
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulatory spin-flip of triplet excitons via diversiform electron-donating units for MR-TADF emitters towards solution-processed narrowband OLEDs†
Shengyu
Li
a,
Zhi
Yang
a,
Yanchao
Xie
a,
Lei
Hua
b,
Shian
Ying
 a,
Yuchao
Liu
*a,
Zhongjie
Ren
c and
Shouke
Yan
*ac
a,
Yuchao
Liu
*a,
Zhongjie
Ren
c and
Shouke
Yan
*ac
aDepartment Key Laboratory of Rubber-Plastics, Ministry of Education/Shandong Provincial Key Laboratory of Rubber-Plastics, School of Polymer Science and Engineering, Qingdao University of Science & Technology, Qingdao 266042, P. R. China. E-mail: liuyc@qust.edu.cn; skyan@qust.edu.cn
bSchool of Materials Science & Engineering, Changzhou University, Changzhou 213164, P. R. China
cState Key Laboratory of Chemical Resource Engineering, College of Materials Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China
First published on 4th October 2024
Abstract
Multiple resonance thermally activated delayed fluorescence (MR-TADF) molecules are emerging as promising candidates for high-resolution organic light-emitting diode (OLED) displays, but MR-TADF emitters always suffer from an unsatisfactory rate constant of reverse intersystem crossing (kRISC) due to inherently low spin orbital coupling strength between excited singlet and triplet states. Herein, we systematically investigate the long-range charge transfer (LRCT) and heavy-atom effects on modulating the excited state natures and energy levels via integrating diversiform electron-donating units with the MR skeleton. Compared with unsubstituted analogues, newly designed MR-TADF emitters exhibit significantly boosted kRISC values and close-to-unity photoluminescence quantum yield especially for tBuCzBN-PXZ (2.5 × 105 s−1) and tBuCzBN-Ph-PSeZ (2.1 × 105 s−1). Leveraging these exceptional properties, the maximum external quantum efficiency values of tBuCzBN-PXZ- and tBuCzBN-Ph-PSeZ-based solution-processed OLEDs can reach 21.3% and 19.4%, which are in the first tier of reported solution-processed MR-TADF binary OLEDs without employing additional sensitizers. This study provides a framework for modulating photoelectrical properties of MR-TADF emitters through fastidiously regulating LRCT and heavy-atom effects.
Introduction
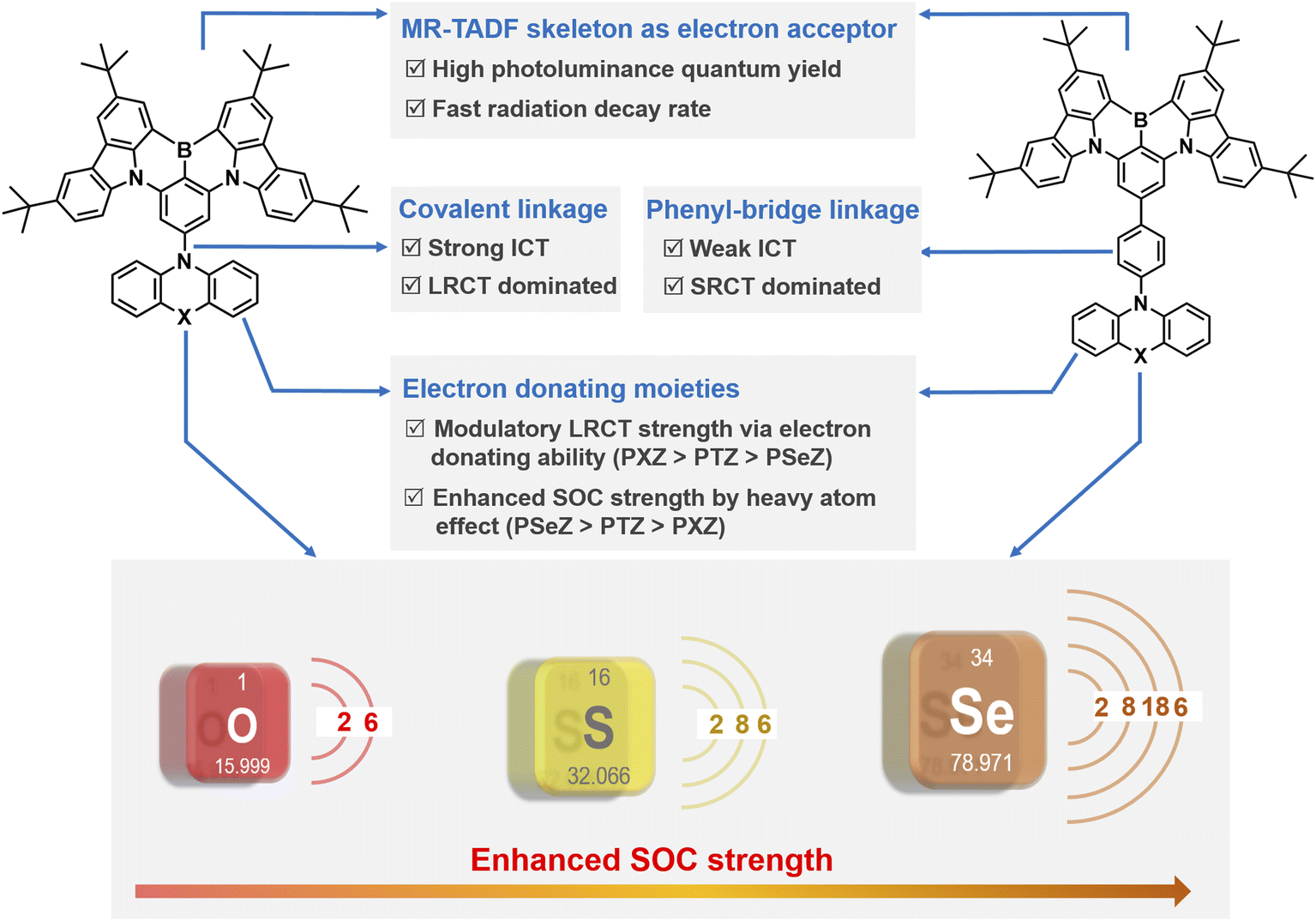
Thermally activated delayed fluorescence (TADF) emitters have been regarded as some of the most promising emitting materials for organic light-emitting diodes (OLEDs) due to simultaneously harvesting electro-generated singlet and triplet excitons and reaching a near-unity internal quantum efficiency.1–6 Traditionally, a small singlet–triplet splitting energy (ΔEST) between the lowest excited singlet state (S1) and triplet state (T1) is a prerequisite to constructing TADF molecules, and thus dark T1 excitons can be effectively harvested via the endothermic reverse intersystem crossing (RISC) process.7–10 Although the spatially separated donor–acceptor (D–A) architectures in conformist TADF molecules can attain small enough ΔEST values by minimizing the overlap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), the inherent intramolecular charge transfer (ICT) characteristic together with inevitable structure relaxation in the excited states undesirably induce broadband emission with full width at half maximum (FWHM) exceeding 70 nm, and thus limiting the further utilization in high-resolution displays.11–15 In this case, Hatakeyama and coworkers initially proposed an innovative molecular designing concept of multiple resonance (MR)-TADF emitters,16 in which the frontier molecular orbitals (FMOs) are atomically separated through rationally positioning electron-donating (nitrogen or oxygen) and electron-accepting (boron) atoms into a polycyclic aromatic skeleton. Consequently, a narrowed emission band (FWHM < 30 nm) and evident TADF character can be achieved simultaneously.17–22However, MR-TADF emitters always suffer from an unsatisfactory rate constant of RISC (kRISC) of the order of only 104 s−1 because of inherently low spin orbital coupling (SOC) strength between S1 and T1 excited states.20,23,24 As a result, the slow RISC process will induce exciton aggregation quenching behavior, such as triplet–triplet annihilation (TTA) and singlet–triplet annihilation (STA), especially under high current density in OLEDs, which can hinder exciton utilization and damage stability of devices. Although the hyperfluorescence mechanism can enable quick consumption of triplet excitons to suppress exciton annihilations via employing a TADF sensitizer with fast kRISC as a mediator,25–27 ternary hyperfluorescence OLEDs comprising host, emitter and sensitizer are confronted with complicated device fabrication procedure and relatively high device operation voltages. In this regard, to facilitate the RISC process is still in high demand for designing superior MR-TADF emitters.
According to the description of Fermi's golden rule, kRISC can be strengthened by reducing ΔEST and enhancing SOC.28,29 Nevertheless, the atomically localized FMOs of MR-TADF molecules hinder the further reduction of ΔEST through optimizing the molecular architecture resembling D–A type TADF compounds.2,3,15,30 That is, the enhancement of SOC is deemed to be a feasible approach to accelerate kRISC, and therefore, state-of-the-art MR-TADF emitters with improved kRISC values have been proposed by delicately modulating excited state natures or resorting to the heavy-atom effect.24,31–37 For instance, Yang and coworkers introduced an extra-long-range charge transfer (LRCT) state by installing a well-chosen electron-donating moiety to a polycyclic aromatic skeleton at the para position to a boron atom.31 A multichannel RISC pathway with large SOC is established without disturbing the MR character of the S1 state, and thus affording significantly increased kRISC values and close-to-unity photoluminescence quantum yield (PLQY). Then they synergistically exploited LRCT and heavy-atom effect to accelerate kRISC in designing MR-TADF emitters, demonstrating an attractive kRISC of 2.2 × 106 s−1 and enabling low-efficiency roll-off in OLEDs.38
Although synergistic LRCT and the heavy-atom effect have been preliminarily investigated by incorporating a phenoselenazine (PSeZ) moiety into the MR framework, the photoelectric properties of MR-TADF emitters are only regulated by manipulating the substitution pattern (para and meta position of boron atoms). From the perspective of chemical structure, the electron-donating ability of additional donor moiety and spacer group can significantly affect the functionality of LRCT. Moreover, donor moieties with other large atomic numbers such as sulfur atom are also crucial to the accomplishment of large SOC strength.39,40 In this context, it is quite indispensable to systematically investigate LRCT and heavy-atom effects on modulating the excited state natures and energy levels for designing high-efficiency MR-TADF emitters. Herein, three MR-TADF emitters were accessed by decorating the MR segment (tBuCzBN) with different donor moieties of phenoxazine (PXZ), phenothiazine (PTZ) and PSeZ, namely tBuCzBN-PXZ, tBuCzBN-PTZ, and tBuCzBN-PSeZ. Then the LRCT strength was delicately manipulated by inserting a phenyl bridge between MR segment and donor moieties, and thus we gained another three MR-TADF emitters denoted as tBuCzBN-Ph-PXZ, tBuCzBN-Ph-PTZ, and tBuCzBN-Ph-PSeZ. The molecular designing concept of these MR-TADF emitters is depicted in Scheme 1. Their photophysical characteristics were systematically studied, and careful quantum chemical calculations were performed, showcasing that the excited state natures and exciton radiative decay avenues are susceptible to the donor moieties. Compared with unsubstituted analogue tBuCzBN, these MR-TADF emitters exhibit significantly boosted kRISC and attractive PLQY values, especially for tBuCzBN-PXZ (2.5 × 105 s−1) and tBuCzBN-Ph-PSeZ (2.1 × 105 s−1). Theoretical calculations together with photophysical studies also unambiguously manifest that the increased LRCT can induce an enhancement of kRISC, in particular with respect to MR-TADF emitters decorated with relatively intense electron-donating units, and the heavy-atom effect can accelerate spin flip of triplet excitons via greatly enhanced SOC in the absence of a modulatory LRCT state. These deductions are also confirmed by EL performance in sensitizer-free solution-processed OLEDs, demonstrating excellent maximum external quantum efficiency (EQEmax) values of 21.3% and 19.4% for tBuCzBN-PXZ and tBuCzBN-Ph-PSeZ, respectively, which are in the first tier of reported solution-processable MR-TADF binary OLEDs.
 | ||
| Scheme 1 Molecular designing concept of MR-TADF emitters in this study. | ||
Results and discussion
The detailed synthetic routes of these emitters are shown in Schemes S1–S12 of the ESI.† The chemical structures of the resultant emitters were characterized by 1H NMR, 13C NMR, and mass spectrometry (Fig. S1–S30†). We conducted thermogravimetric analysis (TGA), and all the emitters exhibited decomposition temperatures (Td) of over 430 °C (Fig. S31†), verifying good thermal stability that benefits fabrication and operation of OLED devices. Firstly, we evaluated the FMOs of electron-donating units (Fig. S32†), and the HOMO values were determined to be −4.78, −5.05 and −5.07 eV for PXZ, PTZ and PSeZ, respectively, revealing the electron-donating ability of these fragments (PXZ > PTZ > PSeZ; Ph-PXZ > Ph-PTZ > Ph-PSeZ), which have been also verified by surface electrostatic distribution analyses as shown in Fig. S33.† Therefore, our design concept follows three considerations: (1) the introduction of LRCT can be accomplished by integrating electron-donating moieties with the MR skeleton; (2) the LRCT strength can be meticulously modulated by diversiform electron-donating units comprising PXZ, PTZ, PSeZ, Ph-PXZ, Ph-PTZ, and Ph-PSeZ, which are in descending order of electron-donating ability; (3) the effects of heavy atom on triplet exciton spin-flip in MR-TADF emitters featuring various LRCT strengths are also evaluated by employing donor moieties with the atomic number from oxygen to sulfur and selenium (Scheme 1).Then quantum chemical simulations were performed to investigate the geometrical and electronic structures of these emitters at the molecular level calculated by density functional theory (DFT) at the M06-2X/6-311G** level of theory using Gaussian 16.41,42 As shown in Fig. 1, these emitters reveal extremely distinct FMO distributions: tBuCzBN-PXZ shows FMO distributions similar to those of conventional D–A-type TADF emitters as expected, in which HOMOs are located on the PSeZ moiety, and the LUMOs are distributed over the MR skeleton. Similarly with tBuCzBN-Ph-PXZ, the HOMOs are located on the PSeZ moiety, while the LUMOs are delocalized on the MR skeleton and phenyl bridge. The extended FMOs not only result in lower energy gap (Eg) between HOMO and LUMO levels of 4.73 eV for tBuCzBN-Ph-PXZ than that of 4.82 eV for tBuCzBN-PXZ, but also can maintain the LRCT effect between donor moiety and MR emitting core. For tBuCzBN-PTZ, the LUMOs are still localized in the MR skeleton, while the HOMOs are mainly located on the PTZ moiety and partly expanded to the MR skeleton, potentially signifying attenuated LRCT between donor moiety and MR emitting core. In contrast, the HOMOs and LUMOs of tBuCzBN-Ph-PTZ are mainly distributed over the MR skeleton except a spot of HOMO distribution on the PTZ unit, featuring a nearly MR-type FMO configuration. In the cases of tBuCzBN-PSeZ and tBuCzBN-Ph-PSeZ, typical MR-type FMO configurations have been demonstrated, in which the HOMOs and LUMOs are almost localized by nitrogen and boron atoms, which can be ascribed to the relatively weak electron-donating ability as mentioned above. We also studied the calculated ground-state dipole moments of these emitters (Fig. S34†), which are all in the range of 2.03–3.11 debyes, with little noticeable changes for different MR-TADF emitters, signifying that ground-state dipole moments hardly affect the exciton decay behaviors. In terms of molecular conformation, for example, tBuCzBN-PXZ displays totally different exciton decay behaviors compared with tBuCzBN-PTZ despite the fact that both MR-TADF emitters exhibit quite similar molecular architecture including molecular conformations and dihedral angles.
 | ||
| Fig. 1 The molecular structures of emitters and the FMO distributions. The corresponding energy levels and Eg values are shown in the diagrams. | ||
The photophysical properties of the obtained emitters were evaluated in dilute toluene (1 × 10−5 M) using ultraviolet-visible (UV-vis) absorption and photoluminescence (PL) spectra at room temperature. As shown in Fig. 2a and b, all emitters exhibit conspicuous signals peaking at 450–470 nm, which experimentally confirm the presence of an intense ICT process. The signals below 400 nm can be assigned to the n–π* and π–π* transitions of the molecular backbone. From the onset band of UV-vis absorption, the Eg values between HOMO and LUMO levels can be determined to be 2.56, 2.56 and 2.59 eV for tBuCzBN-PXZ, tBuCzBN-PTZ and tBuCzBN-PSeZ, while the Eg values tend to be relatively smaller, 2.51, 2.51 and 2.52 eV, for tBuCzBN-Ph-PXZ, tBuCzBN-Ph-PTZ and tBuCzBN-Ph-PSeZ, respectively, after inserting the phenyl bridge between MR segment and donor moieties. Combined with the cyclic voltammetry results (Fig. S35†), the HOMO and LUMO values can be determined to be −5.59/−3.03 eV for tBuCzBN-PXZ, −5.62/−3.06 eV for tBuCzBN-PTZ, −5.57/−2.98 eV for tBuCzBN-PSeZ, −5.36/−2.85 eV for tBuCzBN-Ph-PXZ, −5.54/−3.03 eV for tBuCzBN-Ph-PTZ, −5.50/−2.98 eV for tBuCzBN-Ph-PSeZ, respectively.
 | ||
| Fig. 2 The ultraviolet-visible absorption spectra and PL spectra of (a) tBuCzBN-PXZ, tBuCzBN-PTZ, and tBuCzBN-PSeZ diluted in toluene; (b) tBuCzBN-Ph-PXZ, tBuCzBN-Ph-PTZ, and tBuCzBN-Ph-PSeZ diluted in toluene. | ||
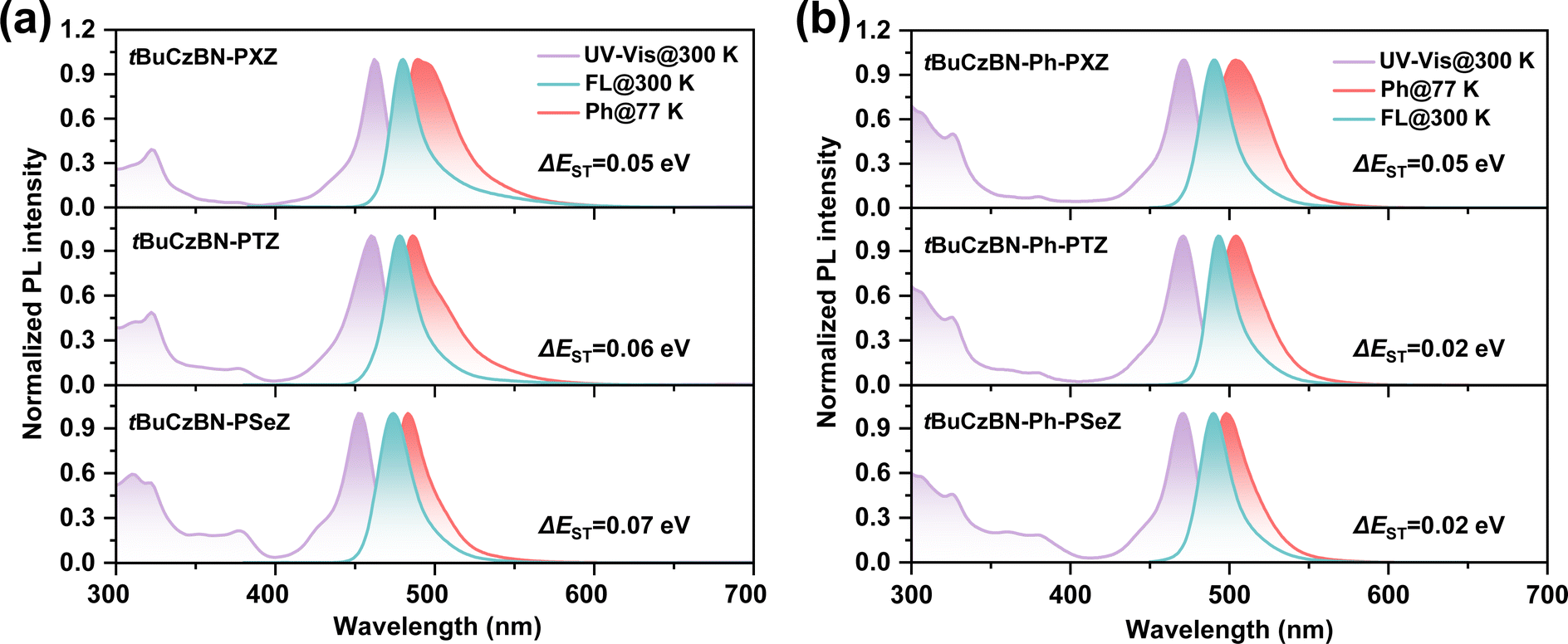
From the PL spectra, all emitters exhibit similar emission profiles with small FWHM of <25 nm. As depicted in Fig. 2a, the maximum emission peaks exhibit discernible hypochromic shift with changing donor moieties from tBuCzBN-PXZ (480 nm) to tBuCzBN-PTZ (478 nm) and tBuCzBN-PSeZ (474 nm), preliminarily revealing the mediator effect of LRCT on excited states. Correspondingly, the emission peaks of tBuCzBN-Ph-PXZ, tBuCzBN-Ph-PTZ and tBuCzBN-Ph-PSeZ are mainly located at ≈490 nm (Fig. 2b). The similar emission peaks of these emitters basically signify limited LRCT strength between MR segment and donor moieties, which provides a platform to investigate the heavy-atom effect on modulating the excited states regardless of the LRCT effect.37,43,44 As the ΔEST value is a prerequisite for maintaining valid spin flip of triplet excitons, we therefore determined the energy splitting of these emitters. The energies of S1 and T1 states can be estimated from the onset of fluorescence and phosphorescence spectra in toluene at 77 K, providing calculated ΔEST values of 0.05, 0.06 and 0.07 eV for tBuCzBN-PXZ, tBuCzBN-PTZ and tBuCzBN-PSeZ, respectively, which represent a gradual increase with increasing atomic number from oxygen to sulfur and selenium in donor moieties. Conversely, ΔEST values are decreasing for tBuCzBN-Ph-PXZ (0.05 eV), tBuCzBN-Ph-PTZ (0.02 eV) and tBuCzBN-Ph-PSeZ (0.02 eV). These small ΔEST values can ensure the establishment of effective channels for the triplet-to-singlet RISC process.
Furthermore, solvatochromic tests were performed to analyze the S1 state properties of these emitters (Fig. 3). According to the solvatochromic effect of tBuCzBN-PXZ, tBuCzBN-PTZ and tBuCzBN-PSeZ (Fig. 3a–c), three emitters display narrow PL spectra at ≈469 nm with FWHM of 19 nm in non-polar hexane, indicating that these emissive states are close in energy level. On increasing the solvent polarity to highly polar dichloromethane, the high-energy emission spectra display a finite bathochromic shift of <28 nm for tBuCzBN-PXZ, <14 nm for tBuCzBN-PTZ and <12 nm for tBuCzBN-PSeZ, revealing the dominant emissive state feature short-range charge transfer (SRCT) characteristics contributed by the MR effect. Besides the high-energy sharp emission band, the PL spectrum of tBuCzBN-PXZ also possesses a low-energy broad emission band even in non-polar hexane. Comparatively, the solvatochromic effect for the low-energy emission peak is more noticeable with a bathochromic shift of ≈100 nm by altering solvent from hexane to dichloromethane, which clearly demonstrates that the strong electron-donating PXZ moiety induces the formation of a low-lying LRCT state. Similarly, the PL spectrum of tBuCzBN-PTZ also manifests dual emission comprising a sharp emission band at 467–481 nm and a perceptible broad emission band at 550–613 nm. Nevertheless, tBuCzBN-PSeZ only displays a single narrowband emission without any appendant LRCT emission, which can be ascribed to the relatively weak electron-donating ability of the PSeZ moiety. In sharp contrast, the PL spectrum of tBuCzBN-Ph-PTZ displays a sharp emission band at 483–495 nm together with faint LRCT emission while tBuCzBN-Ph-PTZ and tBuCzBN-Ph-PSeZ exhibit only a sharp emission band at 479–497 nm (Fig. 3d–f), emphasizing a feeble LRCT effect of these emitters. From the abovementioned fluorescence emission, it is evident that the LRCT effect can not only be modulated by electron-donating moieties (tBuCzBN-PXZ > tBuCzBN-PTZ > tBuCzBN-PSeZ), but also be effectively restrained by inserting a phenyl bridge between MR segment and donor moieties (tBuCzBN-PXZ > tBuCzBN-Ph-PXZ; tBuCzBN-PTZ > tBuCzBN-Ph-PTZ; tBuCzBN-PSeZ > tBuCzBN-Ph-PSeZ). The excited states are also investigated through the analysis of solvation effect by the Lippert-Mataga solvatochromic model (Fig. S36†). For tBuCzBN-PXZ, the hybrid LRCT and SRCT characteristics can be observed with dipole moments of 22.46 and 12.07 debyes, respectively. In addition, there exists only one single linear relationship with calculated dipole moments of 6.55, 5.91, 5.26, 4.75 and 7.23 debyes, respectively, for tBuCzBN-PTZ, tBuCzBN-PSeZ, tBuCzBN-Ph-PXZ, tBuCzBN-Ph-PTZ and tBuCzBN-Ph-PSeZ, representing typical SRCT states. Overall, in terms of the S1 state, all these MR-TADF emitters exhibit dominant SRCT natures, assuredly inheriting narrowband emission of the MR core.
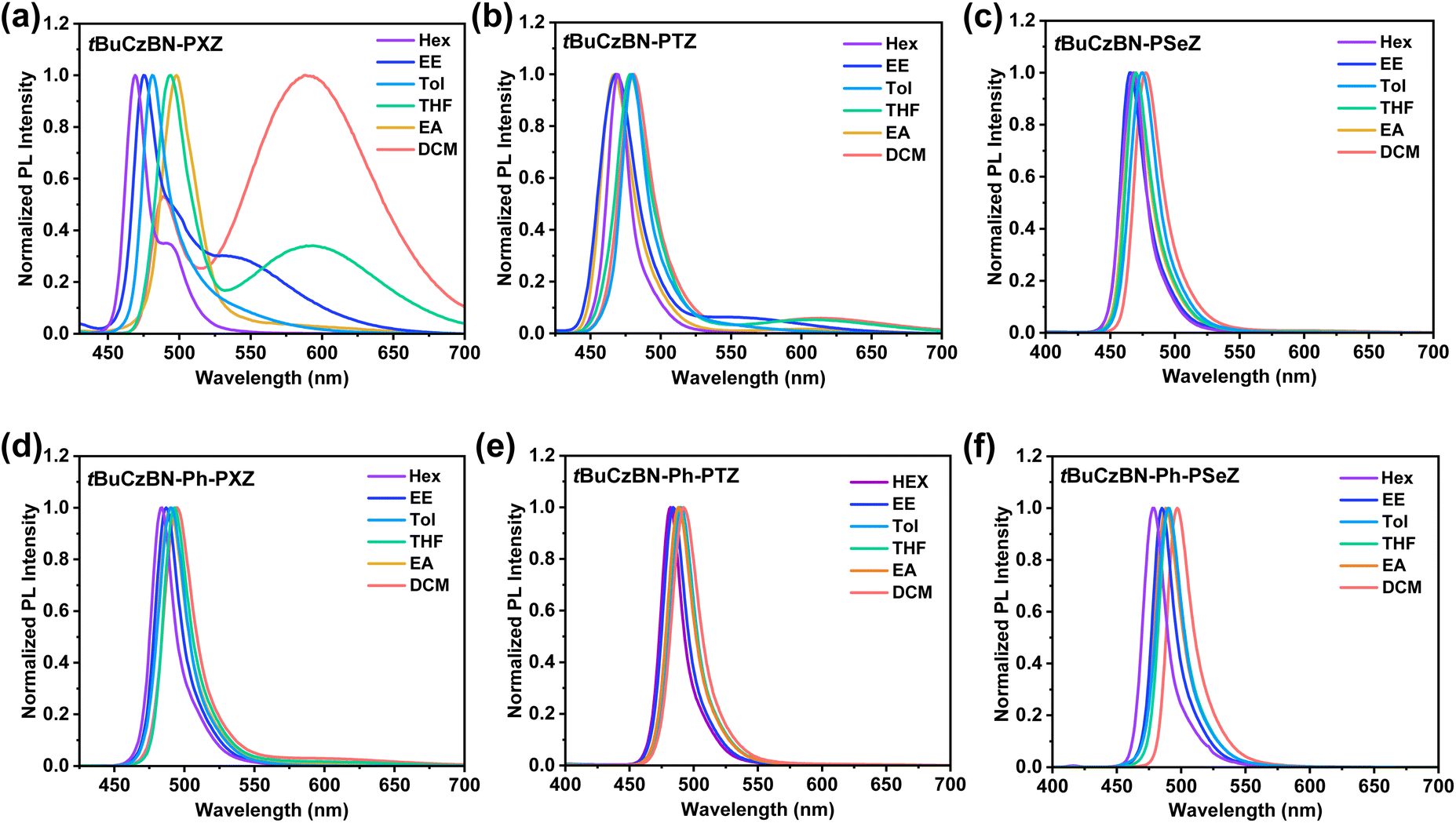 | ||
| Fig. 3 Solvatochromic effect on PL spectra in 10−5 M solution. (a) tBuCzBN-PXZ, (b) tBuCzBN-PTZ, (c) tBuCzBN-PSeZ, (d) tBuCzBN-Ph-PXZ, (e) tBuCzBN-Ph-PTZ and (f) tBuCzBN-Ph-PSeZ. Hex: n-hexane; EE: diethyl ether; Tol: toluene; THF: tetrahydrofuran; EA: ethyl acetate; DCM: dichloromethane. | ||
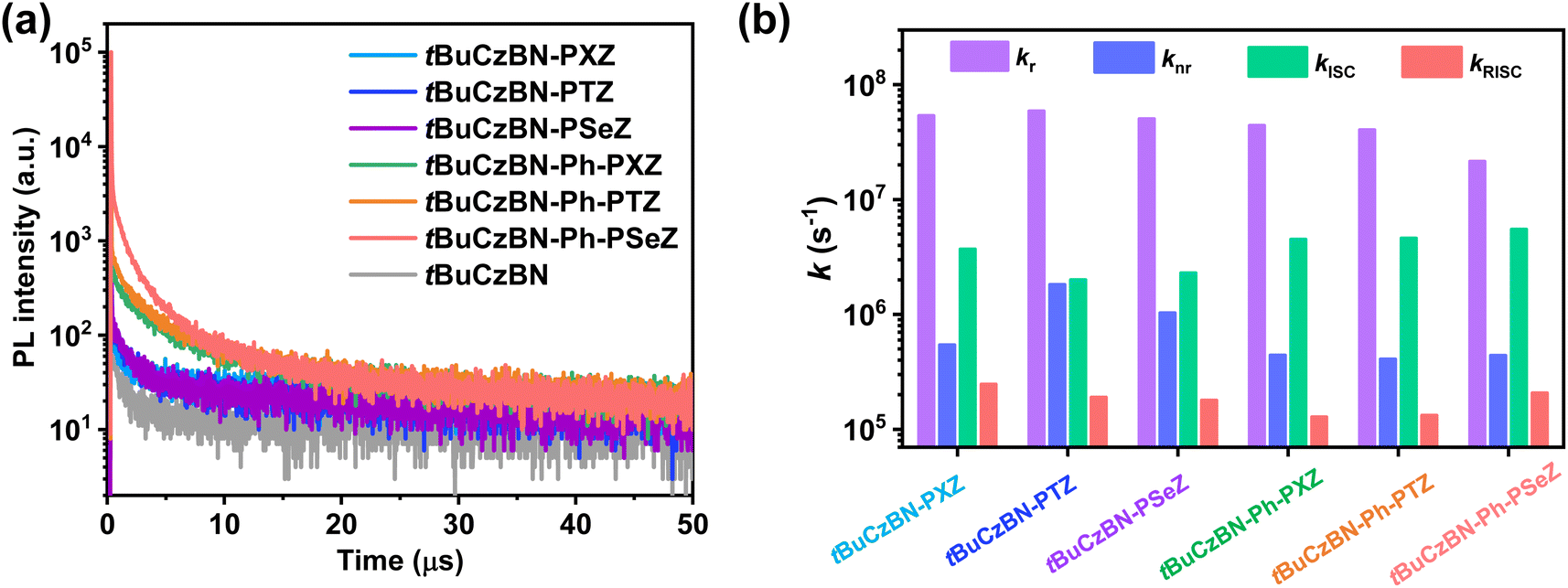
To evaluate the exciton decay kinetics of these emitters, we recorded transient PL decay spectra of emitters blended into 9-(2-(9-phenyl-9H-carbazole-3-yl)phenyl)-9H-3,9′-bicarbazole (PhCzBCz) matrix in vacuum. All these transient PL spectra demonstrate bi-exponential decay, containing nanosecond-scale prompt fluorescence (PF) and microsecond-scale delayed fluorescence (DF) components (Fig. 4a), which afford compelling evidence of typical TADF characteristic for these emitters.45,46 The corresponding prompt (τPF) and delayed (τDF) lifetimes were fitted to be 11.0 ns/6.8 μs for tBuCzBN-PXZ, 11.2 ns/7.7 μs for tBuCzBN-PTZ, 13.7 ns/7.9 μs for tBuCzBN-PSeZ, 11.2 ns/15.8 μs for tBuCzBN-Ph-PXZ, 11.6 ns/16.0 μs for tBuCzBN-Ph-PTZ, and 13.0 ns/16.9 μs for tBuCzBN-Ph-PSeZ, respectively. Additionally, the proportions of DF component (ϕDF) are dynamically tunable by altering the electron-donating moieties: 41% for tBuCzBN-PXZ, 34% for tBuCzBN-PTZ, 31% for tBuCzBN-PSeZ, 51% for tBuCzBN-Ph-PXZ, 53% for tBuCzBN-Ph-PTZ, and 72% for tBuCzBN-Ph-PSeZ. Thus, it can be preliminarily concluded that radiative decay and exciton upconversion avenues are susceptible to the LRCT and heavy-atom effect. Noteworthily, these emitters demonstrate not only significantly shortened τDF and boosted ϕDF values compared with unsubstituted analogue tBuCzBN (65.0 μs and 20%), but also close-to-unity PLQY values (ΦPL). All of these photophysical properties are summarized in Table 1.
 | ||
| Fig. 4 (a) Transient PL decay curves of the emitters blended into matrix in vacuum; (b) comparison between the calculated rate constants. | ||
| Emitter | λ abs [nm] | λ PL [nm] | FWHMc [nm] | ΔESTd [eV] | Φ PL [%] | τ p/τdf [ns]/[μs] | φ DF [%] | k Sr [107 s−1] | k RISC [105 s−1] | k ISC [106 s−1] |
|---|---|---|---|---|---|---|---|---|---|---|
| a Absorption peaks of emitters diluted in toluene. b Maximum emission peaks of emitters diluted in toluene. c Full width at half maximum. d Energy splitting between S1 and T1 determined from fitting the line of temperature-kRISC. e Photoluminescence quantum yields of doped films. f The lifetimes of prompt fluorescence component and the lifetimes of delayed fluorescence component. g The proportion of delayed fluorescence component. h The rate constants of nonradiative decay for singlet excitons by equation kSr = (1 − φDF)/τp. i The rate constants of reverse intersystem crossing process calculated by equation kRISC = ΦPL/[τd(1 − φDF)]. j The rate constants of intersystem crossing process calculated by equation kISC = φDF/(ΦPLτp). | ||||||||||
| tBuCzBN-PXZ | 323, 462 | 480 | 20 | 0.05 | 99 | 11.0/6.8 | 41 | 5.4 | 2.5 | 3.7 |
| tBuCzBN-PTZ | 322, 460 | 478 | 21 | 0.06 | 97 | 11.2/7.7 | 34 | 5.9 | 1.9 | 3.0 |
| tBuCzBN-PSeZ | 310, 453 | 474 | 24 | 0.07 | 98 | 13.7/7.9 | 31 | 5.0 | 1.8 | 2.3 |
| tBuCzBN-Ph-PXZ | 326, 471 | 490 | 23 | 0.05 | 99 | 11.2/15.8 | 51 | 4.4 | 1.3 | 4.5 |
| tBuCzBN-Ph-PTZ | 325, 471 | 492 | 20 | 0.02 | 99 | 11.6/16.0 | 53 | 4.0 | 1.4 | 4.6 |
| tBuCzBN-Ph-PSeZ | 326, 471 | 490 | 23 | 0.02 | 98 | 13.0/16.9 | 72 | 2.2 | 2.1 | 5.5 |
To characterize the spin-flip processes quantitatively, we summarized kRISC and the rate constants of radiative transition (kr), nonradiative transition (knr), and intersystem crossing (kISC) as depicted in Fig. 4b. The kRISC values are 2.5 × 105 s−1, 1.9 × 105 s−1 and 1.8 × 105 s−1 for tBuCzBN-PXZ, tBuCzBN-PTZ and tBuCzBN-PSeZ, respectively, which can be ascribed to the decreased electron-donating ability of donor moieties (PXZ > PTZ > PSeZ) and subsequently decreasing LRCT strength, suggesting that the increased LRCT will induce an enhancement of kRISC despite that heavy atoms can potentially strengthen the SOC between singlet and triplet excited states. Thus, it can be inferred that the LRCT plays a more crucial role than the heavy-atom effect in promoting kRISC, in particular with respect to MR-TADF emitters decorated with relatively intense electron-donating units. Conversely, after inserting a phenyl bridge between MR segment and donor moieties, the kRISC values are significantly increased when integrating heavy atoms into the emitters, especially for tBuCzBN-Ph-PSeZ with kRISC of 2.1 × 105 s−1, which evidently surpasses that of 1.4 × 105 s−1 for tBuCzBN-Ph-PTZ and 1.3 × 105 s−1 for tBuCzBN-Ph-PXZ. When the phenyl bridge is inserted between MR core and electron-donating unit, the LRCT effect will be weakened. Thus, it can be concluded that the heavy-atom effect (PXZ < PTZ < PSeZ) plays a much more vital role than LRCT. Additionally, the kr values of these MR-TADF emitters in blended films are of the order of 107 s−1 and significantly exceed the knr values (of the order of 105 to 106 s−1), possibly due to their large oscillator strength (f) values. Furthermore, it should be noted that, with the reduced proportion of LRCT emission, non-radiative loss can be markedly suppressed, such as from 1.8 × 106 s−1 for tBuCzBN-PTZ to 4.1 × 105 s−1 for tBuCzBN-Ph-PTZ, demonstrating that SRCT state provided by the MR-type skeleton is conducive to strong electronic interactions, which are hardly achieved for a D–A-type TADF emitter due to the inherent separated D–A structure. Moreover, the kISC values are also determined to be (2.3–3.7) × 106 s−1 for tBuCzBN-PXZ, tBuCzBN-PTZ and tBuCzBN-PSeZ, and then further increased for tBuCzBN-Ph-PXZ (4.5 × 106 s−1), tBuCzBN-Ph-PTZ (4.6 × 106 s−1) and tBuCzBN-Ph-PSeZ (5.5 × 106 s−1). The noticeable enhancement of kISC values further proves that the heavy-atom effects can effectively strengthen the SOC between singlet and triplet excited states when inserting a phenyl bridge between MR segment and donor moieties.47 We also note that the kr values are orders of magnitude greater than competitive kISC values, thus reducing multiple spin-flip cycles between singlet and triplet excited states and suppressing the undesired accumulation of excitons. Eventually, STA and TTA can be prevented effectively in these MR-TADF emitters.48,49
We also recorded steady-state PL spectra of 2 wt% and 5 wt% MR-TADF emitters blended with PhCzBCz host materials. As shown in Fig. S37 and S38,† participations of triplet exciton gradually decline with decreasing electron-donating ability of donor moieties. Additionally, increasing doping concentrations of MR-TADF emitters in blended films can also suppress the participations of triplet exciton partly due to a more severe exciton quenching effect. To understand in depth the excited-state properties, we further studied the transient PL decay spectra of these emissive films with different doping concentrations (Fig. S39†). It is found that when the MR core is directly covalently linked with electron-donating moieties in both 2 wt% and 5 wt% MR-TADF emitter blended films, tBuCzBN-PXZ exhibits relatively faster exciton decay dynamic due to favorable LRCT effect. After inserting a phenyl bridge between MR core and donor moieties, tBuCzBN-Ph-PSeZ achieves much higher exciton decay rates and more participations of triplet exciton. Thus, it is concluded that the heavy-atom effect plays a much more vital role in suppressing LRCT strength. Moreover, the calculated kRISC values, especially of tBuCzBN-PXZ in MR-TADF emitter blended films with high doping concentration, are lower than that in 1 wt% MR-TADF emitter blended films, potentially verifying the severe exciton–exciton annihilation. All the photophysical properties are summarized in Tables S1 and S2.†
To gain in-depth insights into the nature of excited states and upconversion process of the excitons, the excited-state energy levels and natural transition orbital (NTO) distributions of S1 and Tn (n = 1, 2) were theoretically evaluated through time-dependent density functional theory (TD-DFT) calculations for these emitters by adopting the M06-2X functional with the 6-311G** basis set.28,50 The SOC matrix elements (SOCME) of 〈S1|ĤSO|Tn〉 between the Tn and S1 states were calculated using the ORCA program. As evidenced by the NTO analysis (Fig. 5), the S1 states of tBuCzBN-PXZ, tBuCzBN-PTZ and tBuCzBN-PSeZ exhibit MR-type FMO configurations, in which particle and hole are localized on separate atoms induced by nitrogen and boron atoms with extremely limited involvement of the donor units. Thus, the ensuing confined FMOs also curtail the vibronic coupling and excited-state structural relaxation, enabling an experimentally narrowed emission. Whereas, for tBuCzBN-Ph-PXZ, tBuCzBN-Ph-PTZ and tBuCzBN-Ph-PSeZ, the electron distributions are partly expanded to the phenyl bridge moieties. Although the delocalized FMOs result in relatively lower-lying S1 energy level and corresponding bathochromic shift of emission peaks as described for PL spectra, the pronounced SRCT characters are expectedly retained. Furthermore, all molecules demonstrate attractive f values of 0.67–0.76, indicative of the SRCT nature of S1 states, and enabling high kr values of these MR-TADF emitters, which are well consistent with photophysical properties.
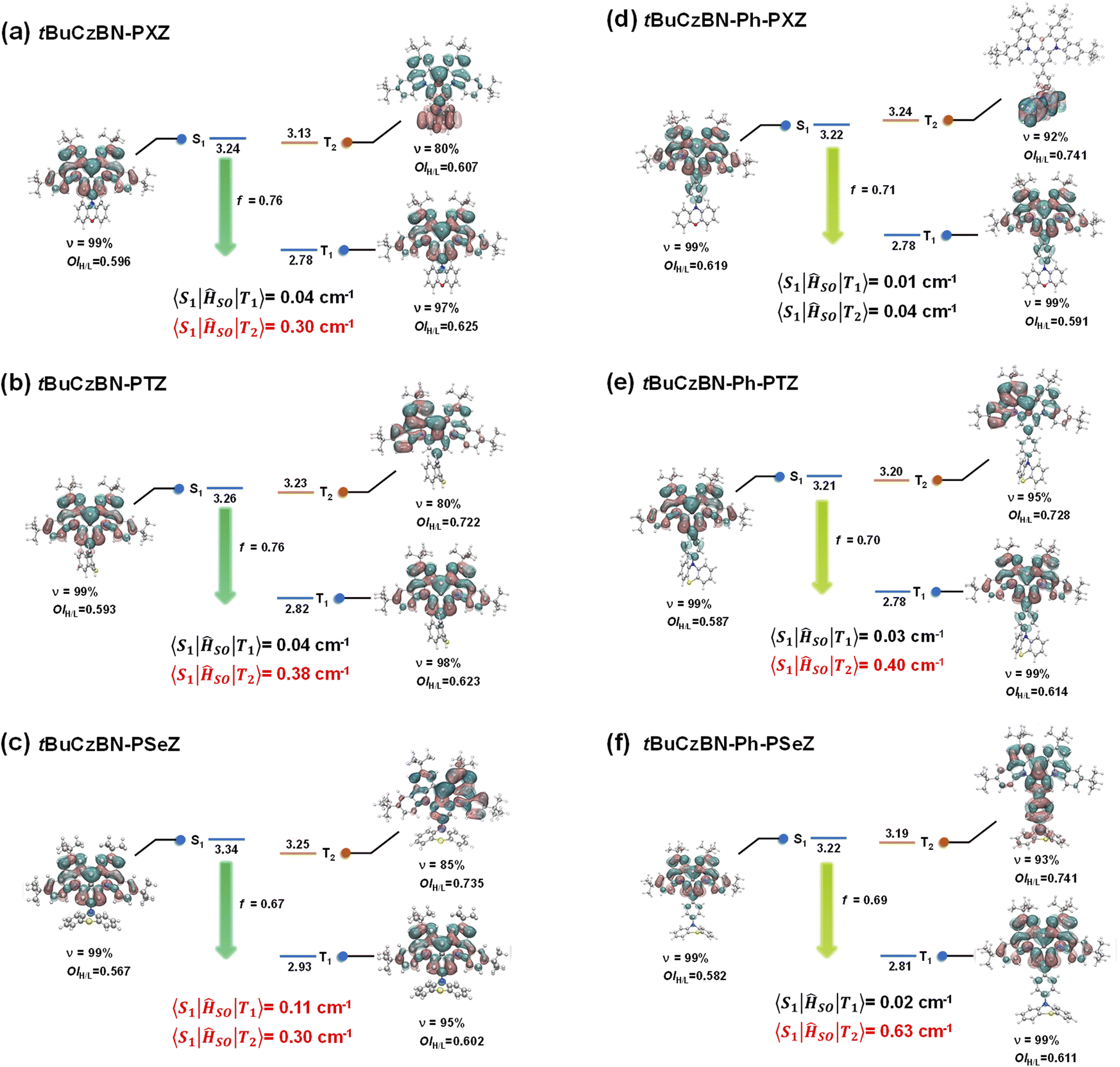 | ||
| Fig. 5 Calculated energy-level diagrams, NTO distributions, and associated spin–orbit coupling (SOC) matrix elements for (a) tBuCzBN-PXZ, (b) tBuCzBN-PTZ, (c) tBuCzBN-PSeZ, (d) tBuCzBN-Ph-PXZ, (e) tBuCzBN-Ph-PTZ and (f) tBuCzBN-Ph-PSeZ. | ||
Notably, the estimated ΔEST values between S1 and T1 exceed 0.4 eV, which are always regarded as too large to implement favorable spin flip of triplet excitons. In addition, the S1 and T1 states of these emitters are accompanied by extremely similar excited-state natures featuring SRCT characteristics, and therefore the corresponding SOCME values 〈S1|ĤSO|T1〉 are as low as 0.01–0.04 cm−1, except for tBuCzBN-PSeZ with relatively high 〈S1|ĤSO|T1〉 of 0.11 cm−1 partly due to the quite limited involvement of the PSeZ unit. A higher-lying T2 close to S1 with promising energy differences of 0.01–0.11 eV is also noted. Considering the energetic proximity of S1 and T2, it can be anticipated that the T2 states serve as intermediate states in the SOC interaction between singlet and triplet excited states, and thus the newly built conversion channels can be engaged synergistically to accelerate the spin-flip of triplet excitons and then to take charge of the unexpectedly excellent RISC dynamics.51–53 More importantly, according to NTO distributions of T2 states, these molecules display a comparatively distinct excited-state nature, in which tBuCzBN-PTZ, tBuCzBN-PSeZ and tBuCzBN-Ph-PTZ have SRCT character but a more locally excited triplet state (3LE) component than relative T1 states, signifying enhanced SOCME values 〈S1|ĤSO|T2〉 of 0.30–0.40 cm−1 compared with S1–T1. In sharp contrast, the T2 state of tBuCzBN-PXZ displays detached NTO distributions in that the hole and particle are individually located on PSeZ donor and MR skeleton, respectively, manifesting a typical LRCT nature. Hence the SOCME value 〈S1|ĤSO|T2〉 of tBuCzBN-PXZ can reach 0.30 cm−1 even without the participation of heavy atoms, in line with excellent kRISC value determined from transient decay spectra. From the above discussions, it is evident that integrating a donor moiety with fair electron-donating ability into the MR skeleton is a prerequisite for formation of a modulatory LRCT state, which plays an even more vital role than the heavy-atom effect in boosting a large SOC effect, and thus facilitating the RISC process.
In the case of the T2 state of tBuCzBN-Ph-PXZ, the SOCME value 〈S1|ĤSO|T2〉 is only 0.04 cm−1. The negligible SOCME value can be interpreted as the insulation of NTO-T2 distribution from the MR skeleton since hole and particle are completely delocalized on the PXZ unit. In particular, the T2 state of tBuCzBN-Ph-PSeZ exhibits an approximately mixed SRCT and LRCT nature as the hole distributions pervade the entire molecule while the particle distributions are mainly located on the MR skeleton and phenyl bridge. The hybrid excited-state nature together with a relatively prominent heavy-atom effect enable a significantly enhanced SOC between S1 and T2 states for tBuCzBN-Ph-PSeZ with 〈S1|ĤSO|T2〉 of as much as 0.63 cm−1. As a consequence, the kRISC value of 2.1 × 105 s−1 for tBuCzBN-Ph-PSeZ is very close to that of 2.5 × 105 s−1 for tBuCzBN-PXZ, disclosing that the heavy-atom effect can undoubtedly accelerate spin flip of triplet excitons via greatly enhanced SOC in the absence of modulatory LRCT state in MR-TADF emitters.
The atom force microscopy (AFM) results of all molecules (1 wt% MR-TADF doped in PhCzBCz) are shown in Fig. S40.† The relatively smooth surface topographies with a root mean square (RMS) surface roughness of 0.35–0.68 nm can be observed. There are no conspicuous voids and aggregates. These results demonstrate that the mixed systems have favorable film-formation abilities in luminous layer during solution-processed fabrication. To verify the potential of these MR-TADF emitters in electroluminescent (EL) devices, solution-processable OLEDs were fabricated with the architecture of ITO/PSS:PEDOT (40 nm)/PVK (10 nm)/emitting layer (40 nm)/TmPyPB (45 nm)/LiF (0.9 nm)/Al (110 nm) shown in Fig. 6a, where PSS:PEDOT, PVK, and TmPyPB are poly(3,4-ethylenedioxythiophene)-poly(styrene sulfonate), poly(9-vinylcarbazole), and 1,3,5-tri(m-pyridin-3-ylphenyl)benzene, respectively. PSS:PEDOT was adopted as hole injecting and transferring layer, and PVK and TmPyPB served as exciton-blocking and electron-transferring layers, respectively. For the emitting layer, the MR-TADF emitters were embedded into host materials of PhCzBCz, which could not only enhance the charge transferring capability, but also restrain the exciton quenching induced by molecular π–π accumulation, and the dopant concentration was optimized to be 1 wt%. From the current density–voltage–luminance (J–V–L) curves shown in Fig. 6b and c, the turn-on voltages (Von) of devices are 3.8–4.4 V, in which the higher Von value of tBuCzBN-Ph-PXZ can be ascribed to the shallower energy levels. Nearly all of the devices exhibit favorable maximum luminance (Lmax) exceeding 1000 cd m−2.
 | ||
| Fig. 6 (a) Device architecture and energy-level diagrams of the functional materials for solution-processed OLEDs. Current density and luminance versus voltage (J–V–L) characteristics for (b) tBuCzBN-PXZ, tBuCzBN-PTZ, and tBuCzBN-PSeZ; (c) tBuCzBN-Ph-PXZ, tBuCzBN-Ph-PTZ, and tBuCzBN-Ph-PSeZ. (d) EL spectra recorded at 6 V. (e) External quantum efficiency–luminance (EQE–L) plots. | ||
As depicted in Fig. 6d, the EL spectra of these devices are in great agreement with the PL profiles, especially for tBuCzBN-PXZ (480 nm), tBuCzBN-PTZ (478 nm) and tBuCzBN-PSeZ (474 nm) with a small FWHM of <30 nm. Slightly bathochromic shifts can be detected for tBuCzBN-Ph-PXZ (496 nm), tBuCzBN-Ph-PTZ (496 nm) and tBuCzBN-Ph-PSeZ (496 nm), and moreover, the FWHM values are unexpectedly increased to 33–36 nm, which should be attributed to the relatively larger transition dipole moment for these emitters and subsequently distinct solid-state solvation effect in high-polarity matrix. Additionally, all these devices exhibit excellent EL spectral stability as shown in Fig. S41.† The maximum current (CEmax) and power (PEmax) efficiencies are 41.9 cd A−1 and 29.9 lm W−1 for tBuCzBN-PXZ, which are correspondingly higher than that of 30.4 cd A−1 and 21.7 lm W−1 for tBuCzBN-PTZ, and 13.8 cd A−1 and 15.2 lm W−1 for tBuCzBN-PSeZ, as illustrated in Fig. S42.† An important observation is that the solution-processable device performances based on these MR-TADF emitters match well with the corresponding photophysical properties, especially kRISC values. Additionally, the CEmax and PEmax values of tBuCzBN-Ph-PSeZ are 50.5 cd A−1 and 26.0 lm W−1, exceeding that of 23.0 cd A−1 and 14.4 lm W−1 for tBuCzBN-Ph-PXZ, and 30.1 cd A−1 and 21.5 lm W−1 for tBuCzBN-Ph-PSeZ, respectively. In line with the kRISC values of these MR-TADF emitters in the film state, the EQEmax value of tBuCzBN-PXZ can reach 21.3%, which is in the first tier of reported solution-processable MR TADF binary OLEDs without employing additional sensitizers (Fig. 6e). On decreasing the electron-donating ability of donor moieties, the EQEmax values are further reduced from 19.0% for tBuCzBN-PTZ to 15.2% for tBuCzBN-PSeZ, rationally validating that LRCT plays a more crucial role than the heavy-atom effect for MR-TADF emitters decorated with strong electron-donating units in promoting EL performance in solution-processable OLEDs. In contrast, the EQEmax value drops dramatically to only 9.6% after inserting a phenyl group between tBuCzBN MR segment and PXZ donor moieties (tBuCzBN-Ph-PXZ). Furthermore, the EQEmax values increase significantly to 11.6% for tBuCzBN-Ph-PTZ and even 19.4% for tBuCzBN-Ph-PSeZ, corroborating that the heavy-atom effect can enable exceptional EL performance when considering relatively limited LRCT and distinct SRCT effects in MR-TADF emitters. The EL properties of solution-processable OLEDs are summarized in Table 2.
| Emitter | V on [V] | EL/FWHMb [nm] | L max [cd m−2] | CEmaxd [cd A−1] | PEmaxe [lm W−1] | EQEmaxf [%] | CIEg |
|---|---|---|---|---|---|---|---|
| a The turn-on voltages. b The electroluminescence spectral peaks and full width at half maximum determined at 6 V. c The maximum luminance. d The maximum current efficiencies. e The maximum power efficiencies. f The maximum external quantum efficiency. g The coordinates of CIE. | |||||||
| tBuCzBN-PXZ | 4.1 | 480/27 | 1255 | 41.9 | 29.9 | 21.3 | (0.15, 0.32) |
| tBuCzBN-PTZ | 3.9 | 478/28 | 1223 | 30.4 | 21.7 | 19.0 | (0.14, 0.24) |
| tBuCzBN-PSeZ | 3.8 | 474/29 | 801 | 20.3 | 13.8 | 15.2 | (0.13, 0.20) |
| tBuCzBN-Ph-PXZ | 4.4 | 496/33 | 1088 | 23.0 | 14.4 | 9.6 | (0.13, 0.50) |
| tBuCzBN-Ph-PTZ | 4.0 | 496/36 | 1301 | 30.1 | 21.5 | 11.6 | (0.14, 0.52) |
| tBuCzBN-Ph-PSeZ | 4.0 | 496/36 | 1677 | 50.5 | 26.0 | 19.4 | (0.13, 0.51) |
We also fabricated solution-processed OLEDs with different doping concentration of 2 wt% and 5 wt% MR-TADF compounds diluted into PhCzBCz host materials. The device performances are summarized in Fig. S43, S44, and Tables S3, S4.† Comparatively, the device performance of 2 wt% MR-TADF-based OLEDs is better than that of 5 wt% MR-TADF-based OLEDs. More importantly, for both 2 wt% MR-TADF- and 5 wt% MR-TADF-based OLEDs, the EQE values are almost declining with decreasing electron-donating ability of donor moieties when directly covalently linking MR core with electron-donating moieties. In contrast, the EQE values will be enhanced on integrating a heavy atom, especially selenium atom, into the MR skeleton when inserting a phenyl bridge. Overall, the MR-TADF solution-processed OLEDs with different dopant concentrations not only prove the superior properties of these compounds, but also demonstrate our designing principle mentioned above. Actually, we also fabricated ternary MR-TADF solution-processable OLEDs with 1 wt% emitter, 10 wt% 5Cz-TRZ and 89 wt% PhCzBCz as emitting layer where 5Cz-TRZ as sensitizer is 9,9′,9′′,9′′′,9′′′′-[6-(4,6-diphenyl-1,3,5-triazin-2-yl)-1,2,3,4,5-benzenepentayl]pentakis-9H-carbazole. The device performances are summarized in Fig. S45 and Table S5.† It is found that the ternary MR-TADF sensitized OLEDs have dramatically improved luminance, but the EQE values are lower than those of non-sensitized binary devices, which can be ascribed to the inefficient energy transfer from sensitizer to MR-TADF emitters or an uncompetitive exciton upconversion process.
Conclusions
In summary, we proposed a series of MR-TADF emitters by integrating diversiform electron-donating units with the MR skeleton to systematically investigate the LRCT and heavy-atom effects on modulating the excited-state natures and energy levels. The photophysical characteristics and careful quantum chemical calculations manifest that radiative decay and exciton upconversion avenues are susceptible to the modulation of electron-donating moieties. For MR-TADF emitters, in which the MR segments are covalently linked with electron-donating moieties, the excited-state characteristics can be delicately manipulated by the electron-donating ability. Especially for tBuCzBN-PXZ, enhanced LRCT can facilitate spin-flip of triplet excitons, and thus dramatically accelerating the RISC process while maintaining narrowed emission of the S1 state. It has also been demonstrated that LRCT represents a more notable advantage than heavy-atom effects in prompting exciton dynamics for MR-TADF emitters decorated with relatively intense electron-donating units. By contrast, the LRCT strength has been further weakened after inserting a phenyl bridge between MR segment and donor moieties. According to the photophysical properties and quantum chemical simulation results, the noticeable enhancement of kRISC values with increased atom numbers indicates that the heavy-atom effect can undoubtedly accelerate spin flip of triplet excitons via greatly enhanced SOC in the absence of modulatory LRCT state in MR-TADF emitters. Thus, both tBuCzBN-PXZ and tBuCzBN-Ph-PSeZ exhibit significantly boosted kRISC values exceeding 2.0 × 105 s−1 and close-to-unity photoluminescence quantum yield. Leveraging these exceptional properties, the EQEmax values of tBuCzBN-PXZ- and tBuCzBN-Ph-PSeZ-based solution-processable OLEDs can reach 21.3% and 19.4% which are in the first tier of reported solution-processable MR TADF binary OLEDs without employing additional sensitizers. We believe that this study provides a framework for modulating photoelectrical properties of MR-TADF emitters through fastidiously regulating LRCT and heavy-atom effects.Data availability
The data (experimental instrumentation, synthetic procedures, structural characterization data including NMR and MS spectra, theoretical calculations, thermodynamics data, and device performance data) that support this article are available in the article itself and its ESI.†Author contributions
Li, S., Yang, Z., Xie, Y., Liu, Y. – conceptualization, investigation, OLED devices, writing – original draft; Li, S., Yang, Z., Xie, Y. – theoretical calculation, investigation; Li, S., Xie, Y. – formal analysis, OLED devices; Li, S. – investigation (thermal properties); Li, S., Hua, L., Xie, Y. – investigation (transient PL properties); Liu, Y., Ren Z., Yan, S., Ying, S. – project administration, supervision; Liu, Y., Ren Z., Yan, S. – writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support of the National Natural Science Foundation of China (52103220 and 52273164), the Shandong Provincial Natural Science Foundation (ZR2022ZD37 and ZR2023QE078), the Science and Technology Support Plan for Youth Innovation of Colleges and Universities in Shandong Province (2023KJ097), and Natural Science Foundation of Qingdao (23-2-1-75-zyyd-jch) is gratefully acknowledged.Notes and references
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS.
- H. Liu, Y. Liu, G. Chen, Y. Meng, H. Peng, J. Miao and C. Yang, Chem. Sci., 2024, 15, 12598–12605 RSC.
- W. Zeng, H. Y. Lai, W. K. Lee, M. Jiao, Y. J. Shiu, C. Zhong, S. Gong, T. Zhou, G. Xie and M. Sarma, Adv. Mater., 2018, 30, 1704961 CrossRef.
- C. Li, A. K. Harrison, Y. Liu, Z. Zhao, C. Zeng, F. B. Dias, Z. Ren, S. Yan and M. R. Bryce, Angew. Chem., Int. Ed., 2022, 134, e202115140 CrossRef.
- G. Meng, H. Dai, Q. Wang, J. Zhou, T. Fan, X. Zeng, X. Wang, Y. Zhang, D. Yang and D. Ma, Nat. Commun., 2023, 14, 2394 CrossRef CAS.
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 1–20 CrossRef.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS.
- X. Tang, L.-S. Cui, H.-C. Li, A. J. Gillett, F. Auras, Y.-K. Qu, C. Zhong, S. T. Jones, Z.-Q. Jiang and R. H. Friend, Nat. Mater., 2020, 19, 1332–1338 CrossRef CAS.
- L.-S. Cui, A. J. Gillett, S.-F. Zhang, H. Ye, Y. Liu, X.-K. Chen, Z.-S. Lin, E. W. Evans, W. K. Myers and T. K. Ronson, Nat. Photon., 2020, 14, 636–642 CrossRef CAS.
- D. Zhang, X. Song, A. J. Gillett, B. H. Drummond, S. T. Jones, G. Li, H. He, M. Cai, D. Credgington and L. Duan, Adv. Mater., 2020, 32, 1908355 CrossRef CAS.
- W. Yuan, H. Yang, C. Duan, X. Cao, J. Zhang, H. Xu, N. Sun, Y. Tao and W. Huang, Chem, 2020, 6, 1998–2008 Search PubMed.
- Y. Liu, L. Hua, Z. Zhao, S. Ying, Z. Ren and S. Yan, Adv. Sci., 2021, 8, 2101326 CrossRef CAS.
- K. Shi, Y. Xie, L. Hua, S. Li, Z. Yang, Y. Yin, Z. Wang, S. Ying, Y. Liu and Z. Ren, ACS Mater. Lett., 2024, 6, 1491–1503 CrossRef CAS.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS.
- X.-C. Fan, K. Wang, Y.-Z. Shi, Y.-C. Cheng, Y.-T. Lee, J. Yu, X.-K. Chen, C. Adachi and X.-H. Zhang, Nat. Photon., 2023, 17, 280–285 CrossRef CAS.
- Y. Zhang, J. Wei, L. Wang, T. Huang, G. Meng, X. Wang, X. Zeng, M. Du, T. Fan and C. Yin, Adv. Mater., 2023, 35, 2209396 CrossRef CAS.
- Y. Zou, J. Hu, M. Yu, J. Miao, Z. Xie, Y. Qiu, X. Cao and C. Yang, Adv. Mater., 2022, 34, 2201442 CrossRef CAS.
- Y. Xu, C. Li, Z. Li, Q. Wang, X. Cai, J. Wei and Y. Wang, Angew. Chem., Int. Ed., 2020, 59, 17442–17446 CrossRef CAS.
- Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai and T. Hatakeyama, Nat. Photon., 2019, 13, 678–682 CrossRef CAS.
- P. Jiang, J. Miao, X. Cao, H. Xia, K. Pan, T. Hua, X. Lv, Z. Huang, Y. Zou and C. Yang, Adv. Mater., 2022, 34, 2106954 CrossRef CAS.
- Y. Zhang, G. Li, L. Wang, T. Huang, J. Wei, G. Meng, X. Wang, X. Zeng, D. Zhang and L. Duan, Angew. Chem., Int. Ed., 2022, 61, e202202380 CrossRef CAS.
- T. Hua, L. Zhan, N. Li, Z. Huang, X. Cao, Z. Xiao, S. Gong, C. Zhou, C. Zhong and C. Yang, Chem. Eng. J., 2021, 426, 131169 CrossRef CAS.
- D. Zhang and L. Duan, Nat. Photon., 2021, 15, 173–174 CrossRef CAS.
- Y. Wada, H. Nakagawa, S. Matsumoto, Y. Wakisaka and H. Kaji, Nat. Photon., 2020, 14, 643–649 CrossRef CAS.
- C.-Y. Chan, M. Tanaka, Y.-T. Lee, Y.-W. Wong, H. Nakanotani, T. Hatakeyama and C. Adachi, Nat. Photon., 2021, 15, 203–207 CrossRef CAS.
- P. K. Samanta, D. Kim, V. Coropceanu and J.-L. Brédas, J. Am. Chem. Soc., 2017, 139, 4042–4051 CrossRef CAS.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS.
- Y. Zhang, Z. Huang, Y. Yang, J. Liu, Y. Tian, Z. Bin and J. You, Chem. Sci., 2023, 14, 5125–5131 RSC.
- Z. Huang, H. Xie, J. Miao, Y. Wei, Y. Zou, T. Hua, X. Cao and C. Yang, J. Am. Chem. Soc., 2023, 145, 12550–12560 CrossRef CAS.
- H.-J. Cheon, S.-J. Woo, S.-H. Baek, J. H. Lee and Y.-H. Kim, Adv. Mater., 2022, 34, 2207416 CrossRef CAS.
- Y. Xu, C. Li, Z. Li, Q. Wang, X. Cai, J. Wei and Y. Wang, Angew. Chem., Int. Ed., 2020, 59, 17442–17446 CrossRef CAS.
- Y. Liu, X. Xiao, Z. Huang, D. Yang, D. Ma, J. Liu, B. Lei, Z. Bin and J. You, Angew. Chem., Int. Ed., 2022, 61, e202210210 CrossRef CAS.
- Y. X. Hu, J. Miao, T. Hua, Z. Huang, Y. Qi, Y. Zou, Y. Qiu, H. Xia, H. Liu and X. Cao, Nat. Photon., 2022, 16, 803–810 CrossRef CAS.
- I. S. Park, H. Min and T. Yasuda, Angew. Chem., Int. Ed., 2022, 61, e202205684 CrossRef CAS.
- X. Cao, K. Pan, J. Miao, X. Lv, Z. Huang, F. Ni, X. Yin, Y. Wei and C. Yang, J. Am. Chem. Soc., 2022, 144, 22976–22984 CrossRef CAS.
- Y. Zou, M. Yu, Y. Xu, Z. Xiao, X. Song, Y. Hu, Z. Xu, C. Zhong, J. He and X. Cao, Chem, 2024, 10, 1485–1501 CAS.
- X. Tong, Z. Zhao, L. Hua, Y. Zhang, B. Xu, Y. Liu, S. Yan and Z. Ren, Adv. Funct. Mater., 2023, 33, 2305324 CrossRef CAS.
- I. S. Park, M. Yang, H. Shibata, N. Amanokura and T. Yasuda, Adv. Mater., 2022, 34, 2107951 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- T. Lu and F. Chen, Acta Chim. Sinica, 2011, 69, 2393 CAS.
- C. L. Kim, J.-M. Kim, H. Jang, D. R. Lee and Y. Lee, ACS Appl. Energy Mater., 2022, 5, 4985–4990 CrossRef CAS.
- D. R. Lee, K. H. Lee, W. Shao, C. L. Kim, J. Kim and J. Y. Lee, Chem. Mater., 2020, 32, 2583–2592 CrossRef CAS.
- S. Wu, W. Li, K. Yoshida, D. Hall, S. Madayanad Suresh, T. Sayner, J. Gong, D. Beljonne, Y. Olivier and I. D. Samuel, ACS Appl. Mater. Interfaces, 2022, 14, 22341–22352 CrossRef CAS.
- Y. Xie, L. Hua, Z. Wang, Y. Liu, S. Ying, Y. Liu, Z. Ren and S. Yan, Sci. China Chem., 2023, 66, 826–836 CAS.
- H. S. Kim, J. Y. Lee, S. Shin, W. Jeong, S. H. Lee, S. Kim, J. Lee, M. C. Suh and S. Yoo, Adv. Funct. Mater., 2021, 31, 2170374 CrossRef.
- P. Han, C. Lin, H. Xu, E. Xia, D. Yang, A. Qin, D. Ma and B. Z. Tang, ACS Mater. Lett., 2022, 4, 1087–1093 CrossRef CAS.
- K. Matsuoka, K. Albrecht, K. Yamamoto and K. Fujita, Sci. Rep., 2017, 7, 41780 CrossRef CAS.
- M. K. Etherington, N. A. Kukhta, H. F. Higginbotham, A. Danos, A. N. Bismillah, D. R. Graves, P. R. McGonigal, N. Haase, A. Morherr and A. S. Batsanov, J. Phys. Chem. C, 2019, 123, 11109–11117 CrossRef CAS.
- H. Noda, X.-K. Chen, H. Nakanotani, T. Hosokai, M. Miyajima, N. Notsuka, Y. Kashima, J.-L. Brédas and C. Adachi, Nat. Mater., 2019, 18, 1084–1090 CrossRef CAS.
- Y. Liu, L. Hua, S. Yan and Z. Ren, Nano Energy, 2020, 73, 104800 CrossRef CAS.
- D. Liu, J. Y. Wei, W. W. Tian, W. Jiang, Y. M. Sun, Z. Zhao and B. Z. Tang, Chem. Sci., 2020, 11, 7194–7203 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05516k |
| This journal is © The Royal Society of Chemistry 2024 |