
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Leveraging ligand-based proton and electron transfer for aerobic reactivity and catalysis
Kate A.
Jesse
 *a and
John S.
Anderson
*b
*a and
John S.
Anderson
*b
aLos Alamos National Laboratory, Los Alamos, NM 87545, USA. E-mail: kjesse@lanl.gov
bDepartment of Chemistry, The University of Chicago, Chicago, Illinois 60637, USA. E-mail: jsanderson@uchicago.edu
First published on 9th September 2024
Abstract
While O2 is an abundant, benign, and thermodynamically potent oxidant, it is also kinetically inert. This frequently limits its use in synthetic transformations. Correspondingly, direct aerobic reactivity with O2 often requires comparatively harsh or forcing conditions to overcome this kinetic barrier. Forcing conditions limit product selectivity and can lead to over oxidation. Alternatively, O2 can be activated by a catalyst to facilitate oxidative reactivity, and there are a variety of sophisticated examples where transition metal catalysts facilitate aerobic reactivity. Many efforts have focused on using metal–ligand cooperativity to facilitate the movement of protons and electrons for O2 activation. This approach is inspired by enzyme active sites, which frequently use the secondary sphere to facilitate both the activation of O2 and the oxidation of substrates. However, there has only recently been a focus on harnessing metal–ligand cooperativity for aerobic reactivity and, especially, catalysis. This perspective will discuss recent efforts to channel metal–ligand cooperativity for the activation of O2, the generation and stabilization of reactive metal–oxygen intermediates, and oxidative reactivity and catalysis. While significant progress has been made in this area, there are still challenges to overcome and opportunities for the development of efficient catalysts which leverage this biomimetic strategy.
1 Introduction
O2 is an abundant, inexpensive, and thermodynamically powerful oxidant. This thermodynamic potency is perhaps most simply illustrated by the formal electrochemical potential of 1.23 V vs. SHE for O2 reduction.1 Importantly, this net reduction of O2 results in water as a benign byproduct. Despite these advantages, O2 is comparatively kinetically inert due to its triplet ground spin state. This triplet ground state leads to inhibited reactivity with many compounds, such as most organic substrates that typically have singlet spin states.2 This kinetic inertness is also illustrated by the bond dissociation free energy (BDFE) of ∼51 kcal mol−1 in solvent for the first proton/electron in the reduction of O2 to water.3 To overcome these spin-forbidden processes, and to participate in oxidative reactivity, O2 must enter the higher energy singlet spin state.2 This means that direct aerobic reactivity often requires forcing conditions, such as the high temperatures used for combustion, or activation via a catalyst. Transition metal complexes feature prominently in aerobic catalysis, which has led to a wide array of elegant examples in which O2 is activated for catalytic transformations. The Wacker oxidation, which was developed in 1956, is one classic example that uses co-catalytic Pd and Cu to perform the oxidation of ethylene to acetaldehyde with O2 as a terminal oxidant.4–6 Cu is essential for this process as it helps with mediating efficient redox exchange between Pd and O2. Alternatively, a supporting ligand can be similarly used to facilitate proton/electron transfer and subsequent O2 activation. For example, Stahl and coworkers have investigated the utility of organic cocatalysts such as aminoxyl or quinone compounds that work in conjunction with metal salts to facilitate aerobic oxidation chemistry.7–16 There has also been extensive research into metal-free aerobic reactivity, primarily using aminoxyl compounds.17–25While there have been many successes in this area, it is noteworthy that Nature activates O2 through elaborate active sites that coordinate transition metal centers with a carefully positioned organic secondary coordination sphere. Secondary coordination spheres in active sites for aerobic catalysis nearly always feature themes such as hydrogen bonding motifs, redox-active cofactors for electron transfer, and proton shuttling machinery, all of which aid in O2 activation.26–33 In many cases this activation can lead to more thermodynamically potent oxidants, such as terminal oxo complexes, through the controlled addition of H-atom equivalents. Thus, the elaborate “ligand” of enzymatic secondary coordination spheres is critical for the activation of O2 and subsequently efficient and selective aerobic oxidations.
Despite the biological ubiquity of coupling transition metals with tailored secondary coordination spheres to facilitate aerobic reactivity, there has only recently been an allied focus on leveraging these same influences in synthetic complexes.13,34 Major advances include hydrogen bonding ligands,35–38 proton shuttling ligands,39–46 and redox-active ligands for cooperative electron transfer.47–60 While these approaches are now broadly used in the design of new synthetic catalysts, their application for O2 activation, particularly aerobic catalysis, is still nascent. Supporting ligand scaffolds that can cooperatively transfer both protons and electrons to activate O2 for aerobic reactivity in a biomimetic manner are even more rare.
This perspective will cover recent advances in leveraging metal–ligand cooperativity for O2 activation and aerobic reactivity (Fig. 1). We will particularly focus on systems that can shuttle both protons and electrons to O2, beginning with examples where this cooperativity enables the characterization of well-defined intermediates which feature O2 binding or reduction. We will then focus on examples which display stoichiometric or catalytic oxidations. Last, we will summarize prospects and challenges, laying out key directions for development and investigation as this field develops. One clear conclusion is that this area holds promise for developing sustainable oxidation reactions, with exciting developments likely coming in the near future.
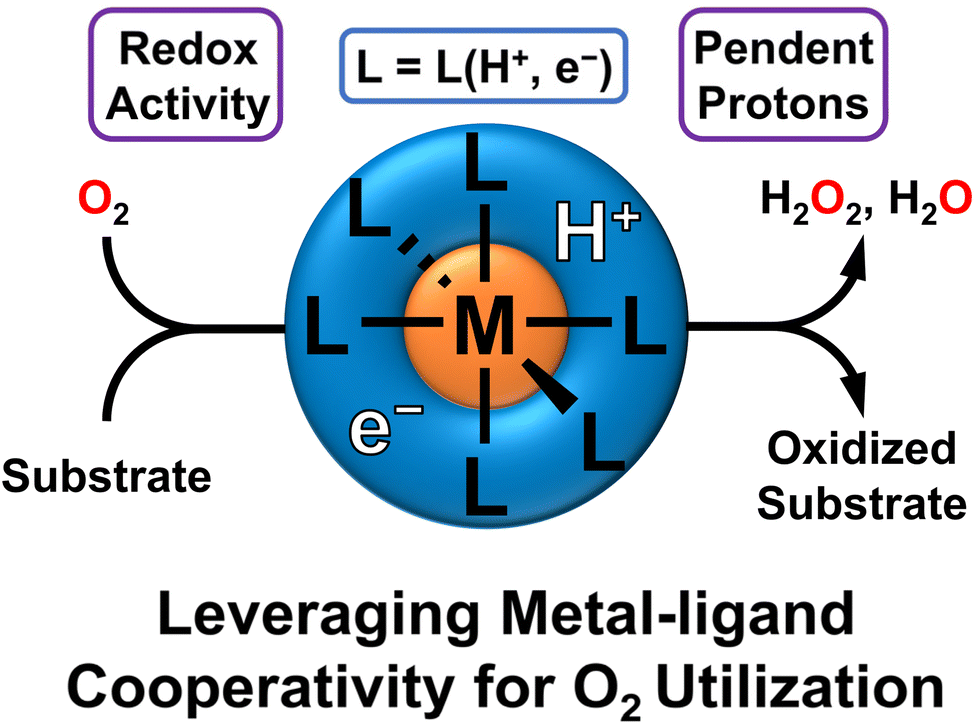 | ||
| Fig. 1 Metal–ligand cooperativity that incorporates redox-active ligands and pendent H-equivalents in the secondary sphere can activate O2 for oxidative reactivity with substrates. This enables the ligand to serve as a proton and electron source. | ||
2 Metal–ligand cooperativity enabling the activation or reduction of O2
Metal–ligand cooperativity involving ligand-stored protons and electrons has been shown to facilitate the formation and subsequent reduction of various metal–oxygen species such as metal-oxos and metal hydroperoxos from O2. Again, this is a common mode of reactivity and O2 activation in biology, but it has also become a useful approach in synthetic systems. Quinoidal motifs are some of the classic building blocks used in redox non-innocent ligands, and ligands with incorporated quinones/quinols can further provide an accessible proton and electron source for O2 reduction. Agapie and coworkers effectively used such a quinone based ligand scaffold with Pd to facilitate O2 reduction to water (Fig. 2A). The diphosphine hydroquinone ligand is able to donate 2 electrons and 2 protons towards O2 reduction. When a Pd atom is coordinated to the ligand, O2 reduction is found to proceed via a side-on η2-peroxo complex, the formation of which can be observed by low temperature ultraviolet-visible (UV-vis) spectroscopy. Low temperature 31P nuclear magnetic resonance (NMR) spectroscopy of this species supports the formation of an asymmetric complex, and low temperature infra-red (IR) spectroscopy shows no indication of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond stretch, indicating that the side on Pd η2-peroxo complex forms prior to the involvement of the backbone quinone. While this first intermediate observed by UV-vis spectroscopy is long-lived at low temperature, the second intermediate generated upon warming decays more rapidly. Spectroscopic data of this second species suggests a Pd2+ complex coordinated to a semiquinone ligand, but concrete assignment of the partially hydrogenated oxygen ligand was not obtained due to the transient nature of this intermediate. The reduction of O2 to H2O as a final product was confirmed by 1H NMR spectroscopic analysis of the reaction volatiles after transferring to a J. Young tube.61
O double bond stretch, indicating that the side on Pd η2-peroxo complex forms prior to the involvement of the backbone quinone. While this first intermediate observed by UV-vis spectroscopy is long-lived at low temperature, the second intermediate generated upon warming decays more rapidly. Spectroscopic data of this second species suggests a Pd2+ complex coordinated to a semiquinone ligand, but concrete assignment of the partially hydrogenated oxygen ligand was not obtained due to the transient nature of this intermediate. The reduction of O2 to H2O as a final product was confirmed by 1H NMR spectroscopic analysis of the reaction volatiles after transferring to a J. Young tube.61
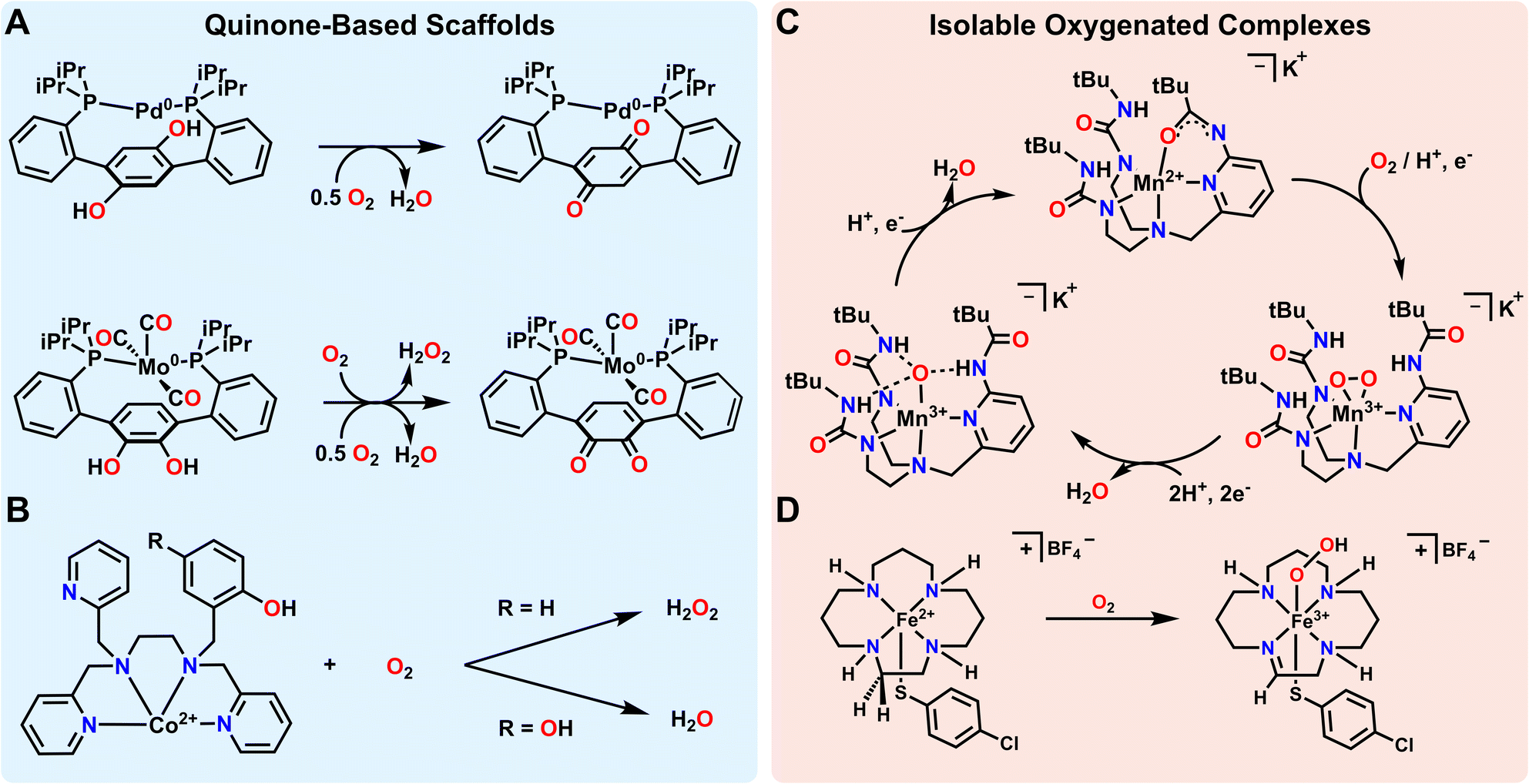 | ||
| Fig. 2 Metal–ligand cooperative transfer of protons and electrons enabling the activation of O2. (A and B) Quinone-based non-innocent ligands that can donate protons and electrons to reduce O2. (C) A non-innocent ligand stabilizes metal–oxygen intermediates and acts as a proton shuttle to reduce O2 to water in the presence of external reductant. (D) Protons and electrons from a non-innocent ligand produce a spectroscopically characterizable Fe–hydroperoxo complex. | ||
Agapie and coworkers have also illustrated the utility of a related isomeric quinone-based ligand scaffold on Mo complexes for a similar reduction of O2 to water (Fig. 2A). In this case it was found that activation of O2 led to initial reduction to H2O2 rather than H2O. H2O2 then putatively interacts with a second equivalent of the Mo complex for the net 4e− reduction to H2O to occur. This illustrates how both the ligand and the metal can influence the selectivity of reductions, even in closely related systems. As a final note, while this perspective focuses on the reduction of O2 with protons and electrons, Mo complexes supported by this general ligand framework, but with Si- or B-functionalized catecholate motifs, make (R2SiO)n and (R2BO)n byproducts rather than H2O as the final reaction product.62 Regardless of the terminal O-accepting reagent, all of these examples illustrate how redox-active quinone-based ligands can facilitate selective reduction of O2 to H2O.
While the above examples rely on the incorporation of quinone/quinol moieties directly into the ligand backbone, the use of a pendent quinol on a ligand arm has also been shown to facilitate the reduction and/or activation of O2. In particular, Goldsmith and coworkers have shown that appending such a quinol group to a chelating amine/pyridine scaffold facilitates superoxide dismutase (SOD) like reactivity with either Mn2+ or Zn2+.63,64 Mn2+ complexes with a ligand featuring one pendent phenol mediate SOD reactivity via an outer-sphere mechanism. However, when the ligand features one or two pendent quinols, the reaction accelerates via an inner-sphere mechanism.63 This quinol assisted inner-sphere pathway can also be leveraged to render a redox-inactive metal ion, Zn2+, active for SOD catalysis.64 Beyond this example, Goldsmith and coworkers have also investigated the electrochemical reduction of O2 using a Co2+ metal center bound to the same ligand framework (Fig. 2B). They found that when a pendent quinol rather than a pendent phenol was incorporated into the ligand scaffold, the product selectivity and mechanism shifts from a 2 electron, 2 proton reduction of O2 to form H2O2 to a 4 electron, 2 proton reduction of O2 to form H2O.65 This switch demonstrates that inclusion of groups that donate both electrons and protons not only enables catalytic reactivity, but also provides determinative control over selectivity in some processes. This conclusion meshes with other observations in biological and synthetic systems. As one example, Machan and coworkers and Stahl and coworkers have both shown the utility of 1,4-hydroquinone/benzoquinone as a cocatalyst in facilitating the reduction of O2 to H2O electrocatalytically.9,66 While the 1,4-hydroquinone/benzoquinone group is not directly incorporated into the ligand backbone in these cases, the electrocatalytic reaction mechanism requires the close association of 1,4-hydroquinone/benzoquinone to produce H2O as the final product. As with the Co example above, omission of 1,4-hydroquinone as a cocatalyst in this example from Stahl and coworkers9 instead results in the preferential reduction of O2 to H2O2.
An alternate strategy for metal–ligand cooperativity is the use of the ligand to create a secondary sphere that stabilizes reactive oxygenated intermediates through hydrogen bonding interactions. The use of H-bonding or proton shuttling ligands has been of great recent interest.67 While these scaffolds do not strictly shuttle electrons, there are several ligand scaffolds that feature conjugated arms that may potentially also be redox-active. As such, some select cases are also highlighted here. As one example among many,35–38,68,69 Borovik and coworkers demonstrated how designing a secondary sphere to stabilize intermediates and shuttle proton equivalents enables the catalytic reduction of O2. They were able to synthesize a side on Mn-peroxo species and Mn-oxo species stabilized by hydrogen bonding interactions (Fig. 2C). Characterization of these intermediates proved crucial to the development of a catalytic cycle converting O2 to H2O at room temperature.70 Catalysis was only observed with the addition of hydrazine or diphenylhydrazine as a terminal reductant. Fout and coworkers utilized a similar strategy to stabilize Fe3+-oxo or Fe3+-hydroxo complexes formed via reactivity between an Fe complex and O2.35 Both complexes are stabilized by hydrogen bonding interactions from the ligand, and the ligand shuttles protons to the O2 substrate.67 Szymczak and coworkers have also used hydrogen bonding motifs to stabilize a Zn-peroxo dimer formed from reactivity with O2 in the presence of cobaltocene. However, we note that neither protons nor electrons are shuttled by the ligand for O2 reduction in this case.69 The fact that no H-equivalents are transferred to O2 makes the formation of the Zn-peroxo dimer reversible with added oxidant, such as ceric ammonium nitrate or iodobenzene dichloride. Another related example comes from Garcia–Bosch and coworkers. They recently reported a Cu complex that uses hydrogen bonding interactions from the ligand to stabilize and reduce O2 to an axial hydroperoxo ligand. However, in this case, the terminal H-atom on the hydroperoxo ligand is abstracted from the ligand of a second Cu complex rather than the oxidized Cu complex itself.71 In all examples, hydrogen bonding interactions help to stabilize reactive intermediates, with some examples also invoking H-atom abstraction and proton shuttling for catalytic reactivity. While definitive evidence for ligand-based redox reactivity is less obvious in most of these cases, it is possible, if not likely, that the conjugated arms in these or related scaffolds might also be non-innocent under the right conditions or with appropriate synthetic modifications.
It is noteworthy that while the quinone/quinol systems discussed above formally transfer H-atoms, the H-bonding examples feature N-based ligands which do not as explicitly exhibit net H-atom reactivity. However, there are other ligands with N–H's that more clearly shuttle reducing equivalents to O2. David Goldberg and coworkers found that they could use a macrocyclic tetraamine ligand combined with electronic effects from a trans thiolate ligand to stabilize an Fe3+-hydroperoxo complex (Fig. 2D).72 Here, the electron and proton equivalents came from the ligand scaffold via desaturation to form an imine. This complex can be generated at −78 °C, and was characterized by UV-visible spectroscopy, electron paramagnetic resonance (EPR) spectroscopy, and electrospray ionization mass spectrometry (EI-MS). Using thiolate ligands for stabilizing Fe–oxygen intermediates has been a topic of some interest due to the presence of this motif in enzyme active sites such as superoxide reductase (SOR) or cytochrome P450.73,74 However, these biomimetic HnOy complexes are rarely generated directly from O2 since their formation requires the movement of both protons and electrons. Instead, Fe-hydroperoxo complexes are often generated from exogenous reducing and acid equivalents.75–84 The reactivity observed in this case would be impossible without metal–ligand cooperativity arising from desaturation of the macrocyclic ligand that facilitates the transfer of both electrons and protons.
Similar in structure to quinols, pendent phenol functionalities on the ligand backbone can act as a proton and electron source for the hydrogenation of O2. Karlin and coworkers used a Cu system with an aliphatic amine pincer ligand and a pendent phenol to capture O2 as a bis(μ-oxo)Cu22+ dimer at −135 °C.85 The pendent phenol group on each ligand then donates one proton and one electron to each O atom, resulting in a bis(μ-hydroxo)Cu22+ dimer where each Cu center features a ligand-based radical on the O atom of the pendent phenol. These ligand-based radicals can be detected with EPR spectroscopy by a characteristic signal at g = 2.00. Upon warming in the presence of excess 1-hydroxy-2,2,6,6-tetramethylpiperidine (TEMPOH) as an H-atom source, this dimer releases two equivalents of H2O and forms a Cu2+ complex with an overall +1 charge, where the Cu center is now coordinated to the NNN amine pincer ligand as well as the O in the pendent phenol (Fig. 3). This reactivity mimics the reactivity observed in the active site of oxy-tyrosinase, which catalyses the regioselective hydroxylation of monophenols with a para substitution to catechols.86,87
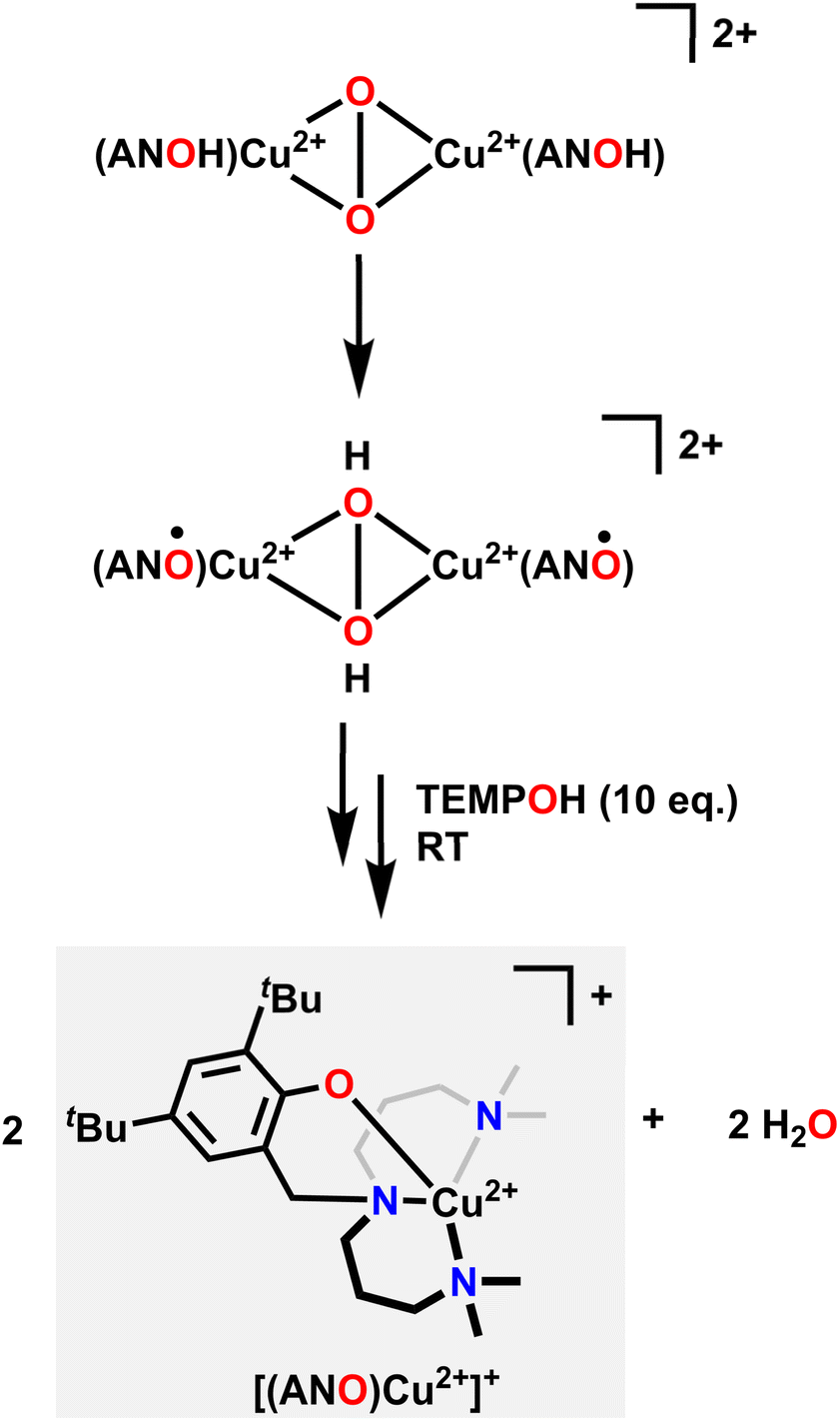 | ||
| Fig. 3 Mechanism of O2 capture, hydrogenation, then release of water upon reacting with TEMPOH. | ||
In each of the examples presented in this section, metal–ligand cooperativity facilitates the shuttling of proton and electron equivalents to O2 resulting in its reduction. Despite this general reactivity paradigm, the products of these reactions are variable depending on the system, with H2O, H2O2, or stabilized metal–oxygen complexes (i.e. hydroperoxo species) that can be spectroscopically characterized all being commonly formed. While these examples demonstrate the utility of incorporating redox active ligands with pendent protons for the hydrogenation of O2, there is still a lack of clarity on what exact design features facilitate the formation of different products. Simple topics of future investigation might reasonably include examining how the positioning of H-donors dictates product selectivity, or how the thermodynamic properties of these donors (BDFE, acidity, etc.) can be tuned for desired reactivity. Beyond these fundamental studies, there is also great potential in harnessing the thermodynamic potential of O2 for subsequent substrate oxidations after activation. Indeed, the next sections of this perspective focus on recent advances in these areas.
3 Metal–ligand cooperativity enabling the activation of O2 for alcohol oxidation
Alcohol oxidation is a widely used transformation in synthetic chemistry. Metal–ligand cooperativity can facilitate alcohol oxidation reactions by either forming an oxygenated intermediate that serves as the active oxidant in the key bond forming/breaking steps or as an inexpensive and benign terminal oxidant for the regeneration of an oxidized transition metal intermediate that instead directly oxidizes the substrate, as seen in Wacker chemistry.4,5 In both instances, O2 acts as a formal H-atom acceptor, but there are key mechanistic differences whether an in situ oxygenated intermediate acts as the oxidant or whether O2 simply acts as a regenerating terminal oxidant.A good example where O2 generates an active oxygenated transition metal intermediate that performs alcohol oxidation comes from Garcia–Bosch and coworkers (Fig. 4).88 They use a ureanyl functionalized 1,2-phenylenediamido Cu2+ platform where the diureanyl ligand can formally store both protons and electrons as well as provide H-bonding interactions. This platform enables the stabilization of a superoxo intermediate via H-bonding interactions from the ligand. This transiently stabilized Cu-superoxo then serves as the active oxidant for the oxidation of primary alcohols to aldehydes. The ligand further facilitates the reaction by mediating the movement of proton and electron equivalents during the dehydrogenation of the alcohol and subsequent H-transfer to the coordinated O2 molecule resulting in both aldehyde and H2O2 as the terminal reaction products. Throughout the course of this reaction mechanism, Cu remains in a 2+ state. This means that the Cu2+ center does not participate in the alcohol oxidation reaction as an electron source, but rather as a docking site for the primary alcohol/O2 to coordinate such that the ligand secondary sphere can mediate the resulting oxidation and reduction respectively. Interestingly, this suggests that transition metal centers, which are frequently considered as redox mediators in many catalytic cycles, can instead be tuned/utilized as simple Lewis acid sites for coordination if a suitably redox-active supporting ligand is used.64,89–91 This is similar to how classic reactions in organic chemistry, such as the Sharpless epoxidation, proceed through use of a catalyst that acts as a Lewis acid.92 Additionally, the reduction of O2 to H2O2 allows for the regeneration of the starting complex and thus allows for a catalytic process. This work is in contrast to the ping-pong mechanism seen in galactose oxidase (GAO), where a primary alcohol is first oxidized to an aldehyde, then O2 is reduced to H2O2 as the terminal regenerating oxidant to reform the active catalyst. The amino acids within the GAO active site act as proton acceptors, whereas Cu plays a more active role in the catalytic cycle of GAO, by acting as an electron acceptor/donor. This ping-pong mechanism has been more closely reproduced by Stack and coworkers (Fig. 5A).93 Here the authors were able to synthesize a Cu complex and related intermediates in the catalytic oxidation of primary alcohols and demonstrate pathways that match those proposed for GAO. In their Cu system, alcohol oxidation occurs first with the ligand scaffold acting as an H-atom acceptor, O2 reduction to H2O2 occurs second, and the Cu2+ center is reversibly reduced to Cu1+ during the course of the catalytic cycle.93 As such, this example from Stack serves as a more faithful biomimetic Cu system for the mechanism observed in GAO where O2 serves as a regenerating oxidant.
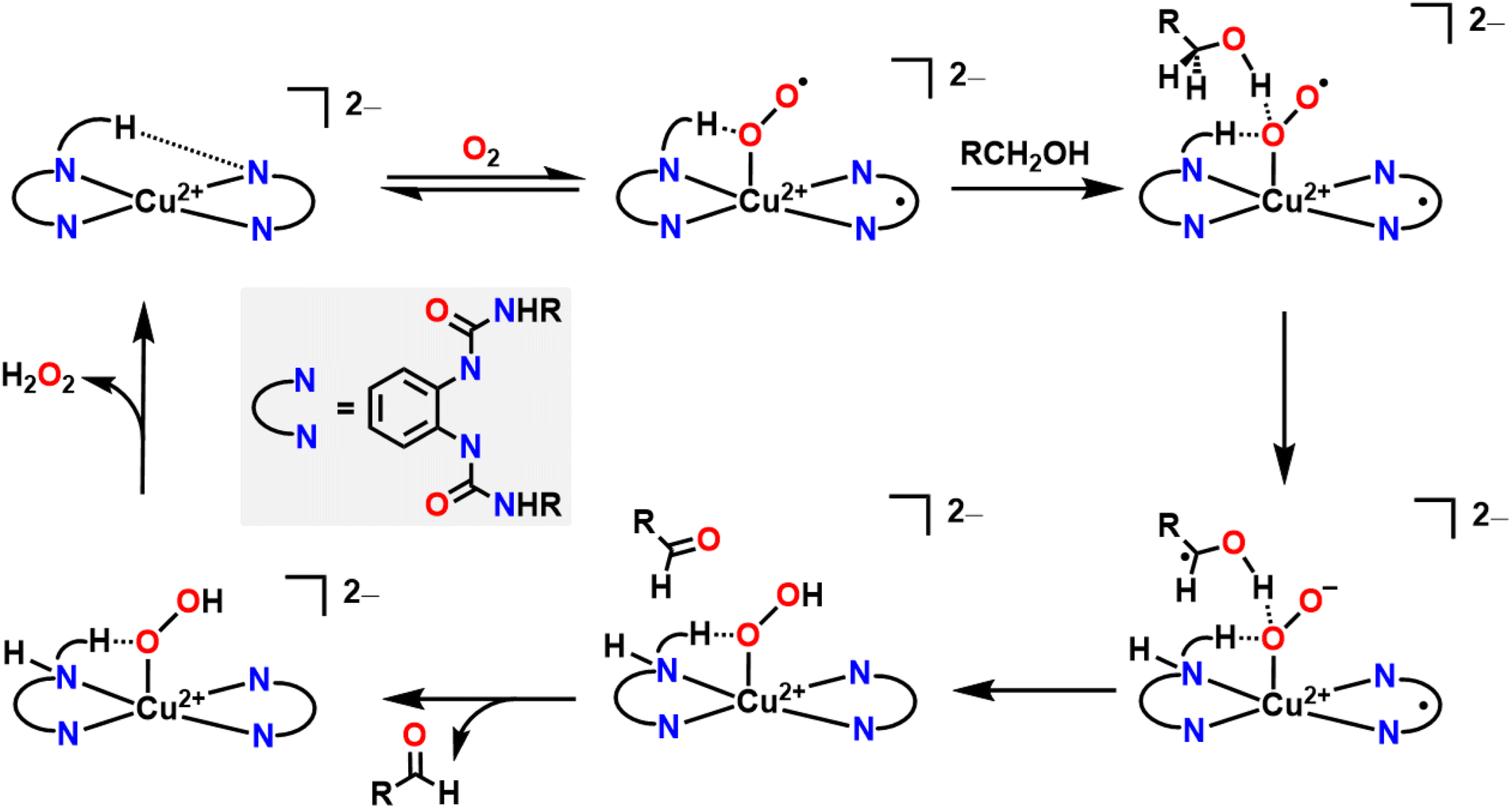 | ||
| Fig. 4 Mechanism of alcohol oxidation where O2 participates in the mechanism in conjunction with ligand non-innocence. O2 generates the Cu-superoxo that enables alcohol oxidation before being reduced via metal–ligand cooperativity. | ||
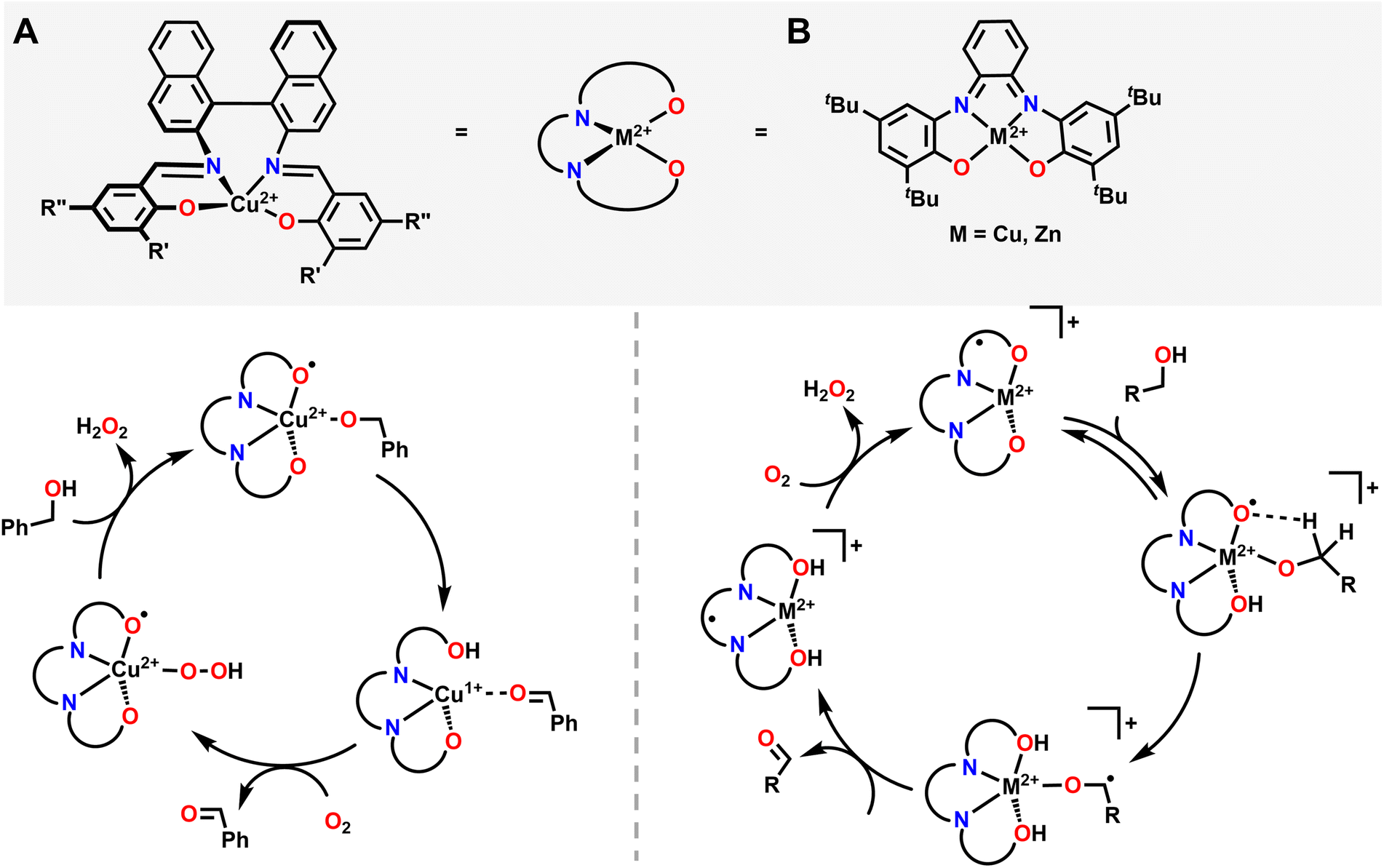 | ||
| Fig. 5 (A) Proposed mechanism of alcohol oxidation reported by Stack and coworkers closely mimicking the ping-pong mechanism observed in GAO. O2 participates in the mechanism in conjunction with ligand non-innocence. O2 generates a Cu-hydroperoxo and displaces the oxidized substrate, before being reduced via metal–ligand cooperativity to H2O2. (B) Proposed mechanism of primary alcohol oxidation reported by Wieghardt and coworkers. In this case O2 serves as a terminal oxidant, allowing the starting metal catalyst to regenerate. | ||
Pioneering work from Wieghardt and coworkers demonstrated that different mononuclear Cu and Zn complexes featuring an ONNO ligand structurally similar the ligand used by Stack and coworkers were also able to facilitate alcohol oxidation of primary and benzylic alcohols using O2 as an oxidant to regenerate the starting metal complex catalytically and release H2O2 (Fig. 5B).94,95 Mechanistic and kinetic studies of their system showed evidence for H-atom abstraction by the ligand from the coordinated alcohol, providing evidence for ligand noninnocence and metal–ligand cooperativity during the alcohol oxidation reaction.
Another early example of a Cu complex facilitating alcohol oxidation in conjunction with O2 reduction comes from Urch, Brown, and coworkers.96,97 In their system, CuCl was mixed with phenanthroline (phen), a substituted azo compound, such as di-tert-butylazodicarboxylate (DBAD), an alcohol, O2, and K2CO3, to form a Cu1+ complex that facilitates alcohol oxidation via hydrogen transfer from the coordinated alcohol to the coordinated azo ligand. Upon release of the aldehyde or ketone, O2 then coordinates to form a Cu2+ dimer with a bridging peroxo ligand. The peroxo ligand is then homolytically cleaved when heated to putatively form a Cu2+-oxyl complex. This then abstracts an H-atom from both the substituted hydrazine ligand and the alcohol substrate to release H2O and restart the catalytic cycle. Interestingly, although this is one of the earliest examples invoking metal–ligand cooperativity for alcohol oxidation with a Cu complex where O2 is used as a terminal oxidant, it is also one of the examples with the widest substrate scope. Urch, Brown, and coworkers found that this system is able to catalytically oxidize primary, allylic, and benzylic alcohols.97
While there are entropic and orientational advantages to having a tethered ligand for H-shuttling, these scaffolds can frequently be difficult to synthesize or tune, which limits their application in organic methodology. An alternative strategy that has been employed to great effect is the use of metal complexes in conjunction with an exogenous organic cocatalyst that acts as a proton/electron reservoir. Stahl and coworkers have been particularly active in this area with extensive applications of Cu bipyridine (bpy) complexes with (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl (TEMPO) as a cocatalyst for alcohol oxidation (Fig. 6A).98 They find that half an equivalent of O2 and one equivalent of TEMPO are necessary to generate a Cu2+ hydroxide complex which can then bind alcohol. TEMPO then serves as an H-atom acceptor in tandem with the Cu2+ center serving as a one e− oxidant to regenerate the starting catalyst and generate the aldehyde product. Other aminoxyl co-catalysts are also competent for this reactivity; when TEMPO is switched out for 9-azabicyclo[3.3.1]nonane N-oxyl (ABNO), the oxidation of both primary and secondary alcohols becomes accessible.99 The transition metal used can also be changed; Stahl and coworkers have also had success using Fe(NOx) catalysts instead of Cu(bpy) in conjunction with aminoxyl compounds.100
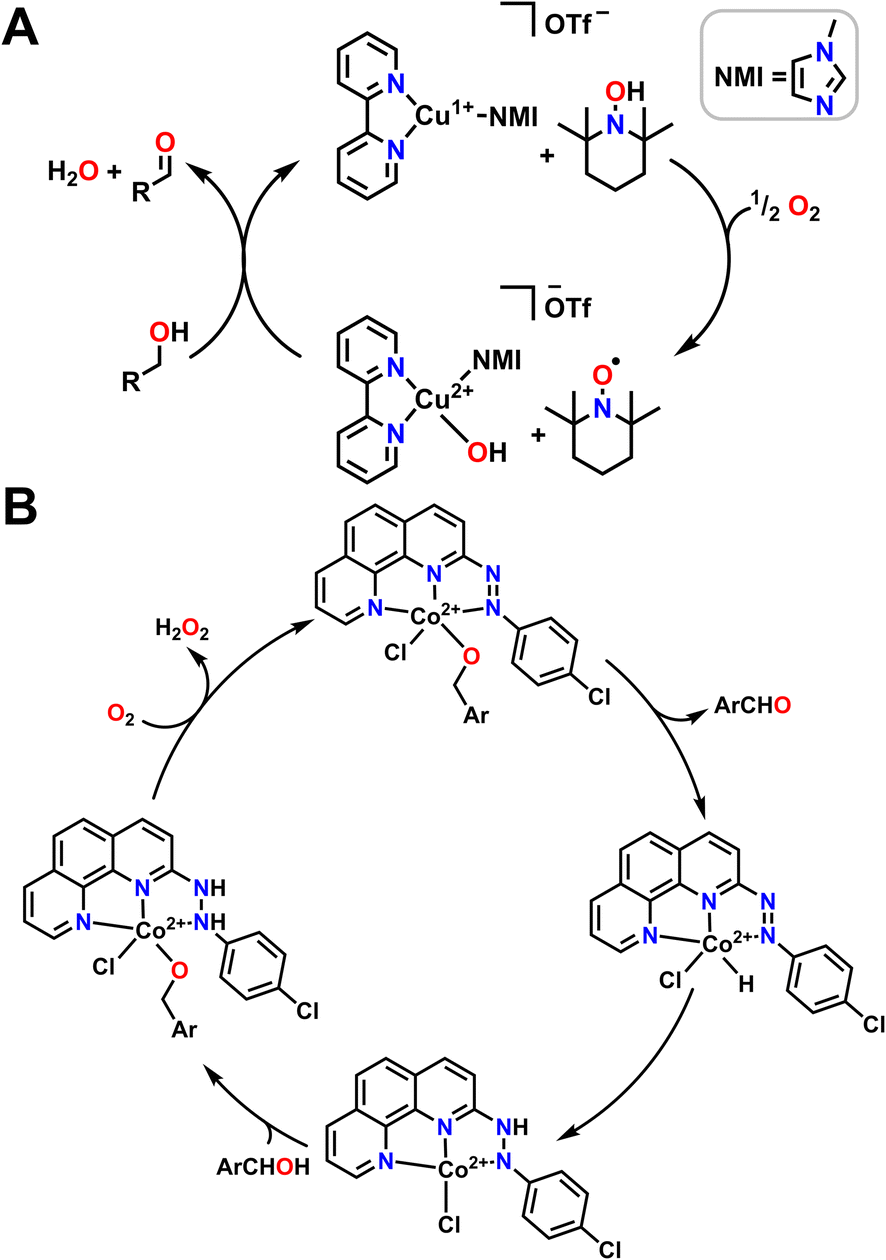 | ||
| Fig. 6 Mechanisms of alcohol oxidation where O2 participates in the mechanism in conjunction with ligand non-innocence. (A) O2 is reduced with an Cu complex and external organic compound that serves as a proton and electron source to regenerate the active molecule for alcohol oxidation. NMI coordinates through the less sterically hindered N atom. (B) O2 regenerates starting metal complex. | ||
While both Cu and Fe are frequently used for oxidative chemistry with O2, particularly in biological or biomimetic contexts, other first row transition metals with redox active ligands and pendent protons can also use O2 for alcohol oxidation. Many of these examples again leverage metal–ligand cooperativity. As one example, the Paul group used 2-(4-chlorophenylazo)-1,10-phenanthroline ligand with Co2+ to catalyse alcohol oxidation with benzyl alcohols (Fig. 6B).101 The proposed reaction mechanism begins by coordinating the alcohol and releasing HCl. The catalytic cycle then proceeds via β-hydride elimination from the bound alcohol to release an aldehyde or a ketone while simultaneously transferring the hydride onto Co2+ to form a transient Co hydride intermediate. The hydride ligand can then transfer onto a diazene fragment of the supporting ligand before a new alcohol equivalent coordinates to Co2+ as an alkoxide, with the ligand accepting a proton. An O2 molecule then oxidizes the ligand, removing a formal H2 equivalent and releasing H2O2 to reform the starting complex in the catalytic cycle (Fig. 6B).
The examples discussed in this section offer some general mechanistic paradigms for catalytic systems featuring both metal–ligand cooperativity and O2. Generally, O2 is either used in the generation of the active oxidant, or it is used to regenerate the active catalyst. In both cases, hydrogenation of O2 occurs in addition to the targeted alcohol oxidation as part of the catalytic cycle. The production of H2O or H2O2 can result in its own challenges since these molecules may participate in side-reactions or competitive coordination, particularly as their relative concentration increases. This is a challenge that will need to be addressed in any future large scale catalytic process, particularly in examples which generate H2O2.
4 Metal–ligand cooperativity enabling the activation of O2 for O-atom transfer and other dehydrogenation reactions
Beyond using O2 as a reagent to drive overall oxidative reactivity, metal–ligand cooperativity can also facilitate using O2 as an O-atom transfer reagent. This typically occurs by first forming an oxygenated metal complex such as metal-superoxo, -hydroperoxo, or -oxo species which then acts as the active O-atom transfer reagent. Several examples where O2 can be used for net O-atom transfer have come from our laboratory using a dihydrazonopyrrole (DHP) based ligand scaffold and various first row transition metals. This ligand scaffold has the ability to reversibly donate or accept two protons and two electrons, while additional electron equivalents can be accessed from the metal center itself.102,103 This unique redox flexibility directly aids in the activation of O2, in many cases through spectroscopically observable intermediates. Metalation with Fe2+ enables the isolation and characterization of a reduced and protonated DHP complex with a formal H2 equivalent stored on the ligand backbone. Assignment of this complex is supported by suite of spectroscopic techniques, including single crystal X-ray diffraction (SXRD) and IR spectroscopy, both of which support the presence of ligand-based N–H protons. In the presence of O2, this complex reacts to form a transiently stable intermediate which can be assigned as an Fe3+-hydroperoxo complex with a ligand based DHP radical (Fig. 7A).104 This reaction can be followed by UV-vis spectroscopy at −40 °C. Isotopic labelling studies with 18O2 show an O–O stretch by Raman spectroscopy that provides good support for the Fe3+-hydroperoxo assignment. Additionally, IR spectroscopy of deuterium labelled N–H complex followed by reaction with O2 shows that the signals associated with the N–H and O–H stretches in the Fe3+-hydroperoxo complex originate from the ligand scaffold and hydroperoxo moiety respectively. This Fe complex can then release H2O2 upon warming. Alternatively, if this complex is instead warmed in the presence of triphenylphosphine (PPh3) one equivalent of triphenylphosphine oxide (OPPh3) can form. It was also found that the Fe3+-hydroperoxo complex will perform a net H-atom abstraction followed by O-atom transfer with the toluene solvent to form benzaldehyde. While only 0.131 equivalents of benzaldehyde could be detected by gas chromatography-mass spectrometry (GC-MS), this reaction demonstrates how the combination of redox-active ligands and pendent protons can enable the reductive activation of O2 for both C–H activation as well as O-atom transfer.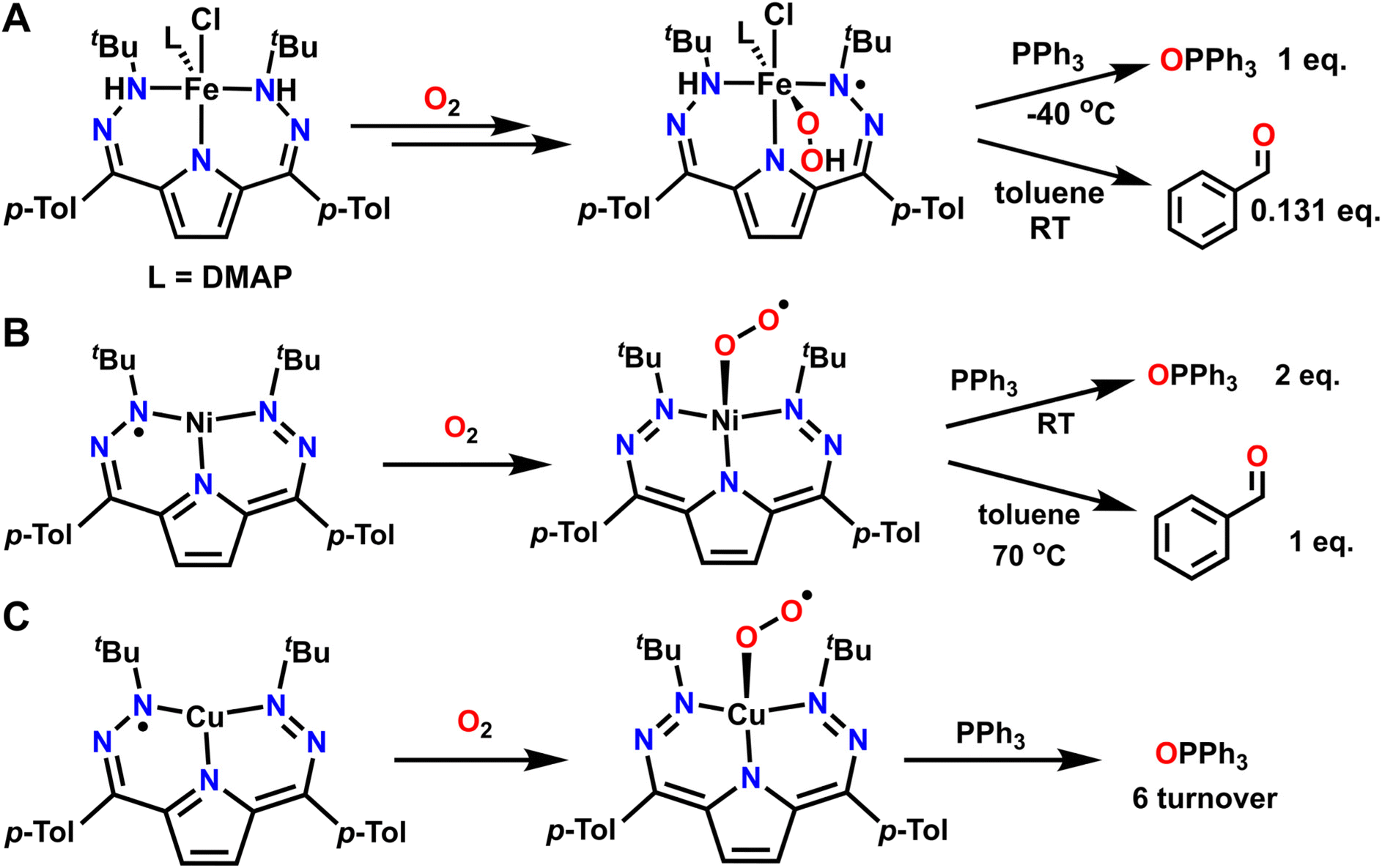 | ||
| Fig. 7 Exposure to O2 forms metal–oxygen complexes that can then facilitate O-atom transfer reactions to phosphines and C–H bonds via metal ligand cooperativity. (A) Reactivity of a fully reduced and hydrogenated Fe(DHP)–H2 complex. (B) Formation and reactivity of a Ni(DHP)-superoxo complex. (C) Formation and reactivity of a Cu(DHP)-superoxo complex. | ||
A second example of O-atom transfer via metal–ligand cooperativity with these DHP systems features a Ni2+ T-shaped complex with a ligand radical (Fig. 7B).105 Exposure of this complex to O2 results in the formation of a Ni2+-superoxo complex with an oxidized DHP ligand. In this case, oxidation occurs at the ligand rather than at the metal center upon O2 binding, paralleling the reactivity proposed in quercetin dioxygenase.106,107 Unlike the above-mentioned Fe3+-hydroperoxo complex with a DHP ligand radical, the Ni2+-superoxo complex utilized ligand-based electron transfer only, instead of both proton and electron transfer. This superoxo intermediate is moderately stable at room temperature. While this Ni2+-superoxo complex can perform stoichiometric O-atom transfer to PPh3, it must be heated to elicit H-atom abstraction from a C–H bond and subsequent O-atom transfer to toluene. The Ni2+-superoxo complex can also perform alcohol oxidation of benzyl alcohol, but once again requires heat for reactivity to occur. This is a stark contrast to the previously discussed Fe complex which could perform these reactions at low temperature or room temperature respectively. Unfortunately, the exact mechanism of oxidation for both the Fe and the Ni complexes is difficult to conclusively determine due to decomposition of the metal complex during reactivity.
The above examples with the DHP scaffold on Fe and Ni resulted in only stoichiometric reactivity. However, moving late in the first-row to Cu results in aerobic catalysis. A related Cu2+-superoxo complex with an oxidized and closed-shell DHP ligand can be synthesized and characterized with a suite of spectroscopic methods (Fig. 7C). This superoxo complex engages in a variety of catalytic aerobic reactivity including deformylation of 2-phenylpropionaldehyde, dehydrogenation of N–H and O–H bonds, and O-atom transfer to PPh3.108 As such, this Cu complex displays the most robust and versatile oxidative reactivity among these DHP ligated complexes. All three examples demonstrate how metal–ligand cooperativity can stabilize metal–oxygen intermediates that serve as potent oxidants. However, only the Fe(DHP) complex has provided direct evidence, via spectroscopy, of how ligand-based hydrogen equivalents can be transferred to O2. Other first row transition metals featuring the DHP ligand, including Ni and Co, have been shown to participate in hydrogenation where a more direct role for the storage of H-equivalents on the ligand is invoked.103,109 An interesting hypothesis is that the DHP ligand plays an active role in accepting protons or H-atoms during the C–H, N–H, or O–H activation step of various oxidations, but additional studies will be required to test this proposal. Regardless, a key conclusion from this series is how different oxidative reactivity is realized with different metal centers all on the same DHP ligand. This highlights how cooperativity between the metal and the ligand is essential beyond the fundamental design of the ligand scaffold itself.
While O-atom transfer to an outside substrate can be extremely challenging, metal–ligand cooperativity can more commonly facilitate intramolecular O-atom transfer, for instance in degradation processes. O-atom transfer to the ligand is often more accessible as the ligand as a substrate is held in close proximity to the activated O-atom equivalents. Unfortunately, it also provides a parasitic pathway that frequently precludes the possibility of performing catalytic oxidations of external substrates. These examples still provide insights into how metal–ligand cooperativity enables the activation of O2 for oxidative chemistry. As such, a few examples of ligand oxidation are described here. As a final note, while initial efforts in this area have focused on designing ligands for controlling the storage/flow of electrons and protons, realizing scaffolds which are oxidatively robust is an increasingly important hurdle for broader catalytic applications (as discussed below).
A few examples of intramolecular O-atom transfer have been demonstrated by Karen Goldberg and coworkers. Here, the authors have explored the reactivity of Pt complexes featuring redox active ligands with O2 and they have observed oxidation of the ligand via C–O bond formation to one O-atom and coordination to the Pt center through the second O-atom to form a 6-member metallocycle ring (Fig. 8A). This Pt–peroxide complex then undergoes O–O bond cleavage to form a 5-membered metallocycle ring with one O-atom and insertion of the other O-atom into a ligand C–H bond. As such, the ligand of this complex stabilizes the coordination of O2 and subsequently engages in O-atom transfer to the ligand via insertion into a C–H bond.110,111 Net C–H activation of the ligand has been previously observed when reactive metal–oxygen complexes are generated.112,113 However, we will not discuss these other examples in detail since the reactive complex in question was not generated directly from O2.
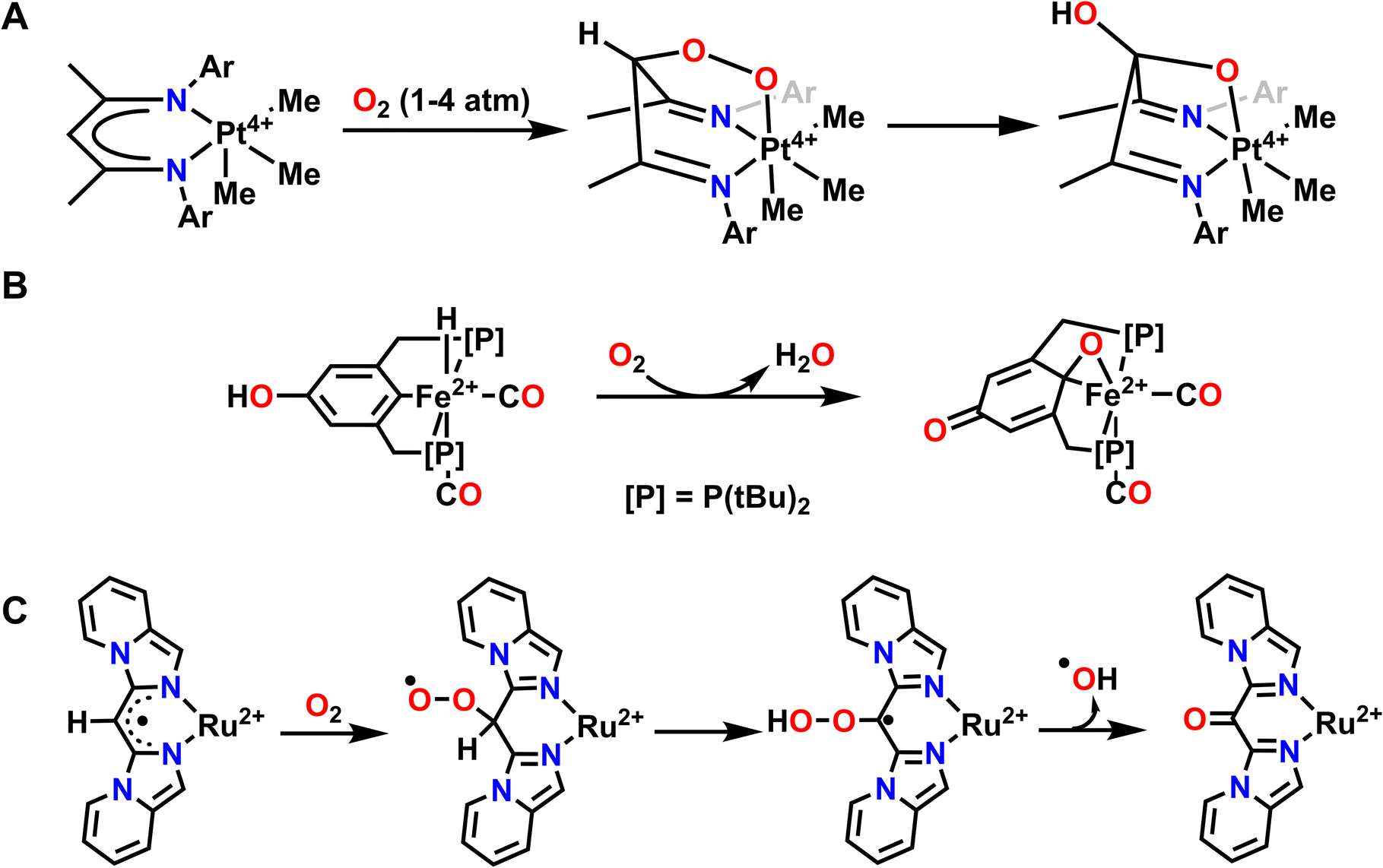 | ||
| Fig. 8 Examples where exposure to O2 results in O-atom transfer to the ligand via metal–ligand cooperativity where the ligand donates protons and electrons for the reduction of O2. (A), ref. 110; (B), ref. 114; (C), ref. 119. | ||
Another example of O2 activation and subsequent reactivity with the ligand comes from Milstein and coworkers. Reaction of an Fe2+ hydride complex supported by a PCP pincer ligand results in hydrogenation of one oxygen equivalent of O2 to H2O and insertion of the second oxygen into the Fe–C ligand bond (Fig. 8B). A proton from the ligand backbone O–H and the hydride ligand are the hydrogen sources for this net hydrogenation reaction to form water. This results in an overall oxidized PCP ligand and one equivalent of H2O from the net reaction with O2.114 This reactivity is reminiscent of extradiol-cleaving catechol dioxygenases, which use the substrate as the ligand when activating O2. In particular, the mechanism for these dioxygenases invokes an Fe–oxygen–ligand metallocycle prior to O-atom insertion into the ligand and the subsequent reduction of the second O-atom to water.115 A similar O-atom insertion into a ligand C–H bond and oxidation of the auxiliary ligand, in this case trimethylphosphine (PMe3) to trimethylphosphine oxide (OPMe3), is also observed with a DHP Ni complex related to those discussed above.116 Abakumov and coworkers have also reported an Sb5+ complex that can coordinate O2, then undergo insertion of the terminal O-atom into the ligand to form a metallocycle and dearomatize the ligand scaffold. This reaction is reversible, and under moderate heating will reform the original Sb5+ complex and O2.117,118
In both the Pt and Fe examples, O2 is first coordinated to the metal center before proceeding through O-atom transfer to the ligand and hydrogenation. However, metal–ligand cooperativity can facilitate other O2 binding events, particularly through interactions with ligand-based radicals. A good example of this interaction can be observed with a Ru2+ complex reported by Lahiri and coworkers. Here, O2 initially reacts with a ligand-based radical to form a C–O–O bond, where a radical primarily resides on the terminal oxygen and effectively dearomatizes the ligand (Fig. 8C). This complex then undergoes a net 1,3-H-atom shift to rearomatize the ligand and provide a hydroperoxo intermediate. Finally, O–O bond homolysis results in the release of hydroxyl radical and overall O-atom transfer to the ligand. Importantly, this net reactivity is again enabled by metal ligand cooperativity between the Ru2+ center and the pincer supporting ligand.119
5 Opportunities and challenges
Metal–ligand cooperativity, particularly with a scaffold capable of storing both electron and proton equivalents, is a powerful tool in synthetic chemistry. This strategy provides a readily available source of formal H-atoms which can facilitate specific catalytic steps or the formation of various intermediates. Combining this approach to metal–ligand cooperativity with O2 in overall aerobic reactivity can allow for oxidative reactivity that otherwise requires significantly more harsh conditions or more oxidizing reagents to realize. While this field holds significant potential, there are also a variety of challenges that must be addressed in to effectively harness this strategy for a wide array of applications.5.1 Positioning of H-atom equivalents to target desired oxygenated intermediates
The proximity of proton, H-atom, or hydride equivalents within the secondary sphere to a bound O2 or substrate molecule can have a dramatic, and even determinative, effect on the selectivity for various activated intermediates. As such, the positioning of these hydrogen equivalents in the secondary sphere or through cocatalysts must be harnessed to achieve the desired selectivity. Lessons from biological systems can be used for initial insights,120–123 but more detailed studies on differential reactivity with various placements of H-equivalents are needed. However, small changes within the secondary sphere can also result in significantly different electronic effects which may also influence the complex's ability to effectively interact with O2 and desired substrates. Careful balancing of ligand properties is essential for robust and effective catalysts.5.2 Balancing and managing the simultaneous flow of reducing and oxidizing equivalents
The activation of O2 requires the addition of reducing (H-atom) equivalents, but these same reducing equivalents may also result in quenching of any active oxidizing intermediates. Thus, finding ways to balance the flow of reducing equivalents is vital to catalytic turnover. Nature circumvents this challenge by gating the delivery of proton and electron equivalents at specific points during turnover. However, this level of control is much more difficult in synthetic systems. There has been some success in this area towards alcohol oxidation, particularly primary alcohol oxidation, where the substrate can also serve as a reductant to activate O2. However, similar progress has been more limited with other oxidative reactions where the substrate is more difficult to oxidize. Management of both reducing and oxidizing equivalents in more challenging oxidative reactions will be vital for expanding the utility of this strategy.5.3 Realizing holistic design principles for broad applicability and scope
While the design of elaborate ligand scaffolds can be useful for a specific reaction, realizing general aerobic catalysts often requires simple and modular systems. Future work will need to balance these two approaches to ensure a useful reaction scope can be achieved. An elaborate ligand design that only catalyses one reaction will rarely be widely useful in industry or academia. In this context, the above-mentioned cases with an external “cooperative” ligand such as aminoxyl radicals are noteworthy. Alternatively, future studies may find success in utilizing more robust and modular “cooperative” ligands that can donate and accept both protons and electrons to enable flexibility and broad scope that are needed for useful synthetic methodologies.5.4 Building in sufficient oxidative stability to enable efficient turnover
Some of the most challenging aerobic oxidations also involve substrates which are similar to ligand scaffolds (i.e. aliphatic C–H groups). Thus, designing appropriate flanking groups on ligands is important to preclude intramolecular decomposition. Examples of this intramolecular decomposition were presented in the final section of this perspective. While those examples provided important evidence for how metal–ligand cooperativity can enable O-atom transfer to C–H bonds, they also demonstrated how intramolecular oxidation of the ligand scaffold precludes the possibility of catalysis. One possible avenue of investigation on this front includes replacing C–H bonds in the ligand framework with more oxidatively robust groups such as C–F bonds or oxygenated groups (i.e. sulfonates, carbonyls, etc.). However, maintaining optimized cooperative reactivity with such oxidatively robust flanking groups is a notable synthetic challenge.6 Conclusions
Significant progress has been made using metal–ligand cooperativity and O2 for oxidative chemistry, but there is still a great deal of space for improvement in this area. In particular, there is a need to build ligand scaffolds that balance stabilizing the active oxidant and providing sufficient activation of the active oxidant to enable catalytic reactivity. Furthermore, a balance between building a ligand scaffold that minimizes intramolecular oxidation while also providing coordination and reaction space for a wide range of substrates to bind and undergo oxidation must be found. Currently, much of the research in this area functions as fundamental chemistry research, which demonstrates the viability of the approach but lacks applicability to a wide substrate scope. Future work should focus on bridging metal–ligand cooperativity in conjunction with O2 with the field of organic methodology to make transition metal complexes incorporating metal–ligand cooperativity an essential part of a synthetic chemist's toolkit. While this will require further fundamental studies on the principles that govern cooperative activation of O2, more applied investigations into tuning these systems for catalysis is also needed.Author contributions
Both authors contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. A. J. would like to thank Los Alamos National Lab and the NNSA for funding (LA-UR-24-25279). We would like to thank Dr Justin Felder and S. J. for helpful discussions. J. S. A. would like to thank the NIGMS (R35GM133470) and the Dreyfus Foundation for a Teacher-Scholar Award (TC-21-064).References
- M. L. Pegis, J. A. S. Roberts, D. J. Wasylenko, E. A. Mader, A. M. Appel and J. M. Mayer, Standard Reduction Potentials for Oxygen and Carbon Dioxide Couples in Acetonitrile and N,N-Dimethylformamide, Inorg. Chem., 2015, 54, 11883–11888 CrossRef CAS PubMed
.
-
A. J. Appleby, Encyclopedia of Electrochemical Power Sources, 2009, pp. 810–852 Search PubMed
- R. G. Agarwal, S. C. Coste, B. D. Groff, A. M. Heuer, H. Noh, G. A. Parada, C. F. Wise, E. M. Nichols, J. J. Warren and J. M. Mayer, Free Energies of Proton-Coupled Electron Transfer Reagents and Their Applications, Chem. Rev., 2022, 122, 1–49 CrossRef CAS PubMed
- J. K. Stille and R. Divakaruni, Mechanism of the Wacker Process. Stereochemistry of the Hydroxypalladation, J. Organomet. Chem., 1979, 169, 239–248 CrossRef CAS
- J. A. Keith and P. M. Henry, The Mechanism of the Wacker Reaction: A Tale of Two Hydroxypalladations, Angew. Chem., Int. Ed., 2009, 48, 9038–9049 CrossRef CAS PubMed
- J. Smidt, W. Hafner, R. Jira, J. Sedlmeier, R. Sieber, R. Rüttinger and H. Kojer, Katalytische Umsetzungen von Olefinen an Platinmetall-Verbindungen Das Consortium-Verfahren zur Herstellung von Acetaldehyd, Angew. Chem., 1959, 71, 176–182 CrossRef CAS
- C. W. Anson, S. Ghosh, S. Hammes-Schiffer and S. S. Stahl, Co(salophen)-Catalyzed Aerobic Oxidation of p-Hydroquinone: Mechanism and Implications for Aerobic Oxidation Catalysis, J. Am. Chem. Soc., 2016, 138, 4186–4193 CrossRef CAS PubMed
- J. S. Bates, S. Biswas, S. E. Suh, M. R. Johnson, B. Mondal, T. W. Root and S. S. Stahl, Chemical and Electrochemical O2 Reduction on Earth-Abundant M-N-C Catalysts and Implications for Mediated Electrolysis, J. Am. Chem. Soc., 2022, 144, 922–927 CrossRef CAS PubMed
- C. W. Anson and S. S. Stahl, Cooperative Electrocatalytic O2 Reduction Involving Co(salophen) with p-Hydroquinone as an Electron-Proton Transfer Mediator, J. Am. Chem. Soc., 2017, 139, 18472–18475 CrossRef CAS PubMed
- J. B. Gerken and S. S. Stahl, High-Potential Electrocatalytic O2 Reduction with Nitroxyl/NOx Mediators: Implications for Fuel Cells and Aerobic Oxidation Catalysis, ACS Cent. Sci., 2015, 1, 234–243 CrossRef CAS PubMed
- S. J. Tereniak, C. R. Landis and S. S. Stahl, Are Phosphines Viable Ligands for Pd-Catalyzed Aerobic Oxidation Reactions? Contrasting Insights from a Survey of Six Reactions, ACS Catal., 2018, 8, 3708–3714 CrossRef CAS
- M. Rafiee, Z. M. Konz, M. D. Graaf, H. F. Koolman and S. S. Stahl, Electrochemical Oxidation of Alcohols and Aldehydes to Carboxylic Acids Catalyzed by 4-Acetamido-TEMPO: An Alternative to ‘anelli’ and ‘pinnick’ Oxidations, ACS Catal., 2018, 8, 6738–6744 CrossRef CAS
-
K. K. Singh and I. Garcia-Bosch, in Redox - Active Ligands, Wiley, 2024, pp. 175–195 Search PubMed
- S. Wertz and A. Studer, Nitroxide-Catalyzed Transition-Metal-Free Aerobic Oxidation Processes, Green Chem., 2013, 15, 3116–3134 RSC
- D. Wang, A. B. Weinstein, P. B. White and S. S. Stahl, Ligand-Promoted Palladium-Catalyzed Aerobic Oxidation Reactions, Chem. Rev., 2018, 118, 2636–2679 CrossRef CAS PubMed
- Z. Ma, K. T. Mahmudov, V. A. Aliyeva, A. V. Gurbanov and A. J. L. Pombeiro, TEMPO in Metal Complex Catalysis, Coord. Chem. Rev., 2020, 423, 213482–213506 CrossRef CAS
- D. Leifert and A. Studer, Organic Synthesis Using Nitroxides, Chem. Rev., 2023, 123, 10302–10380 CrossRef CAS PubMed
- V. A. Schmidt and E. J. Alexanian, Metal-Free, Aerobic Dioxygenation of Alkenes Using Hydroxamic Acids, Angew. Chem., Int. Ed., 2010, 49, 4491–4494 CrossRef CAS PubMed
- X. Wang and D. Z. Wang, Aerobic Oxidation of Secondary Benzylic Alcohols and Direct Oxidative Amidation of Aryl Aldehydes Promoted by Sodium Hydride, Tetrahedron, 2011, 67, 3406–3411 CrossRef CAS
- R. A. Sheldon and I. W. C. E. Arenas, Organocatalytic Oxidations Mediated by Nitroxyl Radicals, Adv. Synth. Catal., 2004, 346, 1051–1071 CrossRef CAS
- V. A. Schmidt and E. J. Alexanian, Metal-Free, Aerobic Ketooxygenation of Alkenes Using Hydroxamic Acids, Chem. Sci., 2012, 3, 1672–1674 RSC
- B. C. Giglio, V. A. Schmidt and E. J. Alexanian, Metal-Free, Aerobic Dioxygenation of Alkenes Using Simple Hydroxamic Acid Derivatives, J. Am. Chem. Soc., 2011, 133, 13320–13322 CrossRef CAS PubMed
- G. Lv, H. Wang, Y. Yang, T. Deng, C. Chen, Y. Zhu and X. Hou, Graphene Oxide: A Convenient Metal-Free Carbocatalyst for Facilitating Aerobic Oxidation of 5-Hydroxymethylfurfural into 2, 5-Diformylfuran, ACS Catal., 2015, 5, 5636–5646 CrossRef CAS
- K. Chen and H. Xie, Selective Aerobic Oxidation Promoted by Highly Efficient Multi-Nitroxy Organocatalysts, Chin. J. Catal., 2017, 38, 625–635 CrossRef CAS
- L. Li, Y. L. Zhao, Q. Wang, T. Lin and Q. Liu, Base-Promoted Oxidative C-H Functionalization of α-Amino Carbonyl Compounds Under Mild Metal-Free Conditions: Using Molecular Oxygen as the Oxidant, Org. Lett., 2015, 17, 370–373 CrossRef CAS PubMed
- E. Romero, J. R. Gómez Castellanos, G. Gadda, M. W. Fraaije and A. Mattevi, Same Substrate, Many Reactions: Oxygen Activation in Flavoenzymes, Chem. Rev., 2018, 118, 1742–1769 CrossRef CAS PubMed
- M. Rolff, J. Schottenheim, H. Decker and F. Tuczek, Copper-O2 Reactivity of Tyrosinase Models Towards External Monophenolic Substrates: Molecular Mechanism and Comparison with the Enzyme, Chem. Soc. Rev., 2011, 40, 4077–4098 RSC
- A. T. Fiedler and A. A. Fischer, Oxygen Activation by Mononuclear Mn, Co, and Ni Centers in Biology and Synthetic Complexes, J. Biol. Inorg. Chem., 2017, 22, 407–424 CrossRef CAS PubMed
- E. I. Solomon, K. M. Light, L. V. Liu, M. Srnec and S. D. Wong, Geometric and Electronic Structure Contributions to Function in Non-Heme Iron Enzymes, Acc. Chem. Res., 2013, 46, 2725–2739 CrossRef CAS PubMed
- S. J. Lange and L. Clue, Oxygen Activating Nonheme Iron Enzymes, Curr. Opin. Chem. Biol., 1998, 2, 159–160 CrossRef CAS
- M. Newcomb, P. F. Hollenberg and M. J. Coon, Multiple Mechanisms and Multiple Oxidants in P450-Catalyzed Hydroxylations, Arch. Biochem. Biophys., 2003, 409, 72–79 CrossRef CAS PubMed
- D. Hamdane, H. Zhang and P. Hollenberg, Oxygen Activation by Cytochrome P450 Monooxygenase, Photosynth. Res., 2008, 98, 657–666 CrossRef CAS PubMed
- A. J. Jasniewski and L. Que, Dioxygen Activation by Nonheme Diiron Enzymes: Diverse Dioxygen Adducts, High-Valent Intermediates, and Related Model Complexes, Chem. Rev., 2018, 118, 2554–2592 CrossRef CAS
- S. Manna, W. J. Kong and J. E. Bäckvall, Adv. Catal., 2021, 69, 1–57 CAS
- Z. Gordon, T. J. Miller, C. A. Leahy, E. M. Matson, M. Burgess, M. J. Drummond, C. V. Popescu, C. M. Smith, R. L. Lord, J. Rodríguez-López and A. R. Fout, Characterization of Terminal Iron(III)-Oxo and Iron(III)-Hydroxo Complexes Derived from O2 Activation, Inorg. Chem., 2019, 58, 15801–15811 CrossRef CAS PubMed
- E. A. Hill, A. C. Weitz, E. Onderko, A. Romero-Rivera, Y. Guo, M. Swart, E. L. Bominaar, M. T. Green, M. P. Hendrich, D. C. Lacy and A. S. Borovik, Reactivity of an FeIV-Oxo Complex with Protons and Oxidants, J. Am. Chem. Soc., 2016, 138, 13143–13146 CrossRef CAS PubMed
- S. A. Cook and A. S. Borovik, Molecular Designs for Controlling the Local Environments around Metal Ions, Acc. Chem. Res., 2015, 48, 2407–2414 CrossRef CAS PubMed
- C. M. Moore and N. K. Szymczak, Redox-Induced Fluoride Ligand Dissociation Stabilized by Intramolecular Hydrogen Bonding, Chem. Commun., 2015, 51, 5490–5492 RSC
- M. L. Pegis, C. F. Wise, D. J. Martin and J. M. Mayer, Oxygen Reduction by Homogeneous Molecular Catalysts and Electrocatalysts, Chem. Rev., 2018, 118, 2340–2391 CrossRef CAS PubMed
- Q. Liao, T. Liu, S. I. Johnson, C. M. Klug, E. S. Wiedner, R. Morris Bullock and D. L. Dubois, Evaluation of Attractive Interactions in the Second Coordination Sphere of Iron Complexes Containing Pendant Amines, Dalton Trans., 2019, 48, 4867–4878 RSC
- N. D. Loewen, E. J. Thompson, M. Kagan, C. L. Banales, T. W. Myers, J. C. Fettinger and L. A. Berben, A Pendant Proton Shuttle on [Fe4N(CO)12]− Alters Product Selectivity in Formate vs. H2 Production via the Hydride [H-Fe4N(CO)12]−, Chem. Sci., 2016, 7, 2728–2735 RSC
- K. C. MacLeod, R. A. Lewis, D. E. DeRosha, B. Q. Mercado and P. L. Holland, C−H and C−N Activation at Redox-Active Pyridine Complexes of Iron, Angew. Chem., Int. Ed., 2017, 129, 1089–1092 CrossRef
- T. Zell and D. Milstein, Hydrogenation and Dehydrogenation Iron Pincer Catalysts Capable of Metal-Ligand Cooperation by Aromatization/Dearomatization, Acc. Chem. Res., 2015, 48, 1979–1994 CrossRef CAS PubMed
- M. Rauch, S. Kar, A. Kumar, L. Avram, L. J. W. Shimon and D. Milstein, Metal-Ligand Cooperation Facilitates Bond Activation and Catalytic Hydrogenation with Zinc Pincer Complexes, J. Am. Chem. Soc., 2020, 142, 14513–14521 CrossRef CAS PubMed
- M. R. Dubois and D. L. Dubois, Development of Molecular Electrocatalysts for CO2 Reduction and H2 Production/Oxidation, Acc. Chem. Res., 2009, 42, 1974–1982 CrossRef
- M. L. Pegis, D. J. Martin, C. F. Wise, A. C. Brezny, S. I. Johnson, L. E. Johnson, N. Kumar, S. Raugei and J. M. Mayer, Mechanism of Catalytic O2 Reduction by Iron Tetraphenylporphyrin, J. Am. Chem. Soc., 2020, 141, 8315–8326 CrossRef
- D. L. J. Broere, N. P. Van Leest, B. De Bruin, M. A. Siegler and J. I. Van Der Vlugt, Reversible Redox Chemistry and Catalytic C(sp3)-H Amination Reactivity of a Paramagnetic Pd Complex Bearing a Redox-Active o-Aminophenol-Derived NNO Pincer Ligand, Inorg. Chem., 2016, 55, 8603–8611 CrossRef CAS PubMed
- C. F. Harris, M. B. Bayless, N. P. Van Leest, Q. J. Bruch, B. N. Livesay, J. Bacsa, K. I. Hardcastle, M. P. Shores, B. De Bruin and J. D. Soper, Redox-Active Bis(phenolate) N-Heterocyclic Carbene [OCO] Pincer Ligands Support Cobalt Electron Transfer Series Spanning Four Oxidation States, Inorg. Chem., 2017, 56, 12421–12435 CrossRef CAS PubMed
- S. Chatterjee and T. K. Paine, Dioxygen Reduction and Bioinspired Oxidations by Non-heme Iron(II)−α-Hydroxy Acid Complexes, Acc. Chem. Res., 2023, 56, 3175–3187 CrossRef CAS PubMed
- O. R. Luca and R. H. Crabtree, Redox-active ligands in catalysis, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC
- J. L. Wong, R. H. Sánchez, J. G. Logan, R. A. Zarkesh, J. W. Ziller and A. F. Heyduk, Disulfide Reductive Elimination from an Iron(iii) Complex, Chem. Sci., 2013, 4, 1906–1910 RSC
- R. Arevalo and P. J. Chirik, Enabling Two-Electron Pathways with Iron and Cobalt: From Ligand Design to Catalytic Applications, J. Am. Chem. Soc., 2019, 141, 9106–9123 CrossRef CAS PubMed
- B. Bagh, D. L. J. Broere, V. Sinha, P. F. Kuijpers, N. P. Van Leest, B. De Bruin, S. Demeshko, M. A. Siegler and J. I. Van Der Vlugt, Catalytic Synthesis of N-Heterocycles via Direct C(sp3 )-H Amination Using an Air-Stable Iron(III) Species with a Redox-Active Ligand, J. Am. Chem. Soc., 2017, 139, 5117–5124 CrossRef CAS PubMed
- S. Sinha, S. Das, R. Sikari, S. Parua, P. Brandaõ, S. Demeshko, F. Meyer and N. D. Paul, Redox Noninnocent Azo-Aromatic Pincers and Their Iron Complexes. Isolation, Characterization, and Catalytic Alcohol Oxidation, Inorg. Chem., 2017, 56, 14084–14100 CrossRef CAS PubMed
- J. C. Ott, D. Bürgy, H. Guan and L. H. Gade, 3d Metal Complexes in T-shaped Geometry as a Gateway to Metalloradical Reactivity, Acc. Chem. Res., 2022, 55, 857–868 CrossRef CAS PubMed
- X. Lu, S. Wang and J. H. Qin, Isolating Fe-O2 Intermediates in Dioxygen Activation by Iron Porphyrin Complexes, Molecules, 2022, 27, 4690–4706 CrossRef CAS PubMed
- T. Liu, Q. Liao, M. O'Hagan, E. B. Hulley, D. L. DuBois and R. M. Bullock, Iron Complexes Bearing Diphosphine Ligands with Positioned Pendant Amines as Electrocatalysts for the Oxidation of H2, Organometallics, 2015, 34, 2747–2764 CrossRef CAS
- V. Lyaskovskyy and B. De Bruin, Redox Non-innocent Ligands: Versatile new Tools to Control Catalytic Reactions, ACS Catal., 2012, 2, 270–279 CrossRef CAS
- W. Kaim, Manifestations of Noninnocent Ligand Behavior, Inorg. Chem., 2011, 50, 9752–9765 CrossRef CAS
- K. A. Jesse, M. C. Chang, A. S. Filatov and J. S. Anderson, Iron(II) Complexes Featuring a Redox-Active Dihydrazonopyrrole Ligand, Z. Anorg. Allg. Chem., 2021, 647, 1415–1420 CrossRef CAS PubMed
- K. T. Horak and T. Agapie, Dioxygen Reduction by a Pd(0)-Hydroquinone Diphosphine Complex, J. Am. Chem. Soc., 2016, 138, 3443–3452 CrossRef CAS PubMed
- J. T. Henthorn and T. Agapie, Combination of Redox-Active Ligand and Lewis Acid for Dioxygen Reduction with π-Bound Molybdenum-Quinonoid Complexes, J. Am. Chem. Soc., 2015, 137, 1458–1464 CrossRef CAS PubMed
- L. Senft, J. L. Moore, A. Franke, K. R. Fisher, A. Scheitler, A. Zahl, R. Puchta, D. Fehn, S. Ison, S. Sader, I. Ivanović-Burmazović and C. R. Goldsmith, Quinol-Containing Ligands Enable High Superoxide Dismutase Activity by Modulating Coordination Number, Charge, Oxidation States and Stability of Manganese Complexes Throughout Redox Cycling, Chem. Sci., 2021, 12, 10483–10500 RSC
- M. B. Ward, A. Scheitler, M. Yu, L. Senft, A. S. Zillmann, J. D. Gorden, D. D. Schwartz, I. Ivanović-Burmazović and C. R. Goldsmith, Superoxide Dismutase Activity Enabled by a Redox-Active Ligand Rather than Metal, Nat. Chem., 2018, 10, 1207–1212 CrossRef CAS PubMed
- S. V. Obisesan, C. Rose, B. H. Farnum and C. R. Goldsmith, Co(II) Complex with a Covalently Attached Pendent Quinol Selectively Reduces O2 to H2O, J. Am. Chem. Soc., 2022, 144, 22826–22830 CrossRef CAS PubMed
- S. L. Hooe, E. N. Cook, A. G. Reid and C. W. Machan, Non-Covalent Assembly of Proton Conors and p-Benzoquinone Anions for Co-Electrocatalytic Reduction of Dioxygen, Chem. Sci., 2021, 12, 9733–9741 RSC
- A. S. Borovik, Bioinspired Hydrogen Bond Motifs in Ligand Design: The Role of Noncovalent Interactions in Metal Ion Mediated Activation of Dioxygen, Acc. Chem. Res., 2005, 38, 54–61 CrossRef CAS PubMed
- M. J. Drummond, C. L. Ford, D. L. Gray, C. V. Popescu and A. R. Fout, Radical Rebound Hydroxylation Versus H-Atom Transfer in Non-Heme Iron(III)-Hydroxo Complexes: Reactivity and Structural Differentiation, J. Am. Chem. Soc., 2019, 141, 6639–6650 CrossRef CAS PubMed
- E. W. Dahl, J. J. Kiernicki, M. Zeller and N. K. Szymczak, Hydrogen Bonds Dictate O2 Capture and Release within a Zinc Tripod, J. Am. Chem. Soc., 2018, 140, 10075–10079 CrossRef CAS PubMed
- R. L. Shook, S. M. Peterson, J. Greaves, C. Moore, A. L. Rheingold and A. S. Borovik, Catalytic Reduction of Dioxygen to Water with a Monomeric Manganese Complex at Room Temperature, J. Am. Chem. Soc., 2011, 133, 5810–5817 CrossRef CAS PubMed
- K. Sagar, M. Kim, T. Wu, S. Zhang, E. L. Bominaar, M. A. Siegler, M. Hendrich and I. Garcia-Bosch, Mimicking the Reactivity of LPMOs with a Mononuclear Cu Complex, Eur. J. Inorg. Chem., 2024, 27, 1–11 CrossRef
- Y. Jiang, J. Telser and D. P. Goldberg, Evidence for the Formation of a Mononuclear Ferric-Hydroperoxo Complex via the Reaction of Dioxygen with an (N4S(thiolate))iron(II) Complex, Chem. Commun., 2009, 6828–6830 RSC
- A. F. Pinto, J. V. Rodrigues and M. Teixeira, Reductive Elimination of Superoxide: Structure and Mechanism of Superoxide Reductases, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 285–297 CrossRef CAS PubMed
- B. Meunier, S. P. de Visser and S. Shaik, Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS PubMed
- S. Kal and L. Que, Activation of a Non-Heme Fe III -OOH by a Second Fe III to Hydroxylate Strong C−H Bonds: Possible Implications for Soluble Methane Monooxygenase, Angew. Chem., 2019, 131, 8572–8576 CrossRef
- E. Nam, P. E. Alokolaro, R. D. Swartz, M. C. Gleaves, J. Pikul and J. A. Kovacs, Investigation of the Mechanism of Gormation of a Thiolate-Ligated Fe(III)-OOH, Inorg. Chem., 2011, 50, 1592–1602 CrossRef CAS PubMed
- T. Kitagawa, A. Dey, P. Lugo-Mas, J. B. Benedict, W. Kaminsky, E. Solomon and J. A. Kovacs, A Functional Model for the Cysteinate-Ligated Non-Heme Iron Enzyme Superoxide Reductase (SOR), J. Am. Chem. Soc., 2006, 128, 14448–14449 CrossRef CAS PubMed
- S. Xu, J. J. Veach, W. N. Oloo, K. C. Peters, J. Wang, R. H. Perry and L. Que, Detection of a Transient FeV(O)(OH) Species Involved in Olefin Oxidation by a Bio-Inspired Non-Haem Iron Catalyst, Chem. Commun., 2018, 54, 8701–8704 RSC
- J. Serrano-Plana, F. Acuña-Parés, V. Dantignana, W. N. Oloo, E. Castillo, A. Draksharapu, C. J. Whiteoak, V. Martin-Diaconescu, M. G. Basallote, J. M. Luis, L. Que, M. Costas and A. Company, Acid-Triggered O−O Bond Heterolysis of a Nonheme FeIII(OOH) Species for the Stereospecific Hydroxylation of Strong C−H Bonds, Chem.–Eur. J., 2018, 24, 5331–5340 CrossRef CAS PubMed
- J. A. Kovacs and L. M. Brines, Understanding how the Thiolate Sulfur Contributes to the Function of the Non-Heme Iron Enzyme Superoxide Reductase, Acc. Chem. Res., 2007, 40, 501–509 CrossRef CAS
- A. J. Simaan, F. Banse, J. J. Girerd, K. Wieghardt and E. Bill, The Electronic Structure of Non-Heme Iron(III)-Hydroperoxo and Iron(III)-Peroxo Model Complexes Studied by Mössbauer and Electron Paramagnetic Resonance Spectroscopies, Inorg. Chem., 2001, 40, 6538–6540 CrossRef CAS PubMed
- F. Li, K. K. Meier, M. A. Cranswick, M. Chakrabarti, K. M. Van Heuvelen, E. Münck and L. Que, Characterization of a High-Spin Non-Heme FeIII-OOH Intermediate and its Quantitative Conversion to an FeIV=O Complex, J. Am. Chem. Soc., 2011, 133, 7256–7259 CrossRef CAS PubMed
- C. Wegeberg, W. R. Browne and C. J. McKenzie, Cis Donor Influence on O-O Bond Lability in Iron(III) Hydroperoxo Complexes: Oxidation Catalysis and Ligand Transformation, Inorg. Chem., 2019, 58, 8983–8994 CrossRef CAS PubMed
- S. Hong, Y. M. Lee, W. Shin, S. Fukuzumi and W. Nam, Dioxygen Activation by Mononuclear Nonheme Iron(II) Complexes Generates Iron-Oxygen Intermediates in the Presence of an NADH Analogue and Proton, J. Am. Chem. Soc., 2009, 131, 13910–13911 CrossRef CAS PubMed
- S. Panda, H. Phan, E. M. Dunietz, M. T. Brueggemeyer, P. K. Hota, M. A. Siegler, A. Jose, M. Bhadra, E. I. Solomon and K. D. Karlin, Intramolecular Phenolic H-Atom Abstraction by a N3 ArOH Ligand-Supported (μ-η2:η2 -Peroxo)dicopper(II) Species Relevant to the Active Site Function of oxy-Tyrosinase, J. Am. Chem. Soc., 2024, 146, 14942–14947 CrossRef CAS PubMed
- I. Kipouros, A. Stanczak, J. W. Ginsbach, P. C. Andrikopoulos, L. Rulısek and E. I. Solomon, Elucidation of the Tyrosinase/O2/Monophenol Ternary Intermediate that Dictates the Monooxygenation Mechanism in Melanin Biosynthesis, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, 1–10 CrossRef PubMed
- I. Kipouros, A. Stańczak, E. M. Dunietz, J. W. Ginsbach, M. Srnec, L. Rulíšek and E. I. Solomon, Experimental Evidence and Mechanistic Description of the Phenolic H-Transfer to the Cu2O2 Active Site of oxy-Tyrosinase, J. Am. Chem. Soc., 2023, 145, 22866–22870 CrossRef CAS PubMed
- K. Rajabimoghadam, Y. Darwish, U. Bashir, D. Pitman, S. Eichelberger, M. A. Siegler, M. Swart and I. Garcia-Bosch, Catalytic Aerobic Oxidation of Alcohols by Copper Complexes Bearing Redox-Active Ligands with Tunable H-Bonding Groups, J. Am. Chem. Soc., 2018, 140, 16625–16634 CrossRef CAS PubMed
- B. J. Charette, J. W. Ziller and A. F. Heyduk, Exploring Ligand-Centered Hydride and H-Atom Transfer, Inorg. Chem., 2021, 60, 5367–5375 CrossRef CAS PubMed
- A. M. Hollas, J. W. Ziller and A. F. Heyduk, Three Oxidation States of the Bis(3,5-di-tert-butyl-2-phenolato)azanido Pincer Ligand on Chromium(III), Polyhedron, 2018, 143, 111–117 CrossRef CAS
- B. J. Charette, J. W. Ziller and A. F. Heyduk, Metal-Ion Influence on Ligand-Centered Hydrogen-Atom Transfer, Inorg. Chem., 2021, 60, 1579–1589 CrossRef CAS PubMed
-
R. A. Johnson and K. B. Sharpless, Comprehensive Organic Synthesis, 1991, vol. 7, pp. 389–436 Search PubMed
- Y. Wang, J. L. Dubois, B. Hedman, K. O. Hodgson and T. D. P. Stack, Catalytic Galactose Oxidase Models: Biomimetic Cu(II)-Phenoxyl-Radical Reactivity, Science, 1998, 279, 537–540 CrossRef CAS PubMed
- P. Chaudhuri, M. Hess, J. Müller, K. Hildenbrand, E. Bill, T. Weyhermüller and K. Wieghardt, Aerobic Oxidation of Primary Alcohols (Including Methanol) by Copper(II)- and Zinc(N)-Phenoxyl Radical Catalysts, J. Am. Chem. Soc., 1999, 121, 9599–9610 CrossRef CAS
- P. Chaudhuri, M. Hess, T. Weyhermüller and K. Wieghardt, Aerobic Oxidation of Primary Alcohols by a New Mononuclear CuII-Radical Catalyst, Angew. Chem., Int. Ed., 1999, 38, 1095–1098 CrossRef CAS PubMed
- I. E. Markó, P. R. Giles, M. Tsukazaki, S. M. Brown and C. J. Urch, Copper-Catalyzed Oxidation of Alcohols to Aldehydes and Ketones: An Efficient, Aerobic Alternative, Science, 1996, 25, 2044–2046 CrossRef PubMed
- I. E. Markó, A. Gautier, I. Chellé-Regnaut, P. R. Giles, M. Tsukazaki, C. J. Urch and S. M. Brown, Efficient and Practical Catalytic Oxidation of Alcohols Using Molecular Oxygen, J. Org. Chem., 1998, 63, 7576–7577 CrossRef
- J. M. Hoover, B. L. Ryland and S. S. Stahl, Mechanism of Copper(I)/TEMPO-Catalyzed Aerobic Alcohol Oxidation, J. Am. Chem. Soc., 2013, 135, 2357–2367 CrossRef CAS PubMed
- R. C. Walroth, K. C. Miles, J. T. Lukens, S. N. MacMillan, S. S. Stahl and K. M. Lancaster, Electronic Structural Analysis of Copper(II)-TEMPO/ABNO Complexes Provides Evidence for Copper(I)-Oxoammonium Character, J. Am. Chem. Soc., 2017, 139, 13507–13517 CrossRef CAS PubMed
- J. E. Nutting, K. Mao and S. S. Stahl, Iron(III) Nitrate/TEMPO-Catalyzed Aerobic Alcohol Oxidation: Distinguishing between Serial versus Integrated Redox Cooperativity, J. Am. Chem. Soc., 2021, 143, 10565–10570 CrossRef CAS PubMed
- S. Sinha, S. Das, R. Mondal, S. Mandal and N. D. Paul, Cobalt Complexes of Redox Noninnocent Azo-Aromatic Pincers. Isolation, Characterization, and Application as Catalysts for the Synthesis of Quinazolin-4(3: H)-ones, Dalton Trans., 2020, 49, 8448–8459 RSC
- M. E. Czaikowski, S. W. Anferov, A. P. Tascher and J. S. Anderson, Electrocatalytic Semihydrogenation of Terminal
Alkynes Using Ligand-Based Transfer of Protons and Electrons, J. Am. Chem. Soc., 2024, 146, 476–486 CrossRef CAS PubMed
- A. J. McNeece, K. A. Jesse, A. S. Filatov, J. E. Schneider and J. S. Anderson, Catalytic Hydrogenation Enabled by Ligand-Based Storage of Hydrogen, Chem. Commun., 2021, 57, 3869–3872 RSC
- K. A. Jesse, S. W. Anferov, K. A. Collins, J. A. Valdez-Moreira, M. E. Czaikowski, A. S. Filatov and J. S. Anderson, Direct Aerobic Generation of a Ferric Hydroperoxo Intermediate Via a Preorganized Secondary Coordination Sphere, J. Am. Chem. Soc., 2021, 143, 18121–18130 CrossRef CAS PubMed
- A. J. Mcneece, K. A. Jesse, J. Xie, A. S. Filatov and J. S. Anderson, Generation and Oxidative Reactivity of a Ni(II) Superoxo Complex via Ligand-Based Redox Non-Innocence, J. Am. Chem. Soc., 2020, 142, 10824–10832 CrossRef CAS PubMed
- J. Jeoung, D. Nianios, S. Fetzner and H. Dobbek, Quercetin-2,4-Dioxygenase Aktiviert Sauerstoff in Einem “side-on” Gebundenen O2 -Ni-Komplex, Angew. Chem., 2016, 128, 3339–3343 CrossRef
- H. Li, X. Wang, G. Tian and Y. Liu, Insights into the Dioxygen Activation and Catalytic Mechanism of the Nickel-Containing Quercetinase, Catal. Sci. Technol., 2018, 8, 2340–2351 RSC
- M. E. Czaikowski, A. J. McNeece, J.-N. Boyn, K. A. Jesse, S. W. Anferov, A. S. Filatov, D. A. Mazziotti and J. S. Anderson, Generation and Aerobic Oxidative Catalysis of a Cu(II) Superoxo Complex Supported by a Redox-Active Ligand, J. Am. Chem. Soc., 2022, 144, 15569–15580 CrossRef CAS PubMed
- S. W. Anferov, A. S. Filatov and J. S. Anderson, Cobalt-Catalyzed Hydrogenation Reactions Enabled by Ligand-Based Storage of Dihydrogen, ACS Catal., 2022, 12, 9933–9943 CrossRef CAS PubMed
- M. L. Scheuermann, U. Fekl, W. Kaminsky and K. I. Goldberg, Metal-Ligand Cooperativity in OO Activation: Observation of a ‘Pt-O-O-C’ Peroxo Intermediate, Organometallics, 2010, 29, 4749–4751 CrossRef CAS
- M. L. Scheuermann, A. T. Luedtke, S. K. Hanson, U. Fekl, W. Kaminsky and K. I. Goldberg, Reactions of Five-Coordinate Platinum(IV) Complexes with Molecular Oxygen, Organometallics, 2013, 32, 4752–4758 CrossRef CAS
- M. K. Goetz, J. E. Schneider, A. S. Filatov, K. A. Jesse and J. S. Anderson, Enzyme-Like Hydroxylation of Aliphatic C-H Bonds from an Isolable Co-Oxo Complex, J. Am. Chem. Soc., 2021, 143, 20849–20862 CrossRef CAS PubMed
- S. Shi, Y. Wang, A. Xu, H. Wang, D. Zhu, S. B. Roy, T. A. Jackson, D. H. Busch and G. Yin, Distinct Reactivity Differences of Metal Oxo and its Corresponding Hydroxo Moieties in Oxidations: Implications from a Manganese(IV) Complex having Dihydroxide Ligand, Angew. Chem., Int. Ed., 2011, 50, 7321–7324 CrossRef CAS PubMed
- A. Dauth, U. Gellrich, Y. Diskin-Posner, Y. Ben-David and D. Milstein, The Ferraquinone-Ferrahydroquinone Couple: Combining Quinonic and Metal-Based Reactivity, J. Am. Chem. Soc., 2017, 139, 2799–2807 CrossRef CAS PubMed
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates, Chem. Rev., 2004, 104, 939–986 CrossRef CAS PubMed
- M. C. Chang, A. J. McNeece, E. A. Hill, A. S. Filatov and J. S. Anderson, Ligand-Based Storage of Protons and Electrons in Dihydrazonopyrrole Complexes of Nickel, Chem.–Eur. J., 2018, 24, 8001–8008 CrossRef CAS PubMed
- G. K. Fukin, E. V. Baranov, A. I. Poddel'Sky, V. K. Cherkasov and G. A. Abakumov, Reversible Binding of Molecular Oxygen to Catecholate and Amidophenolate Complexes of SbV: Electronic and Steric Factors, ChemPhysChem, 2012, 13, 3773–3776 CrossRef CAS PubMed
- G. A. Abakumov, A. I. Poddel’sky, E. V. Grunova, V. K. Cherkasov, G. K. Fukin, Y. A. Kurskii and L. G. Abakumova, Reversible Binding of Dioxygen by a Non-Transition-Metal Complex, Angew. Chem., Int. Ed., 2005, 44, 2767–2771 CrossRef CAS PubMed
- S. Panda, S. K. Bera, P. Goel, A. K. Dutta and G. K. Lahiri, Ruthenium-Chelated Non-Innocent Bis(heterocyclo)methanides: A Mimicked β-Diketiminate, Inorg. Chem., 2019, 58, 11458–11469 CrossRef CAS PubMed
- C. Van Stappen, Y. Deng, Y. Liu, H. Heidari, J. X. Wang, Y. Zhou, A. P. Ledray and Y. Lu, Designing Artificial Metalloenzymes by Tuning of the Environment beyond the Primary Coordination Sphere, Chem. Rev., 2022, 122, 11974–12045 CrossRef CAS PubMed
- M. A. Ehudin, L. B. Gee, S. Sabuncu, A. Braun, P. Moënne-Loccoz, B. Hedman, K. O. Hodgson, E. I. Solomon and K. D. Karlin, Tuning the Geometric and Electronic Structure of Synthetic High-Valent Heme Iron(IV)-Oxo Models in the Presence of a Lewis Acid and Various Axial Ligands, J. Am. Chem. Soc., 2019, 141, 5942–5960 CrossRef CAS PubMed
- Y. Zhou, E. N. Mirts, S. Yook, M. Waugh, R. Martini, Y. S. Jin and Y. Lu, Reshaping the 2-Pyrone Synthase Active Site for Chemoselective Biosynthesis of Polyketides, Angew. Chem., Int. Ed., 2023, 62, 1–9 Search PubMed
- M. A. Ehudin, A. W. Schaefer, S. M. Adam, D. A. Quist, D. E. Diaz, J. A. Tang, E. I. Solomon and K. D. Karlin, Influence of Intramolecular Secondary Sphere Hydrogen-Bonding Interactions on Cytochrome: C Oxidase Inspired Low-Spin Heme-Peroxo-Copper Complexes, Chem. Sci., 2019, 10, 2893–2905 RSC
| This journal is © The Royal Society of Chemistry 2024 |