
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnlocking the chemical environment of nitrogen in perovskite-type oxides†
Shunsuke
Shimizu
 a,
Takeharu
Yoshii
*a,
Ginga
Nishikawa
ab,
Jingwen
Wang
ac,
Shu
Yin
ab,
Eiichi
Kobayashi
d and
Hirotomo
Nishihara
*ab
a,
Takeharu
Yoshii
*a,
Ginga
Nishikawa
ab,
Jingwen
Wang
ac,
Shu
Yin
ab,
Eiichi
Kobayashi
d and
Hirotomo
Nishihara
*ab
aInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai, Miyagi 980-8577, Japan. E-mail: takeharu.yoshii.b3@tohoku.ac.jp; hirotomo.nishihara.b1@tohoku.ac.jp
bAdvanced Institute for Materials Research (WPI-AIMR), Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai, Miyagi 980-8577, Japan
cSchool of Environmental Science and Engineering, State Key Laboratory of Metal Matrix Composites, Shanghai Jiao Tong University, Shanghai, 200240, P. R. China
dKyushu Synchrotron Light Research Center, 8-7 Yayoigaoka, Tosu, Saga, 841-0005, Japan
First published on 27th June 2024
Abstract
Nitrogen (N) doping of perovskite-type oxides is an effective method for enhancing their photocatalytic performance. Quantitative and qualitative analyses of the doped N species are essential for a deeper understanding of the catalytic activity enhancement mechanism. However, examining the N environment in perovskite-type oxides, particularly in the bulk, using conventional analytical techniques, such as X-ray photoelectron spectroscopy (XPS), is challenging. In this study, we propose a new analytical technique, advanced temperature-programmed desorption (TPD) up to 1600 °C, to complement the conventional methods. TPD can quantify all N species in bulk oxides. Moreover, it facilitates chemical speciation of N environments, such as substitutional and interstitial N species. This is verified by XPS, CHN elemental analysis, X-ray absorption spectroscopy, and in situ diffuse reflectance infrared Fourier-transform spectroscopy. This study demonstrates the feasibility of advanced TPD as a new analytical method that offers comprehensive information on the N species within N-doped oxide materials at the bulk level.
Introduction
Perovskite-type oxides are functional materials with excellent physical and chemical properties.1–4 In particular, two-dimensional perovskite-type oxides have recently gained significant attention for solar energy conversion and environmental remediation.5–8 Perovskite-type La2Ti2O7 (LTO) nanoparticles with a layered structure has been regarded as a promising photocatalyst for organic decomposition, water splitting, and CO2 reduction reactions owing to its high charge carrier mobility, high stability, low cost, and low toxicity.6,9–12 However, LTO photocatalysts are inefficient at utilizing visible light because of their wide bandgap of approximately 3.8 eV.13,14 Therefore, N-doping is an effective strategy for narrowing the bandgap.15,16 Generally, the doped N species in LTO belong to two chemical states: (i) substitutional N for oxygen atoms in the lattice (Ti–N bond) or (ii) interstitial N bonding to oxygen in the lattice structure (Ti–O–N bond).17–19 These introduction modes of N species have a significant impact on the resulting band structure. Theoretical studies have revealed a correlation between the N environment and photocatalytic activity.12,20 For substituted N species, mixing of the doped N 2p and O 2p orbitals creates continuous bands above the valence band maximum, thus narrowing the band gap. For interstitial N, the π* NO levels originating from the Ti–O–N bond were in the high-energy region of the band gap, enabling visible light adsorption. However, these π* NO levels also act as recombination centres for charge carriers and may reduce the photocatalytic activity. It has been experimentally shown that an appropriate ratio of substitutional N to the interstitial N species results in high photocatalytic activity.16 Therefore, precisely tuned N-doping is essential for targeted band engineering to develop visible-light-responsive catalysts.Precise N-doping, first of all, requires a reliable analytical technique that can distinguish and quantify the forms of doped N species. In most studies, X-ray photoelectron spectroscopy (XPS) has been used to analyse the N species in LTO and examine the electronic states.9,15,16,21 However, there is an issue with the inconsistent peak assignments of N-doped LTO in the N 1s XPS spectra. Most reports attribute the peak at approximately 399 eV to the Ti–O–N bond,19,22–27 whereas some attribute it to the Ti–N bond.28,29 In addition, XPS measurements can provide only the surface environment; however, to fully understand the doping states of the N species, bulk information is required. CHN elemental analysis can be used to determine the total N content in the bulk but is not suitable for chemical speciation. Therefore, another new technique that can accurately perform both quantitative and qualitative analyses of bulk N species is required.
Temperature-programmed desorption (TPD) is an important thermal analytical method, in which a sample is heated at a constant heating rate, and the desorbed gases from the sample are detected using a mass spectrometer or high-speed gas chromatography.30–33 For example, when the TPD method is applied to carbon materials, each oxygen functional group can be quantified from the information on the desorbed gas species and their desorption temperature. However, the total determination of hydrogen in carbon materials is not possible with commercial equipment because of their low sensitivity under a He flow.34,35 Our group developed an advanced vacuum TPD device with an induction heating system capable of reaching temperatures up to 1800 °C.36–40 This technology enables total hydrogen quantification in carbon.36–39 More recently, the maximum temperature of TPD has been extended to 2100 °C, allowing qualitative and quantitative analysis of N species in bulk carbon materials, with high sensitivity at the 10 ppm level.41 High sensitivity has been achieved by thorough insulation of the sample holder to minimize heat transfer at very high temperatures, which suppresses degassing from the TPD equipment components.41 Owing to these features, advanced TPD paves the way for new frontiers in carbon-edge site chemistry, leading to the development of three-dimensional graphene-based materials.42–48 Here, it is conceivable that the advanced TPD method could be useful not only for carbon materials but also for oxides doped with light elements. An essential feature of the TPD method is that it is a bulk analytical method, complementing the surface-sensitive XPS techniques.
In this study, we propose high-temperature vacuum TPD as a promising evaluation approach for N species in oxides. The measurement targets used in this study were perovskite-type N-doped LTO nanoparticles.16 We conducted a combined study of XPS, X-ray absorption spectroscopy (XAS), CHN elemental analysis, and in situ diffuse reflectance infrared Fourier-transform spectroscopy (DRIFTS). Our findings revealed that advanced TPD can quantify the total N species in bulk materials; more importantly, the features of the TPD spectra reflect the N environment in perovskite-type oxides, such as substitutional and interstitial N.
Experimental
Sample preparation
TPD analysis
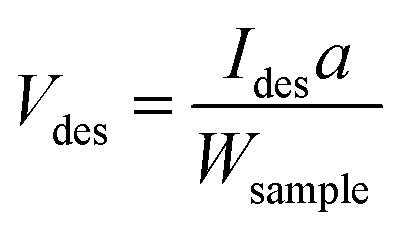 | (1) |
Results and discussion
TPD analysis of N species in La2Ti2O7
For the TPD measurements, perovskite-type N-doped LTO nanoparticles were synthesized as target materials according to a previous report.16 The synthesis procedure is illustrated in Fig. 1a (see the Experimental section for details). Briefly, LTO nanoparticles (LTO_TEA) were synthesized through a hydrothermal method using lanthanum and titanium sources along with triethanolamine (TEA) as an additive. LTO_TEA nanoparticles were white in colour, containing trace amounts of N species derived from TEA on their surface. The prepared LTO_TEA nanoparticles were heated at 650 °C under an NH3 flow to produce grey LTO nanoparticles with N (LTO_N). Nitrogen in the nanoparticles, originating from NH3, was introduced into the matrices by diffusion from the surface to the bulk. A previous study demonstrated that LTO_N has higher photocatalytic activity than LTO_TEA under visible light irradiation because of its narrower band gap caused by N doping via NH3 treatment.16 Note that the PXRD patterns and XPS spectra of LTO_TEA and LTO_N agreed well with the previous study, confirming synthetic reproducibility (see Fig. 1b, c, and S3†).16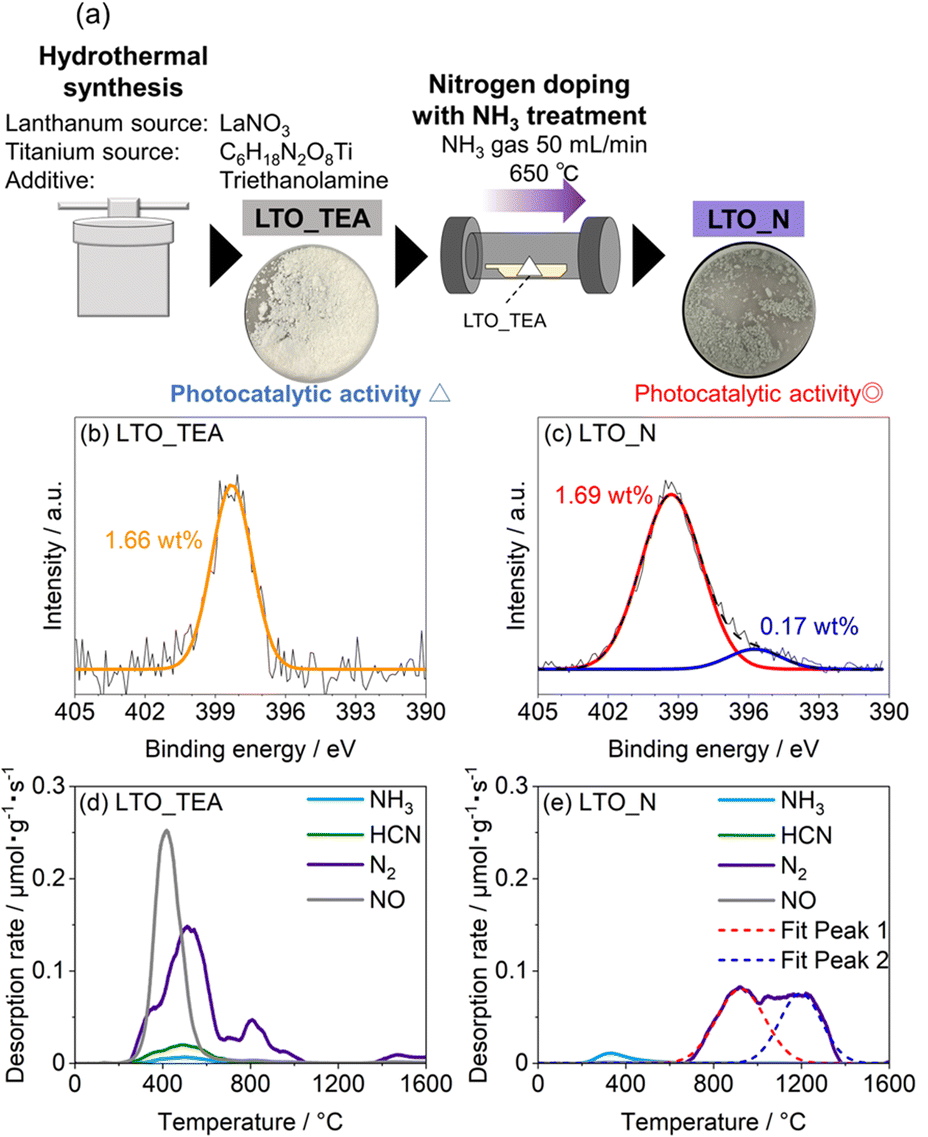 | ||
| Fig. 1 (a) Schematic of the synthesis procedure of LTO_TEA and LTO_N. N 1s XPS spectra of (b) LTO_TEA and (c) LTO_N. TPD profiles of (d) LTO_TEA and (e) LTO_N for N-containing gases. | ||
N 1s XPS measurements were performed to gain insight into the chemical states of the N species. As shown in Fig. 1b, LTO_TEA exhibited a peak at 398.3 eV, corresponding to 1.66 wt% N species even before the NH3 treatment. The presence of a trace amount of N is ascribed to contaminants derived from TEA added during the hydrothermal process. LTO_N, obtained after NH3 treatment, exhibited a peak at approximately 399.3 eV (red), with a small shoulder peak at approximately 395.8 eV (blue) (Fig. 1c). The peak at 399.3 eV can be assigned to Ti–O–N bonds, that is, interstitial N bonding with oxygen in the lattice, based on previous literature.26,27 The peak at 395.8 eV is attributed to Ti–N bonds, that is, substitutional N for oxygen in the framework.6,19,26,53 However, determining the N environment solely from the XPS peak assignments is difficult. First, as mentioned in the Introduction section, various assertions have been presented in the literature concerning the attribution of peaks to particular chemical species. Second, several different N species often have similar binding energies. The major peaks in LTO_TEA and LTO_N were observed at approximately 399 eV (see Fig. S4†). However, these samples, which were synthesized through different processes, may contain different types of N species (discussed in more detail in the next section). Therefore, relying solely on XPS measurements can lead to inappropriate conclusions regarding the N states of the oxide.
In this study, we developed an advanced TPD method for bulk analysis of N-doped metal oxides. We previously developed a highly sensitive TPD system for analysing carbon-based materials (Fig. S1a†). However, graphite sample holders, which have been used for carbon samples, react with metal oxides at high temperatures, even in a vacuum, to produce CO gas.54,55 Thus, we developed a new system that allows the use of tungsten sample holders with high melting and boiling points because they do not react with metal oxides even at high temperatures (Fig. S1b†).56 Minimal gas desorption was confirmed at temperatures up to 1600 °C in the blank test using a tungsten sample holder (Fig. S5†). Fig. 1d shows the TPD profiles of the N-containing gases of LTO_TEA (for analysis details, see the Experimental section and Fig. S2 and S6†). The main desorbed gas species were NO and N2, with the desorption temperatures ranging from 300 to 600 °C and 300 to 1000 °C, respectively. In contrast, in the desorption profile of LTO_N (Fig. 1e), the desorbed gas at 700–1400 °C was primarily N2. These results indicate that the desorption pattern depends on the environment of the N species, suggesting the potential use of TPD for the identification of N species. This has been described in detail in the next section. The quantitative accuracy of TPD was verified by comparison with other analytical methods, and the corresponding data are presented in Table 1. The N contents determined by TPD align closely with the CHN results, providing clear evidence that the N species were fully detected as gas species by TPD. In contrast, XPS revealed larger quantitative values for both samples. This difference is likely due to the surface segregation of N species in the samples, given that XPS is surface-sensitive. This segregation is reasonable because N-containing organic contaminants in LTO_TEA are expected to be anchored to the surface. Moreover, the N source of LTO_N is dissolved in the solid and diffuses from the surface to the bulk during synthesis. The TPD results align well with those obtained from the CHN analysis, in contrast to XPS. This strongly indicates that TPD is a bulk analytical method for oxides. The desorption of all N species at temperatures up to 1600 °C can be verified by the N 1s XPS spectra (Fig. S7†) and CHN elemental analysis results (Table S2†) of post-TPD measurements. Note that CHN elemental analysis overestimates the amount of N species below 0.1 wt%, always showing approximately 0.1 wt%.41
| Sample | TPD/wt% | CHN/wt% | XPS/wt% |
|---|---|---|---|
| LTO_TEA | 1.19 | 1.13 | 1.66 |
| LTO_N | 0.68 | 0.72 | 1.86 |
Commercial instruments generally detect desorbed gases under a He flow rather than under vacuum. Fig. S8† shows the results of the desorbed gas analysis of LTO_TEA using a widely used thermogravimetry-mass spectrometry (TG-MS) system. The peak of m/z = 30 (NO) was observed around 350 °C; however, the signal for m/z = 28, corresponding to N2, continuously decreased, and no clear desorption peak was observed. These results are significantly different from the advanced vacuum TPD results (see Fig. 1d). The commercial system could not detect trace N2 gas desorption from the sample owing to air contamination in the chamber, despite minimizing the air contamination (see also the Experimental section). This highlights the superiority of the advanced TPD method using induction heating under vacuum for N analysis.
Desorption pattern assignments for TPD
To establish a qualitative analytical method, examining the types of N species in the TPD profiles that are responsible for NO and N2 gas desorption from LTO_TEA and LTO_N is important. The TPD profile of LTO_TEA exhibited NO and N2 desorption at low temperatures, along with CO, CO2, and H2O desorption (Fig. 1d and S6a†). The overlap of N-containing gas evolution with those typically found during the thermal decomposition of organic substances strongly suggests that NO and N2 were generated by the decomposition of organic substances derived from the TEA used in the hydrothermal reaction. It has been reported that N species in organic compounds exhibit an N 1s peak at a binding energy of 398–399 eV.57–59 Moreover, the electrostatic attraction between the N of a TEA molecule and the surface![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) TiO− group (N–O bond) also exhibits an N 1s peak at a binding energy of 398 eV.60 These observations are consistent with the peak position of LTO_TEA, suggesting the presence of N–O and/or C–N bonds (Fig. 1b). In contrast, N2 desorption from LTO_N at temperatures above 800 °C was not accompanied by the desorption of other gases (see Fig. 1e and S6b†). Only CO2 desorption, presumably from residual organic substances, was observed at 200–800 °C. Thus, the N2 desorption in a high-temperature region can be ascribed to the strongly bound N species in the oxides. It is noteworthy that both LTO_TEA and LTO_N exhibited peaks at similar binding energies in the N 1s XPS spectra (see Fig. S4†); therefore, discussing the N environment based on XPS alone could not provide accurate inferences. The combination of TPD and XPS facilitates a more reliable chemical structure analysis.
TiO− group (N–O bond) also exhibits an N 1s peak at a binding energy of 398 eV.60 These observations are consistent with the peak position of LTO_TEA, suggesting the presence of N–O and/or C–N bonds (Fig. 1b). In contrast, N2 desorption from LTO_N at temperatures above 800 °C was not accompanied by the desorption of other gases (see Fig. 1e and S6b†). Only CO2 desorption, presumably from residual organic substances, was observed at 200–800 °C. Thus, the N2 desorption in a high-temperature region can be ascribed to the strongly bound N species in the oxides. It is noteworthy that both LTO_TEA and LTO_N exhibited peaks at similar binding energies in the N 1s XPS spectra (see Fig. S4†); therefore, discussing the N environment based on XPS alone could not provide accurate inferences. The combination of TPD and XPS facilitates a more reliable chemical structure analysis.
As mentioned previously, there are two main doped forms of N species in perovskite-type oxides: substitutional (Ti–N bonds) and interstitial (Ti–O–N bonds). If TPD can distinguish between these N species, it could be a useful analytical tool. Thus, we attempted to deconvolute the N2 emission pattern (Fig. 1e) using the depth profiles of the N 1s and C 1s XPS spectra obtained by Ar+ sputtering (Fig. 2 and S9†). LTO_TEA exhibited a peak at almost the same position even after Ar+ sputtering in both the N 1s and C 1s XPS profiles. This indicates that similar species (N-containing organic substances) are present from the surface to the interlayer, considering that LTO is a layered perovskite-type oxide. On the other hand, in the case of LTO_N, the dominant N species at the surface and the interior were different. The concentration of the Ti–O–N species decreased significantly toward the interior of LTO_N compared to the surface, while the Ti–N species, initially present in low concentration at the surface, became the predominant species in the interior. The amount of carbon inside LTO_N was negligible (Fig. S9b†), supporting N bonding within the oxide matrices, unlike LTO_TEA. Further analysis focused on the fact that the N2 desorption pattern of LTO_N can deconvolve into two peaks, one at around 900 °C and the other at around 1200 °C (Fig. 1e). To determine the TPD peak assignments of N species (Ti–O–N and Ti–N), a heat-treated LTO_N sample at 1100 °C was prepared (denoted as LTO_N_HT). At this temperature, the component of Fit Peak 1 was almost desorbed (Fig. 1e). In other words, LTO_N_HT may contain N species corresponding to Fit Peak 2. As shown in the N 1s XPS spectra (Fig. 2c), LTO_N_HT retains the Ti–O–N species at the surface, albeit in a reduced quantity of 0.14 wt% compared to the LTO_N sample prior to heat treatment (Fig. 2b), with their presence nearly negligible in the interior. In contrast, the amounts of the Ti–N species retained by LTO_N_HT at the surface and in the bulk were 0.12 wt% and 0.28 wt%, respectively, indicating no significant change after the heat treatment. These results suggest that the Ti–O–N species decomposed during the heat treatments at temperatures up to 1100 °C, while stronger Ti–N bonds were thermally stable, even at 1100 °C. To verify the thermal stability of the Ti–N species, we performed TPD measurements on titanium nitride powder (Wako Pure Chemicals, TiN), as shown in Fig. S10.† Interestingly, N2 desorption from the TiN matrices can be observed only above 1050 °C, which is in good agreement with the above discussion. The PXRD patterns and SEM images of the LTO_TEA and LTO_N samples after heat treatment (Fig. S3†) show no significant structural or morphological changes up to 1100 °C. On the other hand, after heat treatment at 1600 °C, the crystallinity of La2Ti2O7 increased and the particle size became larger. Thus, interstitial N species desorb below 1100 °C without structural or morphological change, while substitutional N species desorb above 1100 °C with a crystallinity increase of La2Ti2O7. The high thermal stability of La2Ti2O7 up to 1100 °C allows for the sequential desorption of interstitial and substitutional N species, rather than simultaneous desorption and thus enables qualitative analysis by the TPD method.
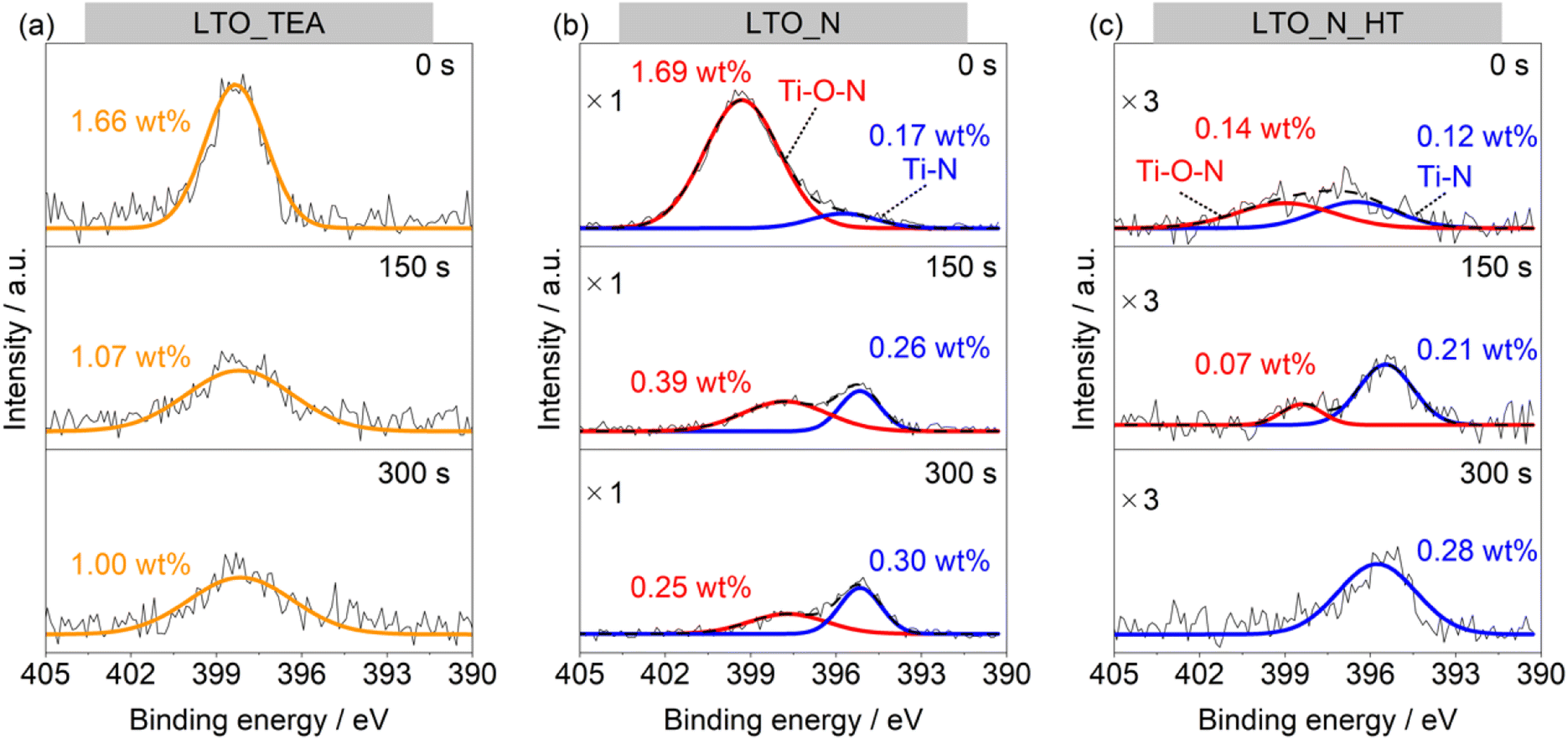 | ||
| Fig. 2 N 1s XPS spectra of (a) LTO_TEA, (b) LTO_N, and (c) LTO_N_HT before and after Ar+ sputtering for 150 s or 300 s. | ||
To further study the N environment at the surface and in the bulk, synchrotron-based XAS analysis was conducted in the total electron yield (TEY) and partial fluorescence yield (PFY) modes. TEY is a surface-sensitive analysis mode, in contrast to PFY, which reveals the internal structure. Thus, XAS can distinguish between the external and internal chemical structures of samples without the Ar+ sputtering process, unlike XPS. Fig. 3a and b show the N K-edge X-ray absorption near-edge (XANES) spectra in the TEY and PFY modes, respectively. LTO_TEA exhibited a peak at 405 eV in both the TEY and PFY modes, which can be attributed to the N–O bond.61,62 The presence of this peak at 405 eV explains the large amount of NO desorption observed in TPD (Fig. 1d) and is consistent with the XPS depth profiles, indicating the presence of a N–O bond. The N–O bond peak at 405 eV was also observed in the TEY XANES spectrum of LTO_N, indicating the presence of Ti–O–N species on the surface (see also Fig. 2b). LTO_N exhibited not only a sharp peak at 405 eV but also broad split peaks around 400 eV, which can be attributed to Ti–N bonds (Fig. 3b).63 The XAS analysis indicated that the spatial distribution of N species in LTO_N is through surface segregation of interstitial N (Ti–O–N) and bulk-substituted N (Ti–N), as described above. More importantly, LTO_N_HT shows a significant decrease in the peak intensity at 405 eV (Ti–O–N bond) in both the TEY and PFY modes. In contrast, the peak corresponding to the substituted N (Ti–N bond) at 400 eV is well maintained, as shown in Fig. 3b, even after heat treatment. This XAS result validates the peak separation and assignment of the N2 profile in TPD (Fig. 1e). Specifically, the low- and high-temperature regions of the N2 desorption peak below and above 1100 °C are attributed to interstitial and substitutional N species, respectively. In addition, the XANES shapes of all the LTO samples differed from that of the reference TiN. This indicates that TiN crystals were not generated inside the LTO samples, and Ti–N bonds were formed by the local substitution of N for O. In summary, the combined study of XPS and XAS elucidated the assignment of N2 desorption in the TPD profiles. This underscores the efficacy of TPD in distinguishing between interstitial and substitutional N species within bulk perovskite-type oxides. According to the peak area ratios derived from TPD results, interstitial N and substitutional N in LTO_N were quantified to be 0.37 wt% and 0.30 wt%, respectively.
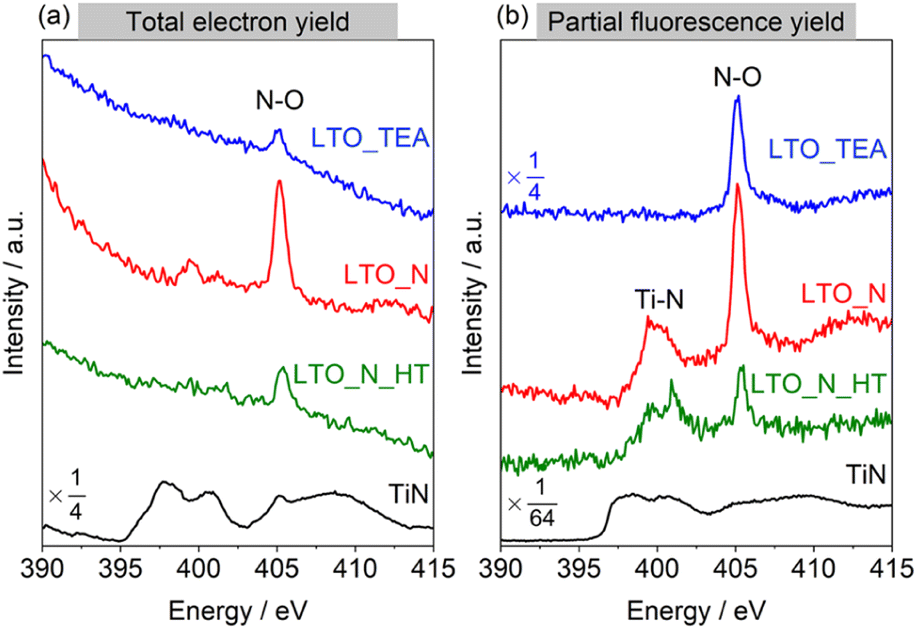 | ||
| Fig. 3 N K-edge XANES spectra of LTO_TEA, LTO_N, LTO_N_HT, and reference TiN recorded in the (a) TEY and (b) PFY modes. | ||
Thermal desorption mechanism of TPD
Finally, the thermal desorption behaviour of the N species from LTO_TEA and LTO_N was analysed by in situ DRIFTS, as shown in Fig. 4a–d, respectively. LTO_TEA displayed absorption bands at 3800–2600 cm−1,64 corresponding to OH bonds, and around 2922 and 2853 cm−1, corresponding to CH bonds (Fig. 4a).65,66 In contrast, LTO_N exhibited significantly smaller bands of OH and CH bonds, which is consistent with the observations concluding that organic-derived N species are present in LTO_TEA but not in LTO_N. These bands decrease with increasing temperature and become negligible around 600 °C, which is nearly consistent with the desorption temperatures of CO2, H2O, NO, and N2 (see Fig. 1d and S6a†). Interestingly, an absorption band around 2175 cm−1, corresponding to the vibration of CO adsorbed on the surface,67,68 emerged during the heat treatment. Moreover, the peak intensity decreased with increasing temperature after reaching a maximum intensity at approximately 600 °C. The absorption behaviour could be related to CO desorption because the second CO desorption peak starts around 600 °C in Fig. S6a.† Similar IR absorption behaviour and CO desorption in the TPD were also observed for LTO_N (Fig. 4d and S6b†). A small absorption band at 2000 cm−1, corresponding to the NO bond,69–71 was observed for LTO_TEA (Fig. 4b and S11†). During the heat treatment, the NO vibration band initially increased up to 300 °C and then decreased with increasing temperature. This absorption behaviour agrees with the TPD profile of LTO_TEA (see Fig. 1d), because the desorption of NO and N2 gases began at approximately 300 °C. In the case of LTO_N (Fig. 4c and d), distinct split bands at approximately 3300 cm−1, which are considered to be NH or OH bonds,65,72,73 and strong absorption at 2000 cm−1 corresponding to the NO bond were observed. The NH or OH vibration appeared at 400 °C, with intensity increasing with increasing temperature up to 800 °C. The absorption intensity of the NO vibration was also enhanced by the heat treatment. These increases in absorption appear to be related to the desorption behaviour of N2 in LTO_N (Fig. 1e). First, the presence of NO bonds in Fig. 4d is consistent with the N2 desorption in the lower temperature range starting at approximately 600 °C (Fit Peak 1 in Fig. 1e), which is attributed to the Ti–O–N species. Because DRIFTS is a surface-sensitive method,74 the growth of the NO vibrations may be related to N migration to the top surface of the sample. The N species bound to the oxide in LTO_N were not desorbed as NO, unlike those in LTO_TEA. Thus, the Ti–O–N species can probably diffuse to the surface at temperatures below 600 °C but cannot desorb (corresponding to increased IR absorption intensity), and only above 600 °C do they desorb as N2 gas, as detected by TPD.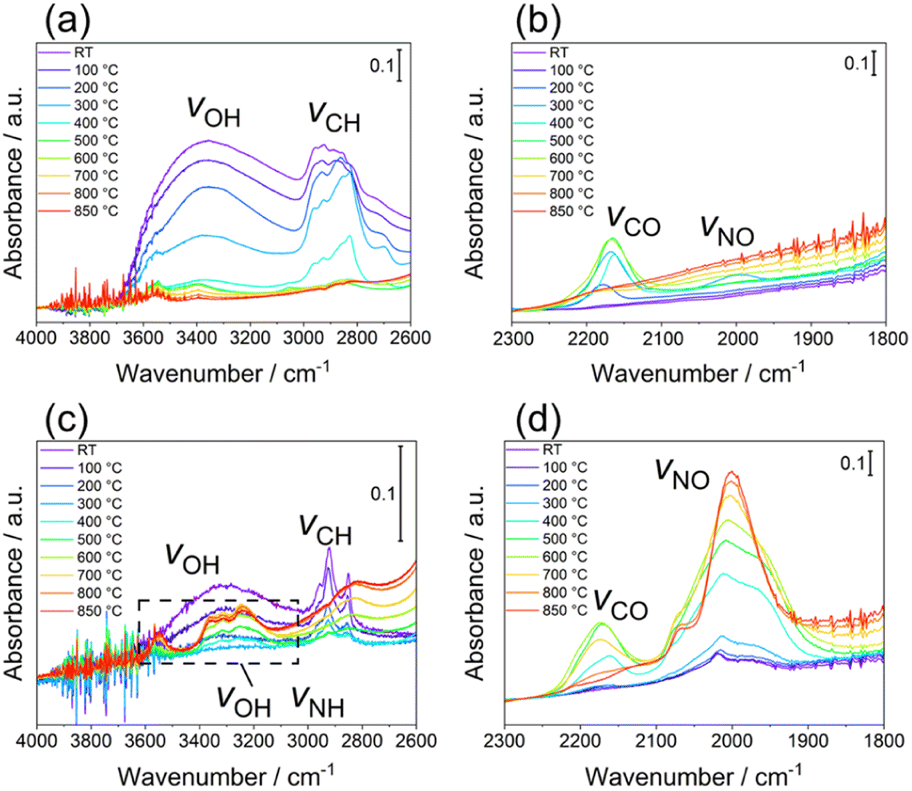 | ||
| Fig. 4 In situ DRIFTS spectra of LTO_TEA in (a) 4000–2600 cm−1 and (b) 2300–1800 cm−1 regions and LTO_N in (c) 4000–2600 cm−1 and (d) 2300–1800 cm−1 regions. | ||
Conclusions
We propose a TPD technique to identify doped N species in perovskite-type oxides. The N 1s XPS spectra of the two types of perovskite-type oxides (LTO_TEA and LTO_N) were similar, although they exhibited different physical and chemical properties. In contrast, TPD successfully distinguished the N species in these oxides based on the desorbed gas species and their desorption temperatures, providing accurate quantification in the bulk. The combined study of TPD results with XPS and XANES analyses revealed that it is possible to distinguish between interstitial and substituted N species using the desorption temperature. The distinction is important for perovskite-type oxide applications. Furthermore, in situ DRIFTS analysis elucidated the desorption mechanism of the N species. This study is the first to demonstrate the usefulness of the TPD technique for analysing doped N species in oxides. TPD, a bulk analysis, complements the surface-sensitive XPS technique; the combination of TPD and XPS will enable discriminative analysis of surface and bulk structures, providing a powerful method for the further development of functional doped oxide materials. Moreover, TPD technology is not limited to the analysis of nitrogen species in perovskite-type oxides, but is expected to be expanded to other dopants such as hydrogen, sulphur, and phosphorus in various inorganic materials, including oxides, carbides, nitrides, and sulphides. Since the high-temperature vacuum TPD system is a lab-scale setup, it will hopefully be commercially available and widely used in the future.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Conceptualization: TY, SY, and HN; methodology: SS, TY, GN, EK, and HN; investigation: SS, GN, and JW; supervision: TY, SY, and HN; writing—original draft: SS; writing—review and editing: TY and HN.Conflicts of interest
The authors declare that they have no competing financial interests or personal relationships that may have influenced the work reported in this study.Acknowledgements
This work was supported by JST PRESTO (Grant No. JPMJPR23QA); JSPS Grant-in-Aid for Scientific Research on Innovative Areas “Mixed anion” (Grant No. 16H06439); JSPS Grant-in-Aid for Scientific Research (A) (Grant No. 20H00297); the “Five-star Alliance” in “NJRC Mater. & Dev.”; JST SICORP (Grant No. JPMJSC2112). XAS experiments using synchrotron radiation were performed at beamline BL12 of the SAGA Light Source, Japan (Proposal No. 137-2301111P). The authors thank S. Ogawa for her kind support with the XPS measurements. The authors also thank S. Hirabuki for her kind support with the CHN elemental analysis. The authors also thank Editage (https://www.editage.jp) for English language editing.References
- J. X. Flores-Lasluisa, F. Huerta, D. Cazorla-Amorós and E. Morallón, J. Colloid Interface Sci., 2019, 556, 658–666 CrossRef CAS.
- Y. Shiroma, H. Mogi, T. Mashiko, S. Yasuda, S. Nishioka, T. Yokoi, S. Ida, K. Kimoto and K. Maeda, J. Mater. Chem. A, 2023, 11, 9485–9492 RSC.
- N. Hou, T. Yao, P. Li, X. Yao, T. Gan, L. Fan, J. Wang, X. Zhi, Y. Zhao and Y. Li, ACS Appl. Mater. Interfaces, 2019, 11, 6995–7005 CrossRef CAS.
- X. Huang, G. Zhao, G. Wang and J. T. S. Irvine, Chem. Sci., 2018, 9, 3623–3637 RSC.
- Y. Lin, Y. Fang, J. Zhao, Y. Shao, S. J. Stuard, M. M. Nahid, H. Ade, Q. Wang, J. E. Shield, N. Zhou, A. M. Moran and J. Huang, Nat. Commun., 2019, 10, 1008 CrossRef PubMed.
- F. Meng, Z. Hong, J. Arndt, M. Li, M. Zhi, F. Yang and N. Wu, Nano Res., 2012, 5, 213–221 CrossRef CAS.
- M. Raciulete, F. Papa, C. Negrila, V. Bratan, C. Munteanu, J. Pandele-Cusu, D. C. Culita, I. Atkinson and I. Balint, Catalysts, 2020, 10, 637 CrossRef CAS.
- H. Fujito, H. Kunioku, D. Kato, H. Suzuki, M. Higashi, H. Kageyama and R. Abe, J. Am. Chem. Soc., 2016, 138, 2082–2085 CrossRef CAS.
- Z. Hua, X. Zhang, X. Bai, L. Lv, Z. Ye and X. Huang, J. Colloid Interface Sci., 2015, 450, 45–53 CrossRef CAS.
- Y. Ao, K. Wang, P. Wang, C. Wang and J. Hou, Appl. Catal., B, 2016, 194, 157–168 CrossRef CAS.
- L. Lv, L. Lei, Q.-W. Chen, C.-L. Yin, H. Fan and J.-P. Zhou, Appl. Catal., B, 2024, 343, 123464 CrossRef CAS.
- J. Zhang, W. Dang, Z. Ao, S. K. Cushing and N. Wu, Phys. Chem. Chem. Phys., 2015, 17, 8994–9000 RSC.
- D. W. Hwang, H. G. Kim, J. S. Lee, J. Kim, W. Li and S. H. Oh, J. Phys. Chem. B, 2005, 109, 2093–2102 CrossRef CAS.
- J. Lian, J. He, X. Zhang and F. Liu, Solid State Sci., 2016, 61, 9–15 CrossRef CAS.
- M. Xia, X. Yan, H. Li, N. Wells and G. Yang, Nano Energy, 2020, 78, 105401 CrossRef CAS.
- J. Wang, Y. Asakura, T. Hasegawa and S. Yin, Chem. Eng. J., 2021, 423, 130220 CAS.
- H. Pan, Y.-W. Zhang, V. B. Shenoy and H. Gao, J. Phys. Chem. C, 2011, 115, 12224–12231 CrossRef CAS.
- J. Ananpattarachai, P. Kajitvichyanukul and S. Seraphin, J. Hazard. Mater., 2009, 168, 253–261 CrossRef CAS.
- A. Panepinto, D. Cornil, P. Guttmann, C. Bittencourt, J. Cornil and R. Snyders, J. Phys. Chem. C, 2020, 124, 17401–17412 CrossRef CAS.
- Z. Ma, K. Wu, R. Sa, Q. Li, C. He and Z. Yi, Int. J. Hydrogen Energy, 2015, 40, 980–989 CrossRef CAS.
- Y. Wang, M. Kim, A. S. Chabungbam, D. Kim, Q. Shao, I. Kymissis and H.-H. Park, Scr. Mater., 2023, 222, 115050 CrossRef CAS.
- Y. Yokosuka, K. Oki, H. Nishikiori, Y. Tatsumi, N. Tanaka and T. Fujii, Res. Chem. Intermed., 2009, 35, 43–53 CAS.
- G. Liu, L.-C. Yin, J. Wang, P. Niu, C. Zhen, Y. Xie and H.-M. Cheng, Energy Environ. Sci., 2012, 5, 9603–9610 RSC.
- W. Zhao, S. Liu, S. Zhang, R. Wang and K. Wang, Catal. Today, 2019, 337, 37–43 CrossRef CAS.
- S. Khan, T. L. Ruwer, N. Khan, A. Köche, R. W. Lodge, H. Coelho-Júnior, R. L. Sommer, M. J. L. Santos, C. F. Malfatti, C. P. Bergmann and J. Alves Fernandes, J. Mater. Chem. A, 2021, 9, 12214–12224 CAS.
- T. Schroeder, G. Lupina, R. Sohal, G. Lippert, C. Wenger, O. Seifarth, M. Tallarida and D. Schmeisser, J. Appl. Phys., 2007, 102, 014103 CrossRef.
- M. Kot, J. Łobaza, F. Naumann, H. Gargouri, K. Henkel and D. Schmeißer, J. Vac. Sci. Technol., 2017, 36, 01A114 CrossRef.
- X. Lu, Q. Wang and D. Cui, J. Mater. Sci. Technol., 2010, 26, 925–930 CrossRef CAS.
- T. C. Jagadale, S. P. Takale, R. S. Sonawane, H. M. Joshi, S. I. Patil, B. B. Kale and S. B. Ogale, J. Phys. Chem. C, 2008, 112, 14595–14602 CrossRef CAS.
- J. P. Boudou, Carbon, 2003, 41, 1955–1963 CrossRef CAS.
- K. Friedel Ortega, R. Arrigo, B. Frank, R. Schlögl and A. Trunschke, Chem. Mater., 2016, 28, 6826–6839 CrossRef CAS.
- E. A. Uslamin, B. Luna-Murillo, N. Kosinov, P. C. A. Bruijnincx, E. A. Pidko, B. M. Weckhuysen and E. J. M. Hensen, Chem. Eng. Sci., 2019, 198, 305–316 CrossRef CAS.
- B. Zsirka, V. Vágvölgyi, K. Győrfi, E. Horváth, R. K. Szilágyi, E. Szabó-Bárdos, S. Balogh and J. Kristóf, Appl. Clay Sci., 2021, 212, 106222 CrossRef CAS.
- J. L. Figueiredo, M. F. R. Pereira, M. M. A. Freitas and J. J. M. Órfão, Carbon, 1999, 37, 1379–1389 CrossRef CAS.
- P. Brender, R. Gadiou, J.-C. Rietsch, P. Fioux, J. Dentzer, A. Ponche and C. Vix-Guterl, Anal. Chem., 2012, 84, 2147–2153 CrossRef CAS.
- T. Ishii, S. Kashihara, Y. Hoshikawa, J. Ozaki, N. Kannari, K. Takai, T. Enoki and T. Kyotani, Carbon, 2014, 80, 135–145 CrossRef CAS.
- T. Ishii, Y. Kaburagi, A. Yoshida, Y. Hishiyama, H. Oka, N. Setoyama, J. Ozaki and T. Kyotani, Carbon, 2017, 125, 146–155 CrossRef CAS.
- R. Tang, K. Taguchi, H. Nishihara, T. Ishii, E. Morallón, D. Cazorla-Amorós, T. Asada, N. Kobayashi, Y. Muramatsu and T. Kyotani, J. Mater. Chem. A, 2019, 7, 7480–7488 RSC.
- T. Ishii and J. Ozaki, Carbon, 2020, 161, 343–349 CrossRef CAS.
- G. B. Choi, S. Hong, J.-H. Wee, D.-W. Kim, T. H. Seo, K. Nomura, H. Nishihara and Y. A. Kim, Nano Lett., 2020, 20, 5885–5892 CrossRef CAS.
- T. Yoshii, G. Nishikawa, V. K. Prasad, S. Shimizu, R. Kawaguchi, R. Tang, K. Chida, N. Sato, R. Sakamoto, K. Takatani, D. Moreno-Rodríguez, P. Škorňa, E. Scholtzová, R. K. Szilagyi and H. Nishihara, Chem, 2024 DOI:10.1016/j.chempr.2024.03.029 , in press.
- W. Yu, Z. Shen, T. Yoshii, S. Iwamura, M. Ono, S. Matsuda, M. Aoki, T. Kondo, S. R. Mukai, S. Nakanishi and H. Nishihara, Adv. Energy Mater., 2024, 14, 2303055 CAS.
- T. Xia, T. Yoshii, K. Nomura, K. Wakabayashi, Z.-Z. Pan, T. Ishii, H. Tanaka, T. Mashio, J. Miyawaki, T. Otomo, K. Ikeda, Y. Sato, M. Terauchi, T. Kyotani and H. Nishihara, Chem. Sci., 2023, 14, 8448–8457 RSC.
- K. Wakabayashi, T. Yoshii and H. Nishihara, Carbon, 2023, 210, 118069 CrossRef CAS.
- K. Pirabul, Z.-Z. Pan, R. Tang, S. Sunahiro, H. Liu, K. Kanamaru, T. Yoshii and H. Nishihara, Bull. Chem. Soc. Jpn., 2023, 96, 510–518 CrossRef CAS.
- W. Yu, T. Yoshii, A. Aziz, R. Tang, Z.-Z. Pan, K. Inoue, M. Kotani, H. Tanaka, E. Scholtzová, D. Tunega, Y. Nishina, K. Nishioka, S. Nakanishi, Y. Zhou, O. Terasaki and H. Nishihara, Adv. Sci., 2023, 10, 2300268 CrossRef CAS PubMed.
- H. Nishihara, T. Simura, S. Kobayashi, K. Nomura, R. Berenguer, M. Ito, M. Uchimura, H. Iden, K. Arihara, A. Ohma, Y. Hayasaka and T. Kyotani, Adv. Funct. Mater., 2016, 26, 6418–6427 CrossRef CAS.
- S. Sunahiro, K. Nomura, S. Goto, K. Kanamaru, R. Tang, M. Yamamoto, T. Yoshii, J. N. Kondo, Q. Zhao, A. G. Nabi, R. Crespo-Otero, D. D. Tommaso, T. Kyotani and H. Nishihara, J. Mater. Chem. A, 2021, 9, 14296–14308 RSC.
- Y. Matsui, T. Yamada, S. Suzuki, T. Yoshii, H. Nishihara and K. Teshima, ACS Appl. Energy Mater., 2021, 4, 2690–2695 CrossRef CAS.
- K. Chida, T. Yoshii, M. Ohwada, Y. Hayasaka, J. Komeda, R. Sakamoto, J. Maruyama, K. Kamiya, M. Inoue, F. Tani and H. Nishihara, Catal. Today, 2023, 411–412, 113830 CrossRef CAS.
- K. Chida, T. Yoshii, N. Hiyoshi, T. Itoh, J. Maruyama, K. Kamiya, M. Inoue, F. Tani and H. Nishihara, Carbon, 2023, 201, 338–346 CrossRef CAS.
- S. C. Chen, K. Y. Sung, W. Y. Tzeng, K. H. Wu, J. Y. Juang, T. M. Uen, C. W. Luo, J.-Y. Lin, T. Kobayashi and H. C. Kuo, J. Phys. Appl. Phys., 2013, 46, 075002 CrossRef CAS.
- F. Zou, Z. Jiang, X. Qin, Y. Zhao, L. Jiang, J. Zhi, T. Xiao and P. P. Edwards, Chem. Commun., 2012, 48, 8514–8516 RSC.
- J. J. Biernacki and G. P. Wotzak, J. Am. Ceram. Soc., 1989, 72, 122–129 CAS.
- M. A. Hunter, J. Am. Chem. Soc., 1910, 32, 330–336 CrossRef CAS.
- C. Miyagawa, T. Kobayashi, T. Taishi and K. Hoshikawa, J. Cryst. Growth, 2013, 372, 95–99 CrossRef CAS.
- W. Sheng, J.-L. Shi, H. Hao, X. Li and X. Lang, Chem. Eng. J., 2020, 379, 122399 CAS.
- J. Duchoslav, M. Kehrer, A. Hinterreiter, V. Duchoslav, C. Unterweger, C. Fürst, R. Steinberger and D. Stifter, Appl. Surf. Sci., 2018, 443, 244–254 CAS.
- S. Golczak, A. Kanciurzewska, M. Fahlman, K. Langer and J. J. Langer, Solid State Ionics, 2008, 179, 2234–2239 CrossRef CAS.
- H. Wang, L. Zheng, G. Liu and Y. Zhou, Colloids Surf., A, 2018, 537, 28–35 CrossRef CAS.
- R. Gago, A. Redondo-Cubero, M. Vinnichenko, J. Lehmann, F. Munnik and F. J. Palomares, Mater. Chem. Phys., 2012, 136, 729–736 CrossRef CAS.
- P. Leinweber, J. Kruse, F. L. Walley, A. Gillespie, K.-U. Eckhardt, R. I. R. Blyth and T. Regier, J. Synchrotron Radiat., 2007, 14, 500–511 CrossRef CAS.
- D. K. Lee, J. I. Choi, G. H. Lee, Y.-H. Kim and J. K. Kang, Adv. Energy Mater., 2016, 6, 1600583 CrossRef.
- X. Cheng, X. Yu, Z. Xing and L. Yang, Arab. J. Chem., 2016, 9, S1706–S1711 CrossRef CAS.
- R. Hayami, N. Endo, T. Abe, Y. Miyase, T. Sagawa, K. Yamamoto, S. Tsukada and T. Gunji, J. Sol-Gel Sci. Technol., 2018, 87, 743–748 CrossRef CAS.
- B. Song, C. Sizemore, L. Li, X. Huang, Z. Lin, K. Moon and C.-P. Wong, J. Mater. Chem. A, 2015, 3, 21789–21796 RSC.
- B. chu, X. Ou, L. Wei, H. Liu, K. chen, Q. Qin, L. Meng, M. Fan, B. Li and L. Dong, Mol. Catal., 2021, 511, 111755 CrossRef CAS.
- S. Royer and D. Duprez, ChemCatChem, 2011, 3, 24–65 CrossRef CAS.
- R. V. Mikhaylov, A. A. Lisachenko, B. N. Shelimov, V. B. Kazansky, G. Martra, G. Alberto and S. Coluccia, J. Phys. Chem. C, 2009, 113, 20381–20387 CrossRef CAS.
- R. V. Mikhaylov, A. A. Lisachenko, B. N. Shelimov, V. B. Kazansky, G. Martra and S. Coluccia, J. Phys. Chem. C, 2013, 117, 10345–10352 CrossRef CAS.
- J. Wu and Y. Cheng, J. Catal., 2006, 237, 393–404 CrossRef CAS.
- R. C. Dante, P. Martín-Ramos, A. Correa-Guimaraes and J. Martín-Gil, Mater. Chem. Phys., 2011, 130, 1094–1102 CrossRef CAS.
- T. Yoshii, K. Tamaki, Y. Kuwahara, K. Mori and H. Yamashita, J. CO2 Util., 2021, 52, 101691 CrossRef CAS.
- A. Urakawa, N. Maeda and A. Baiker, Angew. Chem., Int. Ed., 2008, 47, 9256–9259 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: PXRD patterns, SEM images, N 1s and C 1s XPS spectra, and TPD results of LTO_TEA and LTO_N, MS and in situ DRIFTS spectra of LTO_TEA, TPD result of TiN, the calibration curves for TPD, and a schematic illustration of the advanced TPD. See DOI: https://doi.org/10.1039/d4sc01850h |
| This journal is © The Royal Society of Chemistry 2024 |