229 CAS.
- V. N. Soloviev, A. Eichhöfer, D. Fenske and U. Banin, J. Am. Chem. Soc., 2001, 123, 2354 CrossRef CAS PubMed.
- T. P. Lebold, D. L. B. Stringle, M. S. Workentin and J. F. Corrigan, Chem. Commun., 2003, 1398 RSC.
- R. Stieler, F. Bublitz, E. Schulz Lang and G. Manzoni De Oliveira, Polyhedron, 2012, 31, 596 CrossRef CAS.
- K. Tang, T. Xia, X. Jin and Y. Tang, Polyhedron, 1994, 13, 3023 CrossRef CAS.
- C. Xu, H.-T. Shi, Z.-F. Xin, A.-Q. Jia and Q.-F. Zhang, J. Cluster Sci., 2014, 25, 1353 CrossRef CAS.
- F. D. Da Silva, A. L. Hennemann, R. A. Burrow, E. S. Lang, U. Abram and S. S. Dos Santos, J. Cluster Sci., 2022, 33, 815 CrossRef CAS.
- R.-F. Wang, W.-G. Zhang, J. Fan and S.-L. Wang, Z. Kristallogr., 2005, 220, 61 CAS.
- P. A. W. Dean, N. C. Payne, J. J. Vittal and Y. Yu, Inorg. Chem., 1993, 32, 4632 CrossRef CAS.
- P. A. W. Dean, J. J. Vittal and M. H. Trattner, Inorg. Chem., 1987, 26, 4245 CrossRef CAS.
- J. J. Vittal, P. A. W. Dean and N. C. Payne, Can. J. Chem., 1993, 71, 2043 CrossRef CAS.
- J. Martí-Rujas and M. Cametti, New J. Chem., 2014, 38, 1385 RSC.
- Y. Pérez, A. L. Johnson and P. R. Raithby, Polyhedron, 2011, 30, 284 CrossRef.
- B. Krebs, D. Voelker and K.-O. Stiller, Inorg. Chim. Acta, 1982, 65, L101 CrossRef CAS.
- J. Zhou, Y. Zhang, G.-Q. Bian, C.-Y. Li, X.-X. Chen and J. Dai, Cryst. Growth Des., 2008, 8, 2235 CrossRef CAS.
- C. L. Cahill and J. B. Parise, J. Chem. Soc., Dalton Trans., 2000, 1475 RSC.
- M. Sun, S. Zhang, K.-Y. Wang, J. Wang, L. Cheng, J.-Y. Zhu, Y.-M. Zhao and C. Wang, Inorg. Chem., 2021, 60, 7115 CrossRef CAS PubMed.
- R. J. Wehmschulte and P. P. Power, J. Am. Chem. Soc., 1997, 119, 9566 CrossRef CAS.
- H. Schmidbaur and S. D. Nogai, Z. Anorg. Allg. Chem., 2004, 630, 2218 CrossRef CAS.
- A. Boardman, S. E. Jeffs, R. W. H. Small and I. J. Worrall, Inorg. Chim. Acta, 1984, 83, L39 CrossRef CAS.
- A. Boardman, R. W. H. Small and I. J. Worrall, Inorg. Chim. Acta, 1986, 120, L23 CrossRef CAS.
- K. Wieghardt, M. Kleine-Boymann, B. Nuber and J. Weiss, Inorg. Chem., 1986, 25, 1654 CrossRef CAS.
- J. D. Young, M. A. Khan, D. R. Powell and R. J. Wehmschulte, Eur. J. Inorg. Chem., 2007, 2007, 1671 CrossRef.
- G. Linti and W. Köstler, Chem. Eur J., 1998, 4, 942 CrossRef CAS.
- C. Schnitter, H. W. Roesky, T. Albers, H.-G. Schmidt, C. Röpken, E. Parisini and G. M. Sheldrick, Chem. Eur J., 1997, 3, 1783 CrossRef CAS.
- S. S. Al-Juaid, N. H. Buttrus, C. Eaborn, P. B. Hitchcock, A. T. L. Roberts, J. D. Smith and A. C. Sullivan, J. Chem. Soc., Chem. Commun., 1986, 908 RSC.
- M. Ribes, J. Olivier-Fourcade, E. Philippot and M. Maurin, J. Solid State Chem., 1973, 8, 195 CrossRef CAS.
- B. Eisenmann, J. Hansa and H. Schäfer, Z. Naturforsch. B, 1985, 40, 450 CrossRef.
- B. Eisenmann and H. Schäfer, Z. Anorg. Allg. Chem., 1982, 491, 67 CrossRef CAS.
- B. Krebs and S. Pohl, Z. Naturforsch. B, 1971, 26, 853 CrossRef CAS.
- E. Philippot, M. Ribes and M. Maurin, Rev. Chim. Miner., 1971, 8, 477 CAS.
- S. Pohl and B. Krebs, Z. Anorg. Allg. Chem., 1976, 424, 265 CrossRef CAS.
- B. Krebs and H. Müller, Z. Anorg. Allg. Chem., 1983, 496, 47 CrossRef CAS.
- C. L. Bowes, A. J. Lough, A. Malek, G. A. Ozin, S. Petrov and D. Young, Chem. Ber., 1996, 129, 283 CrossRef CAS.
- K. O. Klepp and M. Zeitlinger, Z. Kristallogr., 2000, 215, 7 CAS.
- G. Eulenberger, Acta Crystallogr., 1976, B32, 3059 CrossRef CAS.
- O. M. Yaghi, Z. Sun, D. A. Richardson and T. L. Groy, J. Am. Chem. Soc., 1994, 116, 807 CrossRef CAS.
- J. Y. Pivan, O. Achak, M. Louer and D. Louer, Chem. Mater., 1994, 6, 827 CrossRef CAS.
- R. Bedard, L. D. Vail, S. T. Wilson and E. N. Flaningen, Crystalline Microporous Metal Sulfide Compositions, US Pat., 4880761, 1989 Search PubMed.
- M. J. MacLachlan, S. Petrov, R. L. Bedard, I. Manners and G. A. Ozin, Angew. Chem., Int. Ed., 1998, 37, 2075 CrossRef PubMed.
- B. Eisenmann and J. Hansa, Z. für Kristallogr. – Cryst. Mater., 1993, 205, 325 CrossRef CAS.
- B. Eisenmann and J. Hansa, Z. für Kristallogr. – Cryst. Mater., 1993, 206, 101 CAS.
- W. S. Sheldrick and B. Schaaf, Z. Naturforsch. B, 1995, 50, 1469 CrossRef CAS.
- W. S. Sheldrick and B. Schaaf, Z. Naturforsch. B, 1994, 49, 655 CrossRef CAS.
- G. Eulenberger, Z. Naturforsch. B, 1981, 36, 521 CrossRef CAS.
- H. Ahari, A. Garcia, S. Kirkby, G. A. Ozin, D. Young and A. J. Lough, J. Chem. Soc., Dalton Trans., 1998, 2023 RSC.
- R. Blachnik and A. Fehlker, Z. Kristallogr., 2000, 215, 95 CAS.
- S. S. Dhingra and R. C. Haushalter, Polyhedron, 1994, 13, 2775 CrossRef CAS.
- K. Tsamourtzi, J.-H. Song, T. Bakas, A. J. Freeman, P. N. Trikalitis and M. G. Kanatzidis, Inorg. Chem., 2008, 47, 11920 CrossRef CAS PubMed.
- A. M. Pirani, H. P. A. Mercier, D. A. Dixon, H. Borrmann and G. J. Schrobilgen, Inorg. Chem., 2001, 40, 4823 CrossRef CAS PubMed.
- J. Campbell, D. P. DiCiommo, H. P. A. Mercier, A. M. Pirani, G. J. Schrobilgen and M. Willuhn, Inorg. Chem., 1995, 34, 6265 CrossRef CAS.
- H. Ahari, A. Lough, S. Petrov, G. A. Ozin and R. L. Bedard, J. Mater. Chem., 1999, 9, 1263 RSC.
- A. Fehlker and R. Blachnik, Z. Anorg. Allg. Chem., 2001, 627, 411 CrossRef CAS.
- A. Fehlker and R. Blachnik, Z. Anorg. Allg. Chem., 2001, 627, 1128 CrossRef CAS.
- K.-Y. Wang, S. Zhang, H.-W. Liu, L. Cheng and C. Wang, Inorg. Chem., 2019, 58, 12832 CrossRef CAS PubMed.
- M. Duchardt, S. Haddadpour, T. Kaib, P. Bron, B. Roling and S. Dehnen, Inorg. Chem., 2021, 60, 5224 CrossRef CAS PubMed.
- C.-W. Park, M. A. Pell and J. A. Ibers, Inorg. Chem., 1996, 35, 4555 CrossRef CAS.
- F. Bonhomme and M. G. Kanatzidis, Chem. Mater., 1998, 10, 1153 CrossRef CAS.
- M. Wachhold and M. G. Kanatzidis, Chem. Mater., 2000, 12, 2914 CrossRef CAS.
- K. K. Rangan and M. G. Kanatzidis, Inorg. Chim. Acta, 2004, 357, 4036 CrossRef CAS.
- M.-S. Wang, W.-T. Chen, L.-Z. Cai, G.-W. Zhou, G.-C. Guo and J.-S. Huang, J. Cluster Sci., 2003, 14, 495 CrossRef CAS.
- J. Lin, Z. Fu, J. Zhang, Y. Zhu, D. Hu, D. Li and T. Wu, Inorg. Chem., 2017, 56, 3119 CrossRef CAS PubMed.
- X.-L. Sun, Q.-Y. Zhu, W.-Q. Mu, L.-W. Qian, L. Yu, J. Wu, G.-Q. Bian and J. Dai, Dalton Trans., 2014, 43, 12582 RSC.
- J. Xu, L.-J. Xue, J.-L. Hou, Z.-N. Yin, X. Zhang, Q.-Y. Zhu and J. Dai, Inorg. Chem., 2017, 56, 8036 CrossRef CAS PubMed.
- X.-L. Sun, Q.-Y. Zhu, L.-W. Qian, L. Yu, Z.-G. Ren, G.-Q. Bian and J. Dai, Inorg. Chem. Commun., 2014, 46, 130 CrossRef CAS.
- F. Danker, C. Näther, F. Pielnhofer and W. Bensch, Eur. J. Inorg. Chem., 2017, 2017, 4317 CrossRef CAS.
- W.-Q. Mu, Q.-Y. Zhu, L.-S. You, X. Zhang, W. Luo, G.-Q. Bian and J. Dai, Inorg. Chem., 2012, 51, 1330 CrossRef CAS PubMed.
- J.-J. Liang, J. Zhao, W.-W. Tang, Y. Zhang and D.-X. Jia, Inorg. Chem. Commun., 2011, 14, 1023 CrossRef CAS.
- F. Zhang, X. Liu, J. Zhou, X.-H. Yin and J. He, Monatsh. Chem., 2011, 142, 763 CrossRef CAS.
- F. Zhang, X.-H. Yin, X. Liu and J. Zhou, Z. Anorg. Allg. Chem., 2011, 637, 1388 CrossRef CAS.
- G.-N. Liu, J.-D. Lin, Z.-N. Xu, Z.-F. Liu, G.-C. Guo and J.-S. Huang, Cryst. Growth Des., 2011, 11, 3318 CrossRef CAS.
- H.-P. Xiao, J. Zhou, X.-L. Wang, H.-H. Zou, R.-Q. Zhao and H. Xiao, Dalton Trans., 2014, 43, 12306 RSC.
- H.-Y. Luo and J. Zhou, Dalton Trans., 2018, 47, 14751 RSC.
- J. Zhou, H. Xiao, H.-P. Xiao, T. Yang, H.-H. Zou, X. Liu, R.-Q. Zhao and Q. Tang, Dalton Trans., 2015, 44, 1350 RSC.
- M.-L. Feng, W.-W. Xiong, D. Ye, J.-R. Li and X.-Y. Huang, Chem.–Asian J., 2010, 5, 1817 CrossRef CAS PubMed.
- T. Van Almsick, A. Kromm and W. S. Sheldrick, Z. Anorg. Allg. Chem., 2005, 631, 19 CrossRef CAS.
- P. Pfeiffer and L. Rügheimer, Ber. Dtsch. Chem. Ges., 1903, 36, 3027 CrossRef CAS.
- J. A. Forstner and E. L. Muetterties, Inorg. Chem., 1966, 5, 552 CrossRef.
- J. C. J. Bart and J. J. Daly, J. Chem. Soc., Dalton Trans., 1975, 2063 RSC.
- M. Unno, Y. Kawai, H. Shioyama and H. Matsumoto, Organometallics, 1997, 16, 4428 CrossRef CAS.
- M. Unno, D. Ishii and H. Matusmoto, Bull. Chem. Soc. Jpn., 1999, 72, 2469 CrossRef CAS.
- N. Rinn, L. Guggolz, H. Y. Hou and S. Dehnen, Chem. Eur J., 2021, 27, 11167 CrossRef CAS PubMed.
- K. Hanau, S. Schwan, M. R. Schäfer, M. J. Müller, C. Dues, N. Rinn, S. Sanna, S. Chatterjee, D. Mollenhauer and S. Dehnen, Angew. Chem., Int. Ed., 2021, 60, 1176 CrossRef CAS PubMed.
- N. W. Rosemann, J. P. Eußner, E. Dornsiepen, S. Chatterjee and S. Dehnen, J. Am. Chem. Soc., 2016, 138, 16224 CrossRef CAS PubMed.
- K. Moedritzer, Inorg. Chem., 1967, 6, 1248 CrossRef CAS.
- R. H. Benno and C. J. Fritchie, J. Chem. Soc., Dalton Trans., 1973, 543 RSC.
- A. Haas, H. -Jürgen Kutsch and C. Krüger, Chem. Ber., 1987, 120, 1045 CrossRef CAS.
- S. Heimann, G. Thiele and S. Dehnen, J. Organomet. Chem., 2016, 813, 36 CrossRef CAS.
- D. Kobelt, E. F. Paulus and H. Scherer, Acta Crystallogr., 1972, B28, 2323 CrossRef.
- M. Komura and R. Okawara, Inorg. Nucl. Chem. Lett., 1966, 2, 93 CrossRef CAS.
- K. Wraage, T. Pape, R. Herbst-Irmer, M. Noltemeyer, H.-G. Schmidt and H. W. Roesky, Eur. J. Inorg. Chem., 1999, 1999, 869 CrossRef.
- A. Blecher, B. Mathiasch and M. Drager, Z. Naturforsch. B, 1981, 36, 1361 CrossRef.
- H. Berwe and A. Haas, Chem. Ber., 1987, 120, 1175 CrossRef CAS.
- N. W. Rosemann, J. P. Eußner, A. Beyer, S. W. Koch, K. Volz, S. Dehnen and S. Chatterjee, Science, 2016, 352, 1301 CrossRef CAS PubMed.
- E. Dornsiepen, F. Dobener, S. Chatterjee and S. Dehnen, Angew. Chem., Int. Ed., 2019, 58, 17041 CrossRef CAS PubMed.
- C. Wagner, C. Raschke and K. Merzweiler, Appl. Organomet. Chem., 2004, 18, 147 CrossRef CAS.
- Z. H. Fard, L. Xiong, C. Müller, M. Hołyńska and S. Dehnen, Chem. Eur J., 2009, 15, 6595 CrossRef CAS PubMed.
- K. Schwedtmann, A. Hepp, K. Schwedtmann, J. J. Weigand and F. Lips, Eur. J. Inorg. Chem., 2019, 2019, 4719 CrossRef CAS.
- M. Zhong, Z. Yang, Y. Yi, D. Zhang, K. Sun, H. W. Roesky and Y. Yang, Dalton Trans., 2015, 44, 19800 RSC.
- K. Merzweiler and L. Weisse, Z. Naturforsch. B, 1990, 45, 971 CrossRef CAS.
- K. Merzweiler and H. Kraus, Z. Naturforsch. B, 1994, 49, 621 CrossRef CAS.
- R. M. Harker, M. F. Mahon and K. C. Molloy, Main Group Met. Chem., 1996, 19, 29 CAS.
- C. Pöhlker, I. Schellenberg, R. Pöttgen and S. Dehnen, Chem. Commun., 2010, 46, 2605 RSC.
- Z. You, J. Bergunde, B. Gerke, R. Pöttgen and S. Dehnen, Inorg. Chem., 2014, 53, 12512 CrossRef CAS PubMed.
- E. Dornsiepen, F. Pieck, R. Tonner and S. Dehnen, J. Am. Chem. Soc., 2019, 141, 16494 CrossRef CAS PubMed.
- E. Dornsiepen, J. P. Eußner, N. W. Rosemann, S. Chatterjee and S. Dehnen, Inorg. Chem., 2017, 56, 11326 CrossRef CAS PubMed.
- C. Gaffney, P. G. Harrison and T. J. King, J. Chem. Soc., Chem. Commun., 1980, 1251 RSC.
- A. Someşan, E. Le Coz, C. I. Raţ, V. Dorcet, T. Roisnel, C. Silvestru and Y. Sarazin, Chem. Eur J., 2019, 25, 16236 CrossRef PubMed.
- E. Wiberg and W. Simmler, Z. Anorg. Allg. Chem., 1955, 282, 330 CrossRef CAS.
- K. Bakthavachalam, K. Yuvaraj, B. Mondal, R. Prakash and S. Ghosh, Dalton Trans., 2015, 44, 17920 RSC.
- P. Kitschke, L. Mertens, T. Rüffer, H. Lang, A. A. Auer and M. Mehring, Eur. J. Inorg. Chem., 2015, 2015, 4996 CrossRef CAS.
- A. Jesser, I. D. S. Vieira and S. Herres-Pawlis, Z. Naturforsch. B, 2013, 68, 653 CrossRef CAS.
- M. L. Walker and J. L. Mills, Synth. React. Inorg. Met.-Org. Chem., 1975, 5, 29 CrossRef CAS.
- K. H. Jost and M. Schneider, Acta Crystallogr., 1981, B37, 222 CrossRef CAS.
- M. H. Möbs and M. Jansen, Z. Anorg. Allg. Chem., 1984, 514, 39 CrossRef.
- M. Jansen and M. Voss, Angew. Chem., Int. Ed., 1981, 20, 100 CrossRef.
- M. L. Walker, D. E. Peckenpaugh and J. L. Mills, Inorg. Chem., 1979, 18, 2792 CrossRef CAS.
- J. Clade, M. Jansen, B. Engels and C. M. Marian, Z. Anorg. Allg. Chem., 1995, 621, 2065 CrossRef CAS.
- T. E. Thorpe and A. E. Tutton, J. Chem. Soc. Trans., 1891, 59, 1019 RSC.
- F. C. Mijlhoff, J. Portheine and C. Romers, Recl. Trav. Chim. Pays-Bas, 1967, 86, 257 CrossRef.
- F. Frick and M. Jansen, Z. für Kristallogr. – Cryst. Mater., 1994, 209, 985 CrossRef CAS.
- M. Jansen and S. Strojek, Z. Anorg. Allg. Chem., 1995, 621, 479 CrossRef CAS.
- J. Clade and M. Jansen, Z. Anorg. Allg. Chem., 1997, 623, 1407 CrossRef CAS.
- J. G. Riess and J. R. Van Wazer, J. Am. Chem. Soc., 1965, 87, 5506 CrossRef CAS.
- J. G. Riess and J. R. Van Wazer, J. Am. Chem. Soc., 1966, 88, 2166 CrossRef CAS.
- E. D. Pierron, P. J. Wheatley and J. G. Riess, Acta Crystallogr., 1966, 21, 288 CrossRef CAS.
- M. Jansen and J. Clade, Acta Crystallogr., 1996, C52, 2650 CAS.
- A. Stock, Ber. Dtsch. Chem. Ges., 1910, 43, 1223 CrossRef CAS.
- A. Vos and E. H. Wiebenga, Acta Crystallogr., 1955, 8, 217 CrossRef CAS.
- A. Vos, R. Olthof, F. Van Bolhuis and R. Botterweg, Acta Crystallogr., 1965, 19, 864 CrossRef CAS.
- A. K. Jami and V. Baskar, Dalton Trans., 2012, 41, 12524 RSC.
- J. Brünig, E. Hupf, E. Lork, S. Mebs and J. Beckmann, Dalton Trans., 2015, 44, 7105 RSC.
- R. D. Rogers, A. H. Bond and M. M. Witt, Inorg. Chim. Acta, 1991, 182, 9 CrossRef CAS.
- L. Salmon, P. Thuéry and M. Ephritikhine, Polyhedron, 2006, 25, 1537 CrossRef CAS.
- M. A. Singh-Wilmot, I. A. Kahwa, A. J. P. White, D. J. Williams and A. J. Lough, Polyhedron, 2010, 29, 270 CrossRef CAS.
- N. Arleth, S. Bestgen, M. T. Gamer and P. W. Roesky, J. Am. Chem. Soc., 2014, 136, 14023 CrossRef CAS PubMed.
- S. T. Tsantis, A. Lagou-Rekka, K. F. Konidaris, C. P. Raptopoulou, V. Bekiari, V. Psycharis and S. P. Perlepes, Dalton Trans., 2019, 48, 15668 RSC.
- S. R. Tamang, A. Singh, D. Bedi, A. R. Bazkiaei, A. A. Warner, K. Glogau, C. McDonald, D. K. Unruh and M. Findlater, Nat. Catal., 2020, 3, 154 CrossRef CAS.
- Q. Yang, L. Ungur, L. F. Chibotaru and J. Tang, Chem. Commun., 2022, 58, 1784 RSC.
- G. Wang, Y. So, K. Wong, K. Au-Yeung, H. H. -Y. Sung, I. D. Williams and W. Leung, Chem. Eur J., 2015, 21, 16126 CrossRef CAS PubMed.
- C. H. Hossack, R. J. Butcher, C. L. Cahill and C. Besson, Inorg. Chem., 2021, 60, 15724 CrossRef CAS PubMed.
- T. V. Balashova, V. A. Ilichev, I. D. Grishin, R. V. Rumyantcev, G. K. Fukin and M. N. Bochkarev, Inorg. Chim. Acta, 2018, 483, 379 CrossRef CAS.
- S.-Y. Lin, B. Sun and Z. Xu, Inorg. Chim. Acta, 2017, 464, 119 CrossRef CAS.
- H. Fink, E. Spundflasche and H.-J. Seifert, Z. für Kristallogr. – Cryst. Mater., 1994, 209, 400 CrossRef CAS.
- G. Stucky and R. E. Rundle, J. Am. Chem. Soc., 1964, 86, 4821 CrossRef CAS.
- H. Vitze, H.-W. Lerner and M. Bolte, Acta Crystallogr., 2011, E67, m1614 Search PubMed.
- A. Decken, H. D. B. Jenkins, C. Knapp, G. B. Nikiforov, J. Passmore and J. M. Rautiainen, Angew. Chem., Int. Ed., 2005, 44, 7958 CrossRef CAS PubMed.
- M. Jura, W. Levason, E. Petts, G. Reid, M. Webster and W. Zhang, Dalton Trans., 2010, 39, 10264 RSC.
- Z. Mazej and E. Goreshnik, Inorg. Chem., 2009, 48, 6918 CrossRef CAS PubMed.
- T. Xing, T. J. Prior, K. Chen and C. Redshaw, Dalton Trans., 2021, 50, 4396 RSC.
- M. K. Mishra, S. P. Kelley, M. Dilip, T. P. Vaid, D. B. Cordes, S. T. Griffin and R. D. Rogers, Inorg. Chem., 2019, 58, 1764 CrossRef CAS PubMed.
- G. Bresciani, M. Bortoluzzi, S. Zacchini, F. Marchetti and G. Pampaloni, Eur. J. Inorg. Chem., 2018, 2018, 999 CrossRef CAS.
- F. A. Cotton, C. A. Murillo and I. Pascual, Inorg. Chem., 1999, 38, 2746 CrossRef CAS.
- S. Trzmiel, J. Langmann, D. Werner, C. Maichle-Mössmer, W. Scherer and R. Anwander, Angew. Chem., Int. Ed., 2021, 60, 20049 CrossRef CAS PubMed.
- T. S. Cameron, T. M. Klapötke, A. Schulz and J. Valkonen, J. Chem. Soc., Dalton Trans., 1993, 659 RSC.
- B. Beagley, C. A. McAuliffe, P. P. M. Rory, P. T. Ndifon and R. G. Pritchard, J. Chem. Soc., Chem. Commun., 1990, 309 RSC.
- B. Beagley, A. G. Mackie, P. P. Matear, C. A. McAuliffe, P. T. Ndifon and R. G. Pritchard, J. Chem. Soc., Dalton Trans., 1992, 1301 RSC.
- O. Guesmi, M. S. M. Abdelbaky, D. Martínez-Blanco, L. Ktari, S. García-Granda and M. Dammak, Inorg. Chim. Acta, 2019, 496, 119033 CrossRef CAS.
- B. T. Kilbourn and J. D. Dunitz, Inorg. Chim. Acta, 1967, 1, 209 CrossRef CAS.
- J. A. Bertrand, Inorg. Chem., 1967, 6, 495 CrossRef CAS.
- N. S. Gill and M. Sterns, Inorg. Chem., 1970, 9, 1619 CrossRef CAS.
- R. C. Dickinson, F. T. Helm, W. A. Baker, T. D. Black and W. H. Watson, Inorg. Chem., 1977, 16, 1530 CrossRef CAS.
- J. Pickardt and N. Rautenberg, Z. Naturforsch. B, 1982, 37, 1355 CrossRef.
- J. T. Guy, J. C. Cooper, R. D. Gilardi, J. L. Flippen-Anderson and C. F. George, Inorg. Chem., 1988, 27, 635 CrossRef CAS.
- W. Clegg, J. R. Nicholson, D. Collison and C. D. Garner, Acta Crystallogr., 1988, C44, 453 CAS.
- R. E. Norman, N. J. Rose and R. E. Stenkamp, Acta Crystallogr., 1989, C45, 1707 CAS.
- S. Brownstein, N. F. Han, E. Gabe and F. Lee, Can. J. Chem., 1989, 67, 551 CrossRef CAS.
- S. Löw, J. Becker, C. Würtele, A. Miska, C. Kleeberg, U. Behrens, O. Walter and S. Schindler, Chem. Eur J., 2013, 19, 5342 CrossRef PubMed.
- W. Hiller, A. Zinn and K. Dehnicke, Z. Naturforsch. B, 1990, 45, 1593 CrossRef CAS.
- A. Erdonmez, J. H. Van Diemen, R. A. G. De Graaff and J. Reedijk, Acta Crystallogr., 1990, C46, 402 CAS.
- H. M. Haendler, Acta Crystallogr., 1990, C46, 2054 CAS.
- F. S. Keij, J. G. Haasnoot, A. J. Oosterling, J. Reedijk, C. J. O'Connor, J. H. Zhang and A. L. Spek, Inorg. Chim. Acta, 1991, 181, 185 CrossRef CAS.
- M. A. El-Sayed, A. Ali, G. Davies, S. Larsen and J. Zubieta, Inorg. Chim. Acta, 1992, 194, 139 CrossRef CAS.
- J. Poitras and A. L. Beauchamp, Can. J. Chem., 1992, 70, 2846 CrossRef CAS.
- V. Pavlenko, V. Kokozay and O. Babich, Z. Naturforsch. B, 1993, 48, 1321 CrossRef CAS.
- K. Yamada, E. Oguma, H. Nakagawa and H. Kawazura, Chem. Pharm. Bull., 1994, 42, 368 CrossRef CAS.
- P. C. M. Duncan, D. M. L. Goodgame, M. A. Hitchman, S. Menzer, H. Stratemeier and D. J. Williams, J. Chem. Soc., Dalton Trans., 1996, 4245 RSC.
- P. Weinberger, R. Schamschule, K. Mereiter, L. Dlhán, R. Boca and W. Linert, J. Mol. Struct., 1998, 446, 115 CrossRef CAS.
- K. Gubina, V. Ovchynnikov and V. Amirkhanov, Acta Crystallogr., 2014, E70, m276 Search PubMed.
- P. F. Kelly, S.-M. Man, A. M. Z. Slawin and K. W. Waring, Polyhedron, 1999, 18, 3173 CrossRef CAS.
- A. M. Atria, A. Vega, M. Contreras, J. Valenzuela and E. Spodine, Inorg. Chem., 1999, 38, 5681 CrossRef CAS.
- R. Becker, J. Weiß, M. Winter, K. Merz and R. A. Fischer, J. Organomet. Chem., 2001, 630, 253 CrossRef CAS.
- C. A. Bolos and P. C. Christidis, Acta Crystallogr., 2002, C58, m29 CAS.
- C. Näther and I. Jeß, Acta Crystallogr., 2002, E58, m4 Search PubMed.
- C. Richardson and P. J. Steel, Dalton Trans., 2003, 992 RSC.
- X. Liu, C. A. Kilner and M. A. Halcrow, Acta Crystallogr., 2003, C59, m100 CAS.
- F. Jian, P. Zhao, H. Xiao and H. Wang, Anal. Sci.: X-Ray Struct. Anal. Online, 2004, 20, X23 CAS.
- A. S. Lyakhov, P. N. Gaponik, M. M. Degtyarik and L. S. Ivashkevich, Acta Crystallogr., 2004, C60, m399 CAS.
- K. Skorda, T. C. Stamatatos, A. P. Vafiadis, A. T. Lithoxoidou, A. Terzis, S. P. Perlepes, J. Mrozinski, C. P. Raptopoulou, J. C. Plakatouras and E. G. Bakalbassis, Inorg. Chim. Acta, 2005, 358, 565 CrossRef CAS.
- P. Cortés, A. M. Atria, M. T. Garland and R. Baggio, Acta Crystallogr., 2006, C62, m311 Search PubMed.
- Z. Fu and T. Chivers, Can. J. Chem., 2007, 85, 358 CrossRef CAS.
- Ž. K. Jaćimović, V. M. Leovac and Z. D. Tomić, Z. Kristallogr., 2007, 222, 246 Search PubMed.
- R. Vafazadeh, A. C. Willis, M. Mehdi Heidari and N. Hasanzade, Acta Chim. Slov., 2015, 62, 122 CrossRef CAS PubMed.
- Z. Cui, Y. Ma, Y. Ling and X. Yang, X-Ray Struct. Anal. Online, 2009, 25, 79 CrossRef CAS.
- S. V. Voitekhovich, P. N. Gaponik, A. S. Lyakhov, J. V. Filipova, A. G. Sukhanova, G. T. Sukhanov and O. A. Ivashkevich, Tetrahedron Lett., 2009, 50, 2577 CrossRef CAS.
- G. A. Van Albada, M. Ghazzali, K. Al-Farhan and J. Reedijk, Inorg. Chem. Commun., 2011, 14, 1149 CrossRef CAS.
- G. A. Bowmaker, C. Di Nicola, F. Marchetti, C. Pettinari, B. W. Skelton, N. Somers and A. H. White, Inorg. Chim. Acta, 2011, 375, 31 CrossRef CAS.
- H. Li, H. Jiang and H. Sun, Acta Crystallogr., 2011, E67, m1372 Search PubMed.
- S. Kashyap, U. P. Singh, A. K. Singh, P. Kumar and S. P. Singh, Transition Met. Chem., 2013, 38, 573 CrossRef CAS.
- V. V. Olijnyk, B. Zarychta and V. Kinzhybalo, Polyhedron, 2014, 69, 234 CrossRef CAS.
- G. Zhang, C. Yang, E. Liu, L. Li, J. A. Golen and A. L. Rheingold, RSC Adv., 2014, 4, 61907 RSC.
- S. Becker, U. Behrens and S. Schindler, Eur. J. Inorg. Chem., 2015, 2015, 2437 CrossRef CAS.
- N. A. Bogachev, D. A. Lyubichev, G. L. Starova, A. B. Nikolskii and M. Yu. Skripkin, Russ. J. Gen. Chem., 2018, 88, 617 CrossRef CAS.
- T. Li and Z. Xing, Z. Kristallogr., 2019, 234, 363 CAS.
- J.-S. Wu, D. G. Shlian, J. H. Palmer and R. K. Upmacis, Acta Crystallogr., 2019, E75, 1057 CrossRef PubMed.
- Y. N. Toikka, A. S. Mikherdov, D. M. Ivanov, T. J. Mooibroek, N. A. Bokach and V. Yu. Kukushkin, Cryst. Growth Des., 2020, 20, 4783 CrossRef CAS.
- R. A. Al Balushi, M. S. Khan, Md. S. H. Faizi, A. Haque, K. Molloy and P. R. Raithby, Acta Crystallogr., 2021, E77, 42 CrossRef PubMed.
- X. Chen, K. Chen, G. Li, C. Huang, Y. Zhang, Y. Feng, N. Qin, J. Luo, W. Chen and L. Mi, Chem. Commun., 2022, 58, 2010 RSC.
- M. Malik, A. Świtlicka, A. Bieńko, U. K. Komarnicka, D. C. Bieńko, S. Kozieł, A. Kyzioł, T. Mazur and B. Machura, RSC Adv., 2022, 12, 27648 RSC.
- V. D. Slyusarchuk, B. J. O'Brien and C. S. Hawes, J. Coord. Chem., 2022, 75, 2039 CrossRef CAS.
- M. R. Churchill, B. G. DeBoer and S. J. Mendak, Inorg. Chem., 1975, 14, 2496 CrossRef CAS.
- A. El-Toukhy, G. Z. Cai, G. Davies, T. R. Gilbert, K. D. Onan and M. Veidis, J. Am. Chem. Soc., 1984, 106, 4596 CrossRef CAS.
- A. Tosik, M. Bukowska-Strzyzewska and J. Mrozinski, J. Coord. Chem., 1991, 24, 113 CrossRef CAS.
- M. Camats, I. Favier, S. Mallet-Ladeira, D. Pla and M. Gómez, Org. Biomol. Chem., 2022, 20, 219 RSC.
- B. M. Kariuki and P. D. Newman, Inorg. Chem., 2018, 57, 9554 CrossRef CAS PubMed.
- M. R. Churchill and F. J. Rotella, Inorg. Chem., 1979, 18, 853 CrossRef CAS.
- K. A. Wheeler, T. R. Helgren and T. W. Clayton, Acta Crystallogr., 2014, C70, 306 Search PubMed.
- S. Becker, M. Dürr, A. Miska, J. Becker, C. Gawlig, U. Behrens, I. Ivanović-Burmazović and S. Schindler, Inorg. Chem., 2016, 55, 3759 CrossRef CAS PubMed.
- J. A. Bertrand and J. A. Kelley, Inorg. Chem., 1969, 8, 1982 CrossRef CAS.
- R. Belford, D. E. Fenton and M. R. Truter, J. Chem. Soc., Dalton Trans., 1972, 2345 RSC.
- R. L. Harlow and S. H. Simonsen, Acta Crystallogr., 1977, B33, 2784 CrossRef CAS.
- G. E. Jackson, A. Voyé and S. A. Bourne, Acta Crystallogr., 1996, C52, 1907 CAS.
- H. Sun, K. Harms and J. Sundermeyer, J. Am. Chem. Soc., 2004, 126, 9550 CrossRef CAS PubMed.
- R. T. Stibrany and J. A. Potenza, J. Chem. Crystallogr., 2012, 42, 199 CrossRef CAS.
- P. De Vreese, N. R. Brooks, K. Van Hecke, L. Van Meervelt, E. Matthijs, K. Binnemans and R. Van Deun, Inorg. Chem., 2012, 51, 4972 CrossRef CAS PubMed.
- D. D. Swank, D. O. Nielson and R. D. Willett, Inorg. Chim. Acta, 1973, 7, 91 CrossRef CAS.
- T. S. Lobana, R. Sultana and R. J. Butcher, Dalton Trans., 2011, 40, 11382 RSC.
- S. Betanzos-Lara, C. Gómez-Ruiz, L. R. Barrón-Sosa, I. Gracia-Mora, M. Flores-Álamo and N. Barba-Behrens, J. Inorg. Biochem., 2012, 114, 82 CrossRef CAS PubMed.
- R. Vafazadeh and A. C. Willis, Acta Chim. Slov., 2016, 63, 186 CrossRef CAS PubMed.
- W. Linert, P. Weinberger, G. Ondrejovic and D. Makanova, Vib. Spectrosc., 1993, 5, 101 CrossRef CAS.
- V. Jorík, M. Koman, D. Makáňová, D. Mikloš, A. Broškovičova and G. Ondrejovič, Polyhedron, 1996, 15, 3129 CrossRef.
- Z. Jiang, G. Tang and L. Lu, Acta Crystallogr., 2008, E64, m958 Search PubMed.
- C. Latouche, R. Gautier, R. Génois and F. Massuyeau, J. Phys. Chem. A, 2018, 122, 4628 CrossRef CAS PubMed.
- M. Asplund, S. Jagner, A. F. Andresen, M. Maeda and H. Ohtaki, Acta Chem. Scand., 1984, 38a, 725 CrossRef.
- S. Chen, J. Gao, J. Chang, Y. Li, C. Huangfu, H. Meng, Y. Wang, G. Xia and L. Feng, ACS Appl. Mater. Interfaces, 2019, 11, 17513 CrossRef CAS PubMed.
- S. Andersson, S. Jagner, I. Grenthe, F. Salvatore, L. Niinistö, H. V. Volden, J. Weidlein and R. A. Zingaro, Acta Chem. Scand., 1986, 40a, 210 CrossRef.
- C. Hasselgren and S. Jagner, Inorg. Chim. Acta, 2002, 336, 137 CrossRef CAS.
- R. J. Murray-Watson and S. D. Pike, Organometallics, 2020, 39, 3759 CrossRef CAS.
- T. Asada, Y. Hoshimoto and S. Ogoshi, J. Am. Chem. Soc., 2020, 142, 9772 CrossRef CAS PubMed.
- W.-H. Fang, L. Zhang and J. Zhang, Chem. Commun., 2017, 53, 3949 RSC.
- R. Gautier, C. Latouche, M. Paris and F. Massuyeau, Sci. Rep., 2017, 7, 45537 CrossRef CAS PubMed.
- X.-M. Zhang, J.-J. Hou, C.-H. Guo and C.-F. Li, Inorg. Chem., 2015, 54, 554 CrossRef CAS PubMed.
- G. Bowmaker, L. Brockliss, C. Earp and R. Whiting, Aust. J. Chem., 1973, 26, 2593 CrossRef CAS.
- G. A. Bowmaker, G. R. Clark and D. K. P. Yuen, J. Chem. Soc., Dalton Trans., 1976, 2329 RSC.
- E. Jalilian, R.-Z. Liao, F. Himo, H. Brismar, F. Laurell and S. Lidin, CrystEngComm, 2011, 13, 4729 RSC.
- E. Jalilian and S. Lidin, CrystEngComm, 2011, 13, 5730 RSC.
- Z.-C. Yang, K.-Y. Song, P.-K. Zhou, L.-L. Zong, H.-H. Li, Z.-R. Chen and R. Jiang, CrystEngComm, 2022, 24, 4940 RSC.
- J. T. Mague, Acta Crystallogr., 1998, C54, IUC9800010 Search PubMed.
- A. Nurtaeva and E. M. Holt, Acta Crystallogr., 1999, C55, 1453 CAS.
- J. A. Rusanova, K. V. Domasevitch, O. Yu. Vassilyeva, V. N. Kokozay, E. B. Rusanov, S. G. Nedelko, O. V. Chukova, B. Ahrens and P. R. Raithby, J. Chem. Soc., Dalton Trans., 2000, 2175 RSC.
- A. V. Artem’ev, M. P. Davydova, A. S. Berezin, D. G. Samsonenko, I. Y. Bagryanskaya, V. K. Brel, X. Hei, K. A. Brylev, O. I. Artyushin, L. E. Zelenkov, I. I. Shishkin and J. Li, ACS Appl. Mater. Interfaces, 2022, 14, 31000 CrossRef PubMed.
- D. Gudat, A. W. Holderberg, N. Korber, M. Nieger and M. Schrott, Z. Naturforsch. B, 1999, 54, 1224 CrossRef.
- N. P. Rath and E. M. Holt, J. Chem. Soc., Chem. Commun., 1985, 665 RSC.
- S.-D. Han, D. Wang, J. Pan, Q. Wei, J.-H. Li and G.-M. Wang, Inorg. Chem., 2018, 57, 11318 CrossRef CAS PubMed.
- F. Bottomley and S. Karslioglu, J. Chem. Soc., Chem. Commun., 1991, 222 RSC.
- F. Bottomley, P. D. Boyle, S. Karslioglu and R. C. Thompson, Organometallics, 1993, 12, 4090 CrossRef CAS.
- K. Adil, M. Leblanc, V. Maisonneuve and J. Fluor, Chem, 2006, 127, 1349 CAS.
- K. Adil, M. Leblanc, V. Maisonneuve and J. Fluor, Chem, 2009, 130, 1099 CAS.
- K. Adil, A. Le Bail, M. Leblanc and V. Maisonneuve, Inorg. Chem., 2010, 49, 2392 CrossRef CAS PubMed.
- A. Eichhöfer, D. Fenske and O. Fuhr, Z. Anorg. Allg. Chem., 1997, 623, 762 CrossRef.
- O. Fuhr and D. Fenske, Z. Anorg. Allg. Chem., 1999, 625, 1229 CrossRef CAS.
- A. Gogoll, L. Toom and H. Grennberg, Angew. Chem., Int. Ed., 2005, 44, 4729 CrossRef CAS PubMed.
- M. L. Miller, S. A. Ibrahim, M. L. Golden and M. Y. Darensbourg, Inorg. Chem., 2003, 42, 2999 CrossRef CAS PubMed.
- J.-K. Cheng, Y.-G. Yao, J. Zhang, Z.-J. Li, Z.-W. Cai, X.-Y. Zhang, Z.-N. Chen, Y.-B. Chen, Y. Kang, Y.-Y. Qin and Y.-H. Wen, J. Am. Chem. Soc., 2004, 126, 7796 CrossRef CAS PubMed.
- M. Wriedt, I. Jess and C. Näther, Acta Crystallogr., 2007, E63, m3145 Search PubMed.
- A. Oppermann, R. Dick, C. Wehrhahn, U. Flörke, S. Herres-Pawlis and G. Henkel, Eur. J. Inorg. Chem., 2016, 2016, 3744 CrossRef CAS.
- V. W.-W. Yam, C.-H. Lam, W. K.-M. Fung and K.-K. Cheung, Inorg. Chem., 2001, 40, 3435 CrossRef CAS PubMed.
- V. Wing-Wah Yam, E. Chung-Chin Cheng and N. Zhu, New J. Chem., 2002, 26, 279 RSC.
- H.-W. Xu, L.-X. Zhang and Y.-H. Li, Inorg. Nano-Met. Chem., 2013, 43, 6 CAS.
- H.-Y. Chao, L. Wu, C.-L. Li, W. Lu, L. Liu and X.-L. Feng, Z. Anorg. Allg. Chem., 2011, 637, 1533 CrossRef CAS.
- S. Bernès, F. Sécheresse and Y. Jeannin, Inorg. Chim. Acta, 1992, 191, 11 CrossRef.
- M.-T. Chen and C.-T. Chen, Dalton Trans., 2017, 46, 10181 RSC.
- H. Binder, W. Diamantikos, K. Dermentzis and H.-D. Hausen, Z. Naturforsch. B, 1982, 37, 1548 CrossRef.
- K. Wolfer, H.-D. Hausen and H. Binder, Z. Naturforsch. B, 1985, 40, 235 CrossRef.
- A. Eichhöfer, J. Eisenmann, D. Fenske and F. Simon, Z. Anorg. Allg. Chem., 1993, 619, 1360 CrossRef.
- J. Eisenmann, D. Fenske and F. Simon, Z. Anorg. Allg. Chem., 1995, 621, 1681 CrossRef CAS.
- R. Dobrovetsky, D. Bravo-Zhivotovskii, B. Tumanskii, M. Botoshansky and Y. Apeloig, Angew. Chem., Int. Ed., 2010, 49, 7086 CrossRef CAS PubMed.
- R. Fischer, H. Görls and M. Westerhausen, Organometallics, 2007, 26, 3269 CrossRef CAS.
- Y. Zhuang, Y. Qian, D. Tu, Y. Li, J. Liu, L. Shen and D. Wu, Eur. J. Inorg. Chem., 2021, 2021, 3443 CrossRef CAS.
- D. Dakternieks, K. Jurkschat, H. Wu and E. R. T. Tiekink, Organometallics, 1993, 12, 2788 CrossRef CAS.
- B. Zobel, M. Schürmann, K. Jurkschat, D. Dakternieks and A. Duthie, Organometallics, 1998, 17, 4096 CrossRef CAS.
- J. Ayari, Tripod-Shaped Organotin Compounds: Complexation Studies towards Lewis Bases and Chalcogenido Clusters of Unprecedented Nuclearity, Technische Universität Dortmund, 2020 Search PubMed.
- I. Rojas-León, J. Christmann, S. Schwan, F. Ziese, S. Sanna, D. Mollenhauer, N. W. Rosemann and S. Dehnen, Adv. Mater., 2022, 34, 2203351 CrossRef PubMed.
- D. Lu, A. D. Rae, G. Salem, M. L. Weir, A. C. Willis and S. B. Wild, Organometallics, 2010, 29, 32 CrossRef CAS.
- M. Nagaoka, H. Tsuruda, M. Amako, H. Suzuki and T. Takao, Angew. Chem., Int. Ed., 2015, 54, 14871 CrossRef CAS PubMed.
- P. Sobota, S. Przybylak, J. Utko and L. B. Jerzykiewcz, Organometallics, 2002, 21, 3497 CrossRef CAS.
- H.-O. Stephan, G. Henkel and M. G. Kanatzidis, Chem. Commun., 1997, 67 RSC.
- J. Lackmann, R. Hauptmann, S. Weißgräber and G. Henkel, Chem. Commun., 1999, 1995 RSC.
- A. Eichhöfer, D. Fenske and J. Olkowska-Oetzel, Eur. J. Inorg. Chem., 2007, 2007, 74 CrossRef.
- C. Xu, J.-J. Zhang, Q. Chen, T. Duan, W.-H. Leung and Q.-F. Zhang, Inorg. Chem. Commun., 2012, 21, 1 CrossRef CAS.
- D. Fuhrmann, T. Severin and H. Krautscheid, Z. Anorg. Allg. Chem., 2017, 643, 932 CrossRef CAS.
- J.-M. Yu, T. Cai, Z.-J. Ma, F. Wang, H. Wang, J.-P. Yu, L.-L. Xiao, F.-F. Cheng and W.-W. Xiong, Inorg. Chim. Acta, 2020, 509, 119698 CrossRef CAS.
- J. F. Corrigan and D. Fenske, Angew. Chem., Int. Ed., 1997, 36, 1981 CrossRef CAS.
- O. Kluge, K. Grummt, R. Biedermann and H. Krautscheid, Inorg. Chem., 2011, 50, 4742 CrossRef CAS PubMed.
- J. F. Corrigan and D. Fenske, Chem. Commun., 1996, 943 RSC.
- E. Nordlander, S. C. Lee, W. Cen, Z. Y. Wu, C. R. Natoli, A. Di Cicco, A. Filipponi, B. Hedman, K. O. Hodgson and R. H. Holm, J. Am. Chem. Soc., 1993, 115, 5549 CrossRef CAS.
- W. Cen, F. M. MacDonnell, M. J. Scott and R. H. Holm, Inorg. Chem., 1994, 33, 5809 CrossRef CAS.
- J. Huang, S. Mukerjee, B. M. Segal, H. Akashi, J. Zhou and R. H. Holm, J. Am. Chem. Soc., 1997, 119, 8662 CrossRef CAS.
- G. Gupta, J. Chaturvedi and S. Bhattacharya, Dalton Trans., 2015, 44, 8932 RSC.
- E. D. Leser, B. C. Noll and R. D. Sommer, Polyhedron, 2010, 29, 2053 CrossRef CAS.
- K. Hegetschweiler, T. Raber, G. J. Reiss, W. Frank, M. Wörle, A. Currao, R. Nesper and T. Kradolfer, Angew. Chem., Int. Ed., 1997, 36, 1964 CrossRef CAS.
- E. K. Brechin, S. G. Harris, S. Parsons and R. E. P. Winpenny, Angew. Chem., Int. Ed., 1997, 36, 1967 CrossRef CAS.
- E. Goreshnik, M. Leblanc and V. Maisonneuve, Z. Anorg. Allg. Chem., 2002, 628, 162 CrossRef CAS.
- G. Fritz, H. Köhler and D. Kummer, Z. Anorg. Allg. Chem., 1970, 374, 54 CrossRef CAS.
- G. Sawitzki and H. G. Von Schnering, Z. Anorg. Allg. Chem., 1976, 425, 1 CrossRef CAS.
- S. S. Dhingra and R. C. Haushalter, J. Am. Chem. Soc., 1994, 116, 3651 CrossRef CAS.
- R. D. Sommer and A. L. Rheingold, Acta Crystallogr., 2006, E62, m74 Search PubMed.
- A. Eichhöfer and S. Lebedkin, Inorg. Chem., 2018, 57, 602 CrossRef PubMed.
- M. Fu, D. Fenske, B. Weinert and O. Fuhr, Eur. J. Inorg. Chem., 2010, 2010, 1098 CrossRef.
- I. G. Dance, J. Am. Chem. Soc., 1980, 102, 3445 CrossRef CAS.
- X. Zeng, X. Yao, J. Zhang, Q. Zhang, W. Wu, A. Chai, J. Wang, Q. Zeng and J. Xie, Inorg. Chem. Front., 2015, 2, 164 RSC.
- D. Craig, I. G. Dance and R. Garbutt, Angew. Chem., Int. Ed., 1986, 25, 165 CrossRef.
- I. G. Dance, R. G. Garbutt, D. C. Craig, M. L. Scudder and T. D. Bailey, J. Chem. Soc., Chem. Commun., 1987, 1164 RSC.
- K. S. Anjali and J. J. Vittal, Inorg. Chem. Commun., 2000, 3, 708 CrossRef.
- E. S. Lang, R. A. Burrow, R. Stieler and M. A. Villetti, J. Organomet. Chem., 2009, 694, 3039 CrossRef CAS.
- E. S. Lang, R. Stieler and G. M. De Oliveira, Polyhedron, 2010, 29, 1760 CrossRef CAS.
- G. A. Casagrande, E. S. Lang, G. M. De Oliveira, M. Hörner and F. Broch, Inorg. Chim. Acta, 2007, 360, 1776 CrossRef CAS.
- E. S. Lang, B. Tirloni, G. M. D. Oliveira and M. A. Villetti, Inorg. Chim. Acta, 2009, 362, 3114 CrossRef CAS.
- S. Finoto, A. Machulek, A. R. L. Caires, E. J. De Arruda, G. A. Casagrande, C. Raminelli, L. H. C. Andrade and S. M. Lima, Polyhedron, 2015, 99, 96 CrossRef CAS.
- E. S. Lang, D. F. Back and G. Manzoni De Oliveira, J. Organomet. Chem., 2010, 695, 1966 CrossRef CAS.
- J. Lin, Y. Dong, Q. Zhang, D. Hu, N. Li, L. Wang, Y. Liu and T. Wu, Angew. Chem., Int. Ed., 2015, 54, 5103 CrossRef CAS PubMed.
- C. Xue, J. Lin, H. Yang, W. Wang, X. Wang, D. Hu and T. Wu, Cryst. Growth Des., 2018, 18, 2690 CrossRef CAS.
- O. M. Yaghi, D. A. Richardson, G. Li, C. E. Davis and T. L. Groy, MRS Proc., 1995, 371, 15 CrossRef CAS.
- O. Achak, J. Y. Pivan, M. Maunaye, M. Louër and D. Louër, J. Alloys Compd., 1995, 219, 111 CrossRef CAS.
- C. L. Bowes, W. U. Huynh, S. J. Kirkby, A. Malek, G. A. Ozin, S. Petrov, M. Twardowski, D. Young, R. L. Bedard and R. Broach, Chem. Mater., 1996, 8, 2147 CrossRef CAS.
- P. N. Trikalitis, K. K. Rangan and M. G. Kanatzidis, J. Am. Chem. Soc., 2002, 124, 2604 CrossRef CAS PubMed.
- N.-N. Xu, L.-W. Qian, Z.-Q. Li, G.-Q. Bian, Q.-Y. Zhu and J. Dai, Inorg. Chem., 2018, 57, 9153 CrossRef CAS PubMed.
- Y. Lin, W. Massa and S. Dehnen, Chem. Eur J., 2012, 18, 13427 CrossRef CAS PubMed.
- S. Santner, A. Wolff, M. Ruck and S. Dehnen, Chem. Eur J., 2018, 24, 11899 CrossRef CAS PubMed.
- Z. H. Fard, R. Clérac and S. Dehnen, Chem. Eur J., 2010, 16, 2050 CrossRef PubMed.
- O. Achak, J. Y. Pivan, M. Maunaye, M. Loüer and D. Loüer, J. Solid State Chem., 1996, 121, 473 CrossRef CAS.
- C. L. Cahill and J. B. Parise, Chem. Mater., 1997, 9, 807 CrossRef CAS.
- J. B. Parise and K. Tan, Chem. Commun., 1996, 1687 RSC.
- K. Tan, A. Darovsky and J. B. Parise, J. Am. Chem. Soc., 1995, 117, 7039 CrossRef.
- K. Tan, Y. Ko, J. B. Parise and A. Darovsky, Chem. Mater., 1996, 8, 448 CrossRef CAS.
- D. M. Nellis, Y. Ko, K. Tan, S. Koch and J. B. Parise, J. Chem. Soc., Chem. Commun., 1995, 541 RSC.
- G. Matsubayashi and A. Yokozawa, J. Chem. Soc., Chem. Commun., 1991, 68 RSC.
- T. Sheng, X. Wu, Q. Wang, X. Gao and P. Lin, Polyhedron, 1998, 17, 4519 CrossRef CAS.
- H. Schwertfeger, A. A. Fokin and P. R. Schreiner, Angew. Chem., Int. Ed., 2008, 47, 1022 CrossRef CAS PubMed.
- A. A. Fokin, M. Šekutor and P. R. Schreiner, The Chemistry of Diamondoids Building Blocks for Ligands, Catalysts, Materials, and Pharmaceuticals, Wiley-VCH, Weinheim, 2024 Search PubMed.
- V. Prelog and R. Seiwerth, Ber. Dtsch. Chem. Ges., 1941, 74, 1769 CrossRef.
- H. Stetter, O. Bänder and W. Neumann, Chem. Ber., 1956, 89, 1922 CrossRef CAS.
- R. C. Fort and P. V. R. Schleyer, Chem. Rev., 1964, 64, 277 CrossRef CAS.
- S. Schwan, A. J. Achazi, F. Ziese, P. R. Schreiner, K. Volz, S. Dehnen, S. Sanna and D. Mollenhauer, J. Comput. Chem., 2023, 44, 843 CrossRef CAS PubMed.
- S. Dehnen, P. R. Schreiner, S. Chatterjee, K. Volz, N. W. Rosemann, W. Pilgrim, D. Mollenhauer and S. Sanna, ChemPhotoChem, 2021, 5, 1033 CrossRef CAS.
- W. K. Weigel, H. T. Dang, A. Feceu and D. B. C. Martin, Org. Biomol. Chem., 2022, 20, 10 RSC.
- I. K. Moiseev, N. V. Makarova and M. N. Zemtsova, Russ. Chem. Rev., 1999, 68, 1001 CrossRef CAS.
- K. Aigami, Y. Inamoto, N. Takaishi, K. Hattori, A. Takatsuki and G. Tamura, J. Med. Chem., 1975, 18, 713 CrossRef CAS PubMed.
- T. M. Gund, P. v. R. Schleyer and C. Hoogzand, Tetrahedron Lett., 1971, 12, 1583 CrossRef.
- G. P. Sollott and E. E. Gilbert, J. Org. Chem., 1980, 45, 5405 CrossRef CAS.
- A. A. Fokin, T. E. Shubina, P. A. Gunchenko, S. D. Isaev, A. G. Yurchenko and P. R. Schreiner, J. Am. Chem. Soc., 2002, 124, 10718 CrossRef CAS PubMed.
- G. L. Baughman, J. Org. Chem., 1964, 29, 238 CrossRef CAS.
- G. S. Lee, J. N. Bashara, G. Sabih, A. Oganesyan, G. Godjoian, H. M. Duong, E. R. Marinez and C. G. Gutiérrez, Org. Lett., 2004, 6, 1705 CrossRef CAS PubMed.
- R. D. Bach and R. C. Badger, Synthesis, 1979, 1979, 529 CrossRef.
- M. Bonsir, C. Davila, A. R. Kennedy and Y. Geerts, Eur. J. Org Chem., 2021, 2021, 5227 CrossRef CAS.
- H. Stetter and M. Krause, Adv. Cycloaddit., 1968, 717, 60 CAS.
- H. Newman, Synthesis, 1972, 692 CrossRef CAS.
- K. M. Patil, M. E. Dickinson, T. Tremlett, S. C. Moratti and L. R. Hanton, Cryst. Growth Des., 2016, 16, 1038 CrossRef CAS.
- R. Mello, L. Cassidei, M. Fiorentino, C. Fusco and R. Curci, Tetrahedron Lett., 1990, 31, 3067 CrossRef CAS.
- S. Gowrisankar, B. Bernhardt, J. Becker and P. R. Schreiner, Eur. J. Org Chem., 2021, 2021, 6806 CrossRef CAS.
- S. K. De, Applied Organic Chemistry: Reaction Mechanisms and Experimental Procedures in Medicinal Chemistry, Wiley, 2021 Search PubMed.
- G. W. Gribble, J. Nat. Prod., 2000, 63, 735 CrossRef CAS.
- K.-W. Yeung, Y. Dong, L. Chen, C.-Y. Tang, W.-C. Law and G. C.-P. Tsui, Nanotechnol. Rev., 2020, 9, 650 CrossRef CAS.
- B. J. Ree, S. Kobayashi, K. Heo, T. J. Lee, T. Satoh, T. Ishizone and M. Ree, Polymer, 2019, 169, 225 CrossRef CAS.
- Z. Miao, J. Shi, T. Liu, P. Li, Z. Su and G. Wei, Appl. Sci., 2019, 9, 881 CrossRef CAS.
- A. Štimac, M. Šekutor, K. Mlinarić-Majerski, L. Frkanec and R. Frkanec, Molecules, 2017, 22, 297 CrossRef PubMed.
- L. Wanka, K. Iqbal and P. R. Schreiner, Chem. Rev., 2013, 113, 3516 CrossRef CAS PubMed.
- W. C. Webber and P. A. Harthoorn, Chlorinated and Brominated Polycyclic Hydrocarbons, Their Preparation and Insecticidal Compositions Containing Them, UK Pat., GB819240, 1959 Search PubMed.
- N. I. Baranov, E. I. Bagrii, R. E. Safir, A. G. Cherednichenko, K. V. Bozhenko and A. L. Maximov, Pet. Chem., 2022, 62, 352 CrossRef CAS.
- K. Spilovska, F. Zemek, J. Korabecny, E. Nepovimova, O. Soukup, M. Windisch and K. Kuca, Curr. Med. Chem., 2016, 23, 3245 CrossRef CAS PubMed.
- A. D. Averin, S. P. Panchenko, A. V. Murashkina, V. I. Fomenko, D. S. Kuliukhina, A. S. Malysheva, A. A. Yakushev, A. S. Abel and I. P. Beletskaya, Catalysts, 2023, 13, 831 CrossRef CAS.
- T. Sasaki, A. Usuki and M. Ohno, J. Org. Chem., 1980, 45, 3559 CrossRef CAS.
- İ. Ün, H. İbişoğlu, A. Kılıç, Ş. Ş. Ün and F. Yuksel, Inorg. Chim. Acta, 2012, 387, 226 CrossRef.
- E. A. Ivleva, M. R. Khamzina, M. S. Zaborskaya and Yu. N. Klimochkin, Russ. J. Org. Chem., 2022, 58, 982 CrossRef CAS.
- T. Sasaki, S. Eguchi and T. Toru, Bull. Chem. Soc. Jpn., 1969, 42, 3613 CrossRef CAS.
- S. M. Islam and R. A. Poirier, J. Phys. Chem. A, 2008, 112, 152 CrossRef CAS PubMed.
- T. Šumanovac, M. Alešković, M. Šekutor, M. Matković, T. Baron, K. Mlinarić-Majerski, C. Bohne and N. Basarić, Photochem. Photobiol. Sci., 2019, 18, 1806 CrossRef PubMed.
- B. L. Adams and P. Kovacic, J. Am. Chem. Soc., 1974, 96, 7014 CrossRef CAS.
- D. Fărcaşiu, J. Slutsky, P. V. R. Schleyer, K. H. Overton, K. Luk and J. B. Stothers, Tetrahedron, 1977, 33, 3265 CrossRef.
- R. W. Murray, S. N. Rajadhyaksha and L. Mohan, J. Org. Chem., 1989, 54, 5783 CrossRef CAS.
- A. Schwenger, W. Frey and C. Richert, Chem. Eur J., 2015, 21, 8781 CrossRef CAS PubMed.
- F. Krupp, S. He, W. Frey and C. Richert, Synlett, 2018, 29, 1707 CrossRef CAS.
- G. W. Smith and H. D. Williams, J. Org. Chem., 1961, 26, 2207 CrossRef CAS.
- A. Bashir-Hashemi, J. Li and N. Gelber, Tetrahedron Lett., 1995, 36, 1233 CrossRef CAS.
- I. Boldog, K. V. Domasevitch, J. Sanchiz, P. Mayer and C. Janiak, Dalton Trans., 2014, 43, 12590 RSC.
- S.-Q. Fu, J.-W. Guo, D.-Y. Zhu, Z. Yang, C.-F. Yang, J.-X. Xian and X. Li, RSC Adv., 2015, 5, 67054 RSC.
- H. Stetter, J. Gärtner and P. Tacke, Chem. Ber., 1965, 98, 3888 CrossRef CAS.
- P. Alexandre, A. Schwenger, W. Frey and C. Richert, Chem. Eur J., 2017, 23, 9018 CrossRef CAS PubMed.
- A. Schwenger, N. Birchall and C. Richert, Eur. J. Org Chem., 2017, 2017, 5852 CrossRef CAS.
- W. Lu, D. Yuan, D. Zhao, C. I. Schilling, O. Plietzsch, T. Muller, S. Bräse, J. Guenther, J. Blümel, R. Krishna, Z. Li and H.-C. Zhou, Chem. Mater., 2010, 22, 5964 CrossRef CAS.
- R. K. Totten, M. H. Weston, J. K. Park, O. K. Farha, J. T. Hupp and S. T. Nguyen, ACS Catal., 2013, 3, 1454 CrossRef CAS.
- C. Shen, H. Yu and Z. Wang, Chem. Commun., 2014, 50, 11238 RSC.
- L. J. Mathias, V. R. Reichert and A. V. G. Muir, Chem. Mater., 1993, 5, 4 CrossRef CAS.
- J. Guo, Y. Wang, L. Feng, X. Zhong, C. Yang, S. Liu and Y. Cui, Polymer, 2013, 37, 437 CAS.
- O. Plietzsch, C. I. Schilling, M. Tolev, M. Nieger, C. Richert, T. Muller and S. Bräse, Org. Biomol. Chem., 2009, 7, 4734 RSC.
- C. Shen and Z. Wang, J. Phys. Chem. C, 2014, 118, 17585 CrossRef CAS.
- N. C. Duncan, B. P. Hay, E. W. Hagaman and R. Custelcean, Tetrahedron, 2012, 68, 53 CrossRef CAS.
- T. Mocanu, L. Pop, N. D. Hădade, S. Shova, L. Sorace, I. Grosu and M. Andruh, Eur. J. Inorg. Chem., 2019, 2019, 5025 CrossRef CAS.
- I. Boldog, K. V. Domasevitch, I. A. Baburin, H. Ott, B. Gil-Hernández, J. Sanchiz and C. Janiak, CrystEngComm, 2013, 15, 1235 RSC.
- T. İslamoğlu, M. Gulam Rabbani and H. M. El-Kaderi, J. Mater. Chem. A, 2013, 1, 10259 RSC.
- M. V. Vasylyev, E. J. Wachtel, R. Popovitz-Biro and R. Neumann, Chem. Eur J., 2006, 12, 3507 CrossRef CAS PubMed.
- M. Tominaga, A. Iekushi, K. Katagiri, K. Ohara, K. Yamaguchi and I. Azumaya, Tetrahedron Lett., 2014, 55, 5789 CrossRef CAS.
- M. Tominaga, A. Iekushi, K. Ohara, M. Kawahata, T. Itoh, K. Yamaguchi and I. Azumaya, Chem. Lett., 2018, 47, 1279 CrossRef CAS.
- K. Zhang, L. Wang, Y. Liang, S. Yang, J. Liang, F. Cheng and J. Chen, Synth. Met., 2012, 162, 490 CrossRef CAS.
- E. Galoppini and R. Gilardi, Chem. Commun., 1999, 173 RSC.
- C. I. Schilling, O. Plietzsch, M. Nieger, T. Muller and S. Bräse, Eur. J. Org Chem., 2011, 2011, 1743 CrossRef.
- H. Tohma, A. Maruyama, A. Maeda, T. Maegawa, T. Dohi, M. Shiro, T. Morita and Y. Kita, Angew. Chem., Int. Ed., 2004, 43, 3595 CrossRef CAS PubMed.
- S. Wang, W. J. Oldham, R. A. Hudack and G. C. Bazan, J. Am. Chem. Soc., 2000, 122, 5695 CrossRef CAS.
- L. Landt, W. Kielich, D. Wolter, M. Staiger, A. Ehresmann, T. Möller and C. Bostedt, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 205323 CrossRef.
- R. C. Fort and P. V. R. Schleyer, Chem. Rev., 1964, 64, 277 CrossRef CAS.
- S. Begam Elavarasi, D. Mariam, M. Ummal Momeen, J. Hu and M. Guin, Chem. Phys. Lett., 2019, 715, 310 CrossRef CAS.
- T. Rander, T. Bischoff, A. Knecht, D. Wolter, R. Richter, A. Merli and T. Möller, J. Am. Chem. Soc., 2017, 139, 11132 CrossRef CAS PubMed.
- T. Rander, M. Staiger, R. Richter, T. Zimmermann, L. Landt, D. Wolter, J. E. Dahl, R. M. K. Carlson, B. A. Tkachenko, N. A. Fokina, P. R. Schreiner, T. Möller and C. Bostedt, J. Chem. Phys., 2013, 138, 024310 CrossRef PubMed.
- G. Molle, S. Briand, P. Bauer and J.-E. Dubois, Tetrahedron, 1984, 40, 5113 CrossRef CAS.
- H. Behringer and G. F. Grunwald, Adv. Cycloaddit., 1956, 600, 23 CAS.
- I. Rojas-León, J. Christmann, S. Schwan, F. Ziese, S. Sanna, D. Mollenhauer, N. W. Rosemann and S. Dehnen, Adv. Mater., 2022, 34, 2203351 CrossRef PubMed.
- E. Dornsiepen, F. Dobener, N. Mengel, O. Lenchuk, C. Dues, S. Sanna, D. Mollenhauer, S. Chatterjee and S. Dehnen, Adv. Opt. Mater., 2019, 7, 1801793 CrossRef.
- S. Schwan, A. J. Achazi, F. Ziese, P. R. Schreiner, K. Volz, S. Dehnen, S. Sanna and D. Mollenhauer, J. Comput. Chem., 2023, 44, 843 CrossRef CAS PubMed.
- M. J. Müller, F. Ziese, J. Belz, F. Hüppe, S. Gowrisankar, B. Bernhardt, S. Schwan, D. Mollenhauer, P. R. Schreiner, K. Volz, S. Sanna and S. Chatterjee, Opt. Mater. Express, 2022, 12, 3517 CrossRef.
- K. Eberheim, C. Dues, C. Attaccalite, M. J. Müller, S. Schwan, D. Mollenhauer, S. Chatterjee and S. Sanna, J. Phys. Chem. C, 2022, 126, 3713 CrossRef CAS.
- D. A. Akimov, M. Schmitt, R. Maksimenka, K. V. Dukel’skii, Y. N. Kondrat’ev, A. V. Khokhlov, V. S. Shevandin, W. Kiefer and A. M. Zheltikov, Appl. Phys. B, 2003, 77, 299 CrossRef CAS.
- R. R. Alfano, The Supercontinuum Laser Source: The Ultimate White Light, Springer, New York, 2016 Search PubMed.
- A. Zheltikov, A. LHuillier and F. Krausz, in Springer Handbook of Lasers and Optics, ed. F. Träger, Springer New York, New York, NY, 2007, p. 157 Search PubMed.
- F. Schmidt, A. Riefer, W. G. Schmidt, A. Schindlmayr, M. Imlau, F. Dobener, N. Mengel, S. Chatterjee and S. Sanna, Phys. Rev. Mater., 2019, 3, 054401 CrossRef CAS.
- C. Dues, M. J. Müller, S. Chatterjee, C. Attaccalite and S. Sanna, Phys. Rev. Mater., 2022, 6, 065202 CrossRef CAS.
- J. Belz, J. Haust, M. J. Müller, K. Eberheim, S. Schwan, S. Gowrisankar, F. Hüppe, A. Beyer, P. R. Schreiner, D. Mollenhauer, S. Sanna, S. Chatterjee and K. Volz, J. Phys. Chem. C, 2022, 126, 9843 CrossRef CAS.
- N. W. Rosemann, H. Locke, P. R. Schreiner and S. Chatterjee, Adv. Opt. Mater., 2018, 6, 1701162 CrossRef.
- B. D. Klee, B. Paulus, J. Link Vasco, S. Hosokawa, J. R. Stellhorn, S. Hayakawa, S. Dehnen and W.-C. Pilgrim, Scr. Mater., 2022, 219, 114851 CrossRef CAS.
- J. R. Stellhorn, S. Hayakawa, B. D. Klee, B. Paulus, J. Link Vasco, N. Rinn, I. Rojas León, S. Dehnen and W.-C. Pilgrim, Phys. Status Solidi B, 2022, 259, 2200088 CrossRef CAS.
- W.-C. Pilgrim, J. R. Stellhorn, B. D. Klee, J. L. Vasco, B. Paulus, A. Zeidler, S. Hosokawa, S. Hayakawa and S. Dehnen, J. Phys. Soc. Jpn., 2022, 91, 091004 CrossRef.
- J. Link Vasco, J. R. Stellhorn, B. D. Klee, B. Paulus, J. Belz, J. Haust, S. Hosokawa, S. Hayakawa, K. Volz, I. Rojas León, J. Christmann, S. Dehnen and W.-C. Pilgrim, J. Phys.: Condens. Matter, 2023, 35, 384001 CrossRef PubMed.
- J. R. Stellhorn, S. Hayakawa, B. D. Klee, B. Paulus, J. Link Vasco, N. Rinn, I. Rojas León, C. A. Hosier, S. Dehnen and W. Pilgrim, Adv. Opt. Mater., 2023, 11, 2201932 CrossRef CAS.
- N. W. Rosemann, J. P. Eußner, E. Dornsiepen, S. Chatterjee and S. Dehnen, J. Am. Chem. Soc., 2016, 138, 16224 CrossRef CAS PubMed.
- S. Schwan, A. J. Achazi, F. Ziese, P. R. Schreiner, K. Volz, S. Dehnen, S. Sanna and D. Mollenhauer, J. Comput. Chem., 2023, 44, 843 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |