
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect conversion of esters to imines/enamines and applications to polyester waste upcycling†
Rebecca A.
Kehner
,
Weiheng
Huang
and
Liela
Bayeh-Romero
 *
*
Department of Chemistry and Biochemistry, Baylor University, One Bear Place 97348, Waco, Texas 76798, USA. E-mail: Liela_Romero@Baylor.edu
First published on 27th September 2024
Abstract
Semi-reductive transformations of esters remain an underdeveloped but valuable class of functional group interconversions. Here, we describe the development of a highly selective method for the interconversion of esters to imines, enamines, aldehydes or amines through an amine-intercepted zirconocene hydride (ZrH)-catalyzed reduction. This protocol employs an inexpensive zirconium catalyst in combination with hydrosilanes and simple unprotected amines. A variety of aryl, benzylic, and aliphatic esters are directly transformed to imines and enamines in up to 99% yield or aldehydes in up to 84% yield, with little-to-no reduction to the corresponding alcohols. The utility of this method for the direct catalytic chemical upcycling of polyester plastic waste is demonstrated through multiple unprecedented depolymerization transformations. Further, the efficient preparation of nitrogen-containing products is also presented, including single-flask multicomponent reactions and the reductive amination of esters.
The direct conversion of esters to imines and enamines remains an elusive functional group interconversion, despite the apparent simplicity of such a transformation. Yet, semi-reductive imination or enamination of esters would serve as a step- and redox-economical alternative to prevailing synthetic strategies necessitating sequential reduction and condensation steps (Scheme 1a).1 Likewise, the continued reduction of these intermediates to amines would provide an unconventional disconnection to traditional reductive aminations using aldehydes, a frequently employed transformation for medicinal and process chemists alike.2 The enhanced stability and wide commercial availability of esters provides further impetus for such a transformation.
 | ||
| Scheme 1 Strategies for the conversion of esters to aldehydes, imines, and enamines and inspiration for a ZrH-catalyzed direct strategy. | ||
Moreover, the partial reduction of esters to aldehydes is a widely desired transformation in organic synthesis. Despite efforts to develop protocols for the direct conversion of esters to aldehydes, chemists still seek safe and reliable procedures for this transformation that do not result in concomitant over-reduction to the corresponding alcohol (Scheme 1a, path 1).3–5 For example, organic chemists most frequently rely on diisobutylaluminum hydride (DIBAL-H) to achieve this semi-reduction.4g However, large scale use of this reductant requires careful handling due to associated pyrophoricity and continuous maintenance of cryogenic reaction temperatures. For these reasons, chemists may instead rely on alternative routes altogether (i.e., utilization of Weinreb amides,6 Rosenmund reduction,7 Fukuyama reduction8) to obtain aldehyde products from precursors at higher oxidation states (Scheme 1a, path 2).
Recently, a renewed interest in zirconocene hydride (ZrH) catalysis has prompted the exploration of new methodologies concerning the functional group interconversion of carbonyl-containing molecules.9 Until now, manifolds employing ZrH reagents in either stoichiometric or catalytic quantities have exclusively resulted in the complete reduction of esters to alcohols.9f,h,j,10,11 Our initial work regarding the ZrH-catalyzed reduction of carbonyls using hydrosilanes showcased this selectivity,9h as did a later report by Cantat and coworkers using an analogous strategy but employing Schwartz's reagent (Cp2ZrHCl) directly.9f In both studies aldehyde intermediates were not observed, even in trace quantities, as these intermediates are more susceptible to reduction than the starting materials themselves.
Among our work in this research area, we recently disclosed the transamination semi-reduction of tertiary amides to imines and enamines (Scheme 1b).9b Key insights from accompanying mechanistic studies suggested the nucleophilic interception of zirconocene hemiaminal intermediates by an exogenous unprotected amine.9b,d Gaining inspiration from these studies, we surmised that this platform could be extended to the partial reduction of esters through interception of zirconocene hemiacetal intermediates by an amine. This would instead promote formation of imines or enamines as a protective trap for the aldehyde oxidation level. This mechanistic distinction would likewise deliver a synthetic handle for the direct conversion of esters to nitrogen-containing products (Scheme 1c).
We began our studies by employing zirconocene dichloride (Cp2ZrCl2) as an inexpensive and stable pre-catalyst in combination with hydrosilanes as the reductant (Table 1). Initial attempts to reduce aryl ester 1a with diethoxy(methyl)silane (DEMS) in the absence of an amine either resulted in over-reduction to the alcohol or unreacted starting material (see Table SI-2†). We then carried out the reduction in the presence of n-butylamine (entry 1). Strikingly, product selectivity completely diverged, favoring the formation of imine 3a in 82% yield. Conversion of 1a was diminished when catalyst loading was decreased or when Cp2ZrHCl was employed as the catalyst (entries 2 and 3). Replacing DEMS with polymethylhydrosiloxane (PMHS) likewise resulted in lower conversion and yield (entry 4). Finally, the yield of 3a was improved upon simply increasing reaction time from 18 to 21 hours (entry 5). Notably, <5% of alcohol was observed throughout the course of these optimization studies.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | Silane (equiv.) | % Yieldb | % Conversionb |
| a Entries 1–4: reactions were carried out under a N2 atmosphere using 0.2 mmol of 1a and 1.7 equiv. n-butylamine in anhydrous PhMe (0.4 M) for a duration of 18 h. Entries 6–10: Reactions were carried out under a N2 atmosphere using 0.2 mmol of 2a and 1.5 equiv. piperidine in anhydrous PhMe (0.4 M) for a duration of 18–23 h. b Yields were determined by 1H NMR spectroscopy of the crude reaction mixture, using mesitylene as an internal standard. c Carried out using 1 mmol 1a for 21 h instead. | ||||
| 1 | Cp2ZrCl2 (5) | DEMS (3 equiv.) | 82 | 85 |
| 2 | Cp2ZrCl2 (2.5) | DEMS (3 equiv.) | 34 | 39 |
| 3 | Cp2ZrHCl (5) | DEMS (3 equiv.) | 42 | 52 |
| 4 | Cp2ZrCl2 (5) | PMHS (5 equiv.) | 23 | 39 |
| 5 | Cp 2 ZrCl 2 (5) | DEMS (3 equiv.) | 91 | 91c |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||

|
||||
| 6 | Cp 2 ZrCl 2 (10) | DEMS (3) | 99 | 100 |
| 7 | Cp2ZrCl2 (5) | DEMS (3) | 75 | 86 |
| 8 | Cp2ZrCl2 (10) | TMDS (5) | 7 | 28 |
| 9 | Cp2ZrCl2 (10) | PMHS (5) | 82 | 83 |
| 10 | Cp2ZrHCl (10) | DEMS (3) | 93 | 93 |

After identifying the optimal reaction conditions for the semi-reductive imination of aryl esters, we turned our attention to aliphatic ester 2a. We supposed that a secondary amine could be used instead to promote the formation of an enamine. Upon exploration of various amine additives (see Table SI-6†), piperidine proved to be the optimal amine, quantitatively furnishing enamine 4a (entry 6). Decreasing the catalyst loading to 5 mol% provided the desired product in a synthetically useful, but lower yield (entry 7). Replacing DEMS with tetramethyldisiloxane (TMDS) proved ineffective; however, we were pleased to find that PMHS promoted the transformation of 2a, albeit with a slightly lower yield of 82% (entries 8 and 9). This hydrosilane, a byproduct of the silicon industry, is an especially appealing reductant due to its low cost and safety profile.12 Lastly, Cp2ZrHCl facilitated the semi-reduction with similar efficiency as Cp2ZrCl2 (entry 10).
With optimized conditions defined, we first investigated the single-step catalytic semi-reductive imination (3) and enamination (4) of esters, a direct functional group interconversion that, to the best of our knowledge, remains unreported (Table 2a). Aryl esters were directly converted to imines using various primary amines (3a–3c), while a cinnamate was transformed into hydrazone 3d when phenylhydrazine was employed as the nucleophile. The enamination of aliphatic esters could be carried out as well when using an assortment of cyclic amines (4a–4d). Notably, when diethylamine was employed instead, enamine 4e was formed in moderate yield. Thiophene, morpholine, and sulfide functionality were all tolerated under these reaction conditions (4f–4h).
| a Reactions were carried out under an atmosphere of N2 at 80 °C on a 0.2–1.0 mmol scale (unless indicated otherwise) using a primary amine (conditions A) or a secondary amine (conditions B) in anhydrous PhMe (0.4 M) for a duration of 18–48 h. b Yield was determined by 1H NMR spectroscopy of the crude reaction mixture, using mesitylene as an internal standard. c Isolated yield on a 1.0 mmol scale. d Carried out using 2.0 equiv. amine instead. e Reactions were carried out using n-butylamine (conditions A) or piperidine (conditions B) and subsequently quenched with 1 N aq. HCl. Yields reflect isolated yields. N.D. = Not Detected. f Carried out using 5 equiv. PMHS instead. g Carried out using 4 equiv. n-butylamine and 6 equiv. DMMS instead. h Carried out on a 0.5–1 mmol scale using conditions A without acidic workup, followed by NaBH4 (1.5–3 equiv.) reduction at 65 °C for 4–9 h. i Carried out using conditions B instead, followed by NaBH4 (3 equiv.) reduction at 65 °C for 9 h. j Carried out using 3 equiv. n-butylamine and 6 equiv. DMMS instead. |
|---|

|
Next, we explored the ZrH-catalyzed semi-reduction of esters to aldehydes through the implementation of a hydrolytic workup. Methyl and ethyl benzoates (1d, 1e) smoothly reacted to afford benzaldehydes in high yields, whereas more sterically encumbered esters either resulted in lower conversion of the starting material (1f) or amidation (1g). The partial reduction of 1e proved to be scalable, furnishing greater than 1 gram of aldehyde 6 on a 10 mmol scale. In general, various esters bearing ether, halide, N-heterocyclic, and sulfide functionality were amenable to the catalytic semi-reduction (7–11). Additionally, reduction of cinnamate 1c provided enal 12 in 71% yield, extending the utility of this protocol to the preparation of α,β-unsaturated aldehydes. Finally, aliphatic aldehydes 13 and 14 were obtained in moderate yields.
Throughout these studies, several notable limitations were observed. Though not detected in our prior studies, at elevated temperatures competitive nitrile reduction becomes problematic under this catalytic manifold. For example, substrate 1l underwent unselective reduction to afford terephthalaldehyde. In accord with our prior observations regarding the steric sensitivity of this catalytic system, esters bearing an α-quaternary or tertiary carbons (e.g.1m and 2k) failed to undergo reduction.
The use of an amine to interrupt traditional metal hydride-mediated ester reduction enables concise entry to valuable reactive intermediates that can be directly telescoped through multi-step synthetic sequences, necessitating only a single purification step.13,14 For example, the interception of imine intermediates with nucleophiles delivers access to α-alkylated secondary amines (Table 2b, 15 and 16). Alternatively, the ester starting material can instead serve as the nucleophilic component through generation of an enamine. This latter strategy was displayed through the single-flask multicomponent synthesis of α-alkylated aldehyde 17 in 59% yield when benzyl bromide was added after enamine formation (Table 2c).
Finally, we sought to illustrate the potential of this catalytic manifold for the two-step single-flask reductive amination of esters when incorporating a NaBH4 reduction prior to workup. An assortment of benzylic, allylic, and aliphatic amines were isolated in 60–72% yield (Table 2d, 18–23). The juxtaposition of imine 3a, aldehyde 5, and amine 18 best exemplify the controllable selectivity attainable through this catalytic manifold.
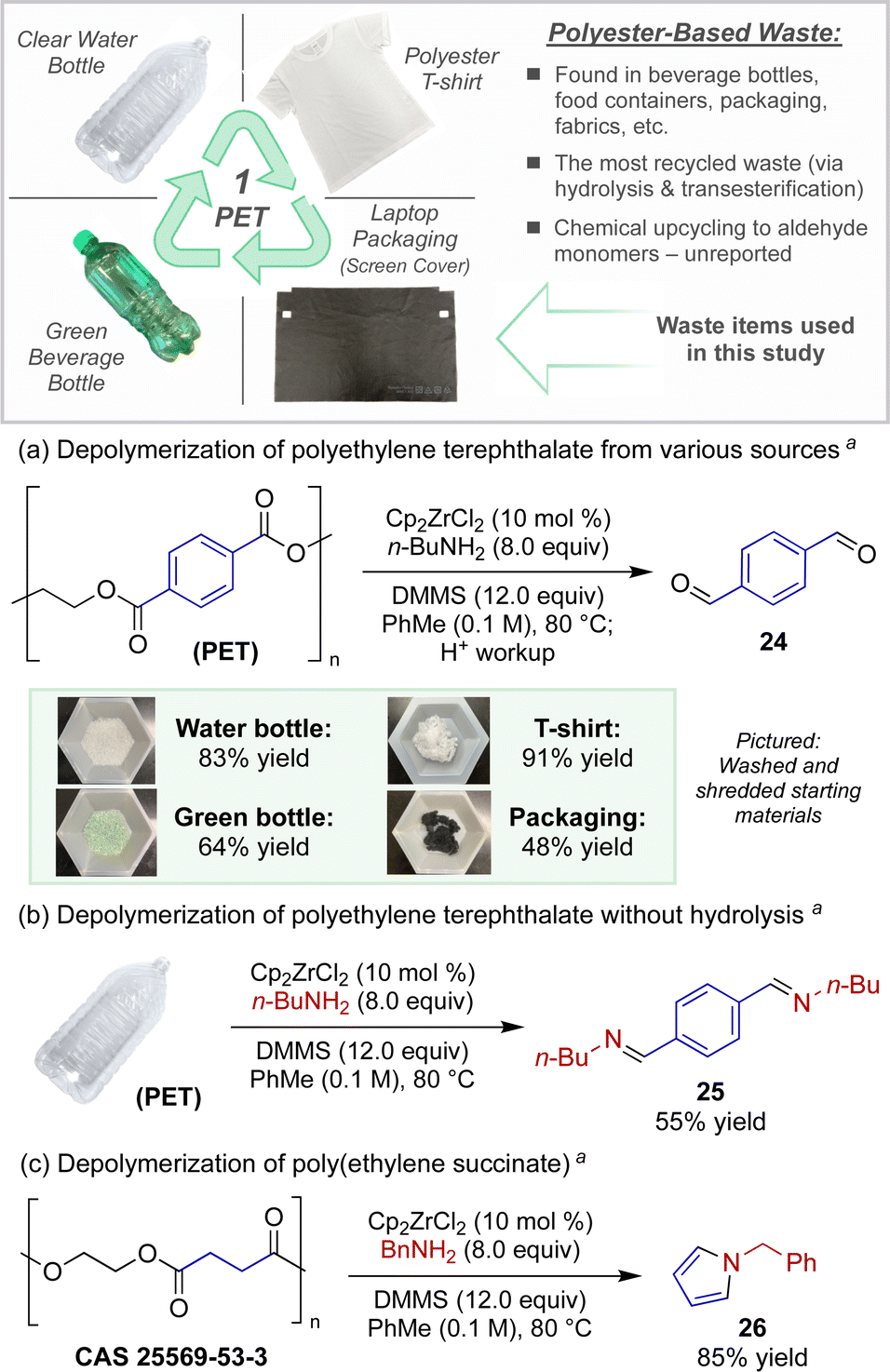
As a direct application toward the chemical recycling of accumulated plastic waste, we sought to leverage this catalytic protocol for new modes of polyester depolymerizations. The catalytic depolymerization of polyester plastics to access versatile chemical feedstocks at an oxidation state in-between that of their original carboxylic acid starting monomers and the fully reduced alcohol monomers would be a powerful and unprecedented form of plastic upcycling.15 Polyethylene terephthalate (PET), often found in plastic beverage bottles and containers, represents the most common polyester recyclable.16
For example, Scheme 2a demonstrates the conversion of an assortment of post-consumer PET wastes to terephthalaldehyde, a versatile building block used for the preparation of small molecules, polymers, and other porous and nonporous materials with wide-ranging uses (ligands, dyes, sensors, liquid crystals, thin films, re-healable thermosets, etc.).17 Utilizing modified semi-reduction conditions, PET plastic pieces obtained directly from a single use water bottle or a green beverage bottle afforded terephthalaldehyde (24) in 83% and 64% yields, respectively. We also chose to investigate the depolymerization of polyester fabric, the most widely produced fiber constituting >50% of total fiber production globally.16d Whether using PET sourced from a 100% polyester t-shirt or dyed fibers belonging to packaging waste from a recent laptop purchase (fabric screen protector insert), the catalytic reduction produced dialdehyde 24 in reasonable quantities (91% and 48% yield respectively).
 | ||
| Scheme 2 Applications to polyester depolymerization. (aReported values are based on the theoretical amount of repeating monomer unit). | ||
The strength of this strategy, however, lies in the ability to directly convert polyester waste to nitrogen-containing building blocks, offering a unique strategy for the repurposing of this plastic. For example, when PET was subjected to the standard depolymerization conditions without hydrolytic workup, diimine 25 was isolated in high purity in 55% yield (Scheme 2b). Further, poly(ethylene) succinate, an aliphatic polyester, underwent a novel depolymerization-cyclization sequence under similar reaction conditions (Scheme 2c). When the catalytic protocol was carried out using benzylamine, depolymerization occurred with concomitant Paal–Knorr-type cyclization to furnish pyrrole 26 in 85% isolated yield.
The profound effect on product chemoselectivity imparted by a simple unprotected amine prompted us to investigate the mechanism of this interrupted ZrH-catalyzed ester reduction. Prior reports regarding the interconversion of esters to amides mediated by Lewis acidic zirconocenes suggested to us that a similar mechanistic pathway might be involved, the products of which would be amenable to ZrH-catalyzed partial reduction.18 Of note, varying quantities of amide byproducts were observed throughout the course of our studies. However, our experimental mechanistic investigations suggest to us that this is unlikely the sole or major route of conversion.19
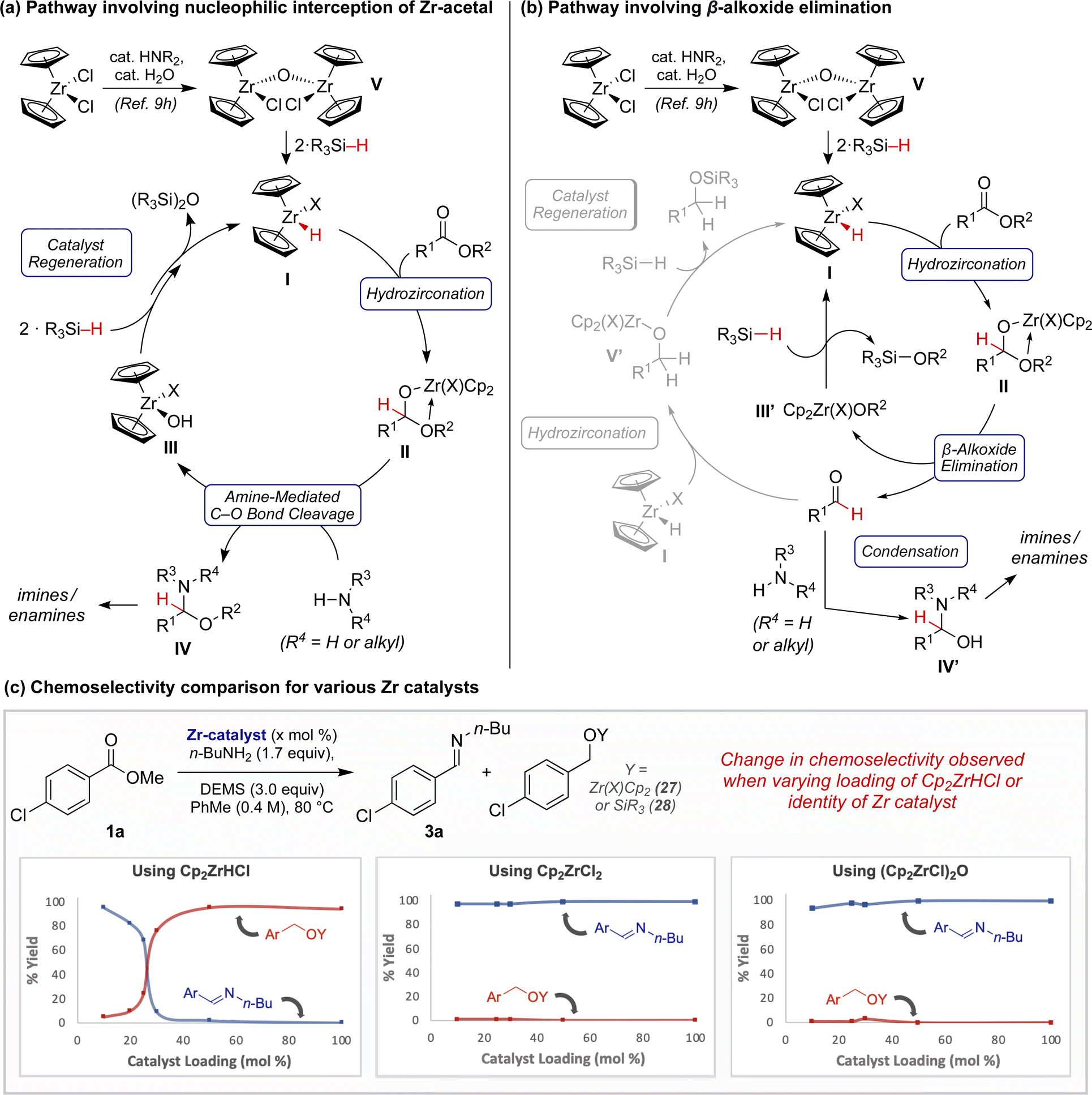
Instead, we hypothesize that the dominant mechanistic pathway leading to product formation could involve zirconocene hemiacetal II (Fig. 1a and b). This species may be directly intercepted by an exogeneous amine, analogous to our prior observations with related hemiaminals (Fig. 1a).9b Alternatively, upon β-alkoxy elimination, interception of the resulting aldehyde by the amine may simply outpace continued reduction to the alcohol (Fig. 1b) (e.g., I to V′),9f,10c In either scenario, the resulting putative hydroxyzirconocene or alkoxyzirconocene species, III/III′, could regenerate active catalyst I through hydrosilane-meditate metathesis.20
 | ||
| Fig. 1 Plausible mechanistic pathways and experimental mechanistic studies. | ||
To gain further mechanistic insight, we performed a series of experiments with varying ZrH sources and loadings. Initial attempts to hydrozirconate 1a using 1.0 equivalent of Cp2ZrHCl in the presence of n-butylamine did not result in the formation of imine 3a, even in trace quantities. Rather, within 30 minutes 1a was iteratively reduced to produce a zirconocene alkoxide, 27 (see Scheme SI–3†). However, while studying the ester reduction at various catalyst loadings, we were perturbed by the distinct differences in reaction outcome (Fig. 1c). Reactions employing ≤25 mol% Cp2ZrHCl exhibited pronounced selectivity for imine 3a, while those employing ≥30 mol% Cp2ZrHCl sharply favored full reduction to 27 and 28. Conversely, this effect was not observed when carrying out an analogous study employing either Cp2ZrCl2 or (Cp2ZrCl)2O (V) pre-catalysts instead. In both studies, the major product observed at all catalyst loadings was imine 3a. These findings suggest that the active catalyst for semi-reduction may involve a ZrH complex I where X ≠ Cl.21 The contrasting product outcomes observed when employing ≥30 mol% of these zirconocene complexes suggests that the identity of the “X” ligand on zirconium may result in impeded rates of catalyst regeneration and/or hydrozirconation.22 Although mechanistic studies are ongoing, at this time we speculate that this “X” ligand could be an alkoxide or siloxide, or that the active catalyst could be dimeric in nature.23
Conclusions
In conclusion, we have developed a series of highly selective and novel reductive transformations of esters enabled by ZrH catalysis. The interrupted catalytic reduction of esters via addition of simple unprotected amines results in the formation of imine and enamine “trapped” intermediates thereby preserving the intermediate oxidation level until workup. The exceptional selectivity of this catalytic strategy was demonstrated through seminal semi-reductive iminations and enaminations of esters in up to 99% yield. Analogously, either monomeric esters or polyester waste materials afforded aldehydes in high yields and with excellent chemoselectivity. α-Alkylated aldehydes and amines are also accessible through single-flask operations via electrophilic or nucleophilic interception of intermediates. Further, we established the reductive amination of esters using primary and secondary amines. Mechanistic studies reveal that the identity of the ZrH-complex has a profound impact on the outcome of these reactions. Additional studies to better understand the mechanism and identity of the active ZrH catalyst are currently underway.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
R. A. Kehner: conceptualization, data curation, investigation, methodology development, and writing the manuscript. W. Huang: data curation, investigation, methodology development, and editing the manuscript. L. Bayeh-Romero: conceptualization, data curation, funding acquisition, investigation, project administration, supervision, and writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support from the Welch Foundation (AA-2077-20210327) and the Cancer Prevention and Research Institute of Texas (CPRIT, RR200039), and for startup funds provided by Baylor University. L. B.-R. is a CPRIT Scholar in Cancer Research. The authors would like to thank R. Loden, S. Phillips and J. Russo for supplying some of the starting materials used in this project. We thank Dr X. Xu (the Center for NMR Spectroscopy, Baylor University, Texas) for technical support. We thank Professors D. Romo and J. L. Wood for access to chemicals and equipment, and Dr J. C. Cooper for insightful discussions regarding this work.Notes and references
- N. Z. Burns, P. S. Baran and R. W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 2854 CrossRef CAS PubMed.
- (a) O. I. Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Chem. Rev., 2019, 119, 11857 CrossRef CAS PubMed; (b) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451 CrossRef CAS PubMed.
- (a) Z. Yang, Org. Chem. Front., 2022, 9, 3908 RSC; (b) R. C. Larock, A. V. Dubrovskiy and N. A. Markina, in Comprehensive Organic Transformations, 2018, DOI:10.1002/9781118662083.cot08-003, p. 1.
- (a) S. Young Kim, Y. Ri Kim, H. Tae Kim, A. Kumar Jaladi and D. Keun An, ChemistrySelect, 2022, 7, e202202351 CrossRef CAS; (b) G. E. Arnott, in Comprehensive Organic Synthesis, ed. P. Knochel, Elsevier, Amsterdam, 2nd edn, 2014, p. 410, DOI:10.1016/B978-0-08-097742-3.00813-2; (c) M. S. Kim, Y. M. Choi and D. K. An, Tetrahedron Lett., 2007, 48, 5061 CrossRef CAS; (d) J. S. Cha and S. S. Kwon, J. Org. Chem., 1987, 52, 5486 CrossRef CAS; (e) M. Muraki and T. Mukaiyama, Chem. Lett., 1975, 4, 215 CrossRef; (f) P. M. Weissman and H. C. Brown, J. Org. Chem., 1966, 31, 283 CrossRef CAS; (g) L. I. Zakharkin and I. M. Khorlina, Tetrahedron Lett., 1962, 3, 619 CrossRef.
- (a) K. Stęsik, A. Franczyk, A. Czapik, I. Kownacki and J. Walkowiak, ChemCatChem, 2023, 15, e202201510 CrossRef; (b) D. Wei, R. Buhaibeh, Y. Canac and J.-B. Sortais, Chem. Commun., 2020, 56, 11617 RSC; (c) S. Pattanaik and C. Gunanathan, Chem. Commun., 2020, 56, 7345 RSC; (d) A. V. Iosub, Š. Moravčík, C.-J. Wallentin and J. Bergman, Org. Lett., 2019, 21, 7804 CrossRef CAS PubMed; (e) V. Rysak, A. Descamps-Mandine, P. Simon, F. Blanchard, L. Burylo, M. Trentesaux, M. Vandewalle, V. Collière, F. Agbossou-Niedercorn and C. Michon, Catal. Sci. Technol., 2018, 8, 3504 RSC; (f) S. Hosokawa, M. Toya, A. Noda, M. Morita, T. Ogawa and Y. Motoyama, ChemistrySelect, 2018, 3, 2958 CrossRef CAS; (g) Y. Corre, V. Rysak, F. Capet, J.-P. Djukic, F. Agbossou-Niedercorn and C. Michon, Chem.–Eur. J., 2016, 22, 14036 CrossRef CAS PubMed; (h) H. Li, L. C. Misal Castro, J. Zheng, T. Roisnel, V. Dorcet, J.-B. Sortais and C. Darcel, Angew Chem. Int. Ed. Engl., 2013, 52, 8045 CrossRef CAS PubMed; (i) C. Cheng and M. Brookhart, Angew. Chem., 2012, 124, 9556 CrossRef; (j) D. Addis, S. Das, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 6004 CrossRef CAS PubMed; (k) D. Depré, A. Horváth, W. Snissaert, L. V. D. Bergh and W. Dermaut, Org. Process Res. Dev., 2008, 12, 96 CrossRef; (l) J. Nakanishi, H. Tatamidani, Y. Fukumoto and N. Chatani, Synlett, 2006, 2006, 869 CrossRef; (m) M. Igarashi, R. Mizuno and T. Fuchikami, Tetrahedron Lett., 2001, 42, 2149 CrossRef CAS; (n) X. Verdaguer, M. C. Hansen, S. C. Berk and S. L. Buchwald, J. Org. Chem., 1997, 62, 8522 CrossRef CAS PubMed; (o) D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440 CrossRef CAS.
- M. P. Sibi, Org. Prep. Proced. Int., 1993, 25, 15 CrossRef CAS.
- E. Mosettig and R. Mozingo, in Organic Reactions, 2011, p. 362, DOI:10.1002/0471264180.or004.07.
- (a) T. Fukuyama and H. Tokuyama, Aldrichimica Acta, 2004, 37, 87 CAS; (b) T. Fukuyama, S. C. Lin and L. Li, J. Am. Chem. Soc., 1990, 112, 7050 CrossRef CAS.
- (a) D. A. Roa and J. J. Garcia, New J. Chem., 2023, 47, 4504 RSC; (b) R. A. Kehner, G. Zhang and L. Bayeh-Romero, J. Am. Chem. Soc., 2023, 145, 4921 CrossRef CAS PubMed; (c) R. A. Kehner, A. E. Lubaev, M. D. Rathnayake, R. Loden, G. Zhang and L. Bayeh-Romero, Tetrahedron, 2023, 133, 133267 CrossRef CAS; (d) J.-T. Tang, Y. Gan, X. Li and B. Ye, Chem, 2023, 9, 869 CrossRef CAS; (e) T. Edlová, A. T. Normand, H. Cattey, S. Brandès, Y. Wu, A. Antonangelo, B. Théron, Q. Bonnin, M. Carta and P. Le Gendre, Organometallics, 2023, 42, 1166 CrossRef; (f) M. Kobylarski, L. J. Donnelly, J.-C. Berthet and T. Cantat, Green Chem., 2022, 24, 6810 RSC; (g) L. J. Donnelly, J.-C. Berthet and T. Cantat, Angew. Chem., Int. Ed., 2022, 61, e202206170 CrossRef CAS PubMed; (h) R. A. Kehner, M. C. Hewitt and L. Bayeh-Romero, ACS Catal., 2022, 12, 1758 CrossRef CAS PubMed; (i) B. Han, J. Zhang, H. Jiao and L. Wu, Chin. J. Catal., 2021, 42, 2059 CrossRef CAS; (j) T. Courant, M. Gavel, R. M. Q. Renard, V. Gandon, A. Y. P. Joosten and T. Lecourt, J. Org. Chem., 2021, 86, 9280 CrossRef CAS PubMed.
- (a) M. Gavel, T. Courant, A. Y. P. Joosten and T. Lecourt, Org. Lett., 2019, 21, 1948 CrossRef CAS PubMed; (b) S. Narasimhan and R. Balakumar, Synth. Commun., 2000, 30, 4387 CrossRef CAS; (c) N. Cénac, M. Zablocka, A. Igau, J.-P. Majoral and A. Skowronska, J. Org. Chem., 1996, 61, 796 CrossRef PubMed.
- For examples using titanium, see: (a) S. C. Berk, K. A. Kreutzer and S. L. Buchwald, J. Am. Chem. Soc., 1991, 113, 5093 CrossRef CAS; (b) M. T. Reding and S. L. Buchwald, J. Org. Chem., 1995, 60, 7884 CrossRef CAS.
- (a) N. M. Hein, Y. Seo, S. J. Lee and M. R. Gagné, Green Chem., 2019, 21, 2662 RSC; (b) Y. Zhang, J. Li, H. Liu, Y. Ji, Z. Zhong and F. Su, ChemCatChem, 2019, 11, 2757 CrossRef CAS; (c) N. J. Lawrence, M. D. Drew and S. M. Bushell, J. Chem. Soc., Perkin Trans. 1, 1999, 1, 3381, 10.1039/a903662h.
- (a) M. Rueping, E. Sugiono, C. Azap, T. Theissmann, A. P. Antonchick, S. Nawaz Khan, N. J. Cho and H.-S. Kim, in Regio- and Stereo- Controlled Oxidations and Reductions, 2007, p. 161, DOI:10.1002/9780470090244.ch4; (b) L. H. Choudhury and T. Parvin, Tetrahedron, 2011, 67, 8213 CrossRef CAS PubMed; (c) P. W. Hickmott, in Enamines, 1994, p. 727, DOI:10.1002/0470024763.ch14; (d) F. Tanaka and C. F. Barbas Iii, in Enantioselective Organocatalysis, 2007, p. 19, DOI:10.1002/9783527610945.ch2a.
- G. Stork, A. Brizzolara, H. Landesman, J. Szmuszkovicz and R. Terrell, J. Am. Chem. Soc., 1963, 85, 207 CrossRef CAS.
- K. V. Khopade, S. H. Chikkali and N. Barsu, Cell Rep. Phys. Sci., 2023, 4, 101341 CrossRef CAS.
- (a) A. Tullo, Chem. Eng. News, 2023, 101, 20 Search PubMed; (b) P. Sarda, J. C. Hanan, J. G. Lawrence and M. Allahkarami, J. Polym. Sci., 2022, 60, 7 CrossRef CAS; (c) M. H. Ghasemi, N. Neekzad, F. B. Ajdari, E. Kowsari and S. Ramakrishna, Environ. Sci. Pollut. Res., 2021, 28, 43074 CrossRef CAS PubMed; (d) Textile Exchange, Preferred Fiber & Materials Market Report, 2022 Search PubMed; (e) R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 Search PubMed.
- For selected examples, see: (a) Z. Ullah, S. Subramanian, H. Lim, N. A. Dogan, J. S. Lee, T. S. Nguyen and C. T. Yavuz, ACS Appl. Mater. Interfaces, 2024 DOI:10.1021/acsami.4c01187; (b) Q. Jiang, X. Xin, S. Zhang, S.-S. Wang, J. Feng and M. Sun, TrAC, Trends Anal. Chem., 2024, 174, 117680 CrossRef CAS; (c) Y. Zhang, G. Lu, D. Zhao and X. Huang, Mater. Chem. Front., 2023, 7, 4782 RSC; (d) M. Yan, Y. Wang, J. Chen and J. Zhou, Chem. Soc. Rev., 2023, 52, 6075 RSC; (e) Q. Guan, L.-L. Zhou and Y.-B. Dong, Chem. Soc. Rev., 2022, 51, 6307 RSC; (f) J. Francis Kurisingal, H. Kim, J. Hyeak Choe and C. Seop Hong, Coord. Chem. Rev., 2022, 473, 214835 CrossRef CAS; (g) E. Troschke, M. Oschatz and I. K. Ilic, Exploration, 2021, 1, 20210128 CrossRef PubMed; (h) K. Liu, L. Wang and R. Dong, J. Mater. Chem. C, 2020, 8, 10696 RSC; (i) P. Taynton, C. Zhu, S. Loob, R. Shoemaker, J. Pritchard, Y. Jin and W. Zhang, Polym. Chem., 2016, 7, 7052 RSC; (j) A. Iwan, Renewable Sustainable Energy Rev., 2015, 52, 65 CrossRef CAS; (k) M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003 RSC.
- D. C. Lenstra, D. T. Nyugen and J. Mecinović, Tetrahedron, 2015, 71, 5547 CrossRef CAS.
- See Supplementary Information† for more details..
- At this time, we have not ruled out the dimerization of III to V via release of H2O as a plausible route to catalyst turnover for a related example, see: E. Samuel, Bull. Soc. Chim. Fr., 1966, 3548 CAS.
- When Cp2ZrH2 was used directly as a catalyst, we failed to observe any appreciable reactivity (see Table SI-4†), suggesting that X ≠ H. This complex has generally been observed to be poorly soluble and less reactive than other zirconocene hydrides see: M. M. Więcław and S. Stecko, Eur. J. Org Chem., 2018, 2018, 6601 CrossRef.
- For an example where an aldehyde intermediate is proposed to undergo condensation at a rate faster than catalyst regeneration and/or hydrozirconation, see ref 9i..
- (a) C. P. Richers, J. A. Bertke and T. B. Rauchfuss, Dalton Trans., 2017, 46, 8756 RSC; (b) P. Perrotin, I. El-Zoghbi, P. O. Oguadinma and F. Schaper, Organometallics, 2009, 28, 4912 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05160b |
| This journal is © The Royal Society of Chemistry 2024 |