
Interfacial charge transfer driven by surface termination-controlled Ti2C MXene for enhanced hydrogen storage in magnesium†
Min Gyu
Kim
a,
ShinYoung
Kang
b,
Brandon C.
Wood
b and
Eun Seon
Cho
 *a
*a
aDepartment of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Yuseong-gu, Daejeon 34141, Republic of Korea. E-mail: escho@kaist.ac.kr; Tel: +82-42-350-3912
bLaboratory for Energy Application for the Future (LEAF), Lawrence Livermore National Laboratory, Livermore, California 94550, USA
First published on 15th August 2024
Abstract
Two-dimensional transition metal carbides and nitrides (MXenes) with layered structure and high conductivity have been effectively utilized in various energy materials, including as a catalytic support of MgH2 for hydrogen storage. However, the terminal groups formed on the surface during the etching step tend to deactivate reactive Mg metal and add dead mass, thereby deteriorating the catalytic role of MXene in hydrogen sorption of Mg. We exploited a molten-salt derived MXene with easily modifiable –Cl terminations, compared to conventional –O, –OH and –F groups, and synthesized a composite of Mg and delaminated Ti2CClx MXene to improve the hydrogen storage performance of Mg through charge transfer. This strategy enables the formation of an intimate interface between Mg and MXene, facilitating charge transfer and thereby boosting the catalytic effect. The resulting composite demonstrates significantly enhanced hydrogen sorption kinetics by modulating Mg–H bond strength. This novel approach of modifying surface terminations leverages the unique properties of MXene as a superior support for active materials, offering broader applications in energy materials.
1. Introduction
With growing apprehensions on the environmental impact of CO2 emission, substituting traditional fossil fuels with renewable energy sources is imperative.1,2 Hydrogen stands out as a promising energy carrier due to its high energy density, clean-burning feature and versatility, thereby enabling us to store a tremendous amount of energy generated from renewable sources. The efficient storage of hydrogen is a crucial aspect that impacts its viability as a widespread energy carrier.3 Solid-state hydrogen storage offers considerable benefits compared to conventional pressurized gas or liquefied hydrogen storage in terms of safety and efficiency.4 Particularly, magnesium hydride (MgH2) has advantages of high theoretical gravimetric capacity (7.6 wt%), good reversibility and abundant natural resources,5–7 while it faces major challenges such as high operating temperature requirements and sluggish reaction kinetics due to a strong Mg–H bonding nature.8,9Introducing catalytic additives to MgH2 such as transition metals10–12 and their corresponding oxides13,14 and carbides15,16 is one of the feasible strategies to improve its hydrogen storage properties by providing additional activation sites or reaction pathways for hydrogen.17 Recently, two-dimensional (2D) transition metal carbides and nitrides (MXenes) have been found to show a great catalytic activity for enhancing hydrogen sorption property of MgH2.18–21 Compared to typical transition metal-based catalysts which are sporadically doped within MgH2 as clusters,18 atomically thin 2D structured MXene offers the potential to effectively provide a number of active catalytic sites at the interface with MgH2. Although previous studies have shown the validity of MXene as a catalyst for MgH2, the underlying mechanism behind the catalytic effect is not adequately understood.22 Particularly, studies on Ti-based MXene have suggested that the metallic Ti acts as a key catalytic component by forming an intermediate titanium hydride phase.18,21,23 However, as metallic Ti is precipitated out of the carbide structure, the 2D structure and properties of a metal–carbon composite might undergo changes, causing it to lose its identity and ceasing its role as a distinct nanomaterial. Apart from the effect of precipitated Ti, the catalytic role of MXene as a metal-carbide has been investigated through computational simulations, yet corresponding experimental observations are currently lacking.24–26
On the other hand, during the MAX phase etching reaction for the synthesis of MXene, terminal groups such as ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, –OH, and –F are inevitably formed on the surface.27,28 However, upon compositing with Mg for the aforementioned catalytic effect, Mg is susceptible to oxidation with these groups, resulting in a possible creation of passivation layers which hinder the reaction kinetics and reduce the active mass.21 To avoid such issues, a post-modification on surface termination of MXene is essential, while the robust Ti–O and Ti–F bonds introduced during the etching step in acid media make it challenging to replace surface groups through a post-synthetic route.29 A newly developed synthetic method produces MXenes with –Cl and –Br terminations by using a molten salt, in place of oxygen or fluorine.29–31 The Ti–Cl and Ti–Br bonds are relatively labile and can be substituted or eliminated to create a variety of MXene derivatives.29,32 Despite such development, studies about elimination of surface termination from MXene remain limited in using LiH as a reducing agent assisted by molten-salt, which involves multiple steps and harsh reaction conditions.29
O, –OH, and –F are inevitably formed on the surface.27,28 However, upon compositing with Mg for the aforementioned catalytic effect, Mg is susceptible to oxidation with these groups, resulting in a possible creation of passivation layers which hinder the reaction kinetics and reduce the active mass.21 To avoid such issues, a post-modification on surface termination of MXene is essential, while the robust Ti–O and Ti–F bonds introduced during the etching step in acid media make it challenging to replace surface groups through a post-synthetic route.29 A newly developed synthetic method produces MXenes with –Cl and –Br terminations by using a molten salt, in place of oxygen or fluorine.29–31 The Ti–Cl and Ti–Br bonds are relatively labile and can be substituted or eliminated to create a variety of MXene derivatives.29,32 Despite such development, studies about elimination of surface termination from MXene remain limited in using LiH as a reducing agent assisted by molten-salt, which involves multiple steps and harsh reaction conditions.29
In this work, we present a facile synthesis of MXene with the removal of the –Cl terminal group (denoted as –Clx) via a molten-salt etching method for boosting the catalytic effect on hydrogen sorption kinetics of Mg upon the formation of a composite (Mg@DL-Ti2CClx) with delaminated Ti2CCl2 MXene (DL-Ti2CCl2). The synthesized composite allows the formation of a Ti–Mg interface which has a catalytic effect on the hydrogen storage property of MgH2. Interestingly, charge redistribution occurs between MgH2 and DL-Ti2CClx, resulting in a weakening of the strength of the Mg–H bond strength, which in turn improves hydrogen sorption properties. Additionally, DFT calculations also show that the strength of the interaction depends on the amount of –Cl present, with a lower –Cl content resulting in a stronger interaction. This implies that controlling the terminal groups of MXene is crucial for further enhancing its catalytic function as a support for MgH2.
2. Results and discussion
The synthesis of Mg@DL-Ti2CClx involves several key steps, as illustrated in Fig. 1a. Multilayer-Ti2CCl2 (ML-Ti2CCl2) was prepared from the MAX phase via a modified molten-salt etching method.29,30 This was followed by delamination with lithium pyrene (LiPy) to form DL-Ti2CCl2, and simultaneous reduction of DL-Ti2CCl2 and the Mg2+ precursor was performed to form Mg@DL-Ti2CClx (see ESI† for the detailed procedure). The scanning electron microscopy (SEM) image of ML-Ti2CCl2 in Fig. 1b shows a distinctive lamellar microstructure formed by the removal of the Al layer from the Ti2AlC MAX phase and the decoration with –Cl terminal groups from the chloride salt. The SEM image (Fig. S1a†) of the Ti2AlC MAX phase displays a combined structure of the Ti2AlC and excess metal (Ti, Al) precursors,30,33 which are eliminated during the etching step. Despite the absence of the Al layer, the ML-Ti2CCl2 MXene synthesized via the molten-salt etching method maintains a laminated structure due to van der Waals forces, posing challenges for good dispersion during solution-based synthesis and full utilization of its large surface area. Thus, a delamination process is employed, exposing the –Cl terminated basal plane on the surface before the composite synthesis. In previous studies, molten-salt etched MXenes were delaminated using n-butyllithium (n-buLi),29,34 which has been widely used for transition-metal dichalcogenides via Li+ ion intercalation.35,36 However, a pyrophoric feature of n-buLi could pose a substantial safety hazard, especially when produced in a large scale for hydrogen storage systems. In this work, we employed lithium pyrene (LiPy) for a safe and more effective delamination procedure, in which Li+ ions are intercalated into ML-Ti2CCl2 layers owing to a lower redox potential, enabling successful delamination via simple ultrasonication.37 The two-dimensional structure of the delaminated-Ti2CCl2 (DL-Ti2CCl2) is shown by transmission electron microscopy (TEM) and SEM images (Fig. 1c and S1b†). The DL-Ti2CCl2 forms a stable colloidal solution in a polar solvent like N-methyl formamide (Fig. S1c†), and its hexagonal crystallinity is retained after the delamination process as shown in the selected area electron diffraction (SAED) pattern (Fig. S2†). | ||
| Fig. 1 Scheme and morphology change of the MXene and Mg composite by each synthetic step. (a) Schematic illustration of Mg@DL-Ti2CClx composite preparation. (b) Scanning electron microscopy (SEM) image of ML-Ti2CCl2. Transmission electron microscopy (TEM) image of (c) DL-Ti2CCl2 and (d) Mg@DL-Ti2CClx. (e) High-angle annular dark-field (HAADF) image of Mg@DL-Ti2CClx and energy-dispersive X-ray spectroscopy (EDS) mapping of Mg, Ti and Cl representing evenly distributed Mg over DL-Ti2CClx. | ||
To synthesize the Mg@DL-Ti2CClx composite, a strong reducing agent – lithium biphenyl (LiBp) – was utilized in this work. In addition to reducing the Mg2+ precursor to Mg nanoparticles,38,39 LiBp also partially eliminates the –Cl terminations from the DL-Ti2CCl2 surface, forming DL-Ti2CClx, which will be discussed in the later section. This removal of –Cl functional groups exposes the titanium carbide surface, facilitating the formation of a direct Ti–Mg catalytic interface. In other words, the synthesis of Mg@DL-Ti2CClx is accomplished through the removal of the –Cl terminal group from the DL-Ti2CCl2 layer along with a simultaneous reduction of Mg2+ precursors using LiBp. High-angle annular dark-field (HAADF) scanning transmission electron microscopy (STEM) with energy-dispersive X-ray spectroscopy (EDS) and bright-field TEM images (Fig. 1d and e) show the composite of Mg and DL-Ti2CClx, demonstrating that Mg is homogeneously decorated on DL-Ti2CClx over the Mg@DL-Ti2CClx composite with residual –Cl termination (Fig. 1e).
To elucidate the effect of LiBp treatment on the elimination of –Cl termination, the MXenes – ML-Ti2CCl2 and DL-Ti2CCl2 – were solely treated with LiBp. The SEM EDS spectra show that the amount of elemental Cl over Ti2CCl2 layers is significantly reduced with the LiBp treatment to form ML-Ti2CClx (Fig. S3 and Table S1†). In the subsequently obtained X-ray diffraction (XRD) pattern (Fig. S4a†), the (11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 0) peak whose position highly depends on the surface termination is shifted from 57.0° to 60.2°, implying the change of terminal groups.29 In addition, the substantially reduced (0001) peak and the shifted (110) peak to 60.2° are consistent with the previously reported result on bare-Ti2C, in which case the terminal groups were found to be completely removed.29 The reduction of the peak assigned to –Cl termination in the Raman spectra also supports that the LiBp treatment is effective for removal of –Cl termination (Fig. S4b†). The c lattice parameters were acquired from the high-resolution TEM (HRTEM) image for both ML-Ti2CCl2 and ML-Ti2CClx, which are c = 8.76 Å and c = 10.46 Å, respectively (Fig. S4c and d†), closely aligned with the reported values in the literature.29 The c lattice parameter of ML-Ti2CCl2 is consistent with the XRD result, as shown in Fig. S4a,† while the c value increases upon the LiBp-treatment because of space group change from
0) peak whose position highly depends on the surface termination is shifted from 57.0° to 60.2°, implying the change of terminal groups.29 In addition, the substantially reduced (0001) peak and the shifted (110) peak to 60.2° are consistent with the previously reported result on bare-Ti2C, in which case the terminal groups were found to be completely removed.29 The reduction of the peak assigned to –Cl termination in the Raman spectra also supports that the LiBp treatment is effective for removal of –Cl termination (Fig. S4b†). The c lattice parameters were acquired from the high-resolution TEM (HRTEM) image for both ML-Ti2CCl2 and ML-Ti2CClx, which are c = 8.76 Å and c = 10.46 Å, respectively (Fig. S4c and d†), closely aligned with the reported values in the literature.29 The c lattice parameter of ML-Ti2CCl2 is consistent with the XRD result, as shown in Fig. S4a,† while the c value increases upon the LiBp-treatment because of space group change from ![[P with combining macron]](https://www.rsc.org/images/entities/i_char_0050_0304.gif) 3m1 to P63/mmc. The change in the Ti–C bond with the loss of the –Cl terminal group was investigated by the X-ray photoelectron spectroscopy (XPS) analysis in DL-Ti2CCl2 which was used during synthesis in delaminated form (Fig. S5†). The peaks for the Ti–C bond are shifted to lower binding energies in both the Ti 2p and C 1s regions as the electronegative element Cl disappears. Additionally, Ti with a higher oxidation state is observed, attributed to the amorphous Ti oxide on the surface, which is reduced by LiBp-treatment. It is noteworthy that the conventional approaches to modifying functional groups over MXene require high temperatures (300–600 °C)21,40 or molten-salt assisted reactions,29 whereas LiBp readily removes functional groups under mild conditions. Even though a non-negligible amount of Cl is detected in the SEM-EDS analysis of ML-Ti2CClx (Fig. S3 and Table S1†) and TEM-EDS mapping of DL-Ti2CClx (Fig. S6†), the –Cl removal is quite evident and is thought to be sufficient to expose a bare Ti surface.
3m1 to P63/mmc. The change in the Ti–C bond with the loss of the –Cl terminal group was investigated by the X-ray photoelectron spectroscopy (XPS) analysis in DL-Ti2CCl2 which was used during synthesis in delaminated form (Fig. S5†). The peaks for the Ti–C bond are shifted to lower binding energies in both the Ti 2p and C 1s regions as the electronegative element Cl disappears. Additionally, Ti with a higher oxidation state is observed, attributed to the amorphous Ti oxide on the surface, which is reduced by LiBp-treatment. It is noteworthy that the conventional approaches to modifying functional groups over MXene require high temperatures (300–600 °C)21,40 or molten-salt assisted reactions,29 whereas LiBp readily removes functional groups under mild conditions. Even though a non-negligible amount of Cl is detected in the SEM-EDS analysis of ML-Ti2CClx (Fig. S3 and Table S1†) and TEM-EDS mapping of DL-Ti2CClx (Fig. S6†), the –Cl removal is quite evident and is thought to be sufficient to expose a bare Ti surface.
The change of the crystalline structure and terminal group over MXene was further investigated. The powder XRD pattern of ML-Ti2CCl2 shows distinct peaks corresponding to Ti2CCl2, indicating the complete etching of the Al layer from the Ti2AlC MAX phase.29,34 Prior to the delamination to form DL-Ti2CCl2, an expansion in interlayer spacing is observed in ML-Ti2CCl2 due to Li+ intercalation, shifting the (0001) peak from c = 8.67 Å to c = 8.88 Å (Fig. S7†).29 The DL-Ti2CCl2 shows a series of (000l) basal plane peaks since the delaminated sheets are horizontally aligned by vacuum-filtration.29,41 The Mg@DL-Ti2CClx shows a clear characteristic metallic Mg pattern with the (110) peak of LiBp-treated Ti2CCl2, indicating the simultaneous partial removal of the –Cl terminal group. The formation of the composite was also confirmed by SAED, which shows a clear single-crystalline pattern from DL-Ti2CClx and the polycrystalline ring from decorated Mg particles (Fig. S8†). The change of surface termination on MXene was also identified through Raman spectroscopy (Fig. 2b). The out-of-plane vibration of Ti–C with –Cl termination is observed at 225 cm−1 (A1g) for ML-Ti2CCl2.34 DL-Ti2CCl2 retains the A1g peak at 225 cm−1 which indicates that –Cl termination is well preserved during the delamination process, while Mg@DL-Ti2CClx shows a peak at 341 cm−1 (A1g) which corresponds to the out-of-plane stretching mode of Ti–C without surface termination,42 and the A1g peak associated with the –Cl termination at 225 cm−1 is significantly diminished. These results suggest that most of the –Cl terminal groups are removed under the highly reductive environment with LiBp, thereby allowing the direct interaction of Mg with Ti atoms in MXene.
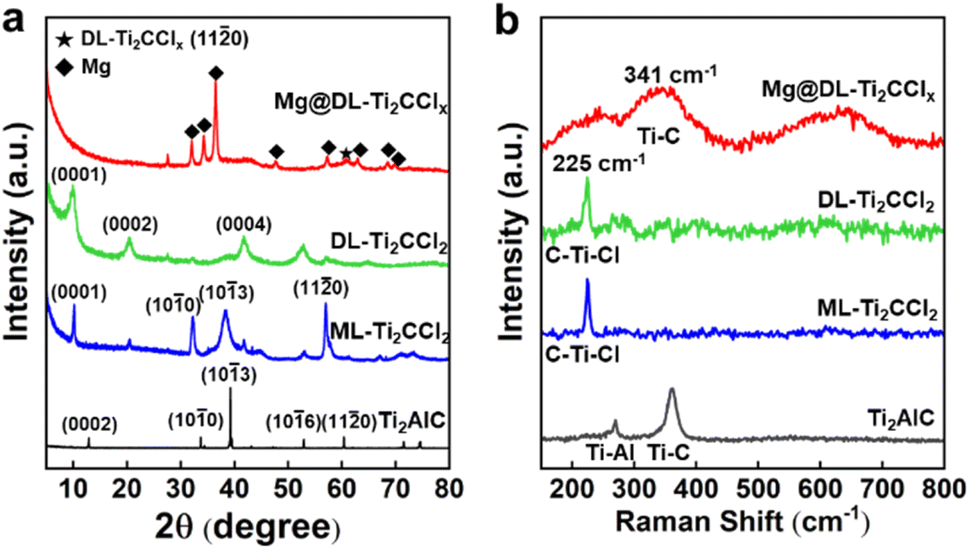 | ||
| Fig. 2 Change of the crystalline and chemical structures of the Ti2AlC MAX phase, ML-Ti2CCl2, DL-Ti2CCl2 and Mg@DL-Ti2CClx composite based on (a) X-ray diffraction (XRD) patterns and (b) Raman spectra. | ||
In order to examine the hydrogen sorption properties of Mg@DL-Ti2CClx, isothermal ab/desorption kinetics was measured with Sievert's apparatus (Fig. 3a and b). In the first cycle, 4.1 wt% of H2 is absorbed within 20 min; however, the kinetics gets sluggish in the subsequent cycle, while its rate is maintained afterwards. On the other hand, the desorption is completed within 20 minutes and maintains a stable rate. To understand these phenomena, we consider the hydrogen sorption process of Mg metal. The H2 absorption follows several sequential steps: (1) dissociation of hydrogen molecule on the surface, (2) diffusion of hydrogen atom into metal and (3) nucleation and growth of hydride phase. Reversely, desorption from MgH2 involves: (1) formation of metallic Mg phase, (2) diffusion of hydrogen atom from hydride phase to metallic Mg phase and (3) recombination of hydrogen atoms to form hydrogen molecule.43,44 It can be assumed that the absorption rate decreases since Mg particles are sintered during the H2 ab/desorption cycle.45,46 Since MgH2 melts at a lower temperature (327 °C) compared to Mg (650 °C), sintering is likely to occur in the hydrogenated state. Increased Mg grain size reduces the surface area for hydrogen dissociation and slows hydrogen atom diffusion to the grain core.47,48 The grain size growth is also evident in the XRD result (Fig. S9a†), in which the initially broad Mg peaks for as-synthesized Mg@DL-Ti2CClx become narrow after H2 sorption cycles, yet the characteristic (110) MXene peak is preserved which shows that DL-Ti2CClx retains its structure (Fig. S9b†). The sintering primarily occurs during the initial cycle, possibly because the DL-Ti2CClx within the composite acts as a physical barrier, preventing further excessive sintering once it reaches a certain equilibrium state. The structural stability is confirmed by preservation of the hexagonal crystallinity of DL-Ti2CClx in the composite after the hydrogen sorption cycle, as shown in the SAED pattern (Fig. S10†).
 | ||
| Fig. 3 Hydrogen sorption kinetics of the Mg@DL-Ti2CClx composite. (a) Isothermal hydrogen absorption at 200 °C under 15 bar of H2 and (b) hydrogen desorption at 300 °C under 0 bar of H2. Activation barrier for (c) absorption and (d) desorption as a function of absorbed and desorbed wt% H2, respectively. | ||
Nevertheless, the desorption rate of Mg@DL-Ti2CClx is not significantly affected as the hydrogen cycle proceeds, compared to the absorption (Fig. 3b). It is assumed that this unusual discrepancy arises from the different reaction mechanism governing the hydrogen ab/desorption process. Typically, MgH2 undergoes a localized charge transfer step between Mg2+ and H− during desorption.49,50 However, due to the lack of free electrons in the conduction band of semi-conductive MgH2 at the fully hydrogenated state, the nucleation of metallic Mg should be preceded which is commonly referred to as an incubation period.50–52 Interestingly, the desorption of Mg@DL-Ti2CClx shows negligible incubation time at the initial desorption stage. We speculate that the high electrical conductivity of DL-Ti2CClx, especially the –Cl removed region,53 allows it to act as a charge transfer intermediate, facilitating hydrogen recombination without a prior incubation period. For comparison, we additionally synthesized the composite of Mg and conventional Ti2CTx (T = –O, –OH and –F) MXene instead of DL-Ti2CCl2 (Mg@Ti2CTx). In the isothermal hydrogen ab/desorption of Mg@Ti2CTx, it is observed that the rate gradually decreases for both cases, along with a typical incubation period at the initial stage of desorption, as opposed to Mg@DL-Ti2CClx (Fig. S11†). This suggests that the catalytic activity of Ti2CTx deteriorates as the hydrogen cycle progresses, which implies the collapse of the Ti2CTx MXene structure. MXene synthesized with an acid solution contains F− ions (LiF/HCl or HF), and it might become structurally unstable because of the formation of atomic defects.27,54 Hence, unlike DL-Ti2CClx, Ti2CTx might be more vulnerable under a hydrogen cycle environment, potentially leading to the loss of its catalytic function and its role as a physical barrier. Furthermore, it was reported that MXene with a terminal group could cause the formation of a passivation layer at the interface of Mg,21 suggesting that the elimination of terminal groups is crucial when creating the composite of Mg and MXene. Despite using LiBp as a reducing agent, the removal of terminal groups from conventional Ti2CTx is difficult to achieve, supported by SEM-EDS analysis and Raman spectroscopy (Fig. S12 and Table S2†). Unlike ML-Ti2CCl2, the terminal groups mostly remain, and both the out-of-plane vibration (A1g) and in-plane vibration of Ti–C (Eg) with terminal groups are observed. Presumably, harsher conditions are required to break the terminal groups attached to Ti2CTx. The key factor for creating an efficient catalytic interface lies in the utilization of –Cl decorated MXene and its subsequent removal through LiBp treatment, as demonstrated in this study.
In order to further probe the hydrogen sorption kinetics of Mg@DL-Ti2CClx, we evaluated activation energies by fitting hydrogen ab/desorption kinetic curves at three different temperatures (Fig. S13†) to the Arrhenius law (Fig. 3c and d) and Johnson–Mehl–Avrami (JMA) equation (Fig. S14 and S15†). The hydrogen ab/desorption kinetic curves and activation barrier plots solely based on the mass of Mg are additionally shown in Fig. S16,† while the overall trend tends to be consistent with the one based on the total composite mass. During absorption, Mg@DL-Ti2CClx displays lower activation energy values than the barriers identified in previous studies on hydrogen dissociation on the Mg surface (Ea ≈ 83.9–101.3 kJ mol−1 H2)55–57 and H atom diffusion through MgH2 (Ea ≈ 95.5 kJ mol−1 H2).58,59 Thus, we anticipate that the energy barriers for both surface and bulk processes are alleviated in the hydrogen absorption kinetics of Mg@DL-Ti2CClx. The activation energy acquired from the JMA equation (Ea ≈ 53.5 kJ mol−1 H2) is also aligned with it, while providing additional information regarding the dimensionality of the growth. The best fitting was obtained with an Avrami exponent (n) of n = 1.0, indicating that the growth of MgH2 occurs in one-dimensional geometry which can be described by hydrogen diffusion through lattice vacancies followed by thickening of the MgH2 layer in one-dimension as proposed in the literature.60,61 Furthermore, the activation energy desorption for Mg@DL-Ti2CClx determined from both the Arrhenius law (Fig. 3d) and JMA equation (Fig. S15d†) is found to be lower than that for other Mg composites (Ea ≈ 140–219 kJ mol−1 H2)59,62–64 which are synthesized through a similar Rieke method.65 This improvement on desorption kinetics is believed to originate from the weakening of the Mg–H bond caused by charge transfer which will be discussed later. A notable characteristic is that the n value of desorption kinetics from the JMA fitting displays a non-integer value (n = 0.5–0.7). This suggests that the desorption process may occur via diffusion-controlled one-dimensional growth with instantaneous nucleation which aligns with a negligible incubation period as described previously.66
To unravel the in-depth catalytic effect of DL-Ti2CClx on the hydrogen storage performance of Mg@DL-Ti2CClx at the interface of Ti–Mg, the change of the chemical state before and after H2 sorption cycles was investigated through XPS analysis (Fig. 4a and b). As-synthesized Mg@DL-Ti2CClx exhibits a comparable binding energy for the Ti–C bond to DL-Ti2CClx in both Ti 2p and C 1s spectra (Fig. S5a and b†); however, the relevant peaks are shifted to the lower binding energy after hydrogen sorption cycling, which clearly shows the charge redistribution in the Mg@DL-Ti2CClx composite which was induced during the cycling with the existence of Mg. The consistent shift of the Ti–C peak to a lower binding energy in the Ti 2p and C 1s XPS results indicates that charge transfer from Mg to Ti–C has occurred. The retention of the transition metal related peak in the C 1s spectra after the hydrogen sorption process indicates that the Ti–C bonding is well preserved. This aligns with the earlier explanation of structural stability. In addition, the hexagonal crystallinity of DL-Ti2CClx in the composite is well maintained after the hydrogen sorption cycle (Fig. S10†). This structural stability is attributed to the molten-salt etching method, wherein hydrofluoric acid (HF) was intentionally excluded from the synthesis process. To the best of our knowledge, our study is the first experimental demonstration of this distinct charge transfer between Mg and Ti–C in the composite of Mg and MXene. This charge transfer effect was further supported by Ti L-edge near edge X-ray absorption fine structure (NEXAFS) and Raman spectroscopy (Fig. 4c and d).67,68 The Ti L-edge spectrum is related to electron excitation from Ti 2p3/2 (L3) and 2p1/2 (L2) core levels to 3d states. As the 3d orbital energy level is split by the crystal field effect, each Ti L3 edge and L2 edge is further divided into t2g and eg subpeaks.69 In the hydrogen cycled sample, the t2g peaks become more intense at the both L2 and L3 edges, accompanied by the loss of the clear splitting and a shift of the pre-edge towards the lower energy, indicating a lower oxidation state of Ti in MXene.70–72 The observation of a blue shift (341 cm−1 to 360 cm−1) in the A1g peak, associated with the Ti–C stretching mode in the Raman spectra of the hydrogen cycled sample (Fig. 4d), suggests a strengthening of the Ti–C bond due to the charge transfer.67 These results confirm the charge transfer between DL-Ti2CClx and Mg, which suggests that DL-Ti2CClx may perturb the Mg–H bonds for the dehydrogenation of MgH2.
 | ||
| Fig. 4 Charge transfer between DL-Ti2CClx and Mg. (a) Ti 2p and (b) C 1s XPS spectra of as-synthesized Mg@DL-Ti2CClx (below) and hydrogen-cycled Mg@DL-Ti2CClx (upper). (c) Ti L-edge NEXAFS and (d) Raman spectra of ML-Ti2CClx, as-synthesized and hydrogen-cycled Mg@DL-Ti2CClx. | ||
To further elucidate the impact of DL-Ti2CClx on the MgH2 desorption kinetics, we conducted density functional theory (DFT) calculations,73,74 in which structures and charge densities were analyzed using the interface models between MgH2 nanoparticle and DL-Ti2CClx sheet (x = 0.0, 0.5, 1.0, and 1.5) that are visualized in Fig. S17.† The Bader change analysis calculates the number of electrons belonging to each atom by defining atomic volume within which the charge density is integrated.75–78 We also computed the Bader charge state of isolated MgH2 nanoparticles free from MXene as a reference. Fig. 5 displays the Bader charge difference in Mg and H atoms before and after forming the interface, i.e., changes in the number of electrons when forming the MgH2@Ti2CClx interface compared to the isolated MgH2 nanoparticles. For Mg, centering from the orange color where the atomic charge is invariant, positive change (reddish color) and negative change (bluish color) indicate oxidation and reduction, respectively. The impact of Ti2CClx is most prominent when the –Cl terminal group is completely removed. Many interface Mg atoms obtain additional electrons, which is from Mg2+ in MgH2 towards the fully desorbed Mg0 metal. Similarly, H− ions in MgH2 oxidize towards H0 near Ti2C, which can easily recombine into H2 molecule. The charge redistribution phenomenon due to Ti2CClx slowly diminishes in the presence of the –Cl terminations, and eventually the Mg and H atoms do not feel the MgH2@Ti2CClx interface at x = 1.5. Besides the charge redistribution within the MgH2 particle, Ti2CClx also participates in the charge transfer (Fig. S18†). However, given that atoms in Ti2CClx undergo both oxidation and reduction due to the complexity in the local environment, it is not clear that the role of Ti2CClx is monolithic in charge transfer. More systematic investigation exploring various contact area, distance, and local environment at the MgH2@Ti2CClx interface is demanded for clarification, which is beyond the scope of this work. Nonetheless, the impact of charge redistribution within MgH2 is clear due to the deformation of its structure. Reduction of Mg and oxidation of H elongate the Mg–H bond from 1.88 Å to 1.91 Å in the MgH2@Ti2C (Fig. S19†), suggesting that Ti2CClx facilitates the initial step of desorption, namely Mg–H bond breakage, and it eventually leads to improved desorption rates as observed in experiments. Our simulation also supports the efficacy of LiBp treatment which helps eliminating the –Cl terminations to maximize the catalytic activity of Ti2CClx.
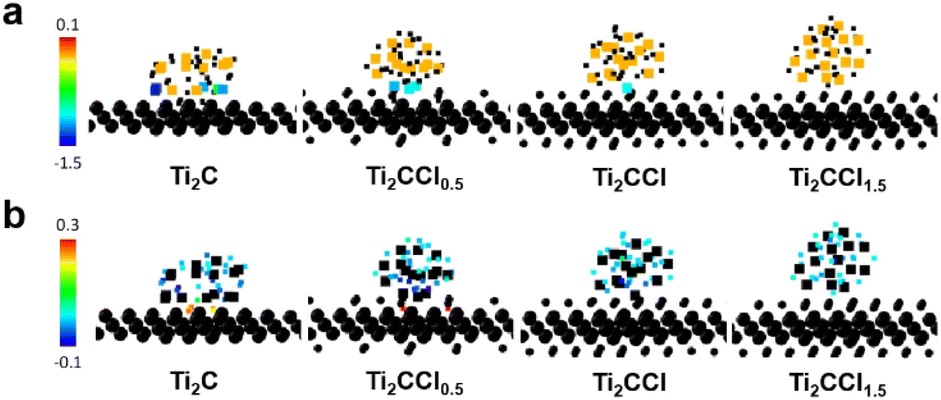 | ||
| Fig. 5 Changes in the Bader charge of (a) Mg atoms and (b) H atoms in the MgH2@DL-Ti2CClx interface models evaluated based on the DFT calculations. Mg and H atoms are denoted by large and small squares, respectively. Circles are Ti, C, and Cl atoms in MXene sheets. Atoms of interest (Mg in (a) and H in (b)) are colored by their valence state change: reddish color indicates electron loss (oxidation), and bluish color indicates electron gain (reduction). Other atoms are colored in black for simplicity. | ||
3. Conclusions
In summary, this study demonstrates the role of DL-Ti2CClx MXene in enhancing the hydrogen storage property of Mg via the formation of an intimate interface with Ti-exposed DL-Ti2CClx. Such interface allows efficient charge transfer between Mg and Ti, attributed to the removal of surface termination groups over MXene, and it is enabled by introducing relatively weak –Cl terminations to the Ti atoms using the molten-salt etching method, in contrast to the previously reported approaches which require harsh conditions. An identical reduction process was employed for incorporating Mg particles, leading to the one-pot synthesis of the Mg@DL-Ti2CClx composite. The composite exhibits improved hydrogen desorption property of MgH2, primarily due to the charge transfer from Mg to DL-Ti2CClx, which reduces the strength of the Mg–H bond at the interface. Furthermore, due to the excellent electrical conductivity of Ti2CClx, it potentially serves as an electron mediator, facilitating electron transfer at the initial stage of desorption and thus reducing the incubation period. This work provides a novel approach for synthesizing Mg–MXene complexes for efficient hydrogen storage medium and elucidates the charge transfer at the Ti–Mg interface. This strategy can be applied to other energy materials, such as those used in energy storage and catalysis, to improve their performance through the formation of a well-defined interface and electronic structure control.Data availability
The authors confirm that the data underlying this article are available within the article and its ESI†.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National R&D Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (NRF-2022K1A4A8A01080242 and NRF-2022M3D1A2095314). A part of this work was supported by the Hydrogen Materials Advanced Research Consortium (HyMARC), established as part of the Energy Materials Network by the U.S. Department of Energy (DOE), Office of Energy Efficiency and Renewable Energy (EERE), Hydrogen and Fuel Cell Technologies Office, and was performed under the auspices of the DOE by Lawrence Livermore National Laboratory (LLNL) under Contract No. DE-AC52-07NA27344. Part of the research was performed using computational resources sponsored by the DOE's EERE and located at the National Renewable Energy Laboratory. We also acknowledge computational resources at Livermore Computing at LLNL.Notes and references
- N. L. Panwar, S. C. Kaushik and S. Kothari, Renewable Sustainable Energy Rev., 2011, 15, 1513–1524 CrossRef.
- J. Rockström, O. Gaffney, J. Rogelj, M. Meinshausen, N. Nakicenovic and H. J. Schellnhuber, Science, 2017, 355, 1269–1271 CrossRef.
- S. Niaz, T. Manzoor and A. H. Pandith, Renewable Sustainable Energy Rev., 2015, 50, 457–469 CrossRef.
- S. McQueen, J. Stanford, S. Satyapal, E. Miller, N. Stetson, D. Papageorgopoulos, N. Rustagi, V. Arjona, J. Adams and K. Randolph, Department of Energy Hydrogen Program Plan, US Department of Energy (USDOE), Washington DC United States, 2020 Search PubMed.
- Y. Wang and Y. Wang, Prog. Nat. Sci.: Mater. Int., 2017, 27, 41–49 CrossRef.
- T. Sadhasivam, H.-T. Kim, S. Jung, S.-H. Roh, J.-H. Park and H.-Y. Jung, Renewable Sustainable Energy Rev., 2017, 72, 523–534 CrossRef CAS.
- D. J. Han, K. R. Bang, H. Cho and E. S. Cho, Korean J. Chem. Eng., 2020, 37, 1306–1316 CrossRef CAS.
- Y. Sun, C. Shen, Q. Lai, W. Liu, D.-W. Wang and K.-F. Aguey-Zinsou, Energy Storage Mater., 2018, 10, 168–198 CrossRef.
- H. Wang, H. Lin, W. Cai, L. Ouyang and M. Zhu, J. Alloys Compd., 2016, 658, 280–300 CrossRef CAS.
- G.-x. Liang, J. Huot, S. Boily, A. Van Neste and R. Schulz, J. Alloys Compd., 1999, 292, 247–252 CrossRef CAS.
- C. Zhou, J. Zhang, R. C. Bowman Jr and Z. Z. Fang, Inorganics, 2021, 9, 36 CrossRef CAS.
- J. Cui, J. Liu, H. Wang, L. Ouyang, D. Sun, M. Zhu and X. Yao, J. Mater. Chem. A, 2014, 2, 9645–9655 RSC.
- L. Dan, H. Wang, X. Yang, J. Liu, L. Ouyang and M. Zhu, ACS Appl. Mater. Interfaces, 2023, 15, 30372–30382 CrossRef.
- M. Song, J.-L. Bobet and B. Darriet, J. Alloys Compd., 2002, 340, 256–262 CrossRef.
- Z. Wang, X. Zhang, Z. Ren, Y. Liu, J. Hu, H. Li, M. Gao, H. Pan and Y. Liu, J. Mater. Chem. A, 2019, 7, 14244–14252 RSC.
- M. Tian and C. Shang, Int. J. Hydrogen Energy, 2019, 44, 338–344 CrossRef.
- X. Zeng, L. Cheng, J. Zou, W. Ding, H. Tian and C. Buckley, J. Appl. Phys., 2012, 111, 093720 CrossRef.
- Y. Liu, H. Du, X. Zhang, Y. Yang, M. Gao and H. Pan, Chem. Commun., 2016, 52, 705–708 RSC.
- W. Zhu, S. Panda, C. Lu, Z. Ma, D. Khan, J. Dong, F. Sun, H. Xu, Q. Zhang and J. Zou, ACS Appl. Mater. Interfaces, 2020, 12, 50333–50343 CrossRef CAS PubMed.
- H. Liu, C. Lu, X. Wang, L. Xu, X. Huang, X. Wang, H. Ning, Z. Lan and J. Guo, ACS Appl. Mater. Interfaces, 2021, 13, 13235–13247 CrossRef CAS PubMed.
- W. Zhu, L. Ren, C. Lu, H. Xu, F. Sun, Z. Ma and J. Zou, ACS Nano, 2021, 15, 18494–18504 CrossRef CAS PubMed.
- J. A. Bolarin, R. Zou, Z. Li, Z. Zhang and H. Cao, Appl. Mater. Today, 2022, 29, 101570 CrossRef.
- W. Ali, X. Li, Y. Yang, N. Li, B. Huang, C. Wu and S. Ding, ACS Appl. Mater. Interfaces, 2023, 15, 36167–36178 CrossRef CAS.
- Z. Huang, Y. Wang and M. Zhang, Int. J. Hydrogen Energy, 2021, 46, 33176–33185 CrossRef CAS.
- C. Lu, H. Liu, L. Xu, H. Luo, S. He, X. Duan, X. Huang, X. Wang, Z. Lan and J. Guo, J. Magnesium Alloys, 2022, 10, 1051–1065 CrossRef CAS.
- H. Gao, R. Shi, Y. Liu, Y. Zhu, J. Zhang, L. Li and X. Hu, J. Magnesium Alloys, 2022, 11, 3724–3735 CrossRef.
- K. R. G. Lim, M. Shekhirev, B. C. Wyatt, B. Anasori, Y. Gogotsi and Z. W. Seh, Nat. Synth., 2022, 1, 601–614 CrossRef.
- A. VahidMohammadi, J. Rosen and Y. Gogotsi, Science, 2021, 372, eabf1581 CrossRef CAS.
- V. Kamysbayev, A. S. Filatov, H. Hu, X. Rui, F. Lagunas, D. Wang, R. F. Klie and D. V. Talapin, Science, 2020, 369, 979–983 CrossRef CAS PubMed.
- M. Li, J. Lu, K. Luo, Y. Li, K. Chang, K. Chen, J. Zhou, J. Rosen, L. Hultman and P. Eklund, J. Am. Chem. Soc., 2019, 141, 4730–4737 CrossRef CAS.
- Y. Li, H. Shao, Z. Lin, J. Lu, L. Liu, B. Duployer, P. O. Persson, P. Eklund, L. Hultman and M. Li, Nat. Mater., 2020, 19, 894–899 CrossRef CAS PubMed.
- C. Zhou, D. Wang, F. Lagunas, B. Atterberry, M. Lei, H. Hu, Z. Zhou, A. S. Filatov, D.-e. Jiang, A. J. Rossini, R. F. Klie and D. V. Talapin, Nat. Chem., 2023, 15, 1722–1729 CrossRef CAS.
- W. Zhou, B. Mei, J. Zhu and X. Hong, Mater. Lett., 2005, 59, 131–134 CrossRef CAS.
- D. Wang, C. Zhou, A. S. Filatov, W. Cho, F. Lagunas, M. Wang, S. Vaikuntanathan, C. Liu, R. F. Klie and D. V. Talapin, Science, 2023, 379, 1242–1247 CrossRef CAS.
- P. Joensen, R. Frindt and S. R. Morrison, Mater. Res. Bull., 1986, 21, 457–461 CrossRef CAS.
- A. Ambrosi, Z. Sofer and M. Pumera, Small, 2015, 11, 605–612 CrossRef CAS.
- X. Zhu, Z. Su, C. Wu, H. Cong, X. Ai, H. Yang and J. Qian, Nano Lett., 2022, 22, 2956–2963 CrossRef CAS PubMed.
- N. S. Norberg, T. S. Arthur, S. J. Fredrick and A. L. Prieto, J. Am. Chem. Soc., 2011, 133, 10679–10681 CrossRef CAS PubMed.
- V. Lomonosov, E. R. Hopper and E. Ringe, Chem. Commun., 2023, 59, 5603–5606 RSC.
- I. Persson, J. Halim, H. Lind, T. W. Hansen, J. B. Wagner, L. Å. Näslund, V. Darakchieva, J. Palisaitis, J. Rosen and P. O. Persson, Adv. Mater., 2019, 31, 1805472 CrossRef PubMed.
- M. Alhabeb, K. Maleski, B. Anasori, P. Lelyukh, L. Clark, S. Sin and Y. Gogotsi, Chem. Mater., 2017, 29, 7633–7644 CrossRef.
- T. Hu, M. Hu, Z. Li, H. Zhang, C. Zhang, J. Wang and X. Wang, J. Phys. Chem. A, 2015, 119, 12977–12984 CrossRef PubMed.
- J. Fernandez and C. Sanchez, J. Alloys Compd., 2002, 340, 189–198 CrossRef.
- C. Zhou, Z. Z. Fang, R. C. Bowman Jr, Y. Xia, J. Lu, X. Luo and Y. Ren, J. Phys. Chem. C, 2015, 119, 22272–22280 CrossRef CAS.
- P. E. De Jongh, Nat. Mater., 2011, 10, 265–266 CrossRef CAS PubMed.
- M. Dornheim, N. Eigen, G. Barkhordarian, T. Klassen and R. Bormann, Adv. Eng. Mater., 2006, 8, 377–385 CrossRef CAS.
- X. Yao, Z. Zhu, H. Cheng and G. Lu, J. Mater. Res., 2008, 23, 336–340 CrossRef CAS.
- C. Zhou, Z. Z. Fang and R. C. Bowman Jr, J. Phys. Chem. C, 2015, 119, 22261–22271 CrossRef CAS.
- S. Kang, T. Ogitsu, S. A. Bonev, T. W. Heo, M. D. Allendorf and B. C. Wood, ECS Trans., 2017, 77, 81 CrossRef.
- I. Gabis, M. Dobrotvorskiy, E. Evard and A. Voyt, J. Alloys Compd., 2011, 509, S671–S674 CrossRef.
- N. Takeichi, Y. Sakaida, T. Kiyobayashi and H. T. Takeshita, Mater. Trans., 2014, 55, 1161–1167 CrossRef.
- A. S. Gangrade, A. A. Varma, N. K. Gor, S. Shriniwasan and S. S. V. Tatiparti, Phys. Chem. Chem. Phys., 2017, 19, 6677–6687 RSC.
- M. Khazaei, A. Ranjbar, M. Arai, T. Sasaki and S. Yunoki, J. Mater. Chem. C, 2017, 5, 2488–2503 RSC.
- X. Sang, Y. Xie, M.-W. Lin, M. Alhabeb, K. L. Van Aken, Y. Gogotsi, P. R. Kent, K. Xiao and R. R. Unocic, ACS Nano, 2016, 10, 9193–9200 CrossRef CAS.
- M. Pozzo and D. Alfe, Int. J. Hydrogen Energy, 2009, 34, 1922–1930 CrossRef CAS.
- T. Vegge, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 035412 CrossRef.
- A. J. Du, S. C. Smith, X. D. Yao and G. Q. Lu, J. Phys. Chem. B, 2006, 110, 21747–21750 CrossRef CAS.
- J. Čermák and L. Král, Acta Mater., 2008, 56, 2677–2686 CrossRef.
- L. F. Wan, E. S. Cho, T. Marangoni, P. Shea, S. Kang, C. Rogers, E. Zaia, R. R. Cloke, B. C. Wood and F. R. Fischer, Chem. Mater., 2019, 31, 2960–2970 CrossRef CAS.
- K.-J. Jeon, H. R. Moon, A. M. Ruminski, B. Jiang, C. Kisielowski, R. Bardhan and J. J. Urban, Nat. Mater., 2011, 10, 286–290 CrossRef CAS PubMed.
- E. S. Cho, A. M. Ruminski, S. Aloni, Y.-S. Liu, J. Guo and J. J. Urban, Nat. Commun., 2016, 7, 10804 CrossRef PubMed.
- D. J. Han, S. Kim and E. S. Cho, J. Mater. Chem. A, 2021, 9, 9875–9881 RSC.
- H. Cho, S. Hyeon, H. Park, J. Kim and E. S. Cho, ACS Appl. Energy Mater., 2020, 3, 8143–8149 CrossRef.
- Y. Cho, S. Kang, B. C. Wood and E. S. Cho, ACS Appl. Mater. Interfaces, 2022, 14, 20823–20834 CrossRef PubMed.
- R. D. Rieke, Science, 1989, 246, 1260–1264 CrossRef CAS PubMed.
- K. Shirzad and C. Viney, J. R. Soc., Interface, 2023, 20, 20230242 CrossRef PubMed.
- Q.-Q. Yan, P. Yin and H.-W. Liang, ACS Mater. Lett., 2021, 3, 1197–1212 CrossRef CAS.
- I. C. Gerber and P. Serp, Chem. Rev., 2019, 120, 1250–1349 CrossRef.
- F. De Groot, J. Fuggle, B. Thole and G. Sawatzky, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 928 CrossRef CAS PubMed.
- A. Al-Temimy, K. Prenger, R. Golnak, M. Lounasvuori, M. Naguib and T. Petit, ACS Appl. Mater. Interfaces, 2020, 12, 15087–15094 CrossRef CAS.
- K. Prenger, Y. Sun, K. Ganeshan, A. Al-Temimy, K. Liang, C. Dun, J. J. Urban, J. Xiao, T. Petit and A. C. Van Duin, ACS Appl. Energy Mater., 2022, 5, 9373–9382 CrossRef.
- A. Al-Temimy, B. Anasori, K. A. Mazzio, F. Kronast, M. Seredych, N. Kurra, M.-A. Mawass, S. Raoux, Y. Gogotsi and T. Petit, J. Phys. Chem. C, 2020, 124, 5079–5086 CrossRef.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, J. Comput. Chem., 2007, 28, 899–908 CrossRef CAS PubMed.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- M. Yu and D. R. Trinkle, J. Chem. Phys., 2011, 134, 064111 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental and theoretical details, and additional characterization experiments. See DOI: https://doi.org/10.1039/d4ta04563g |
| This journal is © The Royal Society of Chemistry 2024 |