
2D anti-MXene boride monolayers: unveiling a promising new family of catalysts for the nitrogen reduction reaction†
Viet Q.
Bui
 *a and
Dinh Quang
Khieu
b
*a and
Dinh Quang
Khieu
b
aAdvanced Institute of Science and Technology, The University of Danang, 41 Le Duan, Danang, Vietnam. E-mail: bqviet@ac.udn.vn; mrbuiquocviet@gmail.com
bUniversity of Sciences, Hue University, 77 Nguyen Hue, Hue city, Vietnam
First published on 3rd July 2024
Abstract
This study presents a comprehensive computational analysis focusing on the electrochemical nitrogen reduction reaction (NRR) catalytic activity of two-dimensional anti-MXene borides. Employing density functional theory (DFT), we systematically assessed the dynamic, mechanical, and thermal stabilities of 23 anti-MXene boride structures, ultimately selecting five: CoB, FeB, IrB, MnB, and OsB for an in-depth investigation of their catalytic efficiency. Additionally, our study included both pristine and boron-deficient variants to elucidate their performance in catalytic processes. Our findings pinpoint FeB and OsB as prime catalysts with notably low limiting potentials of 0.30 eV and 0.48 eV, respectively, when considering solvation effects, compared to 0.32 eV and 0.64 eV without solvation effects. We also observed a reduction in kinetic barriers for the potential-determining step (PDS) with activation energies of 0.15 eV and 0.30 eV on FeB and OsB, respectively. Applied voltage calculations revealed that these materials remain efficient catalysts under operational conditions, with overpotentials of 0.41 eV for FeB and 0.73 eV for OsB. The investigation explored their electronic structures, revealing the distinctive planarity of MnB along with the buckled nature of the others, characterized by active boron sites. Additionally, the impact of boron vacancies on catalysis was examined, highlighting a trade-off between enhanced N2 adsorption and increased energy barriers. A pivotal discovery was the establishment of a linear correlation between the φ descriptor derived from the d-electron count and electronegativity of the transition metals and the limiting potential, offering predictive insights into electrocatalytic performance. This study advances the development of efficient and sustainable anti-MXene boride electrocatalysts.
1. Introduction
The ubiquitous presence of nitrogen, constituting 78% of Earth's atmosphere,1 belies the considerable technical challenge inherent in its direct conversion to ammonia, a process impeded by the formidable stability of the dinitrogen bond (940.95 kJ mol−1).2 Presently, the Haber–Bosch process, with its reliance on elevated temperatures and pressures, stands as the primary industrial methodology for ammonia synthesis.3 Despite its high efficiency, yielding 90% of global ammonia, the ammonia production process is energy-intensive, consuming 1–2% of worldwide energy, and significantly contributes to CO2 emissions, with an estimated ∼2.9 tons of CO2 per ton of NH3 produced, thus accounting for approximately 1.5% of global greenhouse gas emissions.4,5 These factors underscore the imperative for an alternative NH3 synthesis route that combines high catalytic activity with superior selectivity. The electrochemical nitrogen reduction process emerges as a promising candidate, offering direct N2 conversion and the potential for reduced energy consumption, thereby advancing sustainable ammonia production.6–9 However, the current electrochemical NRR is hampered by low efficiency and selectivity, with the hydrogen evolution reaction (HER) predominating in aqueous solutions, thereby impeding ammonia synthesis.10–14 Addressing this challenge necessitates a catalyst capable of enhancing reaction rates and selectivity.In the realm of catalyst development, single-atom transition metal catalysts have garnered attention due to their distinct advantages, such as maximal atomic efficiency and the presence of unsaturated coordination sites. These catalysts facilitate electron interactions with N2, enhancing the SAC–N bond and weakening the triple bond of N2.15 Such attributes enable the optimization of NH3 production from N2 and protons/electrons under conditions that are more benign and cost-effective compared to those demanded by bulk catalysts.16–18 Nonetheless, the performance of single-atom catalysts, particularly in comparison to their precious metal counterparts, necessitates further enhancement, with stability in long-term operation posing a significant challenge.19–22
Transitioning from the atomic scale to the two-dimensional plane, the investigation into anti-MXene borides introduces boron, a non-metal with sp2 hybridization, as a pivotal element in catalysis. The unique electronic configuration of boron enables this element to mimic the behavior of transition metals, offering both electron donation and acceptance capabilities, which are crucial for anchoring N2 molecules.23 This feature, combined with the capacity of boron to reduce hydrogen adsorption under acidic conditions, lessens the competition from the hydrogen evolution reaction and marks a new direction in the development of non-metal electrocatalysts for the NRR.24,25 The catalytic landscape has seen a growing focus on the amalgamation of boron with two-dimensional (2D) materials, spawning a plethora of research into boron-doped graphene,26,27 titanium carbide MXenes,28 and boron-functionalized graphitic carbon nitride layers like g-C3N4,29 g-CN,30 and g-C2N,31 as well as transition metal dichalcogenide (TMD) monolayers such as h-MoS2 (ref. 32 and 33) and ReS2.34 These materials have garnered attention due to their unique catalytic capabilities, notably in facilitating the fixation of nitrogen and its subsequent conversion into ammonia with remarkable selectivity. Extensive empirical evidence has underscored the efficiency of boron atoms in catalyzing the NRR, leading to significant research endeavors. This heightened interest has now extended to two-dimensional transition metal borides, known as MBenes, which capitalize on the synergistic properties of transition metals and boron. These MBenes demonstrate potential for elevated NRR activity, leveraging their extensive surface area and electrical conductivity to facilitate electrocatalytic processes.35–38 The unique attributes of MBenes, augmented by the presence of boron, offer a promising avenue for advancing NRR catalysis.
Anti-MXenes are an emerging class of two-dimensional (2D) materials that are derived from their parent MXenes by replacing the carbon or nitrogen layers with boron. This modification results in boride structures with enhanced stability, tunable electronic properties, and excellent catalytic activity. Unlike traditional MXenes, anti-MXenes offer unique advantages, including increased resistance to surface poisoning and versatility for various applications. The unique electronic configuration of anti-MXenes facilitates efficient electron transfer, making them highly suitable for electrocatalysis, particularly for the nitrogen reduction reaction (NRR). Recent studies, such as those by Jinxing et al.,39 have demonstrated the promising potential of anti-MXenes in catalytic applications. Additionally, Jiawei et al.40 have investigated three anti-MXene borides (FeB, CoB, and IrB) and highlighted their strong correlation effects and magnetic properties, further underscoring their relevance and importance in the field of materials science. In this investigation, we methodically explore the NRR catalysis capabilities of anti-MXene borides (TMBs), postulating that the encapsulation of transition metals by boron layers substantially contributes to the activation process in the NRR. We broaden our analysis to include the impact of boron vacancies (TMBv) on these monolayer structures. Employing a rigorous screening process complemented by density functional theory (DFT) calculations, we scrutinize the stability and catalytic mechanisms of both intact and boron-deficient anti-MXene borides. Our comprehensive analysis reveals that anti-MXene borides, notably FeB and OsB, demonstrate a significant reduction in the Gibbs energy barrier linked to N2 activation and conversion. Moreover, they display a pronounced suppression of the competing HER, thus establishing these materials as compelling candidates for enhanced NRR catalysis. These findings pave the way for future explorations into the tailored design of anti-MXene borides to further harness their full potential as high-efficiency NRR electrocatalysts.
2. Computational methods
The computational framework for this study was founded on spin-polarized density functional theory (DFT), utilizing the Vienna ab initio simulation package (VASP).41–43 Ion-electron interactions were delineated using projector-augmented wave (PAW) pseudopotentials,44 and the exchange–correlation functional was addressed by the generalized gradient approximation (GGA) as parameterized by Perdew, Burke, and Ernzerhof (PBE).45 The van der Waals interactions, pivotal in layered materials, were treated with Grimme's D3 correction method.46 For expanding the plane-wave functions, an energy cutoff of 450 eV was determined to be optimal. Brillouin zone sampling employed the Gaussian smearing technique with a k-mesh of 3 × 3 × 1 for larger supercells and 12 × 12 × 1 for smaller ones.Anti-MXene boride monolayers were modeled using a 3 × 3 periodic supercell, comprising 18 metal and 18 boron atoms. The two configurations, buckled and planar, were exhaustively explored across 23 different structures, as illustrated in Fig. 1a. Only structures demonstrating substantial stability were retained for further analysis. The computational conditions pertinent to the NRR electrocatalytic investigations on these stable structures are elucidated in Fig. 1b. In parallel, the catalytic potential of boron-deficient anti-MXene borides was assessed by simulating boron vacancies in all stable structures, thus designating active catalytic sites.
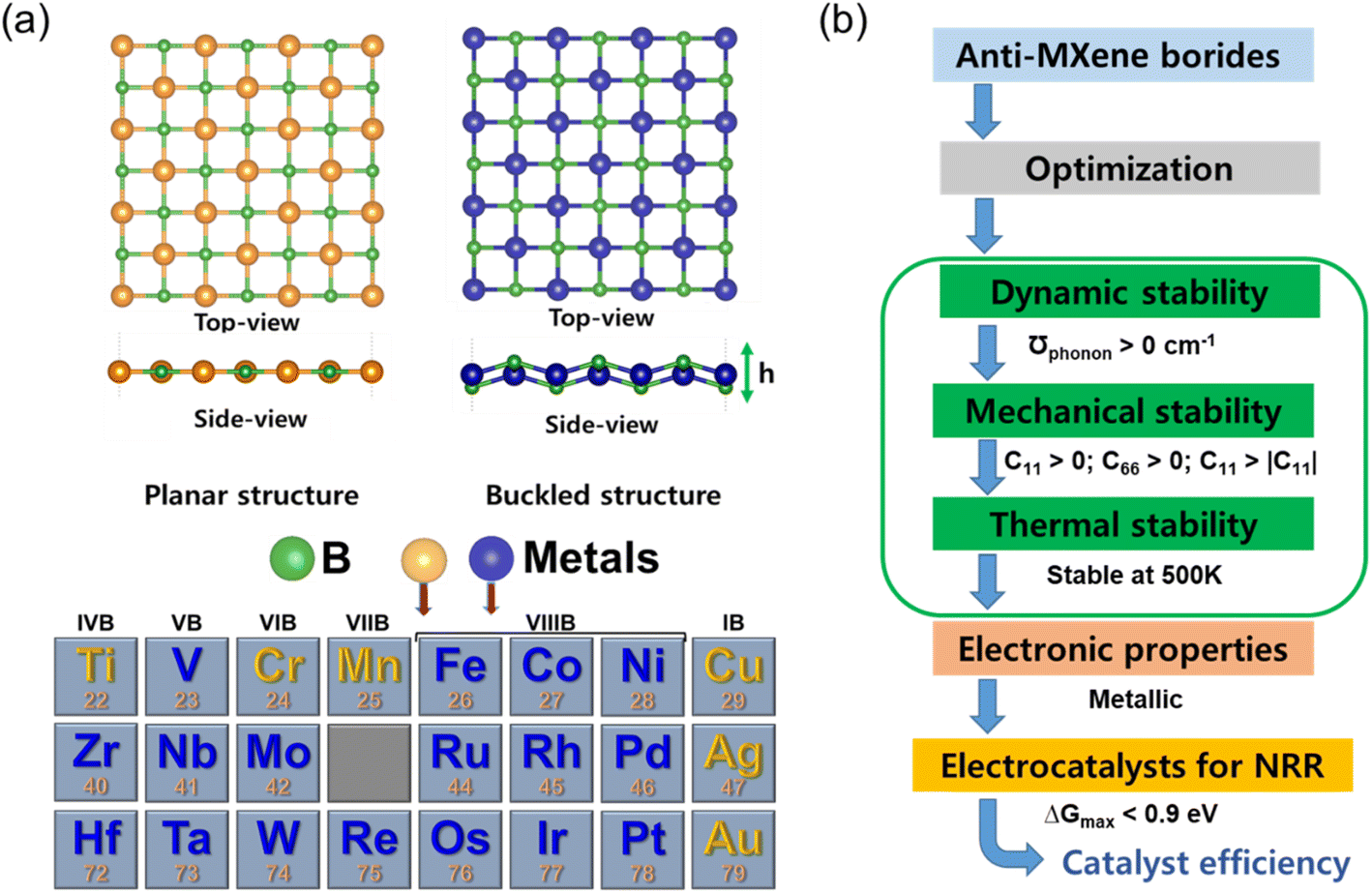 | ||
| Fig. 1 Illustration and criteria for anti-MXene borides as NRR electrocatalysts: (a) anti-MXene borides in planar and buckled topologies (top and side views) with boron (green) and transition metals (blue). (b) Evaluation flowchart for NRR suitability, including dynamic, mechanical, and thermal stability assessments, electronic characterization, and catalytic efficiency. | ||
For geometric optimization, both pristine and defective anti-MXene boride unit cells were allowed to undergo full atomic relaxation until residual forces were attenuated to below 0.01 eV Å−1. A substantial vacuum spacing of 15 Å was imposed to eliminate non-physical interactions due to periodic boundary conditions.
The cohesive energy of the anti-MXene boride monolayer within the monolayer was calculated using a primitive cell model, which includes 2 transition metal atoms and 2 boron atoms, with the equation:
 | (1) |
For thermodynamic considerations of the NRR, we employed the reaction Gibbs free energy (ΔG) framework, integrating the thermal internal energy component as proposed by Nørskov's group:47
| ΔG = ΔE + ΔEZPE − TΔS + ΔGU + ΔGpH | (2) |
Herein, ΔE is the alteration in electronic energy, ΔEZPE is the zero-point energy change, ΔS indicates the change in entropy, ΔGU = −eU reflects the electrode potential with e being the charge transferred and U is the electrode potential. ΔGpH accounts for the pH correction in the electrolyte, which in this study was considered negligible (set to zero). The vibrational entropy (Svib) was factored in, and the individual vibrational frequencies vi were ascertained using the finite difference method to compute ΔEZPE and ΔS for each NRR intermediate.
The zero-point energy was given by:a
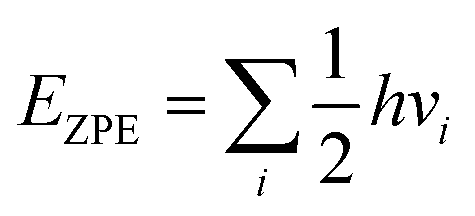 | (3) |
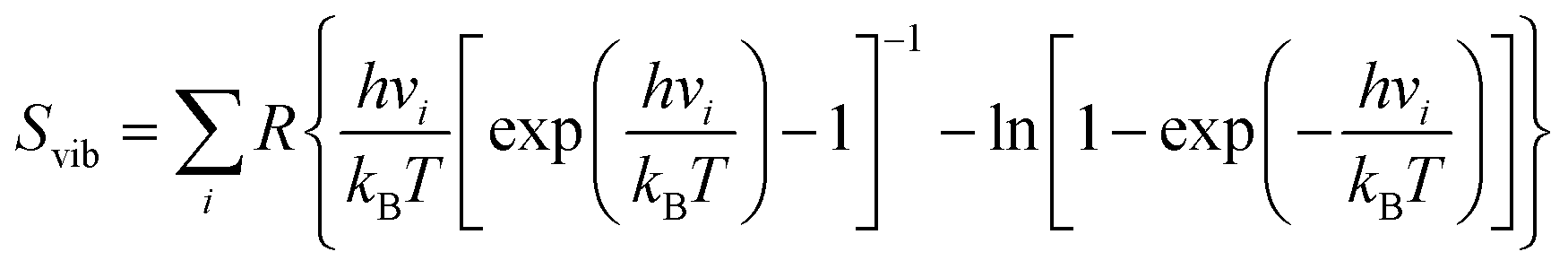 | (4) |
Constants kB and h denote the Boltzmann and Planck constants, respectively, with room temperature T and standard thermodynamic temperature R taken as 298.15 K and 8.314 J mol−1 K−1. The entropic values for gas-phase molecules were sourced from the National Institute of Standards and Technology (NIST) database. It is pertinent to acknowledge that for these calculations, only the surface adsorbed species were allowed to move dynamically during vibrational frequency calculations, while the substrate remained fixed. The overpotential (UL) was computed as UL = −ΔGmax/e to determine the necessary applied potential (U) for NRR activation, where ΔGmax represents the free energy alteration of the rate-determining step, and e denotes the electron transferred in the reaction.
3. Results and discussion
3.1. Structural models of anti-MXene borides
In the realm of two-dimensional (2D) anti-MXenes, a class of materials renowned for their extraordinary versatility and robust stability,39 our investigation embarks on a computational exploration encompassing twenty-three 2D anti-MXene borides. Each structure within this suite consists of transition metal (TM) atoms embedded between layers of boron, resulting in a square lattice configuration. This particular architecture is of significant interest due to its potential in electrocatalytic applications. Upon structural optimization, our findings reveal the existence of two distinct configurations: planar and buckled, as depicted in Fig. 1a. They are not mere morphological variations; they are integral to the nuanced electronic behaviors of these compounds. We have identified six planar structures, specifically in compounds like CuB, MnB, CrB, TiB, AgB, and AuB. These planar configurations may promote the formation of delocalized electronic states, fostering homogeneous catalytic activity across the entire surface of the material. In contrast, the buckled configurations observed in the remaining seventeen anti-MXene borides may introduce asymmetry that could prove advantageous for catalysis, allowing for spatially specific reactivity at electronically distinct active sites.The lattice dynamics detailed in Table S1† exhibit a noteworthy proportional relationship between the TM atomic electron configurations and the resultant lattice constants (a and b), the buckling height (h), and the distance between metal atoms (d). Metals with fewer electron shells trend towards planar geometries or exhibit only slight buckling (0.86–1.3 Å), as seen in group 1B metals like CuB, AgB, and AuB. These metals, characterized by a solitary electron in their outermost s orbital, are predisposed to a flat structure due to the ease with which they can ionize this electron to achieve a stable electronic state akin to that of noble gases. Conversely, metals belonging to groups 7 and 8 typically possess more compact lattice constants (primitive cell: 3.71–3.91 Å), a reflection of the orbital overlaps and interactions from the numerous electrons within their d orbitals. This interaction, particularly the potential for covalent bonding with boron atoms, yields the observable buckled structures. These findings provide an intricate perspective on the interrelation between the structural geometry and the electronic landscape of 2D anti-MXene borides, allowing for the possibility to tailor the properties of these materials through precision synthesis methods. In the context of electrocatalysis, the dynamic, mechanical, and thermal stabilities of catalysts are critical for ensuring their performance and longevity. Dynamic stability ensures resistance to vibrational disturbances, mechanical stability guarantees durability under stress, and thermal stability allows the catalyst to operate effectively at varying temperatures. Together, these stabilities confirm the robustness of the catalyst, making it suitable for long-term and industrial applications. Our comprehensive evaluation of these stabilities for anti-MXene borides ensures that the selected materials can maintain their structural integrity and catalytic efficiency under typical operational conditions. Fig. 1b outlines the critical evaluation criteria to ascertain the candidacy of anti-MXene borides for NRR electrocatalysis. This vetting process commences by verifying dynamic stability, which is indicated by the absence of imaginary phonon frequencies. It extends further to encompass meticulous assessments of mechanical and thermal stability, which are quantified by meeting predefined elastic constant thresholds and demonstrating resilience at 500 K, respectively. Additionally, the confirmation of inherent metallic properties is imperative to facilitate efficient electron transfer during catalytic processes. The critical assessment of catalytic efficiency hinges on the thermodynamic analysis, specifically evaluating the maximum Gibbs free energy change (ΔGmax) for the NRR process. To qualify as an efficient NRR catalyst, an anti-MXene boride must exhibit a ΔGmax value less than 0.9 eV, underscoring its ability to facilitate the NRR over the competing HER process.47,48 Through this multifaceted assessment strategy, only materials satisfying all criteria progress to electrocatalytic NRR testing, ensuring a focused pursuit of the most promising candidates for ammonia synthesis.
3.2. Evaluation of stabilities
In the initial step of evaluating the stability of our structures, we focus on their dynamic stability through phonon dispersion calculations, as illustrated in Fig. 2 and S1.† This analysis segregates the TMBs into two distinct categories; seven structures, namely MnB, FeB, CoB, ZrB, RhB, OsB, and IrB, exhibit dynamic stability, underscored by the absence of negative phonon frequencies, indicative of a stable and robust lattice. These findings not only affirm the structural integrity of these TMB but also earmark them for further in-depth studies, considering their potential resilience under operational conditions. In contrast, the sixteen borides represented in Fig. S1,† displaying regions of imaginary phonon frequencies, reveal a propensity for lattice instability, which precludes them from immediate consideration in the subsequent stages of catalytic property evaluation. This discernment based on dynamic stability is pivotal, as it ensures that only those structures with confirmed lattice robustness are advanced in the quest for efficient and stable electrocatalysts, thereby streamlining the focus towards those candidates with inherent structural soundness for further investigative scrutiny.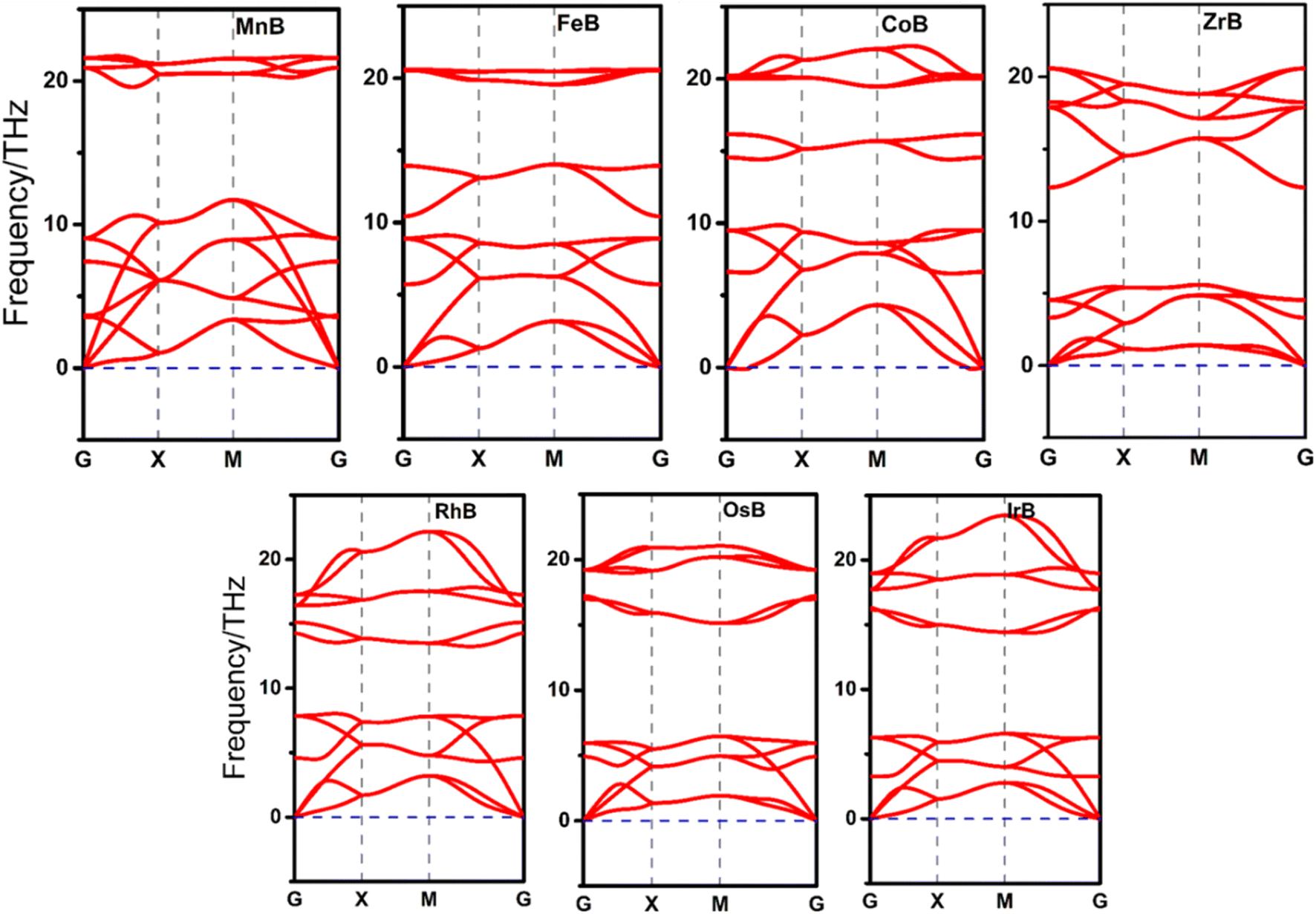 | ||
| Fig. 2 Phonon dispersion curves for seven anti-MXene boride monolayer compounds, illustrating the dynamic stability of MnB, FeB, CoB, ZrB, RhB, OsB, and IrB. The absence of imaginary frequencies (no frequencies dipping below zero, denoted by the blue dashed line) across the entire Brillouin zone (G–X–M–G path) confirms the dynamic stability of these materials. | ||
Upon advancing from dynamic to mechanical stability evaluations, we delve into the mechanical robustness of the seven dynamically stable anti-MXene borides, analyzing their elastic constants to ascertain their resistance to mechanical deformation. This analysis is pivotal, as mechanical integrity underpins the durability of a system in potential electrocatalytic applications. The mechanical stability criteria defined by the inequalities C11C22–C122 > 0 and C66 > 0 are meticulously applied to the data presented in Table 1. Our findings reveal that six TMBs, namely MnB, FeB, CoB, ZrB, IrB, and OsB, satisfactorily meet these criteria, signifying their mechanical fortitude and suggesting their capability to maintain structural integrity under mechanical stresses. However, RhB presents an anomaly with C11C22 < C122, indicating mechanical instability, which raises concerns about its viability under operational stresses, despite its dynamic stability. Further scrutiny of Table 1 allows us to delve into specific mechanical properties such as Young's modulus (Y), Poisson's ratio (ν), and shear moduli (G), enriching our understanding of the mechanical behavior of these materials. Notably, MnB demonstrates a high Young's modulus, indicating substantial stiffness and an ability to withstand significant loads. This characteristic is complemented by its low Poisson's ratio, which suggests minimal lateral deformation under stress, a hallmark of materials with high hardness and strength. However, its relatively low shear modulus (G) implies potential vulnerability to shear forces, which must be considered in its application. Conversely, CoB, with its higher Poisson's ratio, may exhibit more pronounced lateral deformation when subjected to stress, a property that could influence its behavior in certain catalytic environments. The remaining TMBs, satisfying the mechanical stability criteria and exhibiting varied mechanical properties, present a spectrum of potential applications based on their individual mechanical profiles. However, RhB is excluded from further consideration due to its mechanical instability, narrowing our focus to borides that demonstrate both dynamic and mechanical stability. This discernment in mechanical stability further refines our list of candidate materials, underscoring the importance of comprehensive stability assessments in identifying borides that are not only dynamically stable but also mechanically equipped to withstand the rigors of practical applications, thereby streamlining our focus towards those with the most promise for subsequent thermal and electronic property evaluations in the quest for efficient electrocatalysts.
| C11a (N m−1) | C12b (N m−1) | C66 (N m−1) | Y (N m−1) | ν | G (N m−1) | |
|---|---|---|---|---|---|---|
| a C22 = C11. b C21 = C12; Young's moduli Y = Yx = Yy = (C11C22–C12C21)/C11; Poisson's ration ν = C12/C11; shear moduli G = C66. | ||||||
| CoB | 73.683 | 55.521 | 13.184 | 31.847 | 0.754 | 13.184 |
| FeB | 39.442 | 14.516 | 10.608 | 34.100 | 0.368 | 10.608 |
| MnB | 178.258 | 14.622 | 6.300 | 177.059 | 0.082 | 6.300 |
| RhB | 70.362 | 81.278 | 16.123 | −23.526 | 1.155 | 16.123 |
| ZrB | 68.879 | 6.417 | 6.229 | 68.281 | 0.093 | 6.229 |
| IrB | 130.445 | 97.204 | 19.491 | 58.011 | 0.745 | 19.491 |
| OsB | 96.865 | 87.276 | 12.236 | 18.229 | 0.901 | 12.236 |
The cohesive energy calculations, presented in Table S2,† provide crucial insights into their thermodynamic stability. These materials possess more negative cohesive energies compared to some experimentally synthesized two-dimensional materials, such as borophene,49 silicene,50 and phosphorene,51 suggesting a superior level of stability for these newly proposed structures. Particularly, IrB and OsB stand out with their cohesive energies at −6.75 eV and −6.72 eV, respectively, which are significantly more negative than that of borophene at −5.87 eV. These findings suggest a higher degree of binding within the crystal lattices of these materials, indicating their potential for exceptional stability upon synthesis. Notably, ZrB, CoB, and FeB exhibit promising signs of robustness, boasting cohesive energies of −5.56 eV, −5.41 eV, and −4.97 eV, respectively, surpassing the stability benchmarks set by silicene and phosphorene. Moreover, despite possessing the least negative cohesive energy among the investigated borides at −4.50 eV, MnB still aligns with the stability profiles observed in these well-known two-dimensional materials. The observation that MnB is the only structure among the six stable borides to adopt a planar configuration, while the rest exhibit a buckled configuration, could imply that for most elements explored, a buckled configuration might be energetically more favorable. This could be due to the extra degree of freedom it provides for strain relief in the lattice or to more optimal orbital overlap that enhances bonding. This trend toward buckling among the stable borides could indicate a fundamental stabilizing factor intrinsic to their structural makeup. It hints that these new classes of anti-MXene borides might not only achieve stability upon synthesis but could also offer superior properties compared to existing two-dimensional materials. Their strong binding energies could translate into unique and valuable electronic, mechanical, or chemical properties for advanced applications in nanotechnology and materials science.
The electron localization function (ELF) plots shown in Fig. S2† for the six stable TMBs reveal a notable concentration of electrons around the boron atoms. This concentration suggests that there is a high electron density near these atoms and indicates that the metal–boron bonds within these structures possess a significant ionic character. The ELF isosurface is set at a value that enables the visualization of areas where electrons are significantly localized; in this case, the visualization shows that the boron atoms are likely negatively charged. This negative charge suggests that electrons are transferred from the metal atoms to the boron atoms, which is a hallmark of ionic bonding. Ionic bonds are known for their strong electrostatic attractions between positively and negatively charged ions, contributing to the stability of the material. The clear presence of ionic bonding characteristics in the metal–boron bonds as demonstrated by the ELF plots supports the high level of stability indicated by the negative cohesive energy values of these materials, pointing to their ability to maintain structural integrity. This robustness is further evidenced by Fig. S3 and S4† provides snapshots from ab initio molecular dynamics (AIMD) simulations52 of the six anti-MXene borides at 500 K over 5 ps, showcasing their thermal stability. These simulations reveal that, even at elevated temperatures, each anti-MXene boride preserves its geometric configuration without any signs of structural breakdown or significant atomic shifts, attesting to their structural resilience. Such thermal stability is a valuable attribute for high-temperature applications. Consequently, the AIMD results not only reinforce the mechanical and dynamic robustness of these materials but also amplify their potential for practical deployment in environments where thermal resistance is essential.
3.3. Electronic structure and catalytic performance for the NRR
Building upon the cohesive and thermal stability profiles established for the anti-MXene borides, we delve into the intricacies of their electronic structures, pivotal for assessing their catalytic performance. Fig. 3 expertly exhibits the band structures of these stable anti-MXene borides, underscoring the intrinsic metallic nature that underpins their suitability as catalysts in electrochemical processes like the NRR. These band structures highlight bands crossing the Fermi level, indicative of metallic behavior, which is crucial for the mobility and transfer of electrons—key attributes for effective catalysis. Notably, FeB and MnB demonstrate spin polarization, marked by the clear separation of spin-up and spin-down states, alluding to their magnetic properties. This unique feature adds complexity to their electronic structures, which could play a significant role in influencing their catalytic properties. While the ramifications of this spin polarization for catalysis are yet to be fully deciphered within this study, it signals potential avenues for these materials to facilitate or enhance catalytic reactions where spin phenomena are influential.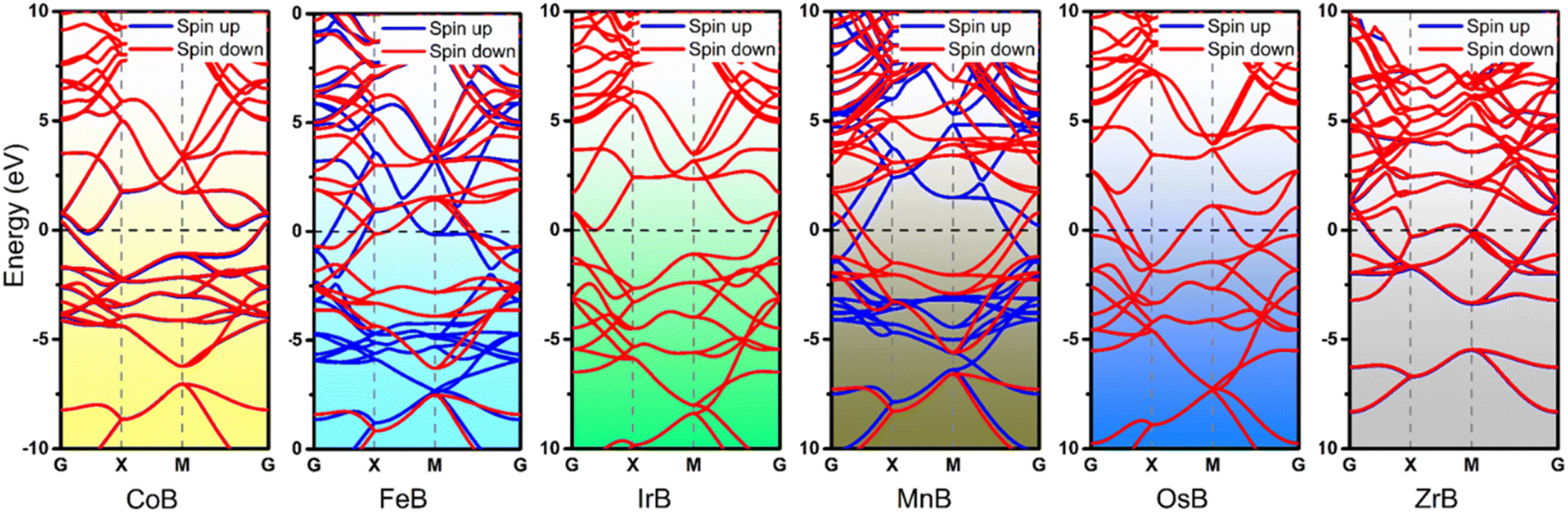 | ||
| Fig. 3 Band structures of six dynamically stable anti-MXene boride monolayers (CoB, FeB, IrB, MnB, OsB, and ZrB) depicting spin-resolved electronic states. The Fermi level is delineated by the dashed horizontal line at zero energy, with spin-up and spin-down states represented by red and blue lines, respectively. The presence of bands crossing the Fermi level indicates metallic behavior in these materials. | ||
In the analysis of NRR pathways on anti-MXene borides, a pivotal stage involves the adsorption and activation of N2, exerting a direct impact on their catalytic efficiency. ZrB, characterized by structural compromises observed during the NRR, has consequently been excluded from subsequent catalytic evaluations. Further investigations have revealed that end-on adsorption exhibits greater stability. MnB showcases active sites located on manganese (Mn) atoms, while the buckled structures of CoB, FeB, IrB, and OsB feature active boron sites, indicative of heightened electron mobility. This observation bridges to the charge density analyses, presented in Fig. 4b and S5,† which unveil a distinct charge transfer from the B and Mn atoms to the adsorbed nitrogen molecules. Such a transfer, indicative of a “σ donation – π backdonation” interaction, involves a dual process of electron exchange crucial for catalytic activation. At both B and Mn sites, the σ donation from nitrogen and the π backdonation to its antibonding orbitals facilitate nitrogen molecule activation, an essential step for effective nitrogen fixation. Simultaneously observed charge accumulation and depletion corroborate this synergistic mechanism, highlighting the potential for boron atoms within the anti-MXene boride structures to act in a manner similar to transition metals and to effectively participate in the NRR. Visualized through Fig. 4a and c, these insights into the electronic interactions at the active sites further accentuate the role of boron atoms in these innovative anti-MXene structures, underscoring their capability to facilitate the NRR.
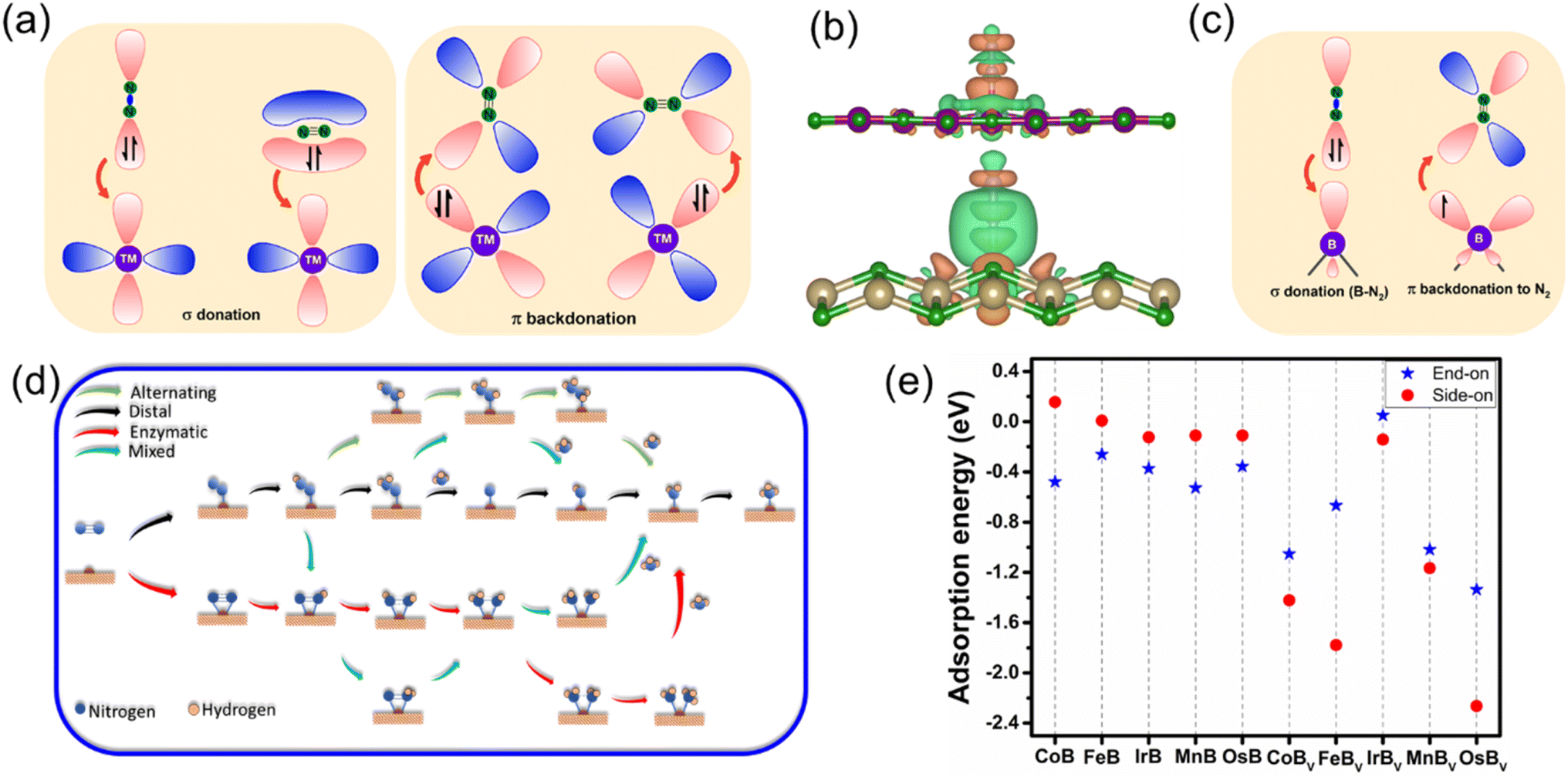 | ||
| Fig. 4 Mechanistic insights and adsorption profiles for N2 on anti-MXene boride surfaces: (a) illustration of σ donation and π backdonation between N2 and transition metal (TM) atoms. (b) Side and top views of N2 adsorption on buckled and planar anti-MXene boride surfaces, showcasing end-on and side-on configurations. (c) Orbital interaction diagrams for σ donation from B–N and π backdonation to N2 on a boron site. (d) Schematic overview of potential N2 reduction reaction mechanisms on anti-MXene boride surfaces. (e) Comparative adsorption energies of N2 on various anti-MXene boride monolayers, highlighting the energetics of end-on versus side-on binding configurations. | ||
Our study on the NRR expands to clarify the catalytic processes in five stable anti-MXene borides, including their variants with boron vacancies (TMBv). The two primary nitrogen adsorption configurations, end-on and side-on, on the active sites are explored in depth; these sites are boron atoms in the pristine anti-MXene borides and vacancies in the defective counterparts. Fig. 4d captures the atomic configurations of NRR intermediates along the reaction routes that transform diatomic nitrogen into ammonia through sequential proton and electron additions. In these illustrations, NRR pathways in the end-on mode involve alternating or distal pathways, whereas the side-on mode aligns with processes that resemble mixed or enzymatic actions, demonstrating the multifaceted nature of the NRR mechanisms on these catalytic surfaces.
Delving into the energetics of the process, Fig. 4e presents the adsorption energies of N2 on the catalysts, revealing a pronounced preference for the end-on adsorption mode at the active boron sites in the four stable, buckled anti-MXene borides, as well as at the active manganese site in the planar MnB. This preference suggests that the end-on mode is energetically more favorable, facilitating stronger interactions with the catalyst and more efficient activation of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond. Conversely, the side-on mode is characterized by relatively lower adsorption energy, which might impede effective N2 activation due to weaker interaction strengths. To gain deep insights into the B–N2 interaction quantitatively, we performed integrated-crystal orbital Hamilton population (ICOHP) analysis53 by integrating the band states up to the highest occupied energy level. Fig. S6† demonstrates the interplay between the electronic structure and catalytic adsorption in anti-MXene borides, excluding MnB due to its distinct orbital characteristics and local geometric environment that affect N2 adsorption energy differently. For CoB, FeB, IrB, and OsB, the iCOHP values for B–N2 bonds, when plotted against the energy levels relative to the Fermi level, showcase a clear trend: more negative iCOHP values are associated with stronger B–N2 bonding. This trend correlates with more favorable N2 end-on adsorption energies, as depicted in Fig. S6b.† This consistent correlation across the anti-MXene borides suggests that stronger B–N2 interactions improve the adsorption process. Notably, FeB exhibits the least negative iCOHP and, consequently, the weakest N2 adsorption energy among the anti-MXene borides, a balance that is paradoxically conducive to NRR catalysis. This moderate bond strength between boron and nitrogen facilitates a balanced adsorption of *N2 and maintains a low Gibbs free energy barrier for the reaction *N2 + H+ + e− → *N2H, favorably positioning FeB for effective NRR catalytic processes.
N bond. Conversely, the side-on mode is characterized by relatively lower adsorption energy, which might impede effective N2 activation due to weaker interaction strengths. To gain deep insights into the B–N2 interaction quantitatively, we performed integrated-crystal orbital Hamilton population (ICOHP) analysis53 by integrating the band states up to the highest occupied energy level. Fig. S6† demonstrates the interplay between the electronic structure and catalytic adsorption in anti-MXene borides, excluding MnB due to its distinct orbital characteristics and local geometric environment that affect N2 adsorption energy differently. For CoB, FeB, IrB, and OsB, the iCOHP values for B–N2 bonds, when plotted against the energy levels relative to the Fermi level, showcase a clear trend: more negative iCOHP values are associated with stronger B–N2 bonding. This trend correlates with more favorable N2 end-on adsorption energies, as depicted in Fig. S6b.† This consistent correlation across the anti-MXene borides suggests that stronger B–N2 interactions improve the adsorption process. Notably, FeB exhibits the least negative iCOHP and, consequently, the weakest N2 adsorption energy among the anti-MXene borides, a balance that is paradoxically conducive to NRR catalysis. This moderate bond strength between boron and nitrogen facilitates a balanced adsorption of *N2 and maintains a low Gibbs free energy barrier for the reaction *N2 + H+ + e− → *N2H, favorably positioning FeB for effective NRR catalytic processes.
Interestingly, the boron-vacancy sites in TMBv structures demonstrate a marked preference for side-on adsorption, where the N2 molecule forms more robust bonds with the neighboring metal atoms, an insight that could be instrumental in tailoring catalysts for improved performance. Our investigation, therefore, bifurcates to probe the NRR on pristine anti-MXene borides via distal and alternating pathways, while the defective structures with B-vacancies are explored through mixed and enzymatic pathways. This approach not only accommodates the inherent adsorption preferences but also opens new avenues to understand the catalytic behavior of these advanced materials, potentially leading to the optimization of catalytic strategies for effective ammonia synthesis.
In our comprehensive examination of the Gibbs free energy diagrams for the NRR on five pristine anti-MXene borides, CoB, FeB, IrB, MnB, and OsB, as depicted in Fig. 5a–e, the diagrams reveal the intricate energetic landscape of the catalytic process. Fig. S7† provides the detailed structures of the reaction intermediates. The initial hydrogenation step, converting N2* to NNH*, varies energetically across the anti-MXene borides, with changes in Gibbs free energy of 0.79, 0.32, 1.18, 0.84, and 0.64 eV, respectively, for CoB, FeB, IrB, MnB, and OsB. This step is vital as it opens the path for the subsequent reaction where a proton and an electron combine with N2H* to form either the NHNH* or NNH2* intermediate.
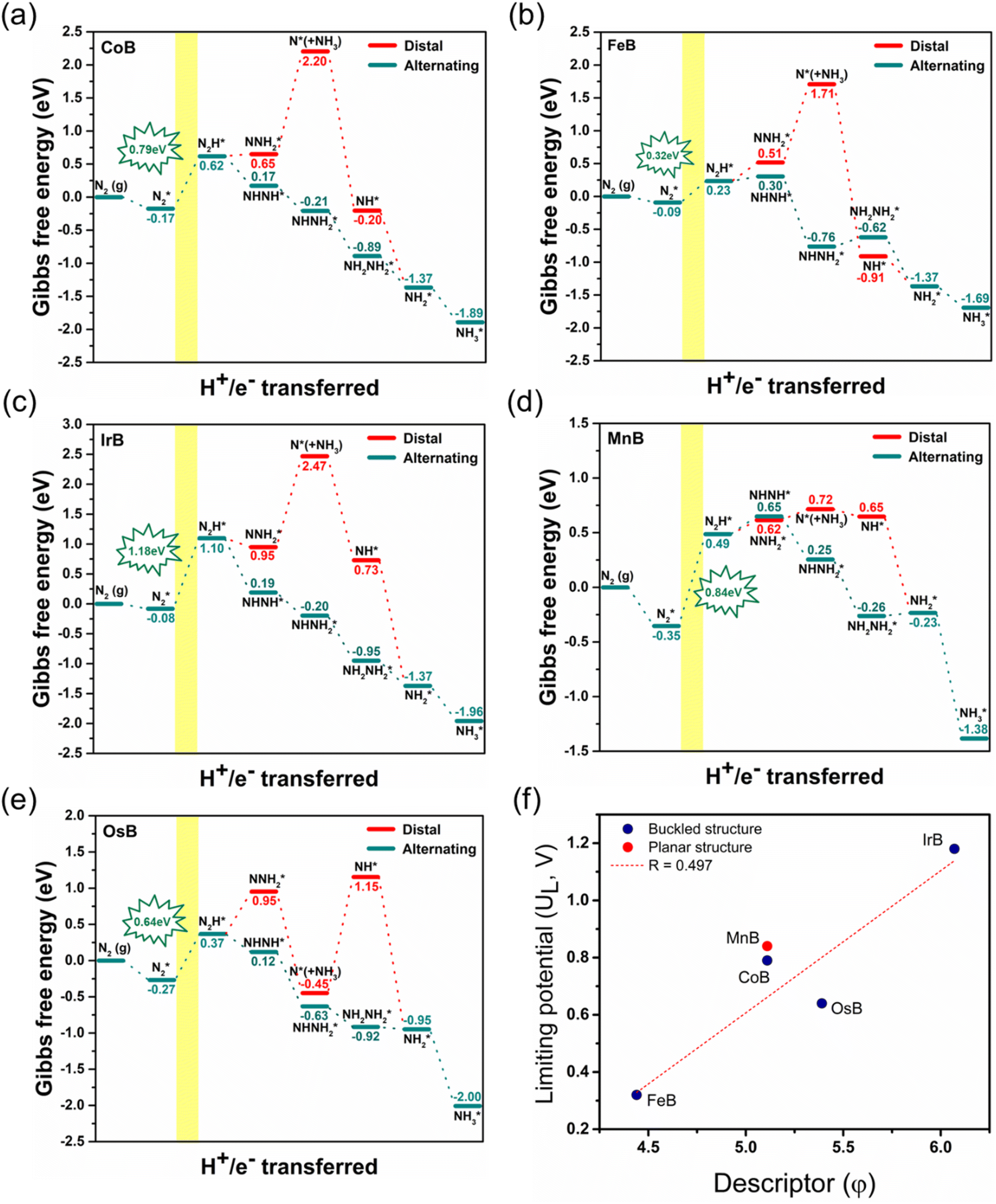 | ||
| Fig. 5 Gibbs free energy diagrams for N2 to NH3 electroreduction on (a) CoB, (b) FeB, (c) IrB, (d) MnB, and (e) OsB monolayers, with distal (solid red) and alternating (dashed red) pathways and their energy values. The optimal NH3 production windows are highlighted in yellow. Panel (f) correlates the calculated descriptor φ with the limiting potential UL for each catalyst, indicating a predictive relationship for their electrocatalytic performance. | ||
Comparing these critical intermediates reveals an energy preference where buckled structures, including CoB, FeB, IrB, and OsB, show thermodynamic favorability towards forming NHNH* at the active boron site, indicating an advantageous pathway for NRR catalysis. Conversely, MnB, with its active manganese site, displays a preference for forming NNH2*. Despite these individual variations, the overall change in Gibbs free energy points to a tendency for the NRR on these borides to favor the alternating pathway, which appears consistently advantageous. Following the initial step, a series of exothermic protonation events lead to the formation of NH3*, with the sequence including NHNH converting to NHNH2 and then to NH* (releasing NH3), proceeding to NH2*, and culminating in NH3*. The first reaction step, creating N2H*, stands out as the potential-determining step (PDS) and critically determines the kinetics and pathway of the reaction. The optimal limiting potentials (UL), which equate to 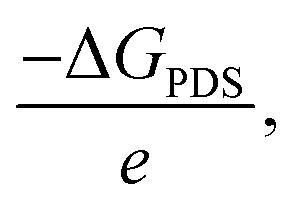 highlight the electrochemical potential needed to surpass the PDS, as illustrated in Fig. S8.† For FeB and OsB, these potentials are −0.32 and −0.64 V, respectively, indicating that applying these potentials sets favorable conditions for NH3 production through the alternating pathway by lowering the energy barrier at the PDS. Furthermore, calculating the overpotentials (η) for each material, where η = Uequilibrium − UPDS, provides further insights into their catalytic efficiency. With Uequilibrium set at +0.09 V, the overpotentials for CoB, FeB, IrB, MnB, and OsB are found to be 0.88, 0.41, 1.27, 0.93, and 0.73 V, respectively. These results confirm that FeB and OsB exhibit the lowest overpotentials, 0.41 V and 0.73 V, respectively, highlighting their superior catalytic efficiency for the NRR. The relatively low energy barriers for the rate-determining step (RDS) in FeB and OsB make these materials particularly promising for ammonia synthesis. This detailed analysis underscores the critical role of overpotential in evaluating and comparing the electrocatalytic performance of the studied anti-MXene borides. In contrast to the NRR occurring under strongly alkaline or high-temperature conditions, where the desorption of NH3* often constitutes the rate-limiting step, electrocatalytic environments, particularly under acidic or neutral conditions, exhibit a distinct behavior. In such cases, the final protonation to form NH4+ from NH3* is not a limiting factor.54 This smooth progression of the last protonation step, without the need for significant overpotential, suggests that catalytic efficiency analyses should primarily focus on the initial, more energetically challenging steps of the reaction pathway, which are critical for the overall efficiency of the NRR on these anti-MXene borides. Moreover, we also examined the kinetics of proton transfer in the potential-determining step at 0.0 V versus the standard hydrogen electrode (SHE), which are are remarkably low with values of 0.15 eV on the FeB catalyst and 0.30 eV on the OsB catalyst. These small barriers suggest that the reduction of NN* to NNH* can be easily surmounted at room temperature or further diminished at more negative applied voltages. In Fig. S9,† the optimized structures and related kinetic barriers for the reduction of NN* to NNH* on FeB and OsB surfaces are depicted. The transition state (TS) structures illustrate the bond distances and activation energies involved in the reaction. For FeB, the transition state occurs with an NN* to NNH* conversion at an activation energy of 0.15 eV, facilitated by the interaction with the solvated proton donor H5O2+. Similarly, on OsB, the activation energy for the same conversion is 0.30 eV. These results underscore the high catalytic efficiency of FeB and OsB, demonstrating their capability to facilitate the NRR with minimal energy input, thereby confirming their potential as efficient electrocatalysts.
highlight the electrochemical potential needed to surpass the PDS, as illustrated in Fig. S8.† For FeB and OsB, these potentials are −0.32 and −0.64 V, respectively, indicating that applying these potentials sets favorable conditions for NH3 production through the alternating pathway by lowering the energy barrier at the PDS. Furthermore, calculating the overpotentials (η) for each material, where η = Uequilibrium − UPDS, provides further insights into their catalytic efficiency. With Uequilibrium set at +0.09 V, the overpotentials for CoB, FeB, IrB, MnB, and OsB are found to be 0.88, 0.41, 1.27, 0.93, and 0.73 V, respectively. These results confirm that FeB and OsB exhibit the lowest overpotentials, 0.41 V and 0.73 V, respectively, highlighting their superior catalytic efficiency for the NRR. The relatively low energy barriers for the rate-determining step (RDS) in FeB and OsB make these materials particularly promising for ammonia synthesis. This detailed analysis underscores the critical role of overpotential in evaluating and comparing the electrocatalytic performance of the studied anti-MXene borides. In contrast to the NRR occurring under strongly alkaline or high-temperature conditions, where the desorption of NH3* often constitutes the rate-limiting step, electrocatalytic environments, particularly under acidic or neutral conditions, exhibit a distinct behavior. In such cases, the final protonation to form NH4+ from NH3* is not a limiting factor.54 This smooth progression of the last protonation step, without the need for significant overpotential, suggests that catalytic efficiency analyses should primarily focus on the initial, more energetically challenging steps of the reaction pathway, which are critical for the overall efficiency of the NRR on these anti-MXene borides. Moreover, we also examined the kinetics of proton transfer in the potential-determining step at 0.0 V versus the standard hydrogen electrode (SHE), which are are remarkably low with values of 0.15 eV on the FeB catalyst and 0.30 eV on the OsB catalyst. These small barriers suggest that the reduction of NN* to NNH* can be easily surmounted at room temperature or further diminished at more negative applied voltages. In Fig. S9,† the optimized structures and related kinetic barriers for the reduction of NN* to NNH* on FeB and OsB surfaces are depicted. The transition state (TS) structures illustrate the bond distances and activation energies involved in the reaction. For FeB, the transition state occurs with an NN* to NNH* conversion at an activation energy of 0.15 eV, facilitated by the interaction with the solvated proton donor H5O2+. Similarly, on OsB, the activation energy for the same conversion is 0.30 eV. These results underscore the high catalytic efficiency of FeB and OsB, demonstrating their capability to facilitate the NRR with minimal energy input, thereby confirming their potential as efficient electrocatalysts.
Fig. 5f displays the relationship between the descriptor φ and the limiting potential (UL) for CoB, FeB, IrB, and OsB. This analysis deliberately omits MnB due to its unique configuration and active site properties. The descriptor φ, calculated as the ratio of the d-electron count (Nd) to the square root of electronegativity 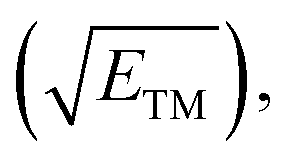 not only sheds light on the indirect influence of the electronic structures on the NRR efficiency of metals but also implies a significant link to the catalytic process. The linear correlation captured in Fig. 5f between φ and the limiting potential (UL) suggests that while the transition metals are not the active sites for the NRR, their electronic characteristics significantly impact the catalytic process. The trend indicates that an increase in φ correlates with an increase in UL, signifying that certain electronic configurations of the transition metals may indirectly influence the ease with which the NRR progresses at the boron sites. This influence is likely due to the modulation of the electronic environment of the boron atoms by the adjacent transition metals, which could affect the adsorption energy and the activation barrier for nitrogen reduction. Descriptor φ serves as an insightful descriptor that captures the subtle interplay between the electronic attributes of transition metals and NRR performance at boron sites. It underscores the indirect but notable role that the electronic structure of transition metals can play in the overall catalytic efficiency, providing a quantitative measure that could guide the design and optimization of boride-based catalysts.
not only sheds light on the indirect influence of the electronic structures on the NRR efficiency of metals but also implies a significant link to the catalytic process. The linear correlation captured in Fig. 5f between φ and the limiting potential (UL) suggests that while the transition metals are not the active sites for the NRR, their electronic characteristics significantly impact the catalytic process. The trend indicates that an increase in φ correlates with an increase in UL, signifying that certain electronic configurations of the transition metals may indirectly influence the ease with which the NRR progresses at the boron sites. This influence is likely due to the modulation of the electronic environment of the boron atoms by the adjacent transition metals, which could affect the adsorption energy and the activation barrier for nitrogen reduction. Descriptor φ serves as an insightful descriptor that captures the subtle interplay between the electronic attributes of transition metals and NRR performance at boron sites. It underscores the indirect but notable role that the electronic structure of transition metals can play in the overall catalytic efficiency, providing a quantitative measure that could guide the design and optimization of boride-based catalysts.
The inclusion of solvation effects in our computational study was critical for providing a more realistic simulation of the electrochemical environment. By utilizing the implicit solvation model implemented in VASPsol, we were able to account for the dielectric properties of the solvent, which significantly influence the electrochemical nitrogen reduction reaction (NRR) processes. The Gibbs free energy diagrams for the NRR on CoB, FeB, IrB, MnB, and OsB monolayers, now including solvation effects, are illustrated in Fig. S10.† These diagrams provide a comprehensive view of the energy changes during the stepwise conversion of N2 to NH3. Notably, except for CoB, the potential-determining steps (PDSs) in the presence of solvation effects exhibit lower energy barriers compared to calculations without considering solvation. Specifically, the PDSs for FeB and OsB are significantly reduced in the presence of solvent effects, with values of 0.30 eV and 0.48 eV, respectively. This reduction is substantial when compared to the corresponding PDS values of 0.32 eV for FeB and 0.64 eV for OsB without solvation effects. The reduction in the PDS values indicates that the solvent environment facilitates the electrochemical reaction by lowering the energy barriers associated with the rate-limiting steps. For FeB, the solvation effects reduce the Gibbs free energy change for the N2* to NNH* transition, which is the PDS, from 0.32 eV to 0.30 eV. Similarly, for OsB, the energy barrier is reduced from 0.64 eV to 0.48 eV. This reduction in the PDS highlights the critical role of the solvent in enhancing the catalytic efficiency of these materials. The solvation environment stabilizes the intermediates and transition states, thereby facilitating smoother electron transfer and protonation steps.
We extend our investigation to the reaction pathways of boron-deficient systems as shown in Fig. 6. The study reveals that the creation of a boron vacancy leads to a side-on adsorption configuration for N2, characterized by each nitrogen atom bonding strongly with a pair of adjacent transition metals. This is attributable to the increased number of available bonding sites around the vacancy, which permits a bidentate mode of attachment for the N2 molecule, a configuration that is bolstered by the overlap of d-orbitals from the surrounding metals with the π* orbitals of the nitrogen molecule. This bonding arrangement provides a robust and distributed interaction, potentially enhancing the stabilization of the adsorbed N2 and offering a configuration that may favor catalysis. The side-on bonded intermediate states facilitate NN bond activation through electron donation from the metal atoms to the antibonding orbitals of the nitrogen molecule, weakening the NN bond and promoting its reduction. These distinctive interactions at the boron vacancy sites alter the reaction pathway and energy profile for the NRR, evidenced by the Gibbs free energy changes and rate-determining steps specific to the boron-deficient systems. Such side-on bonded intermediates, formed between each nitrogen atom and a pair of adjacent metals, are integral to the catalytic properties of these structures. The Gibbs free energy diagrams highlight that the PDSs for these systems are substantially high. Specifically, the rate-determining steps for CoBv, FeBv, MnBv, and OsBv are identified as transitions from NNH2* to NHNH2* (1.25 eV), NH2* to NH3* (1.66 eV), NHNH* to NHNH2* (0.84 eV), and NH2NH3* to NH2* (1.51 eV), respectively. The instability in certain NRR intermediate adsorption configurations leads to the exclusion of IrBv from effective catalytic candidates. This result implies that although boron vacancies can enhance initial N2 binding, the increased strength of this bond may become counterproductive, resulting in higher energy barriers for the transformation of intermediates and potentially trapping them, impeding the overall NRR efficiency. This strong binding, while impeding transformation, elucidates the relative inefficiency of TMBv systems in catalyzing the NRR, highlighting the necessity for substantial energy input to progress beyond the rate-determining steps. In summary, among the ten studied structures, both pristine and boron-deficient FeB and OsB stand out due to their superior NRR activity. This study highlights the exceptional catalytic potential of these two structures, driven by their low limiting potentials of 0.32 eV for FeB and 0.64 eV for OsB, making them promising candidates for sustainable ammonia synthesis, as indicated in Table S3.† This underlines the importance of fine-tuning structural features to harness the most efficient catalysis pathways in these advanced materials.
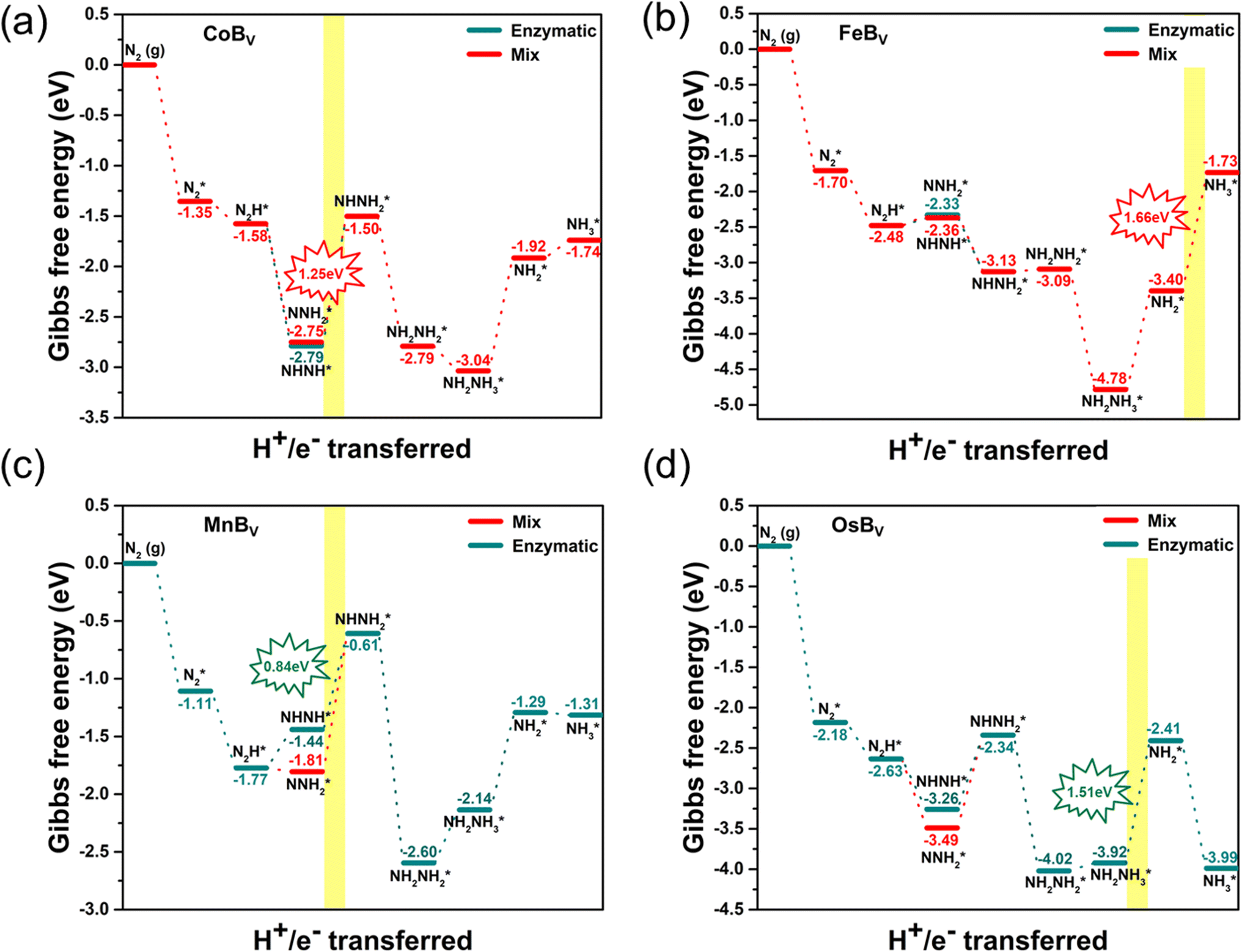 | ||
| Fig. 6 Gibbs free energy diagrams for N2 reduction on boron-deficient anti-MXenes: (a) CoBv, (b) FeBv, (c) MnBv, and (d) OsBv. The diagrams illustrate the enzymatic (dotted blue lines) and mixed (solid red lines) mechanisms. Key reaction intermediates are marked, and the optimal electrochemical window for NH3 synthesis is highlighted in yellow. The absence of the IrBv pathway is due to the disruption of intermediate adsorption, precluding NRR activity. | ||
Fig. 7 shows a nuanced Bader charge analysis across the reaction coordinates for the NRR pathway, providing insight into the intricate electron transfer dynamics that are paramount for assessing the catalytic efficiency of different systems. This analysis divides the reaction intermediates into three charge-defined categories: the transition metal boride substrate without active boron (sub-TMB), the active boron center (B), and the adsorbed nitrogen–hydrogen species (NxHy). In the charge transfer analysis of CoB, FeB, IrB, and OsB, the intricate patterns of electron movement post-initial N2 adsorption are revealed, showcasing the dynamic electron exchange within these systems. The active boron site is observed to be a hive of activity, orchestrating charge transfers to both the adsorbed N2* and the substrate itself. FeB emerges as distinctive, with its substrate actively donating electrons to N2* (0.11|e|), in stark contrast to other systems where the substrate tends to accrue electrons from the active boron site, as indicated by the charge disparities of −0.37, −0.15, and −1.01|e| for CoB, IrB, and OsB, respectively. Remarkably, the boron center in FeB exhibits the most pronounced electron donation between the N2* and NNH* states (0.23|e|), with sub-FeB contributing the lowest electron donation during this transition phase (0.1|e|). This contrasts sharply with the electron transfer from the active B centers and sub-TMB between the N2* and NNH* states in CoB, IrB, and OsB, which are (−0.03, 0.51|e|), (0.14, 0.15|e|), and (−0.56, 0.55|e|), respectively. Such unique electron dynamics could reinforce the superior catalytic activity observed in FeB, particularly in reducing the energy barrier for the N2* → NNH* reaction step, identified as the rate-determining step in the NRR. This suggests that the finely tuned electronic interaction at the active site and substrate plays a crucial role in optimizing electron transfer for efficient NRR. Throughout the NRR, except for the NH3* state, sub-FeB serves as an electron donor, providing charges in the range of 0.11 to 0.3|e| during various intermediate stages. Meanwhile, the B unit functions as the active site for N2 reduction, facilitating the electron transfer between NxHy* intermediates and the TMB substrate. The adsorption state of NH3* exhibits a reverse trend, where it imparts electrons back to the substrate. The higher proportion of hydrogen atoms bonded to nitrogen creates an electron-deficient state for nitrogen, as electrons are held closer to the more electronegative nitrogen, resulting in a tendency for NH3 to donate electrons to the substrate to achieve stability.
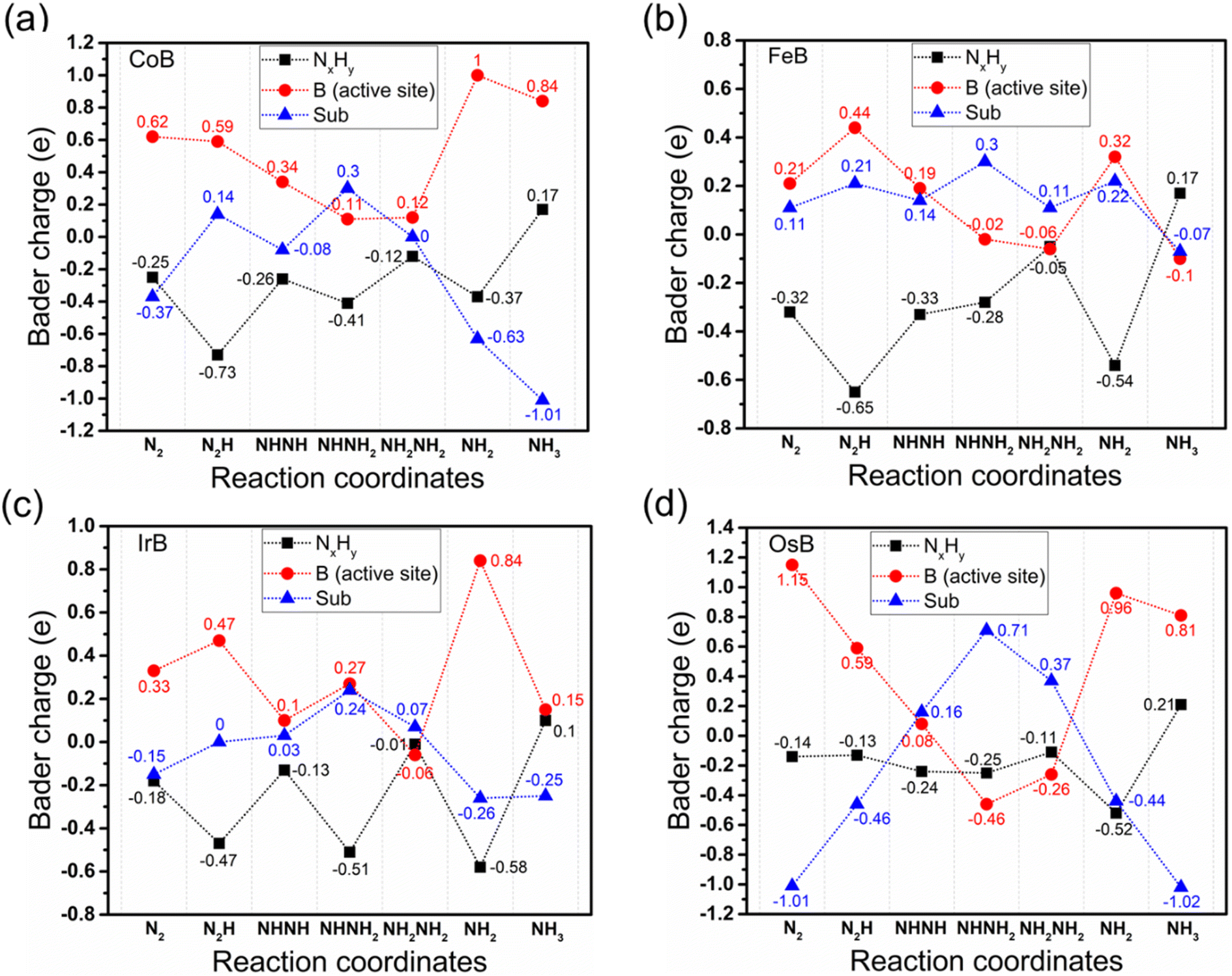 | ||
| Fig. 7 Bader charge analysis along the reaction coordinates for the alternating NRR pathway on (a) CoB, (b) FeB, (c) IrB and (d) OsB. The plots trace the charge variations for NHx intermediates (black dotted lines), the active boron site (red solid lines), and the substrate (blue dashed lines), elucidating the electron transfer dynamics during the catalytic process. | ||
Since the competition between the HER and the NRR inherently influences the efficiency of a catalyst, particular attention must be devoted to the HER tendencies of these transition metal borides. The active sites are prone to H adsorption, which can obstruct the NRR pathway; hence, averting hydrogen adsorption is vital for enhancing the NRR selectivity. In the crucial phase of selectivity determination, we assess the propensity for the NRR based on the comparative adsorption energies of N2* and H*. As shown in Fig. 8, the selectivity plot for anti-MXene borides, both pristine and with boron vacancies, acts as a critical instrument in assessing the tendency of these catalysts to favor the NRR over the HER. This plot is instrumental in discerning the selectivity of these structures, as the active sites are susceptible to occupation by hydrogen, which can hinder the NRR process. In this energetic landscape, the positions of the data points elucidate the competition between hydrogen and nitrogen adsorption; the closer a point is to the bottom-left corner, the stronger the predisposition of the material for facilitating the NRR over the HER. Conversely, data points skewing towards the top-right corner signal a tendency to favor the HER, a less desirable trait when aiming to maximize NRR efficiency. The selectivity plot reveals that, barring IrBv which leans towards the HER, the remaining borides display a marked inclination for the NRR. In particular, for FeB and OsB, the adsorption energy differences between H* and N2* highlight a subtle preference for NRR. FeB exhibits an N2* adsorption energy of −0.26 eV, slightly lower than that of H* at −0.21 eV, indicating marginal selectivity for nitrogen. Meanwhile, OsB presents a stark contrast with adsorption energies at 0.00 eV for H* and −0.36 eV for N2*, suggesting a stronger preference for nitrogen, thus positioning these structures as promising candidates for facilitating the NRR.
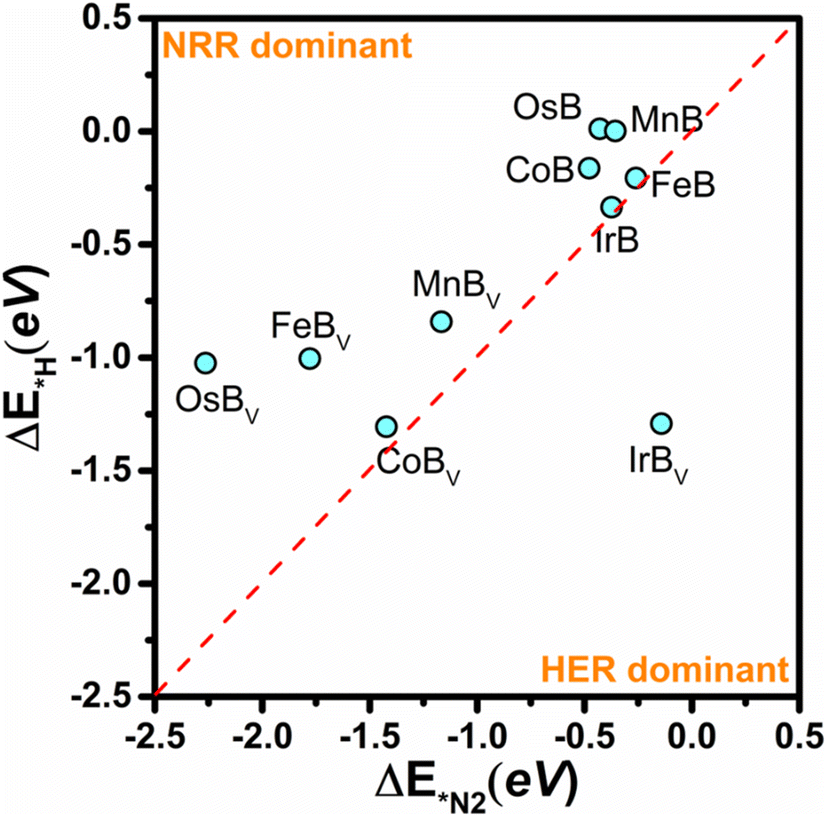 | ||
| Fig. 8 A selectivity plot for anti-MXene borides (pristine and boron-deficient, denoted as Bv) contrasting the adsorption energy of H* (ΔE*H) and nitrogen (ΔE*N2). The compounds are plotted to demonstrate their selectivity towards the NRR over the hydrogen evolution reaction (HER). Points closer to the bottom-left corner favor the NRR, while those closer to the top-right corner indicate a preference for the HER. The optimal catalysts lie near the vertex of the plot, where the selectivity for the NRR is maximized. | ||
Although Fig. 8 provides a clear indication of NRR selectivity for these catalysts, the broader implications that might affect selectivity between the NRR and HER are beyond the scope of this specific analysis. The current study focuses on identifying catalysts with lower limiting potentials conducive to the NRR. Further comprehensive research will be essential to unpack the myriad of factors influencing selectivity, thereby enabling the fine-tuning of these catalysts to optimize their performance for practical applications in the NRR, considering the complex interplay between NRR and HER processes.
4. Conclusions
In summary, this study has conducted a thorough computational examination of a novel family of two-dimensional anti-MXene borides, revealing a set of transition metal borides, specifically CoB, FeB, IrB, MnB, and OsB, with notable promise for catalyzing the electrochemical NRR. A rigorous evaluation of their dynamic, mechanical, and thermal stabilities has verified the robustness of these monolayers, establishing their suitability as durable electrocatalysts. Among the evaluated materials, MnB distinguishes itself with a planar structure and an active site centered on the Mn atom, contrasting with the buckled configurations of its counterparts, where the catalytic action is centered on the boron atoms.In the absence of solvation effects, FeB and OsB have emerged as the leading candidates, characterized by their very low limiting potentials of 0.32 and 0.64 eV, respectively. This underscores their high catalytic efficiency and their potential for sustainable ammonia production. These borides have not only demonstrated superior catalytic activity for the NRR but have also shown marked selectivity against hydrogen adsorption, which is crucial for avoiding the competing HER.
When solvation effects are considered, the limiting potentials for FeB and OsB further decrease to 0.30 eV and 0.48 eV, respectively, highlighting the importance of solvation in enhancing catalytic performance. Additionally, we evaluated the kinetics of proton transfer in the PDS using the Zundel H5O2+ as the solvated proton donor, finding that the activation barriers for FeB and OsB are remarkably low (0.15 and 0.30 eV, respectively), which can be easily surmounted at room temperature or diminished with more negative applied voltages.
The study further uncovered the substantial impact of boron vacancies on adsorption energetics. While these vacancies enable a strong side-on configuration for N2 adsorption, they also induce substantial potential energy barriers, detrimental to catalysis due to the potent interactions between the intermediates and the metal atoms adjacent to the vacancy sites. A pivotal discovery is the linear relationship discerned between the φ descriptor, calculated from the d-electron count and the electronegativity of the transition metals, and the UL. This relationship serves as a predictive measure for the influence of transition metals on electrocatalytic activity and as a guiding principle for the optimization of future catalysts.
Regarding the experimental synthesis of these materials, our findings suggest that the synthesis of two-dimensional anti-MXene borides could be approached similarly to the methods used for other 2D materials like MBenes. Techniques such as chemical vapor deposition (CVD), liquid phase exfoliation, and high-temperature solid-state reactions could potentially be adapted to synthesize these anti-MXenes.55–57 Moreover, the theoretical stability assessments presented in this study provide a solid foundation for their practical realization,39 ensuring that they can maintain structural integrity under typical environmental conditions.
This comprehensive computational inquiry paves the way for the development of next-generation MBene electrocatalysts, setting the stage for advanced synthesis and application of efficient, high-performance materials for sustainable energy conversion processes.
Data availability
The data supporting the findings of this study are available within the article and its ESI† files. Additional data that support the findings of this study are available from the corresponding author upon reasonable request. This includes computational codes and raw data files generated during the study.Author contributions
Viet Q. Bui carried out the DFT calculations and data analysis, wrote the original paper, proposed the concept and supervised the project. All authors actively participated in the review and editing process of the manuscript and the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was financially supported by the Vietnam Ministry of Education and Training, Vietnam, code: B2023. DNA.08.References
- S. B. Carroll and S. D. Salt, Ecology for Gardeners, Timber Press, Portland, Oregon, 2004 Search PubMed.
- N. Zhang, A. Jalil, D. X. Wu, S. M. Chen, Y. F. Liu, C. Gao, W. Ye, Z. M. Qi, H. X. Ju, C. M. Wang, X. J. Wu, L. Song, J. F. Zhu and Y. J. Xiong, J. Am. Chem. Soc., 2018, 140, 9434 CrossRef CAS.
- M. Appl, The Haber–Bosch process and the development of chemical engineering, in A Century of Chemical Engineering, Plenum Press, New York, 1982 Search PubMed.
- R. Y. Hodgetts, A. S. Kiryutin, P. Nichols, H. L. Du, J. M. Bakker, D. R. Macfarlane and A. N. Simonov, ACS Energy Lett., 2020, 5(3), 736–741 CrossRef CAS.
- Y. Kong, Y. Li, X. Sang, B. Yang, Z. Li, S. Zheng, Q. Zhang, S. Yao, X. Yang, L. Lei, S. Zhou, G. Wu and Y. Hou, Adv. Mater., 2022, 34(2), 2103548 CrossRef CAS PubMed.
- V. Kyriakou, I. Garagounis, A. Vourros, E. Vasileiou and M. Stoukides, Joule, 2020, 4(1), 142–158 CrossRef CAS.
- Y. Wu, C. He and W. Zhang, J. Am. Chem. Soc., 2022, 144(21), 9344–9353 CrossRef CAS PubMed.
- X. Xu, Y. Pan, Y. Zhong, R. Ran and Z. Shao, Mater. Horiz., 2020, 7(10), 2519–2565 RSC.
- Y. Liu, L. Wang, L. Chen, H. Wang, A. R. Jadhav, T. Yang, Y. Wang, J. Zhang, A. Kumar, J. Lee, V. Q. Bui, M. G. Kim and H. Lee, Angew. Chem., 2022, 134(50), e202209555 CrossRef.
- S. D. Minteer, P. Christopher and S. Linic, ACS Energy Lett., 2018, 4(1), 163–166 CrossRef.
- B. H. R. Suryanto, H. L. Du, D. Wang, J. Chen, A. N. Simonov and D. R. MacFarlane, Nat. Catal., 2019, 2(4), 290–296 CrossRef CAS.
- X. Qian, C. Ma, U. B. Shahid, M. Sun, X. Zhang, J. Tian and M. Shao, ACS Catal., 2022, 12(11), 6385–6393 CrossRef CAS.
- T. N. Ye, S. W. Park, Y. Lu, J. Li, J. Wu, M. Sasase, M. Kitano and H. Hosono, J. Am. Chem. Soc., 2021, 143(32), 12857–12866 CrossRef CAS.
- H. S. Kim, H. Jin, S. H. Kim, J. Choi, D. W. Lee, H. C. Ham, S. J. Yoo and H. S. Park, ACS Catal., 2022, 12(9), 5684–5697 CrossRef CAS.
- C. Ling, X. Niu, Q. Li, A. Du and J. Wang, J. Am. Chem. Soc., 2018, 140(43), 14161–14168 CrossRef CAS.
- J. Wang, Z. Zhang, Y. Li, Y. Qu, Y. Li, W. Li and M. Zhao, ACS Appl. Mater. Interfaces, 2021, 14(1), 1024–1033 CrossRef.
- C. Liu, Q. Li, J. Zhang, Y. Jin, D. R. MacFarlane and C. Sun, J. Mater. Chem. A, 2019, 7(9), 4771–4776 RSC.
- Z. Chen, J. Zhao, C. R. Cabrera and Z. Chen, Small Methods, 2019, 3(6), 1800368 CrossRef.
- J. Li, C. Chen, L. Xu, Y. Zhang, W. Wei, E. Zhao, Y. Wu and C. Chen, JACS Au, 2023, 3(3), 736–755 CrossRef CAS.
- L. Du, V. Prabhakaran, X. Xie, S. Park, Y. Wang and Y. Shao, Adv. Mater., 2021, 33(6), 1908232 CrossRef CAS.
- X. Liu, D. K. Hoang, Q. A. T. Nguyen, D. P. Do, S. G. Kim, P. C. Nam, A. Kumar, F. Zhang, C. Zhi and V. Q. Bui, Nanoscale, 2024, 16, 13148–13160 RSC.
- F. Abdelghafar, X. Xu, S. P. Jiang and Z. Shao, Mater. Rep. Energy, 2022, 2(3), 100144 CAS.
- M. A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359(6378), 896–900 CrossRef.
- S. Ajmal, A. Rasheed, N. Q. Tran, X. Shao, Y. Hwang, V. Q. Bui, Y. D. Kim, J. Kim and H. Lee, Appl. Catal., B, 2023, 321, 122070 CrossRef CAS.
- W. Qiu, X. Y. Xie, J. Qiu, W. H. Fang, R. Liang, X. Ren, X. Ji, G. Cui, A. M. Asiri, G. Cui and B. Tang, Nat. Commun., 2018, 9(1), 3485 CrossRef PubMed.
- X. Yu, P. Han, Z. Wei, L. Huang, Z. Gu, S. Peng, J. Ma and G. Zheng, Joule, 2018, 2(8), 1610–1622 CrossRef CAS.
- J. Deng and C. Liu, Chem, 2018, 4(8), 1773–1774 CAS.
- T. Leiming, P. Kui, Q. Wen, H. Liming, D. Linhai, Z. Guanhua, L. Qiuye and Y. Changlin, Catal. Sci., Technol., 2023, 13(15), 4517–4524 RSC.
- C. Ling, X. Niu, Q. Li, A. Du and J. Wang, J. Am. Chem. Soc., 2018, 140(43), 14161–14168 CrossRef CAS PubMed.
- X. Lv, W. Wei, F. Li, B. Huang and Y. Dai, Nano Lett., 2019, 19(9), 6391–6399 CrossRef CAS PubMed.
- Y. Cao, S. Deng, Q. Fang, X. Sun, C. Zhao, J. Zheng, Y. Gao, H. Zhuo, Y. Li, Z. Yao, Z. Wei, X. Zhong, G. Zhuang and J. Wang, Nanotechnology, 2019, 30(33), 335403 CrossRef CAS PubMed.
- C. Liu, Q. Li, C. Wu, J. Zhang, Y. Jin, D. R. MacFarlane and C. Sun, J. Am. Chem. Soc., 2019, 141(7), 2884–2888 CrossRef CAS.
- F. Li and Q. Tang, Nanoscale, 2019, 11(40), 18769–18778 RSC.
- T. H. Ho, V. Q. Bui, Q. A. T. Nguyen, Y. Kawazoe, S. G. Kim and P. C. Nam, Phys. Chem. Chem. Phys., 2023, 25(37), 25389–25397 RSC.
- X. Yang, C. Shang, S. Zhou and J. Zhao, Nanoscale Horiz., 2020, 5(7), 1106–1115 RSC.
- S. Qi, Y. Fan, L. Zhao, W. Li and M. Zhao, Appl. Surf. Sci., 2021, 536, 147742 CrossRef CAS.
- M. Yao, Z. Shi, P. Zhang, W. J. Ong, J. Jiang, W. Y. Ching and N. Li, ACS Appl. Nano Mater., 2020, 3(10), 9870–9879 CrossRef CAS.
- Y. Li, L. Li, R. Huang and Y. Wen, Nanoscale, 2021, 13(35), 15002–15009 RSC.
- J. Gu, Z. Zhao, J. Huang, B. G. Sumpter and Z. Chen, ACS Nano, 2021, 15(4), 6233–6242 CrossRef CAS PubMed.
- J. Tang, S. Li, D. Wang, Q. Zheng, J. Zhang, T. Lu, J. Yu, L. Sun, B. Sa, B. G. Sumpter, J. Huang and W. Sun, Nanoscale Horiz., 2024, 9(1), 162–173 RSC.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47(1), 558 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49(20), 14251 CrossRef CAS PubMed.
- G. Kresse, J. Non-Cryst. Solids, 1995, 192, 222–229 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(3), 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77(18), 3865 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed.
- E. Skulason, T. Bligaard, S. Gudmundsdóttir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jónsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2012, 14(3), 1235–1245 RSC.
- J. H. Montoya, C. Tsai, A. Vojvodic and J. K. Norskov, ChemSusChem, 2015, 8, 2180–2186 CrossRef CAS PubMed.
- A. J. Mannix, X. F. Zhou, B. Kiraly, J. D. Wood, D. Alducin, B. D. Myers, X. Liu, B. L. Fisher, U. Santiago, J. R. Guest, M. J. Yacaman, A. Ponce, A. R. Oganov, M. C. Hersam and A. P. Guisinger, Science, 2015, 350(6267), 1513–1516 CrossRef CAS PubMed.
- A. Fleurence, R. Friedlein, T. Ozaki, H. Kawai, Y. Wang and Y. Yamada-Takamura, Phys. Rev. Lett., 2012, 108(24), 245501 CrossRef.
- L. Li, Y. Yu, G. J. Ye, Q. Ge, X. Ou, H. Wu, D. Feng, X. H. Chen and Y. Zhang, Nat. Nanotechnol., 2014, 9(5), 372–377 CrossRef CAS.
- M. E. Tuckerman, J. Phys.: Condens. Matter, 2002, 14(50), R1297 CrossRef CAS.
- S. Maintz, V. L. Deringer, A. L. Tchougreeff and R. Dronskowski, J. Comput. Chem., 2016, 37, 1030–1035 CrossRef CAS.
- H. J. Chun, V. Apaja, A. Clayborne, K. Honkala and J. Greeley, ACS Catal., 2017, 7(6), 3869–3882 CrossRef CAS.
- H. Zhang, H. Xiang, F. Z. Dai, Z. Zhang and Y. Zhou, J. Mater. Sci. Technol., 2018, 34(11), 2022–2026 CrossRef CAS.
- X. Li, F. Ran, F. Yang, J. Long and L. Shao, Trans. Tianjin Univ., 2021, 27, 217–247 CrossRef CAS.
- H. Chen and X. Zou, Inorg. Chem. Front., 2020, 7(11), 2248–2264 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta02284j |
| This journal is © The Royal Society of Chemistry 2024 |