
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unveiling the structural integrity of tunnel-type Na0.44MnO2 cathode for sodium ion battery†
Sankalpita
Chakrabarty‡
ab,
Javeed Ahmad
Dar‡
c,
Akanksha
Joshi
ab,
Arad
Paperni
ab,
Sarah
Taragin
ab,
Ananya
Maddegalla
 ab,
Gopalakrishnan
Sai Gautam
*c,
Ayan
Mukherjee
*ab and
Malachi
Noked
*ab
ab,
Gopalakrishnan
Sai Gautam
*c,
Ayan
Mukherjee
*ab and
Malachi
Noked
*ab
aDepartment of Chemistry, Bar Ilan University, Ramat Gan, Israel. E-mail: ayan.chemical2015@gmail.com; Malachi.Noked@biu.ac.il
bBar-Ilan Institute of Nanotechnology and Advanced Materials, Ramat Gan, Israel
cDepartment of Materials Engineering, Indian Institute of Science, Bengaluru, 560012, India. E-mail: saigautamg@iisc.ac.in
First published on 8th August 2024
Abstract
Tunnel-type Na0.44MnO2 (tt-NMO) is a promising cathode for sodium ion battery having excellent structural stability, diffusion kinetics, and low cost. However, this cathode is reported to suffer from low initial charge capacity (e.g., ≤60 mA h g−1) due to the limited accessibility of sodium ion extraction (0.22–0.24 Na+ per formula unit) from the structure, which hinders the practical viability of this material in a full battery cell. In this study, we report a tailored tt-NMO structure, synthesized using a two-step facile and scalable process, with >95% yield. Our tt-NMO demonstrated a 1st charge capacity of 110 mA h g−1, followed by a discharge capacity of 115 mA h g−1 within the potential window of 4–1.7 V versus Na/Na+. The long-term cycling performance at 0.5C rate and 1C rate (1C = 120 mA h g−1) shows excellent structural integrity for over 400 cycles with >75% capacity retention. We show experimentally and support it with DFT (density functional theory) calculations that the unique microstructure of this tt-NMO, with modulated Na–O bond length and Na–O–Na bond angle, results in open channels along the c-axis in the ab plane, providing a wide pathway for ion diffusion. The Na+ migration barriers (Em) along the two pathways of the c-tunnel are calculated to be within the threshold limit of Na+ migration energy barrier, which renders more sites electrochemically active, enabling the high 1st charge capacity. This novel study opens possibilities to use this unique tt-NMO as an efficient SIB (sodium ion battery) cathode by harnessing the modified structure.
Introduction
Current research in sodium ion battery (SIB) technology focuses heavily on developing cathode materials with high capacity and long-term cycling stability. A few major types of SIB cathode materials have been well studied in recent years, including NaxMPO4F,1–5 NaxMP2O7 [M = V, Fe, Mn…],6–8 NaxVPO4FO2,9–11 Prussian white/blue analogues12 and NaxTMO2 [TM = transition metal; Mn, Fe, Co, Ni…].13–15 Among them, P2-type NaxTMO2 materials (x ≤ 0.8) have attracted significant attention due to their facile synthesis and structural stability. Even though some of these P2-type compositions exhibit promising electrochemical performance, insufficient electronic conductivity, ion deficiency, and undesirable phase transitions limit their practical capacity, rate capability, and structural stability.16–19Among these NaxTMO2 cathodes, sodium manganese oxide (Na0.44MnO2 or NMO) is a unique tunnel-type (tt) structure that can sustain multiple phase changes during the Na electrochemical extraction/insertion process.20 The structure is built up by double and triple linear chains of edge-sharing MnO6 octahedra and a single chain of corner-sharing MnO5 cubic pyramids. For Na ions, there are three binding sites (Na1, Na2 and Na3) within the tt-NMO structure: one-dimensional small tunnels, which are fully occupied by Na atoms (Na1), and two-dimensional S-shaped large tunnels that are partially/half-filled by Na ions (Na2 and Na3 sites).21,22 Na+ ions can reversibly (de)intercalate only through the S-shaped tunnels, conforming to a theoretical specific discharge capacity of 121 mA h g−1 between 2.0 V and 4.0 V, while the sodium ions situated in the one-dimensional tunnels are immobile. The unique large tunnels in tt-NMO provide excellent structural reversibility and hence exhibit robust cycle stability and rate performance in both aqueous and nonaqueous electrolytes.21,23–26
The initial structural arrangement of tt-NMO restricts the amount of sodium extraction to ∼0.22 Na+, which limits the initial charge capacity to 60 mA h g−1.20,22 Although, the extraction of sodium ions occurs from the S-shaped tunnel, where even after the extraction of 0.22 Na+, the S-shaped tunnel is not completely empty but rather partially occupied by Na+, which is theoretically proved by Kim et al.27 Nevertheless, as per previous reports, the initial charge capacity of tt-NMO is reported by many to be ≤60 mA h g−1.24,28–31 The initial capacity is a critical parameter in a full cell and dictates the energy density of practical batteries. Even if the 1st discharge is demonstrated to be high, the demonstration of this capacity in full cell will be profoundly hampered if the 1st charge is low. Increasing the Na content in the cathode is one approach to achieve higher 1st charge capacity, but when the Na content is beyond 0.44, the tunnel-type structure simply transforms to a layered structure. To enhance the sodium diffusion process, shortening the diffusion path by reducing the length-diameter ratio or downsizing the particle size are ways that have been reported so far.24,30 Numerous synthesis processes including hydrothermal method, solid-state method, sol-gel method, glycine nitrate process, and molten-salt method have been proposed to improve the reversible capacity of tt-NMO, but the initial charge capacity is either not mentioned or suffers from a poor value (<60 mA h g−1).21,25,32,33
TM substitution in NMO has also been reported to increase the 1st charge capacity but is still not a satisfying value. For example, Wang et al. have shown that the partial substitution of Mn by Ti in tt-NMO increases the first charge capacity from 45 mA h g−1 to 60 mA h g−1. The authors hypothesized that the modified Mn/Ti arrangement breaks the Na+-vacancy ordering, which increases the charge capacity.34,35 All these previous reports have established a positive improvement in their electrochemical performances though the initial charge capacity does not improve enough. Hence, enhancing the initial charge capacity of tt-NMO beyond 60 mA h g−1 is an important requirement that needs to be fulfilled to increase its practical utility.
Some attempts for chemical sodiation of tt-NMO prior to cell charge were reported and showed some success in increasing the cathode capacity.36 This approach is good to demonstrate that the bottleneck can be overcome through synthesis, yet it is not practical and difficult to upscale.
Herein, we report a two-step solid-state method to synthesize tt-NMO, which exhibits a distorted, tt Pbam structure and an outstanding initial charge capacity of 110 mA h g−1. In order to understand the uniqueness of our tt-NMO, we employed density functional theory (DFT) based calculations to estimate the Na+ migration barrier (Em) and quantify the Na-ion mobility within the NMO framework. Specifically, we evaluated the ease of Na migration within the DFT-calculated ground state configuration of the Na0.22MnO2 composition. Note that the Na0.22MnO2 composition is obtained after the electrochemical extraction of 0.22 Na from tt-NMO. Significantly, we found that both the c-tunnel hops have migration barrier, Em < 525 meV (i.e., ∼460 and 330 meV), emphasizing that both hops should remain electrochemically active at room temperature, and the feasibility of extraction of Na along the two one-dimensional tunnels. Thus, we expect a partial desodiation from Na0.22MnO2 to be quite facile along the c-tunnels, which should facilitate enhanced Na extraction during first charge, corresponding to a higher charge capacity without impeding the structural stability. As per the best of our knowledge, this is the first report on the structural modulation of tt-NMO, which could break the bottleneck of the practical viability of this tunnel-type structure.
Subsequently, we demonstrate a discharge capacity of 115 mA h g−1 at 0.06C rate (1C = 120 mA h g−1) within the potential window of 4–1.7 V vs. Na/Na+. The long cycling performance at 0.5C rate and 1C rate shows excellent stability (90% and 80% capacity retention, respectively) until 200 cycles. The efficient rate capability further confirms the structural stability upon cycling at different current rates ranging from 0.06C to 3C and reverting to 0.2C.
We believe that our findings open an opportunity to utilize tt-NMO cathodes with practical 1st charge capacity and long-term stability.
Results and discussion
The refined X-ray diffraction (XRD) pattern, shown in Fig. 1a, is consistent with the structure of tt-NMO, without any secondary phases.25 The detailed microstructural analysis (see Tables S1–S3 of the ESI†) indicates that lattice distortion occurred as compared to the Pbam space group (JCPDS 27-0750), which accounts for the increase in the Na–O–Na bond angle and the significant decrease of the Na–O bond distance, resulting in open channels along the c-axis in the ab plane, as shown in Fig. 1d and e.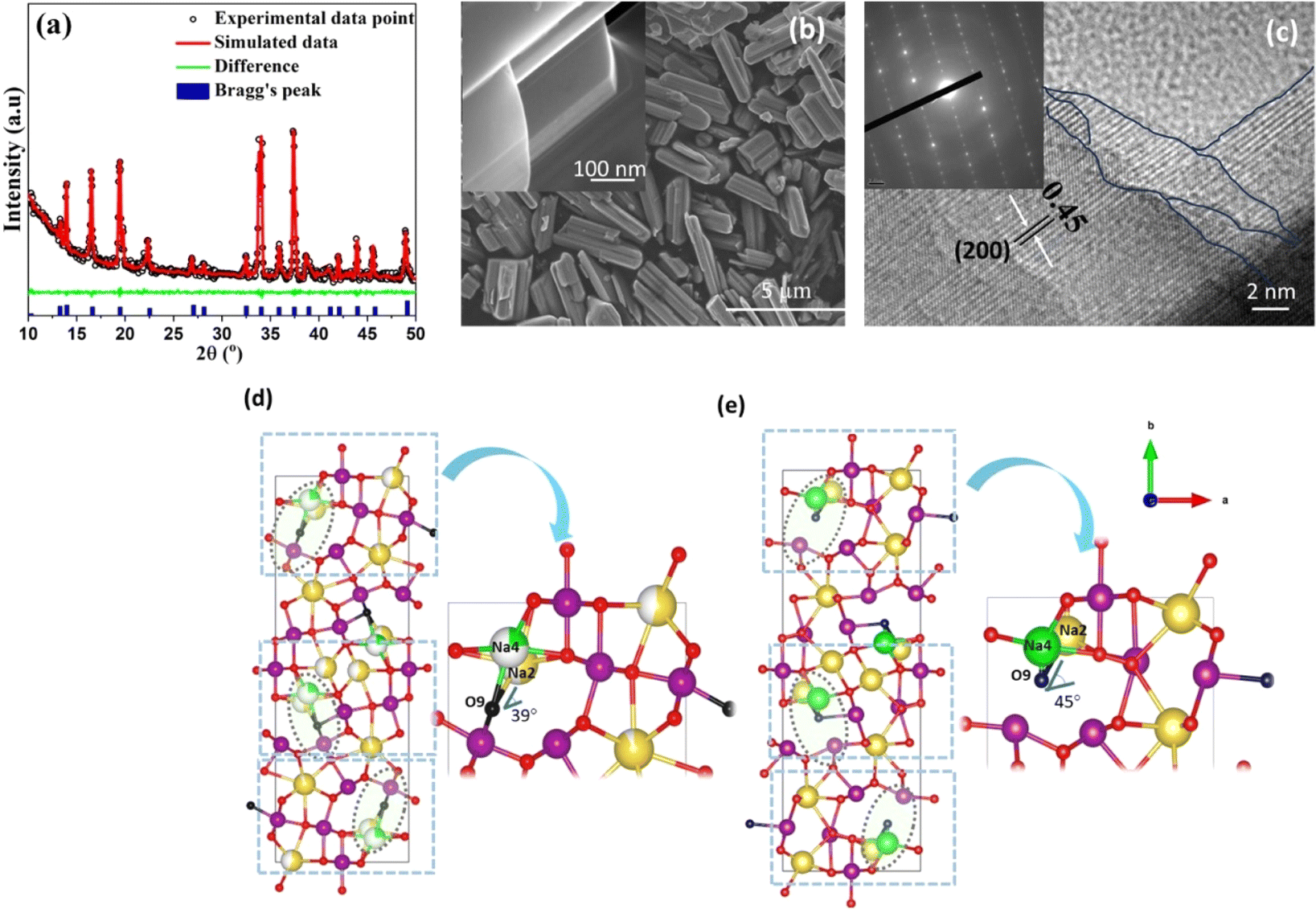 | ||
| Fig. 1 (a) XRD pattern of tt-NMO, (b) low magnification SEM; inset: high magnification HRSEM, and (c) HRTEM images of tt-NMO; inset shows the SAED pattern. (d) Structure of the standard Pbam space group, magnified part of the circulated region shows the Na4–O9–Na2 bond angle. (e) Structure of synthesized tt-NMO; the magnified part of the circulated zone clearly indicates the increasing Na4–O9–Na2 bond angle, which leads to a decrease in the bond length and opens the channel. | ||
The scanning electron microscopy (SEM) image (Fig. 1b) of tt-NMO shows that our synthesized cathode exhibits a rod shape structure. The high magnification (HRSEM; inset Fig. 1b) image shows that the rod shape is formed by several nanoplates bundled together. The well-organized layered surface morphology is clearly visible. The energy dispersive spectra (EDS) and elemental mapping (Fig. S2†) confirm the presence of Na, Mn and O atoms, without any other impurity elements. Fig. 1c shows the high-resolution transmission electron microscopy (HRTEM) image of tt-NMO, with the inset displaying the selected area electron diffraction (SAED). The marked lines in the fringe pattern specify the existence of thin nanoplates at different layers, making the three-dimensional tunnels. The lattice fringes correspond to the [200] planes of the crystal structure,25,30 while the SAED pattern clearly indicates the single-phase crystalline feature of tt-NMO, similar to previous reports.30 We did not notice any detectable impurity phase from the structural analysis. Inductively-coupled plasma atomic emission spectroscopy (ICP-AES) studies further confirmed that the ratio of sodium and manganese of our synthesized material is within 0.442–0.451, which agrees with the expected value.
The high-resolution X-ray photoelectron spectra (XPS) of Mn 2p, as shown in Fig. 2a, displays two major peaks at 642.2 eV and 653.7 eV corresponding to the binding energies of Mn 2p3/2 and Mn 2p1/2 spin orbital doublet, respectively, with a splitting energy of 11.49 eV. The deconvolution of individual peaks shows the coexistence of Mn3+ and Mn4+ oxidation state located at 642 eV and 643.3 eV for Mn 2p3/2 and 653.6 eV and 655 eV for Mn 2p1/2 in that order.26,37 The deconvoluted O 1s spectra (Fig. 2b) exhibits the presence of lattice oxygen centered at 529.9 eV. The O 1s peaks located at 531.4 eV correspond to the surface adsorbed oxygen and oxygen vacancies in the metal oxide, while the peak at 533.3 eV indicates the surface –OH group.37,38 The splitting of the Na 1s spectra gives two peaks positioned at 1071.1 eV and 1071.9 eV, suggesting two Na environments/sites on the surface of the tt-NMO. The peak at the lower binding energy is assigned as the edge shared Na site (Na2 and Na3), whereas the higher binding energy peak signifies the face-sharing Na site (Na1).26,39
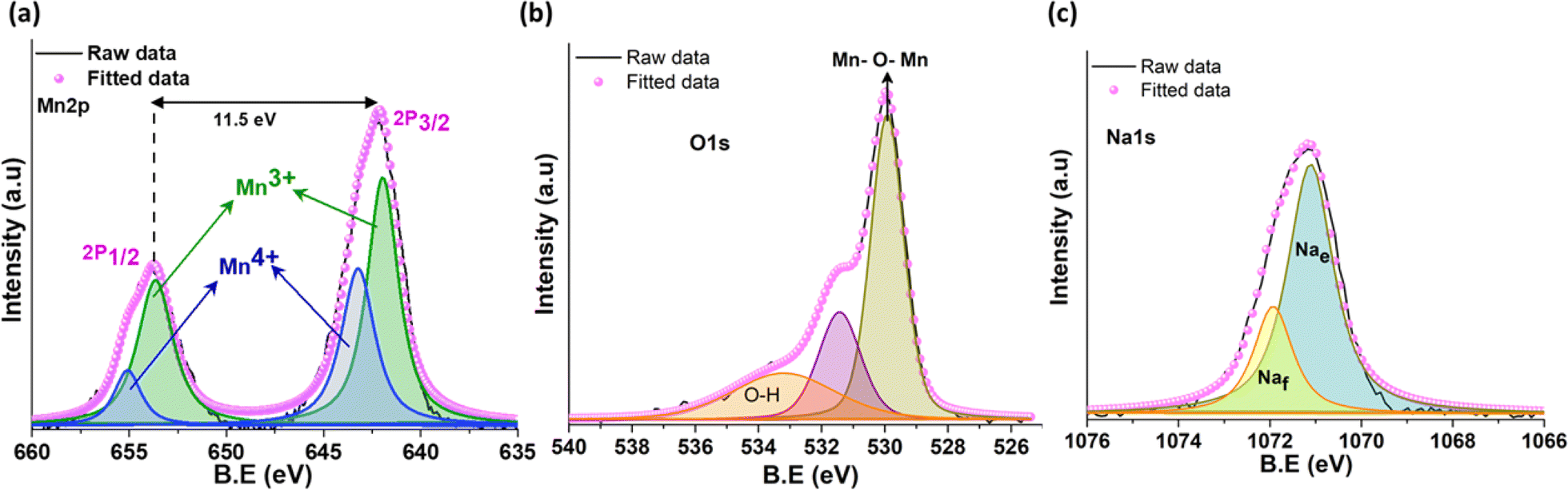 | ||
| Fig. 2 XPS spectra of (a) Mn 2p, (b) O 1s, and (c) Na 1s of tt-NMO. | ||
The initial galvanostatic charge–discharge profile of tt-NMO at 0.06C rate (1C = 120 mA h g−1) within a potential window of 4.0–1.7 V (vs. Na/Na+) is displayed in Fig. 3a. The pristine material delivers 110 mA h g−1 initial charge capacity and 115 mA h g−1 discharge capacity in our electrolyte. The first charge capacity is higher than the previously reported values in the literature (Table S5†), which is attributable to the amount of Na extracted from the pristine structure during the first desodiation process. As per the theoretical capacity of this material (120 mA h g−1), the first charging capacity typically does not cross the limit of 60–70 mA h g−1, corresponding to an extraction of 0.22–0.26 equivalent of Na+ (per formula unit of MnO2) out of the available 0.44 Na.22,30,40 In contrast to the perceived limit of 60–70 mA h g−1, our tt-NMO cathode demonstrates a significantly higher capacity owing to the unique structural modulation, as shown in Fig. 1e and supported by DFT study.
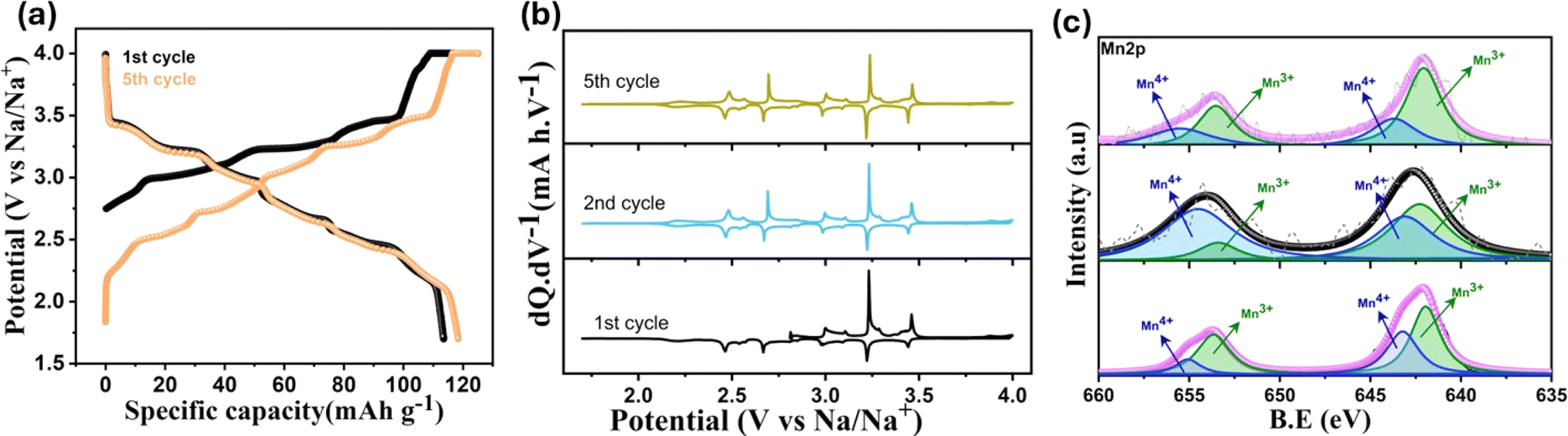 | ||
| Fig. 3 (a) 1st and 5th charge–discharge profile of tt-NMO at 0.06C rate. (b) dQ·dV−1 plot for initial five cycles at 0.06C rate. (c) Mn 2p XPS spectra of the pristine, charged and discharged state. | ||
The derivative of the 1st cycle voltage profile (Fig. 3b) shows sharp anodic peaks centered at 3.0, 3.23 and 3.41 V, and three small peaks at 3.1, 3.28 and 3.46 V versus Na/Na+ are associated with consecutive pair of cathodic peaks. The peaks in the dQ·dV−1 curve are related to the reversible (de)intercalation of sodium ions at a specific voltage range. The sequential ordering of sodium extraction/insertion from the respective crystallite site is depicted in Fig. S4† where Na1 and Na2 signifies the edge-sharing Na site and Na3 is the face-sharing Na site.41 Reversible six distinct redox peaks are sustained in the following cycles, as presented in Fig. 3b, and consistent with the short voltage plateaus in the voltage curve of Fig. 3a. The first cycle redox peaks are exactly overlapping with those of the following cycles, indicating that the material does not need an activation cycle.31 The typical CV curve at different scanning rates shows (Fig. S5a†) six pairs of reversible redox peaks similar to the dQ·dV−1 plot in Fig. 3b. With increasing current rate, both oxidation and reduction peak current intensity increase. The variation of peak current (Ip) with respect to square root of scan rate (v1/2) shows good linear relation (Fig. S5b†), which suggests the diffusion-controlled behavior of the sodiation/desodiation process.41
The Mn 2p XPS spectra of the pristine, 1st charged, and 1st cycled electrode of tt-NMO is depicted in Fig. 3c. The extraction of sodium leads to the oxidation of Mn(III) to Mn(IV) upon charging to 4 V versus Na/Na+, which is distinctly observed from the decreasing of Mn(III)/Mn(IV) peak ratio in the deconvoluted XPS spectra compared to the pristine electrode. After the initial charge, we obtain a Mn(IV)/Mn(III) ratio of ∼0.8, as calculated from the relative deconvoluted peak areas. The following discharge of the electrode (1.7 V) shows a reversible change in the Mn oxidation state, as indicated by the increasing Mn(III)/Mn(IV) peak ratio. During the discharge process after initial charging, the Mn(IV)/Mn(III) reduction occurs at specific Mn sites (i.e., Mn(II) site along the ‘b’ and Mn(III) site along the ‘a’ axis) suggests an asymmetric lattice evolution in the structure. The reduction of Mn(IV) to Mn(III) at specific Mn sites results in a Jahn–Teller distortion of the reduced sites, ensuing different orientations of distorted MnO6 octahedra in the structure.27,42 As reported in the previous work, when the initial adjustment of the Mn(IV)/Mn(III) ratio is near ∼1, it can suppress the Jahn–Teller effect and cause the disproportionation of Mn3+, enhancing the structural stability.29
We further investigated the initial charge/discharge process through in situ XRD. The XRD pattern recorded during the first charge/discharge process is displayed in Fig. 4. The diffraction pattern within the 2θ region from 15° to 45° shows that most of the peaks are consistent with the electrochemical (de)intercalation process and does not lead to any new phases. A closer view in the range of 32° to 38° (bottom panels in Fig. 4) reveals the shifting of the pattern to a higher angle during charging, which signifies lattice contraction due to the extraction of Na. Interestingly, the plane (0 10 0) perpendicular to b axis shows profound shifting in the angles, which might be due to the sodium extraction process along the b axis.25 During the insertion of Na, the shifting of peak towards lower angles suggest the expansion of lattice planes as well as the insertion of Na along the a and c-axis of the structure, which leads to asymmetrical lattice evolution.27 A similar kind of diffraction peak adjustment is observed in the charge/discharge process of the second cycle.
 | ||
| Fig. 4 In situ evolution of XRD pattern recorded at a 12 mA g−1 current rate for the 1st cycle and a 24 mA g−1 current rate for the 2nd cycle. | ||
The initial sodium extraction process is further analyzed through the Na-ion mobility inside the structure and for this purpose, we electrochemically extracted 0.22 moles of Na from our tt-NMO composition by restricting the charge capacity to 60 mA h g−1. Subsequently, we analyzed the structure of the material (Na0.22MnO2) based on experimental XRD refinement (Fig. S3†). To understand the Na-mobility and ease of extra-Na-extraction within the NMO framework, we calculated the Na+Emvia DFT-NEB to quantify the Na-ion mobility at the Na0.22MnO2 composition. We generated the ground state Na-vacancy configuration at Na0.22MnO2 using DFT, where we used the experimental XRD-refined structure of tt-NMO as our template.43,44
Within the Na0.22MnO2 structure, we considered three migration paths, i.e., two along the ‘tunnels’ that are parallel to the c-axis, indicated as ‘hop 1’ and ‘hop 2’ in panels a and b of Fig. S4,† and one path that connects the two tunnels via a ‘diagonal’ path, highlighted as ‘hop 3’ in Fig. S4c.†
Structural information of Na0.22MnO2
The DFT-identified ground state structure of Na0.22MnO2 (Fig. 5a) consists of the square pyramidal MnO5 (brown polyhedra) and octahedral MnO6 polyhedra (purple) that are present in tt-NMO as well (Fig. S3a†). Mn exhibits the oxidation states of +3 and +4 within the MnO5 and MnO6 polyhedra, respectively (see Table S1 and Fig. S3 of the ESI†). Note that DFT estimates the a, b, and c lattice parameters to be 8.96, 25.33, and 2.85 Å, respectively, which agrees well with our experimentally determined parameters of 9.08, 26.31, and 2.82 Å (<3.7% error). Among the lattice angles, DFT estimates do not vary significantly from the experimental values, i.e., 90°. The Na atoms (yellow spheres) in a given Na0.22MnO2 unit cell occupy distinct ac planes; thus, Na atoms continue to occupy tunnels along the c-axis similar to tt-NMO. However, the reduced occupancy of Na in Na0.22MnO2 gives rise to a fully empty c-tunnel in the unit cell (black outlined circle in Fig. 5a), as compared to tt-NMO. The fully empty Na tunnel gives rise to a possible ‘diagonal’ Na migration, apart from the ‘straight’ migrations that can happen along the c-tunnels similar to the tt-NMO structure. We refer to the diagonal migration pathway as ‘hop 3’, while the straight tunnel migrations are referred to as hops 1 and 2 (Fig. S4†).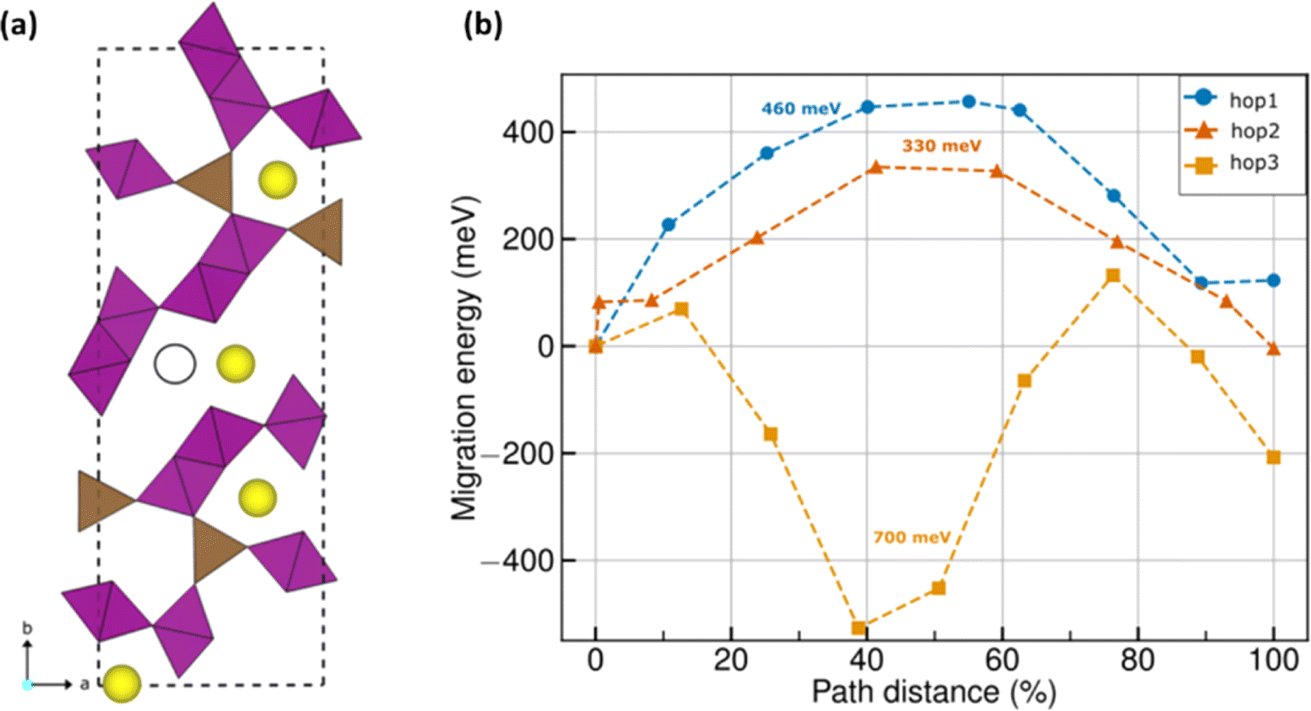 | ||
| Fig. 5 (a) DFT-calculated ground state configuration of Na0.22MnO2. Brown and purple polyhedra indicate MnO5 and MnO6 groups with oxygen atoms occupying the polyhedral vertices. Yellow spheres indicate Na atoms. Black outlined circle indicates a vacant Na site, which is occupied in the case of tt-NMO (see Fig. S3a†). (b) DFT-NEB calculated MEPs of Na+ migration in Na0.22MnO2. Blue, red, and orange symbols indicate hops 1, 2, and 3, respectively. Barrier values for each hop is given by the text annotations of the same color. For hop 3, 700 meV is the effective barrier, where the intermediate images have a lower energy than the endpoint configurations. | ||
Migration barriers
For Na extraction to be facile in Na0.22MnO2, at least one of the three identified migration pathways (i.e., hops 1, 2, and 3) has to be active under typical electrochemical conditions. Note that previous studies have shown that Em magnitudes up to ∼525 meV can be tolerated for reasonable electrochemical performance in micron-sized cathode particles at 298 K.45 Thus, hops for which the calculated Em is less than 525 meV should remain active at typical electrochemical conditions. Fig. 5b displays the DFT-NEB calculated Em for the three different hops considered, with the final elastic bands displayed in Fig. S4 of the ESI.† Blue, red, and orange symbols signify hops 1, 2, and 3, respectively, with the barrier magnitudes indicated as text annotations in Fig. 5b.Importantly, we find that both hops 1 and 2, i.e., both the c-tunnel hops, have Em < 525 meV, namely, ∼460 and 330 meV, highlighting that both hops should remain electrochemically active, and it should be feasible to extract Na along these two tunnels. In the case of hop 3, we find intermediate images that are lower in energy compared to the endpoint configurations, which can indicate the occupation of Na in such interstitial sites under electrochemical conditions. Nevertheless, the effective Em for Na migration across hop 3 includes the negative energies of the intermediate images as well, resulting in a magnitude of ∼700 meV. Thus, the higher Em of 700 meV for hop 3 compared to the threshold 525 meV indicates that Na extraction will be difficult across this pathway. Thus, we expect the partial desodiation from Na0.22MnO2 to be quite facile, along the c-tunnels, in line with our experimental observations.
The galvanostatic cycling stability of the material at different rates is displayed in Fig. 6a. We observe that 90% of the initial discharge capacity at 0.5C rate (100 mA h g−1) is retained after 200 cycles and 80% of initial capacity is retained at 1C up to 200 cycles. Further cycling at 0.5C rate (Fig. S6a†) shows 75% of capacity retention up to 400 cycles, which implies the robust structural stability of our synthesized cathode material with the reversible Na (de)intercalation process over long-term cycling.
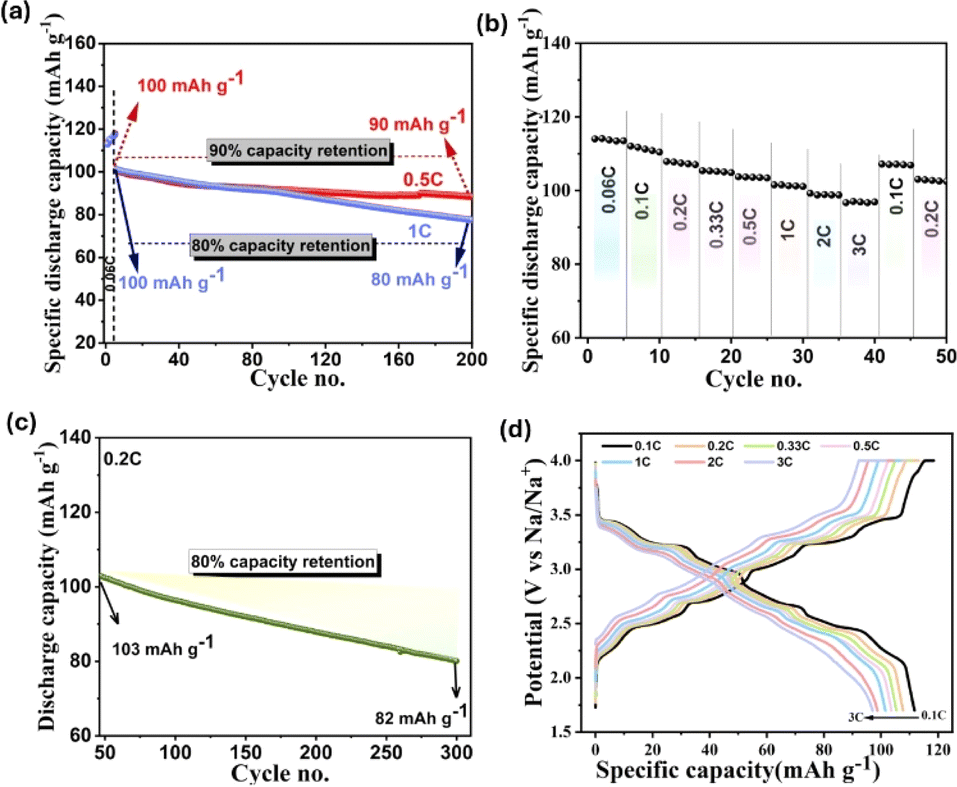 | ||
| Fig. 6 (a) Galvanostatic cycling stability at 0.5C and 1C rate. (b) Rate performance at various current rates. (c) Cycling behavior at 0.2C after the rate study. (d) Capacity–voltage profile at different C-rates. | ||
The distinct plateaus that are well preserved in the capacity–voltage curve even after 400 cycles (Fig. S6b†) confirm the stable reversible redox reaction within the cathode. The HRSEM image of the post-cycled cathode (Fig. S7†) shows that the rod-shape morphology is preserved even after 400 cycles, except for a few cracks. A closer view of the surface indicates that the surface morphology is maintained well, with the marginal degradation of the surface.
The diffraction pattern recorded after 400 cycles illustrates a pattern similar to a pristine electrode (Fig. S8†). Unless a feeble decrease in the intensity (due to the formation of SEI layer) and slight shifting of peaks to a lower angle (reversible extraction/insertion of Na-ion increase the interplanar spacing), respectively, no extra peaks are detected from the post-cycled cathode electrode, which indicates the stability of the phase structure as well.
We evaluated the rate capability of the tt-NMO at different C rates, as presented in Fig. 6b. It is observed that with the stepwise increase in the current rate from 0.06C to 3C and reverting to 0.1C, 97.2% of the initial discharge capacity is retained. Following the rate study, we cycled the same cell for a further 300 cycles at 0.2C, resulting in an 80% capacity retention against the initial value (Fig. 6c). Moreover, the capacity–voltage profile at different C rates (Fig. 6d) distinctly shows the charge/discharge voltage platform even at a high current rate. Despite a decrease in the capacity with a higher C rate, which always affects the internal stability of the cell and lowers the capacity,46 the well-maintained voltage plateaus imply the efficient rate capability of the tt-NMO cathode. Therefore, our tt-NMO exhibits profound structural integrity despite delivering higher specific capacity. The major limitation of tt-NMO is the inferior first charge capacity, which is addressed in this work, and our synthesized tt-NMO can deliver 110 mA h g−1 initial charge capacity, which is a remarkable step towards the practical application of this material. Nevertheless, there are few important issues that need to be accounted for before moving to large-scale application, such as (1) surface protection from direct environmental humidity (to enable the high volume of slurry for electrode printing), (2) preventing the side reaction between the electrode–electrolyte interface, especially when Mn4+ might form during charge, (3) confining the Mn dissolution from the structure to avoid internal lattice collapse. In addition, the high cut-off voltage of tt-NMO is limited to below 4.0 V (versus Na/Na+), which restrained the actual capacity and charging to beyond 4.0 V and caused rapid capacity fading.47 There are several approaches proposed in reported literature for addressing these challenges. Especially, ion doping and surface modification are two key methods to improve the cycling stability of tt-NMO.34,41,48–50 As per previous studies, the surface modification of cathode materials is the most effective way to address this concern in addition to suppressing the side reaction between the electrode and electrolyte and to raise the high cut-off voltage.29,37,47,50 Hence, surface coating technology could be a prospective approach that could be considered in future for this tt-NMO to improve the functionality.
Conclusions
Herein, we have synthesized tt-NMO through a scalable solid-state route, which projects an initial charge capacity of 110 mA h g−1 with a discharge capacity of 115 mA h g−1. 90% of capacity retention up to 200 cycles at 0.5C rate pronounced structural stability upon long charge–discharge cycle. The microstructure analysis of our synthesized tt-NMO presents the modified lattice structure having shorter Na–O bond length compared to conventional Pbam structure created an open channel along the c-axis in the structure. The excess diffusion of sodium ions is analyzed from the migration barrier of Na+ (Em) within the DFT-calculated ground state configuration of Na0.22MnO2, a composition that is obtained after the electrochemical extraction of 0.22 Na from tt-NMO by restricting the charge capacity of 60 mA h g−1. We observed that two of the c-tunnel hops have lower Em (∼460 and 330 meV), which remain electrochemically active to allow partial Na extraction from Na0.22MnO2, while the third diagonal pathway is not electrochemically active (Em ∼700 meV). Hence, the extraction of Na ions is quite possible, which enables to achieve the observed high initial charge capacity without losing the structural stability. According to the above discussions, tt-NMO material could be a promising SIB cathode, and this study creates a new opportunity to design the cathode material through engineering structural chemistry.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Sankalpita Chakrabarty: conceptualization, methodology, formal analysis, validation, writing – original draft; Javeed Ahmad Dar: DFT analysis; Akanksha Joshi: resources; Arad Paperni: resources; Sarah Taragin: resources; Ananya Maddegalla: resources; Gopalakrishnan Sai Gautam: DFT analysis, writing – review & editing; Ayan Mukherjee: conceptualization, validation, investigation, writing – review & editing; Malachi Noked: supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge the financial assistance from the Israel-US Binational Industrial R&D Foundation (BIRD foundation-Grant No. 16489), National Energy Center established by Israel Ministry of Energy, and Grant No. 223-11-066, US-Israel Binational Science Foundation (NSF-BSF; Grant No. 2023-604), Israel Science Foundation (ISF; Grant No. 1211/21). M. N. acknowledges climate prize from JNF (Grant No. 207420).References
- A. Mukherjee, T. Sharabani, I. Perelshtein and M. Noked, Batteries Supercaps, 2020, 1–4 Search PubMed.
- A. Mukherjee, T. Sharabani, R. Sharma, S. Okashy and M. Noked, Batteries Supercaps, 2020, 3, 510–518 CrossRef CAS.
- A. Mukherjee, Rosy, T. Sharabani, I. Perelshtein and M. Noked, J. Mater. Chem. A, 2020, 8, 21289–21297 RSC.
- T. Sharabani, S. Taragin, I. Perelshtein, M. Noked and A. Mukherjee, Small, 2021, 17, 1–9 CrossRef PubMed.
- S. Park, J. N. Chotard, D. Carlier, I. Moog, M. Courty, M. Duttine, F. Fauth, A. Iadecola, L. Croguennec and C. Masquelier, Chem. Mater., 2021, 33, 5355–5367 CrossRef CAS.
- O. A. Drozhzhin, I. V. Tertov, A. M. Alekseeva, D. A. Aksyonov, K. J. Stevenson, A. M. Abakumov and E. V. Antipov, Chem. Mater., 2019, 31, 7463–7469 CrossRef CAS.
- H. Li, Z. Zhang, M. Xu, W. Bao, Y. Lai, K. Zhang and J. Li, ACS Appl. Mater. Interfaces, 2018, 10, 24564–24572 CrossRef CAS PubMed.
- Y. Kee, N. Dimov, A. Staikov, P. Barpanda, Y. C. Lu, K. Minami and S. Okada, RSC Adv., 2015, 5, 64991–64996 RSC.
- X. Sun, Z. Wang, Q. Hu, X. Mei, Y. Zhao, A. Ndahimana, T. Geng and J. Cui, ACS Appl. Energy Mater., 2022, 5, 15799–15808 CrossRef CAS.
- J. Olchowka, R. Fang, R. Bianchini Nuernberg, C. Pablos, D. Carlier, S. Cassaignon and L. Croguennec, Nanoscale, 2022, 14, 8663–8676 RSC.
- S. Chakrabarty, T. Sharabani, S. Taragin, R. Yemini, A. Maddegalla, I. Perelshtein, A. Mukherjee and M. Noked, J. Energy Storage, 2024, 84, 111507 CrossRef.
- J. Peng, W. Zhang, Q. Liu, J. Wang, S. Chou, H. Liu and S. Dou, Adv. Mater., 2022, 34, 2108384 CrossRef CAS PubMed.
- Y. Cao, M. Xiao, X. Sun, W. Dong and F. Huang, Chem.–Eur. J., 2023, 29, 1–20 Search PubMed.
- K. Wang, H. Zhuo, J. Wang, F. Poon, X. Sun and B. Xiao, Adv. Funct. Mater., 2023, 33, 2212607 CrossRef CAS.
- A. Joshi, S. Chakrabarty, S. H. Akella, A. Saha, A. Mukherjee, B. Schmerling, M. Ejgenberg, R. Sharma and M. Noked, Adv. Mater., 2023, 35, 2304440 CrossRef CAS PubMed.
- J. Jiang, H. C. He, C. Cheng, T. Yan, X. Xia, M. Ding, L. He, T. S. Chan and L. Zhang, ACS Appl. Energy Mater., 2022, 5, 1252–1261 CrossRef CAS.
- C. Wang, L. Liu, S. Zhao, Y. Liu, Y. Yang, H. Yu, S. Lee, G. H. Lee, Y. M. Kang, R. Liu, F. Li and J. Chen, Nat. Commun., 2021, 12, 1–9 CrossRef PubMed.
- P. F. Wang, Y. You, Y. X. Yin and Y. G. Guo, Adv. Energy Mater., 2018, 8, 1–23 Search PubMed.
- C. Zhao, Q. Wang, Z. Yao, J. Wang, B. Sánchez-Lengeling, F. Ding, X. Qi, Y. Lu, X. Bai, B. Li, H. Li, A. Aspuru-Guzik, X. Huang, C. Delmas, M. Wagemaker, L. Chen and Y. S. Hu, Science, 2020, 370, 708–712 CrossRef CAS PubMed.
- X. Zhou, A. Zhao, Z. Chen and Y. Cao, Electrochem. Commun., 2021, 122, 106897 CrossRef CAS.
- D. J. Kim, R. Ponraj, A. G. Kannan, H. W. Lee, R. Fathi, R. Ruffo, C. M. Mari and D. K. Kim, J. Power Sources, 2013, 244, 758–763 CrossRef CAS.
- F. Sauvage, L. Laffont, J. M. Tarascon and E. Baudrin, Inorg. Chem., 2007, 46, 3289–3294 CrossRef CAS PubMed.
- M. S. Chae, H. J. Kim, H. Bu, J. Lyoo, R. Attias, B. Dlugatch, M. Oliel, Y. Gofer, S. T. Hong and D. Aurbach, Adv. Energy Mater., 2020, 10, 1–7 Search PubMed.
- Y. Liu, X. Liu, F. Bu, X. Zhao, L. Wang, Q. Shen, J. Zhang, N. Zhang, L. Jiao and L. Z. Fan, Electrochim. Acta, 2019, 313, 122–130 CrossRef CAS.
- X. He, J. Wang, B. Qiu, E. Paillard, C. Ma, X. Cao, H. Liu, M. Cristian, H. Liu, T. Gallash, Y. S. Meng and J. Li, Nano Energy, 2016, 27, 602–610 CrossRef CAS.
- J. Li, L. Li, H. Shi, Z. Zhong, X. Niu, P. Zeng, Z. Long, X. Chen, J. Peng, Z. Luo, X. Wang and S. Liang, ACS Sustain. Chem. Eng., 2020, 8, 10673–10681 CAS.
- H. Kim, D. J. Kim, D. H. Seo, M. S. Yeom, K. Kang, D. K. Kim and Y. Jung, Chem. Mater., 2012, 24, 1205–1211 CrossRef CAS.
- E. Oz, S. Altin and S. Avci, ACS Omega, 2023, 8, 27170–27178 CrossRef CAS PubMed.
- W. Liu, Q. Ren, M. Yang, L. Liu, Y. Zhang, D. Su, J. Wen, Q. Wang, X. Wang and Y. Feng, J. Alloys Compd., 2022, 896, 163087 CrossRef CAS.
- Q. Liu, Z. Hu, M. Chen, Q. Gu, Y. Dou, Z. Sun, S. Chou and S. X. Dou, ACS Appl. Mater. Interfaces, 2017, 9, 3644–3652 CrossRef CAS PubMed.
- C. Ferrara, C. Tealdi, V. Dall’asta, D. Buchholz, L. G. Chagas, E. Quartarone, V. Berbenni and S. Passerini, Batteries, 2018, 4, 1–10 CrossRef.
- L. Rakočević, S. Štrbac, J. Potočnik, M. Popović, D. Jugović and I. S. Simatović, Ceram. Int., 2021, 47, 4595–4603 CrossRef.
- X. Zhang, S. Tang and Y. Du, J. Phys. Chem. C, 2011, 115, 2644–2649 CrossRef CAS.
- Y. Wang, J. Liu, B. Lee, R. Qiao, Z. Yang, S. Xu, X. Yu, L. Gu, Y. S. Hu, W. Yang, K. Kang, H. Li, X. Q. Yang, L. Chen and X. Huang, Nat. Commun., 2015, 6, 1–10 CAS.
- Q. C. Wang, Q. Q. Qiu, N. Xiao, Z. W. Fu, X. J. Wu, X. Q. Yang and Y. N. Zhou, Energy Stor. Mater, 2018, 15, 1–7 Search PubMed.
- X. Zhou, Y. Lai, X. Wu, Z. Chen, F. Zhong, X. Ai, H. Yang and Y. Cao, Chem. Res. Chin. Univ., 2021, 37, 274–279 CrossRef CAS.
- Z. Zhao, X. Huang, Y. Shao, S. Xu, L. Chen, L. Shi, Q. Yi, C. Shang and D. Zhang, Chem. Eng. J. Adv., 2022, 10, 100292 CrossRef CAS.
- J. Gan, X. Lu, J. Wu, S. Xie, T. Zhai, M. Yu, Z. Zhang, Y. Mao, S. C. I. Wang, Y. Shen and Y. Tong, Sci. Rep., 2013, 3, 1–7 Search PubMed.
- P. Kehne, C. Guhl, Q. Ma, F. Tietz, L. Alff, R. Hausbrand and P. Komissinskiy, J. Electrochem. Soc., 2019, 166, A5328–A5332 CrossRef CAS.
- E. Hosono, T. Saito, J. Hoshino, M. Okubo, Y. Saito, D. Nishio-Hamane, T. Kudo and H. Zhou, J. Power Sources, 2012, 217, 43–46 CrossRef CAS.
- X. L. Li, J. Bao, Y. F. Li, D. Chen, C. Ma, Q. Q. Qiu, X. Y. Yue, Q. C. Wang and Y. N. Zhou, Adv. Sci., 2021, 8, 1–10 Search PubMed.
- H. Gwon, D. H. Seo, S. W. Kim, J. Kim and K. Kang, Adv. Funct. Mater., 2009, 19, 3285–3292 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- D. Sheppard, R. Terrell and G. Henkelman, J. Chem. Phys., 2008, 128, 134106 CrossRef PubMed.
- Z. Rong, R. Malik, P. Canepa, G. Sai Gautam, M. Liu, A. Jain, K. Persson and G. Ceder, Chem. Mater., 2015, 27, 6016–6021 CrossRef CAS.
- B. Xu, W. Diao, G. Wen, S. Y. Choe, J. Kim and M. Pecht, J. Power Sources, 2021, 510, 230390 CrossRef CAS.
- Y. Zhang, L. Liu, S. Jamil, J. Xie, W. Liu, J. Xia, S. Nie and X. Wang, Appl. Surf. Sci., 2019, 494, 1156–1165 CrossRef CAS.
- J. Wang, Q. Zhou, J. Liao, X. Ding, Q. Hu, X. He and C. H. Chen, ACS Appl. Energy Mater., 2019, 2, 7497–7503 CrossRef CAS.
- L. Zhao, J. Ni, H. Wang and L. Gao, RSC Adv., 2013, 3, 6650–6655 RSC.
- J. U. Choi, J. H. Jo, C. H. Jo, M. K. Cho, Y. J. Park, Y. Jin, H. Yashiro and S. T. Myung, J. Mater. Chem. A, 2019, 7, 13522–13530 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03034f |
| ‡ These authors are contribute equally. |
| This journal is © The Royal Society of Chemistry 2024 |