
Enhancement effect of oxygen vacancy on photocatalytic CO2 reduction†
Qiang
Yang
a,
Yunyi
Wang
d,
Qingwen
Tian
 *ac,
Xiang
Li
a,
Aixiang
Pan
a,
Mengke
Zhao
a,
Yawei
Zhu
a,
Ting
Wu
*a and
Guigan
Fang
*ab
*ac,
Xiang
Li
a,
Aixiang
Pan
a,
Mengke
Zhao
a,
Yawei
Zhu
a,
Ting
Wu
*a and
Guigan
Fang
*ab
aInstitute of Chemical Industry of Forest Products, Chinese Academy of Forestry, Key Lab. of Biomass Energy and Material, Jiangsu Province, Co-Innovation Center of Efficient Processing and Utilization of Forest Resource, Jiangsu Province, Key Lab. of Chemical Engineering of Forest Products, National Forestry and Grassland Administration, National Engineering Research Center of Low-Carbon Processing and Utilization of Forest Biomass, Nanjing, 210042, P. R. China. E-mail: tianqingwen@icifp.cn; ppfangguigan@163.com; wuting@icifp.cn; Fax: +86 25 85413445; Tel: +86 25 85482542
bNanjing Forestry University, Nanjing 210037, P. R. China
cGuangxi Key Laboratory of Clean Pulp & Papermaking and Pollution Control, College of Light Industry and Food Engineering, Guangxi University, Nanning 530004, P. R. China
dHeilongjiang Provincial Key University Laboratory of Processing Agricultural Products, College of Food and Bioengineering, Qiqihar University, Qiqihar, 161006, China
First published on 2nd February 2024
Abstract
BiOBr was manufactured with various concentrations of oxygen vacancies via a solvothermal method using various alcohols as reducing agents. The reductive alcohol resulted in the fracture of the Bi–O bond, thus producing oxygen vacancies. The less polar solvent and extension of the hydroxyl carbon chain of the alcohol facilitate the generation of oxygen vacancies. Theoretical calculations and experiments demonstrated that oxygen vacancies could act as trapping sites to improve the separation efficiency of photogenerated carriers, accompanied by a decrease in the band gap energy to increase the absorption capacity for visible light. The enriched electrons in the defect band could generate more “hot electrons” that were transported to CO2 molecules adsorbed onto unsaturated coordination centers, lowering the reaction barrier and promoting the activation of CO2via the surface plasmon resonance effect. The resulting catalyst prepared with ethylene glycol exhibited a remarkable CO yield of 122.38 μmol g−1 h−1, which is nearly 7.2 times over that of the catalyst prepared with pure water. The enhanced photocatalytic performance corresponded to changes in oxygen vacancy concentration. Therefore, the change in the band gap structure arising from the variation in the content of oxygen vacancies is the cause of enhanced photocatalytic CO2 reduction.
Introduction
With the acceleration of economic development, a massive amount of fossil fuels is being consumed, resulting in increasing emissions of carbon dioxide (CO2) that induce an intensive greenhouse effect and energy crisis.1–4 Therefore, the capture and recovery of carbon dioxide have become imperative for sustainable development and are of great significance for environmental remediation.5 Photocatalytic CO2 reduction reaction (CO2RR) to produce high value-added chemicals and fuels under moderate conditions has emerged as a prospective strategy to alleviate universal climate warming and energy shortage issues.6 BiOBr has been widely used in the realm of photocatalysis due to its distinctive layered structure consisting of [Bi2O2]2+ plates interleaved with dual slabs of Br atoms, which imparts more active sites and carrier separation efficiency on BiOBr.7 However, the large band gap energy and weak ability to capture visible light inhibit the photocatalytic efficiency of pure BiOBr, limiting its practical applications.8Generally, realizing the practical application of photocatalytic CO2RR suffers from inherent limitations on poor harvesting of visible light and selectivity of carbon products. Numerous studies have revealed that oxygen vacancies (OVs) are of significance in adjusting the photoelectric properties of photocatalysts.9 OVs can narrow the band gap by introducing a defect band below the conduction band to strengthen the trapping capacity for visible light.10,11 Moreover, OVs can act as capturing sites to enhance the transfer of excited electrons to the surface of the catalyst and impede the recombination of photo-generated carriers, thereby generating more reactive oxygen species.12,13 The formation of OVs depends on the application of a reductive atmosphere and formation energy.14 Reducing agents, such as ethanol, acetaldehyde, glucose, and metals, along with high temperature and pressure, are utilized to offer a reductive synthetic atmosphere for OV formation. Kong et al. prepared Bi2WO6 with abundant oxygen vacancies via a simple solvothermal method utilizing ethylene glycol as a reducing reagent.15 During the redox reaction, the ethylene glycol broke the Bi–O bond in the crystal, enabling oxygen atoms to escape from the Bi2WO6 crystalline surface. Liu et al. successfully synthesized OVs–MoO2−x by heat treatment at a high temperature of 550 °C using a 10% H2/Ar mixture.16 High temperatures result in the breakage of Mn–O bonds, and H2 traps the escaping oxygen atoms to produce H2O molecules. In addition, OV sites are more easily regenerated after inducing cationic asymmetric coordination (M1–OVs–M2).17 Brant et al. discovered neutral Cu2+–OVs complexes in Cu-doped TiO2 crystals. First-principle and experimental results showed that OVs were formed near the Cu2+ site with the help of charge compensation.18 Among the numerous methods available, alcohols are the most typically adopted reducing reagents for the preparation of OV-rich catalysts. However, so far, almost no studies have revealed the specific mechanism of alcohol functioning in OV formation.
In previous studies, BiOBr-containing OVs was prepared with ethanol, whereas the formation and role of OVs have not been investigated in detail. Herein, this study aims to elucidate the effect of alcohol reducibility on the concentration of OVs. BiOBr with abundant OVs was manufactured via a facile solvothermal method using different alcohols as reducing solvents. The concentration of OVs was detected by X-ray photoelectron spectroscopy (XPS) and electron paramagnetic resonance (EPR) spectroscopy. The regulation of OVs on band structure was verified by the density functional theory. At the same time, the impact of OVs on the energy band structure was investigated by UV-vis diffuse spectrophotometer (UV-vis DRS), VB-XPS, and Mott–Schottky (MS). This study proposes a rational regulation strategy of OV content, reveals the relationship between alcohol reducibility and oxygen vacancy concentration, clarifies the mechanism of changes in the band structure due to OVs and demonstrates efficient photocatalytic CO2 reduction efficiency.
Experiment
Detailed experimental information is available in the ESI Section.†Results and discussion
Catalyst morphology and characterization
The formation mechanism of micro-flower-shaped catalysts manufactured via a simple solvothermal method is elucidated (Fig. 1a). Notably, the addition of alcohol is essential to the formation of hierarchical micro-flower catalysts. Initially, with the assistance of halides, bismuth precursors react in the solvent to gradually grow into BiOBr nanosheets. Alcohol, acting as a structure-guiding agent, induces these nanosheets to self-assemble into a micro-flower structure. The unique hierarchical structure of microflowers can provide a significant number of active sites and enhance the contact area with CO2 molecules. At the same time, alcohols, being reducing agents, can react with [Bi2O2]2+ layer to produce [Bi2O](4−x)+, thus facilitating the formation of OVs in situ; the alcohol is oxidized into aldehydes and carboxylic acids. As shown in Fig. 1b and S1a,† XRD was utilized to characterize the crystalline phase and crystallinity of all samples. No impurity peaks were detected in the XRD patterns, and the spectra matched well with that of tetragonal BiOBr (JCPDS no. 85-0862). The typical characteristic peaks at 10.9°, 25.2°, 31.7°, and 32.3° corresponded to (001), (101), (102), and (110) planes, respectively. With the increasing proportion of anhydrous ethanol (Fig. S1a†), the characteristic diffraction peaks of all catalysts weakened and broadened slightly, implying a gradual decrease in crystallinity. The peak intensity ratio of (102) to (110) crystal planes changed considerably with the addition of anhydrous ethanol, which may be due to the formation of OVs that affected the growth of (102) crystal faces.19 The diffraction peaks corresponding to (102) to (110) planes in BiOBr also exhibited the same changes when anhydrous ethanol was substituted with other alcohols (Fig. 1b). The crystallite size of all samples calculated via Scherrer's equation is shown in the ESI.†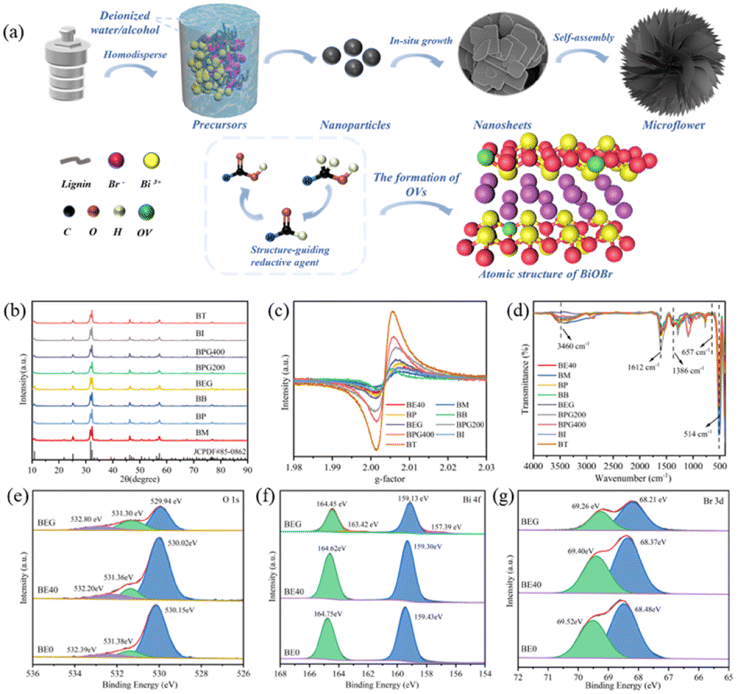 | ||
| Fig. 1 Schematic illustration of the catalyst formation process (a), X-ray diffraction patterns (b), electron paramagnetic resonance spectra (c), in situ FT-IR spectra (d), and X-ray photoelectron spectroscopy spectra (e–g) of BE0, BE40 and BEG. | ||
EPR spectroscopy was utilized to explore the OV content of the photocatalysts, and the spectra are depicted in Fig. 1c and S1b.† The unpaired electrons captured by the OVs were responsible for the characteristic single Lorentzian line seen in all samples, which was centered at g = 2.003.12 Moreover, the relative intensity of EPR signal peaks determined the number of OVs. According to Fig. S1b,† the relative intensities of the EPR peaks were in the following order: BE50 > BE20 > BE30 > BE10 > BE40 > BE0, implying that anhydrous ethanol facilitates the generation of OVs. The BE50 prepared with only anhydrous ethanol as the reaction solvent possessed the largest number of OVs; however, the number of OVs was not proportional to the photocatalytic performance. When anhydrous ethanol was substituted with other alcohols, the relative intensity of the EPR peaks was in the following order: BT > BPG400 > BPG200 > BEG > BG > BP > BE40 > BI > BB. Under the high temperature and pressure atmospheres of the hydrothermal process, the elimination reaction between alcohols and the BiOBr crystal led to the breakage of Bi–O bonds, thus promoting the generation of OVs.20 The polarity of the solvent is not conducive to the elimination reaction; thus, the maximum intensity of OVs could be obtained only when anhydrous ethanol was used as the reaction solvent (BE50) in the BE system. When the reaction solvent is divided into monohydric alcohols and diols, the EPR intensities of OVs for the two groups were in the order of BP > BE40 > BM, BT > BI > BB and BPG400 > BPG200 > BEG, respectively. This result matched well with the increased number of α-C atoms in branched chains, which facilitates the elimination reaction of alcohols, thus promoting the formation of more OVs.
XPS spectra were utilized to elucidate the chemical states and composition of the photocatalysts. The full-scan XPS spectra of BE0, BE20, BE40, BE50 and BEG samples are presented in Fig. S1c,† where the existence of Br, O and Bi elements can be detected. In Fig. 1e and S1d,† the three peaks at 530.15 eV, 531.38 eV, and 532.39 eV can be assigned to the lattice oxygen (OL), oxygen vacancies (OVs), and hydroxyl groups (OH) on the surface of BE0 that may be fitted into the O 1s peak for BEO in the XPS spectra.21 The peak signals of OVs and OH originated from H2O adsorbed onto the surface of the photocatalysts. Notably, the oxygen atoms in metal bonds (Bi–O) have a denser charge cloud distribution compared to those in hydrogen–oxygen covalent bonds, ultimately exhibiting lower binding energies. In addition, the OVs acting as electron traps could attract free electrons, resulting in a higher electron density in the OV region and lower binding energy of OVs than that of OH.22 In Fig. 1f and S1e,† two main peaks at 159.43 eV and 164.75 eV can be identified, which belonged to Bi 4f7/2 and Bi 4f5/2 spin-orbital splitting photoelectrons of Bi3+ chemical state in BE0, respectively.23 Furthermore, two metallic Bi peaks with binding energies of 163.42 eV and 157.39 eV, respectively, were detected.24 A 3d XPS spectrum (Fig. 1g and S1f†) with the binding energy of 68.48 and 69.52 eV could correspond to Br 3d5/2 and Br 3d3/2, respectively. Compared to BE0, the O 1s, Bi 4f and Br 3d peaks of BE20, BE40, BE50 and BEG showed a slight shift, implying electron flow and rearrangement on the surface of other catalysts, which originated from the formation of OVs near Bi3+ in (Bi2O2)2+.24
Raman spectroscopy was employed to characterize the crystal defects and structural deformations in composite materials, and the results are depicted in Fig. S2a.† The Bi–Br stretching mode of A1g external, A1g internal, and E1g internal could be indexed as three main bands of BE0 centered at 35.9, 111.8 and 154.3 cm−1, respectively.25 The D and G bands of carbon can be observed near 1388.9 and 1562.3 cm−1 in all BE catalysts, indicating the presence of sp3-hybridized and sp2-hybridized carbon atoms, respectively.26 Compared to BE0, the peak intensity of other catalysts decreased, implying increased disorder in BiOBr and more OVs. During the hydrothermal reaction process, the hydroxyl groups of alcohol reacted with crystal oxygen and brought about the absconding of crystal oxygen atoms from the lattice, resulting in a decrease in crystallinity and the formation of OVs in situ. These results matched well with the results of XPS, EPR and XRD spectra.22
Based on the three spatial positions of the crystal, OVs could be categorized into surface oxygen vacancies (SOV) and bulk oxygen vacancies (BOV). Generally, the characteristic peaks of OVs in XPS spectra are associated with the intensity of SOV, where ESR signals indicate the intensity of BOV. The relative peak areas under the characteristic peaks of OVs in the XPS spectra can be utilized to explore the trend of SOV content; concrete results are shown in Table S3.†27 More OVs imply more H2O and O2 molecules adsorbed by the catalyst, resulting in higher relative content of SOV. BEG possesses the highest SOV content, implying a tremendous potential for optimal catalytic effects. To further elucidate the synthesis process, an elemental analyzer (CHNS/O) was used to detect the trend of O distribution in the photocatalysts (Fig. S2b†). The results were consistent with trends revealed by XPS calculations.
Combined results from EPR, XPS, Raman, and CHNS/O indicated that increasing OVs does not necessarily lead to better photocatalytic performance. OVs can induce the creation of a defect band beneath the conduction band, which can capture free electrons excited from the conduction band, thus inhibiting the negative recombination of photo-generated electron–hole pairs, prolonging charge carrier lifetime, and promoting their separation. According to previous studies, SOV and BOV can strengthen the absorption capacity of catalysts for visible light. The SOV facilitates the transfer of photo-excited carriers, while an excess of BOV promotes the recombination of photo-excited carriers. The concentrations of SOV and BOV can be controlled by regulating the ratio and type of alcohol. The ultimate positive or negative effect of OVs on photocatalytic efficiency depends on the balance of SOV and BOV.28
In situ FT-IR spectra were employed to detect the general surface chemical composition of catalysts, and the specific results are demonstrated in Fig. 1d and S2c.† The characteristic peaks in the region of 500–1000 cm−1 and 1000–1500 cm−1 were indexed as the Bi–O and Bi–Br bonds in BiOBr, respectively.29 Generally, the peak located at ca. 514 cm−1 corresponded to the stretching vibration of the Bi–O bond, and the fluctuation in peak intensity arose from the shift in the content of oxygen vacancies. The characteristic peak of BE50 at 657 cm−1 could be indexed to the swinging vibration of the aromatic C–H.30 It can be seen that all samples displayed a wide transmittance peak at ca. 3460 and 1612 cm−1, which was ascribed to the O–H tensile and flexural vibration of adsorbed water molecules, respectively. Two peaks at 1367 and 1284 cm−1 indicated the stretching vibrations of the C–O and C-bonds in the aromatic skeleton, respectively.30 The characteristic peak located at 1386 cm−1 corresponded to the asymmetric stretching vibration of –COOH bonds, which is consistent with Fig. 1a, which demonstrated the successful preparation of BiOBr composites.
In Fig. 2a, SEM images show that BE40 consists of a flower-like spherical morphology, and it was assembled by several thin nanosheets with a diameter of 1 μm, which were embedded together with distinct holes in the middle. BE0 exhibited the relatively small size of microflowers loaded on the surface of BiOBr nanosheets (Fig. S3a†). Together Fig. 2a and S3† indicate that the size and thickness of nanosheets become smaller and thinner as the ethanol content increases, and these nanosheets gradually self-assemble into smaller and more compact microflowers. The nucleation and crystal growth process of BiOBr is interrupted by the addition of anhydrous ethanol in the precursor solution, which hides the reaction rate between the precursor solution to suppress the sudden maturation of BiOBr nanoplates.31 Notably, excess anhydrous ethanol destroys the structure of the catalyst and produces more nanosheets with reduced specific surface areas. These results were confirmed by BET experiments. As shown in Fig. 2b, the micro-flower structure of BEG had a smaller size of 250 nm. Furthermore, corresponding EDX images (Fig. S3d†) confirmed the homogeneous distribution of Bi, O, Br, and C elements within the BEG. Oxygen vacancies, as coordination unsaturated centers, can easily adsorb oxygen-containing molecules, such as free water molecules, oxygen and carbon dioxide in the air, resulting in a higher distribution of oxygen atoms in the catalysts.14 In the HRTEM image of BE40 (Fig. 2c), lattice fringes with a spacing of ca. 0.353 nm and 0.811 nm correspond to the (101) and (001) facets of BiOBr, respectively. The lattice space of 0.282 nm (Fig. 2d) in BEG matches well with the (102) plane of BiOBr. Moreover, the inconsecutive crystal fringes observed in Fig. 2c and d (shown as yellow circles) confirm the existence of oxygen vacancies.32 As demonstrated in Fig. 2e and f and Table S5,† N2 adsorption–desorption isotherms and pore size distribution images showed that BE0, BE40, BE50 and BEG manifested a mesoporous structure. BEG possessed the maximal specific surface area and pore volume. The self-assembly growth of the catalyst can be promoted by moderate oxygen vacancies, exhibiting a larger specific surface area and pore volume, which is capable of facilitating the adsorption of pollutants and transmission of electrons and holes at the surface.
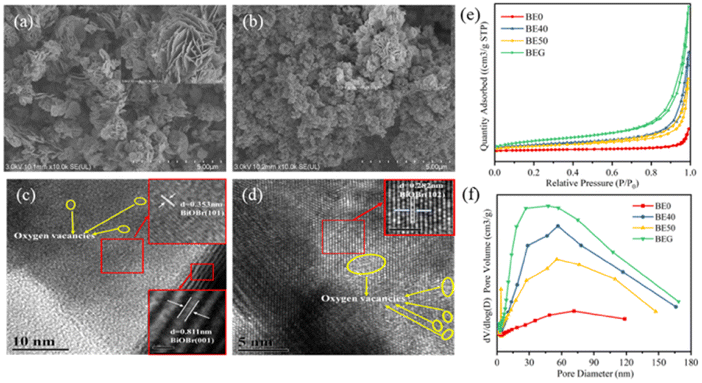 | ||
| Fig. 2 Scanning electron microscope (SEM) images of BE40 (a) and BEG (b), transmission electron microscopy (TEM) images of BE40 (c), BEG (d), N2 adsorption–desorption isotherms (e), and pore size distribution (f). | ||
Optical properties
UV-vis DRS spectroscopy was utilized to evaluate the optical properties of catalysts. As depicted in Fig. 3a and S4a,† BEG exhibited an optimal visible-light absorption capability, which highlighted its tremendous potential for photocatalytic CO2 reduction. Based on UV-vis DRS spectra, corresponding Tauc plots were prepared as demonstrated in Fig. 3b and S4b;† the results of absorption edge and band gap (Eg) are displayed in Table S6.† BEG exhibited the largest absorption edge wavelength, suggesting the enlarged visible-light response region. On the one hand, OVs modified the band structure (e.g., defect states and band position) to expand the visible-light absorption range, which manifested as the redshift of the absorption edge. On the other hand, the absence of crystal oxygen atoms resulted in the accumulation of large numbers of free electrons in the defect band, thus forming charge clouds, which could absorb incident light and undergo intra-band excitation, that is, the local surface plasmon resonance effect (LSPR).28 Nevertheless, excessive OVs could disrupt the band gap structure, while many free electrons enriched in the defect band hindered the incident light energy absorption and intra-band excitation, thus affecting the absorption edge and Eg. It is particularly noteworthy that the extended visible-light absorption range and reduced Eg do not necessarily result in a better photocatalytic performance. The Mott–Schottky plots and VB-XPS spectrum of BE0, BE40 and BEG were evaluated to explore the impact of OVs on the energy band structure. From the VB-XPS spectra (Fig. 3c, S4c and e†), the EVB of BE0, BE40 and BEG were 2.07, 2.09 and 1.93 V, respectively. According to eqn (1), the ECB value based on VB-XPS spectra can be obtained, which were −0.43, −0.35 and −0.46 V, respectively. In Fig. 3d, S4d and f,† the slope of the linear part of the MS curve is positive, indicating that BE0, BE40 and BEG are all n-type semiconductors with flat bands of −0.30 V, −0.37 V and −0.42 V (vs. Ag/AgCl), respectively, the ECB of an n-type semiconductor is approximately 0.2 V negative than Eflat. Hence, the ECB of BE0, BE40 and BEG are −0.5 V, −0.57 V and −0.62 V, respectively. Compared to the ECB obtained from VB-XPS spectra, the ECB revealed by the MS plots is lower.| ECB = EVB − Eg | (1) |
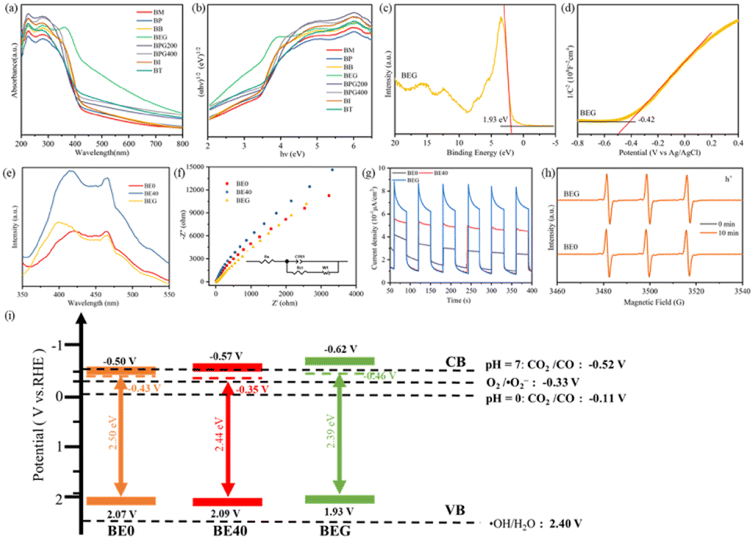 | ||
| Fig. 3 UV-vis absorption spectra (a), band gaps (b), VB-XPS spectra (c), and Mott–Schottky (MS) plots (d) of BEG, photoluminescent spectra (e), EIS spectra (f) and photocurrent intensity (g) of BE0, BE40, and BEG, TEMPO-h+ spin-trapping ESR spectra (h), and band structure of the photocatalysts (i). | ||
Being electron donors, OVs are capable of raising the Fermi level of metal oxide photocatalysts to induce a defect state between the CB and VB, which contributes to the anomalous position of the CB in calculated and measured values. The presence of the OV defect band allows the photocatalyst to absorb less energy than the band gap, resulting in higher light energy utilization and carrier density. The recombination of photogenerated charge carriers can be inhibited by the ability of the defect band to absorb the excited electrons from the VB and drop from the CB. Simultaneously, plenty of electrons enriched in the defect band are capable of undergoing intra-band excitation with the aid of incident light, facilitating the formation of hot electrons and activation of the CO2 molecules.33 However, the OV-induced defect band functions as the recombination site due to its proximity to the center of the forbidden band, which can be unfavorable for the progress of the photocatalytic CO2 reduction reaction.34 The energy band structure of the photocatalysts is depicted in Fig. 3i. The rearrangement of crystal oxygen atoms was dominated by alcohols, which promoted the generation of OVs and controlled their concentrations, thus regulating the band structure of the catalyst.
Photoluminescence spectroscopy (PL), electrochemical analytical impedance spectroscopy (EIS) and photocurrent response were conducted to investigate the impact of OVs on the charge separation efficiency of catalysts. As depicted in Fig. 3e, distinct peaks at 421, 414 and 399 nm assigned as BE0, BE40 and BEG may be intrinsic emission peaks. Meanwhile, peaks located at 464, 466 and 467 nm corresponded to the OVs.35 Due to the limited capability of BE0 to absorb visible light, the concentration of photogenerated carriers over it was lowered as a consequence of the UV-vis DRS spectra; eventually, its PL intensity lowered than that of BE40 and BEG. In the EIS spectra (Fig. 3f), the smaller arc radius was assigned as a more significant separation efficiency of electron pairs. Upon fitting the EIS data with ZView software, the resistances of BE0, BE40 and BEG were calculated to be 6482 Ω, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 317 Ω and 12834 Ω, respectively, which were caused by the excess oxygen vacancies that hindered electron transfer. As per Fig. 3g, the photocurrent density of BEG is the highest, demonstrating a rapid separation efficiency of photogenerated carriers, which agrees well with the result of PL. The OV-induced defect band acted as a trapping site at the CB edge to alleviate unfavorable recombination and facilitate the separation efficiency of photogenerated charge carriers. Nevertheless, the overplus of BOV became the recombination center, which led to excessive electron accumulation, hindering the absorption of incident light and intra-band excitation of free electrons. The final photocatalytic activity depends on the balance between SOV and BOV, while the band structure varying from the OVs is critical for different photocatalytic effects. Benefiting from strong visible-light absorption capacity, more photogenerated carriers and a higher concentration of h+ were generated by BEG (Fig. 3h), which is in agreement with the above analysis.
317 Ω and 12834 Ω, respectively, which were caused by the excess oxygen vacancies that hindered electron transfer. As per Fig. 3g, the photocurrent density of BEG is the highest, demonstrating a rapid separation efficiency of photogenerated carriers, which agrees well with the result of PL. The OV-induced defect band acted as a trapping site at the CB edge to alleviate unfavorable recombination and facilitate the separation efficiency of photogenerated charge carriers. Nevertheless, the overplus of BOV became the recombination center, which led to excessive electron accumulation, hindering the absorption of incident light and intra-band excitation of free electrons. The final photocatalytic activity depends on the balance between SOV and BOV, while the band structure varying from the OVs is critical for different photocatalytic effects. Benefiting from strong visible-light absorption capacity, more photogenerated carriers and a higher concentration of h+ were generated by BEG (Fig. 3h), which is in agreement with the above analysis.
DFT calculations for DOS
To confirm the effect of oxygen vacancies on the electron configuration, molecular configuration, interfacial charge transfer, density of state (DOS) and total density of states (TDOS) were demonstrated in Fig. 4. The band gap of BiOBr without OVs is 1.81 eV, which does not match the commonly reported values for band gaps in the literature due to interactions between electrons.36 The band gaps of BiOBr with one and two OVs are 2.24 eV and 2.06 eV, respectively. The formation of oxygen vacancies was conducive to the reduction of the band gap, which is consistent with our previous analysis. As demonstrated in Fig. 4e and f, the generation of a defect band below the conduction band was induced by the OVs, which was on account of the abundance of localized states due to the OVs. The OV-induced defect band serves as a trapping site to receive the excited electrons arising from the VB and dropping down from the CB, simultaneously enhancing the photo-response region of the catalyst. Compared with BiOBr without OVs, the conduction band of BiOBr with OVs was shifted to a lower energy level, favoring the separation of photogenerated carriers.37 Meanwhile, the DOS of Bi 6p and TDOS at the conduction band edge increased due to the presence of OVs, indicating a high concentration of photogenerated carriers in the conduction band, which favored the formation of reactive oxygen species. The difference in charge distribution between BiOBr without OVs and with one OV is depicted in Fig. 4g and between BiOBr with two OVs and one OV in Fig. 4h. The sections that are yellow and cyan, respectively, show an increase and decrease in the charge density. OVs allow the accumulation of charge around Bi atoms, promoting the occurrence of LSPR and enhancing the visible-light absorption ability, thus strengthening the photooxidative property of catalysts.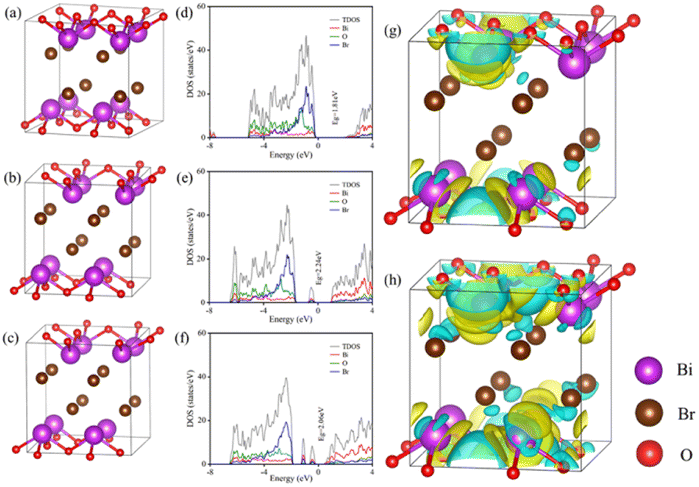 | ||
| Fig. 4 Atomic structure of BiOBr (a), BiOBr with one OV (b), and BiOBr with two OVs (c), density states of BiOBr (d), BiOBr–OVs (e), and BiOBr–2OVs (f), and charge difference distribution (g and h). | ||
Photocatalytic mechanism
Fig. 5a–c and S5† show the performance of all photocatalysts for photocatalytic CO2 reduction. CO was discovered to be the predominant product of photocatalytic CO2 reduction, showing a strong preference for CO. With increasing ethanol content, the rate of CO production increased at first, then dropped. The generation rate of CO over BE40 was 64.76 μmol g−1 h−1, which was nearly four times higher than that of BE0 (17.04 μmol g−1 h−1). When ethanol was replaced by other alcohols, BEG exhibited a remarkable CO yield of 122.38 μmol g−1 h−1, which was much higher than that of BM (25.41 μmol g−1 h−1), BP (42.36 μmol g−1 h−1), BB (33.23 μmol g−1 h−1), BPG200 (84.23 μmol g−1 h−1), BPG400 (45.41 μmol g−1 h−1), BI (37.59 μmol g−1 h−1), and BT (72.58 μmol g−1 h−1). Additionally, a few controlled trials were conducted with different setups. No products were detected when light, CO2, H2O and catalysts were absent, demonstrating that the only way to generate CO is by photocatalytic CO2 reduction. The repeated photocatalytic CO2 reduction tests of BEG proved the durability of photocatalytic, and no discernible loss from it was discovered after five cycles (Fig. S5c†). As depicted in Fig. S6,† the morphology and crystal structure of the catalyst remained unchanged, illustrating good stability. Meanwhile, the crystallinity of the crystal increased after recycling, accounting for the disappearance of oxygen vacancies. Plenty of H2O molecules and other media were absorbed by the catalyst during the reaction, which filled the missing oxygen atoms in the crystal and resulted in increased crystallinity. In situ FT-IR was utilized to identify key intermediates, thereby allowing speculation on the reaction pathway and insight into the photocatalytic CO2 conversion process. As depicted in Fig. S7a,† several characteristic peaks in the range of 985–1490 cm−1 corresponded to CO2 adsorption on the surface of BEG,38 and absorption bands at 1250 and 1620 cm−1 were associated with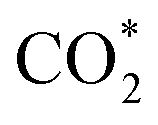 and COOH*, respectively. Furthermore, the weak absorption peak at 2076 cm−1 was attributed to CO* in Fig. S7b†.39 Crucially,
and COOH*, respectively. Furthermore, the weak absorption peak at 2076 cm−1 was attributed to CO* in Fig. S7b†.39 Crucially, 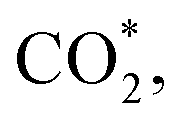 CO* and COOH* are all key intermediates during the photocatalytic reduction of CO2 to CO. The reduction path was speculated to be as follows:
CO* and COOH* are all key intermediates during the photocatalytic reduction of CO2 to CO. The reduction path was speculated to be as follows: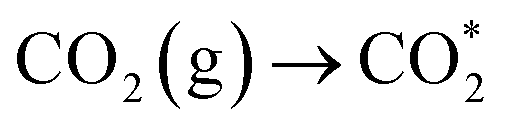 | (2) |
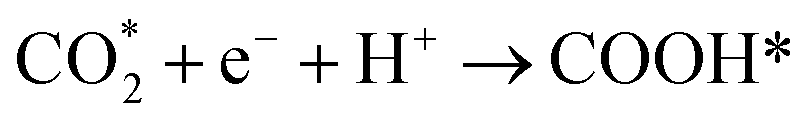 | (3) |
| COOH* + e− + H+ → CO* | (4) |
| CO* → CO(g) | (5) |
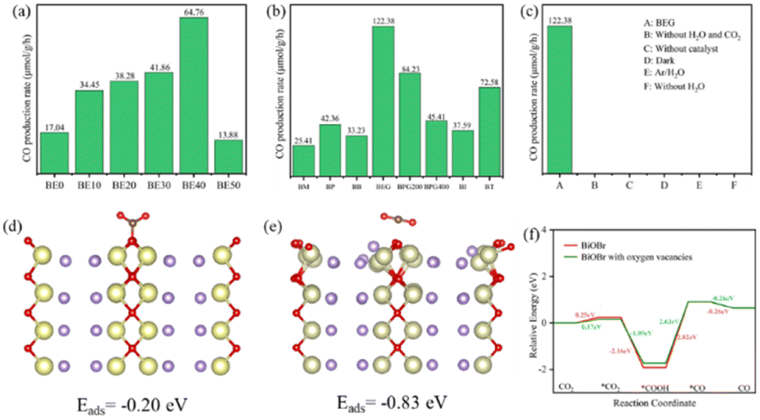 | ||
| Fig. 5 CO production rate (a–c), CO2 adsorption energy of BiOBr (d), and BiOBr with OVs (e), and free energy of CO route (f). | ||
Coordinatively unsaturated metal centers typically arise in tandem with the occurrence of OVs. Such metal centers can serve as ideal capture sites for substrate molecules, which facilitate the establishment of an electron transfer channel between the catalyst and adsorbed CO2 molecules, thus promoting the activation and dissociation of the CO2 molecules for subsequent transformation. To understand the impact of OVs on reactant activation, variations in CO2 molecule adsorption energies (Eads) were examined using various models. As shown in Fig. 5d and e, the Eads of the CO2 molecules are −0.20 eV on BiOBr and −0.83 eV on BiOBr with OVs. As summarized in Table S7,† the C–O bond length and C–O–C angle changed from 1.26 Å and 124.85° on BiOBr to 1.18 Å and 178.77° on BiOBr with OVs. The bond lengths were thus shortened, but bond angles became larger. The DFT simulation was utilized to calculate the Gibbs free energy of BiOBr to examine the pathways of photocatalytic CO2RR. As depicted in Fig. 5f, the enhanced CO2 adsorption on the surface of BiOBr with OVs resulted in a lower energy barrier for *CO2 adsorption than BiOBr with no OVs. The generation of *COOH and *CO intermediates is an essential step in controlling the generation of CO. The *CO2 intermediates could be attacked by proton–electron pairs (H+ + e−) to generate *COOH and *CO active substances in turn. The conversion of *CO2 to *COOH on the BiOBr surface requires a smaller energy barrier than that on the surface of BiOBr with OVs due to the massive adsorption capacity of BiOBr for *COOH. However, the transformation from *COOH to *CO on the surface of BiOBr with OVs was more favorable due to the lower energy barrier. Briefly, the improved photocatalytic CO2 reduction performance is attributed to the enhanced adsorption capacity and decreased energy barrier during the reaction.
Fig. 6 suggests a potential reaction pathway for photocatalytic CO2 reduction over BEG based on analytical results above. When exposed to visible light, the excited electrons from the valence band are transferred to the conduction band and the OV-induced defect band, while the photo-generated holes remain in the valence band. OVs can guide the electron transfer pathway and hinder the unfavorable recombination of the photo-generated charge carriers by the formation of the electric field in the photocatalysts. Meanwhile, the excited electrons are well accumulated and stabilized in the defect band to avoid falling back to the valence band again. Electrons enriched in the OV-induced defect bands can absorb incident light and generate hot electrons with the aid of the LSPR effect, which facilitates a series of subsequent transformations of the CO2 molecules adsorbed at the coordinatively unsaturated metal centers. Additionally, the photocatalytic activity of BEG was evaluated against that of other materials found in the literature. BEG demonstrated a notably higher CO production rate of 122.38 μmol g−1 h−1 in comparison to other materials presented in Table S8,† indicating the remarkable photocatalytic CO2 reduction capabilities of BEG.
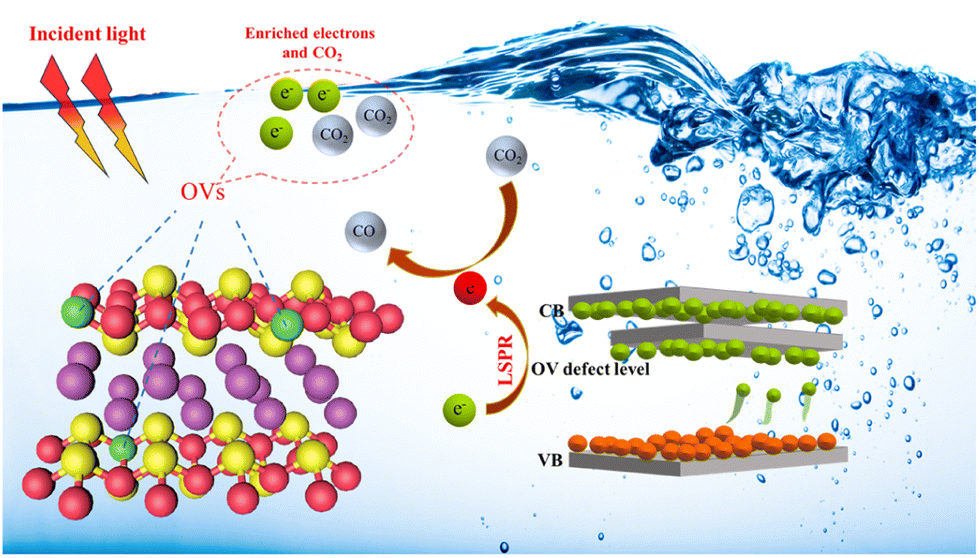 | ||
| Fig. 6 Possible mechanism of photocatalytic CO2 reduction reaction. | ||
Conclusions
In conclusion, a facile solvothermal technique was effectively utilized to manufacture the OV-enriched BiOBr. The OV concentration can be manipulated by adjusting the ratio and type of alcohols of different reducing properties. DFT calculations, UV-vis DRS spectra, VB-XPS and MS plots demonstrated that OVs played a crucial role in enhanced light-enrichment capacity, band position, and electron-transfer pathway, which strongly correlated to photocatalytic activity. Benefiting from the strong electron-withdrawing effect of OVs, electrons captured in the defect band are capable of prolonging the excited-state lifetime and improving the separation efficiency of charge carriers. BEG with a wide range of active sites exhibited an excellent photocatalytic effect on CO2 reduction (CO: 122.38 μmol g−1 h−1). Our study provides a promising strategy for the controlled synthesis of OV-enriched catalysts and for understanding the mechanism of OVs on energy band variation and electron flow.Author contributions
Qiang Yang: conceptualization, data curation, investigation, methodology, project administration, and writing – original draft, writing – review & editing. Yunyi Wang: conceptualization, data curation, and visualization. Qingwen Tian: conceptualization, data curation, investigation, writing – review & editing, and funding acquisition. Xiang Li: investigation and methodology. Aixiang Pan: data curation and resources. Mengke Zhao: investigation and methodology. Yawei Zhu: investigation and methodology. Ting Wu: funding acquisition and writing – review & editing. Guigan Fang: funding acquisition and investigation.Conflicts of interest
No conflicts of interest exist in the submission of this manuscript, and the manuscript is approved by all authors for publication. All authors have read and approved the paper, and it has not been published previously nor is it being considered by any other peer-reviewed journal.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 32301682), the National Key Research and Development Program of China (No. 2022YFC2105505), the Foundation of “Jiebangguashuai” Science and Technology Project of Yibin City (No. 2022JB009)”, and the Taishan Industrial Experts Programme (No. tscy20200213).References
- D. Long, J. Liu, H. Chen, P. Liu, K. Zheng, Y. Zeng, X. Chen, S. Li and M. Lu, Appl. Catal., B, 2023, 330, 122625, DOI:10.1016/j.apcatb.2023.122625.
- R. Zhao, Z. Zhu, T. Ouyang and Z.-Q. Liu, Angew. Chem., Int. Ed., 2023, e202313597, DOI:10.1002/ange.202313597.
- S.-T. Guo, Z.-Y. Tang, Y.-W. Du, T. Liu, T. Ouyang and Z.-Q. Liu, Appl. Catal., B, 2023, 321, 122035, DOI:10.1016/j.apcatb.2022.122035.
- T. Ouyang, Y.-Q. Ye, C. Tan, S.-T. Guo, S. Huang, R. Zhao, S. Zhao and Z.-Q. Liu, J. Phys. Chem. Lett., 2022, 13, 6867–6874, DOI:10.1021/acs.jpclett.2c01509.
- S.-T. Guo, Y.-W. Du, H. Luo, Z. Zhu, T. Ouyang and Z.-Q. Liu, Angew. Chem., Int. Ed., 2024, 63, e202314099, DOI:10.1002/ange.202314099.
- H. Huang, R. Shi, Z. Li, J. Zhao, C. Su and T. Zhang, Angew. Chem., Int. Ed., 2022, 134, e202200802, DOI:10.1002/ange.202200802.
- Y. Xin, Q. Zhu, T. Gao, X. Li, W. Zhang, H. Wang, D. Ji, Y. Huang, M. Padervand, F. Yu and C. Wang, Appl. Catal., B, 2023, 324, 122238, DOI:10.1016/j.apcatb.2022.122238.
- J. Ni, D. Liu, W. Wang, A. Wang, J. Jia, J. Tian and Z. Xing, Chem. Eng. J., 2021, 419, 129969, DOI:10.1016/j.cej.2021.129969.
- Q. Zhu, A. A. Dar, Y. Zhou, K. Zhang, J. Qin, B. Pan, J. Lin, A. O. T. Patrocinio and C. Wang, ACS ES&T Engg, 2022, 2, 1365–1375, DOI:10.1021/acsestengg.1c00479.
- K. Li, K. Gong, J. Liu, Y. Yang, I. Nabi, A.-U.-R. Bacha, H. Cheng, J. Han and L. Zhang, J. Hazard. Mater., 2021, 418, 126207, DOI:10.1016/j.jhazmat.2021.126207.
- Y. Lu, Y. Huang, Y. Zhang, J.-j. Cao, H. Li, C. Bian and S. C. Lee, Appl. Catal., B, 2018, 231, 357–367, DOI:10.1016/j.apcatb.2018.01.008.
- Y. Li, M. Wen, Y. Wang, G. Tian, C. Wang and J. Zhao, Angew. Chem., Int. Ed., 2021, 133, 923–929, DOI:10.1002/ange.202010156.
- L. Hao, H. Huang, Y. Zhang and T. Ma, Adv. Funct. Mater., 2021, 31, 2100919, DOI:10.1002/adfm.202100919.
- W. Jiang, H. Loh, B. Q. L. Low, H. Zhu, J. Low, J. Z. X. Heng, K. Y. Tang, Z. Li, X. J. Loh, E. Ye and Y. Xiong, Appl. Catal., B, 2023, 321, 122079, DOI:10.1016/j.apcatb.2022.122079.
- X. Y. Kong, Y. Y. Choo, A. K. Soh and A. R. Mohamed, Chem. Commun., 2016, 52, 14242–14245, 10.1039/C6CC07750A.
- X. Liu, L. Yang, M. Huang, Q. Li, L. Zhao, Y. Sang, X. Zhang, Z. Zhao, H. Liu and W. Zhou, Appl. Catal., B, 2022, 319, 121887, DOI:10.1016/j.apcatb.2022.121887.
- K. Yu, L. L. Lou, S. Liu and W. Zhou, Adv. Sci., 2020, 7, 1901970, DOI:10.1002/advs.201901970.
- A. T. Brant, S. Yang, N. C. Giles, M. Z. Iqbal, A. Manivannan and L. E. Halliburton, J. Appl. Phys., 2011, 109, 073711, DOI:10.1063/1.3552910.
- C. Hua, X. Dong, Y. Wang, N. Zheng, H. Ma and X. Zhang, J. Mater. Sci., 2019, 54, 9397–9413, DOI:10.1007/s10853-019-03556-y.
- X. Wang, L. Wu, J. Wang, Y. Zhou, Y. Wang, W. D. Wu, W. Li and Z. Wu, Chemosphere, 2022, 307, 135967, DOI:10.1016/j.chemosphere.2022.135967.
- Y. Miao, W. Tian, J. Han, N. Li, D. Chen, Q. Xu and J. Lu, Nano Energy, 2022, 100, 107473, DOI:10.1016/j.nanoen.2022.107473.
- X. Hu, J. Wang, J. Wang, Y. Deng, H. Zhang, T. Xu and W. Wang, Appl. Catal., B, 2022, 318, 121879, DOI:10.1016/j.apcatb.2022.121879.
- L. Zhang, F. Xie, J. Liu, Z. Sun, X. Zhang, Y. Wang, Y. Wang, R. Li and C. Fan, Chem. Eng. J., 2022, 450, 138066, DOI:10.1016/j.cej.2022.138066.
- Y. Xin, Q. Zhu, T. Gao, X. Li, W. Zhang, H. Wang, D. Ji, Y. Huang, M. Padervand and F. Yu, Appl. Catal., B, 2023, 324, 122238, DOI:10.1016/j.apcatb.2022.122238.
- W. An, H. Wang, T. Yang, J. Xu, Y. Wang, D. Liu, J. Hu, W. Cui and Y. Liang, Chem. Eng. J., 2023, 451, 138653, DOI:10.1016/j.cej.2022.138653.
- H. Yu, J. Huang, L. Jiang, Y. Shi, K. Yi, W. Zhang, J. Zhang, H. Chen and X. Yuan, Chem. Eng. J., 2020, 402, 126187, DOI:10.1016/j.cej.2020.126187.
- Y. Bu, H. Li, W. Yu, Y. Pan, L. Li, Y. Wang, L. Pu, J. Ding, G. Gao and B. Pan, Environ. Sci. Technol., 2021, 55, 2110–2120, DOI:10.1021/acs.est.0c07274.
- W. Jiang, H. Loh, B. Q. L. Low, H. Zhu, J. Low, J. Z. X. Heng, K. Y. Tang, Z. Li, X. J. Loh and E. Ye, Appl. Catal., B, 2022, 321, 122079, DOI:10.1016/j.apcatb.2022.122079.
- C. Guan, T. Hou, W. Nie, Q. Zhang, L. Duan and X. Zhao, J. Colloid Interface Sci., 2023, 633, 177–188, DOI:10.1016/j.jcis.2022.11.106.
- C. Liang, J. Ma, Y. Cao, T. Zhang, C. Yang, Y. Wu, H. Li, H. Xu, Y. Hua and C. Wang, Chemosphere, 2022, 304, 135320, DOI:10.1016/j.chemosphere.2022.135320.
- M. Z. Shahid, R. Mehmood, M. Athar, J. Hussain, Y. Wei and A. Khaliq, ACS Appl. Nano Mater., 2020, 4, 746–758, DOI:10.1021/acsanm.0c03042.
- H. Qin, J. Sun, D. Xia, H. Xu, Q. Yu, Y. Zheng and Y. Shi, Chem. Eng. J., 2022, 435, 134847, DOI:10.1016/j.cej.2022.134847.
- A. Agrawal, S. H. Cho, O. Zandi, S. Ghosh, R. W. Johns and D. J. Milliron, Chem. Rev., 2018, 18, 3121–3207, DOI:10.1021/acs.chemrev.7b00613.
- S. Khan, T. L. Ruwer, N. Khan, A. Köche, R. W. Lodge, H. Coelho-Júnior, R. L. Sommer, M. J. L. Santos, C. F. Malfatti and C. P. Bergmann, J. Mater. Chem. A, 2021, 9, 12214–12224, 10.1039/D0TA11494D.
- W. Ji, M.-H. Lee, L. Hao, X. Xu, S. Agathopoulos, D. Zheng and C. Fang, Inorg. Chem., 2015, 54, 1556–1562, DOI:10.1021/ic502568s.
- J. Lee, A. Seko, K. Shitara, K. Nakayama and I. Tanaka, Phys. Rev. B, 2016, 93, 115104, DOI:10.1103/PhysRevB.93.115104.
- X. Dong, L. Xu, J. Ma, Y. Li, Z. Yin, D. Chen, Q. Wang, J. Han, J. Qiu and Z. Yang, Chem. Eng. J., 2023, 459, 141557, DOI:10.1016/j.cej.2023.141557.
- Q. Yang, Q. Tian, X. Li, Y. Zhu and G. Fang, Sep. Purif. Technol., 2024, 332, 125775, DOI:10.1016/j.seppur.2023.125775.
- X. Li, Y. Sun, J. Xu, Y. Shao, J. Wu, X. Xu, Y. Pan, H. Ju, J. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690–699, DOI:10.1038/s41560-019-0431-1.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta07981c |
| This journal is © The Royal Society of Chemistry 2024 |