
Recent advances of bifunctional electrocatalysts and electrolyzers for overall seawater splitting
Xiaoyan
Wang
ab,
Meiqi
Geng
b,
Shengjun
Sun
b,
Qian
Xiang
a,
Shiyuan
Dong
a,
Kai
Dong
 b,
Yongchao
Yao
c,
Yan
Wang
c,
Yingchun
Yang
b,
Yongsong
Luo
b,
Dongdong
Zheng
b,
Qian
Liu
d,
Jianming
Hu
*a,
Qian
Wu
*f,
Xuping
Sun
*bc and
Bo
Tang
*be
b,
Yongchao
Yao
c,
Yan
Wang
c,
Yingchun
Yang
b,
Yongsong
Luo
b,
Dongdong
Zheng
b,
Qian
Liu
d,
Jianming
Hu
*a,
Qian
Wu
*f,
Xuping
Sun
*bc and
Bo
Tang
*be
aCollege of Physics and Electronic Engineering, Chongqing Normal University, Chongqing 401331, China. E-mail: hujianming@cqnu.edu.cn
bCollege of Chemistry, Chemical Engineering and Materials Science, Shandong Normal University, Jinan 250014, Shandong, China. E-mail: tangb@sdnu.edu.cn; xpsun@uestc.edu.cn; xpsun@sdnu.edu.cn
cInstitute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu 610054, Sichuan, China
dInstitute for Advanced Study, Chengdu University, Chengdu 610106, Sichuan, China
eLaoshan Laboratory, Qingdao 266237, Shandong, China
fDepartment of Chemistry and Chemical Engineering, Weifang University, Weifang 261061, Shandong, China. E-mail: qianwu@wfu.edu.cn
First published on 2nd December 2023
Abstract
As a promising method for hydrogen (H2) production, seawater electrolysis has gained increasing attention as seawater is the most abundant water resource on Earth. The development of high-performance bifunctional electrocatalysts that facilitate both hydrogen evolution reaction and the oxygen evolution reaction and efficient electrolyzers are the key factors for H2 production from seawater. This review endeavors to provide a comprehensive analysis of the progress and challenges associated with bifunctional electrocatalysts for seawater splitting, along with efficient electrolyzers. We start with a brief overview of the fundamental aspects, including the involved reaction mechanisms and the evaluation parameters relevant to bifunctional electrocatalysts for seawater splitting. Subsequently, recent advancements in bifunctional electrocatalysts and electrolyzers designed for overall seawater splitting are summarized and discussed. Finally, we propose perspectives for the future development of highly efficient bifunctional electrocatalysts and electrolyzers for seawater splitting.
 Xuping Sun | Xuping Sun received his Ph.D. degree from the Changchun Institute of Applied Chemistry (CIAC), Chinese Academy of Sciences in 2006. During 2006–2009, he carried out postdoctoral research studies at Konstanz University, University of Toronto, and Purdue University. In 2010, he started his independent research career as a full Professor at CIAC, and then moved to Sichuan University in 2015. In 2018, he joined University of Electronic Science and Technology of China, where he found the Research Center of Nanocatalysis & Sensing. Now, he is also a professor in Shandong Normal University. He was recognized as a highly cited researcher (2018–2020) in both areas of chemistry and materials science by Clarivate Analytics. He published over 600 papers with total citations of 70 |
1. Introduction
As the global demand for clean energy continues to rise, hydrogen (H2) is increasingly recognized as the optimal energy option in the low-carbon era due to its high energy density of approximately 284 kJ mol−1 and environmentally friendly characteristic.1,2 At present, the dominant industrial approaches to H2 production, including natural gas reforming, methanol reforming, and coal gasification, pose substantial environmental challenges due to their substantial carbon dioxide (CO2) emissions and the generation of other detrimental byproducts.3–6 Water electrolysis offers a promising alternative for green H2 production. Currently, the commercial water splitting technology includes proton exchange membrane (PEM) water electrolysis and alkaline (ALK) water electrolysis, which both rely on fresh water as the electrolyte.7,8 Considering the lack of fresh water, the study of seawater (about 96.5% of the world's water resource)9 electrolysis is receiving increasing attention. By utilizing the abundant seawater resources and integrating with renewable energy sources, such as solar and wind power, seawater electrolysis holds significant potential in facilitating the transition to a sustainable and environmentally friendly energy landscape.10 Nevertheless, the complex composition of natural seawater presents some formidable challenges when it comes to realizing large-scale industrial applications of its electrolysis process.Seawater electrolysis faces challenges similar to electrolysis using fresh water in facilitating both hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) simultaneously. HER in seawater can be hindered by impurities like dissolved cations (e.g., Mg2+ and Ca2+), bacteria, microorganisms, and particles that may clog or damage electrodes, reducing durability. OER faces challenges from electrochemically active anions (Cl−), which can interfere with the reaction. Interestingly, Qiao's team recently reported a positive effect of Cl− in alkaline seawater electrolysis, rather than the commonly perceived negative effect.11,12 They found that the adsorption sites for Cl− on the surface of NiFe layered double hydroxide (LDH) catalysts are Fe sites rather than Ni sites. This means that Cl− can inhibit the leaching of Fe while creating more active Ni sites, thereby increasing the activity and stability of the catalyst. This is a novel and interesting point of view that provides a new perspective and idea to understand and optimize processes and catalysts for alkaline seawater electrolysis. Nevertheless, the limitation is that it only explores one specific catalyst, NiFe LDH, which may not be applicable to other types of catalysts or other ions in seawater. In general, both HER and OER reactions often suffer from limited rates and voltage requirements.13 In particular, a higher voltage range of 1.8–2.0 V is required, exceeding the theoretical voltage of 1.23 V.14
In order to realize the large-scale H2 production from seawater electrolysis, electrocatalysts and electrolyzers are two key factors. Most reported catalysts are used for HER15–19 or OER,20–24 so two different electrocatalysts must be applied in the seawater electrolysis process. If there are efficient bifunctional electrocatalysts, which have the ability to promote the reaction of HER and OER simultaneously, the overall seawater splitting process will be easier to carry out. Bifunctional catalysts not only enhance reaction rates and reduce the required voltage, but also contribute to achieving higher electrolysis efficiency, potentially reducing the cost of H2 production. Meanwhile, the study of bifunctional catalysts is conducive to the exploration of novel catalytic mechanisms and principles, and provides guidance for the design and synthesis of more efficient catalysts. Therefore, the development of bifunctional catalysts with high activity and low cost is key to achieve efficient H2 production from seawater.
Efficient seawater splitting also relies on the design of efficient, high-performance, and cost-effective seawater electrolyzers. In addition to PEM and ALK electrolyzers that are more mature in the commercial market, there are anion exchange membrane electrolyzers25,26 and solid oxide electrolytic cells (SOEC).27 They are relatively new and costly compared to the above two technologies. However, when these electrolyzers are used directly for the seawater electrolysis, the complex natural composition of seawater significantly impacts the process, such as the physical or chemical clogging of the ion exchange membranes and corrosion of the metal components. Effectively addressing these issues is critical for the widespread adoption of seawater electrolysis technology. Research and development efforts are primarily dedicated to improving the durability of ion exchange membranes to prevent clogging and exploring alternative corrosion-resistant materials for the metal components. Moreover, exploring new materials and engineering approaches to withstand the harsh conditions inherent in seawater will facilitate improving the overall performance and durability of seawater electrolyzers. In conclusion, the selection and design of catalyst materials can influence the structure and performance of the electrolyzer. A well-planned electrolyzer design can maximize the utilization of the catalyst activity and enhance the electrolysis efficiency.
Several recent reviews have discussed the electrocatalysts for seawater splitting from different perspectives, mainly single-function OER catalysts28,29 and design strategies of electrocatalysts.30,31 Recently, our team completed a review covering both lab-scale fundamental research and pilot-scale reactor levels, as well as future trends in the field of seawater electrolysis.32 However, there are few reviews that are particularly focused on bifunctional electrocatalysts and electrolyzers for overall seawater splitting. In this review, we begin by introducing the fundamentals of seawater electrolysis, including the principles of the reactions involved and the parameters used to evaluate the performance of bifunctional catalysts. Subsequently, we present a comprehensive overview of recently reported bifunctional catalysts for seawater splitting, focusing on their activity and stability. Common bifunctional catalysis materials for seawater electrolysis encompass noble metal-based catalysts and transition metal-based catalysts such as transition metal phosphides, borides, chalcogenides, nitrides, oxides, and hydroxides. Furthermore, we delve into recent advancements in electrolyzers, encompassing both membrane and membrane-free systems, and provide a detailed analysis of these developments. Finally, we discuss future research directions and highlight opportunities to promote the practical application of seawater electrolysis systems and technologies.
2. Principles of electrolytic seawater
2.1 Reaction mechanisms
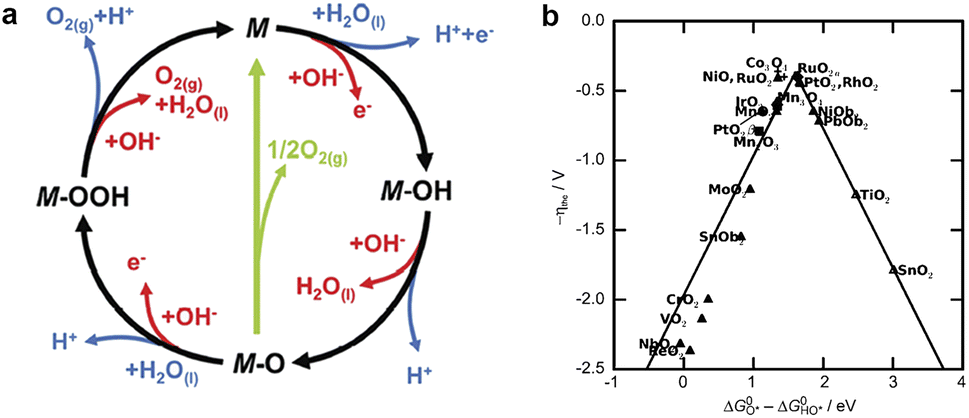 | ||
| Fig. 1 (a) The mechanism of OER on an electrode surface. Reprinted with permission.35 Copyright 2017, The Royal Society of Chemistry. (b) Activity trends for OER as a function of ΔGO* − ΔGOH* for rutile and anatase oxides. The activity is expressed by the value of the overpotential to achieve a certain value of current density. Reprinted with permission.36 Copyright 2011, Wiley-VCH. | ||
Acidic:
| 2H2O → O2 + 4e− + 4H+ | (1) |
Alkaline:
| 4OH− → O2 + 2H2O + 4e− | (2) |
Fig. 1b compares the effectiveness of the first row of transition metal oxides as oxygen generation catalysts under acidic or alkaline conditions based on their redox properties.36 The figure shows that RuO2 and IrO2 are the best catalysts for oxygen generation due to their low redox potentials and high electrical conductivity. However, they are costly and do not perform well in hydrogen generation reactions. Therefore, some more economical and sustainable bifunctional catalysts, such as Co phosphates/phosphides and Ni–S composites, have been developed, which are highly active in both oxygen generation and hydrogen generation reactions and have good electrochemical stability.
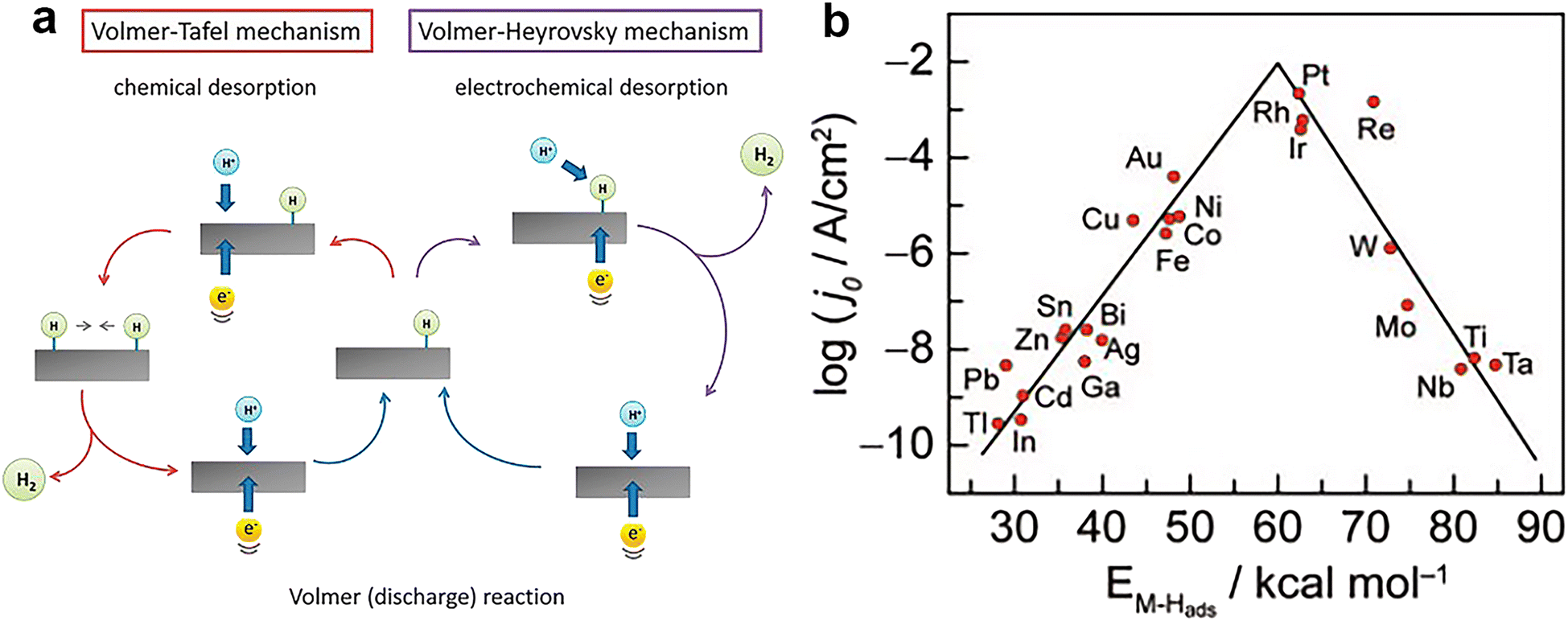 | ||
| Fig. 2 (a) The mechanism of HER on the surface of an electrode in acidic solutions. Reprinted with permission.37 Copyright 2014, The Royal Society of Chemistry. (b) Volcano plot for the HER on metal electrodes in acidic media. The log of the exchange current density j0 is plotted versus the M–H bond energy for each metal surface. Reprinted with permission.38 Copyright 1972, Elsevier. | ||
Acidic:
| 2e− + 2H+ → H2 | (3) |
Alkaline:
| 2H2O + 2e− → H2 + 2OH− | (4) |
Fig. 2b shows the curve relating the logarithm of the HER exchange current density (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) j0) for different metals to the strength of the metal–hydrogen (M–H) bond in an acidic environment.38 This volcano curve allows a quick comparison of the activity of different metals. However, this volcano curve only considers the effect of the M–H bond energy (or the properties of the metal) on the HER kinetics, and does not take into account other factors, such as the pH of the solution. Perhaps, it seems more convincing that this plot becomes a 3D volcano plot obtained with pH as the third axis.
j0) for different metals to the strength of the metal–hydrogen (M–H) bond in an acidic environment.38 This volcano curve allows a quick comparison of the activity of different metals. However, this volcano curve only considers the effect of the M–H bond energy (or the properties of the metal) on the HER kinetics, and does not take into account other factors, such as the pH of the solution. Perhaps, it seems more convincing that this plot becomes a 3D volcano plot obtained with pH as the third axis.
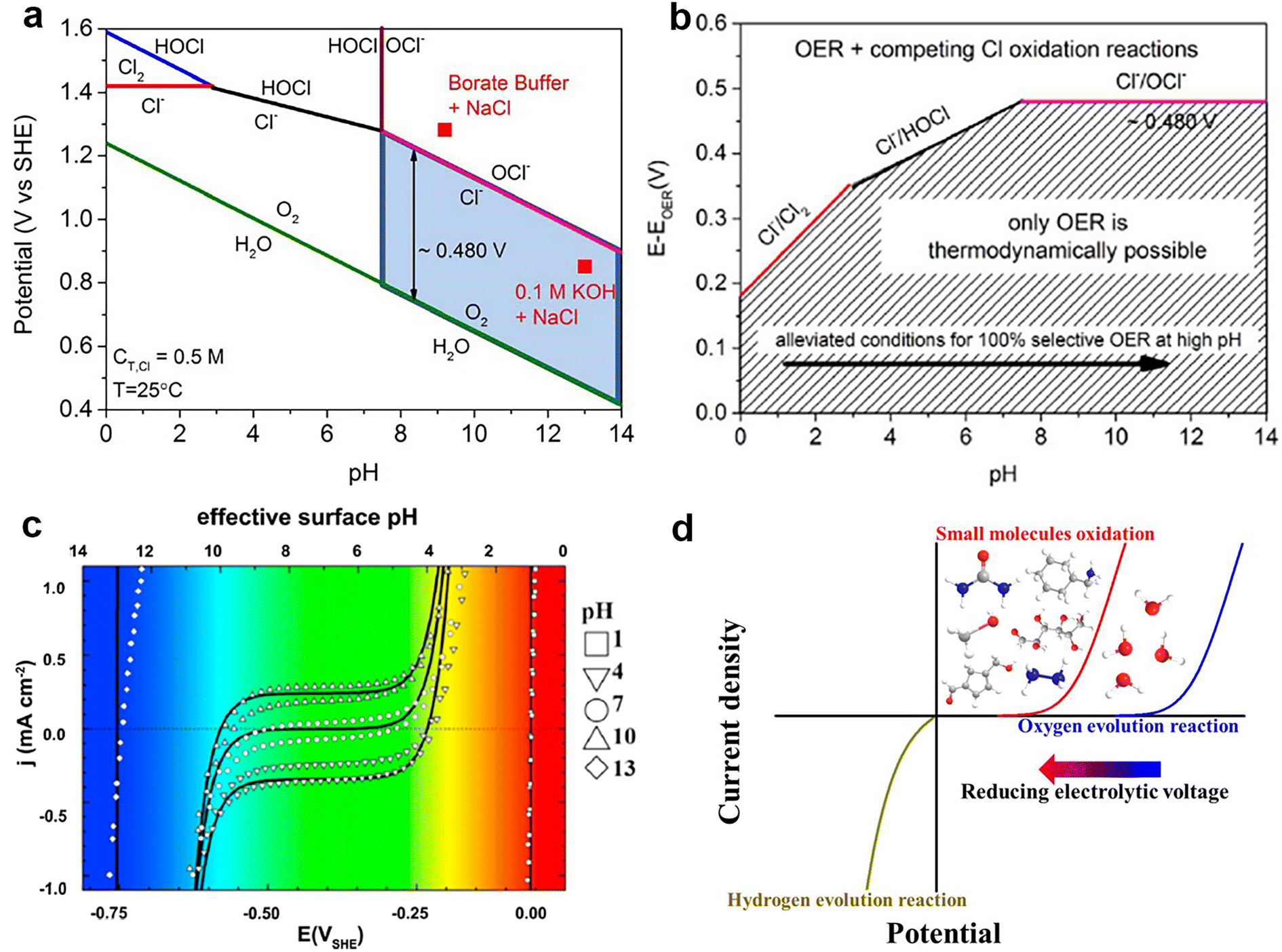 | ||
| Fig. 3 (a) Pourbaix diagram for the artificial seawater model. A chlorine system, in the case of dissolved 0.5 M NaCl aqueous solution and no other chlorine sources, with a total chlorine species (cT,Cl) of 0.5 M. The electrode potential for OER is also included (assuming oxygen partial pressure of 0.21 atm = 0.021 MPa). Two red square points show the operating potentials (vs. SHE) after 1 h constant current electrolysis (10 mA cm−2) with the NiFe layered double hydroxide (NiFe LDH) catalyst in 0.1 M KOH + 0.5 M NaCl (pH 13) and 0.3 M borate buffer + 0.5 M NaCl (pH 9.2) electrolyte. The light blue box highlights our proposed design criterion. (b) The maximum allowed overpotential of the OER electrolyzer catalysts to ensure 100% selective water splitting. Values are obtained as the difference between the standard electrode potentials of the 3 relevant chloride oxidation reactions (chlorine, hypochlorous acid, and hypochlorite formation) and the OER versus pH. The dashed area corresponds to oxygen-selective overpotential–pH conditions where the thermodynamics point to 100% selective oxygen evolution. Above the limiting lines, chlorine-based reaction products are thermodynamically feasible, yet may be limited by kinetic overpotentials. Reprinted with permission.49 Copyright 2016, Wiley-VCH. (c) The current density–surface pH relation obtained by cyclic voltammetry in H2-saturated, unbuffered solutions of bulk pH 1–13. Reprinted with permission.50 Copyright 2011, Elsevier. (d) Comparison of water electrolysis with the OER and with small-molecule oxidation reactions. Reprinted with permission.48 Copyright 2022, Wiley-VCH. | ||
2.2 Evaluation parameters for catalysts
485 C mol−1), the number of electrons transferred in the reaction, the concentration of oxidation products, and the concentration of reduction products, respectively. Then, the overpotential can be expressed by eqn (6).
Nernst equation:
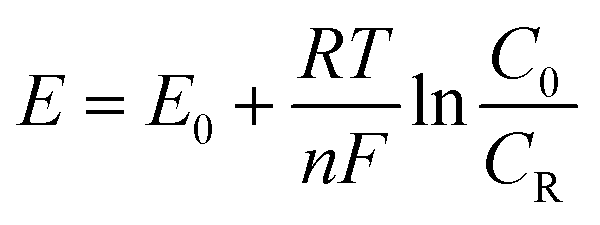 | (5) |
Overpotential formula:
| η = Ei − Et | (6) |
The presence of an overpotential means that more energy than thermodynamics is required to drive the related reaction. With a lower overpotential of the catalyst at the specified current density, a better catalytic ability of the catalyst for the target reaction can be achieved. For overall water splitting, the overpotential is the part of the cell voltage that is over 1.23 V vs. RHE.
Butler–Volmer formula:
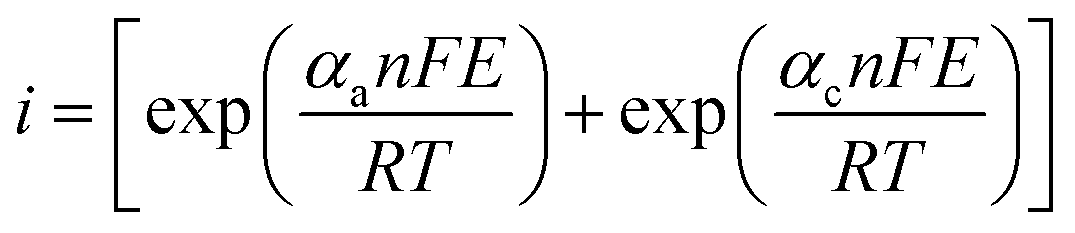 | (7) |
Tafel formula:
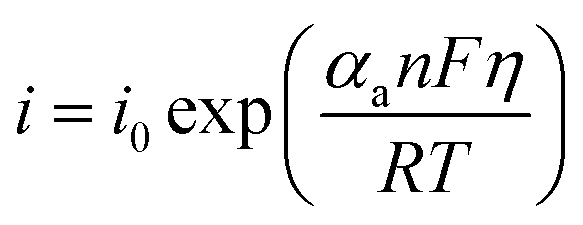 | (8) |
Logarithm of both sides of the Tafel formula:
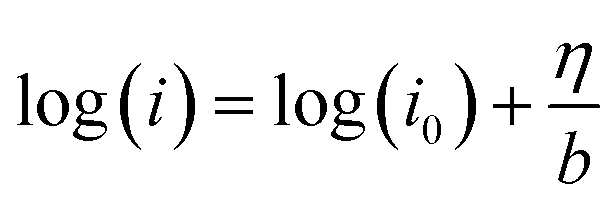 | (9) |
Tafel slope formula:
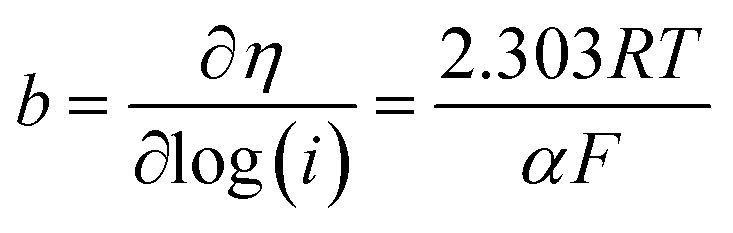 | (10) |
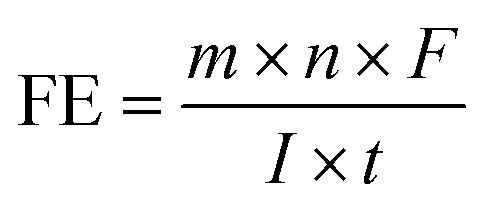 | (11) |
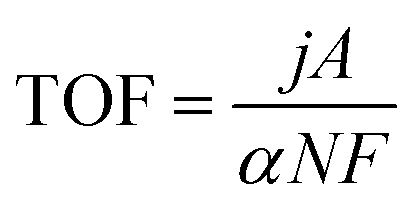 | (12) |
However, it is difficult to determine the number of active sites, resulting in subtle differences in the calculation of TOF for different catalytic materials. The active sites (those parts of the catalyst that really play the catalytic role, often also called active centre) differs for each catalyst. It can be an atom, a cluster of surface atoms, a coordination complex, an unsaturated atom on a solid surface or others, which makes it difficult to obtain TOF accurately. For the OER reaction, the ring current collected in the rotating ring-disk electrode (RRDE) can be used to calculate the TOF.55 Also, for some single transition metal materials (Ni, Co, Fe, etc.) undergoing electrocatalysis and having a known number of redox-transferred electrons, the number of active sites can be determined by calculating the redox charge Q (peak area).56,57 In any case, the characteristics of different materials are distinct, and the calculation of TOF should be combined with specific analysis of each work.
3. Seawater treatment
Seawater treatment is an important step in realizing H2 production from seawater electrolysis, which can effectively remove harmful substances in seawater and improve the efficiency and selectivity of electrolysis. There are various methods for seawater treatment, which mainly include the following:3.1 Filtration
Separate solid impurities, microorganisms, organics, etc. from seawater by physical or chemical means, so as to reduce the contamination of electrodes and membranes. The advantages of the filtration method are simplicity and low cost, but the disadvantages are that it cannot remove calcium and magnesium ions in seawater, and it may cause clogging or depletion of filter materials. Guo et al.58 filtered out solid impurities and removed microbes in natural seawater (pH = 7.9) before electrolysis.3.2 Alkali addition method
By adding alkaline substances such as sodium hydroxide (NaOH) and potassium hydroxide (KOH) to seawater, the pH of the seawater is raised, thus inhibiting the oxidation reaction of Cl− and increasing the generation of oxygen.59 The advantages of the alkali addition method are that it can improve the selectivity of electrolysis and has a low electrolytic voltage, but the disadvantages are that it requires high corrosion resistance of the materials used in electrolytic device and will increase the conductivity of seawater, resulting in an increase in the energy consumption of electrolysis, and may cause the waste of alkaline substances or the problem of seawater discharge.3.3 Acid addition method
By adding acidic substances, such as sulfuric acid and hydrochloric acid, to seawater, the pH value of seawater is lowered, thus inhibiting the precipitation reaction of calcium ions (Ca2+) and magnesium ions (Mg2+), and reducing the scaling of electrodes and membranes. The advantages of the acid addition method are that it can improve the efficiency of electrolysis and avoid possible CER side reactions in alkaline electrolytes, but the disadvantages are that it also requires high corrosion resistance of the materials used in electrolytic device and will increase the corrosiveness of seawater, leading to the damage of electrodes and membranes, and may cause the waste of acid or the problem of seawater discharge. However, under acidic conditions, the difference in overpotential between HER and OER is not significant.3.4 Salt addition method
Wu et al. used 0.68 g Na2CO3 to remove Mg and Ca salts before application.46 This treatment reduces deposition and corrosion problems during electrolysis and improves the efficiency of electrolysis; this also changes the pH of the seawater and increases the cost of the electrolyte, which needs to be replaced or treated regularly.The method of seawater treatment should be selected on the basis of different electrolyzers and electrolysis conditions, while the cost and effectiveness of seawater treatment, as well as seawater monitoring and discharge issues, should also be considered. The aim of seawater treatment is to improve the feasibility and sustainability of seawater electrolysis for hydrogen production, contributing to energy and environmental development.
4. Bifunctional electrocatalysts for seawater splitting
So far, various kinds of catalysts have been reported to catalyze both HER and OER efficiently in alkaline electrolyte, including noble-metal-based catalysts and transition-metal-based catalysts. Some bifunctional transition-metal-based electrocatalysts have been demonstrated to be highly efficient toward HER-OER, with some even surpassing the performance of benchmark noble metal catalysts. The following sections will give a detailed discussion on their synthesis, performance, and the real active sites.4.1 Noble metal-based compounds
Platinum (Pt) is widely recognized as the benchmark catalyst for the HER, while iridium (Ir) and ruthenium (Ru) oxides are respectively acknowledged as benchmark catalysts for the OER.60,61 This implies that Pt exhibits exceptional catalytic activity in HER, whereas Ir and Ru oxides demonstrate outstanding catalytic performance in OER. Given the scarcity and high cost of precious metals, some researchers have adopted strategies to reduce the usage of these precious metals, while preserving their excellent catalytic performance. Mu et al.62 introduced an electrocatalyst for water electrolysis known as ultralow Ru-incorporated amorphous cobalt-based oxides (Ru–CoOx/NF), which exhibited exceptional bifunctional electrocatalytic capabilities in both alkaline water and seawater environments. As a bifunctional catalyst for overall seawater splitting, it exhibited a lower overpotential of 1390 mV to achieve a current density of 1 A cm−2 compared to Pt/C/NF‖RuO2/NF (1530 mV) under the same conditions (Fig. 4a), and the FE was almost 100% (Fig. 4b). According to the content of the article, the authors asserted that this catalyst's high activity can be attributed to several factors. The amorphous structure provides additional active sites and the incorporation of Ru modulates the electronic structure of cobalt-based oxides, facilitating charge transfer. Additionally, surface electronic restructuring of cobalt-based oxides and the formation of β-CoO(OH) occur during the OER process. Recently, Wu and colleagues63 employed an electroless deposition technique to synthesize Ru-incorporated amorphous-crystalline Ni(Fe)P2 nanosheets directly on a nickel foam substrate (Ru–Ni(Fe)P2/NF), as shown in Fig. 4c. These nanosheets were utilized as a bifunctional electrocatalyst for alkaline seawater electrolysis. In alkaline seawater, this catalyst exhibited overpotentials of 295 mV for the HER and 375 mV for the OER to attain the large current density of 1 A cm−2, surpassing the performance of commercial Pt/C and RuO2 catalysts. The assembled electrolyzer achieved the same current density at a low voltage of 1.63 V (Fig. 4d) and demonstrated remarkable stability, sustaining continuous operation at 1 A cm−2 for 50 hours with less than 10% reduction in performance (Fig. 4e). The unique amorphous-crystalline structure of the catalyst provided additional active sites, expanded the electrochemical active surface area, and reduced the adsorption free energy of the hydrogen-containing intermediates, resulting in enhanced HER catalytic efficiency. Additionally, the incorporation of Ru into the catalyst adjusted the electronic structure of Ni(Fe)P2, facilitating charge transfer and reducing the adsorption free energy of the oxygen-containing intermediates, thus improving the OER catalytic efficiency.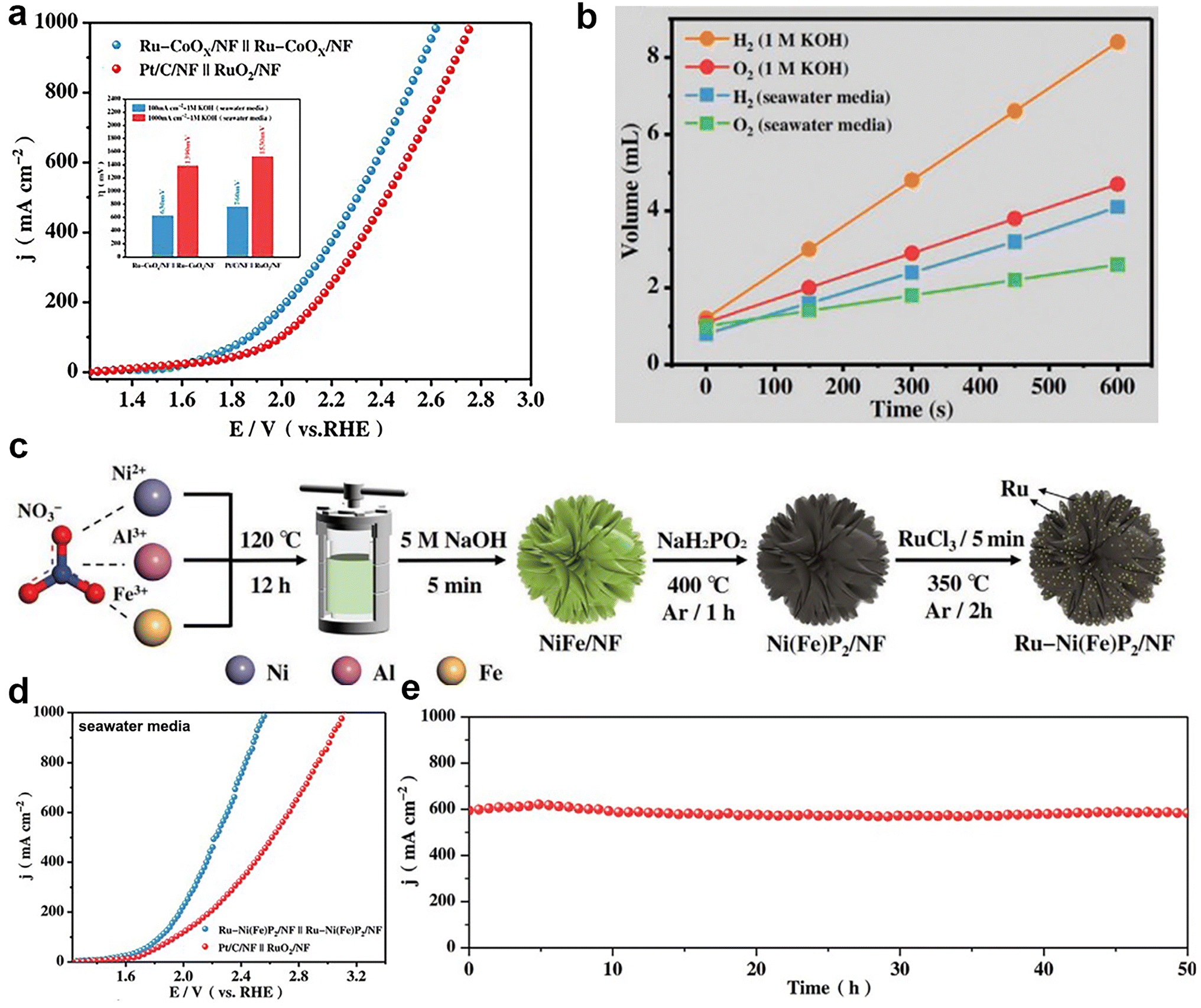 | ||
| Fig. 4 (a) Polarization curves of Ru–CoOx/NF‖Ru–CoOx/NF and Pt/C/NF‖RuO2/NF toward overall water splitting in seawater media. Inset: the value of ΔE. (b) Diagram of the amount of H2 and O2 released over time in 1 M KOH and seawater media. Reprinted with permission.62 Copyright 2021, Wiley-VCH. (c) Construction route of Ru–Ni(Fe)P2/NF. (d) Polarization curves of Ru–Ni(Fe)P2/NF‖Ru–Ni(Fe)P2/NF and Pt/C/NF‖RuO2/NF toward water splitting in seawater media. (e) 50 h constant current stability test plot of Ru–Ni(Fe)P2/NF in seawater media. Reprinted with permission.63 Copyright 2023, Wiley-VCH. | ||
The incorporation of small amounts of noble metals to enhance the catalyst performance is regarded as an effective and practical strategy. By doping trace amounts of noble metals into cost-effective non-noble metal catalysts, catalyst performance can be effectively improved while reducing costs. This approach not only enhances the catalytic activity but also improves stability and selectivity, making it more suitable for practical applications. However, this strategy also presents several challenges. Firstly, achieving the effective and uniform dispersion of noble metals within non-noble metal catalysts for efficient utilization remains a technical challenge. Secondly, optimizing the doping concentration of noble metals for optimal performance requires further investigation. Moreover, for applications involving water electrolysis in complex media such as seawater, the impact of side reactions like chlorine evolution needs to be considered. In summary, this is a promising field worthy of further research. Through continuous refinement and improvement, an efficient, stable, and cost-effective seawater electrolysis technology is anticipated.
4.2 Transition metal phosphides
Generally, transition metal phosphides have good HER properties because they can provide the appropriate electron transfer capacity, active sites, adjustable structure, and reduce energy barriers to facilitate the reduction of H+ to H2.64 At present, there are mainly monometallic phosphides and bimetallic phosphides for seawater electrolysis, of which the latter has been reported relatively more. Recently, Hao's group65 deposited cobalt phosphorus-based species on a robust nickel net (Co–P@NN) by the one-step mild electroless plating method (Fig. 5a). With different active sites including P for HER and Co2+ for OER, as a bifunctional catalyst, Co–P@NN required overpotentials of 298 and 235 mV to attain current densities of 500 and 10 mA cm−2 for HER and OER in 1 M KOH with 0.5 M NaCl solution, respectively. Moreover, the Tafel slopes of 61.5 and 70 mV dec−1 for HER and OER were lower than that for the Pt foil electrode (76.4 mV dec−1) and IrO2@NN (78.2 mV dec−1), exhibiting faster kinetics. During the overall seawater splitting process, the two-electrode system with Co–P@NN as the anode and cathode showed better performance than Pt foil‖IrO2@NN in 1 M KOH and 0.5 M NaCl electrolyte, accompanied with ultra-stability of 2880 h at 500 mA cm−2 without obvious decay (6.95%) (Fig. 5b and c). Feng et al.66 constructed a cobalt-based catalyst rich in phosphorus vacancies (denoted as CoxPv@NC). According to the paper, this bifunctional catalyst had two different active sites, including Co(OH)2 coated on CoxPv for HER and the new formed active species of CoO(OH) for OER. The introduction of the P vacancy could optimize the adsorption energy of active intermediates for HER and accelerate the reconstruction of active species for OER (Fig. 5d and e). A two-electrode system used CoxPv@NC as the anode and the cathode with 1.0 M KOH seawater as the electrolyte, requiring a low voltage of 1.88 V to afford 1.0 A cm−2 and exhibiting no significant decrease over the 100 h test. Fu et al.67 synthesized a class of catalysts by growing robust Ni–P nanospheres on flexible corrosion-resistant hydrophobic asbestos (HA) (Fig. 5f). The Ni–P and NiOxHy compounds are the real catalytically-active species during the HER and OER process (Fig. 5g). During the overall seawater splitting process, the ratio of H2 to O2 produced is close to 2:1, meaning that the FE of this system is close to 100% (Fig. 5h). Moreover, the overall electrolysis system composed of this catalyst as the cathode and anode can work stably for 40 days at a current density of 500 mA cm−2 (Fig. 5i).
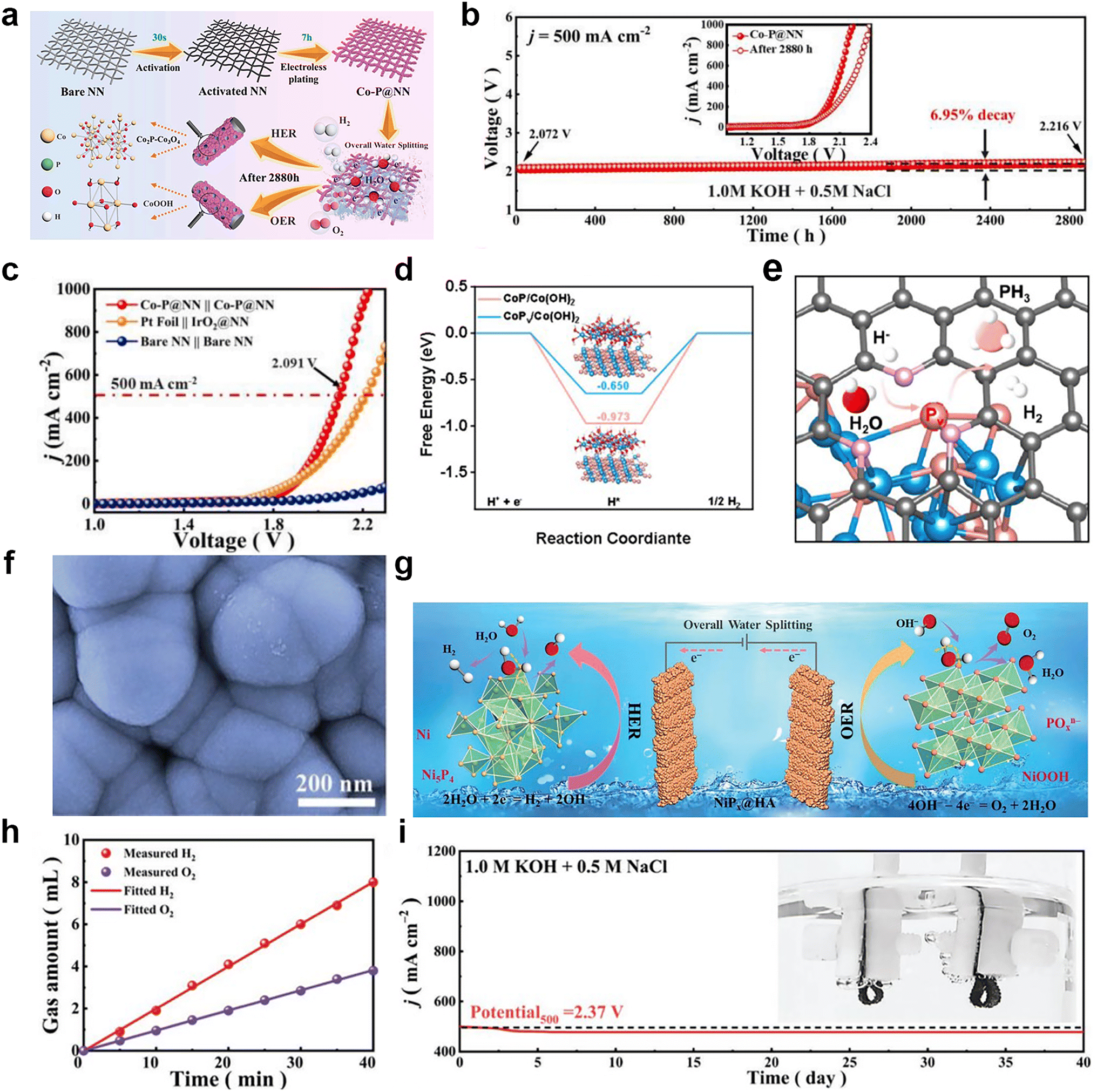 | ||
| Fig. 5 (a) Illustration of the synthesis process of the Co–P@NN electrode for the overall water splitting. (b) CP response curves for the overall seawater splitting device at 500 mA cm−2 (inset: LSV curves of Co–P@NN‖Co–P@NN before and after 2880 h overall seawater splitting). (c) LSV curves of Co–P@NN‖Co–P@NN, Pt foil‖IrO2@NN and bare NN‖bare NN for overall seawater splitting. Reprinted with permission.65 Copyright 2023, Elsevier. (d) Hydrogen evolution of CoPv/Co(OH)2 and CoP/Co(OH)2. (e) Schematic illustration of P vacancies in CoxPv@NC. Reprinted with permission.66 Copyright 2023, Elsevier. (f) SEM image of NiPx@HA. (g) Schematic illustration showing the HER and OER mechanisms of the NiPx@HA bifunctional electrode in alkaline simulated seawater. (h) FE curve for the NiPx@HA bifunctional electrode. (i) CP measurement of NiPx@HA‖NiPx@HA at 500 mA cm−2 for 960 h. Reprinted with permission.67 Copyright 2023, Wiley-VCH. | ||
In addition, bimetal phosphides can improve the catalyst activity and stability through a synergistic effect of the bimetal. Ren et al.68 reported a bifunctional catalyst Ni2P–Fe2P/NF with bi-active neutral P atoms for HER and Ni-/Fe-oxides or -hydroxides for OER. Moreover, the self-support effect and from the alloying Ni and Fe with P atoms enhanced the stability and corrosion resistance of the catalyst. In the two-electrode system with 1 M KOH seawater electrolyte, Ni2P–Fe2P/NF required voltages of 1.811 V to attain a current density of 100 mA cm−2 for the overall seawater splitting that was superior to the benchmark of IrO2‖Pt/C, and can operate continuously for 48 h. In a recent study, Luo et al.69 designed a high-performance bifunctional catalyst doping of Mn atoms in bimetallic compounds (Ni2P/Fe2P) by a facile hydrothermal-phosphorylation method. Owing to the increased number of active sites, this catalyst exhibited excellent catalytic performance. It only needed an overpotential of 358 mV and 470 mV to drive a large current density of 1000 mA cm−2 for HER and OER in alkaline seawater, respectively. Moreover, the two-electrode system with it as cathode and anode required a tank voltage of 2.02 V to achieve the current density of 500 mA cm−2 and could operate steadily for 120 h.
Transition metal phosphides have received much attention in the study of bifunctional catalysts for the electrolysis of seawater due to their unique structures and properties. They represent an important research direction in the field of catalysts, and may play an important role in energy conversion and other related applications in the future.
4.3 Transition metal oxides and hydroxides
Transition metal oxides and hydroxides have been extensively studied as catalysts for seawater splitting because of their unique advantages, including rich electronic structure, electrochemical activity, and low cost. Especially for OER, they can lower the energy barrier and promote the desorption of O2.70 Iron oxides, nickel oxides, and cobalt oxides are the three most common of the transition metal oxide catalyst family. NiFe LDH has excellent electrocatalytic activity, especially OER. However, its stability is often unsatisfactory. In order to make it suitable for seawater splitting, Enkhtuvshin and colleagues71 reported a surface reconstructed NiFe LDH (Fig. 6a and b) by RF-plasma treatment under reducing conditions, which transformed it into a nanostructure consisting of a crystalline Ni3Fe alloy and a low-crystalline NiFe (oxy)hydroxide phase. The Ni3Fe alloy phase as a Cl− blocking layer was able to effectively inhibit the adsorption and corrosion of Cl− on the surface of NiFe LDH, thus improving the selectivity and stability of seawater electrolysis. Meanwhile, the NiFe alloy had efficient reduction reaction (HER) activity, while the NiFe (oxy)hydroxide phase was responsible for the oxidation reaction (OER) activity, realizing the bifunctional catalysis. This material has the structure of nanosheets that is beneficial for catalytic activity (Fig. 6c and d). According to the data, this bifunctional catalyst could steadily work for 100 h at current density of 100 mA cm−2 in concentrated alkaline solution (30 wt% KOH and 0.5 M NaCl) during overall water splitting. Our group also had found that benzoate anions-intercalated NiFe LDH nanosheet on carbon cloth could enhance the stability during the process of alkaline seawater electrolysis.72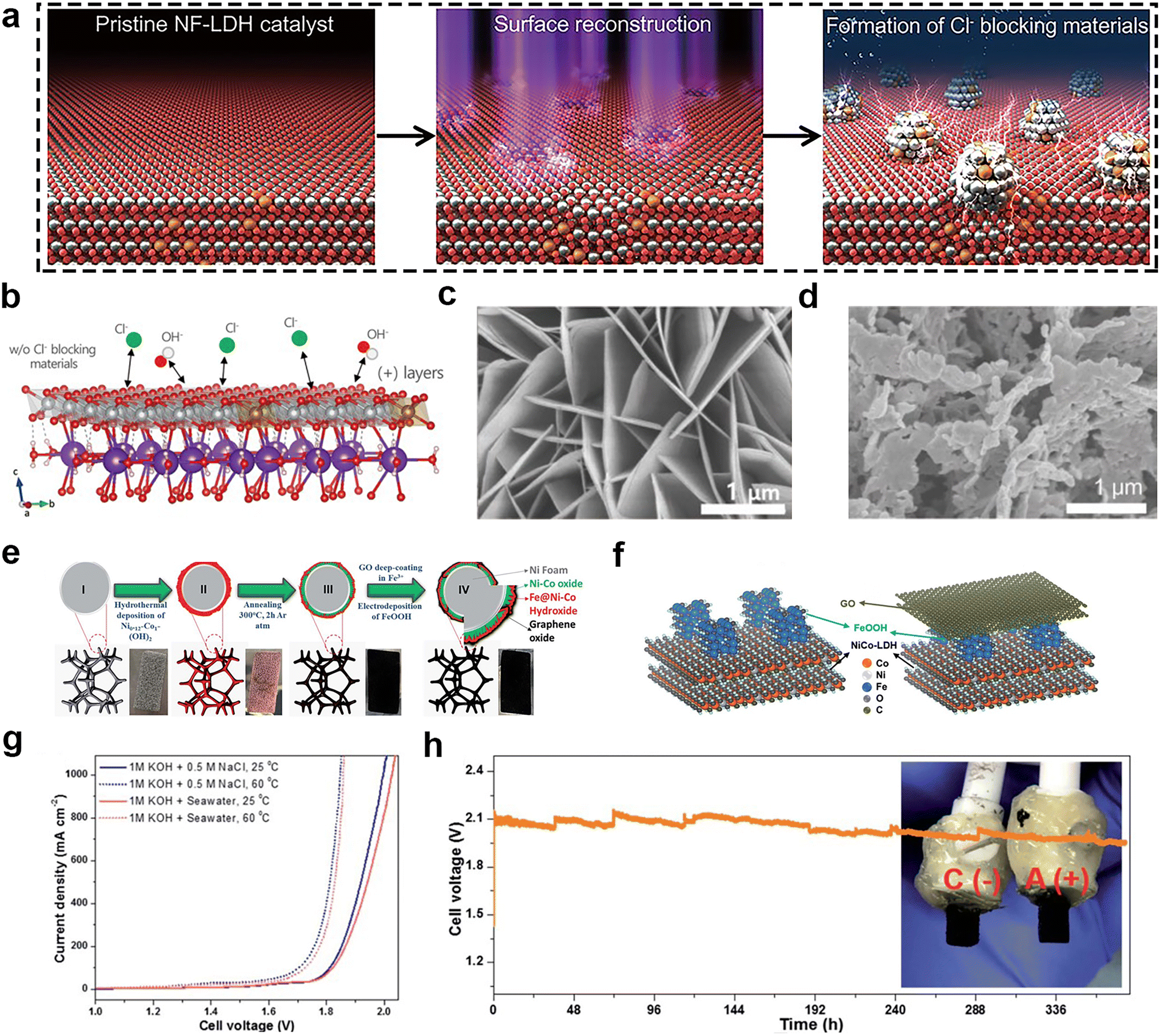 | ||
| Fig. 6 (a) Schematic illustration of the surface reconstruction process of pristine NiFe LDH catalyst transforming to a mixture of NiFe (oxy) hydroxide and metallic alloy phases via the RF-plasma process. (b) Positively charged surface layer of Ni0.9Fe0.1(OH)2 with intercalated potassium and water molecules. Gray, brown, red, white, purple, and green balls indicate Ni, Fe, O, H, K, and Cl atoms, respectively. SEM images of (c) pristine NF-LDH and (d) c-NF//a-NF-LDH NS. Reprinted with permission.71 Copyright 2023, Wiley-VCH. (e) Schematic illustration of the synthesis procedure for a free-standing GO@Fe@Ni–Co@NF electrode with corresponding digital images of the electrode in intermediate steps [II: Ni–Co hydroxide@NF, III: Ni–Co@NF, IV: GO@Fe@Ni–Co@NF]. (f) Model of the outermost layer structure of the Fe@Ni–Co@NF catalyst (left) and the GO@Fe@Ni–Co@NF catalyst with a GO overlayer (right). (g) Overall alkaline seawater splitting performance of the GO@Fe@NiCo@NF(+)‖GO@Fe@NiCo@NF(−) electrolyzer in different electrolytes and temperature. (h) Durability tests (378 h) recorded at a constant current density of 1000 mA cm−2 of the seawater splitting electrolyzer under 1 M KOH + 0.5 M NaCl (inset images; a digital photograph of anode and cathode after 378 h of seawater splitting testing under 1 M KOH + 0.5 M NaCl). Reprinted with permission.73 Copyright 2023, The Royal Society of Chemistry. | ||
Moreover, it is also possible to design a catalyst with different components in which an oxide can act as a resistance barrier to chloride ions. For instance, in order to deal with the challenge caused by Cl−, Lee's group73 proposed a method of using an interfacial Cl− blocking layer on the catalyst surface. They reported an active material in which FeOOH deposited β-Ni–Co hydroxide with an outer graphene oxide (GO) layer. This catalyst had a multilayer structure (Fig. 6e and f). The GO layer formed nanochannels (0.3–0.7 nm), acting as a sieve to block the ions movement of Cl− and Na+ and allow HO− and H2 gas penetration. The o-tolidine test suggested that there was no chloride oxidation product after 378 h during the stability test (Fig. 6h), which means this catalyst had great selectivity. Moreover, the metal oxide layer underneath helped to lower the charge transfer resistance of the catalyst. Using it as the anode and the cathode to form the alkaline seawater electrolyzer, a large current density of 1000 mA cm−2 was driven at a low voltage of 2.02 during the overall seawater splitting (Fig. 6g).
Both anion electrostatic repulsion and catalyst surface reconstruction to form a chloride ion barrier are effective response strategies to the OER challenge (CER). The efficiency and stability of seawater electrolysis can be significantly improved by changing the surface structure and composition of the catalyst. These provide the possibility of developing new and more efficient technologies for seawater electrolysis.
4.4 Transition metal chalcogenides
As mentioned before, the CER competes with the OER, so the design of catalysts applied to seawater electrolysis must take this into account. Recently, based on CoFe LDH with good performance for OER, Zhang's group74 designed a bifunctional catalyst by partial selenitization (Se–CoFe-LDH), as shown in Fig. 7a. This article argued that the introduction of Se during electrolysis can create an ionic aggregation state around the electrodes (selenate), thus achieving resistance to Cl−. The introduction of selenium atoms does not usually act as active sites for the catalyst. Rather, it is used to modify the internal structure of the precursor, thereby increasing the active surface area, and ultimately enhancing the activity of the catalyst. According to the data, this catalyst both as the anode and the cathode for overall seawater splitting could work for 245 h steadily (Fig. 7b). However, its true active sites were the oxides/(oxy)hydroxides of Fe and Co. | ||
| Fig. 7 (a) Schematic illustration of the synthetic process and seawater electrolysis of Se–FeCo-LDH. (b) The stability test (at 10 mA cm−2) of alkaline seawater splitting for 245 h. Reprinted with permission.74 Copyright 2023, Elsevier. (c) I–t curves of the S–NiFe-Pi/NFF electrolyzer at a constant voltage of 1.68 and 1.80 V for overall seawater electrolysis, respectively. (d) Corrosion polarization curve in pure natural seawater. Reprinted with permission.76 Copyright 2023, Elsevier. | ||
As a homologous element, sulfur also has the same properties as selenium. Tanveer ul Haq and Yousef Haik75 found that the introduced S enhanced the hydrophilicity aerophobicity and ECSA of the free-standing electrode material. Song and coworkers76 reported a bifunctional catalyst denoted as S–NiFe-Pi/NFF for overall seawater splitting. They found that the doping of the S atom distorted the lattice of NiFe-phosphate and regulated the local electronic environment around the Ni/Fe active metal, which enhanced the electrocatalytic activity. As a result, S–NiFe-Pi/NFF‖S–NiFe-Pi/NFF required voltage of 1.8 V to attain a large current density of 500 mA cm−2 and could continuously work for 100 h (Fig. 7d) because the phosphate groups repelled Cl−. The values of the corrosion potential and corrosion current also reflect the reason for good stability. As shown in Fig. 7d, S–NiFe-Pi/NFF had higher corrosion potential and lower corrosion current, meaning that it has better resistance to seawater corrosion. Furthermore, a nickel–iron sulfide nanosheet array on nickel foam (NiFeS/NF)77 was a superb bifunctional catalyst for seawater splitting that had been proven by our group. Thus, NiFeS/NF can drive the industrially demanded current density of 500 mA cm−2 at overpotentials of 300 and 347 mV cm−2 for HER and OER in alkaline seawater. Additionally, our group reported a bifunctional catalyst Fe–NiS/NF,78 which could attain a current density of 1000 mA cm−2 at 1.88 V during the overall seawater splitting.
According to these reported literature studies, the role of S is not as a true active site. However, it is worth noting that as the electrolysis process proceeds, sulfur or selenium being an anion is electronegative with Cl− and appears to be more suitable for real seawater in terms of chlorine corrosion resistance. Also, S could combine with Mg2+ and Ca2+ to solve the problem of HER caused by the complexity of seawater.79
4.5 Transition metal borides
Transition metal borides can be used as electrocatalysts for the OER and HER to produce high-purity H2 and O2. There are more than 100 different crystalline phases of transition metal borides, in which the boron atoms can exhibit a rich variety of covalent bonding modes, affecting the electronic structure and catalytic activity.80 There have also been a lot of reports written about it. For instance, Mandavkar and coworkers81 reported on a ternary nickel–molybdenum–boron (NiMoB) electrocatalyst with multi-sphere morphology (Fig. 8a), which was fabricated by the incorporation of B atoms into the NiMo system. The B atoms boosted the catalytic property by modifying the electronic structure to lower the kinetic barriers with the electron enriched metallic sites and improving the stability of electrodes. As a bifunctional catalyst, the two-electrode system using NiMoB as the anode and the cathode could be applied to a variety of environments, i.e., 1 M KOH (pH ∼ 14), 0.5 M H2SO4 (pH ∼ 1), 1 M phosphate-buffered saline (pH ∼ 7), 1 M KOH + seawater, 1 M KOH + river water, and 6 M KOH at 60 °C, as shown in Fig. 8b. However, the stability of the two-electrode system used in those different solutions was not desirable, which hindered its practical application. In contrast, Fan et al.82 reported that amorphous CoB was successfully “interspersed” on MOF-based CC via mild one-step and electroless plating. The stability performance was better than that of NiMoB, and the catalyst has a nanosheet structure (Fig. 8c). This bifunctional catalyst has different active sites for HER and OER. The XPS results showed that the boron oxides (BOx) with the “chimney effect” were beneficial to the HER, and the Co atoms on the surface oxidized to CoO(OH) were beneficial for OER. Moreover, the electrochemical tests suggested that it required low overpotentials of 266 and 423 mV for HER and OER to attain the large current density of 500 mA cm−2 in simulated seawater (1 M KOH + 0.5 M NaCl), and could steadily run for about 2500 h during the overall seawater splitting process (Fig. 8d). Transition metal borides have other morphologies, such as those shown in Fig. 8e,83Fig. 8f,84 and Fig. 8g.85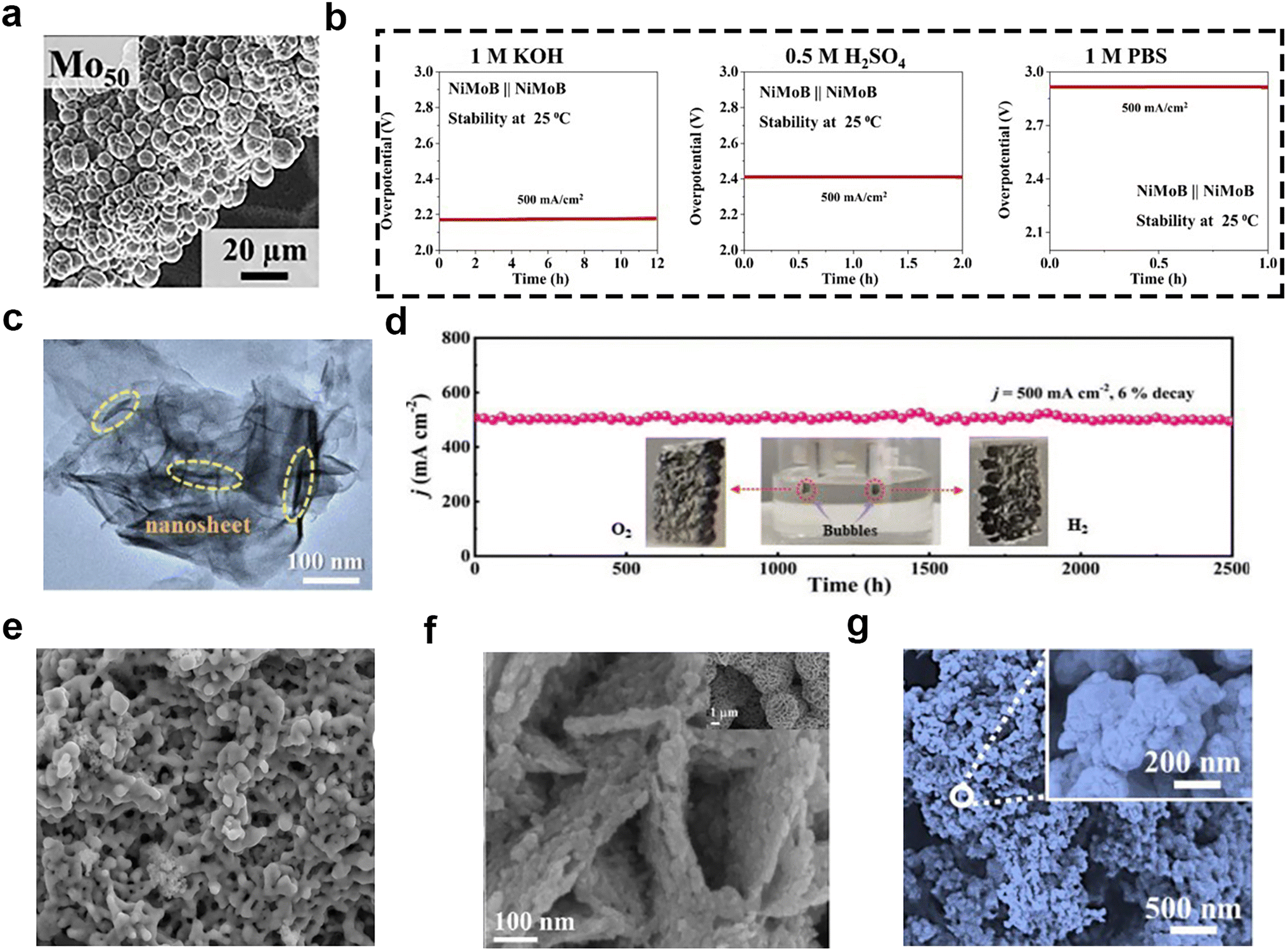 | ||
| Fig. 8 (a) SEM image of the NiMoB electrode, where Mo50 indicates 50% of Mo and 50% Ni. (b) Two-electrode stability in 1 M KOH, 0.5 M H2SO4 and 1 M PBS at 500 mA cm−2. Reprinted with permission.81 Copyright 2022, Elsevier. (c) High-resolution TEM image of the CoB@MOF@CC electrode. (d) Chronopotentiometry curve (V–t) of CoB@MOF@CC for overall water splitting at 500 mA cm−2 (inset: photograph of CoB@MOF@CC for overall water splitting). Reprinted with permission.82 Copyright 2022, Wiley-VCH. (e) SEM image of the crystalline Co2B* catalyst. Reprinted with permission.83 Copyright 2023, Wiley-VCH. (f) SEM images of Co–Mo–B/CoMoO4−x/CF; the insets show the morphology at low magnifications. Reprinted with permission.84 Copyright 2021, The Royal Society of Chemistry. (g) SEM image of Fe–B–O@FeBx/HC. Reprinted with permission.85 Copyright 2023, Elsevier. | ||
Transition metal borides show excellent HER and OER activities. However, in practical applications, higher energy inputs may be required, which may increase the overall energy costs and efficiency issues. Although some transition metal borides show good stability, our understanding of these catalysts may be incomplete; for example, how it responds to HER and OER challenges. Seawater is a very complex electrolyte, so more research may be needed to fully understand and optimize the performance of these catalysts.
4.6 Transition metal nitrides
Transition metal nitrides (TMNs) are a class of two-dimensional materials that exhibit high conductivity, high catalytic activity, and high catalytic stability.86 These characteristics endow them with a wide range of applications in electrocatalytic energy conversion. The formation of TMNs results in a narrower d-band filled state and a wider unfilled state of the metal, which results in the electronic structure of the TMNs being similar to that of the noble metal and improves the rate of the electrocatalytic reaction. For the HER, the change of the electronic states on the surface of the TMNs brings the Gibbs free energy of adsorbed hydrogen closer to zero, which improves the catalytic activity. For the OER, the metal bonding in the TMNs improves the electrical conductivity and accelerates the electron transport, thus increasing the catalytic activity.87 Although TMNs show good activity in HER or OER, the activity remains limited when used as bifunctional catalysts. In order to construct efficient bifunctional catalysts, an effective strategy is to selectively couple materials with HER activity and OER activity into a heterogeneous structure, which results in the formation of new active sites to achieve the simultaneous promotion of HER and OER to realize total hydrolysis.Wang et al.88 synthesized a cobalt molybdenum nitride supported on nitrogen-doped carbon nanosheets catalyst (MoN–Co2N) using a metal–organic framework (ZIF-67) template (Fig. 9a), which consisted of cobalt molybdenum nitride with HER activity and nitrogen-doped carbon nanosheets with OER activity, creating new active sites and enhancing the catalytic efficiency of alkaline seawater electrolysis. During its use as the anode and cathode to assemble the two-electrode system for overall seawater splitting, it required 1.70 V to attain the current density of 100 mA cm−2 (Fig. 9b) and could steadily operate over 62 h. They found the introduction of molybdenum facilitates the performance of HER and OER (Fig. 9c and d). Ojha's team89 reported a bifunctional catalyst: Ni3N and Fe3N nanoparticles encapsulated in a nitrogen-doped carbon (NC) layer were prepared by a one-pot method under ambient conditions using polyacryloylhydrazine (PAHz) as a reductant and encapsulant, followed by high-temperature treatment under nitrogen ambient to obtain Ni3N and Fe3N nanoparticles. The NC–Ni3N and NC–Fe3N nanocomplexes were coated on the surface of nickel foam (NF), on which a FeOOH layer was grown to form a FeOOH–NC–Ni3N/Fe3N heterostructure electrode (Fig. 9e). The results showed that the presence of metal nitrides improved the conductivity and charge transfer ability of the electrodes, which reduced the overpotentials for the OER and HER (Fig. 9f). In particular, the efficiency of the OER and HER increased when the content of Ni3N is increased.
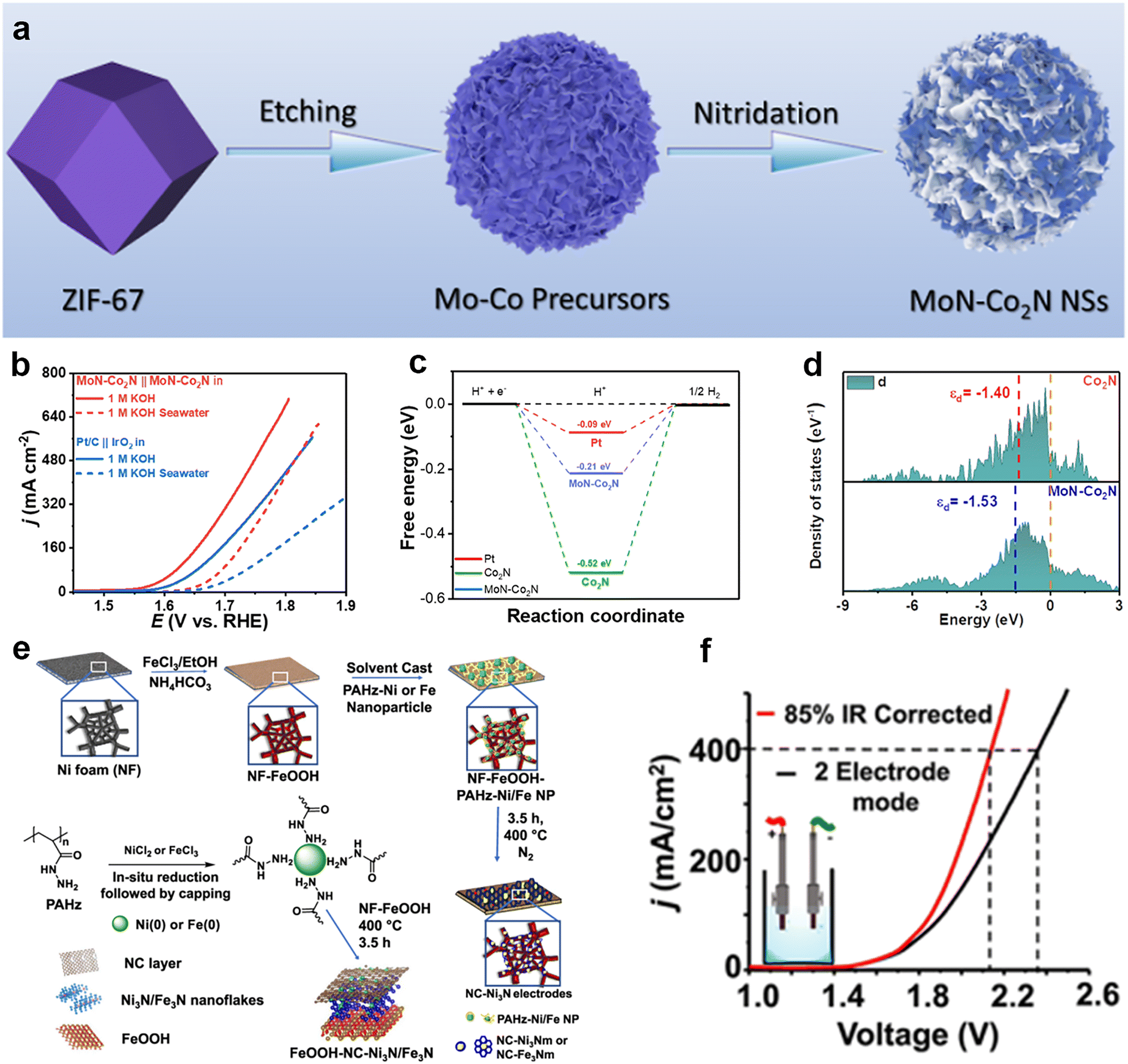 | ||
| Fig. 9 (a) Schematic illustration of the synthesis of MoN–Co2N nanosheets. (b) Overall water/seawater splitting performance of MoN–Co2N/NF‖MoN–Co2N/NF and Pt/C‖IrO2 pair in 1 M KOH and 1 M KOH seawater. (c) Calculated free energy diagram of H* adsorption on the surface of Pt, Co2N, and MoN–Co2N. (d) The d-band centers of Co 3d partial DOS for Co2N and MoN–Co2N. Reprinted with permission.88 Copyright 2022, American Chemical Society. (e) Schematics showing the FeOOH coating of the NF, followed by the in situ growth of the NC–Ni3N or NC–Fe3N nanocomposite layer and synthesis of PAHz–Ni/Fe nanoparticles, followed by NC–Ni3N/Fe3N nanocomposite. (f) IR-uncorrected and corrected LSV traces of NC–Ni3N21 recorded under two-electrode mode in seawater possessing 2 M KOH. Reprinted with permission.89 Copyright 2023, American Chemical Society. | ||
These works make use of heterogeneous structures to improve the performance of the catalysts, whereby the kinetic and thermodynamic properties of the reaction can be optimized by forming interfaces between different materials. This is an effective strategy for designing bifunctional catalysts by combining components that can act on HER and OER separately.
4.7 Other compounds
Increasing the number of active sites and improving the intrinsic activity of the catalyst are two effective methods to enhance the electrocatalytic performance of catalytic materials. As the size of the nanoparticles decreases, the number of atoms exposed on the surface usually increases, resulting in changes in the atomic structure of the surface, the electronic structure and the surface defects. The number of ligand-unsaturated atoms can increase as the size of the particle decreases, thus improving the catalytic activity. Single-atom catalysts (SACs) have been promising candidates for bifunctional catalysts for seawater splitting. Recently, Feng et al.90 reported a bifunctional catalyst for efficient water/seawater splitting, which consisted of Ru single atoms stabilized by flower-like amorphous MoO3−x nanoflakes (Fig. 10a–d). The catalyst exhibited excellent activity and stability for both HER and OER in alkaline media (Fig. 10e and f), and achieved low voltage and high yield for overall water/seawater splitting. The high performance was attributed to the large surface area and abundant active sites of the amorphous structure, as well as the efficient utilization and synergistic effect of Ru single atoms. They successfully stabilized single-atom Ru on an amorphous MoO3−x substrate, harnessing the advantages of the amorphous matrix to enhance both the performance and stability of the catalyst.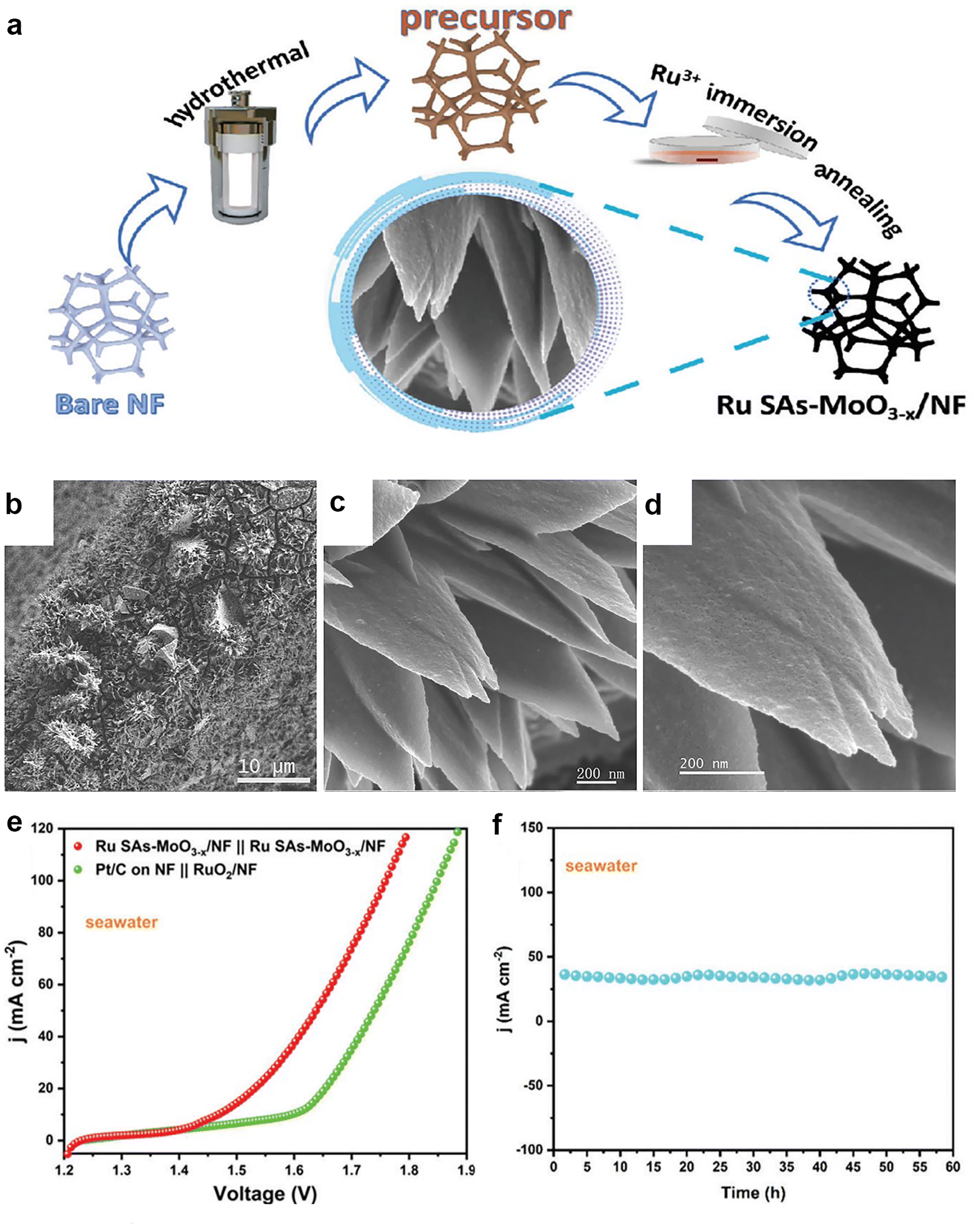 | ||
| Fig. 10 (a) Schematic illustration for the fabrication of Ru SAs–MoO3−x/NF. (b–d) FESEM images of Ru SAs–MoO3−x/NF. (e) Polarization curves of the Ru SAs–MoO3−x/NF‖Ru SAs–MoO3−x/NF couple and commercial catalyst couple toward overall water splitting in alkaline seawater media. (f) Stability test of the Ru SAs–MoO3−x/NF‖Ru SAs–MoO3−x/NF couple in alkaline seawater media. Reprinted with permission.90 Copyright 2023, Wiley-VCH. | ||
Single-atom catalysts offer the highest degree of atomic efficiency, leveraging unique quantum size effects and tailor-made active sites to significantly enhance the catalytic activity and selectivity. This presents a novel avenue for the development of cost-effective catalysts. Furthermore, the amorphous matrix, characterized by its distinct structure and properties, offers the advantage of additional active sites and oxygen vacancies. This feature proves advantageous in enhancing the catalyst's conductivity and reaction kinetics. It introduces a novel avenue for designing highly efficient catalysts.
5. Electrolyzers design
In addition to the development of bifunctional catalysts, the design of the efficient electrolyzer also served as a pivotal factor in increasing the seawater electrolysis efficiency, and achieving the sustainable, cost-effective production of H2 from seawater. At present, there are two ways to achieve H2 production from seawater, including direct and indirect seawater electrolysis. They differ in that indirect seawater electrolysis adds an extra step of desalination. After desalination, seawater splitting is equivalent to freshwater electrolysis, which is already in used industry. As mentioned for ALK and PEM, they are relatively mature in freshwater electrolysis applications, while SOCE is still in the laboratory stage. A comprehensive comparison of these three electrolysis techniques is shown in Table 1. However, it is relatively difficult to use these electrolyzers directly for seawater electrolysis, so it is necessary to make corresponding improvements on their basis. Park and his group91 proposed a different type electrolyser coupled with anion exchange membranes, cation exchange membranes (CEM) and bipolar membranes (BPMs), as shown in Fig. 11a. Anion exchange membranes and CEM could achieve unidirectional transport of Cl− and Na+ to attain the purpose of desalination of brine. And the BPMs could continual supply OH− and H+ to maintain the pH of anolyte (pH ∼ 14) and catholyte (pH ∼ 0.5), which was beneficial for reducing the overpotential of OER. Coupling NiFe LDH as the anode and NiMo as the cathode, this electrolysis system could achieve the FE above 95% for O2 and H2 production at a current density of 100 mA cm−2 over 20 h. Meanwhile, the direct seawater splitting has two types of electrolyzers, including membrane electrolyzers and membrane-free electrolyzers. In the following presentation, we collect the most recent developments in electrolyzer design for direct seawater splitting.| Type | ALK | PEM | SOEC |
|---|---|---|---|
| Electrolyte used | 20–40 wt% KOH (aqueous) | Perfluorinated sulfonic acid (PFSA) | Yttria-stabilized zirconia (YSZ) Sc2O3–ZrO2 |
| MgO–ZrO2 | |||
| CaO–ZrO2 | |||
| Current density (conventional, A cm−2) | 0.2–0.6 | 0.0–3.0 (up to 20) | 0.0–2.0 |
| Cell voltage (V) | 1.8–2.4 | 1.8–2.2 | 0.95–1.3 |
| Operating temperature (°C) | 60–90 | 70–80 | 600–1000 |
| Hydrogen production purity | >99.8% | >99.99% | — |
| Electrolyte quality requirements | Stabilized power supply | Stabilized power supply | Stabilized power supply |
| Dynamic responsiveness | Relatively strong | Stronger | Weaker |
| Lifetime (h) | 60000–90000 |
30000–90000 |
10000–30000 |
| Maintainability | Strong alkali corrosion, low cost | Non-corrosive media; low cost | — |
| Technology maturity level | Commercialization stage | Preliminary commercialization | Lab-stage |
| Operating pressure (bar) | 10–30 | 20–50 | 1–15 |
| System efficiency (%) | 60–75 | 70–90 | 85–100 |
| Energy consumption (kW h N−1 m−3) | 4.5–5.5 | 3.8–5.0 | 2.6–3.6 |
| Advantages | Mature technology, low cost | Good renewable energy applicability | High conversion efficiency |
| Disadvantage | Low energy efficiency; weak applicability to intermittent energy sources; high operation and maintenance costs | Requires high purity water, precious metal catalysts | Requires high temperature; poor durability; easy to deteriorate |
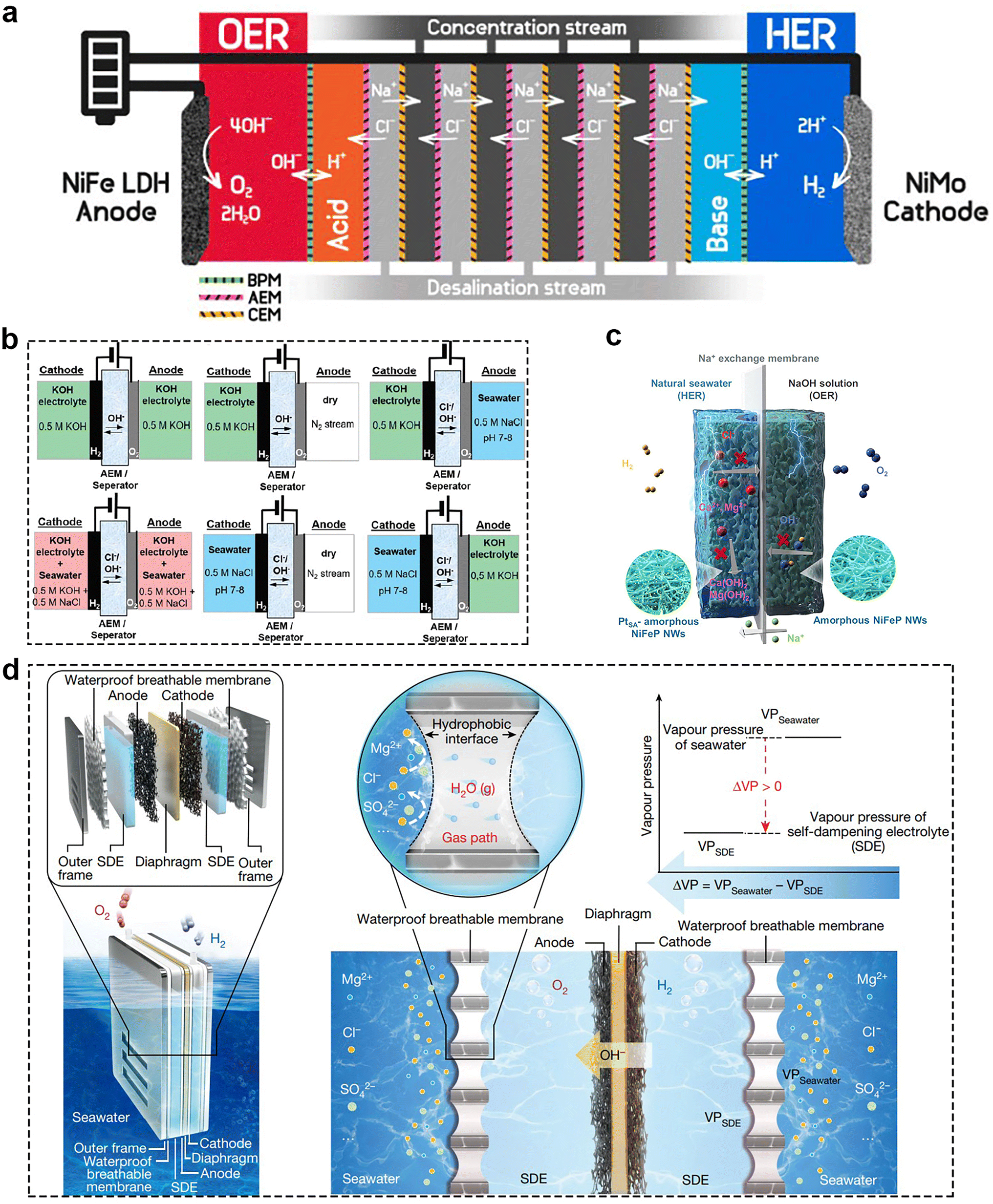 | ||
| Fig. 11 (a) Desalination-coupled electrocatalytic stack device. The five desalination cells are stacked by alternately inserting anion exchange membranes and CEMs between acid and base cells. Sandwiched cells between adjacent two desalination cells are denoted as concentration cells. Reprinted with permission.91 Copyright 2023, Elsevier. (b) Schemes for anion exchange membranes electrolyzers using independent electrolyte feeds with different electrolyte composition. Reprinted with permission.92 Copyright 2020, The Royal Society of Chemistry. (c) Scheme for the asymmetric electrolyzer with sodium ions exchange membrane. Reprinted with permission.93 Copyright 2023, Springer Nature. (d) Schematic diagram of a typical SES (left) and the liquid–gas–liquid phase transition-based migration mechanism of the water purification and migration process and the driving force. Reprinted with permission.94 Copyright 2022, Springer Nature. | ||
5.1 Membrane electrolyzer
As mentioned before, seawater has many complex compositions, which caused difficulties for direct seawater electrolysis. At present, there are several main strategies to deal with this problem. Firstly, the use of asymmetric electrolyzers means that the anode and cathode chambers have different electrolytic media. In 2020, Strasser et al.95 proposed the concept of asymmetric electrolyzers. They systematically studied six different feeding methods (Fig. 11b) to demonstrate the promise for direct seawater splitting. Recently, Li's96 team designed a pH asymmetric electrolyzer containing a sodium ion exchange membrane for direct seawater electrolysis (Fig. 11c), which prevented both chloride corrosion and calcium/magnesium ion precipitation problems and captured the chemical potential between the different electrolytes, thus reducing the required voltage. PtSA–Ni6.6–Fe0.4P3 was chosen as the cathode and Ni5Fe2P3 as the anode, while the electrolyte for the anode was 4 M NaOH and that for the anode was 4 M NaCl to achieve overall seawater splitting. In this way, the asymmetric electrolyzer exhibits a large current density of 400 mA cm−2 at low voltage of 1.66 V at 80 °C, corresponding to the electricity price of US$1.36 per kg of H2, which is lower than the Department of Energy (DOE) 2025 target of US$1.4 per kg of H2. Secondly, designing some special membranes. Xie et al.97 developed a phase change migration-driven in situ direct electrolytic H2 production from seawater without desalination. They constructed a micron-scale gas phase isolation zone in seawater by using a waterproof breathable membrane and permeable layer (Fig. 11d). This design leveraged the inherent pressure difference caused by the self-humidifying electrolyte and seawater, facilitating the movement of water vapor. As a result, water vapor spontaneously transitioned from the seawater side, diffused through the membrane, and was absorbed and liquefied on the electrolyte side. According to their report, the designed electrolyzer could operate stably in natural seawater for more than 72 h. Moreover, using bipolar membrane-based devices is also a great method. For example, Marin et al.98 introduced a novel bipolar membrane electrolyser for seawater electrolysis to produce H2 and O2. It consists of a cation-exchange layer and an anion-exchange layer, which can effectively control the ion transport and reaction environment. Compared with PEM, the bipolar membrane electrolyzer significantly inhibits the penetration and oxidation of chloride ions, thus avoiding the generation of harmful free chlorine species at the anode. Furthermore, the bipolar membrane electrolyzer has higher stability and selectivity for seawater electrolysis over a long period of time, whereas the PEM electrolyzer fails quickly.5.2 Membrane-less electrolyzer
Because the membrane has the disadvantage of high cost and limited lifespan, the membrane-less electrolyzers offer an alternative approach. Membrane-less electrolyzers can be mainly classified into two types based on the type of electrodes employed, including membrane-less flow-by electrodes and flow-through electrodes (Fig. 12a).99 Among them, the microfluidic electrolyzer caught the attention of some researchers. In 2013, the use of the microfluidic electrolyzer for energy conversion was first demonstrated,100 and could attain the current density of 100 mA cm−2 at 2.5 V. Recently, some researchers applied it for seawater splitting. They proposed that a microfluidic alkaline membrane-less electrolyzer (μAEM)101 (Fig. 12b) may resolve the challenges of conventional membrane-based electrolyzers, and minimize the number of components and condense the membrane-based electrolyzers to a single chip, marking a new paradigm for water splitting. In order to make the microfabrication of the electrolyzer simple, the bifunctional catalyst is important for this type of electrolyzers. Psaltis et al.102 reported a novel membrane-less electrochemical reactor (Fig. 12c) that could perform brine electrolysis at room temperature and atmospheric pressure without the use of ion-conducting membranes or separators. Using the 3D printing technology, they integrated flow plates and fluid ports into a single component and used the inertial fluid forces to separate the generated gases. The results unequivocally demonstrated the reactor's remarkable capacity to yield exceedingly high-purity (>99%) and impressive FE (>90%) in the production of hydrogen, oxygen, chlorine, and sodium hydroxide. These findings are commensurate with the performance of commercial brine electrolyzers, thus underscoring the reactor's potential as a groundbreaking technology in this scientific domain.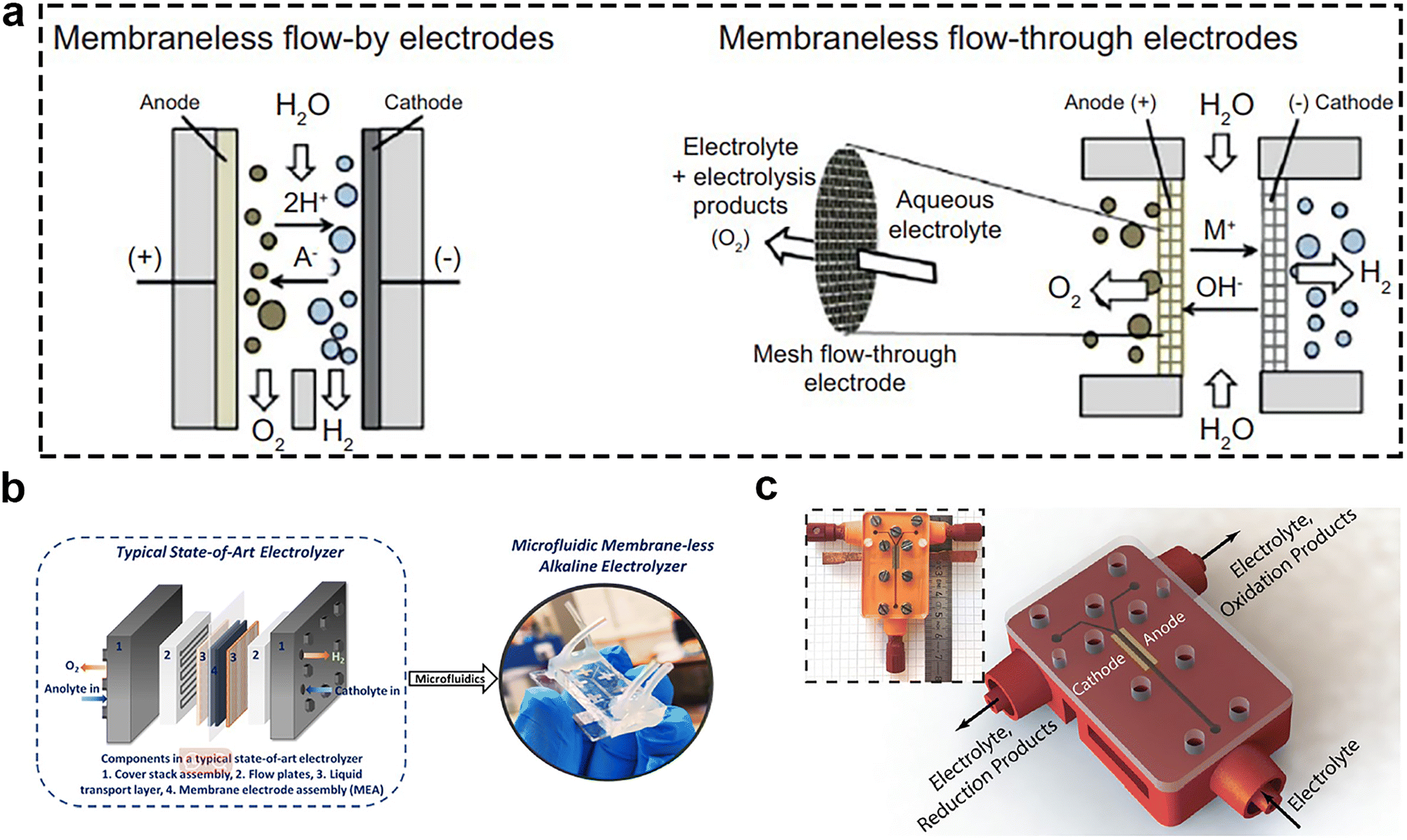 | ||
| Fig. 12 (a) Emerging membrane-less electrolyzers based on flow-by electrodes (left) and flow-through electrodes (right). Reprinted with permission.103 Copyright 2017, Elsevier. (b) Stacked, multilayer structure of a typical state-of-art electrolyzer can be implemented on a single chip using microfluidics. Reprinted with permission.98 Copyright 2021, American Chemical Society. (c) Schematic illustration of the electrolysis cell: the main body (in red) is 3D printed with stereolithography technology. The electrodes (in yellow) are pressed into the devised slots and the assembly is covered by a transparent PMMA plate. A flexible sealing film is sandwiched in between this plate and the 3D printed part to prevent leaking. The inset shows the final assembled device with attached fluidic connectors. Reprinted with permission.102 Copyright 2019, The Royal Society of Chemistry. | ||
6. Conclusions and perspectives
Seawater electrolysis holds the chance to be a cost-effective and sustainable approach for the production of H2 in the future owing to the demand of clean energy power and the advancement of related technologies. With growing academic research results, electrolysis of seawater has become a major topic for research within the field of H2 production. Nevertheless, it is important to note that research in seawater electrolysis remains in its early stages and has encountered several significant challenges. In view of this, this review highlighted the current advances in bifunctional catalysts capable of converting both anodic and cathodic reactants, as well as the experimental findings in nicely designed electrolytic system, with the goal of helping scientists find feasible bifunctional catalysts for industrial-scale H2 product in seawater. A wide range of bifunctional materials (noble metal-based material, transition metal phosphides, chalcogenides, borides, nitrides, oxides, and hydroxides) as efficient electrocatalysts toward overall seawater splitting is reviewed (Table 2).| Electrocatalyst | Electrolyte | Cell voltage (V @ mA cm−2) | Pt/C‖IrO2 or RuO2 (V @ mA cm−2) | Stability (h @ mA cm−2) | Overpotentials of HER and OER (mV @ cm−2) | Tafel slopes of HER and OERa (mV dec−1) | Reference |
|---|---|---|---|---|---|---|---|
| a The Tafel slopes of these bifunctional catalysts were obtained using 1 M KOH as the electrolyte. | |||||||
| FCNP@CQDs | 1 M KOH + seawater | 1.61 @ 10 | — | 50 @ 20 | 183 @ 50; 268 @ 20 | 87.0; 45.2 | 104 |
| Ni3Se2@MoO3/CF | 1 M KOH + seawater | 1.84 @ 100 | 2.05 @ 100 | 200 @ 100 | 426 @ 500; 280 @ 100 | 39.6; 74.7 | 105 |
| Ni2P–Fe2P/NF | 1 M KOH + seawater | 2.004 @ 500 | — | 48 @ 100 | 389 @ 1000; 431 @ 1000 | 86.0; 58.0 | 68 |
| Ru–CoFe2O4/NF | 1 M KOH + seawater | 1.778 @ 100 | 1.85 @ 100 | 100 @ 30 | — | 44.49; 47.81 | 106 |
| S–NiMoO4@NiFe LDH | 1 M KOH + seawater | 1.73 @ 100 | 1.81 @ 100 | 20 @ 60 | 220 @ 100; 315 @ 100 | 69; 90 | 107 |
| Er–MoO2 | 1 M KOH + seawater | 1.52 @ 10 | — | 24 @ 10 | 94 @ 10; 173 @ 10 | — | 108 |
| MnCo2O4@NiFe-LDH | 1 M KOH + 0.5 M NaCl | 1.54 @ 10 | — | 100 @ 100 | 97 @ 10; 460 @ 500 | —; 45.4 | 109 |
| Fe–B–O@FeBx/HC | 1 M KOH + 0.5 M NaCl | 2.06 @ 200 | 2.31 @ 200 | 168 @ 400 | 135 @ 100; 358 @ 100 | 57.62; 65.44 | 85 |
| FMCO/NF | 1 M KOH + seawater | 1.58 @ 10 | 1.61 @ 10 | 180 @ 100 | 250 @ 50; 365 @ 50 | 147.1; 52.1 | 110 |
| NRAHM–NiO | 1 M KOH + 0.5 M NaCl | 2.01 @ 500 | 2.31 @ 500 | 168 @ 240 (60 °C) | 478 @ 500; 540 @ 500 | 116; 73 | 111 |
| NiFe/Fe–MoO2 | 1 M KOH + seawater | 1.73 @ 100 | — | 50 @ 50 | 38 @ 10; 219 @ 20 | 47; 65 | 112 |
| Mo–CoPx/NF | 1 M KOH + seawater | 2.16 @ 100 | — | 100 @ 10 | 158 @ 10; 260 @ 10 | 89; 35 | 113 |
| Co–Fe2P/NF | 1 M KOH + 0.5 M NaCl | 1.69 @ 100 | 1.97 @ 100 | 22 @ 100 | 221 @ 100; 266 @ 100 | 57; 45 | 114 |
| NiFe–P@NC | 1 M KOH + seawater | 1.93 @ 500 | — | 12 @ 500 | 149 @ 100; 280 @ 100 | 58; — | 115 |
| F-FeCoPv@IF | 1 M KOH + seawater | 1.92 @ 1000 | — | 100 @ 1000 | 210 @ 1000; 370 @ 1000 | 108; 118 | 116 |
| C@CoP–FeP/FF | 1 M KOH + 0.5 M NaCl | 1.73 @ 100 | — | 28 @ 100 | 192 @ 100; 297 @ 100 | 67.23; 58.48 | 117 |
| NiPS/NF | 1 M KOH + seawater | 1.79 @ 200 | — | 60 @ 200 | 275 @ 500; 392 @ 500 | 89; 23 | 118 |
| S,P-(Ni,Mo,Fe)OOH/NiMoP/wood aerogel | 1 M KOH + seawater | 1.861 @ 500 | — | 10 @ 100 | 258 @ 500; 297 @ 500 | 91.7; 45.5 | 119 |
| MoN–Co2N | 1 M KOH + seawater | 1.7 @ 100 | — | 62 @ 100 | 351 @ 500; 432 @ 500 | 82.1; 35.6 | 88 |
| CoSe2-NCF | 1 M KOH + seawater | 1.842 @ 500 | 1.929 @ 500 | 50 @ 500 | 134 @ 10; 245 @ 10 | 67; 93 | 120 |
| CdFe-BDC | Seawater | 1.68 @ 10 | — | 12 @ 9.5 | 148 @ 10; 290 @ 100 | 108.71; 44.57 | 121 |
| NiFe-PBA-gel-cal | 1 M KOH + 0.5 M NaCl | 1.66 @ 100 | — | 60 @ 50 | /; 427 @ 500 | 82.4; 68.7 | 122 |
| NiCoHPi@Ni3N/NF | 1 M KOH + 0.5 M NaCl | 1.86 @ 100 | — | 40 @ 200 | 174 @ 100; 365 @ 100 | 77.5; 79.8 | 123 |
| NiFe LDH/FeOOH | 1 M KOH + 0.5 M NaCl | 1.55 @ 10 | — | 105 @ 100 | 181.8 @ 10; 286.2 @ 100 | — | 124 |
| Ti@NiB 1.5 h | 1 M KOH + 0.5 M NaCl | 1.75 @ 20 | — | 40 @ 100 | 149 @ 10; 397 @ 50 | 118; 34.2 | 125 |
| Ni@CNTs-MoxC/Ni2P | 1 M KOH + seawater | 1.56 @ 10 | 1.59 @ 10 | 50 @ 50 | 65.9 @ 10; 230 @ 10 | 54; 43 | 126 |
| FeNiCoMnRu@CNT | 1 M KOH + seawater | 2.15 @ 100 | 2.2 @ 100 | 30 @ 10 | 286 @ 100; 490 @ 100 | 50; 107 | 127 |
| GDY/RhOx/GDY | 1 M KOH + 0.5 M NaCl | 1.49 @ 100 | 1.71 @ 100 | 50 @ 500 | 249 @ 1000; 261 @ 100 | 42; 68 | 128 |
| Pt–CoFe(II)LDHs (2.2 wt%) | 1 M KOH + seawater | 1.651 @ 100 | 1.99 @ 100 | 20 @ 500 | 224 @ 500; 375 @ 500 | 45.3; 43.6 | 129 |
| cRu-Ni3N/NF | 1 M KOH + seawater | 1.73 @ 100 | 1.84 @ 100 | — | 36 @ 10; 280 @ 50 | 26.2; 40.4 | 130 |
| RuNi–Fe2O3/IF | 1 M KOH + seawater | 1.73 @ 100 | 2.03 @ 100 | 100 @ 100 | 353 @ 1000; 497 @ 1000 | 85.08; 60.85 | 131 |
| Ir0.05–Co2P/Co2P2O7 NWs/NF | Seawater | 1.97 @ 30 | 2.32 @ 30 | — | 205 @ 10; 280 @ 20 | 30; 138 | 132 |
| Ag2Se–Ag2S–CoCH/NF | 1 M KOH + seawater | 1.69 @ 100 | — | 24 @ 100 | 367 @ 200; 440 @ 100 | 185.59; 144.87 | 133 |
| Ru–CoV-LDH/NF | 1 M KOH + seawater | 1.809 @ 100 | 1.973 @ 100 | — | 147 @ 300; 440 @ 300 | 49.6; 53.5 | 134 |
| P–Fe2O3–CoP | Seawater | 1.65 @ 10 | — | 12 @ 10 | 275 @ 50; 270 @ 10 | 95; 59 | 135 |
| MnCo/NiSe/NF | 1 M KOH + seawater | 2.11 @ 1000 | — | 200 @ 500 | 270.1 @ 1000; 460.2 @ 1000 | 45.05; 69.71 | 136 |
| Fe2P/Ni1.5Co1.5N/Ni2P | 1 M KOH + seawater | 1.742 @ 500 | — | 40 @ 50 | 141 @ 500; 307 @ 500 | 43.4; 29.1 | 137 |
| Au–Gd–Co2B@TiO2 | 1 M KOH + seawater | 1.68 @ 500 | — | 200 @ 500 | 194 @ 500 | 65; 43 | 138 |
| Cu2O–CoO/CF | 1 M KOH + seawater | 1.82 @ 100 | — | 50 @ 100 | 359 @ 500; 410 @ 500 | 83.0; — | 139 |
These bifunctional transition metal compounds all exhibit high activity towards HER and OER. However, they also have some improvements: the electronic structure and valence states of oxides are difficult to regulate and have low conductivity; sulfides, nitrides, phosphides, and borides are relatively complex to synthesize and are easily oxidized, leading to a decrease in their catalytic activity and stability. Despite considerable progress made in recent years of seawater electrolysis, research in the following sub-areas should be prioritized in future work:
(1) For seawater electrolysis to become more productive and stable, the catalyst design is crucial. For example, the catalytic performance can be improved by investigating state-of-the-art materials, tailoring compositions, and optimizing active sites. Developing protective outer/inner-coatings, robust catalyst structures, and knowing degradation mechanisms are all key considerations for ensuring long-term operation.
(2) More powerful bifunctional nano-architectures with increased well-adjusted catalytic sites should be constructed in order to facilitate large-scale seawater to H2 electrolysis.
(3) Efficiency, scalability, and durability of electrolyzers can all be improved by upgrading cell configurations, electrodes materials, and mass transport control.
(4) Only renewable energy sources (e.g., wind power)-enabled electrolysis represents the carbon-neutral method to reduce the use of fossil fuels, which makes the H2 production truly sustainable. Researchers should thus create integration strategies to improve energy management systems, allowing for the efficient exploitation of clean and plentiful renewable energy.
(5) Although different types of bifunctional catalysts exist in alkaline media, such as metal–sulfur compounds, borides, oxides, carbide, and phosphides, which can make the necessary changes in electronic properties and morphology, very few of them can be directly applied in natural seawater. Researchers should simulate or even use real seawater for use as an electrolyte.
(6) Substituting the conventional OER with alternative oxidation reaction, such as the Selective Methanol Oxidation Reaction (SMOR),140 holds the promise of significantly enhancing both energy efficiency and sustainability. This forward-looking approach not only paves the way for reduced energy consumption, but also unlocks intriguing possibilities for simultaneous production of valuable chemicals. This dual benefit not only enhances the economic viability of the process, but also aligns with environmental sustainability goals. As we look ahead, the adoption of such innovative strategies is poised to play a pivotal role in shaping the landscape of clean energy production and resource utilization.
(7) At present, research on direct seawater electrolysis is still insufficient. In the future, we should pay more attention to and study the technology of in situ direct electrolysis of seawater without desalination for H2 production. This will have significant implications for the commercialization of seawater electrolysis for H2 production. Such a research direction can not only promote technological progress, but also help solve energy problems and achieve sustainable development.
(8) In the majority of research literature on bifunctional catalysts for seawater electrolysis, the chosen electrolyte (e.g., 1 M KOH + seawater, 1 M KOH + 0.5 M NaCl) deviates significantly from the conditions of natural seawater. This does not contribute significantly to addressing the challenges of cathodic reactions, such as calcium and magnesium precipitation. Therefore, future research should focus more on conditions that closely resemble natural seawater, or even under actual seawater conditions. This will have significant implications for achieving the goal of large-scale seawater electrolysis for hydrogen production. Such a research direction can not only promote technological progress, but also help solve energy problems and achieve sustainable development.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22072015 and 21927811) and the Free Exploration Project of Frontier Technology for Laoshan Laboratory (No. 16-02).References
- H. Yu, J. Wan, M. Goodsite and H. Jin, One Earth, 2023, 6, 267–277 CrossRef.
- L. Zhang, H. Zhao, S. Xu, Q. Liu, T. Li, Y. Luo, S. Gao, X. Shi, A. M. Asiri and X. Sun, Small Struct., 2021, 2, 2000048 CrossRef.
- X. Xiao, L. Yang, W. Sun, Y. Chen, H. Yu, K. Li, B. Jia, L. Zhang and T. Ma, Small, 2022, 18, 2105830 CrossRef PubMed.
- L. Mosca, J. A. M. Jimenez, S. A. Wassie, F. Gallucci, E. Palo, M. Colozzi, S. Taraschi and G. Galdieri, Int. J. Hydrogen Energy, 2020, 45, 7266–7277 CrossRef.
- X. C. S. Rivera, E. Topriska, M. Kolokotroni and A. Azapagic, J. Cleaner Prod., 2018, 196, 863–879 CrossRef.
- J. D. Holladay, J. Hu, D. L. King and Y. Wang, Catal. Today, 2009, 139, 244–260 CrossRef.
- K. Zhang, X. Liang, L. Wang, K. Sun, Y. Wang, Z. Xie, Q. Wu, X. Bai, M. S. Hamdy, H. Chen and X. Zou, Nano Res. Energy, 2022, 1, e9120032 CrossRef.
- P. Haug, B. Kreitz, M. Koj and T. Turek, Int. J. Hydrogen Energy, 2017, 42, 15689–15707 CrossRef CAS.
- S. Ahmadvand, B. Abbasi, B. Azarfar, M. Elhashimi, X. Zhang and B. Abbasi, Water, 2019, 11, 696 CrossRef CAS.
- M. Elimelech and W. A. Phillip, Science, 2011, 333, 712–717 CrossRef CAS PubMed.
- H. Liu, W. Shen, H. Jin, J. Xu, P. Xi, J. Dong, Y. Zheng and S. Qiao, Angew. Chem., Int. Ed., 2023, 62, e202311674 CrossRef CAS PubMed.
- L. Huang, P. Wang, Y. Jiang, K. Davey, Y. Zheng and S.-Z. Qiao, J. Am. Chem. Soc., 2023, 145, 15565–15571 CrossRef CAS.
- H. Zhang, Y. Luo, P. K. Chu, Q. Liu, X. Liu, S. Zhang, J. Luo, X. Wang and G. Hu, J. Alloys Compd., 2022, 922, 166113 CrossRef CAS.
- H. Sun, Z. Yan, F. Liu, W. Xu, F. Cheng and J. Chen, Adv. Mater., 2020, 32, 1806326 CrossRef CAS PubMed.
- T. Marimuthu, R. Yuvakkumar, P. Senthil Kumar, G. Ravi, X. Xu, G. Xu and D. Velauthapillai, Int. J. Hydrogen Energy, 2022, 47, 37171–37182 CrossRef CAS.
- Q. Liu, S. Sun, L. Zhang, Y. Luo, Q. Yang, K. Dong, X. Fang, D. Zheng, A. A. Alshehri and X. Sun, Nano Res., 2022, 15, 8922–8927 CrossRef CAS.
- J. Li, J. Sun and X. Meng, Int. J. Hydrogen Energy, 2023, 48, 29583–29592 CrossRef CAS.
- T. Marimuthu, R. Yuvakkumar, P. Senthil Kumar, G. Ravi, X. Xu, D. Velauthapillai and D. V. N. Vo, Int. J. Hydrogen Energy, 2022, 47, 30819–30829 CrossRef CAS.
- X. Fang, X. Wang, L. Ouyang, L. Zhang, S. Sun, Y. Liang, Y. Luo, D. Zheng, T. Kang, Q. Liu, F. Huo and X. Sun, Molecules, 2022, 27, 7617 CrossRef CAS.
- H. You, D. Wu, D. Si, M. Cao, F. Sun, H. Zhang, H. Wang, T.-F. Liu and R. Cao, J. Am. Chem. Soc., 2022, 144, 9254–9263 CrossRef CAS.
- N. Wang, P. Ou, S. Hung, J. E. Huang, A. Ozden, J. Abed, I. Grigioni, C. Chen, R. K. Miao, Y. Yan, J. Zhang, Z. Wang, R. Dorakhan, A. Badreldin, A. Abdel-Wahab, D. Sinton, Y. Liu, H. Liang and E. H. Sargent, Adv. Mater., 2023, 35, 2210057 CrossRef CAS.
- M. Chen, N. Kitiphatpiboon, C. Feng, Q. Zhao, A. Abudula, Y. Ma, K. Yan and G. Guan, Appl. Catal., B, 2023, 330, 122577 CrossRef CAS.
- J. Tang, S. Sun, X. He, H. Zhang, C. Yang, M. Zhang, M. Yue, H. Wang, Y. Sun, Y. Luo, S. Alfaifi, A. Farouk, M. S. Hamdy, X. Sun, H. Wang and B. Ying, Nano Res., 2023 DOI:10.1007/s12274-023-6087-y.
- D. Jin, H. Woo, S. Prabhakaran, Y. Lee, M. H. Kim, D. H. Kim and C. Lee, Adv. Funct. Mater., 2023, 33, 2301559 CrossRef CAS.
- J. Xing, Z. Zeng, W. Best, Z. Liu, L. Bonville, R. Maric and S. Bliznakov, J. Power Sources, 2023, 558, 232564 CrossRef CAS.
- G. A. Lindquist, Q. Xu, S. Z. Oener and S. W. Boettcher, Joule, 2020, 4, 2549–2561 CrossRef CAS.
- Z. Liu, B. Han, Z. Lu, W. Guan, Y. Li, C. Song, L. Chen and S. C. Singhal, Appl. Energy, 2021, 300, 117439 CrossRef CAS.
- Z. Qin, W. Liu, W. Que, J. Feng, W. Shi, F. Wu and X. Cao, ChemPhysMater, 2023, 2, 185–196 CrossRef.
- Q. Wu, Q. Gao, B. Shan, W. Wang, Y. Qi, X. Tai, X. Wang, D. Zheng, H. Yan, B. Ying, Y. Luo, S. Sun, Q. Liu, M. S. Hamdy and X. Sun, Acta Phys.-Chim. Sin., 2023, 39, 2303012 Search PubMed.
- B. Xu, J. Liang, X. Sun and X. Xiong, Green Chem., 2023, 25, 3767–3790 RSC.
- C. Wang, H. Shang, L. Jin, H. Xu and Y. Du, Nanoscale, 2021, 13, 7897–7912 RSC.
- J. Liang, Z. Li, X. He, Y. Luo, D. Zheng, Y. Wang, T. Li, B. Ying, S. Sun, Z. Cai, Q. Liu, B. Tang and X. Sun, Mater. Today, 2023, 69, 193–235 CrossRef.
- S. Liu, Y. Chang, N. He, S. Zhu, L. Wang and X. Liu, ACS Appl. Mater. Interfaces, 2023, 15, 20563–20570 CrossRef PubMed.
- X. Wang, S. Xi, P. Huang, Y. Du, H. Zhong, Q. Wang, A. Borgna, Y.-W. Zhang, Z. Wang, H. Wang, Z. G. Yu, W. S. V. Lee and J. Xue, Nature, 2022, 611, 702–708 CrossRef CAS PubMed.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- C. G. Morales-Guio, L.-A. Stern and X. Hu, Chem. Soc. Rev., 2014, 43, 6555 RSC.
- S. Trasatti, J. Electroanal. Chem., 1972, 39, 163–184 CrossRef CAS.
- S. Dresp, F. Dionigi, M. Klingenhof and P. Strasser, ACS Energy Lett., 2019, 4, 933–942 CrossRef CAS.
- J. Bennett, Int. J. Hydrogen Energy, 1980, 5, 401–408 CrossRef CAS.
- K. S. Exner, J. Anton, T. Jacob and H. Over, Angew. Chem., Int. Ed., 2014, 53, 11032–11035 CrossRef CAS PubMed.
- X.-Y. Yang, Photo-Driven Seawater Splitting for Hydrogen Production, Springer, Singapore, Berlin, 2023 Search PubMed.
- N. Wang, P. Ou, S. Hung, J. E. Huang, A. Ozden, J. Abed, I. Grigioni, C. Chen, R. K. Miao, Y. Yan, J. Zhang, Z. Wang, R. Dorakhan, A. Badreldin, A. Abdel-Wahab, D. Sinton, Y. Liu, H. Liang and E. H. Sargent, Adv. Mater., 2023, 35, 2210057 CrossRef CAS.
- H. Komiya, K. Obata, M. Wada, T. Nishimoto and K. Takanabe, ACS Sustainable Chem. Eng., 2023, 11, 12614–12622 CrossRef CAS.
- M. J. Kenney, M. Gong, Y. Li, J. Z. Wu, J. Feng, M. Lanza and H. Dai, Science, 2013, 342, 836–840 CrossRef CAS.
- L. Wu, L. Yu, B. McElhenny, X. Xing, D. Luo, F. Zhang, J. Bao, S. Chen and Z. Ren, Appl. Catal., B, 2021, 294, 120256 CrossRef CAS.
- S. Khatun, H. Hirani and P. Roy, J. Mater. Chem. A, 2021, 9, 74–86 RSC.
- T. Wang, X. Cao and L. Jiao, Angew. Chem., Int. Ed., 2022, 61, e202213328 CrossRef CAS PubMed.
- F. Dionigi, T. Reier, Z. Pawolek, M. Gliech and P. Strasser, ChemSusChem, 2016, 9, 962–972 CrossRef CAS.
- I. Katsounaros, J. C. Meier, S. O. Klemm, A. A. Topalov, P. U. Biedermann, M. Auinger and K. J. Mayrhofer, Electrochem. Commun., 2011, 13, 634–637 CrossRef CAS.
- H. Zhu, J. Zhang, R. Yanzhang, M. Du, Q. Wang, G. Gao, J. Wu, G. Wu, M. Zhang, B. Liu, J. Yao and X. Zhang, Adv. Mater., 2015, 27, 4752–4759 CrossRef CAS PubMed.
- A.-S. Feiner and A. J. McEvoy, J. Chem. Educ., 1994, 71, 493 CrossRef CAS.
- G. Zhao, K. Rui, S. X. Dou and W. Sun, Adv. Funct. Mater., 2018, 28, 1803291 CrossRef.
- S. Enthaler, J. V. Langermann and T. Schmidt, Energy Environ. Sci., 2010, 3, 1207 RSC.
- H. Jiang, Q. He, X. Li, X. Su, Y. Zhang, S. Chen, S. Zhang, G. Zhang, J. Jiang, Y. Luo, P. M. Ajayan and L. Song, Adv. Mater., 2019, 31, 1805127 CrossRef.
- S. Anantharaj, P. E. Karthik and S. Noda, Angew. Chem., Int. Ed., 2021, 60, 23051–23067 CrossRef CAS PubMed.
- S. Anantharaj and S. Kundu, ACS Energy Lett., 2019, 4, 1260–1264 CrossRef CAS.
- J. Guo, Y. Zheng, Z. Hu, C. Zheng, J. Mao, K. Du, M. Jaroniec, S.-Z. Qiao and T. Ling, Nat. Energy, 2023, 8, 264–272 CAS.
- M. Zhang, X. He, K. Dong, H. Zhang, Y. Yao, C. Yang, M. Yue, S. Sun, Y. Sun, D. Zheng, Y. Luo, Q. Liu, N. Li, B. Tang, J. Liu and X. Sun, Chem. Commun., 2023, 59, 9750–9753 RSC.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef.
- D. Wu, D. Chen, J. Zhu and S. Mu, Small, 2021, 17, 2102777 CrossRef PubMed.
- D. Wu, B. Liu, R. Li, D. Chen, W. Zeng, H. Zhao, Y. Yao, R. Qin, J. Yu, L. Chen, J. Zhang, B. Li and S. Mu, Small, 2023, 19, 2300030 CrossRef PubMed.
- J. Liu, X. Liu, H. Shi, J. Luo, L. Wang, J. Liang, S. Li, L.-M. Yang, T. Wang, Y. Huang and Q. Li, Appl. Catal., B, 2022, 302, 120862 CrossRef.
- R. Liang, J. Fan, F. Lei, P. Li, C. Fu, Z. Lu and W. Hao, J. Colloid Interface Sci., 2023, 645, 227–240 CrossRef PubMed.
- X. Wang, X. Liu, S. Wu, K. Liu, X. Meng, B. Li, J. Lai, L. Wang and S. Feng, Nano Energy, 2023, 109, 108292 CrossRef.
- C. Fu, W. Hao, J. Fan, Q. Zhang, Y. Guo, J. Fan, Z. Chen and G. Li, Small, 2023, 19, 2205689 CrossRef CAS PubMed.
- L. Wu, L. Yu, F. Zhang, B. McElhenny, D. Luo, A. Karim, S. Chen and Z. Ren, Adv. Funct. Mater., 2021, 31, 2006484 CrossRef CAS.
- Y. Luo, P. Wang, G. Zhang, S. Wu, Z. Chen, H. Ranganathan, S. Sun and Z. Shi, Chem. Eng. J., 2023, 454, 140061 CrossRef CAS.
- H. Zhang, Z. Bi, P. Sun, A. Chen, T. Wågberg, X. Hu, X. Liu, L. Jiang and G. Hu, ACS Nano, 2023, 17, 16008–16019 CrossRef CAS.
- E. Enkhtuvshin, S. Yeo, H. Choi, K. M. Kim, B.-S. An, S. Biswas, Y. Lee, A. K. Nayak, J. U. Jang, K.-H. Na, W.-Y. Choi, G. Ali, K. H. Chae, M. Akbar, K. Y. Chung, K. Yoo, Y.-C. Chung, T. H. Shin, H. Kim, C.-Y. Chung and H. Han, Adv. Funct. Mater., 2023, 33, 2214069 CrossRef CAS.
- L. Zhang, J. Liang, L. Yue, K. Dong, J. Li, D. Zhao, Z. Li, S. Sun, Y. Luo, Q. Liu, G. Cui, A. Ali Alshehri, X. Guo and X. Sun, Nano Res. Energy, 2022, 1, e9120028 CrossRef.
- A. R. Jadhav, A. Kumar, J. Lee, T. Yang, S. Na, J. Lee, Y. Luo, X. Liu, Y. Hwang, Y. Liu and H. Lee, J. Mater. Chem. A, 2020, 8, 24501–24514 RSC.
- Y. Gong, H. Zhao, Y. Sun, D. Xu, D. Ye, Y. Tang, T. He and J. Zhang, J. Colloid Interface Sci., 2023, 650, 636–647 CrossRef CAS.
- T. U. Haq and Y. Haik, Catal. Today, 2022, 400–401, 14–25 CrossRef CAS.
- S. Song, Y. Wang, X. Tian, F. Sun, X. Liu, Y. Yuan, W. Li and J. Zang, J. Colloid Interface Sci., 2023, 633, 668–678 CrossRef CAS.
- J. Chen, L. Zhang, J. Li, X. He, Y. Zheng, S. Sun, X. Fang, D. Zheng, Y. Luo, Y. Wang, J. Zhang, L. Xie, Z. Cai, Y. Sun, A. A. Alshehri, Q. Kong, C. Tang and X. Sun, J. Mater. Chem. A, 2023, 11, 1116–1122 RSC.
- C. Yang, K. Dong, L. Zhang, X. He, J. Chen, S. Sun, M. Yue, H. Zhang, M. Zhang, D. Zheng, Y. Luo, B. Ying, Q. Liu, A. M. Asiri, M. S. Hamdy and X. Sun, Inorg. Chem., 2023, 62, 7976–7981 CrossRef CAS PubMed.
- L. Zhao, S. Zhou, Z. Lv, W. Xu, J. Liu, Z. Liu, Q. Zhang, J. Lai and L. Wang, Appl. Catal., B, 2023, 338, 122996 CrossRef CAS.
- Y. Yao, Z. Zhang and L. Jiao, Energy Environ. Mater., 2021, 5, 470–485 CrossRef.
- R. Mandavkar, M. A. Habib, S. Lin, R. Kulkarni, S. Burse, J.-H. Jeong and J. Lee, Appl. Mater. Today, 2022, 29, 101579 CrossRef.
- J. Fan, C. Fu, R. Liang, H. Lv, C. Fang, Y. Guo and W. Hao, Small, 2022, 18, 2203588 CrossRef CAS.
- R. Silviya, Y. Vernekar, A. Bhide, S. Gupta, N. Patel and R. Fernandes, ChemCatChem, 2023, 15, e202300635 CrossRef CAS.
- Y. Ren, J. Wang, W. Hu, H. Wen, Y. Qiu, P. Tang, M. Chen and P. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 42605–42612 CrossRef CAS.
- C. Fu, J. Fan, Y. Zhang, H. Lv, D. Ji and W. Hao, J. Colloid Interface Sci., 2023, 634, 804–816 CrossRef CAS.
- H. Jin, T. Song, U. Paik and S.-Z. Qiao, Acc. Mater. Res., 2021, 2, 559–573 CrossRef CAS.
- R. Qin, P. Wang, C. Lin, F. Cao, J. Zhang, L. Chen and S. Mu, Acta Phys.-Chim. Sin., 2021, 37, 2009099 Search PubMed.
- X. Wang, X. Han, R. Du, C. Xing, X. Qi, Z. Liang, P. Guardia, J. Arbiol, A. Cabot and J. Li, ACS Appl. Mater. Interfaces, 2022, 14, 41924–41933 CrossRef CAS.
- A. Tewary, S. Mandal, Z. Alam, A. S. K. Sinha and U. Ojha, ACS Sustainable Chem. Eng., 2023, 11, 6556–6566 CrossRef CAS.
- D. Feng, P. Wang, R. Qin, W. Shi, L. Gong, J. Zhu, Q. Ma, L. Chen, J. Yu, S. Liu and S. Mu, Adv. Sci., 2023, 10, 2300342 CrossRef CAS PubMed.
- B.-J. Kim, H. K. Shon, D. S. Han and H. Park, Desalination, 2023, 551, 116431 CrossRef CAS.
- A. Buttler and H. Spliethoff, Renewable Sustainable Energy Rev., 2018, 82, 2440–2454 CrossRef CAS.
- H. Jin, J. Xu, H. Liu, H. Shen, H. Yu, M. Jaroniec, Y. Zheng and S.-Z. Qiao, Sci. Adv., 2023, 9, eadi7755 CrossRef CAS PubMed.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- S. Dresp, T. N. Thanh, M. Klingenhof, S. Brückner, P. Hauke and P. Strasser, Energy Environ. Sci., 2020, 13, 1725–1729 RSC.
- H. Shi, T. Wang, J. Liu, W. Chen, S. Li, J. Liang, S. Liu, X. Liu, Z. Cai, C. Wang, D. Su, Y. Huang, L. Elbaz and Q. Li, Nat. Commun., 2023, 14, 3934 CrossRef CAS PubMed.
- H. Xie, Z. Zhao, T. Liu, Y. Wu, C. Lan, W. Jiang, L. Zhu, Y. Wang, D. Yang and Z. Shao, Nature, 2022, 612, 673–678 CrossRef CAS PubMed.
- D. H. Marin, J. T. Perryman, M. A. Hubert, G. A. Lindquist, L. Chen, A. M. Aleman, G. A. Kamat, V. A. Niemann, M. B. Stevens, Y. N. Regmi, S. W. Boettcher, A. C. Nielander and T. F. Jaramillo, Joule, 2023, 7, 765–781 CrossRef CAS.
- A. Malek, X. Lu, P. R. Shearing, D. J. Brett and G. He, Green Energy Environ., 2023, 8, 989–1005 CrossRef.
- M. A. Modestino, C. A. Diaz-Botia, S. Haussener, R. Gomez-Sjoberg, J. W. Ager and R. A. Segalman, Phys. Chem. Chem. Phys., 2013, 15, 7050 RSC.
- B. S. De, P. Kumar, N. Khare, J.-L. Luo, A. Elias and S. Basu, ACS Appl. Energy Mater., 2021, 4, 9639–9652 CrossRef.
- S. M. H. Hashemi, P. Karnakov, P. Hadikhani, E. Chinello, S. Litvinov, C. Moser, P. Koumoutsakos and D. Psaltis, Energy Environ. Sci., 2019, 12, 1592–1604 RSC.
- D. V. Esposito, Joule, 2017, 1, 651–658 CrossRef.
- S. Lv, Y. Deng, Q. Liu, Z. Fu, X. Liu, M. Wang, Z. Xiao, B. Li and L. Wang, Appl. Catal., B, 2023, 326, 122403 CrossRef.
- S. Feng, C. Gu, Y. Yu, P. Rao, P. Deng, J. Li, Z. Kang, X. Tian and Z. Wu, J. Mater. Chem. A, 2023, 11, 11740–11747 RSC.
- Y.-T. Zeng, M.-Y. Xu, T. Wang, S.-Y. Wu, J. Zhang, S.-C. Mu and J. Yu, Electrochim. Acta, 2023, 444, 142004 CrossRef.
- H. Wang, L. Chen, L. Tan, X. Liu, Y. Wen, W. Hou and T. Zhan, J. Colloid Interface Sci., 2022, 613, 349–358 CrossRef PubMed.
- T. Yang, H. Lv, Q. Quan, X. Li, H. Lu, X. Cui, G. Liu and L. Jiang, Appl. Surf. Sci., 2023, 615, 156360 CrossRef.
- N. Kitiphatpiboon, M. Chen, C. Feng, Y. Zhou, C. Liu, Z. Feng, Q. Zhao, A. Abudula and G. Guan, J. Colloid Interface Sci., 2023, 632, 54–64 CrossRef PubMed.
- W. Liu, W. Que, R. Yin, J. Dai, D. Zheng, J. Feng, X. Xu, F. Wu, W. Shi, X. Liu and X. Cao, Appl. Catal., B, 2023, 328, 122488 CrossRef.
- K. Hemmati, A. Kumar, A. R. Jadhav, O. Moradlou, A. Z. Moshfegh and H. Lee, ACS Catal., 2023, 13, 5516–5528 CrossRef CAS.
- W. Shi, J. Zhu, L. Gong, D. Feng, Q. Ma, J. Yu, H. Tang, Y. Zhao and S. Mu, Small, 2022, 18, 2205683 CrossRef CAS PubMed.
- Y. Yu, J. Li, J. Luo, Z. Kang, C. Jia, Z. Liu, W. Huang, Q. Chen, P. Deng, Y. Shen and X. Tian, Mater. Today Nano, 2022, 18, 100216 CrossRef CAS.
- S. Wang, P. Yang, X. Sun, H. Xing, J. Hu, P. Chen, Z. Cui, W. Zhu and Z. Ma, Appl. Catal., B, 2021, 297, 120386 CrossRef CAS.
- Z. Chen, Q. Li, H. Xiang, Y. Wang, P. Yang, C. Dai, H. Zhang, W. Xiao, Z. Wu and L. Wang, Inorg. Chem. Front., 2023, 10, 1493–1500 RSC.
- J. Zhu, J. Chi, T. Cui, L. Guo, S. Wu, B. Li, J. Lai and L. Wang, Appl. Catal., B, 2023, 328, 122487 CrossRef CAS.
- J. Li, Y. Hu, X. Huang, Y. Zhu and D. Wang, Small, 2023, 19, 2206533 CrossRef CAS.
- H.-Y. Wang, J.-T. Ren, L. Wang, M.-L. Sun, H.-M. Yang, X.-W. Lv and Z.-Y. Yuan, J. Energy Chem., 2022, 75, 66–73 CrossRef CAS.
- H. Chen, Y. Zou, J. Li, K. Zhang, Y. Xia, B. Hui and D. Yang, Appl. Catal., B, 2021, 293, 120215 CrossRef CAS.
- H. Chen, S. Zhang, Q. Liu, P. Yu, J. Luo, G. Hu and X. Liu, Inorg. Chem. Commun., 2022, 146, 110170 CrossRef.
- Y. Luo, X. Yang, L. He, Y. Zheng, J. Pang, L. Wang, R. Jiang, J. Hou, X. Guo and L. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 46374–46385 CrossRef PubMed.
- H. Zhang, S. Geng, M. Ouyang, H. Yadegari, F. Xie and D. J. Riley, Adv. Sci., 2022, 9, 2200146 CrossRef PubMed.
- H. Sun, J. Sun, Y. Song, Y. Zhang, Y. Qiu, M. Sun, X. Tian, C. Li, Z. Lv and L. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 22061–22070 CrossRef PubMed.
- K. Jiang, W. Liu, W. Lai, M. Wang, Q. Li, Z. Wang, J. Yuan, Y. Deng, J. Bao and H. Ji, Inorg. Chem., 2021, 60, 17371–17378 CrossRef PubMed.
- Y. Zhang, C. Fu, J. Fan, H. Lv and W. Hao, J. Electroanal. Chem., 2021, 901, 115761 CrossRef.
- J. Wang, D. T. Tran, K. Chang, S. Prabhakaran, J. Zhao, D. H. Kim, N. H. Kim and J. H. Lee, Nano Energy, 2023, 111, 108440 CrossRef CAS.
- Q. Zhang, K. Lian, Q. Liu, G. Qi, S. Zhang, J. Luo and X. Liu, J. Colloid Interface Sci., 2023, 646, 844–854 CrossRef CAS.
- Y. Gao, Y. Xue, F. He and Y. Li, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2206946119 CrossRef CAS PubMed.
- J. Wu, Z. Nie, R. Xie, X. Hu, Y. Yu and N. Yang, J. Power Sources, 2022, 532, 231353 CrossRef CAS.
- J. Zhu, R. Lu, W. Shi, L. Gong, D. Chen, P. Wang, L. Chen, J. Wu, S. Mu and Y. Zhao, Energy Environ. Mater., 2023, 6, e12318 CrossRef CAS.
- T. Cui, X. Zhai, L. Guo, J.-Q. Chi, Y. Zhang, J. Zhu, X. Sun and L. Wang, Chin. J. Catal., 2022, 43, 2202–2211 CrossRef CAS.
- V. H. Hoa, M. Austeria, H. Thi Dao, M. Mai and D. H. Kim, Appl. Catal., B, 2023, 327, 122467 CrossRef CAS.
- L. Yang, C. Feng, C. Guan, L. Zhu and D. Xia, Appl. Surf. Sci., 2023, 607, 154885 CrossRef CAS.
- Q. Ma, H. Jin, F. Xia, H. Xu, J. Zhu, R. Qin, H. Bai, B. Shuai, W. Huang, D. Chen, Z. Li, J. Wu, J. Yu and S. Mu, J. Mater. Chem. A, 2021, 9, 26852–26860 RSC.
- Z. Cui, Z. Yan, J. Yin, W. Wang, M.-E. Yue and Z. Li, J. Colloid Interface Sci., 2023, 652, 1117–1125 CrossRef CAS PubMed.
- R. Andaveh, A. Sabour Rouhaghdam, J. Ai, M. Maleki, K. Wang, A. Seif, G. Barati Darband and J. Li, Appl. Catal., B, 2023, 325, 122355 CrossRef CAS.
- F. Zhang, Y. Liu, F. Yu, H. Pang, X. Zhou, D. Li, W. Ma, Q. Zhou, Y. Mo and H. Zhou, ACS Nano, 2023, 17, 1681–1692 CrossRef CAS.
- T. U. Haq, M. Pasha, Y. Tong, S. A. Mansour and Y. Haik, Appl. Catal., B, 2022, 301, 120836 CrossRef CAS.
- L. Xie, Q. Liu, X. He, Y. Luo, D. Zheng, S. Sun, A. Farouk, M. S. Hamdy, J. Liu, Q. Kong and X. Sun, Chem. Commun., 2023, 59, 10303–10306 RSC.
- X. Du, M. Tan, T. Wei, H. Kobayashi, J. Song, Z. Peng, H. Zhu, Z. Jin, R. Li and W. Liu, Chem. Eng. J., 2023, 452, 139404 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |