
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular brush-based ultrathin polymer electrolytes with stable interfaces for high-voltage large-areal-capacity lithium metal batteries†
Rongfeng
Liao
a,
Congping
Li
a,
Minghong
Zhou
b,
Ruliang
Liu
 c,
Shaohong
Liu
*a and
Dingcai
Wu
*a
c,
Shaohong
Liu
*a and
Dingcai
Wu
*a
aKey Laboratory for Polymeric Composite and Functional Materials of Ministry of Education, School of Chemistry, Sun Yat-sen University, Guangzhou 510006, P. R. China. E-mail: liushh27@mail.sysu.edu.cn; wudc@mail.sysu.edu.cn
bMedical Research Institute, Guangdong Provincial People's Hospital (Guangdong Academy of Medical Sciences), Southern Medical University, Guangzhou 510080, P. R. China
cSchool of Chemistry and Materials Science, Guangdong University of Education, Guangzhou 510303, P. R. China
First published on 17th October 2024
Abstract
Polymer electrolytes hold great promise for long-cycling lithium metal batteries, but their unsatisfactory ionic conductivities and unstable interfacial contacts with electrodes greatly limit their practical applications under high cut-off voltage and large areal capacity conditions. Herein, a super-structured multifunctional molecular brush, BC-g-P(CCMA-co-TFEMA) (BC = bacterial cellulose; CCMA = (2-oxo-1,3-dioxolan-4-yl) methyl methacrylate; TFEMA = 2,2,2-trifluoroethyl methacrylate), has been designed to develop an ultrathin polymer electrolyte with superior ionic conductivity and stable electrolyte/electrode interfaces. The cyclic carbonate group in CCMA can weaken the binding of solvents and anions with lithium ions, thereby enhancing ionic transport. Meanwhile, the fluorine-containing group in TFEMA is beneficial for simultaneously constructing LiF-rich electrolyte/anode and electrolyte/cathode interfaces with enhanced stability. Moreover, the robust BC backbone provides the polymer electrolyte with outstanding mechanical properties. With such polymer electrolytes, a remarkable capacity retention of 83% has been demonstrated for Li/LiFePO4 cells at 1C after 1000 cycles. Remarkably, the solid-state full cell with a high-loading LiNi0.8Mn0.1Co0.1O2 cathode delivers a high discharge specific capacity of 204 mA h g−1 for more than 400 cycles at a high cut-off voltage of 4.5 V. This work provides a novel design principle for advanced electrolytes of high-voltage and large-areal-capacity lithium metal batteries.
1 Introduction
Lithium metal is considered as one of the most promising anode materials due to its high theoretical capacity (3860 mA h g−1) and low electrochemical redox potential (−3.04 V vs. the standard hydrogen electrode).1 However, the uncontrolled dendrite growth and significant volume expansion of lithium metal anodes during cycling can cause rapid battery failure and even internal short circuits with severe safety hazards, which greatly hinders the practical applications of lithium metal batteries.2–5 In recent years, various strategies have been explored to enhance the stability and safety of lithium metal batteries, including modification of electrolyte and separator,6–9 as well as employment of artificial solid electrolyte interphase (SEI) films10–14 and solid-state electrolytes.15–20 In particular, replacing a liquid electrolyte with a nonflammable and mechanically robust solid electrolyte is effective in suppressing lithium dendrite growth to enhance the safety of lithium metal batteries. Among various types of solid electrolytes, polymer electrolytes have attracted great attention because of their unique advantages of excellent flexibility, processability and good electrode/electrolyte interface compatibility.21To date, poly(ethylene oxide) (PEO) and poly(vinylidene fluoride) (PVDF) have emerged as crucial categories of the polymer electrolyte community for lithium metal batteries.22–25 Unfortunately, the low ionic conductivity and unstable interfaces between the electrolyte and anode/cathode greatly limit their practical applications in lithium metal batteries (Fig. 1a). In this context, various efforts have been made to enhance the overall performance of polymer electrolytes. On the one hand, the introduction of plasticizers (e.g., succinonitrile (SN),26,27 fluoroethylene carbonate (FEC),28 and N,N-dimethylformamide (DMF)29) and even liquid electrolytes30 can enhance the ionic conductivity of polymer electrolytes. However, these additives are either prone to reactions with lithium metal anodes or easily oxidized by high-voltage cathodes, leading to shortened battery life. On the other hand, some strategies, such as designing asymmetric solid electrolytes31 and utilizing in situ copolymerization of fluorine/nitrogen-containing monomers,32 have been employed to improve the electrolyte/electrode interfacial stability. Unfortunately, the mechanical properties of the resulting polymer electrolytes are unsatisfactory, which inevitably increases the thickness of the membranes (≥100 μm) and thus impedes their further application in high-energy-density lithium metal batteries. Although crosslinked polymer networks can enhance the strength of polymer electrolytes to a certain content, the ionic conductivity is compromised. What is worse, the stable and long-cycling battery performance of conventional polymer electrolytes can only be accomplished at relatively low areal capacities or low cut-off voltages. Up to now, polymer electrolytes that adapt to high areal capacities (>1 mA h cm−2) and high cut-off voltages (>4.3 V) have rarely been reported. Therefore, it is extremely crucial, yet remains a great challenge, to design and fabricate polymer electrolytes that simultaneously possess high ionic conductivity, high interfacial stability and superior mechanical properties to enable long-cycle operation for practical lithium metal batteries.
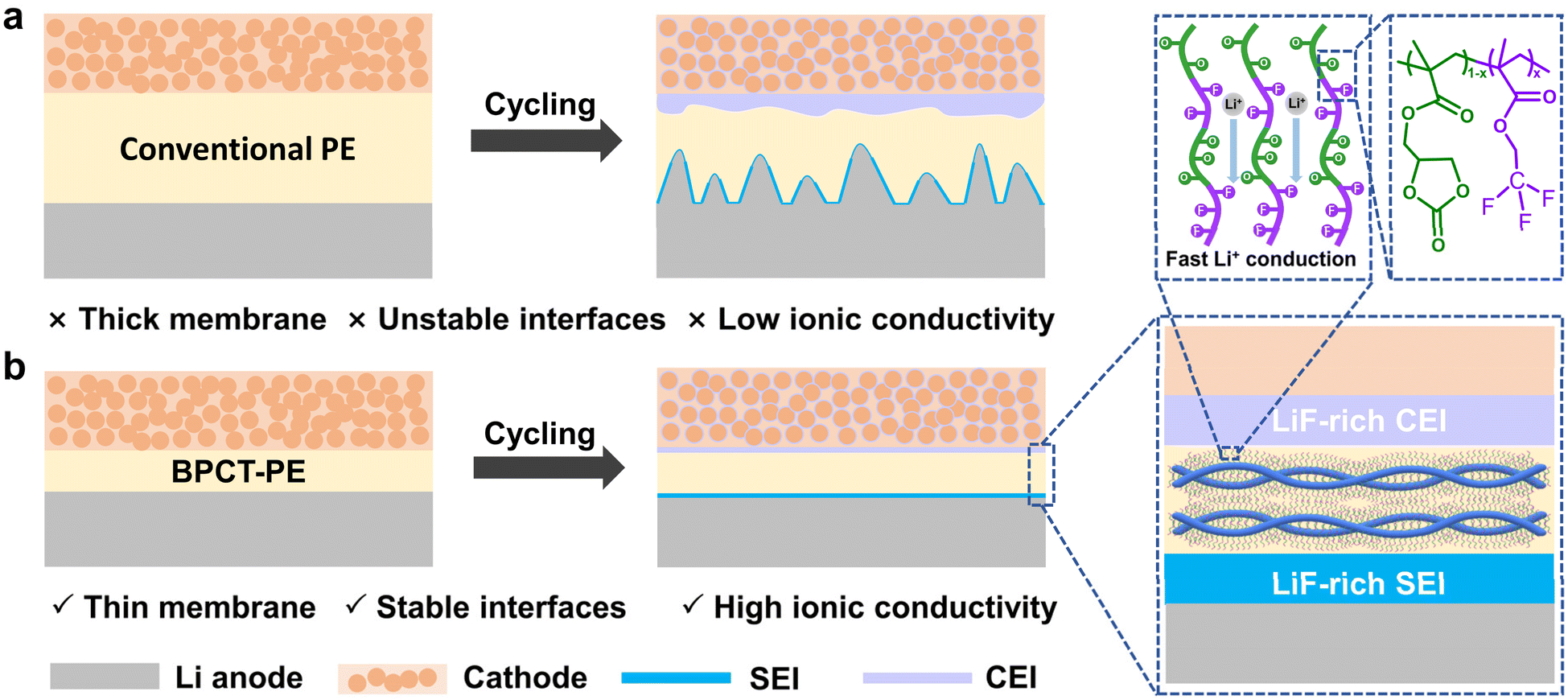 | ||
| Fig. 1 Schematic illustration of lithium plating/stripping behaviors for solid-state lithium metal batteries with (a) conventional PE and (b) our polymer electrolyte (BPCT-PE) with stable electrolyte/anode and electrolyte/cathode interfaces. | ||
Herein, a class of ultrathin polymer electrolytes with outstanding mechanical properties, superior ionic conductivity and stable electrolyte/electrode interfaces is successfully developed to achieve long-cycling high-voltage lithium metal batteries with high areal capacities (Fig. 1b). The key to this novel polymer electrolyte is the employment of multifunctional poly((2-oxo-1,3-dioxolan-4-yl) methyl methacrylate-co-2,2,2-trifluoroethyl methacrylate)-grafted bacterial cellulose (BC-g-P(CCMA-co-TFEMA)) molecular brushes as building blocks. The CCMA units in BC-g-P(CCMA-co-TFEMA) molecular brushes provide abundant cyclic carbonate groups to facilitate the dissociation and motion of lithium ions; the TFEMA units with fluorine-containing groups enable the formation of LiF-rich ultrathin electrolyte/electrode interfaces, which simultaneously suppress dendrite growth on the lithium anode and side reactions on cathodes; the robust BC backbones can substantially increase the mechanical strength of polymer electrolyte. Benefiting from these synergistic advantages, the polymer electrolyte based on BC-g-P(CCMA-co-TFEMA) (denoted as BPCT-PE) exhibits an ionic conductivity of up to 7.6 × 10−4 S cm−1 at 30 °C. Remarkably, the Li/LiFePO4 cell with BPCT-PE exhibits an excellent cycling performance of 1000 cycles with a capacity retention of 83% at 1C. More importantly, with BPCT-PE as the solid-state electrolyte, the high-loading LiNi0.8Mn0.1Co0.1O2 (NCM811) cathode can deliver a high discharge specific capacity of 204 mA h g−1 at 1C, and stably operate over 400 cycles with a high capacity retention of 71% at 4.5 V.
2 Results and discussion
The synthetic process of the BC-g-P(CCMA-co-TFEMA) membrane is depicted in Fig. S1.† The BC nanofibers are first modified using 2-bromoisobutyryl bromide to yield Br-containing initiation sites on their surface. The synthesized CCMA monomer is identified by 1H nuclear magnetic resonance (NMR) spectroscopy (Fig. S2†). Subsequently, P(CCMA-co-TFEMA) random copolymers are grafted from BC nanofibers via surface-initiated atom transfer radical polymerization (SI-ATRP) of CCMA and TFEMA monomers, leading to the formation of BC-g-P(CCMA-co-TFEMA) molecular brushes. As shown in the Fourier transform infrared (FTIR) spectra of Fig. 2a, BC-g-P(CCMA-co-TFEMA) molecular brushes exhibit a characteristic peak of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of the five-membered ring at 1792 cm−1 and a characteristic peak of the C–F bond at 1282 cm−1,33,34 suggestive of the successful grafting of P(CCMA-co-TFEMA) side-chains from BC. X-ray photoelectron spectroscopy (XPS) results confirm the existence of CO (288.6 eV) and C–F (292.6 eV) groups35,36 in BC-g-P(CCMA-co-TFEMA) molecular brushes (Fig. S3†).
O bond of the five-membered ring at 1792 cm−1 and a characteristic peak of the C–F bond at 1282 cm−1,33,34 suggestive of the successful grafting of P(CCMA-co-TFEMA) side-chains from BC. X-ray photoelectron spectroscopy (XPS) results confirm the existence of CO (288.6 eV) and C–F (292.6 eV) groups35,36 in BC-g-P(CCMA-co-TFEMA) molecular brushes (Fig. S3†).
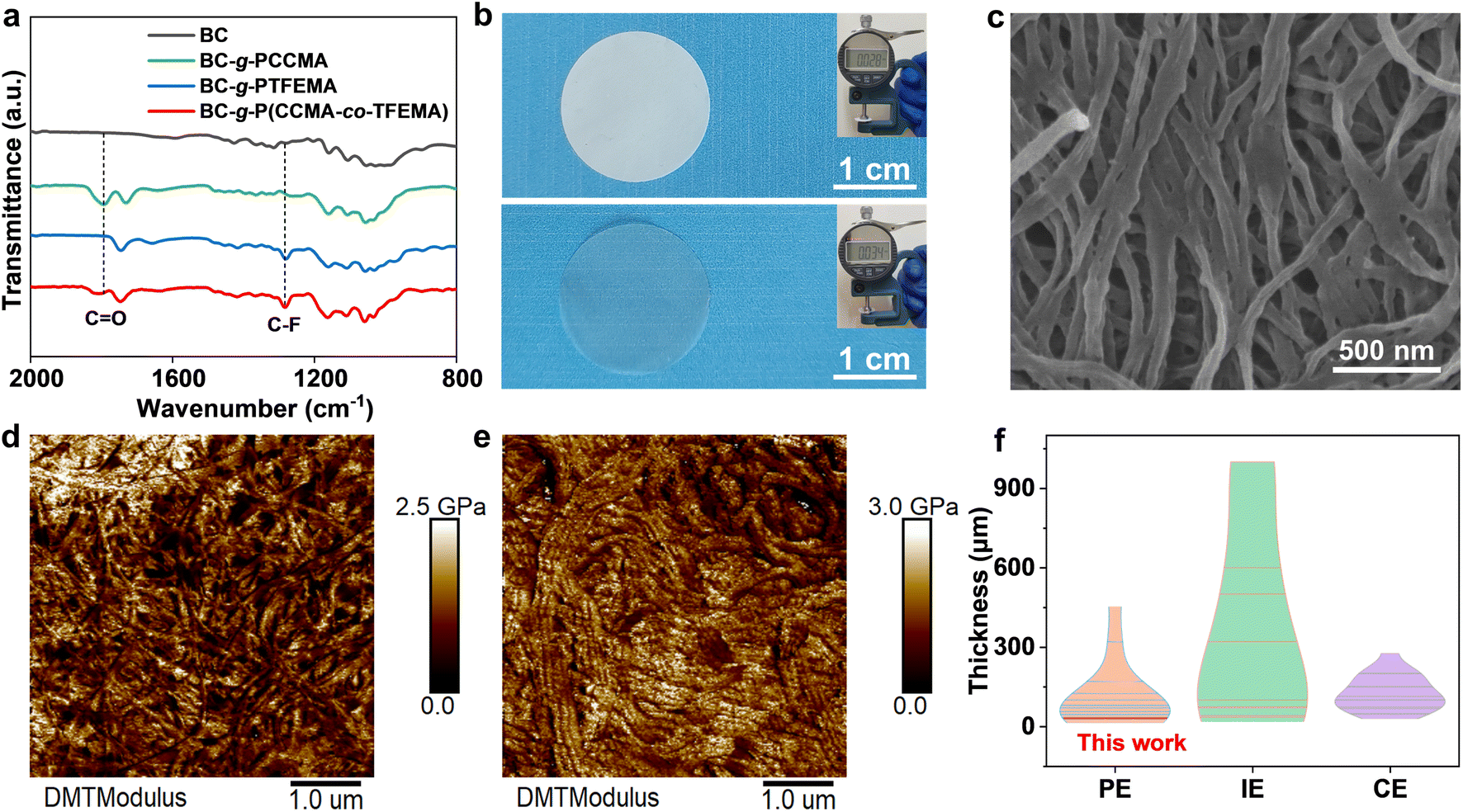 | ||
| Fig. 2 (a) FTIR spectra of BC, BC-g-PCCMA, BC-g-PTFEMA and BC-g-P(CCMA-co-TFEMA). (b) Digital photos of the BC-g-P(CCMA-co-TFEMA) membrane and BPCT-PE (inset: digital photos of thickness measurement). (c) Top-view SEM image of the BC-g-P(CCMA-co-TFEMA) membrane. AFM Young's modulus mappings of (d) the BC/P(CCMA-co-TFEMA) membrane and (e) the BC-g-P(CCMA-co-TFEMA) membrane. (f) Thickness comparison of BPCT-PE with reported PE, IE and CE. | ||
According to thermogravimetric analysis, the weight percentage of P(CCMA-co-TFEMA) side-chains for BC-g-P(CCMA-co-TFEMA) molecular brushes is up to 73 wt% (Fig. S4†). As shown in Fig. S5,† the number average molecular weight of P(CCMA-co-TFEMA) chains is 8.6 × 104 (PDI = 1.27).
Subsequently, a free-standing and robust membrane with a thickness of only ∼28 μm can be obtained via simple vacuum filtration of BC-g-P(CCMA-co-TFEMA) molecular brushes (Fig. 2b and S6†). The top-view SEM images reveal that the as-obtained BC-g-P(CCMA-co-TFEMA) membrane presents a characteristic nanofiber-based 3D porous network (Fig. 2c), similar to BC, BC-g-PCCMA and BC-g-PTFEMA membranes (Fig. S7a–c†). Elemental mapping analysis reveals that fluorine and oxygen elements distribute uniformly on the BC-g-P(CCMA-co-TFEMA) membrane (Fig. S8†). This homogeneously porous structure is beneficial for efficient electrolyte storage and homogeneous and fast lithium ion conduction. In contrast, obvious phase separation can be observed for the composite membrane composed of BC nanofibers and the conventional non-grafted linear polymer P(CCMA-co-TFEMA) (denoted as BC/P(CCMA-co-TFEMA) membrane), indicating the critical role of molecular brushes in homogeneous assembly (Fig. S7d†). Benefiting from the homogeneous distribution of P(CCMA-co-TFEMA) side-chains, the BC-g-P(CCMA-co-TFEMA) membrane shows a higher Young's modulus (1.3 GPa) than the BC/P(CCMA-co-TFEMA) membrane (1.0 GPa) (Fig. 2d and e). Additionally, the dynamic ultra-micro hardness test reveals that the elasticity modulus of the BC-g-P(CCMA-co-TFEMA) membrane is up to 591 MPa, much higher than that of the BC/P(CCMA-co-TFEMA) membrane (390 MPa), suggestive of excellent mechanical strength (Fig. S9†). Thermal stability of polymeric membranes is a critical factor for ensuring the safety of lithium metal batteries. As shown in Fig. S10,† the commercial PP separator begins to shrink at 90 °C and completely deforms at 150 °C. In contrast, the BC-g-P(CCMA-co-TFEMA) membrane shows no distinct shrinkage during the whole heating process (30–150 °C). These results reveal that the BC-g-P(CCMA-co-TFEMA) membrane possesses superior thermal stability, indicating its promising contribution to improving the thermal safety of solid-state lithium-metal batteries.
BPCT-PE can be further obtained by swelling the BC-g-P(CCMA-co-TFEMA) membrane in a liquid electrolyte. Notably, Young's modulus of BPCT-PE is nearly five times higher than that of B/PCT-PE obtained from the BC/P(CCMA-co-TFEMA) membrane (Fig. S11†). Owing to its robust structure, the thickness of BPCT-PE increases slightly from 28 μm (the BC-g-P(CCMA-co-TFEMA) membrane) to 34 μm (Fig. 2b). To the best of our knowledge, the robust BPCT-PE is thinner than most previous solid-state electrolytes (normally 100–1000 μm), including polymer electrolytes (PE), inorganic electrolytes (IE) and composite electrolytes (CE) (Fig. 2f and Table S1†). The ionic conductivity of BPCT-PE is tested by electrochemical impedance spectroscopy (EIS) at 30 °C. As shown in Fig. S12,† BPCT-PE displays an ionic conductivity of 7.6 × 10−4 S cm−1, much higher than B/PCT-PE (2.0 × 10−4 S cm−1). Notably, the ionic conductivity of BPCT-PE is nearly two times higher than that of BPT-PE (4.0 × 10−4 S cm−1) based on the BC-g-PTFEMA membrane, and is also higher than that of BPC-PE (6.9 × 10−4 S cm−1) obtained from the BC-g-PCCMA membrane, indicating that the cyclic carbonate groups in CCMA units provide lithium ion transport pathways and are beneficial for improving the ionic conductivity. As displayed in Fig. 3a and S13,† the relationship between ionic conductivity and temperature of BPCT-PE fits the typical Arrhenius linear equation. The corresponding activation energy (Ea) of ion migration in BPCT-PE is only 0.14 eV, lower than those of BPC-PE (0.17 eV), BPT-PE (0.20 eV) and B/PCT-PE (0.18 eV).
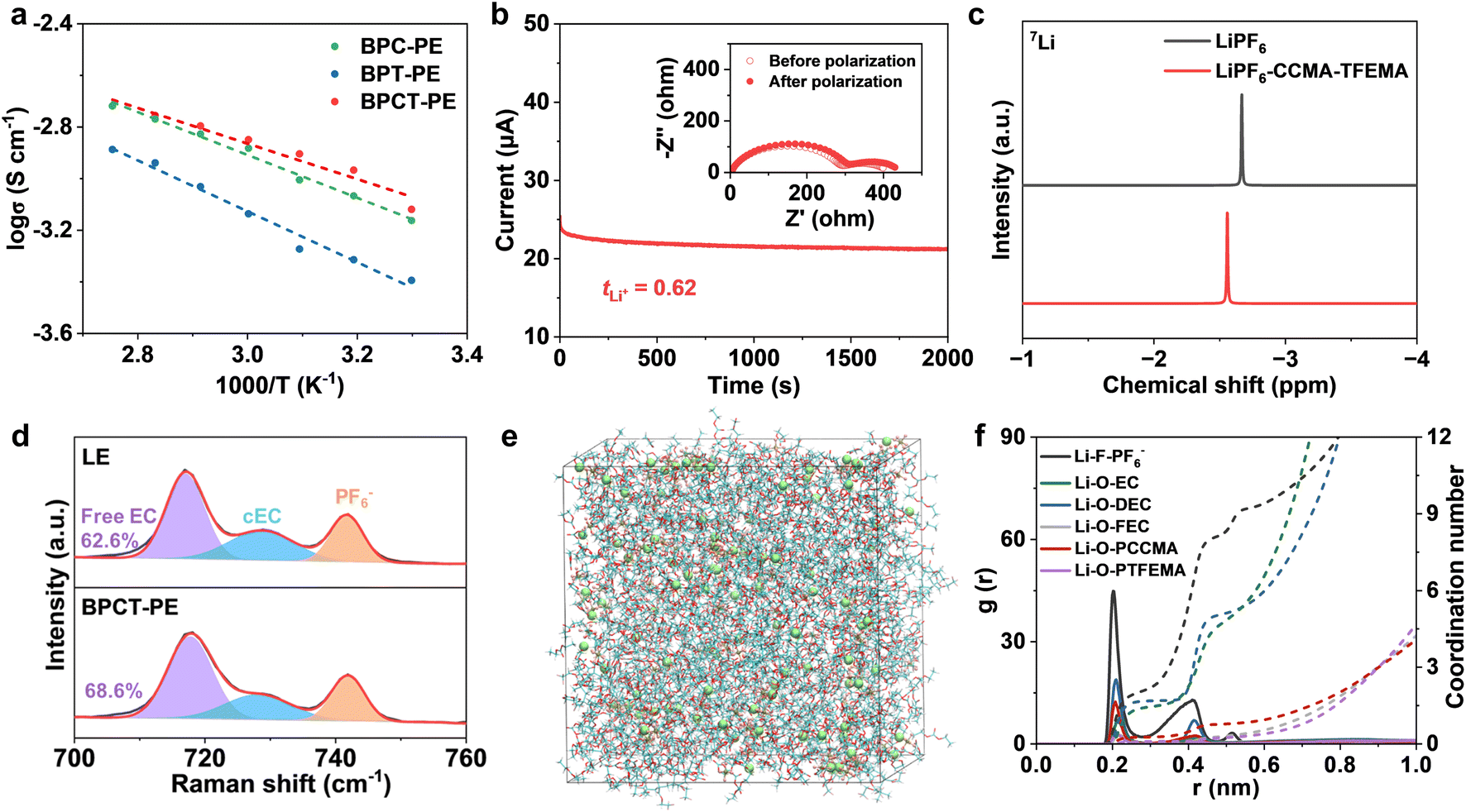 | ||
| Fig. 3 (a) Ionic conductivity fitted using Arrhenius plots for BPC-PE, BPT-PE and BPCT-PE. (b) Current–time profile for the Li|BPCT-PE|Li cell at 10 mV of polarization (inset: Nyquist plots before and after polarization). (c) 7Li NMR spectra of LiPF6 and LiPF6–CCMA–TFEMA in CD3CN. (d) Raman spectra of LE and BPCT-PE. (e) A snapshot of the MD simulation box for BPCT-PE. (f) Radial distribution functions (solid lines) and corresponding coordination numbers (dashed lines) of BPCT-PE. | ||
Single lithium ion conducting ability has a significant effect on the performance of lithium metal batteries. A high lithium ion transference number is beneficial for decreasing the lithium ion concentration polarization at the interface and alleviating the growth of lithium dendrites.37 As shown in Fig. 3b and S14,† the lithium ion transference number for BPCT-PE is measured to be 0.62, which is significantly higher than those of the BC membrane with liquid electrolyte (BC/LE, 0.32) and B/PCT-PE (0.41), indicating the critical role of polymer grafting in efficient lithium ion transport. It is worth noting that the lower lithium ion transference numbers of BPC-PE (0.45) and BPT-PE (0.51) clearly demonstrate the synergistic effect between CCMA and TFEMA units in facilitating lithium ion transport. The electrochemical stability window of BPCT-PE is further investigated by linear sweeping voltammetry (LSV) on Li/stainless-steel cells. Owing to the excellent electrochemical stability of TFEMA units, BPCT-PE has a higher oxidation voltage (4.8 V) than BPC-PE (4.6 V) (Fig. S15†).
The effect of CCMA units on the enhanced ionic conductivity of BPCT-PE is further analyzed by 7Li NMR and Raman spectroscopy. Compared with pure LiPF6 salt, LiPF6–CCMA–TFEMA exhibits significant positive shifts in 7Li NMR, suggestive of less electron density around lithium ions due to weaker binding with solvents and/or anions38 (Fig. 3c). Notably, BPC-PE exhibits more positive shifts than BPT-PE, confirming that CCMA units play a dominant role in facilitating the dissociation and motion of lithium ions (Fig. S16†). According to the Raman spectrum of BPCT-PE, the peaks at 717, 729 and 742 cm−1 are assigned to free ethylene carbonate (EC), coordinated EC (cEC) and PF6− anions, respectively39,40 (Fig. 3d). The higher proportion of free EC in BPCT-PE (68.6%) than that in LE (62.6%) indicates that the molecular brushes can weaken the interaction of EC with lithium ions and thus facilitate ionic transport.41 Meanwhile, BPC-PE exhibits the highest proportion of free EC molecules (73.5%), further demonstrating that the weaker solvent binding is mainly contributed by CCMA units (Fig. S17†). Molecular dynamics (MD) simulations are performed to illustrate the lithium ion solvation structures in LE and BPCT-PE. The molecular configurations are shown in Fig. S18.† The MD simulation snapshots of LE and BPCT-PE are shown in Fig. 3e and S19a.† The radial distribution functions and corresponding coordination numbers are displayed in Fig. 3f and S19b.† In the lithium ion solvation sheath structure of BPCT-PE, the coordination numbers of Li–F–PF6−, Li–O–EC and Li–O–DEC are 2.20, 1.46 and 1.68, respectively, which are lower than those in the lithium ion solvation sheath structure of LE (2.53, 1.59 and 1.81). Meanwhile, density functional theory (DFT) calculations demonstrate that CCMA units have stronger binding with lithium ions than EC, DEC, FEC or PF6−(Fig. S20†). These results confirm that P(CCMA-co-TFEMA) side-chains are beneficial for weakening the binding of solvents and anions with lithium ions, thereby enhancing the ionic transport and increasing the lithium ion transference number.42
The critical current density serves as an important indicator of the stability of Li metal anodes and reveals many intrinsic behaviors at high current densities in solid-state lithium metal batteries.43 The Li/Li symmetric cell with BPCT-PE exhibits a substantially higher critical current density of 1.4 mA cm−2 than that with B/PCT-PE (0.5 mA cm−2), suggestive of fast lithium ion migration kinetics and superior interface stability (Fig. S21†). The rate performance of Li/Li symmetric cells further reveals that BPCT-PE shows a relatively low overpotential (32 mV) when the current density is increased to 1 mA cm−2 (Fig. S22†). In sharp contrast, the overpotential of BC/LE is up to 112 mV at 1 mA cm−2, and an obvious short circuit is observed for B/PCT-PE at 0.75 mA cm−2. Similarly, the Li/Li symmetric cell with BPCT-PE exhibits very stable voltage plateaus for over 500 hours of cycling without increased polarization or short circuits at a current density of 0.5 mA cm−2 with a cycling capacity of 0.5 mA h cm−2 (Fig. 4a). In contrast, the Li/Li symmetric cells with BPC-PE, BPT-PE, and B/PCT-PE display much larger voltage hysteresis and fail within only 44, 201, and 19 h, respectively. The results demonstrate that the homogeneous P(CCMA-co-TFEMA) side-chains on BC substrates are crucial for accelerating Li ion migration and stabilizing the electrolyte/anode interface. As depicted in Fig. S23,† the Li/Li symmetric cell with BPCT-PE exhibits a charge transfer resistance of 319 Ω, obviously lower than those with B/PCT-PE (585 Ω), BPC-PE (370 Ω), and BPT-PE (463 Ω).
 | ||
| Fig. 4 (a) Voltage profiles of Li/Li symmetric cells with BPCT-PE and control samples, including B/PCT-PE, BPC-PE and BPT-PE at 0.5 mA cm−2 and 0.5 mA h cm−2. (b–e) The high-resolution C 1s and F 1s spectra obtained from XPS depth measurement of the SEI formed in (b and c) BPC-PE and (d and e) BPCT-PE at 0.1 mA cm−2 and 0.1 mA h cm−2 after 100 cycles. | ||
The surface morphologies of Li anodes after symmetric cell cycles are investigated. As shown in Fig. S24a and c,† the Li/Li symmetric cell with BPC-PE exhibits uneven Li deposition and massive Li dendrites on the surface of the cycled Li anode. In contrast, the Li/Li symmetric cell with BPCT-PE exhibits a relatively dense and smooth morphology without distinct Li dendrites (Fig. S24b and d†). Moreover, the SEI components of Li anodes obtained from symmetric Li|BPC-PE|Li and Li|BPCT-PE|Li cells after 100 cycles are investigated through XPS with different Ar-ion sputtering times. Notably, the SEI of the cycled Li|BPCT-PE|Li cell shows higher contents of Li and F as well as lower contents of C and O, compared to the cycled Li|BPC-PE|Li cell (Fig. S25†). The high-resolution C 1s spectra reveal that the SEI of the cycled Li|BPCT-PE|Li cell exhibits less content of Li2CO3 (289.7 eV) and C–O (286.9 eV) than that of the cycled Li|BPC-PE|Li cell,34 suggestive of fewer organic decomposition products (Fig. 4b and d). In addition, much higher content of LiF (684.9 eV)44 is detected in the SEI of the cycled Li|BPCT-PE|Li cell (Fig. 4c and e). The results clearly indicate that the TFEMA units can simultaneously suppress the solvent decomposition and facilitate the formation of a LiF-rich SEI, thereby leading to a stabilized electrolyte/anode interface and enabling superior cycling performance.
To evaluate the potential practical application of BPCT-PE, the electrochemical performances of Li/LiFePO4 cells with BPCT-PE are investigated. Benefiting from the accelerated lithium ion migration, the Li/LiFePO4 cell with BPCT-PE shows excellent rate capability. As shown in Fig. 5a, the Li/LiFePO4 cell with BPCT-PE delivers specific capacities of 158, 151, 140, and 124 mA h g−1 at 0.2, 0.5, 1.0, and 2.0C, respectively. When the current density is increased to 5.0C, a high specific capacity of 97 mA h g−1 can still be maintained, which is obviously higher than those of the cells with PP/LE (88 mA h g−1), BPC-PE (82 mA h g−1), and BPT-PE (90 mA h g−1). Remarkably, long-term cycling tests reveal that a specific capacity of up to 114 mA h g−1 can be achieved for the Li/LiFePO4 cell with BPCT-PE after 1000 cycles at 1C, corresponding to an excellent capacity retention of 83%. Nevertheless, owing to unstable electrolyte/electrode interfaces, the cell with BPC-PE experiences a short circuit after about 120 cycles and the capacity of the cell with PP/LE suddenly degrades after 550 cycles; meanwhile, a specific capacity of only 72 mA h g−1 is maintained for the cell with BPT-PE after 1000 cycles (Fig. 5b and S26†). The superior cycling performance could be ascribed to the synergetic effect of CCMA and TFEMA units in BC-g-P(CCMA-co-TFEMA) molecular brushes. Upon increasing the current density to 2C, the Li/LiFePO4 cell with BPCT-PE still maintains a decent cycling behavior with a specific capacity of 92 mA h g−1 after 400 cycles (Fig. S27†). It is worth noting that the cycling performance of the Li/LiFePO4 cell with BPCT-PE is highly competitive, compared to previously reported solid-state electrolytes (Table S2†).
 | ||
| Fig. 5 (a) Rate performance of Li/LiFePO4 cells with PP/LE, BPC-PE, BPT-PE and BPCT-PE. (b) Cycling performance of Li/LiFePO4 cells with PP/LE, BPC-PE, BPT-PE and BPCT-PE at 1C. (c) Cycling performance of Li/NCM811 cells with a high-loading NCM811 of 7.5 mg cm−2 at charging/discharging rates of 0.2/1C and cut-off voltages of 4.3 and 4.5 V. (d) Performance comparison between Li/NCM811 cells in this work and those with reported solid-state electrolytes in terms of the cycle number, cut-off voltage and areal capacity. | ||
BPCT-PE demonstrates superior stability when paired with the high-voltage LiNi0.8Co0.1Mn0.1O2 (NCM811) cathode. As shown in Fig. 5c and S28,† the Li|BPCT-PE|NCM811 cell with a high-loading cathode (7.5 mg cm−2) shows an initial discharge specific capacity of 193 mA h g−1 (1.44 mA h cm−2) and achieves a remarkable capacity retention of 70% after long-term cycling (600 cycles) at charging/discharging rates of 0.2/1C and a cut-off voltage of 4.3 V, much better than the Li|PP/LE|NCM811 cell (24% after 400 cycles). When the cut-off voltage is increased to 4.5 V, the initial discharge specific capacity of Li|BPCT-PE|NCM811 cell increases to 204 mA h g−1 (1.53 mA h cm−2), and its capacity retention is 71% after long-term cycling (400 cycles) at charging/discharging rates of 0.2/1C. To the best of our knowledge, such long-term stable high-voltage solid-state lithium metal batteries with large areal capacities are superior to those based on previously reported solid-state electrolytes (Fig. 5d and Table S3†), including polymer electrolytes (for example, 150 cycles with an areal capacity of 0.58 mA h cm−2 at 4.3 V)34,45–52 and composite electrolytes (for example, 360 cycles with an areal capacity of 0.64 mA h cm−2 at 4.3 V).38,53–58
The microstructural evolution of cycled NCM811 cathodes is characterized by SEM and TEM. Obvious structural cracks and damage are observed in the cycled NCM811 cathode with PP/LE, and the thickness of the corresponding cathode electrolyte interphase (CEI) layer is up to 11 nm (Fig. S29a and c†). In sharp comparison, the cycled NCM811 cathode with BPCT-PE maintains excellent structural integrity with a thin CEI layer of only 3 nm (Fig. S29b and d†). The chemical components of cycled NCM811 cathodes are characterized by XPS. High-resolution C 1s spectra reveal that the peak area ratio of C–O to C–C is decreased from 0.40 for the CEI of the cycled NCM811 cathode with PP/LE to 0.24 for that with BPCT-PE (Fig. S30a and c†), suggestive of suppressed electrolyte side reactions in BPCT-PE. Moreover, the high-resolution F 1s spectra show a substantially higher ratio of LiF component in the CEI of the cycled NCM811 cathode with BPCT-PE than that with PP/LE (Fig. S30b and d†). The presence of a LiF-rich CEI on the NCM811 cathode can lead to a robust electrolyte/cathode interface with reduced side reactions, eventually leading to improved cycling stability.59
3 Conclusions
In summary, a super-structured multifunctional molecular brush, BC-g-P(CCMA-co-TFEMA), has been elaborately prepared by grafting random copolymers from BC nanofibers to construct an ultrathin polymer electrolyte with superior ionic conductivity as well as stable electrolyte/anode and electrolyte/cathode interfaces. Cyclic carbonate groups in BC-g-P(CCMA-co-TFEMA) can facilitate the transportation of lithium ions. Meanwhile, the fluorine-containing groups contribute to the formation of LiF-rich electrolyte/anode and electrolyte/cathode interfaces, which simultaneously suppress dendrite growth on the lithium anode and side reactions on the cathodes. Besides, the robust BC backbones can enhance the mechanical strength of the polymer electrolyte. As a result, BPCT-PE delivers superior electrochemical performances in terms of high ionic conductivity at 30 °C and excellent cycling stability. It is worth noting that the Li/LiFePO4 cell with BPCT-PE displays a superior cycling performance of 1000 cycles with a capacity retention of 83% at 1C. Moreover, the high-loading Li/NCM811 cell (7.5 mg cm−2) with BPCT-PE delivers an initial discharge specific capacity of up to 204 mA h g−1 at 1C with a capacity retention of 71% after 400 cycles at 4.5 V. This work provides an efficient method to obtain a novel polymer electrolyte via controllable polymerization, and could play a significant role in constructing stable interfaces toward high-performance solid-state lithium metal batteries.Data availability
Preparation and characterization of samples, electrochemical tests of batteries, and additional data are available in the ESI.†Author contributions
D. W. conceived the concept and supervised the research. S. L. and D. W. designed the project. R. Liao carried out material synthesis. R. Liao, C. L., and M. Z. performed material characterization and electrochemical tests. R. Liao, S. L., and D. W. wrote the paper. R. Liu gave advice on the research. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2021YFF0500600), the National Natural Science Foundation of China (51925308), the Guangdong Major Project of Basic and Applied Basic Research (2023B0303000025), the Natural Science Foundation of Guangdong (2024B1515020023), the Fundamental Research Funds for the Central Universities, Sun Yat-sen University (23yxqntd002 and 23lgbj004), and the Guangdong Basic Research Center of Excellence for Functional Molecular Engineering.References
- W. Wu, W. Luo and Y. Huang, Chem. Soc. Rev., 2023, 52, 2553–2572 RSC.
- H. Xu, J. Zhang, H. Zhang, J. Long, L. Xu and L. Mai, Adv. Energy Mater., 2023, 13, 2204411 CrossRef CAS.
- F. N. Jiang, X. B. Cheng, S. J. Yang, J. Xie, H. Yuan, L. Liu, J. Q. Huang and Q. Zhang, Adv. Mater., 2023, 35, 2209114 CrossRef CAS.
- L. Zhang, S. Wang, Q. Wang, H. Shao and Z. Jin, Adv. Mater., 2023, 35, 2303355 CrossRef CAS.
- R. M. Gao, H. Yang, C. Y. Wang, H. Ye, F. F. Cao and Z. Guo, Angew. Chem., Int. Ed., 2021, 60, 25508–25513 CrossRef CAS.
- L. Sheng, Q. Wang, X. Liu, H. Cui, X. Wang, Y. Xu, Z. Li, L. Wang, Z. Chen, G. L. Xu, J. Wang, Y. Tang, K. Amine, H. Xu and X. He, Nat. Commun., 2022, 13, 172 CrossRef CAS.
- M. M. Rahman, S. Tan, Y. Yang, H. Zhong, S. Ghose, I. Waluyo, A. Hunt, L. Ma, X. Q. Yang and E. Hu, Nat. Commun., 2023, 14, 8414 CrossRef CAS.
- R. He, K. Deng, D. Mo, X. Guan, Y. Hu, K. Yang, Z. Yan and H. Xie, Angew. Chem., Int. Ed., 2024, 63, e202317176 CrossRef CAS.
- Y. Liu, X. Tao, Y. Wang, C. Jiang, C. Ma, O. Sheng, G. Lu and X. W. Lou, Science, 2022, 375, 739–745 CrossRef CAS.
- J. M. Kim, M. H. Engelhard, B. Lu, Y. Xu, S. Tan, B. E. Matthews, S. Tripathi, X. Cao, C. Niu, E. Hu, S. M. Bak, C. Wang, Y. S. Meng, J. G. Zhang and W. Xu, Adv. Funct. Mater., 2022, 32, 2207172 CrossRef CAS.
- S. Li, J. Huang, Y. Cui, S. Liu, Z. Chen, W. Huang, C. Li, R. Liu, R. Fu and D. Wu, Nat. Nanotechnol., 2022, 17, 613–621 CrossRef CAS.
- J. Pokharel, A. Cresce, B. Pant, M. Y. Yang, A. Gurung, W. He, A. Baniya, B. S. Lamsal, Z. Yang, S. Gent, X. Xian, Y. Cao, W. A. Goddard, K. Xu and Y. Zhou, Nat. Commun., 2024, 15, 3085 CrossRef CAS.
- Y. Sun, J. Li, S. Xu, H. Zhou and S. Guo, Adv. Mater., 2024, 36, 2311687 CrossRef CAS.
- Q. Zhang, X. Zhang, J. Wan, N. Yao, T. Song, J. Xie, L. Hou, M. Zhou, X. Chen, B. Li, R. Wen, H. Peng, Q. Zhang and J. Huang, Nat. Energy, 2023, 8, 725–735 CrossRef CAS.
- W. Zhang, V. Koverga, S. Liu, J. Zhou, J. Wang, P. Bai, S. Tan, N. K. Dandu, Z. Wang, F. Chen, J. Xia, H. Wan, X. Zhang, H. Yang, B. L. Lucht, A. Li, X. Yang, E. Hu, S. R. Raghavan, A. T. Ngo and C. Wang, Nat. Energy, 2024, 9, 386–400 CrossRef CAS.
- J. Yang, R. Li, P. Zhang, J. Zhang, J. Meng, L. Li, Z. Li and X. Pu, Energy Storage Mater., 2024, 64, 103088 CrossRef.
- S. Lv, X. He, Z. Ji, S. Yang, L. Feng, X. Fu, W. Yang and Y. Wang, Adv. Energy Mater., 2023, 13, 2302711 CrossRef CAS.
- Y. He, C. Wang, P. Zou, R. Lin, E. Hu and H. L. Xin, Angew. Chem., Int. Ed., 2023, 62, e202308309 CrossRef CAS.
- S. Liu, L. Zhou, J. Han, K. Wen, S. Guan, C. Xue, Z. Zhang, B. Xu, Y. Lin, Y. Shen, L. Li and C. W. Nan, Adv. Energy Mater., 2022, 12, 2200660 CrossRef CAS.
- P. Shi, J. Ma, M. Liu, S. Guo, Y. Huang, S. Wang, L. Zhang, L. Chen, K. Yang, X. Liu, Y. Li, X. An, D. Zhang, X. Cheng, Q. Li, W. Lv, G. Zhong, Y. B. He and F. Kang, Nat. Nanotechnol., 2023, 18, 602–610 CrossRef CAS.
- G. Xi, M. Xiao, S. Wang, D. Han, Y. Li and Y. Meng, Adv. Funct. Mater., 2021, 31, 2007598 CrossRef CAS.
- Y. Su, X. H. Rong, A. Gao, Y. Liu, J. W. Li, M. L. Mao, X. G. Qi, G. L. Chai, Q. H. Zhang, L. M. Suo, L. Gu, H. Li, X. J. Huang, L. Q. Chen, B. Y. Liu and Y. S. Hu, Nat. Commun., 2022, 13, 4181 CrossRef CAS.
- H. Liu, L. Xu, H. Tu, Z. Luo, F. Zhu, W. Deng, G. Zou, H. Hou and X. Ji, Small, 2023, 19, 2301275 CrossRef CAS.
- M. Li, H. An, Y. Song, Q. Liu, J. Wang, H. Huo, S. Lou and J. Wang, J. Am. Chem. Soc., 2023, 145, 25632–25642 CrossRef CAS.
- K. Yang, L. Chen, J. Ma, C. Lai, Y. Huang, J. Mi, J. Biao, D. Zhang, P. Shi, H. Xia, G. Zhong, F. Kang and Y. B. He, Angew. Chem., Int. Ed., 2021, 60, 24668–24675 CrossRef CAS.
- W. Zha, Y. Ruan and Z. Wen, Chem. Eng. J., 2022, 429, 132506 CrossRef CAS.
- W. Zha, J. Li, W. Li, C. Sun and Z. Wen, Chem. Eng. J., 2021, 406, 126754 CrossRef CAS.
- Q. Sun, S. Wang, Y. Ma, D. Song, H. Zhang, X. Shi, N. Zhang and L. Zhang, Adv. Mater., 2023, 35, 2300998 CrossRef CAS.
- Y. F. Huang, J. P. Zeng, S. F. Li, C. Dai, J. F. Liu, C. Liu and Y. B. He, Adv. Energy Mater., 2023, 13, 2203888 CrossRef CAS.
- Q. Kang, Z. Zhuang, Y. Liu, Z. Liu, Y. Li, B. Sun, F. Pei, H. Zhu, H. Li, P. Li, Y. Lin, K. Shi, Y. Zhu, J. Chen, C. Shi, Y. Zhao, P. Jiang, Y. Xia, D. Wang and X. Huang, Adv. Mater., 2023, 35, 2303460 CrossRef CAS.
- M. Yao, Q. Ruan, S. Pan, H. Zhang and S. Zhang, Adv. Energy Mater., 2023, 13, 2203640 CrossRef CAS.
- S. Qi, M. Li, Y. Gao, W. Zhang, S. Liu, J. Zhao and L. Du, Adv. Mater., 2023, 35, 2304951 CrossRef CAS.
- Z. Lin, X. Guo, R. Zhang, M. Tang, P. Ding, Z. Zhang, L. Wu, Y. Wang, S. Zhao, Q. Zhang and H. Yu, Nano Energy, 2022, 98, 107330 CrossRef CAS.
- Y. Wang, S. Chen, Z. Li, C. Peng, Y. Li and W. Feng, Energy Storage Mater., 2022, 45, 474–483 CrossRef.
- R. Hu, H. Qiu, H. Zhang, P. Wang, X. Du, J. Ma, T. Wu, C. Lu, X. Zhou and G. Cui, Small, 2020, 16, 1907163 CrossRef CAS.
- P. Wang, B. He, B. Wang, L. Wang, H. Yu, S. Liu, Q. Ye and F. Zhou, Prog. Org. Coat., 2022, 165, 106706 CrossRef CAS.
- D. Chen, T. Zhu, M. Zhu, S. Yuan, P. Kang, W. Cui, J. Lan, X. Yang and G. Sui, Energy Storage Mater., 2022, 53, 937–945 CrossRef.
- H. X. Yang, Z. K. Liu, Y. Wang, N. W. Li and L. Yu, Adv. Funct. Mater., 2023, 33, 2209837 CrossRef CAS.
- S. Woo, E. Hwang, H. Kang, H. Lee, J. Lee, H. Kim, G. Jeong, D. Yoo, J. Lee, S. Kim, J. Yu and J. W. Choi, Energy Environ. Sci., 2021, 14, 1420–1428 RSC.
- J. L. Allen, O. Borodin, D. M. Seo and W. A. Henderson, J. Power Sources, 2014, 267, 821–830 CrossRef CAS.
- F. Cheng, W. Zhang, Q. Li, C. Fang, J. Han and Y. Huang, ACS Nano, 2023, 17, 24259–24267 CrossRef CAS.
- G. Jiang, J. Liu, Z. Wang and J. Ma, Adv. Funct. Mater., 2023, 33, 2300629 CrossRef CAS.
- Y. Lu, C. Z. Zhao, H. Yuan, X. B. Cheng, J. Q. Huang and Q. Zhang, Adv. Funct. Mater., 2021, 31, 2009925 CrossRef CAS.
- R. Deng, F. Chu, F. Kwofie, Z. Guan, J. Chen and F. Wu, Angew. Chem., Int. Ed., 2022, 61, e202215866 CrossRef CAS.
- Z. Wen, Z. Zhao, L. Li, Z. Sun, N. Chen, Y. Li, F. Wu and R. Chen, Adv. Funct. Mater., 2021, 32, 2109184 CrossRef.
- T. Zhu, G. Liu, D. Chen, J. Chen, P. Qi, J. Sun, X. Gu and S. Zhang, Energy Storage Mater., 2022, 50, 495–504 CrossRef.
- Y. Du, L. Zhao, C. Xiong, Z. Sun, S. Liu, C. Li, S. Hao, W. Zhou and H. Li, Energy Storage Mater., 2023, 56, 310–318 CrossRef.
- C. Wang, H. Liu, Y. Liang, D. Li, X. Zhao, J. Chen, W. Huang, L. Gao and L. Z. Fan, Adv. Funct. Mater., 2022, 33, 2209828 CrossRef.
- L. Tang, B. Chen, Z. Zhang, C. Ma, J. Chen, Y. Huang, F. Zhang, Q. Dong, G. Xue, D. Chen, C. Hu, S. Li, Z. Liu, Y. Shen, Q. Chen and L. Chen, Nat. Commun., 2023, 14, 2301 CrossRef CAS.
- B. Yang, C. Deng, N. Chen, F. Zhang, K. Hu, B. Gui, L. Zhao, F. Wu and R. Chen, Adv. Mater., 2024, 36, 2403078 CrossRef CAS.
- S. Kim, H. K. Jung, P. L. Handayani, T. Kim, B. M. Jung and U. H. Choi, Adv. Funct. Mater., 2023, 33, 2210916 CrossRef CAS.
- C. Jing, K. Dai, D. Liu, W. Wang, L. Chen, C. Zhang and W. Wei, Sci. Bull., 2024, 69, 209–217 CrossRef CAS.
- J. Pan, Y. C. Zhang, J. Wang, Z. C. Bai, R. G. Cao, N. N. Wang, S. X. Dou and F. Q. Huang, Adv. Mater., 2022, 34, 2107183 CrossRef CAS.
- D. Xu, J. Su, J. Jin, C. Sun, Y. Ruan, C. Chen and Z. Wen, Adv. Energy Mater., 2019, 9, 1900611 CrossRef.
- Y. Zhai, W. Hou, M. Tao, Z. Wang, Z. Chen, Z. Zeng, X. Liang, P. Paoprasert, Y. Yang, N. Hu and S. Song, Adv. Mater., 2022, 34, 2205560 CrossRef CAS.
- H. Cheng, J. Cao, F. Li, X. Geng, D. Li, Y. Wei, X. Lin, H. Xu and Y. Huang, Adv. Funct. Mater., 2024, 34, 2307677 CrossRef CAS.
- K. Mu, D. Wang, W. Dong, Q. Liu, Z. Song, W. Xu, P. Yao, Y. Chen, B. Yang, C. Li, L. Tian, C. Zhu and J. Xu, Adv. Mater., 2023, 35, 2304686 CrossRef CAS.
- C. Shen, W. Feng, Y. Yu, H. Wang, Y. Cheng, C. Dong, J. Gu, A. Zheng, X. Liao, X. Xu and L. Mai, Adv. Energy Mater., 2024, 14, 2304511 CrossRef CAS.
- J. Han, M. J. Lee, J. H. Min, K. H. Kim, K. Lee, S. H. Kwon, J. Park, K. Ryu, H. Seong, H. Kang, E. Lee, S. W. Lee and B. J. Kim, Adv. Funct. Mater., 2024, 34, 2310801 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04454a |
| This journal is © The Royal Society of Chemistry 2024 |