
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTricyclononenes and tricyclononadienes as efficient monomers for controlled ROMP: understanding structure–propagation rate relationships and enabling facile post-polymerization modification†
Landon J.
Kilgallon
 a,
Timothy P.
McFadden
b,
Matthew S.
Sigman
b and
Jeremiah A.
Johnson
*acd
a,
Timothy P.
McFadden
b,
Matthew S.
Sigman
b and
Jeremiah A.
Johnson
*acd
aDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
bDepartment of Chemistry, University of Utah, Salt Lake City, Utah 84112, USA
cKoch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, 500 Main Street, Cambridge, MA 02139, USA. E-mail: jaj2109@mit.edu
dBroad Institute of MIT and Harvard, Cambridge, MA 02142, USA
First published on 22nd May 2024
Abstract
Grubbs 3rd-generation (G3) pre-catalyst-initiated ring-opening metathesis polymerization (ROMP) remains an indispensable tool in the polymer chemist's toolbox. Tricyclononenes (TCN) and tricyclononadienes (TCND) represent under-explored classes of monomers for ROMP that have the potential to both advance fundamental knowledge (e.g., structure-polymerization kinetics relationships) and serve as practical tools for the polymer chemist (e.g., post-polymerization functionalization). In this work, a library of TCN and TCND imides, monoesters, and diesters, along with their exo-norbornene counterparts, were synthesized to compare their behaviors in G3-initiated ROMP. Real-time 1H NMR was used to study their polymerization kinetics; propagation rates (kp) were extracted for each monomer. To understand the relationships between monomer structure and ROMP propagation rates, density functional theory methods were used to calculate a variety of electronic and steric parameters for each monomer. While electronic parameters (e.g., HOMO energy levels) correlated positively with the measured kp values, steric parameters generally gave improved correlations, which indicates that monomer size and shape are better predictors for kp than electronic parameters for this data set. Furthermore, the TCND diester—which contains an electron-deficient cyclobutene that is resistant to ROMP—and its polymer p(TCND) are shown to be highly reactive toward DBU-catalyzed conjugate addition reactions with thiols, providing a protecting- and activating-group free strategy for post-polymerization modification.
Introduction
Ruthenium-initiated ring-opening metathesis polymerization (ROMP) is an invaluable tool in the polymer chemist's repertoire due to the commercial availability of fast-initiating ruthenium initiators and various strained olefin monomers, its wide functional group tolerance, and the ease in which complex polymers may be synthesized in a rapid and controlled manner. Grubbs 3rd-generation pyridine-ligated pre-catalysts (G3) are most frequently used for living ROMP due to their fast initiation rates.1 Moreover, exo-norbornenes (NB) are often used as monomers due to their fast propagation rates, especially in comparison to their endo counterparts.2,3Exo-NBs are, however, often more difficult to synthesize and more expensive than their endo isomers. Recent detailed studies of the mechanism4 and the effects of the exo-norbornene monomer structure on the rate of propagation and degree of “livingness” of these polymerizations5–8 have led to new insights that may guide the design of next-generation initiator and monomer classes. For example, Matson and coworkers5–8 have suggested that the HOMO energy levels of exo-NBs correlate with their G3-initiated ROMP propagation rates. Inspired by this work, and seeking synthetically accessible, functional monomers that do not compromise polymerization kinetics and controlled polymerization behavior, we identified tricyclononenes (TCN) and tricyclononadienes (TCND) as monomers ripe for further study (Fig. 1).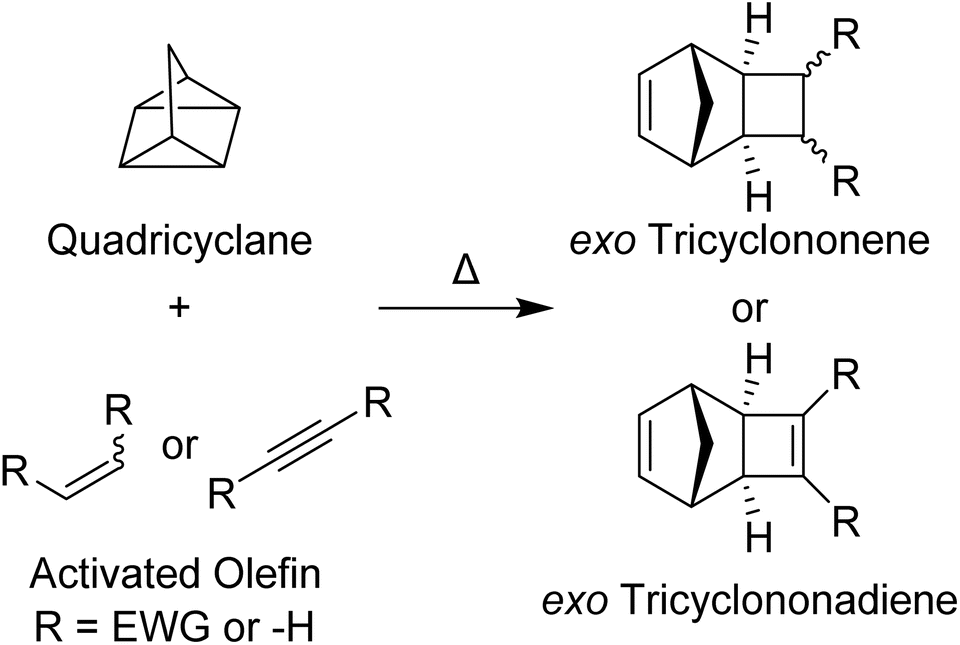 | ||
| Fig. 1 General syntheses and structures of exo-tricyclononene and exo-tricyclononadiene analogs. | ||
TCNs and TCNDs are tricyclic, strained olefins with fused cyclobutane and cyclobutene rings, respectively, connected to the two exo positions of the NB skeleton. Compared to other exo-substituted NB monomers, which often require high temperatures9–11 and/or precious metal catalysts8,12 for their synthesis, exo-TCNs and TCNDs can be conveniently assembled by thermal cycloadditions of activated olefins and quadricyclane with perfect exo selectivity.13,14 While TCN and TCND-based polymers have been synthesized using Mo (Schrock-type initiators)15 and Ru (Grubbs 1st and 2nd generation initiators) initiated ROMP16,17 and studied for applications in gas separations,18–25 no studies of their performance under “living” ROMP conditions initiated by G3 or post-polymerization functionalization have been reported. We hypothesized that they may be competitive with commonly used exo-NBs in terms of ROMP propagation rates, but that they could offer advantages of facile synthesis and, in some cases, post-polymerization modification. Moreover, by accessing a training set of monomers, we sought to investigate if the HOMO level was primarily responsible for propagation rate as proposed by Matson and coworkers, or whether other molecular features (e.g., steric parameters) of the monomers may also impact performance.
Here, we synthesize a library of TCNs and TCNDs that incorporates a broad functional group scope, including imides, diesters, and monoesters. We assess their structures and properties, investigate their ROMP behavior using G3, and compare the results to analogous exo-NB derivatives. We identify trends in the relative propagation rates across these monomers, showing that while their HOMO energies are good predictors of reactivity, readily computed steric parameters (e.g., volume, solvent accessible surface area (SASA), and Sterimol B5) offer improved structure–reactivity correlations across all exo-NB, TCN, and TCND monomer classes. Moreover, we demonstrate that the cyclobutene pendants of p(TCND)s can be excellent substrates for conjugate addition reactions with thiol nucleophiles (i.e., “thiol-Michael-type additions”),26–28 offering a straightforward methodology for efficient post-polymerization modification without the need for protecting groups or monomer activation (e.g., activated esters). These studies demonstrate that TCNs and TCNDs are attractive next-generation monomers for ROMP as (a) they are easy to synthesize in high stereochemical purity in as few as two simple steps from commercial starting materials; (b) they display propagation kinetics similar to or faster than many typical exo-NBs; and (c) they offer facile routes to post-polymerization modification that are not possible for common exo-NBs. Finally, this study presents the first application of computed steric parameters to correlate monomer structure with propagation rates in ROMP, providing insights into the predictive design of future ROMP monomers.
Results/discussion
TCN and TCND monomer synthesis
Quadricyclane was prepared by photochemical isomerization of norbornadiene in batches of up to 17 g and yields up to >90% (ESI† Section 2). Heating quadricyclane in the presence of maleic anhydride gave TCN exo-anhydride 1 as a mixture of anti and syn diastereomers (67![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :33 anti:syn) in quantitative yield (Fig. 2a).29,30 Crystallization led to enrichment of the anti-1 to 78:22 anti:syn (69% yield); further isolation of pure diastereomers was conducted following a subsequent synthetic step. TCN diester TCN-(CO2Me)2 was prepared by acid-catalyzed esterification of 1 with methanol.29 The TCN imides TCN-NBnanti, TCN-NCy, and TCN-NHex were prepared by condensation of 1 with benzylamine, cyclohexylamine, and n-hexylamine, respectively, in an analogous fashion to Warrener's procedure for preparing the N-methyl TCN imide from 1 and methylamine.30 All of these TCNs were crystalline solids wherein crystallization afforded the anti-isomer in a >95:5 anti:syn ratio in each case.
:33 anti:syn) in quantitative yield (Fig. 2a).29,30 Crystallization led to enrichment of the anti-1 to 78:22 anti:syn (69% yield); further isolation of pure diastereomers was conducted following a subsequent synthetic step. TCN diester TCN-(CO2Me)2 was prepared by acid-catalyzed esterification of 1 with methanol.29 The TCN imides TCN-NBnanti, TCN-NCy, and TCN-NHex were prepared by condensation of 1 with benzylamine, cyclohexylamine, and n-hexylamine, respectively, in an analogous fashion to Warrener's procedure for preparing the N-methyl TCN imide from 1 and methylamine.30 All of these TCNs were crystalline solids wherein crystallization afforded the anti-isomer in a >95:5 anti:syn ratio in each case.
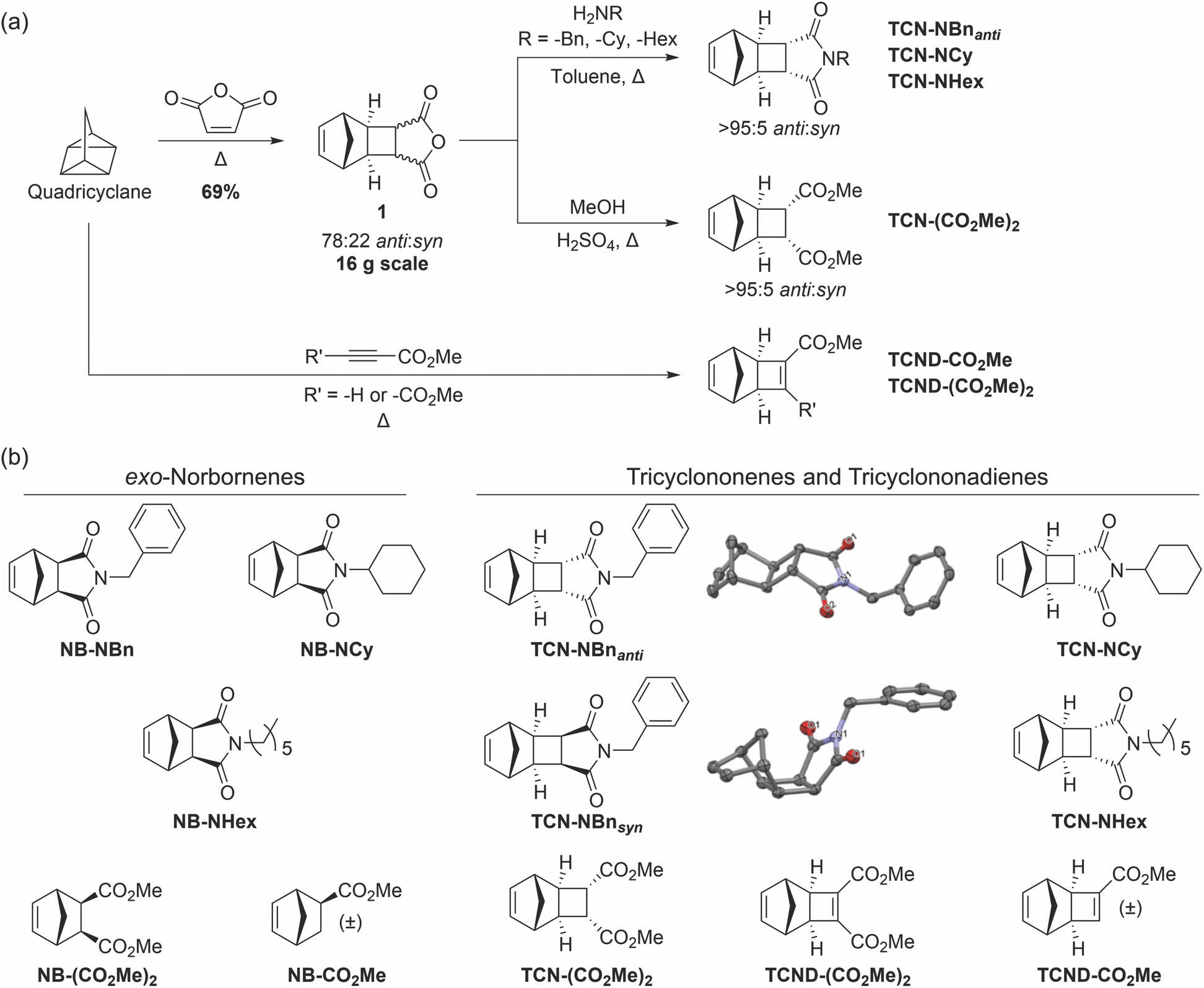 | ||
| Fig. 2 (a) Synthetic pathways toward TCN and TCND monomers. (b) Library of exo-NBs, TCNs and TCNDs synthesized in this study. The crystal structures of TCN-NBnanti and TCN-NBnsyn are shown with ellipsoids drawn at the 50% probability level. Hydrogen atoms are omitted for clarity; black = C; red = O; blue = N. | ||
We also sought TCN-NBnsyn for comparison to its anti-diastereomer (Fig. 2b). While TCN-NBnsyn and TCN-NBnanti may potentially be separable by chromatography,30 we identified a kinetic resolution method that leveraged the large difference in imidation rates between the syn (slow) and anti (fast) amic acids formed upon initial ring-opening opening of 1, ultimately giving TCN-NBnsyn in 4% yield over 2 steps with >98:2 d.r. (ESI† Section 2). The relative configurations of TCN-NBnsyn and TCN-NBnanti were unambiguously determined by single-crystal X-ray diffraction (Fig. 2b). TCND derivatives TCND-(CO2Me)2 and TCND-CO2Me were synthesized by direct cycloaddition of quadricyclane with dimethylacetylenedicarboxylate and methyl propiolate, respectively (Fig. 2a).13,31
We note that upon storage under ambient atmosphere for extended periods, TCND-(CO2Me)2 yellows and a new product is observed via1H NMR spectroscopy. A small sample of this impurity was isolated chromatographically; its 1H NMR spectrum contained no olefinic resonances and its high-resolution mass spectrum suggested a mass 16 Da (i.e., 1 oxygen atom) larger than TCND-(CO2Me)2. Single-crystal X-ray analysis of the saponified derivative of this impurity revealed its identity to be the exo-epoxidized variant of TCND-(CO2Me)2 (ESI† Section 11). Thus, we recommend storage of uninhibited TCND-(CO2Me)2 under inert atmosphere, as is often done for olefinic monomers, to avoid its oxidative decomposition.
Finally, for comparison of ROMP propagation rates (vide infra), exo-NB analogs of each TCN and TCND were synthesized following previously reported procedures.32–34 All monomers and intermediates were isolated non-chromatographically, and the key intermediates towards TCNs (1) and TCNDs (quadricyclane) were synthesized on the 16 g and 17 g scales, respectively. Notably, the melting temperatures for all but one (TCND-CO2Me) of the TCNs and TCNDs synthesized in this study were greater than their exo-NB analogs (from 10 °C to >150 °C higher), presumably due to their multicyclic, comparably rigid structures. In particular, all of the TCNs studied here have melting points >40 °C above room temperature, making them generally easy to handle and purify (crystallization).
ROMP of TCN and TCND monomers
Each TCN and TCND described above (200 equiv.) was exposed to G3 (1 equiv.) in CDCl3 solvent to initiate ROMP. Reactions of TCNs were allowed to proceed for 15 minutes before quenching and precipitating twice from methanol, giving p(TCN)s in good yields (Table 1). Size exclusion chromatography (SEC) revealed low dispersities (Đ) and number-average molar masses (Mn) close to theoretical values for all p(TCN) samples (Fig. 3a–d).| Polymer | M n (theo.) | M n (SEC) | Đ | Yield | E/Z ratiod |
|---|---|---|---|---|---|
| a Monomer conversion was >98% in each case. b Theoretical Mn values were calculated assuming 100% monomer conversion. c SEC conditions: DMF (0.025 M LiBr) mobile phase, 60 °C, with differential refractive index and multi-angle light scattering detectors. A 100% mass recovery method was used to calculate Mn. d E/Z ratio was determined by the relative integrations of the trans (downfield) and cis (upfield) backbone olefinic protons in the polymers' 1H NMR spectra.35,36 | |||||
| p(TCN-NBnanti) | 56.0 kDa | 52.2 kDa | 1.02 | 97% | 37:63 |
| p(TCN-NCy) | 54.4 kDa | 54.2 kDa | 1.02 | 95% | 34:66 |
| p(TCN-NHex) | 54.8 kDa | 52.2 kDa | 1.02 | 92% | 37:63 |
| p(TCN-(CO2Me)2) | 47.4 kDa | 47.8 kDa | 1.02 | 97% | 33:67 |
| p(TCND-(CO2Me)2) | 47.0 kDa | 49.5 kDa | 1.04 | 92% | 30:70 |
| p(TCND-CO2Me) | 35.3 kDa | 59.3 kDa | 1.97 | 75% | 37:63 |
 | ||
| Fig. 3 (a–d) SEC traces of p(TCN)s. (e) Overlaid SEC traces of p(TCND-(CO2Me)2) when quenched after 15 min, 2 min, or after carefully distilling the monomer and quenching after 2 min (f) SEC trace of p(TCND-CO2Me) after working up the polymer in the presence of excess pyridine. | ||
SEC analysis of p(TCND-(CO2Me)2) prepared under the same conditions (i.e., 15 min reaction time) showed a minor high molecular weight shoulder peak at 2 × Mn indicative of cross-linking, as was also reported for Mo-initiated ROMP of this monomer (Fig. 3e).15 Quenching the ROMP reactions after a shorter time (2 min) using the same batch of TCND-(CO2Me)2 or a freshly distilled sample of TCND-(CO2Me)2 (discarding the first and last ∼10–15% of the distillate) substantially decreased the size of this shoulder peak. Notably, conversion was unaffected by decreasing the reaction time to 2 min, as evidenced by the similar SEC traces, suggesting that this monomer is highly reactive. Moreover, these results indicate that the cyclobutene component of TCND-(CO2Me)2 remains largely intact under these ROMP conditions, providing an opportunity for further modification post-polymerization (vide infra).
While the synthesis of p(TCND-CO2Me) appeared to be no different than for the other samples prepared here, the polymer product did not re-dissolve after the first precipitation. We hypothesized that polymer-associated residual Ru led to cross-linking reactions upon precipitation; the addition of excess pyridine (∼2000 equiv. relative to G3) to the quenched ROMP reaction suppressed this cross-linking (ESI† Section 3), enabling NMR and SEC analysis of p(TCND-CO2Me); however, SEC analysis showed a significant high molecular weight shoulder (Fig. 3f). Notably, while cyclobutene 1-carboxylic esters, like the cyclobutene fragment of TCND-CO2Me, cannot homopolymerize via G3-initiated ROMP,37 they are known to slowly form eneoic carbenes that are kinetically stabilized by intramolecular chelation.38 Thus, we propose that ROMP of TCND-CO2Me occurs first through rapid polymerization of its NB-like alkene followed by slow cross-linking of its cyclobutene pendants, which are more sterically accessible than for TCND-(CO2Me)2 above.
1H NMR spectroscopy was used to characterize the structures of these p(TCN)s and p(TCND)s in more detail (Fig. 4). Resonances associated with each of the monomer protons could be identified in the polymers. Compared to p(exo-NB) analogs (ESI† Section 3), the backbone olefins of p(TCN)s and p(TCND)s were slightly skewed toward the Z configuration: E:Z ranged from 48:52 to 37:63 for p(exo-NB)s while p(TCN)s and p(TCND)s ranged from 37:63 to 30:70 (Table 1). The spectrum of p(TCND-CO2Me) contained an extra downfield resonance at 6.8 ppm that we attribute to the olefinic proton of the cyclobutene. Otherwise, the spectra generally resembled those of their p(exo-NB) counterparts, with the addition of two proton resonances in the carbocyclic region (∼1.4 ppm–3.5 ppm) for the p(TCN)s.
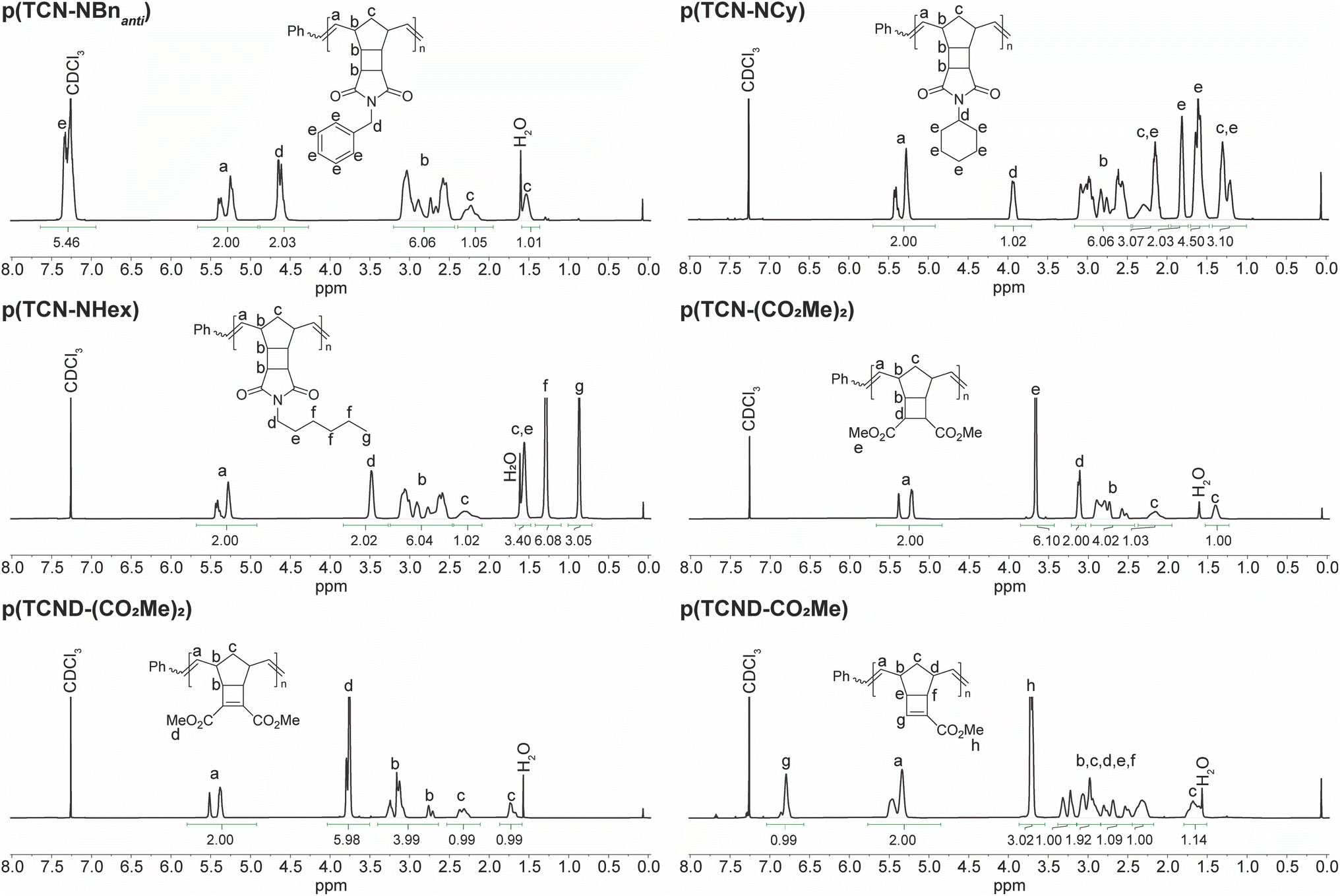 | ||
| Fig. 4 1H NMR spectra for the p(TCN)s and p(TCND)s prepared in this study. | ||
We anticipated that TCN and TCND monomers would display controlled polymerization behavior due to their high ring strain and low likelihood of backbiting/chain transfer. In support of this hypothesis, Mn,SEC values for p(TCN-NBnanti) agreed well with the theoretical values and increased linearly as a function of conversion up to a degree of polymerization (DP) of 500 (Fig. 5a). Moreover, a plot of ln([TCN-NBnanti]0/[TCN-NBnanti]) was linear as a function of time (Fig. 5b), and Đ remained low throughout the polymerization (Fig. 5a), suggesting minimal irreversible termination or chain transfer. Finally, to demonstrate the active nature of the p(TCN-NBnanti) Ru-alkylidene chain ends, a block copolymer p(TCN-NBnanti)100-b-p(TCN-NHex)100 was synthesized via sequential monomer addition (Fig. 5c). SEC analysis showed a clear peak shift to shorter retention time upon addition of TCN-NHex, suggesting complete chain extension; Đ for the block copolymer remained low (1.02).
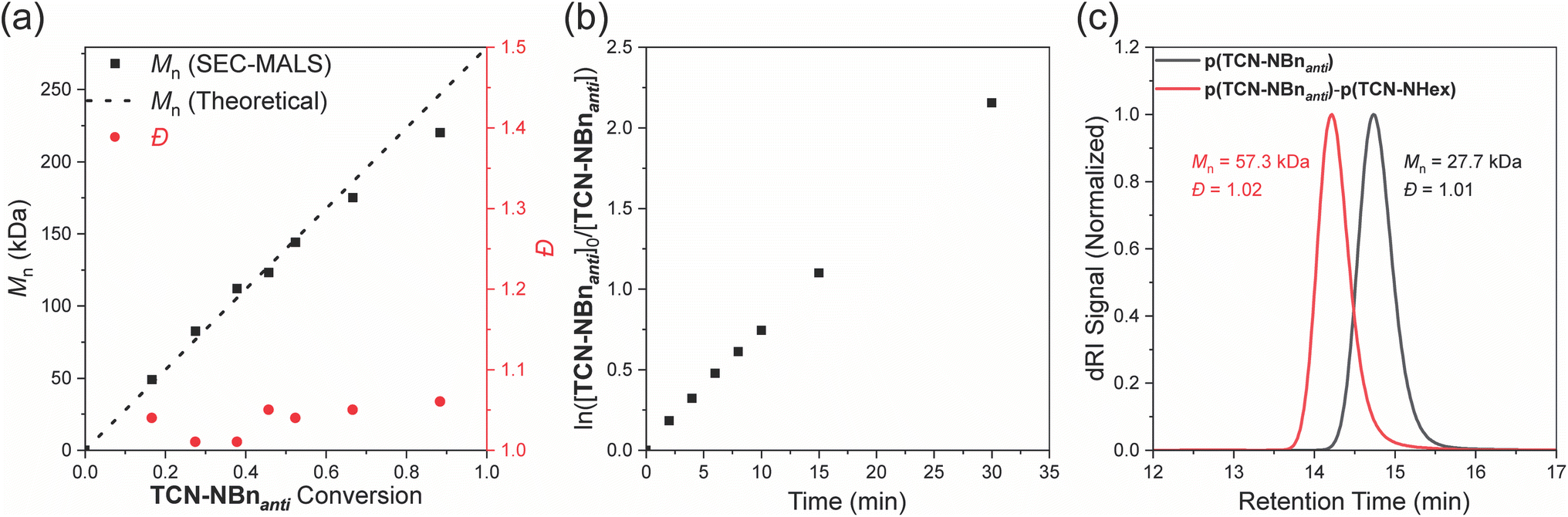 | ||
| Fig. 5 (a) Plot of Mn,SEC as a function of conversion for the polymerization of TCN-NBnanti with G3. Conditions: 500 equiv. TCN-NBnanti, 5 equiv. pyridine, 1 equiv. G3, 500 mM in CDCl3, RT. (b) Plot of ln([TCN-NBnanti]/[TCN-NBnanti]0) versus time for the polymerization described in panel a. (c) SEC analysis of the first and second blocks of the block copolymerization of TCN-NBnanti (DP 100) with TCN-NHex (DP 100). Conditions: 100 equiv. TCN-NBnanti, G3, 100 mM CDCl3, RT, then 100 equiv. TCN-NHex (100 mM soln. in CDCl3). | ||
1H NMR spectroscopic kinetic investigations of TCN and TCND ROMP reactions
Real-time 1H NMR spectroscopy was used to evaluate the ROMP propagation kinetics for each monomer shown in Fig. 2 in the presence of pyridine (5 equiv. relative to G3; note: pyridine was added to retard the polymerization reactions sufficiently for 1H NMR analysis). For all monomers except TCND-CO2Me, conversion was measured by comparison of the monomer signal to an added internal standard (mesitylene). The additional downfield olefin resonance of p(TCND-CO2Me) overlapped with that of the internal standard; monomer conversion was measured by relative integration of the monomer signal to the backbone olefin signals in this case. Each polymerization followed pseudo-first order kinetics up to ≥95% conversion, indicative of a controlled process (Fig. 6a–f). The propagation rates (kp), obtained by linear fitting of ln([M]0/[M]) vs. time plots (Fig. 6, insets, and ESI† Section 4), spanned a factor of ∼13 (kp = 0.094–1.259 min−1) for the monomers analyzed in this study (Table 2).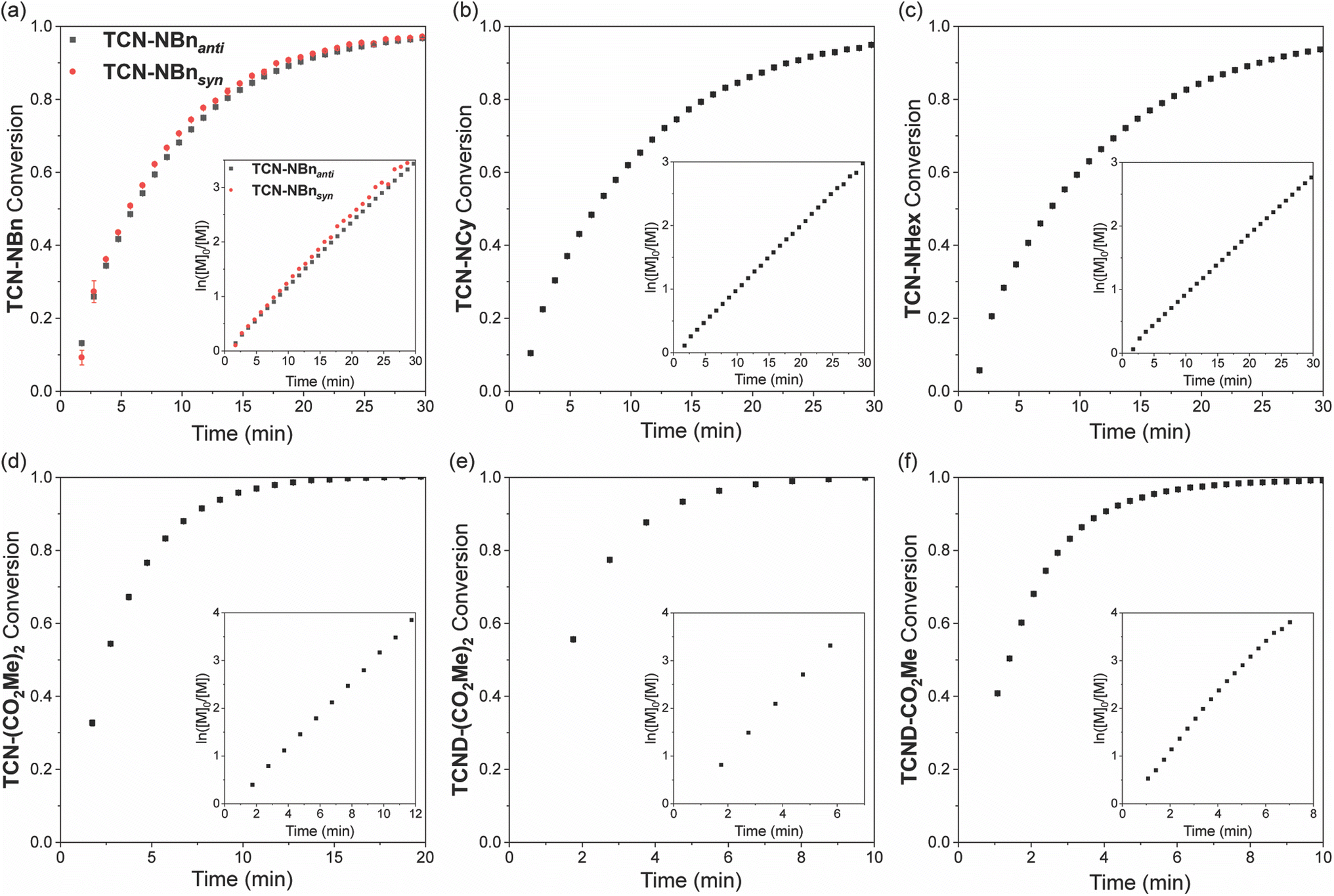 | ||
| Fig. 6 (a–f) ROMP propagation kinetics of all TCN and TCND monomers in this study. The raw data are presented with an inset plot of ln([M]0/[M]) versus time. Each was run in triplicate, and error bars are plotted as ± SD in the raw data plots. Conditions: 95 mM monomer (200 equiv.), 2.4 mM pyridine (5 equiv.), 0.48 mM G3 (1 equiv.) and internal standard (mesitylene) in CDCl3 at 25 °C. | ||
| Monomer | k p (min−1) | t 1/2 (min) |
|---|---|---|
| a Conditions: 95 mM monomer (200 equiv.), 2.4 mM pyridine (5 equiv.), 0.48 mM G3 (1 equiv.) and internal standard (mesitylene) in CDCl3 at 25 °C. | ||
| NB-NBn | 0.157 ± 0.001 | 4.4 |
| TCN-NBnanti | 0.117 ± 0.001 | 5.9 |
| TCN-NBnsyn | 0.122 ± 0.001 | 5.7 |
| NB-NCy | 0.183 ± 0.001 | 3.8 |
| TCN-NCy | 0.101 ± 0.001 | 6.9 |
| NB-NHex | 0.140 ± 0.001 | 5.0 |
| TCN-NHex | 0.094 ± 0.001 | 7.4 |
| NB-(CO2Me)2 | 0.225 ± 0.001 | 3.1 |
| TCN-(CO2Me)2 | 0.342 ± 0.001 | 2.0 |
| TCND-(CO2Me)2 | 0.622 ± 0.007 | 1.1 |
| NB-CO2Me | 1.259 ± 0.014 | 0.6 |
| TCND-CO2Me | 0.564 ± 0.010 | 1.2 |
The kp values for imides TCN-NBnanti, TCN-NBnsyn, TCN-NCy, and TCN-NHex were within ∼20% of each other and were slightly lower than their exo-NB imide counterparts. A similar trend was observed for the monoesters, but to a larger extent: kp for NB-CO2Me was 2.2-fold higher than for TCND-CO2Me. Notably, however, the opposite trend was observed for the diester monomers; kp for TCND-(CO2Me)2 was greater than TCN-(CO2Me)2, which was greater than NB-(CO2Me)2.
Comparing across the imides, diesters, and monoesters, the imides had the lowest kp values without exception; the fastest imide (NB-NCy) had a ∼19% lower kp than the slowest diester/monoester (NB-(CO2Me)2). This result agrees well with the trends reported for imide versus ester anchor groups for both small molecule monomers and macromonomers.5–7 Comparison of the monoesters and diesters shows that while the monoester TCND-CO2Me and diester TCND-(CO2Me)2 propagate at nearly the same rate, NB-CO2Me propagates 5.6 times faster than its diester counterpart NB-(CO2Me)2. Altogether, these results demonstrate that TCN and TCND monomers possess similar living behavior and G3-initiated ROMP kp values compared to traditional exo-NBs.
Density functional theory (DFT) calculations of monomer electronic and steric parameters to predict kp
Matson and coworkers have proposed that the DFT-calculated HOMO energy levels of exo-NB monomers correlate well with their kp values in G3-initiated ROMP.5–8 To determine if TCNs and TCNDs follow this trend, the HOMO energies for each monomer shown in Fig. 2b were calculated using the M06-2X functional and dev2-TZVP basis set (with a conductor-like polarizable continuum model for chloroform) and plotted against the measured ln(kp) values (Fig. 7b). The HOMO energies ranged from −183 kcal mol−1 to −194 kcal mol−1, which falls within the range reported for other ROMP monomers (−174 kcal mol−1 to −197 kcal mol−1) calculated with the same functional and basis set.5,6,8 In agreement with Matson and coworkers, a positive correlation between kp values and HOMO energies was observed; however, NB-CO2Me stands out as having a much larger kp value than its HOMO energy would predict. Notably, NB-CO2Me is the only monosubstituted norbornene (only one of the two exo positions is substituted) of the monomers studied here.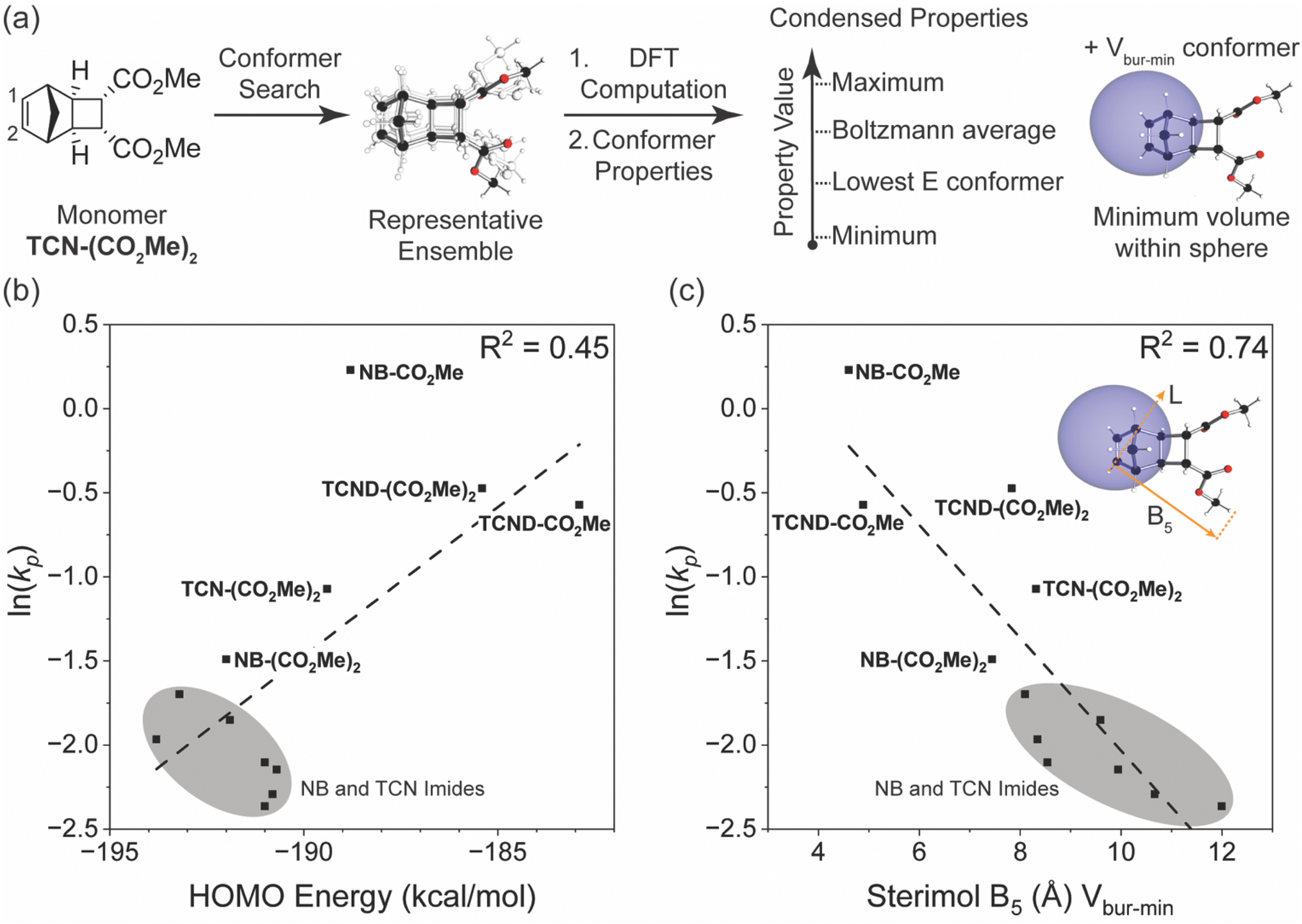 | ||
| Fig. 7 (a) Modeling workflow for steric parameters. AQME39 (rd-kit40) was used to convert monomer SMILES strings into a representative conformer ensemble.44 The ensemble was subjected to DFT computation and conformer properties were obtained. For each conformer the maximum, Boltzmann average, lowest energy (E) conformer, minimum, property value was obtained. Property values for the Vbur-min conformer at C1 were obtained as well to further probe steric effects. (b) Plot of measured monomer ln(kp) versus their HOMO energy levels calculated from their M06-2X/def2-TZVP optimized geometries. (c) Plot of measured monomer ln(kp) versus their calculated Sterimol B5 length from the minimum buried volume conformer of the adjacent active site olefinic carbon C2 (B3LYP/def2SVP/M06-2X/def2TZVP). | ||
Grubbs3 and Matson also found that monosubstituted exo-NBs polymerize faster than disubstituted variants; Matson attributed this difference either to the possibility that monosubstituted monomers can form regioisomers where their substituents point away from the ruthenium center (a steric effect),7 or to electronic effects (HOMO level and HOMO/LUMO gap).5–7 While the chelating ability of NB monomer substituents has also been shown to significantly affect the activation parameters of polymerization and may slow the polymerization rate by ground-state stabilization in certain cases,3 the chelating ability of exo-NB monomers seems to have little correlation with their propagation rates.5 We note that NB-CO2Me has a ∼4-fold larger kp than TCN-(CO2Me)2 despite having nearly the same calculated HOMO energy (0.6 kcal difference), and that TCND-(CO2Me)2 and TCND-CO2Me both have higher-lying HOMOs (by ∼4 and ∼6 kcal mol−1, respectively) than NB-CO2Me but significantly lower kp values. Overall, while HOMO energy is a useful tool for predicting ROMP reactivity for exo-NB-like scaffolds, other effects, such as steric effects near the propagation site, may impact the rate as well.
These findings led us to consider alternative readily-computed structural descriptors that may provide better correlations with ROMP monomer performance over a wider structure range, and may then also provide greater predictive power for the design of novel monomers. We began by collecting DFT-derived parameters from an AQME derived conformer ensemble39 using RDKit40 for each monomer. From the conformer ensemble, five representative conformers were selected to extract a range of steric and electronic parameters (Fig. 7a).41,42 The ensemble conformer properties were condensed to include the lowest energy conformer (low E), minimum (min), maximum (max), and Boltzmann derived parameter values. Additionally, we also extracted condensed properties from the conformer presenting the minimum percent buried volume (Vbur-min) parameter value at the active site olefinic carbon (C1), as we hypothesized that steric effects may be a driver of the observed rate differences. Percent buried volume probes the proximal steric environment by placing a 3 Å sphere at the site of interest and measuring the resulting overlap.43 The library of parameters was regressed against ln(kp) to reveal a range of good correlations with various parameters describing steric effects (molar volume (MV), solvent accessible surface area (SASA) and Sterimol B5). These correlations all resulted from the Vbur-min conformer values located at the active site olefinic carbon C1. A representative correlation using Sterimol B5 is depicted in Fig. 7c. This parameter describes the display of steric information for how the adjacent olefinic carbon C2 may present itself to the propagating Ru chain end (Fig. 7a). The other two strong correlations, MV and SASA, represent the shape of the monomer while accounting for the local environment defined by the Vbur-min conformer. Notably, the Sterimol B5 and the MV Vbur-min conformers were found to be collinear, and the same parameters derived from other conformers (i.e., low E, min, and max values) do not perform nearly as well (ESI† Section 13). Overall, these data suggest that the overall size and shape of the monomer has a significant impact on polymerization kinetics.
Further derivatization of TCND-(CO2Me)2 and post-polymerization functionalization of p(TCND-(CO2Me)2)
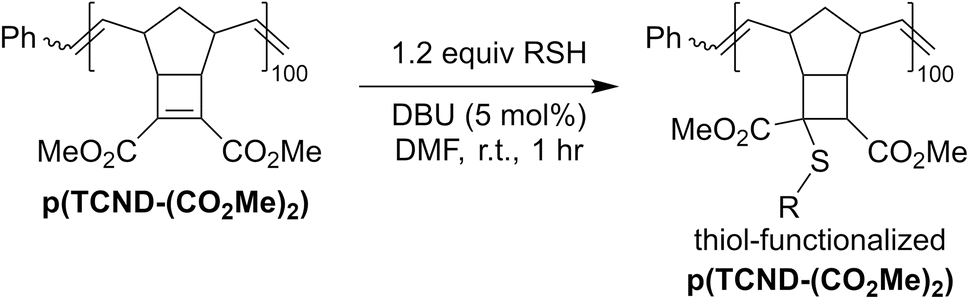
In addition to their straightforward syntheses and good kp values, TCND-(CO2Me)2 and TCND-CO2Me, and their corresponding polymers p(TCND-(CO2Me)2) and p(TCND-CO2Me), offer unique opportunities for further derivatization and post-polymerization functionalization, respectively, compared to exo-NBs. While their cycloaddition reactivity has been explored,45–47 we suspected that their strained, electron-deficient cyclobutene substituents would make them good electrophiles for conjugate addition reactions (e.g., “thiol-Michael” addition). To test this idea, TCND-(CO2Me)2 was exposed to dodecanethiol (1.0 equiv.) in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (10 mol%) in CDCl3 for 30 min (Table 3, entry 1); 47% conversion to the conjugate addition product was observed under these conditions, while 0% conversion was observed in the absence of DBU. Polar solvents are known to accelerate conjugate addition reactions that involve polar mechanisms;48 when DMF-d7 was used as the solvent for this reaction, 87% conversion was achieved in 30 min using only 1 mol% DBU (Table 3, entry 2).|
|
|||||
|---|---|---|---|---|---|
| Entry | Conjugate acceptor | LUMO energy (kcal mol−1) | Solvent | DBU (mol%) | Conversion |
| 1 | TCND-(CO2Me)2 | −30 | CDCl3 | 10% | 47% |
| 2 | TCND-(CO2Me)2 | −30 | DMF-d7 | 1% | 87% |
| 3 | DDM | −8 | CDCl3 | 10% | 0% |
| 4 | TCND-CO2Me | −10 | CDCl3 | 10% | 3% |
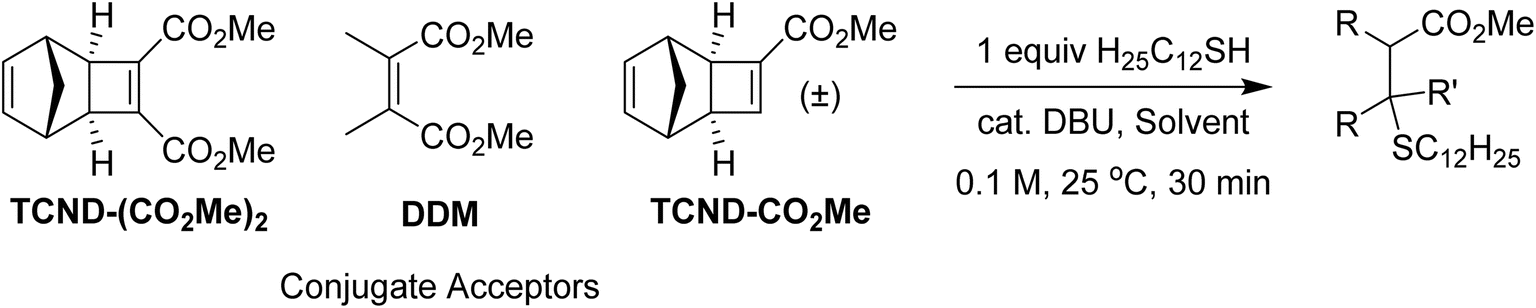
Notably, dimethyl 2,3-dimethylmaleate (DDM), a less-strained electrophile with a similar substitution pattern, did not react under the CDCl3 conditions described above (Table 3, entry 3), and TCND-CO2Me reached only 3% conversion (Table 3, entry 4). These results correlate with the DFT calculated LUMO energies (M06-2X/def-TZVP) for each molecule (Table 3), suggesting that the rigid structure of TCND-(CO2Me)2 paired with its two electron-withdrawing ester substituents make it a better electrophile for conjugate addition. Meanwhile, as discussed above, these structural features are also key for (1) ensuring a high ROMP kp (by avoiding steric hindrance associated with diesters appended directly to the exo-NB) and (2) avoiding undesired cross-linking during ROMP (as observed for TCND-CO2Me), making TCND-(CO2Me)2 a particularly promising monomer for further exploration.
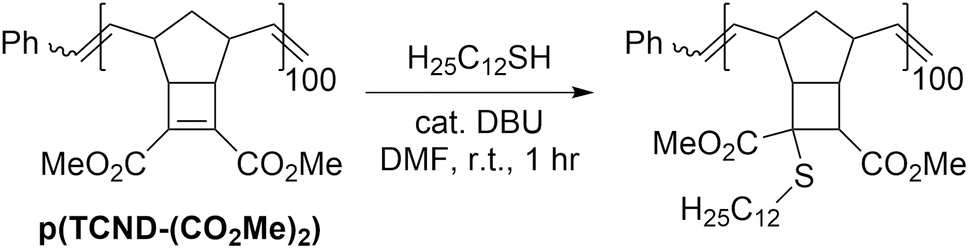
We hypothesized that the electrophilicity of TCND-(CO2Me)2 would not change significantly after ROMP, rendering p(TCND-(CO2Me)2) a similarly useful substrate for post-polymerization conjugate addition reactions. This hypothesis was supported by DFT calculations (M06-2X/def-TZVP); the LUMO energy of a model substrate designed to mimic the p(TCND-(CO2Me)2) backbone—the ethenolysis product of TCND-(CO2Me)2—was very similar (−32 kcal mol−1) to that of TCND-(CO2Me)2 (−30 kcal mol−1). Moreover, nearly quantitative conjugate addition was observed when p(TCND-(CO2Me)2) was exposed to dodecanethiol (5 equiv.) and DBU (20 mol%) in DMF solvent for 1 h under ambient atmosphere (Table 4, entry 1). The reaction solution became cloudy almost immediately after addition of the DBU, suggesting rapid conjugate addition and limited solubility of the functionalized polymer product in DMF. Decreasing the number of equivalents of thiol to 2 (Table 4, entry 2) and lowering the catalyst loading to 5 mol% (Table 4, entry 3) did not affect the conversion. Reduction of the equivalents of thiol to 1.5 (Table 4, entry 4) did not affect conversion either; however, further decreasing the amount of thiol to 1.1 equiv. gave slightly reduced conversion (95%, Table 4, entry 5), which was reduced further with a lower catalyst loading of 2.5 mol% (93%, Table 4, entry 6). Over 95% conversion was achieved with 1.2 equiv. of thiol and 5 mol% DBU (Table 4, entry 7); these conditions were chosen as optimal for further studies.
|
|
|||
|---|---|---|---|
| Entry | H25C12SH equiv. | DBU equiv. | Conversionb (1H NMR) |
|
a Conditions: p(TCND-(CO2Me)2) in DMF (50 mg mL−1) at room temperature. Dodecanethiol was added, followed by the addition of DBU as a stock solution in DMF (1:9 v/v DBU:DMF) and was allowed to react for 1 hour.
b Conversion was assessed by comparing the integration of the backbone olefinic protons (4.8 ppm – 5.7 ppm) versus the integration of the terminal –CH3 of the dodecanethiol side-chain (0.9 ppm).
|
|||
| 1 | 5 | 20 mol% | >95% |
| 2 | 2 | 20 mol% | >95% |
| 3 | 2 | 5 mol% | >95% |
| 4 | 1.5 | 5 mol% | >95% |
| 5 | 1.1 | 5 mol% | 95% |
| 6 | 1.1 | 2.5 mol% | 93% |
| 7 | 1.2 | 5 mol% | >95% |
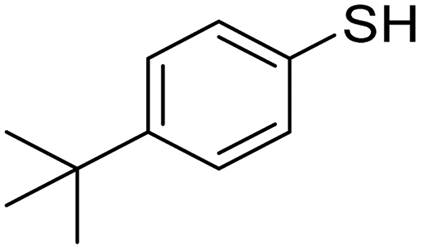
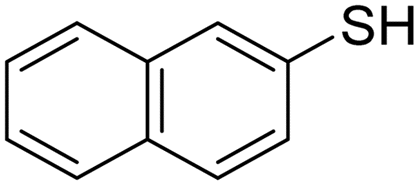
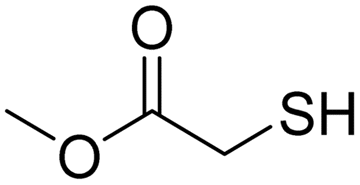
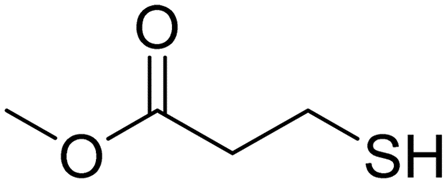
A variety of thiol nucleophiles was explored using these optimized conditions (Table 5); 1H NMR and SEC were used to measure conversion of each reaction, with the two methods showing agreement within ∼15% in most cases. Incomplete conversion was observed for 4-tert-butylthiophenol and 2-naphthalenethiol under ambient (Table 5, entries 1 and 3) or inert (N2) atmosphere (Table 5, entries 2 and 4), which we attribute to the steric bulk of these thiols. Nearly quantitative functionalization was observed for methyl thioglycolate (Table 5, entries 5 and 6) regardless of atmosphere; however, the SEC peak shape for the product showed distinct broadening and tailing compared to the initial p(TCND-(CO2Me)2), which may be due to possible deleterious side-reactions promoted by the relatively acidic α-protons of the α-mercapto ester (ESI† Section 9).49,50 By contrast, 1-methyl-3-mercaptoproprionate, a β-mercapto ester, underwent high conversion to give a polymer product with no significant change in peak shape (Table 5, entry 7). Finally, hexanethiol also gave high conversion under these conditions (Table 5, entry 8).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Thiol | Atmosphere | M n initial | M n final | Conversion (SEC) | Conversion (1H NMR) |
| 1 |
|
Air | 27.0 kDa | 33.4 kDa | 33% | 45% |
| 2 | N2 | 27.0 kDa | 39.7 kDa | 66% | 64% | |
| 3 |
|
Air | 27.0 kDa | 32.4 kDa | 29% | 16% |
| 4 | N2 | 27.0 kDa | 33.2 kDa | 34% | 36% | |
| 5 |
|
Air | 27.0 kDa | 41.4 kDa | Quant. | N/A |
| 6 | N2 | 27.0 kDa | 40.0 kDa | Quant. | N/A | |
| 7 |
|
Air | 27.0 kDa | 39.4 kDa | Quant. | N/A |
| 8 |
|
Air | 27.0 kDa | 45.4 kDa | Quant. | >95% |
Conclusions
This work represents the first detailed study of the polymerization behavior of TCN and TCND monomers in G3-initiated ROMP. A variety of functional (imides, monoesters, and diesters) TCNs, TCNDs, and exo-NBs were synthesized and their ROMP propagation rates were measured to span a factor of ∼13. To understand the relationship between monomer structure and propagation rate, the monomers were featurized using DFT to extract a variety of electronic and steric parameters and search for correlations with measured propagation rates. While simple electronic parameters (e.g., HOMO levels) positively correlated with monomer kp, steric parameters that described the overall size and shape of the monomer were more strongly correlated with kp. TCND-(CO2Me)2—which contains an electron-deficient cyclobutene—was discovered to be highly active towards DBU-catalyzed conjugate addition of dodecanethiol. This conjugate addition reaction was optimized on p(TCND-(CO2Me)2), and a variety of thiol nucleophiles were used to functionalize p(TCND-(CO2Me)2) in this post-polymerization modification reaction.Overall, this study revealed that TCNs and TCNDs are a synthetically accessible class of ROMP monomers that do not compromise on activity or controlled polymerization behavior in comparison to other commonly used monomers (e.g. exo-NBs). In particular, TCND-(CO2Me)2 is a uniquely promising monomer due to its ease of synthesis (2 steps from commercial materials), fast ROMP kinetics, and its innate activity towards post-polymerization functionalization with thiols; we envision its future widespread use for functional polymer design. Finally, our experimental and computational study revealed that monomer steric parameters correlate most strongly with their measured propagation rates; these results offer the polymer chemist both the intuitive framework and computational tools for developing future novel ROMP monomers.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
Conceptualization: all authors. Data curation: L. K and T. M. formal analysis: L. K. and T. M. funding acquisition: J. J and M. S. investigation: L. K and T. M. methodology: L. K. and T. M. project administration: all authors. Resources: all authors. Software: T. M. and M. S. supervision: J. J. and M. S. validation: L. K. and T. M. visualization: L. K. and T. M. writing – original draft: L. K. and J. J. writing – review and editing: all authors.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported as part of the Center for Regenerative Energy-Efficient Manufacturing of Thermoset Polymeric Materials (REMAT), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under award #DE-SC0023457. Thanks to Dr Peter Müller of the MIT Chemistry Department X-Ray Diffraction Facility for crystal structure determination.References
- J. A. Love, J. P. Morgan, T. M. Trnka and R. H. Grubbs, A Practical and Highly Active Ruthenium-Based Catalyst that Effects the Cross Metathesis of Acrylonitrile, Angew. Chem., Int. Ed., 2002, 41(21), 4035–4037, DOI:10.1002/1521-3773(20021104)41:21<4035::AID-ANIE4035>3.0.CO;2-I.
- H. L. Cater, I. Balynska, M. J. Allen, B. D. Freeman and Z. A. Page, User Guide to Ring-Opening Metathesis Polymerization of endo-Norbornene Monomers with Chelated Initiators, Macromolecules, 2022, 55(15), 6671–6679, DOI:10.1021/acs.macromol.2c01196.
- W. J. Wolf, T.-P. Lin and R. H. Grubbs, Examining the Effects of Monomer and Catalyst Structure on the Mechanism of Ruthenium-Catalyzed Ring-Opening Metathesis Polymerization, J. Am. Chem. Soc., 2019, 141(44), 17796–17808, DOI:10.1021/jacs.9b08835.
- M. G. Hyatt, D. J. Walsh, R. L. Lord, J. G. Andino Martinez and D. Guironnet, Mechanistic and Kinetic Studies of the Ring Opening Metathesis Polymerization of Norbornenyl Monomers by a Grubbs Third Generation Catalyst, J. Am. Chem. Soc., 2019, 141(44), 17918–17925, DOI:10.1021/jacs.9b09752.
- S. J. Scannelli, A. Paripati, J. R. Weaver, C. Vu, M. Alaboalirat, D. Troya and J. B. Matson, Influence of the Norbornene Anchor Group in Ru-Mediated Ring-Opening Metathesis Polymerization: Synthesis of Linear Polymers, Macromolecules, 2023, 56(11), 3848–3856, DOI:10.1021/acs.macromol.3c00172.
- S. J. Scannelli, M. Alaboalirat, D. Troya and J. B. Matson, Influence of the Norbornene Anchor Group in Ru-Mediated Ring-Opening Metathesis Polymerization: Synthesis of Bottlebrush Polymers, Macromolecules, 2023, 56(11), 3838–3847, DOI:10.1021/acs.macromol.3c00214.
- S. C. Radzinski, J. C. Foster, R. C. Chapleski Jr, D. Troya and J. B. Matson, Bottlebrush Polymer Synthesis by Ring-Opening Metathesis Polymerization: The Significance of the Anchor Group, J. Am. Chem. Soc., 2016, 138(22), 6998–7004, DOI:10.1021/jacs.5b13317.
- S. J. Scannelli, M. Alaboalirat, D. Troya and J. B. Matson, Ring-opening metathesis polymerization of norbornene–benzoladderene (macro)monomers, Polym. Chem., 2023, 14(41), 4726–4735, 10.1039/D3PY00981E.
- L. T. Birchall, S. Shehata, C. J. Serpell, E. R. Clark and S. C. G. Biagini, Himic Anhydride: A Retro Diels–Alder Reaction for the Organic Laboratory and an Accompanying NMR Study, J. Chem. Educ., 2021, 98(12), 4013–4016, DOI:10.1021/acs.jchemed.1c00661.
- C. M. Bates, A. B. Chang, N. Momčilović, S. C. Jones and R. H. Grubbs, ABA Triblock Brush Polymers: Synthesis, Self-Assembly, Conductivity, and Rheological Properties, Macromolecules, 2015, 48(14), 4967–4973, DOI:10.1021/acs.macromol.5b00880.
- A. M. Spring, F. Yu, F. Qiu, K. Yamamoto and S. Yokoyama, The preparation of well-controlled poly(N-cyclohexyl-exo-norbornene-5,6-dicarboximide) polymers, Polym. J., 2014, 46(9), 576–583, DOI:10.1038/pj.2014.26.
- B. J. Sundell, J. A. Lawrence III, D. J. Harrigan, S. Lin, T. P. Headrick, J. T. O'Brien, W. F. Penniman and N. Sandler, Exo-selective, Reductive Heck Derived Polynorbornenes with Enhanced Molecular Weights, Yields, and Hydrocarbon Gas Transport Properties, ACS Macro Lett., 2020, 9(9), 1363–1368, DOI:10.1021/acsmacrolett.0c00555.
- C. D. Smith, Cycloaddition Reactions of “Quadricyclanes”1, J. Am. Chem. Soc., 1966, 88(18), 4273–4274, DOI:10.1021/ja00970a038.
- V. A. Petrov and N. V. Vasil'ev, Synthetic Chemistry of Quadricyclane, Curr. Org. Synth., 2006, 3(2), 215–259 CrossRef CAS.
- R. S. Saunders, New Polymers from Ring-Opening Metathesis Polymerization of Quadricyclane Adducts, Macromolecules, 1995, 28(12), 4347–4349, DOI:10.1021/ma00116a044.
- E. S. Finkelshtein, P. P. Chapala, M. L. Gringolts and Y. V. Rogan, Polymerization of Tricyclononenes, Polym. Sci., Ser. C, 2019, 61(1), 17–30, DOI:10.1134/S1811238219010077.
- M. L. Gringolts, M. V. Bermeshev, Y. V. Rogan, M. V. Moskvicheva, M. P. Filatova, E. S. Finkelshtein and G. N. Bondarenko, Comparative Reactivity of Me3Si-substituted Norbornene Derivatives in Ring-Opening Metathesis Polymerization, Silicon, 2015, 7(2), 107–115, DOI:10.1007/s12633-014-9238-7.
- P. Chapala, M. Bermeshev, L. Starannikova, I. Borisov, V. Shantarovich, V. Lakhtin, V. Volkov and E. Finkelshtein, Synthesis and Gas-Transport Properties of Metathesis Polytricyclononenes Bearing Three Me3Si Groups per Monomer Unit, Macromol. Chem. Phys., 2016, 217(17), 1966–1976, DOI:10.1002/macp.201600232.
- M. V. Bermeshev, L. E. Starannikova, S. R. Sterlin, A. A. Tyutyunov, A. N. Tavtorkin, Y. P. Yampolskii and E. S. Finkelshtein, Synthesis and gas-separation properties of metathesis poly(3-fluoro-3-pentafluoroethyl-4,4-bis(trifluoromethyl)tricyclonene-7), Pet. Chem., 2015, 55(9), 753–758, DOI:10.1134/S0965544115050035.
- G. O. Karpov, M. V. Bermeshev, I. L. Borisov, S. R. Sterlin, A. A. Tyutyunov, N. P. Yevlampieva, B. A. Bulgakov, V. V. Volkov and E. S. Finkelshtein, Metathesis-type poly-exo-tricyclononenes with fluoroorganic side substituents: Synthesis and gas-transport properties, Polymer, 2018, 153, 626–636, DOI:10.1016/j.polymer.2018.08.055.
- I. L. Borisov, T. R. Akmalov, A. O. Ivanov, V. V. Volkov, E. S. Finkelshtein and M. V. Bermeshev, A new cycloadduct based on quadricyclane and perfluorocyclohexene: synthesis, metathesis polymerization and gas-transport properties of the obtained polymer, Mendeleev Commun., 2016, 26(2), 124–126, DOI:10.1016/j.mencom.2016.03.013.
- M. Bermeshev, B. Bulgakov, D. Demchuk, M. Filatova, L. Starannikova and E. Finkelshtein, Metathesis and addition polymerization of novel Me3Si- and Me3Ge-substituted tricyclononenes, Polym. J., 2013, 45(7), 718–726, DOI:10.1038/pj.2012.211.
- D. A. Alentiev, E. S. Egorova, M. V. Bermeshev, L. E. Starannikova, M. A. Topchiy, A. F. Asachenko, P. S. Gribanov, M. S. Nechaev, Y. P. Yampolskii and E. S. Finkelshtein, Janus tricyclononene polymers bearing tri(n-alkoxy)silyl side groups for membrane gas separation, J. Mater. Chem. A, 2018, 6(40), 19393–19408, 10.1039/C8TA06034G.
- A. Y. Kanatieva, D. A. Alentiev, V. E. Shiryaeva, A. A. Korolev and A. A. Kurganov, Impact of the Polymer Backbone Structure on the Separation Properties of New Stationary Phases Based on Tricyclononenes, Polymers, 2022, 14(23), 5120, DOI:10.3390/polym14235120.
- A. Kanateva, M. Bermeshev, D. Alentiev, A. A. Korolev and A. Kurganov, Chromatographic Method for Evaluation of Polymeric GC Stationary Phases Ageing Using the Novel Non-Cross-Linked Poly(3-(Tributoxysilyl)Tricyclononene-7) as the Model Stationary Phase, Polymers, 2021, 13(11), 1899, DOI:10.3390/polym13111899.
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Synthesis of Functional Polymers by Post-Polymerization Modification, Angew. Chem., Int. Ed., 2009, 48(1), 48–58, DOI:10.1002/anie.200801951.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Thiol-click chemistry: a multifaceted toolbox for small molecule and polymer synthesis, Chem. Soc. Rev., 2010, 39(4), 1355–1387, 10.1039/B901979K.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, The Thiol-Michael Addition Click Reaction: A Powerful and Widely Used Tool in Materials Chemistry, Chem. Mater., 2014, 26(1), 724–744, DOI:10.1021/cm402180t.
- I. Tabushi, K. Yamamura and Z. Yoshida, Regio- and stereospecific [2.pi. + 2.sigma. + 2.sigma.] cycloaddition reaction of quadricyclane, J. Am. Chem. Soc., 1972, 94(3), 787–792, DOI:10.1021/ja00758a018.
- P. S. Kearns, B. K. Wells, R. N. Warrener and D. Margetić, The Preparation of Stereoisomeric Tricyclo[4.2.1.02,5]nona-7-ene-3,4-dicar-boximides and Anhydrides: Literature Corrections and New Products, Synlett, 2014, 25(11), 1601–1605, DOI:10.1055/s-0033-1339031.
- R. N. Warrener, Dimethyl (1α,2β, 5β,6α) tricyclo[4.2.1.02,5] nona-3,7-dien-3,4-dicarboxylate, in Encyclopedia of Reagents for Organic Synthesis (EROS), 2004 Search PubMed.
- T.-W. Wang, P.-R. Huang, J. L. Chow, W. Kaminsky and M. R. Golder, A Cyclic Ruthenium Benzylidene Initiator Platform Enhances Reactivity for Ring-Expansion Metathesis Polymerization, J. Am. Chem. Soc., 2021, 143(19), 7314–7319, DOI:10.1021/jacs.1c03491.
- K. E. L. Husted, A. Herzog-Arbeitman, D. Kleinschmidt, W. Zhang, Z. Sun, A. J. Fielitz, A. N. Le, M. Zhong and J. A. Johnson, Pendant Group Modifications Provide Graft Copolymer Silicones with Exceptionally Broad Thermomechanical Properties, ACS Cent. Sci., 2023, 9(1), 36–47, DOI:10.1021/acscentsci.2c01246.
- A. B. Chang, T.-P. Lin, N. B. Thompson, S.-X. L. Luo, A. L. Liberman-Martin, H.-Y. Chen, B. Lee and R. H. Grubbs, Design, Synthesis, and Self-Assembly of Polymers with Tailored Graft Distributions, J. Am. Chem. Soc., 2017, 139(48), 17683–17693, DOI:10.1021/jacs.7b10525.
- Y.-Z. Ke, S.-L. Huang, G. Lai and T.-Y. Luh, Selective ring-opening metathesis polymerization (ROMP) of cyclobutenes. Unsymmetrical ladderphane containing polycyclobutene and polynorbornene strands, Beilstein J. Org. Chem., 2019, 15, 44–51, DOI:10.3762/bjoc.15.4.
- J. Suriboot, C. E. Hobbs, W. Guzman, H. S. Bazzi and D. E. Bergbreiter, Polyethylene as a Cosolvent and Catalyst Support in Ring-Opening Metathesis Polymerization, Macromolecules, 2015, 48(16), 5511–5516, DOI:10.1021/acs.macromol.5b01141.
- A. Song, K. A. Parker and N. S. Sampson, Synthesis of Copolymers by Alternating ROMP (AROMP), J. Am. Chem. Soc., 2009, 131(10), 3444–3445, DOI:10.1021/ja809661k.
- A. Song, J. C. Lee, K. A. Parker and N. S. Sampson, Scope of the Ring-Opening Metathesis Polymerization (ROMP) Reaction of 1-Substituted Cyclobutenes, J. Am. Chem. Soc., 2010, 132(30), 10513–10520, DOI:10.1021/ja1037098.
- J. V. Alegre-Requena, S. Sowndarya, R. Pérez-Soto, T. M. Alturaifi and R. S. Paton, AQME: Automated quantum mechanical environments for researchers and educators, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2023, 13(5), e1663, DOI:10.1002/wcms.1663.
- RDKit: Open-source cheminformatics, https://www.rdkit.org accessed.
- M. S. Sigman, K. C. Harper, E. N. Bess and A. Milo, The Development of Multidimensional Analysis Tools for Asymmetric Catalysis and Beyond, Acc. Chem. Res., 2016, 49(6), 1292–1301, DOI:10.1021/acs.accounts.6b00194.
- C. B. Santiago, J.-Y. Guo and M. S. Sigman, Predictive and mechanistic multivariate linear regression models for reaction development, Chem. Sci., 2018, 9(9), 2398–2412, 10.1039/C7SC04679K.
- A. C. Hillier, W. J. Sommer, B. S. Yong, J. L. Petersen, L. Cavallo and S. P. Nolan, A Combined Experimental and Theoretical Study Examining the Binding of N-Heterocyclic Carbenes (NHC) to the Cp*RuCl (Cp* = η5-C5Me5) Moiety: Insight into Stereoelectronic Differences between Unsaturated and Saturated NHC Ligands, Organometallics, 2003, 22(21), 4322–4326, DOI:10.1021/om034016k.
- T. Gensch, G. dos Passos Gomes, P. Friederich, E. Peters, T. Gaudin, R. Pollice, K. Jorner, A. Nigam, M. Lindner-D’Addario and M. S. Sigman, et al., A Comprehensive Discovery Platform for Organophosphorus Ligands for Catalysis, J. Am. Chem. Soc., 2022, 144(3), 1205–1217, DOI:10.1021/jacs.1c09718.
- R. N. Warrener and D. N. Butler, Dimethyl tricyclo[4.2.1.02,5]nona-3,7-diene-3,4-dicarboxylate: A Versatile Ambident Dienophile, Aldrichimica Acta, 1997, 30(4), 119–130 CAS.
- G. Jenner, High pressure sequential (π2 + π2 + π2) and (π2 + σ2 + σ2) cycloaddition reactions. Synthesis of polycyclic bisadducts from C7H8 hydrocarbons, Tetrahedron Lett., 1987, 28(34), 3927–3930, DOI:10.1016/S0040-4039(00)96422-0.
- D. Margetic, D. N. Butler and R. N. Warrener, Photolysis of Fused Cyclobutatriazolines: An Adventitious Route to 3,6-Di(2-pyridyl)pyridazonorbornadiene, Aust. J. Chem., 2001, 53(12), 959–963, DOI:10.1071/CH00129.
- G.-Z. Li, R. K. Randev, A. H. Soeriyadi, G. Rees, C. Boyer, Z. Tong, T. P. Davis, C. R. Becer and D. M. Haddleton, Investigation into thiol-(meth)acrylate Michael addition reactions using amine and phosphine catalysts, Polym. Chem., 2010, 1(8), 1196–1204, 10.1039/C0PY00100G.
- F. G. Bordwell, Equilibrium acidities in dimethyl sulfoxide solution, Acc. Chem. Res., 1988, 21(12), 456–463, DOI:10.1021/ar00156a004.
- X. M. Zhang, F. G. Bordwell, M. Van Der Puy and H. E. Fried, Equilibrium acidities and homolytic bond dissociation energies of the acidic carbon-hydrogen bonds in N-substituted trimethylammonium and pyridinium cations, J. Org. Chem., 1993, 58(11), 3060–3066, DOI:10.1021/jo00063a026.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2326835, 2326836 and 2351614. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01986e |
| This journal is © The Royal Society of Chemistry 2024 |