
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe development of cage phosphine ‘DalPhos’ ligands to enable nickel-catalyzed cross-couplings of (hetero)aryl electrophiles
Kathleen M.
Morrison
and
Mark
Stradiotto
 *
*
Department of Chemistry, Dalhousie University, 6274 Coburg Road, P.O. 15000, Halifax, Nova Scotia B3H 4R2, Canada. E-mail: mark.stradiotto@dal.ca
First published on 25th April 2024
Abstract
Nickel-catalyzed cross-couplings of (hetero)aryl electrophiles with a diversity of nucleophiles (nitrogen, oxygen, carbon, and others) have evolved into competitive alternatives to well-established palladium- and copper-based protocols for the synthesis of (hetero)aryl products, including (hetero)anilines and (hetero)aryl ethers. A survey of the literature reveals that the use of cage phosphine (CgP) ‘DalPhos’ (DALhousie PHOSphine) bisphosphine-type ligands operating under thermal conditions currently offers the most broad substrate scope in nickel-catalyzed cross-couplings of this type, especially involving (hetero)aryl chlorides and phenol-derived electrophiles. The development and application of these DalPhos ligands is described in a ligand-specific manner that is intended to serve as a guide for the synthetic chemistry end-user.
 Kathleen M. Morrison | Kathleen M. Morrison received her BSc (Hons.) in Chemistry (Minor in Psychology, Summa Cum Laude, 2019) from Mount Allison University (Canada), during which time she was also a varsity athlete (basketball). She completed her PhD in 2023 as both a Natural Sciences and Engineering Research Council of Canada Postgraduate PhD Scholar and a Killam Predoctoral Scholar in the Stradiotto group at Dalhousie University (Canada). Kathleen's doctoral research was focused on the development of new and synthetically useful nickel-catalyzed C–O cross-coupling chemistry, with a focus on ligand design and new chemoselective methodology development. |
 Mark Stradiotto | Prof. Mark Stradiotto received his BSc (Hons.) in Applied Chemistry (1995) and PhD in Organometallic Chemistry (1999) from McMaster University (Canada). After conducting research as an NSERC Postdoctoral Fellow at the University of California at Berkeley (1999–2001), Mark moved to Dalhousie University where he is the Alexander McLeod Professor of Chemistry. In 2020 he was named a Dalhousie University Arthur B. McDonald Research Chair. Mark has received several awards in recognition of excellence in research and teaching, including the 2023 Government of Canada Governor General's Innovation Award in recognition of the impact of his commercialized ‘DalPhos’ family of ligands/catalysts. |
1. Introduction
The capacity of coordinated ligands to influence the steric and electronic properties of a metal centre, and thus the overall reactivity characteristics of the coordination complex, is well-known.1 As a result, ‘ligand design’2 informed by knowledge of the associated catalytic mechanism is exploited to great benefit in the rational development of new and useful homogeneous metal catalysts for the transformation of organic molecules, including with industrial applications.3 Indeed, the 2001 (asymmetric catalysis), 2005 (olefin metathesis), and 2010 (palladium-catalyzed C–C cross-coupling) Nobel Prizes in Chemistry collectively acknowledge the way in which mechanistically guided ligand design can accelerate advances in transition metal-mediated synthesis.The rapid progression of palladium-catalyzed C–N4 (i.e., Buchwald–Hartwig amination) and C–O5 cross-coupling chemistry, for example, can be attributed to the systematic development of typically bulky and relatively electron-rich ligands,6 exemplified by Buchwald's biarylmonophosphines;4b,7 these promote the formation of low-coordinate LnPd0 species that are pre-disposed towards challenging (hetero)aryl-X oxidative additions (Fig. 1). Such ligands enable access to broader substrate scope versus existing methodologies (e.g., SNAr,8 copper catalysis9), including the use of inexpensive and abundant (hetero)aryl chlorides and phenol-derived electrophiles. As a result, palladium-catalyzed C–N/O cross-couplings quickly evolved into widely used synthetic methodologies, including for the discovery and production of the active ingredients in pharmaceuticals and agrochemicals.3 From a practical perspective, end-user uptake is facilitated by the commercialization of some effective ligand classes.
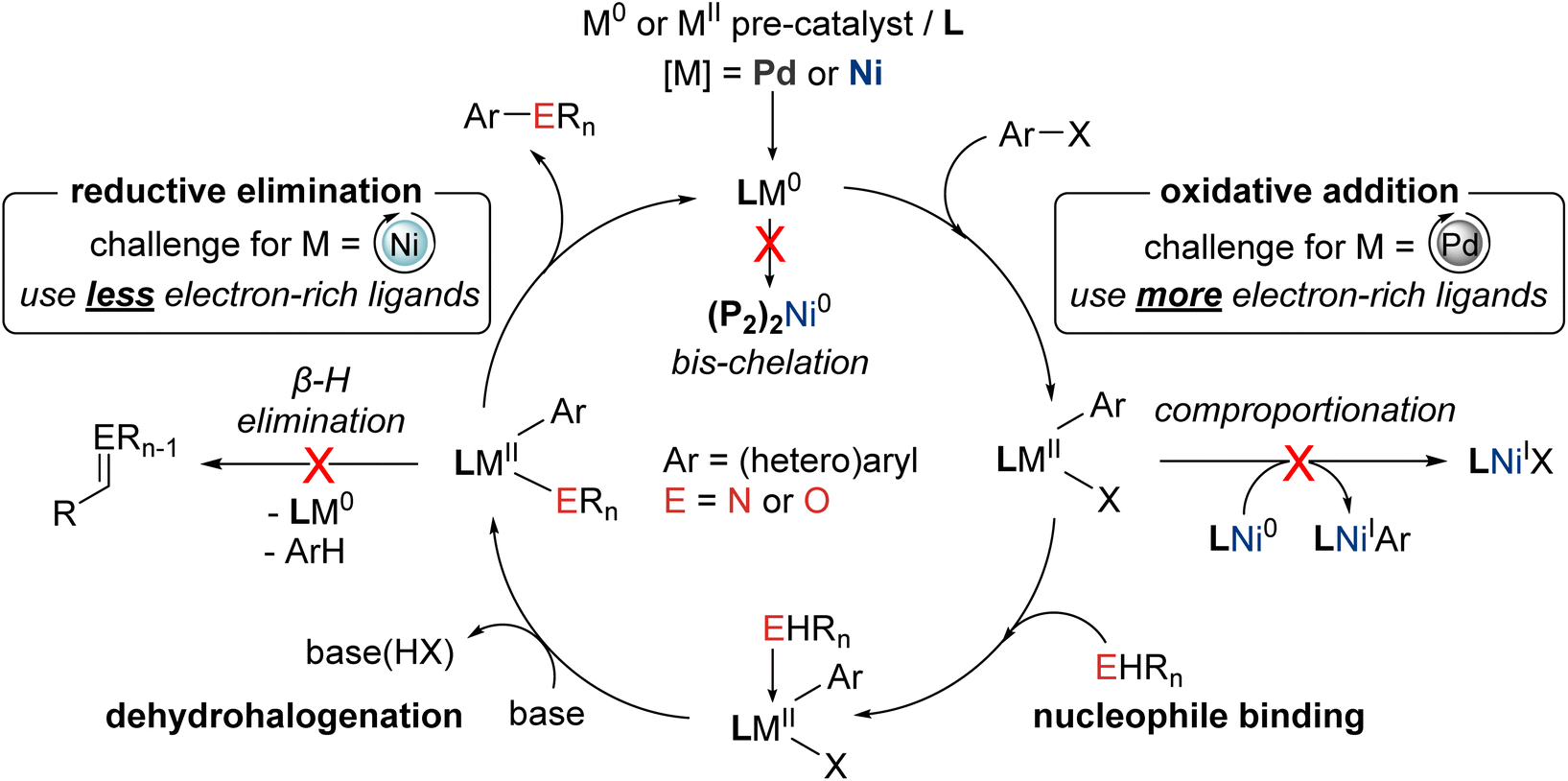 | ||
| Fig. 1 Generic mechanism for prototypical palladium- and nickel-catalyzed C–N/O cross-couplings involving a M(0/II) mechanism, highlighting some unwanted side reactions, and ligand design considerations (P2 = bisphosphine ligand). | ||
Despite the utility of palladium-catalyzed cross-coupling chemistry, as exemplified by such transformations involving nitrogen or oxygen nucleophiles, the cost and supply limitations associated with this precious metal, and the quest to establish new or improved substrate transformations, have created motivation for developing competitive and more sustainable cross-coupling methodologies involving base metal catalysts.10
Nickel represents a promising target in developing C–N/O and other cross-coupling chemistry, conceptually merging the benefits of established protocols based on palladium (e.g., broad scope) or copper (e.g., relatively inexpensive and abundant).11 The lower electronegativity of nickel (1.91) versus palladium (2.20) renders oxidative addition reactions inherently more facile at nickel, including with (hetero)aryl chlorides and phenol derivatives,12 while the expanded oxidation state range (commonly 0 to III) and propensity for single-electron transfer associated with nickel leads to more complex, but potentially more versatile, mechanistic landscapes versus conventional Pd(0/II) C–N/O cross-coupling cycles (Fig. 1).13 In this context, two parallel strategies have emerged in the quest to promote efficient turnover in nickel-catalyzed C–N/O cross-coupling cycles: (a) application of photochemical, electrochemical, or alternative redox strategies within putative Ni(I/III) catalytic cycles,14 typically involving nickel species bearing nitrogen donor ligands; and (b) the use of soft ancillary ligands such as phosphines15 under thermal conditions within putative Ni(0/II) cycles. Whereas the former ‘redox’ approaches have attracted considerable attention both in terms of reaction development and mechanistic elucidation, the demonstrated substrate scope to date has proven rather limited, with (hetero)aryl chlorides and phenol-derived electrophiles generally proving unsuitable. Mixed results were also obtained initially in the application of phosphorus-based ligands: while chelating bisphosphines (P2) such as 1,1′-bis(diphenylphosphino)ferrocene (DPPF16) proved useful in some nickel-catalyzed C–N/O cross-couplings including with challenging (hetero)aryl electrophiles, specialized phosphine ligands optimized for use in palladium-catalyzed C–N/O cross-couplings (e.g., Buchwald's biarylmonophosphines) were generally ineffective. The latter observation, in retrospect, is not entirely surprising, given the divergent properties (e.g., size, electronegativity, redox behavior, etc.) and associated mechanistic requirements of nickel versus palladium.13
Building on our interest in the development of useful ligands of relevance to metal-catalyzed cross-coupling chemistry (e.g., Mor-DalPhos17), in 2015 we initiated what at the time was the first ligand design program of which we were aware focused on promoting Ni(0/II)-catalyzed C–N/O cross-coupling cycles (Fig. 1). Notwithstanding our prior success in developing the first nickel-catalyzed monoarylation of ammonia employing the electron-rich JosiPhos ligand CyPF-Cy,18 in our new program we targeted P2 ligands featuring the sterically demanding yet not overly electron-rich cage phosphine donor group, 1,3,5,7-tetramethyl-2,4,8-trioxaphosphaadamantane (CgP19), paired with a pendant phosphorus donor fragment possessing variable steric and electronic properties, and spanned by an ortho-phenylene linker to enforce ligand rigidity. Unlike ligands designed for use with palladium, we anticipated that these new CgP-based DalPhos ligands would function well in promoting Ni(0/II) C–N, C–O, and other cross-coupling cycles (e.g., C–C) by driving product-forming reductive elimination, while discouraging unwanted bis-chelation (i.e., (P2)2Ni)20 and/or comproportionation21 leading to Ni(I) (Fig. 1). We envisioned that the success of such a ligand design strategy would give rise to (P2)Ni(0/II) catalytic species that could provide broad scope in both the nucleophile and electrophile, thereby overcoming the key limitations of Ni(I/III) redox approaches and affording a complementary, and potentially superior, alternative to palladium-based methods.
In this perspective we summarize our development of CgP-based DalPhos ligands, and their use in enabling a broad collection of nickel-catalyzed C–N/O cross-coupling reactions, as well as C–C cross-couplings, with performance that is commonly competitive with, and in many cases superior to, the best catalysts known for such transformations (i.e., Pd, Cu, Ni, or other). In focusing our discussion mainly on five complementary and particularly high-performing members of this family (i.e., the commercialized ligands PAd-DalPhos, CyPAd-DalPhos, PhPAd-DalPhos, and PAd2-DalPhos, as well as the more recently developed CgPhen-DalPhos; Fig. 2) spanning literature from 2016 to the first half of 2024, we hope to provide a practical guide for end-users regarding the scope and applicability of each of these CgP-based DalPhos ligands in challenging nickel-catalyzed C–N/O/C cross-coupling applications. While our understanding of the mechanistic landscape traversed by such nickel catalysts is still emerging, we provide a summary of our findings obtained to date. For a broader survey of nickel-catalyzed cross-coupling catalysis, including catalyst development and mechanistic analysis beyond the scope of this discussion, the reader is directed toward a selection of published review articles.11,13,14b,15,22
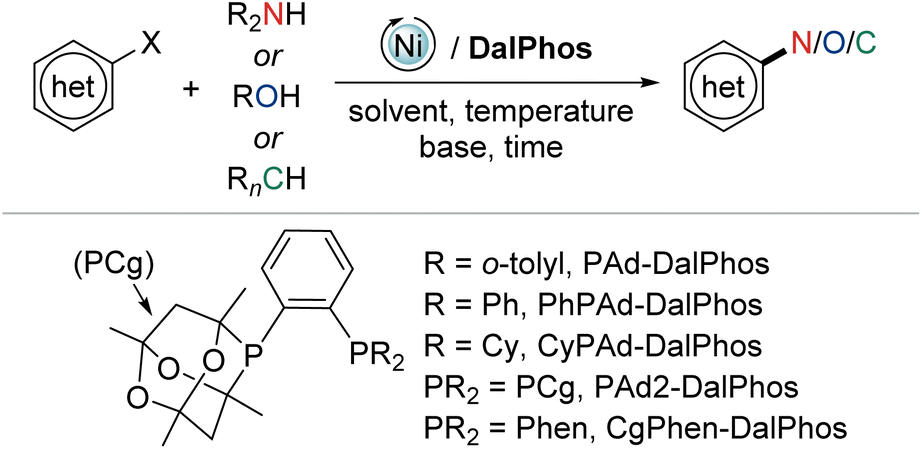 | ||
| Fig. 2 The CgP-based DalPhos/nickel-catalyzed C–N/O/C cross-couplings featured in this perspective. | ||
2. DalPhos ligand/pre-catalyst synthesis
In the course of developing the CgP-based DalPhos family in 2015 for use in nickel-catalyzed C–N/O cross-couplings and beyond, the ligand evolution process was driven by our motivation to solve what at the time were numerous unmet reactivity challenges, including: (i) accommodating difficult and in some cases unprecedented nucleophile (e.g., ammonia, fluoroalkylamines, hindered α,α,α-branched primary alkylamines, heteroarylamines, amides, sulfonamides, sulfinamides, primary/secondary/tertiary aliphatic alcohols, phenols, etc.) and electrophile (e.g., (hetero)aryl chlorides and phenol derivatives) pairings; and (ii) enabling mild reaction conditions (e.g., low loadings, <1 mol%, and/or room temperature), chemoselective transformations, and reactions employing soluble organic bases in place of typically used inorganic bases. Notably, each of these reactivity goals were achieved by use of the DalPhos ligands depicted in Fig. 2.In developing the CgP-containing DalPhos ligand family for use in nickel catalysis, we prioritized practicality: air-stable ligands that could be prepared easily and cost-effectively from a single precursor were strategically targeted, as were derived air-stable Ni(II) pre-catalysts that could be activated under catalytic conditions. The general strategy used for the synthesis of the CgP-based DalPhos ligand family is presented in Fig. 3, which makes use of the now commercially available precursor CgP(o-C6H4Br); this precursor itself can be prepared easily from the commercial reagents 2-bromoiodobenzene and air-stable CgPH.23 Lithium–halogen exchange involving CgP(o-C6H4Br), followed by quenching with R2PCl, affords access to most DalPhos ligands of interest in synthetically useful isolated yields.24 In many instances we find the performance of air-stable pre-catalysts of the type (P2)Ni(aryl)Cl22c (P2 = DalPhos, aryl = o-tolyl or p-CNPh) to be superior to DalPhos/Ni admixtures (e.g., DalPhos/Ni(COD)2) including enabling lower loadings and more mild conditions as noted in our initial study of ammonia monoarylation,24 and we commonly use such pre-catalysts. A representative synthesis of (DalPhos)Ni(o-tolyl)Cl pre-catalysts using Jamison's method25 is presented in Fig. 3. These (P2)Ni(aryl)Cl complexes represent ‘oxidative addition intermediates’ in a putative Ni(0/II) catalytic cycle and thus are naturally predisposed, following nucleophile binding, dehydrohalogenation, and reductive elimination, to generate key (P2)Ni intermediates under cross-coupling conditions without the need for exogenous activating agents (Fig. 1). Given that the CgP fragment itself is chiral (racemic), the presence of one or more additional stereochemical elements (e.g., a second CgP group as in PAd2-DalPhos; hindered Ni-(o-tolyl) bond rotation in (DalPhos)Ni(o-tolyl)Cl pre-catalysts, beyond cis/trans isomerism) can result in the formation of multiple isomeric forms of the ligand/pre-catalyst.
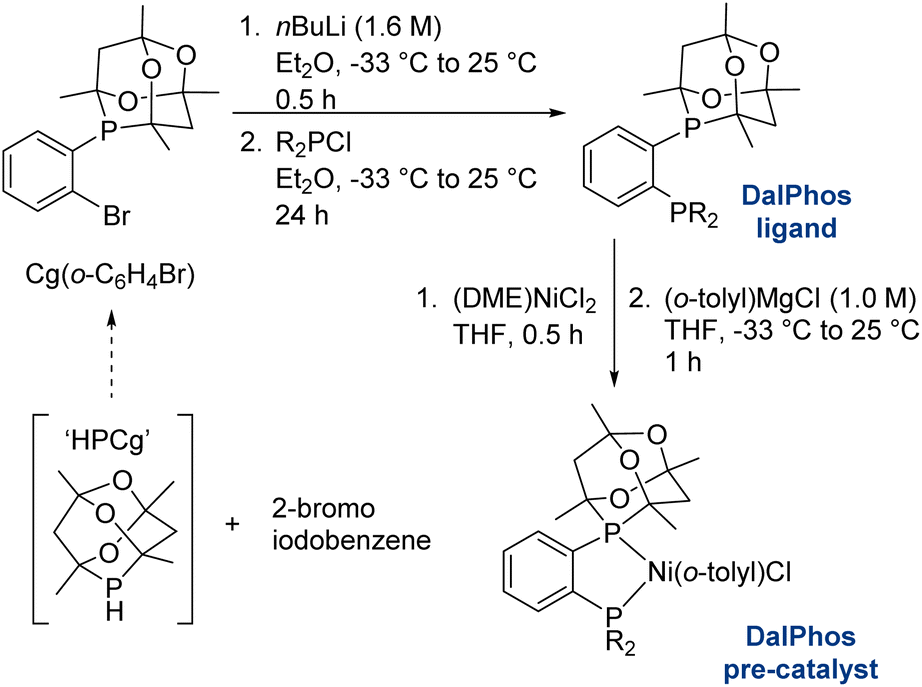 | ||
| Fig. 3 Representative CgP-based DalPhos ligand and nickel pre-catalyst synthesis (DME = 1,2-dimethoxyethane). | ||
3. What can these DalPhos ligands do?
As noted above, use of the CgP-based DalPhos ligands depicted in Fig. 2 has provided access to useful, and in some cases previously unknown, nickel-catalyzed C–N/O/C cross-coupling reactions, including those involving inexpensive and abundant (hetero)aryl chloride and phenol-derived electrophiles for which most alternative nickel catalysts based on photochemical, electrochemical, or other redox strategies are typically ineffective. In the following sections we provide a ligand-by-ligand summary of this research progress, so as to inform and guide the end-user.3.1 PAd-DalPhos
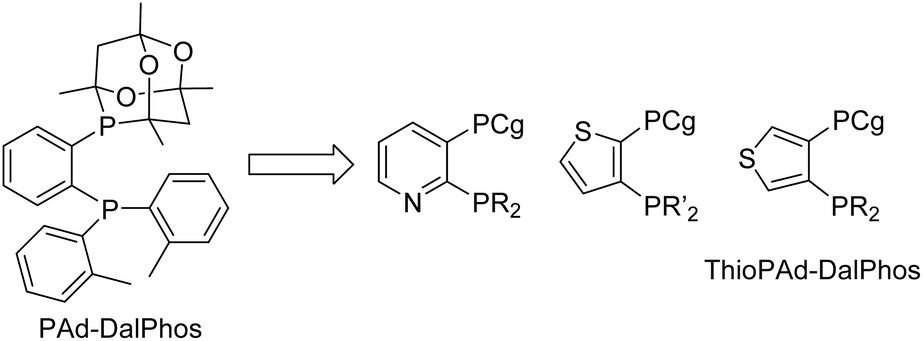
The monoarylation of ammonia to afford primary (hetero)aniline derivatives was selected initially as a challenging testing ground for our new CgP-containing DalPhos ligands.24 This reaction type is highly sought-after, yet has proven difficult, due to possible uncontrolled di- and triarylation arising from the primary (hetero)aniline products competing with ammonia as the nucleophile coupling partner.26 Prior work by our group (vide supra)18 and Hartwig and co-workers,27 independently disclosed in 2015, established the first examples of nickel-catalyzed ammonia monoarylation using relatively electron-rich JosiPhos-type ligands (e.g., in our work, CyPF-Cy18). Notably, other bulky electron-rich ligands used in palladium-catalyzed ammonia monoarylation (e.g., Mor-DalPhos,17 Bippy-Phos,28 JosiPhos CyPF-tBu29) were not suitable when re-purposed in corresponding nickel catalysis, thus emphasizing the importance of new ligand design tailored to the unique properties of nickel as a strategic means of advancing the catalytic state-of-the-art.In 2016, we reported on the initial development and screening of our DalPhos ligand variants featuring CgP/P(o-tolyl)2 (PAd-DalPhos), CgP/PPh2 (PhPAd-DalPhos), CgP/PCy2 (CyPAd-DalPhos), or CgP/PiPr2 donor pairings, and for comparison P(o-tolyl)2/P(tBu)2 where CgP was replaced with P(tBu)2, in the selective monoarylation of ammonia with 4-chlorobiphenyl using NaOtBu as base at 110 °C.24 This preliminary ligand/Ni(COD)2 test reaction established PAd-DalPhos as being uniquely effective in such transformations relative to the other ligands surveyed, offering performance that was competitive with CyPF-Cy. In estimating the % buried volumes (%Vbur) of PAd-DalPhos and CyPF-Cy by use of the SambVca web tool30 values of 80.4% (Fig. 4) and 80.7% are found, thereby confirming the similar steric profiles of these ligands. Previously reported structural analyses by the Pringle group determined the CgP donor group to be as sterically encumbering as a P(tBu)2 group, with electronics comparable to a relatively electron-poor P(OR)2 fragment.19 While this steric trend is consistent with the similar % buried volume of the PAd-DalPhos variant, where the CgP was replaced with P(tBu)2 (81.0%, versus 80.4% in PAd-DalPhos), the inferior performance of this P(o-tolyl)2/P(tBu)2 cousin of PAd-DalPhos suggests that sterics alone do not predict useful catalytic performance in a given nickel-catalyzed transformation, such as ammonia monoarylation catalysis. This conjecture is supported by our observation that while members of the CgP-based DalPhos ligand family do not vary significantly in terms of % buried volume (∼79 ± 3%, vide infra), and are sterically comparable to more conventionally employed cis-chelating P2 ligands such as DPPF (79.8%), PAd-DalPhos emerged from this study as being uniquely suited to this ammonia monoarylation catalysis. Primary alkylamines also proved to be suitable substrates with PAd-DalPhos. Given the difficulty in predicting ligand-based reactivity trends a priori, we have found that access to a relatively small family of DalPhos ligands (e.g., Fig. 2) provides several ‘keys’ that have proven successful in ‘unlocking’ new nickel-catalyzed cross-coupling reactivity.
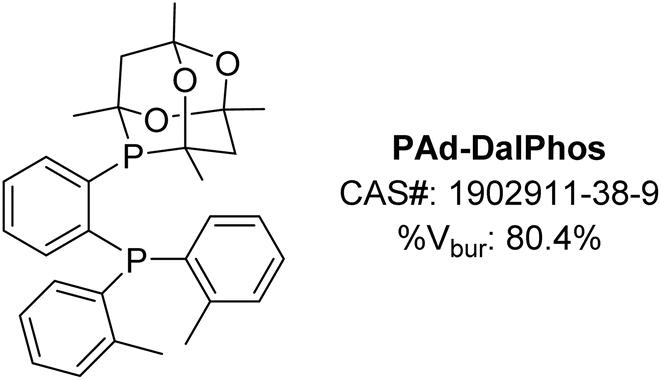 | ||
| Fig. 4 PAd-DalPhos ancillary ligand with associated CAS number and % buried volume. | ||
The scope of reactivity achieved by use of air-stable (PAd-DalPhos)Ni(o-tolyl)Cl in the monoarylation of ammonia encompasses unprecedented diversity in the aryl electrophile coupling partner (i.e., chlorides, bromides, iodides, mesylates, tosylates, triflates, and imidazolylsulfonates) with several examples demonstrated under mild reaction conditions (i.e., room temperature; 1 mol% catalyst loadings). The use of short reaction times and high temperatures (160 °C) under microwave irradiation was also tolerated, as was the use of gaseous ammonia (>100 psi), both of which represent conditions that are not tolerated by a majority of reported metal catalysts for ammonia monoarylation (Fig. 5).
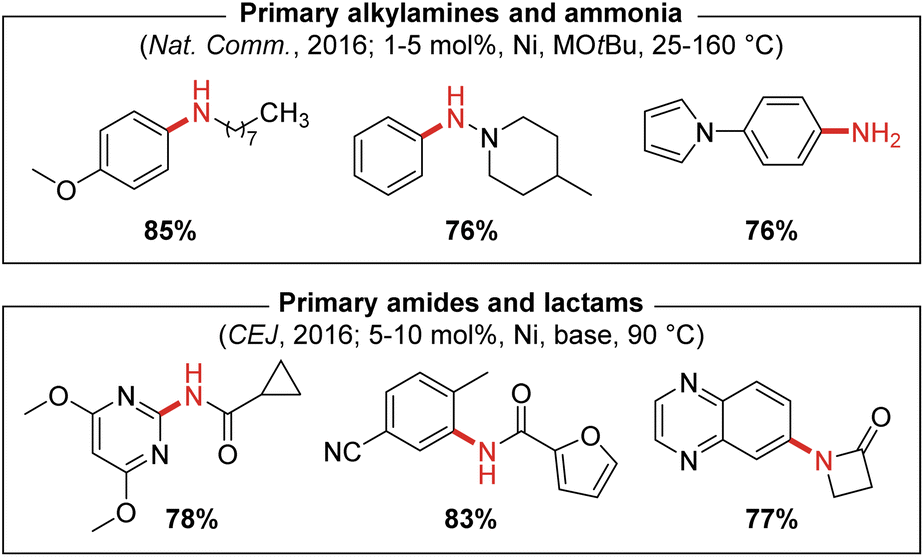 | ||
| Fig. 5 Representative nickel-catalyzed N-arylations enabled by catalyst systems based on the CgP-containing ancillary ligand PAd-DalPhos (M = Li or Na; base = NaOtBu, Cs2CO3, or K3PO4). | ||
In 2016 we also reported the first nickel-catalyzed N-arylation of amides with (hetero)aryl (pseudo)halides which was also achieved using (PAd-DalPhos)Ni(o-tolyl)Cl pre-catalyst (Fig. 5).31 In this report, comparisons were made to other prominent ancillary ligands (XantPhos, JackiePhos,32 BippyPhos,28 DPPF,33rac-BINAP,20 SIPr,34 and JosiPhos CyPF-Cy18) that had proven effective in palladium- and/or nickel-catalyzed C–N cross-couplings, in a test reaction involving benzamide and 4-chlorobenzonitrile using Ni(COD)2 (10 mol%) at 90 °C. Poor reactivity was observed for all ligands except CyPF-Cy and PAd-DalPhos, and subsequent screening of these two ligands by use of (L)Ni(o-tolyl)Cl pre-catalysts in the C–N cross-coupling of nicotinamide and 1-chloronapthalene revealed the superiority of PAd-DalPhos. Given the differing challenges associated with the use of ammonia (e.g., monoarylation selectivity, possible inhibition by highly nucleophilic ammonia) and primary amides (e.g., poorly nucleophilicity, possible inhibition via κ2-N,O chelation), the efficacy of PAd-DalPhos in these divergent chemistries provided early evidence of the potentially privileged nature of the CgP-based DalPhos ligand class in nickel-catalyzed C–N cross-coupling. A notable exception to this trend is the cross-coupling of secondary amine nucleophiles such as morpholine, whereby DPPF proved superior to both CyPF-Cy and PAd-DalPhos.35
While not examined widely in the CgP-based DalPhos ligand family, we have demonstrated that heteroaryl analogues of PAd-DalPhos based on a pyridine or thiophene backbone can in some instances function well in nickel-catalyzed cross-couplings. In examining C–N cross-couplings of primary alkylamines at room temperature, the use of low catalyst loadings (0.25–0.50 mol% Ni) proved possible when using a thiophene-based analogue of PAd-DalPhos (i.e., ThioPAd-DalPhos, Fig. 6).36
 | ||
| Fig. 6 Examples of PAd-DalPhos ligand variants featuring a pyridine or thiophene backbone (R = o-tolyl). | ||
In seeking to test our hypothesis that CgP-based DalPhos ligands might accelerate rate-limiting reductive elimination within putative Ni(0/II) catalytic cycles (Fig. 1), in 2017 we disclosed the results of a combined experimental and computational study probing the influence of ancillary ligand and oxidation state of nickel on the nickel-catalyzed C–N cross-coupling of aryl chlorides. In this work, (L)NiCl and (L)Ni(o-tolyl)Cl pre-catalysts incorporating either DPPF or PAd-DalPhos ancillary ligands were examined.37 Computational analyses confirmed the viability of Ni(0/II) and Ni(I/III) mechanistic cycles in this chemistry with reductive elimination being rate-limiting in Ni(0/II) cycles, and oxidative addition being rate-determining in Ni(I/III) cycles. In 2024 we disclosed the results of an analogous study focusing instead on C–O cross-coupling, that is discussed in the following section.
3.2 CyPAd-DalPhos
In our initial 2016 report describing the development of the CgP-based DalPhos ligand family,24 several variants were disclosed, including CyPAd-DalPhos (Fig. 7). Although relatively poor in comparison to PAd-DalPhos in the monoarylation of ammonia (vide supra), this cyclohexyl variant was subsequently proven by our group to be highly effective in other challenging nickel-catalyzed cross-coupling reactions involving (hetero)aryl (pseudo)halides.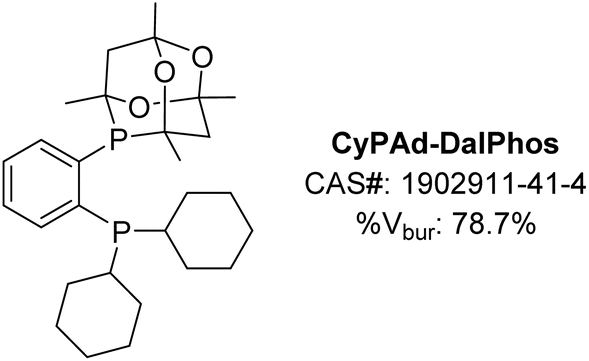 | ||
| Fig. 7 CyPAd-DalPhos ancillary ligand with associated CAS number and % buried volume. | ||
Our success in 2016 related to the use of PAd-DalPhos in nickel-catalyzed ammonia monoarylation inspired further investigation of this and other CgP-based DalPhos ligands in addressing unmet challenges regarding the N-arylation of amine nucleophiles. As part of this initiative, the C–N cross-coupling of cyclopropylamines with (hetero)aryl electrophiles for the synthesis of biologically relevant N-arylcyclopropylamines was targeted by our group in 2017.38 Beyond seeking to address limitations in metal-catalyzed procedures of this type that existed at the time, we also viewed cyclopropylamines as being potentially challenging test reaction partners, given the possibility that nickel might engage in radical chemistry leading to unwanted ring opening. In an initial reaction comparing (PAd-DalPhos)Ni(o-tolyl)Cl and (CyPF-Cy)Ni(o-tolyl)Cl, only modest conversion to the desired N-(hetero)arylcyclopropylamine products was achieved; conversely, the new air-stable pre-catalyst (CyPAd-DalPhos)Ni(o-tolyl)Cl provided excellent reactivity, in the absence of detectable cyclopropyl ring-opening. A useful substrate scope was then established by use of (CyPAd-DalPhos)Ni(o-tolyl)Cl, spanning a diverse range of (hetero)aryl electrophile coupling partners (i.e., chlorides, bromides, tosylates, sulfamates, carbamates, triflates, mesylates), with several examples demonstrated at room temperature (Fig. 8).
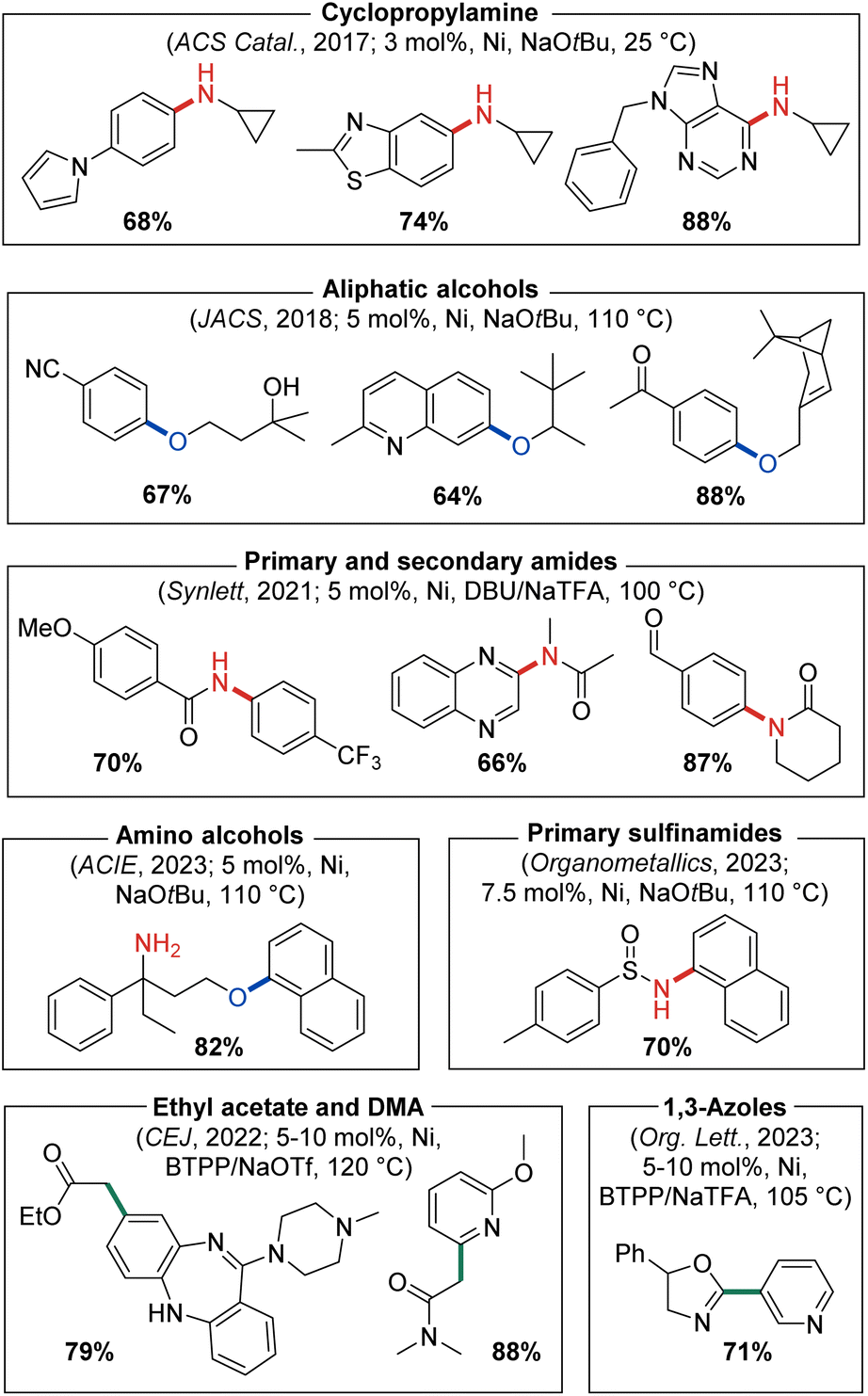 | ||
| Fig. 8 Representative nickel-catalyzed C–N, C–O, and C–C cross-coupling products enabled by catalyst systems based on CyPAd-DalPhos. | ||
MacMillan and co-workers established the first nickel-catalyzed C–O cross-coupling of primary and secondary aliphatic alcohols with (hetero)aryl bromides using iridium/nickel photoredox catalysis.39 Following this seminal report, our group, driven by successes in nickel-catalyzed C–N cross-coupling under thermal conditions, achieved the first complementary ligand-enabled nickel-catalyzed C–O cross-coupling of aliphatic alcohols with (hetero)aryl chlorides and phenol-derived electrophiles.40 Our initial reaction optimization included pre-catalysts (DPPF)Ni(o-tolyl)Cl, (PAd-DalPhos)Ni(o-tolyl)Cl, (CyPAd-DalPhos)Ni(o-tolyl)Cl, (IPr)Ni(styrene)2, and (PAd-DalPhos)NiCl, where CyPAd-DalPhos was determined to be the optimal ligand for such C–O bond forming reactions. Notably, DPPF and IPr-based catalysts gave negligible conversion to the desired alkyl aryl ether products, thus highlighting the utility of the DalPhos ligand class generally. Using (CyPAd-DalPhos)Ni(o-tolyl)Cl as an optimized pre-catalyst ((PAd-DalPhos)Ni(o-tolyl)Cl also proved useful in some transformations), a broad range of primary and secondary aliphatic alcohols were accommodated, using NaOtBu or Cs2CO3 as base at 110 °C (Fig. 8). Moreover, an unprecedented electrophile scope for any catalyst system was demonstrated, spanning (hetero)aryl chlorides, bromides, mesylates, triflates, pivalates, and tosylates. A related report from our group examining the efficacy of Ni(I) versus Ni(II) pre-catalysts featuring either PAd-DalPhos or CyPAd-DalPhos, both empirically and by use of computational methods, appeared in 2024.41 In this work comparing Ni(0/II) and Ni(I/III) cycles, both ligands and pre-catalyst classes (i.e., Ni(I) and Ni(II)) proved competent in the C–O cross-coupling of aliphatic alcohols and (hetero)aryl electrophiles;40 it was computationally determined that these reactions likely occur within a Ni(0/II) catalytic cycle with a turnover-limiting C–O bond-forming reductive elimination step.
As discussed above, our use of (PAd-DalPhos)Ni(o-tolyl)Cl enabled the first broadly effective nickel-catalyzed N-arylation of primary amides with (hetero)aryl (pseudo)halides (Fig. 5). While several inorganic bases proved effective in this chemistry (i.e., NaOtBu, Cs2CO3, K3PO4), they come with important limitations. The use of mild carbonate or phosphate bases can lead to poor solubility in organic media and inconsistent reaction kinetics due to variable particle size. Furthermore, strong inorganic bases exhibit poor substrate functional group tolerance, and in the case of NaOtBu can react as a competitive nucleophile in cross-coupling chemistry.42 In 2019, Simmons and co-workers reported a so-called ‘dual-base’ system using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and halide scavenger sodium trifluoroacetate (NaTFA) to circumvent the limitations of inorganic bases in palladium-catalyzed C–N cross-couplings of primary amides or anilines with (hetero)aryl chlorides.43 Inspired by this strategy, in 2021 we targeted the nickel-catalyzed N-arylation of amides using a DBU/NaTFA through an initial ligand screen that included PAd-DalPhos, CyPAd-DalPhos, PhPAd-DalPhos (vide infra), PAd2-DalPhos (vide infra), XantPhos, DPPF, JosiPhos CyPF-Cy, IPr, and SIPr. The CyPAd-DalPhos/Ni(COD)2 catalyst mixture outcompeted all other ligands in the test cross-coupling of 2-furamide and 4-chloroquinalidine.44 Interestingly, the corresponding pre-catalyst (CyPAd-DalPhos)Ni(o-tolyl)Cl was not effective in this chemistry; the substrate scope was therefore developed using CyPAd-DalPhos/Ni(COD)2 (5 mol%) where primary and selected secondary amides/lactams/oxazolidones were suitable coupling partners. Using an analogous approach, in 2022 we reported the nickel-catalyzed mono-α-arylation of ethyl acetate and dimethylacetamide enabled by use of (CyPAd-DalPhos)Ni(4-CN-Ph)Cl with tert-butylimino-tri(pyrrolidino)phosphorane (BTPP) and sodium triflate (NaOTf) as the optimal dual-base system,45 and in 2023 we disclosed the C2–H arylation of 1,3-azoles using (CyPAd-DalPhos)Ni(o-tolyl)Cl and BTPP/NaTFA (Fig. 8).46
Through a systematic evaluation of C–N versus C–O cross-coupling selectivity differences between CgP-based DalPhos ligands, we reported in 2023 that (CyPAd-DalPhos)Ni(o-tolyl)Cl enables challenging chemoselective O-arylations of amino alcohols with (hetero)aryl chlorides (Fig. 8).47 Under the optimized conditions, C–O cross-coupling was favoured for substrates featuring potentially contending branched primary and secondary alkylamine groups, whereas the orthogonal C–N cross-coupling was observed in amino alcohols featuring less-hindered alkylamines and aniline functionalities. The same year we disclosed C–N cross-couplings of a related nucleophile class, amino acid esters, where (CyPAd-DalPhos)Ni(o-tolyl)Cl was found to be effective in the synthesis of a variety of N-heteroaryl amino acid esters in high yield and retained enantiomeric purity.48
In 2023 we also targeted the nickel-catalyzed C–N cross-coupling of sulfinamides, an underdeveloped transformation of interest to both synthetic and medicinal chemists. In a comparative ligand/Ni(COD)2 screen involving 4-chloroquinaldine and tert-butanesulfinamide (i.e., Ellman's sulfinamide), PAd-DalPhos, DPPF, and DPEPhos exhibited negligible conversion to the desired cross-coupled product, thereby emphasizing the importance of having available a small library of DalPhos ligand variants for targeted reaction development: CyPAd-DalPhos and PhPAd-DalPhos (vide infra) in particular proved useful in such transformations.49 Using (CyPAd-DalPhos)Ni(o-tolyl)Cl and (PhPAd-DalPhos)Ni(o-tolyl)Cl (vide infra), useful substrate scope was established, focusing primarily on tert-butanesulfinamide as a test nucleophile with a variety of (hetero)aryl chlorides (Fig. 8).
3.3 PhPAd-DalPhos
The modular nature of the CgP-based DalPhos ligand synthesis (Fig. 3) allows for subtle tuning of steric and electronic properties. This has proven useful, as we have observed that PhPAd-DalPhos (Fig. 9), the less hindered diphenylphosphino variant of ‘parent’ PAd-DalPhos, offers excellent catalytic performance in a range of sought-after and otherwise challenging cross-coupling applications.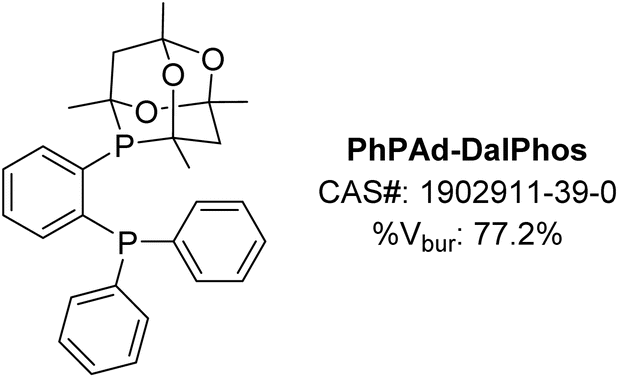 | ||
| Fig. 9 PhPAd-DalPhos ancillary ligand with associated CAS number and % buried volume. | ||
The C–N cross-coupling of α,α,α-trisubstituted primary alkylamines, such as 1-adamantylamine, is a class of transformations of interest to the pharmaceutical industry as it provides a means of introducing large lipophilic hydrocarbon groups into active pharmaceutical ingredients (APIs) that can allow for manipulation of absorption or membrane permeability properties.50 To address synthetic limitations in the metal-catalyzed N-arylation of so-called ‘bulky primary alkylamines’ of this type, our group in 2019 examined several P2-based nickel pre-catalysts including JosiPhos CyPF-Cy, DPPF, and CgP-containing DalPhos ligands.51 Efficient C–N cross-coupling was achieved specifically when using PhPAd-DalPhos (Fig. 10) in the test cross-couplings of 1-adamantylamine with (hetero)aryl chloride electrophiles. In identifying (PhPAd-DalPhos)Ni(o-tolyl)Cl as a suitable pre-catalyst for the C–N cross-coupling of α,α,α-trisubstituted primary alkylamines, a substrate scope was developed comprising (hetero)aryl chlorides, as well as (hetero)aryl bromide and tosylate electrophiles, with several room temperature transformations of this type being demonstrated for the first time (Fig. 10).
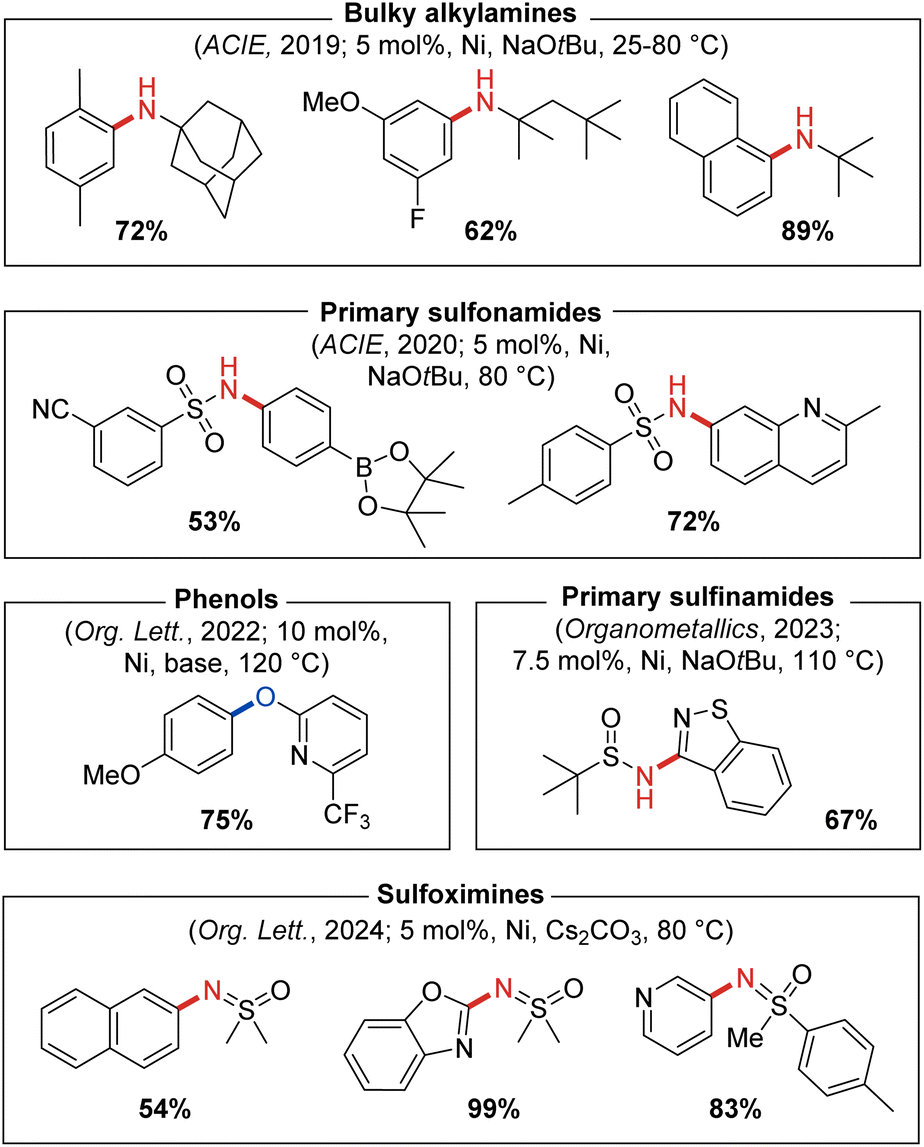 | ||
| Fig. 10 Representative nickel-catalyzed C–N and C–O cross-coupling products enabled by catalyst systems based on PhPAd-DalPhos. | ||
In 2020 our group reported the first thermal nickel-catalyzed cross-coupling of primary sulfonamides with (hetero)aryl chlorides, enabled by using (PhPAd-DalPhos)Ni(o-tolyl)Cl.52 In a preliminary screen that revealed the efficacy of PhPAd-DalPhos in such transformations, all other tested carbene and P2 ligands (i.e., IPr, DPEPhos, DPPF, and PAd-DalPhos ligand variants) gave negligible conversion to the desired N-arylated sulfonamide product. The reported substrate scope featured several aryl electrophile classes (i.e., chloride, bromide, iodide, carbamate, and tosylate) paired with a breadth of primary sulfonamides (Fig. 10).
The optimized conditions for the N-arylation of bulky alkylamines51 and primary sulfonamides52 with (hetero)aryl electrophiles use NaOtBu as base and (PhPAd-DalPhos)Ni(o-tolyl)Cl as the pre-catalyst (vide supra). Such nucleophile classes span more than 20 pKa units in NH acidity, yet the same catalyst system is effective in both transformations. To better understand such divergent C–N cross-couplings enabled by use of PhPAd-DalPhos, we conducted an experimental and computational mechanistic study of C–N reductive elimination, analogous to processes depicted in Fig. 1.53 Our 2022 study provided evidence in support of a previously undocumented, base-promoted C–N reductive elimination process proceeding from anionic imido/nitrene species [(PhPAd-DalPhos)Ni(aryl)(NSO2(aryl′))]−, which arise via deprotonation of sulfonamido intermediates of the type (PhPAd-DalPhos)Ni(aryl)(NHSO2(aryl′)) by the NaOtBu base present during catalysis. While such an unprecedented pathway for C–N reductive elimination is apparently preferred in cross-couplings of primary sulfonamides, the more conventional ‘direct’ reductive elimination from the amido complex (PhPAd-DalPhos)Ni(aryl)(NHtBu) was shown computationally to be favoured for the N-arylation of tert-butylamine.
Publications from our group appearing in 2023 and 2024 demonstrated (PhPAd-DalPhos)Ni(o-tolyl)Cl to be an effective catalyst system for related nickel-catalyzed cross-coupling of sulfinamides49 as well as sulfoximines54 with (hetero)aryl chlorides (Fig. 10). In sulfinamide cross-couplings, C–N reductive elimination was found to proceed via putative anionic nitrene-type species [(L)Ni(o-tolyl)(NS(O)tBu)]−, in keeping with sulfonamide N-arylation (vide supra).53 Beyond demonstrating broad electrophile scope in sulfoximine cross-couplings, including the successful N-arylation of the pharmaceutical Clozapine, competition experiments confirmed a marked reactivity preference of sulfoximine > sulfonamide > sulfinamide when using (PhPAd-DalPhos)Ni(o-tolyl)Cl as a pre-catalyst.
Following our successful application of DalPhos ligands in the nickel-catalyzed C–O cross-coupling of aliphatic alcohols,40,55 in 2022 we expanded such methodologies to the O-(hetero)arylation of phenols with chloropyridine-type electrophiles,56 inspired by the prevalence of pyridyl-O-aryl frameworks in natural products and APIs.57 Catalytic screening involving JosiPhos CyPF-Cy, DPPF, PAd-DalPhos, CyPAd-DalPhos, and PhPAd-DalPhos in the test cross-coupling of 4-chloroquinaldine and phenol revealed PhPAd-DalPhos/Ni(COD)2 mixtures to be particularly effective in affording the targeted unsymmetrical diaryl ether product (Fig. 10). The corresponding (PhPAd-DalPhos)Ni(o-tolyl)Cl pre-catalyst afforded negligible turnover in such transformations, an observation that parallels findings by our group in the context of amide N-arylation by use of a dual-base system (vide supra).44
3.4 PAd2-DalPhos
Encouraged by the success of ‘single-cage’ DalPhos ligands including PAd-DalPhos, CyPAd-DalPhos, and PhPAd-DalPhos (vide supra), in 2019 we wondered if two cages might be better than one in addressing unmet reactivity challenges in nickel-catalyzed cross-coupling. On this basis PAd2-DalPhos (Fig. 11) and related ligands were developed by our group in 2019.58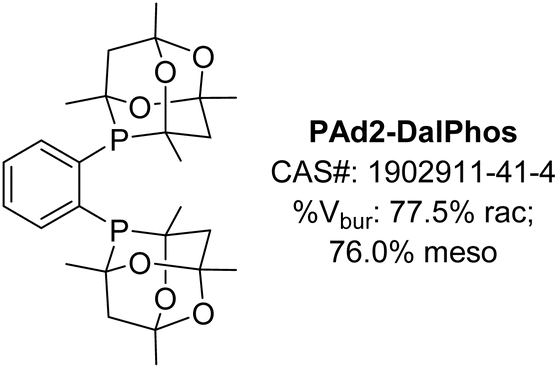 | ||
| Fig. 11 PAd2-DalPhos ancillary ligand with associated CAS number and % buried volumes for rac and meso diastereomers. | ||
In our initial report on this new ‘double cage’ ligand type, (CgP)2(hetero)arene variants of PAd-DalPhos incorporating phenyl (PAd2-DalPhos), pyridyl, or quinoxalyl backbone linkages were disclosed.58 Due to the chiral (racemic) nature of the HPCg precursor (Fig. 3), ∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric mixtures of meso (RS,SR) and rac (RR,SS) double cage ligands are formed in the course of ligand synthesis involving, for example in the case of PAd2-DalPhos, the P–C cross-coupling of CgP(o-C6H4Br) with HPCg. These diastereomers can be separated in air using column chromatography, and can exhibit differing catalytic efficacy. However, to date we have commonly employed PAd2-DalPhos as the prepared ∼1:1 meso/rac mixture. The synthesis of corresponding (L)Ni(o-tolyl)Cl pre-catalysts for these ligands follows the same steps outlined in Fig. 3. Initial testing of our new (CgP)2(hetero)arene ligand variants involved the use of derived (L)Ni(o-tolyl)Cl pre-catalysts in room temperature nickel-catalyzed C–N cross-couplings of furfurylamine with aryl chloride electrophiles at low catalyst loadings (0.25–2.5 mol%). The impressive reactivity displayed by PAd2-DalPhos (with meso out-performing rac) in these reactions encouraged our examination of its utility in the nickel-catalyzed cross-coupling of primary five- or six-membered ring heteroarylamines with (hetero)aryl chlorides for the synthesis of unsymmetrical di(hetero)arylamines. Such heteroatom-dense cross-couplings, while of relevance to the synthesis of bioactive compounds such as APIs, are challenging owing to the inhibiting effect of such substrates/products on catalytic turnover. In exploring the test cross-coupling of 2-aminooxazole and 4-chlorobenzophenone (2.5 mol% Ni, 80 °C), negligible catalytic turnover was achieved by use of pre-catalysts featuring JosiPhos CyPF-Cy, DPPF, or XantPhos; conversely, (PAd2-DalPhos)Ni(o-tolyl)Cl afforded quantitative conversion to the desired C–N cross-coupling product. In examining the scope of nickel-catalyzed C–N cross-coupling of primary heteroarylamines with (hetero)aryl electrophiles (Fig. 12), it was determined that PAd2-DalPhos offers catalytic performance competitive with state-of-the-art palladium catalysis in the synthesis of unsymmetrical di(hetero)arylamines.58 The successful use of (PAd2-DalPhos)Ni(o-tolyl)Cl for the N-arylation of indoles59 and N-methylsulfonamides52 with (hetero)aryl chlorides was also demonstrated in subsequent reports from our group.
:1 diastereomeric mixtures of meso (RS,SR) and rac (RR,SS) double cage ligands are formed in the course of ligand synthesis involving, for example in the case of PAd2-DalPhos, the P–C cross-coupling of CgP(o-C6H4Br) with HPCg. These diastereomers can be separated in air using column chromatography, and can exhibit differing catalytic efficacy. However, to date we have commonly employed PAd2-DalPhos as the prepared ∼1:1 meso/rac mixture. The synthesis of corresponding (L)Ni(o-tolyl)Cl pre-catalysts for these ligands follows the same steps outlined in Fig. 3. Initial testing of our new (CgP)2(hetero)arene ligand variants involved the use of derived (L)Ni(o-tolyl)Cl pre-catalysts in room temperature nickel-catalyzed C–N cross-couplings of furfurylamine with aryl chloride electrophiles at low catalyst loadings (0.25–2.5 mol%). The impressive reactivity displayed by PAd2-DalPhos (with meso out-performing rac) in these reactions encouraged our examination of its utility in the nickel-catalyzed cross-coupling of primary five- or six-membered ring heteroarylamines with (hetero)aryl chlorides for the synthesis of unsymmetrical di(hetero)arylamines. Such heteroatom-dense cross-couplings, while of relevance to the synthesis of bioactive compounds such as APIs, are challenging owing to the inhibiting effect of such substrates/products on catalytic turnover. In exploring the test cross-coupling of 2-aminooxazole and 4-chlorobenzophenone (2.5 mol% Ni, 80 °C), negligible catalytic turnover was achieved by use of pre-catalysts featuring JosiPhos CyPF-Cy, DPPF, or XantPhos; conversely, (PAd2-DalPhos)Ni(o-tolyl)Cl afforded quantitative conversion to the desired C–N cross-coupling product. In examining the scope of nickel-catalyzed C–N cross-coupling of primary heteroarylamines with (hetero)aryl electrophiles (Fig. 12), it was determined that PAd2-DalPhos offers catalytic performance competitive with state-of-the-art palladium catalysis in the synthesis of unsymmetrical di(hetero)arylamines.58 The successful use of (PAd2-DalPhos)Ni(o-tolyl)Cl for the N-arylation of indoles59 and N-methylsulfonamides52 with (hetero)aryl chlorides was also demonstrated in subsequent reports from our group.
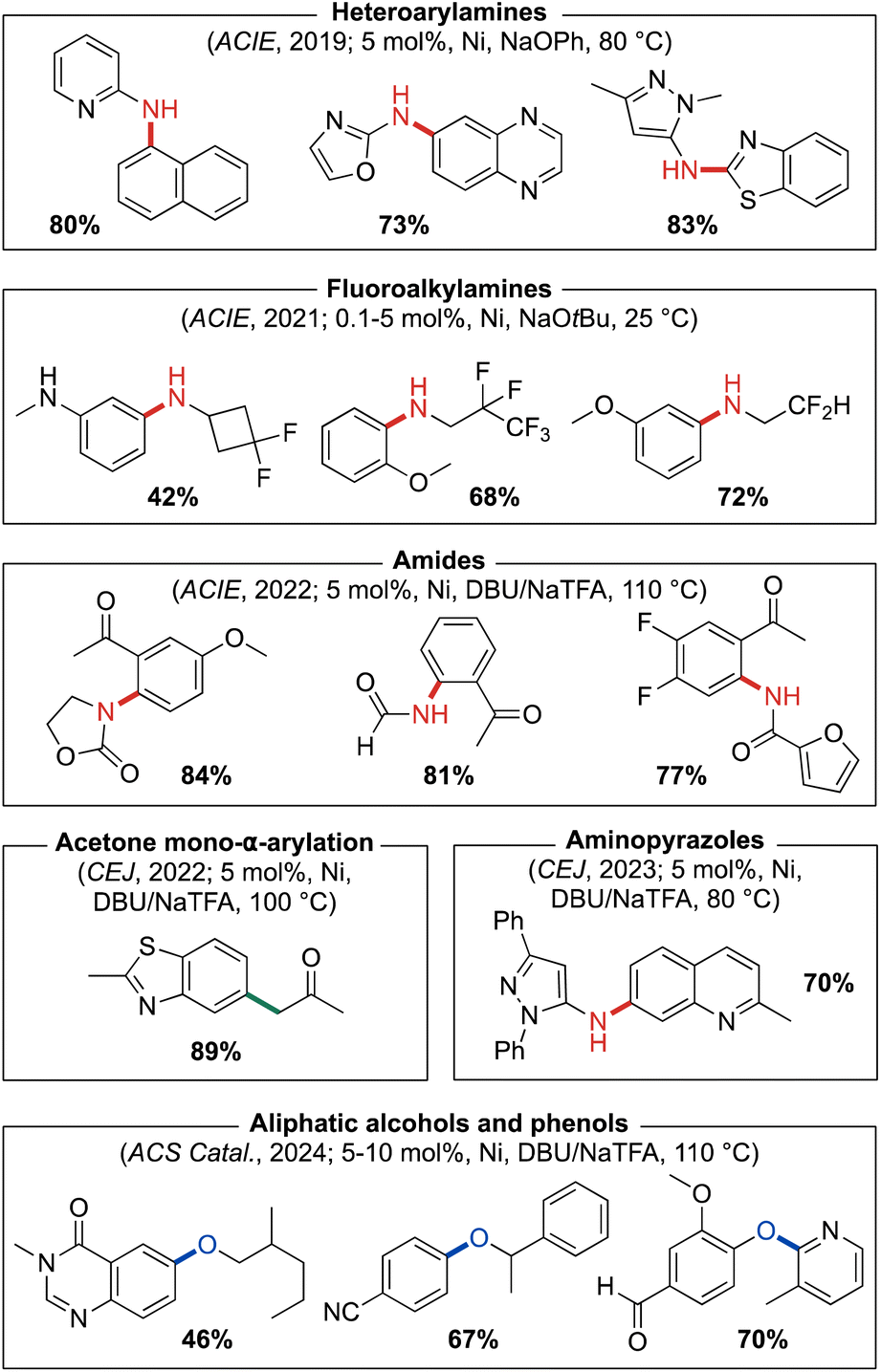 | ||
| Fig. 12 Representative nickel-catalyzed cross-coupling products enabled by catalyst systems based on PAd2-DalPhos. | ||
The incorporation of fluorinated addenda into APIs provides means of controlling adsorption, distribution, and metabolism.60 While the N-arylation of fluoroalkylamines is a reaction of interest in this context, such transformations have proven challenging. In 2021 we developed the first broadly useful nickel-based catalyst system for C–N cross-couplings of β-fluoroalkylamines with (hetero)aryl chlorides and phenol derivatives, using (PAd2-DalPhos)Ni(o-tolyl)Cl;61 this catalyst system complements, and in some cases improves upon, existing copper-62 and palladium-based63 protocols. We initially screened (L)Ni(o-tolyl)Cl pre-catalysts featuring PAd-DalPhos, PhPAd-DalPhos, CyPAd-DalPhos, PAd2-DalPhos, Phen-DalPhos (vide infra), NHP-DalPhos (vide infra), or DPPF ancillary ligands in the room temperature coupling of 2,2-difluoroethylamine with (hetero)aryl chlorides. Only (PAd2-DalPhos)Ni(o-tolyl)Cl afforded high conversion to the desired N-(β-fluoroalkyl)aniline products, and was subsequently deployed for a more thorough investigation of substrate scope (Fig. 12). Under the optimized conditions (i.e., 0.5–5.0 mol% Ni, 25 °C, NaOtBu), a broad range of linear and branched primary β-fluoroalkylamines were coupled with (hetero)aryl (pseudo)halides (i.e., chloride, bromide, iodide, tosylate, triflate). To accommodate base-sensitive functionalities in our electrophile scope, a soluble organic dual-base system (i.e., DBU/NaOTf, 110 °C) was successfully employed as an alternative to NaOtBu.
Previous work by Tlili, Amgoune, and co-workers64 focused on the mono-α-arylation of acetone using Ni/JosiPhos (CyPF-Ph) with CsF as base. Encouraged by this work, in 2022 we sought to further develop nickel-catalyzed α-arylation chemistry of small-molecule carbonyl compounds (e.g., acetone, ethyl acetate, dimethylacetamide, etc.), where in place of poorly soluble inorganic bases we targeted the use of soluble organic base systems as a means of addressing unmet synthetic limitations.45 Initially, we surveyed a series of CgP-based DalPhos and other commercially available ligands (i.e., DPEPhos, DPPF, and IPr) in nickel-catalyzed acetone mono-α-arylation with 4-chlorotoluene using DBU as base and NaTFA as the putative halide scavenger. Under these screening conditions (10 mol% Ni(COD)2, 15 mol% L, 100 °C), the highest conversion to product was achieved using PAd2-DalPhos. Further optimization determined (PAd2-DalPhos)Ni(4-CN-Ph)Cl (5 mol%) to be the optimal pre-catalyst (meso > rac) using DBU/NaTFA as the dual-base system for the mono-α-arylation of acetone (Fig. 12). Furthermore, a PAd2-DalPhos (12 mol%)/Ni(COD)2 (8 mol%) catalyst mixture also proved viable using DBU/NaOTf for this class of transformation. In related transformations of ethyl acetate and acetamide, CyPAd-DalPhos offered optimal performance (Fig. 8).
In a separate 2022 report, we detailed the use of a dual-base-enabled methodology in the nickel-catalyzed C–N cross-coupling of amides with 2′-(pseudo)halide-substituted acetophenones for application in the synthesis of pharmaceutically relevant 4-quinolones.65 In the pursuit of a suitable catalyst system for such reactions, a series of ancillary ligands were screened with Ni(COD)2 in the amidation of 2′-chloroacetophenone using DBU/NaTFA. Negligible conversion to product was achieved when using IPr, DPEPhos, or DPPF; while other DalPhos ligands demonstrated moderate reactivity, PAd2-DalPhos in particular proved highly effective (meso > rac). Following optimization of this reaction, a broad substrate scope of N-(ketoaryl)-amides (Fig. 12) was achieved through the N-arylation of amides with 2′-(pseudo)haloacetophenones using (PAd2-DalPhos)Ni(4-CN-Ph)Cl (5 mol%) and DBU/NaTFA, including room temperature reactions. Select examples of the formation of 4-quinolones through exposure to Camps cyclization conditions were reported.
Given the dearth of mechanistic information regarding the use of organic amine bases in thermally promoted nickel-catalyzed C–N cross-couplings, as part of this 2022 study we completed a stoichiometric examination of catalytic steps that support a putative Ni(0/II) cycle.65 Also disclosed therein was an assessment of possible Ni(I) involvement; however, the poor catalytic performance of ((PAd2-DalPhos)NiCl)2 relative to (PAd2-DalPhos)Ni(aryl)X pre-catalysts indicated that this Ni(I) complex, potentially generated via comproportionation of (PAd2-DalPhos)Ni(II) and (PAd2-DalPhos)Ni(0) species in catalysis, is likely not a key catalytic intermediate. The role that the DBU/NaTFA dual-base plays in the proposed mechanistic cycle was also investigated. Halide exchange involving (PAd2-DalPhos)Ni(aryl)Cl to give (PAd2-DalPhos)Ni(aryl)(TFA) proved viable, but not required, for catalysis. Moreover, our observation that catalyst inhibition occurs in the presence of added DBU·HCl supports the view that NaTFA serves to sequester chloride off-cycle, as invoked by Simmons and co-workers43 in Pd catalysis. Our work also established the feasibility of (PAd2-DalPhos)Ni(L′)n resting states in this catalytic system, where L′ is the C–N cross-coupling product or some other Lewis base. Given that catalytic turnover may require loss of L′, in a follow-up report in 2023 we probed whether the addition of a Lewis acid may facilitate L′ loss and regeneration of putative ‘(PAd2-DalPhos)Ni’.66 In this study we established that the addition of AlOTf3 (5 mol%) to amide C–N cross-coupling reactions employing (PAd2-DalPhos)Ni(4-CN-Ph)Cl (1 mol%) can in some instances greatly facilitate catalytic turnover.
As is evident from our discussion thus far, our development of CgP-based DalPhos ligands has been an evolution centered on addressing synthetic limitations in the field of metal-catalyzed cross-coupling, with a particular emphasis on N-arylation processes. In this journey since 2016, we developed over time several effective new reaction methodologies and DalPhos ligands for nickel-catalyzed C–N cross-coupling, but lacked comparative catalyst screening data that placed the performance of these various ligands in context. As such, in 2023 we conducted a systematic ‘head-to-head’ reactivity study employing representative reaction classes (known and new transformations) as a means of directly comparing the performance of these DalPhos ligands in nickel-catalyzed C–N cross-coupling.42 In this study, 672 individual experiments were conducted under mild conditions (3 mol% Ni, 60 °C), with various reaction parameters examined in the C–N cross-coupling of NH nucleophiles with (hetero)aryl chlorides, including: base (i.e., NaOtBu, K2CO3, or DBU/NaTFA); solvent (i.e., toluene or 1,4-dioxane); and pre-catalyst (i.e., (L)Ni(o-tolyl)Cl, where L = PAd-DalPhos, CyPAd-DalPhos, PhPAd-DalPhos, ThioPAd-DalPhos, PAd2-DalPhos, Phen-DalPhos (vide infra), or DPPF, the latter as a non-DalPhos comparison). Among the various reactivity trends observed, the successful cross-coupling of otherwise challenging aminopyrazole-type nucleophiles was achieved by use of (PAd2-DalPhos)Ni(o-tolyl)Cl when using DBU/NaTFA in 1,4-dioxane solvent; a diverse substrate scope pairing pyrazole-containing nucleophiles with (hetero)aryl chlorides was in turn established with this reaction system (Fig. 12).
Building on our successful development of thermal nickel-catalyzed C–O cross-coupling chemistry22e encompassing (hetero)aryl chloride and other electrophile classes, and enabled by use of DalPhos ligation (vide supra),40,55,56 we sought to further improve on such methodologies. In 2024 we reported on our program directed toward the use of soluble organic base in such C–O cross-couplings, rather than traditionally used inorganic bases; the benefits of such an approach have been highlighted throughout this perspective. With this focus, we achieved the first thermal, nickel-catalyzed O-arylation of aliphatic alcohols and phenols with (hetero)aryl chlorides proceeding using organic base, without the requirement of photochemical or electrochemical activation.67 Using catalyst systems based on PAd2-DalPhos (variation in performance observed for meso versus rac) and DBU/NaTFA or BTPP/NaTFA, we reported a scope of reactivity (Fig. 12) that is competitive with best-in-class metal-catalyzed (i.e., Cu, Pd, Ni, or other) C–O cross-coupling strategies. The use of PAd2-DalPhos/Ni(COD)2 in a C-to-O atom-swapping reaction sequence has been reported by Luu and Li.68
3.5 CgPhen-DalPhos
To frame our discussion of the final CgP-based DalPhos ligand to be presented in this perspective (i.e., CgPhen-DalPhos, vide infra), it is helpful if we briefly discuss our examination of two DalPhos ligands of relevance that do not feature the CgP motif: NHP-DalPhos and Phen-DalPhos (Fig. 13). As noted in the Introduction, our initial choice of the CgP fragment for incorporation into new P2 DalPhos ancillary ligands was made on the basis that we envisioned these would promote Ni(0/II) C–N, C–O, and other cross-coupling cycles by facilitating product-forming reductive elimination, while circumventing bis-chelation and/or comproportionation processes. Having successfully translated these concepts into productive catalysis by use of ligands such as PAd-DalPhos (Fig. 4), we wondered to what extent other non-CgP ligands might be similarly useful, if their design adhered to similar principles (i.e., sterically demanding, not overly electron-donating, within an ortho-phenylene-bridged P2 structure). In 2018 we disclosed that pairing a saturated and bulky N-heterocyclic phosphine (NHP) donor fragment with a diphenylphosphino donor afforded NHP-DalPhos (Fig. 13), which proved useful in nickel-catalyzed C–N cross-couplings of (hetero)aryl chlorides with primary alkylamines at room temperature,69 although not in a manner that improved upon the reactivity achieved with PAd-DalPhos. Our coordination chemistry studies revealed that cyclometallation involving an ortho-methyl of the mesityl group in NHP-DalPhos is facile, thereby representing a possible limitation of this ligand type.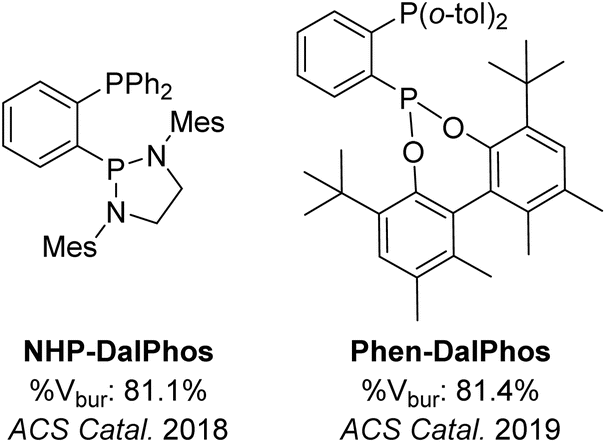 | ||
| Fig. 13 Non-CgP-based DalPhos ligands with associated % buried volumes (Mes = mesityl). | ||
In the continued pursuit of useful and complementary classes of non-CgP DalPhos ligands, we turned our attention to developing ligands in which a modestly donating and sterically encumbering phosphonite donor group replaces the CgP fragment of PAd-DalPhos.59 As part of this program, in 2019 we disclosed the preparation of Phen-DalPhos (Fig. 13), via lithiation of 1-P(o-tolyl)2-2-bromobenzene with n-BuLi, followed by quenching with the chlorophosphonite derived from commercially available racemic 5,5′,6,6′-tetramethyl-3,3′-di-tert-butyl-1,1′-biphenyl-2,2′-diol (herein rac-Phen-PCl). When using the derived pre-catalyst, (Phen-DalPhos)Ni(o-tolyl)Cl, two unique patterns of nickel-catalyzed C–N cross-coupling reactivity were observed versus CgP-type DalPhos ligands: selective diarylation of ammonia with (hetero)aryl chlorides under conditions whereby clean monoarylation is achieved by use of (PAd-DalPhos)Ni(o-tolyl)Cl; and remarkable efficacy in cross-couplings of (hetero)anilines with (hetero)aryl chlorides, including high NH2 arylation selectivity in cross-couplings of substrates such as 5-aminoindole under conditions whereby (PAd2-DalPhos)Ni(o-tolyl)Cl afforded clean indole NH arylation.59
Encouraged by the unique and useful reactivity behaviour achieved by use of Phen-DalPhos, we posited that the combination of Phen and CgP groups might give rise to a new ligand offering beneficial properties in nickel catalysis. We reported on such a conceptual merging of the CgP-containing PAd-DalPhos and the phosphonite Phen-DalPhos in our 2021 report on CgPhen-DalPhos (Fig. 14).55 CgPhen-DalPhos was prepared via the lithiation of CgP(o-C6H4Br) with n-BuLi, followed by quenching with rac-Phen-PCl, similar to the method outlined in Fig. 3. In this manner, the ligand is prepared as a mixture of diastereomers, owing to the presence of chiral (racemic) CgP and phosphonite groups.
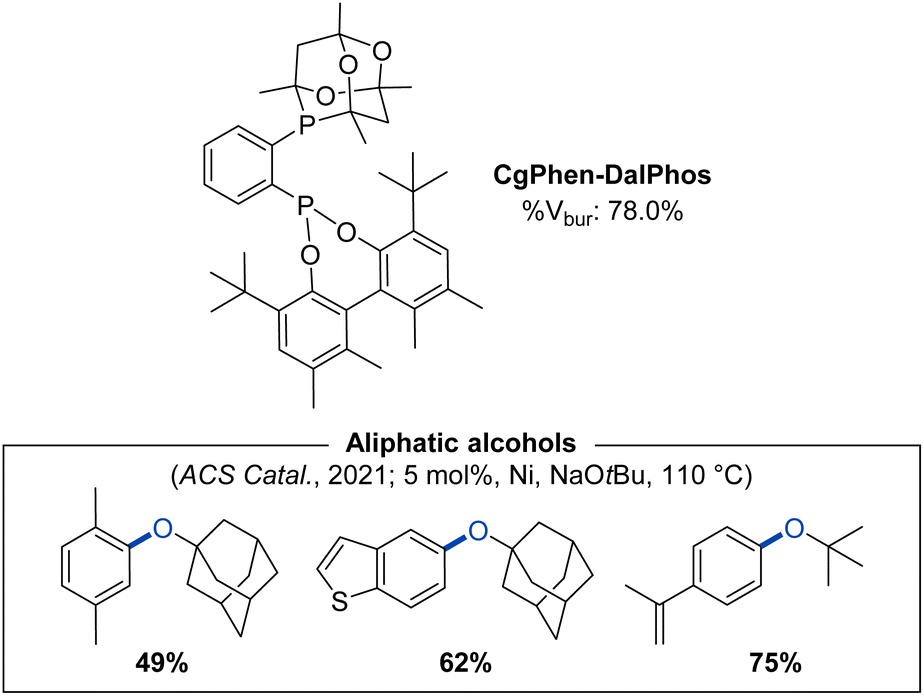 | ||
| Fig. 14 CgPhen-DalPhos ancillary ligand with associated % buried volume and representative nickel-catalyzed C–O cross-coupling products enabled by use of (CgPhen-DalPhos)Ni(o-tolyl)Cl. | ||
Notably, CgPhen-DalPhos proved successful in addressing what at the time was an important limitation in the field of nickel-catalyzed C–O cross-coupling: the efficient O-arylation of tertiary aliphatic alcohols.22e,55 In an initial screening campaign involving 1-adamantanol and a series of (hetero)aryl chlorides, we probed the utility (L)Ni(o-tolyl)Cl pre-catalysts comprising CgPhen-DalPhos, Phen-DalPhos, CyPAd-DalPhos,40 PhPAd-DalPhos,51 or DPPF.70 Despite the competence of these established ligands in various nickel-catalyzed C–O cross-couplings, only the new ligand CgPhen-DalPhos afforded synthetically useful conversion to product across the electrophile test set. (CgPhen-DalPhos)Ni(o-tolyl)Cl in turn displayed unprecedentedly broad scope in the O-arylation of tertiary aliphatic alcohols with (hetero)aryl (pseudo)halides (Fig. 14).
3.6 DalPhos ‘on demand’
Notwithstanding the efficacy of the DalPhos preparative synthetic protocols presented in Fig. 3, we recognized that the repeated low-temperature generation of CgP(o-C6H4Li) from CgP(o-C6H4Br), unreliable commercial access to R2PCl (or (RO)2PCl), and/or variability in the efficiency with which such P–Cl reagents react with CgP(o-C6H4Li), collectively create a barrier to preparing and applying our CgP-based DalPhos ligands by end-users. In response, we have recently developed an expedient means of producing both known and new DalPhos ligands of this type efficiently at room temperature from a single precursor, CgP(o-C6H4PCl2), as shown in Fig. 15. In a 2024 report, we demonstrate the successful use of CgP(o-C6H4PCl2) in cleanly generating libraries of known and new CgP-based DalPhos ligand variants (in situ, without requiring purification), thereby accelerating reaction screening and discovery of useful new DalPhos variants.71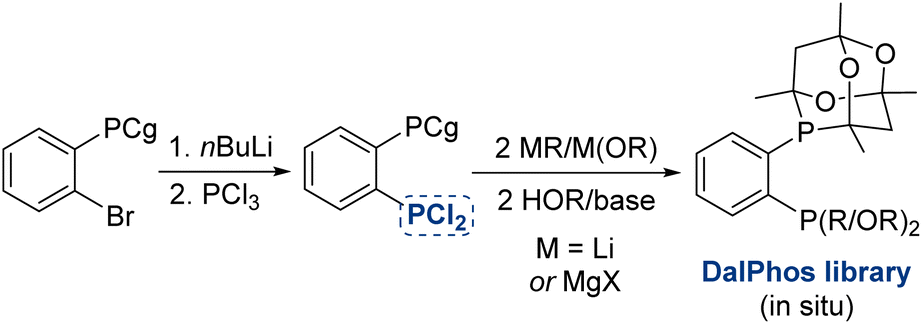 | ||
| Fig. 15 Expedited approach to DalPhos ligand synthesis for use in catalyst reaction screening. | ||
4. Conclusions and outlook
Over the past several years our research group has developed a family of new cage phosphine (CgP) ‘DalPhos’ (DALhousie PHOSphine) ligands that when deployed in nickel catalysis have advanced the state-of-the-art in metal-catalyzed cross-couplings of (hetero)aryl chloride or phenol-derived electrophiles with nitrogen-, oxygen-, or carbon-based nucleophiles (Fig. 16). Our progress in this regard in terms of ligand design and reaction development, including use of organic base in place of more conventionally used inorganic bases with potential applications in high-throughput reaction screening and flow methodologies, was outlined in this perspective. The experimental evidence presented herein collectively establishes that the sterically demanding and modestly electron-donating CgP fragment paired with the tunability of an adjacent phosphorus donor fragment (spanned by a phenylene linker) affords a privileged ligand architecture in this chemistry. In our research program we aim to develop operationally simple, high-performing, and sustainable nickel cross-coupling catalysts, amenable for use under mild conditions that can be adopted by end-users in place of precious metal-based catalysis. Our current and future commercialization of these DalPhos ligands and pre-catalysts is carried out with the intention of facilitating such end-user uptake.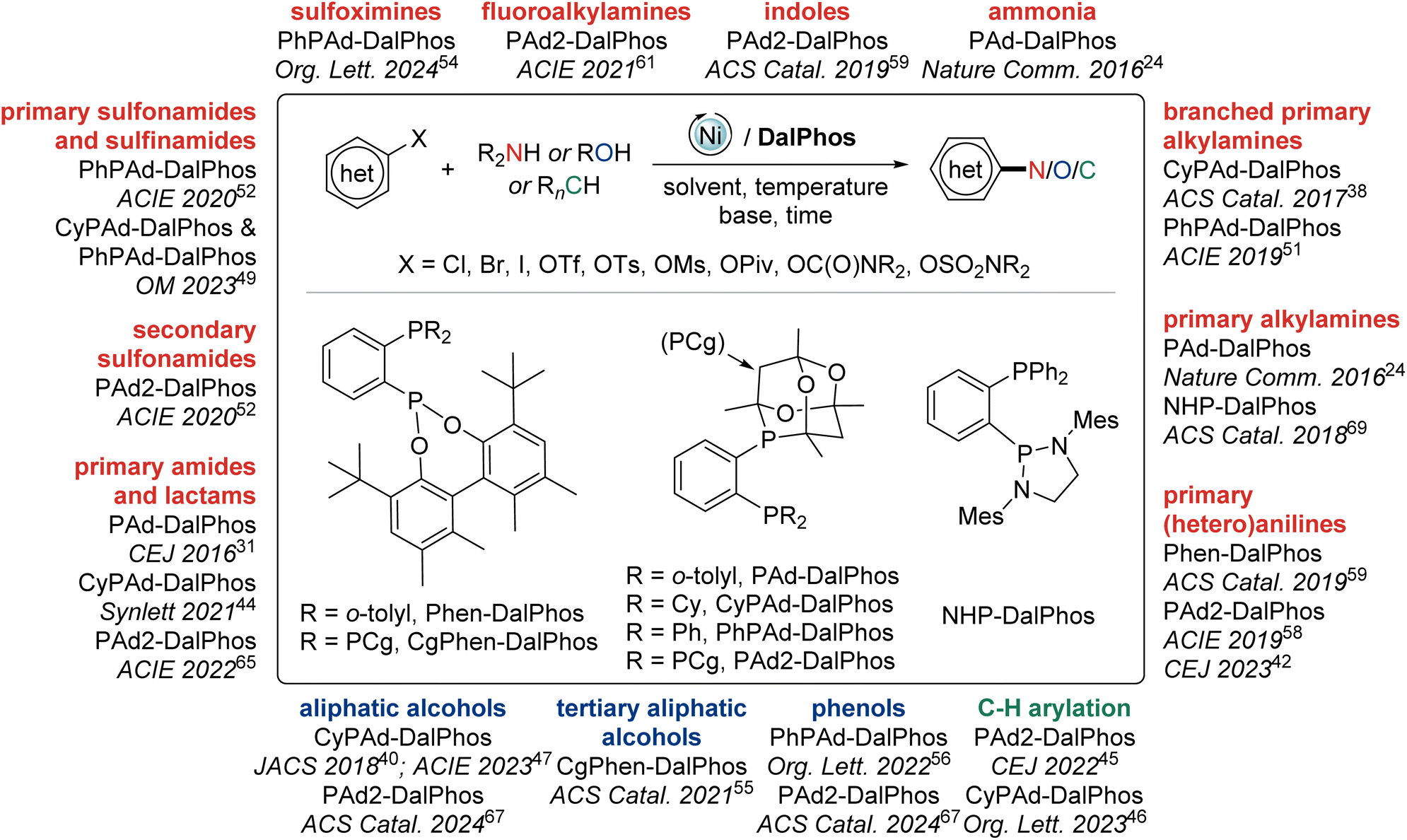 | ||
| Fig. 16 Overview of DalPhos ligands outlined in this perspective and their application in nickel-catalyzed C–N/O/C cross-coupling reactions with key lead references provided. | ||
These CgP-based DalPhos ligands were designed with the intent of accelerating key elementary reaction steps in Ni(0/II) catalytic cycles (e.g., product-forming reductive elimination), while discouraging what we anticipated would be deleterious side-reactions (e.g. unwanted redox chemistry). While we have made some progress in elucidating the mechanistic underpinnings of such cross-couplings, including the discovery of unexpected pathways both empirically and computationally (e.g., base-promoted C–N reductive elimination from anionic imido/nitrene species), more work needs to be done to confirm the way our CgP-based DalPhos ligands give rise to observed superlative catalytic performance. Such insights will inform the development of new DalPhos ligands and associated catalytic protocols needed to address remaining synthetic limitations and challenges, including expanding to other mechanistically distinct metal-catalyzed reaction manifolds.
Author contributions
K. M. M. and M. S. conceived and collaboratively drafted the manuscript.Conflicts of interest
The authors declare the following competing financial interests: Dalhousie University has filed patents on the DalPhos ancillary ligands and derived nickel pre-catalysts used in this work, from which royalty payments may be derived.Acknowledgements
We are grateful to the NSERC of Canada (Discovery Grant for M. S. RGPIN-2019-04288; PGS-D for K. M. M.), the province of Nova Scotia (including a Nova Scotia Graduate Scholarship for K. M. M.), and Dalhousie University (including a Killam Scholarship for K. M. M.) for their support of this work. M. S. would like to personally thank the student and postdoctoral researchers, academic and industrial collaborators, and other colleagues whose collective insight, focus, and determination enabled the development of the chemistry summarized in this manuscript.Notes and references
-
J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Mill Valley, California, USA, 2009 Search PubMed
.
-
M. Stradiotto and R. J. Lundgren, Ligand Design in Metal Chemistry: Reactivity and Catalysis, John Wiley & Sons, Ltd, Chichester, West Sussex, UK, 2016 Search PubMed
-
(a) C. A. Busacca, D. R. Fandrick, J. J. Song and C. H. Senanayake, Adv. Synth. Catal., 2011, 353, 1825–1864 CrossRef CAS
-
(a) J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534–1544 CrossRef CAS PubMed
-
(a) S. Enthaler and A. Company, Chem. Soc. Rev., 2011, 40, 4912–4924 RSC
- R. J. Lundgren and M. Stradiotto, Chem.–Eur. J., 2012, 18, 9758–9769 CrossRef CAS
- D. S. Surry and S. L. Buchwald, Chem. Sci., 2011, 2, 27–50 RSC
-
F. Terrier, Modern Nucleophilic Aromatic Substitution, Wiley-VCH, Weinheim, 2013 Search PubMed
-
(a) C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525–3550 RSC
-
(a)
R. M. Bullock, Catalysis Without Precious Metals, Wiley-VCH, Weinheim, Germany, 2010 CrossRef
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed
- V. V. Grushin and H. Alper, Chem. Rev., 1994, 94, 1047–1062 CrossRef CAS
-
(a) V. P. Ananikov, ACS Catal., 2015, 5, 1964–1971 CrossRef CAS
-
(a) E. B. Corcoran, M. T. Pirnot, S. S. Lin, S. D. Dreher, D. A. DiRocco, I. W. Davies, S. L. Buchwald and D. W. C. MacMillan, Science, 2016, 353, 279–283 CrossRef CAS PubMed
- C. M. Lavoie and M. Stradiotto, ACS Catal., 2018, 8, 7228–7250 CrossRef CAS
-
(a) J. P. Wolfe and S. L. Buchwald, J. Am. Chem. Soc., 1997, 119, 6054–6058 CrossRef CAS
- R. J. Lundgren, B. D. Peters, P. G. Alsabeh and M. Stradiotto, Angew. Chem., Int. Ed., 2010, 49, 4071–4074 CrossRef CAS PubMed
- A. Borzenko, N. L. Rotta-Loria, P. M. MacQueen, C. M. Lavoie, R. McDonald and M. Stradiotto, Angew. Chem., Int. Ed., 2015, 54, 3773–3777 CrossRef CAS PubMed
-
P. G. Pringle and M. B. Smith, in Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, ed. P. C. J. Kamer and P. W. N. M. van Leeuwen, John Wiley & Sons Ltd, Chichester, UK, 2012, ch. 13, pp. 391–404 Search PubMed
- S. Z. Ge, R. A. Green and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 1617–1627 CrossRef CAS
- C. S. Day and R. Martin, Chem. Soc. Rev., 2023, 52, 6601–6616 RSC
-
(a) M. Marín, R. J. Rama and M. C. Nicasio, Chem. Rec., 2016, 16, 1819–1832 CrossRef PubMed
- M. Epstein and S. A. Buckler, J. Am. Chem. Soc., 1961, 83, 3279–3282 CrossRef CAS
- C. M. Lavoie, P. M. MacQueen, N. L. Rotta-Loria, R. S. Sawatzky, A. Borzenko, A. J. Chisholm, B. K. V. Hargreaves, R. McDonald, M. J. Ferguson and M. Stradiotto, Nat. Commun., 2016, 7, 11073 CrossRef PubMed
- E. A. Standley, S. J. Smith, P. Müller and T. F. Jamison, Organometallics, 2014, 33, 2012–2018 CrossRef CAS PubMed
- J. Schranck and A. Tlili, ACS Catal., 2018, 8, 405–418 CrossRef CAS
- R. A. Green and J. F. Hartwig, Angew. Chem., Int. Ed., 2015, 54, 3768–3772 CrossRef CAS PubMed
- S. M. Crawford, C. B. Lavery and M. Stradiotto, Chem.–Eur. J., 2013, 19, 16760–16771 CrossRef CAS
-
(a) Q. L. Shen and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 10028–10029 CrossRef CAS
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed
- C. M. Lavoie, P. M. MacQueen and M. Stradiotto, Chem.–Eur. J., 2016, 22, 18752–18755 CrossRef CAS
- J. D. Hicks, A. M. Hyde, A. M. Cuezva and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 16720–16734 CrossRef CAS
- W. C. Shakespeare, Tetrahedron Lett., 1999, 40, 2035–2038 CrossRef CAS
-
(a) L. Hie, S. D. Ramgren, T. Mesganaw and N. K. Garg, Org. Lett., 2012, 14, 4182–4185 CrossRef CAS PubMed
- J. S. K. Clark, C. M. Lavoie, P. M. MacQueen, M. J. Ferguson and M. Stradiotto, Organometallics, 2016, 35, 3248–3254 CrossRef CAS
- J. S. K. Clark, R. T. McGuire, C. M. Lavoie, M. J. Ferguson and M. Stradiotto, Organometallics, 2019, 38, 167–175 CrossRef CAS
- C. M. Lavoie, R. McDonald, E. R. Johnson and M. Stradiotto, Adv. Synth. Catal., 2017, 359, 2972–2980 CrossRef CAS
- J. P. Tassone, P. M. MacQueen, C. M. Lavoie, M. J. Ferguson, R. McDonald and M. Stradiotto, ACS Catal., 2017, 7, 6048–6059 CrossRef CAS
- J. A. Terrett, J. D. Cuthbertson, V. W. Shurtleff and D. W. C. MacMillan, Nature, 2015, 524, 330–334 CrossRef CAS PubMed
- P. M. MacQueen, J. P. Tassone, C. Diaz and M. Stradiotto, J. Am. Chem. Soc., 2018, 140, 5023–5027 CrossRef CAS PubMed
- K. M. Morrison, N. J. Roberts, S. L. Dudra, J. P. Tassone, M. J. Ferguson, E. R. Johnson and M. Stradiotto, J. Org. Chem., 2024 DOI:10.1021/acs.joc.1023c01584
- N. Martinek, K. M. Morrison, J. M. Field, S. A. Fisher and M. Stradiotto, Chem.–Eur. J., 2023, 29, e202203394 CrossRef CAS PubMed
- G. L. Beutner, J. R. Coombs, R. A. Green, B. Inankur, D. Lin, J. Qiu, F. Roberts, E. M. Simmons and S. R. Wisniewski, Org. Process Res. Dev., 2019, 23, 1529–1537 CrossRef CAS
- T. Lundrigan, J. P. Tassone and M. Stradiotto, Synlett, 2021, 32, 1665–1669 CrossRef CAS
- J. W. M. MacMillan, R. T. McGuire and M. Stradiotto, Chem.–Eur. J., 2022, 28, e202200764 CrossRef CAS
- N. E. Bode and M. Stradiotto, Org. Lett., 2023, 25, 8809–8813 CrossRef CAS PubMed
- K. M. Morrison, C. S. Yeung and M. Stradiotto, Angew. Chem., Int. Ed., 2023, 62, e202300686 CrossRef CAS
- T. Lundrigan, J. P. Tassone and M. Stradiotto, Can. J. Chem., 2023, 101, 275–283 CrossRef CAS
- C. M. Simon, K. N. Robertson, P. L. DeRoy, A. A. Yadav, E. R. Johnson and M. Stradiotto, Organometallics, 2023, 42, 1704–1710 CrossRef CAS
-
(a) J. Liu, D. Obando, V. Liao, T. Lifa and R. Codd, Eur. J. Med. Chem., 2011, 46, 1949–1963 CrossRef CAS PubMed
- J. P. Tassone, E. V. England, P. M. MacQueen, M. J. Ferguson and M. Stradiotto, Angew. Chem., Int. Ed., 2019, 58, 2485–2489 CrossRef CAS PubMed
- R. T. McGuire, C. M. Simon, A. A. Yadav, M. J. Ferguson and M. Stradiotto, Angew. Chem., Int. Ed., 2020, 59, 8952–8956 CrossRef CAS PubMed
- C. M. Simon, S. L. Dudra, R. T. McGuire, M. J. Ferguson, E. R. Johnson and M. Stradiotto, ACS Catal., 2022, 12, 1475–1480 CrossRef CAS
- S. A. Fisher, C. M. Simon, P. L. Fox, M. J. Cotnam, P. L. DeRoy and M. Stradiotto, Org. Lett., 2024, 26, 1326–1331 CrossRef CAS PubMed
- K. M. Morrison, R. T. McGuire, M. J. Ferguson and M. Stradiotto, ACS Catal., 2021, 11, 10878–10884 CrossRef CAS
- N. E. Bode, R. T. McGuire and M. Stradiotto, Org. Lett., 2022, 24, 8986–8989 CrossRef CAS PubMed
- K. A. Scott, P. B. Cox and J. T. Njardarson, J. Med. Chem., 2022, 65, 7044–7072 CrossRef CAS
- J. S. K. Clark, M. J. Ferguson, R. McDonald and M. Stradiotto, Angew. Chem., Int. Ed., 2019, 58, 6391–6395 CrossRef CAS PubMed
- R. T. McGuire, J. F. J. Paffile, Y. Q. Zhou and M. Stradiotto, ACS Catal., 2019, 9, 9292–9297 CrossRef CAS
-
(a) K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed
- R. T. McGuire, A. A. Yadav and M. Stradiotto, Angew. Chem., Int. Ed., 2021, 60, 4080–4084 CrossRef CAS
- S. Chen, H. Wang, W. Jiang, P. X. Rui and X. G. Hu, Org. Biomol. Chem., 2019, 17, 9799–9807 RSC
- A. T. Brusoe and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 8460–8468 CrossRef CAS PubMed
- S. Abou Derhamine, T. Krachko, N. Monteiro, G. Pilet, J. Schranck, A. Tlili and A. Amgoune, Angew. Chem., Int. Ed., 2020, 59, 18948–18953 CrossRef PubMed
- R. T. McGuire, T. Lundrigan, J. W. M. MacMillan, K. N. Robertson, A. A. Yadav and M. Stradiotto, Angew. Chem., Int. Ed., 2022, 61, e202200352 CrossRef CAS PubMed
- M. J. Cotnam, N. Martinek and M. Stradiotto, Eur. J. Org Chem., 2023, e202301004 CrossRef CAS
- K. M. Morrison, N. E. Bodé, S. M. E. Knight, J. Choi and M. Stradiotto, ACS Catal., 2024, 14, 566–573 CrossRef CAS
- Q. H. Luu and J. Li, Chem. Sci., 2022, 13, 1095–1100 RSC
- A. V. Gatien, C. M. Lavoie, R. N. Bennett, M. J. Ferguson, R. McDonald, E. R. Johnson, A. W. H. Speed and M. Stradiotto, ACS Catal., 2018, 8, 5328–5339 CrossRef CAS
- G. Mann and J. F. Hartwig, J. Org. Chem., 1997, 62, 5413–5418 CrossRef CAS
- J. W. M. MacMillan, R. T. McGuire, A. M. McMahon, T. S. Anderson, K. N. Robertson and M. Stradiotto, ACS Catal., 2024, 14, 4074–4081 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2024 |