DOI:
10.1039/D4TB00572D
(Paper)
J. Mater. Chem. B, 2024,
12, 7848-7857
A small-molecule probe to decipher stress-induced ER microenvironments and ER-Golgi communication†
Received
19th March 2024
, Accepted 21st May 2024
First published on 22nd May 2024
Abstract
Cellular stress is a crucial factor in regulating and maintaining both organismal and microenvironmental homeostasis. It induces a response that also affects the micropolarity of specific cellular compartments, which is essential for early disease diagnosis. In this contribution, we present a quantitative study of micropolarity changes inside the endoplasmic reticulum (ER) during the G1/S and G2/M phases, using a biocompatible small-molecule fluorophore called ER-Oct. This probe is selectively driven to the ER by its hydrophobicity, and it has the fastest diffusion properties among a series of analogous probes. We found that induced ER stress caused cell cycle arrests leading to an increase in ER micropolarity which is well supported by lambda scanning experiments and fluorescence lifetime imaging microscopy (FLIM) as well. ER-Oct is a versatile staining agent that could effectively stain the ER in various living/fixed mammalian cells, isolated ER, Caenorhabditis elegans, and mice tissues. Furthermore, we used this probe to visualize a well-known biological event, ER to Golgi transport, by live-cell fluorescence microscopy. Our exhaustive investigation of micropolarity using ER-staining dye provides a new way to study ER stress, which could provide a deeper understanding of proteostasis in model systems and even in fixed patient samples.
Introduction
Cellular organelles are delimited by membrane-lined structures which are responsible for compartmentalization of individual organelles in terms of their respective biochemical functions.1,2 These functions act in harmony to maintain organismal homeostasis. In addition, sub-cellular compartmentalization allows for the developing of unique microenvironments within the cellular milieu. Therefore, inter-organelle communication is crucial for cellular responses that require the coordination of these functions.3 The term ‘inter-organelle communication’ is often fancied by biologists as it involves sophisticated biological experiments to establish the communication between any pair of organelles. This communication has several modes to operate i.e., inter-organelle signaling, intracellular trafficking, membrane contact sites, and inter-organelle transient connections.4 Now, in terms of connectivity, the endoplasmic reticulum and Golgi apparatus make one of the most important pairs owing to their synchronous role in protein synthesis, sorting, and trafficking.5,6 Therefore, we shall extend this discussion to better understand their functions, stress-induced responses, and microenvironmental alterations.
The endoplasmic reticulum (ER) is one of the most important and largest multifunctional organelles present in eukaryotic cells associated with the synthesis and transport of secretaries as well as membrane proteins, their folding, and lipid metabolism.7–11 The post-translational modification along with the glycosylation of proteins is also regulated by ER.12 The diverse role of ER also includes the regulation of cytosolic calcium levels and carbohydrate metabolism.13–15 The ER also undergoes cell cycle-dependent morphological transformations.16 In the mammalian cell, the usual polygonal network-like structure of the ER becomes much more partitioned and fragmented during cell division. However, such ER morphogenesis during cell cycles is also specific to each cell line.17 The cell cycle is a combination of biochemical events that result in the accurate duplication of cellular components into genetically identical daughter cells.18 In the eukaryotic cell, the synthesis phase or S-phase involves DNA replication while segregation of genetic materials occurs in the mitosis phase or M-phase. These phases are separated by two gap phases – G1 and G2, assigned to protein synthesis, cellular growth, and cell division preparation. Now, while this discussion is aimed to provide a basic idea of the cell cycle process, it is also important to take note of the fact that these complex biochemical processes would have an impact on the micropolarity of the ER. The cell cycle arrest, induced by small molecule drugs, is also associated with unfolded protein response, commonly known as ER stress.19–22 The unsettlement of homeostasis due to the accumulation of the misfolded and unfolded proteins in ER during the cell cycle arrest leads to several fatal symptoms and diseases like neurodegeneration, cardiovascular aberration, diabetes, and cancer.23–26 On that account, the micropolarity inside the ER is expected to be a fundamental physical parameter for monitoring cellular health.27–29
Now, the transport between ER and Golgi bodies is the primary step in the secretory pathway.30,31 This transport mechanism between them is particularly operated by COP II (coat protein complex II) and COP I-coated transport vesicles.32 The newly synthesized proteins are exported from ER to Golgi while recycled proteins are sent back from cis-Golgi to ER.5 A handful of studies have been reported to show this transport phenomenon in yeasts and mammalian cells.33–35 Sophisticated experimental procedures and expertise are some of the main criteria for performing such kind of experiment. Nevertheless, the current literature is yet to have a report with a small molecule fluorescent reporter as a chemical tool to visualize this phenomenon using optical microscopy. The literature has been enriched with reports demonstrating several small-molecule ER-targeting fluorescent probes for measuring micropolarity in live cells and animal models.36–38 Unfortunately, the quantified approach is missing in most of the cases, and no report mentions how micropolarity changes in G1 and G2 cell cycle arrest phases by induced ER stress.
In this contribution, we present a series of three alkylated nitrobenzoxadiazole-based fluorescent probes, developed by straightforward synthetic procedures, manifesting excellent environment sensitivity and exclusive ER targeting abilities (Scheme 1). We further judiciously selected ER-Oct among the three probes gleaned from their efficiency of ER localization and diffusion properties by fluorescence recovery after photobleaching experiment. The specificity of the ER labeling of ER-Oct was also validated in isolated ER obtained from subcellular fractionation. Moreover, the fluorescent turn-on feature of these probes, moving from polar to non-polar media, was employed to perform wash-free organelle labeling. Wash-free bioimaging of live cells and tissues is a matter of increasing interest thanks to its simplified analytical procedure and significantly faster detection time while the washing step is practically non-feasible in the case of in vivo imaging.39–41 Therefore, developing a wash-free imaging technique using a small organic fluorophore is highly desirable.
 |
| | Scheme 1 Schematic representation of the brief overview of this work shows several applications using the probe ER-Oct. | |
Moving forward, the micropolarity of the ER was quantified in several cancerous and non-cancerous cell lines in homeostatic conditions as well as G1/S and G2/M cell cycle arrest phases. The stress-induced ER micropolarity was found to be significantly higher as compared to homeostasis. The fluorescence lifetime imaging microscopy provides another quantitative aspect of micropolarity alteration during G1 arrest in live cells. Additionally, the long-term imaging of ER stained with ER-Oct led us to visualize another interesting phenomenon – ER to Golgi transport (Scheme 1). Pearson's correlation coefficients, measured with commercially available ER and Golgi trackers at different times, suggested a reduction in exclusive ER localization and allocation in both ER and Golgi. Finally, we have demonstrated ER-Oct to be useful for staining 3D tumoroids, Caenorhabditis elegans, and mice tissue sections.
Results and discussion
Development and spectroscopic characterization of tailored ER staining dyes
Solvatochromic fluorescent probes with organelle targeting capability are always been a popular choice for monitoring intracellular polarity in homeostatic, stress-induced, and apoptotic conditions.42–46 Substituted nitrobenzoxadiazole (NBD) fluorescent probes exhibit excellent sensitivity to solvent polarity, large Stokes shift, and appreciable cellular uptake efficiency.47,48 Hence, we have synthesized a series of NBD-based fluoroprobes with varying alkyl chain lengths to have enhanced hydrophobicity. An interesting point of concern can be the choice of chain length for these synthesized derivatives. We have considered a few key aspects here i.e., cellular uptakability, photostability, and water solubility. Even though the longer alkyl chains account for enhanced hydrophobicity, the water solubility and cellular uptakability due to bigger size would take a hit. We focused on using probes with alkyl chains no longer than C8 for bioimaging applications. This was to avoid increasing their lipophilicity, which would have reduced their water solubility. The synthetic procedure, which was previously reported, required either elevated temperatures or longer reaction times.49–51 We present here a significantly faster synthesis time of 10 min at 298 K with high yields (70–88%) (Scheme S1, ESI†). The rate of the substitution reaction was impacted by the addition of triethylamine to the reaction medium which enhanced the nucleophilicity of the alkyl amines. The three probes, ER-Bu, ER-Hex, and ER-Oct were well characterized by 1H NMR, 13C NMR, and mass spectrometry (Fig. S1–S9, ESI†). Furthermore, the exact overlap of their absorption spectra and excitation spectra monitored at different wavelengths confirmed the spectroscopic purities of these probes (Fig. S10, ESI†). The probes consist of a π-conjugated linker that connects the electron-rich secondary amine (–NH–) moiety with the electron-deficient nitro (–NO2) group. The electron-rich nitrogen atom is attached to the alkyl chains of varying lengths. The structures of these probes are perfect for exhibiting intra-molecular charge transfer (ICT) behavior thereby showing solvent polarity-dependent photophysical properties (Tables S1–S3, ESI†). The absorption and emission spectra of all three probes were recorded in solvents with varying polarity. As per our anticipation, the probes show gradual redshift with increasing polarity, both in the ground and excited states (Fig. 1A, B and Fig. S11, ESI†). The emission maxima versus solvent polarity parameter, ET(30) yielded a linear plot suggesting these probes to be sensitive to the environmental polarity (Fig. 1C). Moreover, we observed higher absolute quantum yield values in solution phase while moving from ER-Bu to ER-Oct. With all these results in hand, we further moved on to our subsequent exploration of live-cell imaging and quantification of intracellular micropolarity using these probes.
 |
| | Fig. 1 Solvent-dependent (A) UV-visible and (B) emission spectra of the ER-Oct probe in different polar solvents, (C) the linear variation (R2 = 0.92) of the emission maxima with the solvent polarity parameter (ET(30) in kcal mol−1), and (D) solvent-dependent fluorescence lifetimes of the ER-staining probes (5 μM). | |
Intracellular localization of the ER-staining dyes
Based on our design principle of the probes and inspired by their environment-sensing abilities, we turned to study intracellular localization. Prior to that, the cytotoxic effect of the probes, if any, was monitored using standard MTT [3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide] assay in BHK-21 (baby hamster kidney cells), A549 (human lung cancer cells), MDA-MB-231 (human breast cancer cells), and HeLa (human cervical cancer cells) cell lines for 24 h. The half-maximal inhibitory concentrations, i.e., IC50 values in all cell lines were calculated to be 20–30 μM (Fig. S13 and S14, ESI†). Therefore, to perform all the live-cell imaging experiments, 2 μM of the probe concentration was used keeping ∼80–90% of cells viable, and hence these probes are safe to be termed biocompatible. The optimal incubation time for efficient cellular Once these experiments provided us with a basic yet foremost understanding of the probes’ properties, we moved on to fluorescence colocalization microscopy looking for the specific compartmentalization in live cells. BHK-21 cells were incubated with all three staining probes for 10 min and imaged under a confocal microscope. In the FITC channel, beautiful inter-connected micro-networks were visualized all over the cellular structure except the nucleus (Fig. S15, ESI†). We reckoned these to be endoplasmic reticulum (ER) and therefore co-stained the cells with commercial ER labeling dye – ER Tracker Red (Fig. 2A–C). Pearson's colocalization coefficient (PCC), even though above 0.8 for all (Fig. S16, ESI†), was highest for ER-Oct (0.96) having the longest alkyl chain among all three (Fig. 2D). It is to be noted that all the PCC values were calculated considering the whole frame (whole field of view) and not any single cell.
 |
| | Fig. 2 Investigation of subcellular localization of ER-Oct in live BHK-21 cells using fluorescence colocalization microscopy; stained with (A) 2.0 μM of ER-Oct, (B) 0.3 μM of ER-Tracker Red, (C) is the merge image of (A) and (B), (D) shows the DIC image. The corresponding scatter plot in the inset of (D) shows a Pearson's correlation co-efficient value of 0.94 ± 0.02 (scale bar = 10 μm); (E) an extended ER network in a single cell visualized using 100× objective lens [λex/λem = 488 nm/500–540 nm, laser power: 0.2%; λex/λem = 561 nm/575–620 nm, laser power: 0.5%; dwell time: 8 μs per pixel, confocal aperture: 1 AU]. | |
Now, it is well demonstrated that all three probes are efficient ER markers while ER-Oct came up with the best results. The large inter-connected tubular ER network structure of a single cell could be visualized by ER-Oct staining (Fig. 2E). Furthermore, to confirm the specificity for the ER, the colocalization with other organelle markers i.e., Mito Tracker Red, LysoTracker Red, and LipidSpot were performed which yielded much lower PCC values of 0.43, 0.41, and 0.36 for respective cases (Fig. S17, ESI†). As such lipophilic probes are known to have strong hydrophobic interaction with lipid bilayers,52,53 we suppose that the alkylated probes bind to the phospholipid bilayer of the ER membrane. The rapid internalization (within a minute) in live cells of the longest chain congener ER-Oct was also demonstrated by time-lapse imaging (Fig. S18, ESI†). A quantified approach to understanding the relationship between the length of the side chain and the cellular uptakability of the probes can be obtained from their respective clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values using the DataWarrior program.54 The clogP values ER-Bu and ER-Hex are 1.65 and 2.55 while ER-Oct has a comparatively higher clogP value of 3.47. Now, even though a higher clogP accounts for greater affinity to the lipid phase, the cell permeability is eventually affected. For example, the commercial ER Tracker has a clogP value of 6.79 and it takes a minimum of 20 minutes to label the ER. Therefore, the balance between lipophilicity and cell permeability is influenced by the length of the alkyl chain.
P values using the DataWarrior program.54 The clogP values ER-Bu and ER-Hex are 1.65 and 2.55 while ER-Oct has a comparatively higher clogP value of 3.47. Now, even though a higher clogP accounts for greater affinity to the lipid phase, the cell permeability is eventually affected. For example, the commercial ER Tracker has a clogP value of 6.79 and it takes a minimum of 20 minutes to label the ER. Therefore, the balance between lipophilicity and cell permeability is influenced by the length of the alkyl chain.
Understanding dynamics using fluorescence recovery after photobleaching (FRAP)
Recent findings indicate that probes containing only hydrophobic moiety tethered to the fluorescent reporter, devoid of any specific targeting group, have the propensity to localize in the ER.55,56 As demonstrated in our previous experiments, the synthesized dyes are not exceptional in this aspect as their localization in ER has been well represented by the colocalization experiments. However, a detailed study of living cells often requires long-term imaging involving continuous exposure to excitation laser sources. Photobleaching is one of the most common phenomena that limit the use of a fluorophore for such a purpose.57 Fluorescence recovery after the photobleaching (FRAP) experiment is a useful technique to judge the suitability of a probe to image in living cells for a longer period.58,59 This method is also followed to understand the mobility of the labeling dye and analyze the diffusion properties, and spatial distribution in a specific region of a cell or tissue sample. As the set of synthesized molecules has different chain lengths, we anticipated them to exhibit distinct diffusibility after cellular uptake, and the FRAP experiment was performed in live BHK-21 cells (Fig. 3A–F). The imaging parameters including scanning speed and area, bleaching duration, laser power, and stimulation duration were optimized to avoid any consequential photobleaching in the area of interest. The obtained FRAP data were fitted and analyzed using the Soumpasis equation,60
where, rn = radius of the uniformly bleaching spot or sampling region radius, τ1/2 = half-time for recovery. The diffusion during photobleaching is assumed to be negligible. Now, from the FRAP analysis, the diffusion coefficient was calculated to be highest for ER-Oct followed by its lower congeners (Fig. 3G). Hence, ER-Oct is the superior one for bioimaging applications owing to its fastest dynamics and recovery time compared to ER-Hex and ER-Bu. The photostability of ER-Oct in the imaging condition was also examined by the time-lapse imaging in live cells and found that only 10% of photobleaching occurred over the time course (Fig. S19, ESI†). More importantly, ER-Oct was less susceptible to being photobleached as compared to both ER Tracker Green and ER Tracker Red (Fig. S20 and S21, ESI†). Henceforth, considering the colocalization microscopy results, FRAP analysis, and photostability in the biological system we have performed all the imaging experiments on live cells and animal models with ER-Oct only.
 |
| | Fig. 3 Fluorescence recovery after photobleaching measurement performed in BHK-21 cell line stained with 2 μM ER-Oct. The results show the bleaching and recovery phenomena and the superiority of the ER-Oct derivative compared to the other two. The circular region (1S) represents the bleached region; (A) and (B) are pre-bleach cycles, (C) shows the bleaching, (D), (E), and (F) show the recovery of fluorescence after 1 s, 5 s, and 10 s post-bleaching, respectively, (G) the FRAP curve showing the comparative results with all three ER-staining probes [λex/λem = 488 nm/500–540 nm, imaging laser power: 0.2%, bleaching laser power: 5%, bleaching duration: 2 s, dwell time: 8 μs per pixel, confocal aperture: 1 AU]. | |
Staining isolated ER, fixed cells, and wash-free imaging
Sub-cellular fractionation and separation have gained popularity for studying specific intra-cellular structures, trafficking, and functions of low-abundance proteins.61,62 Even after the separation of the organelles, most of their original biochemical properties are retained. Hence, a biocompatible and photostable small organic fluorescent probe that can stain the isolated ER would attract researchers. Therefore, once the exclusive and efficacious ER staining potency of ER-Oct in live cells was thoroughly validated, we were interested in the possibility of staining the isolated ER using our probe. The subcellular fractionation was performed using live BHK-21 cells and the details procedure has been provided in the ESI† (Scheme S2). Briefly, live BHK-21 cells were lysed and centrifuged at 3000 rpm to pellet down nuclei. The supernatant was further centrifuged at 12000 rpm to remove heavier organelles i.e., mitochondria, lysosomes, endosomes, and any remaining nuclei. Finally, membrane buffer was added to the supernatant, and ultra-centrifugation @70000 rpm gave the pellet of isolated ER. Now, for imaging purposes, the ER pellet was resuspended in membrane buffer followed by co-staining with ER-Oct and ER Tracker Red for 10 min. Finally, the solution was ultra-centrifuged again and the stained ER pellet was imaged under the confocal microscope. Much to our delight, the same fibrous microstructures were visualized in both green and red emission channels indicating successful staining of the isolated ER using ER-Oct (Fig. 4A and B). The probe was also able to stain the ER in fixed cells as demonstrated in Fig. 4C. The live BHK-21 cells were fixed using 4% paraformaldehyde followed by a 10-minute incubation with ER-Oct. Moreover, the possibilities of wash-free imaging with this probe were also explored. The non-polar ER microenvironment compared to the bulk polarity of an aqueous medium suggested that the fluorescence turn-on behavior of the probe in a polar to the non-polar medium can be utilized for wash-free imaging. The probe accounts for zero background i.e., the residual staining solution does not interfere with imaging and an excellent signal-to-noise ratio could be obtained even when no post-staining washing was performed (Fig. 4D). Such wash-free property of the probe is advantageous for in vivo imaging applications.
 |
| | Fig. 4 Isolated ER obtained by subcellular fractionation of live BHK-21 cells stained with (A) 2 μM ER-Oct and (B) 0.3 μM ER Tracker Red (scale bar: 2 μm); (C) shows staining with ER-Oct after formaldehyde fixation, and (D) shows a wash-free image of BHK-21 cells stained with ER-Oct (scale bar: 10 μm). | |
Micropolarity inside ER in homeostatic and non-homeostatic conditions
Building upon the combined spectroscopic and microscopic results obtained until now, the probe ER-Oct can be an ideal candidate to quantify the micropolarity inside ER. We anticipated ER micropolarity to act as a marker for ER stress induced by the accumulation of misfolded and unfolded proteins which would play a role in affecting the local environment of the ER. The impairment of protein glycosylation or disulfide-bond formation along with protein overexpression in the secretory pathway is considered to trigger ER stress.63 The different stages of the cell cycle would also have an impact on such protein accumulation. Consequently, we have used ER-Oct for monitoring ER micropolarity in homeostatic conditions, G1/S, and G2/M cell cycle arrest phases. For these experiments, we have taken four different cell lines – BHK-21, A549, MDA-MB-231, and HepG2. Initially, the cells were stained with 2 μM ER-Oct, imaged, and spectral scanning in lambda mode was performed in a confocal laser scanning microscope (Fig. 5A–H). For scanning, several regions of interest (ROI) were chosen for each case to have better confidence in the obtained dataset. The ER network gave completely different emission spectra as compared to the aqueous media. The emission spectrum of ER-Oct in water was also recorded in the lambda scanning mode to maintain the same experimental condition for accurate comparison. In all four cell lines, significant blue shifts were observed while the ER in MDA-MB-231 showed the most non- polar environment (Fig. 5E–H).
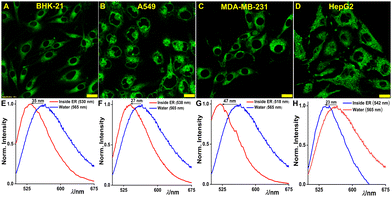 |
| | Fig. 5 Micropolarity inside ER in homeostatic conditions. CLSM images of (A) BHK-21, (B) A549, (C) MDA-MB-231, and (D) HepG2 cells incubated with 2 μM ER-Oct; (E)–(H) show the corresponding emission spectra recorded inside live cells in lambda scanning mode (scale bar: 20 μm). The spectra recorded inside ER were compared with bulk water spectrum to understand the spectral shift. The blue line denotes the spectra of ER-Oct in water and red line inside ER [λex/λem = 488 nm/500–540 nm, laser power: 0.2%, dwell time: 8 μs per pixel, confocal aperture: 1 AU; for lambda scanning: λex/λem = 488 nm/500–675 nm]. | |
Moreover, the disparate nature of the emission spectra inside the ER is indicative of the fact that the ER microenvironment is unique to each cell line (Table 1). This discovery indicates that ER-Oct can distinguish between the ER microenvironment in various cell lines. To further explore if there were any variations in the microenvironment during different cell cycle arrest phases (G1/S and G2/M), we conducted similar experiments to the ER micropolarity studies in homeostatic conditions. The same cell lines were exposed to the 24-hour treatment with 100 ng mL−1 tunicamycin – a well-known ER stress inducer that causes the activation of unfolded protein response (UPR). It eventually leads to the accumulation of cell cycle regulator protein p27 and G1/S cell cycle arrest.64 The spectral scanning experiment in the cells after G1 arrest revealed substantial enhancement of ER micropolarity while MDA-MB-231 incurred the highest change with an 18 nm red-shifted spectrum (Fig. S22, ESI†). Likewise, the DNA synthesis blocker molecule 5-fluorouracil (5-FU) was used as the inducer for G2/M cell cycle arrest in those cell lines.65 5-FU is an antimetabolite drug that is widely used for the treatment of cancer. Following a 24-hour treatment with 100 ng mL−1 5-FU, the cells were stained with ER-Oct and spectral scanning was performed. Also, in the G2/M arrest phase, the spectra inside cells were found to be red-shifted as compared to the drug untreated cells (Fig. S23, ESI†). Hence, it can be concluded that in both cell cycle arrest phases the micropolarity is higher than it was in the homeostatic condition possibly due to the accumulation of polar proteins.
Table 1 Emission properties of ER-Oct inside the ER in different cells before and after cell cycle arrests
| Cell line |
λ
maxem (nm) inside ER |
| Before arrest |
After G1 arrest |
After G2 arrest |
| BHK-21 |
530 |
540 |
545 |
| A549 |
538 |
546 |
545 |
| MDA-MB-231 |
518 |
536 |
534 |
| HepG2 |
542 |
552 |
560 |
Filming the micropolarity alteration during G1 arrest
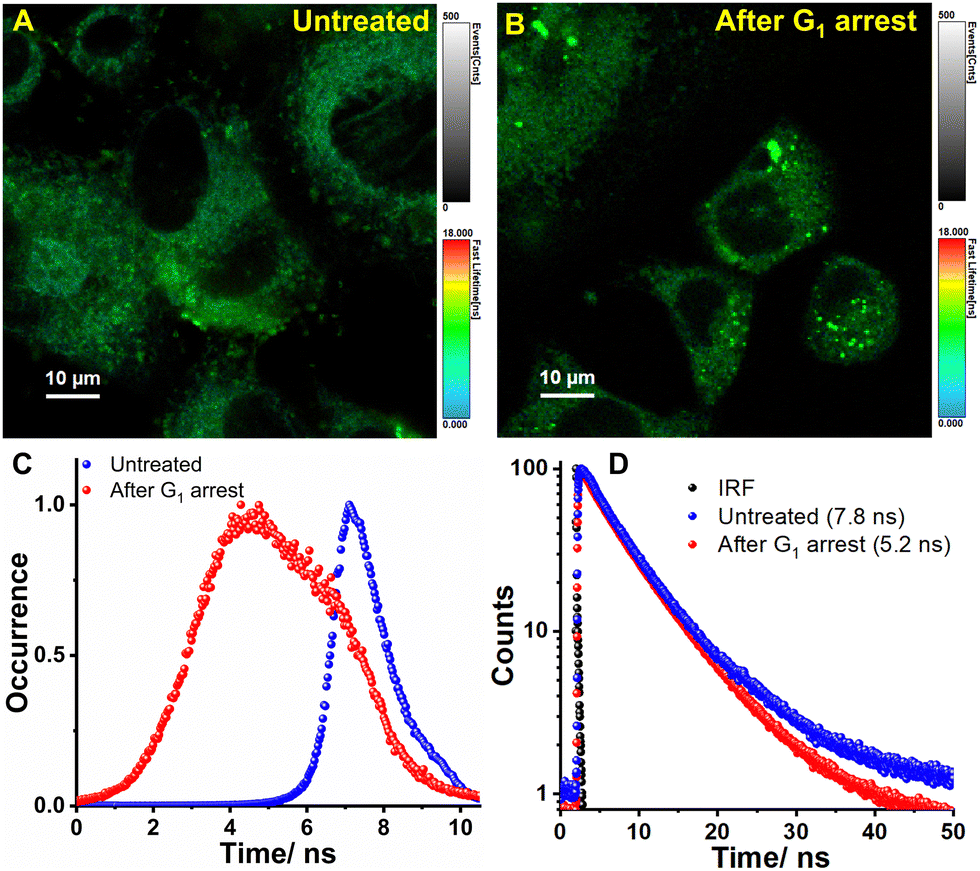
The environment-sensitive fluorophores are particularly interesting in combination with fluorescence lifetime imaging microscopy (FLIM), as their fluorescence lifetime can be directly correlated to the micropolarity of the surroundings. FLIM is also regarded for its high accuracy in quantifying the fluctuations of various signals in living cells since the fluorescence lifetime of fluorophores are dependent on the environment rather than on concentration. Thus, FLIM can sensitively monitor any subtle change in the intra-ER micropolarity upon induced stress. Therefore, live BHK-21 cells were subjected to G1 arrest using tunicamycin for 24 h (Fig. 6A and B). The arrest state is indicated by significant lipid accumulation which occurs due to diacylglycerol acyltransferase-dependent lipid droplet formation to sequester excessive polyunsaturated fatty acids that accumulate in arrested cells.66 The heterogeneity in the lifetime distribution histograms arises from the response of individual cells to external stimuli (Fig. 6C). The fitted data analysis yielded fluorescence lifetime values of 7.8 ns and 5.2 ns for untreated and G1 arrested cells, respectively (Fig. 6D). Our research findings reaffirm our previous discovery that the micropolarity levels within the ER increase during cell cycle arrest.
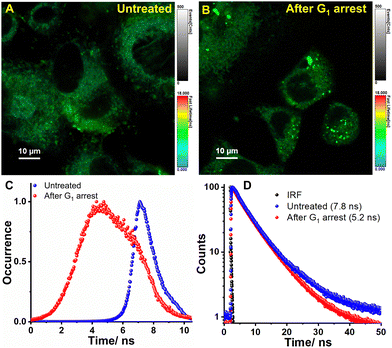 |
| | Fig. 6 FLIM images of ER-Oct stained BHK-21 cells (A) before and (B) after cell cycle arrest at G1. The colors of the pixels describe the τ values according to the color bar shown at the side of the images. (C) the corresponding lifetime distribution histogram and (D) decay profile show that lifetime decreases after inducing G1 arrest. | |
Imaging 3D tumoroids, Caenorhabditis elegans, and mice tissues
Optical microscopy has become an indispensable tool in contemporary biomedical research. The recent advancements in imaging techniques have significantly improved the choices available for in vivo imaging of animal research. Even though, the two-dimensional (2D) cell culture has been the most used research method, in vivo imaging provides compelling evidence suggesting that way more advanced experiments can be performed to come up with valuable insights. Hence, the applicability of the probe ER-Oct as a versatile staining agent was further validated by imaging formaldehyde-fixed cells, three-dimensional (3D) spheroidal cell models, Caenorhabditis elegans, and animal tissue samples. Initially, we considered the 3D cell culture environment, known as 3D tumoroids or spheroids, which allows cells to grow and interact with the surrounding extracellular framework in three dimensions.67,68 The tumoroid culture is a comparatively satisfactory model to mimic behavior and organization in terms of morphology and physiology. The spheroids were grown in a scaffold-free technique using live BHK-21 cells and treated with ER-Oct for 10 min. The confocal microscopy images indicate significant uptake of the probe by the tumoroids (Fig. 7A), while the corresponding Z-scans show spherical morphologies of the individual cells grown in the cluster (Fig. S24, ESI†). The spheroid imaging experiments were followed by staining Caenorhabditis elegans which is the simplest and most frequently chosen model organism to study human diseases.69–71 This model is regarded as a bridge between in vitro and in vivo models that provide physiologically relevant results being compliant with high-throughput technologies. Hence, for imaging, the wild-type N2 hermaphrodite worms were synchronized to the young adult stage and grown in the nematode growth media (NGM). The NGM itself contained 2 μM ER-Oct and the worms were grown at 20 °C for 60 h. The stained young adults were initially paralyzed by 5 mM levamisole treatment and then transferred to agar pads on glass slides. The confocal microscopy images show the whole tissue section is stained by the probe, rather than any specific region which corroborates with the reports suggesting the presence of a continuous ER lumen throughout the Caenorhabditis elegans structure (Fig. 7B).72 The beautifully stained worms indicated the possibility of the probe for tissue imaging. Hence, to demonstrate further, wild-type mice liver tissue section was taken and incubated with the same probe concentration for 15 min. Next, we explored the applicability of the probe ER-Oct to samples that may be difficult to image live such as human patient samples or several mouse model samples. As a proof of concept, we used formalin-fixed wild-type mice liver tissue section and incubated with the same probe concentration for 15 min. As demonstrated in Fig. 7C–K, this probe exhibits fast uptakability in the tissue sample and demonstrates applicability for large-scale animal and even human tissue imaging.
 |
| | Fig. 7 CLSM images of (A) 3D tumoroids, (B) Caenorhabditis elegans, and (C)–(J) wild-type mice liver tissues stained with 2 μM ER-Oct; (D)–(I) show tissue section images at different z-depth, (J) shows the extended focus image (EFI) of the tissue section with enhanced SNR, and (K) shows the plot of mean intensity vs. z-slice as obtained from the tissue z-sectioning [λex/λem = 488 nm/500–540 nm, laser power: 0.2%, dwell time: 8 μs per pixel, confocal aperture: 1 AU]. | |
Communication between ER to Golgi bodies: visualization of the ER-Golgi transport
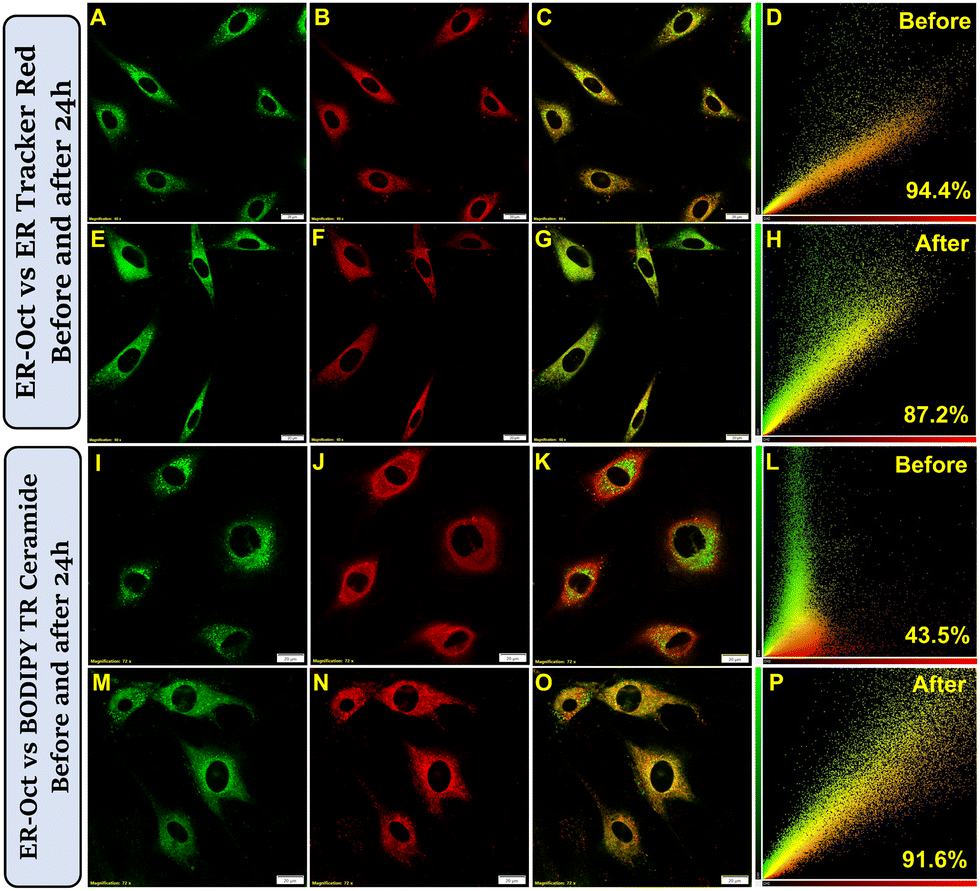
Alongside validating the versatility of ER-Oct to image tumoroids, model organisms, and animal tissues, we took a specific interest in visualizing the fate of the probe after cellular uptake in longer times. In our earlier experimental results, we demonstrated that ER-Oct exclusively localizes in the endoplasmic reticulum of the cell. However, until this point, we did not have an idea of how long the probe is retained inside the ER after localization. Even though probe retention was our subject of interest, another important biological phenomenon that we kept in mind is ER to Golgi transport.30,31 As the probe localizes in the ER driven by its hydrophobicity, there is every chance that it would bind to some ER protein and later get transported out of the ER lumen. Hence, we planned for two sets of experiments – co-staining with ER-Oct/ER Tracker Red, and ER-Oct/BODIPY TR ceramide, and subsequently, image them immediately and after 24 h. For the first set of experiments, the observation was – exclusive ER localization and very little Golgi localization, the corresponding Pearson's correlation coefficient values (PCC) being 0.94 and 0.43, respectively (Fig. 8A–D and I–L). The next set came up with exciting findings i.e., while the ER localization was associated with a slightly lesser PCC value of 0.87 (Fig. 8E–H), there was a massive enhancement in the Golgi localization indicated by a PCC value of 0.91 (Fig. 8M–P). The same sets of experiments were performed in the HeLa cell line to further consolidate the obtained results in the normal cell line. In this case, too, we obtained a high PCC value of 0.88 for ER-Octvs. BODIPY TR ceramide after 24 h (Fig. S25, ESI†). Therefore, it can be concluded that ER-Oct, alongside being a specific marker of ER, also enables the visualization of the ER to Golgi transport in the live cells. Once the probe was transported to the Golgi apparatus, we also performed lambda scanning and found that the Golgi environment is comparatively more polar than the ER (Fig. S26, ESI†). Now, these promising results led us to visualize the inhibition of ER to Golgi protein transport in live cells using confocal microscopy. For this purpose, we have taken Brefeldin A (BFA) which blocks the transport of secreted and membrane proteins from the ER to the Golgi apparatus.73,74 The live BHK-21 cells were stained with ER-Oct/BODIPY TR ceramide and followed by immediate treatment of 5 μM BFA for 24 h. The confocal images recorded thereafter did not show a good correlation between green/red channels, unlike BFA-untreated cells – the corresponding PCC value being 0.62 (Fig. S27, ESI†). However, it is possible to vary the extent of inhibition by changing the BFA concentration as well as the treatment time. Thus, the effective trafficking inhibition in living cells by BFA treatment could be monitored by the straightforward microscopic approach. These findings are suggestive of studying inter-organelle communication between ER and Golgi using the developed probe.
 |
| | Fig. 8 CLSM images show the transport from ER to Golgi in BHK-21 cells. The first and third rows show images taken immediately after 10 min staining. The second and fourth rows show images taken after 24 h of 10 min staining. Images (A), (E), (I) and (M) show cells stained with 2 μM ER-Oct, (B) and (F) show cells stained with 0.3 μM ER Tracker Red, (J) and (N) show cells stained with BODIPY TR Ceramide; (C), (G), (K) and (O) are merge images, (D), (H), (L) and (P) are the corresponding scatter plots (scale bar = 20 μm) [λex/λem = 488 nm/500–540 nm, laser power: 0.2%; λex/λem = 561 nm/575–620 nm, laser power: 0.5%; dwell time: 8 μs per pixel, confocal aperture: 1 AU]. | |
Conclusions
This contribution introduces a series of biocompatible nitrobenzoxadiazole probes as wash-free markers of the endoplasmic reticulum in live cells. The probes diffused nicely in live cells with one of the probes, ER-Oct exhibiting a diffusion coefficient value of 1.4 × 10−2 μm2 s−1. Additionally, ER-Oct showed efficient staining possibilities in isolated ER, 3D tumoroids, Caenorhabditis elegans, and mice tissue sections. The probe works on the principle of intramolecular charge transfer that enables the environment sensitivity based on the solvent polarity. We have taken advantage of this bulk polarity-sensitive behavior to quantitatively decipher the micropolarity changes in different cell cycle arrest phases. The study also shows that, even though cancer cells are more polar compared to normal cells, the ER micropolarity is unique for each cell line. The cell cycle arrests in the G1/S and G2/M phases cause a significant increment in the ER micropolarity. Finally, the ER-Golgi transport in live cells has been visualized through optical microscopy using ER-Oct as the staining agent. The long-term imaging revealed that Pearson's correlation coefficient increases corresponding to the Golgi localization after 24 h of staining indicating the probe getting partitioned in both ER and Golgi. We believe, this probe alongside being cost-effective and easy to develop, comes up with a plethora of bioimaging applications and provides a quantitative idea of endoplasmic reticulum micropolarity and its response to induced stress.
Author contributions
TD: synthesis, spectroscopic measurements, live-cell imaging, tumoroid and C. elegans imaging, data curation and analysis, manuscript writing; BC: FLIM experiment & data curation; AN & SM: tissue samples preparation, ALK: data curation, project design.
Conflicts of interest
A provisional Indian patent application (App. no. 202321033584) has been filed.
Acknowledgements
We thank the Department of Chemistry and Central Instrumentation Facility at the Indian Institute of Science Education and Research Bhopal (IISERB) for the instrumentation and infrastructural facility. TD thanks IISERB for his doctoral fellowship. We also thank Dr Ishaan Gupta (IIT Delhi) for his suggestions. We acknowledge the Department of Science and Technology (DST), India, for the FIST support (SR/FST/LSI-643/2015) for the live-cell imaging facility. We also thank Dr Yogesh Hooda (University of Cambridge, UK) for helping with the microsome separation protocol.
References
- A. Jain and R. Zoncu, Mol. Metab., 2022, 60, 101481 CrossRef CAS PubMed.
- M. Petkovic, C. E. O'Brien and Y. N. Jan, Genes. Dev., 2021, 35, 449–469 CrossRef CAS PubMed.
- D. E. Gottschling and T. Nyström, Cell, 2017, 169, 24–34 CrossRef CAS PubMed.
- P. Lujan, J. Angulo-Capel, M. Chabanon and F. Campelo, Curr. Opin. Cell Biol., 2021, 71, 95–102 CrossRef CAS PubMed.
- N. Gomez-Navarro and E. Miller, J. Cell Biol., 2016, 215, 769–778 CrossRef CAS PubMed.
- M. C. Lee, E. A. Miller, J. Goldberg, L. Orci and R. Schekman, Annu. Rev. Cell Dev. Biol., 2004, 20, 87–123 CrossRef CAS PubMed.
- D. S. Schwarz and M. D. Blower, Cell. Mol. Life Sci., 2016, 73, 79–94 CrossRef CAS PubMed.
- L. M. Westrate, J. E. Lee, W. A. Prinz and G. K. Voeltz, Annu. Rev. Biochem., 2015, 84, 791–811 CrossRef CAS PubMed.
- F. J. Stevens and Y. Argon, Semin. Cell Dev. Biol., 1999, 10, 443–454 CrossRef CAS PubMed.
- J. Yoon, E. E. Nekongo, J. E. Patrick, T. Hui, A. M. Phillips, A. I. Ponomarenko, S. J. Hendel, R. M. Sebastian, Y. M. Zhang, V. L. Butty, C. B. Ogbunugafor, Y.-S. Lin and M. D. Shoulders, PLoS Biol., 2022, 20, e3001569 CrossRef CAS PubMed.
- H. Ma, Y. Lu, Z. Huang, S. Long, J. Cao, Z. Zhang, X. Zhou, C. Shi, W. Sun, J. Du, J. Fan and X. Peng, J. Am. Chem. Soc., 2022, 144, 3477–3486 CrossRef CAS PubMed.
- L. Ellgaard, N. McCaul, A. Chatsisvili and I. Braakman, Traffic, 2016, 17, 615–638 CrossRef CAS PubMed.
- M. Csala, G. Banhegyi and A. Benedetti, FEBS Lett., 2006, 580, 2160–2165 CrossRef CAS PubMed.
- D. Mekahli, G. Bultynck, J. B. Parys, H. De Smedt and L. Missiaen, Cold Spring Harb. Perspect. Biol., 2011, 3, 1–32 Search PubMed.
- J. Rieusset, Biochim. Biophys. Acta, Mol. Cell Res., 1864, 2017, 865–876 Search PubMed.
- M. Puhka, H. Vihinen, M. Joensuu and E. Jokitalo, J. Cell Biol., 2007, 179, 895–909 CrossRef CAS PubMed.
- C. Vedrenne and H. P. Hauri, Traffic, 2006, 7, 639–646 CrossRef CAS PubMed.
- K. Vermeulen, D. R. Van Bockstaele and Z. N. Berneman, Cell Proliferation, 2003, 36, 131–149 CrossRef CAS PubMed.
- K. Bourougaa, N. Naski, C. Boularan, C. Mlynarczyk, M. M. Candeias, S. Marullo and R. Fahraeus, Mol. Cell, 2010, 38, 78–88 CrossRef CAS PubMed.
- C. Han, L. Jin, Y. Mei and M. Wu, Cell Signal., 2013, 25, 144–149 CrossRef CAS PubMed.
- N. Song, Y. Song, B. Hu, X. Liu, X. Yu, H. Zhou, J. Long and Z. Yu, Adv. Healthcare Mater., 2022, 2202039 Search PubMed.
- Y. Li, X. Zhang, X. Wan, X. Liu, W. Pan, N. Li and B. Tang, Adv. Funct. Mater., 2020, 30, 2000532 CrossRef CAS.
- C. Hetz, E. Chevet and H. P. Harding, Nat. Rev. Drug Discov., 2013, 12, 703–719 CrossRef CAS PubMed.
- S. Y. Wang and R. J. Kaufman, J. Cell Biol., 2012, 197, 857–867 CrossRef CAS PubMed.
- M. Wang and R. J. Kaufman, Nature, 2016, 529, 326–335 CrossRef CAS PubMed.
- H. Yoshida, FEBS J., 2007, 274, 630–658 CrossRef CAS PubMed.
- T. Dutta, K. Pal and A. L. Koner, Chem. Rec., 2022, e202200035 CrossRef CAS PubMed.
- K. Pal, T. Dutta and A. L. Koner, ACS Omega, 2021, 6, 28–37 CrossRef CAS PubMed.
- W. Xu, Z. B. Zeng, J. H. Jiang, Y. T. Chang and L. Yuan, Angew. Chem., Int. Ed., 2016, 55, 13658–13699 CrossRef CAS PubMed.
- P. Watson and D. J. Stephens, Biochim. Biophys. Acta, Mol. Cell Res., 1744, 2005, 304–315 Search PubMed.
- J. McCaughey and D. J. Stephens, Trends Cell Biol., 2019, 29, 940–953 CrossRef CAS PubMed.
- D. Tang and Y. Wang, Trends Cell Biol., 2013, 23, 296–304 CrossRef CAS PubMed.
- Y. Suda, K. Kurokawa and A. Nakano, Front. Cell Dev. Biol., 2018, 5, 1–6 Search PubMed.
- P. T. A. Linders, C. van der Horst, M. ter Beest and G. van den Bogaart, Cells, 2019, 8 Search PubMed.
- E. C. Gaynor, T. R. Graham and S. D. Emr, Biochim. Biophys. Acta, Mol. Cell Res., 1998, 1404, 33–51 CrossRef CAS PubMed.
- W. Shu, S. P. Zang, C. Wang, M. X. Gao, J. Jing and X. L. Zhang, Anal. Chem., 2020, 92, 9982–9988 CrossRef CAS PubMed.
- S. J. Park, V. Juvekar, J. H. Jo and H. M. Kim, Chem. Sci., 2020, 11, 596–601 RSC.
- K. Pal, I. Samanta, R. K. Gupta, D. Goswami and A. L. Koner, Chem. Commun., 2018, 54, 10590–10593 RSC.
- J. Y. Wu, A. Y. Bi, F. Zheng, S. Huang, Y. J. Li, J. P. Ding, D. X. Xiang and W. B. Zeng, Chem. Commun., 2021, 57, 801–804 RSC.
- L. Hu, B. S. Xu, H. Chen and H. Wang, Sens. Actuators, B, 2021, 340, 129950 CrossRef CAS.
- H. R. Jia, Y. X. Zhu, K. F. Xu, G. Y. Pan, X. Y. Liu, Y. Qiao and F. G. Wu, Chem. Sci., 2019, 10, 4062–4068 RSC.
- W. H. Song, B. L. Dong, Y. R. Lu, X. Q. Kong, A. H. Mehmood and W. Y. Lin, New J. Chem., 2019, 43, 12103–12108 RSC.
- H. B. Xiao, P. Li and B. Tang, Coord. Chem. Rev., 2021, 427 Search PubMed.
- J. Yin, W. Quan, X. Kong, C. Liu, B. Lu and W. Lin, ACS Appl. Bio Mater., 2021, 4, 3630–3638 CrossRef CAS PubMed.
- Q. Q. Bai, C. J. Yang, M. J. Yang, Z. Q. Pei, X. B. Zhou, J. X. Liu, H. W. Ji, G. Li, M. M. Wu, Y. L. Qin, Q. Wang and L. Wu, Anal. Chem., 2022, 94, 2901–2911 CrossRef CAS PubMed.
- P. Alam, W. He, N. L. C. Leung, C. Ma, R. T. K. Kwok, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, K. S. Wong and B. Z. Tang, Adv. Funct. Mater., 2020, 30, 1909268 CrossRef CAS.
- C. Y. Jiang, H. J. Huang, X. Y. Kang, L. Yang, Z. Xi, H. Y. Sun, M. D. Pluth and L. Yi, Chem. Soc. Rev., 2021, 50, 7436–7495 RSC.
- T. Yamaguchi, M. Asanuma, S. Nakanishi, Y. Saito, M. Okazaki, K. Dodo and M. Sodeoka, Chem. Sci., 2014, 5, 1021–1029 RSC.
- S. K. Das, A. S. Patra, D. Jose and M. Sarkar, Chem. Phys. Lett., 2012, 528, 11–15 CrossRef CAS.
- W. Q. Chen, H. C. Luo, X. J. Liu, J. W. Foley and X. Z. Song, Anal. Chem., 2016, 88, 3638–3646 CrossRef CAS PubMed.
- N. C. P. Araujo, R. Afonso, A. Bringela, M. L. Cancela, M. L. S. Cristiano and R. B. Leite, Parasitol. Int., 2013, 62, 575–582 CrossRef CAS PubMed.
- L. F. Guo, C. Y. Li, H. Shang, R. Y. Zhang, X. C. Li, Q. Lu, X. Cheng, Z. Q. Liu, J. Z. Sun and X. Q. Yu, Chem. Sci., 2020, 11, 661–670 RSC.
- R. Yang, F. F. Meng, G. L. Niu, J. Niu, Q. H. Hao, C. Zong, P. Gao and X. Q. Yu, Sens. Actuators, B, 2022, 372, 132639 CrossRef CAS.
- T. Sander, J. Freyss, M. von Korff and C. Rufener, J. Chem. Inf. Model., 2015, 55, 460–473 CrossRef CAS PubMed.
- Z. Yang, Y. He, J. H. Lee, W. S. Chae, W. X. Ren, J. H. Lee, C. Kang and J. S. Kim, Chem. Commun., 2014, 50, 11672–11675 RSC.
- H. Lee, Z. Yang, Y. Wi, T. W. Kim, P. Verwilst, Y. H. Lee, G. I. Han, C. Kang and J. S. Kim, Bioconjugate Chem., 2015, 26, 2474–2480 CrossRef CAS PubMed.
- A. P. Demchenko, Methods Appl. Fluoresc., 2020, 8, 022001 CrossRef CAS PubMed.
- E. A. J. Reits and J. J. Neefjes, Nat. Cell Biol., 2001, 3, E145–E147 CrossRef CAS PubMed.
- A. Kitamura and M. Kinjo, Biophys. Physicobiol., 2018, 15, 1–7 CrossRef CAS PubMed.
- M. Kang, C. A. Day, A. K. Kenworthy and E. DiBenedetto, Traffic, 2012, 13, 1589–1600 CrossRef CAS PubMed.
- J. B. Harford and J. S. Bonifacino, Curr. Protoc. Cell Biol., 2011, 52, 301–308 Search PubMed.
-
B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular biology of the cell, Garland Science, 4th edn, 2002 Search PubMed.
- N. Cherepanova, S. Shrimal and R. Gilmore, Curr. Opin. Cell Biol., 2016, 41, 57–65 CrossRef CAS PubMed.
- B. Durand, F. B. Gao and M. Raff, EMBO J., 1997, 16, 306–317 CrossRef CAS PubMed.
- R. Yoshikawa, M. Kusunoki, H. Yanagi, M. Noda, J. I. Furuyama, T. Yamamura and T. Hashimoto-Tamaoki, Cancer Res., 2001, 61, 1029–1037 CAS.
- H. Lee, A. Horbath, L. Kondiparthi, J. K. Meena, G. Lei, S. Dasgupta, X. Liu, L. Zhuang, P. Koppula, M. Li, I. Mahmud, B. Wei, P. L. Lorenzi, K. Keyomarsi, M. V. Poyurovsky, K. Olszewski and B. Gan, Nat. Commun., 2024, 15, 79 CrossRef CAS PubMed.
- S. A. Langhans, Front. Pharmacol., 2018, 9, 1–14 CrossRef PubMed.
- N. E. Ryu, S. H. Lee and H. Park, Cells, 2019, 8, 1–13 Search PubMed.
- P. M. Meneely, C. L. Dahlberg and J. K. Rose, Curr. Protoc. Essent. Lab. Tech., 2019, 19, 1–35 Search PubMed.
- M. B. Veldman and S. Lin, Pediatr. Res., 2008, 64, 470–476 CrossRef PubMed.
- T. Kaletta and M. O. Hengartner, Nat. Rev. Drug Discov., 2006, 5, 387–398 CrossRef CAS PubMed.
- Z. Y. Lee, M. Prouteau, M. Gotta and Y. Barral, J. Cell Biol., 2016, 214, 665–676 CrossRef CAS PubMed.
- A. Colanzi, G. Grimaldi, G. Catara, C. Valente, C. Cericola, P. Liberali, M. Ronci, V. S. Lalioti, A. Bruno, A. R. Beccari, A. Urbani, A. De Flora, M. Nardini, M. Bolognesi, A. Luini and D. Corda, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9794–9799 CrossRef CAS PubMed.
- T. Fujiwara, K. Oda, S. Yokota, A. Takatsuki and Y. Ikehara, J. Biol. Chem., 1988, 263, 18545–18552 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization (NMR, mass, UV-vis, fluorescence), photostability, cytotoxicity, and other control experiments. See DOI: https://doi.org/10.1039/d4tb00572d |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab,
Barsha
Chakraborty
*ab,
Barsha
Chakraborty