
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMaking an inverted Keggin ion lacunary†
Lu-Lu
Liu
a,
Zi-Yu
Xu
a,
Peng
Yi
a,
Chao-Qin
Chen
a,
Zhong-Ling
Lang
 *b and
Peng
Yang
*a
*b and
Peng
Yang
*a
aCollege of Chemistry and Chemical Engineering, Advanced Catalytic Engineering Research Center of the Ministry of Education, Hunan University, Changsha 410082, P. R. China. E-mail: pengyang216@hnu.edu.cn
bKey Laboratory of Polyoxometalate and Reticular Material Chemistry of Ministry of Education, Faculty of Chemistry, Northeast Normal University, Changchun 130024, P. R. China. E-mail: langzl554@nenu.edu.cn
First published on 16th September 2024
Abstract
The century-old inverted Keggin ion has been revisited in an effort to unleash its potential in the structural engineering and functional development of polyoxomolybdates (POMos). Over the past hundred years, attempts to program the metal–oxo scaffold of inverted Keggins have been conducted continually but without any success. In this work, a structurally inert, inverted Keggin-type POMo could finally be altered by means of a binary heterogroup-templated approach, resulting in the successful isolation of two lacunary species. The local structure and charge distribution of these species are adjustable, and hence they serve as available building blocks for the subsequent controlled assembly of a CeIII-incorporated derivative. From the plenary to the lacunary, the enclosed structure of the inverted Keggin has been opened up significantly, resulting in less steric hindrance, along with a transition from an electron neutral species to a negatively charged species. Owing to these beneficial properties, the emerging defect-containing polyanions demonstrated outstanding Lewis acid–base catalytic activity in the high efficiency production of pyrazoles.
Introduction
The study of polyoxometalates (POMs) dates back nearly two centuries to when Berzelius first reported the ammonium salt of [PMo12O40]3− in 1826.1 This dodecanuclear archetype of hetero POMs was then named after J. F. Keggin for his great contribution to its structural determination in 1933.2 To this day, the so-called Keggin ion, [(XO4)M12O36]n− (X = heteroatom; M = addenda atom), remains at the forefront of research into POM chemistry. The role of “X” can be played by nearly all of the elements (noble gases excluded) across the periodic table. For “M”, besides the conventional metal ions, MoVI and WVI, the oxides of several p-block metals (e.g., AlIII, SnIV, SbV)3–6 and d-block metals (e.g., TiIV, FeIII, PdII)7–10 are also able to adopt the Keggin scaffold, as demonstrated by very recent studies. Owing to their diverse library of compositions, Keggin-type POMs with a remarkable range of physicochemical properties have gained wide acceptance in many important fields.11 By stark contrast, the structurally related inverted Keggin ions of the general formula [(RXO3)4M12O34]n− (X = AsV or PV; M = MoVI; R = oxygen or organic groups) have seldom been in the spotlight, despite their discovery being communicated by H. Debray as early as 1874.12 Over the past hundred years, despite the sustained efforts from generations of chemists worldwide, the progress in the fundamental and applied research of inverted Keggin-type POMs has not matched that of their Keggin counterparts.13–27The principal advantage of the classical Keggin ions, in particular the polyoxotungstates (POWs) with a [XW12O40]3− archetype, lies in their structural editability. Driven mainly by pH fluctuations, a varying number of {WO6} octahedra at specific locations can be selectively removed to yield mono-, di-, or tri-lacunary derivatives with versatile coordination modes (Scheme 1).28 The exposed oxygens around the openings of these derivatives are able to bond entities such as transition metals, lanthanides, and organometallic fragments with catalytic, magnetic, or biomedical functions.29–31 As a result, the birth of lacunary Keggin ions has provided an enriched reservoir of molecular building blocks for the design and engineering of POM-based materials.32,33 The structures of inverted Keggin ions, in contrast, has been more difficult to change. In contrast to the Keggin ions, the four groups of the edge-sharing {M3O13} moieties in the inverted Keggin archetype are turned inside-out and are joined together via their corners, resulting in a tetrahedral conformation with an idealized Td symmetry.13 Instead of being positioned at the center of the molecule, the heterogroups (e.g., organo-arsonates and -phosphonates) are attached peripherally to the four faces confined by the adjacent {M3O13} motifs.14–21,24–27 There is, in turn, a central cavity with a radius of ca. 2.9 Å. Relying on the protection from the increased number of heterogroups with hydrophobic aryl or alkyl pendent arms, the preparation of lacunary inverted Keggin species is rather challenging and has yet to be achieved.28
 | ||
| Scheme 1 Structural representation of Keggin and inverted Keggin ions as well as their lacunary derivatives. Color code: {WO6}, plum octahedra; {MoO6}, dark blue octahedra; X, green; R, turquoise; O, red. | ||
Of particular note is that, compared with lacunary POWs, the number of lacunary polyoxomolybdates (POMos) is rather limited.28 Most POMos are self-assembled via a bottom-up approach, so that the structures of the in situ formed species are often poorly controlled and are hard to predict. Moreover, the isolation of lacunary POMos is also tough, as a series of complex dynamic equilibria in the mixture of Mo–oxo clusters that appear in solution is usually involved.34 In an effort to maintain the metastable frameworks of lacunary POMos, several strategies have been tried, such as the surface passivation by interesting organic ligands.35–37 However, the synthesis of lacunary POMos is achieved at the expense of performance, as they almost lose the ability to combine with other electrophiles as most of the active coordination sites have been occupied. Therefore, maximizing the coordination ability of lacunary POMos to make them useful precursors represents a key issue of future research in POM chemistry.
To tap the potential of the inverted Keggin ion, the structural synthesis of its lacunary derivatives has been carried out in the current work. By virtue of a binary heterogroup-templated assembly, the structural evolution of di-vacant POMos bearing the inverted Keggin topology has been achieved in a controlled manner. The resulting open framework and tunable charge distribution of the polyanions meet the requirements of a useful synthetic precursor and allow coordination with CeIII ions to extend the structural diversity further. Apart from the structural interest of the as-made compounds, their significantly enhanced Lewis acid–base catalytic activity for the preparation of pyrazoles has been evaluated and analyzed in detail.
Results and discussion
Based on the findings from reviewing previous works, almost all of the known inverted Keggin POMos were built with organo-arsonates/phosphonates as the heterogroups, where the sterically hindered organic parts prohibit the formation of Keggin structures.14–21,24–27 Moreover, a strong acidic environment (pH ≤ 1) has been reported to be essential for their assemblies. In consideration of the above, ligands of the type R–As/PO3H2 (R = C6H5, C6H4-4-NH2, C6H4-4-COOH) were reacted with (NH4)6Mo7O24 in one-pot reactions. Upon increasing the pH stepwise from 1.0 to 6.0, none of the desired lacunary inverted Keggin species were obtained, indicating that the commonly used pH-dependent strategy might not work here. In this context, a binary heterogroup-templated approach has been proposed, as demonstrated in our previous research.38 Specifically, a combination of tetrahedral {R–As/PO3} and trigonal pyramidal {SeO3} species was afforded simultaneously. The differences in their structural and electronic configurations were expected to steer the assembly process of the inverted Keggin ions towards a low symmetry route.In an acidic aqueous solution containing (NH4)6Mo7O24 (pH = 0.9), the reaction of SeO2 and p-H2NC6H4AsO3H2 in a stoichiometric ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 resulted in a decanuclear Mo–oxo cluster, [(SeO3)(p-H3NC6H4AsO3)3Mo10O29]3− (SeAs3Mo10); see the ESI† for synthetic details. As shown in Fig. 1a and b, the structure of SeAs3Mo10 can be described as a di-lacunary derivative of the saturated inverted Keggin [(p-H3NC6H4AsO3)4Mo12O34] (As4Mo12), which was reported by Pope et al. in 1981.15 Interestingly, one of the four faces of the As4Mo12 tetrahedron is capped by a {SeO3} function instead. Accordingly, the {(p-H3NC6H4AsO3)Mo2O5} moiety has been removed, resulting in a di-vacant species. With the proportion of SeO2 increased from 2:1 to 3:1, the {p-H3NC6H4AsO3} cap close to the opening could be further replaced by a {SeO3} heterogroup but without changing the Mo–oxo framework, yielding another di-vacant species, [(SeO3)2(p-H3NC6H4AsO3)2Mo10O29]4− (Se2As2Mo10, Fig. 1c). To the best of our knowledge, SeAs3Mo10 and Se2As2Mo10 represent the first examples of lacunary inverted Keggin ions.28 It is noteworthy that no new terminal oxygen atoms are present in SeAs3Mo10 and Se2As2Mo10 and that even some addenda atoms have been removed, which is quite different from the typical lacunary POWs. This might be attributed to the special atomic arrangement in the inverted Keggin ions, where two terminal metal–oxygen bonds are located on each addenda. Therefore, the formation of new terminal oxygen atoms in SeAs3Mo10 would require the removal of the {SeO3} heterogroup, and the expected three terminal oxygen atoms would be arranged in a mer fashion (Fig. S1†). However, such behavior does violate the famous Lipscomb rule, which has been explained in terms of the strong trans influence of the terminal metal–oxygen bonds.39 For a typical {MO6} octahedron, the three terminal metal–oxygen bonds could arrange only in a fac but not in a mer fashion. Theoretically speaking, it is not possible to have new terminal oxygen atoms next to the lacuna in SeAs3Mo10 and Se2As2Mo10. Meanwhile, the central heterogroup is responsible for connecting the four {M3O13} moieties, which plays a crucial role in the stabilization of the metal–oxo framework of the Keggin structure. However, in the absence of a heterogroup, the lacunary species of, for example, the Keggin-type species [H2W12O40]9− could not be obtained. With regards to the inverted Keggin structure, the four {M3O13} moieties are bridged by the four heterogroups from the outside. It is believed that if one of the heterogroups (e.g., {SeO3}) is missing, there might be a risk that the whole cluster will be highly unstable and dissociate thereafter. Obviously, the previous definition of lacunary POMs is mostly based on the Keggin and Wells-Dawson archetypes. For these “non-classical” lacunary inverted Keggin ions reported here, the above-mentioned structural factors have to be considered.
:1 resulted in a decanuclear Mo–oxo cluster, [(SeO3)(p-H3NC6H4AsO3)3Mo10O29]3− (SeAs3Mo10); see the ESI† for synthetic details. As shown in Fig. 1a and b, the structure of SeAs3Mo10 can be described as a di-lacunary derivative of the saturated inverted Keggin [(p-H3NC6H4AsO3)4Mo12O34] (As4Mo12), which was reported by Pope et al. in 1981.15 Interestingly, one of the four faces of the As4Mo12 tetrahedron is capped by a {SeO3} function instead. Accordingly, the {(p-H3NC6H4AsO3)Mo2O5} moiety has been removed, resulting in a di-vacant species. With the proportion of SeO2 increased from 2:1 to 3:1, the {p-H3NC6H4AsO3} cap close to the opening could be further replaced by a {SeO3} heterogroup but without changing the Mo–oxo framework, yielding another di-vacant species, [(SeO3)2(p-H3NC6H4AsO3)2Mo10O29]4− (Se2As2Mo10, Fig. 1c). To the best of our knowledge, SeAs3Mo10 and Se2As2Mo10 represent the first examples of lacunary inverted Keggin ions.28 It is noteworthy that no new terminal oxygen atoms are present in SeAs3Mo10 and Se2As2Mo10 and that even some addenda atoms have been removed, which is quite different from the typical lacunary POWs. This might be attributed to the special atomic arrangement in the inverted Keggin ions, where two terminal metal–oxygen bonds are located on each addenda. Therefore, the formation of new terminal oxygen atoms in SeAs3Mo10 would require the removal of the {SeO3} heterogroup, and the expected three terminal oxygen atoms would be arranged in a mer fashion (Fig. S1†). However, such behavior does violate the famous Lipscomb rule, which has been explained in terms of the strong trans influence of the terminal metal–oxygen bonds.39 For a typical {MO6} octahedron, the three terminal metal–oxygen bonds could arrange only in a fac but not in a mer fashion. Theoretically speaking, it is not possible to have new terminal oxygen atoms next to the lacuna in SeAs3Mo10 and Se2As2Mo10. Meanwhile, the central heterogroup is responsible for connecting the four {M3O13} moieties, which plays a crucial role in the stabilization of the metal–oxo framework of the Keggin structure. However, in the absence of a heterogroup, the lacunary species of, for example, the Keggin-type species [H2W12O40]9− could not be obtained. With regards to the inverted Keggin structure, the four {M3O13} moieties are bridged by the four heterogroups from the outside. It is believed that if one of the heterogroups (e.g., {SeO3}) is missing, there might be a risk that the whole cluster will be highly unstable and dissociate thereafter. Obviously, the previous definition of lacunary POMs is mostly based on the Keggin and Wells-Dawson archetypes. For these “non-classical” lacunary inverted Keggin ions reported here, the above-mentioned structural factors have to be considered.
 | ||
| Fig. 1 (a)–(c) Structural evolution from the plenary As4Mo12 species to the di-lacunary species, SeAs3Mo10 and Se2As2Mo10. Color code: {MoO6}, dark blue octahedra; As, green; Se, pink; C, gray; N, blue; O, red. (d) Structural symmetry of As4Mo12 and the as-made polyanions. (e) FT-IR spectra of As4Mo12, SeAs3Mo10 and Se2As2Mo10. (f) and (g) EDX analysis of SeAs3Mo10 and Se2As2Mo10. | ||
It has been noted that the raw material, (NH4)6Mo7O24·4H2O, could not be replaced by Na2MoO4·2H2O in the synthesis, indicating that the counter cations (NH4+) might play a key role in the crystallization of such polyanions. Meanwhile, only the {SeO3} and {p-H3NC6H4AsO3} couple could cooperate to shape the defect polyanions. This might be attributed to (i) the pseudo-zwitterionic nature of the {p-H3⊕NC6H4AsO3−} ligands, of which the positively charged quaternary ammonium and negatively charged arsonate group could withstand the pH fluctuation over the course of the crystallization;15 (ii) the similar bond lengths and tri-dentate coordination mode of {SeO3} as compared to those of the {p-H3NC6H4AsO3} function;40 and (iii) {p-H3NC6H4AsO3} cannot be replaced by {p-H3NC6H4PO3}, possibly because of the longer As–O bond distance (about 1.60 Å), as compared to that of the P–O bonds (about 1.50 Å), which meets the requirement for the connection of the {Mo3O13} moieties. Due to the loss of addenda and the partial substitution of the heterogroups, the symmetry of these molecules is reduced from Td (As4Mo12) to Cs (SeAs3Mo10) and C2v (Se2As2Mo10). Taking the heteroatoms as vertices, the standard tetrahedron of {As4} (As4Mo12) has been gradually distorted by the increased number of {SeO3} groups in {SeAs3} (SeAs3Mo10) and {Se2As2} (Se2As2Mo10), as displayed in Fig. 1d. This might be explained by the lone pair of electrons on the {SeO3} groups, which point at the center of the molecules. Possibly because of the twisted inner space and intense electrostatic repulsion, the surroundings are no longer suitable to support the presence of the addenda. Therefore, the {Mo2O5} unit departs from the plenary inverted Keggin scaffold, yielding the di-vacant species. Furthermore, the addition of even more SeO2 into the reaction system did not change the structure of Se2As2Mo10 further. In addition to the crystallographic characterization, the structural changes of the as-made polyanions were also validated via FT-IR spectroscopy. As depicted in Fig. 1e, the broad bands located at 670 and 583 cm−1 (As4Mo12) are split into several peaks in the spectra of SeAs3Mo10 and Se2As2Mo10, corresponding to the changed vibrations of the O–Mo–O and Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups in the defect-containing frameworks.41 Furthermore, the appearance of the {SeO3} heterogroups was also unequivocally verified using EDX analysis (Fig. 1f and g). The valence states of the addenda and oxygen atoms were determined through XPS characterization (Fig. S2 and S3†) and BVS calculations42 (Table S1†), showcasing the presence of MoVI and indicating that none of the oxygen atoms are protonated.
O groups in the defect-containing frameworks.41 Furthermore, the appearance of the {SeO3} heterogroups was also unequivocally verified using EDX analysis (Fig. 1f and g). The valence states of the addenda and oxygen atoms were determined through XPS characterization (Fig. S2 and S3†) and BVS calculations42 (Table S1†), showcasing the presence of MoVI and indicating that none of the oxygen atoms are protonated.
To gather more information with regard to the solution behaviors of the as-made polyanions, UV-vis spectroscopy and electrospray-ionization mass spectrometry (ESI-MS) were carried out. Re-dissolution of the respective SeAs3Mo10 and Se2As2Mo10 species in water displayed an absorption band located at ca. 254 nm, which can be ascribed to the pπ–dπ charge transfer of the O → Mo bands as well as the π–π* charge transfer of the aromatic groups (Fig. 2a and b).43 Over a time span of 24 hours, the intensity of the band declined slowly within the first 12 hours, indicating the gradual decomposition of their lacunary structures. After that, the intensity was nearly unchanged for the rest of the analysis time, suggesting that a dissociation equilibrium might be reached (Fig. S4†).44 The kinetically stable nature of the species was then verified using ESI-MS study. Apart from the dissociated fragments of different charges and compositions, the envelopes belonging to the intact polyanions were clearly identified. For SeAs3Mo10, the peaks centered at m/z = 822.90 and 1234.42 can be assigned to the −3 charged {SeAs3Mo10(H2O)15}3− and −2 charged {(NH4)SeAs3Mo10(H2O)14}2−, respectively (Fig. 2c). For Se2As2Mo10, the peak situated at m/z = 1207.55 corresponds to the −2 charged species of {(NH4)2Se2As2Mo10(H2O)15}2− (Fig. 2d). Additional MS assignments for the major peaks are summarized in Table S2.† In addition, the pH-dependent UV-vis spectra showcased that the position and intensity of the bands were shifted, suggesting the rapid deformation of SeAs3Mo10 and Se2As2Mo10 upon a rise in pH (Fig. 2e and f). The data from the above studies suggest that the lacunary SeAs3Mo10 and Se2As2Mo10 species may have adequate kinetic stability for being used as precursors for successive reactions at specific pH. Furthermore, the crystallinity of SeAs3Mo10 and Se2As2Mo10 is well retained in many kinds of organic solvents, as shown by the powder XRD patterns (Fig. 2g and h). Meanwhile, the frameworks of the lacunary species remain intact up to 220 °C, as indicated by the thermogravimetric analysis (Fig. 2i). The outstanding chemical and thermal stability of SeAs3Mo10 and Se2As2Mo10 indicate their potential as heterogeneous catalysts for various organic syntheses.
 | ||
| Fig. 2 (a) and (b) UV-vis spectra of aqueous solutions containing SeAs3Mo10 and Se2As2Mo10 at various time intervals. (c) and (d) Negative-ion mass spectra of SeAs3Mo10 and Se2As2Mo10. (e) and (f) UV-vis spectra of the aqueous solution containing SeAs3Mo10 and Se2As2Mo10 at different pH conditions. (g) and (h) Powder XRD patterns of SeAs3Mo10 and Se2As2Mo10 after soaking in different organic solvents for 24 h. (i) Thermogravimetric curves of SeAs3Mo10 and Se2As2Mo10. | ||
To examine the coordination ability of SeAs3Mo10, CeIII ions were added into the reaction system followed by the successful isolation of a dimeric assembly, [{Ce(H2O)6(SeO3)(p-H3NC6H4AsO3)3Mo10O29}2] (Se2As6Mo20Ce2). It should be noted that the synthesis of Se2As6Mo20Ce2 could be achieved using SeAs3Mo10 as the precursor or via an in situ self-assembly. As shown in Fig. 3a, the structure of Se2As6Mo20Ce2 is comprised of two polyanionic SeAs3Mo10 moieties, which are bridged by two CeIII cations in the belt position, resulting in a rare electroneutral Mo–oxo cluster. Every CeIII center is coordinated with three oxygen atoms from the terminal MoO groups together with six water molecules. Of particular interest, the CeIII ions did not fill the lacuna of SeAs3Mo10, which is in complete contrast to the lacunary POW counterparts. This might also be attributed to the effect of the lone pair electrons on the {SeO3} group next to the opening. Such rare behaviors have been encountered before, which demonstrate again the “non-classical” nature of lacunary inverted Keggin ions.45,46 According to previous findings, the plenary As4Mo12 is difficult to react with electrophiles because of its electroneutrality. More importantly, the protruding organic arms with protonated aniline groups would obstruct cation access to a large extent (Fig. 3b). In contrast, the anionic SeAs3Mo10 with an open-shell configuration allows sufficient contact with metal ions, demonstrating its coordination ability (Fig. 3c). The successful combination of SeAs3Mo10 and CeIII was also verified using FT-IR spectroscopy. In the spectrum of Se2As6Mo20Ce2, the split of the peak centered at 914 cm−1 (SeAs3Mo10) signals the change in vibrations of Mo–oxo framework (Fig. 3d).41 Meanwhile, the presence of the CeIII ions was verified through EDX and XPS analysis as well (Fig. 3e and f). The peaks at 904.6 and 901.1 eV as well as at 886.6 and 881.8 eV are, respectively, located in the Ce 3d3/2 and Ce 3d5/2 energy region, which are in line with the CeIII oxidation state.47–49
 | ||
| Fig. 3 (a) Structural representation of Se2As6Mo20Ce2. Color code: {MoO6}, dark blue octahedra; Ce, yellow; As, green; Se, pink; C, gray; N, blue; O, red. (b) Inhibition of coordination caused by the sterically hindered organic arms with positively charged quaternary ammonium groups in As4Mo12. (c) Anionic SeAs3Mo10 with an open-shell configuration. (d) FT-IR spectra of SeAs3Mo10 and Se2As6Mo20Ce2. (e) EDX analysis of Se2As6Mo20Ce2. (f) XPS spectrum of Se2As6Mo20Ce2. | ||
Based on the enriched metallic centers, outstanding chemical and thermal stability as well as their pseudo-liquid behaviors, POMs are widely used as heterogeneous catalysts in the fabrication of pharmaceutical intermediates.29 Pyrazole-based medicines, in particular, hold high clinical value and are extensively used as anti-inflammatory drugs, for sedation, pain relief, and other applications.50 The ever increasing demand for pyrazoles has promoted the rapid development of their synthetic techniques.51 Recently, the potential of POMs as Lewis acid–base catalysts in the dehydration condensation reaction of hydrazines and 1,3-dicarbonyls has been tapped, which has created an efficient route for the preparation of pyrazoles.38,52–59 In this context, the catalytic performance of the as-made polyanions and As4Mo12 has been systematically evaluated and compared to reveal the structure–activity relationships of this inverted Keggin family.
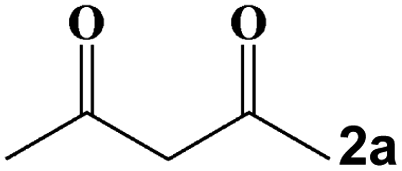
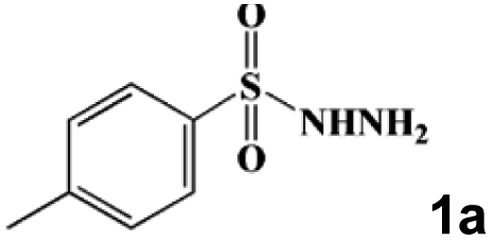
Taking the condensation–cyclization of p-toluenesulfonyl hydrazide (1a) and acetylacetone (2a) as the model reaction, the optimal conditions were investigated (Table S3†). Follow-up experiments were performed at room temperature (RT) for 10 min to screen the activity of different catalysts. As shown in Fig. 4a and Table S4,† the yield of the desired product reached 28% without any catalyst. Using the starting materials (NH4)6Mo7O24·4H2O, p-H2NC6H4AsO3H2, SeO2, and their mixture as catalysts, yields of 47%, 45%, 54%, and 51% were attained, respectively. Meanwhile, a yield of 56% was observed using As4Mo12 as the catalyst. In contrast, the lacunary SeAs3Mo10 and CeIII-incorporated Se2As6Mo20Ce2 have been found to effectively promote the yield to 61% and 68%, respectively. Surprisingly, a high yield of 99% was attained using Se2As2Mo10 under mild conditions, which represents one of the best POM catalysts for the preparation of pyrazoles (Table S5†).38,52–59 The desired product was obtained in 98% yield using Se2As2Mo10 as the catalyst during the first 5 min of the reaction (Fig. 4b). However, for SeAs3Mo10 and Se2As6Mo20Ce2, a similar yield of ≥98% was achieved after 90 and 40 min, respectively, indicating that Se2As2Mo10 has the best activity of the inverted Keggin POMos. The heterogeneous catalytic nature of the as-made compounds was validated using a filtration test (Fig. 4c and S5†). As exemplified by Se2As2Mo10, the yield (81%) barely changed after the removal of catalysts from the reaction at the third min. Moreover, SEM and BET characterizations were carried out on the as-made catalysts. As shown in Fig. S6,† there was no obvious difference in the size distribution of the three catalysts, with an average diameter of ca. 3 μm. Their specific surface areas were analyzed through N2 adsorption experiments at 77 K. As depicted in Fig. S7,† all three compounds displayed type II adsorption isotherms, indicating their non-porous nature. After activation, the total adsorption capacities of SeAs3Mo10, Se2As2Mo10 and Se2As6Mo20Ce2 are 6.06, 6.15 and 4.65 cm3 g−1, respectively, with specific surface areas of 4.41, 4.68 and 3.07 m2 g−1, respectively. Therefore, there is not much difference in their specific surface areas. Based on the above, the different catalytic activities of the as-made compounds might be mainly attributed to the differences in their molecular structures. Due to the pseudo-liquid behavior of POMs as heterogeneous catalysts, substrates can react not only on the surface of the POM catalysts but deep in their crystal lattices also.29 Therefore, the structural and physicochemical features of the catalysts play a crucial role in the catalytic reactions. Additionally, these catalysts could be easily separated and reused for at least six cycles, and the average yield is still above 95% (Fig. 4d–f). No obvious changes were observed in the powder XRD patterns (Fig. 4g–i) and FT-IR spectra (Fig. S8†) of these compounds before and after the catalytic reactions. On the basis of the above mentioned findings, the as-made polyanions, particularly Se2As2Mo10, are found to be heterogeneous catalysts of outstanding activity and stability.
 | ||
| Fig. 4 (a) Catalytic efficiency of the different catalysts in the model reaction ((A) SeAs3Mo10; (B) Se2As2Mo10; (C) Se2As6Mo20Ce2; (D) As4Mo12; (E) (NH4)6Mo7O24·4H2O; (F) p-H2NC6H4AsO3H2; (G) SeO2; (H) mixture; (I) no catalyst). (b) Catalytic efficiency of SeAs3Mo10 (A), Se2As2Mo10 (B), and Se2As6Mo20Ce2 (C) as a function of time. (c) Results of the filtration test conducted on the Se2As2Mo10 heterogeneous catalyst. (d)–(f) Catalytic stability of SeAs3Mo10 (d), Se2As2Mo10 (e), and Se2As6Mo20Ce2 (f) (error bar ± 1%). (g)–(i) Powder XRD patterns of SeAs3Mo10, Se2As2Mo10, and Se2As6Mo20Ce2 before and after catalysis. | ||
Next, a cross combination of different kinds of hydrazines/hydrazides (1a–1d) and 1,3-diketones (2a–2c) containing various substituent groups were employed to evaluate the catalytic activity of the as-made compounds. As recorded in Table 1, satisfactory catalytic efficiency (yield ≥ 99%) was obtained. In terms of the reaction time and temperature, Se2As2Mo10 still demonstrated the best performance of the three catalysts. Moreover, from an industrial point of view, the feasibility of mass production is a critical element to be considered. Therefore, the catalytic performance at 10 times scale was examined for the reaction of 1d and 2a. The gram-scale production of 3j (yield ≥ 99%) went smoothly in the presence of SeAs3Mo10 (RT, 90 min), Se2As2Mo10 (RT, 10 min), and Se2As6Mo20Ce2 (RT, 90 min). Regarding all the reactions mentioned above, the structure and purity of the targeted products were validated using NMR spectroscopy (Fig. S9 and S20†).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Hydrazine/hydrazide | 1,3-Diketone | SeAs3Mo10 | Se2As2Mo10 | Se2As6Mo20Ce2 | Product | Yieldb (%) |
| a Reaction conditions: hydrazine/hydrazide 1 (0.2 mmol), 1,3-diketone 2 (0.2 mmol), catalyst (1.5 mol%), and DMC (dimethyl carbonate, 0.5 mL) at different reaction temperatures and times. b Isolated yields. | |||||||
| 1 |

|
|
RT, 90 min | RT, 10 min | RT, 30 min | 3a | 99 |
| 2 |
|
|
60 °C, 120 min | 60 °C, 30 min | 60 °C, 60 min | 3b | 99 |
| 3 |

|

|
80 °C, 60 min | 60 °C, 120 min | 80 °C, 30 min | 3c | 99 |
| 4 |
|

|
60 °C, 90 min | 60 °C, 30 min | 60 °C, 60 min | 3d | 99 |
| 5 |
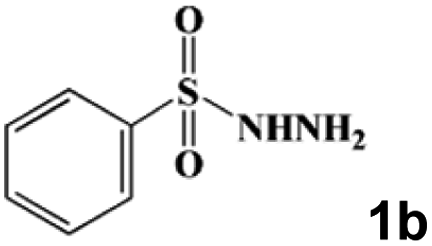
|

|
60 °C, 120 min | 60 °C, 60 min | 60 °C, 90 min | 3e | 99 |
| 6 |

|

|
80 °C, 120 min | 60 °C, 120 min | 80 °C, 60 min | 3f | 99 |
| 7 |
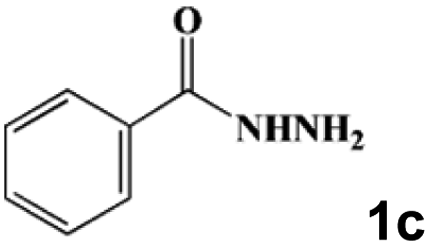
|

|
60 °C, 90 min | 60 °C, 30 min | 60 °C, 60 min | 3g | 99 |
| 8 |

|
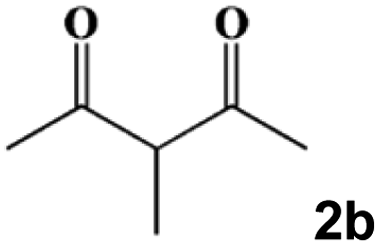
|
80 °C, 30 min | 60 °C, 60 min | 80 °C, 30 min | 3h | 99 |
| 9 |
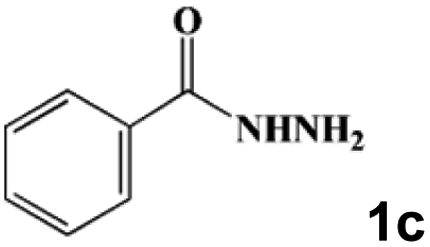
|

|
100 °C, 120 min | 80 °C, 120 min | 100 °C, 60 min | 3i | 99 |
| 10 |

|

|
RT, 90 min | RT, 10 min | RT, 90 min | 3j | 99 |
| 11 |

|
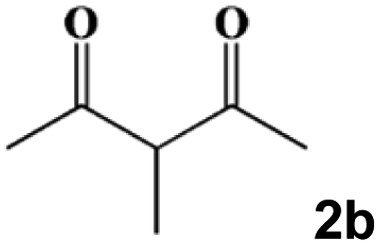
|
60 °C, 120 min | 60 °C, 30 min | 60 °C, 60 min | 3k | 99 |
| 12 |

|
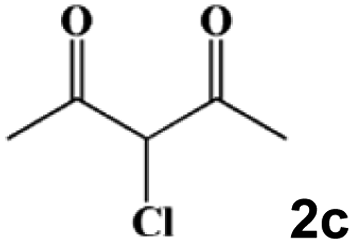
|
80 °C, 120 min | 80 °C, 30 min | 80 °C, 60 min | 3l | 99 |

To understand the advantages that the Se2As2Mo10 catalyst has, compared with the other inverted Keggin POMos employed in this work, several structural factors must be considered. First of all, from As4Mo12viaSeAs3Mo10 to Se2As2Mo10, the enclosed structure has been gradually opened up mainly due to the loss of {p-H3NC6H4AsO3} groups with significant steric hindrance (Fig. 5a–c). As a consequence, Se2As2Mo10 has the most open framework, which is accessible for the contact of substrates.56 Secondly, it is noted that the charge of the Mo–oxo clusters evolves from electroneutrality (As4Mo12) to electronegativity (SeAs3Mo10 and Se2As2Mo10). Accordingly, the molecular electrostatic potential (MEP) maps of the clusters have been calculated and are displayed in Fig. 5d–f. For As4Mo12, an obviously reduced nucleophilicity was detected as compared with the other two species. Due to the replacement of {p-H3NC6H4AsO3}− by {SeO3}2−, the increased charges are mainly concentrated on the lacuna of the clusters. Owing to the enlarged electronegative area, the exposed μ-oxo groups could aid the contact with electrophilic hydrazine substrates.56 Finally, despite As4Mo12 and Se2As6Mo20Ce2 having the same electroneutral features, the catalytic activity of the latter was significantly enhanced as compared to that of the former. Meanwhile, the catalytic performance of Se2As6Mo20Ce2 is also better than that of the SeAs3Mo10 precursor. This could be attributed to the incorporation of the CeIII ions, of which the large size and positively charged nature provide suitable environments for the access of substrates (Fig. 5g).59 To further verify the advantages of the as-made compounds as Lewis-acid–base catalysts, the preparation of another medical intermediate, 2-phenyl-2,3-dihydroquinazolin-4(1H)-one, via the acetalization of 2-aminobenzamide and benzaldehyde has been carried out. Based on the data listed in Table S6 and Fig. S21,† the as-made compounds could catalyze the reaction in a more cost-effective way, saving time and energy. In particular, Se2As2Mo10 still behaved the best of the three compounds and the other reported catalysts, which is in agreement with the findings of the pyrazole syntheses.60,61 To sum up, the combination of an open-shell configuration and electronegative character are mainly responsible for the superior catalytic activity of Se2As2Mo10, demonstrating the structural advantages of the lacunary inverted Keggin archetype.
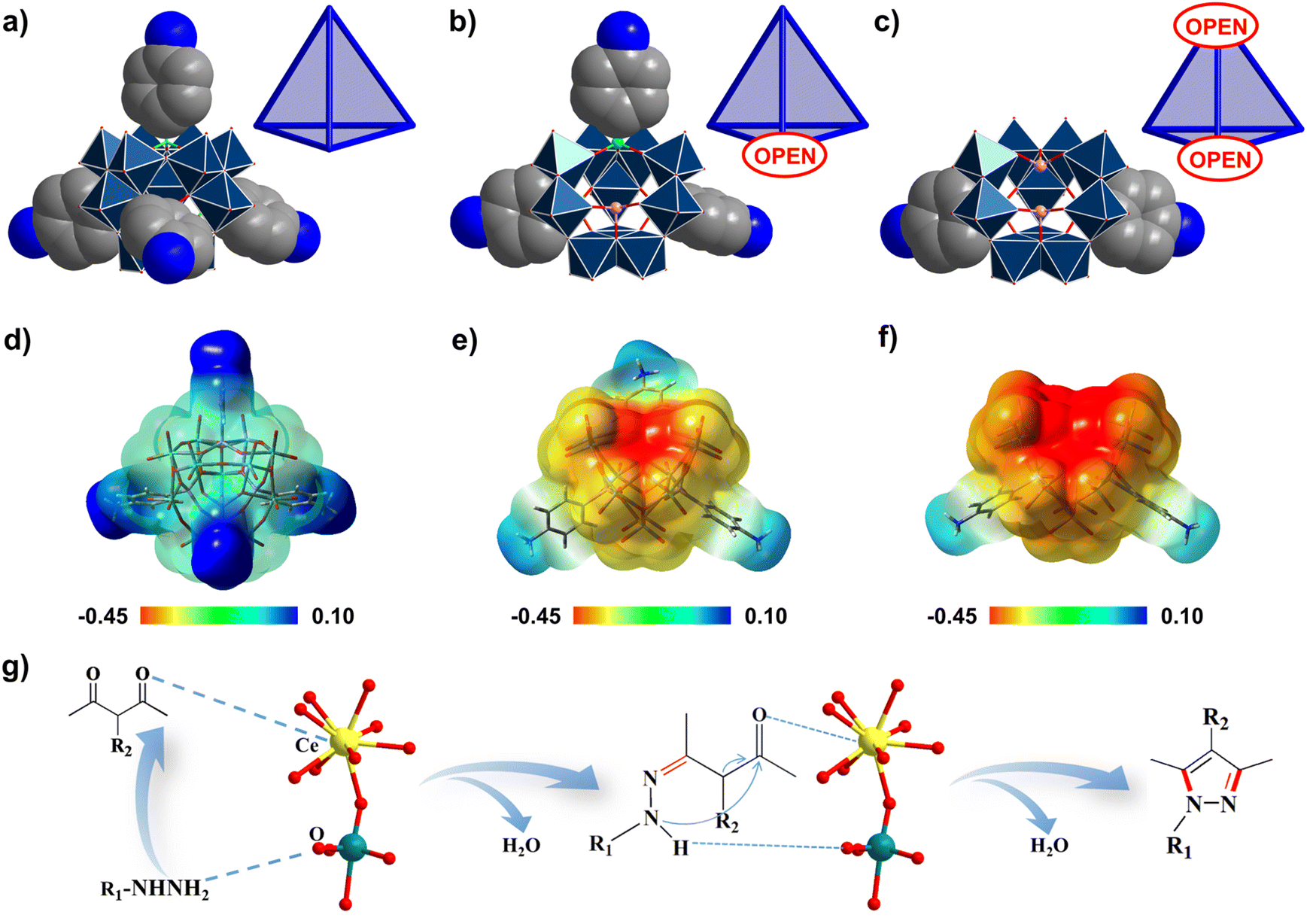 | ||
| Fig. 5 (a)–(c) Transition of the Mo–oxo frameworks from fully closed (As4Mo12 (a)) to gradually open structures (SeAs3Mo10 (b); Se2As2Mo10 (c)). (d)–(e) Molecular electrostatic potential (MEP) distribution (isovalue 0.02) for As4Mo12 (d), SeAs3Mo10 (e), and Se2As2Mo10 (f). The red areas are more nucleophilic regions. (g) Proposed catalytic synergistic effect of the CeIII ions in Se2As6Mo20Ce2. Color code: Ce, yellow; Mo, cyan; O, red. | ||
Conclusions
After a hundred years of waiting, the use of a synthesis that relies on binary heterogroups has succeeded in making an inverted Keggin ion lacunary. In addition to the removal of addenda, the enclosed Mo–oxo framework of the inverted Keggin has been opened up, thus allowing further development of the structure. These kinetically stable vacant species are suitable precursors to advance the synthetic chemistry of POMos via a building block approach. Aside from the breakthrough relating to structural control, a combination of advantageous properties, including reduced steric hindrance, tunable charge distribution, and diversified composition, makes these polyanions behave as Lewis-acid–base catalysts. Fine chemicals such as pyrazoles could be produced in large quantity with ultrahigh efficiency. It can be foreseen that, as more efforts are devoted to the emerging area of lacunary inverted Keggin ions, the field of POM research shall be rejuvenated.Data availability
Experimental details (e.g., synthesis of compounds and procedure of catalytic experiments), characterization of the compounds (e.g., XPS spectra, FT-IR spectra, NMR spectra, and TG curves), and tables (e.g., crystallographic and refinement data, values of BVS calculations, assignments for the main peaks in the ESI-MS spectra).Author contributions
P. Y. conceived the project and designed the experiments. L.-L. L., Z.-Y. X. and P. Y. performed the experiments and analyzed the data. C.-Q. C. helped with the elemental analysis. Z.-L. L. carried out the MEP calculations. L.-L. L. drafted the manuscript. All the authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was financially supported by the Natural Science Foundation of Hunan Province (2022JJ20007), the Science and Technology Innovation Program of Hunan Province (2022RC1115), the Natural Scientific Foundation of Jilin Province Science and Technology Department (20230101032JC), and the Foundation of Jilin Educational Committee (JJKH20231298KJ). Single-crystal X-ray diffraction data were acquired at the Analytical Instrumentation Center of Hunan University.References
- J. Berzelius, Ann. Phys., 1826, 6, 369–380 CrossRef.
- J. F. Keggin, Nature, 1933, 131, 908–909 CrossRef CAS.
- S. E. Smart, J. Vaughn, I. Pappas and L. Pan, Chem. Commun., 2013, 49, 11352–11354 RSC.
- J. Roswell and L. F. Nazar, J. Am. Chem. Soc., 2000, 122, 3777–3778 CrossRef.
- D. C. Hutchison, R. D. Stern, M. R. Olsen, L. N. Zakharov, K. A. Perssonb and M. Nyman, Dalton Trans., 2018, 47, 9804–9813 RSC.
- V. Baskar, M. Shanmugam, M. Helliwell, S. J. Teat and R. E. P. Winpenny, J. Am. Chem. Soc., 2007, 129, 3042–3043 CrossRef CAS PubMed.
- N. Li, J. Liu, J.-J. Liu, L.-Z. Dong, S.-L. Li, B.-X. Dong, Y.-H. Kan and Y.-Q. Lan, Angew. Chem., Int. Ed., 2019, 58, 17260–17264 CrossRef CAS PubMed.
- O. Sadeghi, L. N. Zakharov and M. Nyman, Science, 2015, 347, 1359–1362 CrossRef CAS PubMed.
- E. V. Chubarova, M. H. Dickman, B. Keita, L. Nadjo, F. Miserque, M. Mifsud, I. W. C. E. Arends and U. Kortz, Angew. Chem., Int. Ed., 2008, 47, 9542–9546 CrossRef CAS PubMed.
- P. Yang and U. Kortz, Acc. Chem. Res., 2018, 51, 1599–1608 CrossRef CAS PubMed.
- J.-X. Liu, X.-B. Zhang, Y.-L. Li, S.-L. Huang and G.-Y. Yang, Coord. Chem. Rev., 2020, 414, 213260 CrossRef CAS.
- H. Debray, C. R. Hebd. Seances Acad. Sci., 1874, 78, 1411 Search PubMed.
- T. Nishikawa and Y. Sasaki, Chem. Lett., 1975, 4, 1185–1186 CrossRef.
- M. Filowitz and W. G. Klemperer, J. Chem. Soc., Chem. Commun., 1977, 7, 201–202 RSC.
- K. M. Barkigia, L. M. Rajkovic-Blazer, M. T. Pope and C. O. Quicksall, Inorg. Chem., 1981, 20, 3318–3323 CrossRef CAS.
- B. J. S. Johnson, R. C. Schroden, C. Zhu and A. Stein, Inorg. Chem., 2001, 40, 5972–5978 CrossRef CAS PubMed.
- B. J. S. Johnson, R. C. Schroden, C. Zhu, V. G. Young and A. Stein, Inorg. Chem., 2002, 41, 2213–2218 CrossRef CAS PubMed.
- B. J. S. Johnson, S. A. Geers, W. W. Brennessel, V. G. Young and A. Stein, Dalton Trans., 2003, 24, 4678–4681 RSC.
- E. Burkholder, S. Wright, V. Golub, C. J. O'Connor and J. Zubieta, Inorg. Chem., 2003, 42, 7460–7471 CrossRef CAS PubMed.
- E. Burkholder and J. Zubieta, Inorg. Chim. Acta, 2004, 357, 301–304 CrossRef CAS.
- T. Ueda, T. Yonemura, M. Shiro, M. Fukudome and M. Hojo, Inorg. Chem. Commun., 2007, 10, 1301–1303 CrossRef CAS.
- B. Liu, Z.-T. Yu, J. Yang, W. Hua, Y.-Y. Liu and J.-F. Ma, Inorg. Chem., 2011, 50, 8967–8972 CrossRef CAS PubMed.
- J. Fielden, K. Quasdorf, L. Cronin and P. Kögerler, Dalton Trans., 2012, 41, 9876–9878 RSC.
- T. Ueda, K. Machida, M. Shiro and D. Kaneno, Inorg. Chem. Commun., 2013, 38, 123–126 CrossRef CAS.
- M.-S. Liu, W.-D. Yu, Q.-W. Yan and J. Yan, Eur. J. Inorg. Chem., 2017, 2017, 1947–1950 CrossRef CAS.
- X.-X. Zhang, H. Yuan, W.-D. Yu, Y.-Y. Gu and J. Yan, Inorg. Chem. Commun., 2018, 91, 77–80 CrossRef CAS.
- X. Ma, S. Bhattacharya, D. H. Taffa, T. Nisar, M. Wark, V. Wagner and U. Kortz, Inorg. Chem., 2023, 62, 1813–1819 CrossRef CAS PubMed.
- L.-L. Liu, L. Wang, X.-Y. Xiao, P. Yang, J. Zhao and U. Kortz, Coord. Chem. Rev., 2024, 506, 215687 CrossRef CAS.
- S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- J.-C. Liu, J.-W. Zhao, C. Streb and Y. F. Song, Coord. Chem. Rev., 2022, 471, 214734 CrossRef CAS.
- M. Aureliano, N. I. Gumerova, G. Sciortino, E. Garribba, C. C. McLauchlan, A. Rompel and D. C. Crans, Coord. Chem. Rev., 2022, 454, 214344 CrossRef CAS.
- H. N. Miras, J. Yan, D.-L. Long and L. Cronin, Chem. Soc. Rev., 2012, 41, 7403–7430 RSC.
- D. E. S. Marcano, N. D. Savić, K. Declerck, S. A. M. Abdelhameed and T. N. Parac-Vogt, Chem. Soc. Rev., 2024, 53, 84–136 RSC.
- L. A. Combs-Walker and C. L. Hill, Inorg. Chem., 1991, 30, 4016–4026 CrossRef CAS.
- J. Hao, J. Zhang, P. C. Yin, Z. C. Xiao, F. P. Xiao and Y. G. Wei, Chem.–Eur. J., 2012, 18, 2503–2506 CrossRef CAS PubMed.
- C. Li, N. Mizuno, K. Yamaguchi and K. Suzuki, J. Am. Chem. Soc., 2019, 141, 7687–7692 CrossRef CAS PubMed.
- C. Li, K. Yamaguchi and K. Suzuki, Angew. Chem., Int. Ed., 2021, 60, 6960–6964 CrossRef CAS PubMed.
- L.-L. Liu, Y.-H. Wang, X.-Y. Xiao, K.-W. Tong, Y. Zhao, C.-Q. Chen, J. Du and P. Yang, Rare Met., 2023, 42, 3345–3353 CrossRef CAS.
- W. N. Lipscomb, Inorg. Chem., 1965, 4, 132–134 CrossRef CAS.
- L. Liu, J. Jiang, X. Liu, G. Liu, D. Wang, L. Chen and J. Zhao, Inorg. Chem. Front., 2020, 7, 4640–4651 RSC.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds-Part A: Theory and Applications in Inorganic Chemistry, Wiley and Sons, New York, 5th edn, 1997 Search PubMed.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244–247 CrossRef.
- H. So and M. T. Pope, Inorg. Chem., 1972, 11, 1441–1443 CrossRef CAS.
- P. R. Wu, C. H. Wang, Y. W. Chen, Y. J. Lin, T. Ide, Y. Tsuchido, Y. Seid and M. Horie, J. Mater. Chem. C, 2022, 10, 4306–4316 RSC.
- L. Li, Q. Shen, G. Xue, H. Xu, H. Hu, F. Feng and J. Wang, Dalton Trans., 2008, 5698–5700 RSC.
- M. Xu, T. Wang, F. Li, W. Xu, Y. Zheng and L. Xu, Chem. Commun., 2020, 56, 1097–1100 RSC.
- P. W. Park and J. S. Ledford, Langmuir, 1996, 12, 1794–1799 CrossRef CAS.
- H.-X. Sheng, B.-Y. Lin, C.-Q. Chen, J. Du and P. Yang, Polyoxometalates, 2024, 3, 9140060 CrossRef.
- Y. Zhao, C.-L. Li, C.-Q. Chen, J. Du, U. Kortz, T. Gong and P. Yang, Inorg. Chem. Front., 2024, 11, 1413–1422 RSC.
- Z. Xu, C. Gao, Q.-C. Ren, X.-F. Song, L.-S. Feng and Z.-S. Lv, Eur. J. Med. Chem., 2017, 139, 429–440 CrossRef CAS PubMed.
- S. Fustero, M. Sánchez-Roselló, P. Barrio and A. Simón-Fuentes, Chem. Rev., 2011, 111, 6984–7034 CrossRef CAS PubMed.
- G.-P. Yang, X.-L. Zhang, Y.-F. Liu, D.-D. Zhang, K. Li and C.-W. Hu, Inorg. Chem. Front., 2021, 8, 4650–4656 RSC.
- K. Li, Y.-F. Liu, X.-L. Lin and G.-P. Yang, Inorg. Chem., 2022, 61, 6934–6942 CrossRef CAS PubMed.
- M.-Y. Yao, Y.-F. Liu, X.-X. Li, G.-P. Yang and S.-T. Zheng, Chem. Commun., 2022, 58, 5737–5740 RSC.
- Y.-F. Liu, C.-W. Hu and G.-P. Yang, Chin. Chem. Lett., 2023, 34, 108097 CrossRef CAS.
- G.-P. Yang, X. He, B. Yu and C. W. Hu, Appl. Organomet. Chem., 2018, 32, e4532 CrossRef.
- G. Yang, Y. Liu, X. Lin, B. Ming, K. Li and C. Hu, Chin. Chem. Lett., 2022, 33, 354–357 CrossRef CAS.
- G.-P. Yang, S.-X. Shang, B. Yu and C.-W. Hu, Inorg. Chem. Front., 2018, 5, 2472–2477 RSC.
- M. Cheng, Y. Liu, W. Du, J. Shi, J. Li, H. Wang, K. Li, G. Yang and D. Zhang, Chin. Chem. Lett., 2022, 33, 3899–3902 CrossRef CAS.
- V. Jeevananthan, G. C. Senadi, K. Muthu, A. Arumugam and S. Shanmugan, Inorg. Chem., 2024, 63, 5446–5463 CrossRef CAS PubMed.
- K. Li, Y. F. Liu, G. P. Yang, Z. J. Zheng, X. L. Lin, Z. B. Zhang, S. J. Li, Y. H. Liu and Y. G. Wei, Green Chem., 2024, 26, 6454–6460 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2367552–2367554. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04634j |
| This journal is © The Royal Society of Chemistry 2024 |