
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dichlorine–pyridine N-oxide halogen-bonded complexes†
Niklas
Limberg
a,
J. Mikko
Rautiainen
 b,
Jan
Lundell
b,
Sebastian
Riedel
*a,
Kari
Rissanen
*b and
Rakesh
Puttreddy
*b
b,
Jan
Lundell
b,
Sebastian
Riedel
*a,
Kari
Rissanen
*b and
Rakesh
Puttreddy
*b
aDepartment of Chemistry and Biochemistry, Freie Universität Berlin, Fabeckstr. 34/36, 14195 Berlin, Germany. E-mail: s.riedel@fu-berlin.de
bDepartment of Chemistry, University of Jyvaskyla, P.O. BOX 35, FI-40014, Jyväskylä, Finland. E-mail: kari.t.rissanen@jyu.fi; rakesh.r.puttreddy@jyu.fi
First published on 21st October 2024
Abstract
A new Cl–Cl···−O–N+ halogen-bonded paradigm has been demonstrated, using dichlorine as a halogen bond (XB) donor and N-oxide as an XB acceptor. Their crystalline complexes were formed during the warm-up process from −196 °C to −80 °C for X-ray diffraction analysis. They exhibit high instability in the crystalline state, even at these low temperatures, leading to rapid decomposition and the formation of Cl⋯H–O–N hydrogen-bonded complexes. The normalized XB interaction ratio (RXB) of Cl⋯O interactions in the solid-state demonstrates affinity comparable to traditional I⋯O interactions observed in I–I···−O–N+ halogen-bonded systems. The Cl–Cl⋯O XB angles vary from 172° to 177°, manifesting the structure-guiding influence of the electronegative chlorine atom's σ-hole on these XB interactions.
Introduction
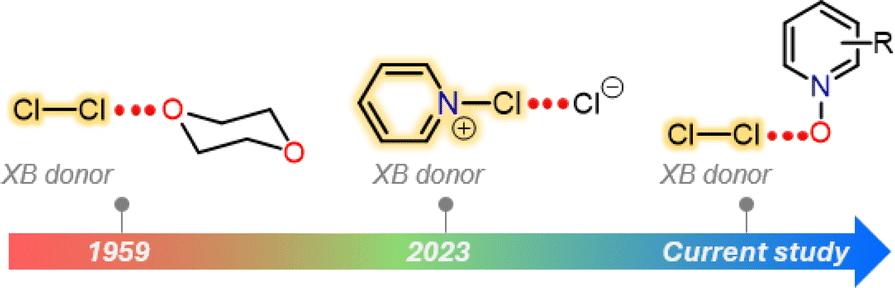
Halogen bonding is an attractive interaction of the type R–X⋯B, where X is a halogen atom, and B can be any kind of Lewis base (e.g., N, O, and S).1 The basis of this interaction is the occurrence of an anisotropic charge distribution around the X-atom, which leads to the formation of a so-called σ-hole, a region of decreased electron density on the extension of a R–X bond.2 The size of the σ-hole has been shown to define the directionality of the XB interaction. This preference was explained by natural bond order analysis of alkyl halides by Clark et al., who described an approximate s2px2py2pz1 configuration (where z is the orientation of the R–X bond) and a deficient electron density site on the halogen atom.3 In general, the heavier iodine and bromine atoms possess larger σ-holes than the lighter chlorine and fluorine atoms enabling the heavier halogen atoms to more effectively accept electron density from a Lewis base. In view of an MO picture the Lewis base donates electron density to a σ* orbital of R−X species. This characteristic feature has been exploited in a palette of I/Br⋯N/O/S XBs for a wide range of applications from crystal engineering to biology.4–6 In contrast, the CI⋯B XBs were exclusively studied using computational approaches.7–9 Their X-ray crystallography data are extremely rare.10,11 In 1950s, Hassel and Strømme solved the X-ray crystal structure of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Cl2·dioxane11 adduct featuring Cl⋯O contacts (ca. 2.678 Å) that are below the sum of the van der Waals radii of Cl- and O-atoms (3.27 Å), indicating an attractive interaction. Almost 70 years later, we have reported the X-ray crystal structures of a series of Cl⋯N XB complexes, including [pentafluoropyridine-Cl]+[AsF6]−, [bis(pyridine)Cl]+[BF4]−, [(lutidine-Cl)+](lutidine)[Cl3]− and a 1:1 [(pyridine-Cl)+][Cl]− adduct (Fig. 1).10 Notably, in all these complexes, the Cl-atom resides extremely close to the pyridinic nitrogen, with Cl⋯N distances ranging between 1.754(2) and 2.232(6) Å.
:1 Cl2·dioxane11 adduct featuring Cl⋯O contacts (ca. 2.678 Å) that are below the sum of the van der Waals radii of Cl- and O-atoms (3.27 Å), indicating an attractive interaction. Almost 70 years later, we have reported the X-ray crystal structures of a series of Cl⋯N XB complexes, including [pentafluoropyridine-Cl]+[AsF6]−, [bis(pyridine)Cl]+[BF4]−, [(lutidine-Cl)+](lutidine)[Cl3]− and a 1:1 [(pyridine-Cl)+][Cl]− adduct (Fig. 1).10 Notably, in all these complexes, the Cl-atom resides extremely close to the pyridinic nitrogen, with Cl⋯N distances ranging between 1.754(2) and 2.232(6) Å.
 | ||
| Fig. 1 X-ray crystal structures of Cl⋯O/N halogen-bonded complexes reported to date. | ||
Pyridine N-oxides (PyNOs) are versatile ambivalent species, allowing them to react with both nucleophilic and electrophilic reagents.12 Consequently, they have received recognition as highly valuable synthetic intermediates in the preparation of pyridines.13 Despite their lower basicity compared to pyridines, their ability to activate various Lewis acids is often adequate to catalyze numerous organic reactions. Thus, the research in PyNO chemistry has centred on organic synthesis, leaving their potential underexplored in supramolecular chemistry. In the context of XB chemistry, considerable efforts have been made in understanding the I⋯O XBs formed by perfluoroiodoalkanes/aromatics and PyNOs by the research groups of Resnati,14,15 Rosokha,16,17 Jin,18–20 and Pennington.21 Although less common, reports of Br⋯O XBs22,23 involving aromatic XB donors and PyNOs also exist. Apart from the above mentioned XBs, complexes involving PyNOs and dihalogens of the type X2·PyNO (X = I) are exclusively known with iodine and have been studied by X-ray crystallography.16,18,24 In contrast, the high affinity of N-oxides for the water content in the atmosphere has hampered the isolation of X2·PyNO (X = Br, Cl, F) complexes for X-ray diffraction studies. For instance, attempts to isolate the highly reactive Br2·PyNO have typically resulted in the formation of [PyNO-H]+[Br]− and [(PyNO)2-H]+[Br]− type hydrogen-bonded complexes25,26 as a result of the reaction with the solvent molecules. Therefore, there is an overwhelming lack of knowledge about the chemistry of X2·PyNO (X = Br, Cl, F) complexes in the solid state.
Here, we report the synthesis, X-ray crystallography, and Density Functional Theory (DFT) studies of dichlorine–PyNO complexes, demonstrating the potential of electronegative chlorine's σ-hole in forming strong Cl–Cl···−O–N+ XBs comparable in strength to I–I···−O–N+ XBs.
Results and discussion
The dichlorine–PyNO halogen-bonded complexes are formed by condensing one millimole of elemental Cl2 onto one millimole of appropriate PyNO dissolved in propionitrile at −196 °C in a Schlenk tube with a PTFE valve. The solutions were warmed to −80 °C and maintained at that temperature to form crystals suitable for X-ray diffraction analysis. Despite our rigorous attempts to broaden the scope, only two crystal structures, Cl2-PyNO and Cl2-26DiMePyNO (26DiMePyNO = 2,6-dimethylpyridine N-oxide) could be isolated and characterized by X-ray diffraction analysis. Due to the polydentate coordination mode of N-oxide oxygen and its strong affinity for protonation and formation of hydrogen-bonded complexes, the Cl⋯O XB complexes exhibited high instability, transforming into hydrogen-bonded complexes of the type [PyNO-H]+[Cl]− and [(PyNO)2-H]+[Cl3]− (Fig. 1 and S1–S8†). For instance, when the Schlenk tube containing Cl2-PyNO and Cl2·(26DiMePyNO) complexes was opened in an argon environment to select crystals for X-ray crystallography, their bulk samples immediately reacted with water traces from air to form [PyNO-H]+[Cl]− and [26DiMePyNO-H]+[Cl]− complexes, respectively (Fig. 2). Furthermore, it is important to note that the chlorine complex of 3-methoxypyridine N-oxide exhibited strong reactivity during the warming process from −196 to −80 °C, resulting in shattering of the Schlenk tubes. These aspects prompted us to limit our current investigation to X-ray crystallography of two XB complexes and DFT studies.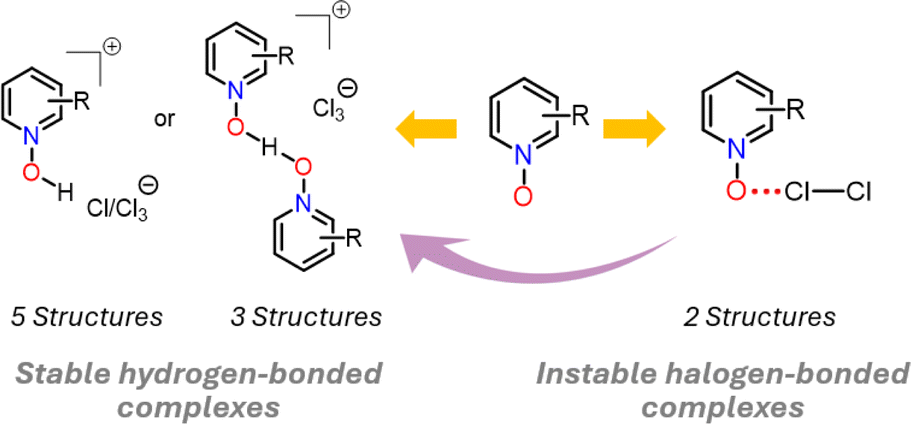 | ||
| Fig. 2 A summary of X-ray crystal structures of halogen- and hydrogen-bonded complexes. | ||
Electrostatic potential (ESP, Vs,max) maps were computed at the PBE0-D3/def2-TZVP27–35 level of theory (Fig. 3a–e) to compare the σ-hole strength of Cl2 with a more common I2 XB donor. Despite the large electronegativity differences, the σ-hole strength of dichlorine and diiodine differs only by 30 kJ mol−1. Furthermore, the Vs,max for Cl2 (+108 kJ mol−1) exceeds that of aromatic XB donors, e.g., C6H5Cl (+20 kJ mol−1) and C6F5Cl (+74 kJ mol−1), as well as non-aromatic N-chlorosuccinimide (NCS, +92 kJ mol−1). The Vs,max values follow the XB donor strength order: I2 > Cl2 > NCS > C6F5Cl > C6H5Cl, suggesting that Cl2 is the strongest XB donor among the Cl-donors from an electrostatic perspective.
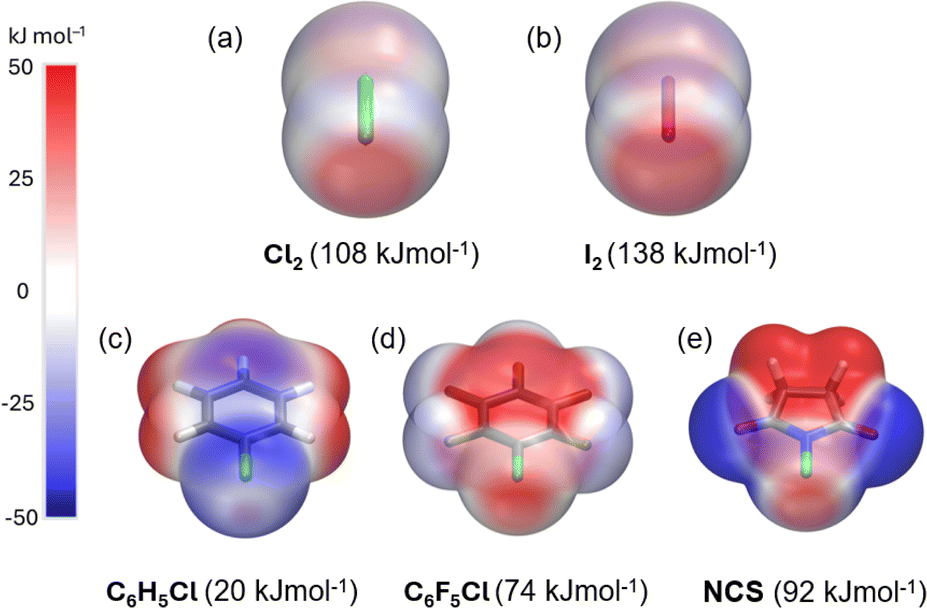 | ||
| Fig. 3 Computed electrostatic potential surface (ESP) at PBE0-D3/def2-TZVP level projected on the 0.001 au electron density surfaces of XB donors with VS,max values, (a) Cl2, (b) I2, (c) C6H5Cl, (d) C6F5Cl, and (e) NCS. | ||
The 1:1 donor:acceptor complexes of Cl2-PyNO and Cl2-26DiMePyNO are shown in Fig. 4a and b. The asymmetric unit of Cl2-PyNO consists of two molecules of Cl2 and two molecules of PyNO, whereas the asymmetric unit of Cl2-26DiMePyNO consists of half a molecule of Cl2 and a full 26DiMePyNO. Their packing structures revealed that both complexes form infinite 1D polymeric chains via short Cl⋯O XB contacts (Fig. 1). The bidentate N-oxide oxygen atoms connect the dichlorine molecules. The interatomic distances between Cl and O atoms in Cl2-PyNO range from 2.567(5) Å to 2.627(5) Å, while in Cl2-26DiMePyNO, it is 2.5676(8) Å.
 | ||
| Fig. 4 The X-ray crystal structures of (a) Cl2-PyNO and (b) Cl2-26DiMePyNO with the thermal displacement parameter at the 50% probability level. Disordered parts in Cl2-PyNO are omitted for clarity. | ||
Their normalized interaction ratio (RXB)36,37 values are between 0.79–0.80. Notably, the RXB values of Cl⋯O contacts in Cl2-PyNO are similar to those of I⋯O interactions (2.684(8)–2.791(8) Å, RXB = 0.77–0.80) in the reported l2-PyNO complex.17 The Cl–Cl⋯O angles vary between 171.98(5)° and 177.09(18)°. These characteristics collectively indicate a strong interaction between chlorine and N-oxide oxygen in the form of XBs.
The Cl–Cl···−O–N+ XB interaction energies (ΔEXB) in Cl2-PyNO and Cl2-26DiMePyNO complexes with a 1:1 donor:acceptor ratio were calculated and compared with the I–I···−O–N+ energies in l2-PyNO and l2-26DiMePyNO complexes, respectively (Table S1†). The optimized Cl⋯O distances in Cl2-PyNO and Cl2-26DiMePyNO are 2.446 and 2.410 Å, respectively, which are 0.152 Å and 0.158 Å shorter than those observed in their crystal structures. The ΔEXB values of Cl⋯O contacts are smaller than those of I⋯O XBs, with the ΔEXB values of Cl/I···−O–N+ XBs of unsubstituted PyNO complexes being smaller than those of 26DiMePyNO complexes (Fig. 5). This difference can be attributed to the larger nucleophilic character of the 26DiMePyNO's oxygen due to the electron-donating ortho-methyl groups. In comparison to the Cl⋯O XBs in the reported Cl2-dioxane11 complex, which has an energy of −20 kJ mol−1, the Cl···−O–N+ interaction energies are larger, varying between −29 and −36 kJ mol−1. This can be attributed to the efficient overlap of the negatively charged N-oxide oxygen atom lone pair and p-orbital of the donor chlorine as compared to the uncharged ether oxygen atom.
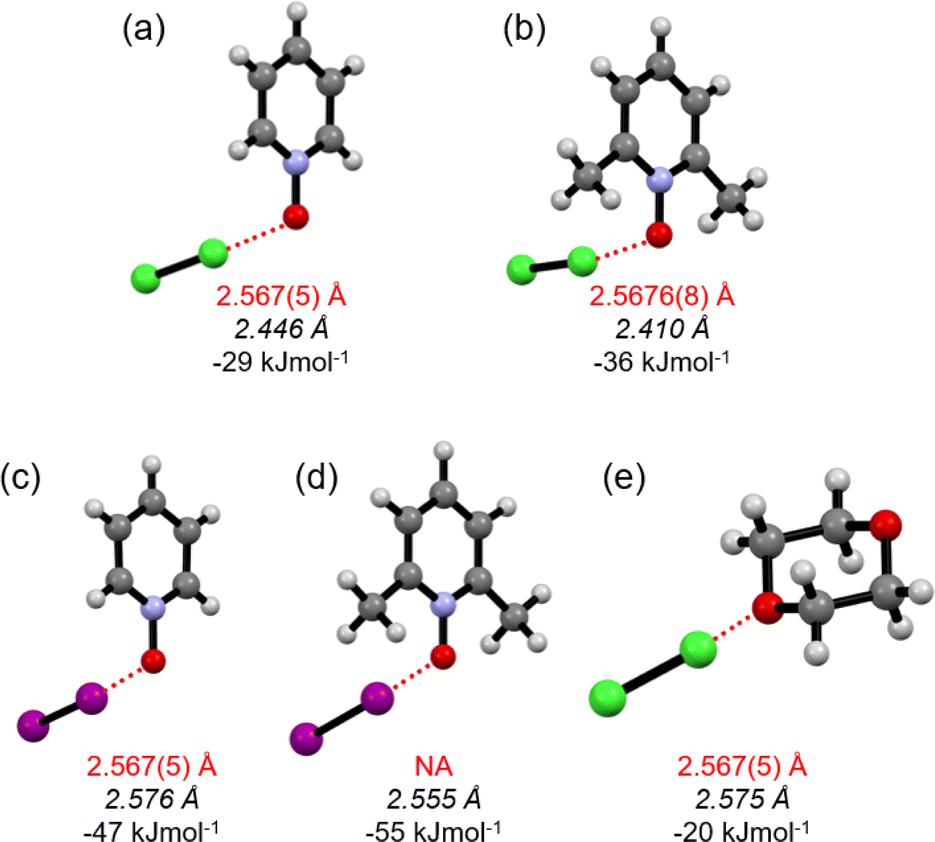 | ||
| Fig. 5 The DFT structures of (a) Cl2-PyNO (b) Cl2-26DiMePyNO, (c) I2-PyNO (d) I2-26DiMePyNO and (e) Cl2-dioxane optimized at the PBE0-D3/def2-TZVP level of theory. Red broken lines represent XBs. The red font represents XB distances of the crystal structure and the italicized black font represents XB distances of DFT optimized structures. NA: crystal structure is not available. | ||
The protonated N-oxide stabilized by the chloride or trichloride counter anion is the common and stable outcome of these crystallizations. Attempts to model the Cl⋯H–−O–N+ hydrogen-bonded structures of [PyNO-H]+[Cl]− and [(26DiMePyNO)-H]+[Cl]− in the gas phase resulted in non-charge separated 1:1 adducts of HCl and PyNO and HCl and 26DiMePyNO with Cl–H···−O–N+ energies of −59 and −71 kJ mol−1, respectively. The Cl–H···−O–N+ energies are roughly twice as high as the Cl–Cl···−O–N+ XB interaction energies. An estimation of the Cl−···H+–−O–N+ interaction energy can be made by single point calculation of the [(26DiMePyNO)-H]+[Cl]− in the crystal structure environment showing −435 kJ mol−1 as the interaction energy between charged species.
The 15N nitrogen chemical shifts in nitrogen-metal/halogen-bonded complexes are lower compared to their non-coordinated forms. The lower 15N nitrogen chemical shifts in complexes can be attributed to the decrease in the paramagnetic deshielding term on the pyridinic nitrogen during metal38,39/halogen-bond40,41 complexation, caused by the replacement of n–π* electron transitions with σ–π* transitions.42,43 In principle, this can be experimentally determined by 15N NMR spectroscopy by estimating the coordination shift Δδ(15N), which is the difference in δ(15N) chemical shift between a complex (δ15Ncompl) and its corresponding pyridine ligand (δ15Nlig). However, owing to the difficulties in handling dichlorine-PyNO samples for NMR studies, we used the DFT calculations to analyse 15N NMR chemical shifts of N-oxides in their uncomplexed and complexed forms to estimate Δδ(15N) values (Table 1 and S2†). The isotropic shielding constants were computed at the relativistic level using the ZORA–PBE0/TZ2P46,47 method using a COSMO48 model to treat the solvation effects. The chemical shifts were determined by calibrating the NIS shift to the experimental shift and scaling the chemical shifts of N-oxide ligands and their complexes correspondingly. To determine the coordination power of N-oxides in Cl2-PyNO and Cl2-26DiMePyNO, their 15N NMR chemical shifts were compared to the calculated 15N chemical shifts of I2-PyNO, I2-26DiMePyNO, NIS-PyNO and NIS-26DiMePyNO.
| 15N Chemical shift (ppm) | Δδ(15N) = δ15Ncompl − δ15Nlig (ppm) | |
|---|---|---|
| a The values in parentheses represent experimental 15N NMR data acquired in CDCl3 (ref. 44 and 45) | ||
| PyNO | −90 (−90)a | — |
| 26DiMePyNO | −95 (−95)a | — |
| Cl2-PyNO | −106 | −16 |
| Cl2-26DiMePyNO | −111 | −16 |
| I2-PyNO | −111 | −21 |
| I2-26DiMePyNO | −116 | −21 |
| NIS-PyNO | −110 | −20 (−9)a |
| NIS-26DiMePyNO | −116 | −21 (−13)a |
The 15N chemical shifts of PyNO and 26DiMePyNO are −90 ppm and −95 ppm, respectively. Additionally, their chlorine and iodine/NIS complexes exhibit shifts ranging between −106 and −116 ppm (Table 1). The observed trend in Δδ(15N) values indicates similar Δδ(15N) values for both PyNO and 26DiMePyNO complexes, regardless of the XB interaction type. The difference in coordination powers suggested by the ΔEXB values of PyNO and 26DiMePyNO complexes appears not to be significant enough to affect the Δδ(15N) values. The magnitude of the Δδ(15N) values of chlorine complexes is slightly smaller than that of the iodine/NIS complexes, indicating that the Cl–Cl⋯−O–N+ halogen-bonded systems formed by Cl2 and N-oxides demonstrate the n(O) → σ*(Cl) electron donation strength almost to the same extent as the N–I/I–I⋯−O–N+ systems. The comparison between this small set of solid-state and DFT results and our comprehensive study on N/C−I⋯−O–N+ XBs44,49 suggests that the structure directing element of the Cl⋯O interactions is the chlorine σ-hole.
Experimental
Materials
Pyridine N-oxide (>95%) was purchased from Sigma-Aldrich, whereas 3-phenylpyridine N-oxide, 3-methoxypyridine N-oxide, 2-methyl-4-nitropyridine N-oxide, 2,6-dimethylpyridine N-oxide, 2,4-dimethylpyridine N-oxide, 2-methyl-4-methoxy pyridine N-oxide, 2,4,6-trimethylpyridine N-oxide, and 4-tert-butylpyridine N-oxide were synthesized using the literature method.50 Chlorine was purchased from Linde. Propionitrile (>90%) was purchased from Sigma-Aldrich. Prior to crystallization experiments, the N-oxides were vacuum-dried overnight, and SPS propionitrile was dried over 4 Å molecular sieves.X-ray crystallography
The X-ray crystal structure data of all crystal structures were collected at 100 K, using a Bruker D8 Venture diffractometer equipped with a CMOS area detector and Mo-Kα (λ = 0.71073 Å) radiation. APEX5 (version v2023.9-2) was used for the data collection and reduction. The intensities were absorption corrected using a multi-scan absorption correction method. All structures were solved by intrinsic phasing (SHELXT)51 and refined by full-matrix least squares on F2 using the OLEX2 (ref. 52) and utilizing the SHELXL-2015 (ref. 51) module. Anisotropic displacement parameters were assigned to non-H atoms and isotropic displacement parameters for all H atoms were constrained to multiples of the equivalent displacement parameters of their parent atoms with Uiso(H) = 1.2 Ueq(parent atom).DFT calculations
To be consistent with our earlier halogen bonding studies on N-oxide systems,37,44 DFT calculations were carried out with the PBE0 hybrid functional28,30,31 employing def2-TZVP basis sets34,35 and treating dispersion interactions with the empirical D3 model by Grimme that includes Becke–Johnson damping.32,33 The counterpoise method was used to derive basis set superposition error corrected complexation energies.53 The Gaussian 16 program package54 was used for DFT optimizations. Calculations for isotropic 15N chemical shielding constants55 were carried out with the ADF 2022.1 program56 at the relativistic two-component ZORA46,47 level using the PBE0 hybrid functional and TZ2P57 basis sets. The chloroform solvent (dielectric constant ε = 4.81) environment was simulated using the implicit COSMO48 solvent model (for more details, see the ESI†).Synthesis of dichlorine–N-oxide complexes
Caution! The reaction mixtures can react violently during the warming process from −196 to −80 °C. Care must be taken when handling the samples.
Conclusions
The combined findings from X-ray crystallography and theoretical investigations of dichlorine-pyridine N-oxide and dichlorine-2,6-dimethylpyridine N-oxide halogen-bonded complexes indicate that dichlorine serves as a moderately strong XB donor and it's σ-hole exerts a guiding influence on the halogen-bonded systems. The unsubstituted pyridine N-oxide forms a weaker Cl⋯O interaction (−29 kJ mol−1) in contrast to the 2,6-dimethylpyridine N-oxide (−36 kJ mol−1) with two electron-donating methyl groups proximal to the N–O group. The Cl–CI···−O–N+ halogen-bonded systems exhibit structural and energetic similarities to I–I···−O–N+ systems and comparable energy levels to (imide) N–I···−O–N+ systems, yet the chlorine complexes display high instability. Thus, broadening the Cl⋯O scope, while highly desirable, poses a formidable challenge. Nevertheless, the role of the σ-hole on the electronegative chlorine should not be underestimated when investigating interactions between chlorinated species and Lewis bases.Data availability
The ESI data† for this article include X-ray crystallography data and DFT optimized structures as xyz files. Crystal structures are deposited in The Cambridge Crystallographic Data Centre (CCDC). Deposition numbers 2383807 (for Cl2-PyNO), 2383799 (for Cl2-26DiMePyNO), 2383800 (for [PyNOH][Cl]), 2383803 (for H[2PhPyNO][Cl3]) 2383804 (for H[246TriMePyNO]2[Cl3]), 2383802 (for H[24DiMePyNO]2[Cl3]·2[24DiMePyNO]), 2383801 (for H[2Me4OMePyNO]2[Cl3]), 2383798 (for [26DiMePyNOH][Cl]), 2383806 (for [2Me4OMePyNOH][Cl]), and 2383805 (for [4tBuPyNOH][Cl]) contain the ESI† crystallography data for this paper.Author contributions
R. P. designed the project and wrote the manuscript. R. P. and N. L. carried out the crystallization experiments and chlorination reactions. N. L. did the X-ray crystallography. J. M. R. carried out the computational studies. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the Research Council of Finland [grant number 351121 (KR), 332023 (JL), and 338733 (JMR)]. The work was also funded by the European Research Council (ERC) Project HighPotOx (ID: 818862) (SR). We gratefully acknowledge the assistance of the Core Facility BioSupraMol at FU Berlin which is supported by the DFG.References
- G. R. Desiraju, P. Shing Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178–11189 RSC.
- T. Clark and A. Heßelmann, Phys. Chem. Chem. Phys., 2018, 20, 22849–22855 RSC.
- M. R. Scholfield, C. M. Vander Zanden, M. Carter and P. S. Ho, Protein Sci., 2013, 22, 139–152 CrossRef CAS PubMed.
- R. Wilcken, M. O. Zimmermann, A. Lange, S. Zahn and F. M. Boeckler, J. Comput. Aided Mol. Des., 2012, 26, 935–945 CrossRef CAS PubMed.
- P. Auffinger, F. A. Hays, E. Westhof and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16789–16794 CrossRef CAS PubMed.
- D. Sutradhar and A. K. Chandra, ChemistrySelect, 2020, 5, 554–563 CrossRef CAS.
- Q. Li, Q. Lin, W. Li, J. Cheng, B. Gong and J. Sun, ChemPhysChem, 2008, 9, 2265–2269 CrossRef CAS.
- J. W. Zou, Y. J. Jiang, M. Guo, G. X. Hu, B. Zhang, H. C. Liu and Q. Sen Yu, Chem.–Eur. J., 2005, 11, 740–751 CrossRef CAS PubMed.
- P. Pröhm, W. Berg, S. M. Rupf, C. Müller and S. Riedel, Chem. Sci., 2023, 14, 2325–2329 RSC.
- O. Hassel, K. O. Strømme, E. Hammarsten, C.-G. Hedén, B. Malmgren and H. Palmstierna, Acta Chem. Scand., 1959, 13, 1775–1780 CrossRef CAS.
- P. E. Alford, in Progress in Heterocyclic Chemistry, ed. G. W. Gribble and J. A. Joule, Elsevier, 2011, vol. 23, pp. 329–369 Search PubMed.
- I. Habib, K. Singha and M. Hossain, ChemistrySelect, 2023, 8, e202204099 CrossRef CAS.
- C. Cavallotti, P. Metrangolo, F. Meyer, F. Recupero and G. Resnati, J. Phys. Chem. A, 2008, 112, 9911–9918 CrossRef CAS PubMed.
- A. Forni, P. Metrangolo, T. Pilati and G. Resnati, Cryst. Growth Des., 2004, 4, 291–295 CrossRef CAS.
- Y. P. Nizhnik, A. Sons, M. Zeller and S. V Rosokha, Cryst. Growth Des., 2018, 18, 1198–1207 CrossRef CAS.
- W. Borley, B. Watson, Y. P. Nizhnik, M. Zeller and S. V Rosokha, J. Phys. Chem. A, 2019, 123, 7113–7123 CrossRef CAS PubMed.
- W. X. Wu, H. C. Liu and W. J. Jin, Chem.–Eur. J., 2022, 28, e202103336 CrossRef CAS PubMed.
- W. J. Liang, H. Wang, X. Chen, T. T. Zhang, Y. F. Bai, F. Feng and W. J. Jin, Chempluschem, 2021, 86, 252–258 CrossRef CAS PubMed.
- W. X. Wu, H. Wang and W. J. Jin, Cryst. Growth Des., 2018, 18, 6742–6747 CrossRef CAS.
- C. W. Padgett, R. Dean, A. Cobb, A. Miller, A. Goetz, S. Bailey, K. Hillis, C. McMillen, S. Toney, G. L. Guillet, W. Lynch and W. T. Pennington, Cryst. Growth Des., 2024, 24, 2425–2438 CrossRef CAS PubMed.
- C. B. Aakeröy, P. D. Chopade and J. Desper, Cryst. Growth Des., 2013, 13, 4145–4150 CrossRef.
- T. Shirman, M. Boterashvili, M. Orbach, D. Freeman, L. J. W. Shimon, M. Lahav and M. E. Van Der Boom, Cryst. Growth Des., 2015, 15, 4756–4759 CrossRef CAS.
- K. Wzgarda-Raj, A. J. Rybarczyk-Pirek, S. Wojtulewski, E. Pindelska and M. Palusiak, Struct. Chem., 2019, 30, 827–833 CrossRef CAS.
- V. V. Romanov, Y. P. Nizhnik and A. D. Fofanov, J. Struct. Chem., 2015, 56, 365–369 CrossRef CAS.
- A. V. Ryzhakov, V. P. Andreev, P. S. Sobolev and V. A. Tafeenko, Russ. J. Gen. Chem., 2017, 87, 215–218 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, M. Häser, H. Patzelt and R. Ahlrichs, Chem. Phys. Lett., 1998, 294, 143–152 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- R. Puttreddy, J. M. Rautiainen, T. Mäkelä and K. Rissanen, Angew. Chem., Int. Ed., 2019, 58, 18610–18618 CrossRef CAS PubMed.
- L. Pazderski, Magn. Reson. Chem., 2008, 46, S3–S15 CrossRef PubMed.
- L. Pazderski, 15N NMR coordination shifts in transition metal complexes and organometallics with heterocycles containing nitrogen—Update for 2012–20, Annual Reports on NMR Spectroscopy, Academic Press, 2020, vol. 101, pp. 151–284 Search PubMed.
- R. Kleinmaier, S. Arenz, A. Karim, A.-C. C. Carlsson and M. Erdélyi, Magn. Reson. Chem., 2013, 51, 46–53 CrossRef CAS.
- D. Sethio, G. Raggi, R. Lindh and M. Erdélyi, J. Chem. Theory Comput., 2020, 16, 7690–7701 CrossRef CAS PubMed.
- R. Marek, in Encyclopedia of Spectroscopy and Spectrometry, ed. J. C. Lindon, G. E. Tranter and D. W. Koppenaal, Academic Press, Oxford, 3rd edn, 2017, pp. 110–116 Search PubMed.
- R. Marek and A. Ly Ka, N NMR Spectroscopy in Structural Analysis, 2002, vol. 6 Search PubMed.
- R. Puttreddy, J. M. Rautiainen, S. Yu and K. Rissanen, Angew. Chem., Int. Ed., 2023, 62, e202307372 CrossRef CAS PubMed.
- R. Puttreddy, O. Jurček, S. Bhowmik, T. Mäkelä and K. Rissanen, Chem. Commun., 2016, 52, 2338–2341 RSC.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef CAS.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS.
- E. van Lenthe, A. Ehlers and E.-J. Baerends, J. Chem. Phys., 1999, 110, 8943–8953 CrossRef CAS.
- J. M. Rautiainen, A. Valkonen, J. Lundell, K. Rissanen and R. Puttreddy, Adv. Sci., 2024, 11, 2403945 CrossRef CAS PubMed.
- I. Prat, D. Font, A. Company, K. Junge, X. Ribas, M. Beller and M. Costas, Adv. Synth. Catal., 2013, 355, 947–956 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- M. Krykunov, T. Ziegler and E. van Lenthe, Int. J. Quantum Chem., 2009, 109, 1676–1683 CrossRef CAS.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- E. Van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2383798–2383807. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc06270a |
| This journal is © The Royal Society of Chemistry 2024 |