
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA changeable critical state for a switchable photocurrent direction via the photo-electrochemical photocurrent polarity switching effect in BiFeO3 nanoparticulate films†
Ajay
,
Jyoti
Saroha
and
Pravin Popinand
Ingole
 *
*
Department of Chemistry, Indian Institute of Technology Delhi, Hauz Khas, New Delhi, India-110016. E-mail: ppingole@chemistry.iitd.ac.in; Fax: +91 11 2658 1102; Tel: +91 11 2659 7547
First published on 7th October 2024
Abstract
The photoelectrochemical photocurrent switching (PEPS) effect to change the photocurrent direction from cathodic to anodic via external bias is an important phenomenon. Specifically, tuning the critical state or the potential corresponding to a switchable photocurrent direction through easily controllable parameters is crucial for developing efficient photo-electrocatalyst systems. Although the PEPS effect has been reported in quite a few recently published studies, the changeable critical state has not yet been demonstrated. In this study, for the first time, we present a photoelectrochemical (PEC) system based on bismuth ferrite (BiFeO3) nanoparticulate films that demonstrates a changeable critical state controlled via the composition of an electrolyte medium. In particular, the ionic strength (tuned via addition of inert salt) and the concentration of dissolved oxygen in an electrolyte medium are noted to dictate the potential corresponding to the critical state. Importantly, we demonstrate that this potential can be predicted using the Nernst equation by considering electrolyte energy level rearrangement and the kinetic theory of semiconductor electrodes. This study enhances the understanding of carrier transport in PEC activities and enables precise control over the reversal of the photocurrent direction that may pave the way for developing sophisticated multifunctional photoelectric devices and efficient photo-electrocatalyst systems.
1 Introduction
Photoelectrochemical (PEC) systems with bipolar photocurrent, i.e., with both the cathodic and anodic polarity, are important for a variety of applications including the sensing technology and water splitting.1–4 The ability to manipulate the photocurrent direction in bipolar PEC systems, via the photoelectrochemical photocurrent switching (PEPS) effect, represents a significant advancement across technologies.5–8 The emergence of this effect in semiconductor materials is contingent upon specific electronic band structures compared to the dynamic Fermi levels of electrolytes that delineate the conditions for change in PEC reactions at the semiconductor/electrolyte interface. The degree and type of PEC reaction can be significantly influenced by varying both the composition of electrolytes and the surface electronic properties of semiconductors as per the theory of semiconductor electrode dynamics.5 To realize practical PEC switching devices, semiconductor electrode materials with appropriate band edge positions, narrow band gaps for visible light absorption, and ease of fabrication using earth-abundant materials are highly desirable. The presence of electrolytes in PEC systems offers additional and distinct advantages, encompassing both physical processes, such as excitation, diffusion, and carrier drift reminiscent of those found in solid-state transistors in semiconductors, and chemical processes, such as interfacial ion diffusion, migration, redox reactions, and PEC reactions that are absent in solid-state transistors.9Currently, the prevailing strategies for switching the photocurrent polarity involve changing either the excitation wavelength or the bias voltage or intensity of incident light, among which the use of external bias is more convenient and advantageous.4,10–13 Predominantly, these methods are targeted towards tailoring the surface electronic properties of semiconductor nanostructures and modulating the electronic effects in molecular hybrids.8,14–17 This is because conventional semiconductor materials, such as BiVO4, SrTiO3,18 Fe2O3, etc. typically display unidirectional photocurrent behaviour due to their electrochemical diode-like properties.19–21 Recently, switchable photocurrent behavior has been reported in TiO2 nanosheets layered with polyaniline,22 as well as in thin film electrodes comprising a blend of n-type N-doped TiO2 and p-type CuI12 and in CuPc/β-Ga2O3 p–n junctions,23 demonstrating diverse approaches for achieving photocurrent switching. However, such configurations generally involve multi-material systems, making it unusual for single-phase metal oxide photo-electrodes to exhibit both cathodic and anodic photocurrent polarity. Only a limited number of studies have reported the bipolar photo-response (i.e., PEPS effect) in single-phase metal oxides, such as Fe2O3, TiO2, and BiFeO3. Besides, attributions to extrinsic factors such as the presence of alkaline electrolytes and dissolved oxygen are often lacking.24–26 Thus, the discovery of single-phase metal oxides capable of intrinsic switchable photocurrents and comprehensive elucidation of the PEPS effect presents intriguing prospects for advanced PEC devices.
Another important aspect of the PEPS effect is that the electrolyte properties also have a significant impact on the photocurrent as well as the polarity reversal. Unfortunately, the influence of electrolyte on PEC detection devices and tuning of electrolyte properties for the PEPS effect has been overlooked in terms of its impact on both the photocurrent polarity and the photocurrent density. Nevertheless, the effect of electrolyte pH on the direction of photocurrent in MnPS3 photodetectors has been recently reported by Geng et al.,5 where the photocurrent polarity was noted to reverse at an alkaline pH of 11. Below pH ∼ 11, the photocurrent is cathodic, while anodic photocurrent is observed above pH 11, marking pH 11 as the critical state. However, variation of the critical state or controlling the potential corresponding to the critical state for the PEPS effect, through easily and externally controllable parameters, is essential for further development of advanced photodetection and PEC devices. In this context, Guo et al. introduced strategies to regulate the switching potential in photoelectrochemical systems by varying the β-Ga2O3 shell thickness in α/β-Ga2O3 structures,27 altering Ti3C2Tx modification levels on nanorod arrays28 and controlling crystalline phases in Ga2O3.29 These approaches underscore the significance of structural modifications in tuning photocurrent switching behavior, highlighting the need to further explore external parameters for optimizing the PEPS effect in PEC devices.
In this study, for the first time, we demonstrate a tuneable critical state controlled through the ionic strength of an electrolyte over a wide range of switching potentials from 0.78 to 0.89 V (ΔE = 110 mV). BiFeO3 (BFO) was chosen due to its promising candidature for a range of applications, owing to its room-temperature multiferroic, ferroelectric, and magnetic properties. Due to the unique band structure, suitable Fermi level position, and mid-gap states, it possesses the unique ability to switch the photocurrent polarity under applied bias.30–33 Recent research has highlighted the potential of BFO as a photocatalyst, leveraging the spontaneous dipole moment to enhance carrier separation and to accelerate chemical reaction rates. Another important consideration behind selecting BFO as an electrode material for studying the PEPS effect is the requirement of an appropriate band structure for redox reactions in electrolytes under illumination.24
We hypothesize that altering the composition of electrolyte via changing the ionic strength through the addition of an inert electrolyte and the concentration of dissolved oxygen would alter the energy level in an electrolyte without changing its pH. This adjustment influences solvent-ion interaction and the concentration of oxidized and reduced species at the electrode–electrolyte interface. Consequently, it alters the dynamics of charge carriers before and after equilibrium, as the position of the electrolyte Fermi level significantly impacts the width of the space charge layer of the semiconductor at the interface. The variable critical state in single phase BFO controlled through externally adjustable parameters such as modification of the electrolyte without affecting the pH of the medium presents a unique approach to harness the potential of technologically important BFO-based materials for PEC applications.
2 Results and discussion
In order to observe the PEPS effect in a bipolar metal oxide nanomaterial without p–n junction hybrids, it has been reported that the phase-purity is crucial. Therefore, single-phase BFO was synthesized via a modified protocol based on controlled thermal decomposition of a Bi–Fe glycolate single molecular precursor to give precise control over stoichiometry (Scheme S1†).34,35 The detailed methodology and the relevant details regarding synthesis and material characterization are given in the ESI.† Typically, 20 mL of ethylene glycol (EG) was transferred into a pre-cleaned glass beaker and bismuth nitrate pentahydrate (1 mmol) was added to it at room temperature under continuous stirring. Then, 1 mmol of iron nitrate nonahydrate was dissolved in the above transparent solution under stirring. The resultant solution with a pH of ∼5 was heated at 180 °C for 3 h under continuous stirring and the obtained slurry was cooled down to room temperature and then precipitated out using ethanol. The precipitate was centrifuged, washed with ethanol, and dried in an oven at 60 °C overnight to get Bi–Fe glycolate.The thermogravimetric analysis (TGA) of Bi–Fe glycolate as shown in Fig. S1† was performed to decide the calcination temperature, which displays a characteristic weight loss comprising two distinct steps. An initial weight loss (∼4%) below 100 °C is attributed to the desorption of physisorbed water molecules, while a subsequent weight loss (∼54%) within 100 to 420 °C is ascribed to the decomposition of organic moieties and the release of chemically bonded EG. Notably, no further weight loss beyond 420 °C was noted implying the completion of the decomposition process. Therefore, Bi–Fe glycolate was subjected to air calcination at 500 °C for 3 hours with a heating rate of 2 °C min−1 inside a muffle furnace and the obtained sample was named BFO NPs. Finally, the as-synthesized BFO NPs were ground for 30 min and stored for further characterization studies.
The powder X-ray diffraction (pXRD) pattern of BFO depicted in Fig. 1(a) exhibits well-defined and intense diffraction peaks at 2θ of 22.4°, 31.9°, 32.1°, 39.5°, 45.7°, 51.8°, and 45.7° corresponding to the crystallographic planes (101), (012), (110), (021), (202), (113), and (122), respectively, that match with monoclinic BiFeO3 (JCPDS card no. #00-020-0169). The absence of any discernible secondary phases (within the limits of detection) indicates the highly crystalline nature and phase purity of the BFO NPs. The FTIR analysis (Fig. 1(b)) depicts characteristic absorption bands at around 459 and 555 cm−1 (ν1) for Fe–O stretching vibrational mode of octahedral FeO6 groups supporting the formation of the BiFeO3 phase.36 The appearance of a distinct Fe–O peak at 786 cm−1 (ν2) is infrequent in amorphous materials because of their lack of short-range order, further supporting the formation of phase-pure BiFeO3 as required for the PEPS effect.37
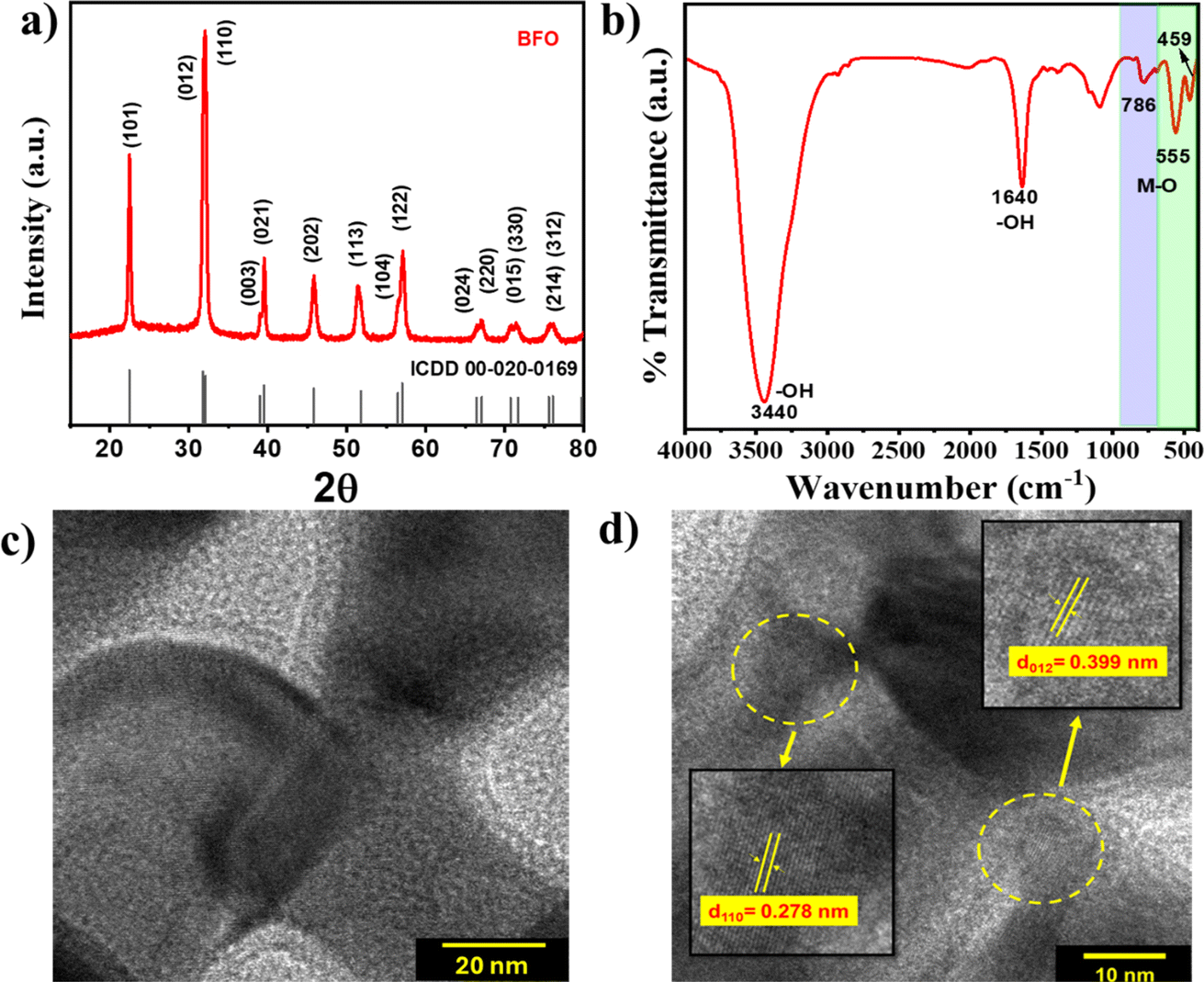 | ||
| Fig. 1 (a) XRD pattern, (b) FTIR spectrum, and (c & d) HRTEM images with different magnifications of BFO NPs. | ||
High resolution transmission electron microscopy (HR-TEM) images of BFO NPs are shown in Fig. 1(c) and (d) that depict lattice fringes with an interplanar spacing of 0.278 and 0.399 nm corresponding to lattice planes (110) and (012), respectively. Low-resolution TEM and field-emission scanning electron microscopy images depicted in Fig. S2(a) and (c),† respectively, show nanoparticles of elongated spherical symmetry with an average length of 47 ± 5 nm [Fig. S2(b)†]. The diffraction rings corresponding to various lattice planes are evident from the selected area electron diffraction (SAED) pattern shown in Fig. S2(d),† which also corroborates the findings from XRD analysis indicating the polycrystalline nature of BFO NPs.
X-ray photoelectron spectroscopy (XPS) was performed to identify the elemental composition and energy states within the BFO NPs. The XPS survey spectra provided in Fig. S3† revealed the presence of Bi, Fe, O, and C elements. Carbon was detected as a common impurity resulting from sample handling and exposure to air during preparation. The Bi 4f core level (CL) spectrum shown in Fig. 2(a) is deconvoluted into a Bi 4f7/2 peak at 158.5 eV and Bi 4f7/2 peak at 163.0 eV, indicating the presence of Bi3+.38,39 Notably, no features related to metallic Bi and/or Bi2O3, which typically appear at binding energies of 157.0 and/or 159.3 eV, respectively, were seen. The deconvoluted Fe 2p CL XPS spectrum displayed in Fig. 2(b) exhibited peaks corresponding to Fe2+ (Fe 2p3/2 ∼709.4 eV and Fe 2p1/2 ∼721.5 eV) and Fe3+ (Fe 2p3/2 ∼711.4 eV and Fe 2p1/2 ∼724.5 eV) and satellite peaks at ∼718.5 eV and 732.6 eV.39 The O 1s CL spectra [Fig. 2(c)] were deconvoluted into three peaks: a peak at 529.5 eV represents lattice oxygen from BFO (OL) and a peak at 531.0 eV was attributed to oxygen adjacent to an oxygen vacancy (Ov) (also referred to as dangling oxygen bonds). The peak at the highest binding energy (532.4 eV) indicates adsorbed oxygen species, possibly originating from vacancy healing under ambient conditions.39 The presence of oxygen vacancies is known to be an important factor in the PEPS effect.
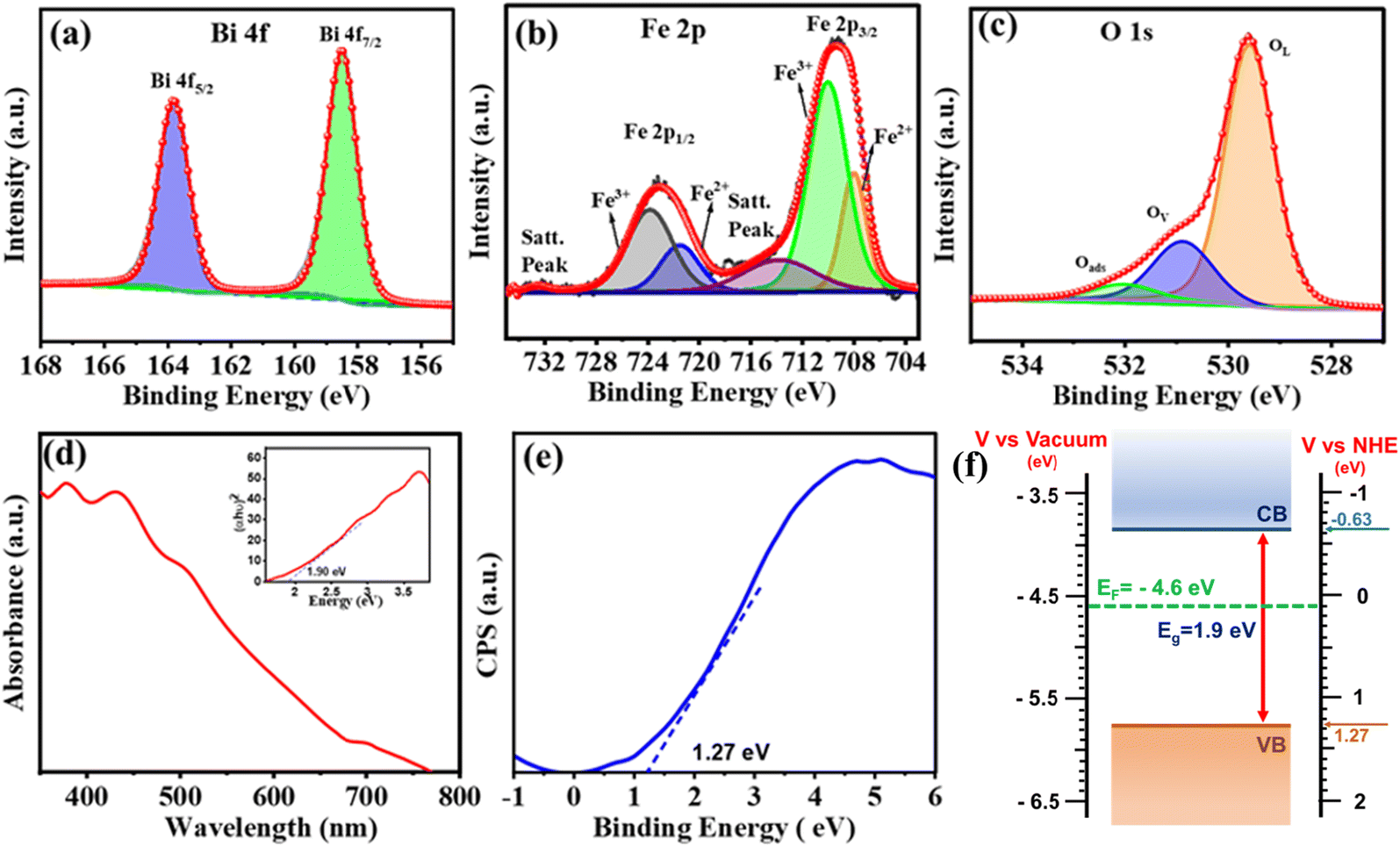 | ||
| Fig. 2 High-resolution CL XPS spectra for Bi 4f (a), Fe 2p (b), and O 1s (c) elements of BFO NPs. (d) UV-Vis absorption spectrum with the corresponding Tauc plot in the inset, (e) valence band edge position determined using XPS, and (f) band edge alignment with a Fermi level diagram. | ||
The presence of defect states, particularly those associated with oxygen vacancies within the BFO structure, is also demonstrated using the UV-Vis absorption spectrum of BFO (Fig. 2(d)) that depicts a gradual increase in absorbance starting from 650 nm, followed by a pronounced increase at around 460 nm, due to sub-bandgap transitions.40 The band gap energy (∼1.9 eV) estimated from the Tauc plot (inset, Fig. 2(d)) is governed by the orbital overlap between O 2p and Fe 3d levels. This indicates BFO as a charge-transfer semiconductor.26 Furthermore, the electronic band structure of BFO (Fig. 2(f)) was established by determining its valence band maxima (VBM) using the XPS valence band spectrum depicted in Fig. 2(e). The VBM of ∼−5.77 eV vs. vacuum was then subtracted from an optical band gap of 1.9 eV to estimate the conduction band minima (CBM) that was found to be at ∼−3.87 eV vs. vacuum. The ultraviolet photoelectron spectroscopy (UPS) spectra, as shown in Fig. S4,† revealed a secondary electron cutoff energy of 17.60 eV. Using eqn (1) (ESI†), the work function (Φ) was calculated to be 4.5 eV. The Fermi level (Ef) was determined to be at −4.5 eV, indicating its position between the CBM (−3.87 eV vs. vacuum) and VBM (−5.77 eV vs. vacuum), confirming the mid-gap alignment and providing critical insight into the electronic properties of the material.
2.1 Photoelectrochemical characterization towards the PEPS effect
To verify if the as-synthesized BFO NPs would demonstrate the PEPS effect, a BFO nanoparticulate film was deposited on an FTO quartz substrate and studied for photocurrent generation as a function of applied potential under the intermittent chopping of incidence light. The PEC measurements were performed using a Zahner Zennium Pro 212 equipped with an LED driver (450 nm, 0.65 A) in a standard three electrode single-compartment cell assembly using Ag/AgCl/Cl− (saturated with KCl) as a reference electrode, a Pt mesh as a counter electrode and BFO nanoparticulate film coated FTO as a working electrode. The working electrode was fabricated by drop casting catalyst ink (10 μL) prepared by mixing 1 mg of BFO NPs with 100 μL isopropanol followed by sonication for ca. 30 minutes on an FTO quartz substrate (with dimensions of 1.0 cm × 1.0 cm) and then drying under ambient conditions before using it for PEC measurements. Fig. 3(a) illustrates photocurrent vs. applied potential (Vapp) curves measured for the BFO nanoparticulate film electrode in Na2SO4 under simulated solar (AM 1.5) light illumination with intermittent chopping. A clear switch in the photocurrent direction from cathodic to anodic was noted. An enlarged portion of the plot [Fig. 3(b)] clearly illustrates this switchable photocurrent direction at a switching potential (Vswitch) of 0.78 V vs. RHE. Fig. 3(c) illustrates the transient photo-response of the material at Vswitch, i.e., at 0.78 V vs. RHE, and the potentials slightly above and below Vswitch that clearly represent the differences in the dynamics of photogenerated charge carriers inside the space charge region of the BFO semiconductor. At Vapp > Vswitch (at 0.85 V vs. RHE), the photocurrent direction is anodic, while it is cathodic at Vapp < Vswitch (at 0.75 V vs. RHE). At Vapp = Vswitch (0.78 V vs. RHE), a unique critical or confusing state exists: when light is switched to ON, the photocurrent first increases and then decreases, whereas when the light is switched to OFF, the photocurrent first decreases and then increases. In other words, below 0.78 V RHE, the behaviour of BFO resembles that of a p-type photocathode with the predominance of cathodic photocurrent, while beyond 0.78 V RHE, the anodic photocurrent becomes dominant representing n-type photoconductivity.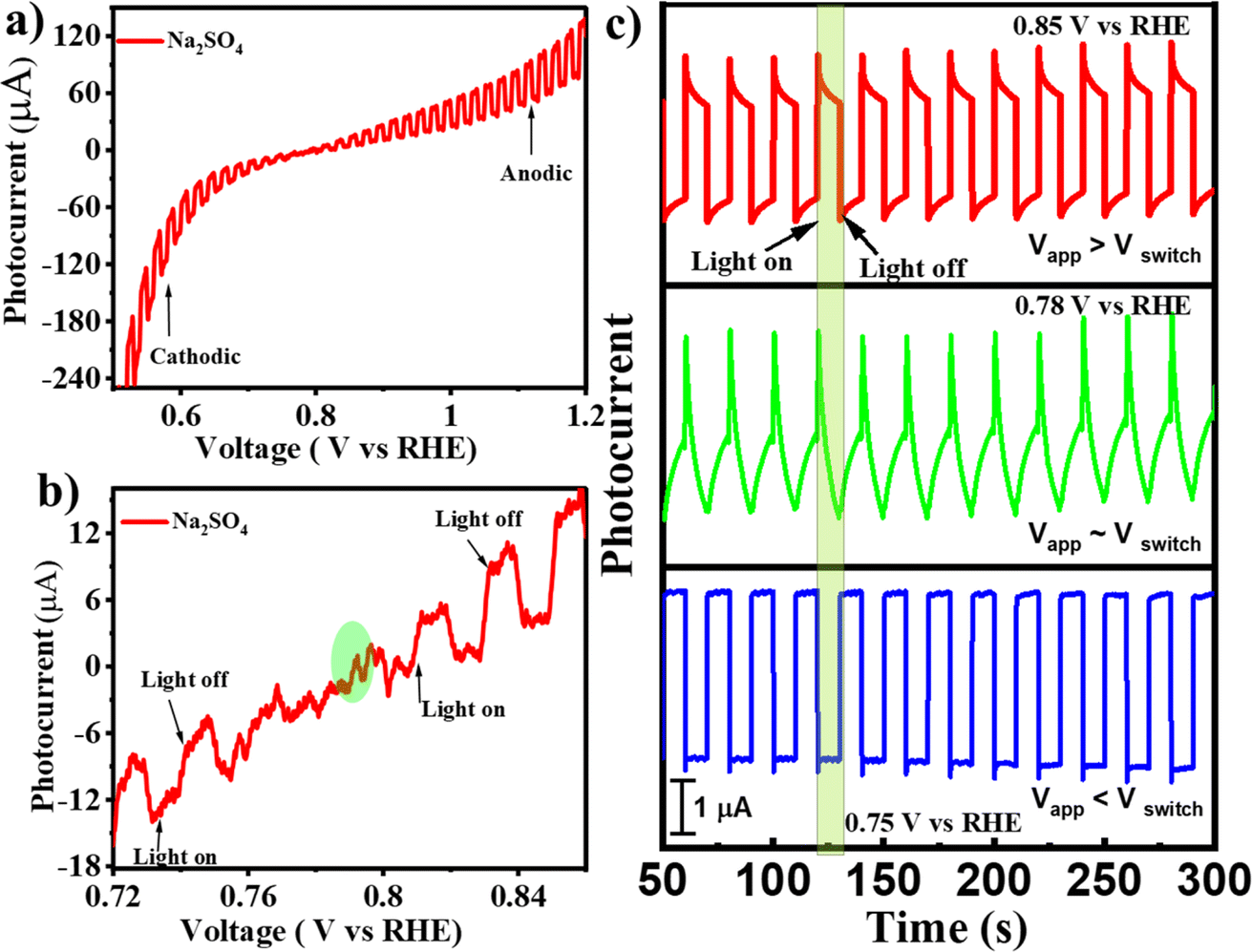 | ||
| Fig. 3 (a) Photocurrent vs. voltage curves for the BFO photoelectrode in Na2SO4, (b) enlarged portion showing the switching potential, and (c) transient photocurrent at switching potential and the potential above or below the switching potential. | ||
The existence of such a critical state has been reported for MnPS3 in an electrolyte with pH of 11.5 Unfortunately, such harsh electrolyte conditions are not conducive for device fabrication. Recently Wang et al. reported the effect of the H2O2 hole scavenger on the photocurrent density of BiFeO3 that also changes the onset of photocurrent.26 Therefore, the effect of externally added neutral species on the switching potential without changing the pH of electrolyte is important to explore. To study this effect, an inert electrolyte, NaNO3, was added to the electrolytic solution as it is known not to participate in electrochemical or photo-electrochemical charge transfer within the operational potential range because of a relatively negative reduction potential for the NO3−/NO2− couple (Eo = 0.42 V vs. RHE) compared to water (Eo = 1.23 V vs. RHE) for the O2/H2O couple.41,42 It is worth mentioning that the addition of NaNO3 to Na2SO4 electrolyte induces only a slight change in pH (from 7.16 to 7.01), which has been duly considered in the conversion of the potential scale vs. RHE. However, the addition of NaNO3 leads to the excessive presence of NO−3 ions at the electrode/electrolyte interface that becomes the dominant factor influencing the energetics of the semiconductor/electrolyte interface. The Fermi level of the electrolyte establishes a Nernstian equilibrium with the O2/OH− redox couple, and the Nernst equation (eqn (1)) defines the apparent Fermi level, E0F.
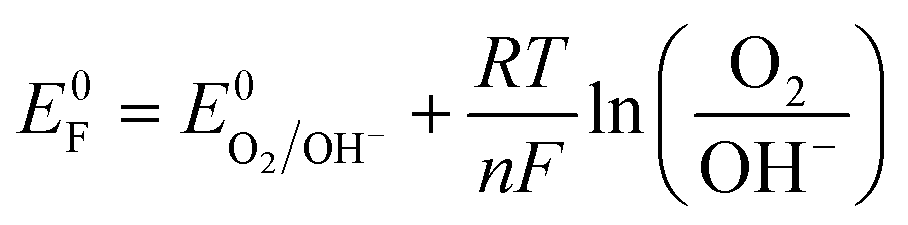 | (1) |
This change in E0F due to interference of NO−3 ions would alter the charge transfer dynamics at the interface that might change the switching potential. To verify the hypothesis, photocurrent measurements were performed under simulated solar light (AM 1.5) with intermittent chopping. Fig. 4(a) illustrates photocurrent against Vapp plots measured for the BFO electrode in Na2SO4 with and without addition of NaNO3. Upon increasing the NaNO3 concentration the switching potential shifts anodically from 0.8 V RHE (in 0.1 M Na2SO4) to ∼0.9 V RHE (in 0.1 M Na2SO4 + 0.4 M NaNO3). An enlarged portion of the plot [Fig. 4(b)] clearly illustrates this switch from photocathodic to photoanodic polarity as well as anodic shift in the switching potential. A distinct critical state manifests at the switching potential in all these systems, as depicted in Fig. S5,† which is further verified by chronoamperometry at that potential under chopping of light. To verify that the hypothesis holds true for similar types of anions, we repeated the photocurrent vs. voltage measurements by replacing NO3− anions with ClO4−, keeping the cation fixed (i.e., NaClO4 was used in place of NaNO3). A varying concentration of NaClO4 was added in the Na2SO4 electrolyte. Consistent with our initial findings, the anodic shift in switching potential from 0.8 V RHE (in 0.1 M Na2SO4) to ∼0.86 V RHE (in 0.1 M Na2SO4 + 0.4 M NaClO4) as shown in Fig. S6† was observed. This further supports the impact of specific anion interactions at the semiconductor–electrolyte interface, as predicted using eqn (1). To further verify the validity of eqn (1) in predicting the change in the Fermi level, we conducted a chopped LSV experiment in 0.1 M Na2SO4 under two different conditions: (i) in the absence of O2 and (ii) in O2-saturated (O2 was purged for 60 minutes in 20 mL of Na2SO4) electrolyte solution. As depicted in Fig. S7,† the switching potential shifted anodically from 0.78 V vs. RHE in Na2SO4 without O2 saturation to 0.88 V vs. RHE in O2-saturated solution as per eqn (1).
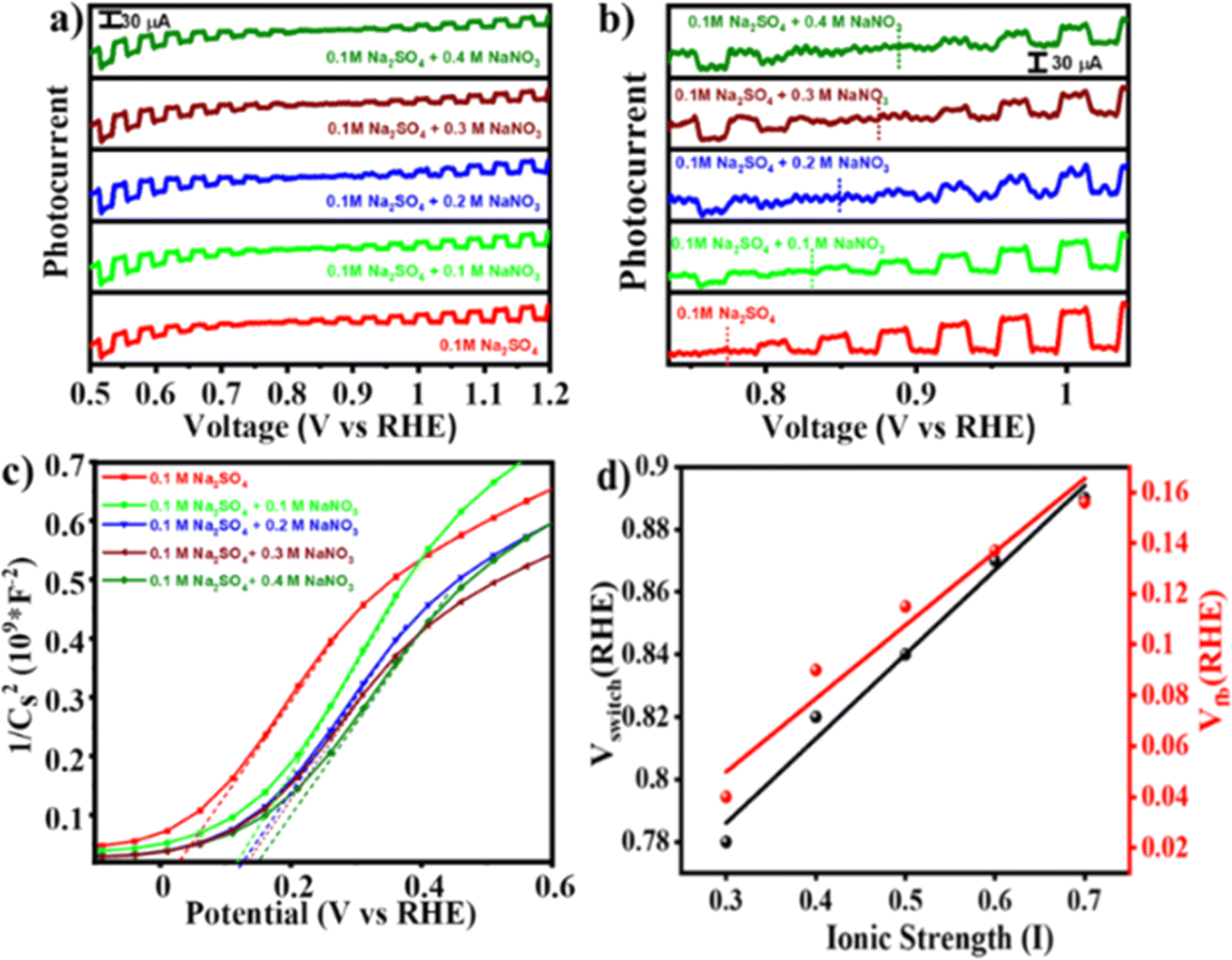 | ||
| Fig. 4 (a) Photocurrent vs. voltage curves for the BFO photoelectrode in different electrolyte compositions, and (b) enlarged portion of (a) showing the switching potential and shift in the onset of photocurrent. Plots of (c) Mott–Schottky, and (d) switching potential vs. ionic strength. | ||
To understand the origin of the PEPS effect and to explore the impact of variation in electrolyte composition on the electrochemical properties, Mott–Schottky (M–S) analysis was done across all compositions and under O2 saturated conditions utilizing a three-electrode configuration in a potential range of −1.0 to +1.0 V vs. the Ag/AgCl/Cl− reference electrode at an applied frequency of 1 kHz. As depicted in Fig. 4(c) and S8,† the M–S plot of BFO exhibited a positive slope, indicative of an n-type semiconducting nature, without the formation of any heterojunction with the substrate. Electrochemical impedance spectroscopy (EIS) studies were also performed within a frequency range of 1000 kHz to 100 mHz. Moreover, the flat-band potential (VFB) can be derived from the intercept of the linear portion of M–S curves at the X-axis, as determined using eqn (2).42
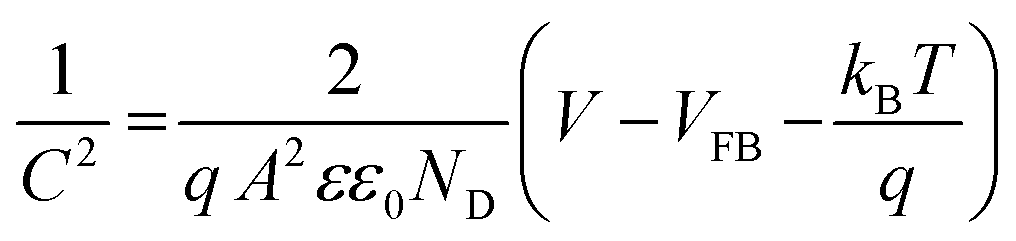 | (2) |
The VFB obtained using eqn (2) was measured to be 0.03 V vs. RHE and consistently exhibited an anodic shift in a trend similar to that of the switching potential when both the inert electrolyte was added and when electrolyte was saturated with O2. On the absolute potential scale, VFB lies between the conduction band edge and the valence band edge of a non-degenerate semiconductor under the flat band conditions. While VFB is typically considered the onset potential of photocurrent, the onset potential tends to be slightly higher than VFB due to slow interfacial charge transfer kinetics necessitating a slight overpotential.44 With an increase in the concentration of inert electrolyte, both the flat-band potential and the onset of photocurrent shift anodically. Fig. S9† illustrates EIS spectra obtained at OCP (open circuit potential) aimed at assessing the impact of inert electrolyte addition on electrolyte conductivity. The spectra reveal a decrease in solution resistance and a simultaneous increase in ionic conductivity because of inert electrolyte addition.
2.2 PEPS effect and shift in switching potential
Elucidating the precise mechanisms underlying charge transfer proves challenging due to the complex interplay between redox couples in the electrolyte and the solvent. The anodic shift in the photocurrent polarity reversal potential can be explained with the support of the quasi-Fermi level method, band bending and traditional chemical reaction kinetics to describe the various charge transfer pathways. In a photodetector based on BFO, the O2/OH− redox couple plays a pivotal role as per eqn (1). In a solution containing 0.1 M Na2SO4, during and after the onset of photocurrent (approximately at 0.8 V vs. RHE) a positive trend in photocurrent (Iph) is favoured. Consequently, the positive trend in Iph outweighs the negative trend, resulting in an apparent positive signal in Iph.5 From eqn (1), it can be predicted that changing the O2/OH− concentration at the interface would change the Fermi level of the electrolyte. The subsequent additions of NaNO3 and NaClO4 result in an excessive presence of NO3− and ClO4− ions, relatively lowering the OH− concentration at the semiconductor–electrolyte interface that changes the Fermi level of the electrolyte system to a more positive value.This upward shift in the Fermi level of the electrolyte would alter the charge transfer dynamics at the interface as it reduces the width of the space charge layer (W) according to eqn (3) for establishing thermodynamic equilibrium at the interface (Fig. 5). Consequently, the degree of band bending decreases following NaNO3 and NaClO4 addition, and with this, lowering in W continues as more salt is introduced. This argument is supported by the observation of an anodic shift in the flat band potential in M–S plots after NaNO3 addition, indicating a decrease in the width of space charge layer, W, calculated at 0.78 V vs. RHE according to eqn (3) and as demonstrated in Fig. S10†.45
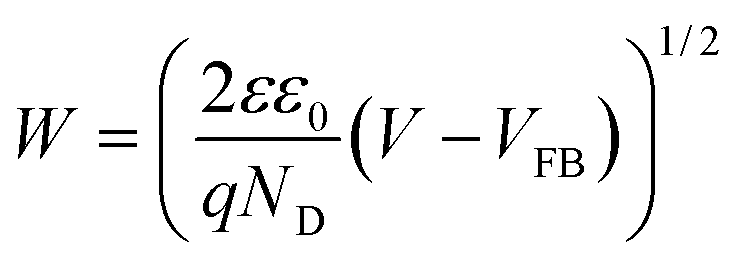 | (3) |
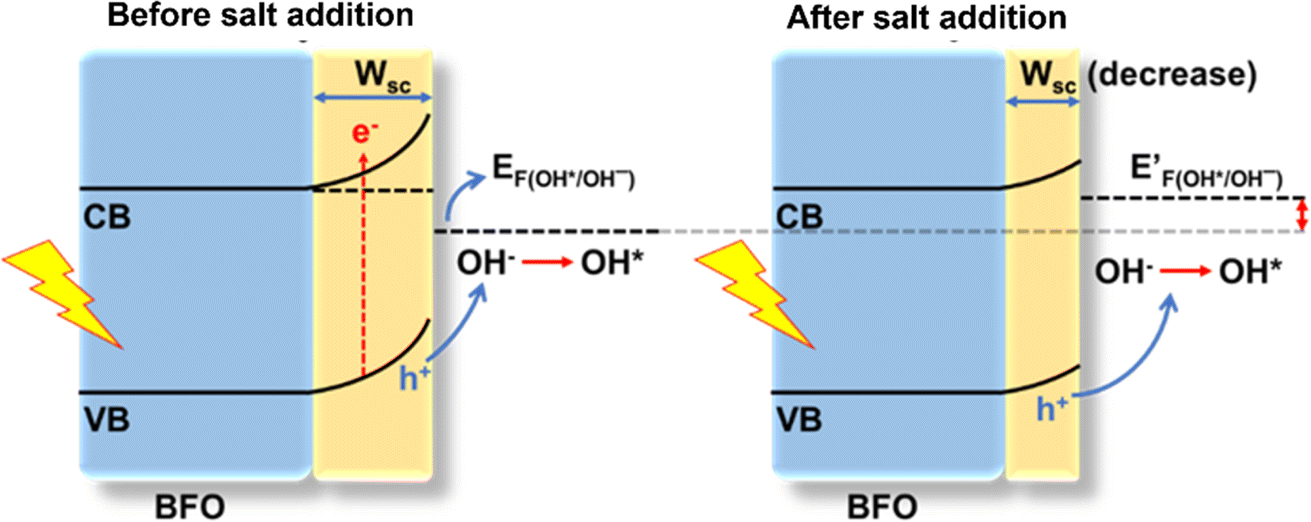 | ||
| Fig. 5 Mechanism diagram of carrier transport between BFO working electrodes with electrolytes. | ||
Furthermore, a compact double layer at the interface impedes the mobility of OH− and OH radicals, necessitating a higher overpotential for the completion of reaction pathways. As a result, there is an anodic increase in the photocurrent onset. The composition and concentration of electrolytes along with their respective redox couple concentrations and carrier pathways play a critical role in this process. Thus, a change in the types and concentrations of electrolytes has a significant impact on both the magnitude and polarity of the apparent photocurrent (Iph). Additionally, by altering the electrolytes, the polarity of the output signal from PEC-type photodetectors at a specific potential can be reversed as shown in eqn (4) and (5).
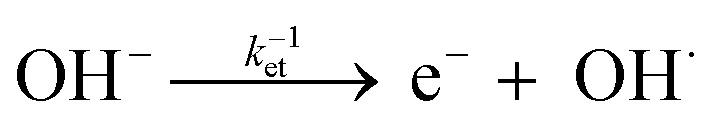 | (4) |
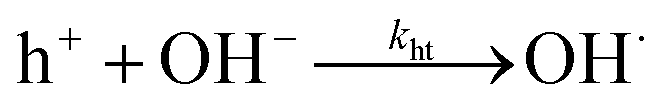 | (5) |
Similarly, increasing the concentration of O2, an electron acceptor molecule, in Na2SO4 solution alters the E0F, shifting it to a more positive value. This shift slows the tendency of hydroxyl oxidation, which reduces the space charge layer width and impacts the charge transfer dynamics at the semiconductor–electrolyte interface. As a result, this shift in the E0F leads to an anodic shift in the photocurrent switching potential and an increase in the photocurrent in the cathodic region. The observed changes are further supported by Mott–Schottky plots showing an anodic shift in flatband potential, analogous to the NaNO3 and NaClO4 cases. In PEC systems, not only the concentration of electrons and holes in semiconductors but also the species and concentration of redox couples in electrolytes regulate carrier pathways and trigger the PEPS effect.
3 Conclusion
In summary, a novel, cost-effective, and scalable approach has been shown to synthesize BFO NPs that exhibit a 1.9 eV bandgap, suitable for photoelectrochemical devices. BFO photoelectrodes have been shown to exhibit reversible photocurrent behaviour, which is explained by their distinct bandgap structure, paving the way for the formation of either cathodic or anodic photocurrents based on applied bias. The effect of inert electrolyte on photocurrent switching potential has been investigated in detail and it was found that electrolyte composition may be precisely tailored to provide accurate tuning of photocurrent polarity switching, hence opening a new avenue in the field of photoconversion applications. This study has ramifications not just for BFO but also for other metal oxide perovskites, which might lead to the development of promising switchable photoelectrode materials in the future, especially for compensated semiconductors.Data availability
All underlying data are available in the published article itself and its ESI.†Author contributions
Ajay: performed all the experiments, conceptualization, data curation and analysis, and writing of the original draft of the manuscript. JS: data analysis, photo-electrochemical measurements, writing – review and editing. PPI: conceptualization, methodology, writing – review & editing, supervision, and funding acquisition.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors are grateful to the Central Research Facility (CRF) and Nanoscale Research Facility (NRF) of IIT Delhi for assistance in material characterization. The authors are also thankful to the Advanced Materials Research Center (AMRC), IIT Mandi, for UPS measurements. Ajay is thankful to the Ministry of Human Resource Development (MHRD) for the research fellowships. PPI is thankful to SERB, Department of Science and Technology (DST), India, for the financial support under the grant sanction order CRG/2022/009352 and EEQ/2020/000558.References
- K. Sivula, R. Zboril, F. L. Formal, R. Robert, A. Weidenkaff, J. Tucek, J. Frydrych and M. Grätzel, J. Am. Chem. Soc., 2010, 132(21), 7436–7444 CrossRef CAS PubMed.
- L. Meng, Y. Li, R. Yang, X. Zhang, C. Du and J. Chen, Chem. Commun., 2019, 55, 2182–2185 RSC.
- D. K. Neethipathi, H. S. Ryu, M. S. Jang, S. Yoon, K. M. Sim, H. Y. Woo and D. S. Chung, ACS Appl. Mater. Interfaces, 2019, 11(23), 21211–21217 CrossRef CAS PubMed.
- C. Fan, J. Lai, Z. Shao, X. Zhou, Y. Liu, Y. Lin, L. Ding and K. Wang, Anal. Chem., 2023, 95(40), 15049–15056 CrossRef CAS PubMed.
- X. Geng, Y. Cai, M. Gao, X. Ma, L. Yu, Y. Xu, W. Shan and M. Qiu, ACS Appl. Mater. Interfaces, 2023, 15(48), 55938–55947 CrossRef CAS PubMed.
- A. Efrati, O. Yehezkeli, R. Tel-Vered, D. Michaeli, R. Nechushtai and I. Willner, ACS Nano, 2012, 6(10), 9258–9266 CrossRef CAS PubMed.
- M. Warzecha, M. Warzecha, M. Oszajca, M. Oszajca, K. Pilarczyk, K. Pilarczyk, K. Szaciłowski and K. Szaciłowski, Chem. Commun., 2015, 51, 3559–3561 RSC.
- B. Seger, J. McCray, A. Mukherji, X. Zong, Z. Xing and L. Wang, Angew. Chem., Int. Ed., 2013, 52, 6400–6403 CrossRef CAS PubMed.
- D. K. Singh, P. Prajapat, J. Saroha, R. K. Pant, S. N. Sharma, K. K. Nanda, S. B. Krupanidhi and G. Gupta, ACS Appl. Electron. Mater., 2023, 5(3), 1394–1400 CrossRef CAS.
- A. Yucknovsky, Y. Shlosberg, N. Adir and N. Amdursky, Angew. Chem., Int. Ed., 2023, 62, e202301541 CrossRef CAS PubMed.
- A. Podborska, M. Suchecki, K. Mech, M. Marzec, K. Pilarczyk and K. Szaciłowski, Nat. Commun., 2020, 11, 854 CrossRef CAS PubMed.
- R. Beranek and H. Kisch, Angew. Chem., Int. Ed., 2008, 47, 1320–1322 CrossRef CAS PubMed.
- W. S. Bourée, M. S. Prévot, X. A. Jeanbourquin, N. Guijarro, M. Johnson, F. L. Formal and K. Sivula, Adv. Mater., 2016, 28, 9440 CrossRef.
- H. Chen, G. Liu, L. Wang, H. Chen, G. Liu and L. Wang, Sci. Rep., 2015, 5, 12368 CrossRef PubMed.
- C. V. Hoang, K. Hayashi, Y. Ito, N. Gorai, G. Allison, X. Shi, Q. Sun, Z. Cheng, K. Ueno, K. Goda and H. Misawa, Nat. Commun., 2017, 8, 771 CrossRef PubMed.
- Y. Nishikata, A. Morikawa, M.-A. Kakimoto, Y. Imai, Y. Hirata, K. Nishiyama and M. Fujihira, J. Chem. Soc., Chem. Commun., 1989, 1772–1774 RSC.
- S. Wojtyła and T. Baran, J. Inorg. Organomet. Polym. Mater., 2017, 27, 436–445 CrossRef.
- J. Puerres, S. Polanía, A. F. Pérez-Torres, E. A. Erazo, M. T. Cortés and P. Ortiz, ACS Appl. Nano Mater., 2023, 6(15), 14029–14039 CrossRef CAS.
- L. Yan, W. Zhao and Z. Liu, Dalton Trans., 2016, 45, 11346–11352 RSC.
- I. Cesar, K. Sivula, A. Kay, R. Zboril and M. Grätzel, J. Phys. Chem. C, 2009, 113, 772–782 CrossRef CAS.
- A. Kumar, P. G. Santangelo and N. S. Lewis, J. Phys. Chem., 2002, 96(2), 834–842 CrossRef.
- B. Seger, J. McCray, A. Mukherji, X. Zong, Z. Xing and L. Wang, Angew. Chem., Int. Ed., 2013, 52, 6400–6403 CrossRef CAS PubMed.
- T. Zhao, H. He, C. Wu, L. Lai, Y. Ma, H. Yang, H. Hu, A. Liu, D. Guo and S. Wang, ACS Appl. Nano Mater., 2023, 6(5), 3856–3862 CrossRef CAS.
- M. P. Dare-Edwards, J. B. Goodenough, A. Hamnett and P. R. Trevellick, J. Chem. Soc., Faraday Trans. 1, 1983, 79, 2027–2041 RSC.
- P. Salvador, J. Phys. Chem., 2002, 89, 3764–3773 Search PubMed.
- Y. Wang, M. Daboczi, M. Zhang, J. Briscoe, J.-S. Kim, H. Yan and S. Dunn, Mater. Horiz., 2023, 10, 5892–5897 RSC.
- J. Ye, S. Jin, Y. Cheng, H. Xu, C. Wu, F. Wu and D. Guo, ACS Appl. Mater. Interfaces, 2024, 16(20), 26512–26520 CrossRef CAS PubMed.
- H. Xu, L. Deng, Y. Cheng, C. Wu, K. Chen and D. Guo, ACS Appl. Nano Mater., 2024, 7(2), 2359–2369 CrossRef CAS.
- Y. Cheng, J. Ye, L. Lai, S. Fang and D. Guo, Adv. Electron. Mater., 2023, 9, 2201216 CrossRef CAS.
- P. Banoth, C. Kandula, P. K. Lavudya, S. Akaram, L. D. L. S. Valladares, R. Ammanabrolu, G. K. Mamidipudi and P. Kollu, ACS Omega, 2023, 8(21), 18653–18662 CrossRef CAS PubMed.
- S. Yang, G. Ma, L. Xu, C. Deng and X. Wang, RSC Adv., 2019, 9, 29238–29245 RSC.
- T.-J. Park, G. C. Papaefthymiou, A. J. Viescas, a. A. R. Moodenbaugh and S. S. Wong, Nano Lett., 2007, 7(3), 766–772 CrossRef CAS PubMed.
- J. Qi, N. Ma, X. Ma, R. Adelung and Y. Yang, ACS Appl. Mater. Interfaces, 2018, 10(16), 13712–13719 CrossRef CAS PubMed.
- D. Tomar and P. Jeevanandam, J. Magn. Magn. Mater., 2022, 564, 170092 CrossRef.
- Ajay, V. Tanwar, A. A. Gujare and P. P. Ingole, Adv. Sustainable Syst., 2024, 2400244 CrossRef.
- X. Shi, S. Quan, L. Yang, C. Liu and F. Shi, J. Mater. Sci., 2019, 54, 12424–12436 CrossRef CAS.
- K. P. Remya, D. Prabhu, R. J. Joseyphus, A. C. Bose, C. Viswanathan and N. Ponpandian, Mater. Des., 2020, 192, 108694 CrossRef CAS.
- A. H. Ibrahim, Y. M. Abbas, M. H. Ali, H. A. Ayoub and M. Aldoori, J. Mater. Sci.: Mater. Electron., 2024, 35, 676 CrossRef CAS.
- N. P. Prasad, M. Rohnke, M. A. Verheijen, J. M. Sturm, J. P. Hofmann, E. J. M. Hensen and A. Bieberle-Hütter, ACS Appl. Energy Mater., 2023, 6(24), 12237–12248 CrossRef CAS.
- S. J. Clark and J. Robertson, Appl. Phys. Lett., 2007, 90, 132903 CrossRef.
- K.-C. Pan, C.-S. Chuang, S.-H. Cheng and Y. O. Su, J. Electroanal. Chem., 2001, 501, 160–165 CrossRef CAS.
- K. Sivula, ACS Energy Lett., 2021, 6(7), 2549–2551 CrossRef CAS.
- J. Lu, A. Günther, F. Schrettle, F. Mayr, S. Krohns, P. Lunkenheimer, A. Pimenov, V. D. Travkin, A. A. Mukhin and A. Loidl, Eur. Phys. J. B, 2010, 75, 451–460 CrossRef CAS.
- A. Hankin, F. E. Bedoya-Lora, J. C. Alexander, A. Regoutz and G. H. Kelsall, J. Mater. Chem. A, 2019, 7, 26162–26176 RSC.
- K. K. Dey, S. Gahlawat and P. P. Ingole, J. Mater. Chem. A, 2019, 7, 21207–21221 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04504a |
| This journal is © The Royal Society of Chemistry 2024 |