
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical and redox non-innocence in low-valent molybdenum β diketonate complexes: novel pathways for CO2 and CS2 activation†
Fabio
Masero
and
Victor
Mougel
 *
*
Laboratory of Inorganic Chemistry (LAC), Department of Chemistry and Applied Biosciences (D-CHAB), ETH Zurich, Vladimir-Prelog Weg 2, 8093 Zurich, Switzerland. E-mail: mougel@inorg.chem.ethz.ch
First published on 18th September 2024
Abstract
The investigation of fundamental properties of low-valent molybdenum complexes bearing anionic ligands is crucial for elucidating the molybdenum's role in critical enzymatic systems involved in the transformation of small molecules, including the nitrogenase's iron molybdenum cofactor, FeMoco. The β-diketonate ligands in [Mo(acac)3] (acac = acetylacetonate), one of the earliest low-valent Mo complexes reported, provide a robust anionic platform to stabilize Mo in its +III oxidation state. This complex played a key role in demonstrating the potential of low-valent molybdenum for small molecule activation, serving as the starting material for the preparation of the first reported molybdenum dinitrogen complex. Surprisingly however, given this fact and the widespread use of β-diketonate ligands in coordination chemistry, only a very limited number of low-valent Mo β-diketonate complexes have been reported. To address this gap, we explored the redox behavior of homoleptic molybdenum tris-β-diketonate complexes, employing a tertiary butyl substituted diketonate ligand (dipivaloylmethanate, tBudiket) to isolate and fully characterize the corresponding Mo complexes across three consecutive oxidation states (+IV, +III, +II). We observed marked reactivity of the most reduced congener with heterocumulenes CE2 (E = O, S), yet with very distinct outcomes. Specifically, CO2 stoichiometrically carboxylates one of the β-diketonate ligands, while in the presence of excess CS2, catalytic reductive dimerization to tetrathiooxalate occurs. Through the isolation and characterization of reaction products and intermediates, we demonstrate that the observed reactivity results from the chemical non-innocence of the β-diketonate ligands, which facilitates the formation of a common ligand-bound intermediate, [Mo(tBudiket)2(tBudiket·CE2)]1− (E = O, S). The stability of this proposed intermediate dictates the specific reduction products observed, highlighting the relevance of the chemically non-innocent nature of β-diketonate ligands.
Introduction
Molybdenum (Mo) plays a central role in a variety of enzymatic systems involved in the conversion of small molecules, particularly within the iron-molybdenum cofactor (FeMoco) of nitrogenases, and within the active site of nitrate reductase, formate dehydrogenase, carbon monoxide dehydrogenase, and sulfite oxidase enzymes.1–4 Most of these enzymatic systems operate in the higher oxidation states of Mo (+IV to +VI), and the coordination chemistry as well as the corresponding reactivities of high oxidation state Mo complexes have been extensively studied,5–13 and applied for the design of bioinspired electrocatalysts.14,15 In this context, the redox non-innocence of the molybdopterin ligand plays a key role to stabilize both high and low-valent forms of the complex16 and enable complex, multi-electron transformations.The preparation and reactivity of low-valent molybdenum compounds (+III and lower) has been comparatively underexplored, despite promising activities in a multitude of reductive transformations. Pioneering studies demonstrated the ability of tris(amide) supported Mo(III) complexes to promote dinitrogen activation and reduction.17,18 Pincer ligands were most widely employed for stabilizing low-valent oxidation states of Mo and the corresponding complexes were exploited as catalysts for hydrogenation of alkynes19 and nitriles,20 dinitrogen reduction,21–23 olefin isomerization,24 and CO2 reduction,25,26 among others. In the context of reactivity with CO2, dianionic molybdenum carbonyl – (M2[Mo(CO)6] (M = Li, Na, K)) as well as phosphine-supported complexes (e.g. cis-[Mo(N2)2(PMe3)4]) were notably found to enable the stoichiometric reductive disproportionation of CO2 to carbonate (CO32−) and metal-bound CO,27–29 while on the other hand, the reverse comproportionation reaction of Mo-bound CO and CO32− is catalyzed by a bisphosphine supported Mo complex.30 Recently, the electrocatalytic reduction of CO2 to CO mediated by [Mo(CO)6]31 and later on by bipyridine (bpy) supported molybdenum carbonyl complexes (e.g. [Mo(bpy)(CO)4],32, [Mo(η3-allyl)(NCS)(bpy)(CO)2]33) has been investigated. Reactivity studies of related Mo complexes were extended to the coupling of CO2 with ethylene under formation of acrylic acid.34–36 Bisphosphine-arene complexes were employed as a platform to study the Fischer–Tropsch related reductive coupling of CO to multi-carbon products.37,38
In most of these examples, strong π-acceptor ligands such as CO and phosphines were used to stabilize Mo in its lower oxidation states, yet also decreasing its overall reducing power. Surprisingly, the reactivity of octahedral low-valent Mo complexes with CO2 and small molecules in the absence of such strong π-acidic ligands, of high relevance to better understand and replicate the activity of Mo-based enzymatic systems,39 has not been extensively documented (Scheme 1).
 | ||
| Scheme 1 Coordination of neutral vs. anionic ligands in low-valent molybdenum complexes. | ||
Our group has recently focused on using β-diketonate supported Mo compounds as a platform to investigate the fundamental chemistry of mid- to low-valent, octahedral Mo complexes,40 building on the early report of one of the simplest homoleptic Mo(III) complexes, [MoIII(Mediket)3] (1) (Mediket = acetylacetonate), first prepared in the 1960s.41 Compared to the generally well-explored chemistry of transition metal complexes supported by β-diketonate ligands, 1 so far did not attract a lot of attention in literature. However, it played a key role in establishing the potential of low-valent molybdenum for small molecule activation by serving as the starting material for the preparation of the first reported molybdenum dinitrogen complex.42–44 Interestingly, despite the recent observation of the redox non-innocence of β-diketonate ligands in chromium diketonate derivatives,45,46 the use of β-diketonate ligands to facilitate electron transfer through non-innocent behavior, has not been extensively explored.
In the present work, we have explored the chemical and electrochemical properties of a series of homoleptic Mo β-diketonate complexes, [Mo(Rdiket)3]z (R = Me, R = tBu in oxidation states +IV, +III and +II (z = +1, 0, −1)). The latter constitutes a very rare example of a formal Mo(II) complex uniquely supported by anionic ligands.
This highly reduced complex exhibits reactivity towards both CO2 and CS2 yet with a very distinct outcome: it undergoes exclusively a stoichiometric reaction with CO2, whereas it catalytically promotes the reduction of CS2, selectively yielding tetrathiooxalate. Based on the isolation of reaction intermediates using the more soluble tBudiket ligand, we propose a reaction mechanism involving the (dithio)carboxylation of one of the diketonate ligands, demonstrating that β-diketonate ligands can exhibit both redox and chemical non-innocence.
Results and discussion
We developed here a simple, multigram scale synthesis of [Mo(Mediket)3] (1), inspired by reported syntheses,41,47 in two steps from inexpensive ammonium molybdate and under exclusively aqueous conditions (see Fig. 1a and ESI Section 2.1.1† for details). Analytical data (elemental analysis, UV-vis, magnetic susceptibility) of the obtained brown-purple crystalline solid matches previous literature reports.48 In addition, we noted that despite its paramagnetism, 1 can be easily characterized by 1H NMR (e.g. in MeCN-d3), displaying two signals with relative integral intensities of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at δ = 126.6 ppm (CH3) and 41.3 ppm (CH) respectively (see ESI Fig. S16†). The cyclic voltammetry (CV) of 1 (Fig. 2, see ESI Fig. S51–S53† for details) displays two reversible redox waves at E1/2 = −2.09 V and E1/2 = −0.21 V (all potentials are given vs. [FeIICp2]/[FeIIICp2]+ (Fc/Fc+), unless otherwise noted), which were assigned to the one electron reduction- and oxidation of 1 to the corresponding homoleptic Mo(II) and Mo(IV) complexes, respectively.
:1 at δ = 126.6 ppm (CH3) and 41.3 ppm (CH) respectively (see ESI Fig. S16†). The cyclic voltammetry (CV) of 1 (Fig. 2, see ESI Fig. S51–S53† for details) displays two reversible redox waves at E1/2 = −2.09 V and E1/2 = −0.21 V (all potentials are given vs. [FeIICp2]/[FeIIICp2]+ (Fc/Fc+), unless otherwise noted), which were assigned to the one electron reduction- and oxidation of 1 to the corresponding homoleptic Mo(II) and Mo(IV) complexes, respectively.
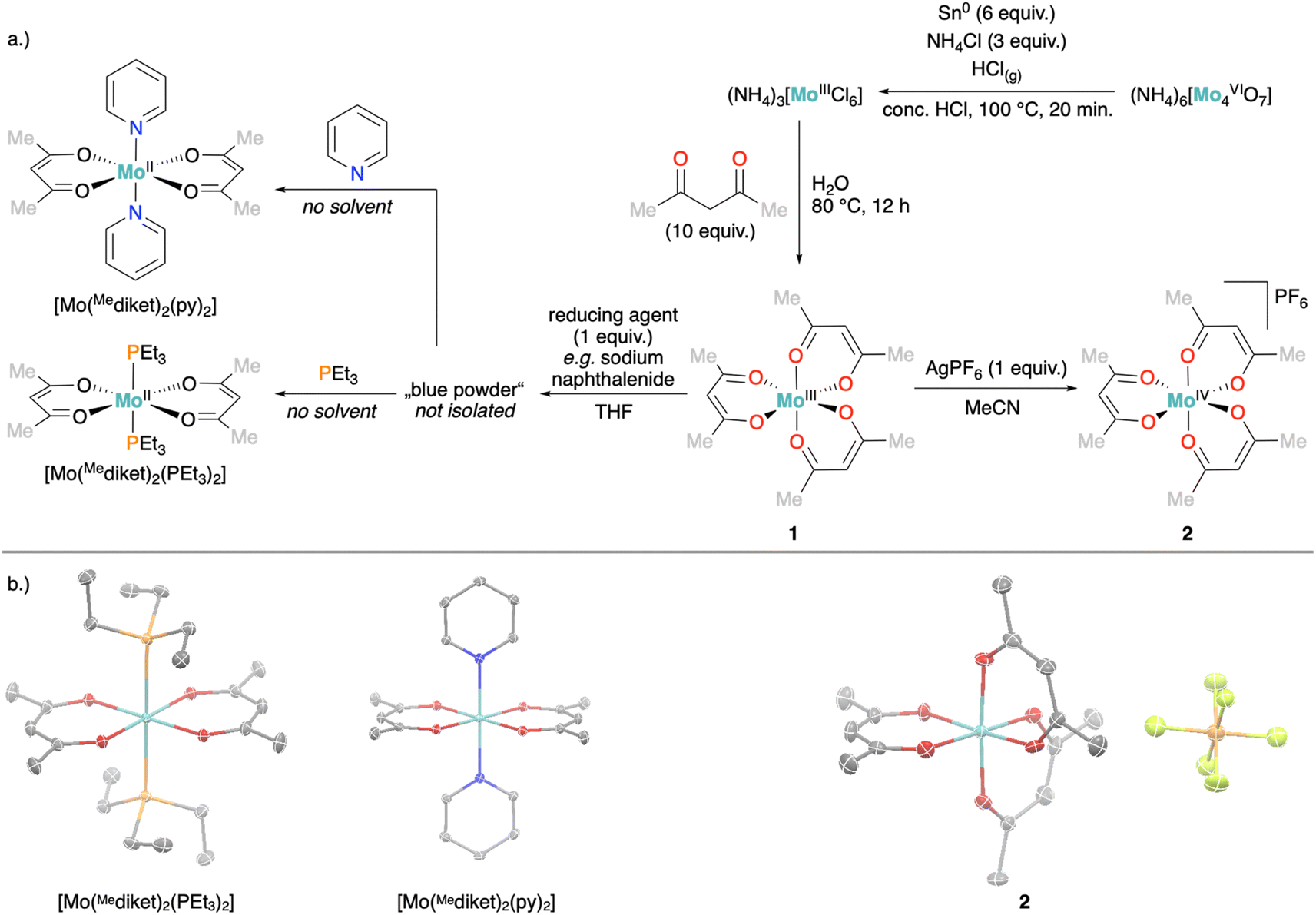 | ||
| Fig. 1 (a) Synthesis of 1 followed by its oxidation (right arrow) and reduction (left arrow), respectively. (b) Corresponding solid state structures. Thermal ellipsoids at 50% probability level are shown, co-crystallized solvent and hydrogen atoms were removed for clarity. | ||
 | ||
| Fig. 2 CV of [Mo(Mediket)3] (1) (1 mM) in TBAPF6/MeCN (0.1 M) at a scan rate of ν = 100 mV s−1. | ||
The reversibility of the redox waves in the CV studies prompted us to isolate the oxidized and reduced complexes by chemical means.
Oxidation of 1 with either [FeIIICp2][PF6] or AgPF6 afforded a dark red crystalline solid. Single crystal X-ray diffraction (XRD) analysis confirmed the successful oxidation to the expected product [Mo(Mediket)3]PF6 (2, Fig. 1b and ESI Section S4†) and the CV of 2 results in an identical trace as for 1 (see ESI Section S2.2† for details on synthesis and analytical data, and Fig. S56† for CV). On the other hand, the reduction of 1 with several alkali-metal based reducing agents (Na-naphthalene, K-naphthalene, KC8) afforded blue to green precipitates with very limited solubility and stability. Elemental analysis suggested formulations of reduced products in between M[Mo(Mediket)3] (with M = Na+ or K+) and [Mo(Mediket)2] of which the latter is known for its chromium analogue [Cr(Mediket)2].49 Dissolving the solids obtained after reduction in neat pyridine (Py), or alternatively, conducting the reduction in pyridine as solvent, resulted in an abrupt color change to deep purple. Slow diffusion of either diethyl ether or npentane led to the precipitation of a crystalline solid, which was identified as trans-[Mo(Mediket)2(py)2], further confirming the +II formal oxidation state of the reduced complex (Fig. 1). The same reactivity was observed with neat triethyl phosphine (PEt3), affording purple crystals of trans-[Mo(Mediket)2(PEt3)2] (see ESI Section 2.4† for details on synthesis and analytical data).
Aiming at increasing the solubility of the complexes in apolar solvents, we carried out an analogous set of reactions with the 2,2,6,6-tetramethylheptadionate (tBudiket) ligand, where both terminal methyl groups are permethylated. The homoleptic Mo(III) complex [Mo(tBudiket)3] (3) was prepared by an alternative synthetic route, from [Mo(CO)6] and excess ligand in refluxing toluene, due to the complete immiscibility of tBudiketH with aqueous solutions (see Fig. 4a and ESI Section S2.1.2†).
After recrystallization, 3 was isolated with a yield of 59% and its identity was confirmed by standard analytical methods (elemental analysis, single crystal XRD). In the 1H NMR spectrum of 3 (in benzene-d6), the methine protons (CH) of the ligand backbone appear at a similar chemical shift (δ = 44.1 ppm) as for 1, whereas the protons of the tBu groups show up at δ = 7.0 ppm (br) (see ESI Fig. S17 and S18†). 3 displays a similar CV as 1 with two reversible redox waves, yet shifted cathodically by 0.2 V, in line with the stronger σ-donating nature of the tBu groups (Fig. 3 and S57–S59†).
 | ||
| Fig. 3 CV of [Mo(tBudiket)3] (3) (1 mM, solid line) and [Mo(Mediket)3] (1 mM, dashed line) in TBAPF6/MeCN (0.1 M) at a scan rate of ν = 100 mV s−1. | ||
In analogy to the synthetic routes described above for 1, we were able to synthesize both the oxidized Mo(IV) complex [Mo(tBudiket)3]PF6 (4), and the reduced Mo(II) complex K[Mo(tBudiket)3] (5) (Fig. 4a).
 | ||
| Fig. 4 (a) Synthesis of 3 followed by its oxidation and reduction, respectively. (b) Corresponding solid state structures. Thermal ellipsoids at 50% probability level are shown, co-crystallized solvent and hydrogen atoms were removed for clarity. | ||
5 was isolated as a dark green crystalline solid, soluble in most common organic solvents including ethers and alkanes. Its 1H NMR spectrum shows multiple broad signals in the range of −5–13 ppm and its magnetic moment was determined to be μeff = 2.70 B.M., suggesting an overall S = 1 spin state (see ESI Fig. S33†). X-ray diffraction analysis revealed the solid-state structure displayed in Fig. 4b: the Mo-center is surrounded by three tBudiket ligands and the presence of one potassium cation per Mo suggests the formation of an anionic Mo(II) complex. In combination with elemental analysis, the obtained product can thus be described as K[Mo(tBudiket)3] (5). Interestingly, the potassium counter cation is strongly bound to the oxygen atoms of a distorted tBudiket ligand, and could not be displaced upon addition of crown ethers, while addition of [2,2,2]-cryptand did not afford any crystalline material suitable for X-ray diffraction studies (see ESI Section S4† for solid state structures involving 15-crown-5 and 18-crown-6). The use of Na-based reducing agents did not lead to clean reductions and isolation of the reduced complex, highlighting the important role of the alkali counter cation.
To the best of our knowledge, 5 only represents the second report of a divalent Mo complex bearing only anionic ligands that do not display a strong π-accepting character.50 However, the solid state structure of 5 (Fig. 4b) reveals that one of the tBudiket ligands changed its binding mode upon reduction, with one of the carbonyl groups bound in an η2-mode via its oxygen atom (dMo–O1 = 2.0401(8) Å), and carbonyl carbon (dMo–C1 = 2.1780(12) Å). The C–O bond is tilted from the plane spanned by the two adjacent carbon atoms and the second oxygen atom of the tBudiket ligand by 53.17(9)°. In addition to significantly elongated carbon–oxygen bond lengths (dC1–O1 = 1.3737(14) Å, dC3–O2 = 1.3509(14) Å), this deviation from planarity within the diketonate ligand may be regarded as an indication for its redox non-innocent behavior; the geometrical change allows for accommodation of additional electron density onto the ligand by increasing its π-accepting abilities. This metallaoxirane, or oxymetallacyclopropane motif is to the best of our knowledge unprecedented in transition metal β-diketonate coordination chemistry, but redox non-innocence of β-diketonate ligands has been recently observed for chromium diketonate derivatives e.g. [CrII(Mediket)2(py)2],45,46 and η2-CO bound ketone and aldehyde ligands have been reported in a handful of Mo complexes.51–54 While redox non-innocence has not been extensively investigated for β-diketonate ligands, it has been documented in multiple instances for closely related β-diketiminate (nacnac) transition metal complexes.55 In analogy to the metallaoxirane feature present in 5, Schrock and co-workers, structurally characterized a Mo(IV) complex supported by an η1:η2-nacnac ligand metallaaziridine structure.56 More generally, the formation of metallaoxiranes has been observed upon oxygen atom transfer to a carbene ligand,57,58via CO insertion into metal-alkyl bonds,59 or as in the present case via the direct coordination of a ketone to a low-valent metal center, mostly under reducing conditions.60–68
5 reacts with excess neutral ligands such as pyridine or tertiary phosphines (e.g. PEt3) to afford the corresponding [MoII(tBudiket)2(L2)] complexes (L = pyridine, PEt3), in exact analogy to what was observed with the Mediket complexes (see ESI Section 2.4† for details). This, together with the reversible reduction to Mo(II) observed with both 1 and 3 in CV studies, suggests that analogous anionic [MoII(Rdiket)3]− complexes are formed regardless of the ligand used.
The formation of such stable Mo(II) complexes prompted us to investigate their reactivity with CO2. When the CV of 1 is recorded in CO2-purged electrolyte, the reversibility of the Mo(III)/Mo(II) redox wave is lost and accompanied by the appearance of a small increase in peak current  at low scan rates (ν = 20 mV s−1, Fig. 5a and S54†), suggesting that the electrogenerated Mo(II) complex reacts with CO2. However, the characterization of the reaction products appeared non straightforward. Analyses of the head space and the cathodic solution after controlled potential electrolysis (CPE, see Fig. 5b and ESI Section S10.2†) did not allow for the detection of any expected CO2 reduction products, in particular no CO was observed in the gas phase, and no formic acid was detected in solution by 1H NMR, despite a ca. 2.7-fold enhancement in the passed charge in the presence of CO2. However, the strong paramagnetic nature of solutions combined with the presence of a 20-fold excess of supporting electrolyte prevented the unambiguous identification of formed products by a multitude of analytical methods (see ESI Section S10.2.1† for details).
at low scan rates (ν = 20 mV s−1, Fig. 5a and S54†), suggesting that the electrogenerated Mo(II) complex reacts with CO2. However, the characterization of the reaction products appeared non straightforward. Analyses of the head space and the cathodic solution after controlled potential electrolysis (CPE, see Fig. 5b and ESI Section S10.2†) did not allow for the detection of any expected CO2 reduction products, in particular no CO was observed in the gas phase, and no formic acid was detected in solution by 1H NMR, despite a ca. 2.7-fold enhancement in the passed charge in the presence of CO2. However, the strong paramagnetic nature of solutions combined with the presence of a 20-fold excess of supporting electrolyte prevented the unambiguous identification of formed products by a multitude of analytical methods (see ESI Section S10.2.1† for details).
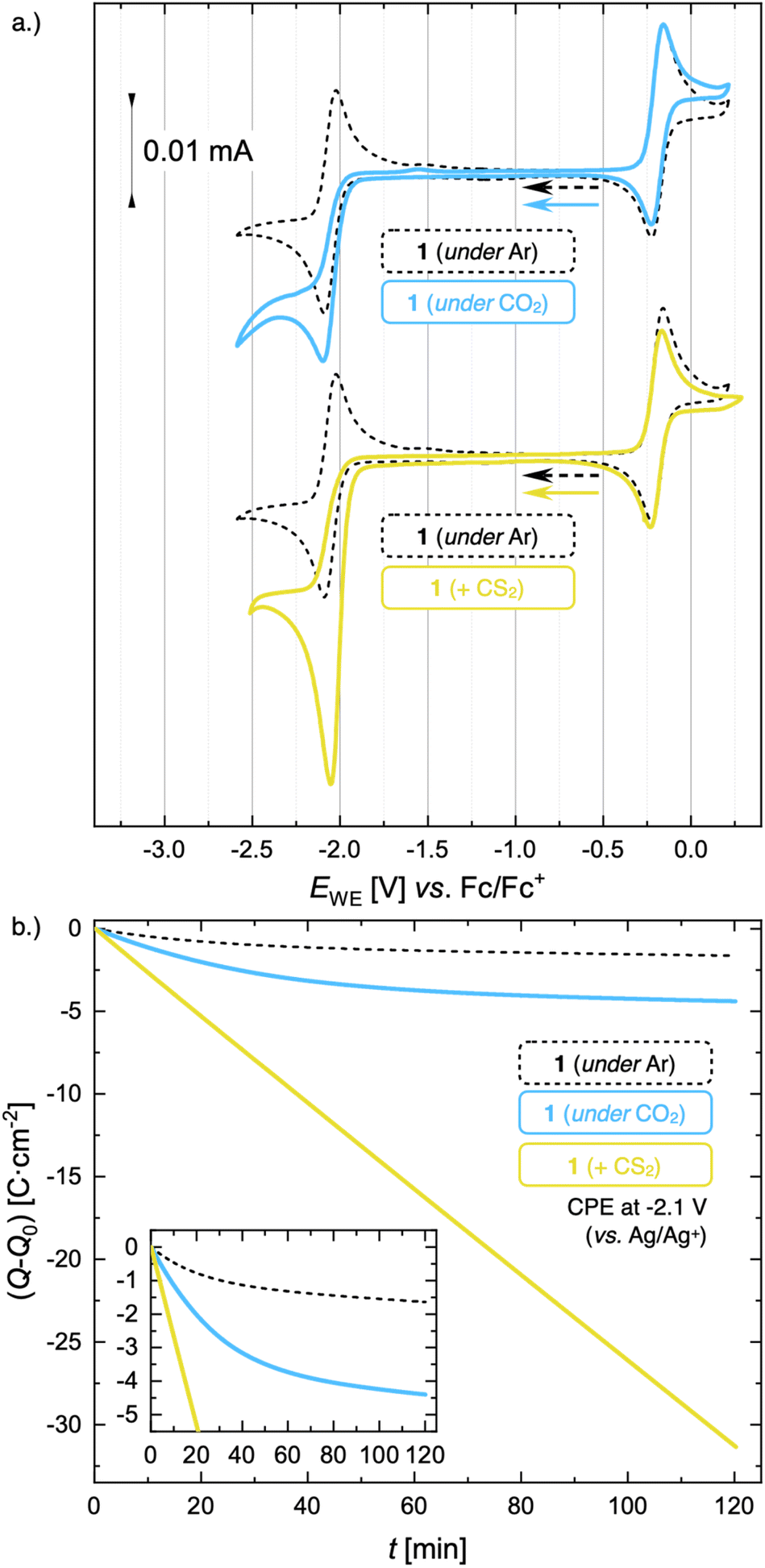 | ||
| Fig. 5 (a) CV of 1 (1 mM) under Ar (black, dashed line), under CO2 (light blue, solid line), and in the presence of CS2 (1 equiv.) in TBAPF6/MeCN (0.1 M) at a scan rate ν = 20 mV s−1. (b) Passed charge (Q − Q0) vs. time of CPE for 2 hours of 1 (5 mM) under Ar (black, dashed line), under CO2 (light blue, solid line) and in the presence of excess CS2 (56 equiv.). | ||
To gain a deeper understanding on this reactivity, we investigated the reactivity of chemically reduced 1 with CO2 (see ESI Section 3.1† for details). Analysis by 13C NMR spectroscopy (in methanol-d4) revealed two major signals at δ = 176.0 & 161.4 ppm, both of which remarkably increased in intensity when the same experiments were conducted with 13C-labelled 13CO2 (see ESI Fig. S32†), suggesting that C- or O-carboxylation of the Mediket ligand has occurred, but the functionalized complex could not be crystallized. The observed deuteration of the Mediket ligand in deuterated solvent, which has already been reported for Mediket complexes of vanadium, cobalt and aluminum,69,70 further supports the reactive nature of the ligand in the reduced complex (see ESI Section S3.3† for details).
Compared to the absence of catalytic turnover of 1 under CO2, a very different behavior was observed in the presence of CS2. The CV trace of 1 after addition of 1 equiv. of CS2 (Fig. 5a) displays a large cathodic current at −2.08 V (vs. Fc/Fc+), together with the loss of reversibility of the Mo(III)/Mo(II) redox wave, while the reversibility of the Mo(III)/Mo(IV) redox couple remains unchanged. This suggest that 1 catalyzes the reduction of CS2. Preparative electrolysis of 1 at a constant potential of −2.1 V in presence of excess CS2 (56 equiv.) resulted in a linear charge consumption of 33 C, suggesting multiple turnover. Product analysis by 13C NMR spectroscopy showed the formation of tetrathiooxalate [C2S4]2− as the main reduction product (see ESI Section S10.3† for details). Using tetraethyl ammonium tetrafluoroborate (TEABF4) as the supporting electrolyte [TEA][C2S4] was obtained as a pure product with a faradaic yield of 42%.
To better understand the different behavior of 1 in presence of either CO2versus CS2 under reducing conditions, we focused on chemical reactivity studies employing 3, given the increased solubility and stability of its reduced congener, 5. In exact analogy to 1, the 3 CV reduction wave to Mo(II) becomes irreversible in the presence of CO2 (ESI Fig. S60†), together with a small peak current enhancement (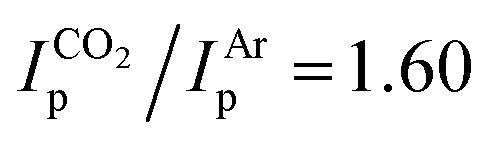 at a scan rate of ν = 20 mV s−1). However, at the difference of the less soluble Mediket analogue, the increased solubility of 5 facilitated the identification of the products resulting from its reactivity with CO2. Exposing a solution of 5 to CO2 in diethyl ether led to an abrupt color change from green to brown/red. Upon extraction with npentane, a dark green precipitate could be separated from the brown/red filtrate by centrifugation. 3 was identified as the main species in the filtrate, while a very distinct broad feature at δ = 2.39 ppm in the 1H NMR spectrum of the dark green solid (see ESI Fig. S22 and S23†) indicated the formation of a new paramagnetic species. Crystallization of this solid from saturated THF or DME solutions at ambient temperature revealed the formation of K2[Mo(tBudiket·CO2)(tBudiket)2] (6), crystallizing as a tetramer and where the CO2 moiety has been added to the central carbon of the tBudiket ligand via a new C–C bond (dC–C = 1.541(10) Å) (Fig. 6b). The modified ligand now consists of a β,β′-diketo-carboxylate. Since the negative charge of the carboxylated ligand [tBudiket·CO2]− is located on the carboxylate group, the diketo group is formally neutral. This induced a further change in its binding mode; tBudiket·CO2 coordinates as a diketone ligand to the Mo-center in an η2-η2-fashion, with both keto groups pointing into the same direction, leading to a formally 8-coordinate Mo-complex (Fig. 7c). The IR spectrum of 6 displays distinct vibrations assigned to the carboxylate group (νC–O = 1556 cm−1) and the newly formed C–C single bond (νC–C = 1371 cm−1), respectively, both of which exhibited a bathochromic shift when 13CO2 was employed during the preparation of the Mo(I) complex (13C6) (Fig. 6c, see ESI Fig. S45 and S46† for full spectra).
at a scan rate of ν = 20 mV s−1). However, at the difference of the less soluble Mediket analogue, the increased solubility of 5 facilitated the identification of the products resulting from its reactivity with CO2. Exposing a solution of 5 to CO2 in diethyl ether led to an abrupt color change from green to brown/red. Upon extraction with npentane, a dark green precipitate could be separated from the brown/red filtrate by centrifugation. 3 was identified as the main species in the filtrate, while a very distinct broad feature at δ = 2.39 ppm in the 1H NMR spectrum of the dark green solid (see ESI Fig. S22 and S23†) indicated the formation of a new paramagnetic species. Crystallization of this solid from saturated THF or DME solutions at ambient temperature revealed the formation of K2[Mo(tBudiket·CO2)(tBudiket)2] (6), crystallizing as a tetramer and where the CO2 moiety has been added to the central carbon of the tBudiket ligand via a new C–C bond (dC–C = 1.541(10) Å) (Fig. 6b). The modified ligand now consists of a β,β′-diketo-carboxylate. Since the negative charge of the carboxylated ligand [tBudiket·CO2]− is located on the carboxylate group, the diketo group is formally neutral. This induced a further change in its binding mode; tBudiket·CO2 coordinates as a diketone ligand to the Mo-center in an η2-η2-fashion, with both keto groups pointing into the same direction, leading to a formally 8-coordinate Mo-complex (Fig. 7c). The IR spectrum of 6 displays distinct vibrations assigned to the carboxylate group (νC–O = 1556 cm−1) and the newly formed C–C single bond (νC–C = 1371 cm−1), respectively, both of which exhibited a bathochromic shift when 13CO2 was employed during the preparation of the Mo(I) complex (13C6) (Fig. 6c, see ESI Fig. S45 and S46† for full spectra).
 | ||
| Fig. 6 (a) Reaction of 5 upon exposure to CO2. (b) XRD solid state structure of 6. (c) IR-spectrum of 6 (green), 13C6 (dark blue) and the corresponding subtraction spectrum (light blue). | ||
 | ||
| Fig. 7 Different coordination modes of the β-diketonate ligand of (a) 3, (b) 5, (c) 6, and (d) 7: connectivity of Lewis-structures (top) and corresponding fragments of solid-state structures obtained from XRD studies (counter cations and co-crystallized solvent molecules are omitted for clarity). | ||
Noteworthy, the subtraction of IR spectra of chemically reduced 1 exposed to either 12CO2 or 13CO2 reveals nearly identical bathochromic shifts (Fig. S47†), implying analogous reactivity of 5 and in situ reduced 1 with CO2. Due to the paramagnetic nature of 6, no signal in 13C NMR could be detected, even if the labelled compound 13C6 was employed. Interestingly, despite several reports of similar ligand-based reactivity with β-diketiminate, pyridine diimine as well as PNP-pincer transition metal complexes,71–75 the formation of 6 represents to the best of our knowledge the first example of carboxylation of a β-diketonate bound to a transition metal center.
The presence of two potassium cations per Mo-center, supported by three anionic ligands, suggests a formal oxidation state of +I for Mo. This is in good agreement with its measured μeff of 1.7 B.M., suggesting an overall spin state of S = ½. The electron paramagnetic resonance spectrum of 6 displays a broad, nearly axial feature with g values amounting to 1.994, 1.98 and 1.93, as well as weak 95Mo and 97Mo (15.8 and 9.5% natural abundance, respectively) hyperfine coupling (Fig. S49†), in good agreement with a Mo-centered S = ½ configuration. This assignment is corroborated by the measured μeff for 6 of 1.7 B.M., as well as by the qualitative molecular orbital diagram and corresponding spin density plot (Fig. S50†) obtained by DFT calculations, which further shows that the bulk spin density is centered at the metal and not on the ligand. The formation of 3 and 6 from 5 implies that exposure to CO2 triggers a formal disproportionation of Mo(II) to Mo(III) and Mo(I) (Fig. 6a). This overall 2 electron reduction of 5 to 6 is in good agreement with the ca. 2.7-fold enhancement in the charge passed in CPE experiments in the presence of CO2 introduced above. We hypothesize that the slight excess of charge passed (a 2-fold enhancement only would be expected to afford the carboxylated product quantitatively) results from the limited stability of the carboxylated product in solution. We indeed observed that 6 slowly decomposes upon long term storage in solution. Likewise, from a synthetic standpoint, the stepwise addition of a second equiv. of KC8 allows to increase the isolated yield of 6 (with respect to 5) from 22% (1 equiv. of KC8) to 48% (2 equiv. of KC8, see ESI Section S2.6† for details). Worthy of note, a few crystals of K[Mo(tBudiket)2(tBudiket·H)] (7), where the carboxylate is replaced by a proton, could be isolated from these reaction mixtures. The exact origin of the proton in this decomposition product is unclear and 7 could not be isolated upon reaction of 6 with proton sources. We believe that this structure is however worth mentioning due to the unique coordination mode of the tBudiket·H ligand, which adopts a zig-zag geometry with the two keto groups pointing into opposite directions (Fig. 7d). To the best of our knowledge, such binding modes have not been documented in transition metal β-diketonate coordination chemistry.
Most importantly, as highlighted by the plateau observed in the charge passed in CPE experiments, the carboxylated complex is not catalytically active for the reduction of CO2. A similar situation has been observed by Kubiak and co-workers with a pyridine monoimine supported Mo-complex74 and the potential disadvantages of redox-active ligands for electrocatalysis were recently more generally summarized by Queyriaux.76
However, this appears in sharp contrast with the electrocatalytic reduction of CS2 mediated by 1, given its analogy with CO2. To further analyze this divergence in reactivity, we explored the chemical reactivity of 5 with CS2. Exposing a solution of 5 to CS2 in THF led to an instant color change from dark green to brown/red, and the precipitation of a light brown solid. At the difference to the reaction with CO2, potassium tetrathiooxalate (K2[C2S4]) was identified and isolated as the main reaction product, while 3 is cleanly regenerated as the sole Mo complex (see Scheme 2a, and ESI Section S3.2†).
 | ||
| Scheme 2 (a) Reactivity of 5 with CS2 in the absence of 18-crown-6 and (b) on a preparative scale in the presence of 18-crown-6 (aisolated yield after recrystallization, bspectroscopic yield by UV-vis). (c) Thiocarboxylation of K[tBudiket] with CS2 and concomitant formation of potassium methyl xanthate. | ||
To probe the role of 5 in this reaction, and to investigate the possible intermediacy of a Mo-bound dithiocarboxylated ligand, we turned to preparative scale reactions using 5 or potassium metal (K0) as reducing agents for CS2 (ESI Section S2.7†). Equimolar amounts of 18-crown-6 were added to increase the solubility of the potassium salt of tetrathiooxalate and hence facilitate its analysis. Significantly higher yields of tetrathiooxalate were observed in the presence of 5 than in the presence of K0, suggesting a catalytic role 3 in the formation of tetrathiooxalate (Scheme 2b). To attempt observing the formation of the thiocarboxylated ligand, we performed the same reaction, using 5 in the presence of an excess of CS2, and quenched the crude extract with MeOH.
Methoxy dithiocarbonate (potassium methyl xanthate, K[S2COMe], Scheme 2a) was identified as a by-product in the reaction mixture. K[S2COMe] is not observed upon mixing tetrathiooxalate or CS2 with methanol, but was observed when reacting MeOH with K[tBudiket·13CS2] (observed upon mixing K[tBudiket] and 13CS2 (10 equiv.) in THF-d8 see Scheme 2c and ESI Section S3.5† for details). The formation of K[S2COMe] hence suggests the presence of a Mo-complex coordinated to a dithiocarboxylated ligand [tBudiket·CS2]. In direct analogy, potassium methyl carbonate (K[O2COCD3]) was quantitatively observed upon reaction of 6 with MeOH (see ESI Section S3.4† for details).
The reactivity and isolated complexes observed allow to propose a general reaction mechanism for the reduction of CO2 and CS2 by the Mo(II) β-diketonate complexes reported here. The nucleophilic addition recognized upon reacting CS2 and CO2 on K[tBudiket]− (see ESI† for details), as well as reported carboxylation reactions of (di-)ketones in the presence of strong bases (e.g. DBU),77 suggests that the first step of the reaction to be redox neutral and involves the formation of a transient Mo(II) (dithio)carboxylated complex, [Mo(tBudiket)2(tBudiket·CE2)]1− (E = O, S), in the reaction mixture (Scheme 3). We propose that the less electron withdrawing nature of the formed dithiocarboxylate moiety relative to a carboxylate moiety accounts for the difference in reactivity between CO2 and CS2. The more anodic redox potential associated to the oxidation of 6 to [Mo(tBudiket)2(tBudiket·CO2)]1− at Eox = −1.80 V (with respect to 5/3, E1/2 = −2.29 V) observed in the CV of 6 (see ESI Fig. S61†) and in good agreement with the lower electron donating properties of the carboxylated ligand, highlights that [Mo(tBudiket)2(tBudiket·CO2)]1− is more oxidizing than the parent 5 complex and is consequently further reduced by an additional equivalent of 5 (see ESI Scheme S2†). This corroborates well with the increased yield of 6 when additional KC8 was added to the reaction mixture (vide supra). Comparatively, the [tBudiket·CS2]− ligand is expected to be a better donor than [tBudiket·CO2]−. This prevents the formation of the Mo(I) complex observed in the presence of CO2 (red arrow), and instead results in the formation of tetrathiooxalate (yellow arrow), in analogy with what observed by Murray and co-workers, with tris(β-diketiminate) cyclophane ligands.75 The quantitative formation of 3 in the presence of CS2 further supports this hypothesis, suggesting that the observed difference in activity in the presence of CS2 and CO2 arises from the reducibility of the (dithio)carboxylated ligand (see ESI Scheme S2†). Mo has no direct role in the dimerization of CS2, yet it is essential here, enabling a redox-non innocent character of the β-diketonate ligand, consequently increasing its nucleophilicity as well as facilitating electron transfer from the electrode (or chemical reducing agent) towards the substrate. These two roles are well exemplified by the metallaoxirane formation observed for 5 as well as by the reversible electrochemistry of [Mo(Rdiket)3] (R = Me, tBu) complexes in the absence of substrate, which turns irreversible in the presence of CO2 or CS2. Note that, while non-catalytic, this property could also be exploited to synthetize the free carboxylated ligand [tBudiket·CO2]−, generally requiring demanding synthetic conditions to be synthetized with low yields.77–80 We found that the addition of tBudiketH (2 equiv.) to 13C6, cleanly afforded 3, while releasing the carboxylated ligand [tBudiket·CO2]−. Isolation of [tBudiket·CO2]− is however made challenging due to the known lability of the carboxylate group located in β-position to two carbonyls, prone to decarboxylation (see ESI Section S3.4† for details).81
 | ||
| Scheme 3 Generalized mechanistic pattern for the reactivity of [Mo(Rdiket)3]−1 with CO2 (red arrow) vs. CS2 (yellow arrow). | ||
Overall, the [Mo(Rdiket)3] reductive carboxylation and (electro)catalyzed reductive dimerization of CS2 to tetrathiooxalate allow to illustrate another aspect of the non-innocence of the β-diketonate ligands. Here, bond forming- and breaking events with the substrate exclusively occur at the ligand, acting as a chemically non-innocent ligand.82 This cooperative character of the ligand is a consequence of its redox-non innocent nature when coordinated to low-valent Mo, highlighting the synergistic roles of Mo and the β-diketonate ligand in facilitating CO2 and CS2 activation.
Conclusions
In summary, we have introduced here new synthetic protocols for the preparation of β-diketonate supported Mo(III) complexes (1 and 3), as well as their transformation to the corresponding oxidized (+IV) and reduced (+II) species. This allowed for a detailed study of the rich redox-chemistry of such low-valent octahedral molybdenum complexes bearing exclusively anionic ligands by electrochemical methods combined with chemical synthesis. X-ray crystallographic studies revealed a clear redox non-innocent behavior of the β-diketonate ligands, expressed by an unprecedented binding mode switch from κ-O to η2-CO upon reduction. 5 represents a rare example of a formal Mo(II) complex exclusively supported by anionic ligands, giving it a sufficiently strong reducing power to react with both CO2 and CS2, yet with distinct outcomes: Exposure of 5 to CO2 resulted in a formal disproportionation reaction yielding 3 and 6, while quantitative formation of 3 along with the formation of tetrathiooxalate was observed in the presence of CS2. Reactivity studies suggest a mechanism involving an analogous intermediate for both substrates, resulting from the formation of a C–C bond between the central carbon of a β-diketonate ligand and CO2/CS2 to yield a Mo-bound (dithio)carboxylated ligand ([Mo(tBudiket)2(tBudiket·CE2)]1−). Although this ligand-centered reactivity undermines the use of [Mo(Rdiket)3] (R = Me, tBu) for the catalytic reduction of CO2, it however does not prevent the reductive dimerization of CS2 to tetrathiooxalate, for which [Mo(Rdiket)3] was found to be a suitable (electro)catalyst. This work emphasizes the redox and chemical non-innocent nature of the β-diketonate ligands, highlighting that this non-innocent character can be either beneficial or detrimental to catalytic turnover, depending on the nature of the reactive substrate studied.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 16 structurally characterized complexes has been deposited at the CCDC under 2352819–2352834 and can be obtained from https://www.ccdc.cam.ac.uk/structures/.Author contributions
V. M. designed and supervised the project. F. M. carried the synthetic work and analytical characterizations. All authors contributed to the writing of the manuscript and validated it before submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 853064). We deeply thank Dr Daniel Klose for recording the EPR spectrum of 6. We thank Dr Michael Wörle and Dr René Verel, for their help with XRD and NMR studies, respectively.Notes and references
- R. Hille, J. Hall and P. Basu, The Mononuclear Molybdenum Enzymes, Chem. Rev., 2014, 114, 3963–4038 CrossRef CAS PubMed.
- R. Hille, The Mononuclear Molybdenum Enzymes, Chem. Rev., 1996, 96, 2757–2816 CrossRef CAS PubMed.
- G. Schwarz, R. R. Mendel and M. W. Ribbe, Molybdenum cofactors, enzymes and pathways, Nature, 2009, 460, 839–847 CrossRef CAS PubMed.
- A. Majumdar and S. Sarkar, Bioinorganic chemistry of molybdenum and tungsten enzymes: A structural–functional modeling approach, Coord. Chem. Rev., 2011, 255, 1039–1054 CrossRef CAS.
- B. Spivack and Z. Dori, Structural aspects of molybdenum(IV), molybdenum(V) and molybdenum(VI) complexes, Coord. Chem. Rev., 1975, 17, 99–136 CrossRef CAS.
- R. H. Holm, The biologically relevant oxygen atom transfer chemistry of molybdenum: from synthetic analogue systems to enzymes, Coord. Chem. Rev., 1990, 100, 183–221 CrossRef CAS.
- J. H. Enemark, J. J. A. Cooney, J.-J. Wang and R. H. Holm, Synthetic Analogues and Reaction Systems Relevant to the Molybdenum and Tungsten Oxotransferases, Chem. Rev., 2004, 104, 1175–1200 CrossRef CAS PubMed.
- S. Groysman and R. H. Holm, Biomimetic chemistry of iron, nickel, molybdenum, and tungsten in sulfur-ligated protein sites, Biochemistry, 2009, 48, 2310–2320 CrossRef CAS PubMed.
- K. Heinze, Bioinspired functional analogs of the active site of molybdenum enzymes: Intermediates and mechanisms, Coord. Chem. Rev., 2015, 300, 121–141 CrossRef CAS.
- S. Patsch, J. V. Correia, B. J. Elvers, M. Steuer and C. Schulzke, Inspired by Nature-Functional Analogues of Molybdenum and Tungsten-Dependent Oxidoreductases, Molecules, 2022, 27, 1–31 CrossRef PubMed.
- B. R. Williams, Y. Fu, G. P. A. Yap and S. J. N. Burgmayer, Structure and Reversible Pyran Formation in Molybdenum Pyranopterin Dithiolene Models of the Molybdenum Cofactor, J. Am. Chem. Soc., 2012, 134, 19584–19587 CrossRef CAS PubMed.
- J.-P. Porcher, T. Fogeron, M. Gomez-Mingot, L.-M. Chamoreau, Y. Li and M. Fontecave, Synthesis and Reactivity of a Bio-inspired Dithiolene Ligand and its Mo Oxo Complex, Chem.–Eur. J., 2016, 22, 4447–4453 CrossRef CAS PubMed.
- T. Fogeron, P. Retailleau, L. M. Chamoreau, Y. Li and M. Fontecave, Pyranopterin Related Dithiolene Molybdenum Complexes as Homogeneous Catalysts for CO2 Photoreduction, Angew. Chem., Int. Ed., 2018, 57, 17033–17037 CrossRef CAS PubMed.
- J.-P. Porcher, T. Fogeron, M. Gomez-Mingot, E. Derat, L.-M. Chamoreau, Y. Li and M. Fontecave, A Bioinspired Molybdenum Complex as a Catalyst for the Photo- and Electroreduction of Protons, Angew. Chem., Int. Ed., 2015, 54, 14090–14093 CrossRef CAS PubMed.
- A. Mouchfiq, T. K. Todorova, S. Dey, M. Fontecave and V. Mougel, A bioinspired molybdenum–copper molecular catalyst for CO2 electroreduction, Chem. Sci., 2020, 11, 5503–5510 RSC.
- S. J. Nieter Burgmayer and M. L. Kirk, The Role of the Pyranopterin Dithiolene Component of Moco in Molybdoenzyme Catalysis, Struct. Bonding, 2019, 179, 101–152 CrossRef CAS.
- C. E. Laplaza and C. C. Cummins, Dinitrogen Cleavage by a Three-Coordinate Molybdenum(III) Complex, Science, 1995, 268, 861–863 CrossRef CAS PubMed.
- D. V. Yandulov and R. R. Schrock, Catalytic reduction of dinitrogen to ammonia at a single molybdenum center, Science, 2003, 301, 76–78 CrossRef CAS PubMed.
- N. F. Both, A. Spannenberg, K. Junge and M. Beller, Low-Valent Molybdenum PNP Pincer Complexes as Catalysts for the Semihydrogenation of Alkynes, Organometallics, 2022, 41, 1797–1805 CrossRef CAS PubMed.
- T. Leischner, A. Spannenberg, K. Junge and M. Beller, Synthesis of Molybdenum Pincer Complexes and Their Application in the Catalytic Hydrogenation of Nitriles, ChemCatChem, 2020, 12, 4543–4549 CrossRef CAS.
- K. Arashiba, Y. Miyake and Y. Nishibayashi, A molybdenum complex bearing PNP-type pincer ligands leads to the catalytic reduction of dinitrogen into ammonia, Nat. Chem., 2011, 3, 120–125 CrossRef CAS PubMed.
- G. A. Silantyev, M. Forster, B. Schluschass, J. Abbenseth, C. Wurtele, C. Volkmann, M. C. Holthausen and S. Schneider, Dinitrogen Splitting Coupled to Protonation, Angew. Chem., Int. Ed., 2017, 56, 5872–5876 CrossRef CAS PubMed.
- Q. Liao, N. Saffon-Merceron and N. Mezailles, Catalytic dinitrogen reduction at the molybdenum center promoted by a bulky tetradentate phosphine ligand, Angew. Chem., Int. Ed., 2014, 53, 14206–14210 CrossRef CAS PubMed.
- R. Castro-Rodrigo, S. Chakraborty, L. Munjanja, W. W. Brennessel and W. D. Jones, Synthesis, Characterization, and Reactivities of Molybdenum and Tungsten PONOP Pincer Complexes, Organometallics, 2016, 35, 3124–3131 CrossRef CAS.
- Y. Zhang, P. G. Williard and W. H. Bernskoetter, Synthesis and Characterization of Pincer-Molybdenum Precatalysts for CO2 Hydrogenation, Organometallics, 2016, 35, 860–865 CrossRef CAS.
- Y. Zhang, B. S. Hanna, A. Dineen, P. G. Williard and W. H. Bernskoetter, Functionalization of Carbon Dioxide with Ethylene at Molybdenum Hydride Complexes, Organometallics, 2013, 32, 3969–3979 CrossRef CAS.
- G. R. Lee, J. M. Maher and N. J. Cooper, Reductive disproportionation of carbon dioxide by dianionic carbonylmetalates of the transition metals, J. Am. Chem. Soc., 1987, 109, 2956–2962 CrossRef CAS.
- J. Chatt, M. Kubota, G. J. Leigh, F. C. March, R. Mason and D. J. Yarrow, A possible carbon dioxide complex of molybdenum and its rearrangement product di-μ-carbonato-bis{carbonyltris(dimethylphenylphosphine)molybdenum}: X-ray crystal structure, J. Chem. Soc., Chem. Commun., 1974, 1033–1034, 10.1039/C39740001033.
- R. Alvarez, J. L. Atwood, E. Carmona, P. J. Perez, M. L. Poveda and R. D. Rogers, Formation of carbonyl-carbonate complexes of molybdenum by reductive disproportionation of carbon dioxide. X-ray structure of Mo4(μ4-CO3)(CO)2(O)2(μ2-O)2(μ2-OH)4(PMe3)6, Inorg. Chem., 1991, 30, 1493–1499 CrossRef CAS.
- L. Contreras, M. Paneque, M. Sellin, E. Carmona, P. J. Pérez, E. Gutiérrez-Puebla, A. Monge and C. Ruiz, Novel carbon dioxide and carbonyl carbonate complexes of molybdenum. The X-ray structures of trans-[Mo(CO2)2{HN(CH2CH2PMe2)2}(PMe3)] and [Mo3(μ2-CO3)(μ2-O)2(O)2(CO)2(H2O)(PMe3)6]·H2O, New J. Chem., 2005, 29, 109–115 RSC.
- K. A. Grice and C. Saucedo, Electrocatalytic Reduction of CO2 by Group 6 M(CO)6 Species without “Non-Innocent” Ligands, Inorg. Chem., 2016, 55, 6240–6246 CrossRef CAS PubMed.
- M. L. Clark, K. A. Grice, C. E. Moore, A. L. Rheingold and C. P. Kubiak, Electrocatalytic CO2 reduction by M(bpy-R)(CO)4 (M = Mo, W; R = H, tBu) complexes. Electrochemical, spectroscopic, and computational studies and comparison with group 7 catalysts, Chem. Sci., 2014, 5, 1894–1900 RSC.
- J. O. Taylor, F. L. P. Veenstra, A. M. Chippindale, M. J. Calhorda and F. Hartl, Group 6 Metal Complexes as Electrocatalysts of CO2 Reduction: Strong Substituent Control of the Reduction Path of [Mo(η3-allyl)(CO)2(x,x′-dimethyl-2,2′-bipyridine)(NCS)] (x = 4–6), Organometallics, 2019, 38, 1372–1390 CrossRef CAS.
- R. Alvarez, E. Carmona, D. J. Cole-Hamilton, A. Galindo, E. Gutierrez-Puebla, A. Monge, M. L. Poveda and C. Ruiz, Formation of acrylic acid derivatives from the reaction of carbon dioxide with ethylene complexes of molybdenum and tungsten, J. Am. Chem. Soc., 1985, 107, 5529–5531 CrossRef CAS.
- R. Alvarez, E. Carmona, A. Galindo, E. Gutierrez, J. M. Marin, A. Monge, M. L. Poveda, C. Ruiz and J. M. Savariault, Formation of carboxylate complexes from the reactions of carbon dioxide with ethylene complexes of molybdenum and tungsten. X-ray and neutron diffraction studies, Organometallics, 1989, 8, 2430–2439 CrossRef CAS.
- W. H. Bernskoetter and B. T. Tyler, Kinetics and Mechanism of Molybdenum-Mediated Acrylate Formation from Carbon Dioxide and Ethylene, Organometallics, 2011, 30, 520–527 CrossRef CAS.
- J. A. Buss and T. Agapie, Four-electron deoxygenative reductive coupling of carbon monoxide at a single metal site, Nature, 2016, 529, 72–75 CrossRef CAS PubMed.
- J. A. Buss and T. Agapie, Mechanism of Molybdenum-Mediated Carbon Monoxide Deoxygenation and Coupling: Mono- and Dicarbyne Complexes Precede C-O Bond Cleavage and C-C Bond Formation, J. Am. Chem. Soc., 2016, 138, 16466–16477 CrossRef CAS PubMed.
- L. C. Seefeldt, Z. Y. Yang, D. A. Lukoyanov, D. F. Harris, D. R. Dean, S. Raugei and B. M. Hoffman, Reduction of Substrates by Nitrogenases, Chem. Rev., 2020, 120, 5082–5106 CrossRef CAS PubMed.
- F. Masero and V. Mougel, Molybdenum(IV) β-diketonate complexes as highly active catalysts for allylic substitution reactions, Chem. Commun., 2023, 59, 4636–4639 RSC.
- M. L. Larson and F. W. Moore, Synthesis and Properties of Molybdenum(III) Acetylacetonate, Inorg. Chem., 1962, 1, 856–859 CrossRef CAS.
- T. M. Harris and C. M. Harris, Carboxylation of β-Dicarbonyl Compounds through Dicarbanions. Cyclizations to 4-Hydroxy-2-pyrones, J. Org. Chem., 1966, 31, 1032–1035 CrossRef CAS.
- M. Hidai, K. Tominari, Y. Uchida and A. Misono, A molybdenum complex containing molecular nitrogen, J. Chem. Soc. D, 1969, 37, 814 RSC.
- M. Hidai, K. Tominari and Y. Uchida, Preparation and properties of dinitrogen-molybdenum complexes, J. Am. Chem. Soc., 1972, 94, 110–114 CrossRef CAS.
- M. G. Vinum, L. Voigt, C. Bell, D. Mihrin, R. W. Larsen, K. M. Clark and K. S. Pedersen, Evidence for Non-Innocence of a β-Diketonate Ligand, Chem.–Eur. J., 2020, 26, 2143–2147 CrossRef CAS PubMed.
- M. G. Vinum, L. Voigt, S. H. Hansen, C. Bell, K. M. Clark, R. W. Larsen and K. S. Pedersen, Ligand field-actuated redox-activity of acetylacetonate, Chem. Sci., 2020, 11, 8267–8272 RSC.
- T. Shibahara and M. Yamasaki, A New Syntheic Route to Hexachloromolybdate(III). An X-Ray Structure of (NH4)3[MoCl6], Bull. Chem. Soc. Jpn., 1990, 63, 3022–3023 CrossRef CAS.
- M. L. Larson and F. W. Moore, Characterization of the Brown Molybdenyl Bisacetylacetonate, Inorg. Chem., 1963, 2, 881–882 CrossRef CAS.
- F. A. Cotton, C. E. Rice and G. W. Rice, The crystal and molecular structures of bis(2,4-pentanedionato)chromium, Inorg. Chim. Acta, 1977, 24, 231–234 CrossRef CAS.
- K. M. Wampler and R. R. Schrock, Molybdenum tris(2,5-dimethylpyrrolide), a rare homoleptic molybdenum(III) monomer, Inorg. Chem., 2008, 47, 10226–10228 CrossRef CAS PubMed.
- H. Brunner, J. Wachter, I. Bernal and M. Creswick, Synthesis and X-Ray Structure Analysis of a Mo Complex with η2-Coordinated Benzaldehyde; Transformation of a CO Ligand by Double α-Addition, Angew. Chem., Int. Ed., 1979, 18, 861–862 CrossRef.
- S. Gambarotta, C. Floriani, A. Chiesi-Villa and C. Guastini, Carbon dioxide and formaldehyde coordination on molybdenocene to metal and hydrogen bonds of the C1 molecule in the solid state, J. Am. Chem. Soc., 1985, 107, 2985–2986 CrossRef CAS.
- N. J. Christensen, P. Legzdins, J. Trotter and V. C. Yee, Reactivities of representative cyclopentadienyl η4-trans-diene nitrosyl complexes of molybdenum toward acetone, Organometallics, 1991, 10, 4021–4030 CrossRef CAS.
- Y.-C. Tsai, M. J. A. Johnson, D. J. Mindiola, C. C. Cummins, W. T. Klooster and T. F. Koetzle, A Cyclometalated Resting State for a Reactive Molybdenum Amide: Favorable Consequences of β-Hydrogen Elimination Including Reductive Cleavage, Coupling, and Complexation, J. Am. Chem. Soc., 1999, 121, 10426–10427 CrossRef CAS.
- C. Camp and J. Arnold, On the non-innocence of “Nacnacs”: ligand-based reactivity in β-diketiminate supported coordination compounds, Dalton Trans., 2016, 45, 14462–14498 RSC.
- Z. J. Tonzetich, A. J. Jiang, R. R. Schrock and P. Müller, Molybdenum Imido Alkylidene Complexes that Contain a β-Diketiminate Ligand, Organometallics, 2007, 26, 3771–3783 CrossRef CAS.
- L. E. Doyle, W. E. Piers and J. Borau-Garcia, Ligand Cooperation in the Formal Hydrogenation of N2O Using a PCsp2P Iridium Pincer Complex, J. Am. Chem. Soc., 2015, 137, 2187–2190 CrossRef CAS PubMed.
- A. Dauth, U. Gellrich, Y. Diskin-Posner, Y. Ben-David and D. Milstein, The Ferraquinone–Ferrahydroquinone Couple: Combining Quinonic and Metal-Based Reactivity, J. Am. Chem. Soc., 2017, 139, 2799–2807 CrossRef CAS PubMed.
- M. H. P. Rietveld, H. Hagen, L. van de Water, D. M. Grove, H. Kooijman, N. Veldman, A. L. Spek and G. van Koten, Tantalacyclobutane Complexes Containing the Potentially C,N,N‘ -Coordinating Ligand [C6H4(CH2 N(Me)CH2CH2NMe2)-2]- (CNN) and Their Reactivity with Carbon Monoxide and tert-Butyl Isocyanide, Organometallics, 1997, 16, 168–177 CrossRef CAS.
- W. D. Harman, D. P. Fairlie and H. Taube, Synthesis, characterization, and reactivity of the (η2-acetone)pentaammineosmium(II) complex, J. Am. Chem. Soc., 1986, 108, 8223–8227 CrossRef CAS.
- M. A. Lockwood, P. E. Fanwick and I. P. Rothwell, Reactivity of a tungsten(II) aryloxide with imines, ketones and aldehydes, Chem. Commun., 1996, 2013, 10.1039/cc9960002013.
- D. J. Mindiola, R. Waterman, D. M. Jenkins and G. L. Hillhouse, Synthesis of 1,2-bis(di-tert-butylphosphino)ethane (dtbpe) complexes of nickel: radical coupling and reduction reactions promoted by the nickel(I) dimer [(dtbpe)NiCl]2, Inorg. Chim. Acta, 2003, 345, 299–308 CrossRef CAS.
- T. J. Hadlington, T. Szilvási and M. Driess, Metal nitrene-like reactivity of a Si-N bond towards CO2, Chem. Commun., 2018, 54, 9352–9355 RSC.
- D. G. A. Verhoeven, M. A. C. van Wiggen, J. Kwakernaak, M. Lutz, R. J. M. Klein Gebbink and M.-E. Moret, Periodic Trends in the Binding of a Phosphine-Tethered Ketone Ligand to Fe, Co, Ni, and Cu, Chem.–Eur. J., 2018, 24, 5163–5172 CrossRef CAS PubMed.
- K. C. MacLeod, I. M. DiMucci, E. P. Zovinka, S. F. McWilliams, B. Q. Mercado, K. M. Lancaster and P. L. Holland, Masked Radicals: Iron Complexes of Trityl, Benzophenone, and Phenylacetylene, Organometallics, 2019, 38, 4224–4232 CrossRef CAS PubMed.
- L. E. Helberg, T. B. Gunnoe, B. C. Brooks, M. Sabat and W. D. Harman, Rhenium(I) Terpyridine π-Bases: Reversible η2-Coordination of Ketones, Aldehydes, and Olefins in the Terpyridine Plane, Organometallics, 1999, 18, 573–581 CrossRef CAS.
- D. S. Williams, M. H. Schofield, J. T. Anhaus and R. R. Schrock, Synthesis and reactions of tungsten(IV) bis(imido) complexes: relatives of bent metallocenes, J. Am. Chem. Soc., 1990, 112, 6728–6729 CrossRef CAS.
- B. W. H. Saes, D. G. A. Verhoeven, M. Lutz, R. J. M. Klein Gebbink and M.-E. Moret, Coordination of a Diphosphine–Ketone Ligand to Ni(0), Ni(I), and Ni(II): Reduction-Induced Coordination, Organometallics, 2015, 34, 2710–2713 CrossRef CAS.
- W. Egan, G. Gunnarsson, T. E. Bull and S. Forsen, A nuclear magnetic resonance study of the intramolecular hydrogen bond in acetylacetone, J. Am. Chem. Soc., 1977, 99, 4568–4572 CrossRef CAS.
- D. E. Ryan and K. Nakanishi, Methine-selective deuteration of V(acac)3, [Co(acac)2], and Al(acac)3, J. Labelled Compd. Radiopharm., 1995, 36, 595–598 CrossRef CAS.
- M. D. Anker, M. Arrowsmith, P. Bellham, M. S. Hill, G. Kociok-Köhn, D. J. Liptrot, M. F. Mahon and C. Weetman, Selective reduction of CO2 to a methanol equivalent by B(C6F5)3-activated alkaline earth catalysis, Chem. Sci., 2014, 5, 2826–2830 RSC.
- F. A. LeBlanc, A. Berkefeld, W. E. Piers and M. Parvez, Reactivity of Scandium β-Diketiminate Alkyl Complexes with Carbon Dioxide, Organometallics, 2012, 31, 810–818 CrossRef CAS.
- M. Vogt, M. Gargir, M. A. Iron, Y. Diskin-Posner, Y. Ben-David and D. Milstein, A new mode of activation of CO2 by metal-ligand cooperation with reversible C-C and M-O bond formation at ambient temperature, Chem.–Eur. J., 2012, 18, 9194–9197 CrossRef CAS PubMed.
- D. Sieh, D. C. Lacy, J. C. Peters and C. P. Kubiak, Reduction of CO2 by Pyridine Monoimine Molybdenum Carbonyl Complexes: Cooperative Metal-Ligand Binding of CO2, Chem.–Eur. J., 2015, 21, 8497–8503 CrossRef CAS PubMed.
- M. V. L. Ocampo and L. J. Murray, Metal-Tuned Ligand Reactivity Enables CX2 (X = O, S) Homocoupling with Spectator Cu Centers, J. Am. Chem. Soc., 2024, 146, 1019–1025 CrossRef CAS PubMed.
- N. Queyriaux, Redox-Active Ligands in Electroassisted Catalytic H+ and CO2 Reductions: Benefits and Risks, ACS Catal., 2021, 11, 4024–4035 CrossRef CAS.
- K. I. Assaf, A. K. Qaroush, I. K. Okashah, F. a. M. Al-Qaisi, F. Alsoubani and A. a. F. Eftaiha, Activation of β-diketones for CO2 capture and utilization, React. Chem. Eng., 2021, 6, 2364–2375 RSC.
- T. Baran, A. Dibenedetto, M. Aresta, K. Kruczała and W. Macyk, Photocatalytic Carboxylation of Organic Substrates with Carbon Dioxide at Zinc Sulfide with Deposited Ruthenium Nanoparticles, ChemPlusChem, 2014, 79, 708–715 CrossRef CAS.
- A. Dibenedetto, J. Zhang, M. Trochowski, A. Angelini, W. Macyk and M. Aresta, Photocatalytic carboxylation of C H bonds promoted by popped graphene oxide (PGO) either bare or loaded with CuO, J. CO2 Util., 2017, 20, 97–104 CrossRef CAS.
- S. Saini, R. S. Das, A. Kumar and S. L. Jain, Photocatalytic C–H Carboxylation of 1,3-Dicarbonyl Compounds with Carbon Dioxide Promoted by Nickel(II)-Sensitized α-Fe2O3 Nanoparticles, ACS Catal., 2022, 12, 4978–4989 CrossRef CAS.
- G. Bartoli, M. Bosco, A. Guerrieri, R. Dalpozzo, A. De Nino, E. Iantorno and G. Palmieri, Reaction of dianions of acyclic β-enamino ketones with electrophiles. vii. synthesis of 5-(monoalkylamino)-3-oxo γ,δ-unsaturated acids and esters and of 3-(monoalkylamino)-5-oxo β,γ-unsaturated esters, Gazz. Chim. Ital., 1996, 126, 25–29 CAS.
- F. Schneck, M. Finger, M. Tromp and S. Schneider, Chemical Non-Innocence of an Aliphatic PNP Pincer Ligand, Chem.–Eur. J., 2017, 23, 33–37 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2352819–2352834. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03496a |
| This journal is © The Royal Society of Chemistry 2024 |