
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced ORR performance with biomass-derived freestanding catalyst layers: advancing mass transport in gas diffusion electrodes†
Mengnan
Wang
 a,
Jiaguang
Zhang
b,
Simon
Kellner
a,
Ifan E. L.
Stephens
c and
Maria-Magdalena
Titirici
*a
a,
Jiaguang
Zhang
b,
Simon
Kellner
a,
Ifan E. L.
Stephens
c and
Maria-Magdalena
Titirici
*a
aDepartment of Chemical Engineering, Imperial College London, South Kensington Campus, SW7 2AZ London, UK. E-mail: m.titirici@imperial.ac.uk
bDepartment of Chemistry, University of Lincoln, Bayford Pool, Lincoln LN6 7TS, UK
cDepartment of Materials, Imperial College London. White City Campus, 80 Wood Ln, London, W12 7TA, UK
First published on 9th September 2024
Abstract
Proton Exchange Membrane Fuel Cells (PEMFCs) are increasingly recognized as a pivotal technology for transitioning the energy sector towards net-zero emissions. The efficiency of PEMFCs largely hinges on the development of catalysts, especially for the Oxygen Reduction Reaction (ORR) at the cathode. Currently, commercial PEMFCs predominantly utilize Pt-based catalysts for their exceptional activity and stability, highlighting the need for high catalytic efficiency to mitigate Pt scarcity issues. However, beyond intrinsic activity enhancements in catalyst development, the architecture of the catalyst layer—typically carbon-based—emerges as a critical, yet underexplored, factor. Here, we introduce a novel catalyst layer derived from lignin, a plentiful biomass resource in woody materials, crafted through a dual templating strategy to yield an interconnected hierarchical structure. Our evaluation of this unique catalyst layer within a gas diffusion electrode setup revealed significant improvements in mass transport. These advancements offer a sustainable and effective pathway for next-generation catalyst layer innovations in PEMFCs, potentially accelerating the widespread commercialization of this green technology to decarbonize the energy sector.
Introduction
Catalysts, particularly for the ORR at the cathode, are pivotal for the efficiency of PEMFCs, which are crucial for achieving net-zero emissions in the energy sector. Despite their importance, current PEMFC designs predominantly rely on Pt-based catalysts for their exceptional activity and stability.1 Not only is it essential to use high-quality active materials such as nanostructured Pt2,3 and Pt based alloys,4–6 achieving optimal efficiency in PEMFCs also depends on the strategic organization and integration of active materials within the electrode structure.7 The architecture of the catalyst layer, often overlooked, is as critical as the catalysts themselves.8Traditional PEMFC catalysts layers combine carbon-supported platinum catalysts and an ionomer, serving both as proton conductor and binder. These components are combined to form an ink slurry that is subsequently deposited onto a membrane or a gas diffusion layer, resulting in a porous electrode structure. This ‘top-down’ methodology produces a stochastic electrode architecture, characterized by a network of ionomer and pore channels with a high degree of tortuosity.9 Such a configuration poses considerable challenges for mass transport, and consequently, results in sub-optimal catalyst utilization. Remarkably, the structure of electrodes used in today's commercial PEMFCs shows a significant degree of continuity with the original design introduced by Wilson and Gottesfeld, dating back over three decades.10,11 This persistence of conventional designs, despite their inherent limitations, underscores the challenge of implementing radical innovations in electrode architecture. A critical limitation in the preparation of these catalyst layers is the control over their microscopic architecture, particularly with respect to pore structures within the catalyst layer. The dielectric constant of the solvent and ionomer content in the catalyst ink profoundly influence the microstructure of PEMFC catalyst layers by affecting particle agglomeration and ink rheology, which are critical for optimizing layer porosity and integrity.12,13 In response to these challenges, a number of investigations have been dedicated to advancing alternative electrode structures.14–16
Enabled by breakthroughs in microfabrication and nanofabrication techniques, these studies exemplify the ongoing efforts in pioneering innovative electrode designs that could surpass traditional MEA architectures. One significant family of such structures developed is inverse opals, which offers outstanding interconnected structures and low tortuosity.17,18 However, this kind of structure usually suffers from the lack of a hierarchy to host and protect the active catalytic site, as well as to facilitate the multi scale transportation. Particularly, mesopores around 4–6 nm have been demonstrated to enhance catalyst–ionomer interactions by preventing the detrimental direct contact between them, while still facilitating efficient local transport near the active sites.19 This concept of utilizing mesopores has also been endorsed by industry advancements. For instance, Toyota has innovatively adopted mesoporous carbon nano dendrites (MCND), a form of carbon with inherent small mesopores that are impermeable to the ionomer. This strategic choice ensures that a significant portion of the Pt catalyst is embedded within the MCND, thereby minimizing the direct interaction between Pt and the ionomer and reducing the risk of sulfonic acid cover poisoning. By integrating this novel material with a platinum–cobalt (PtCo) alloy catalyst, Toyota has reported a commendable increase in catalytic activity, estimated at around 50%.7 Hence, the development of a hierarchical structure within the electrode catalysts layer holds great importance by interconnecting the catalytic sites, ensuring a rapid delivery of reactants and a swift removal of products.
Moreover, assessing the performance gains due to enhanced mass transport cannot be effectively achieved with the conventional rotating disk/ring electrode technique, hindered by the low oxygen solubility in the electrolyte. This results in a significant deviation in catalytic efficiency from that observed in membrane electrode assemblies (MEAs) within actual PEMFCs, where the operational current is substantially higher, up to a hundredfold, with the transport of oxygen, protons, electrons, and water being essential. The considerable amount of catalyst needed and the extensive time required for MEA evaluations have limited their application in the development and refinement of new catalyst layers. Nonetheless, advancements in the use of gas diffusion electrode (GDE) half-cell setups have proven to be a promising approach for investigating the effects of catalyst layer attributes on ORR performance, offering a closer approximation to the conditions within fuel cells and enhancing our comprehension of catalyst durability in practical electrochemical energy conversion systems.20
Last but not least, although carbon is commonly used as the main support for electrocatalysts due to its widespread availability and superior electrical conductivity, the production of standard carbon supports like carbon black and activated carbon predominantly depends on petroleum-based resources.21 This dependence stands in contrast to the broader objective of achieving decarbonization in future fuel cell technologies. Consequently, there is an urgent requirement to source carbon materials from more renewable resources, with biomass presenting itself as a viable and plentiful option. Lignin, a residual product from the paper and bio-refinery sectors, represents one such biomass source. Currently, only a minimal portion (<10% of 500–3600 million tonnes) of the industry's total lignin output is employed for chemical and material applications.22 Therefore, leveraging lignin as a precursor for electrode material synthesis not only promotes the utilization of biomass but also supports the PEMFC advancements, creating a mutually beneficial scenario for both sectors.
This study introduces a novel catalyst layer derived from lignin, utilizing a dual templating strategy to enhance mass transport and catalytic efficiency, aiming to address the limitations of current PEMFC catalyst designs.
Results and discussion
Nanostructure development
In the quest to develop nanostructured carbons, the study employed a dual templating process involving two distinct techniques to introduce mesoporosity and macroporosity in the carbon structure (Fig. 1). Initially, soft templating, using block copolymers, specifically Pluronic™ F127, was adopted to induce mesoporous structures into the synthesized carbons. This choice was guided by previous studies that have demonstrated the effectiveness of mesopores around 4 nm, templated by Pluronic™ F127, in improving the interactions between catalysts and ionomers, crucially eliminating harmful direct contact while still ensuring effective local transport around active sites.19,23 The process commenced with dissolving the polymer template in methanol, followed by the addition of organosolv lignin, phloroglucinol, and glyoxal to the dispersion.24 These carbon precursors, replete with hydroxy moieties, interact non-covalently with the PEO sequence of the template via hydrogen bonding, governing the ensuing organic–organic self-assembly process. The formation of micelles, guided by the incompatibility between hydrophilic and hydrophobic polymer blocks, led to the creation of a robust mesophase. The evaporation induced self-assembly (EISA) process then facilitated microphase separation within the block copolymers. Subsequent thermopolymerization at 85 °C stabilizes this ordered mesophase through cross-linking reactions with glyoxal, creating a three-dimensional network of covalently bridged phenols. This robust network supports the successful removal of the template, without compromising the structural integrity of the mesopores. The process concludes with pyrolysis at 1000 °C to yield the final mesoporous carbon. | ||
| Fig. 1 Schematic synthetic procedure for lignin derived carbon with mesoporous, macroporous and hierarchical structure. | ||
To further enhance mass transport throughout the catalyst layer, the strategic incorporation of macropores is essential. These macropores are designed to facilitate efficient flow and access within the layer. We identified polystyrene particles as particularly effective for this purpose due to their availability in various sizes and the simplicity of their removal by thermal decomposition during pyrolysis. The synthesis of polystyrene was undertaken employing an emulsion method,25 utilizing a 5% styrene solution in water. Following synthesis, the resulting emulsion was subjected to DLS measurements and SEM imaging after drying. Fig. S1† conspicuously illustrates that the synthesized polystyrene has narrow distributed size around 500 nm, exhibiting a polydispersity of merely 1.4%. This controlled synthesis underscores the suitability of polystyrene as a macropore template, aligning with the targeted design criteria for enhanced mass transport within the catalyst layer. To fabricate the inverse opal structure utilizing polystyrene, the precursor mixtures were prepared as described previously, with in-house synthesized polystyrene nanospheres being dispersed into the solution via probe sonication. This method produced a stable and homogeneous suspension, conducive to subsequent cross-linking and carbonization processes, ultimately resulting in the formation of a carbon structure embedded with both mesopores and macropores. Additionally, a control sample with only macropores was prepared using a similar approach but without employing soft templating.
Characterisation of the carbon films
After obtaining the 3 different types of freestanding carbon films, gas adsorption analysis using N2 was applied to evaluate the porosity of their structures. From the results shown in Fig. 2, it was observed that all three carbon structures shared a similar microporous volume, for pores smaller than 2 nm. This commonality underlined a consistent formation of microporosity across the various templating processes employed. A noteworthy distinction emerged when studying the mesoporosity. The pore size distribution plots, complemented by the characteristic isotherm hysteresis, clearly delineated the presence of ∼4 nm mesopores for both the hierarchical carbon and the mesoporous-only carbon. These features are consistent with the previously identified optimal pore size for ionomer interactions.19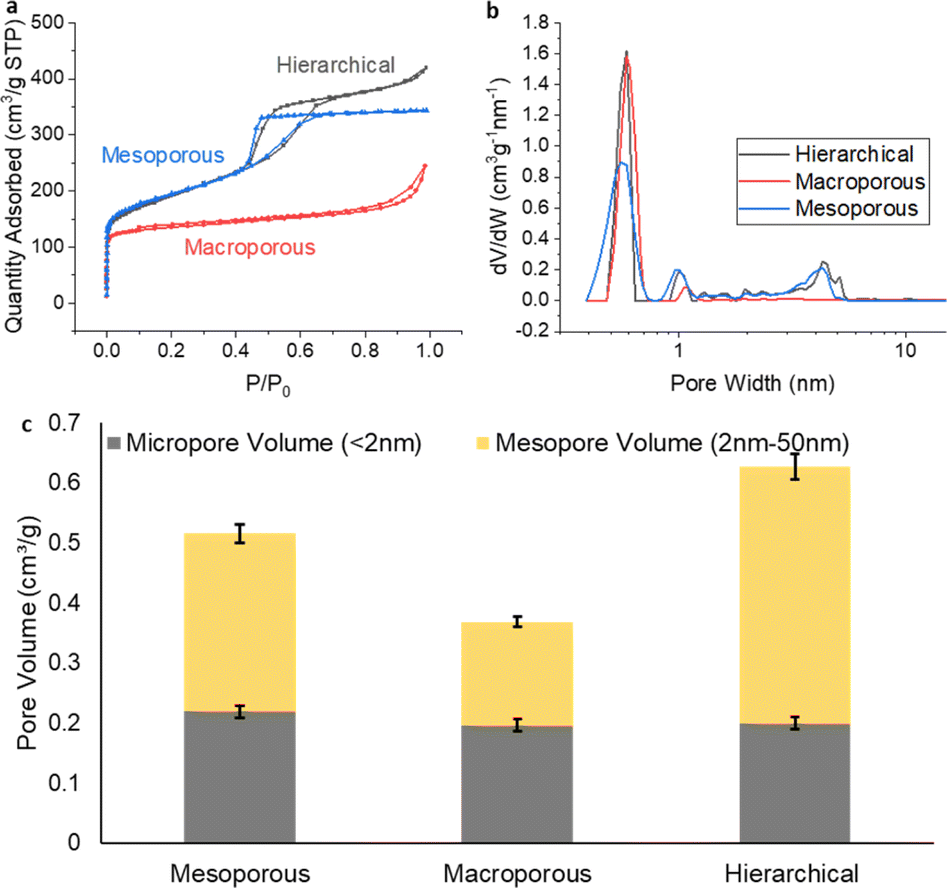 | ||
| Fig. 2 (a) N2 sorption isotherm, (b) NLDFT pore size distribution and (c) micropore and mesopore volume comparation for the three different carbon film. | ||
While the BET method could not resolve macropores explicitly, indirect evidence of their existence was inferred from the adsorption trends observed at high relative pressures (P/P0). This observation hinted at the presence of macropores within the carbon structure, specifically for the hierarchical and macroporous-only carbon. To verify the meso- and macropore structure established with the gas sorption results, SEM and TEM were used to image the morphology of the carbon materials (Fig. 3). Mesoporous channels are clearly shown on the TEM images for hierarchical (Fig. 3e and S2†) and mesoporous-only sample (Fig. 3d), while macropores with size of about 400 nm are observed in the SEM images for hierarchical (Fig. 3b and S3†) and macroporous-only carbon (Fig. 3c), supporting the conclusions from gas sorption analysis. It is worth noting that for the hierarchical materials, the bimodal porous structure is interconnected to form a continuous architecture, posing great potential for the mass transportation.
 | ||
| Fig. 3 (a–c) SEM and (d–f) TEM images display yielded carbons after pyrolysis at 1000 °C. | ||
Understanding the electrochemical accessibility of the carbon film
To further probe the electrochemical attributes of the distinct carbon structures developed in this research, we employed CV analysis using the carbon films as gas diffusion electrodes, in a 1 M HClO4 solution tested in GDE half-cell. As depicted in Fig. 4a, CV currents were normalized to the mass of each specific nanostructured carbon substrate. This standardization enables an effective comparative assessment between our carbon samples and benchmark data from the existing body of literature. An interesting observation emerged from the analysis – both the hierarchical and mesoporous-only carbon structures displayed comparable capacitance values. Contrarily, the material with only macroporous structure demonstrated a notably diminished capacitance.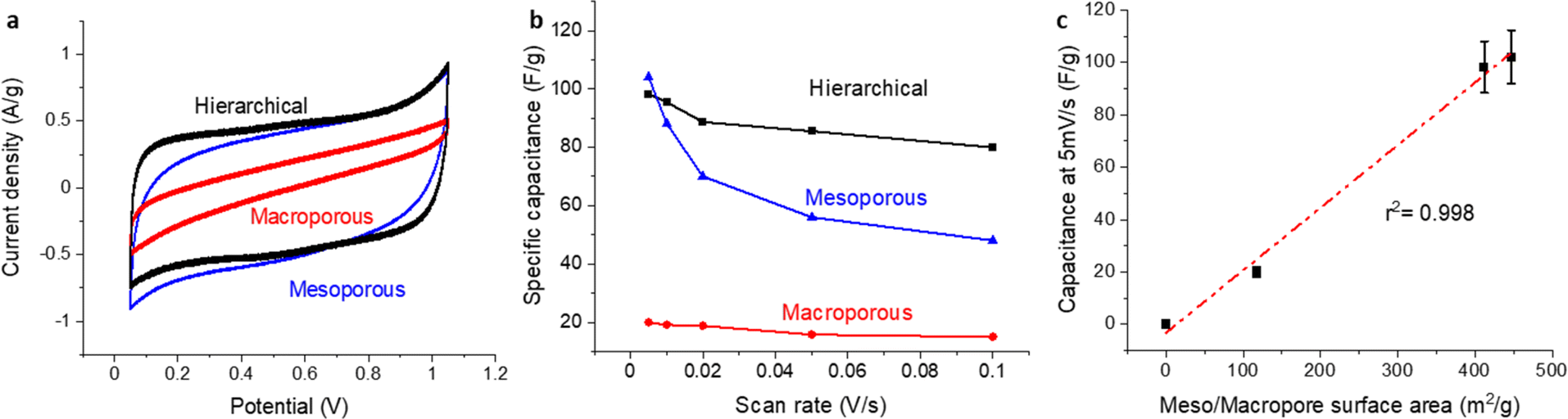 | ||
| Fig. 4 (a) CVs at a scan rate of 10 mV s−1 for various carbon-based materials, recorded in a gas diffusion half-cell setup using a 1 M HClO4 electrolyte at room temperature with 200 mL per min N2. Pt wire was used as the counter electrode in the same electrolyte chamber without membrane separation, RHE was used as the reference electrode in a different electrolyte chamber connected with Luggin capillary. Potential was corrected for IR compensation post measurements. (b) Their capacitance retention as a function of the scan rate. (c) The correlation between meso/macropore surface area and capacitance at 5 mV s−1. | ||
In a further effort to explore the electrochemical accessibility of these materials, we scrutinized the capacity retention at high scan rates, an investigative approach commonly employed within the field. Significantly, materials incorporating macroporous structures, namely, the hierarchical and macroporous-only materials, exhibited outstanding capacity retention. A marginal loss of 20% (Fig. 4b) implies a swift ion transport rate within the macropores, a testament to the advantageous characteristics intrinsic to macroporous structures.
Attempting to correlate capacitance to surface area for these materials, a definitive conclusion remained elusive when employing the BET surface area. Conversely, upon eliminating the microporous surface area, a strong correlation was achieved with an r2 of 0.998 (Fig. 4c). This result is in agreement with the literature which suggests that electrochemical accessibility can be challenging for microporous structures below 2 nm.26
In conclusion, our investigations suggest the potential of hierarchical carbon materials for future applications. These materials boast an ideal, ultra-low tortuosity microstructure, devoid of binders, and exhibit superior electrochemical accessibility. The findings of this study highlight the promise of these carbon materials as effective substrates for the deposition of Pt NPs, heralding a significant leap forward in the development of innovative catalyst layers for use in PEMFCs.
Improved ORR performance for catalysts layer with hierarchical architecture
Pt NPs were introduced to the free-standing electrodes as the active materials to investigate the effect of the catalyst architecture onto the mass transport for ORR. The resulting catalyst layers, with their deposited Pt, were then analysed via TEM, demonstrating successful preparation of Pt NPs on all three carbon substrates. The findings presented in Fig. 5 and S4† reveal that the Pt NPs, when deposited onto the hierarchical carbon material, exhibit remarkable dispersion. Meanwhile, a moderate degree of aggregation is observed on the alternative carbon substrates. These observations further underscore the beneficial influence of the hierarchical structure in enhancing diffusion phenomena. | ||
| Fig. 5 TEM images of the three catalyst layers with Pt nanoparticles deposited. | ||
The loading of Pt was regulated by the volume of precursor solution applied to the carbon film. The precise Pt loading was determined by extracting a circular 0.5 cm2 sample from the catalyst layer, followed by microwave-assisted digestion using aqua regia, with subsequent quantification via ICP-MS.
Prior to incorporating the previously characterized catalyst layers into the GDE half-cell (Fig. 6a), the electrode preparation entailed the application of a dilute Nafion ionomer solution (0.05 wt% in IPA) onto the catalyst layer, achieving a balanced 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ionomer to carbon mass ratio. Subsequently, polyvinylidene fluoride (PVDF) dissolved in N-methyl-2-pyrrolidone (NMP) was dispensed onto the rear of the catalysts layers. This treatment endowed the back of the electrode with hydrophobic properties to prevent electrolyte flooding, an essential feature that boosts the overall performance of the catalyst layer.
:1 ionomer to carbon mass ratio. Subsequently, polyvinylidene fluoride (PVDF) dissolved in N-methyl-2-pyrrolidone (NMP) was dispensed onto the rear of the catalysts layers. This treatment endowed the back of the electrode with hydrophobic properties to prevent electrolyte flooding, an essential feature that boosts the overall performance of the catalyst layer.
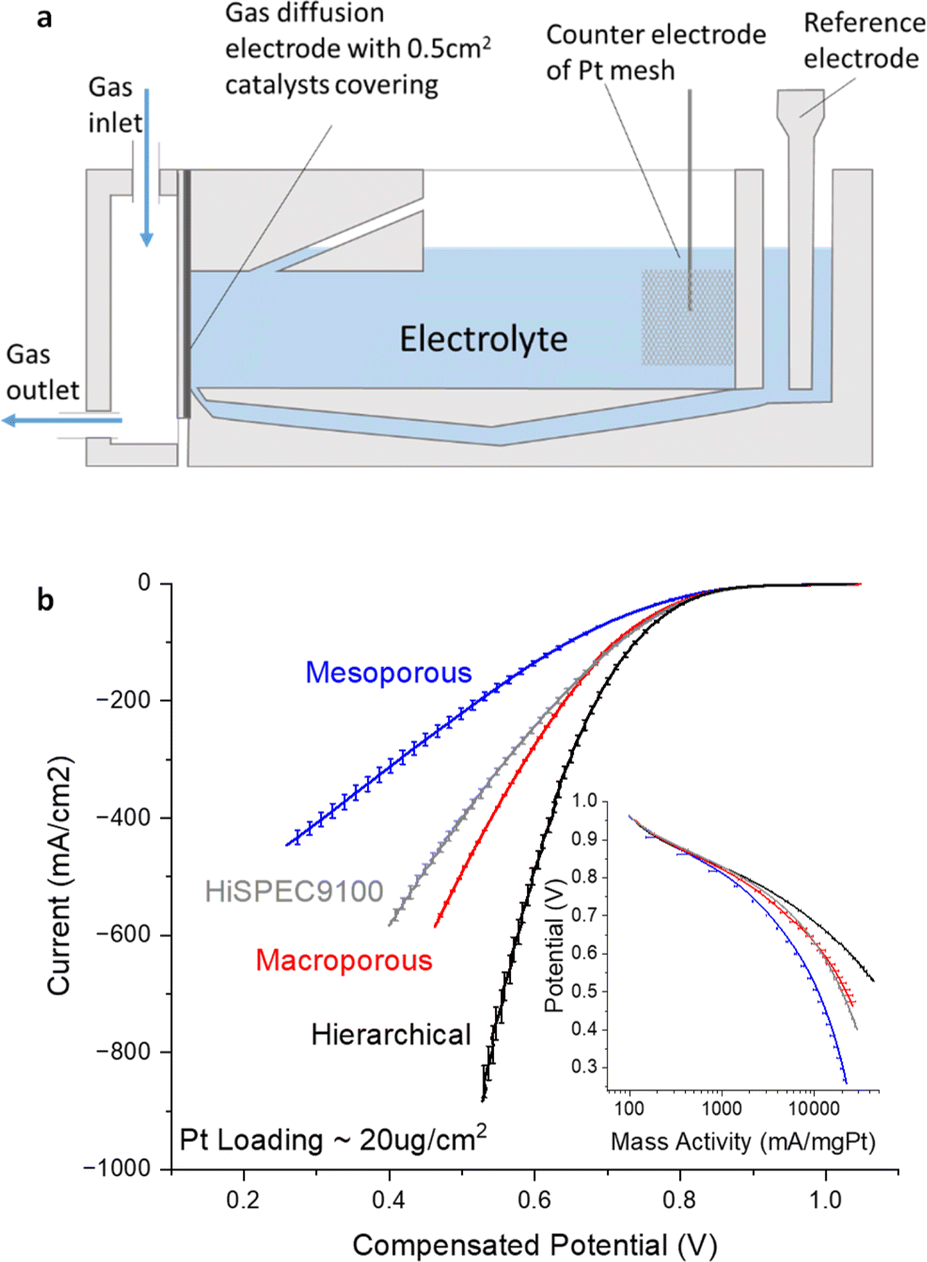 | ||
| Fig. 6 (a) The cross-sectional diagram of the GDE half-cell modified based on commercial Gaskatel FlexCell. (b) ORR polarization curves for all three catalyst layers. Inset: plot of logarithm of mass activity vs. potential, highlighting the similarity at low current densities. Measurements were done in 1 M HClO4 electrolyte at room temperature with 200 mL per min O2 and 10 mV s−1 scanning rate for LSV. Pt wire was used as the counter electrode in the same electrolyte chamber without membrane separation, RHE was used as the reference electrode in a different electrolyte chamber connected with Luggin capillary. Potential was corrected for IR compensation after measurements. Two electrodes were prepared and measured with ∼20 μgPt cm−2 loading to generate the polarization curves. | ||
Prior to evaluating the freestanding catalysts layers, the electrochemical cell was benchmarked using HiSPEC4000 commercial Pt/C catalysts as a standard reference. The results obtained from our experiments aligned closely with findings documented in existing literature (Fig. S5 and S6†).
When subjecting the prepared electrodes to the GDE half-cell under N2, the CVs presented in Fig. S7† displayed distinct hydrogen underpotential deposition (HUPD) peaks for all three catalysts representing the successful loading of Pt. The Electrochemical Surface Area (ECSA), determined HUPD, yielded values of 48.81 m2 g−1 for Pt/hierarchicalC, 43.51 m2 g−1 for Pt/macroporousC, and 21.91 m2 g−1 for Pt/mesoporousC. The notably lower ECSA observed for the Pt supported on mesoporous carbon is attributed to the aggregation of Pt nanoparticles within the mesoporous channels, which significantly diminishes the accessibility of Pt active sites. The double-layer capacitance aligns with that of the pristine carbon film without Pt introduction. The changes of specific capacitance across three samples are all below 13% suggesting that the integration of Pt did not significantly alter the accessibility of the nanostructures (Table S2†) Furthermore, Fig. 6b delineates the ORR performance of the various catalyst layers, highlighting pronounced disparities in the polarization curves. Given that at low current densities, where kinetics will dominate, the polarisation curves all coincide as illustrated in Fig. 6b inset, we attribute the differences at high current densities transport effects within the distinct porous architectures of the carbon materials.
The design of hierarchical structures, incorporating both meso- and macropores, promotes superior ion and mass transport, thereby enhancing electrochemical performance. On the other end of the spectrum, the electrode composed solely of mesoporous structures displayed pronounced transport limitations at high current region, exhibiting the least efficient ORR activity. The mesoporous structure likely poses hindrances to ion transport and the accessibility of Pt, essential for catalysing the ORR. The restrictive nature of the mesopores, coupled with their higher length-to-diameter ratio, may slow down the diffusion process and impede effective contact with the catalyst sites.
Conclusions
In conclusion, this study has demonstrated a significant advancement in electrochemical energy systems by developing a freestanding hierarchical electrode catalyst layer using lignin and a dual templating approach, emphasizing the critical role of electrode design in efficiency. The introduction of meso- and macroporosity, confirmed by SEM, TEM, and N2 sorption analysis, facilitated efficient mass transport. Electrochemical analysis showed that hierarchical and mesoporous structures had similar capacitance, with macroporous materials showcasing superior capacity retention, indicating faster ion transport. By the incorporation of Pt nanoparticles onto the freestanding electrode catalyst layers, improved mass transport capability of the hierarchical cathode catalyst layer was demonstrated in a GDE half-cell setup. These findings highlight the importance of hierarchical structures in improving catalyst layer efficiency and suggest future research should focus on optimizing catalytic site distribution and electrochemical performance.Experimental section
Preparation of polystyrene nanoparticles
Polystyrene nanoparticles were synthesized through an emulsion polymerization technique. The first step was to purify the styrene (purity ≥ 99%, Sigma-Aldrich) from the stabilizer by washing it using a solution of 2 M NaOH. After the complete removal of the stabilizer, the styrene was wash with DI water to ensure a neutral pH. A 3-neck round flask equipped with a stirrer, a reflux condenser, and a thermometer, was first charged with 180 mL DI water. And 0.08 g KPS (potassium persulfate, purity ≥ 99%, Sigma-Aldrich) was dissolved in 20 mL DI water and was deoxidized for 15 minutes. After the temperature in the flask reaching 80 °C, the batch was quickly charged with 11 mL styrene. The polymerization process was initiated by adding KPS. The batch temperature was maintained at 80 °C for 24 hours under argon and a white suspension containing polystyrene was obtained.To collect and wash the polystyrene nanoparticles, the suspension was sent to centrifuge at 10000 rpm for 15 minutes. The resulting precipitation was resuspended in DI water and centrifuged again at the same condition. The washing was repeated for 3 times and white polystyrene particles was obtained, which was subsequently dried in 80 °C for 24 hours.
Preparation of hierarchical carbon film via dual templating
To generate a hierarchical and continuous catalyst layer, a dual template consisting of soft template and hard template was incorporated and then removed. Polystyrene microparticles were used as hard templates here while Pluronic F-127 as soft template.In a typical synthesis, 0.335 g lignin, 0.335 g phloroglucinol (purity ≥ 99%, Sigma-Aldrich), 1.1675 g Pluronic® F-127 (Sigma-Aldrich) were dissolved in 32.5 mL methanol and along with 0.6 mL of glyoxal solution (40 wt% in H2O, Sigma-Aldrich).24 Afterwards, the mixture was sent to sonication bath until the Pluronic® F-127 was dissolved completely to present a brown suspension. Then 25 mL of suspension was transferred to a container and 1.015 g polystyrene nanoparticles was added into the suspension. Probe sonication was applied to ensure the polystyrene was well dispersed and a brown milky slurry was obtained.
The original slurries were then divided into two parts, one was left to slow evaporate and the self-assembly is induced during the evaporation, and the other part was air sprayed on the surface of carbon fibre paper (Freudenberg H23C8) using airbrush (Iwata CN gravity feed, 0.35 mm nozzle). And slurries with additives were all left to evaporated on the carbon fibre paper. After that, all of the samples were put under 85 °C for 24 hours to induce crosslinking, after which a polymeric film was obtained. In the end, all dried samples were sent to a tube furnace in 300 mL per min N2 flow with a heating rate of 1 °C min−1 from room temperature to 1000 °C, followed by maintenance at 1000 °C for 2 hours to carbonise the film and remove all the templates. Afterwards, the samples were cooled to near-ambient temperature in N2 flow and a catalyst layer of hierarchal mesopores was then obtained.
Preparation of macroprous-only and mesoporous-only carbon film
To produce carbon films with exclusively macropores or mesopores as control samples, the method described above was employed with slight modifications. For the sample containing only macropores, Pluronic® F-127 was omitted, whereas for the mesoporous-only sample, the use of PS as hard templates was excluded.Preparation of Pt/C catalysts on hierarchical carbon film
After obtaining the hierarchical carbon film from previous step, the preparation of Pt/C electrocatalysts on this freestanding hierarchical carbon film commenced with the formulation of a platinum precursor solution at a concentration of 1 mgPt mL−1 using H2PtCl6·xH2O (∼38% Pt basis, Sigma-Aldrich). This solution was meticulously sprayed onto the carbon film using an airbrush (Iwata CN gravity feed, 0.35 mm nozzle), taking great care to ensure a comprehensive penetration of the precursor into the structure of the carbon film. The spraying process was performed gradually, allowing the precursor to dry with each application, ensuring an even distribution across the surface and within the pores of the carbon film.Following the application and impregnation process, the carbon films were subjected to a reduction treatment to convert the platinum precursor into metallic platinum. This reduction was conducted at 350 °C for a duration of 2 hours, utilizing a controlled atmosphere with a 5% hydrogen in nitrogen gas flow.
Materials characterization
Nitrogen isothermal adsorption/desorption was conducted on a Micromeritics 3flex system at 77 K. Before measurements, ∼50 mg samples were degassed at 200 °C for 16 h under vacuum. Gases used were nitrogen (N5.2 CP grade) for adsorption/desorption and He (N5.0 CP grade) for the free-space measurement. The BET equation was used to estimate the total surface area, where the fitting region was determined by the Rouquerol method on 3Flex software version 5.02.27 The pore size distribution was calculated using slit geometry and heterogeneous surface-2D-NLDFT model in-built with 3Flex software. Regularization level was selected at the interception point of the error of fit plot and roughness of distribution plot. Scanning Electron Microscope (LEO Gemini 1252 FEG-SEM) was used to study the morphologies of samples operating at accelerating voltage of 3 keV and working distance around 3 mm. Transmission Electron Microscopy (TEM) was carried out for the direct imaging of Pt/C catalysts in a JEOL JEM-2100F at 200 kV and the images analysed with the Gatan software. The freestanding materials were grounded into powders before being dispersed in ethanol by sonicating for 15 min and left settling for 30 min. The upper solution was collected and deposited onto the TEM grids (Holey Carbon Films on 300 Mesh Copper Grids, agar scientific). The grid was then left dry overnight and stored in dry conditions before imaging. Dynamic light scattering measurements were performed using a Malvern Zetasizer MicroV, to determine the polystyrene size synthesized. Since the resulted emulsion were too concentrated for the instrument specifications, they were diluted 100 times. The measurements were repeated at least 3 times. In general, all the suspensions were found to be stable for the duration of the experiment. Before the Inductively Coupled Plasma Mass Spectrometry (ICP-MS) analysis with the Agilent 7850 ICP-MS, freestanding catalyst layers were extracted using a piece punch (ID 8 mm) to obtain an area of 0.5 cm2 for determining the geometrical Pt mass loading. These samples were subsequently digested in 69% HNO3 (Certified AR, Eur.Ph., for analysis Fisher Chemical™, Fisher Scientific) using a MARS 6 microwave operating at 1800 W for 25 minutes at a temperature of 215 °C. Upon digestion, the solutions were diluted to 2% HNO3 to prepare them for analysis. Calibration standards containing platinum (Pt) concentrations of 0, 25, 50, 100, 250, and 400 ppb were utilized to establish a calibration curve. To ensure accuracy and reproducibility, two duplicate measurements were taken for each sample, and the average value was computed. This rigorous methodology enabled a thorough assessment of the Pt content within the analysed samples.Electrochemical measurements
Gas diffusion electrode (GDE) measurement was carried out in an in-house modified commercial three-electrode half-cell from Gasketel. The half-cell was modified and benchmarked with commercial catalysts HiSPEC4000 (40 wt% Pt/C, Johnson Matthey) prior to in-house catalysts layer testing (Fig. S5 and S6†).The main and reference chamber was connected with a Haber-Luggin capillary channel and filled up with respective electrolyte for different measurements. A Pt wire was used as the counter electrode. An in-house built RHE was used as the reference electrode. Electrochemical tests were performed with 100% post measurement iR correction employing an AUTOLAB PGSTAT204 coupled with 10A booster in N2 (≥99.99998% BIP® Plus, Air Products) and O2 (≥99.9998% UltraPure Plus, Air Products) and saturated 1 M HClO4 (Suprapur®, Merck) electrolyte.
Before each test, N2 gas was purged to the electrolyte at 200 mL min−1 and the main chamber was sealed with tapes. Cyclic voltammetry was then measured for 50 cycles with N2 supply in the gas chamber in the range of 0.05–1 V vs. RHE at 500 mV s−1 to electrochemically clean the catalysts surface. The gas was then switched to O2 for 10 minutes and the impedance spectroscopy was measured at open-circuit potential (OCP) in the frequency range of 10 kHz to 100 Hz to obtain the resistance for iR correction. Lastly the linear sweep voltammetry (LSV) was performed from 1 V to 0.05 V vs. RHE at 10 mV s−1. The polarisation curve was plotted after compensating the working electrode potential with equation E = Emeasured − iR. All tests were repeated 2 times, reported in the paper are the average and the standard deviation (for LSV measurements).
ECSA calculation for platinum-based electrocatalysts was conducted using the HUPD method. CV scans were performed on the Pt/C electrodes (∼20 μgPt cm−2) and the hydrogen adsorption peaks were integrated from the CV curve (0.05 V to 0.4 V versus RHE), after subtracting the double layer charging current. This charge was then converted to ECSA by applying a specific capacity of 210 μC cm2, assuming monolayer hydrogen adsorption on platinum.
Data availability
The raw data supporting the findings of this study are summarized in an Excel file named “rawdata.Enhanced ORR Performance with Biomass-Derived Freestanding Catalyst Layers”, which is submitted alongside the manuscript. This dataset includes all relevant data necessary to understand and replicate the research findings reported in our article. No additional data processing analysis scripts or computational software code were developed or used in the preparation of this study. ESI accompanying this article includes additional data supporting the results presented in the main text and figures.† This ESI is available directly within the article's ESI files.†Author contributions
M. W. contributed to the conceptualization, investigation, methodology, data curation, formal analysis and writing. J. Z. contributed to the validation, visualization and editing. S. K. contributed to the validation. IELS and M. M. T. contributed to the resources, supervision and reviewing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by Programme Grant ‘Bio-derived and Bio-inspired Advanced Materials for Sustainable Industries’ (grant number EP/W031019/1). M. W would like to thank EPSRC Centre for Doctoral Training in the Advanced Characterisation of Materials (grant number EP/L015277/1). M.-M. T. thanks financial support from RAEng (CiET1819\2\60).References
- Y. Sun, S. Polani, F. Luo, S. Ott, P. Strasser and F. Dionigi, Nat. Commun., 2021, 12, 1–14 CrossRef PubMed.
- M. Li, Z. Zhao, T. Cheng, A. Fortunelli, C. Y. Chen, R. Yu, Q. Zhang, L. Gu, B. V. Merinov, Z. Lin, E. Zhu, T. Yu, Q. Jia, J. Guo, L. Zhang, W. A. Goddard, Y. Huang and X. Duan, Science, 2016, 354, 1414–1419 CrossRef CAS PubMed.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed.
- X. Tian, X. Zhao, Y. Q. Su, L. Wang, H. Wang, D. Dang, B. Chi, H. Liu, E. J. M. Hensen, X. W. Lou and B. Y. Xia, Science, 2019, 366, 850–856 CrossRef CAS PubMed.
- C. Kim, F. Dionigi, V. Beermann, X. Wang, T. Möller and P. Strasser, Adv. Mater., 2019, 31, 1805617 CrossRef PubMed.
- S. Mizuno, T. Hayashi*1, H. Kubo, M. Okumura, T. Kurihara and K. Mori, Toyota Tech. Rev., 2021, 66, 22 CAS.
- J. Fan, M. Chen, Z. Zhao, Z. Zhang, S. Ye, S. Xu, H. Wang and H. Li, Nat. Energy, 2021, 6, 475–486 CrossRef CAS.
- H. Wang, R. Wang, S. Sui, T. Sun, Y. Yan and S. Du, Automot. Innov., 2021, 4, 144–164 CrossRef.
- M. S. Wilson and S. Gottesfeld, J. Electrochem. Soc., 1992, 139, L28–L30 CrossRef CAS.
- C. Zawodzinski, M. S. Wilson and S. Gottesféld, ECS Proc. Vol., 1995, 1995–23, 57 CrossRef.
- K. B. Hatzell, M. B. Dixit, S. A. Berlinger and A. Z. Weber, J. Mater. Chem. A, 2017, 5, 20527–20533 RSC.
- Y. Guo, D. Yang, B. Li, D. Yang, P. Ming and C. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 27119–27128 CrossRef CAS PubMed.
- A. D. Taylor, E. Y. Kim, V. P. Humes, J. Kizuka and L. T. Thompson, J. Power Sources, 2007, 171, 101–106 CrossRef CAS.
- J. K. Koh, Y. Jeon, Y. Il Cho, J. H. Kim and Y. G. Shul, J. Mater. Chem. A, 2014, 2, 8652–8659 RSC.
- C. H. Lee, W. J. M. Kort-Kamp, H. Yu, D. A. Cullen, B. M. Patterson, T. A. Arman, S. Komini Babu, R. Mukundan, R. L. Borup and J. S. Spendelow, Nat. Energy, 2023, 8, 685–694 CrossRef CAS.
- O. H. Kim, Y. H. Cho, S. H. Kang, H. Y. Park, M. Kim, J. W. Lim, D. Y. Chung, M. J. Lee, H. Choe and Y. E. Sung, Nat. Commun., 2013, 4, 2473 CrossRef PubMed.
- M. Atwa, X. Li, Z. Wang, S. Dull, S. Xu, X. Tong, R. Tang, H. Nishihara, F. Prinz and V. Birss, Mater. Horiz., 2021, 8, 2451–2462 RSC.
- M. Wang, J. Zhang, S. Favero, H. Luo, L. Higgins, I. E. L. Stephens and M. Titirici, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-t446q.
- K. Ehelebe, N. Schmitt, G. Sievers, A. W. Jensen, A. Hrnjić, P. Collantes Jiménez, P. Kaiser, M. Geuß, Y. P. Ku, P. Jovanovič, K. J. J. Mayrhofer, B. Etzold, N. Hodnik, M. Escudero-Escribano, M. Arenz and S. Cherevko, ACS Energy Lett., 2022, 7, 816–826 CrossRef CAS.
- C. M. Long, M. A. Nascarella and P. A. Valberg, Environ. Pollut., 2013, 181, 271–286 CrossRef CAS PubMed.
- W. Zhang, X. Qiu, C. Wang, L. Zhong, F. Fu, J. Zhu, Z. Zhang, Y. Qin, D. Yang and C. C. Xu, Carbon Res., 2022, 11(1), 1–39 Search PubMed.
- V. Yarlagadda, M. K. Carpenter, T. E. Moylan, R. S. Kukreja, R. Koestner, W. Gu, L. Thompson and A. Kongkanand, ACS Energy Lett., 2018, 3, 618–621 CrossRef CAS.
- S. Herou, M. C. Ribadeneyra, R. Madhu, V. Araullo-Peters, A. Jensen, P. Schlee and M. Titirici, Green Chem., 2019, 21, 550–559 RSC.
- A. B. D. Nandiyanto, A. Suhendi, T. Ogi, R. Umemoto and K. Okuyama, Chem. Eng. J., 2014, 256, 421–430 CrossRef CAS.
- B. Kastening and M. Heins, Electrochim. Acta, 2005, 50, 2487–2498 CrossRef CAS.
- J. Rouquerol, P. Llewellyn and F. Rouquerol, Stud. Surf. Sci. Catal., 2007, 160, 49–56 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03864a |
| This journal is © The Royal Society of Chemistry 2024 |