
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLeveraging ordered voids in microporous perovskites for intercalation and post-synthetic modification†
Connor W. Dalton ,
Paige M. Gannon,
Werner Kaminsky and
Douglas A. Reed*
,
Paige M. Gannon,
Werner Kaminsky and
Douglas A. Reed*
Department of Chemistry, University of Washington, Seattle, WA 98195, USA. E-mail: dreed4@uw.edu
First published on 16th October 2024
Abstract
We report the use of porous organic layers in two-dimensional hybrid organic–inorganic perovskites (HOIPs) to facilitate permanent small molecule intercalation and new post-synthetic modifications. While HOIPs are well-studied for a variety of optoelectronic applications, the ability to manipulate their structure after synthesis is another handle for control of physical properties and could even enable use in future applications. If designed properly, a porous interlayer could facilitate these post-synthetic transformations. We show that for a series of copper-halide perovskites, a crystalline arrangement of designer ammonium groups allows for permanently porous interlayer space to be accessed at room temperature. Intercalation of the electroactive molecules ferrocene and tetracyanoethylene into this void space can be performed with tunable loadings, and these intercalated perovskites are stable for months. The porosity also enables reactivity at the copper-halide layer, allowing for facile halide replacement. Through this, we access previously unobserved reactivity with halogens to perform halide substitution, and even replace halides with pseudohalides. In the latter case, the porous structure allows for stabilization of new phases, specifically a novel copper–thiocyanate perovskite phase, only accessible through post-synthetic modification. We envision that this broad design strategy can be expanded to other industrially relevant HOIPs to create a new class of highly adjustable perovskites.
Introduction
Two-dimensional hybrid organic–inorganic perovskites (HOIPs) have been studied extensively for applications in solar cells and light-emitting devices due to their optoelectronic properties.1–7 Composed of metal-halide sheets and organic spacers, typically alkylammonium groups, every facet of these structures can be changed to tune these features.8–12 Post-synthetic modification of HOIPs has also emerged as an active area of research to further control various physical characteristics.13,14 However, modifications are generally limited to the surface, and direct interactions with the interior bulk lattice are more challenging to engineer.15Intercalation is one possible route for post-synthetically modifying the bulk perovskite lattice. Simply exposing certain perovskites to guest molecules can lead to encapsulation of new species. In one of the first reports, Mitzi et al. used halogen bonding between aromatic C–H bonds of phenethylammonium (PEA) spacers and C–F groups of perfluorobenzene to induce intercalation in (PEA)2[SnI4].16 Smith et al. further showed that exposure of ((CH2I)(CH2)5NH3)2[PbI4] to I2 vapor can accommodate I2 in the interlayer.17 In both cases, modifying this interlayer impacted the structural and electronic properties of the inorganic layer. While successful, these intercalations are highly unstable. Removal from solvent or I2 atmosphere resulted in rapid deintercalation. Permanent encapsulation of guest molecules within perovskites can be achieved by introducing these molecules during synthesis. Recent examples of HOIPs synthesized with donor–acceptor moieties in the organic layer have led to stable intercalations.18–20 Neutral molecules with proper intermolecular interactions with the organic cation spacers can also be incorporated during perovskite formation.21 However, these guests need to be introduced during synthesis, and are highly specific for a particular molecule. General post-synthetic routes toward stable intercalated perovskites are not currently available.
Altering the metal-halide lattice itself is also a way to post-synthetically tune specific properties. For example, the halides can be exchanged after synthesis of the material, typically by adding a halide-containing reagent and relying on ion migration through the lattice. This has been well reported in 3D perovskites, as it allows for alteration of optical properties but retains the original morphology of the perovskite.22–25 Halide exchange in 2D perovskites has also been demonstrated, although this has been comparatively less studied.26 Current halide exchange methods are effective for particular form factors, like thin films and nanocrystals, but have not been demonstrated in larger morphologies like bulk single crystals. Furthermore, performing any halide substitution that requires approaches beyond ion migration, which would be necessary for nearly any type of halide to pseudohalide exchange, are difficult with current approaches. If designed properly, the organic interlayers could facilitate new exchange reactions at this metal-halide layer.
Porosity is a promising method to enable new types of chemical transformations in HOIPs. For other relatively unstable materials, intrinsic permanent porosity enables both small molecule intercalation and post-synthetic modification.27 The open space allows for molecules to be introduced in a rational fashion and increases the reactive surface area of the material. Researchers have explored introducing porosity in 3D perovskites. Chemical etching of bulk lead-halide perovskites has been shown to create mesoporous phases.28 Additionally, the introduction of large quantities of metal defects to create “hollow” perovskites has also been demonstrated.29,30 However, while these methods increase the surface area, they do not rationally allow interaction between guest molecules and the interior of the perovskite lattice, necessitating a different approach.
In 2D HOIPs, organic spacers can be used to introduce porosity. Recent work by Kataoka et al. demonstrated layered perovskite phases templated by mixing octylammonium polyhedral oligomeric silsesquioxane chloride (APOSSCl8, Si8O12((CH2)3NH3)8Cl8) and MCl2 salts.31 Certain metals, specifically copper chloride and palladium chloride, generated a material with measurable porosity of >200 m2 g−1. However, without structural details the pore environment remains insufficiently characterized. Elucidating the exact nature of the porosity is critical for rational introduction of guest molecules in this materials class. Furthermore, fundamentally the use of this porous interlayer like other porous materials could be challenging, as perovskites are more dynamic than traditional porous materials.32–36 Recent reports on other types of layered metal-halide species show that even with cage-like interlayers, these phases are inaccessible to standard gas molecules like N2 and CO2.37,38 It is therefore crucial to understand how this porous interlayer can be used, and to determine whether this porosity observed at cryogenic temperatures would be accessible to intercalate molecules or allow for post-synthetic modification at higher temperatures.
Here, we characterize the porous perovskite (APOSS)[CuCl4]4 that can permanently host small molecules in its interlayer and undergo facile post-synthetic halide exchange. We use the void spaces in this lattice to permanently host electronically active small molecules like ferrocene (Fc) and tetracyanoethylene (TCNE) to create highly tunable interlayer environments, with the intercalants showing direct contact with the metal-halide layer. We also demonstrate porosity-enabled post-synthetic exchange reactions at the metal-halide layer, specifically chloride to bromide halide exchange using Br2. As expected with porosity, the reactivity of guest species differs from reports in nonporous systems, facilitating new types of transformations. We lastly show that the porosity enables us to access entirely new perovskites phases. Specifically, we leverage post-synthetic thiocyanate exchange to report the first copper–thiocyanate perovskite. The intrinsic porosity of these copper-halide materials enables new perovskite chemistries to be more thoroughly explored.
Results and discussion
Synthesis and crystal structure
X-ray crystallography was used to understand the nature of porosity in these copper halide perovskites. Yellow single crystals of (APOSS)[CuCl4]4 suitable for diffraction were grown by diffusing ethanol into an aqueous solution of CuCl2 and APOSSCl8. The plate-like crystals displayed Fmmm symmetry, consistent with a layered perovskite structure (Fig. 1). Similar to previous examples of copper halide perovskites, the inorganic layer adopts an antiferrodistortive arrangement of Jahn–Teller distorted [CuCl4]2− octahedra (Fig. 1).39–42 The average Cu–Cl bond distance is 2.80 Å for the in-plane long axis, 2.33 Å for the in-plane short axis, and 2.30 Å for the out-of-plane axis.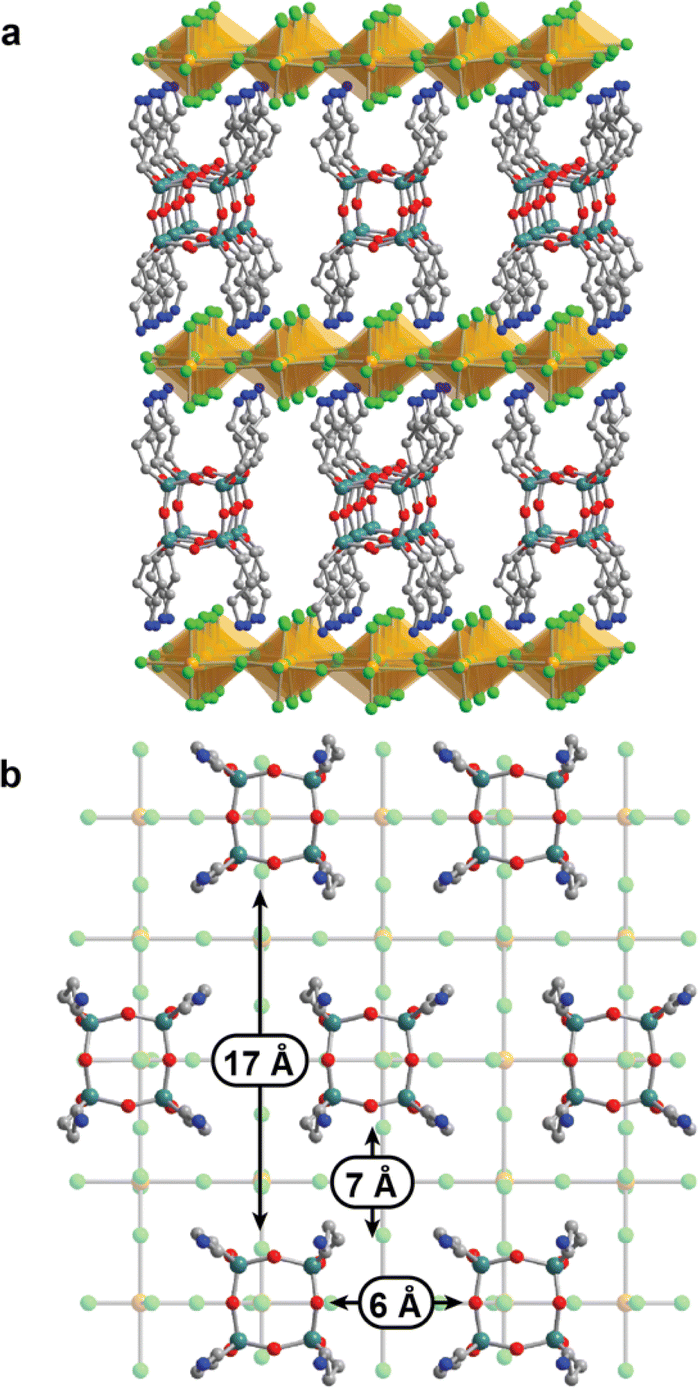 | ||
| Fig. 1 (a) Portion of the structure of (APOSS)[CuCl4]4 as determined by analysis of single crystal X-ray diffraction data. (b) Detail of the arrangement of APOSS groups within organic interlayer, with selected pore dimensions highlighted. Yellow, green, gray, blue, red, and teal spheres represent Cu, Cl, C, N, O, and Si, respectively. H atoms and disorder are omitted for clarity. | ||
Immediately apparent in interlayer spacing is the ordered siloxane cores (Fig. 1b). Adjacent rows of POSS groups are offset to create large pores, where the APOSS ligands serve as pillars and the space in between can be accessed by guest molecules (Fig. S2†). Between different rows the siloxane groups are 7 Å apart, and the siloxane groups in each individual row are 6 Å apart. The offset alignment between each row gives large spaces of up to 17 Å between siloxane groups. The interlayer spacing of 17 Å results in a height of 12 Å for all pores, with terminal halide groups on the top and bottom of the pores, suggesting that reactive chemistry can occur at these positions.
Powder crystalline samples can also be prepared. Dropwise addition of an aqueous solution of CuCl2 and APOSSCl8 into ethanol produces a microcrystalline powder.31 Pawley refinement of the powder X-ray diffraction pattern produces similar unit cell parameters to the single crystal data, although a slightly different unit cell was obtained with I4 symmetry (Fig. S5†). The difference in unit cell could potentially be due to the higher temperature at which this measurement was obtained (Fig. S7†), as is seen in other 2D HOIPs.43 Despite the different symmetry, a permanent porosity of 276 m2 g−1 was obtained through N2 adsorption at 77 K, indicating that this material, at low temperature, demonstrated accessible void space in the APOSS layers (Fig. S8†). This is consistent with previous reports.31
Mixing various amounts of CuBr2 or APOSSBr8 during the synthesis leads to variable bromide incorporation in the lattice to form (APOSS)[CuCl4−xBrx]4. Bromide substitution results in a shift of peak positions in powder X-ray diffraction measurements (Fig. S9 and S25†). In the fully brominated case x = 4, analysis of the unit cell parameters reveals that the average Cu–X distance increases from 2.60 (Cu–Cl) in (APOSS)[CuCl4]4 to 2.73 Å (Cu–Br) in (APOSS)[CuBr4]4. The change in bond length, 0.13 Å, is consistent with the change in atomic radii from Cl to Br and the Cu–Br bond length is similar to previous literature reports.39,40 The interlayer direction, which is dominated by the APOSS spacer, remains relatively unchanged, consistent with previous examples mixed-halide Cl/Br systems in other copper perovskites.41,42 The disappearance of the peak at 15.7° and the growth of a peak at 16.3° (Fig. S9 and S10†) are indicative of a change in symmetry to the orthorhombic P222 space group in the fully brominated sample.
Permanent small molecule intercalation
The pore size seen in the single crystal data indicates that several molecules should be amenable to intercalation. While in traditional porous materials held together entirely by covalent bonds this intercalation might be viewed as trivial, perovskites are highly dynamic.33–35 The interlayer POSS groups, particularly at room temperature, could be moving in ways that prevent guest molecule introduction. In other 2D metal-halide materials with void spaces in the interlayers, small guest molecules like N2 or CO2 cannot be accommodated.37,38 Despite these concerns, we demonstrate that even at room temperature, (APOSS)[CuCl4−xBrx]4 can use its porosity to host many types of molecules which can be introduced in different methods. We show this with two specific molecules: tetracyanoethylene (TCNE) and ferrocene Fe(C5H5)2 (Fc) (Fig. 2a). | ||
| Fig. 2 (a) Scheme of gas-phase or liquid-phase intercalation in (APOSS)[CuCl4]4 to make (APOSS)[CuCl4]4·1.22Fc and (APOSS)[CuCl4]4·xTCNE, respectively. (b and c) EDX mapping data for Fe (b, orange dots) and Cu (c, yellow dots) of representative flake of Fc-intercalated (APOSS)[CuCl4]4, showing even dispersion of Fe throughout the material. White bars are 25 μm. (d) Powder X-ray diffraction patterns for (APOSS)[CuCl4]4 (black), (APOSS)[CuCl4]4·xTCNE (green), and (APOSS)[CuCl4]4·1.22Fc (orange), showing that perovskite phase remains intact during intercalation. (e) Infrared spectra of (APOSS)[CuCl4]4 (black), TCNE (purple), and (APOSS)[CuCl4]4·xTCNE (green), showing the presence of intercalated TCNE. | ||
Facile gas-phase intercalation of Fc is accomplished by exposing activated (APOSS)[CuCl4]4 to Fc vapor at 75 °C for one week. A color change to a darker yellow was observed, which is retained after washing the material thoroughly with isopropyl alcohol. A loading of 1.22 equivalents of Fc per APOSS group, or just over one molecule per pore, was determined through NMR spectroscopy of the digested material (Fig. S12†), indicating a substantial loading of Fc inside the material that only can be possible by having accessible void space. This loading amount is tunable, where exposure to Fc vapor for shorter amounts of time results in variable amounts of Fc (Fig. S13†). We can further confirm the presence of Fc through infrared (IR) spectroscopy (Fig. S14†). The series of peaks from 1600–1800 cm−1 is observed in the IR spectra of Fc as well as (APOSS)[CuCl4]4·1.22Fc. These peaks have been previously assigned to overtones and combination bands of C–H bending modes in the cyclopentadienyl rings of Fc.44 Additional evidence of Fc intercalation can be seen by the slight increase in intensity at 1408 cm−1 corresponding to the asymmetric C–C stretching vibration in Fc. Finally, thermogravimetric analysis of (APOSS)[CuCl4]4·0.45Fc shows a weight loss of 5.6% at 155 °C (Fig. S15†). Not only is this consistent with the loading determined by 1H NMR of the digested material, the temperature at which the ferrocene molecules deintercalated are consistent with other intercalated materials,45 rather than surface-adsorbed species.
As further evidence that intercalation is occurring in the bulk lattice, we demonstrated that ferrocene can be introduced in larger crystal morphologies. Microcrystalline (0.001–0.005 mm2) flakes of (APOSS)[CuCl4]4 were subjected to Fc intercalation under the same conditions as the powder samples. Energy-dispersive X-ray spectroscopy (EDX) maps were collected of three flakes which all show a homogeneous distribution of Fc within the flake (Fig. 2b, c and S16†). This confirms that Fc diffuses through the interlayer void space as it intercalates.
The introduction of so many ferrocene molecules should create large structural differences in nonporous perovskites.16,17 In this porous material, however, the unit cell remains largely unchanged, with an increase of ∼0.1 Å in the interlayer spacing parameter (Fig. 2d and S18†). This further confirms that the porosity is used to host these Fc units instead of some other possible mechanism. Full loading of 1.22 equivalents of Fc should take up the majority of the void space and might require rearrangement of the POSS groups. Indeed, the powder X-ray diffraction data show the presence of new peaks. These peaks index within the same I4 space group as the parent material but are suggestive of ordered electron density in new areas. We attribute this to the Fc molecules forcing the POSS groups themselves to be ordered. Despite remaining a perovskite structure, the intercalated material shows a negligible surface area of <10 m2 g−1 (Fig. S19†), consistent with the void spaces being fully occupied by the ferrocene molecules.
Unlike other examples of intercalation into 2D perovskites, which undergo rapid deintercalation once removed from the vapor or solution of intercalant,16,17 (APOSS)[CuCl4]4·1.22Fc was stable for months under ambient conditions on our benchtop. Ferrocene molecules remained intercalated within (APOSS)[CuCl4]4 even under more intense conditions like exposure to vacuum. To measure the EDX positional mapping in Fig. 2b and c, the intercalated perovskite are placed under vacuum at 10 μbar during sample preparation, indicating that Fc remains intercalated under low vacuum conditions for at least 1 hour. To our knowledge, this is the first example of a perovskite permanently encasing a molecule that can rationally be introduced post-synthetically. This could represent a new method with which to alter perovskite properties.
While ferrocene can be introduced in the gas phase, we also attempted to observe liquid-phase intercalation. We chose TCNE, which needs to be introduced as a solution. Soaking (APOSS)[CuCl4]4 in a tetrahydrofuran (THF) solution of TCNE at 50 °C for one week results in a material with a large signal in its infrared spectrum at 2222 cm−1 and 2194 cm−1 (Fig. 2e). Compared to free TCNE, these values are redshifted by an average of 38 cm−1, indicating some change in local environment which we attribute to be inside the pores. Even subtle changes in molecular orbital population46 or dielectric constant around the TCNE could cause this shift in stretching frequency. While exact loading of TCNE is difficult to determine through normal methods, comparing the relative integrated absorbance to several different samples obtained by physically mixing TCNE and (APOSS)[CuCl4]4 in various ratios suggest that the TCNE is intercalated on the order of 1 molecule per pore (Fig. S22†). Similar analysis through thermogravimetric analysis shows a weight loss of 5.9% at 172 °C, which corresponds to a loading of 0.83 equivalents per POSS unit (Fig. S15†). We also confirm the presence of TCNE through diffuse reflectance spectroscopy (DRS). While the transitions of TCNE at 260 and 390 nm coincidentally overlap with the Cl 2p to Cu dx2−y2 ligand-to-metal charge transfer (LMCT) bands in (APOSS)[CuCl4]4 (282 and 385 nm), intercalating TCNE in (APOSS)[CuBr4]4 allows us to clearly observe the features of TCNE (Fig. S23†).
Similar to Fc intercalation, the powder X-ray diffraction data of (APOSS)[CuCl4]4·xTCNE show nearly identical features to the unintercalated material (Fig. 2d), indicating the porosity is responsible for the large uptake of TCNE. Unlike in the case of Fc, there does not appear to be an additional ordering of POSS groups upon intercalation of TCNE, which is expected based on the smaller size of TCNE compared to Fc. The TCNE-intercalated material is also stable under ambient conditions for months. The different methods of introducing molecules between Fc and TCNE demonstrates the new possibilities that can be explored in post-synthetic modification of perovskites by utilizing porous interlayers.
While the stability of intercalation in these materials under ambient conditions is dramatically different than in previous nonporous perovskites, the overall intercalation is reversible. For example, soaking the ferrocene-intercalated perovskite in THF removes these Fc molecules to result in pristine (APOSS)[CuCl4]4. This allows for even further post-synthetic modification, as in our case we can exchange the Fc molecules for TCNE directly (Fig. S24†).
Post-synthetic halide exchange
After demonstrating that the interlayer spacing is accessible to various molecules, we then looked to use the porosity to modify the inorganic layer. Porosity gives unprecedented access to the inorganic layer in these materials, allowing for new types of rational post-synthetic modification. We demonstrate that we can change these chlorides through simple exposure to bromine vapor or bromine solution.Many previous examples at post-synthetic halide exchanges in both 2D and 3D perovskites have been demonstrated.22–25 Exposure to halogen gasses Br2 and Cl2 was demonstrated by Solis-Ibarra et al. and Smith et al. to show facile exchange in thin films in lead perovskites.13,26 However, this uses the oxidizing potential of the halogen gas to facilitate this exchange. In two-dimensional copper perovskites, exposure to Cl2 could convert a bromide-containing perovskite to a chloride lattice, as Cl2 is more oxidizing than Br2, but the reverse reactivity of changing a chloride for bromide using Br2 was not observed.26 Our hypothesis is that the pores in (APOSS)[CuCl4]4 would allow for much more ready exchange at the halide positions, given that pores can be used to enhance the reactivity of any intercalated substrates.
Exposure of (APOSS)[CuCl4]4 powder to Br2, either in the gas phase or as a solution in CH2Cl2 produced a color change on the order of hours (Fig. 3a). Analysis by powder X-ray diffraction showed a retention of crystallinity in a perovskite phase (Fig. 3b). Additionally, the expected shifts in peak positions were observed.41,42 Through gas-phase introduction of Br2 vapor, we see a conversion to 18.5% bromide, as determined by EDX (Fig. S26†). In the liquid cases, exact equivalents of Br2 could be introduced. This allows for precising tuning of the halide composition, with various Cl/Br ratios shown in Fig. 3. Comparison of these peak positions matches that of mixed halide species synthesized ex situ (Fig. S9 and S10†). Even after halide exchange, the sample remains highly porous. Analysis of N2 adsorption measurements at 77 K for the fully brominated sample (APOSS)[CuBr4]4 reveals a surface area of 166 m2 g−1 (Fig. S27†), which is consistent with substitution of the heavier bromide ion.
 | ||
| Fig. 3 (a) Scheme of post-synthetic halide exchange using Br2, either in the liquid or gas phase. The specific bromide content, as determined by EDX, for these 14 samples are 0.0, 5.8, 10.4, 11.0, 21.7, 27.8, 39.5, 42.3, 43.3, 55.8, 59.2, 79.8, 90.0, and 100.0% (expressed as a gradient). Black lines on either side represent x = 0 or x = 4. (b) Powder X-ray diffraction patterns for (APOSS)[CuCl4−xBrx]4. (c) Infrared spectra of (APOSS)[CuCl4−xBrx]4, highlighting the symmetric N–H vibration around 1490 cm−1. (d and e) Diffuse reflectance spectra of (APOSS)[CuCl4−xBrx]4, highlighting two LMCT transitions. | ||
Both DRS and IR spectroscopy were used to confirm the presence of bromide in the lattice, as this exchange produces gradual changes in these spectra as a function of bromine incorporation. In infrared spectroscopy measurements (Fig. 3c), the symmetric N–H vibration in the ammonium group also redshifts from 1496 cm−1 to 1482 cm−1 when bromide is incorporated, as this stretching frequency is dependent on the donation of electron density into the N–H σ* orbital.42 The more electropositive bromide increases this donation, decreasing the bond order. The DRS data show a redshift of LMCT transitions of the halide p orbitals to the Cu dx2−y2 orbital, consistent with the higher energy of bromide p orbitals compared to those of chloride (Fig. 3d and e). In the all-chloride case, these values are 272 nm and 384 nm, which gradually shift to 354 nm and 545 nm in the all-bromide case. Both methods are consistent with trends seen in other copper perovskite systems.41,42
To confirm that porosity is essential for the halide exchange with Br2, similar substitution attempts were made with (PEA)2[CuCl4]. The interlayer spacing in this framework is similar to that in (APOSS)[CuCl4]4, however the interlayer spacing is nonporous.47 Exposure of this material to excess Br2 solution did not result in any noticeable changes by PXRD or IR spectroscopy (Fig. S29†). Assessing the bromine content by EDX showed a small incorporation of bromine (Fig. S30†), which we attribute to defect sites in the material.
We also show that porosity can facilitate halide exchange in single crystals. Single crystals of (APOSS)[CuCl4]4 ∼0.5 cm in scale were exposed to Br2 solution, and similar to powder crystalline samples showed a drastic color change (Fig. S31†). Analysis of these products through PXRD demonstrated that the lattice did indeed expand as expected based on bromine incorporation, showing 100% exchange of Cl− (Fig. S31†). The ability to easily perform single-crystal-to-single-crystal changes in perovskites is again enabled by the porosity.
Post-synthetic thiocyanate exchange
We envisioned that the porous interlayers could allow for further modification of the metal-halide layer, enabling post-synthetic synthesis of previously unknown perovskite phases. Currently, many halide substitution reactions involve ion migration,22–25 limiting their use beyond halide-to-halide replacement. In contrast, porosity could allow for larger ions like pseudohalides to be introduced, all while retaining the initial perovskite structure. We leverage this property to create a novel copper–thiocyanate structure. Pseudohalides like thiocyanates have been used as ligands in perovskite frameworks,48,49 particularly in mixed pseudohalide-halide systems like (MA)2[Pb(SCN)2I2] (MA = methylammonium) where all terminal halides in the lead-halide layer are replaced with the larger thiocyanate unit.50,51 The wide variety of pseudohalides gives yet another handle to modify the optoelectronic properties of perovskites. However, copper–thiocyanate perovskites have not been observed, as addition of SCN− to copper salts under standard perovskite-forming conditions results in a different structure entirely (see details in ESI Fig. S32†).52,53 We demonstrate that, to the best of our knowledge, evidence for the first example of a copper–thiocyanate perovskite can be observed by utilizing pores to post-synthetically install thiocyanate.Starting from (APOSS)[CuCl4]4, exposure to a solution of NH4SCN in isopropyl alcohol for one week results in a new stretch in the infrared spectra at 2087 cm−1 (Fig. 4a and b). This stretch is blueshifted relative to that of free thiocyanate, and consistent with terminal metal binding through the nitrogen.54,55 It is also different than stretches observed in common copper–thiocyanate phases Cu(SCN) or Cu(SCN)2 (Table S4†), indicating that this is a new material that we postulate is (APOSS)[CuCl4−x(SCN)x]4 with the thiocyanates exclusively replacing terminal chlorides. This stretch grows in gradually, reaching a maximum thiocyanate composition of x = 0.44 as determined by EDX. The substantial thiocyanate incorporation indicates that this is not just surface halide substitution, but is mixed throughout the material. Additionally, the material shows a new peak in DRS measurements that gradually grows in at 309 nm (Fig. S35†). This is similar to known LMCT transitions that occur in copper–thiocyanate complexes,56 supporting that thiocyanate is binding to the copper centers. X-ray photoelectron spectroscopy (XPS) data was collected for (APOSS)[CuCl3.79(SCN)0.21]4. There are two sulfur species present, each with diagnostic S 2p1/2 and 2p3/2 features that are consistent with copper–bound thiocyanates (Fig. 4c) and greatly shifted from ammonium thiocyanate (Fig. S36†).57 We attribute the existence of two different sulfur species to both bulk copper–thiocyanate interactions inside the perovskite lattice as well as surface copper–thiocyanate impurities, for which XPS would be extremely sensitive. Measurements that characterize the bulk lattice, such as IR, indicate that the vast majority of copper–thiocyanate species are in one environment with the thiocyanate clearly bound to copper, as the integrated absorbance correlates well to the elemental composition of sulfur as determined by EDX (Fig. S33†).
 | ||
| Fig. 4 (a) Scheme of thiocyanate exchange to make (APOSS)[CuCl4−x(SCN)x]4 with variable thiocyanate loadings. The specific thiocyanate loading, as determined through sulfur composition in EDX, for these 8 samples are x = 0.03, 0.05, 0.11, 0.16, 0. 21, 0.27, 0.34, and 0.44 (expressed as a gradient). (b) Infrared spectra of (APOSS)[CuCl4−x(SCN)x]4, overlaid with that of NH4SCN. (c) X-ray photoelectron spectroscopy of S 2p1/2 and 2p3/2 in (APOSS)[CuCl3.79(SCN)0.21]4. Black crosses are experimental data, and the lines represent the total fit (teal) and background (gray). The 2p1/2 fits (dashed lines) and 2p3/2 fits (solid lines) for two sites (purple and yellow lines) are shown. (d) Powder X-ray diffraction patterns for (APOSS)[CuCl4−x(SCN)x]4, showing the perovskite structure is retained. | ||
Importantly, even with the introduction of a new pseudohalide ligand, PXRD data confirm that at these loadings, the perovskite phase persists (Fig. 4d). The ability to stabilize a terminal copper–thiocyanate inside a new phase is a unique characteristic of porous materials, and could be utilized in new ways for a variety of different metal-pseudohalide combinations. Additionally, the unit cells for each loading remain nearly unchanged with increasing thiocyanate. This again suggests that only the terminal chloride positions of the copper-halide layer are replaced, as substitution of the bridging chlorides would be expected to induce a change in unit cell parameters given the different ionic radii of thiocyanate and chloride. Given these observations, the final structure should remain porous, which was confirmed through N2 adsorption isotherms at 77 K which indicates a surface area of 146 m2 g−1 (Fig. S39†).
We do note that at higher equivalents of thiocyanate, the perovskite phase is gradually replaced by a new phase. At molar thiocyanate loadings of x = 0.61, a peak grows in at 2102 cm−1 in the IR spectra (Fig. S40†). At x = 0.81, peaks at 9.75° and 12.12° appear in the PXRD pattern (Fig. S42†). These peaks are predicted in the I4 space group and could be indicative ordered siloxane groups that accommodate the bulkier SCN− ligands, analogous to intercalated Fc. However, at x = 1.27 a peak at 29.2° is clearly indicative of a new phase, and eventually this phase dominates both PXRD and IR measurements. Given the surface sensitivity of EDX used to determine thiocyanate loading, we are unable to definitively quantify the exact amount of thiocyanate where this material converts from a perovskite phase to this new phase. However, the spectroscopic differences between the phases observed at high and low thiocyanate loadings suggest that, at low loadings, a perovskite structure featuring some degree of direct copper–thiocyanate interactions may remain intact.
Conclusions
Porosity has been used to enable new chemistries in 2D HOIPs that were previously inaccessible or unstable in nonporous variants. This work demonstrates that nearly every aspect of the perovskite (APOSS)[CuCl4]4, particularly the interlayer composition and the metal-halide layer, can be precisely changed due to these open channels. The porosity allows these modifications in a variety of form factors, ranging from powders to single crystals. While this study focused on the copper-chloride variant, we believe these methods should translate to other HOIPs with different metals, where the interlayer composition could have substantial impacts on optoelectronic properties, or halide-to-pseudohalide replacement could provide another way to access new physical properties or new phases entirely. As perovskites continue to be studied as one of the most promising materials classes for many optoelectronics applications, endowing these materials with access to several new modes of reliable post-synthetic transformations could allow for use in further applications yet to be explored.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for (APOSS)[CuCl4]4 has been deposited at the CCDC under accession number 2354173.Author contributions
C. W. D. and D. A. R. formulated the project. C. W. D. synthesized and characterized all compounds, and collected and analyzed spectroscopic and powder X-ray diffraction data. P. M. G. and W. K. collected and analyzed single crystal X-ray diffraction data. C. W. D. and D. A. R. wrote the manuscript, and all authors approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the University of Washington for startup funding support. C. W. D. was supported in part by the state of Washington through the University of Washington Clean Energy Institute Graduate Fellowship. The authors acknowledge the use of instrumentation at the Molecular Analysis Facility, a National Nanotechnology Coordinated Infrastructure (NNCI) site at the University of Washington, which is supported in part by funds from the National Science Foundation (Awards NNCI-2025489, NNCI-1542101), the Molecular Engineering & Sciences Institute, and the Clean Energy Institute.Notes and references
- B. Saparov and D. B. Mitzi, Chem. Rev., 2016, 116, 4558–4596 CrossRef CAS PubMed.
- Y. Chen, Y. Sun, J. Peng, J. Tang, K. Zheng and Z. Liang, Adv. Mater., 2018, 30, 1703487 CrossRef PubMed.
- M. D. Smith, E. J. Crace, A. Jaffe and H. I. Karunadasa, Annu. Rev. Mater. Res., 2018, 48, 111–136 CrossRef CAS.
- L. Mao, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2019, 141, 1171–1190 CrossRef CAS PubMed.
- X. Li, J. M. Hoffman and M. G. Kanatzidis, Chem. Rev., 2021, 121, 2230–2291 CrossRef CAS PubMed.
- J.-C. Blancon, J. Even, C. C. Stoumpos, M. G. Kanatzidis and A. D. Mohite, Nat. Nanotechnol., 2020, 15, 969–985 CrossRef CAS PubMed.
- G. K. Grandhi, D. Hardy, M. Krishnaiah, B. Vargas, B. Al-Anesi, M. P. Suryawanshi, D. Solis-Ibarra, F. Gao, R. L. Z. Hoye and P. Vivo, Adv. Funct. Mater., 2023, 2307441 CrossRef.
- Y. Gao, E. Shi, S. Deng, S. B. Shiring, J. M. Snaider, C. Liang, B. Yuan, R. Song, S. M. Janke, A. Liebman-Peláez, P. Yoo, M. Zeller, B. W. Boudouris, P. Liao, C. Zhu, V. Blum, Y. Yu, B. M. Savoie, L. Huang and L. Dou, Nat. Chem., 2019, 11, 1151–1157 CrossRef CAS PubMed.
- Y.-H. Kim, Y. Zhai, H. Lu, X. Pan, C. Xiao, E. A. Gaulding, S. P. Harvey, J. J. Berry, Z. V. Vardeny, J. M. Luther and M. C. Beard, Science, 2021, 371, 1129–1133 CrossRef CAS PubMed.
- C. Yang, W. Chen, Y. Ding, J. Wang, Y. Rao, W. Liao, Y. Tang, P. Li, Z. Wang and R. Xiong, Adv. Mater., 2019, 31, 1808088 CrossRef PubMed.
- J. V. Passarelli, D. J. Fairfield, N. A. Sather, M. P. Hendricks, H. Sai, C. L. Stern and S. I. Stupp, J. Am. Chem. Soc., 2018, 140, 7313–7323 CrossRef CAS PubMed.
- C. Pareja-Rivera and D. Solis-Ibarra, Adv. Opt. Mater., 2021, 9, 2100633 CrossRef CAS.
- I. C. Smith, M. D. Smith, A. Jaffe, Y. Lin and H. I. Karunadasa, Chem. Mater., 2017, 29, 1868–1884 CrossRef CAS.
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Chem. Rev., 2019, 119, 3296–3348 CrossRef CAS PubMed.
- S. M. Park, M. Wei, J. Xu, H. R. Atapattu, F. T. Eickemeyer, K. Darabi, L. Grater, Y. Yang, C. Liu, S. Teale, B. Chen, H. Chen, T. Wang, L. Zeng, A. Maxwell, Z. Wang, K. R. Rao, Z. Cai, S. M. Zakeeruddin, J. T. Pham, C. M. Risko, A. Amassian, M. G. Kanatzidis, K. R. Graham, M. Grätzel and E. H. Sargent, Science, 2023, 381, 209–215 CrossRef CAS PubMed.
- D. B. Mitzi, D. R. Medeiros and P. R. L. Malenfant, Inorg. Chem., 2002, 41, 2134–2145 CrossRef CAS PubMed.
- M. D. Smith, L. Pedesseau, M. Kepenekian, I. C. Smith, C. Katan, J. Even and H. I. Karunadasa, Chem. Sci., 2017, 8, 1960–1968 RSC.
- N. Marchal, W. Van Gompel, M. C. Gélvez-Rueda, K. Vandewal, K. Van Hecke, H.-G. Boyen, B. Conings, R. Herckens, S. Maheshwari, L. Lutsen, C. Quarti, F. C. Grozema, D. Vanderzande and D. Beljonne, Chem. Mater., 2019, 31, 6880–6888 CrossRef CAS.
- W. T. M. Van Gompel, R. Herckens, K. Van Hecke, B. Ruttens, J. D'Haen, L. Lutsen and D. Vanderzande, Chem. Commun., 2019, 55, 2481–2484 RSC.
- J. V. Passarelli, C. M. Mauck, S. W. Winslow, C. F. Perkinson, J. C. Bard, H. Sai, K. W. Williams, A. Narayanan, D. J. Fairfield, M. P. Hendricks, W. A. Tisdale and S. I. Stupp, Nat. Chem., 2020, 12, 672–682 CrossRef CAS PubMed.
- A. M. Najarian, M. Vafaie, R. Sabatini, S. Wang, P. Li, S. Xu, M. I. Saidaminov, S. Hoogland and E. H. Sargent, J. Am. Chem. Soc., 2023, 145, 27242–27247 CrossRef CAS PubMed.
- Q. A. Akkerman, V. D'Innocenzo, S. Accornero, A. Scarpellini, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2015, 137, 10276–10281 CrossRef CAS PubMed.
- G. Li, J. Y.-L. Ho, M. Wong and H. S. Kwok, J. Phys. Chem. C, 2015, 119, 26883–26888 CrossRef CAS.
- J. B. Hoffman, A. L. Schleper and P. V. Kamat, J. Am. Chem. Soc., 2016, 138, 8603–8611 CrossRef CAS PubMed.
- S. E. Creutz, E. N. Crites, M. C. De Siena and D. R. Gamelin, Chem. Mater., 2018, 30, 4887–4891 CrossRef CAS.
- D. Solis-Ibarra, I. C. Smith and H. I. Karunadasa, Chem. Sci., 2015, 6, 4054–4059 RSC.
- M. Kalaj and S. M. Cohen, ACS Cent. Sci., 2020, 6, 1046–1057 CrossRef CAS PubMed.
- S. Zhuo, J. Zhang, Y. Shi, Y. Huang and B. Zhang, Angew. Chem., Int. Ed., 2015, 54, 5693–5696 CrossRef CAS PubMed.
- I. Spanopoulos, W. Ke, C. C. Stoumpos, E. C. Schueller, O. Y. Kontsevoi, R. Seshadri and M. G. Kanatzidis, J. Am. Chem. Soc., 2018, 140, 5728–5742 CrossRef CAS PubMed.
- A. Senocrate, I. Spanopoulos, N. Zibouche, J. Maier, M. S. Islam and M. G. Kanatzidis, Chem. Mater., 2021, 33, 719–726 CrossRef CAS.
- S. Kataoka, S. Banerjee, A. Kawai, Y. Kamimura, J. C. Choi, T. Kodaira, K. Sato and A. Endo, J. Am. Chem. Soc., 2015, 137, 4158–4163 CrossRef CAS PubMed.
- S. Barman, N. V. Venkataraman, S. Vasudevan and R. Seshadri, J. Phys. Chem. B, 2003, 107, 1875–1883 CrossRef CAS.
- X. Wu, M. T. Trinh and X.-Y. Zhu, J. Phys. Chem. C, 2015, 119, 14714–14721 CrossRef CAS.
- M. J. Schilcher, P. J. Robinson, D. J. Abramovitch, L. Z. Tan, A. M. Rappe, D. R. Reichman and D. A. Egger, ACS Energy Lett., 2021, 6, 2162–2173 CrossRef CAS.
- R. L. Kingsford, S. R. Jackson, L. C. Bloxham and C. G. Bischak, J. Am. Chem. Soc., 2023, 145, 11773–11780 CrossRef CAS PubMed.
- J. Kang and L.-W. Wang, J. Phys. Chem. Lett., 2017, 8, 3875–3880 CrossRef CAS PubMed.
- A. Azmy, S. Li, G. K. Angeli, C. Welton, P. Raval, M. Li, N. Zibouche, L. Wojtas, G. N. M. Reddy, P. Guo, P. N. Trikalitis and I. Spanopoulos, Angew. Chem., Int. Ed., 2023, 62, e202218429 CrossRef CAS PubMed.
- A. Azmy, X. Zhao, G. K. Angeli, C. Welton, P. Raval, L. Wojtas, N. Zibouche, G. N. Manjunatha Reddy, P. N. Trikalitis, J. Cai and I. Spanopoulos, ACS Appl. Mater. Interfaces, 2023, 15, 42717–42729 CrossRef CAS PubMed.
- R. Willett, H. Place and M. Middleton, J. Am. Chem. Soc., 1988, 110, 8639–8650 CrossRef CAS.
- A. Jaffe, S. A. Mack, Y. Lin, W. L. Mao, J. B. Neaton and H. I. Karunadasa, Angew. Chem., Int. Ed., 2020, 59, 4017–4022 CrossRef CAS PubMed.
- D. Cortecchia, H. A. Dewi, J. Yin, A. Bruno, S. Chen, T. Baikie, P. P. Boix, M. Grätzel, S. Mhaisalkar, C. Soci and N. Mathews, Inorg. Chem., 2016, 55, 1044–1052 CrossRef CAS PubMed.
- M. Bochalya, P. K. Kanaujia, G. Vijaya Prakash and S. Kumar, J. Solid State Chem., 2019, 273, 219–225 CrossRef CAS.
- T. Sheikh, A. Shinde, S. Mahamuni and A. Nag, ACS Energy Lett., 2018, 3, 2940–2946 CrossRef CAS.
- W. K. Winter, B. Curnutte and S. E. Whitcomb, Spectrochim. Acta, 1959, 15, 1085–1102 CrossRef CAS.
- S. Liu, J. Xu, E. Dai, J. Qiu and Y. Liu, Microporous Mesoporous Mater., 2018, 264, 133–138 CrossRef CAS.
- J. S. Chappell, A. N. Bloch, W. A. Bryden, M. Maxfield, T. O. Poehler and D. O. Cowan, J. Am. Chem. Soc., 1981, 103, 2442–2443 CrossRef CAS.
- S. Kataoka, Y. Kamimura and A. Endo, Langmuir, 2018, 34, 4166–4172 CrossRef CAS PubMed.
- B. Walker, G. Kim and J. Y. Kim, Adv. Mater., 2019, 31, 1807029 CrossRef PubMed.
- W. Li, Z. Wang, F. Deschler, S. Gao, R. H. Friend and A. K. Cheetham, Nat. Rev. Mater., 2017, 2, 16099 CrossRef.
- Q. Jiang, D. Rebollar, J. Gong, E. L. Piacentino, C. Zheng and T. Xu, Angew. Chem., Int. Ed., 2015, 54, 7617–7620 CrossRef CAS PubMed.
- D. Umeyama, Y. Lin and H. I. Karunadasa, Chem. Mater., 2016, 28, 3241–3244 CrossRef CAS.
- J. A. Hunter, W. H. S. Massie, J. Meiklejohn and J. Reid, Inorg. Nucl. Chem. Lett., 1969, 5, 1–4 CrossRef CAS.
- M. J. Cliffe, J. Lee, J. A. M. Paddison, S. Schott, P. Mukherjee, M. W. Gaultois, P. Manuel, H. Sirringhaus, S. E. Dutton and C. P. Grey, Phys. Rev. B, 2018, 97, 144421 CrossRef CAS.
- R. H. Toeniskoetter and S. Solomon, Inorg. Chem., 1968, 7, 617–620 CrossRef CAS.
- K. Nakamoto, Infrared and Raman spectra of inorganic and coordination compounds, Wiley, Hoboken, NJ, 6th edn., 2007 Search PubMed.
- L. Casella, M. Gullotti, G. Pallanza and M. Buga, Biol. Met., 1990, 3, 137–140 CrossRef CAS.
- R. A. Walton, Inorganica Chim. Acta, 1979, 37, 237–240 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2354173. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04378b |
| This journal is © The Royal Society of Chemistry 2024 |