
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The aquastatin biosynthetic gene cluster encodes a versatile polyketide synthase capable of synthesising heteromeric depsides with diverse alkyl side chains†
Nicolau
Sbaraini
 *a,
Andrew
Crombie
b,
John A.
Kalaitzis
c,
Daniel
Vuong
b,
Joe
Bracegirdle
ab,
Fraser
Windsor
a,
Ashli
Lau
a,
Rachel
Chen
b,
Yu Pei
Tan
de,
Alastair
Lacey
b,
Ernest
Lacey
bc,
Andrew M.
Piggott
*c and
Yit-Heng
Chooi
*a
*a,
Andrew
Crombie
b,
John A.
Kalaitzis
c,
Daniel
Vuong
b,
Joe
Bracegirdle
ab,
Fraser
Windsor
a,
Ashli
Lau
a,
Rachel
Chen
b,
Yu Pei
Tan
de,
Alastair
Lacey
b,
Ernest
Lacey
bc,
Andrew M.
Piggott
*c and
Yit-Heng
Chooi
*a
aSchool of Molecular Sciences, The University of Western Australia, Perth, WA 6009, Australia. E-mail: nicolau.sbarainioliveira@uwa.edu.au; yitheng.chooi@uwa.edu.au
bMicrobial Screening Technologies Pty. Ltd, Smithfield, NSW 2164, Australia
cSchool of Natural Sciences, Macquarie University, Sydney, NSW 2109, Australia. E-mail: andrew.piggott@mq.edu.au
dDepartment of Agriculture and Fisheries, Plant Pathology Herbarium, Dutton Park, QLD 4102, Australia
eCentre for Crop Health, University of Southern Queensland, Toowoomba, QLD 4350, Australia
First published on 24th October 2024
Abstract
Depsides have garnered substantial interest due to the diverse biological activities exhibited by members of this class. Among these are the antibacterial aquastatins, glycosylated heteromeric depsides formed through the condensation of orsellinic acid with corticiolic acid. In this work, we isolated aquastatins and the recently described geministatins, along with several novel aquastatin-related depsides with different alkyl side chains from the fungus Austroacremonium gemini MST-FP2131. The structures were determined through comprehensive spectroscopic analysis and chemical degradation. Genome mining and heterologous expression in Aspergillus nidulans and Saccharomyces cerevisiae revealed that aquastatin biosynthesis requires only two genes: a non-reducing polyketide synthase (SAT-KS-AT-PT-ACP-TE) and a glycosyltransferase. We demonstrated that the single polyketide synthase can synthesise an acetyl-primed orsellinic acid and alkylresorcylate with various chain lengths (C14, C16, or C18) by incorporating different long-chain acyl-CoAs as starter units, and then join these as heteromeric depsides. Using chemical degradation, we generated a series of analogues and showed that several aglycone depsides exhibit antibacterial activity against Staphylococcus aureus and methicillin-resistant S. aureus (MRSA), as well as antifungal and cytotoxic activities. Interestingly, heterologous expression of the aquastatin gene cluster in A. nidulans produced higher levels of geministatins with Δ15,16 and Δ18,19 double bonds, which have superior bioactivities compared to the aquastatins but are only present as minor compounds in the native fungus A. gemini.
Introduction
Depsides are a class of compounds produced by the condensation of two or more phenolic carboxylic acids, linked by ester groups. These compounds are commonly found in lichens, but higher plants and free-living fungi can also produce them.1 Over the years, these compounds have attracted significant scientific and commercial interest due to the intriguing bioactive properties that some of them exhibit.1,2 For example, aspergiside A and MS-3 possess antibacterial activity,3,4 while the arenicolins and lecanoric acid demonstrated antitumour activity.5,6 Thielavin W exhibited antifouling properties,7 and atranorin showcased anti-inflammatory, analgesic, and wound healing capabilities without displaying toxicity in in vivo assays.8The biosynthetic mechanisms for depside bond formation were recently elucidated.9–11 The initial step in depside biosynthesis is typically catalysed by an iterative non-reducing polyketide synthase (NR-PKS).9,10 The resulting molecule is further modified by various enzymes such as oxygenases methyltransferases, glycosyltransferases, and others.
Aquastatin A (1a) is a glycosylated heteromeric depside that was initially isolated from Fusarium aquaeductuum as an inhibitor of mammalian adenosine triphosphatases.12 In later studies, 1a was also found to inhibit HIV-1 integrase and fatty acid synthase II (FAS II).13,14 This molecule, along with its aglycone, aquastatin B (1b), demonstrated antibacterial activity against Staphylococcus aureus and methicillin-resistant S. aureus (MRSA).13 The glycosylated phenolic monomer, aquastatin C (1c), was isolated from Sporothrix sp. FN611 along with 1a.15 Unlike 1a, 1b and 1c do not demonstrate FAS II inhibitory or antibacterial activities.15 Furthermore, 1a was patented in 1994 for the treatment of gastric ulcers, demonstrating its diverse therapeutic potential.16
Recently, we described geministatins A and B (2a and 2b) isolated from Austroacremonium gemini MST-FP2131.17 These fungal depsides are closely related to the aquastatins, with both classes consisting of an orsellinic acid unit and a glycosylated alkylresorcylic acid unit, differing only in the length and saturation of the alkyl chain. Moreover, bioactivity assays on the geministatins demonstrated the importance of an unmodified carboxylic acid for antibiotic activity and that the isolated aglycones (2b and dehydromerulinic acid A) exhibited superior bioactivity.17 In this study, we delved further into A. gemini MST-FP2131, which led to isolation of the previously described aquastatins (1a and 1b), geministatins (2a and 2b) and a suite of related molecules with different alkyl chain length and saturation. The chemodiversity of this group of compounds led us to investigate their biosynthesis, which revealed a versatile NR-PKS that can accept diverse long-chain acyl-CoAs, in addition to acetyl and malonyl-CoA, to generate a range of heteromeric depsides. The compounds were shown to exhibit varying levels of antibacterial activities, including anti-MRSA.
Results and discussion
Isolation and structural characterisation of heteromeric depsides from A. gemini MST-FP2131
Depsides related to the aquastatins were detected in the acetone extract of A. gemini cultivated on jasmine rice, as described previously (Fig. S1†).17 The evaporated acetone extract was partitioned between ethyl acetate and water, then fractionated by silica gel chromatography and purified by reversed-phase preparative HPLC (Fig. S1†) to yield the previously reported aquastatins (1a and 1b) and geministatins (2a and 2b), along with two putative novel analogues, which we named ariestatin A (3a) and capricostatin A (4a). The structures of the compounds were determined as described previously,17 using a combination of comprehensive 1H, 13C and 2D NMR analyses, and ozonolysis (Fig. 1). Compound 1a, the major compound in A. gemini (Fig. S2†), along with its aglycone (1b), have been previously described from Fusarium aquaeductuum as aquastatins A and B, respectively.15 We have also recently reported the related metabolites 2a and 2b, which are aquastatin analogues containing a longer (C17) unsaturated (Δ15,16 and Δ18,19) alkyl side chain.17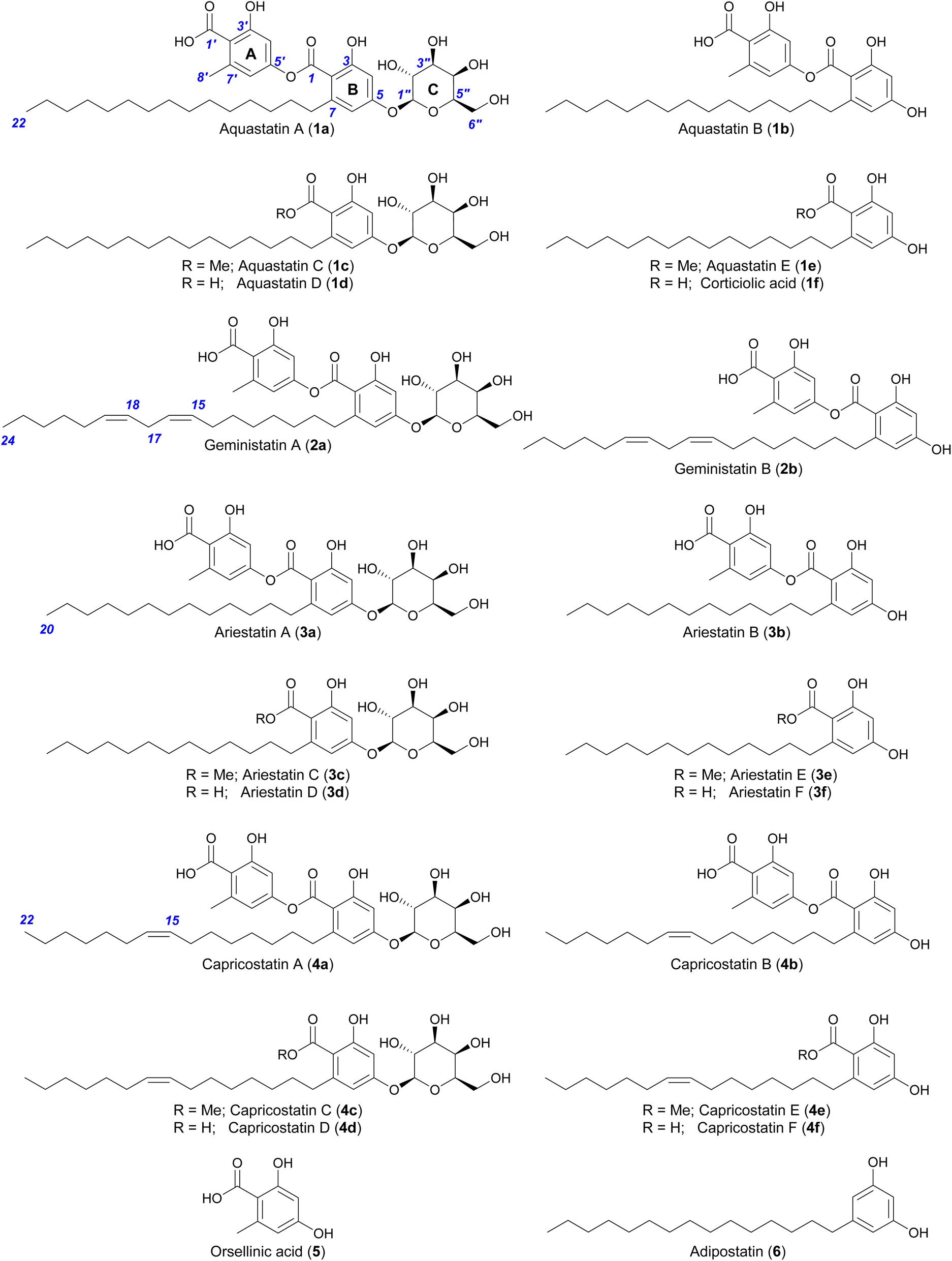 | ||
| Fig. 1 Structures of compounds described in this study. Compounds 1a–b, 2a–b, 3a and 4a were isolated from A. gemini, while the others were generated by chemical degradation. | ||
HR-ESI(−)-MS analysis of ariestatin A (3a) revealed a molecular formula C34H48O12, containing two carbon and four hydrogen atoms (C2H4) fewer than 1a. The 1H and 13C NMR spectra of 3a were essentially identical to those of 1a, with only differences being in the overlapping regions corresponding to the methylene envelope of the alkyl side chain. Close examination of 2D NMR data confirmed 3a is a lower homologue of 1a containing a saturated C15 alkyl side chain (Table S17 and Fig. S26–S31†). HR-ESI(−)-MS analysis of the structurally related capricostatin A (4a) revealed a molecular formula C36H50O12, containing two hydrogen atoms fewer than 1a. The 1H and 13C NMR spectra of 4a were also very similar to those of 1a, with the main difference being the presence of additional signals associated with an isolated double bond (δH 5.29; δC 129.6) in the alkyl side chain. A Z geometry was assigned based on the carbon chemical shifts of the methylene groups adjacent to the double bond (δC 26.5) with literature values for E- (δC 34.4) and Z- (δC 27.0) palmitoleic acid.18 Ozonolysis of 4a in MeOH17 confirmed the position of the double bond to be Δ15,16 and secured the structure of capricostatin A as 4a (Table S20 and Fig. S46–S51†).
In light of the discovery of the novel metabolites 3a and 4a from A. gemini as well as 1a, 1b, 2a and 2b, we set about exploring the possibility that a single biosynthetic gene cluster (BGC) encodes a versatile PKS capable of assembling all of these and possibly other minor analogues not readily observed in organic extracts of A. gemini cultures.
Heterologous expression of aqu biosynthetic gene cluster in Aspergillus nidulans led to production of aquastatin-like molecules
In our endeavour to identify the BGC involved in the biosynthesis of aquastatin-related molecules, we sequenced the genome of A. gemini MST-FP2131. While not all glycosylated polyketides possess integrated glycosyltransferases (GTs) within their BGCs,19 we oriented our search towards BGCs containing a non-reducing polyketide synthase (NR-PKS) and a GT. We located a potential gene cluster (named aqu) comprising four genes, which included a NR-PKS gene (named aquA) with the following domain organisation: starter unit acyltransferase (SAT) domain, keto-synthase (KS) domain, acyltransferase (AT) domain, product template (PT) domain, acyl carrier protein (ACP) domain, and thioesterase (TE) domain. Additionally, the cluster includes a potential resistance to 7-aminocholesterol (RTA1)-like domain-containing protein gene (named aquB), a potential GT from family 28 gene (named aquC), and a potential ABC transporter gene (named aquD) (Fig. 2A). Since no genetic manipulation tool or transformation method has been standardised for A. gemini, we decided to explore this potential gene cluster through heterologous reconstruction of the pathway in Aspergillus nidulans. For heterologous expression, genes aquA, aquB, and aquC were cloned into AMA1-based plasmids, replacing the native promoters with alc promoters, which can be induced using alcohols or ketones and are repressed by glucose.20,21 The heterologous expression of these genes in A. nidulans L08030 (ref. 22) led to the production of several additional compounds, which were characterised as 1a–b, 2a–b and orsellinic acid (5) (Fig. 2B). Interestingly, unlike in A. gemini MST-FP2131, 2a is more abundant than 1a in A. nidulans L08030 (Fig. 2B and S2†). Heterologous expression of aquAB resulted in the biosynthesis of only the aglycones 1b and 2b, with complete abolition of the glycosylated products 1a and 2a, confirming AquC as the GT responsible for attaching galactose to the depside core structure (Fig. 2B).With the function of AquC established, we then set about determining the role of aquB. In the absence of aquB, A. nidulans expressing only aquA yielded 1b and 2b. Similarly, A. nidulans expressing aquAC resulted in the production of the major metabolites 1a and 2a. Taken together, these results suggested that AquB has no catalytic role in the formation of the heteromeric depsides and the genes aquA and aquC are sufficient to generate the glycosylated depsides 1a and 2avia heterologous expression in A. nidulans. More importantly, it suggests that the NR-PKS AquA alone can generate heteromeric depsides, incorporate different acyl-CoAs to generate an alkylresorcylic unit (ring B) and use acetyl-CoA as starter unit to generate an orsellinic acid unit (ring A; Fig. 1). Furthermore, no homomeric depsides were observed in either A. gemini or A. nidulans by LC-MS and HR-ESI-MS analysis (Tables S1 and S2†), suggesting that AquA selectively synthesises heteromeric depsides. Moreover, using extracted ion chromatograms (EIC) and comparison with standards, we could also detect 3a and 4a along with their aglycones (3b and 4b) in A. nidulans strains expressing the aqu BGC (Tables S3 and S4†).
 | ||
| Fig. 2 Heterologous expression of the aqu biosynthetic gene cluster in A. nidulans. (A) The aqu BGC in A. gemini is depicted. Gene names from the aqu cluster are indicated, as well as the backbone gene domain organisation. (B) LC-DAD (254 nm) chromatograms of A. nidulans cultures expressing different combinations of the aqu genes leading to the production of 1a, 1b, 2a and 2b as well as 5 (Fig. 1), as well as A. gemini MST-FP2131 grown on jasmine rice. | ||
Heterologous expression of NR-PKS aquA and precursor feeding in Saccharomyces cerevisiae led to production of geministatin B
A. nidulans is known to produce 5 and lecanoric acid23 and can encode other potential enzymes that modify such products. In order to rule out the potential effect of other enzymes in the biosynthesis of heteromeric depsides and to prove that AquA is the only enzyme required for the biosynthesis of these compounds, we expressed the aquA gene in Saccharomyces cerevisiae, which is not known to produce such aromatic polyketides. cDNA was prepared from an A. nidulans strain expressing aquA, and the intron-free gene was cloned into the plasmid pXW55 (ref. 24) to generate the plasmid pXW55::aquA, which was then transformed into S. cerevisiae BJ5464-NpgA.25–27 Notably, yeast cells expressing aquA were able to produce the heteromeric depsides 1b, 3a and 4a (Fig. 3), thus confirming no involvement of endogenous A. nidulans enzymes in the biosynthesis of these compounds. Like the A. nidulans transformants, no homomeric depsides were detected from these expression cultures through LC-MS and HR-ESI-MS analysis (Table S1†), confirming that the biosynthetic products of AquA are solely heteromeric depsides.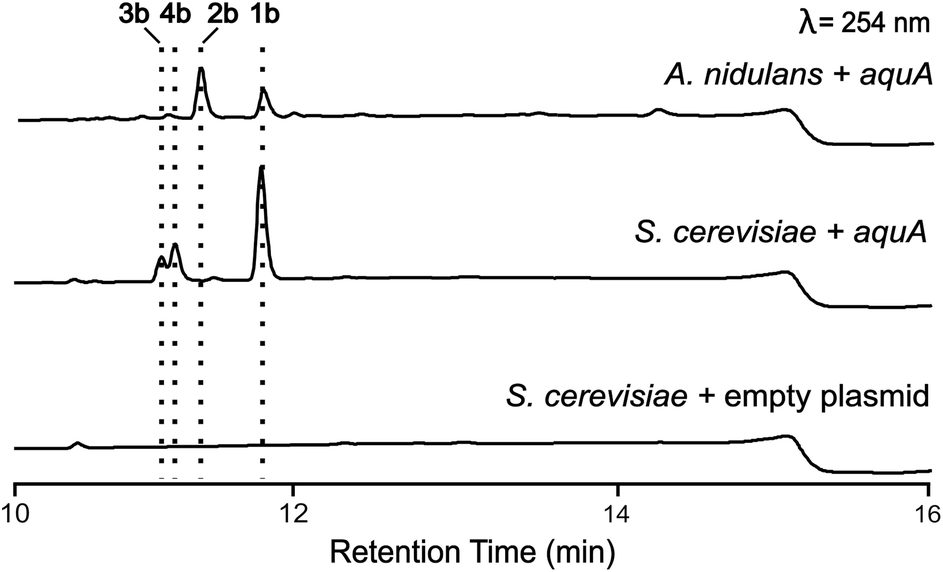 | ||
| Fig. 3 Heterologous expression of the aquA gene in S. cerevisiae. Expansions of LC-DAD (254 nm) chromatograms of A. nidulans and S. cerevisiae cultures expressing aquA. Compounds 1b and 2b can be found in A. nidulans, while 2b was not produced in S. cerevisiae. | ||
The absence of 2b in the above expression (Fig. 3) led us to investigate the assembly of the geministatins, which are related metabolites possessing an unsaturated (Δ15,16 and Δ18,19) alkyl side chain. We hypothesised that the alkyl side chain observed in the geministatins could be derived from linoleic acid (C18:2) given its abundance in A. nidulans.28 Moreover, S. cerevisiae, was previously shown to be incapable of synthesising polyunsaturated fatty acids,29 suggesting that the absence of 2b in S. cerevisiae expressing aquA could be due to the non-availability of the proposed linoleic acid precursor. To investigate this hypothesis, we carried out feeding assays by supplementing the yeast culture medium with linoleic acid. As predicted, this led to an accumulation of 2b (Fig. 4). This outcome suggested that linoleic acid provided to the yeast can be converted into linoleoyl-CoA, which AquA subsequently utilises as a biosynthetic building block to produce the geministatins.
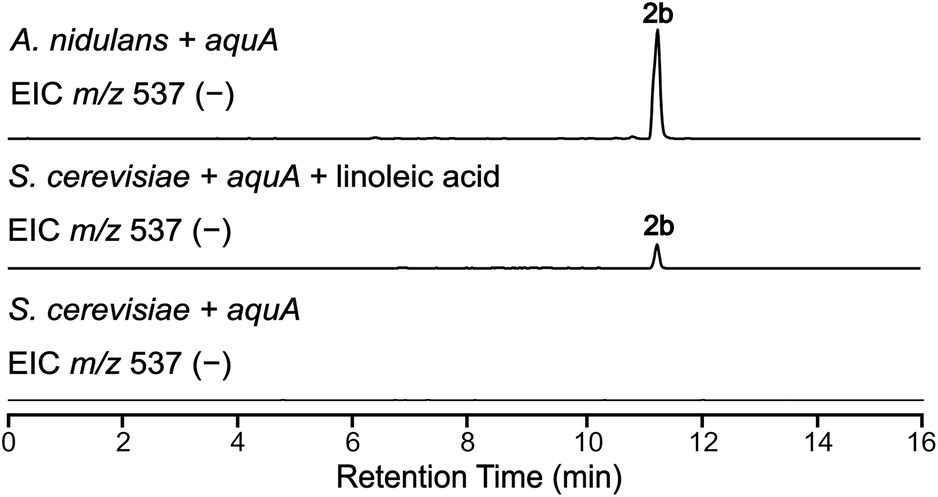 | ||
| Fig. 4 Feeding linoleic acid to S. cerevisiae. EIC(−)MS of S. cerevisiae expressing aquA, both with and without linoleic acid feeding, compared to A. nidulans. | ||
Based on this result, we deduced that the alkyl chain of aquastatins is likely derived from palmitic acid/palmitoyl-CoA (C16:1), while the alkyl chains of 3a and 4a are likely derived from myristic acid/myristoyl-CoA (C14:0) and palmitoleic acid/palmitoleoyl-CoA (C16:1), respectively. Combining the current knowledge of depside biosynthesis with the results obtained above, we postulated possible mechanisms for the biosynthesis of aquastatin-related depsides, which involved incorporation of available long-chain fatty acyl-CoAs that are formed through enzymes encoded by genes that are not clustered within the aqu BGC. The varying abundance of 1a/1b and 2a/2b between A. gemini MST-FP2131, A. nidulans L08030, and S. cerevisiae BJ5464-NpgA suggested that the difference could be attributed to the efficiency of the acyl-CoA ligases in converting various long chain fatty acids to corresponding acyl-CoAs.
NR-PKSs are known to incorporate different acyl-CoAs besides acetyl-CoA. For instance, the norsolorinic acid synthase (PksA) incorporates a hexanoyl-CoA starter unit via the starter-unit acyltransferase (SAT) domain.30 For depside biosynthesis, Wei et al.9 initially demonstrated the thioesterase (TE) domain of AN7909 (OrsA) catalysed depside bond formation to generate lecanoric acid and subsequent Smiles rearrangement to form the diaryl ether, diorcinolic acid. More recently, the TE domain of ThiA was shown to be solely responsible for depside bond formation in the biosynthesis of the trimeric depside, thielavin.31 On the other hand, Chen et al.10 demonstrated that in the duricamidepside biosynthetic pathway, the DrcA SAT domain, instead of TE, is responsible for the depside bond formation. Subsequently, Liu et al.11 used domain swapping and fusions to demonstrate that the PKS Preu6 requires SAT and TE domain–domain interactions to form the depside bond in lecanoric acid biosynthesis.
To gain further insights into the potential mechanism of heteromeric depside formation by AquA, we used phylogenetic analysis to explore the evolutionary proximity of the SAT and TE domains to characterised domains from NR-PKSs producing diorcinolic acid, lecanoric acid, thielavin, and duricamidepside. In the characterisation of thielavin biosynthesis, Ji et al.31 demonstrated through phylogenetic analysis that the TE domains of ThiA and AN7909 group together, while the TE domains of depside-forming NR-PKSs that use the SAT domain for depside bond formation are distantly related to ThiA/AN7909. Additionally, phylogenetic analyses have been used to propose the potential depside bond formation mechanism in the biosynthesis of exophilic acid.32 Interestingly, for both AquA domains, a similar phylogenetic distribution was revealed where the domains of depside-forming NR-PKSs were grouped into two distantly related clades based on whether the depside bond formation involved SAT or TE (Fig. 5B, S118 and S119†). One group includes depside-forming PKS enzymes AN7909 (lecanoric/diorinolic acid), ThiA (thielavin), GyrPKS (gyrophoric acid), ExoA (exophilic acid) as well as aquastatin (AquA), where the TE domains of AN7909 and ThiA have been shown to catalyse depside bond formation (Fig. 5B, S118 and S119†). The other group includes DrcA (duricamidepside) and Preu6 (lecanoric acid), where the SAT domains have been shown to be essential for depside bond formation. This group also includes depside-forming NR-PKSs Atr1 (atranorin), MollE (mollicelin), and DepH (nidulin) (Fig. 5A, S118 and S119†). Moreover, both catalytic residues of the depside bond-forming TE domain identified in ThiA (Ser1937 and His2100) are also conserved in the AquA TE domain.31 In particular, His2100 is only conserved in enzymes where the TE domain is believed to catalyse depside bond formation (Fig. S120†).
 | ||
| Fig. 5 The biosynthesis of aquastatin-related heteromeric depsides. (A) Phylogenetic analysis of TE domain sequences from selected fungal NR-PKSs. TE domain sequences from depside-forming NR-PKSs that utilise the SAT domain for depside bond formation are distantly related to NR-PKSs that employ the TE domain for this purpose. TE-releasing mechanisms are indicated. A more extensive phylogenetic analysis is presented in Fig. S119†. (B) The proposed biosynthetic pathway to the aquastatins. | ||
To explore the function of TE and SAT domains of AquA in vivo, we performed domain deletions on aquA. Heterologous expression of aquA-ΔTE in S. cerevisiae completely abolished compound production, with neither the depside product 1b nor the monomers 1f, and 5 detected. This is consistent with the phylogenetic analysis, suggesting that the TE domain is not only involved in depside bond formation, but also in the hydrolysis of the nascent compound (Fig. S121†). Similarly, no compounds were detected in strains expressing aquA-ΔSAT (Fig. S121†), suggesting that the SAT domain is also required for biosynthesis of 1b. As such, we propose that the SAT domain can load the fatty acyl-CoA as starter unit to the ACP for synthesis of the alkylresorcylic acid unit, while acting as a gatekeeper for selecting a specific range of substrates. Based on these results, we propose the biosynthetic mechanism for heteromeric depside biosynthesis by AquA (Fig. 5B). Additional in vitro experiments using recombinant proteins are required to shed light on how AquA controls the alternating starter unit selectivity and order to yield the two different aromatic monomers.
Recently, Yang et al.33 described how DepH synthesises both heteromeric and homomeric depsides in the biosynthesis of nornidulin and emeguisin A.33 However, in this pathway, heteromeric depside biosynthesis requires an auxiliary highly-reducing (HR) PKS, DepD. In the absence of this auxiliary HR-PKS, the NR-PKS DepH produces lecanoric acid.33 In the case of AquA, it appears that the single NR-PKS can incorporate different long chain acyl-CoAs as starter units to generate an alkylresorcylic unit (ring B) and can also use acetyl-CoA as an alternative starter unit to generate an orsellinic acid unit (ring A), and assemble the two phenolic units in a specific order via an ester bond to form a heteromeric depside selectively. To the best of our knowledge, such an NR-PKS is unprecedented.
MS/MS-based molecular networking and fragment ion search uncovered an even greater diversity of heteromeric depsides
Given that AquA can utilise C14, C16, and C18 substrates (saturated or unsaturated), as evidenced by the production of 1a, 2a, 3a, and 4a, we next explored the extent of this substrate promiscuity using HR-LC-ESI-Orbitrap-MS. Our hypothesis was that other minor compounds with modifications in the alkyl side chain might also be present. Using the Molecular Networking tool in the Thermo Fisher Compound Discoverer software (Fig. S3†) and analysis of the MS/MS spectra, several potential analogues were identified. A characteristic diagnostic fingerprint pattern emerged when we examined the MS/MS spectra of 1a–4a, where the parent ions were fragmented into the decarboxylated ions (−44 u), aglycone ions (1b–4b; −162 u), the glycosylated alkylresorcylate ions (1c–4c; −150 u) and the orsellinate ion (5; m/z 167.0350 [M–H]−) (Fig. S5–S8†). Common to the 1a–4a MS/MS spectra are the [M–H]− ions further fragmented from 5 (m/z 105.0346, 123.0451, and 149.0244) (Fig. S4†), which are also common MS/MS fragments observed for lichen depsides in a previous study.34 This characteristic fragmentation pattern helped to locate several orsellinate-containing depside peaks and to identify other possible structural variations present in the alkyl side chains.Using the above methods, we uncovered a range of putative aquastatin-like heteromeric depsides with alkyl chain lengths ranging from C14 to C18 (Fig. S9–S13†). Interestingly, molecules with alkyl side chain lengths shorter than C14 were not detected by LC-MS and HR-ESI-MS analysis, indicating that myristoyl-CoA is potentially the smallest substrate that can be accepted (Tables S3 and S4†). Additionally, glycosylated heteromeric depsides with structures potentially similar to 2a (linoleoyl-derived), but containing oxygenated unsaturated alkyl chains, were also identified (Fig. S9 and S10†). Compounds 7a (m/z 715.3330 [M–H]−; C38H52O13) and 8a (m/z 731.3278 [M–H]−; C38H52O14) are potentially analogues of 2a with monooxygenated and dioxygenated unsaturated alkyl side chains, respectively (Fig. S9 and S10†). Oxygenated saturated alkyl side chain compounds were also located, including 9a (m/z 691.3332 [M–H]−; C36H52O13), potentially derived from palmitoyl-CoA (C16:0), and 10a (m/z 719.3643 [M–H]−; C38H56O13), potentially derived from stearoyl-CoA (C18:0) (Fig. S11 and S12†). The modifications in these molecules could mirror the oxygenation seen in oxylipins previously identified in Aspergillus spp., which are known to regulate fungal branching and differentiation.35 Moreover, a compound potentially featuring an alkyl side chain derived from linolenic acid (C18:3) was also discernible (11a; m/z 697.3224 [M−H]−; C38H50O12) (Fig. S13†).
It is also noteworthy that several alkylresorcylic acids with variable alkyl side chain lengths, and their corresponding methylated derivatives, were also detected in A. nidulans expressing aqu BGC and S. cerevisiae expressing aquA (Tables S4–S7 and S9†). These monomers could be generated by abortive cycles of AquA or as degradation products, and could be further modified by host enzymes, such as O-methyltransferases or orsellinate decarboxylase36 (to give rise to 6 observed in A. gemini).
Chemical degradation and bioassays reveal the structure–activity relationships of aquastatin-like depsides
The impressive array of bioactivities previously reported for the aquastatins and geministatins,12–14,16 as well as for other depsides,37 and the detection of a range of putative analogues in our MS/MS analysis above, prompted us to generate additional semisynthetic analogues of 1a–4avia chemical degradation to generate a structure–activity relationship for these molecules. This led to production of the corresponding heteromeric depsides (3b and 4b), glycosylated alkylresorcylic acids (1d–4d) and their methyl ester derivatives (1c–4c), as well as the alkylresorcylic acid monomers (1f–4f) and their methyl esters (1e–4e). The structures of the semisynthetic analogues of aquastatins (1c–1f), ariestatins (3b–3f), and capricostatins (4b–4f) were all confirmed using extensive NMR analysis (Tables S15–S19, S21–S25, and Fig. S18–S25, S32–S45 and S52–S81†) and HRMS measurements (Fig. S84–S87, S89–S92 and S94–S98†).We evaluated all the isolated parent molecules and semisynthetic derivatives in bioassays against the Gram-positive bacteria Bacillus subtilis, S. aureus, and MRSA, the yeast S. cerevisiae, mouse myeloma NS-1, and neonatal foreskin fibroblast (NFF) mammalian cell lines (Tables 1 and S13†). Geministatins 2a and 2b have previously displayed antibiotic activity against B. subtilis, S. aureus, and MRSA, and our results here show that 2a and 2b have superior antibacterial activities when compared to the aquastatin depsides (1a and 1b), but with similar cytotoxicity (Table 1). Interestingly, ariestatin (3a and 3b) and capricostatin (4a and 4b) depsides also displayed antibiotic activities. Similar to geministatins, where the aglycone (2b) exhibited increased bioactivity compared to the glycosylated form (2a), aquastatin B (1b), ariestatin B (3b), and capricostatin B (4b) also displayed stronger inhibitory activities in bioassays than their glycosylated forms (Table 1). In particular, the capricostatin aglycone (4b) displayed stronger cytotoxic activity, showing a half-maximal inhibitory concentration (IC50) of 0.05 μM after 96 h against mouse myeloma NS-1 cells (Table 1). These results are similar to those of the related 4f, which also displayed a similar MIC against the same cell line. Interestingly, while 3c did not display antibiotic activity, cytotoxicity was observed against NS-1 cells, while no activity was detected against NFF cells (Table 1). Previously, we reported that geministatin D (the glycosylated alkylresorcylic acid derivative) displayed antifungal activity against S. cerevisiae. Notably, similar molecules 1d, 3d, and 4d exhibited even better antifungal activity than that reported for geministatin D (Table 1). Moreover, these molecules did not exhibit cytotoxicity against either of the mammalian cell lines tested.
| Compound | Minimum inhibitory concentration (MIC; μg mL−1) | IC50 (μM) | ||||
|---|---|---|---|---|---|---|
| B. subtilis | S. aureus | MRSA | S. cerevisiae | NS-1 | NFF | |
| a Controls: B. subtilis ATCC 6633 = tetracycline; S. aureus ATCC 25923 and MRSA ATCC 33592 = ampicillin; S. cerevisiae ATCC 9763 = blasticidin S HCl; NS-1 ATCC TIB-18 and NFF TCC PCS-201 = sparsomycin. * Results from Crombie et al.17 | ||||||
| Aquastatin A (1a) | 1.6 | 12.5 | 1.6 | >200 | >100 | >100 |
| Aquastatin B (1b) | 1.6 | >100 | >50 | >200 | >50 | >100 |
| Aquastatin D (1d) | 3.1 | 12.5 | 6.3 | 3.1 | >50 | >100 |
| Geministatin A (2a)* | 1.6 | 6.3 | 6.3 | >200 | >50 | >100 |
| Geministatin B (2b)* | 0.4 | 3.1 | 1.6 | >200 | >50 | >100 |
| Ariestatin A (3a) | 1.6 | 3.1 | 6.3 | >200 | >100 | >100 |
| Ariestatin B (3b) | 1.6 | 12.5 | 12.5 | >200 | >50 | >50 |
| Ariestatin C (3c) | >100 | >100 | >100 | >200 | 0.2 | >100 |
| Ariestatin D (3d) | 25 | >50 | >50 | 3.1 | >50 | >50 |
| Capricostatin A (4a) | 1.6 | 3.1 | 6.3 | >200 | >100 | >100 |
| Capricostatin B (4b) | 1.6 | 12.5 | 12.5 | >200 | 0.05 | >100 |
| Capricostatin D (4d) | 25 | >50 | >50 | 3.1 | >50 | >100 |
| Capricostatin F (4f) | 0.8 | 6.3 | 6.3 | >200 | 0.05 | >100 |
| Controls | 6.3 | 3.1 | >100 | 3.1 | 1.7 | 1.7 |
In summary, our results suggest that molecules derived from the aqu BGC are attractive targets for further therapeutic investigation, with a myriad of activities. Moreover, our findings demonstrate how modifications in the alkyl side chain and glycosylation can significantly affect the bioactivity of such compounds.
Conclusions
Naturally occurring depsides have garnered significant scientific and commercial interest due to their diverse biological activities, and unsurprisingly, the biosynthetic pathways to these compounds have also attracted considerable recent attention. Here, we have investigated the biosynthetic pathway to the heteromeric fungal depside, aquastatin A. Through compound isolation, heterologous expression, and feeding assays, we have demonstrated the versatility of AquA, a single PKS that can generate a series of heteromeric depsides consisting of orsellinate and alkylresorcylate subunits with variable alkyl side chain lengths. Future experiments could investigate how the PKS selectively couples alkylresorcylic acids with orsellinate in a specific order, without generating any homomeric depsides, and how the PKS utilises an acetyl-CoA starter unit to generate the orsellinate while utilising a range of fatty acyl-CoAs to generate diverse alkylresorcylic acids. Furthermore, bioassays have highlighted the potential of these compounds as antibacterial agents against S. aureus, including MRSA, and demonstrated the antifungal and cytotoxic effects of selected molecules. Given their demonstrated bioactivity efficacy, aquastatin-like molecules could serve as a promising foundation for future modifications. The biosynthesis knowledge uncovered here could lead to biosynthetic approaches to generate novel analogues, which may facilitate the development of novel anti-infectives and anticancer compounds.Data availability
All experimental procedures, and additional experimental and characterisation data are available in the ESI.† The DNA sequence of the aqu BGC has been deposited in NCBI GenBank with the accession number PQ095580.Author contributions
Conceptualisation – NS, EL, AMP, and YHC; investigation – NS, AC, JAK, DV, JB, FW, AL, RC, and YPT; data curation – NS, AC, and JAK; formal analysis – NS, AC, JAK, AMP, and YHC; funding acquisition – AL, EL, AMP, and YHC; supervision – EL, AMP, and YHC; writing – original draft – NS, AC, JAK, and YHC; writing – review & editing – EL, AMP, and YHC.Conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.Acknowledgements
This research was funded by the Australian Research Council (ARC) (LP210100458) and CRC-P (CRCPFIVE119). Y.-H. C. was supported by an ARC Future Fellowship (FT160100233). We would like to thank Prof Berl R. Oakley (University of Kansas) for providing A. nidulans strain LO8030. HR-MS and NMR data is acquired at the UWA Separation Science & Mass Spectrometry Platform (ARC LE190100183) and the UWA's Centre for Microscopy Characterisation and Analysis (CMCA) (LE190100183), respectively. Preparative LC used here was supported by the Western Australia Future Health Research and Innovation Infrastructure Fund (FHRIFGCOVID19/14).Notes and references
- S. R. M. Ibrahim, A. Sirwi, B. G. Eid, S. G. A. Mohamed and G. A. Mohamed, Metabolites, 2021, 11, 683 CrossRef CAS
.
- I. Ureña-Vacas, E. González-Burgos, P. K. Divakar and M. P. Gómez-Serranillos, J. Fungi, 2023, 9, 116 CrossRef
- P. Phainuphong, V. Rukachaisirikul, S. Phongpaichit, J. Sakayaroj, P. Kanjanasirirat, S. Borwornpinyo, N. Akrimajirachoote, C. Yimnual and C. Muanprasat, Tetrahedron, 2018, 74, 5691–5699 CrossRef CAS
- P. Aqueveque, C. L. Céspedes, J. Becerra, M. Dávila and O. Sterner, Z. Naturforsch. C, 2015, 70, 97–102 CrossRef CAS
- B. Perlatti, N. Lan, C. E. Earp, S. AghaAmiri, S. H. Vargas, A. Azhdarinia, G. F. Bills and J. B. Gloer, J. Nat. Prod., 2020, 83, 668–674 CrossRef CAS
- L. A. Roser, P. Erkoc, R. Ingelfinger, M. Henke, T. Ulshöfer, A.-K. Schneider, V. Laux, G. Geisslinger, I. Schmitt, R. Fürst and S. Schiffmann, Biomed. Pharmacother., 2022, 148, 112734 CrossRef CAS
- Z. Han, Y.-X. Li, L.-L. Liu, L. Lu, X.-R. Guo, X.-X. Zhang, X.-Y. Zhang, S.-H. Qi, Y. Xu and P.-Y. Qian, Mar. Drugs, 2017, 15, 128 CrossRef
- E. Studzińska-Sroka, A. Galanty and W. Bylka, Mini-Rev. Med. Chem., 2017, 17, 1 CrossRef
- Q. Wei, Z.-P. Wang, X. Zhang and Y. Zou, J. Am. Chem. Soc., 2022, 144, 9554–9558 CrossRef CAS PubMed
- L. Chen, X. Wei and Y. Matsuda, J. Am. Chem. Soc., 2022, 144, 19225–19230 CrossRef CAS PubMed
- Q. Liu, D. Zhang, S. Gao, X. Cai, M. Yao, Y. Xu, Y. Gong, K. Zheng, Y. Mao, L. Yang, D. Yang, I. Molnár and X. Yang, Angew. Chem., Int. Ed., 2023, 62, e202214379 CrossRef CAS
- K. Hamano, M. Kinoshita-Okami, K. Minagawa, H. Haruyama, T. Kinoshita, T. Hosoya, K. Furuya, K. Kinoshita, K. Tabata, A. Hemmi and K. Tanzawa, J. Antibiot., 1996, 46, 1648–1657 CrossRef PubMed
- Y.-J. Kwon, Y. Fang, G.-H. Xu and W.-G. Kim, Biol. Pharm. Bull., 2009, 32, 2061–2064 CrossRef CAS PubMed
- J. G. Ondeyka, D. L. Zink, A. W. Dombrowski, J. D. Polishook, P. J. Felock, D. J. Hazuda and S. B. Singh, J. Antibiot., 2003, 56, 1018–1023 CrossRef CAS
- Y.-J. Kwon, M.-J. Sohn and W.-G. Kim, J. Antibiot., 2011, 64, 213–216 CrossRef CAS
-
(a)
K. Hamano, K. Tanzawa, T. Hosoya, K. Tabata and T. Kinoshita, JP Pat., JP3775393A, Sankyo Co Ltd, 1993 Search PubMed
- A. Crombie, J. A. Kalaitzis, R. Chen, D. Vuong, A. E. Lacey, E. Lacey, R. G. Shivas, Y. P. Tan, N. Sbaraini, Y.-H. Chooi and A. M. Piggott, J. Antibiot., 2024, 77, 639–646 CrossRef CAS PubMed
- M. S. F. Lie Ken Jie and C. C. Lam, Chem. Phys. Lipids, 1995, 78, 1–13 CrossRef
- L. Xie, L. Zhang, C. Wang, X. Wang, Y. Xu, H. Yu, P. Wu, S. Li, L. Han, A. A. L. Gunatilaka, X. Wei, M. Lin, I. Molnár and Y. Xu, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, E4980–E4989 CAS
- J. Hu, F. Sarrami, H. Li, G. Zhang, K. A. Stubbs, E. Lacey, S. G. Stewart, A. Karton, A. M. Piggott and Y.-H. Chooi, Chem. Sci., 2019, 10, 1457–1465 RSC
-
I. Roux and Y. H. Chooi, in Engineering Natural Product Biosynthesis: Methods and Protocols, ed. E. Skellam, Springer US, New York, NY, 2022, pp. 75–92 Search PubMed
- Y.-M. Chiang, M. Ahuja, C. E. Oakley, R. Entwistle, A. Asokan, C. Zutz, C. C. C. Wang and B. R. Oakley, Angew. Chem., Int. Ed., 2016, 55, 1662–1665 CrossRef CAS
- V. Schroeckh, K. Scherlach, H.-W. Nützmann, E. Shelest, W. Schmidt-Heck, J. Schuemann, K. Martin, C. Hertweck and A. A. Brakhage, Proc. Natl. Acad. Sci. U.S.A., 2009, 106, 14558–14563 CrossRef CAS
- W. Xu, X. Cai, M. E. Jung and Y. Tang, J. Am. Chem. Soc., 2010, 132, 13604–13607 CrossRef CAS
- S. M. Ma, J. W.-H. Li, J. W. Choi, H. Zhou, K. K. M. Lee, V. A. Moorthie, X. Xie, J. T. Kealey, N. A. Da Silva, J. C. Vederas and Y. Tang, Science, 2009, 326, 589–592 CrossRef CAS PubMed
- H. D. Mootz, K. Schörgendorfer and M. A. Marahiel, FEMS Microbiol. Lett., 2002, 213, 51–57 CAS
- K. Michael Lee and N. A. DaSilva, Yeast, 2005, 22, 431–440 CrossRef
- A. M. Calvo, H. W. Gardner and N. P. Keller, J. Biol. Chem., 2001, 276, 25766–25774 CrossRef CAS
- T. C. Ferreira, L. M. P. de Moraes and É. G. Campos, FEMS Yeast Res., 2011, 11, 408–417 CrossRef CAS PubMed
- J. M. Crawford, B. C. R. Dancy, E. A. Hill, D. W. Udwary and C. A. Townsend, Proc. Natl. Acad. Sci. U.S.A., 2006, 103, 16728–16733 CrossRef CAS
- Q. Ji, H. Xiang, W.-G. Wang and Y. Matsuda, Angew. Chem., Int. Ed., 2024, 63, e202402663 CrossRef CAS PubMed
- H. Yang, Z. Shang, Y. Chen, F. Li, K. Li, H. Zhu, M. Peng, J. Yang, C. Cai and J. Ju, Org. Lett., 2024, 26, 8317–8322 CrossRef CAS
- J. Yang, Z. Zhou, Y. Chen, Y. Song and J. Ju, Acta Pharm. Sin. B, 2023, 13, 3919–3929 CrossRef
- S. G. Musharraf, N. Kanwal, V. M. Thadhani and M. I. Choudhary, Anal. Methods, 2015, 7, 6066–6076 RSC
- M. Niu, B. N. Steffan, G. J. Fischer, N. Venkatesh, N. L. Raffa, M. A. Wettstein, J. W. Bok, C. Greco, C. Zhao, E. Berthier, E. Oliw, D. Beebe, M. Bromley and N. P. Keller, Nat. Commun., 2020, 11, 5158 CrossRef CAS
- K. Mosbach and J. Schultz, Eur. J. Biochem., 1971, 22, 485–488 CrossRef CAS
- D. Chen, Z. Song, J. Han, J. Liu, H. Liu and J. Dai, J. Am. Chem. Soc., 2024, 146, 9614–9622 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: General experimental procedures, chemical degradation studies, compound characterisation, heterologous expression, molecular networking analysis, bioactivity screening, phylogenetic analysis and spectroscopic data. See DOI: https://doi.org/10.1039/d4sc05557h |
| This journal is © The Royal Society of Chemistry 2024 |