
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring efficient and air-stable d2 Re(V) alkylidyne catalysts: toward room temperature alkyne metathesis †
Mingxu
Cui‡
 a,
Jie
Huang‡
a,
Long Yiu
Tsang
a,
Herman H. Y.
Sung
a,
Ian D.
Williams
*a and
Guochen
Jia
*ab
a,
Jie
Huang‡
a,
Long Yiu
Tsang
a,
Herman H. Y.
Sung
a,
Ian D.
Williams
*a and
Guochen
Jia
*ab
aDepartment of Chemistry, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, P. R. China. E-mail: chjiag@ust.hk
bHKUST Shenzhen Research Institute, 518057 Shenzhen, P. R. China
First published on 7th October 2024
Abstract
Transition metal-catalyzed alkyne metathesis has become a useful tool in synthetic chemistry. Well-defined alkyne metathesis catalysts comprise alkylidyne complexes of tungsten, molybdenum and rhenium. Non-d0 Re(V) alkylidyne catalysts exhibit advantages such as remarkable tolerance to air and moisture as well as excellent functional group compatibility. However, the known Re(V) alkylidynes with a pyridine leaving ligand require harsh conditions for activation, resulting in lower catalytic efficiency compared to d0 Mo(VI) and W(VI) alkylidynes. Herein, we report the first non-d0 alkylidyne complex capable of mediating alkyne metathesis at room temperature, namely, the Re(V) aqua alkylidyne complex Re(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH2Ph)(PhPO)2(H2O) (14). The aqua complex readily dissociates a water ligand in solution, confirmed by ligand substitution reactions with other σ-donor ligands. The aqua complex can be readily prepared on a large scale, and is stable to air and moisture in the solid state and compatible with a variety of functional groups. The versatile ability of the catalyst has been demonstrated through examples of alkyne cross-metathesis (ACM), ring-closing alkyne metathesis (RCAM), and acyclic diyne metathesis macrocyclization (ADIMAC) reactions. All in all, this work presents a solution for an efficient and air-stable alkyne metathesis catalytic system based on d2 Re(V)-alkylidynes.
CCH2Ph)(PhPO)2(H2O) (14). The aqua complex readily dissociates a water ligand in solution, confirmed by ligand substitution reactions with other σ-donor ligands. The aqua complex can be readily prepared on a large scale, and is stable to air and moisture in the solid state and compatible with a variety of functional groups. The versatile ability of the catalyst has been demonstrated through examples of alkyne cross-metathesis (ACM), ring-closing alkyne metathesis (RCAM), and acyclic diyne metathesis macrocyclization (ADIMAC) reactions. All in all, this work presents a solution for an efficient and air-stable alkyne metathesis catalytic system based on d2 Re(V)-alkylidynes.
Introduction
Transition metal-catalyzed alkyne metathesis has emerged as a highly valuable synthetic tool in constructing molecules with a C–C triple bond,1 finding increasing applications in both organic synthesis2 and material science.3–7 Highly efficient alkyne metathesis catalysts are usually 4-coordinate tetrahedral alkylidyne complexes of Mo(VI)8,9 and W(VI)10 supported by alkoxide, aryloxide, or silyloxide ligands (Chart 1, top). The low-coordinate and electron-deficient natures of these catalysts make them highly sensitive to moisture, requiring particularly vigorous techniques/conditions for handling and storage. | ||
| Chart 1 Selected structures of d0 Mo/W alkylidyne complexes for alkyne metathesis. | ||
An ideal catalyst should possess three key characteristics:11 (1) it can be easily prepared from cheap commercially available materials; (2) it should be active enough to effect catalytic reactions at ambient or slightly elevated temperatures in a reasonable time period; (3) it should be reasonably stable toward air & moisture and has high functional-group tolerance. Recent advancements in alkyne metathesis catalyst development have shifted focus toward enhancing catalyst stability, crucial for enabling practical handling in synthetic labs without the need for strict air/moisture-free equipment.12 Strategies include utilizing strong σ donating NHC ligands (e.g.5)13 or polydentate ligands (e.g.6,14–167![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17,18) to stabilize low-coordinate and electron-deficient centers of Mo or W. Encapsulation of active species in paraffin wax provided an alternative way to extend their lifetime in air (7).17 Another strategy toward air-stable Mo alkylidynes involves associating N-based sigma-donors like pyridine and 1,9-phenanthroline with the 4-coordinated metal center to form 5- or 6-coordinated alkylidyne complexes (e.g.8),9,19 at the expense of lowering or losing activity. Activation of complex 8 is required (e.g. with a Lewis acid such as MnCl2 or ZnCl2 at 80 °C) prior to use. Achieving both high activity and stability simultaneously is challenging. A recent advancement is the invention of the pyridine-supported catalyst 9, which closely aligns with these goals. With fine-tuning of a tripodal ligand, the pyridine could dissociate under mild conditions.20
17,18) to stabilize low-coordinate and electron-deficient centers of Mo or W. Encapsulation of active species in paraffin wax provided an alternative way to extend their lifetime in air (7).17 Another strategy toward air-stable Mo alkylidynes involves associating N-based sigma-donors like pyridine and 1,9-phenanthroline with the 4-coordinated metal center to form 5- or 6-coordinated alkylidyne complexes (e.g.8),9,19 at the expense of lowering or losing activity. Activation of complex 8 is required (e.g. with a Lewis acid such as MnCl2 or ZnCl2 at 80 °C) prior to use. Achieving both high activity and stability simultaneously is challenging. A recent advancement is the invention of the pyridine-supported catalyst 9, which closely aligns with these goals. With fine-tuning of a tripodal ligand, the pyridine could dissociate under mild conditions.20
On the other hand, the chemistry of low valent middle or late transition metal alkylidyne/carbyne complexes has been well-established21 and some of these complexes exhibit remarkable stabilities toward air and moisture. [2 + 2] cycloaddition reactions of non-d0 metal carbyne complexes with alkynes,22–25 olefins26 or allenes27 have been documented and a few stoichiometric alkyne metathesis reactions of W(IV)28 and Re(V)29 alkylidynes have been observed, implying an alternative approach toward user-friendly alkyne metathesis catalysts. Indeed, our group recently reported a series of well-defined d2 Re(V) alkylidyne catalysts30–32(e.g.10a–c, Scheme 1). These Re(V) alkylidyne complexes, bearing two bulky phosphinophenolate (PO) ligands, induce the coordinated alkyne to align parallel to the alkylidyne ligand due to steric, thus facilitating [2 + 2] cycloaddition.33 These d2 Re(V) alkylidyne catalysts exhibit remarkable advantages such as stability in air, ease of handling, and excellent functional group compatibility. Despite these advantages, their catalytic activities are relatively low. They are inactive at ambient temperature and requires harsh conditions (100–110 °C) for activation, placing this series notably behind d0 Mo(VI) and W(VI) alkylidyne catalysts in terms of efficiency. In this work, we report new Re(V) alkylidyne-based catalysts that show significantly enhanced catalytic activity and can promote catalytic alkyne metathesis reactions at room temperature.
 | ||
| Scheme 1 Examples of d2 Re(V) alkylidyne complexes serving as catalyst precursors for alkyne metathesis and the “bulky PO ligand strategy”. | ||
Results and discussion
Synthesis of Re alkylidyne complexes
Alkyne metathesis reactions catalyzed by 10 involve initial dissociation of a pyridine ligand from 10 to form the 5-coordinated active species Re(CCH2Ph)(RPO)2 (10’). The catalytic activity or the overall kinetic barrier of the catalytic reactions are expected to be influenced by both pyridine dissociation and the metathesis reaction of the active species Re(CCH2Ph)(RPO)2 (10’). In this regard, it is appealing to isolate 5-coordinated complexes Re(CR)(RPO)2, which should be catalytically more active than the pyridine complexes 10. In addition, the availability of such complexes will allow us to reveal the intrinsic barrier for metathesis reaction of active species Re(CCH2Ph)(RPO)2 (10’).
Experimentally, we first attempted to isolate the 5-coordinated species Re(CCH2Ph)(PhPO)2 (10a’) from the readily available complex Re(CCH2Ph)(PhPO)2(PMePh2) (11a), which can be easily prepared in large scale from commercially available phosphine (see ESI†). Despite the fact that ligand substitution of 11a with pyridine can be successfully achieved using CuI as a phosphine scavenger, yielding Re(CCH2Ph)(PhPO)2(py) (10a), essentially no reaction occurred between the complex 11a and CuI in the absence of pyridine. Therefore, a more efficient phosphine scavenger was sought.
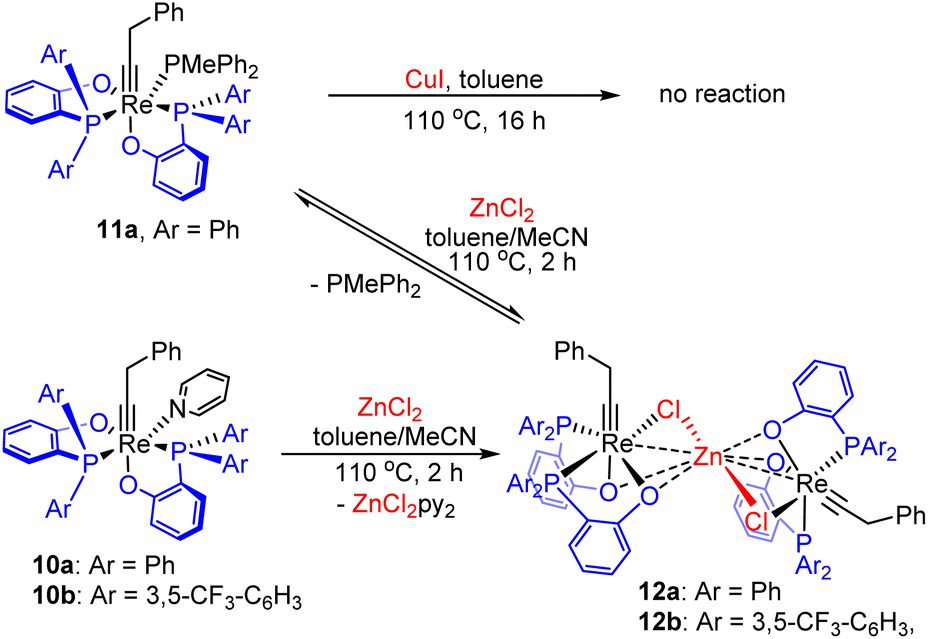
Promising results were obtained with ZnCl2. Heating a mixture of 11a and anhydrous ZnCl2 in a mixed solvent of toluene and acetonitrile (v/v = 3:1) at 110 °C for 2 hours, PMePh2 dissociation occurred, together with the formation of the new Re alkylidyne complex 12a (Scheme 2), as evidenced by the appearance of two broad singlet 31P{1H} signals at 37.6 and 26.2 ppm. However, attempts to isolate the complex failed as a result of the reaction with the released PMePh2 to regenerate 11a during workup. Interestingly, when a mixture of the pyridine complex 10a and ZnCl2 was heated, the same Re alkylidyne complex 12a was also produced (see Fig S4 and S5†). Similarly, the reaction of 10b with ZnCl2 produced analogous complex 12b. It is the formation of stable ZnCl2(pyridine)2 adduct from the dissociated pyridine ligand and ZnCl2 that prevents the reaction from reversing, enabling the isolation of complexes 12. Pale yellow single crystals of 12b were obtained, enabling the determination of its structure by single crystal X-ray diffraction.
 | ||
| Scheme 2 Attempted extraction of PMePh2 from Re(CCH2Ph)(ArPO)2(PMePh2) with 3d transition metal salts. | ||
The solid-state structure of 12b reveals a confacial trioctahedral Re–Zn–Re complex (Fig. 1). This structure can be regarded as two Re(CCH2Ph)(ArPO)2 fragments stabilized by ZnCl2 through chloride coordination. The Re(1)–Cl(1) and Re(2)–Cl(2) bond distances are 2.4798(13) Å and 2.5031(13) Å, respectively. The central Zn atom is 6-coordinated, with an average Zn–O bond length of 2.109 Å. The Zn(1)–Cl(1) and Zn(1)–Cl(2) bonds (2.4356(16) Å and 2.4377(16) Å) are significantly elongated from normal Zn–Cl bonds.34 The chelating effect of phenolates together with a strong affinity of Zn for O, stabilize the ZnCl2 bridge, allowing the isolation of 12. However, it was found experimentally that the catalytic activity of 12a in metathesis reactions is very low (entry 2, Table 1), consistent with the strong coordination of ZnCl2 by the chelating oxygen ligands.
 | ||
| Fig. 1 Top: ORTEP diagram of the confacial trioctahedral complex 12b with thermal ellipsoids at the 40% probability level. The solvent molecule as well as all hydrogen atoms are omitted for clarity. Bottom: selected bond lengths (Å) and angles (deg) of 12b. Only the core atoms are shown for clarity. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | T/°C | Time/h | Yield/% |
| a Standard condition: 1-methoxy-4-(1-propyn1-yl)benzene (17, 0.3 mmol), catalyst (5 mol%), 5 Å MS (450 mg), dry toluene (3 mL). The yields were determined by 1H NMR using CH2Ph2 as an internal standard. b 0.5 mol% catalyst loading. | ||||
| 1 | 10a | 60 | 16 | Trace |
| 2 | 12a | 100 | 8 | 14 |
| 3 | 14 | 24 | 16 | 92 |
| 4 | 14 | 60 | 1 | >95 |
| 5 | 14 | 60 | 3 | 84 |
| 6 | 15 | 24 | 16 | 85 |
| 7 | 16 | 60 | 16 | 58 |
| 8 | 15 | 24 | 1 | 95 |
| 9 | 16 | 60 | 1 | 92 |
Taking a lesson from the formations of 12, we searched for more efficient phosphine scavengers among coordinatively unsaturated 4d and 5d late transition metal complexes, as they possess a stronger affinity for phosphorous but a weaker affinity for oxygen. This may ensure efficient trapping of the PMePh2 ligand without forming stable adducts with Re(V) alkylidynes, as observed in compounds 12.
In dry toluene or benzene, complex 11a reacted with one equivalent of [(p-cymene)RuCl2]2, resulting in the formation of the expected complex (p-cymene)RuCl2(PMePh2) together with a rhenium alkylidyne complex. The latter exhibits two singlet 31P{1H} signals at 40.1 and 30.5 ppm, as well as characteristic doublets of triplets 1H signals at 2.40 and 2.92 ppm for a ReCCH2 group and four characteristic doublet 1H signals of coordinated p-cymene (see Fig. S6 and S7† for details). Based on the NMR data and reactivity (see below), we tentatively assigned the new alkylidyne complex as the dinuclear complex 13, wherein the Re(CCH2Ph)(PhPO)2 fragment is stabilized by (p-cymene)RuCl2 through chloride coordination (Fig. 2).
 | ||
| Fig. 2 (a) Synthesis of the aqua complex 14. (b) Ligand substitution reactions of 14 with methyl thioether and 2-picoline. (c) Photograph of the aqua complex 14 stored under air atmosphere for 3 months. | ||
Efforts to isolate pure complex 13 were unsuccessful due to its high moisture sensitivity. The species reacted readily with a trace amount of water, yielding the aqua complex Re(CCH2Ph)(PhPO)2(H2O) (14, Fig. 2). After allowing an NMR tube containing 13 to stand at room temperature overnight, yellow crystals of the aqua complex 14 deposited on the wall due to the involvement of trace moisture. Alternatively, gram-scale preparation of the aqua complex 14 can be achieved with a high yield by heating the complex 11a with [(p-cymene)RuCl2]2 and water in toluene. The pure product of 14 was obtained as an air and moisture stable yellow powder (Fig. 2c).
The structure of 14 has been confirmed by NMR, X-ray diffraction, and elemental analysis. As shown in Fig. 3, the complex adopts a distorted octahedral geometry bearing two cis PO ligands, similar to complexes 11a and 10a. The aqua ligand is cis to the alkylidyne ligand and trans to a phosphine. The Re(1)–P(2) bond (trans to the Re–OH2 bond) has a bond length of 2.3553(11) Å, which is shorter than the corresponding bonds in 11a (2.3722(5) Å) and 10a (2.4109(6) Å), implying that the trans influence of OH2 is less than that of PMePh2 and pyridine, hinting that the Re–OH2 bond might be relatively weak. Consistent with the solid-state structure, the 31P{1H} NMR spectrum of 14 showed two singlets at 23.4 and 38.2 ppm; the 1H NMR spectrum shows two multiplet signals at 2.65 and 1.32 ppm for the ReCCH2 group and a singlet at 7.84 ppm for the bound H2O. The compound 14 is only sparingly soluble in non-coordinating organic solvents such as benzene and dichloromethane, preventing the recording of the 13C NMR spectrum.
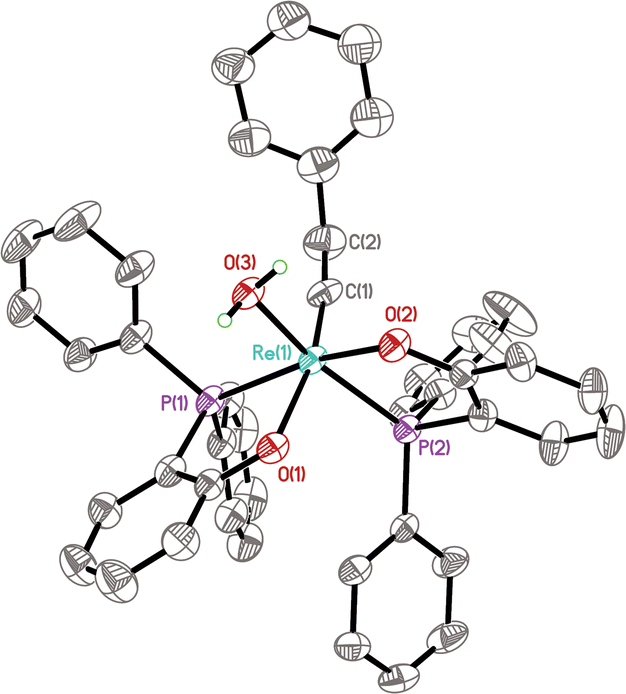 | ||
| Fig. 3 ORTEP diagram of the aqua complex 14 with thermal ellipsoids at the 40% probability level. All hydrogen atoms except those of the water ligand are omitted for clarity. Selected bond lengths (Å) and angles (deg): Re(1)–C(1) 1.759(5), Re(1)–O(3) 2.173(3), Re(1)–P(1) 2.3312(11), Re(1)–P(2) 2.3553(11), O(3)–Re(1)–P(2) 161.32(8). | ||
The isolation of the aqua complex 14 confirms that Re(V) alkylidynes are indeed robust toward water. It's worth noting that structurally characterized aqua–alkylidyne complexes are still rare.35 The Re(VII) aqua alkylidyne complex [Re(C-tBu)(CH2-tBu)3(H2O)]+[BArF4]− has been previously reported by Schrock and his co-workers, although the structure of this complex was not confirmed by X-ray diffraction.36
The isolation of the ZnCl2-bridged complexes 12, the aqua complex 14 as well as the observation of the bimetallic complex 13 suggests that Re(V) alkylidynes bearing two PO ligands have a strong tendency to be 6-coordinated, even by incorporating a chloride salt or complex. As a result, attempts to isolate the 5-coordinated active species 10’ have been so far unsuccessful.
Echoing the weak Re–OH2 bond indicated by the X-ray data, the aqua ligand of 14 is quite labile and can be easily substituted by other two-electron σ donors at room temperature. Utilizing this advantage, we can obtain other adducts of Re(CCH2Ph)(PhPO)2 that cannot be synthesized before. For instance, the SMe2 adduct 15 could be quantitatively formed by treating the aqua complex 14 with 3 equivalents of methyl thioether at room temperature. Analogously, stirring a suspension of 14 with excess 2-picoline in toluene led to the quantitative formation of the 2-picoline complex 16, which was isolated as a yellow solid in high yield (Fig. 2). Stirring 14 in O-donor solvents like ethers or acetone resulted in messy mixtures, and their structures remain unidentified.
New complexes 15 and 16 have been characterized by multinuclear NMR spectroscopy. The structure of the SMe2 complex 15 has also been characterized by single crystal X-ray diffraction analysis (Fig. 4).
 | ||
| Fig. 4 ORTEP diagram of the SMe2 complex 15 with thermal ellipsoids at the 40% probability level. All hydrogen atoms as well as solvent atoms are omitted for clarity. Selected bond lengths (Å) and angles (deg): Re(01)–C(1) 1.7690(18), Re(01)–S(1) 2.4524(4), Re(01)–P(1) 2.3370(4), Re(01)–P(2) 2.3811(4), S(1)–Re(01)–P(2) 164.018(15). | ||
Benchmarking of alkyne metathesis
The lower affinity of OH2 compared with pyridine and PMePh2 for the Re(V) center suggests that the aqua complex 14 might be a more active alkyne metathesis catalyst than the phosphine complex 11a and the pyridine complex 10a. To compare the activity of the new complex 14 with those of the phosphine complex 11a and the pyridine complex 10a, we conducted kinetic studies on the model reaction, the homometathesis of 1-methoxy-4-(1- propyn-1-yl)benzene (17), catalyzed by 5 mol% complexes 14, 11a and 10a at 100 °C, respectively, with 5 Å molecular sieves (MS) added as butyne scavenger.The kinetic plots are presented in Fig. 5. Surprisingly, the aqua complex 14 exhibits significantly improved catalytic activity compared to 11a and 10a and the model reaction catalyzed by 14 was completed within 5 min (red line). In contrast, the reaction with the phosphine complex 11a (olive line) and the pyridine complex 10a (blue line) achieved conversions of less than 5% and ca. 50%, respectively, after 2 h.
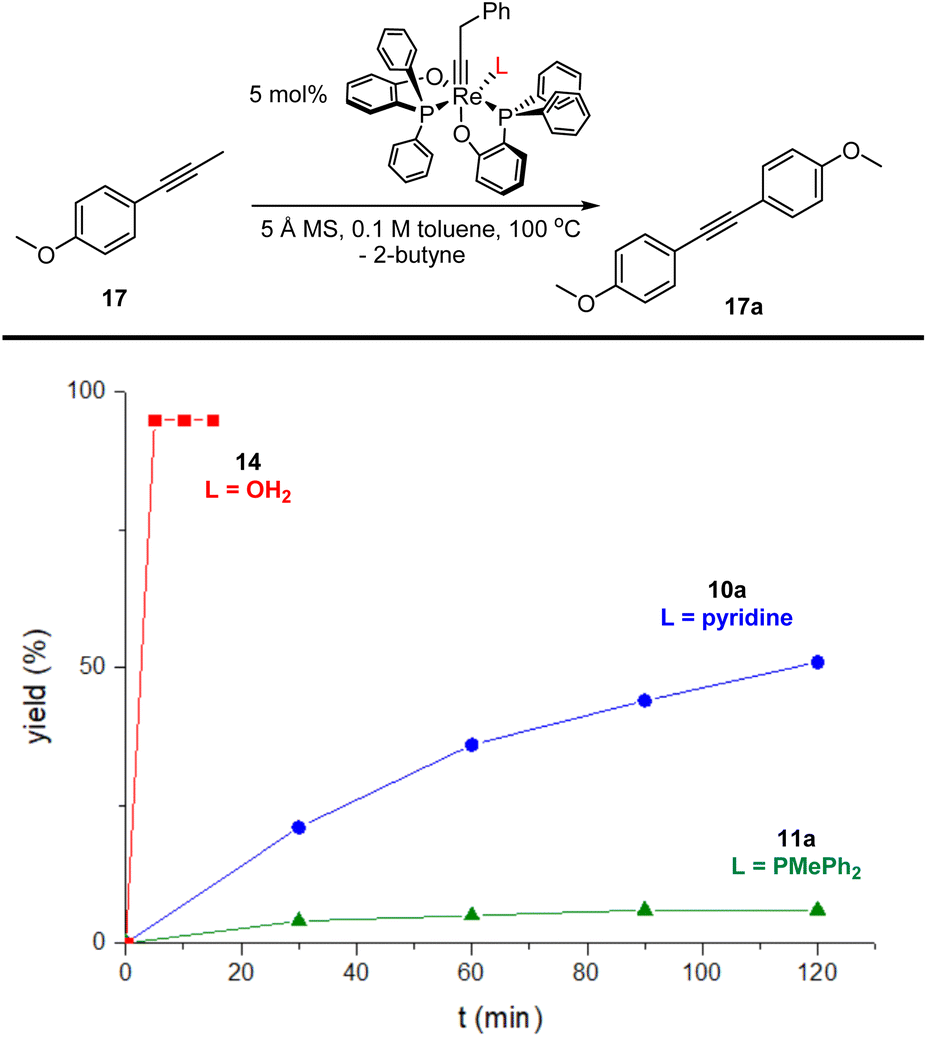 | ||
| Fig. 5 Kinetic plots of homometathesis of 1-methoxy-4-(1-propyn-1-yl)benzene (17) mediated by catalysts 10a (blue line), 11a (olive line) and 14 (red line). Condition: substrate (17, 0.3 mmol), catalyst (5 mol%), 5 Å MS (450 mg), dry toluene (3 mL), 100 °C. The yields were determined by 1H NMR using CH2Ph2 as an internal standard. | ||
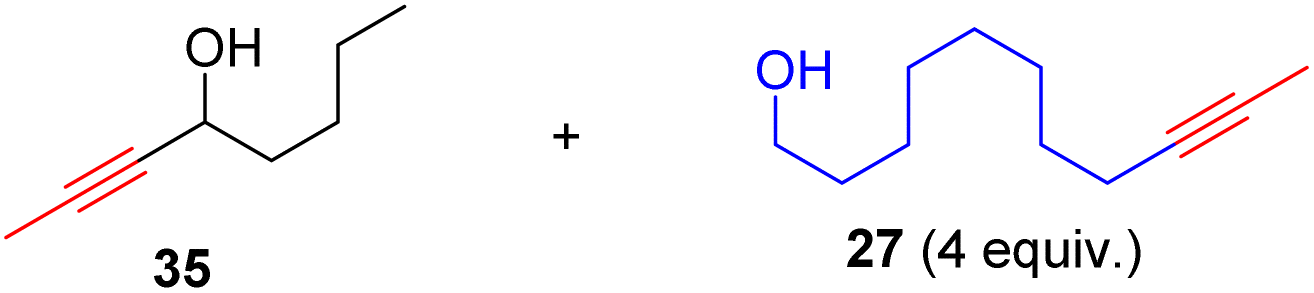
To our delight, the aqua complex 14 is catalytically active even at room temperature (Table 1, entry 3): The model reaction promoted by 5 mol% of aqua complex 14 proceeded to 92% conversion in 16 hours at room temperature (24 °C). This result is interesting as it represents the first demonstrated room temperature alkyne metathesis reaction catalyzed by a non-d0 alkylidyne complex. The activity of 14 increased significantly when slightly heated; the model reaction achieved full conversion within 1 hour at 60 °C (entry 4). With a loading of 0.5 mol% of the catalyst, the reaction at 60 °C achieved a conversion of 84% in 3 hours (entry 5), indicating that 14 is an catalyst with sufficiently high TON. The SMe2 adduct 15 and the 2-picoline adduct 16 are also active at ambient temperature. The model reaction mediated by 15 and 16 yielded homometathesis product in 58% and 85% yields after 16 h at 24 °C. The results indicated that 15 and 16 are also efficient catalysts with activity similar to or slightly less than the aqua complex 14.
Substrate scope
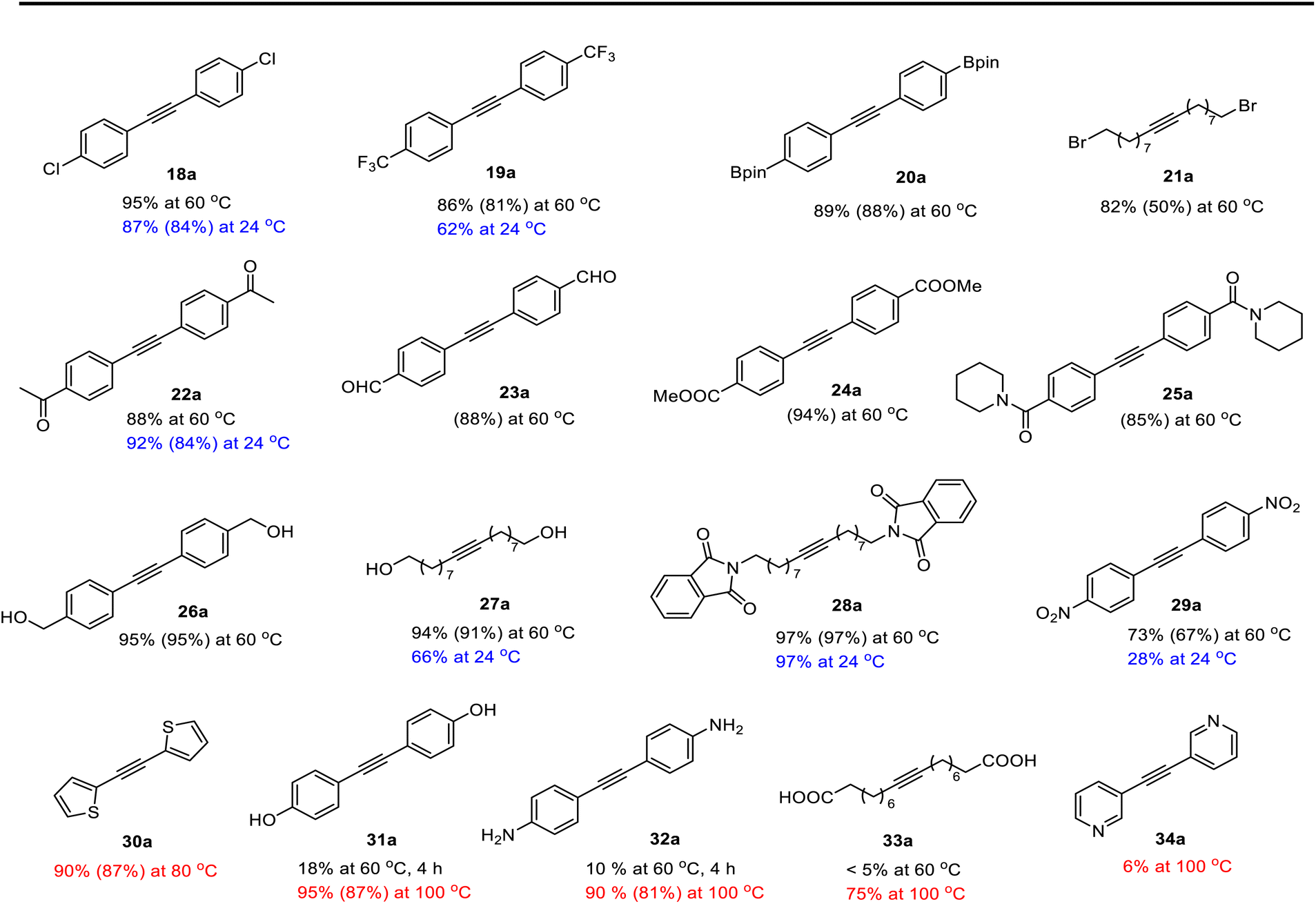
To evaluate the functional group tolerance of the readily-available and user-friendly Re(V) catalyst 14, we have investigated its capability in homometathesis reactions of a series of functionalized methyl-capped alkynes, as illustrated in Table 2. Metathesis reactions were typically carried out under an inert atmosphere with a 2 mol% catalyst loading. 5 Å molecular sieves (MS) were added as a 2-butyne abstractor to facilitate the reactions to go completion. The reactions were usually carried out at 60 °C. For some of the substrates, the reactions were also carried out at 24, 80 or 100 °C, which are color-labeled. Noteworthy is that those reactions could also proceed well in unpurified solvent by applying a higher catalyst loading (5 mol%) (see General Procedure B in the ESI†).|
|
|---|
| a Standard condition: substrate (0.3 mmol), catalyst (2–5 mol%), 5 Å MS (450 mg), dry toluene (3 mL). The yields were determined by 1H NMR using CH2Ph2 as an internal standard. Isolated yields are given in parathesis. Reaction temperatures are color-labeled: blue: 24 °C, 16 h; black: 60 °C, 1–12 h; red: 80 °C or 100 °C, 4–16 h. |
|
As anticipated, the new catalyst demonstrates outstanding compatibility with diverse functional groups. Substrates featuring halide, ether, trifluoromethyl, ketone, ester, boric ester, amine, and amide functionalities exhibit favorable reactivity. Notably, substrates containing aldehyde groups (e.g.23), known to present difficulties in many catalytic systems,9,14,15,20 undergo successful metathesis with the complex 14. The catalyst also works well for substrates with protic hydrogen groups. Primary alcohols (e.g.26, 27) undergo smooth metathesis, and even more acidic functional groups such as phenol (31) and carboxylic acid (33) are well-tolerated. This tolerance can be attributed to the low affinity of the Re(V) center for oxygen donors. Electron-deficient alkynes, such as 29, are known to form more stable alkylidyne complexes with Re(V) centers,29 leading to slower reaction rates at ambient temperature. However, when the reaction was conducted at 60 °C, it proceeded more efficiently and achieved promising yield.
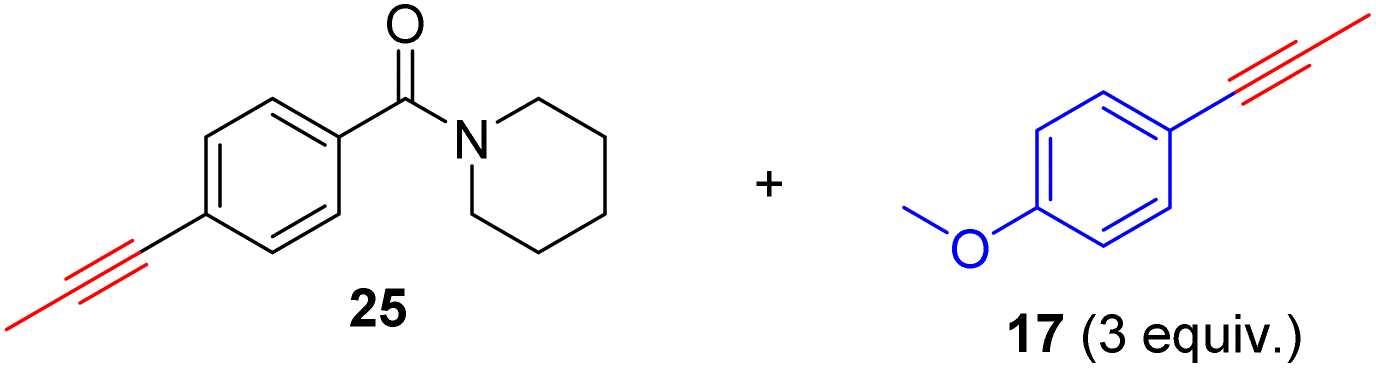
As previously mentioned, alkyne metathesis reactions mediated by 14 presumably proceed though alkyne-alkylidyne intermediates Re(CCH2Ph)(PhPO)2(η2-alkyne) formed by displacement of the labile H2O ligand in 14. Consequently, the rates or kinetic barriers of alkyne metathesis reactions are closely related to or governed by the coordination ability of functional groups present in the substrates. Thus, reactions involving substrates with a weakly coordinating functional group (e.g., Table 2, 18–28) can achieve almost full conversion either at room temperature (24 °C) in 16 hours or at 60 °C within 1–8 hours. In contrast, substrates with a strongly coordinating functional group have reduced activity, due to competing coordination of the alkyne and the coordinating functional group. A higher temperature is required for such substrates to achieve a high conversion in a reasonably short period of time. For example, the homometathesis of 2-thiophenyl-1-propyne (30) progressed slowly at 60 °C due to competing S coordination, but can complete at 80 °C within 8 hours to produce 30a in 90% yield. As additional examples, homometathesis of 4-propynyl aniline (32) proceeded slowly at 60 °C (10% in 4 h) and requires a reaction temperature of 100 °C to go completion in 8 h, due to competing NH2 coordination; homometathesis of the sterically unshielded pyridine substrate (34) yields only 6% of compound 34a even after heating at 100 °C for 16 hours due to the coordination of pyridine. In contrast, sterically hindered amides, such as 25 and 28, readily undergo metathesis, even at room temperature. Substrates with strong acidic functional groups also slow down metathesis reactions.
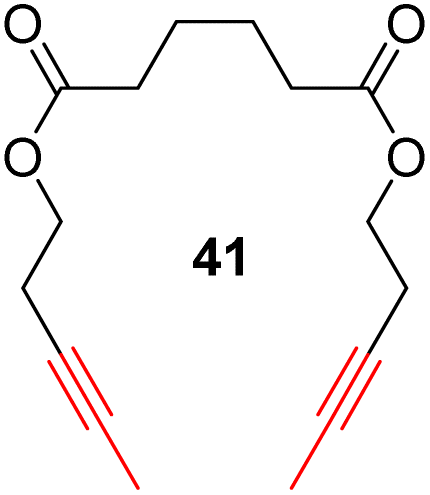
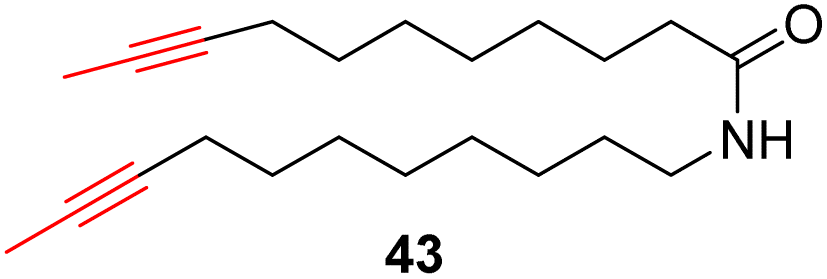
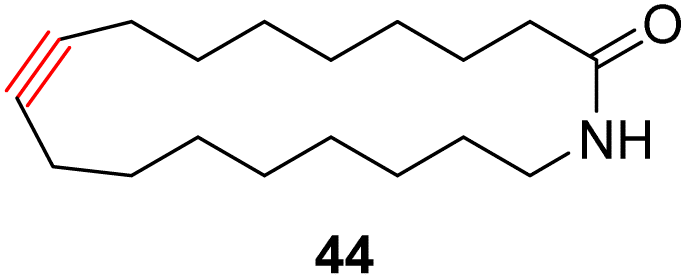
With the well-behaved alkyne metathesis catalyst 14 in hand, we have performed a series of representative alkyne metathesis reactions to show its applications in diverse scenarios (Table 3). The complex 14 is able to catalyze cross-metathesis (ACM) between two aliphatic alkynes (entries 1 and 4), two aromatic alkynes (entry 2), as well as an aliphatic and an aromatic alkynes (entry 3) to give crossed products in high yields. Entry 4 shows that the catalyst works well for a bio-active substrate, providing a new route to aliphatic alkyne modification. The catalyst also works well in ring-closing alkyne metathesis (RCAM) reactions, giving cyclic products in good to excellent yields (entries 5 and 6). The catalyst can also mediate acyclic diyne metathesis macrocyclization (ADIMAC). For example, the bispropynylated carbazole derivative 45 was efficiently converted to the macrocycle 46 (entry 7), demonstrating the potential usage of the catalytic system in constructing molecular rings and cages.

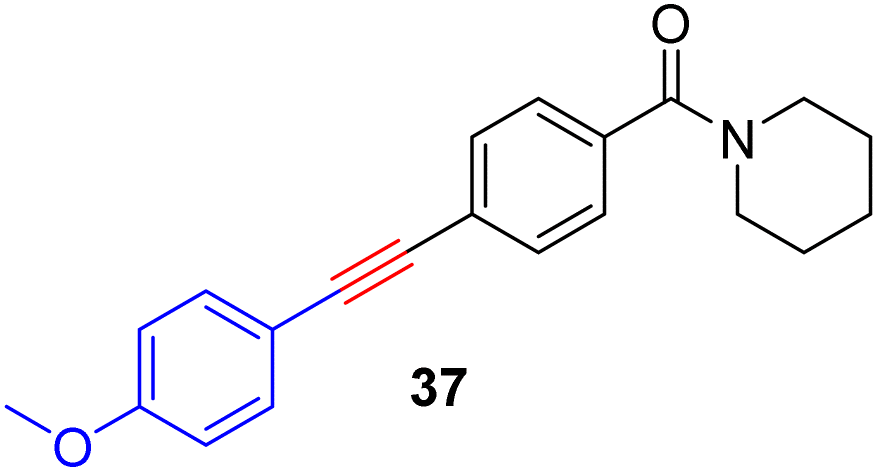
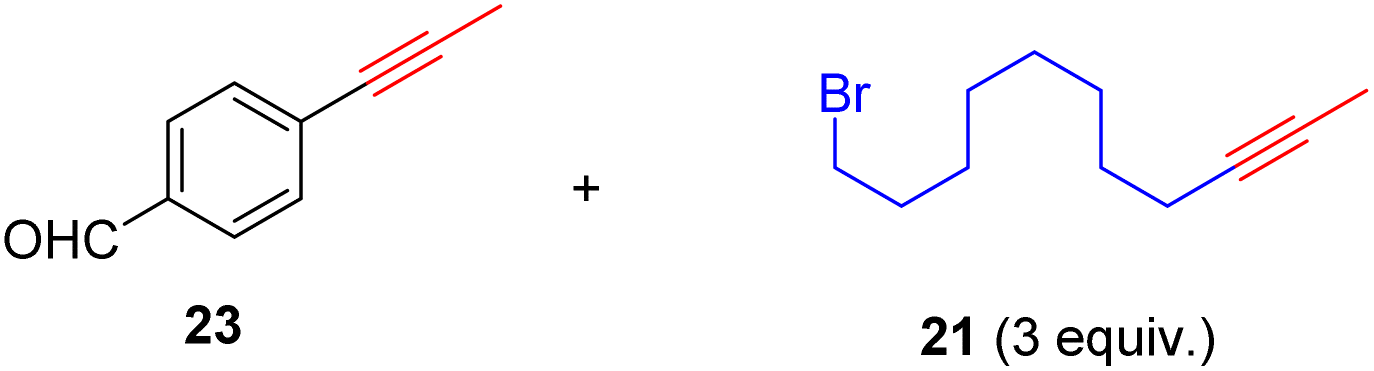
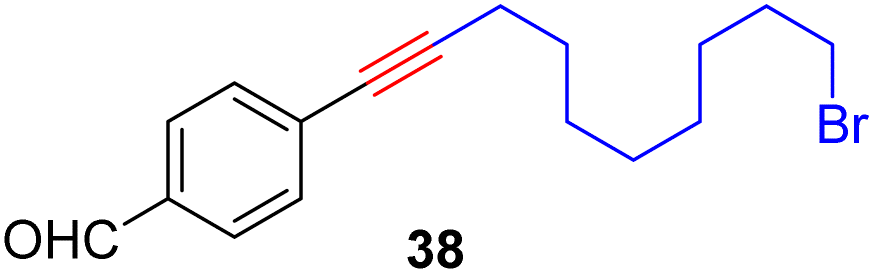

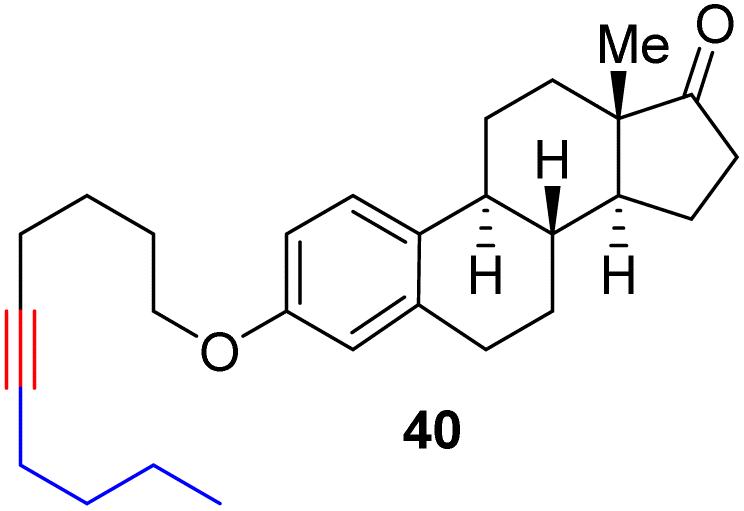



Conclusions
In this work, we have presented the first non-d0 alkylidyne complex capable of effecting alkyne metathesis at room temperature, namely, the Re(V) alkylidyne complex Re(CCH2Ph)(PhPO)2(H2O) (14), featuring a H2O leaving ligand. Analogous complexes with labile ligands such as SMe2 (15) and 2-picoline (16) also show remarkable activity in mediating alkyne metathesis at moderate temperatures. The d2 Re(V)-alkylidyne catalysts now exhibits activity comparable with or approaching to those of d0 Mo(VI)/W(VI) alkylidyne catalytic systems. This study also implies that the impact of metal d electrons on the kinetic barrier of [2 + 2] cycloaddition of a non-d0 alkylidyne and an alkyne can be significantly diminished by employing suitable ligands.
The aqua complex 14 is stable to air and moisture in the solid state, enabling storage and handling in air. It can efficiently promote alkyne homometathesis, cross-metathesis (ACM), ring-closing alkyne metathesis (RCAM), as well as acyclic diyne metathesis macrocyclization (ADIMAC) reactions, and tolerate a broad range of functional groups. Last but not least, the complex 14 can be readily prepared on a large scale from commercially available starting materials. Thus it represents a practical alkyne metathesis catalytic system based on d2 Re(V)-alkylidynes.
Data availability
Crystallographic data for 12b·2CHCl3 (CCDC no. 2360919), 14·0.5C7H8 (CCDC no. 2289258), and 15 (CCDC no. 2360920) have been deposited at The Cambridge Crystallographic Data Centre viahttps://www.ccdc.cam.ac.uk/structures/.Author contributions
G. J. and M. C. conceived the project. M. C. and J. H. performed synthetic experiments. L. Y. T. contributed to scope studies. H. H. Y. S. and I. D. W. collected and refined the X-ray data. M. C. and G. J. wrote the manuscript. G. J. supervised the project. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare the following competing financial interest: A provisional patent application has been filed.Acknowledgements
This work was supported by the National Natural Science Foundation of China (project no.: 21971218) and the Hong Kong Research Grants Council (project no.: 16308721, 16302322).Notes and references
- Selected reviews:
(a) M. Cui and G. Jia, J. Am. Chem. Soc., 2022, 144, 12546 CrossRef CAS PubMed
; (b) A. Fürstner, J. Am. Chem. Soc., 2021, 143, 15538 CrossRef PubMed
- Recent examples:
(a) J. L. Sutro and A. Fürstner, J. Am. Chem. Soc., 2024, 146, 2345 CrossRef CAS
- Selected reviews on diyne metathesis polymerization:
(a) H. Yang, Y. Jin, Y. Du and W. Zhang, J. Mater. Chem. A, 2014, 2, 5986 RSC
- Selected examples of ring-opening alkyne metathesis polymerization:
(a) S. von Kugelgen, R. Sifri, D. Bellone and F. R. Fischer, J. Am. Chem. Soc., 2017, 139, 7577 CrossRef CAS
- H. Jeong, S. von Kugelgen, D. Bellone and F. R. Fischer, J. Am. Chem. Soc., 2017, 139, 15509 CrossRef CAS PubMed
- Selected examples of acyclic diyne metathesis macrocyclization:
(a) S. Huang, Z. Lei, Y. Jin and W. Zhang, Chem. Sci., 2021, 12, 9591 RSC
- Z. Lei, H. Chen, S. Huang, L. J. Wayment, Q. Xu and W. Zhang, Chem. Rev., 2024, 124, 7829 CrossRef CAS PubMed
- Selected pioneering works:
(a) L. G. McCullough and R. R. Schrock, J. Am. Chem. Soc., 1984, 106, 4067 CrossRef CAS
-
(a) J. Heppekausen, R. Stade, R. Goddard and A. Fürstner, J. Am. Chem. Soc., 2010, 132, 11045 CrossRef CAS PubMed
- Selected pioneering works:
(a) J. H. Wengrovius, J. Sancho and R. R. Schrock, J. Am. Chem. Soc., 1981, 103, 3932 CrossRef CAS
- U. H. F. Bunz, Science, 2005, 308, 216 CrossRef CAS
-
(a) Y. Ge, Y. Hu, G. Duan, Y. Jin and W. Zhang, Trends Chem., 2022, 4, 540 CrossRef CAS
-
(a) J. V. Musso, V. Gramm, S. Stein, W. Frey and M. R. Buchmeiser, Eur. J. Inorg. Chem., 2022, 26, e202200649 CrossRef
-
(a) J. Hillenbrand, M. Leutzsch, E. Yiannakas, C. P. Gordon, C. Wille, N. Nöthling, C. Copéret and A. Fürstner, J. Am. Chem. Soc., 2020, 142, 11279 CrossRef CAS PubMed
-
(a) R. R. Thompson, M. E. Rotella, X. Zhou, F. R. Fronczek, O. Gutierrez and S. Lee, J. Am. Chem. Soc., 2021, 143, 9026 CrossRef CAS PubMed
- Other examples with a tripodal ligand:
(a) J. Hillenbrand, M. Leutzsch, C. P. Gordon, C. Copéret and A. Fürstner, Angew. Chem., Int. Ed., 2020, 59, 21758 CrossRef CAS
- Y. Ge, S. Huang, Y. Hu, L. Zhang, L. He, S. Krajewski, M. Ortiz, Y. Jin and W. Zhang, Nat. Commun., 2021, 12, 1136 CrossRef CAS PubMed
-
(a) Y. Du, H. Yang, C. Zhu, M. Ortiz, K. D. Okochi, R. Shoemaker, Y. Jin and W. Zhang, Chem.–Eur. J., 2016, 22, 7959 CrossRef CAS PubMed
- M. Bindl, R. Stade, E. K. Heilmann, A. Picot, R. Goddard and A. Fürstner, J. Am. Chem. Soc., 2009, 131, 9468 CrossRef CAS PubMed
- J. N. Korber, C. Wille, M. Leutzsch and A. Fürstner, J. Am. Chem. Soc., 2023, 145, 26993 CrossRef CAS PubMed
- Selected reviews see:
(a) H. P. Kim and R. J. Angelici, Adv. Organomet. Chem., 1987, 27, 51 CrossRef CAS
-
(a) C. Zhu, J. Zhu, X. Zhou, Q. Zhu, Y. Yang, T. B. Wen and H. Xia, Angew. Chem., Int. Ed., 2018, 57, 3154 CrossRef CAS PubMed
-
U. Das, PhD thesis, University of Bonn Mensch & Buch Verlag, Berlin, ISBN: 978-3-86387-551-0, 2014
- H. Fischer and C. Troll, J. Chem. Soc. Chem. Commun., 1994, 457 RSC
- A. Mayr, K. S. Lee and B. Kahr, Angew. Chem., Int. Ed., 1988, 27, 1730 CrossRef
- M. A. Esteruelas, A. I. González, A. M. López and E. Oñate, Organometallics, 2004, 23, 4858 CrossRef CAS
- J. Rao, S. Dong, C. Yang, Q. Liu, X. Leng, D. Wang, J. Zhu and L. Deng, J. Am. Chem. Soc., 2023, 145, 25766 CrossRef CAS
-
(a) L. M. Atagi, S. C. Critchlow and J. M. Mayer, J. Am. Chem. Soc., 1992, 114, 9223 CrossRef CAS
-
(a) W. Bai, K.-H. Lee, H. H. Y. Sung, I. D. Williams, Z. Lin and G. Jia, Organometallics, 2016, 35, 3808 CrossRef CAS
- M. Cui, H. H. Y. Sung, I. D. Williams and G. Jia, J. Am. Chem. Soc., 2022, 144, 6349 CrossRef CAS PubMed
- M. Cui, W. Bai, H. H. Y. Sung, I. D. Williams and G. Jia, J. Am. Chem. Soc., 2020, 142, 13339 CrossRef CAS PubMed
- L. Y. Tsang, W. Bai, L. C. Kong, H. H. Y. Sung, I. D. Williams and G. Jia, Organometallics, 2024, 43, 849 CrossRef CAS
- For computational studies, see: M. Tomasini, M. Gimferrer, L. Caporaso and A. Poater, Inorg. Chem., 2024, 63, 5842 CrossRef CAS PubMed
-
(a) J. Qin, N. Su, C. Dai, C. Yang, D. Liu, M. W. Day, B. Wu and C. Chen, Polyhedron, 1999, 18, 3461 CrossRef CAS
- Examples see:
(a) W. Bai, K.-H. Lee, W. Y. Hung, H. H. Y. Sung, I. D. Williams, Z. Lin and G. Jia, Organometallics, 2017, 36, 657 CrossRef CAS
- A. M. LaPointe and R. R. Schrock, Organometallics, 1995, 14, 1875 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2360919, 2289258 and 2360920. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05369a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |