
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
6,6′-Biindeno[1,2-b]fluorene: an open-shell indenofluorene dimer†
Himanshu
Sharma
 a,
Palash
Jana
b,
Dibyendu
Mallick
c,
Subhajit
Bandyopadhyay
b and
Soumyajit
Das
*a
a,
Palash
Jana
b,
Dibyendu
Mallick
c,
Subhajit
Bandyopadhyay
b and
Soumyajit
Das
*a
aDepartment of Chemistry, Indian Institute of Technology Ropar, Rupnagar 140001, Punjab, India. E-mail: chmsdas@iitrpr.ac.in
bDepartment of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur 741246, West Bengal, India
cDepartment of Chemistry, Presidency University, Kolkata 700073, West Bengal, India
First published on 18th November 2024
Abstract
Nakano et al. reported that the antiaromatic indenofluorene (IF) isomers are diradicaloid molecules having varying degrees of open-shell character, with indeno[1,2-b]fluorene displaying a weaker diradical character index (y0 = 0.072). Unlike 6,12-trimethylsilylethynyl disubstituted [1,2-b]IF, the 6,12-aryl disubstituted [1,2-b]IF derivatives did not show any experimental evidence of diradical properties. This raised the question of whether a [1,2-b]IF dimer would prefer a closed-shell or an open-shell ground state. To address this, herein we report the synthesis of a 6,6′-biindeno[1,2-b]fluorene derivative, which is a [1,2-b]IF dimer, constructed by linking two [1,2-b]IF units with a C–C single bond at carbons 6 and 6′ bearing the largest orbital coefficients for the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO). The C6–C6′ linkage effectively narrowed the HOMO–LUMO gap while the strong desire to avoid s-indacene antiaromaticity restored two Clar sextets in two proaromatic para-quinodimethane subunits, resulting in an open-shell bifluorenylidene-type diradicaloid (y0 = 0.268) ground state with minor tetraradical character index (y1 = 0.007). The open-shell nature was confirmed by single crystal X-ray and electron paramagnetic resonance analyses, and supported by theoretical calculations.
Introduction
Open-shell (OS) singlet diradicaloid1 polycyclic hydrocarbons (PHs) have received widespread attention in the fields of organic synthesis,2 physical organic,3 structural organic,4 and organic materials5 chemistry. The recovery of aromaticity in proaromatic quinoidal PHs6 is a major driving force toward the OS ground state for higher order zethrenes,7 π-extended quinodimethanes (QDMs),8 anthenes,9 benzenoid10 and non-benzenoid acenes,11peri-acenoacenes,12 and formally antiaromatic indenofluorene (IF) isomers.13 The five IF isomers are predicted to be OS diradicaloids,13c,13f but the degree of diradical character (y0) for indeno[1,2-a]fluorene and indeno[2,1-b]fluorene is larger than that of the other three isomers including indeno[1,2-b]fluorene (y0 = 0.072). Indeed, 6,12-aryl and 6,12-triisopropylsilylethynyl (TIPSE) disubstituted [1,2-b]IFs14 didn't show any experimental evidence of OS diradicaloid features despite carrying a proaromatic para-QDM (p-QDM shown in red, Fig. 1a) subunit in the π-backbone. Antiaromatic molecules are regarded as delocalized diradicals.15 Zhao reported that replacing bulkier TIPSE with a trimethylsilylethynyl group in [1,2-b]IF resulted in cyclo-oligomeric products due to diradical contribution of the [1,2-b]IF,16 clearly indicating its OS character.13c,17 | ||
| Fig. 1 (a) Antiaromatic indeno[1,2-b]fluorene derivatives 1-M, 1-MF and 2; (b) Zimmerman and Stępień's tetrafluorenofulvalene (TFF) 3; (c) our reported 3,3′-biindeno[1,2-b]fluorene derivative 4; (d) closed-shell structure of targeted mesityl disubstituted 6,6′-biindeno[1,2-b]fluorene 5 and its representative open-shell (OS) diradicaloid bifluorenylidene-type structure 5-OS1 and tetraradicaloid biindenofluorene-type structure 5-OS2. | ||
Aryl substitutions on the apical carbons (C6 and C12, Fig. 1a) of [1,2-b]IF for either symmetrical14b1-M or unsymmetrical181-MF didn't allow any diradical properties to be observed experimentally, despite lowering of the highest occupied molecular orbital (HOMO)-lowest unoccupied molecular orbital (LUMO) energy gap (hereafter denoted as HLG) in 1-MF than 1-M due to a smaller dihedral angle between the C6F5 unit and IF backbone enabling a greater π-delocalization. Quinoidal π-extension of the s-indacene core in the form of 2,6-anthraquinodimethane (2,6-AQDM) may result in an OS π-extended [1,2-b]IF,19 similar to non-benzenoid bis(phenalenyl)20 and benzenoid nonazethrene7b bearing a 2,6-AQDM core. While the 2,6-naphthaquinodimethane (2,6-NQDM)-embedded π-extended [1,2-b]IF was originally reported to be closed-shell (CS), its OS properties were accessed subsequently only above 200 °C.21 Notably, benzenoid octazethrene containing a 2,6-NQDM subunit is an OS molecule.22
Alongside aromaticity recovery, a small HLG is crucial23 for PHs displaying OS ground state. Extension of the π-delocalization path is the key to decrease the HLG. In this regard, Yamashita's work on anthryl disubstituted [1,2-b]IF 2 is noteworthy (Fig. 1a).24 Molecule 2 displayed a CS ground state, though one can draw structure 2-OS with three additional aromatic benzene rings (Clar sextets7d). Presumably, the large dihedral angle (∼76°) between the anthryl groups and IF backbone for 2 inhibits efficient π-delocalization, and thus recovery of the three Clar sextets in 2-OS has negligible contribution to the ground state structure. Sterically overcrowded alkene bridged PHs have lately gained attention as novel OS diradicaloids as they release steric strain and recover Clar sextets when switching from the Kekulé-type CS quinoidal form to OS diradical form.25 Tetrafluorenofulvalene (TFF)263 is the latest example among them bearing two π-extended [1,2-b]IF units connected through an apical olefinic C–C linkage (Fig. 1b). TFF 3 was viewed26 as a diradicaloid 3-OS bearing diindeno-fused 6,6′-biindeno[1,2-b]fluorene (6,6′-[1,2-b]BIF unit shown in blue for 3). While 3 clearly exhibited an OS singlet ground state with a small singlet(S)–triplet(T) energy gap (ΔES–T) due to the release of steric strain and recovery of Clar sextets as it switches from its non-aromatic olefinic form to polyradical forms, what remained unknown is whether the 6,6′-[1,2-b]BIF motif without diindeno-radical (shown in black for 3-OS) fusion exhibits OS properties.
We envisaged the design of 12,12′-dimesityl-6,6′-biindeno[1,2-b]fluorene 5 that can recover two additional Clar sextets in its OS diradical 5-OS1 and tetraradical 5-OS2 forms (Fig. 1d). The 5-OS2 and 5 forms are viewed as biindenofluorene (BIF) motifs while 5-OS1 is a diindeno-fused bifluorenylidene (BF) diradical. BF usually prefers a twisted form over the folded form,25a and BF-based materials are promising hole and electron transporters in organic photovoltaics.27 Interestingly, an extended quinoidal form of 6,6′-[1,2-b]BIF was theoretically shown to contribute to a poly-IF which is a π-elongated BF-type structure.28 However, synthesis of a hypothetical smaller quinoidal fragment like 5, which majorly contributes to the ground state properties of Scherf's poly-IF,28 remained unknown thus far. Our recently reported BIF 4 (Fig. 1c) displayed a CS ground state, which was formed by dimerizing two [1,2-b]IF units through carbons 3 and 3′ with appreciable HOMO and LUMO coefficients.29 Because the apical carbons 6/6′(12) contain larger HOMO and LUMO distributions than those of carbons 3/3′(9),29 we hypothesized that dimerizing two [1,2-b]IF units by apical carbons 6 and 6′ may significantly influence both HOMO and LUMO energy levels in 5.
Moreover, the large alternation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C–C bonds of the p-QDM subunit of s-indacene makes 5 a polyene-like system considering the apical π-conjugation path resembling a dodecahexaene subunit (shown in red for 5, Fig. 1d). This polyene subunit may act as a plausible π-delocalization path due to a smaller inter-IF torsional angle. It is known that the C–C double bond for ethylene is quite short, while in longer polyene chain, an increase of the C–C double bond and shortening of the adjacent C–C single bond may occur, leading to an extension of π-conjugation and a decrease of HLG.30 A small torsional angle may lead to a greater electronic communication between the IF units for 5, resulting in a smaller HLG. The hypotheses of a smaller HLG for the BIF 5 than that of 4 and recovery of two Clar sextets in OS forms for 5 enthused us to explore the ground state properties of 5 by computational and experimental approaches.
C/C–C bonds of the p-QDM subunit of s-indacene makes 5 a polyene-like system considering the apical π-conjugation path resembling a dodecahexaene subunit (shown in red for 5, Fig. 1d). This polyene subunit may act as a plausible π-delocalization path due to a smaller inter-IF torsional angle. It is known that the C–C double bond for ethylene is quite short, while in longer polyene chain, an increase of the C–C double bond and shortening of the adjacent C–C single bond may occur, leading to an extension of π-conjugation and a decrease of HLG.30 A small torsional angle may lead to a greater electronic communication between the IF units for 5, resulting in a smaller HLG. The hypotheses of a smaller HLG for the BIF 5 than that of 4 and recovery of two Clar sextets in OS forms for 5 enthused us to explore the ground state properties of 5 by computational and experimental approaches.
Results and discussion
Since a para-quinoidal 6,6′-[1,2-b]BIF-type π-extended structure is reported to dominate the ground state of BF-type poly-IF to favor mutual distortion,28 we first conducted density functional theory (DFT) calculations to examine if 5 is OS in the ground state. The DFT calculations of 5, using the same level of theory ((U)CAM-B3LYP/6-31G(d,p)) as that used for 3,26 suggested that the energy of the OS singlet state is 4.85 kcal mol−1 lower than that of the triplet state (i.e. ΔES–T = −4.85 kcal mol−1, Table S1†). The frontier molecular orbital (FMO) profiles of singly occupied molecular orbitals (SOMO) display a characteristic disjointed nature of the alpha (SOMO-α) and beta (SOMO-β) spins for the ground state singlet diradical (Fig. 2b and c), while the spin densities are found to be delocalized over the π-conjugated IF backbone with four apical carbons bearing the larger spin densities (Fig. 2a). The large spin densities on the four apical carbons imply singlet diradical and tetraradical characters, which were found to be 26.8% and 0.7%, respectively, based on the natural orbital occupancy number (NOON) calculations (see ESI†).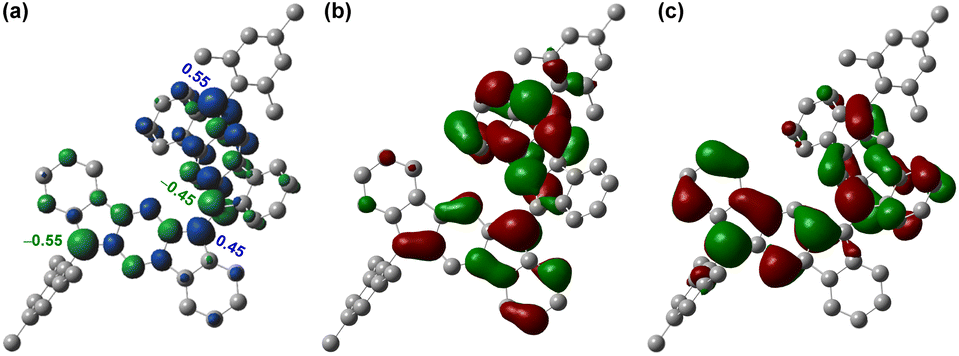 | ||
| Fig. 2 (a) Spin density map of 5. Isovalue for surfaces: MO = 0.02, density = 0.005; frontier molecular orbital profiles for the (b) α-spin and (c) β-spin of 5. | ||
The negligible tetraradical character of 5 may be attributed to the minor driving force (no additional Clar sextet recovery; only a minor inter-IF steric clash) toward 5-OS2 from 5-OS1 (Fig. 1d). However, the strong desire6a to avoid s-indacene antiaromaticity through recovery of two Clar sextets may drive quinoidal 5 to become diradicaloid 5-OS1 in the singlet ground state by enhancing the C6–C6′ π-bond character while locating radicals at apical carbons 12 and 12′ bearing the largest spin densities (Mulliken spin density of 0.55). As molecules with intermediate diradical character are important for non-linear optics, singlet fission and molecular electronics,3,5 we designed a sterically promoted synthetic approach to construct 5 and study its ground state characteristics experimentally.
BIF 5 was synthesized in multiple steps as illustrated in Scheme 1. 2,2′-Dibromo-9,9′-bifluorenylidene 6 was synthesized as a diastereomeric mixture following a literature method31 (Fig. S1†), and then it was subjected to a two-fold Suzuki coupling with (2-formylphenyl)boronic acid to afford dialdehyde 8 in 57% yield. Treatment of 8 with an excess of 2-mesitylmagnesium bromide at room temperature gave dicarbinol 9, which, without purification, was treated with BF3·Et2O in dichloromethane (DCM) to afford dihydro derivative 10 (46% yield over two steps) by utilizing the steric crowding of bulky mesityl (Mes) groups.18 Treatment of dihydro precursor 10 with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in 1,2-dichloroethane (1,2-DCE) for 2 h at 80 °C produced 5 as a blue solid in 75% yield after silica gel column chromatographic purification. While nuclear magnetic resonance (NMR, see ESI†) and high-resolution mass spectrometry (HRMS) analyses indicated the formation of desired product 5, unambiguous structural confirmation was obtained from single crystal X-ray diffractometry (SCXRD) analysis (Fig. 3a).
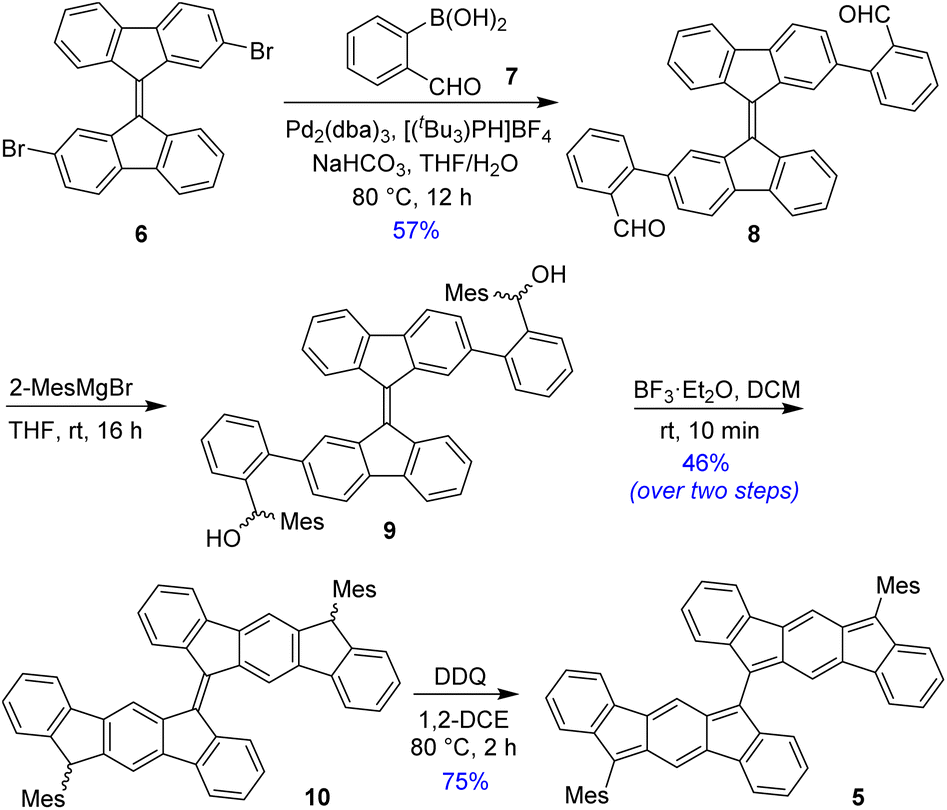 | ||
| Scheme 1 Synthesis of BIF 5. | ||
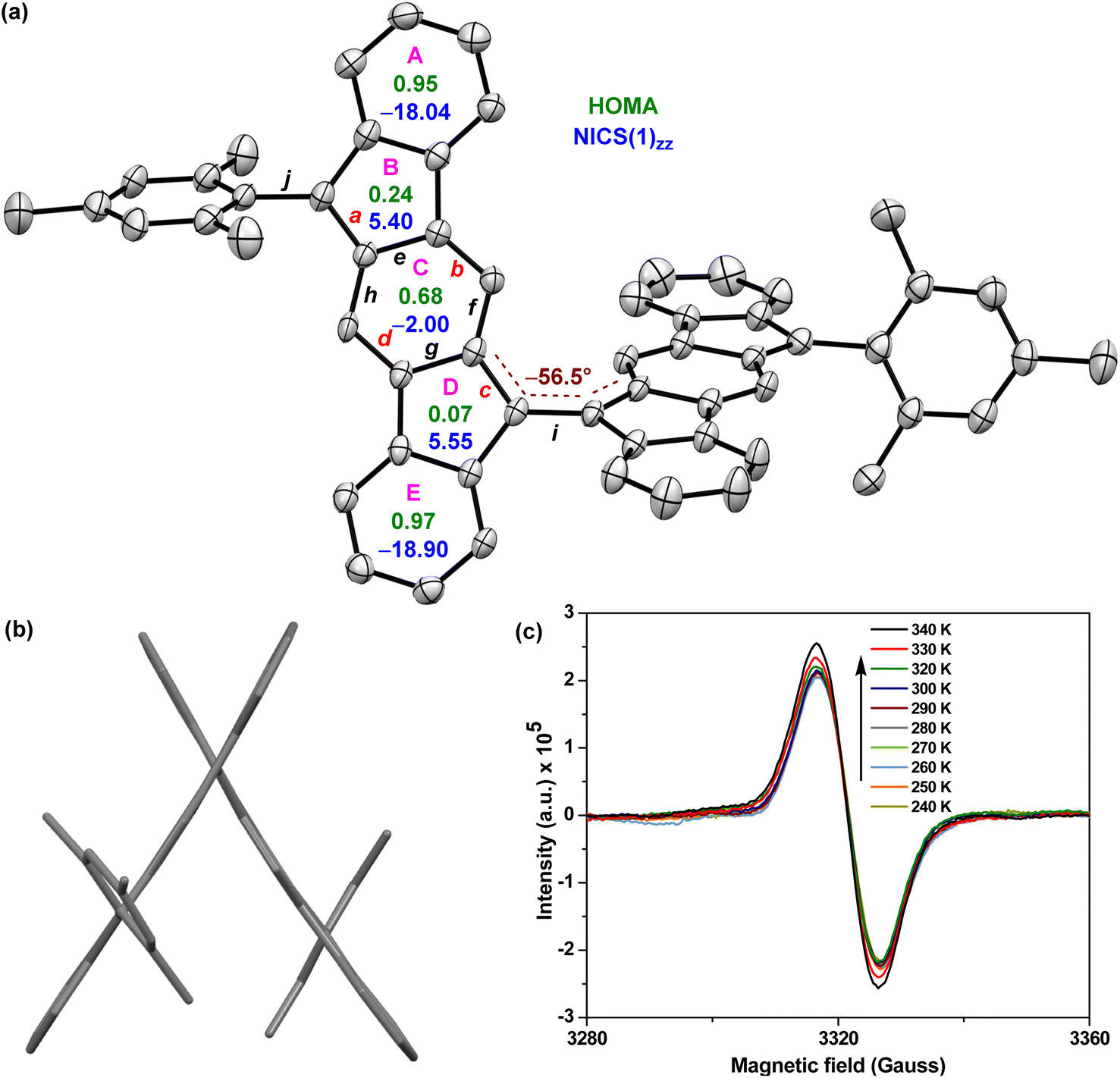 | ||
| Fig. 3 (a) X-ray crystallographic structure of 5 with the ellipsoids drawn at 30% probability level (hydrogens omitted), including NICS(1)zz (blue) and HOMA (green) indices; (b) side-view of 5; (c) EPR spectra of 5 recorded at variable temperatures for the powder form. | ||
Single crystals32 of 5 were obtained by slow solvent diffusion of methanol into DCM/carbon disulfide (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solution. As depicted in Fig. 3a and b, the two [1,2-b]IF units of twisted BIF 5 form a torsional angle of ∼56.5° which is much smaller than that of anthryl disubstituted [1,2-b]IF 2,24 suggesting a greater π-delocalization/electronic communication between two IF units. The bond lengths (bonds a to j are labelled in Fig. 3a) of 5 from SCXRD analyses are in line with those from DFT analyses, and are summarized in Table 1. The π-delocalization is evident from the C–C bond length analyses (Table 1; see the ESI† for full bond length analyses including e.s.d values), as an increase of the C–C double bond lengths (Csp2Csp2 bonds a, b, c, d) for the p-QDM subunit of 5 was clearly observed when compared to those of symmetrical 1-M (a/c and b/d) and unsymmetrical 1-MF. At the same time, the C–C single bond lengths (Csp2–Csp2 bonds e, f, g, h) of the p-QDM subunit of 5 were also found to be shorter than those of the 1-M (e/g and f/h). Moreover, the bond length for central C–C single bond i connecting the IF units for 5 was found to be 1.452 Å (DFT: 1.414 Å), which corresponds to a formal single bond in the CS configuration. However, it is shorter than the C–C single bond j (crystal: 1.501 Å; DFT: 1.480 Å) connecting the mesityl group and [1,2-b]IF core, and clearly shorter than the C–C single bond connecting the anthryl and [1,2-b]IF rings in 2 (1.492 Å).24 The distance is comparable to BIF 4 (1.451 Å),29 suggesting some π-bond character for the central C–C bond i in 5 which is strengthened due to π-electron delocalization.
:1) solution. As depicted in Fig. 3a and b, the two [1,2-b]IF units of twisted BIF 5 form a torsional angle of ∼56.5° which is much smaller than that of anthryl disubstituted [1,2-b]IF 2,24 suggesting a greater π-delocalization/electronic communication between two IF units. The bond lengths (bonds a to j are labelled in Fig. 3a) of 5 from SCXRD analyses are in line with those from DFT analyses, and are summarized in Table 1. The π-delocalization is evident from the C–C bond length analyses (Table 1; see the ESI† for full bond length analyses including e.s.d values), as an increase of the C–C double bond lengths (Csp2Csp2 bonds a, b, c, d) for the p-QDM subunit of 5 was clearly observed when compared to those of symmetrical 1-M (a/c and b/d) and unsymmetrical 1-MF. At the same time, the C–C single bond lengths (Csp2–Csp2 bonds e, f, g, h) of the p-QDM subunit of 5 were also found to be shorter than those of the 1-M (e/g and f/h). Moreover, the bond length for central C–C single bond i connecting the IF units for 5 was found to be 1.452 Å (DFT: 1.414 Å), which corresponds to a formal single bond in the CS configuration. However, it is shorter than the C–C single bond j (crystal: 1.501 Å; DFT: 1.480 Å) connecting the mesityl group and [1,2-b]IF core, and clearly shorter than the C–C single bond connecting the anthryl and [1,2-b]IF rings in 2 (1.492 Å).24 The distance is comparable to BIF 4 (1.451 Å),29 suggesting some π-bond character for the central C–C bond i in 5 which is strengthened due to π-electron delocalization.
| Bondsa | 5 | 5 | 1-MF | 1-M |
|---|---|---|---|---|
| a a–j bonds are labelled in Fig. 3a. b SCXRD data. c DFT data. d SCXRD data from ref. 18. e SCXRD data from ref. 14b. | ||||
| a | 1.392 | 1.403 | 1.372 | 1.380 |
| b | 1.364 | 1.368 | 1.349 | 1.356 |
| c | 1.392 | 1.429 | 1.373 | 1.380 |
| d | 1.367 | 1.371 | 1.346 | 1.356 |
| e | 1.456 | 1.441 | 1.433 | 1.467 |
| f | 1.415 | 1.416 | 1.456 | 1.433 |
| g | 1.459 | 1.436 | 1.435 | 1.467 |
| h | 1.416 | 1.414 | 1.457 | 1.433 |
| i | 1.452 | 1.414 | — | — |
| j | 1.501 | 1.480 | 1.482 | 1.484 |
The above findings clearly indicate the major contribution of a diradicaloid structure 5-OS1 in the electronic ground state of 5, which is in line with the computational analyses and the broad NMR signals observed for core protons due to thermally populated triplet species (Fig. S8†). Electron paramagnetic resonance (EPR) studies of solid 5 displayed a featureless broad EPR signal (Fig. 3c), and when the temperature dropped, the signal intensity dropped as well. This can be explained by the fact that triplet biradical species have smaller populations at lower temperatures, thus further confirming the OS singlet ground state for 5 with ΔES–T = −4.35 ± 0.6 kcal mol−1, as obtained by fitting of the EPR data using the Bleaney–Bowers equation (Fig. S14†).23b,33
The harmonic oscillator model of aromaticity (HOMA, Fig. 3a)34 for the ground state structure of 5 suggested insignificant bond length alternation (BLA) for rings A (0.95) and E (0.97) and large BLA for rings B (0.24) and D (0.07), similar to 1-MF.18 However, a not so large BLA was found for the benzenoid ring C (0.68), implying its moderate aromatic character according to the geometrical criterion of aromaticity. Nucleus independent chemical shift [NICS(1)zz, Fig. 3a]35 calculations for the ground state structure of 5 using the BHandHLYP functional36 suggested aromaticity for rings A (−18.04) and E (−18.90) and moderate antiaromaticity for rings B (5.40) and D (5.55). A weak aromatic character for the central benzenoid ring C (−2.0) for 5, which is in line with the HOMA analysis, further indicated a major contribution of the OS diradical form to the electronic ground state. To support the NICS analysis, we conducted anisotropy of the induced current density37 (ACID) calculation for 5 and current density vectors were plotted onto the ACID isosurfaces (Fig. 4). The ACID plot clearly exhibited strong diatropicity (clockwise ring current) for rings A and E, and a diatropic ring current with low intensity over ring C, signifying strong to weak aromaticity for six-membered rings A/E to C. The five-membered rings B and D were found to be atropic (nonaromatic), implying the diatropic ring currents of fused benzene rings could induce some paratropic effect.38 The aromaticity indices overall suggested that the contribution of the diradical structure 5-OS1 is much more important for 5 in the singlet ground state.
 | ||
| Fig. 4 Current–density vectors plotted onto the ACID isosurface of 0.02 for the π system of 5. | ||
The ultraviolet-visible-near infrared (UV-vis-NIR) spectrum (Fig. 5a) of teal colored 5 in chloroform displayed a broad absorption band in the low energy region at λmax = 661 nm (ε = 48000 M−1 cm−1), which may be attributed to the HOMO → LUMO transition according to time dependent-DFT (TD-DFT: λmax(TD) = 582 nm, oscillator strength (f) = 0.7125) calculations (Table S3†), with the absorption tail extended to ∼825 nm in the NIR region. The lowest energy band for 5 was found to be 98 nm red-shifted than that of 4, implying a greater electronic communication in 5, which is also reflected by an enhancement of the molar extinction coefficient (ε). The optical HLG of 5 is 1.50 eV, as roughly estimated from the absorption onset, which is 0.10 eV and 0.52 eV smaller than those of its BIF 4 and bipentacene (BP)39 counterparts, respectively. BIF 5 was found to be non-emissive by the naked eye, similar to the monomer,40 and quite stable under ambient conditions with a half-life of 19 days in toluene (Fig. S13†).
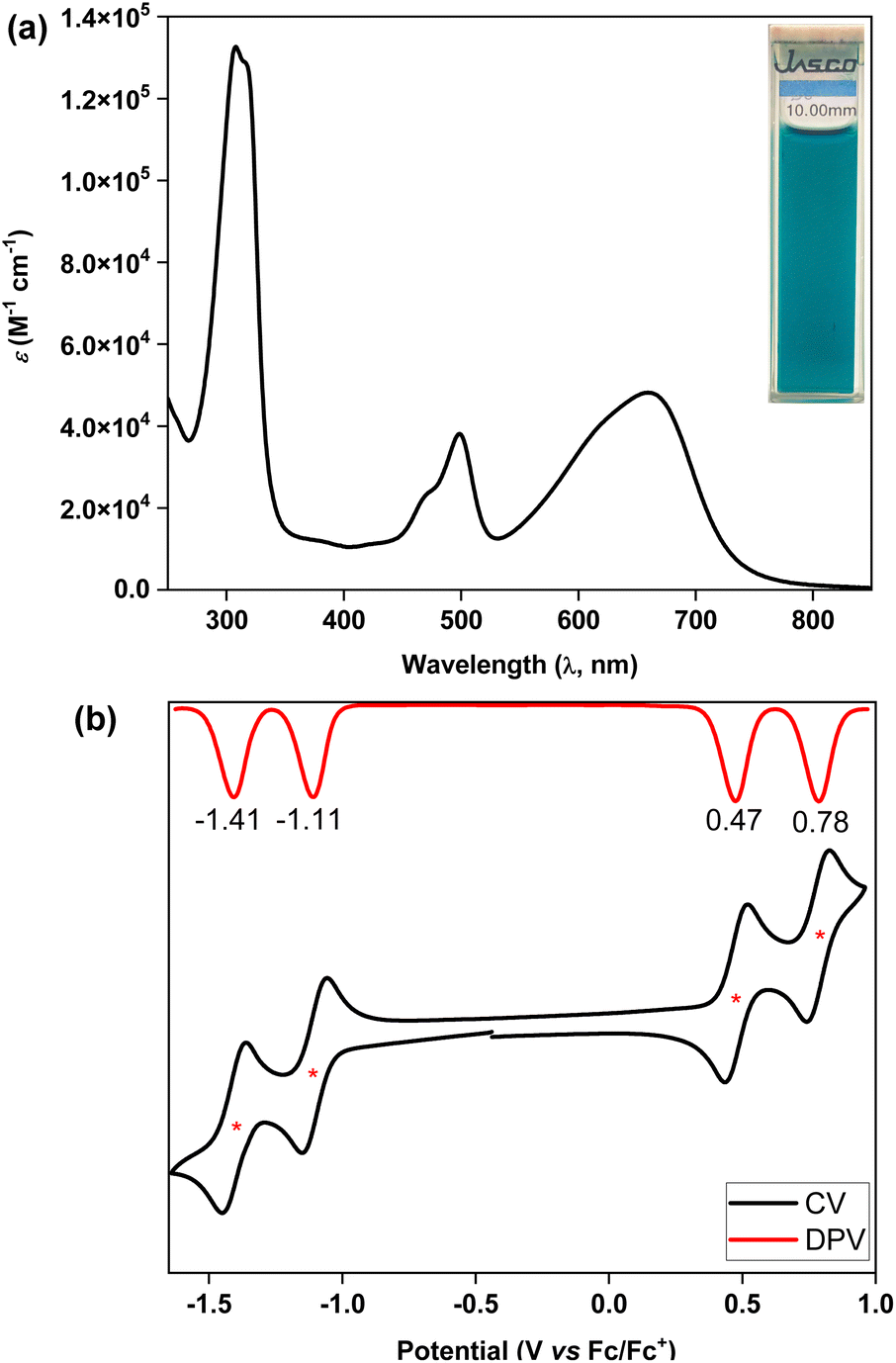 | ||
| Fig. 5 (a) UV-vis-NIR spectrum of 5 in chloroform; (b) CV and DPV of 5. | ||
Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) analyses of 5 in the DCM/Bu4NPF6 solvent/electrolyte couple exhibited four-stage redox amphotericity (Fig. 5b), displaying two reversible reduction waves with half-wave potentials Ered11/2 = −1.11 V and Ered21/2 = −1.41 V and two reversible oxidation waves at Eox11/2 = 0.47 V and Eox21/2 = 0.78 V (vs. ferrocene/ferrocenium (Fc/Fc+)). The excellent reversibility of the redox waves for 5 may be attributed to the dominance of an OS ground state, suggesting promise for application in organic electronics.5c,41 The HOMO and LUMO energy levels of 5 were found to be −5.18 and −3.80 eV, respectively, based on the onset redox potentials, affording an electrochemical HLG of 1.38 eV, which is 0.36 eV smaller than that of 4. Notably, the low-lying LUMO of BIF 5 is remarkably stabilized by 0.82 eV when compared to that of its aromatic BP counterpart with the same solubilizing group.39
Conclusions
In summary, we have designed and synthesized a stable diradicaloid BIF 5, as demonstrated by computational and experimental analyses, that exhibits four-stage redox amphotericity and improved optoelectronic properties compared to the known BIF and BP counterparts.29,39 Our BIF gains a sterically hindered BF-type OS diradicaloid ground state through gaining of partial double bond character for the inter-IF C–C single bond, which, by design, is opposite to the conventionally designed sterically overcrowded alkene-type switchable di/tetra-radicaloids.25,26 A shorter Csp2–Csp2 single bond length linking the two [1,2-b]IF units was found in the SCXRD analysis of 5, which is attributed to the better π-electron delocalization between two IF cores owing to a reduced inter-IF torsional angle as they are π-extended through carbons 6 and 6′ of [1,2-b]IF units bearing large HOMO and LUMO distributions. Our study showed that the oligomerization of antiaromatic IF units through appropriate carbon positions may significantly influence the optoelectronic properties, as the electronic ground state was effectively tuned. Syntheses of other regioisomeric BIF diradicaloids for organic electronics and photonics studies are presently underway.Data availability
The data that support the findings of this study are available in the ESI† of this article.Author contributions
H. S. performed the synthesis and characterization of all compounds. S. D. and H. S. performed DFT calculations. P. J. and S. B. performed EPR measurements. D. M. performed ACID calculations. S. D. conceived the idea, supervised the project, secured funding, and wrote the manuscript together with H. S. All authors gave approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. D. acknowledges the financial support from the science and engineering research board (SERB), India (CRG/2022/003012). H. S. and P. J. thank IIT Ropar and CSIR, respectively, for the senior research fellowships. D. M. thanks SERB for funding (SRG/2019/001461) and DST India's National Supercomputing Mission for DST/NSM/R&D_HPC_Applications/2021/8 for providing computing resources of “PARAM Shakti” at IIT Kharagpur and of “PARAM Brahma” at IISER Pune. S. B. also thanks SERB for funding (CRG/2022/006776). The authors thank Kamlesh Satpute (IIT Ropar) for the single crystal data of 5.Notes and references
- J. Wu, Diradicaloids, Jenny Stanford Publishing, New York, 2022 Search PubMed.
- (a) J. Hu, Q. Xiang, X. Tian, L. Ye, Y. Wang, Y. Ni, X. Chen, Y. Liu, G. Chen and Z. Sun, J. Am. Chem. Soc., 2024, 146, 10321–10330 CrossRef CAS PubMed; (b) F. Kuriakose, M. Commodore, C. Hu, C. J. Fabiano, D. Sen, R. R. Li, S. Bisht, Ö. Üngör, X. Lin, G. F. Strouse, A. E. DePrince III, R. A. Lazenby, F. Mentink-Vigier, M. Shatruk and I. V. Alabugin, J. Am. Chem. Soc., 2022, 144, 23448–23464 CrossRef CAS PubMed; (c) A. Borissov, P. J. Chmielewski, C. J. Gómez García, T. Lis and M. Stępień, Angew. Chem., Int. Ed., 2023, 62, e202309238 CrossRef CAS PubMed; (d) P. Ravat, T. Šolomek, M. Rickhaus, D. Häussinger, M. Neuburger, M. Baumgarten and M. Juríček, Angew. Chem., Int. Ed., 2016, 55, 1183–1186 CrossRef CAS PubMed; (e) M. Feofanov, V. Akhmetov, D. I. Sharapa and K. Amsharov, Org. Lett., 2020, 22, 5741–5745 CrossRef CAS PubMed.
- (a) S. Muhammad, M. Nakano, A. G. Al-Sehemi, Y. Kitagawa, A. Irfan, A. R. Chaudhry, R. Kishi, S. Ito, K. Yoneda and K. Fukuda, Nanoscale, 2016, 8, 17998–18020 RSC; (b) M. Nakano, Chem. Rec., 2017, 17, 27–62 CrossRef CAS PubMed; (c) Z. Zhou, K. Yang, L. He, W. Wang, W. Lai, Y. Yang, Y. Dong, S. Xie, L. Yuan and Z. Zeng, J. Am. Chem. Soc., 2024, 146, 6763–6772 CrossRef CAS PubMed; (d) M. Nakano, R. Kishi, T. Nitta, T. Kubo, K. Nakasuji, K. Kamada, K. Ohta, B. Champagne, E. Botek and K. Yamaguchi, J. Phys. Chem. A, 2005, 109, 885–891 CrossRef CAS PubMed; (e) T. Minami and M. Nakano, J. Phys. Chem. Lett., 2012, 3, 145–150 CrossRef CAS.
- (a) C. Liu, M. E. Sandoval-Salinas, Y. Hong, T. Y. Gopalakrishna, H. Phan, N. Aratani, T. S. Herng, J. Ding, H. Yamada, D. Kim, D. Casanova and J. Wu, Chem, 2018, 4, 1586–1595 CrossRef CAS; (b) S. Das, T. S. Herng, J. L. Zafra, P. M. Burrezo, M. Kitano, M. Ishida, T. Y. Gopalakrishna, P. Hu, A. Osuka, J. Casado, J. Ding, D. Casanova and J. Wu, J. Am. Chem. Soc., 2016, 138, 7782–7790 CrossRef CAS PubMed; (c) K. Horii, R. Kishi, M. Nakano, D. Shiomi, K. Sato, T. Takui, A. Konishi and M. Yasuda, J. Am. Chem. Soc., 2022, 144, 3370–3375 CrossRef CAS PubMed.
- (a) T. Jousselin-Oba, M. Mamada, J. Marrot, A. Maignan, C. Adachi, A. Yassar and M. Frigoli, J. Am. Chem. Soc., 2019, 141, 9373–9381 CrossRef CAS PubMed; (b) C. Zong, X. Zhu, Z. Xu, L. Zhang, J. Xu, J. Guo, Q. Xiang, Z. Zeng, W. Hu, J. Wu, R. Li and Z. Sun, Angew. Chem., Int. Ed., 2021, 60, 16230–16236 CrossRef CAS PubMed; (c) X. Hu, W. Wang, D. Wang and Y. Zheng, J. Mater. Chem. C, 2018, 6, 11232–11242 RSC.
- (a) Z. Zeng, X. Shi, C. Chi, J. T. L. Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578–6596 RSC; (b) S. Das and J. Wu, Phys. Sci. Rev., 2017, 2, 20160109 Search PubMed.
- (a) Z. Sun, Z. Zeng and J. Wu, Acc. Chem. Res., 2014, 47, 2582–2591 CrossRef CAS PubMed; (b) R. Huang, H. Phan, T. S. Herng, P. Hu, W. Zeng, S. Dong, S. Das, Y. Shen, J. Ding, D. Casanova and J. Wu, J. Am. Chem. Soc., 2016, 138, 10323–10330 CrossRef CAS PubMed; (c) S. Das, S. Lee, M. Son, X. Zhu, W. Zhang, B. Zheng, P. Hu, Z. Zeng, Z. Sun, W. Zeng, R. Li, K. Huang, J. Ding, D. Kim and J. Wu, Chem.–Eur. J., 2014, 20, 11410–11420 CrossRef CAS PubMed; (d) Z. Sun, S. Lee, K. H. Park, X. Zhu, W. Zhang, B. Zheng, P. Hu, Z. Zeng, S. Das, Y. Li, C. Chi, R. Li, K. Huang, J. Ding, D. Kim and J. Wu, J. Am. Chem. Soc., 2013, 135, 18229–18236 CrossRef CAS PubMed.
- (a) Z. Zeng, Y. M. Sung, N. Bao, D. Tan, R. Lee, J. L. Zafra, B. S. Lee, M. Ishida, J. Ding, J. T. L. Navarrete, Y. Li, W. Zeng, D. Kim, K. Huang, R. D. Webster, J. Casado and J. Wu, J. Am. Chem. Soc., 2012, 134, 14513–14525 CrossRef CAS PubMed; (b) Z. Zeng, M. Ishida, J. L. Zafra, X. Zhu, Y. M. Sung, N. Bao, R. D. Webster, B. S. Lee, R. Li, W. Zeng, Y. Li, C. Chi, J. T. L. Navarrete, J. Ding, J. Casado, D. Kim and J. Wu, J. Am. Chem. Soc., 2013, 135, 6363–6371 CrossRef CAS PubMed.
- (a) A. Konishi, Y. Hirao, M. Nakano, A. Shimizu, E. Botek, B. Champagne, D. Shiomi, K. Sato, T. Takui, K. Matsumoto, H. Kurata and T. Kubo, J. Am. Chem. Soc., 2010, 132, 11021–11023 CrossRef CAS PubMed; (b) A. Konishi, Y. Hirao, K. Matsumoto, H. Kurata, R. Kishi, Y. Shigeta, M. Nakano, K. Tokunaga, K. Kamada and T. Kubo, J. Am. Chem. Soc., 2013, 135, 1430–1437 CrossRef CAS PubMed.
- (a) M. Bendikov, H. M. Duong, K. Starkey, K. N. Houk, E. A. Carter and F. Wudl, J. Am. Chem. Soc., 2004, 126, 7416–7417 CrossRef CAS PubMed; (b) B. Purushothaman, M. Bruzek, S. R. Parkin, A. Miller and J. E. Anthony, Angew. Chem., Int. Ed., 2011, 50, 7013–7017 CrossRef CAS PubMed.
- (a) T. Maekawa, H. Ueno, Y. Segawa, M. M. Haley and K. Itami, Chem. Sci., 2016, 7, 650–654 RSC; (b) H. Sharma, P. K. Sharma and S. Das, Chem. Commun., 2020, 56, 11319–11322 RSC; (c) T. Xu, X. Hou, Y. Han, H. Wei, Z. Li and C. Chi, Angew. Chem., Int. Ed., 2023, 62, e202304937 CrossRef CAS PubMed.
- (a) T. Jousselin-Oba, M. Mamada, K. Wright, J. Marrot, C. Adachi, A. Yassar and M. Frigoli, Angew. Chem., Int. Ed., 2022, 61, e202112794 CrossRef CAS PubMed; (b) M. Desroches and J.-F. Morin, Chem.–Eur. J., 2018, 24, 2858–2862 CrossRef CAS PubMed.
- (a) A. Shimizu, R. Kishi, M. Nakano, D. Shiomi, K. Sato, T. Takui, I. Hisaki, M. Miyata and Y. Tobe, Angew. Chem., Int. Ed., 2013, 52, 6076–6079 CrossRef CAS PubMed; (b) J. J. Dressler, Z. Zhou, J. L. Marshall, R. Kishi, S. Takamuku, Z. Wei, S. N. Spisak, M. Nakano, M. A. Petrukhina and M. M. Haley, Angew. Chem., Int. Ed., 2017, 56, 15363–15367 CrossRef CAS PubMed; (c) K. Fukuda, T. Nagami, J. Fujiyoshi and M. Nakano, J. Phys. Chem. A, 2015, 119, 10620–10627 CrossRef CAS PubMed; (d) C. K. Frederickson, B. D. Rose and M. M. Haley, Acc. Chem. Res., 2017, 50, 977–987 CrossRef CAS PubMed; (e) Y. Tobe, Top. Curr. Chem., 2018, 375, 107–168 Search PubMed; (f) M. D. Giovannantonio, K. Eimre, A. V. Yakutovich, Q. Chen, S. Mishra, J. I. Urgel, C. A. Pignedoli, P. Ruffieux, K. Müllen, A. Narita and R. Fasel, J. Am. Chem. Soc., 2019, 141, 12346–12354 CrossRef PubMed.
- (a) D. T. Chase, B. D. Rose, S. P. McClintock, L. N. Zakharov and M. M. Haley, Angew. Chem., Int. Ed., 2011, 50, 1127–1130 CrossRef CAS PubMed; (b) D. T. Chase, A. G. Fix, S. J. Kang, B. D. Rose, C. D. Weber, Y. Zhong, L. N. Zakharov, M. C. Lonergan, C. Nuckolls and M. M. Haley, J. Am. Chem. Soc., 2012, 134, 10349–10352 CrossRef CAS PubMed.
- M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed.
- X. Fu and D. Zhao, Org. Lett., 2015, 17, 5694–5697 CrossRef CAS PubMed.
- S. Moles Quintero, M. M. Haley, M. Kertesz and J. Casado, Angew. Chem., Int. Ed., 2022, 61, e202209138 CrossRef CAS PubMed.
- H. Sharma, N. Bhardwaj and S. Das, Org. Biomol. Chem., 2022, 20, 8071–8077 RSC.
- G. E. Rudebusch, J. L. Zafra, K. Jorner, K. Fukuda, J. L. Marshall, I. Arrechea-Marcos, G. L. Espejo, R. P. Ortiz, C. J. Gómez-García, L. N. Zakharov, M. Nakano, H. Ottosson, J. Casado and M. M. Haley, Nat. Chem., 2016, 8, 753–759 CrossRef CAS PubMed.
- A. Shimizu, Y. Hirao, K. Matsumoto, H. Kurata, T. Kubo, M. Uruichib and K. Yakushib, Chem. Commun., 2012, 48, 5629–5631 RSC.
- (a) J. E. Barker, C. K. Frederickson, M. H. Jones, L. N. Zakharov and M. M. Haley, Org. Lett., 2017, 19, 5312–5315 CrossRef CAS PubMed; (b) H. Hayashi, J. E. Barker, A. C. Valdivia, R. Kishi, S. N. MacMillan, C. J. Gómez-García, H. Miyauchi, Y. Nakamura, M. Nakano, S. Kato, M. M. Haley and J. Casado, J. Am. Chem. Soc., 2020, 142, 20444–20455 CrossRef CAS PubMed.
- Y. Li, W. Heng, B. S. Lee, N. Aratani, J. L. Zafra, N. Bao, R. Lee, Y. M. Sung, Z. Sun, K. Huang, R. D. Webster, J. T. L. Navarrete, D. Kim, A. Osuka, J. Casado, J. Ding and J. Wu, J. Am. Chem. Soc., 2012, 134, 14913–14922 CrossRef CAS PubMed.
- (a) W. Zeng, Q. Qi and J. Wu, Eur. J. Org Chem., 2018, 2018, 7–17 CrossRef CAS; (b) P. K. Sharma, P. Jana, S. Bandyopadhyay and S. Das, Chem. Commun., 2024, 60, 7319–7322 RSC.
- J.-i. Nishida, S. Tsukaguchi and Y. Yamashita, Chem.–Eur. J., 2012, 18, 8964–8970 CrossRef CAS PubMed.
- (a) Y. Ishigaki, T. Harimoto, T. Shimajiri and T. Suzuki, Chem. Rev., 2023, 123, 13952–13965 CrossRef CAS PubMed; (b) X. Xu, S. Takebayashi, H. Hanayama, S. Vasylevskyi, T. Onishi, T. Ohto, H. Tada and A. Narita, J. Am. Chem. Soc., 2023, 145, 3891–3896 CrossRef CAS PubMed; (c) C. Wentrup, M. J. Regimbald-Krnel, D. Müller and P. Comba, Angew. Chem., Int. Ed., 2016, 55, 14600–14605 CrossRef CAS PubMed.
- B. Prajapati, M. D. Ambhore, D. Dang, P. J. Chmielewski, T. Lis, C. J. Gómez-García, P. M. Zimmerman and M. Stępień, Nat. Chem., 2023, 15, 1541–1548 CrossRef CAS PubMed.
- (a) K. Rakstys, M. Saliba, P. Gao, P. Gratia, E. Kamarauskas, S. Paek, V. Jankauskas and M. K. Nazeeruddin, Angew. Chem., Int. Ed., 2016, 55, 7464–7468 CrossRef CAS PubMed; (b) H. U. Kim, J.-H. Kim, H. Suh, J. Kwak, D. Kim, A. Grimsdale, S. C. Yoon and D.-H. Hwang, Chem. Commun., 2013, 49, 10950–10952 RSC.
- H. Reisch, U. Wiesler, U. Scherf and N. Tuytuylkov, Macromolecules, 1996, 29, 8204–8210 CrossRef CAS.
- H. Sharma, Ankita, V. Mittal, U. K. Pandey and S. Das, Org. Lett., 2024, 26, 2617–2622 CrossRef CAS PubMed.
- (a) N. Suaud, N. B. Amor, N. Guihéry and J. Malrieu, Theor. Chem. Acc., 2021, 140, 117 Search PubMed; (b) T. Kupka, A. Buczek, M. A. Broda, M. Stachów and P. Tarnowski, J. Mol. Model., 2016, 22, 101 CrossRef PubMed.
- J. Xu, A. Takai, A. Bannaron, T. Nakagawa, Y. Matsuo, M. Sugimoto, Y. Matsushita and M. Takeuchi, Mater. Chem. Front., 2018, 2, 780–784 RSC.
- CCDC 2362686† for 5.
- (a) P. Jana, S. Koppayithodi, S. Mahato, S. Molla and S. Bandyopadhyay, J. Phys. Chem. Lett., 2023, 14, 7433–7439 CrossRef CAS PubMed; (b) B. Bleaney and K. D. Bowers, Proc. R. Soc. London, Ser. A, 1952, 214, 451–465 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- H. Fallah-Bagher-Shaidaei, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Org. Lett., 2006, 8, 863–866 CrossRef CAS PubMed.
- (a) S. Lehtola, M. Dimitrova, H. Fliegl and D. Sundholm, J. Chem. Theory Comput., 2021, 17, 1457–1468 CrossRef CAS PubMed; (b) H. K. Saha, D. Mallick and S. Das, Chem. Commun., 2022, 58, 8492–8495 RSC.
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed.
- P. K. Sharma, A. Babbar, D. Mallick and S. Das, J. Org. Chem., 2023, 88, 5473–5482 CrossRef CAS PubMed.
- S. Lukman, A. J. Musser, K. Chen, S. Athanasopoulos, C. K. Yong, Z. Zeng, Q. Ye, C. Chi, J. M. Hodgkiss and J. Wu, Adv. Funct. Mater., 2015, 25, 5452–5461 CrossRef CAS.
- B. D. Rose, L. E. Shoer, M. R. Wasielewski and M. M. Haley, Chem. Phys. Lett., 2014, 616–617, 137 CrossRef CAS.
- T. Y. Gopalakrishna, W. Zeng, X. Lua and J. Wu, Chem. Commun., 2018, 54, 2186–2199 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization data for new compounds; NMR spectra; single crystal data of 5; details of DFT calculations. CCDC 2362686. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03996c |
| This journal is © The Royal Society of Chemistry 2024 |