
Manganese-based A-site high-entropy perovskite oxide for solar thermochemical hydrogen production†
Cijie
Liu‡
a,
Dawei
Zhang‡
b,
Wei
Li
 *a,
Jamie A.
Trindell
c,
Keith A.
King
c,
Sean R.
Bishop
d,
Joshua D.
Sugar
c,
Anthony H.
McDaniel
c,
Andrew I.
Smith
d,
Perla A.
Salinas
d,
Eric N.
Coker
d,
Arielle L.
Clauser
c,
Murugesan
Velayutham
ef,
Joerg C.
Neuefeind
g,
Jingjing
Yang
b,
Héctor A.
De Santiago
a,
Liang
Ma
a,
Yi
Wang
a,
Qiang
Wang
h,
Wenyuan
Li
i,
Qingsong
Wang
j,
Qingyuan
Li
a,
Hanchen
Tian
a,
Ha Ngoc
Ngan Tran
i,
Xuemei
Li
i,
Brandon
Robinson
i,
Angela M.
Deibel
a,
Gregory
Collins
a,
Nhat Anh
Thieu
a,
Jianli
Hu
i,
Valery V.
Khramtsov
ef,
Jian
Luo
*bk and
Xingbo
Liu
*a
*a,
Jamie A.
Trindell
c,
Keith A.
King
c,
Sean R.
Bishop
d,
Joshua D.
Sugar
c,
Anthony H.
McDaniel
c,
Andrew I.
Smith
d,
Perla A.
Salinas
d,
Eric N.
Coker
d,
Arielle L.
Clauser
c,
Murugesan
Velayutham
ef,
Joerg C.
Neuefeind
g,
Jingjing
Yang
b,
Héctor A.
De Santiago
a,
Liang
Ma
a,
Yi
Wang
a,
Qiang
Wang
h,
Wenyuan
Li
i,
Qingsong
Wang
j,
Qingyuan
Li
a,
Hanchen
Tian
a,
Ha Ngoc
Ngan Tran
i,
Xuemei
Li
i,
Brandon
Robinson
i,
Angela M.
Deibel
a,
Gregory
Collins
a,
Nhat Anh
Thieu
a,
Jianli
Hu
i,
Valery V.
Khramtsov
ef,
Jian
Luo
*bk and
Xingbo
Liu
*a
aDepartment of Mechanical and Aerospace Engineering, Benjamin M. Statler College of Engineering and Mineral Resources, West Virginia University, Morgantown, WV 26506, USA. E-mail: wei.li@mail.wvu.edu; xingbo.liu@mail.wvu.edu
bProgram in Materials Science and Engineering, University of California San Diego, La Jolla, CA 92093, USA
cSandia National Laboratories, Livermore, CA 94551, USA
dSandia National Laboratories, Albuquerque, NM 87123, USA
eIn Vivo Multifunctional Magnetic Resonance Center, Robert C. Byrd Health Sciences Center, West Virginia University, Morgantown, WV 26506, USA
fDepartment of Biochemistry and Molecular Medicine, School of Medicine, West Virginia University, Morgantown, WV 26506, USA
gChemical and Engineering Materials Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
hShared Research Facilities, West Virginia University, Morgantown, WV 26506, USA
iDepartment of Chemical and Biomedical Engineering, Benjamin M. Statler College of Engineering and Mineral Resources, West Virginia University, Morgantown, WV 26506, USA
jDepartment of Chemistry, Bavarian Center for Battery Technology (BayBatt), University of Bayreuth, Universitätsstrasse 30, Bayreuth, 95447, Germany
kDepartment of Nano and Chemical Engineering, University of California San Diego, La Jolla, CA 92093, USA. E-mail: jluo@alum.mit.edu
First published on 18th December 2023
Abstract
Non-stoichiometric perovskite oxides have been studied as a new family of redox oxides for solar thermochemical hydrogen (STCH) production owing to their favourable thermodynamic properties. However, conventional perovskite oxides suffer from limited phase stability and kinetic properties, and poor cyclability. Here, we report a strategy of introducing A-site multi-principal-component mixing to develop a high-entropy perovskite oxide, (La1/6Pr1/6Nd1/6Gd1/6Sr1/6Ba1/6)MnO3 (LPNGSB_Mn), which shows desirable thermodynamic and kinetics properties as well as excellent phase stability and cycling durability. LPNGSB_Mn exhibits enhanced hydrogen production (∼77.5 mmol moloxide−1) compared to (La2/3Sr1/3)MnO3 (∼53.5 mmol moloxide−1) in a short 1 hour redox duration and high STCH and phase stability for 50 cycles. LPNGSB_Mn possesses a moderate enthalpy of reduction (252.51–296.32 kJ (mol O)−1), a high entropy of reduction (126.95–168.85 J (mol O)−1 K−1), and fast surface oxygen exchange kinetics. All A-site cations do not show observable valence changes during the reduction and oxidation processes. This research preliminarily explores the use of one A-site high-entropy perovskite oxide for STCH.
1 Introduction
Hydrogen fuels are promising, environmentally-friendly energy sources with high efficiency and renewability.1–5 To realize large-scale hydrogen production and store intermittent solar energy, solar-driven thermochemical hydrogen (STCH) attracts considerable attention because it utilizes the full spectrum of solar energy through a two-step redox cycle.6–10 (1) The first step of the process involves the reduction of metal oxide under high temperatures (e.g., T > 1473 K) and low PO2 (partial pressure of oxygen), resulting in the production of oxygen vacancies. (2) The second step involves oxidizing the metal oxide with steam at low temperatures (e.g., T < 1373 K) to produce hydrogen.11,12Reduction step:
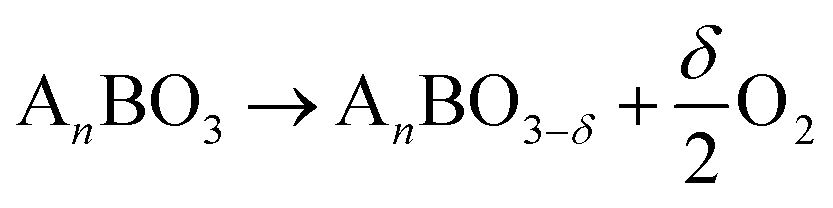 | (1) |
H2O splitting:
| AnBO3−δ + δH2O(g) → AnBO3 + δH2(g) | (2) |
The active redox metal oxides that have been investigated for STCH include stoichiometric oxides (e.g., SnO2/SnO13 and ZnO/Zn14) and non-stoichiometric oxides (e.g., ceria and perovskite-type oxide). Stoichiometric oxides usually require high reduction temperatures (≥1873 K) to reduce, and the reduction–oxidation rates are low.15 Additionally, the reduced oxide is in the gas phase, which increases the collection and separation cost. Ceria (CeO2−δ, where δ represents oxygen non-stoichiometry) materials are well-investigated for STCH reactions due to their thermal stability and fast reaction kinetics. However, ceria suffers from high reduction temperatures (1823 K or above), limiting the theoretical hydrogen yield.16–19 Perovskite-type oxide materials (ABO3−δ) are considered a promising candidate for STCH reaction because they have a low standard partial molar reduction enthalpy resulting in high Δδ. Additionally, the Δδ value of perovskite can be tuned to achieve desirable thermodynamic and kinetic properties, which have been widely studied.20–23 For instance, the Δδ value of the perovskite family LaxSr1−xMnyAl1−yO3−δ (0 ≤ x ≤ 1, 0 ≤ y ≤ 1) is tuneable and increases with the increasing of Sr concentration.24 However, the issue of phase instability and poor cycling performance, as well as sintering and slow kinetics in the reduction step, remains a challenge. For instance, La and, Gd dopants in BaFeO3−δ have been investigated in STCH, but they suffer from the problem of phase instability after the thermochemical H2O splitting reaction. Notably, the poor cycling stability of La- and Gd-doped BaFeO3−δ system also limits the potential for development in STCH. Additionally, the time required for one redox cycle is usually longer than 1.5 hours, which is unfavourable for energy utilization. Therefore, searching for materials with desirable phase stability and kinetics during short-time redox cycling reactions is important.
High-entropy ceramics (HECs) include a wide number of materials such as high-entropy oxides, borides, carbides, silicides, sulfides, phosphides, and phosphates.25–28 Specifically, high-entropy perovskite oxides (HEPOs) were reported in 2018,29 and subsequently attracted great interest because of their catalytic, dielectric, ferroelectric, magnetic, thermoelectric, magnetocaloric, & electrocaloric properties, as well as promising applications in solid oxide fuel cells, batteries, and supercapacitors.30–33 Recent studies found high-entropy oxides with favourable kinetics and stability in the cycling redox reaction.34–38 For example, the (FeMgCoNi)O1.2@SiC composite demonstrated a hydrogen yield that surpassed the thermodynamic limits of cutting-edge materials, including spinel ferrites and ceria.39 Also, (FeMgCoNi)O1.2 showed no performance degradation for 10 cycles. Recently, HECs have been further extended to compositionally complex ceramics (CCCs) where maximizing the configurational entropy is not always needed or beneficial.28,40 Notably, a recent report showed that compositionally complex perovskite oxides (CCPOs) with non-equimolar B-site mixing exhibit excellent STCH performance with Co as the redox-active element.41
Herein, we reported a Mn-based HEPO for STCH reaction via A-site mixing with the chemical formula (La1/6Pr1/6Nd1/6Gd1/6Sr1/6Ba1/6Mn)O3, denoted as LPNGSB_Mn for brevity, for STCH reaction with further improved performance (and are Co-free). LPNGSB_Mn possesses excellent phase stability even after 50 cycling reactions. LPNGSB_Mn exhibits a favourable kinetic property (oxygen exchange coefficient), desirable thermodynamic parameters (Δδ, enthalpy, and entropy of reduction), and high H2 production (∼77.5 mmol moloxide−1) within 1 h. The conclusion that Mn is the only redox centre in this material is supported by X-ray photoelectron spectroscopy (XPS) and electron energy loss spectroscopy (EELS) results. Moreover, LPNGSB_Mn shows excellent phase stability and cycling durability even after over 50 long cycles. This work also provides a new pathway for utilizing A-site HEPO in thermochemical applications such as chemical looping and explores the potential of HEPOs for use in two-step seawater splitting. In this study, we explore both kinetic and thermodynamic aspects in a high-entropy system. Additionally, our extended testing over 50 cycles indicates long-term stability and potential for application, thereby enriching the current understanding beyond the state-of-the-art redox oxides in this field.39,42,43
2 Results and discussion
2.1 Characterizations and experiments
The nominal compositions and sample abbreviations for perovskite oxides developed in this work are shown in Table 1.| Sample abbreviations | Nominal composition |
|---|---|
| LS21_Mn | (La2/3Sr1/3)MnO3 |
| LPNGSB_Mn | (La1/6Pr1/6Nd1/6Gd1/6Sr1/6Ba1/6)MnO3 |
X-ray diffraction (XRD) patterns revealed that samples exhibited rhombohedral crystal structures (with space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c) without any detectable secondary phase (Fig. 1a). Rietveld refinements confirm all compositions are Rc phase by assuming random A site occupation of La, Pr, Nd, Gd, Sr, and Ba (Fig. S1 and S2†). Table S1† displays the refined lattice parameters for samples. In addition, the neutron total scattering conducted at Oak Ridge National Laboratory (ORNL) Spallation Neutron Source (SNS) Nanoscale-Ordered Materials Diffractometer (NOMAD) further confirmed that LS21_Mn has a rhombohedral crystal structure, as shown in Fig. S3.† The fitting results of the NOMAD data are presented in Table S2.† The comparison between the XRD and NOMAD neutron total scattering results shows excellent agreement, which further validates the crystal structure and space group. The crystal structure is shown in Fig. 1b. Additionally, the tolerance factor of LPNGSB_Mn can be calculated by eqn (3).
c) without any detectable secondary phase (Fig. 1a). Rietveld refinements confirm all compositions are Rc phase by assuming random A site occupation of La, Pr, Nd, Gd, Sr, and Ba (Fig. S1 and S2†). Table S1† displays the refined lattice parameters for samples. In addition, the neutron total scattering conducted at Oak Ridge National Laboratory (ORNL) Spallation Neutron Source (SNS) Nanoscale-Ordered Materials Diffractometer (NOMAD) further confirmed that LS21_Mn has a rhombohedral crystal structure, as shown in Fig. S3.† The fitting results of the NOMAD data are presented in Table S2.† The comparison between the XRD and NOMAD neutron total scattering results shows excellent agreement, which further validates the crystal structure and space group. The crystal structure is shown in Fig. 1b. Additionally, the tolerance factor of LPNGSB_Mn can be calculated by eqn (3).
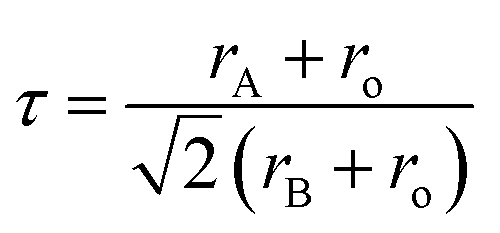 | (3) |
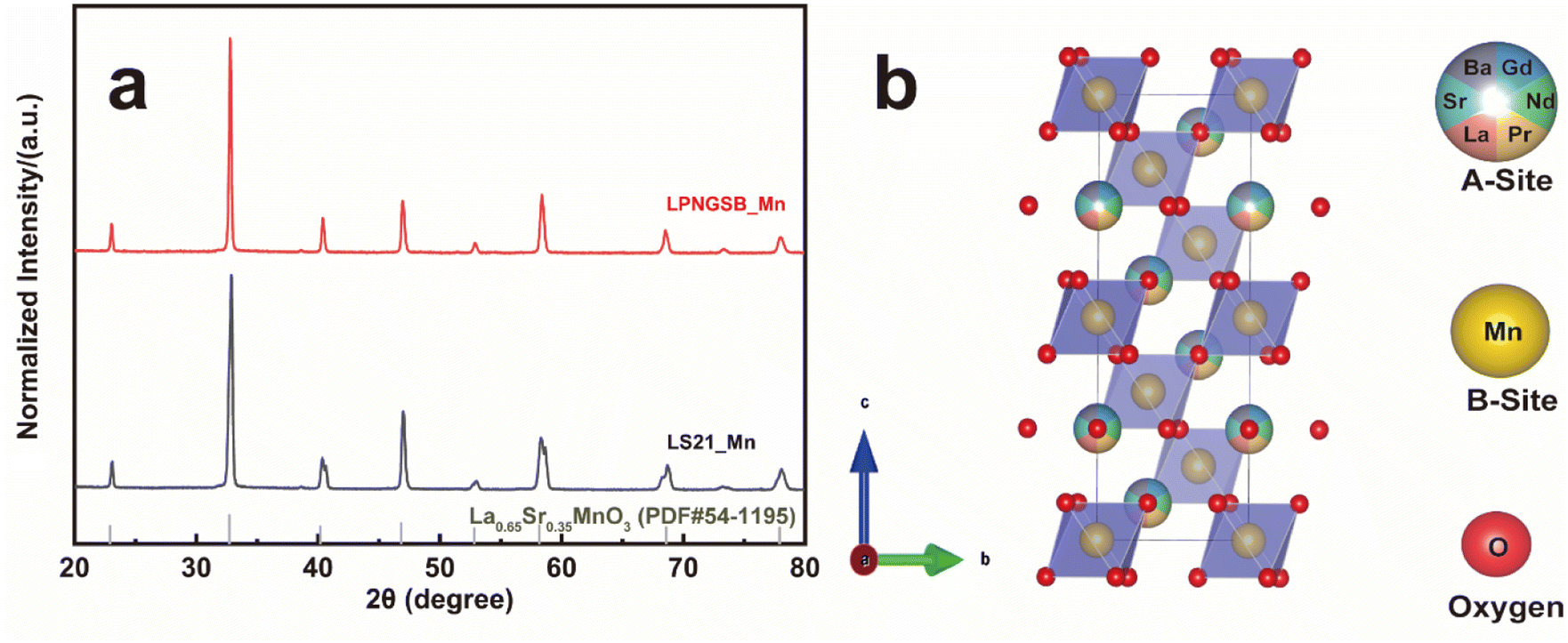 | ||
| Fig. 1 (a) XRD patterns of LPNGSB_Mn and LS21_Mn. The standard XRD card for La0.65Sr0.35MnO3 with structure is shown for reference. (b) Crystal structure of LPNGSB_Mn. | ||
The morphologies, microstructure, and elemental distribution of LPNGSB_Mn were characterized using scanning electron microscopy (SEM) in combination with energy-dispersive X-ray (EDX) spectroscopy (Fig. 2). The fresh sample showed irregularly connected spherical particles with an average diameter of approximately 5–15 μm. The EDX mappings showed a homogeneous distribution of elements.
 | ||
| Fig. 2 SEM images and corresponding EDX elemental maps showing the distribution of La (cyan), Pr (purple), Nd (orange), Gd (blue), Sr (green), Ba (red), and Mn (yellow) in the fresh (La1/6Pr1/6Nd1/6Gd1/6Sr1/6Ba1/6)MnO3 (LPNGSB_Mn) perovskite sample. | ||
The scanning transmission electron microscopy (STEM) with EDX mapping (Fig. 3) was used to characterize the atomic microstructure and elemental distribution of LPNGSB_Mn, which revealed a uniform distribution of all elements. These results show a homogenous occupancy of A sites between the A site dopants, but that B-sites are occupied by Mn. STEM-EDX quantitative analysis displayed the average composition for all metal percentages as shown in Table S3.†
 | ||
| Fig. 3 Pristine LPNGSB_Mn (a) drift corrected frame integrated (DCFI) HAADF image showing A and B-site atoms as white and gray points of contrast respectively. (b) HAADF image (c) bright-field (BF) image (d) well-ordered Mn EDX map corresponding to the gray points of contrast in the HAADF image. (e) La EDX map (f) Pr EDX map (g) Nd EDX map (h) Gd EDX map (i) Sr EDX map and (j) Ba EDX map. (e–j) Show atomic signals corresponding to the brightest white points in the HAADF images indicating a mixed species A-site occupation. | ||
To assess the redox capability, the LPNGSB_Mn series samples were tested by temperature programmed reduction (TPR).45 Fig. S4† displays that LPNGSB_Mn has a larger Δδ than LS21_Mn. LPNGSB_Mn shows a Δδ value of 0.0822, which is approximately 2.6 times greater than that of LS21_Mn (Δδ = 0.032).
EPR technique was used to study the magnetic properties of perovskite oxides. The formation of oxygen vacancy in LPNGSB_Mn was investigated using EPR spectroscopy. Room temperature X-band EPR spectra were recorded for the powder samples of fresh LPNGSB_Mn and reduced LPNGSB_Mn (thermally reduced at 1350 °C under nitrogen atmosphere). EPR spectra of fresh LPNGSB_Mn and reduced LPNGSB_Mn samples are shown in Fig. S5.† Both fresh LPNGSB_Mn and reduced LPNGSB_Mn samples exhibited a broad and symmetric signal of high-spin Mn4+ (t32ge0g, S = 3/2) located in the cubic symmetry of the perovskite B site.46–48 The broad spectrum is due to isotropic ferromagnetic exchange coupling arises from the hopping of electrons between Mn4+ and nearest neighbour Mn3+ ions in the crystal lattice.48–50 The calculated g value for both spectra is g = 1.98. No EPR signal was observed below 2000 G (data not shown). The linewidth (ΔHpp) values of fresh LPNGSB_Mn and reduced LPNGSB_Mn samples, calculated from the maximum and minimum values of the spectrum, were 704 and 680 G, respectively. As compared to the ΔHpp of fresh LPNGSB_Mn, the ΔHpp value of and reduced LPNGSB_Mn sample was significantly decreased (from 704 G to 680 G). EPR spectroscopy has been used to study the oxygen vacancy defects in the perovskite oxides.47,48,51 Under a high external thermal energy, some oxygen ions deviated from the original sites and oxygen vacancies are generated in perovskite oxides. The increased sites of oxygen vacancies increase the electron concentration inside the crystals. Our EPR study shows that the g (=1.98) value of the reduced LPNGSB_Mn sample is same as the fresh LPNGSB_Mn. It shows that the lattice structure and coordination environment of Mn4+ in LPNGSB_Mn sample is not changed even after heated at 1350 °C under nitrogen atmosphere. The formation of oxygen vacancies results in defect induced free electron spins. EPR studies have shown that the electrons trapped in the formation of oxygen vacancy sites exhibits a sharp signal around g = 2.003–2.004.52,53 In our study, due to the broad EPR spectra that lack any resolved fine structure, the characteristic g-value of oxygen vacancy cannot be extracted from the spectrum. Importantly, the change in ΔHpp is attributed to the changes of magnetic interaction between the Mn4+ and newly formed oxygen vacancies in reduced LPNGSB_Mn sample. The decreased ΔHpp in reduced LPNGSB_Mn sample demonstrates that there is an increased exchange/magnetic interaction between Mn4+ and neighbouring electrons with the free spins induced by the oxygen vacancies. Overall, the EPR study clearly demonstrates that LPNGSB_Mn under external thermal energy and nitrogen atmosphere resulted in oxygen vacancies.
To reveal the redox-active elements in LPNGSB_Mn, in situ electron energy loss spectroscopy (EELS) was conducted to investigate their redox activity by heating LPNGSB_Mn to 700 °C under a vacuum (Fig. S6†). The EELS spectra of Mn, O, La, Pr, Nd, Gd, and Ba are shown in Fig. S6a–g.† For the A-site elements, no cation peak shift was observed, indicating no valence change in the A-site elements.41 For the B-site elements, Mn is the only redox-active element, as evidenced by the shift in the Mn L-edge to lower energies (Fig. S6a†). The XPS analysis of LPNGSB_Mn shows the same trend, with a shift in the peak edge to lower energies, further demonstrating that Mn is the redox-active element (Fig. S6h†). The Mn 2p peak has two split spin–orbit components of 2p3/2 (∼642.3 eV) and 2p1/2 (∼653.5 eV). The differences in the binding energy of split (ΔBE) were 11.2 eV in fresh LPNGSB_Mn and increased to 11.7 eV in reduced LPNGSB_Mn. The results show a shift of the Mn 2p3/2 and Mn 2p1/2 peaks toward lower binding energies, indicating that Mn has been reduced.54 The absence of the satellite peak at 648 eV indicates that Mn2+ does not dominate in this oxide.55 Thus, Mn3+, and Mn4+ are the predominant valence states in this perovskite-type oxide.
The XPS results also confirmed that rare earth elements (La, Pr, Nd, Gd) remained stable without any change in LPNGSB_Mn before and after the reduction process (Fig. S7 and S8†). One observable change, shown in Fig. S8e,† is that the Sr signal increases in intensity after reduction at 1350 °C. This change in Sr signal can be attributed to Sr segregation, which is commonly observed in perovskite oxides like LaSrMnO3 series.56,57
Note that direct water thermolysis catalysed by the samples and an alumina tube at 1100 °C produces a small amount of H2, which is also reported in the literature.58–62 The H2 production from direct water thermolysis was measured at 800–1100 °C (Fig. S9†). Thus, the amount of hydrogen background produced through direct thermolysis was subtracted from the total hydrogen production to obtain the net H2 production from the STCH redox reaction. To evaluate the water-splitting capabilities of LPNGSB_Mn and LS21_Mn in terms of hydrogen production and reversibility, these materials were subjected to consecutive H2O-splitting cycles. Fig. 4a shows the cumulative hydrogen production for both samples under different protocols in terms of different oxidation temperatures and reduction/oxidation durations at different cycles. Fig. 4b presents the corresponding oxygen production under these protocols. The protocols implemented are detailed in Table S4.† For all materials under different protocols, the molar ratio of O2 to H2 produced in the reduction and oxidation step respectively, was approximately 0.5 (Fig. S10 and S11†), indicating that the splitting of H2O is realized by the redox reaction in a stepwise way. As shown in Fig. 4a and Table 2, hydrogen production increases with the number of cycles. Notably, LPNGSB_Mn exhibits the highest H2 production (∼77.5 mmol moloxide−1) at 900 °C in the oxidation step. The H2 production of LPNGSB_Mn is comparable with the reported oxides tested by the same reactor at Sandia National Laboratories, as shown in Table S5.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 63,64
63,64
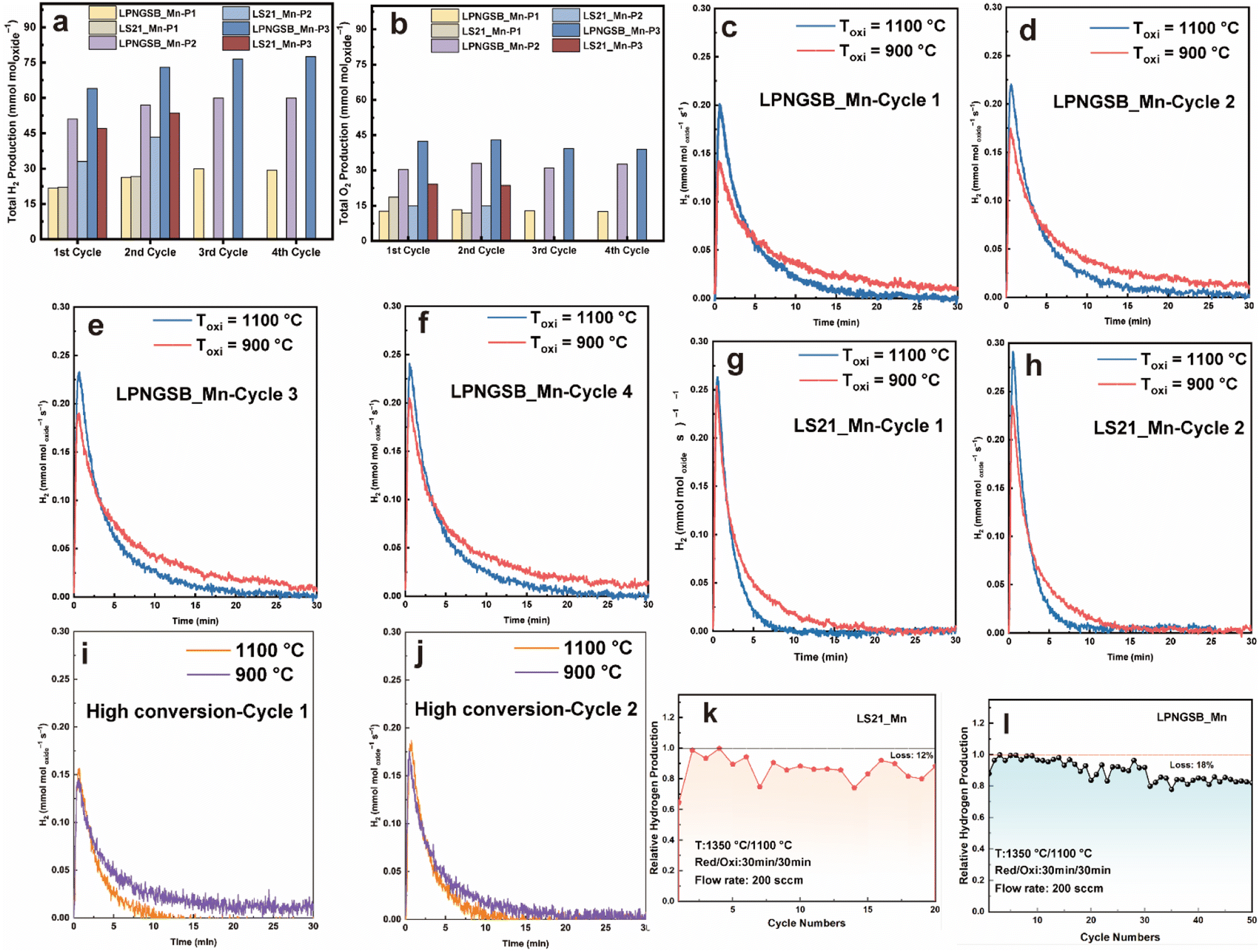 | ||
| Fig. 4 H2 (a) and O2 (b) production for LS21_Mn and LPNGSB_Mn under different protocols and cycles, respectively. The plots of molar H2 production rate versus time for LPNGSB_Mn under protocols P2 and P3 in different cycles (c–f). The plots of molar H2 production rate versus time for LS21_Mn under protocols P2 and P3 in different cycles (g and h). The plots of molar H2 production rate versus time for LPNGSB_Mn under protocols P4 and P5 in different cycles (i and j). Cycling stability of H2 production of LPNGSB_Mn (k) and LS21_Mn (l) under protocol P2. Detailed testing protocols are summarized in Table S4.† Results of (a–j) were obtained in a STCH reactor at Sandia National Laboratories, while results of (k) and (l) were obtained in another homemade reactor. | ||
| Condition | T red/Tox (°C) | t red/tox (min) | Flow rate (sccm) | Cumulative H2 production (mmol moloxide−1) | Steam to hydrogen conversion (%) |
|---|---|---|---|---|---|
| a In all cases, the cycle numbers are designated as C1 to C4. For example, C1 represents the first cycle. The protocol numbers are designed as P1 to P5 indicating different conditions which are detailed in Table S4. Tred and Tox are the reduction and oxidation temperature, respectively. tred and tox are the reduction and oxidation time, respectively. | |||||
| C1-LPNGSB_Mn-P1 | 1350/1100 | 5/20 | 200 | 21.8 | 0.45 |
| C2-LPNGSB_Mn-P1 | 1350/1100 | 5/20 | 200 | 26.3 | 0.54 |
| C3-LPNGSB_Mn-P1 | 1350/1100 | 5/20 | 200 | 30 | 0.61 |
| C4-LPNGSB_Mn-P1 | 1350/1100 | 5/20 | 200 | 29.3 | 0.60 |
| C1-LS21_Mn-P1 | 1350/1100 | 5/20 | 200 | 22 | 0.55 |
| C2-LS21_Mn-P1 | 1350/1100 | 5/20 | 200 | 26.7 | 0.67 |
| C1-LPNGSB_Mn-P2 | 1350/1100 | 30/30 | 200 | 51 | 0.70 |
| C2-LPNGSB_Mn-P2 | 1350/1100 | 30/30 | 200 | 57 | 0.78 |
| C3-LPNGSB_Mn-P2 | 1350/1100 | 30/30 | 200 | 60 | 0.82 |
| C4-LPNGSB_Mn-P2 | 1350/1100 | 30/30 | 200 | 60 | 0.82 |
| C1-LS21_Mn-P2 | 1350/1100 | 30/30 | 200 | 32.95 | 0.55 |
| C2-LS21_Mn-P2 | 1350/1100 | 30/30 | 200 | 43.35 | 0.71 |
| C1-LPNGSB_Mn-P3 | 1350/900 | 30/30 | 200 | 64 | 0.87 |
| C2-LPNGSB_Mn-P3 | 1350/900 | 30/30 | 200 | 73 | 1.0 |
| C3-LPNGSB_Mn-P3 | 1350/900 | 30/30 | 200 | 76.5 | 1.05 |
| C4-LPNGSB_Mn-P3 | 1350/900 | 30/30 | 200 | 77.5 | 1.05 |
| C1-LS21_Mn-P3 | 1350/900 | 30/30 | 200 | 47 | 0.78 |
| C2-LS21_Mn-P3 | 1350/900 | 30/30 | 200 | 53.5 | 0.89 |
| C2-LPNGSB_Mn-P4 | 1350/1100 | 30/30 | 200 | 27.9–51 | — |
| C2- LS21_Mn-P4 | 1350/1100 | 30/30 | 200 | 34.15–46.1 | — |
| C2-LPNGSB_Mn-P5 | 1350/900 | 30/30 | 200 | 33.6–41.95 | — |
Initially, the hydrogen production of LS21_Mn (∼26.7 mmol moloxide−1) and LPNGSB_Mn (∼26.3 mmol moloxide−1) is similar when both thermally are reduced for 5 min (protocol P1). However, the reduction and oxidation time increases, the hydrogen production of LPNGSB_Mn and LS21_Mn is 57 and 43.35 mmol moloxide−1 under protocol P2, respectively. In a comparison between conditions of protocols P2 and P3, with the reduction temperature fixed at 1350 °C, the oxidation temperatures are set at 1100 and 900 °C, respectively. A lower oxidation temperature of 900 °C is favourable, yielding a hydrogen production of ∼73 mmol moloxide−1 for LPNGSB_Mn and ∼53.5 mmol moloxide−1 for LS21_Mn, respectively, as shown in Fig. 4a. The hydrogen production of LPNGSB_Mn in the 4th cycle at 900 °C (∼77.5 mmol moloxide−1) is higher than 1100 °C (∼60 mmol moloxide−1), suggesting a greater extent of the reaction at 900 °C, indicating a more thermodynamically favourable condition compared to 1100 °C. The oxidation reaction kinetics are favoured at higher temperatures, as shown in the plots of molar hydrogen production rate versus time (Fig. 4c–h). The oxidative water splitting step is an exothermic reaction and thus thermodynamically favoured at a lower temperature. In contrast, the H2 production kinetics at 1100 °C are higher than at 900 °C, as indicated by the higher peak rates at 1100 °C. The detailed values can be found in Table S6.† The favoured thermodynamics at 900 °C likely cause the higher H2 production within 30 min. A similar effect of lower oxidation temperature on the increased H2 production has been reported in La0.6Ca0.4MnO3.65
Fig. 4i and j shows the rate curve for LPNGSB_Mn during high-conversion water-splitting cycles (with a molar ratio of H2O to H2 at 1000:1) at temperatures of 1100 and 900 °C with detailed conditions described in Table S4.† The plots of gas molar production rates versus time for high conversion water-splitting cycles are shown in Fig. S12.† LPNGSB_Mn exhibits similar peak rates in the presence of H2 at 1100 and 900 °C, as shown in Fig. 4i and j. Fig. S13† compares LS21_Mn and LPNGSB_Mn in high-conversion water-splitting cycles, where LPNGSB_Mn shows a higher H2 production peak rate than LS21_Mn. Moreover, hydrogen production can be detected in LPNGSB_Mn at 900 °C under protocol P5, while LS21_Mn shows negligible H2 production under the same conditions. After STCH cycles, the sample of LPNGSB_Mn was kept as the reduced state and characterized by the HAADF-STEM. The atomic-level crystal structure of reduced LPNGSB_Mn remains intact, and the distribution of elements is homogenous (Fig. S14†), indicating its phase stability. The STEM-EDX quantitative analysis of reduced LPNGSB_Mn is shown in Table S7.†
In addition, it delivers similar H2 production under the seawater vapor feeding condition. Therefore, this STCH technology can flexibly function for splitting low-grade water without the reliance on the costly deionized water due to the use of H2O vapor feeding, which is a promising technological complement to proton exchange membrane and anion exchange membrane water electrolysis that need to use deionized water.66–71 Furthermore, the surface oxygen exchange kinetics for a dense LPNGSB_Mn pellet (Fig. S15†) were measured by the electrical conductivity relaxation (ECR) method. The ECR result (Fig. S16†) can be fitted to obtain the surface oxygen exchange coefficient (Ks).72,73 The Ks of LPNGSB_Mn is 7.992 × 10−5 cm s−1.
Table 2 summarizes the STCH production results under different conditions/protocols. To investigate the correlation between H2 production and material properties, we used excess steam to react with a small quantity of perovskite oxide samples to ensure the sufficient solid–gas contact and mass transfer for analysing the thermodynamic limit situation. Therefore, the steam-to-H2 conversion percentages under all conditions are lower than 1.1%.
We investigated the cycling ability of LS21_Mn for 20 cycles and LPNGSB_Mn for 50 cycles. Fig. 4k and l shows the cycling test results. LPNGSB_Mn and LS21_Mn maintained stable hydrogen production, respectively. The cycling stability of LPNGSB_Mn under harsh cycling conditions is comparable to that reported for other STCH oxides.41,63 LS21_Mn exhibits a degradation of 12% after 20 cycles. In contrast, LPNGSB_Mn shows a degradation of 18% after 50 cycles. The gradually decreased hydrogen production may be attributed to the particle coarsening of samples, which may decrease both the heat transfer rate and the oxygen release rate. The morphologies and EDX maps of pristine LPNGSB_Mn and LPNGSB_Mn cycled for 50 cycles are shown in Fig. 2 and S17–S19.† After 50 cycles, LPNGSB_Mn had an average diameter of approximately 10–25 μm and did not show obvious element segregation. Regarding the texture of the samples, there was a considerable difference in the structural variation. The fresh LPNGSB_Mn sample had a rough surface with particle dispersion (Fig. 2), compared to the 50-cycled sample, which had a relatively smoother surface (Fig. S17d†) with some particle aggregation and material sintering. The larger grain-size observed after the 50-cycling experiment could be attributed to the LPNGSB_Mn being exposed to high temperatures (1100–1350 °C) for a longer time. The distribution of different elements was analysed (Fig. 2, S18 and S19†). The elements of La, Pr, Nd, Gd, and Mn were distributed evenly in both the pristine and cycled LPNGSB_Mn, while the elements of Ba and Sr displayed small segregation in the cycled LPNGSB_Mn. This small difference can be attributed to the sample being subjected to high-temperature sintering. Therefore, the negligible element aggregations are consistent with the phase stability observed in the XRD pattern (Fig. S20†). This phenomenon commonly occurs in perovskite oxides containing alkaline earth elements like Sr, which have a significant influence on material activity and stability.57
The pristine and 50-cycled LPNGSB_Mn was characterized by Raman spectroscopy. The Raman spectra are shown in Fig. S21,† and a three-peak model was used to fit the spectra. The fitting results were summarized in Table 3 below:
| Sample | Peak 1 (cm−1) | Peak 2 (cm−1) | Peak 3 (cm−1) |
|---|---|---|---|
| LPNGSB_Mn pristine | 218 | 497 | 620 |
| LPNGSB_Mn cycled | 207 | 490 | 613 |
The observed vibrational band close to 600 cm−1 is probably due to Mn–O vibrational modes coupled with the MnO6 octahedral structure, which are associated with the existence of oxygen vacancies nearby.74,75 The small peak shift between two samples could be attributed to the variation in the heavy ion content. The peaks close to 200 cm−1 and 490 cm−1 could be attributed to the A1g mode from La vibration and the Eg mode from oxygen bending vibration, respectively. The peak around 620 cm−1 can be assigned to the B2g mode, representing the in-phase stretching of LPNGSB_Mn.76–79
The structural stability of LPNGSB_Mn was further demonstrated based on the result of Raman.77 Additionally, the red-shift is observed in the Raman spectra of 50-cycled LPNGSB_Mn indicating the increasing defects in the material structure.80 Collectively, the results of XRD, SEM, EDX and Raman spectra indicate LPNGSB_Mn maintained the relatively stable composition, structure and phase.
Two thermodynamic properties, reduction enthalpy and entropy, are key factors that determine the H2 production of STCH materials.81,82 Therefore, we measured the δ over a range of PO2 values and temperatures for LS21_Mn and LPNGSB_Mn through a reported TGA method.83–85Fig. 5a and b show the oxygen non-stoichiometry of LPNGSB_Mn and LS21_Mn, respectively, as a function of temperatures at different oxygen partial pressures PO2.
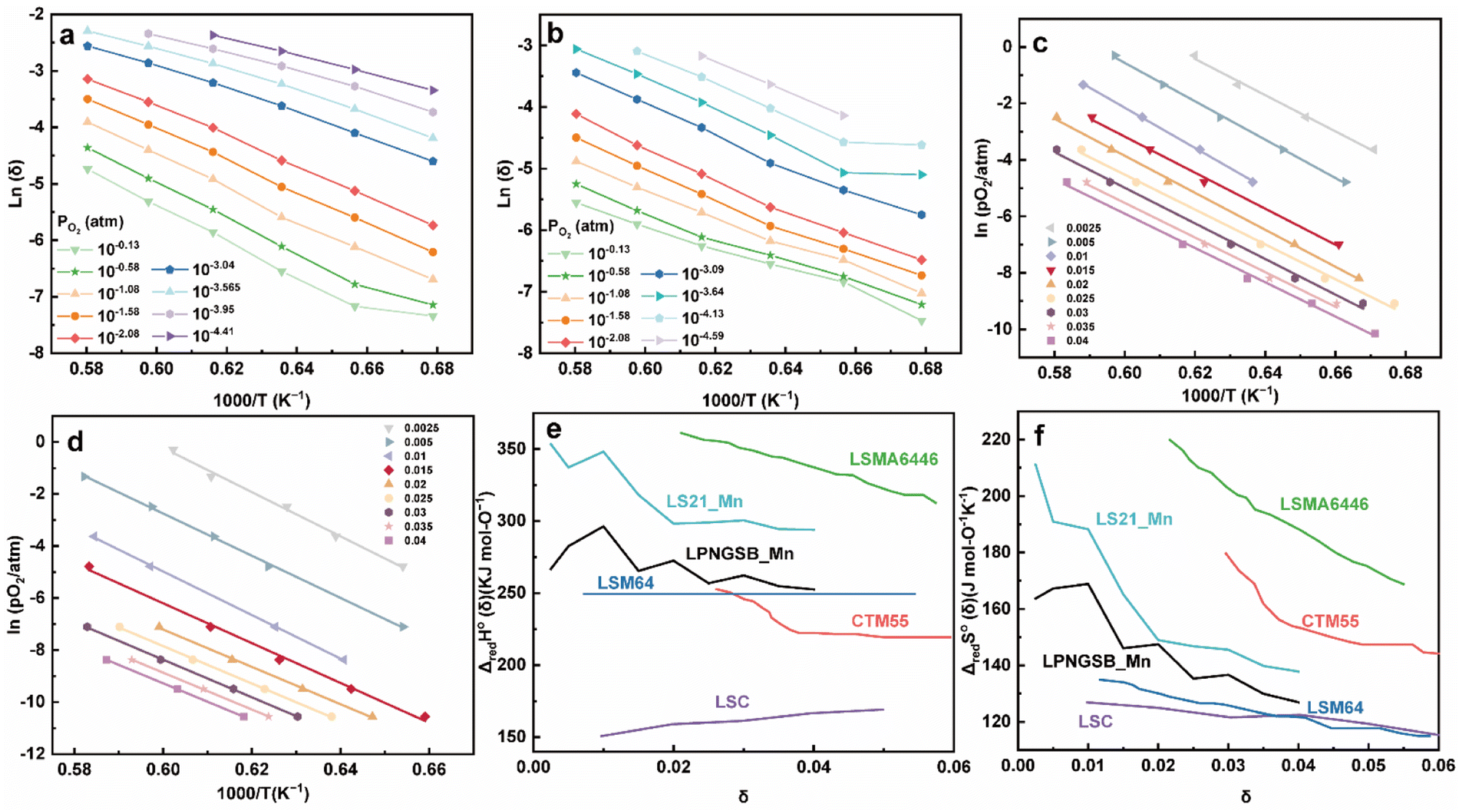 | ||
| Fig. 5 Oxygen non-stoichiometry of (a) LPNGSB_Mn and, (b) LS21_Mn as a function of temperatures at different oxygen partial pressures PO2. Arrhenius representation of (c) LPNGSB_Mn and (d) LS21_Mn in 1000/T and ln(PO2) for obtaining reduction enthalpy and entropy by the van't Hoff method at specific δ range from 0.0025–0.04. (e) Standard enthalpy and (f) entropy for LS21_Mn, LPNGSB_Mn, and some redox oxide materials reported in literatures (e.g., CTM55,19 LSMA6446,24 LSC,86 and LSM64 (ref. 87)). The oxygen non-stoichiometry of (a) LPNGSB_Mn measured at 1400 °C was reprinted with permission from ref. 88. Copyright 2023 The Electrochemical Society. | ||
According to the van't Hoff method,84 the standard entropy change 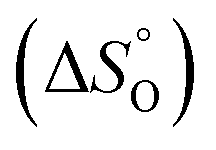 and the standard enthalpy change
and the standard enthalpy change 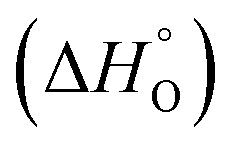 can be acquired from the correlation with PO2 and temperature as the following equation.83,85,86
can be acquired from the correlation with PO2 and temperature as the following equation.83,85,86
| ΔG°(T) = −RTlnKeqred | (4) |
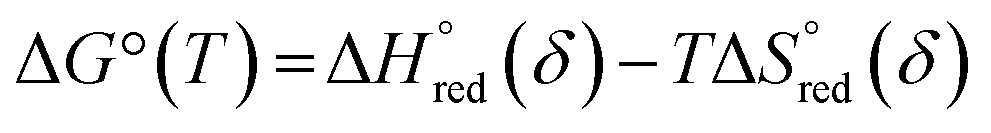 | (5) |
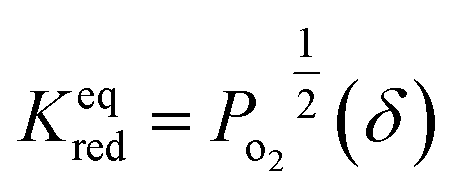 | (6) |
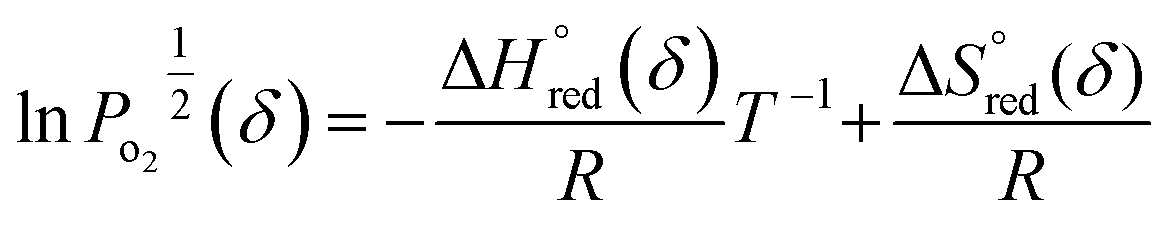 | (7) |
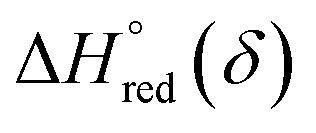 and
and 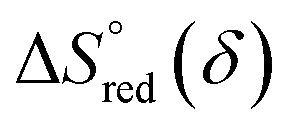 are the standard enthalpy and entropy, respectively, assuming
are the standard enthalpy and entropy, respectively, assuming 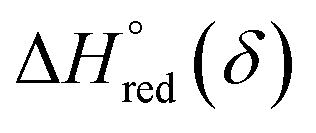 and
and 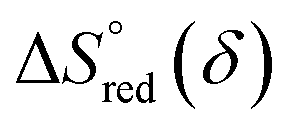 are irrelevant with temperature.
are irrelevant with temperature.
An Arrhenius plot of 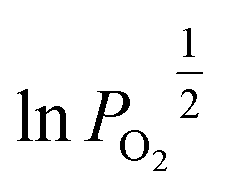 vs. 1000/T (at a specific δ) with linear fitting can be acquired. Fig. 5c and d show the plots for LPNGSB_Mn and LS21_Mn, respectively. The value of
vs. 1000/T (at a specific δ) with linear fitting can be acquired. Fig. 5c and d show the plots for LPNGSB_Mn and LS21_Mn, respectively. The value of 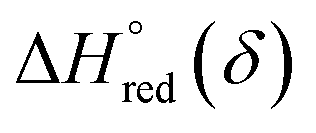 and
and 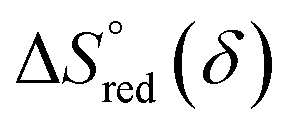 can be obtained from the slope and the intercept of the straight line, respectively. Consequently, the enthalpy and entropy of LS21_Mn and LPNGSB_Mn as a function of oxygen non-stoichiometry are obtained (Fig. 5e and f), respectively and compared with those of some reported redox materials such as La0.8Sr0.2CoO3 (LSC),86 Ca(Mn0.5Ti0.5)O3−δ (CTM55), La0.6Sr0.4MnO3−δ (LSM64),87 La0.6Sr0.4Mn0.4Al0.6O3−δ (LSMA6446).24 LPNGSB_Mn is in a favourable window of
can be obtained from the slope and the intercept of the straight line, respectively. Consequently, the enthalpy and entropy of LS21_Mn and LPNGSB_Mn as a function of oxygen non-stoichiometry are obtained (Fig. 5e and f), respectively and compared with those of some reported redox materials such as La0.8Sr0.2CoO3 (LSC),86 Ca(Mn0.5Ti0.5)O3−δ (CTM55), La0.6Sr0.4MnO3−δ (LSM64),87 La0.6Sr0.4Mn0.4Al0.6O3−δ (LSMA6446).24 LPNGSB_Mn is in a favourable window of 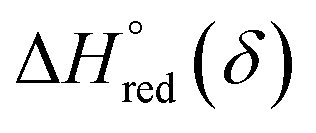 and
and 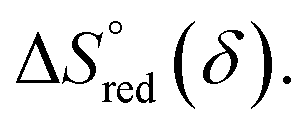 The reduction enthalpy and entropy of reduction are in the range of 252.51–296.32 kJ (mol O)−1 and 126.95–168.85 J (mol O)−1 K−1, respectively.
The reduction enthalpy and entropy of reduction are in the range of 252.51–296.32 kJ (mol O)−1 and 126.95–168.85 J (mol O)−1 K−1, respectively.
3 Conclusions
In summary, we reported a new A-site high-entropy HPEOs based on LS21_Mn as the parent perovskite-type oxide for solar-driven thermochemical water splitting. Notably, (La1/6Pr1/6Nd1/6Gd1/6Sr1/6Ba1/6)MnO3 (LPNGSB_Mn) shows an excellent balance between fast kinetics and suitable thermodynamics as well as extraordinary phase stability during the redox reaction. It exhibits moderate enthalpy (ranging between 252.51 and 296.32 kJ (mol O)−1) and entropy (ranging between 126.95 and 168.85 J (mol O)−1 K−1) of reduction. These traits of LPNGSB_Mn enable a maximum hydrogen production of ∼77.5 mmol moloxide−1 within a short 1 hour duration under optimized STCH conditions. We further showed that Mn is the only redox-active element in this HEPO. LPNGSB_Mn shows excellent phase stability and cycling durability over 50 cycles. This STCH technology can be also extended to thermochemical seawater splitting, eliminating the strict requirements of high-purity water. This study opens new avenues for screening redox materials for STCH and other possible chemical looping reactions. We suggest future studies focusing on testing this high entropy oxide in a reticulated porous ceramic structure for practical applications and exploring in-depth mechanisms.4 Experimental section
4.1 Materials synthesis
All perovskite-type oxide powders were synthesized using a solid-state method. The chemical precursors were used without further purification. Stoichiometric amounts of La2O3 (Alfa Aesar, 99.99%), Pr6O11 (Alfa Aesar, 99.99%), Nd2O3 (Sigma Aldrich, 99.99%), Gd2O3 (Alfa Aesar, 99.99%), SrCO3 (Alfa Aesar, 99.99%), BaCO3 (Sigma Aldrich, 99.98%) and MnO2 (Alfa Aesar 99.9%) were placed in a poly(methyl methacrylate) high-energy ball mill (HEBM) vial with endcaps and millings balls made of tungsten carbide, and dry milled for 100 min (SPEX 8000D, SPEX SamplePrep, USA). The mixed powder was heated from room temperature (RT) to 1300 °C with a heating rate of 5 °C min−1 and annealed in air at 1300 °C for 10 hours. Subsequently, the annealed powder was cooled to RT at a rate of 5 °C min−1. After that, the synthesized powder was ground with a pestle and mortar to achieve uniform mixing and prevent local non-uniformities. Then, the powder was further annealed at 1400 °C with a heating/cooling rate of 5 °C min−1 for 10 hours to enhance the homogeneity of the final product.4.2 Phase characterization
The phases were confirmed by X-ray diffraction (XRD) using a powder diffractometer (Rigaku Miniflex) at 30 kV and 15 mA, with Cu Kα radiation. All XRD data were collected with a step size of 0.02° and a scan rate of 2° min−1. The crystal structures of powders and specific crystallographic parameters were analysed by Rietveld refinement (Crystallography data analysis software: GSAS-II). The crystallographic information file (CIF) of La0.65Sr0.35MnO3 (Rc) from the Inorganic Crystal Structure Database (#54-1195) was used as an initial structure.
4.3 Pair distribution function
Pair distribution function (PDF) G(r) analysis was obtained via neutron total scattering at Nanoscale-Ordered Materials Diffractometer (NOMAD BL-1B) at the Spallation Neutron Source (SNS) of the Oak Ridge National Laboratory (ORNL). All perovskite powder specimens (around 0.6 g) were loaded into quartz capillaries (Hampton Research, HR6-150, 2 mm diameter and 8 cm length). The total scattering experiments were carried out at 290 K for a total accelerator proton charge of 8 C, corresponding to about 92 min acquisition time at the full power. To produce the total scattering function S(Q), the background was subtracted for the data from six detector banks and the data were normalized to the intensity of vanadium. This function was Fourier-transformed with a sliding Qmax to obtain the neutron weighted pair distribution functions, G(r) = r[g(r) − 1]. The shortest distance correlations were transformed with a Qmax = 31.4 Å−1. The momentum transfer, Q, is given as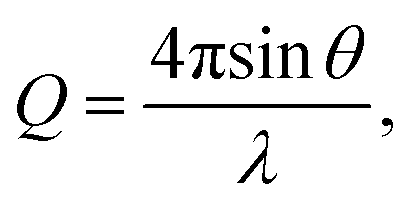 where θ and λ are the scattering angle and the neutron wavelength, respectively. The analysis of PDF data was done with small-box modeling by the PDFgui software. Rhombohedral (Rc) structure was used to model the data. The refined parameters include lattice parameters (a and c), scale factor, linear correlated motion factor (Delta 2 in PDFgui), isotropic atomic displacement parameters (ADP) for A-, B- and O sites, and the number of oxygen vacancies. Multiple elements occupying A or B site were refined with the same XYZ position and ADP.
where θ and λ are the scattering angle and the neutron wavelength, respectively. The analysis of PDF data was done with small-box modeling by the PDFgui software. Rhombohedral (Rc) structure was used to model the data. The refined parameters include lattice parameters (a and c), scale factor, linear correlated motion factor (Delta 2 in PDFgui), isotropic atomic displacement parameters (ADP) for A-, B- and O sites, and the number of oxygen vacancies. Multiple elements occupying A or B site were refined with the same XYZ position and ADP.
4.4 Temperature programmed reduction experiment
Temperature programmed reduction (TPR) characterization was conducted using a simultaneous thermal analyzer (STA 449 F3 Jupiter, NETZSCH Germany). Around 30 mg of powder sample was heated up to 1350 °C with a heating rate of 30 °C min−1 and held isothermally for 45 min in ultrahigh-purity (UHP) Ar atmosphere. Subsequently, the sample was cooled down to 1100 °C with a cooling rate of 30 °C min−1. Then, 21% O2 mixed with 79% Ar was introduced to oxidize the sample. A blank crucible was tested to calibrate the mass loss curve of samples. The extent of reduction (Δδ) was then calculated by measuring the mass change of the sample,89 which should remain constant to confirm complete oxidation under these conditions. The initial cycle usually displayed activation behaviours because of surface adsorption. Thus, the stabilized cycle 2 for all LPNGSB_Mn samples was reported for comparison.4.5 Electron paramagnetic resonance (EPR) spectroscopy
EPR spectra were recorded using a Bruker ELEXSYS spectrometer (Bruker BioSciences, Billerica, MA, USA) operating at X-band with a 100 kHz modulation frequency as described previously.90,91 Solid/powder samples were loaded directly in to an EPR quartz tube (O.D 4 mm). The quartz tube was positioned inside the resonator/cavity and the sample was adjusted to the middle of the resonator. EPR spectra were recorded at room temperature. Data acquisition was performed using Bruker Xepr software. The following settings were used: microwave frequency, 9.86 GHz; microwave power, 15 mW; modulation amplitude, 4.91 G; modulation frequency, 100 kHz; receiver gain, 60 dB; conversion time, 29.3 ms; sweep time, 120 s; number of scans, (1) data processing was performed using GraphPad Prism 10 program.4.6 Scanning transmission electron microscopy (STEM)
Electron transparent specimens were prepared using a Helios Nanolab 660 dual beam focused ion beam (FIB)/scanning electron microscope (Thermo Fisher Scientific). The procedure for preparing the FIB TEM lamella, including using a protective Pt coating on the surface of the samples. Bulk material removal and thinning were performed at 30 kV. The final thinning was performed at 5 kV to prevent further incidental Ga beam damage and remove previously damaged. STEM analysis was performed with a probe-corrected Titan Themis Z electron microscope operated at 300 kV (Thermo Fisher Scientific). The TEM is equipped with a monochromator (0.14 eV fwhm ZLP@300 kV), large-solid angle SDD EDX detector (Super X, solid angle ≈ 0.7 sr), and Quantum 969 EELS spectrometer (Gatan, Inc.), which has both a conventional CCD and a direct-electron CMOS K2 sensor for electron counting mode EELS analysis.4.7 In situ scanning transmission electron microscopy (STEM) combined with electron energy loss spectroscopy (EELS)
In situ heating was performed with a Gatan double-tilt model 652 heating holder. The sample was heated in a microscope vacuum (approximately 5.6 × 10−8 Torr; therefore, less than 4 × 10−8 bar O2) to the desired temperature at a ramp rate of approximately 10 °C min−1 and then held for approximately 15 min to allow time for drift to settle and the sample to equilibrate. EELS spectra of LPNGSB_Mn samples were collected at 25, 250, 400, 500, 600, and 700 °C. EELS spectra were collected in identical sample locations at a given temperature before and after the sample was heated to the next target temperature. For monochromated EELS, the convergence angle was approximately 25 mrad. The collection angle was approximately 35 mrad, and typical beam currents were 100−200 pA. To avoid a strong contribution from the electron beam during heating, the beam was quickly scanned over an area of interest during spectrum acquisition so that the electron dose was spread out over an area and not focused on one point. Additionally, EELS spectra were collected using Digital Micrograph's “time series” mode. Individual EELS spectra were collected with a 20–50 ms integration time while the beam scanned over a selected region of the sample, and then 5000–8000 spectra were summed up to increase signal-to-noise. The K2 sensor was used for these measurements. The regions of the sample that were selected were always within a single phase and compositionally uniform region of the sample.4.8 X-ray photoelectron spectroscopy
XPS measurements were performed using a PHI VersaProbe 5000 Scanning X-ray Photoelectron Spectrometer (ULVAC-PHI, Inc.) at room temperature and under a vacuum better than 1 × 10−6 Pascal. All measurements were performed using a focused Al K-Alpha X-ray source with a photon energy of 1486 eV and power of 25 W, along with an X-ray spot size of 100 μm. To correct sample charging effects, a primary electron neutralizer and a secondary Ar+ ion neutralizer were used. The take-off angle of the photoelectron was set at 45°. Compositional survey scans were obtained using a pass energy of 117.4 eV and an energy step of 0.5 eV. High-resolution detailed scans for each element were acquired using a pass energy of 23.5 eV and an energy step of 0.1 eV. The C 1s peak signal at 284.8 eV served as a reference for calibrating the binding energies.4.9 Thermochemical water splitting measurements
LS21_Mn and LPNGSB_Mn compositions were also tested in Sandia National Laboratories' (SNL) stagnation flow reactor (SFR) equipped with a 500 W near-IR continuous-wave laser for rapid sample heating. A mass spectrometer was positioned downstream of the SFR for gas analysis. All systems were operated under a sub-ambient pressure of 75 Torr. For calibration of the mass spectrometer, aliquots of oxygen and hydrogen were utilized. These were regulated using mass flow controllers and sourced from verified gas mixture standards, specifically 25% oxygen in argon (Ar) and 5% hydrogen in argon. Approximately 100 mg of powder samples were evenly distributed on a shallow platinum crucible supported by a zirconia fibreboard holder to form a thin bed. The reactor was then brought to the oxidation temperature (Tox) under an ultra-high purity argon atmosphere. The samples were reduced by laser-heating from Tox to the reduction temperature (Tred) at a rate of 10 °C s−1, then returned to Tox after a fixed reduction duration. To initiate the reoxidation process, a gas flow containing 40 vol% water vapour in Ar was introduced. The total quantities of released oxygen and produced hydrogen were determined by integrating the signals from the mass spectrometer, which were adjusted for baseline corrections, across the entire period of gas evolution. Further details about the testing conditions have been published elsewhere.63,64 Table S4† shows the detailed protocols. The STCH cycling experiments were conducted by a homemade flow reactor. The powder (approximately 0.1 g) was loaded into the middle of an alumina tube (AdValue Technology, OD 1/4′′ × ID 3/16′′ × L 24′′) and placed horizontally near the thermocouple within a programmed heating and cooling electric furnace. The powder was uniformly dispersed in the tube to ensure sufficient gas–solid contact. There was some space over the powder to prevent a sudden pressure drop and avoid powder blowing off. N2 (Matheson UHP) was used as a carrier gas, and a mass flow controller (Alicat Scientific) was used to control the flow rate. The steam (40 vol% H2O) was generated by a water evaporator (Fuel Cell Technologies, Inc. humidity bottle). N2 was used to balance the steam and create the desired oxidation conditions. The outlet was connected to a cold trap, which condensed steam into water. The total amount of hydrogen produced was collected using the Restek Sample Gas Bag (10 L) for a given duration. Then, the H2 product concentration in the gas bag was detected using the gas chromatography (GC, Shimadzu GC-2014) with a thermal conductivity detector (TCD) which was calibrated with a series of standard H2 gases (GASCO, Cal Gas Direct Inc.) with different known and certified concentrations balanced by N2. The direct water thermolysis part at different temperatures was subtracted from the total H2 production.5 Measurements of thermodynamic properties
The thermodynamic properties (the enthalpy and entropy of reduction) of LPNGSB_Mn and LS21_Mn were measured up to 1450 °C under different PO2 conditions spanning from 10−0.58 to 10−4.59 atm. About 500 mg of sample was used in the TGA measurements, which was tested using a NETZSCH STA 449 F1. Oxygen partial pressure was controlled by mixing oxygen, argon, and pre-mixed 1100 ppm O2 in argon and measured on the exhaust side of the instrument using a Zirox ZR5 oxygen sensor. Corrections for buoyancy were performed using a crucible filled with coarse alumina powder to approximate the same volume of powder as measured samples.Author contributions
C. L. and D. Z. contributed equally to this work. C. L., D. Z. and W. L. (Wei Li) designed the experiments and methodology. C. L. contributed to STCH materials screening and cycling stability tests. D. Z. synthesized materials and conducted most material characterizations. C. L. and D. Z. analysed the data and prepared the figures and plots. W. L. (Wei Li) conceived the work and sample composition. J. A. T., J. D. S., and A. L. C. contributed the STEM, EELS, and a part of SEM characterizations. K. A. K. and A. H. M. performed the STCH measurements with the reactor at Sandia National Laboratories. S. R. B., A. I. S., P. A. S. and E. N. C. contributed to the TGA measurements under different temperatures and PO2 for acquiring enthalpy and entropy of reduction. M. V. and V. V. K. conducted the EPR measurements and analysed the EPR data. J. C. N. contributed to neutron total scattering and PDF characterizations. Q. W. (Qiang Wang) assisted in Raman and XPS characterizations. W. L. (Wenyuan Li) and H. T. assisted in the ECR characterization. J. Y. assisted in the synthesis and material characterization. Q. W. (Qingsong Wang) and Q. L. assisted in investigating the redox process. H. N. N. T., B. R., J. H. and A. M. D. assisted in STCH testing. H. A. D. S. contributed to the sample synthesis and assisted in STCH testing. L. M., Y. W. and G. C. contributed to the STCH reactor construction. X. L. (Xuemei Li) and N. A. T. T. assisted in SEM characterization. C. L., D. Z. and W. L. (Wei Li) wrote the original draft. All authors reviewed and commented the manuscript. Project administration, funding acquisition, and supervision, W. L. (Wei Li), J. L. and X. L. (Xingbo Liu).Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work is supported by the U.S. Department of Energy (DOE), Office of Energy Efficiency and Renewable Energy (EERE), under the Agreement Number DE-EE0008839, managed by the Hydrogen and Fuel Cell Technologies Office in the Fiscal Year 2019 H2@SCALE program. The UCSD team also acknowledges partial support from the NSF (DMR-2026193) after July 2023. Sandia National Laboratories is a multi-mission laboratory managed and operated by National Technology and Engineering Solutions of Sandia, LLC, a wholly owned subsidiary of Honeywell International, Inc., for the U.S. Department of Energy's National Nuclear Security Administration under contract DE-NA0003525. This article has been authored by an employee of National Technology & Engineering Solutions of Sandia, LLC under Contract No. DE-NA0003525 with the U.S. Department of Energy (DOE). The employee owns all right, title and interest in and to the article and is solely responsible for its contents. The publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this article or allow others to do so, for United States Government purposes. The DOE will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan. Any subjective views or opinions that might be expressed in the paper do not necessarily represent the views of the U.S. Department of Energy or the United States Government. The Alliance for Sustainable Energy, LLC, operates and manages the National Renewable Energy Laboratory (NREL) for DOE under contract no. DE-AC36-08GO28308. A part of this research used resources at the Spallation Neutron Source (SNS), a Department of Energy Office of Science User Facility operated by the Oak Ridge National Laboratory. The use of the WVU Shared Research Facilities is acknowledged. We thank Dr Xin Qian at Northwestern University for the technical discussion of direct water thermolysis, Dr J. Madhusudhan Naik at University of Zurich for the technical discussion of thermodynamics and configurational entropy, and Dr Yue Qi and Boyuan Xu at Brown University for the theoretical computation discussion.Notes and references
- J. D. Holladay, J. Hu, D. L. King and Y. Wang, Catal. Today, 2009, 139, 244–260 CrossRef CAS.
- W. Lubitz and W. Tumas, Chem. Rev., 2007, 107, 3900–3903 CrossRef CAS PubMed.
- Z. X. Cai, Z. L. Wang, Y. J. Xia, H. Lim, W. Zhou, A. Taniguchi, M. Ohtani, K. Kobiro, T. Fujita and Y. Yamauchi, Angew. Chem., Int. Ed., 2021, 60, 4747–4755 CrossRef CAS PubMed.
- M. Luo, J. T. Yang, X. G. Li, M. Eguchi, Y. Yamauchi and Z. L. Wang, Chem. Sci., 2023, 14, 3400–3414 RSC.
- Z. Y. Zhang, H. Tian, L. Bian, S. Z. Liu, Y. Liu and Z. L. Wang, J. Energy Chem., 2023, 83, 90–97 CrossRef CAS.
- A. Onigbajumo, P. Swarnkar, G. Will, T. Sundararajan, A. Taghipour, S. Couperthwaite, T. Steinberg and T. Rainey, J. Cleaner Prod., 2022, 371, 133303 CrossRef CAS.
- A. Bayon, A. de la Calle, E. B. Stechel and C. Muhich, Energy Technol., 2022, 10, 2100222 CrossRef CAS.
- C. L. Muhich, B. D. Ehrhart, V. A. Witte, S. L. Miller, E. N. Coker, C. B. Musgrave and A. W. Weimer, Energy Environ. Sci., 2015, 8, 3687–3699 RSC.
- J. E. Miller, A. H. McDaniel and M. D. Allendorf, Adv. Energy Mater., 2014, 4, 1300469 CrossRef.
- L. Zhou, X. Li, Q. Li, A. Kalu, C. Liu, X. Liu and W. Li, ACS Catal., 2023, 13, 15087–15106 CrossRef CAS.
- A. Steinfeld, Sol. Energy, 2005, 78, 603–615 CrossRef CAS.
- Z. P. Chen, Q. Q. Jiang, F. Cheng, J. H. Tong, M. Yang, Z. X. Jiang and C. Li, J. Mater. Chem. A, 2019, 7, 6099–6112 RSC.
- S. Abanades, P. Charvin, F. Lemont and G. Flamant, Int. J. Hydrogen Energy, 2008, 33, 6021–6030 CrossRef CAS.
- P. G. Loutzenhiser, A. Meier and A. Steinfeld, Materials, 2010, 3, 4922–4938 CrossRef PubMed.
- I. E. Yigiter and B. Piskin, JOM, 2022, 74, 4682–4694 CrossRef CAS.
- S. Abanades and G. Flamant, Sol. Energy, 2006, 80, 1611–1623 CrossRef CAS.
- W. C. Chueh and S. M. Haile, ChemSusChem, 2009, 2, 735–739 CrossRef CAS PubMed.
- H. Kaneko, T. Miura, H. Ishihara, S. Taku, T. Yokoyama, H. Nakajima and Y. Tamaura, Energy, 2007, 32, 656–663 CrossRef CAS.
- P. Singh and M. S. Hegde, Chem. Mater., 2010, 22, 762–768 CrossRef CAS.
- A. H. Bork, M. Kubicek, M. Struzik and J. L. M. Rupp, J. Mater. Chem. A, 2015, 3, 15546–15557 RSC.
- M. Orfila, M. Linares, R. Molina, J. A. Botas, R. Sanz and J. Marugan, Int. J. Hydrogen Energy, 2016, 41, 19329–19338 CrossRef CAS.
- L. L. Wang, M. Al-Mamun, Y. L. Zhong, L. X. Jiang, P. R. Liu, Y. Wang, H. G. Yang and H. J. Zhao, Sustainable Energy Fuels, 2017, 1, 1013–1017 RSC.
- X. Qian, J. G. He, E. Mastronardo, B. Baldassarri, W. Z. Yuan, C. Wolverton and S. M. Haile, Matter, 2021, 4, 688–708 CrossRef CAS.
- M. Ezbiri, M. Takacs, D. Theiler, R. Michalsky and A. Steinfeld, J. Mater. Chem. A, 2017, 5, 4172–4182 RSC.
- D. B. Miracle, J. D. Miller, O. N. Senkov, C. Woodward, M. D. Uchic and J. Tiley, Entropy, 2014, 16, 494–525 CrossRef CAS.
- S. Akrami, P. Edalati, M. Fuji and K. Edalati, Mater. Sci. Eng., R, 2021, 146, 100644 CrossRef.
- C. Oses, C. Toher and S. Curtarolo, Nat. Rev. Mater., 2020, 5, 295–309 CrossRef CAS.
- A. J. Wright and J. Luo, J. Mater. Sci., 2020, 55, 9812–9827 CrossRef CAS.
- S. C. Jiang, T. Hu, J. Gild, N. X. Zhou, J. Y. Nie, M. D. Qin, T. Harrington, K. Vecchio and J. Luo, Scr. Mater., 2018, 142, 116–120 CrossRef CAS.
- A. Amiri and R. Shahbazian-Yassar, J. Mater. Chem. A, 2021, 9, 782–823 RSC.
- Z. Q. Li, B. Guan, F. Xia, J. Y. Nie, W. Y. Li, L. Ma, W. Li, L. F. Zhou, Y. Wang, H. C. Tian, J. Luo, Y. Chen, M. Frost, K. An and X. B. Liu, ACS Appl. Mater. Interfaces, 2022, 14, 24363–24373 CrossRef CAS PubMed.
- X. Yang, R. K. Guo, R. Cai, Y. J. Ouyang, P. P. Yang and J. F. Xiao, Int. J. Hydrogen Energy, 2022, 47, 13561–13578 CrossRef CAS.
- D. W. Zhang, J. Park, B. Y. Xu, C. J. Liu, W. Li, X. B. Liu, Y. Qi and J. Luo, Dalton Trans., 2023, 52, 1082–1088 RSC.
- A. Le Gal, M. Valles, A. Julbe and S. Abanades, Catalysts, 2022, 12, 1116 CrossRef CAS.
- S. Zhai, J. Rojas, N. Ahlborg, K. Lim, M. F. Toney, H. Y. Jin, W. C. Chueh and A. Majumdar, Energy Environ. Sci., 2018, 11, 2172–2178 RSC.
- M. Biesuz, L. Spiridigliozzi, G. Dell'Agli, M. Bortolotti and V. M. Sglavo, J. Mater. Sci., 2018, 53, 8074–8085 CrossRef CAS.
- R. Djenadic, A. Sarkar, O. Clemens, C. Loho, M. Botros, V. S. K. Chakravadhanula, C. Kubel, S. S. Bhattacharya, A. S. Gandhif and H. Hahn, Mater. Res. Lett., 2017, 5, 102–109 CrossRef CAS.
- A. Sarkar, R. Djenadic, D. Wang, C. Hein, R. Kautenburger, O. Clemens and H. Hahn, J. Eur. Ceram. Soc., 2018, 38, 2318–2327 CrossRef CAS.
- Y. B. Gao, Y. P. Mao, Z. L. Song, X. Q. Zhao, J. Sun, W. L. Wang, G. F. Chen and S. Y. Chen, Appl. Energy, 2020, 279, 115777 CrossRef CAS.
- A. J. Wright, Q. Y. Wang, C. Y. Huang, A. Nieto, R. K. Chen and J. Luo, J. Eur. Ceram. Soc., 2020, 40, 2120–2129 CrossRef CAS.
- D. W. Zhang, H. A. De Santiago, B. Y. Xu, C. J. Liu, J. A. Trindell, W. Li, J. Park, M. A. Rodriguez, E. N. Coker, J. D. Sugar, A. H. McDaniel, S. Lany, L. Ma, Y. Wang, G. Collins, H. C. Tian, W. Y. Li, Y. Qi, X. B. Liu and J. Luo, Chem. Mater., 2023, 35, 1901–1915 CrossRef CAS.
- Y. J. Han, M. Tian, C. J. Wang, T. Zong and X. D. Wang, Appl. Catal., B, 2023, 339, 123096 CrossRef CAS.
- Y. B. Gao, M. M. Zhang, Y. P. Mao, H. Cao, S. J. Zhang, W. L. Wang, C. G. Sun, Z. L. Song, J. Sun and X. Q. Zhao, Energy Convers. Manage., 2022, 252, 115125 CrossRef CAS.
- D. R. Munazat, B. Kurniawan and IOP, J. Phys.: Conf. Ser., 2019, 1170, 012051 CrossRef CAS.
- M. D. Sanders, A. M. Bergeson-Keller, E. N. Coker and R. P. O'Hayre, Front. Energy Res., 2022, 10, 856943 CrossRef.
- C. Zhao, C. Li, H. Liu, Q. Qiu, F. S. Geng, M. Shen, W. Tong, J. X. Li and B. W. Hu, J. Am. Chem. Soc., 2021, 143, 18652–18664 CrossRef CAS PubMed.
- R. A. Eichel, Phys. Chem. Chem. Phys., 2011, 13, 368–384 RSC.
- D. Cordischi, M. Faticanti, G. Minelli, M. Occhiuzzi and P. Porta, Phys. Chem. Chem. Phys., 2003, 5, 1467–1473 RSC.
- V. A. Ivanshin, J. Deisenhofer, H. A. K. von Nidda, A. Loidl, A. A. Mukhin, A. M. Balbashov and M. V. Eremin, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 6213–6219 CrossRef CAS.
- M. T. Causa, M. Tovar, A. Caneiro, F. Prado, G. Ibanez, C. A. Ramos, A. Butera, B. Alascio, X. Obradors, S. Pinol, F. Rivadulla, C. Vazquez-Vazquez, M. A. Lopez-Quintela, J. Rivas, Y. Tokura and S. B. Oseroff, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 3233–3239 CrossRef CAS.
- Y. Zhao, J. P. Zhu, H. L. Wang, Z. Ma, L. H. Gao, Y. B. Liu, Y. Liu, Y. C. Shu and J. L. He, Ceram. Int., 2021, 47, 5549–5558 CrossRef CAS.
- Z. Li, W. J. Yang, L. B. Xie, Y. Li, Y. Liu, Y. Sun, Y. F. Bu, X. Y. Mi, S. H. Zhan and W. P. Hu, Appl. Surf. Sci., 2021, 549, 149278 CrossRef.
- Z. Wei, M. Zhao, Z. Yang, X. Duan, G. Jiang, G. Li, F. Zhang and Z. Hao, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2217148120 CrossRef CAS PubMed.
- M. K. Dalai, R. Kundu, P. Pal, M. Bhanja, B. R. Sekhar and C. Martin, J. Alloys Compd., 2011, 509, 7674–7676 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- Y. F. Li, W. Q. Zhang, Y. Zheng, J. Chen, B. Yu, Y. Chen and M. L. Liu, Chem. Soc. Rev., 2017, 46, 6345–6378 RSC.
- B. Koo, K. Kim, J. K. Kim, H. Kwon, J. W. Han and W. Jung, Joule, 2018, 2, 1476–1499 CrossRef CAS.
- Q. Q. Jiang, Z. P. Chen, J. H. Tong, M. Yang, Z. X. Jiang and C. Li, Chem. Commun., 2017, 53, 1188–1191 RSC.
- Z. P. Chen, Q. Q. Jiang, J. H. Tong, M. Yang, Z. X. Jiang and C. Li, Sol. Energy, 2017, 144, 643–659 CrossRef CAS.
- C. L. Muhich, K. C. Weston, D. Arifin, A. H. McDaniel, C. B. Musgrave and A. W. Weimer, Ind. Eng. Chem. Res., 2015, 54, 4113–4122 CrossRef CAS.
- T. C. Davenport, M. Kemei, M. J. Ignatowich and S. M. Haile, Int. J. Hydrogen Energy, 2017, 42, 16932–16945 CrossRef CAS.
- J. R. Scheffe, A. H. McDaniel, M. D. Allendorf and A. W. Weimer, Energy Environ. Sci., 2013, 6, 963–973 RSC.
- D. R. Barcellos, M. D. Sanders, J. H. Tong, A. H. McDaniel and R. P. O'Hayre, Energy Environ. Sci., 2018, 11, 3256–3265 RSC.
- R. B. Wexler, G. S. Gautam, R. T. Bell, S. Shulda, N. A. Strange, J. A. Trindell, J. D. Sugar, E. Nygren, S. Sainio, A. H. McDaniel, D. Ginley, E. A. Carter and E. B. Stechel, Energy Environ. Sci., 2023, 16, 2550–2560 RSC.
- L. L. Wang, M. Al-Mamun, P. R. Liu, Y. L. Zhong, Y. Wang, H. G. Yang and H. J. Zhao, Acta Metall. Sin., 2018, 31, 431–439 CrossRef CAS.
- W. Li, H. C. Tian, L. Ma, Y. Wang, X. B. Liu and X. F. Gao, Mater. Adv., 2022, 3, 5598–5644 RSC.
- W. M. Tong, M. Forster, F. Dionigi, S. Dresp, R. S. Erami, P. Strasser, A. J. Cowan and P. Farras, Nat. Energy, 2020, 5, 367–377 CrossRef CAS.
- Y. Yu, C. Y. Cao, W. Li, P. Li, J. Qu and W. G. Song, Nano Res., 2012, 5, 434–442 CrossRef CAS.
- W. Li, X. F. Gao, D. H. Xiong, F. Wei, W. G. Song, J. Y. Xu and L. F. Liu, Adv. Energy Mater., 2017, 7, 1602579 CrossRef.
- Y. Wang, W. Y. Li, L. Ma, W. Li and X. B. Liu, J. Mater. Sci. Technol., 2020, 55, 35–55 CrossRef CAS.
- W. Li, D. H. Xiong, X. F. Gao, W. G. Song, F. Xia and L. F. Liu, Catal. Today, 2017, 287, 122–129 CrossRef CAS.
- Y. P. Cao, M. J. Gadre, A. T. Ngo, S. B. Adler and D. D. Morgan, Nat. Commun., 2019, 10, 1346 CrossRef PubMed.
- H. Qi, Y. L. Lee, T. Yang, W. Y. Li, W. Li, L. Ma, S. S. Hu, Y. H. Duan, G. A. Hackett and X. B. Liu, ACS Catal., 2020, 10, 5567–5578 CrossRef CAS.
- T. Runka and M. Berkowski, J. Mater. Sci., 2012, 47, 5393–5401 CrossRef CAS.
- T. Scherban, R. Villeneuve, L. Abello and G. Lucazeau, J. Raman Spectrosc., 1993, 24, 805–814 CrossRef CAS.
- M. C. Weber, J. Kreisel, P. A. Thomas, M. Newton, K. Sardar and R. I. Walton, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 054303 CrossRef.
- M. N. Iliev and M. V. Abrashev, J. Raman Spectrosc., 2001, 32, 805–811 CrossRef CAS.
- H. C. Gupta, M. K. Singh and L. M. Tiwari, J. Raman Spectrosc., 2002, 33, 67–70 CrossRef CAS.
- N. Dilawar, U. Chandra, G. Parthasarathy and A. K. Bandyopadhyay, J. Raman Spectrosc., 2008, 39, 1765–1771 CrossRef CAS.
- X. Yu, C. W. Hu, P. X. Ji, Y. M. Ren, H. Y. Zhao, G. Liu, R. Xu, X. D. Zhu, Z. Q. Li, Y. Q. Ma and L. Ma, Appl. Catal., B, 2022, 310, 121301 CrossRef CAS.
- K. J. Warren, J. T. Tran and A. W. Weimer, Energy Environ. Sci., 2022, 15, 806–821 RSC.
- J. Park, B. Y. Xu, J. Pan, D. W. Zhang, S. Lany, X. B. Liu, J. Luo and Y. Qi, npj Comput. Mater., 2023, 9, 29 CrossRef CAS.
- X. Qian, S. M. Haile, T. C. Davenport and E. Mastronardo, J. Am. Ceram. Soc., 2022, 105, 4375–4386 CrossRef CAS.
- R. J. Panlener, R. N. Blumenthal and J. E. Garnier, J. Phys. Chem. Solids, 1975, 36, 1213–1222 CrossRef CAS.
- Y. Hao, C. K. Yang and S. M. Haile, Chem. Mater., 2014, 26, 6073–6082 CrossRef CAS.
- J. Mizusaki, Y. Mima, S. Yamauchi, K. Fueki and H. Tagawa, J. Solid State Chem., 1989, 80, 102–111 CrossRef CAS.
- M. J. Ignatowich, A. H. Bork, T. C. Davenport, J. L. M. Rupp, C. K. Yang, Y. Yamazaki and S. M. Haile, MRS Commun., 2017, 7, 873–878 CrossRef CAS.
- S. R. Bishop, D. W. Zhang, C. J. Liu, W. Li, P. A. Salinas, A. I. Smith, E. N. Coker, H. A. De Santiago, X. B. Liu, A. H. McDaniel and J. Luo, ECS Trans., 2023, 111, 1135 CrossRef.
- A. Riaz, T. Tsuzuki, F. Kremer, S. Sattayaporn, M. U. Ali, W. Lipinski and A. Lowe, ACS Catal., 2020, 10, 8263–8276 CrossRef CAS.
- X. J. Chen, W. Li, Y. B. Xu, Z. P. Zeng, H. C. Tian, M. Velayutham, W. Y. Shi, W. Y. Li, C. M. Wang, D. Reed, V. V. Khramtsov, X. L. Li and X. B. Liu, Nano Energy, 2020, 75, 104869 CrossRef CAS.
- N. Majumder, W. T. Goldsmith, V. K. Kodali, M. Velayutham, S. A. Friend, V. V. Khramtsov, T. R. Nurkiewicz, A. Erdely, P. C. Zeidler-Erdely, V. Castranova, J. R. Harkema, E. E. Kelley and S. Hussain, Redox Biol., 2021, 46, 102092 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta03554a |
| ‡ These authors (C. L. and D. Z.) contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |