
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnnatural enzyme activation by a metal-responsive regulatory protein†
Olga
Halfin
a,
Liat
Avram
 b,
Shira
Albeck
c,
Tamar
Unger
c,
Leila
Motiei
a and
David
Margulies
*a
b,
Shira
Albeck
c,
Tamar
Unger
c,
Leila
Motiei
a and
David
Margulies
*a
aDepartment of Chemical and Structural Biology, Weizmann Institute of Science, Rehovot, Israel. E-mail: david.margulies@weizmann.ac.il
bDepartment of Chemical Research Support, Weizmann Institute of Science, Rehovot, Israel
cDepartment of Life Sciences Core Facilities, Weizmann Institute of Science, Rehovot, Israel
First published on 5th August 2024
Abstract
As a result of calcium ion binding, the calcium-dependent regulatory protein calmodulin (CaM) undergoes a conformational change, enabling it to bind to and activate a variety of enzymes. However, the detoxification enzyme glutathione S-transferase (GST) is notably not among the enzymes activated by CaM. In this study, we demonstrate the feasibility of establishing, in vitro, an artificial regulatory link between CaM and GST using bifunctional chemical transducer (CT) molecules possessing binders for CaM and GST. We show that the CTs convert the constitutively active GST into a triggerable enzyme whose activity is unnaturally regulated by the CaM conformational state and consequently, by the level of calcium ions. The ability to reconfigure the regulatory function of CaM demonstrates a novel mode by which CTs could be employed to mediate artificial protein crosstalk, as well as a new means to achieve artificial control of enzyme activity by modulating the coordination of metal ions. Within this study, we also investigated the impact of covalent interaction between the CTs and the enzyme target. This investigation offers further insights into the mechanisms governing the function of CTs and the possibility of rendering them isoform specific.
Introduction
Owing to the critical role irregular enzyme activity plays in disease, significant efforts have been devoted to creating synthetic agents that can interact with enzyme active sites and disrupt their function.1 Although this mode of regulation has proven successful in the development of various small-molecule-based drugs, in recent years, an alternative strategy for controlling enzyme function has emerged. Rather than disrupting enzyme activity, chemists have become fascinated by the idea of artificially activating them.2–20 The motivation for developing artificially triggered enzymes lies in the potential to apply them across various research fields, such as sensing,2,3 cell signaling regulation,4,20 artificial biology,5–8 catalyst development,5,10,11 multi-step biocatalytic reactions,9 and therapy.12–19 Furthermore, in the context of biomimicry, these systems contribute to our ability to understand and thereby imitate and exploit natural enzyme activation mechanisms such as the ones underlying the activation of apoenzymes,5,10,11 zymogens3–5,7,12–16 or allosteric enzymes.2,8,17,19An essential role that activable enzymes play in cells is mediating signaling transduction pathways. In these pathways, enzymes are remotely activated by a network of protein–protein interactions that transduce the response of cells to signaling molecules and ions.21 Inspired by the pivotal role these interactions play in the activation of signaling enzymes, we have been developing artificially triggered enzymes that respond to the binding of non-native protein partners (Fig. 1a).19,20 This unnatural activation mode, which imitates the way enzymes are activated by ligands or effectors, does not require that the protein scaffold constituting the unnaturally triggered enzymes be chemically or genetically modified. Instead, our approach is based on bifunctional chemical transducer (CT) molecules that possess binders for two proteins of interest (POIs): the enzyme of choice (POI-1) and an unnatural, activating protein partner (POI-2) (Fig. 1a). As illustrated, the transducer binds to POI-1 and inhibits it (Fig. 1a, state I). However, when the unnatural activating partner is present, it interacts with an excess of the unbound CT molecules in the solution, reducing their affinity for POI-1. This decrease in affinity, which often results from proximity-induced, non-specific interactions between the CT and the surface of POI-2,20 shifts the equilibrium toward the dissociation of the CT-POI-1 complex, ultimately leading to an unnatural activation of POI-1 by POI-2 (state II). In the context of enzyme mimicry, the enzyme-bound CTs emulate the function of natural inhibitory domains (IDs) that govern the function of various activable enzymes.22 Similar to IDs, the binding of the synthetic ID (sID) to the non-native protein effector (POI-2) leads to the liberation of the sID from the active site of POI-1 and subsequently to enzyme activation (Fig. 1a, state II).
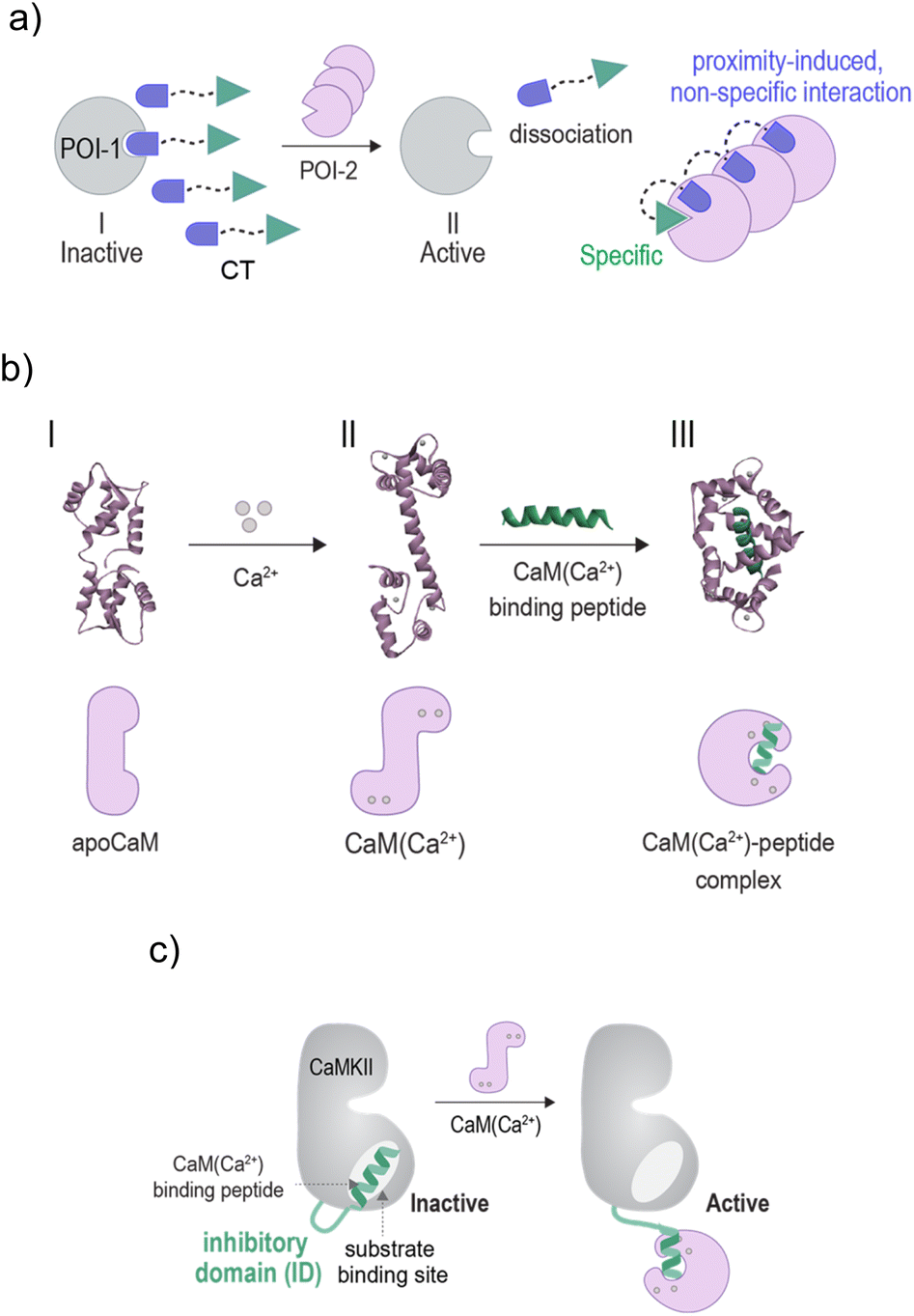 | ||
| Fig. 1 Schematic illustration of the (a) mechanism underlying CT-mediated artificial protein crosstalk. (b) Top: crystal structures indicating the conformational changes that occur on CaM upon binding to calcium ions (I → II) and subsequently, to a mastoparan peptide (II → III) (PDB: 1CFC,1CLL, and 1CDL). Bottom: representation of the different structural states. (c) A schematic illustration showing how the binding of CaM(Ca2+) to the inhibitory domain (ID) of CaMKII leads to the activation of CaMKII. | ||
In our previous research, two classes of CT molecules were developed. With the first,19 we demonstrated the feasibility of establishing unnatural protein effector-mediated enzyme activation in vitro, as well as showcased its potential for prodrug activation by non-enzyme disease markers. A similar approach was recently used to create a triggerable biopharmaceutical release system.18 With the second transducer class,20 such artificial enzyme activation was established within native (non-engineered) cells. Although these developments bring us closer to rewiring intracellular signaling events, the artificial CT-mediated enzyme activation demonstrated thus far19,20 was triggered by an increase in the concentration19 or the expression levels20 of the non-native protein activators. Although important, a limitation of this activating mode is that it represents just one, relatively slow mechanism by which signals are transduced within cells. Moreover, when considering the application of artificially regulated enzymes in an extracellular environment,9 achieving reversibility becomes challenging due to the difficulty of removing the unnatural protein ‘effector’ (POI-2) from the solution.
To broaden the scope of CT-mediated enzyme activation, and potentially allow faster response rates and reversible control over enzyme function, we examined additional ways by which enzymes are activated during cellular signaling.23 A common activation mode, which does not rely on changes in the protein expression levels, is based on regulatory proteins that act as conformational switches.23,24 In response to upstream signals, these proteins undergo rapid conformational changes that enable them to bind to downstream signaling enzymes and activate them. We postulated that endowing a regulatory switch with the ability to activate an unrelated enzyme would demonstrate the wide applicability of CTs in controlling the function of proteins by creating artificial regulatory links between them. Furthermore, given our recent demonstration of expression-dependent, artificial protein crosstalk in cells,20 it would demonstrate an initial step towards utilizing CTs to dynamically reconfigure rapidly evolving cell signaling pathways.
Herein, we present a third class of chemical transducers, denoted as CT-3, which mediate, in vitro, an unnatural regulatory connection between the calcium sensor calmodulin (CaM) and glutathione S-transferase (GST). We show that this artificial linkage makes GST's activity unnaturally dependent on the CaM conformational state and consequently, on the calcium ion levels. Given that CaM activates a variety of intracellular enzymes in a calcium-dependent manner, and that the activity of GST does not naturally rely on calcium ions or calcium-dependent CaM structural changes, these findings highlight the versatile nature of the CT technology and, in particular, its potential use in rewiring the regulatory function of enzyme modulating proteins. By switching the activity of GST on and off using Ca2+ and an EDTA chelator, we also demonstrated how reversible, metal-dependent artificial enzyme regulation can be uniquely achieved, without introducing unnatural metal binding sites to the enzyme, cofactor, inhibitor, or substrates. Finally, within this study, we also investigate the impact of covalent interaction between the CTs and the enzyme target. This investigation offers further insights into the mechanisms governing the function of CTs and the possibility of establishing a new mode by which artificial enzyme activation could be rendered isoform dependent.7
Results and discussion
To demonstrate the possibility of reconfiguring the enzyme modulation abilities of regulatory proteins, we aimed to establish unnatural communication between CaM and GST. CaM is a signaling protein found in all eukaryotic cells, functioning as a primary calcium sensor. In the presence of calcium ions (Ca2+), CaM adopts a dumbbell-like conformation (Fig. 1b, I → II) and exposes a hydrophobic patch, enabling it to bind to distinct peptides like mastoparan25 (II → III), as well as activate various enzymes as a result of these interactions. For example, the binding of CaM(Ca2+) (calcium-bound CaM) to the ID of calcium/calmodulin-stimulated protein kinase II (CaMKII) liberates the ID from the active site, resulting in CaMKII activation26,27 (Fig. 1c). Enzyme activation by CaM(Ca2+) plays major role in the regulation of various cellular processes such as proliferation and apoptosis.28 GST is a detoxification enzyme29 that operates independently of CaM or calcium. Its primary function is to catalyze the conjugation of electrophilic substrates to glutathione, facilitating their neutralization and excretion. Its activity can be disrupted by small-molecule inhibitors such as bis-ethacrynic amides (bis-EAs).30,31Based on these properties, namely, the constitutive activity of GST, the calcium-dependent conformation of CaM, the ability of CaM to activate enzymes by binding to their IDs (Fig. 1c),26,27 the unrelated functions of CaM and GST, and the fact that these proteins can be targeted by small-molecule or peptide binders, we thought that these proteins would serve as excellent candidates for testing the concept of rewiring enzyme activation by regulatory proteins. Specifically, we anticipated that linking bis-EA to a CaM(Ca2+) binding peptide (Fig. 2a) could result in a new class of CTs (CT-3a and CT-3b) serving as sIDs of GSTs (Fig. 2b, step I). This would make the activity of GSTs unnaturally dependent on calcium-induced CaM conformational changes (Fig. 2b, step II). Another reason for choosing GST as the artificially regulated enzyme is the ability of EAs to distinctly inhibit GST-M1 and the thiol-containing GST-P1 isozymes. Whereas EAs non-covalently bind GST-M1, GST-P1 is covalently inhibited by EAs.32 We anticipated that this difference would provide a powerful tool for investigating the mechanism underlying the CTs function, while also demonstrating a way to render it isoform specific.
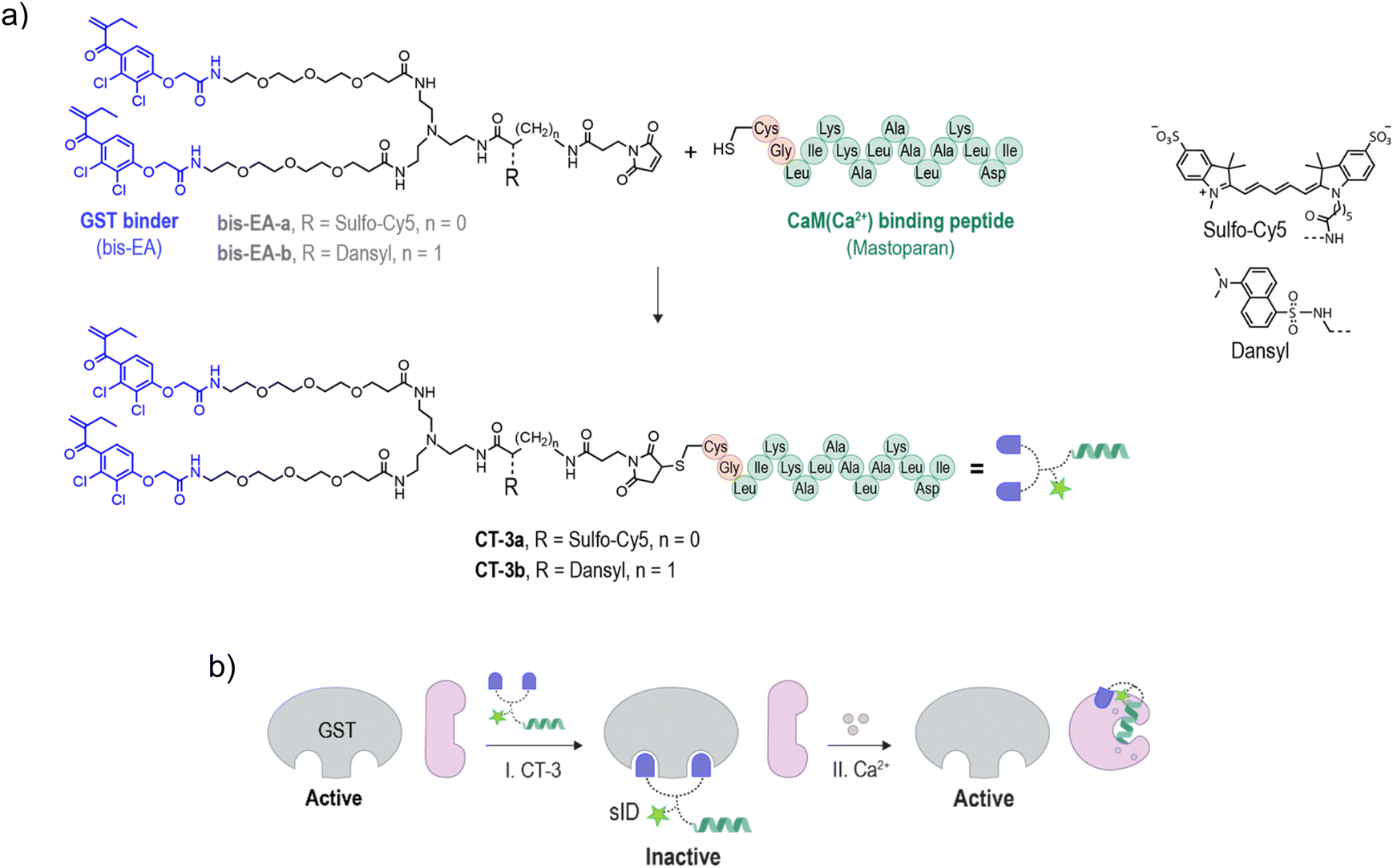 | ||
| Fig. 2 (a) Structures of CT-3a and CT-3b, generated by conjugating a GST binding molecule (bis-EA-a and bis-EA-b) to a CaM(Ca2+) binding peptide (Cys-Gly-modified mastoparan). (b) Operating principles of the CTs: upon binding to GST, CT-3 acts as a synthetic inhibitory domain (sID) (step I) with which the constitutively active enzyme is transformed into an activable enzyme whose activity is triggered by a calcium-induced CaM conformational change (step II). | ||
The structure of the CTs (Fig. 2a, CT-3a and CT-3b) integrates a bis-EA derivative and a 14-mer mastoparan peptide, which have been shown to bind with low nanomolar affinities to GST30,31 and CaM(Ca2+),33 respectively. Mastoparan has been elongated with two additional amino acids, namely, glycine (Gly) and cysteine (Cys), to enable its linkage to a maleimide-modified bis-EA inhibitor. To facilitate the investigation of their binding interactions, the CTs were also modified with distinct fluorescent reporters. CT-3a bears an ‘always on’ sulfo-cyanine 5 (sCy5) dye. sCy5 is one of the most popular dyes for studying biomolecular interactions, owing to its water solubility compatibility with various analytical instrumentations, such as plate readers and imagers. To probe proximity-induced interactions between the CT and the surface of CaM(Ca2+), CT-3b was equipped with an environmental responsive solvatochromic dansyl dye, previously used by our group to sense proximity-induced interactions with CaM(Ca2+).34 Another reason for choosing these dyes is the polarity difference between them. In our previous study, we demonstrated that hydrophobic groups contribute to the formation of proximity-induced interactions with CaM(Ca2+), whereas negatively charged groups disrupt these interactions.34 Therefore, we hypothesized that the difference between the negatively charged sCy5 and the hydrophobic dansyl might impact the ability of the CTs to mediate CaM-GST crosstalk (Fig. 2b). In this study, the bis-EA units used to construct the CTs (Fig. 2a, bis-EA-a and bis-EA-b) also served as control compounds. These compounds lack the mastopapran peptide; therefore, they are not expected to mediate a CaM-GST crosstalk.
Following the synthesis of CT-3a and CT-3b (ESI, Section 2†), a GST activity assay that follows the conjugation of glutathione to 1-chloro-2,4-dinitrobenzene (ESI, Section 3†) was used to determine whether the CTs bind to the GST-M1 isoform (Fig. 3a). Incubation of GST-M1 with increasing concentrations of CT-3a (Fig. 3a, left) or CT-3b (right) resulted in dose-dependent inhibition curves, from which the inhibitory constants for the CT-GST-M1 complexes were determined as Ki = 2.7 ± 0.4 nM and 1.6 ± 0.5 nM, respectively. Notably, the CTs exhibit greater inhibitory potency towards the enzyme than the does the monovalent EA inhibitor (Ki = 1–5 μM)30,31 (ESI, Fig. S1†), as expected from a bivalent CT-GST interaction (Fig. 2b).
 | ||
| Fig. 3 (a) Representative inhibition curves that follow changes in the activity of GST-M1 (10 nM) upon incubation with increasing concentrations of CT-3a (left) or CT-3b (right). (b) Fluorescence anisotropy curve generated by subjecting CT-3a (50 nM) to increasing concentrations of CaM(Ca2+) or apoCaM. (c) Fluorescence response of CT-3b (400 nM) to the addition of increasing concentrations of CaM(Ca2+) or apoCaM. Excitation: 340 nm. (d) Fluorescence emission spectra of CT-3b (400 nM) before (black line) and after the addition of apoCaM (1 μM, grey line), CaCl2 (0.3 mM, dashed grey line), or both (green line). | ||
To determine the affinity of CT-3a for CaM(Ca2+), it was incubated with increasing concentrations of CaM in both the absence and presence of CaCl2, and the change in anisotropy was measured (Fig. 3b). The resulting binding curve (black line) revealed a Kd value of 31 ± 8 nM for the CT-3a-CaM(Ca2+) interaction. This experiment also confirmed that CT-3a does not interact with apoCaM, namely, CaM without bound calcium ions (Fig. 3b, blue). To determine the affinity of CT-3b for CaM(Ca2+) (Fig. 3c), we monitored the changes in the fluorescence emission spectra of its dansyl dye (Fig. 3d). Inspecting the emission spectrum of CT-3b following the addition of CaM(Ca2+) revealed an enhancement and a blue shift in the emission signal (Fig. 3d, green line), indicating the creation of a hydrophobic environment for the solvatochromic dye.35 Control experiments in which CT-3b was incubated with only apoCaM (grey line) or Ca2+ (dashed grey line), did not significantly impact the fluorescence signal of CT-3b. The fact that dansyl dye, despite its distance from the mastoparan peptide, is in contact with the surface of CaM(Ca2+) (Fig. 3d), supports the hypothesis underlying CT function. Specifically, it shows that the binding of the CT to its protein target could promote secondary, non-specific interactions with this protein, which could ultimately disrupt the CT's binding to a second protein. The fluorescence response of CT-3b to CaM(Ca2+) was used to construct a binding curve (Fig. 3c, black line), from which a Kd approx value of 16 ± 7 nM was deduced. Collectively, these measurements confirm that the transducers bind to the individual protein targets. It should be noted that, owing to the engagement of the CTs in non-specific, proximity-induced interactions, such as those observed for the dansyl dye, the binding affinities for the CT-protein interactions themselves cannot be applied to accurately predict the efficiencies of different CTs.
Next, we determined whether the CTs can mediate artificial, CaM-GST crosstalk, in which GST is unnaturally activated by the addition of calcium ions (Fig. 2b, step II). To this end, the activity of GST-M1 was measured before and after incubation with either CT-3a (Fig. 4a, left) or CT-3b (Fig. 4a, right), apoCaM, Ca2+, and a combination thereof. The results indicate that in the presence of the CTs (grey solid line), or CTs and apoCaM (black line), GST-M1 activity was significantly inhibited. However, upon introducing Ca2+ to the inhibited GST-M1 (CT-bound GST-M1) in the presence of apoCaM, a substantial recovery of enzymatic activity was observed (blue line), with the CT-3b-inhibited enzyme showing a remarkable 100% reactivation (Fig. 4a, right). The partial reactivation of CT-3a-inhibited GST-M1 (67%), compared with the full reactivation of CT-3b-inhibited GST-M1, may be due to a preferable proximity-induced interaction between the more hydrophobic CT-3b and CaM(Ca2+). To support our hypothesis that the bifunctionality of the CTs is essential for achieving Ca2+-induced activation of GST-M1, we performed a similar experiment with bis-EA-a and bis-EA-b, which lack the mastoparan peptide (Fig. 4b). As expected, almost no change in the activity of bis-EA-inhibited GST-M1 was observed upon the addition of apoCaM or apoCaM and calcium, with the slight increase in activity likely resulting from non-specific interactions.
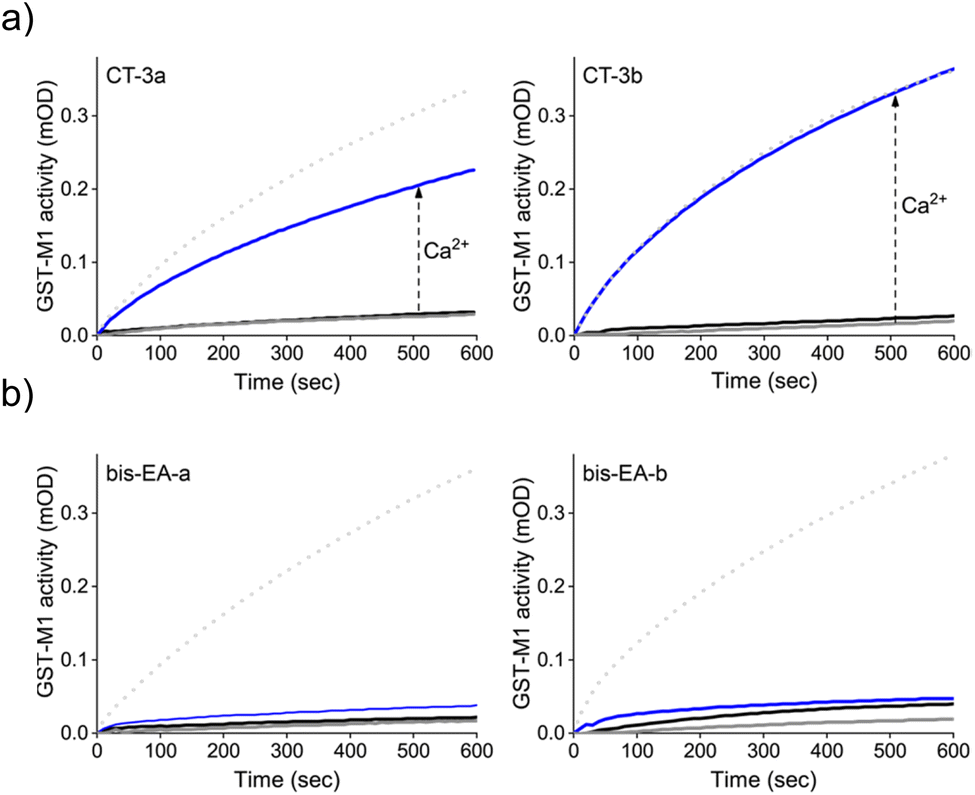 | ||
| Fig. 4 (a) Activity of GST-M1 (10 nM) before (dotted line) and after (grey line) the addition of 200 nM of CT-3a (left) or CT-3b (right) and after subsequent additions, of first, apoCaM (1 μM, black line) and then, Ca2+ (0.3 mM, blue line). (b) Results of a similar experiment performed with control compounds, bis-EA-3a (left) and bis-EA-3b (right), which lack the CaM(Ca2+) binding peptide (mastoparan). | ||
To further validate the mechanism underlying the CT-mediated regulation of GST by CaM, we also followed the binding of the CTs to the thiol-containing GST-P1 isoform and determined the effect of CaM(Ca2+) on this interaction (Fig. 5). EA-based inhibitors have been shown to form covalent bonds with GST-P1.32 Therefore, we anticipated that this covalent interaction would prevent the dissociation of the CT-GST-P1 complexes and the consequent GST activation. Initially, we determined the ability of the CTs to bind to and inhibit this isoform by measuring GST-P1 activity following incubation with increasing concentrations of the CTs (Fig. 5a). The resulting inhibition curves show that CT-3a and CT-3b inhibit GST-P1 with IC50 approx values of 140 ± 20 nM, and 144 ± 17, respectively. To confirm that the CTs form a covalent bond with GST-P1, we measured the mass of GST-P1 before and after incubation with CT-3a (Fig. 5b, top). As a control, we also measured the mass of GST-M1 following treatment with the same CT (Fig. 5b, bottom). The spectra reveal that whereas the mass of GST-M1 remained unchanged, the mass of GST-P1 (monomer or dimer) increased by 3625 Da, corresponding to the calculated mass of the CT-3a adduct. In the next step, we determined the effect of CaM(Ca2+) on the activity of CT-bound GST-P1 (Fig. 5c). The results show that GST-P1 activity was not regenerated following incubation with CaM and Ca2+. Thus, this control experiment provides additional evidence that the previously observed reactivation of GST-M1 (Fig. 4a) by calcium resulted from a CaM(Ca2+)-mediated dissociation of the CTs from GST-M1.
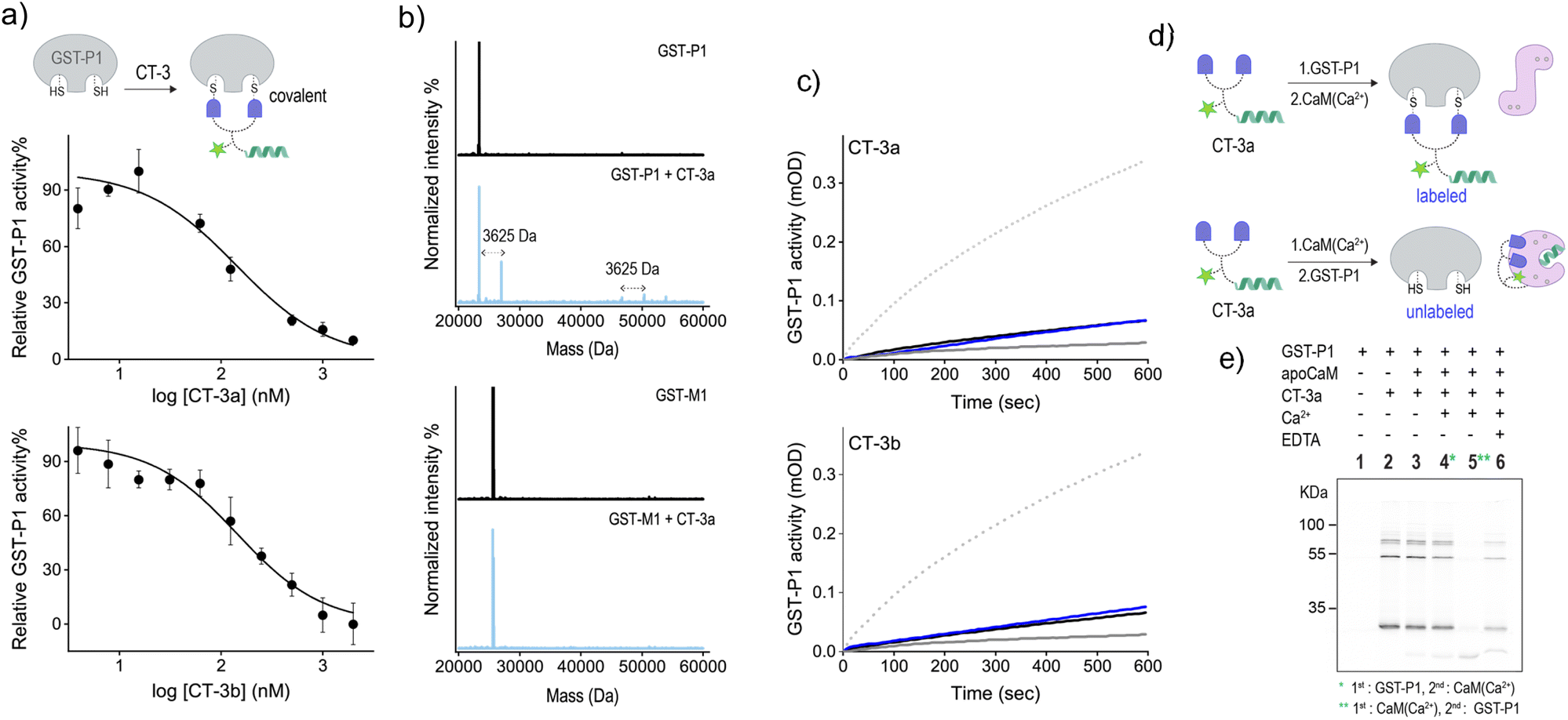 | ||
| Fig. 5 (a) Activity of GST-P1 (20 nM) in the presence of increasing concentrations of CT-3a (top) or CT-3b (bottom). (b) MS deconvoluted spectrum for GST-P1 (1.25 μM) (top) or GST-M1 (bottom) before (black) or after (pale blue) the addition of CT-3a (2.5 μM). (c) Activity of GST-P1 (20 nM) before (dotted line) and after (grey line) the addition of 400 nM of CT-3a (top) or CT-3b (bottom) and after subsequent additions of apoCaM (1 μM. black line) and Ca2+ (0.3 mM, blue line). (d) Schematic illustration of the expected labelling of GST-P1 following incubation with first CT-3a and then with CaM(Ca2+) (top) or following incubation with first CaM(Ca2+) and then GST-P1 (bottom). (e) In-gel fluorescence imaging of the bands formed following the incubation of CT-3a (300 nM) with different combinations of GST-P1 (300 nM), apoCaM (300 nM), Ca2+ (0.09 mM) and EDTA (9 mM). The image was captured by exciting sCy5 at 635 nm. | ||
Besides serving as a control isoform, an advantage of using GST-P1 to investigate CaM-GST communication is the ability to track the formation of GST-P1-CT complexes using fluorescence gel imaging. Unlike the previous experiments, in which the CT-GST interactions were determined indirectly by observing inhibition of the catalytic activity, SDS-PAGE visualization provides the means to follow the formation of the CT-GST-P1 complex without using colorimetric substrates. We anticipated that, in line with enzymatic activity measurements (Fig. 4a and 5c), incubating the CT with GST-P1 first and then with CaM(Ca2+) would result in covalent fluorescent labeling of GST-P1 (Fig. 5d, top), which would enable visualizing the CT-GST-P1 complex using in-gel fluorescence imaging. However, if CaM(Ca2+) indeed disrupts the CT-GST interaction, then subjecting the transducer to CaM(Ca2+) before incubating it with GST-P1 (Fig. 5d, bottom) should prevent the covalent labeling of GST-P1 and, consequently, the visualization of gel bands.
Fig. 5e shows the fluorescence imaging of the bands formed by mixing CT-3a with different combinations of GST-P1, CaM, Ca2+ and EDTA. The covalent binding of the transducer to GST-P1 was verified by the appearance of bands at around 25 kDa and 50 kDa (lane 2), indicating the binding of CT-3a to a GST-P1 monomer or dimer, respectively. A weaker band at 75 kDa also appeared. This may indicate that two CT molecules connect a dimer and a monomer by reacting with an additional cysteine located outside the catalytic site. The in-gel imaging also showed that the same fluorescence pattern was obtained following the addition of apoCaM (lane 3) or apoCaM and Ca2+ (lane 4) to a mixture containing CT-3a and GST-P1. This indicates that the calcium-induced conformational change of apoCaM into CaM(Ca2+) did not trigger the dissociation of CT-3a from GST-P1, as expected from the covalent interaction between the two (Fig. 5d, top). However, when CT-3a was incubated with apoCaM and Ca2+ prior to the addition of GST-P1, the bands corresponding to the CT-3a-GST-P1 complex disappeared (Fig. 5e, lane 5). This observation confirms that the binding of CaM(Ca2+) to CT-3a prevents the CT from binding to GST-P1, in accordance with our hypothesis (Fig. 5d, bottom). This last experiment was then repeated in the presence of EDTA (Fig. 5e, lane 6). Specifically, after subjecting CT-3a to apoCaM, Ca2+ and then to GST-P1, EDTA was added. EDTA has been shown to bind to the Ca2+ ions of CaM(Ca2+) and to shift the equilibrium toward the formation of an apoCaM conformation.36 Thus, we expected that in the presence of EDTA, CaM would no longer be able to bind to the CT, leading to CT release and its covalent binding to GST-P1. The reappearance of a band corresponding to a CT-3a-GST-P1 complex (lane 6) confirmed this hypothesis, providing additional evidence that the CT-GST interaction depends on the Ca2+-induced conformational change in CaM. Collectively, the gel mobility data support a fundamental aspect of the CT function, namely, that although a CT can strongly bind to each of its targets, it cannot bind to them at the same time.
The simplicity by which metal–ligand interactions can be reversibly controlled, for example, by adding a competing chelator or changing oxidation states, makes metal ions attractive chemical signals for achieving unnatural enzyme regulation.8,11 The observation that the addition of EDTA triggers the liberation of CT-3a from calmodulin and the subsequent reformation of a CT-3a-GST-P1 complex (Fig. 5e, lane 6) suggests the potential for establishing a novel method to artificially regulate enzyme activity by modulating the coordination of metal ions. Specifically, the gel mobility data (Fig. 5e) indicate that, unlike other artificially regulated enzymes where metal-dependent control over enzyme activity was achieved by modifying an enzyme8 or an unnatural cofactor11 with a metal chelating agent, with our approach such control might be attained by rewiring of metal-responsive regulatory proteins.
To determine whether the CT-3-mediated activation of GST could be dynamically controlled by subjecting the system to Ca2+ and EDTA, we first determined the effect of EDTA on the activity of an unnaturally activated GST-M1, namely, GST-M1 subjected to CT-3b and then CaM(Ca2+) (Fig. 6a). The results indicate that, in accordance with the observed exchange of binding partners (Fig. 5e), the addition of EDTA led to re-inhibition of GST-M1. In the next step, we determined whether the indirect response of GST-M1 to Ca2+ and EDTA could also be observed in real time, specifically by introducing them while monitoring the activity of GST (Fig. 6b). To this end, GST-M1 was placed in two wells and incubated with CT-3b, CaM, colorimetric substrates and the catalytic activity of the enzyme in both wells was followed (Fig. 6b, top). After 210 s, CaCl2 was added to only one of the wells and the measurement continued. The results show that the addition of calcium ions led to an immediate GST reactivation. Similarly, we have shown that the addition of EDTA after 210 s rapidly reversed the unnatural, calcium-mediated GST-M1 activation (Fig. 6b, bottom). The reversibility of the system was further demonstrated by monitoring the response of GST-M1 to the sequential additions of Ca2+ and the more effective Ca2+ chelator, EGTA, resulting in an inhibition–activation cycle (Fig. 6c). Mastoparan binding has been shown to increase the affinities of CaM to Ca2+.37 Therefore, we anticipated that EGTA would more efficiently remove calcium from the CaM(Ca2+)-CT complex compared to EDTA. Only a few activation-inhibition steps were obtained (ESI, Fig. S2†) due to the accumulation of CaM's modulating signals (Ca2+ and EGTA) in the solution, a phenomenon that does not occur in cell signaling. At high concentrations (step VIII), these species begin to affect the activity of GST (ESI, Fig. S3†) and possibly other components in the system, hindering GST reactivation and a direct evaluation of the CT-mediated GST response. Notably, this unnatural regulation of GST activity was achieved without introducing chemical or genetic modifications to GST or CaM, demonstrating the feasibility of reversibly controlling enzyme function by establishing an artificial link with a metal-responsive regulatory protein.
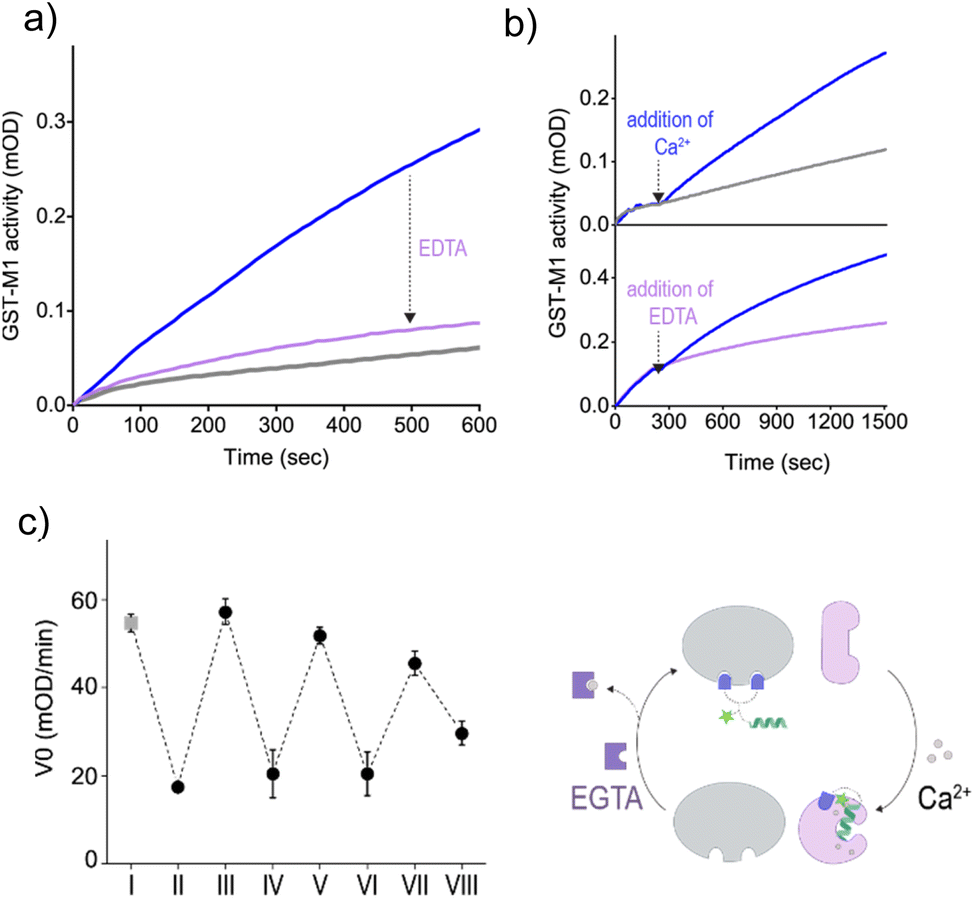 | ||
| Fig. 6 (a) Activity of GST-M1 (10 nM) in the presence of CT-3b (200 nM) and apoCaM (1 μM) before (grey line) and after the addition of CaCl2 (0.3 mM, blue line), and after subsequent addition of EDTA (3 mM. purple line). (b) Changes in the activity of CT-3b-inhibited GST-M1 upon the addition of CaCl2 (0.3 mM) and EDTA (3 mM) at t = 210 s. Concentrations: 10 nM GST-M1, 200 nM CT-3b (c) initial velocity (V0) measured before (I) and after (II) the addition of CT-3b to GST-M1 in the presence of CaM, followed by sequential additions of (III) Ca2+ (20 μM), (IV) EGTA (80 μM), (V) Ca2+ (100 μM), (VI) EGTA (400 μM), (VII) Ca2+ (500 μM), and (VIII) EGTA (3 mM). | ||
Experimental
All the experimental details are provided in the ESI.†Conclusions
To summarize, we have demonstrated the feasibility of establishing, in vitro, an artificial regulatory link between a GST enzyme and the calcium sensor protein CaM through the rational design of suitable CT molecules. It was shown that in the presence of CT-3 the constitutively active GST is converted into an activatable enzyme. Concurrently, the CaM is transformed into an unnatural effector protein, rendering GST activity artificially dependent on the Ca2+-mediated CaM's conformational state. Given that in nature, GST is not among the enzymes activated by CaM, these results demonstrate a potential new mode of artificial enzyme modulation which is based on reconfiguring the enzyme activation abilities of regulatory proteins.Accordingly, we believe that this development could contribute to the future development of artificially activated enzymes in various ways. First, it aligns well with our vision of utilizing CTs to mediate non-natural cell signaling processes.19,20 Given our recent demonstration of CT-mediated artificial protein crosstalk within native cells,20 the unnatural regulatory link between CaM and GST indicates the possibility of creating alternative classes of artificial enzyme activation steps in which one protein's activity would be artificially linked to the conformational states of another. This should expand the choice of proteins that could serve as unnatural effectors, as well as accelerate artificial protein crosstalk in cells by bypassing the dependence on slow effector expression.20 Another unique feature of the CTs is their ability to afford artificial, metal-dependent control of enzyme activity without introducing an unnatural metal binding site to the components that play a role in the catalysis. Considering the prevalence of protein switches that mediate signal transduction steps in a metal-dependent manner,38,39 this approach should provide the means to unnaturally activate proteins by other metal ions, such as copper or zinc. This versatility could open the way to parallelly controlling the unnatural activation of distinct enzymes as well as establishing more robust enzyme activation systems capable of undergoing multiple activation-inhibition cycles. Finally, the inability to establish communication between CaM and GST-P1 isoform indicates the feasibility of introducing isoform specificity to CT-mediated artificial protein crosstalk. Interestingly, although covalent modification of catalytic cysteines has been demonstrated as a powerful method for converting enzymes into activable enzymes (i.e., chemical zymogens),7 with the CTs, this covalent interaction yields the opposite effect. Specifically, only isoforms lacking a catalytic cysteine can be converted into artificially triggered enzymes using the CTs.
Collectively, we expect that these features—rewiring enzyme activation by regulatory proteins, obtaining a novel mode of artificial, metal-dependent enzyme regulation, and demonstrating isoform-dependent artificial protein crosstalk—could expand the current chemical toolbox used to create artificially activated enzymes and, consequently, broaden their application in various research fields.
Data availability
Data for this paper, including synthetic procedures, compound characterization, and experimental details, are available at the ESI section.†Author contributions
The manuscript was written through contributions of all authors. All authors have approved the final submitted version.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This research was supported by the Minerva Foundation, grant no. 714437.References
- K. Singh, B. Bhushan, N. Mittal, A. kushwaha, K. C. Raikwar, K. A. Sharma, K. D. Chanchal, S. Kumar and M. Agrawal, Recent advances in enzyme inhibition: A pharmacological review, Curr. Enzym. Inhib., 2024, 20, 1–18 CrossRef.
- A. Saghatelian, K. M. Guckian, D. A. Thayer and M. R. Ghadiri, DNA detection and signal amplification via an engineered allosteric enzyme, J. Am. Chem. Soc., 2003, 125, 344–345 CrossRef CAS PubMed.
- R. Singh, W. A. Blattler and A. R. Collinson, An amplified assay for thiols based on reactivation of papain, Anal. Biochem., 1993, 213, 49–56 CrossRef CAS PubMed.
- E. Arbely, J. Torres-Kolbus, A. Deiters and J. W. Chin, Photocontrol of tyrosine phosphorylation in mammalian cells via genetic encoding of photocaged tyrosine, J. Am. Chem. Soc., 2012, 134, 11912–11915 CrossRef CAS PubMed.
- Z. Liu, V. Lebrun, T. Kitanosono, H. Mallin, V. Köhler, D. Häussinger, D. Hilvert, S. Kobayashi and T. R. Ward, Upregulation of an artificial zymogen by proteolysis, Angew. Chem., Int. Ed., 2016, 55, 11587–11590 CrossRef CAS PubMed.
- D. V. D. W. Kankanamalage, J. H. T. Tran, N. Beltrami, K. Meng, X. Zhou, P. Pathak, L. Isaacs, A. L. Burin, M. F. Ali and J. Jayawickramarajah, DNA strand displacement driven by host–guest interactions, J. Am. Chem. Soc., 2022, 144, 16502–16511 CrossRef CAS PubMed.
- M. C. Montasell, P. Monge, S. Carmali, L. M. Dias Loiola, D. G. Andersen, K. B. Løvschall, A. B. Søgaard, M. M. Kristensen, J. M. Pütz and A. N. Zelikin, Chemical zymogens for the protein cysteinome, Nat. Commun., 2022, 13, 4861 CrossRef CAS PubMed.
- Y. S. Zubi, K. Seki, Y. Li, A. C. Hunt, B. Liu, B. Roux, M. C. Jewett and J. C. Lewis, Metal-responsive regulation of enzyme catalysis using genetically encoded chemical switches, Nat. Commun., 2022, 13, 1864 CrossRef CAS PubMed.
- C. Claaßen, T. Gerlach and D. Rother, Stimulus-responsive regulation of enzyme activity for one-step and multi-step syntheses, Adv. Synth. Catal., 2019, 361, 2387–2401 CrossRef PubMed.
- H. J. Davis and T. R. Ward, Artificial metalloenzymes: Challenges and opportunities, ACS Cent. Sci., 2019, 5, 1120–1136 CrossRef CAS PubMed.
- D. J. Raines, J. E. Clarke, E. V. Blagova, E. J. Dodson, K. S. Wilson and A.-K. Duhme-Klair, Redox-switchable siderophore anchor enables reversible artificial metalloenzyme assembly, Nat. Catal., 2018, 1, 680–688 CrossRef CAS.
- X. Li, Y. Wei, Y. Wu and L. Yin, Hypoxia-induced pro-protein therapy assisted by a self-catalyzed nanozymogen, Angew. Chem., Int. Ed., 2020, 59, 22544–22553 CrossRef CAS PubMed.
- D. Y. W. Ng, M. Arzt, Y. Wu, S. L. Kuan, M. Lamla and T. Weil, Constructing hybrid protein zymogens through protective dendritic assembly, Angew. Chem., Int. Ed., 2014, 53, 324–328 CrossRef CAS PubMed.
- M. Wang, S. Sun, C. I. Neufeld, B. Perez-Ramirez and Q. Xu, Reactive oxygen species-responsive protein modification and its intracellular delivery for targeted cancer therapy, Angew. Chem., Int. Ed., 2014, 53, 13444–13448 CrossRef CAS PubMed.
- J. Chang, W. Cai, C. Liang, Q. Tang, X. Chen, Y. Jiang, L. Mao and M. Wang, Enzyme-instructed activation of pro-protein therapeutics in vivo, J. Am. Chem. Soc., 2019, 141, 18136–18141 CrossRef CAS PubMed.
- J. Lu, H. Wang, Z. Tian, Y. Hou and H. Lu, Cryopolymerization of 1,2-dithiolanes for the facile and reversible grafting-from synthesis of protein–polydisulfide conjugates, J. Am. Chem. Soc., 2020, 142, 1217–1221 CrossRef CAS PubMed.
- P. Mukherjee, L. J. Leman, J. H. Griffin and M. R. Ghadiri, Design of a DNA-programmed plasminogen activator, J. Am. Chem. Soc., 2018, 140, 15516–15524 CrossRef CAS PubMed.
- H. Shigemitsu, R. Kubota, K. Nakamura, T. Matsuzaki, S. Minami, T. Aoyama, K. Urayama and I. Hamachi, Protein-responsive protein release of supramolecular/polymer hydrogel composite integrating enzyme activation systems, Nat. Commun., 2020, 11, 3859 CrossRef PubMed.
- R. Peri-Naor, T. Ilani, L. Motiei and D. Margulies, Protein–protein communication and enzyme activation mediated by a synthetic chemical transducer, J. Am. Chem. Soc., 2015, 30, 9507–9510 CrossRef PubMed.
- O. Suss, O. Halfin, Z. Porat, Y. Fridmann Sirkis, L. Motiei and D. Margulies, Artificial protein crosstalk with a molecule that exchanges binding partners, Angew. Chem., Int. Ed., 2024, 63, e202312461 CrossRef CAS PubMed.
- G. M. Cooper, The Cell: A Molecular Approach, Sinauer Associates, Sunderland, MA, 2nd edn, 2000 Search PubMed.
- J. Monod, J.-P. Changeux and F. Jacob, Allosteric proteins and cellular control systems, J. Mol. Biol., 1963, 6, 306–329 CrossRef CAS PubMed.
- A. Nair, P. Chauhan, B. Saha and K. F. Kubatzky, Conceptual evolution of cell signaling, Int. J. Mol. Sci., 2019, 20, 3292 CrossRef CAS PubMed.
- J.-H. Ha and S. N. Loh, Protein conformational switches: From nature to design, Chem. –Eur. J., 2012, 18, 7984–7999 CrossRef CAS PubMed.
- N. Matsushima, Y. Izumi, T. Matsuo, H. Yoshino, T. Ueki and Y. Miyake, Binding of both Ca2+ and mastoparan to calmodulin induces a large change in the tertiary structure, J. Biochem., 1989, 105, 883–887 CrossRef CAS PubMed.
- R. Sloutsky, N. Dziedzic, M. J. Dunn, R. M. Bates, A. P. Torres-Ocampo, S. Boopathy, B. Page, J. G. Weeks, L. H. Chao and M. M. Stratton, Heterogeneity in human hippocampal CaMKII transcripts reveals allosteric hub-dependent regulation, Sci. Signal., 2020, 13, eaaz0240 CrossRef CAS PubMed.
- P. Pellicena and H. Schulman, CaMKII inhibitors: from research tools to therapeutic agents, Front. Pharmacol, 2014, 5, 21 CrossRef PubMed.
- M. W. Berchtold and A. Villalobo, The many faces of calmodulin in cell proliferation, programmed cell death, autophagy, and cancer, Biochim. Biophys. Acta Mol. Cell Res., 2014, 1843, 398–435 CrossRef CAS PubMed.
- A. M. A. Mazari, L. Zhang, Z.-W. Ye, J. Zhang, K. D. Tew and D. M. Townsend, The Multifaceted Role of Glutathione S-Transferases in Health and Disease, Biomolecules, 2023, 13, 688 CrossRef CAS PubMed.
- D. Y. Maeda, S. S. Mahajan, W. M. Atkins and J. A. Zebala, Bivalent inhibitors of glutathione S-transferase: The effect of spacer length on isozyme selectivity, Bioorg. Med. Chem. Lett., 2006, 16, 3780–3783 CrossRef CAS PubMed.
- L. Unger-Angel, B. Rout, T. Ilani, M. Eisenstein, L. Motiei and D. Margulies, Protein recognition by bivalent, ‘turn-on’ fluorescent molecular probes, Chem. Sci., 2015, 6, 5419–5425 RSC.
- J. H. Ploemen, B. van Ommen, J. J. Bogaards and P. J. van Bladeren, Ethacrynic acid and its glutathione conjugate as inhibitors of glutathione S-transferases, Xenobiotica, 1993, 23, 913–923 CrossRef CAS PubMed.
- D. A. Malencik and S. R. Anderson, High affinity binding of the mastoparans by calmodulin, Biochem. Biophys. Res. Commun., 1983, 114, 50–56 CrossRef CAS PubMed.
- Y. Nissinkorn, N. Lahav-Mankovski, A. Rabinkov, S. Albeck, L. Motiei and D. Margulies, Sensing protein surfaces with targeted fluorescent receptors, Chem. –Eur. J., 2015, 21, 15981–15987 CrossRef CAS PubMed.
- R. F. Chen, Fluorescence of dansyl amino acids in organic solvents and protein solutions, Arch. Biochem. Biophys., 1967, 120, 609–620 CrossRef CAS.
- M. Nirschl, J. Ottl and J. Vörös, Conformational changes of calmodulin on calcium and peptide binding monitored by film bulk acoustic resonators, Biosensors, 2011, 1, 164–176 CrossRef CAS PubMed.
- J. B. Sperry, R. Y. Huang, M. M. Zhu, D. L. Rempel and M. L. Gross, Hydrophobic peptides affect binding of calmodulin and Ca2+ as explored by H/D amide exchange and mass spectrometry, Int. J. Mass Spectrom., 2011, 302, 85–92 CrossRef CAS PubMed.
- H. Reyes-Caballero, G. C. Campanello and D. P. Giedroc, Metalloregulatory proteins: metal selectivity and allosteric switching, Biophys. Chem., 2011, 156, 103–114 CrossRef CAS PubMed.
- Z. Ma, F. E. Jacobsen and D. P. Giedroc, Coordination chemistry of bacterial metal transport and sensing, Chem. Rev., 2009, 109, 4644–4681 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02635g |
| This journal is © The Royal Society of Chemistry 2024 |