
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA cocktail of Cu2+- and Zn2+-peptoid-based chelators can stop ROS formation for Alzheimer's disease therapy†
Anastasia E.
Behar
 and
Galia
Maayan
*
and
Galia
Maayan
*
Schulich Faculty of Chemistry, Technion – Israel Institute of Technology, Technion City, 3200008 Haifa, Israel. E-mail: gm92@technion.ac.il
First published on 24th October 2024
Abstract
The formation of reactive oxygen species (ROS) in the brain is a major cause of neuropathologic degradation associated with Alzheimer's Disease (AD). It has been suggested that the copper (Cu)–amyloid-β (Aβ) peptide complex can lead to ROS formation in the brain. An external chelator for Cu that can extract Cu from the CuAβ complex should inhibit the formation of ROS, making Cu chelation an excellent therapeutic approach for AD. Such a chelator should possess high selectivity for Cu over zinc (Zn), which is also present within the synaptic cleft. However, such selectivity is generally hard to achieve in one molecule due to the similarities in the binding preferences of these two metal ions. As an alternative to monotherapy (where Cu extraction is performed using a single chelator), herein we describe a variation of combination therapy – a novel cocktail approach, which is based on the co-administration of two structurally different peptidomimetic chelators, aiming to target both Cu2+ and Zn2+ ions simultaneously but independently from each other. Based on rigorous spectroscopic experiments, we demonstrate that our peptidomimetic cocktail allows, for the first time, the complete and immediate inhibition of ROS production by the CuAβ complex in the presence of Zn2+. In addition, we further demonstrate the high stability of the cocktail under simulated physiological conditions and its resistance to proteolytic degradation by trypsin and report the water/n-octanol partition coefficient, initially assessing the blood–brain barrier (BBB) permeability potential of the chelators.
Introduction
Copper (Cu) is an essential co-factor in biology,1 and disruption of its regulation within living organisms is a cause for the development of several conditions, including neurodegenerative disorders such as Alzheimer's disease (AD).2 AD is a multifactorial disease, and its mechanisms and causes are still unclear; nevertheless, one of the hypotheses developed in recent decades suggests that Cu can induce abnormal extracellular accumulation of a peptide called amyloid-β (Aβ),3 leading to the formation of insoluble fibrils and senile plaques, which are among the pathological hallmarks of AD.4 In addition, the ability of the Cu–Aβ complex to catalyze the incomplete reduction of dioxygen in the presence of a natural reducing agent such as ascorbate, leads to the formation of reactive oxygen species (ROS) that are toxic to neurons and directly impact their viability.5 Therefore, extraction of Cu from Cu–Aβ is a promising approach and an excellent strategy towards the development of therapeutics for AD.6Despite the growing interest in the last decade regarding AD-associated Cu-chelation,4a there are still many challenges to be addressed for any proposed chelators to be utilized as potential drugs. The reasons for this lie in the highly demanding criteria that potential chelators should meet in order to be administered as efficient drugs: in addition to the general requirements for drug candidates, i.e. good kinetic and stability properties under physiological conditions, the ability to cross the blood–brain-barrier (BBB) and low toxicity,4a a chelator in the context of AD should bind Cu more strongly than Aβ,4,6e,f,7 forming a Cu–chelator complex that does not produce ROS by itself.4a Along with sufficient affinity for Cu over other metal ions, high selectivity for Cu over Zn is extremely significant because Zn is co-present in the synaptic cleft at a higher concentration than Cu, thus it is more available for coordination by Aβ.8 Moreover, as the affinity of Aβ to Zn2+ is lower than its affinity to Cu,7a,9 it is possible that the Cu-chelator will bind Zn2+ ions rather than extracting Cu2+ ions from the toxic Cu–Aβ complex (due to the similarities in the binding preferences of these two metals),4a,10 thus hindering the inhibition of ROS production. Despite the critical importance of selective chelation of Cu over Zn, the vast majority of currently reported ligands for AD-associated chelation have not been studied in the presence of zinc at all,4a leading to the conclusion that the issue of co-presence of Zn and Cu in the synaptic cleft is currently highly overlooked. It is important to mention, that achieving exclusive chelation of Cu in the presence of Zn is a challenging task; many potential Cu chelators that can extract Cu from Cu–Aβ under non-biological conditions in the absence of Zn, fail to perform in more advanced biochemical in vitro experiments simulating physiological conditions when Zn ions are co-present,4a,11 and do not qualify for in vivo studies.4a,11a The only four examples of the chelators that demonstrated some selectivity for Cu over Zn, either targeted Cu+ rather than Cu2+,12 couldn't achieve either complete13 or immediate10a,12a arrest of Cu–Aβ mediated ROS production in the presence of Zn2+, or their stability was insufficient.12 Complete and immediate arrest of ROS even in initial biochemical assays is highly important because any kinetic effects can preclude the ability of the chelator to stop ROS formation under subsequent biological conditions during in vitro or in vivo studies.
Overcoming the Cu-over-Zn selectivity issue via the utilization of a single chelator is yet to be achieved, but the utilization of a cocktail of chelators, called combination therapy, was studied in the context of AD.14 Traditional combination therapy in AD involves the co-administration of multiple drugs for enhanced efficacy, and typically targets a singular pathological mechanism, such as acetylcholinesterase inhibition.14,15 Notably, only one study attempted to target simultaneous chelation of Cu and Zn, and this is towards the inhibition of Aβ aggregation, and not for ROS inhibition.16 In this study, the proposed chelator combination comprised two small molecule ligands: clioquinol (for Zn) and phanquinone (for Cu), and their effect on the reduction of metal-induced Aβ aggregation when injected together was higher than when administered separately; however, the overall effect on Aβ-aggregation was rather moderate (50–60% inhibition).16 Therefore, the cocktail strategy is still far from being fully developed. Thus, it is desirable to develop a chelating cocktail system that will enable highly selective extraction of Cu from Cu–Aβ in the presence of Zn, which will lead to complete inhibition of ROS formation. In such systems, the Cu-targeting peptoid will extract Cu from the Cu–Aβ complex, thus stopping the redox cycle and ROS formation, while the Zn-targeting chelator will bind to the co-present Zn,(Scheme 1). Furthermore, Zn has recently been shown to promote Aβ oligomer formation;17 therefore its chelation from the Aβ-complex could also be beneficial in the context of AD-targeted chelation.
 | ||
| Scheme 1 Schematic representation of a cocktail of two structurally different peptoid-based chelators, designed to selectively bind Cu or Zn ions from the Aβ-peptide complex, stopping the production of ROS in the context of AD. | ||
As an alternative to small molecules and peptide-based ligands, peptoids, peptidomimetic oligomers of N-substituted glycines, are an excellent platform for the development of metal chelators as potential therapeutics for AD. In recent years, peptoids18 have been extensively studied for biological applications including protein–protein interactions,19 metal binding and recognition,20 particularly in the context of AD21 and catalysis.22 The interest in peptoids stems from their advantageous properties over both small molecules and peptides: (i) they can be easily synthesized on a solid support by the solid-phase “submonomer” method,23 which utilizes primary amines as building blocks rather than amino acids, thus allowing the introduction of a variety of functional side-chains, such as metal-binding ligands (MBLs); (ii) they can fold into a well-defined secondary structure in solution, when bulky-chiral side-chains are incorporated within their sequences;24 (iii) they can be employed in various biological activities, as they exhibit high proteolytic resistance25 (thus high stability towards proteases and peptidases), high membrane permeability,26 and tolerance towards high temperatures,24a excess salt concentrations and various pH conditions.22g,h
Capitalizing on the advantages of peptoids, here we present a new cocktail of two different water-soluble peptoid-based chelators: a new helical peptoid chelator for Cu2+ (AD2), which is fully soluble under biological conditions, in combination with the unstructured chelator PT1, which has been previously reported by our group as an excellent chelator for Zn2+ ions that can extract Zn from a peptide motif (the Zn finger motif).27 We demonstrate here for the first time, how we applied this cocktail of Cu- and Zn-peptoid chelators for the complete and immediate inhibition of the ROS formation by the Cu–Aβ complex, in an aqueous medium in the presence of Zn2+. In addition, we report the biocompatibility properties of our proposed chelators, including their stability at physiological pH and temperature, as well as their resistance to proteolytic degradation by trypsin; we also report the water/n-octanol partition coefficient, initially assessing the blood–brain barrier (BBB) permeability potential of the chelators.
Results and discussion
Peptoid design and solubility properties
We have shown that the main prerequisite for the successful extraction of Cu from the CuAβ complex and the subsequent inhibition of ROS formation, is the stabilization of Cu in its +2 oxidation-state.21 The stabilization of Cu2+ by P3 was attributed to its helical structure, which was unable to stabilize Cu+, thus prohibiting peptoid-bound Cu from participating in the redox cycle. Thus, we wished to design a peptoid-chelator that can adopt a stable helical structure to ensure the successful extraction of Cu2+ from the Aβ complex, followed by Cu2+ stabilization, while also improving water-solubility in buffer solutions at physiological pH. To this aim, we have identified N,N-diisopropylethylenediamine (NiPr2ae) as a potential peptoid side-chain that can induce high water-solubility. As was shown recently,28 peptoids containing NiPr2ae moieties form stable secondary structures in water when the majority of peptoid's side-chains consist of NiPr2ae groups. Due to this reason, we anticipated, that incorporation of NiPr2ae won't affect the peptoid's overall helical structure, while it would likely improve the peptoid's solubility (since NiPr2ae tertiary nitrogen is usually protonated at neutral pH).28 In addition, the presence of positively charged groups within peptoid scaffolds could enhance their permeability, which is crucial for the peptoid's ability to cross the BBB. However, in this work, the position and number of NiPr2ae groups required to obtain peptoid sequences with high water-solubility have not been studied.Based on the requirements for obtaining water-soluble peptoids using piperazine as a side-chain,29 we initially designed the peptoid-heptamer AD1 (Fig. 1A), in which we incorporated one NiPr2ae group as a side chain at the N-terminal of the sequence.
 | ||
| Fig. 1 (A) Chemical structures of peptoid oligomers AD1–AD2 and PT1. (B) Solubilities of peptoid AD1 and AD2 in un-buffered water (pH = 7.0) and HEPES buffer (50 mM, pH = 7.4). | ||
Peptoid AD1 was synthesized via the sub-monomer approach on a solid support, purified by preparative HPLC (>99% purity) and characterized by analytical HPLC and ESI-MS (Fig. S1 and S4†). Next, it's solubility in un-buffered water and in HEPES buffer was evaluated.21 Surprisingly, peptoid AD1 appeared to be completely insoluble in both un-buffered water and HEPES buffer (Fig. 1B), implying that having only one NiPr2ae side chain is not enough to ensure sufficient solubility of the helical sequence. Thus, we decided to increase the number of NiPr2ae side chains from one to two and designed the peptoid AD2 (Fig. 1A).
Peptoid AD2 was synthesized via the sub-monomer approach on the solid support, purified by preparative HPLC (>99% purity) and characterized by analytical HPLC and ESI-MS (Fig. S2 and S5†). The molecular weight measured by ESI-MS was consistent with the expected mass. Next, it's solubility was evaluated. AD2 exhibits high solubility in both un-buffered water (1.914 × 105 mg L−1 at pH = 7.0) and HEPES buffer (1.492 × 105 mg L−1 at pH = 7.4) (Fig. 1B) with no signs of aggregation at μM concentrations.
The UV/Vis titration of AD2 in HEPES buffer (50 mM, pH = 7.4) revealed that upon addition of Cu2+ ions, the two bands at λ = 245 and 277 nm diminished simultaneously, and new absorption bands appeared near λ = 259, 317 and 330 nm (Fig. 2A). A metal-to-peptoid ratio plot, constructed from this titration, suggested a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 AD2 to Cu2+ ratio and the formation of an intramolecular CuAD2 complex (Fig. S7A,† inset). To further support the stoichiometry of the CuAD2 complex as suggested by the UV/Vis titration experiments, we conducted a Job plot experiment. The total molar concentration of a mixture solution containing both Cu(II) and AD2 was maintained constant at 26 μM, while their mole fractions were varied, and the UV/Vis spectra of these mixtures were recorded. The absorbance proportional to CuAD2 complex formation at 330 nm was plotted against the mole fraction (Fig. 2A, inset), and from the intersection point at χ = 0.503 the stoichiometric ratio was determined to be 1.01,30 supporting the metal-to-peptoid ratio obtained from the UV/Vis titrations. HR-MS analysis of the mixture of 1 equiv. each of Cu2+ and AD2 exclusively resulted in a mass of m/z 1639.9048, and the isotopic distribution of this mass corresponded to the calculated mass of the intramolecular 1:1 CuAD2 complex (m/z = 1639.5380, Fig. S9 and S10†). Notably, HR-MS analysis did not show any other masses that could be assigned to full or half-masses of species with different stoichiometric ratios Cu(II):AD2 1:2 or 2:2 (Fig. S11 and S12†).31 Overall, the results from the UV/Vis titration, Job plot and HR-MS confirmed the formation of the intramolecular CuAD2 complex.
:1 AD2 to Cu2+ ratio and the formation of an intramolecular CuAD2 complex (Fig. S7A,† inset). To further support the stoichiometry of the CuAD2 complex as suggested by the UV/Vis titration experiments, we conducted a Job plot experiment. The total molar concentration of a mixture solution containing both Cu(II) and AD2 was maintained constant at 26 μM, while their mole fractions were varied, and the UV/Vis spectra of these mixtures were recorded. The absorbance proportional to CuAD2 complex formation at 330 nm was plotted against the mole fraction (Fig. 2A, inset), and from the intersection point at χ = 0.503 the stoichiometric ratio was determined to be 1.01,30 supporting the metal-to-peptoid ratio obtained from the UV/Vis titrations. HR-MS analysis of the mixture of 1 equiv. each of Cu2+ and AD2 exclusively resulted in a mass of m/z 1639.9048, and the isotopic distribution of this mass corresponded to the calculated mass of the intramolecular 1:1 CuAD2 complex (m/z = 1639.5380, Fig. S9 and S10†). Notably, HR-MS analysis did not show any other masses that could be assigned to full or half-masses of species with different stoichiometric ratios Cu(II):AD2 1:2 or 2:2 (Fig. S11 and S12†).31 Overall, the results from the UV/Vis titration, Job plot and HR-MS confirmed the formation of the intramolecular CuAD2 complex.
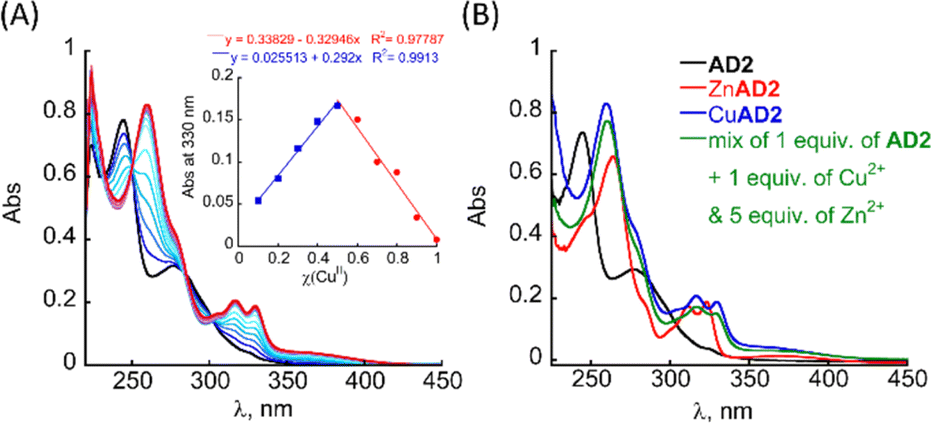 | ||
| Fig. 2 (A) UV/Vis titration of AD2 (33 μM) with Cu2+ ions in HEPES buffer (50 mM, pH = 7.4). Inset: Job-plot of AD2 with Cu2+ (26 μM total concentration). (B) UV-Vis spectra of AD2 (33 μM), it's Cu2+ and Zn complexes, and the complexes formed upon addition of a mixture of 1 equiv. of Cu2+ and 5 equiv. of Zn2+. | ||
The binding affinity of AD2 to Cu2+ was estimated by a competition experiment with 2,2′,2′′,2′′′-(ethane-1,2-diyldinitrilo)tetraacetic acid (EDTA).32 The mixture of AD2 and EDTA (16.7 μM each, in water, at pH = 7.0) was titrated with Cu2+ followed by UV/Vis spectroscopy. The obtained data were analyzed according to a previously reported method.32 The slope obtained from plotting ([AD2]total/[CuAD2]-1) against ([EDTA]total/[CuEDTA]-1) represents the dissociation constant KD(CuAD2), which was found to be 1.43 × 10−16 M (Fig. S8†). From this, we calculated the association constant KA(CuAD2) to be 7.00 × 1015 M−1. This value is 106 times higher than the binding affinity of Aβ1–16 for Cu2+ under the same conditions (Table S2†),33 and it reflects a strong binding affinity, suggesting that AD2 will be able to compete with Aβ1–16 for Cu2+, thus supporting our peptoid design.21
Circular Dichroism (CD) spectra of the metal-free peptoid were recorded in HEPES buffer at pH = 7.4. AD2 exhibits characteristic double-minima at 205 nm and 220 nm, indicating the peptoid's folding into a helical structure (Fig. S7D†). Upon the addition of 1 equiv. of Cu2+ ions, the intensity of the double minima at 205 and 220 nm further increases, and an exciton couplet CD peak appears, with a minimum at 274 nm, crossing ε = 0 near 260 nm (Fig. S7D†). This exciton couplet corresponds to the π–π* transitions of the HQ ligand within the peptoid.21 These results confirm that AD2 adopts a helical structure in aqueous HEPES buffer and that the secondary structure of AD2 is further stabilized upon binding to Cu2+.
To test the selectivity of AD2 to Cu over Zn we added 1 equiv. of AD2 to a mixture solution containing 1 equiv. of Cu2+ and 1 equiv. of Zn2+ in HEPES buffer (50 mM, pH = 7.4), stirred it for 5 min, and recorded a UV/Vis spectrum of this solution. The obtained UV/Vis spectrum was identical to that of CuAD2 only, suggesting that AD2 binds exclusively to Cu2+ over Zn2+ (Fig. S7C†).34 We repeated the experiment, increasing the excess of Zn2+ up to 5 equiv., while keeping the Cu2+ amount at 1 equiv., and monitored mixing by UV/Vis until no changes in the UV/Vis spectrum were observed, indicating the formation of the thermodynamic product (Fig. S7C† and 2B). Interestingly, increasing the excess of Zn2+ ions led to an increase in the time required for the formation of the CuAD2 complex: initially, after mixing, the UV/Vis spectrum of the mixture resembled that of CuAD2 and ZnAD2 complexes, but over time it changed to resemble the spectrum of CuAD2 after 10 min (for Cu2+:Zn2+ = 1:2), 15 min (for Cu2+:Zn2+ = 1:3) and 60 min (for Cu2+:Zn2+ = 1:5). These observations suggest, that the selectivity of AD2 to Cu2+ over Zn2+ is thermodynamically driven and requires some preincubation time, while kinetic products such as Zn(AD2)2 might form first,21 and the time needed for preincubation increases with the increase in excess of Zn ions.21 Overall, introducing water-solubilising groups at the N-terminus of the peptoid not only allowed high solubility in HEPES buffer under physiological conditions but also improved the selectivity for Cu2+ over Zn2+ from 1:1 to 1:5.
ROS production by the Cu,Zn–Aβ complex in the presence of the AD2 and PT1 cocktail
The influence of AD2 on ROS production by the CuAβ complex was probed by a previously described method,21 monitoring the consumption of ascorbate (the reductant that fuels the formation of ROS H2O2 or HO˙) by UV/Vis at 265 nm (ε = 14500 M−1 cm−1).35 As the metal-ion binding sites of Aβ to Cu and Zn lie within residues 1–16,36 and the fibrillary forms of Aβ are found to be less active than monomeric peptides by at least one order of magnitude,37 we have focused our study on the monomeric peptide Aβ1–16 complex as the suitable model for investigations.
At first, we checked the ability of AD2 and PT1 (the highly selective chelator for Zn2+ ions previously reported by our group,27Fig. 1A) to extract Cu2+ from the CuAβ complex and inhibit ROS formation in the absence of Zn2+. To a UV/Vis cuvette containing 1 equiv. of the preformed CuAβ1–16 complex, 1 equiv. of either AD2 or PT1 was added, and the solution was stirred for 10 min before ascorbate was added. The addition of the peptoid to the sample led to an increase in the intensity of the absorbance at 265 nm, which is attributed to the formation of the metal–peptoid complex (UV/Vis spectra of Cu and Zn complexes with each of the peptoid are shown in Fig. 2A, S7A, B and S13†). We anticipated that the addition of ascorbate to the mixture, aiming to start the catalytic redox cycle, would result in no change in the intensity of the absorbance at 265 nm if no ascorbate is consumed, indicating no ROS production (ROS inhibition), or a decrease in intensity, which would suggest the consumption of ascorbate and formation of ROS (no ROS inhibition).
Typically, unbound copper leads to rapid consumption of ascorbate, which slows down when copper is bound to Aβ, regardless of the co-presence of Zn2+.10,11a,35,37 Indeed, under our experimental conditions full consumption of ascorbate by unbound copper occurred within 8.5 min, while in the presence of the Cu2+–Aβ1–16 complex the ascorbate was fully consumed within 29 min (see Table 1). In the absence of Zn2+, addition of 1 equiv. of AD2 led to 2.0% ascorbate consumption in 30 min, allowing for a 98% reduction in ROS production by CuAβ. 1 equiv. of PT1, on the other hand, is unable to stop ROS production, as its addition led to 71.0% ascorbate consumption in 30 min. When repeating the experiments by mixing the Aβ peptide with Cu2+ and Zn2+, the ascorbate consumption in 30 min increased to 20.4% when 1 equiv. of AD2 was added to the mixture and to 97.2% when 1 equiv. of PT1 was added (with full consumption of ascorbate observed within 43 min). However, when the ascorbate consumption experiment was repeated and both AD2 and PT1 were added simultaneously as a cocktail, a 99% reduction in ROS production was observed in 30 min.
| Experimental conditions | Degree of helicity | ROS production | ||
|---|---|---|---|---|
| Cu-peptoid | Zn-peptoid | Molar ellipticity per residue at 219 nm, [θ] × 104 (deg cm2 dmol−1) | % of Asc consumption in 30 min | % of ROS arrest |
| Full consumption of ascorbate happens within:a 8.5 min (free Cu2+).b 29 min (CuAβ1–16).c 21.5 min (CuZnAβ1–16 + 1 equiv. EDTA).d 18.5 min (CuZnAβ1–16 + 2 equiv. EDTA).e 43 min (CuZnAβ1–16 + 1 equiv. PT1).f 44 min (CuZnAβ1–16 + 2 equiv. PT1).g Conditions: [PT1, AD2, EDTA (1 equiv.)] = [Aβ1–16] = [Zn2+] = 10 μM, [PT1, AD2, EDTA (2 equiv.)] = 20 μM, [Cu2+] = 9 μM, [Asc] = 100 μM, [HEPES] = 50 mM, pH 7.4, with background subtraction of the signal at 800 nm. | ||||
| In the absence of Zn 2+ | ||||
| Control Cu2+ onlya | — | 100 | 0 | |
| Control Cu–Aβ1–16b | — | 100 | 0 | |
| EDTA (1 equiv.) | Not helical | 11.3 | 88.7 | |
| AD2 (1 equiv.) | — | −1.9651 | 2.0 | 98.0 |
| — | PT1 (1 equiv.) | Not helical | 71.0 | 29.0 |
|
||||
| In the presence of Zn 2+ | ||||
| EDTAc (1 equiv.) | Not helical | 100 | 0 | |
| EDTAd (2 equiv.) | Not helical | 100 | 0 | |
| AD (1 equiv.) | — | −1.9651 | 20.4 | 79.6 |
| AD2 (2 equiv.) | — | −1.9651 | 66.9 | 33.1 |
| — | PT1 (1 equiv.) | Not helical | 97.2 | 2.8 |
| — | PT1 (2 equiv.) | Not helical | 94.1 | 5.9 |
| AD2 (1 equiv.) | PT1 (1 equiv.) | −1.9651/not helical | 1.0 | 99.0 |
To test the synergistic effect of the cocktail, we repeated the ascorbate consumption assays but adding either AD2 or PT1 as single chelators to Cu2+–Aβ1–16 in the presence of Zn2+, in a 2:1 ratio. This ratio was used to imitate the ratio between the peptoids in the cocktail and Cu2+–Aβ1–16 in our initial experiments. The results showed 66.9% and 94.1% ascorbate consumption in 30 min, when either 2 equiv. of AD2 or 2 equiv. of PT1 were used, respectively, indicating that each chelator separately cannot stop ROS production (see Table 1). These results demonstrate that using a single peptoid-chelator at an increased concentration does not have a positive effect on ROS arrest in the presence of Zn2+.
Furthermore, we sought to compare the activity of the peptoid-cocktail to that of a known Cu-chelator. For this purpose, we performed ascorbic consumption assays using EDTA as the chelator. EDTA is a small-molecule chelator well-known for its high affinity for many metal ions, including Cu2+ (KA = 6.31 × 1018 M−1) and Zn2+ (KA = 3.16 × 1016 M−1). As can be seen from Table 1, in the absence of Zn2+, 1 equiv. of EDTA significantly decreases ROS production, with only 11.3% ascorbate consumption in 30 min. However, in the presence of Zn2+, EDTA completely loses its ability to arrest ROS production and shows full ascorbic consumption within ∼20 min, regardless of whether 1 or 2 equiv. of EDTA were added to the cuvette. This illustrates the lack of selectivity of EDTA for Cu2+ in the presence of Zn2+, and again supports the unique function of our cocktail method.
Taking together, the control experiments show that the complete and immediate arrest of ROS production provided by peptoid-cocktail arises from the synergy between the two chelators, rather than from the increase in the overall chelator concentration, and that the rational design of this cocktail system, where each peptoid chelator is selective to a specific target ion, is a crucial element in Cu extraction in the presence of Zn2+. This is in contrast to the activity of a single chelator, such as AD2 or EDTA, which, despite having higher binding affinity to Cu than to Zn, lack selectivity for Cu2+ in the presence of Zn2+.
To further support the synergy between AD2 and PT1, we wished to show that their ability to selectively bind Cu or Zn, respectively, when they are not present in the solution together as a cocktail, is limited. To this aim, we studied the interaction of each chelator separately with the CuZn–Aβ complex by ESI-MS, EPR, and CD and compared it to the interaction of the cocktail of chelators with the CuZn–Aβ complex.
Cu2+ and Zn2+ extraction from the Cu,Zn–Aβ complex by either AD2 or PT1 separately
At first, we wanted to characterize the ability of each chelator AD2 and PT1 separately to compete with the CuZnAβ complex for their relevant targeted metal ion (Cu2+ for AD2, and Zn2+ for PT1). For this, we monitored the interaction of CuZnAβ with either AD2 or PT1 by EPR and HR-MS spectroscopy.The EPR spectra of a mixture containing CuAβ1–16 in the presence of 1 equiv. of Zn2+ and 1 equiv. of either AD2 or PT1 were recorded and compared to the EPR spectra of CuZnAβ and CuAD2 or CuPT1, respectively. CuAD2 is characterized by the following Hamiltonian parameters obtained from experimental spectra: g∥ = 2.26, g⊥ = 2.060 and A∥ = 160 G (Fig. S14† and 3, green), which are identical to those obtained from the CuP3 EPR spectrum reported previously.21 In contrast, the Hamiltonian parameters for CuPT1 were found to be g∥ = 2.26, g⊥ = 2.10 and A∥ = 146 G (Fig. S16† and Fig. 3, cyan). Both spectra are typical for the Cu2+ center in an axial environment and consistent with a square pyramidal coordination geometry.38 The signature of CuAβ1–16 is identical to that previously reported with two species co-existing at pH 7.4 in the presence of Zn (Fig. S15, S17† and 3, black).36,39 Hyperfine lines of CuAD2 (or CuPT1) and CuAβ1–16 are shown as plain and dotted lines, respectively.
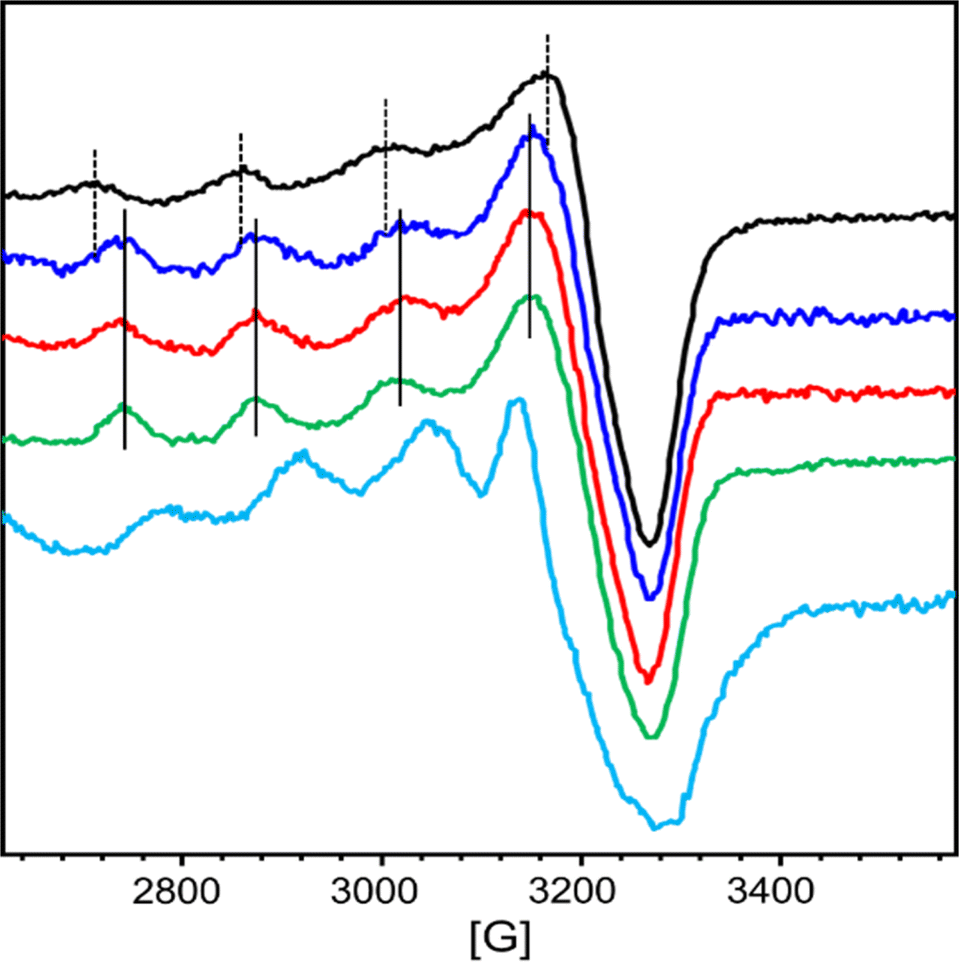 | ||
| Fig. 3 EPR spectra of Cu2+ + AD2 (green), Aβ1–16 + Cu2+ + Zn2+ (black), Aβ1–16 + Cu2+ + Zn2+ + AD2 + PT1 EPR tube frozen asap (blue), Aβ1–16 + Cu2+ + Zn2+ + AD2 + PT1 EPR tube frozen after four hours (red), Cu2+ + PT1 (cyan). Conditions: [PT1] = [AD2] = [Aβ1–16] = [Zn2+] = 280 μM, [Cu2+] = 250 μM, [HEPES] = 50 mM (pH = 7.4). Recording conditions: T = 203 K, ν = 9.5 GHz, modulation amplitude = 3 G, microwave power: 20 mW. | ||
In the presence of Zn, AD2 and PT1 alone cannot fully extract Cu2+ from CuAβ – in both cases, the EPR spectra recorded one hour after the addition of peptoid resemble the overlap of CuAβ and Cu-peptoid signals (Fig. S15, brown and S17, pink†). However, while the spectrum of the CuZnAβ mixture with AD2 appears closer to that of CuAD2 (Fig. S15, brown†), suggesting that most of Cu was extracted from Aβ and is bound by AD2, the spectrum of the CuZnAβ mixture with PT1 suggests that both CuAβ and CuPT1 are co-present in similar amounts (Fig. S17, pink†). The formation of the ternary species Aβ-Cu-PT1 is ruled out by the subsequent HR-MS analysis of the mixture (Fig. S20†). The isotopic distribution comparison of the peak at 744.0536 m/z suggests the formation of a mixture of CuPT1 and ZnPT1 (Fig. S21†), while detailed analysis of fragmentation showed a major peak at 1954.5327 m/z corresponding to full mass of free Aβ1–16, and few minor peaks corresponding to CuAβ and ZnPT1 complexes (see details in Fig. S20 and Table S3†). Notably, we did not observe any peaks that could be assigned to Aβ-Cu-PT1 species, suggesting these complexes are not formed. These observations support the results obtained from the EPR studies and suggest that PT1 alone (not in a cocktail with AD2) is unable to extract Zn from CuZnAβ complex selectively; instead it forms a mixture of CuPT1 and ZnPT1, leaving Aβ partially coordinated to the remaining Cu and Zn. A possible reason for the lack of selectivity of PT1 for Zn over Cu could arise from the rather small difference in the binding affinities of PT1 to Cu2+ and Zn2+, which is only one order of magnitude,27 while both affinities are 2–6 orders of magnitude higher than the affinity of Aβ to Cu2+ and Zn2+ (Table S2†).9a,33 Hence, under these conditions, the coordination of PT1 to both Cu2+ and Zn2+ is plausible leading to its low selectivity to Zn2+.
Cu2+ and Zn2+ extraction from the Cu,Zn–Aβ complex using a cocktail mixture of AD2 and PT1
The ability of AD2 and PT1 to act together to remove Cu2+ and Zn2+ from Aβ1–16 was studied first by EPR. Fig. 3 shows the monitoring of Cu2+ extraction from CuZnAβ using the cocktail mixture: green and cyan lines represent the EPR signature of CuAD2 and CuPT1, black line corresponds to CuAβ in the presence of 1 equiv. of Zn2+ and red and blue lines represent the resulting species after the addition of a cocktail mixture of AD2 and PT1. In the presence of Zn2+, the spectrum recorded immediately after cocktail addition was already similar to that of CuAD2 (Fig. 3, blue), while after four hours, the whole signature of CuAD2 was detected (Fig. 3, red). The EPR experiment perfectly demonstrates that the cocktail mixture enables fast extraction of Cu by AD2 from Aβ1–16 even in the presence of Zn.These observations were further confirmed by the ESI-MS technique, which is routinely used to investigate the interactions between Aβ peptides and metal ions.40 The ESI-MS chromatogram of Aβ1–16 in HEPES buffer (50 mM, pH = 7.4) exhibits a multitude of intense signals, with the most pronounced signals at m/z 652.4 (assigned to the triply charged ion [M + 3H]3+) and m/z 978.1 (assigned to the doubly charged ion [M + 2H]2+), suggesting that Aβ1–16 contains many sites which can accommodate protons (Fig. S22A†).41 Next, 1 equiv. of Aβ1–16 was treated with 1 equiv. of Cu2+ and 1 equiv. of Zn2+ ions, and the mixture was stirred for 5 minutes to ensure complex formation, and the ESI-MS spectrum was measured again. In the obtained spectrum, in addition to peaks at m/z 652.4 and m/z 978.1 assigned to metal-free Aβ1–16, new peaks appeared, indicating the formation of various Aβ1–16–metal complexes, with the signal at m/z 1041.1 assigned to [M + Cu(II) + Zn(II)]2+ (Fig. S22B and C†). This mixture was further treated with a cocktail solution containing 1 equiv. each of AD2 and PT1, stirred for one hour before it was analyzed again by ESI-MS. The measured MS spectrum showed only the peaks assigned to [M + 3H]3+ and [M + 2H]2+ at m/z 652.4 and 978.0 while the peaks assigned to metal–Aβ1–16 complexes diminished, indicating the presence of metal-free Aβ1–16 (Fig. S22D and E†). In addition, two new peaks at m/z 1017.3822 and 1639.9246 appeared, and isotopic distribution analysis of these two peaks confirmed the formation of ZnPT1 and CuAD2 complexes, respectively (Fig. S23 and 24†). Notably, we did not observe peaks that could be assigned to CuPT1 or ZnAD2, suggesting that these complexes are not formed.
Our conclusions from the EPR and ESI-MS experiments were further examined by the CD study under the same experimental conditions. The results obtained from this study (Fig. S25†) matched perfectly with those obtained from EPR and ESI-MS, supporting the ability of AD2 to selectively extract Cu2+ from the CuAβ complex in the presence of Zn2+ and PT1.
The restored ability of AD2 and PT1 to bind selectively to Cu2+ and Zn2+ when added together in a cocktail correlate perfectly with the differences in binding affinities of these peptoids for these metal ions: binding affinity of AD2 for Cu2+ is 4 orders of magnitude higher than the affinity of PT1 to Cu2+ under the same conditions. This difference allows AD2 to win the competition with PT1 for Cu2+, while PT1 binds the remaining Zn2+. Overall, EPR, ESI-MS, and CD experiments support the ability of the AD2/PT1 cocktail to extract Cu and Zn ions from the Aβ peptide complex simultaneously in a selective manner, where AD2 helps PT1 and vice versa, further highlighting the uniqueness of the tandem of cocktail peptoids.
Metabolic stability of peptoid cocktail
Among biochemical requirements, for a molecule to be considered for in vitro and in vivo models, metabolic stability and stability to proteolytic degradation are crucial parameters, together with the molecule's ability to cross the BBB.4a The general ability of peptoids to withstand different ranges of pH and temperatures, as well as the stability towards proteolytic degradation has been studied before.22g,h,24a,25 However, these features have not been previously studied for the peptoid oligomers bearing metal-binding ligands.First, the stability of peptoids AD2 and PT1 at physiologically relevant pH and temperature was studied by UV-Vis spectroscopy. The stability experiments were performed at pH 7.4 and 4.0, representing two main physiological pH for a potential oral drug candidate (simulating body fluids such as blood or stomach juice). In a typical experiment, an aliquot of peptoid AD2 or PT1 was added to the buffer, mixed, and its spectrum was recorded at RT (25 °C). Next, the sample holder was heated to 37 °C, and the spectrum of the mixture was recorded again. The temperature was maintained constant at 37 °C and the spectra were recorded at 1, 24, and 72 hours after heating (Fig. 4A, B and S26†). It is seen that under both pH conditions, at 37 °C, there are no significant changes in the spectra with respect to time, and both AD2 and PT1 are stable under these conditions for the entire duration of 72 hours (3 days).
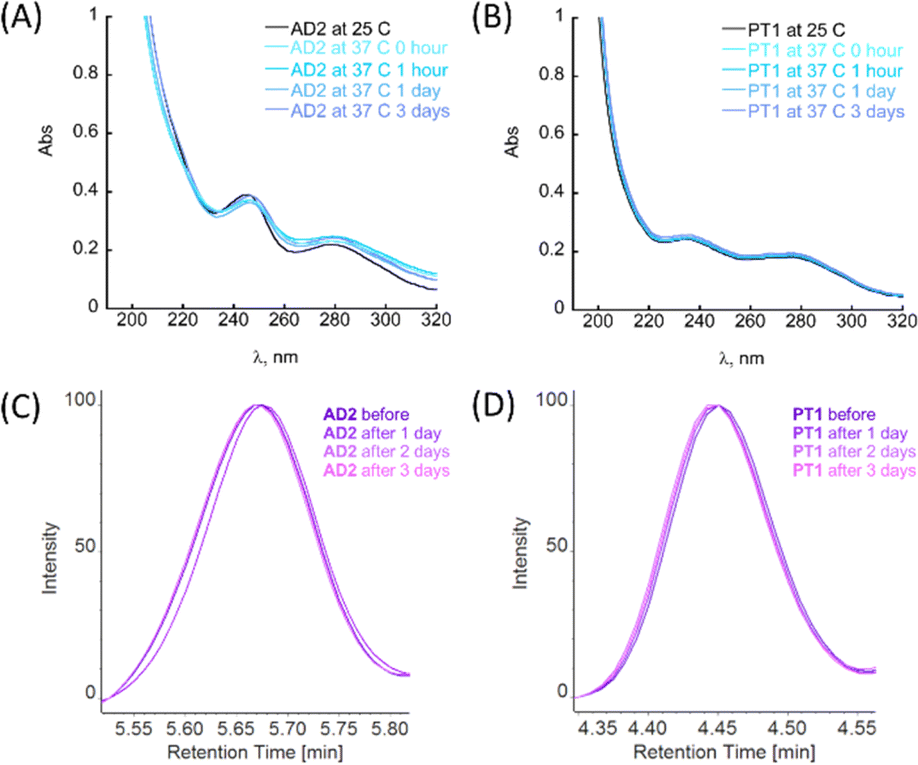 | ||
| Fig. 4 Stability of (A) AD2 and (B) PT1 under physiological conditions (pH = 7.4 and T 37 °C), as monitored by UV\Vis. Conditions: [PT1] = [AD2] = 100 μM, [PBS] = 50 mM. Proteolysis experiments: overlap of the retention peaks of (C) AD2 and (D) PT1 from the corresponding HPLC chromatograms (λ = 214 nm, a linear gradient of 5–95% ACN/H2O 0.1% TFA over 10 min, at a flow rate of 0.7 min mL−1 on the analytical C18 column), taken at different times from the proteolytic stability experiment. Conditions: [PT1] = [AD2] = 0.1 mg mL−1, [trypsin] = 0.1 μM, [PBS] = 50 mM (pH = 7.4), at the constant temperature of 37 °C. | ||
To evaluate the relative proteolytic stability of the peptoids, we performed an enzymatic degradation study. For this assay, we chose trypsin, which is a key digestive enzyme and is known to be frequently used in cell culture applications.42 In a typical experiment, each of the peptoids AD2 or PT1 was exposed to 0.1 μM trypsin in PBS buffer (50 mM, pH = 7.4) and the reaction was maintained at a constant temperature of 37 °C. The aliquots were taken once a day and prepared for the chromatographic analysis (for more details see ESI†).43
The maximum absorbance of each aliquot sample was normalized against its respective control (peptoid sample in buffer under the same conditions but without trypsin) at fixed retention times, and the results are depicted in Fig. 4C and D. From the obtained HPLC spectra of each peptoid, it is clear that the retention peaks of the aliquots taken at different time scales overlap and remain consistently intact throughout the experiment (3 days). Based on these results, we can conclude that AD2 and PT1 maintained the stability towards proteolytic degradation by trypsin.
Assessing the ability of the peptoids to cross the blood–brain barrier (BBB)
Recently, some studies have reported peptoid oligomers capable of crossing the BBB.44 The initial assessment of the ability to cross the BBB could be evaluated by determining the lipophilicity of the proposed molecules.45 For this purpose, the n-octanol/water partition coefficient (Pow) is widely used in bioactivity predictive studies, where an immiscible mixture of octanol and water serves as a model for biological lipid and aqueous phases in partition processes.46 Generally, the distribution of the compound between the two phases is illustrated by the log to base ten of the ratio of its concentration in both phases at the equilibrium, where:
Based on the interpretation of Lipinski's rules for drug development in the context of neurodegenerative diseases, a molecule should have positive value of logPow in the range of 0.3 to 3,47 or less than 5 (ref. 48) in order to be able to cross the BBB. Molecules, that showed too low logPow (below 0.3) are too hydrophilic and therefore their uptake is not sufficient;48,49 too high values of logPow indicate that the molecule is too hydrophobic to maintain the ability to cross the BBB.
The n-octanol/water partition coefficient (Pow) for peptoids AD2 and PT1 was determined by the shake-flask method.50 In a typical experiment, a peptoid solution (10 μM) in PBS buffer (50 mM, pH = 7.4) was added to an equal volume of n-octanol and the system was allowed to equilibrate under gentle agitation for 1 week (∼150 hours) to ensure the partitioning of the peptoid between two immiscible phases.51 Next, the phases were separated by centrifugation, and the UV-Vis spectrum of each phase was recorded. From the absorbance at λmax the concentration of peptoid in each phase was established, and logPow was calculated (Fig. S28†). The values of logPow obtained for AD2 and PT1 (1.05 and 0.52, respectively) are in the prediction range of 0.3 to 3, suggesting that both peptoids could potentially cross the BBB.
Overall, the stability experiments demonstrate the peptoids' ability to withstand physiological conditions (pH and body temperature), as well as their high stability towards proteolysis by enzymes and potential to cross the BBB, and therefore the proposed peptoids could be good candidates for further therapeutic applications.
Conclusions
Cu-chelation is one of the known and extensively studied approaches in the context of AD. Among all the prerequisites for potential drug candidates, their selectivity for Cu over Zn is a crucial factor; the ionic pool of the synaptic cleft, where harmful ROS production takes place, contains a higher concentration of Zn over Cu, which can potentially hinder Cu binding by a proposed chelator due to the similar binding preferences of Cu and Zn. Nevertheless, the selectivity of a chelator for Cu over Zn, does not guarantee full inhibition of ROS production because the selectivity can be thermodynamic and/or insufficient. To overcome this, herein we implemented another approach: the co-administration of a cocktail containing two structurally different chelators that aim to target two different metal ions, i.e. Cu2+ and Zn2+, simultaneously, but independently from each other. In contrast to commonly known combination therapy, where two or more different drugs aim at the same target, in our strategy two different chelators aim at the binding of two different metal ions simultaneously, towards complete and immediate inhibition of ROS formation. The chelators we develop are based on peptoids, which are more versatile than small molecule-based chelators and more stable than peptide-based chelators. As a Cu-targeting chelator, we utilized a helical sequence containing the metal-binding ligands terpyridine and hydroxyquinoline pre-organized at the same site of the helix for enhanced metal-binding, and two water-solubility-inducing side-chains which enable the peptoid's solubility in HEPES buffer under physiological conditions (the peptoid AD2). We demonstrated that AD2 is highly selective for Cu2+ and is able to extract Cu from the CuAβ complex and slow down ROS production under physiological conditions in a reducing environment and in the co-presence of excess of Zn2+ ions. Furthermore, we illustrated, that when AD2 is co-administrated in a cocktail that contains PT1, a peptoid-chelator for Zn2+ previously developed in our lab, the ROS production by the CuAβ1–16 complex in the presence of Zn2+ is immediately and fully prevented (>99% inhibited). To support the application of these peptoids as drug candidates in the context of AD, we further evaluated the stability and blood–brain barrier (BBB) penetrability of AD2 and PT1. We found that both AD2 and PT1 are stable under conditions that simulate physiological pH and temperature and invulnerable to proteolytic degradation by enzymes like trypsin, as well as show potential to cross the BBB.We believe that our cocktail approach, demonstrated here for the first time towards ROS inhibition in the context of AD, opens up an excellent and new avenue for alternative AD-associated chelation therapy.‡
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
Conceptualization, A. E. B. and G. M.; methodology, A. E. B. and G. M.; software, A. E. B.; formal analysis, A. E. B.; investigation, A. E. B.; data curation, A. E. B.; writing—original draft preparation, A. E. B. and G. M.; writing—review and editing, A. E. B. and G. M.; project administration, G. M.; funding acquisition, G. M. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Mrs Larisa Panz and Dr Oleg Zgadzai for their assistance with MS and EPR measurements. A. E. B. thanks the Schulich Foundation for part of her PhD fellowship.Notes and references
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114(7), 3659–3853 CrossRef CAS PubMed.
- A. I. Bush, Curr. Opin. Chem. Biol., 2000, 4, 184 CrossRef CAS PubMed.
- (a) P. Faller, C. Hureau and O. Berthoumieu, Inorg. Chem., 2013, 52(21), 12193–12206 CrossRef CAS PubMed; (b) A. Tiiman, P. Palumaa and V. Tõugu, Neurochem. Int., 2013, 62, 367–378 CrossRef CAS.
- (a) C. Esmieu, D. Guettas, A. Conte-daban, L. Sabater, P. Faller and C. Hureau, Inorg. Chem., 2019, 58(20), 13509–13527 Search PubMed; (b) K. P. Kepp, N. K. Robakis, P. F. Hoilund-Carlsen, S. L. Sensi and B. Vissel, Brain, 2023, awad159 Search PubMed.
- C. Cheignon, M. Tomas, D. Bonnefont-Rousselot, P. Faller, C. Hureau and F. Collin, Redox Biol., 2018, 14, 450–464 Search PubMed.
- (a) S. Ayton, P. Lei and A. I. Bush, Neurotherapeutics, 2015, 12, 109–120 Search PubMed; (b) V. Chiurchiù, A. Orlacchio and M. Maccarrone, Oxid. Med. Cell. Longevity, 2016, 2016, 7909380 Search PubMed; (c) N. Xia and L. Liu, Mini-Rev. Med. Chem., 2014, 14(3), 271–281 Search PubMed; (d) P. J. Crouch and K. J. Barnham, Acc. Chem. Res., 2012, 45(9), 1604–1611 Search PubMed; (e) K. J. Barnham and A. I. Bush, Chem. Soc. Rev., 2014, 43, 6727–6749 Search PubMed; (f) A. Robert, Y. Liu, M. Nguyen and B. Meunier, Acc. Chem. Res., 2015, 48(5), 1332–1339 CrossRef CAS PubMed; (g) C. Hureau and P. Faller, Biochimie, 2009, 91(10), 1212–1217 CrossRef CAS PubMed.
- (a) C. Hureau in Encyclopedia of Inorganic and Bioinorganic Chemistry, ed. R. A. Scott, John Wiley & Sons Ltd, 2018, pp. 1–14 Search PubMed; (b) P. Faller and C. Hureau, Dalton Trans., 2009, 1080–1094 Search PubMed; (c) M. A. Santos, K. Chand and S. Chaves, Coord. Chem. Rev., 2016, 327–328, 287–303 Search PubMed.
- (a) J. Kardos, I. Kovfics, F. Hajs, M. Kfilmfin and M. Simonyi, Neurosci. Lett., 1989, 103, 139–144 Search PubMed; (b) D. E. Hartter and A. Barnea, Synapse, 1988, 2, 412–415 CrossRef CAS PubMed; (c) C. J. Frederickson, Int. Rev. Neurobiol., 1989, 31, 145–238 Search PubMed.
- (a) E. Atrián-Blasco, P. Gonzalez, A. Santoro, B. Alies, P. Faller and C. Hureau, Coord. Chem. Rev., 2018, 371, 38–55 CrossRef; (b) I. Zawisa, M. Rozga and W. Bal, Coord. Chem. Rev., 2012, 256(19–20), 2297–2307 Search PubMed; (c) S. Noël, S. Bustos, S. Sayen, E. Guillon, P. Faller and C. Hureau, Metallomics, 2014, 6, 1220–1222 CrossRef PubMed; (d) B. Alies, E. Renaglia, M. Rozga, W. Bal, P. Faller and C. Hureau, Anal. Chem., 2013, 85(3), 1501–1508 CrossRef CAS PubMed.
- (a) A. Conte-Daban, A. Day, P. Faller and C. Hureau, Dalton Trans., 2016, 45, 15671–15678 Search PubMed; (b) E. Atrian-Blasco, A. Conte-Daban and C. Hureau, Dalton Trans., 2017, 46, 12750–12759 RSC.
- (a) M. G. Savelieff, G. Nam, J. Kang, H. J. Lee, M. Lee and M. H. Lim, Chem. Rev., 2019, 119(2), 1221–1322 CrossRef CAS PubMed; (b) J. L. Arbiser, S. K. Kraeft, R. van Leeuwen, S. J. Hurwitz, M. Selig, G. R. Dickersin, A. Flint, H. R. Byers and L. B. Chen, Mol. Med., 1998, 4(10), 665–670 CrossRef CAS PubMed; (c) M. K. Lawson, M. Valko, M. T. D. Cronin and K. Jomová, Curr. Pharmacol. Rep., 2016, 271–280 CrossRef CAS.
- (a) E. Atrián-Blasco, E. Cerrada, A. Conte-Daban, D. Testemale, P. Faller, M. Laguna and C. Hureau, Metallomics, 2015, 7(8), 1229–1232 CrossRef PubMed; (b) C. Rulmont, J.-L. Stigliani, C. Hureau and C. Esmieu, Inorg. Chem., 2024, 63(5), 2340–2351 CrossRef CAS PubMed.
- W. Zhang, Y. Liu, C. Hureau, A. Robert and B. Meunier, Chem.–Eur. J., 2018, 24, 7825 CrossRef CAS PubMed.
- F. Gervais and F. Bellini, WO2007049098A3, 2007 CAS.
- L. Patel and G. T. Grossberg, Drugs Aging, 2011, 28, 539–546 CrossRef CAS.
- M. Xilinas, WO2006117660A2, 2006.
- L.-L. Chen, Y.-G. Fan, L. X. Zhao, Q. Zhang and Z.-Y. Wang, Bioorg. Chem., 2023, 131, 106301 CrossRef CAS PubMed.
- J. Seo, B.-C. Lee and R. N. Zuckermann, in Comprehensive Biomaterials, ed. P. Ducheyne, K. E. Healy, D. W. Hutmacher, D. W. Grainger, C. J. Kirkpatrick, Elsevier, 2011, vol. 2, pp. 53–76 Search PubMed.
- (a) J. T. Nguyen, C. W. Turck, F. E. Cohen, R. N. Zuckermann and W. A. Lim, Science, 1998, 282(5396), 2088–2092 Search PubMed; (b) T. Hara, S. R. Durell, M. C. Myers and D. H. Appella, J. Am. Chem. Soc., 2006, 128, 1995–2004 Search PubMed; (c) D. G. Udugamasooriya, S. P. Dineen, R. A. Brekken and T. A. Kodadek, J. Am. Chem. Soc., 2008, 130, 5744–5752 CrossRef CAS PubMed.
- (a) B. C. Lee, T. K. Chu, K. A. Dill and R. N. Zuckermann, J. Am. Chem. Soc., 2008, 130, 8847–8855 CrossRef CAS PubMed; (b) G. Maayan, M. D. Ward and K. Kirshenbaum, Chem. Commun., 2009, 56–58 RSC; (c) M. Baskin and G. Maayan, Chem. Sci., 2016, 7, 2809–2820 Search PubMed; (d) A. D'Amato, P. Ghosh, C. Costabile, G. Della Sala, I. Izzo, G. Maayan and F. De Riccardis, Dalton Trans., 2020, 49, 6020–6029 Search PubMed; (e) P. Ghosh and G. Maayan, Chem.–Eur. J., 2021, 27, 1383 CrossRef CAS PubMed; (f) P. Ghosh, I. Rosenberg and G. Maayan, J. Inorg. Biochem., 2021, 217, 111388 CrossRef CAS PubMed; (g) A. E. Behar and G. Maayan, Chem.–Eur. J., 2023, e202301118 CrossRef CAS PubMed.
- A. E. Behar, L. Sabater, M. Baskin, C. Hureau and G. Maayan, Angew. Chem., Int. Ed., 2021, 60, 24588 CrossRef CAS.
- (a) G. Maayan, M. D. Ward and K. Kirshenbaum, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13679–13684 CrossRef CAS; (b) G. Della Sala, B. Nardone, F. De Riccardis and I. Izzo, Org. Biomol. Chem., 2013, 11, 726–731 RSC; (c) R. Schettini, B. Nardone, F. De Riccardis, G. Della Sala and I. Izzo, Eur. J. Org. Chem., 2014, 7793–7797 CrossRef CAS; (d) K. J. Prathap and G. Maayan, Chem. Commun., 2015, 51, 11096–11099 RSC; (e) R. Schettini, F. De Riccardis, G. Della Sala and I. Izzo, J. Org. Chem., 2016, 81, 2494–2505 CrossRef CAS; (f) C. M. Darapaneni, P. Ghosh, T. Ghosh and G. Maayan, Chem.–Eur. J., 2020, 26, 9573–9579 CrossRef CAS PubMed; (g) T. Ghosh, P. Ghosh and G. Maayan, ACS Catal., 2018, 8, 10631–11064 CrossRef CAS; (h) G. Ruan, L. Engelberg, P. Ghosh and G. Maayan, Chem. Commun., 2021, 57, 939–942 RSC; (i) G. Ruan, P. Ghosh, N. Fridman and G. Maayan, J. Am. Chem. Soc., 2021, 143(28), 10614–10623 CrossRef CAS PubMed.
- R. N. Zuckermann, J. M. Kerr, W. H. Moos and S. B. H. Kent, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS.
- (a) K. Kirshenbaum, A. E. Barron, R. A. Goldsmith, P. Armand, E. K. Bradley, K. T. V. Truong, K. A. Dill, F. E. Cohen and R. N. Zuckermann, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4303–4308 CrossRef CAS PubMed; (b) C. W. Wu, K. Kirshenbaum, T. J. Sanborn, J. A. Patch, K. Huang, K. A. Dill, R. N. Zuckermann and A. E. Barron, J. Am. Chem. Soc., 2003, 125, 13525–13530 CrossRef PubMed; (c) J. R. Stringer, J. A. Crapster, I. A. Guzei and H. E. Blackwell, J. Am. Chem. Soc., 2011, 133, 15559–15567 CrossRef; (d) J. A. Crapster, I. A. Guzei and H. E. Blackwell, Angew. Chem., Int. Ed., 2013, 52, 5079–5084 CrossRef CAS; (e) O. Roy, G. Dumonteil, S. Faure, L. Jouffret, A. Kriznik and C. Taillefumier, J. Am. Chem. Soc., 2017, 139, 13533–13540 CrossRef CAS PubMed; (f) A. E. Behar, P. Ghosh and G. Maayan in Copper Bioinorganic Chemistry, ed. A. J. Simaan and M. Réglier, World Scientific, 2023, pp. 211–249, DOI:10.1142/9789811269493_0007.
- S. M. Miller, R. J. Simon, S. Ng, R. N. Zuckermann, J. M. Kerr and W. H. Moos, Drug Dev. Res., 1995, 35, 20–32 CrossRef CAS.
- Y. Kwon and T. Kodadek, J. Am. Chem. Soc., 2007, 129(6), 1508–1509 CrossRef CAS PubMed.
- P. Ghosh and G. Maayan, Chem. Sci., 2020, 11, 10127–10134 RSC.
- A. W. Wijaya, A. I. Nguyen, L. T. Roe, G. L. Butterfoss, R. K. Spencer, N. K. Li and R. N. Zuckermann, J. Am. Chem. Soc., 2019, 141(49), 19436–19447 CrossRef CAS PubMed.
- C. M. Darapaneni, P. J. Kaniraj and G. Maayan, Org. Biomol. Chem., 2018, 16, 1480–1488 RSC.
- M. Baskin and G. Maayan, Dalton Trans., 2018, 47, 10767–10774 RSC.
- The HR-MS analysis was also conducted for a mixture of 1 equiv. of Cu(II) and 2 equiv. of AD2. Despite preincubation in different stoichiometric ratios, the obtained spectrum exhibits the presence of only 1:1 intramolecular CuAD2 species (see Fig. S10 and S11†).
- (a) Z. Xiao and A. G. Wedd, Nat. Prod. Rep., 2010, 27, 768–789 RSC; (b) L. Zhang, M. Koay, M. J. Maher, Z. Xiao and A. G. Wedd, J. Am. Chem. Soc., 2006, 128, 5834–5850 CrossRef CAS.
- B. Alies, E. Renaglia, M. Rózga, W. Bal, P. Faller and C. Hureau, Anal. Chem., 2013, 85(3), 1501–1508 CrossRef CAS.
- For more details of AD2 binding to Zn2+ and ZnAD2 UV-Vis signature see Fig S7B.† UV-Vis titration of AD2 with Zn2+ under similar conditions to those with Cu2+, resulted in appearance of new bands at λ = 264, 311 and 323 nm. The metal-to-peptoid ratio plot suggested a 1:0.5 AD2 to Zn2+ ratio and formation of the intermolecular 1:2 Zn(AD2)2 complex.
- B. Alies, I. Sasaki, O. Proux, S. Sayen, E. Guillon, P. Faller and C. Hureau, Chem. Commun., 2013, 49, 1214–1216 RSC.
- C. Hureau, Coord. Chem. Rev., 2012, 256, 2164–2174 CrossRef CAS.
- J. T. Pedersen, S. W. Chen, C. B. Borg, S. Ness, J. M. Bahl, N. H. H. Heegaard, C. M. Dobson, L. Hemmingsen, N. Cremades and K. Teilum, J. Am. Chem. Soc., 2016, 138, 3966–3969 CrossRef CAS.
- (a) B. Lucchese, K. J. Humphreys, D.-H. Lee, C. D. Incarvito, R. D. Sommer, A. L. Rheingold and K. D. Karlin, Inorg. Chem., 2004, 43(19), 5987–5998 CrossRef CAS; (b) N. Wei, N. N. Murthy and K. D. Karlin, Inorg. Chem., 1994, 33, 6093–6100 CrossRef CAS.
- S. C. Drew and K. J. Barnham, Acc. Chem. Res., 2011, 44(11), 1146–1155 CrossRef CAS PubMed.
- M. Jureschi, A. V. Lupaescu, L. Ion, B. A. Petre and G. Drochioiu in Advancements of Mass Spectrometry in Biomedical Research. Advances in Experimental Medicine and Biology, ed. A. Woods and C. Darie, Springer, Cham, 2019, vol. 1140 Search PubMed.
- L. Habasescu, M. Jureschi, B. A. Petre, M. Mihai, R. V. Gradinaru, M. Murariu and G. Drochioi, Int. J. Pept. Res. Ther., 2020, 26, 2529–2546 CrossRef CAS.
- J. M. Chen, E. S. Radisky and C. Férec, Handb. Proteolytic Enzymes, 2013, 3, 2600–2609 Search PubMed.
- H. C. Schunk, M. J. Austin, B. Z. Taha, M. S. McClellan, L. J. Suggs and A. M. Rosales, Mol. Syst. Des. Eng., 2023, 8, 92 RSC.
- (a) Z. Zhao, L. Zhu, H. Li, P. Cheng, J. Peng, Y. Yin, Y. Yang, C. Wang, Z. Hu and Y. Yang, Small, 2017, 13, 1602857 CrossRef; (b) K. Pradhan, G. Das, V. Gupta, P. Mondal, S. Barman, J. Khan and S. Ghosh, ACS Chem. Neurosci., 2019, 10(3), 1355–1368 CrossRef CAS.
- (a) W. A. Banks and A. J. Kastin, Brain Res. Bull., 1985, 15, 287–292 CrossRef CAS; (b) X. Liu, B. Testa and A. Fahr, Pharm. Res., 2011, 28, 962–977 CrossRef CAS.
- (a) A. Leo, C. Hansch and D. Elkins, Chem. Rev., 1971, 71, 525 CrossRef CAS; (b) M. S. Tute, Adv. Drug Res., 1971, 6, 1 CAS.
- (a) N. Gulyaeva, A. Zaslavsky, P. Lechner, M. Chlenov, O. McConnell, A. Chait, V. Kipnis and B. Zaslavsky, Eur. J. Med. Chem., 2003, 38(4), 391–396 CrossRef CAS PubMed; (b) W. P. Walters, Expert Opin. Drug Discovery, 2012, 7(2), 99–107 CrossRef CAS PubMed.
- M. Dichiara, B. Amata, R. Turnaturi, A. Marrazzo and E. Amata, ACS Chem. Neurosci., 2020, 11(1), 34–44 CrossRef CAS PubMed.
- T. S. Carpenter, D. A. Kirshner, E. Y. Lau, S. E. Wong, J. P. Nilmeier and F. C. Lightstone, Biophys. J., 2014, 107(3), 630–641 CrossRef CAS PubMed.
- H. L. Bolt, C. E. J. Williams, R. V. Brooks, R. N. Zuckermann, S. L. Cobb and E. H. C. Bromley, Biopolymers, 2017, 108, e23014 CrossRef PubMed.
- OECD Guideline 107, July 1995 Search PubMed.
- (a) G. Maayan, B. Yoo and K. Kirshenbaum, Tetrahedron Lett., 2008, 49, 335–338 CrossRef CAS; (b) M. Baskin and G. Maayan, Biopolymers, 2015, 104, 577–584 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04313h |
| ‡ Additional references are cited in the ESI.†52 |
| This journal is © The Royal Society of Chemistry 2024 |