
Nordihydroguaiaretic acid microparticles are effective in the treatment of osteoarthritis†
Kaamini M.
Dhanabalan‡
 a,
Bhagyashree
Padhan‡
a,
Ameya A.
Dravid
a,
Smriti
Agarwal
a,
Nicholas M.
Pancheri
c,
Angela
Lin
c,
Nick J.
Willet
c,
Ashok Kumar
Padmanabhan
b and
Rachit
Agarwal
*a
a,
Bhagyashree
Padhan‡
a,
Ameya A.
Dravid
a,
Smriti
Agarwal
a,
Nicholas M.
Pancheri
c,
Angela
Lin
c,
Nick J.
Willet
c,
Ashok Kumar
Padmanabhan
b and
Rachit
Agarwal
*a
aDepartment of Bioengineering, Indian Institute of Science, Bengaluru, India 560012. E-mail: rachit@iisc.ac.in
bDepartment of Orthopaedics, MS Ramaiah Medical College, Bengaluru, India 560054
cPhil and Penny Knight Campus for Accelerating Scientific Impact, Department of Bioengineering, University of Oregon, Eugene, OR, USA 97403
First published on 28th September 2024
Abstract
Several disease-modifying osteoarthritis (OA) drugs have emerged, but none have been approved for clinical use due to their systemic side effects, short half-life, and rapid clearance from the joints. Nordihydroguaiaretic acid (NDGA), a reactive oxygen species (ROS) scavenger and autophagy inducer, could be a potential treatment for OA. In this report, we show for the first time that sustained delivery of NDGA through polymeric microparticles maintains therapeutic concentrations of drug in the joint and ameliorates post-traumatic OA (PTOA) in a mouse model. In vitro treatment of oxidatively stressed primary chondrocytes from OA patients using NDGA-loaded poly(lactic-co-glycolic acid) (PLGA) microparticles (NDGA-MP) inhibited 15-lipoxygenase, induced autophagy, prevented chondrosenescence, and sustained matrix production. In vivo intra-articular delivery of NDGA-MP was non-toxic and had prolonged retention time (up to 35 days) in murine knee joints. Intra-articular therapy using NDGA-MP effectively reduced cartilage damage and reduced pain in the surgery-induced PTOA mouse model. Our studies open new avenues to modulate the immune environment and treat post-traumatic OA using ROS quenchers and autophagy inducers.
Introduction
Osteoarthritis (OA) is a multifactorial degenerative joint disease that has remained a growing problem with rising incidence since the 20th century.1,2 Current treatments for OA include administering nonsteroidal anti-inflammatory drugs (NSAIDs) to alleviate pain and inflammation.3,4 However, no disease-modifying OA drug is available to date, as current treatments fail to prevent cartilage degradation and have several side effects.5 One of the critical causative factors for OA is the accumulation of oxidative stress.6,7 Chondrocytes, the only resident cells of articular cartilage, produce reactive oxygen species (ROS) at basal levels via mitochondrial respiration and the NADPH oxidase system, vital for maintaining cartilage homeostasis.8,9Joint trauma or aging leads to the overproduction of reactive oxygen species (ROS), altering intracellular signalling pathways.6,9,10 ROS mediates chondrocyte senescence and apoptosis, degradation of extracellular matrix, synovial inflammation, and erosion of the subchondral bone.6,11 Clinical data of OA patients aligns with these observations, showing elevated oxidative stress due to ROS and accumulation of lipid peroxidation products with decreased antioxidant enzymes.12,13 Excessive ROS can damage other cellular organelles resulting in cellular senescence or apoptosis.14 Moreover, free radicals released from dysfunctional mitochondria can lead to telomere instability, pushing the chondrocytes into senescence.15 Once the chondrocytes start becoming senescent, they secrete senescence-associated secretory phenotype (SASP) factors, which attract immune cells and lead to chronic inflammation and destruction of joint tissue.16,17
Nordihydroguaiaretic acid (NDGA), a phenolic lignan extensively studied for its anti-cancer properties, emerges as a promising therapeutic to address these processes.18–20 NDGA prevents lipoxygenase (LOX) activity by breaking the redox cycle of Fe3+ to Fe2+ conversion. This maintains the iron in the Fe2+ state to inactivate LOX and keeps it from generating ROS through its downstream products, thereby mitigating cellular damage.21 NDGA is also a potent in vitro scavenger of several ROS species such as hypochlorous acid (HOCl), peroxynitrite (ONOO−), hydroxyl molecules (OH−), and nascent oxygen ([–O–]) and can prevent ROS-mediated cellular damage.22 NDGA modulates the nuclear factor erythroid 2-related factor 2 (Nrf2) anti-oxidant pathway, helps reduce the intracellular oxidative stress damage to delay senescence and helps prevent age-related diseases.23,24
Autophagy, another critical aspect of OA, represents a cellular homeostasis mechanism that ensures cellular stress survival by balancing the catabolic and anabolic pathways.25–29 In the context of OA, autophagy is an emerging area of therapeutic research that involves the upregulation of autophagy by mTOR inhibition, thereby recycling ROS-damaged organelles and preventing cellular damage.27,30–34 Autophagy upregulation via mTOR inhibition prevents further cellular damage by recycling ROS-damaged organelles.35
A key study by the National Institute on Aging's Interventions Testing Program identified NDGA as a treatment to prolong the life span of mice by acting as an autophagy modulator and preventing cellular aging.23,36 EP300 acetyltransferase is one of the essential enzymes causing raptor acetylation leading to mTORC1 activation and autophagy inhibition.37–39 NDGA induces autophagy via EP300-mediated mTORC1 inhibition to recycle the ROS-damaged and dysfunctional organelles, avoiding their accumulation and can prevent senescence onset in cells.23,40 NDGA also partially inhibited MMP-13 production in chondrocytes caused by fibronectin fragments and reduced nuclear factor-B and MAP kinase phosphorylation, thereby preventing the development of the self-perpetuating inflammation-resolution cycle that contributes to the development of OA.41 NDGA also has been shown to prevent ionizing radiation-mediated oxidative stress-induced senescence in articular chondrocytes in vitro.42 Since OA is a disease with multiple contributing factors such as increased oxidative stress, imbalanced autophagy, and chondrosenescence, NDGA has the potential to act as a DMOAD candidate.21 However, NDGA has not been used to treat OA in any animal model, perhaps due to its low molecular weight, which poses the challenge of low residence time in the joint.
Novel drugs and biologics such as tanezumab and anakinra that were successful in OA preclinical studies, have not been successful in human trials; a major reason is attributed to systemic toxicity, rapid clearance, and sub-therapeutic concentrations in the joints.43–47 Administering frequent intra-articular injections can help sustain therapeutic concentrations but does not offer a patient-compliant therapy. Microparticle-mediated drug delivery using sustained-release formulations is a promising approach to overcome this challenge. The clinical success of sustained-release formulations in several disease conditions is supported by studies involving drugs such as doxycycline (NCT02487186), triamcinolone acetonide (NCT04094298), and Zilretta®, risperidone (Risperdal Consta®), and naltrexone (Vivitrol®). Hence, given NDGA's potential benefits, we hypothesized that biomaterial microparticle-based intra-articular delivery of NDGA would result in sustained delivery and improved therapeutic efficacy for the treatment of OA. Although few reports have explored sustained-release formulations of NDGA48,49 none have investigated its application for OA treatment.
Herein, we report that the NDGA retained bioactivity after encapsulation in microparticle formulation, prevented senescence, and induced autophagy in human primary chondrocytes obtained from OA patients. Microparticle formulations of NDGA resulted in sustained release of NDGA for several weeks. Both preventive and curative regimens with NDGA-microparticle formulations effectively ameliorated post-traumatic OA in a mouse model by inducing autophagy, inhibiting LOX production, and preventing senescence in articular chondrocytes. These results suggest a promising avenue for treating OA using NDGA microparticle formulations.
Results
Chondrocytes from OA patients have a high burden of oxidative stress and senescence with reduced basal autophagy near OA lesions
Studies have shown that diminished autophagy and a high burden of senescence are associated with OA.27,46 Hence, to determine the ROS levels, autophagy signaling, and senescence burden of articular chondrocytes at the end-stage OA in humans, we examined the sections from explanted cartilage obtained from human patients undergoing knee replacement surgery.The articular surfaces that showed rough fibrillated surfaces with visible underlying subchondral bone were classified as OA lesions, while those that appeared intact with healthy smooth surfaces were identified as non-lesioned areas (Fig. S1, ESI† top panel). We stained the sections for the markers of ROS mediator (15-LOX), autophagy (LC3BII (microtubule-associated protein 1B-Light chain 3)), and senescence (ARF (alternative reading frame product of cyclin-dependent kinase inhibitor 2A)). The lesioned areas had a qualitatively higher number of cells positive for 15-LOX and ARF and fewer positive cells for LC3B (Fig. S1, ESI† bottom panel A, C, and E). These results indicate that OA lesioned cartilage exhibited a highly active 15-LOX mediating ROS production and high senescence burden coupled with low autophagy. The non-lesioned surfaces had lower oxidative stress and senescence burden (low expression levels of 15-LOX and ARF) with active basal autophagy (higher number of cells positive for LC3B) (Fig. S1, ESI† bottom panel B, D, and F). These results encouraged us to design therapies that can selectively target ROS and senescence while inducing autophagy to treat OA.
Tunable NDGA delivery platform using PLGA microparticles
A sustained release formulation of NDGA can result in a lower frequency of drug administration for the long-term treatment of OA. PLGA has been widely used as sustained drug delivery formulation due to its excellent biocompatibility and ease of tunability to encapsulate and sustainably deliver a wide variety of drugs.50,51 We hypothesised that, since NDGA is a hydrophobic drug with a low water solubility, encapsulating it in the PLGA platform using the single emulsion technique can create a slow-release platform via PLGA degradation to deliver higher therapeutic cargo to the target site. This method of NDGA encapsulation in PLGA matrix has not been evaluated before.Our previous studies have shown that the microparticles (MPs) with 1 μm diameter exhibit prolonged retention inside mice and rodent knee joints.33,34,52 Hence, we prepared PLGA particles (molecular weight: 10–15 kDa; encapsulation) with an average hydrodynamic diameter of 939.8 ± 335.3 nm (Fig. 1(A) and Table S1, ESI†). The fluorescence image of Cy3 loaded PLGA MP is shown in Fig. 1(B).
 | ||
| Fig. 1 Characterization of NDGA microparticles. (A) Size distribution of 10–15 kDa PLGA particles measured by Dynamic Light Scattering (DLS). (B) Fluorescence microscopy image of Cy3 loaded PLGA particles (molecular weight 10–15 kDa), Scale bar, 3 μm. (C) In vitro release profiles of NDGA from microparticles synthesized from different molecular weight PLGA polymers (n = 3 per group at each time point). Data in the graph were fitted with non-linear regression (least square method) two-phase exponential decay curves. | ||
Next, we determined whether the release rate of NDGA could be varied with PLGA microparticles of different molecular weights (Mw). The size and NDGA encapsulation efficiency in various PLGA MPs formulations are listed in Table S1 (ESI†). From the NDGA release profiles, it was evident that within 48 h, 10–15 kDa PLGA MPs released all the drug, while the 85–100 kDa PLGA MPs of Mw released the drug for 45 days. With variation in the PLA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PGA ratio of the PLGA from 50:50 to 85:15 and using higher molecular weight PLGA (190–240 kDa), we could sustain the NDGA release up to 120 days (Fig. 1(C)). Thus, adjusting the molecular weights and the PLA:PGA ratio allowed NDGA release in a controlled and sustained manner, providing a robust and versatile platform. For 48 h in vitro experiments, the shorter releasing particles (10–15 kDa) were used.
:PGA ratio of the PLGA from 50:50 to 85:15 and using higher molecular weight PLGA (190–240 kDa), we could sustain the NDGA release up to 120 days (Fig. 1(C)). Thus, adjusting the molecular weights and the PLA:PGA ratio allowed NDGA release in a controlled and sustained manner, providing a robust and versatile platform. For 48 h in vitro experiments, the shorter releasing particles (10–15 kDa) were used.
NDGA – microparticles (NDGA-MP) and NDGA reduce 15(S)-HETE, the primary metabolite of 15-LOX, and prevent oxidative stress in human chondrocytes
Before beginning to assess the NDGA and NDGA-MP therapeutic effect on chondrocytes, we sought to test the chondrocytes’ metabolic activity in the presence of various concentrations of NDGA to establish a safe range for our in vitro experiments. We treated the human chondrocytes cell line (C28/I2) to different concentrations of NDGA for 24 and 48 h (Fig. S2A and B, ESI†). Cellular metabolic activity did not reduce for concentrations up to 30 μM at the end of 48 h. Therefore, for all future experiments, we decided to work with the range of 200 nM to 30 μM.Several studies have documented that 15-LOX and its metabolites are associated with inflammatory diseases, such as OA.53,54 15(S)-HETE, the primary metabolite of 15-LOX from the arachidonic acid pathway, breaks down into potent ROS molecules (Fig. 2(A)), damaging several organelles inside the cells.21,53–55 NDGA has been known to inhibit action of 15-LOX in several studies.53,54 Therefore, we hypothesized that NDGA-MP containing the active NDGA molecules should be capable of inhibiting 15-LOX production in oxidatively stressed chondrocytes and preventing the production of 15(S)-HETE. As expected, H2O2 (200 μM) treatments increased the levels of 15(S)-HETE in chondrocytes. Treatments with free NDGA (3 μM) and NDGA-MP (equivalent NDGA dose of 3 μM) significantly reduced the levels of 15(S)-HETE to almost two log orders, thereby preventing the accumulation of oxidative stress in the cells (Fig. 2(B)). Blank microparticles (Blank-MP) did not cause any reduction in 15(S)-HETE levels. This experiment validates that NDGA prevented ROS production by inhibiting 15-LOX levels.
 | ||
| Fig. 2 NDGA-MP lowered the levels of 15(S)-HETE in chondrocytes exposed to oxidative stress and induced autophagy. (A) Key products and pathways related to 15-LOX-activity. (B) 15(S)-HETE levels in chondrocytes under oxidative stress conditions along with NDGA and NDGA-MP co-treatment (n = 3 per group). Fluorescence microscopy images of HACs stained with DAPI and LC3B after (C) no treatment, (D) free NDGA (3 μM), (E) Blank-MPs treatment and, (F) NDGA-MP (3 μM NDGA). (G) Quantification of LC3B puncta per 100 mm2 using ImageJ software (n = 3 per group). Images and graphs were representatives of data collected from three OA patients. Data in graphs represent the mean ±, s.d. and p values were determined by one-way analysis of variance (ANOVA) and Tukey's post hoc tests. p-Value < 0.05 was considered significant. HACs – human articular chondrocytes, Blank–MP – Blank microparticles, NDGA-MP – NDGA loaded microparticles. ****p < 0.0001, ns – nonsignificant. Scale bar, 20 μm. | ||
Autophagy upregulation in primary human articular chondrocytes (HACs) using NDGA-MP
NDGA is a well-known autophagy inducer that inhibits the p300 acetyltransferase enzyme, thus indirectly acting on the mTOR pathway.21,23 During autophagy signalling, LC3B protein in the cytoplasm, visualized as a low diffuse signal, gets converted from the LC3B-I isoform into the phosphatidylethanolamine conjugated LC3B-II isoform followed by insertion into the autophagosome membranes.56 Thus, a low diffuse signal from the cytoplasm and nucleus gets converted into a punctate signal visualized by immunocytochemical staining.Cells were treated with NDGA formulations, and LC3B puncta per 100 mm2 were quantified in all the images using an automated macros algorithm using ImageJ. The untreated cells had an average of fewer than 100 puncta per 100 mm2 (in representative image), indicative of basal autophagy. On the contrary, free NDGA and NDGA-MP treated groups exhibited ≥5000 puncta per 100 mm2 (in representative image) indicative of upregulated autophagy (Fig. 2(D) and (F)). The results from NDGA-MP were comparable to that of the free NDGA treated groups (Fig. 2(D)), suggesting that NDGA was active in our MP formulations. Our findings in the primary HACs (Fig. 2(C)–(G)) as well as in C28/I2 cell line (Fig. S3A–E, ESI†) indicate that both free NDGA (3 μM) and NDGA-MP (equivalent NDGA dose of 3 μM) can successfully induce autophagy.
NDGA-MP prevented senescence in HACs
We hypothesized that NDGA, a potent ROS quencher and autophagy up regulator, can prevent oxidative stress build-up in the chondrocytes. We treated the HACs under oxidative stress conditions by externally adding H2O2 (200 μM) to test this hypothesis. The concentrations of stress agents such as H2O2 and BrdU were previously optimised.33 The experimental results indicated that NDGA could prevent senescence. The lower doses of NDGA below 3 μM were non-efficacious to prevent senescence (Fig. S4, ESI†). Hence, 3 μM NDGA concentration was used for all further in vitro studies.To evaluate the senescence prevention capability of NDGA as a microparticle formulation, we co-treated HACs exposed to H2O2 (200 μM) with NDGA-MP (NDGA equivalent to 3 μM). The H2O2 (200 μM) treatment increased the percentage of senescent cells to 35.67 ± 5.44%, while the free NDGA and NDGA-MP co-treated groups decreased the percentage of senescent cells to 6.88 ± 2.91% and 6.66 ± 0.82% respectively compared to untreated cells, which showed 6.55 ± 2.08% senescent cells (Fig. 3(A)–(E)).
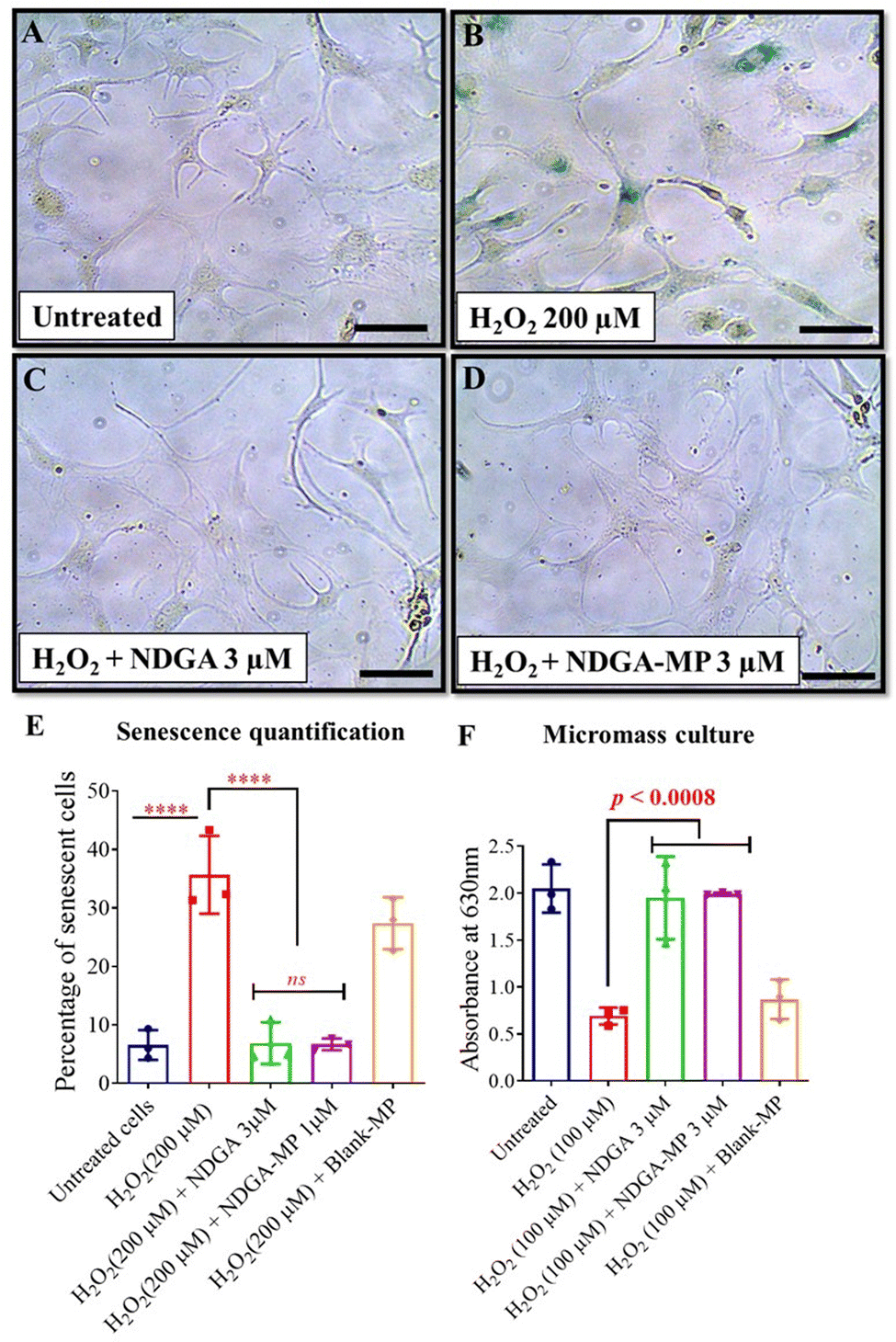 | ||
| Fig. 3 NDGA-MP prevented senescence in primary HACs and sustained sGAG production in micromass cultures exposed to oxidative stress. SA-β Gal-stained images of primary HACs exposed to (A) no treatment, (B) oxidative (H2O2) stress, (C) oxidative (H2O2) stress with free NDGA (3 μM), and (D) oxidative (H2O2) stress with NDGA-MP (3 μM). (E) Percentage of senescent HACs post oxidative (H2O2) stress condition for 48 h with various treatment conditions (n = 3 per group). (F) Absorbance of Alcian blue stain from stressed micromass cultures (n = 3 per group). Images and graphs were representatives of data collected from three OA patients. Data in graphs represent the mean ±, s.d. and p values were determined by one-way analysis of variance (ANOVA) and Tukey's post hoc tests. p-Value < 0.05 was considered significant. HACs – human articular chondrocytes, Blank-MP – Blank microparticles, NDGA-MP – NDGA loaded microparticles. ****p < 0.0001, ns – non significant. Scale bar, 20 μm. | ||
Similar results were also obtained with C28/I2 cells under oxidative (H2O2) or genotoxic stress (BrdU) (Fig. S5 and S6, ESI†). Collectively, these results indicate that NDGA MPs prevented cellular senescence under different stress conditions in HACs and C28/I2 cell lines.
NDGA-MP treatment sustained sGAG production in stressed micromass cultures
We next wanted to evaluate whether chondrocyte function can be restored under oxidative stress. To evaluate this, we performed functional assays to evaluate the sulfated glycosaminoglycans (sGAG) production in the oxidatively stressed chondrocytes. When seeded at very high densities in the presence of the growth factor (TGF-β), chondrocytes form 3D micromasses with high deposition of extracellular matrix components including sGAG.57 There was a 2.6-fold decrease in the sGAG production by the micro masses with H2O2 treatment compared to the vehicle-treated groups (Fig. 3(F)). The free NDGA or NDGA-MP treatment both sustained the sGAG production at levels comparable with untreated groups. We performed similar experiments on the micromasses formed from C28/I2 cells by exposing them to genotoxic (BrdU, 600 μM) stress or oxidative (H2O2, 100 μM) for 48 h, and the results correlated with our findings from HACs (Fig. S7A and B, ESI†).To evaluate the long-term sGAG production, we treated micromass formed from C28/I2 cells for eight days with BrdU (600 μM) or H2O2 (100 μM) as individual experiments along with co-treatment groups containing NDGA or NDGA-MP at 3 μM dose. In the BrdU or H2O2 treated group, the absorbance values dropped almost three-fold compared to untreated, while the NDGA-MP treated groups sustained the sGAG production on par with untreated and free NDGA treated groups (Fig. S7C and D, ESI†). In summary, these results suggest that NDGA and NDGA-MP sustained sGAG production in stressed chondrocytes for long durations under oxidative and genotoxic stress.
PLGA MPs of high molecular weight exhibit extended residence time in mice knee joints
For our initial studies, we hypothesized that a monthly once injection regimen would be an acceptable therapy. Such timelines are also followed in OA clinical trials for administering NSAID, SI-613 (NCT03209362), and viscosupplementation using hyaluronic acid (NCT00669032). Our animal experiments were designed as a 2-month model of OA, and since the 85–100 kDa PLGA MPs displayed a sustained release pattern for more than a month (Fig. 1(C)), we chose this Mw PLGA formulation for all subsequent mice experiments. Since the longer releasing particles (190–240 kDa) released NDGA for about 120 days, we did not employ it in this study. To determine the residence time of PLGA MPs in the knee joint, fluorescent Cy7 amine dye encapsulated in PLGA MPs was administered intra-articularly, and the fluorescence signal was monitored using an in vivo imaging system – PerkinElmer IVIS® Spectrum. The contralateral legs of mice were injected with an equal amount of Cy7 free dye. On day 3, the free dye injected joints only had 10% of the initial signal, whereas the Cy7 MPs injected group exhibited fluorescent signal even on day 35 (Fig. 4(A)–(B)). Collectively, the results demonstrate the feasibility of using microparticles for prolonged delivery of molecules into the intra-articular space. We had previously reported similar retention times for 1 μm PLGA particles (75–85 kDa).33,34 | ||
| Fig. 4 PLGA MPs exhibit prolonged residence time in mice knee joints. (A) In vivo images of mice, knee joints imaged at day 0, 1, 3, 7, 14, 21, 28, and 35 after intra-articular injection of free Cy7 dye (top panel) and Cy7-labeled 85–100 kDa PLGA MPs (bottom panel). (B) Percentage of fluorescent intensity remaining from injected formulations for days (n = 5 mice per group). Data in the graph were fitted with non-linear regression (least square method) one-phase exponential decay curve. Data in the graph represent the mean ± s.d., and p values were determined by unpaired t-test or Mann–Whitney test. p-Value < 0.05 was considered significant. $ – 0.0159, $$ ≤ 0.0079, $$$ – 0–0006, $$$$ ≤ 0.0001. | ||
NDGA-MP prevented post-traumatic OA (PTOA) disease in mice
To assess the potential of NDGA-MP formulation on a functional model of OA, we employed the widely used PTOA mouse model obtained by surgical destabilization of the medial meniscus (DMM surgery).58–60 PTOA induced via DMM surgery causes joint damage in both female and male mice. However, it has been reported that the severity of joint damage is generally less pronounced in female mice61,62 Therefore, we chose male mice for all surgery-based OA studies to ensure a robust and consistent model of joint damage and to facilitate the assessment of therapeutic interventions.We administered free NDGA (4 μg per joint at each time point), NDGA-MP (1 μg per joint at each time point), and Blank-MPs (MPs equivalent to the injected particle dose of NDGA-MP) intra-articularly as three injections, starting at day 7 post the DMM surgery to assess the preventive capability of our formulation (Fig. 5(A)). The residence time of free drug in mice joints is very less (Fig. 5(B)), therefore, we used 4 times higher dose of free drug as compared to microparticle formulation for all in vivo studies.
 | ||
| Fig. 5 NDGA-MP prevented the development of OA in a murine post-traumatic model of OA. (A) Graphic illustration of treatment timeline. (B) Characteristic Safranin-O and Fast Green–stained images of the medial tibial plateau (MTP) of mice knee joints collected from the different treatment groups. (C) OARSI scores indicate the extent of injury in mice after receiving different treatments. (D) Pain threshold (using von Frey filaments) of different groups of mice treated with a preventive dosing regimen at 9 weeks post DMM surgery and dotted line representing pain threshold of unoperated mice. (E) Representative images of 15-LOX IHC staining (F) Quantification of 15-LOX positive cells per 100 mm2. (G) Representative images of LC3B IHC staining. (H) Quantification of LC3B positive cells per 100 mm2 (I) Representative images of p19ARF IHC staining. (J) Quantification of p19ARF positive cells per 100 mm2. The data in the graphs represent the mean ±, s.d., and p values were determined using one-way analysis of variance (ANOVA) or Kruskal Wallis test and Tukey's post hoc analysis. DMM (n = 7), sham (n = 7), NDGA (high dose, n = 7; low dose n = 7), NDGA-MP (n = 7), Blank-MPs (n = 7). ns – nonsignificant; scale bar, 50 μm. | ||
Preventive dosing regimen
To determine an optimal dose of NDGA, we assessed two different doses (4 μg and 400 ng per joint at each time point) based on our in vitro results. NDGA's in vitro release curve from PLGA particles (85–100 kDa PLGA, Fig. 1(C)) showed that about 90% of the drug gets released in about 30 days. Hence, we set the injection time points that were 21 days apart so that at any time point before the next dose, the therapeutic levels of NDGA are not entirely depleted from the knee joints.The DMM-operated groups that received Blank-MPs, free NDGA, or no injections had a rough cartilage surface with many empty lacunas. There were visible changes such as chondrocyte hypertrophy and depletion of safranin O staining indicating cartilage loss (Fig. 5(B)). The NDGA-MP (1 μg per joint) effectively prevented OA, evident by smooth cartilage surface and minimal loss of safranin stain (Fig. 5(B) and (C)). Histomorphometry was performed and scored using OARSI scoring guidelines. The OARSI scores of NDGA-MP (1 μg dose) (p = 0.001) treated mice were more than two-fold lower when compared to free NDGA (4 μg dose) treated and DMM mice groups. The NDGA (4 μg per joint) failed to prevent cartilage damage as a free drug and as MP formulation (100 ng per joint) (Fig. 5(C)). So, for all our subsequent studies, we utilized the dose of 1 μg NDGA in NDGA-MP per joint. We also monitored the drug toxicity-related weight loss. All the animals showed a steady weight gain throughout the study period (Fig. S8A–C, ESI†). We also performed serum alanine aminotransferase assay (ALT) in mice to look for any hepatotoxicity. ALT measurements analysis indicated no signs of drug-related hepatotoxicity at the administered dosage. The serum was collected on day 43, 24 hours after the 3rd intra-articular injection on day 42 (Fig. S9, ESI†).
Pathological pain (allodynia) is one of the leading clinical symptoms of OA.63 To test if NDGA-MP formulations can reduce mechanical allodynia associated with OA, we tested the pain threshold of mice using Von Frey filaments. We observed that administration of NDGA-MP effectively alleviated the allodynia than free NDGA and untreated groups. While the untreated and free drug administered group had a low pain threshold (<2 g), NDGA-MP injected animals showed higher pain thresholds up to 4 g, closer to surgical and healthier controls (Fig. 5(D)). Furthermore, NDGA MPs treated knee joints exhibited reduced synovial inflammation as seen by fewer inflammatory cells infiltration in the synovium compared to the untreated and free -NDGA treated groups (Fig. S10A–D, ESI†). The synovial membrane also showed reduced thickness compared to the other untreated and free NDGA-treated groups (Fig. S10E, ESI†), indicating that the MPs also helped mitigate OA-related synovial inflammation.
Immunohistochemical staining of the mice knee joints for catabolic proteases (ADAMTS-5 and MMP-13), ROS mediator (15-LOX), autophagy marker (LC3B), and senescence marker (p19ARF) was carried out on the mice knee joint sections. The progression of DMM-induced OA in mice has been linked to increased expression of 15-LOX in cartilage.55 15-LOX expression was reduced in the NDGA-MP (1 μg) treated groups (p = 0.0001 compared to all other groups) while the untreated, free NDGA treated, and Blank-MP treated groups expressed a higher number of cells positive for 15-LOX (Fig. 5(E) and (F)). The results correlated with in vitro data where the metabolite of 15-LOX, 15(S)-HETE was reduced in NDGA-MP, and NDGA treated groups (Fig. 2(B)). Hence, we show that the sustained release of NDGA from NDGA-MP inhibited 15-LOX expression, which decreased 15(S)-HETE levels and reduced ROS production.
In alignment with in vitro results, the articular cartilage surface of NDGA-MP (1 μg) treated mice exhibited a higher number of LC3B positive cells (p = 0.0001 compared to all groups), indicative of increased autophagy in the articular cartilage cells (Fig. 5(G) and (H)) while, the untreated or free NDGA treated groups showed fewer LC3B cells. The cells positive for p19ARF, a senescence marker, were lower in the mice joints treated with NDGA-MP (1 μg) than the DMM group (p = 0.0298) (Fig. 5(I) and (J)). The NDGA-MP decreased ROS production by inhibiting 15-LOX and effectively upregulated autophagy to recycle damaged molecules. Autophagy upregulation prevented the cells from turning senescent and mediated OA disease prevention in the preventive treatment regimen.
When chondrocytes are mechanically stressed due to trauma, they generate ROS that creates a redox imbalance to produce several cytokines and inflammatory proteases such as matrix metalloproteases (MMPs).55 These MMPs and cytokines affect cellular health and cause degradation of the extracellular matrix, consequently leading to chondrocyte death and cartilage degradation.28,64,65 The increase in MMPs is attributed to the autophagic imbalance due to ROS-mediated oxidative stress.28 Hence, to evaluate the expression of the cartilage degradation enzymes, we assessed the expression levels of MMP-13 and ADAMTS-5, which are widely implicated in OA. The expression levels of MMP-13 and ADAMTS-5 in untreated, free NDGA treated, and Blank-MP treated groups were much higher than the NDGA-MP treated or sham groups in the preventive dosing study (Fig. S11A and C, ESI†). This suggests that sustained presence of NDGA can reduce the production of cartilage catabolic proteases.
The curative regimen of NDGA-MP reduces the severity of OA in a murine post-traumatic model of OA
Orthopedic clinics receive patients who arrive with severe symptoms due to progressive damage of the existing knee joint.66 Hence, to hold more relevance for treating the patients at the clinics, we wanted to evaluate whether our NDGA formulations would be effective after the disease has progressed to early OA.Studies have shown that OA hallmarks such as proteoglycan loss, chondrocyte hypertrophy, and cartilaginous osteophyte formation are detectable as early as two weeks post-surgery in DMM mice models.58 Therefore, we administered the DMM-operated mice groups with NDGA-MP formulation three weeks post-DMM surgery to assess if the formulation can cure the early OA damage. The frequency of injections was reduced from three injections in the preventive regimen to two injections spaced 21 days apart (Fig. 6(A)).
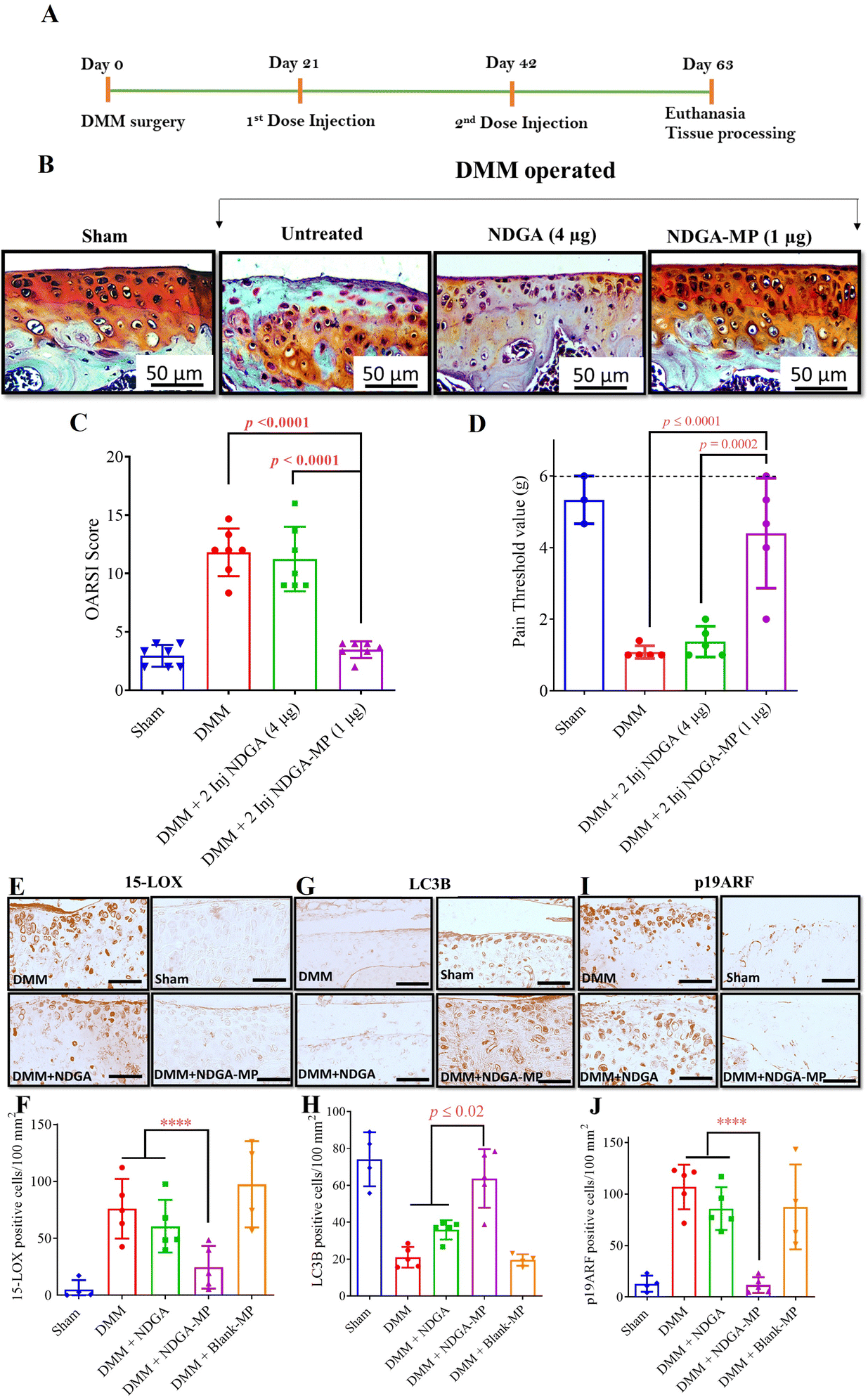 | ||
| Fig. 6 NDGA-MP as a curative regimen reduced the severity of OA in the murine post-traumatic model of OA (A) Graphic illustration of treatment timeline. (B) Characteristic Safranin-O and Fast Green–stained images of medial tibial plateau (MTP) of mice knee joints collected from the different treatment groups. (C) OARSI scores indicate the extent of injury in mice after receiving different treatments. (D) Pain threshold of different groups of mice treated with a curative two dosing regimen at 9 weeks post DMM surgery and dotted line representing pain threshold of unoperated mice. (E) Characteristic images of 15-LOX IHC staining (F) Quantification of 15-LOX positive cells per 100 mm2. (G) Characteristic images of LC3B IHC staining. (H) Quantification of LC3B positive cells per 100 mm2 (I) Characteristic images of p19ARF IHC staining. (J) Quantification of p19ARF positive cells per 100 mm2. The data in the graphs represent the mean ±, s.d., and p values were determined using one-way analysis of variance (ANOVA) or Kruskal Wallis test and Tukey's post hoc analysis. DMM (n = 7), sham (n = 7), free NDGA (n = 7), NDGA-MP (n = 7), Blank-MPs (n = 7). ns – non significant; scale bar, 50 μm. | ||
DMM operated groups with no treatment, Blank-MPs treatment, and free NDGA treatment showed cartilage surface irregularities with significant cartilage thinning. There was diminished safranin O staining of proteoglycans in the cartilage along with hypertrophic chondrocytes exhibiting focal perichondral staining or empty lacunae, indicating chondrocyte death (Fig. 6(B)). The OARSI scores in mice receiving two-dose injections of NDGA-MP were three times lower than those treated with free NDGA (p < 0.0001) and DMM only (p < 0.0001) (Fig. 6(C)). Similar to 3-dose experiment, we observed that administration of NDGA-MP effectively alleviated the allodynia than free NDGA and untreated groups. Untreated and free drug administered group had a low pain threshold (<2 g) whereas NDGA-MP injected animals showed similar pain thresholds to surgical and healthier controls (Fig. 6(D)).
The immunohistochemical studies provide additional confirmation of our in vitro findings and the outcomes of the preventive treatment regimen. The cells positive for 15-LOX (Fig. 6(E) and (F)) and p19ARF (Fig. 6(I) and (J)) were 3-fold lower in the mice joints treated with 1 μg NDGA-MP than free NDGA (p = 0.0001) or DMM only (p = 0.0001) groups. There were fewer senescent cells in the DMM and Blank-MP-treated groups but were not statistically different from the free NDGA-treated group. NDGA-MP treatment also induced an upregulation of basal autophagy in the chondrocytes on the surface of the articular cartilage in mice treated with NDGA-MP (1 μg), evidenced by an increased number of LC3B-positive cells. In contrast, the free NDGA and Blank-MP treated groups exhibited fewer LC3B positive cells indicative of low basal autophagy (Fig. 6(G) and (H)).
Consistent with our preventive dosing studies, the DMM and Blank-MP treated groups’ articular surface showed more MMP-13 and ADAMTS-5 positive cells, indicative of inflammation and cartilage degradation (Fig. S11B and D, ESI†). In contrast, the NDGA-MP treated groups exhibited very few cells positive for these markers with lower damage and uniform Safranin O staining. Overall, our results suggests that the sustained presence of NDGA prevents the cells from secreting degradative proteases and can be used to treat OA.
Single intervention study
Finally, we wanted to evaluate if we can further reduce the dosing frequency of our particles to achieve therapeutic benefits. Hence, we designed a single injection dosing regimen (Fig. 7(A)). Under the single intervention regimen, mice treated with NDGA-MP exhibited OARSI scores that were 2-fold lower than those treated with free NDGA (p < 0.0001) and DMM only (p = 0.027) (Fig. 7(B) and (C)). Thus, NDGA-MP at a dose of 1 μg per joint effectively improved overall cartilage health. We also checked for pain threshold which did not improve with NDGA-MP or free NDGA injections compared to DMM group (Fig. 7(D)). μCT analysis was performed to determine changes in subchondral bone and osteophytes (Fig. 7(E)–(H)). Single dose curative treatments did not reduce osteophytes volume compared to DMM in medial tibial plateau (Fig. 7(J)). | ||
| Fig. 7 Top Panel: NDGA-MP as a curative single-dose regimen reduced the severity of OA in the murine post-traumatic model of OA. (A) Graphic illustration of treatment timeline. (B) Characteristic Safranin-O and Fast Green–stained images of the medial tibial plateau (MTP) of mice knee joints collected from the different treatment groups. (C) OARSI scores indicate the extent of injury in mice after receiving different treatments. (D) Pain threshold of different groups of mice treated with a curative single dosing regimen at 9 weeks post DMM surgery. The data in the graphs represent the mean ±, s.d., and p values were determined using one-way analysis of variance (ANOVA) or Kruskal Wallis test and Tukey's post hoc analysis. DMM (n = 9), sham (n = 9), free NDGA (n = 9), NDGA-MP (n = 9), Blank-MP (n = 9). ns – non significant; scale bar, 50 μm. Bottom panel: μCT analysis of joints obtained after single dose regimen in the murine post-traumatic model of OA. Representative 3D posterior whole joint reconstructions from μCT analyses with medial tibial plateau marginal tissues (osteophytes) highlighted in red collected from (E) sham, (F) DMM, (G) NDGA and (H) NDGA-MP. (I) Medial subchondral bone volume in mice treated with a curative single dosing regimen showed no statistical difference among any groups. (J) Medial osteophyte bone volume in mice treated with a curative single dosing regimen did not decrease the osteophyte volume compared to the DMM group and were all significantly higher compared to the sham group in statistical analysis. The data in the graphs represent the mean ±, s.d., and p values were determined using one-way analysis of variance (ANOVA) followed by Tukey's post hoc analysis or Kruskal–Wallis test followed by Dunn's post hoc analysis. n = 4 for all groups. | ||
Discussion
Given the progressive nature of OA and the lifelong morbidity, treating joints post-trauma with disease-modifying drugs can prevent severe OA. New emerging agents targeting specific etiologies of OA, such as the Interleukin-1 receptor antagonist (IL-1 RA), MMP–inhibitor PG-116800 (NCT00041756), anti-IL-1α/β antibody – Lutikizumab (NCT02087904), and novel DMOAD drugs such as UBX0001 (NCT04129944), Enbrel (NCT02722772), kartogenin and tanezumab, did not work efficiently in clinical trials,45,67,68 possibly due to rapid joint clearance.Intra-articular drug injection via a sustained release carrier could represent a more practical and safer alternative. Our study identified a novel DMOAD, NDGA, which has not been previously used in animal models for OA research. NDGA induces autophagy that recycles the damaging ROS molecules and damaged organelles to restore cellular health.23,69,70 Despite its great curative potential, NDGA can cause hepatotoxicity and nephrotoxicity if administered at high doses.69,71 Hence, a slow and a carrier mediated sustained local delivery will help prevent these toxic effects. Using this novel DMOAD, we have successfully engineered a sustained release microparticle formulation that maintained therapeutic concentrations in the knee joint. Our study treated the PTOA mouse model by administering IA injection once every three weeks and eventually showing that a single injection over two month OA model is sufficient to provide curative benefit. Such dosing frequencies have been clinically acceptable such as Hyalgan® administration (NCT00669032, 3 cycles of weekly intra-articular injections for 5 weeks) or the current clinical trials for bioactive agents such as DA-5202 (NCT02554240, weekly intra-articular injection for 3 weeks).
The excellent biocompatibility and biodegradability of PLGA make them a suitable carrier for NDGA delivery. In addition, PLGA encapsulated formulations, like Zilretta®, Lupron®, and Zoladex®, have been approved by the US Food and Drug Administration (FDA) as a safe formulation with no adverse effects for OA and other clinical therapies. Our previous studies have demonstrated that particles in the 900 nm – 1 μm size range are appropriate for retention in mice or rodent knee joints via intra-articular administration.33,34,52 This size range prevents the MPs from being cleared away from the joint space through lymphatics. The cartilage penetration typically requires very low size of the particles (<50 nm) and there is a risk of rapid clearance from the lymphatics at this size range.72 To evaluate the retention time of these particles in vivo, we used microparticles synthesized from 85–100 kDa molecular weight PLGA. The MPs exhibited extended residence time (∼35 days) in murine knee joints (Fig. 4). Further investigation into PLGA particles made from even higher molecular weight polymer may exhibit longer residence time in the knee joints for effective translation into larger animal models and eventually humans. For the in vitro experiments, which typically lasted 48 hours, we chose PLGA MPs that released the drug within this timeframe to achieve the maximum therapeutic response in chondrocytes in culture. In contrast, the high molecular weight particles used for the in vivo studies were designed to release the drug efficiently over a more extended period (∼30 days) and maintain therapeutic concentration in the knee joints.
The various etiologies of OA are trauma due to sudden impact, and aging that can cause a surge in ROS production and diminished autophagy.27,73–77 Previous studies have demonstrated that 15-LOX was upregulated in OA under trauma and hypoxic conditions, and its prime metabolite 15(S)-HETE is implicated in mediating the surge in ROS production.55 Autophagy plays an essential role by actively recycling the ROS molecules and the dysfunctional mitochondria and stressed endoplasmic reticulum.25–27,35 With trauma to the knee joints, the chondrocytes undergo autophagic imbalance leading to cellular senescence and impairment of the cartilage repair process.27,78
NDGA is a cell-permeable anti-oxidant, autophagy up regulator, and selective LOX inhibitor. It prevents the accumulation of damaging ROS species and helps maintain cellular homeostasis.21,53,79 Our in vitro studies have shown that NDGA as a free drug and a microparticle formulation reduced levels of 15(S)-HETE in oxidatively stressed chondrocytes (Fig. 2(B)) induced autophagy (Fig. 2(C)–(G)). In our in vivo studies, the NDGA-MP treated groups showed a higher number of cells positive for LC3B and reduced the number of cells positive for 15-LOX expression (Fig. 5(E)–(H) and 6(E)–(H)), which contributed to the prevention of OA damage in mice knee joints, corroborating our in vitro results and the literature available.
Chondrosenescence is another important contributing factor of OA disease due to ROS accumulation, dysfunctional organelles, and poorly upregulated autophagy.80 For the first time, we provide evidence in primary HACs that NDGA-MP can prevent oxidative stress-induced senescence (Fig. 3(A)–(E)) and sustain sGAG production (Fig. 3(F)). Similarly, in vivo studies showed a reduced number of p19ARF in the NDGA-MP treated group, indicating fewer senescent cells (Fig. 5(I), (J) and 6(I), (J)). Thus, NDGA-MP administration into mouse knee joints effectively attenuated surgery-induced cartilage damage (Fig. 5(B), (C) and 6(B), (C)) and reduced clinical manifestation of pain (Fig. 5(D) and 6(D)). Additionally, the H&E staining of the mice knee joint synovium showed that our MPs mitigated osteoarthritic changes in knee cartilage and effectively reduced synovial inflammation associated with OA. Although the primary therapeutic focus was maintaining and improving chondrocyte health within the cartilage, we attribute the reduced synovial inflammation observed could be an indirect benefit of improved joint health due to the MP's intervention (Fig. S10A–E, ESI†).
We also attempted a single-dose regimen administered a month after the surgery and observed continued protection against cartilage damage (Fig. 7(B) and (C)), however, delayed treatment with NDGA-MPs failed to significantly improve pain scores or reduce osteophyte volume (Fig. 7(D)–(J)). Unfortunately, we did not have access to μCT during the early stages of our research, which prevented us from including μCT data for the preventive and curative two-dose regimen animal studies. These findings suggest that a single administration of the NDGA-MP formulation may be insufficient to fully address bone degeneration in more advanced cases of established OA. Further investigation is required in larger animal models where the disease development is slower compared to mice. Several potent and safer analogues of NDGA are also described and can also be tested for the treatment of OA.48,69,81 Additional studies are needed to evaluate the dosing regimens and effectiveness in higher animal models for progression into the clinics.
In summary, we report NDGA in a sustained release formulation has the potential to be a DMOAD to ameliorate post-traumatic OA. The relevance of our results to human disease was corroborated using chondrocytes isolated from osteoarthritic patients. Although the inclusion of human samples strengthens the translational potential of our study, we acknowledge that achieving an equal number of male and female donors would have further enhanced its robustness. However, due to the limited availability of male donors during the sample collection period, we were unable to achieve this balance. Overall, this study open up avenues of using autophagy inducers, ROS quenchers and senescence prevention for the treatment of OA.
Experimental
A brief description of the experimental methods used in the study is presented here. A detailed description of the methods is provided in the ESI.†Synthesis of PLGA microparticles
PLGA microparticles were synthesized using a single emulsion technique with PLGA polymers of different molecular weights (10–15 kDa, 85–100 kDa, and 190–240 kDa). The release studies were performed in 1× PBS. For in vitro studies, NDGA (3 mg) encapsulated in 10–15 kDa PLGA was used and for in vivo studies, NDGA (5 mg) encapsulated in 85–100 kDa PLGA was used for therapeutic delivery.Isolation of human articular chondrocytes from human knee joint surfaces
Knee joints excised during total knee arthroplasty surgery were immediately brought to the IISc tissue culture facility and processed for articular chondrocyte isolation. The isolated cells were cultured and used within the first 3 passages for most of the experiments.WST assay
Chondrocytes (C28/I2) plated in a 96-well plate were treated with increasing concentrations of NDGA for 24 h or 48 h. Post incubation timepoint, WST reagent was added, and absorbance readings were measured at 470 nm.Senescence induction assay
Primary human articular chondrocytes (HACs) or immortal chondrocytes (C28/I2) were treated for 48 h with genotoxic stress agent-BrdU (200 μM) or oxidative stress agent-hydrogen peroxide (H2O2) (200 μM) to induce senescence. Colorimetric SA-β Gal activity was used to stain senescent cells as previously described.82 Bright-field images were taken from each treatment group and a macros algorithm was used to count the total number of senescent cells in ImageJ.NDGA-MP treatment in senescence induction assays (micromass culture)
The HACs were seeded as a 15 μL suspension in growth media in a 24-well plate, allowed to adhere, and treated with BrdU (600 μM) or H2O2 (100 μM) and co-treated with free NDGA (3 μM) or NDGA-MP (final concentration 3 μM) for 48 h. Post incubation, Alcian Blue staining was used to quantify the sGAG produced by the different treatment groups.Immunocytochemistry
Post 48 h of treatment, HACs were fixed and stained using LC-3 polyclonal antibody and DAPI. The LC3B puncta in the images were quantified using ImageJ using a custom-macros algorithm.Measurement of 15(S)-HETE metabolite by enzyme-linked immunosorbent assay (ELISA)
The production level of 15(S)-HETE in chondrocytes (C28/I2) was measured using 15(S)-HETE EIA Kit, post 48 h of treatment of with (H2O2) (200 μM).The residence time of PLGA MPs in mouse knee joint
Ten μL of PLGA microparticles (85 kDa–100 kDa with Cy7 dye) were injected in mice's left knee joints by intra-articular injections on day 0 and the contralateral legs received free dye injection. Fluorescence imaging was done using IVIS® Spectrum starting at day 0 until day 35 to evaluate the residence time of PLGA MPs in mice knee joints.PTOA mice model
The widely used DMM surgery to induce PTOA in mice was used in this study. The DMM-operated mice were treated with NDGA as a free drug or in MP formulation administered as an intra-articular injection at predetermined time points to maintain therapeutic levels.Histopathology
The fixed mice knee joints were embedded in paraffin and five-micron sections were stained with Safranin O staining, Hematoxylin and Eosin staining or immunohistochemical staining for several antibodies such as MMP-13, ADAMTS-5, LC3B, 15-LOX, and p19ARF antibodies.MicroCT experiments
Mice knee joints were mounted in Bruker SKYSCAN 1272 and scanned at preset voltage/current as 10 μm voxels to obtain tomographs that were reconstructed using Scanco Medical evaluation software and further evaluated for several morphometric parameters.Assessment of pain
Mechanical sensitivity of knee joints post DMM surgery was evaluated before and after treatments to assess the functional evaluation of the knee joints using Von Frey Anesthesiometer.Author contributions
Conceptualization: RA; methodology: RA, KMD, BP, AKP, NMP, AL, NJW; investigation: RA, KMD, BP, AAD, SA, NMP, AL, NJW; visualization: RA, KMD, BP; Supervision: RA; writing – original draft: KMD; writing – review & editing: RA, KMD, BP, AAD, SA, NMP, AL, NJW.Data availability
All data are available in the main text or the ESI.†Conflicts of interest
All other authors declare they have no competing interests.Acknowledgements
We thank Central Animal Facility (CAF) for breeding and maintaining mice and access to IVIS facilities which Ms Lekha Kandasami facilitated. Dr G. Dhanusha and Dr Dinesh M. B. performed the OARSI scoring as a blinded trained veterinarian. We also extend our thanks to Dr Pallavi Raj Sharma for help in editing the manuscript. Early Career Research Award (Science and Engineering Research Board, India, ECR/2017/002178). Har Gobind Khorana Innovative Young Biotechnologist Award (Department of Biotechnology, India, BT/12/IYBA/2019/04). Funding from Dr Vijaya and Rajagopal Rao for Biomedical Engineering research at the Department of Bioengineering is also acknowledged.References
- D. J. Hunter, L. March and M. Chew, Lancet, 2020, 396, 1711–1712 CrossRef PubMed.
- H. Long, Q. Liu, H. Yin, K. Wang, N. Diao, Y. Zhang, J. Lin and A. Guo, Arthritis Rheum., 2022, 74, 1172–1183 CrossRef PubMed.
- W. Zhang, H. Ouyang, C. R. Dass and J. Xu, Bone Res., 2016, 4, 15040 CrossRef CAS PubMed.
- G. L. Matthews and D. J. Hunter, Expert Opin. Emerging Drugs, 2011, 16, 479–491 CrossRef PubMed.
- A. Latourte, M. Kloppenburg and P. Richette, Nat. Rev. Rheumatol., 2020, 16, 673–688 CrossRef PubMed.
- K. Yudoh, T. van Nguyen, H. Nakamura, K. Hongo-Masuko, T. Kato and K. Nishioka, Arthritis Res. Ther., 2005, 7, R380–R391 CrossRef CAS PubMed.
- F. J. Blanco, I. Rego and C. Ruiz-Romero, Nat. Rev. Rheumatol., 2011, 7, 161–169 CrossRef CAS PubMed.
- Y. E. Henrotin, P. Bruckner and J. P. Pujol, Osteoarthritis Cartilage, 2003, 11, 747–755 CrossRef CAS PubMed.
- M. Koike, H. Nojiri, Y. Ozawa, K. Watanabe, Y. Muramatsu, H. Kaneko, D. Morikawa, K. Kobayashi, Y. Saita, T. Sasho, T. Shirasawa, K. Yokote, K. Kaneko and T. Shimizu, Sci. Rep., 2015, 5, 11722 CrossRef CAS PubMed.
- M. Z. Miao, Q. P. Su, Y. Cui, E. M. Bahnson, G. Li, M. Wang, Y. Yang, J. A. Collins, D. Wu, Q. Gu, S. Chubinskaya, B. O. Diekman, K. M. Yamada and R. F. Loeser, Sci. Signaling, 2023, 16, eadf8299 CrossRef CAS PubMed.
- M. L. Tiku, R. Shah and G. T. Allison, J. Biol. Chem., 2000, 275, 20069–20076 CrossRef CAS PubMed.
- C. Rathakrishnan, K. Tiku, A. Raghavan and M. L. Tiku, J. Bone Miner. Res., 1992, 7, 1139–1148 CrossRef CAS PubMed.
- Y. Henrotin, G. Deby-Dupont, C. Deby, M. De Bruyn, M. Lamy and P. Franchimont, Br. J. Rheumatol., 1993, 32, 562–567 CrossRef CAS PubMed.
- C. L. Fattman, L. M. Schaefer and T. D. Oury, Free Radical Biol. Med., 2003, 35, 236–256 CrossRef CAS PubMed.
- M. Y. Ansari, N. M. Khan, I. Ahmad and T. M. Haqqi, Osteoarthritis Cartilage, 2017, 26, 1087–1097 CrossRef PubMed.
- H. J. Faust, H. Zhang, J. Han, M. T. Wolf, O. H. Jeon, K. Sadtler, A. N. Peña, L. Chung, D. R. Maestas, Jr., A. J. Tam, D. M. Pardoll, J. Campisi, F. Housseau, D. Zhou, C. O. Bingham, III and J. H. Elisseeff, J. Clin. Invest., 2020, 130, 5493–5507 CrossRef CAS PubMed.
- C. Vinatier, E. Domínguez, J. Guicheux and B. Caramés, Front. Physiol., 2018, 9, 1–25 Search PubMed.
- J. Ghosh and C. E. Myers, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 13182–13187 CrossRef CAS PubMed.
- I. M. Avis, M. Jett, T. Boyle, M. D. Vos, T. Moody, A. M. Treston, A. Martínez and J. L. Mulshine, J. Clin. Invest., 1996, 97, 806–813 CrossRef CAS PubMed.
- T. W. Friedlander, V. K. Weinberg, Y. Huang, J. T. Mi, C. G. Formaker, E. J. Small, A. L. Harzstark, A. M. Lin, L. Fong and C. J. Ryan, Oncol. Rep., 2012, 27, 3–9 CAS.
- G. Manda, A. I. Rojo, E. Martínez-Klimova, J. Pedraza-Chaverri and A. Cuadrado, Front. Pharmacol., 2020, 11, 151 CrossRef CAS PubMed.
- E. Floriano-Sánchez, C. Villanueva, O. N. Medina-Campos, D. Rocha, D. J. Sánchez-González, N. Cárdenas-Rodríguez and J. Pedraza-Chaverrí, Free Radical Res., 2006, 40, 523–533 CrossRef PubMed.
- T. Tezil, M. Chamoli, C.-P. Ng, R. P. Simon, V. J. Butler, M. Jung, J. Andersen, A. W. Kao and E. Verdin, npj Aging Mech. Dis., 2019, 5, 7 CrossRef PubMed.
- S. Guzmán-Beltrán, S. Espada, M. Orozco-Ibarra, J. Pedraza-Chaverri and A. Cuadrado, Neurosci. Lett., 2008, 447, 167–171 CrossRef PubMed.
- M. K. Lotz and B. Carames, Nat. Rev. Rheumatol., 2011, 7, 579–587 CrossRef CAS PubMed.
- N. Mizushima, B. Levine, A. M. Cuervo and D. J. Klionsky, Nature, 2008, 451, 1069–1075 CrossRef CAS PubMed.
- B. Carames, N. Taniguchi, S. Otsuki, F. J. Blanco and M. Lotz, Arthritis Rheum., 2010, 62, 791–801 CrossRef CAS PubMed.
- H. Sasaki, K. Takayama, T. Matsushita, K. Ishida, S. Kubo, T. Matsumoto, N. Fujita, S. Oka, M. Kurosaka and R. Kuroda, Arthritis Rheum., 2012, 64, 1920–1928 CrossRef CAS PubMed.
- J. K. Meckes, B. Carames, M. Olmer, W. B. Kiosses, S. P. Grogan, M. K. Lotz and D. D. D’Lima, Osteoarthritis Cartilage, 2017, 25, 1880–1889 CrossRef CAS PubMed.
- B. Carames, A. Hasegawa, N. Taniguchi, S. Miyaki, F. J. Blanco and M. Lotz, Ann. Rheum. Dis., 2012, 71, 575–581 CrossRef CAS PubMed.
- Y. Zhang, F. Vasheghani, Y. H. Li, M. Blati, K. Simeone, H. Fahmi, B. Lussier, P. Roughley, D. Lagares, J. P. Pelletier, J. Martel-Pelletier and M. Kapoor, Ann. Rheum. Dis., 2015, 74, 1432–1440 CrossRef CAS PubMed.
- U. Nogueira-Recalde, I. Lorenzo-Gomez, F. J. Blanco, M. I. Loza, D. Grassi, V. Shirinsky, I. Shirinsky, M. Lotz, P. D. Robbins, E. Dominguez and B. Carames, EBioMedicine, 2019, 45, 588–605 CrossRef PubMed.
- K. M. Dhanabalan, A. A. Dravid, S. Agarwal, R. K. Sharath, A. K. Padmanabhan and R. Agarwal, Bioeng. Transl. Med., 2023, 8, e10298 CrossRef CAS PubMed.
- K. M. Dhanabalan, V. K. Gupta and R. Agarwal, Biomater. Sci., 2020, 8, 4308–4321 RSC.
- G. Filomeni, D. De Zio and F. Cecconi, Cell Death Differ., 2015, 22, 377–388 CrossRef CAS PubMed.
- H. Fuhrmann-Stroissnigg, Y. Y. Ling, J. Zhao, S. J. McGowan, Y. Zhu, R. W. Brooks, D. Grassi, S. Q. Gregg, J. L. Stripay, A. Dorronsoro, L. Corbo, P. Tang, C. Bukata, N. Ring, M. Giacca, X. Li, T. Tchkonia, J. L. Kirkland, L. J. Niedernhofer and P. D. Robbins, Nat. Commun., 2017, 8, 422 CrossRef PubMed.
- I. H. Lee and T. Finkel, J. Biol. Chem., 2009, 284, 6322–6328 CrossRef CAS PubMed.
- S. M. Son, S. J. Park, E. Stamatakou, M. Vicinanza, F. M. Menzies and D. C. Rubinsztein, Nat. Commun., 2020, 11, 3148 CrossRef CAS PubMed.
- S. M. Son, S. J. Park, H. Lee, F. Siddiqi, J. E. Lee, F. M. Menzies and D. C. Rubinsztein, Cell Metab., 2019, 29, 192–201 CrossRef CAS PubMed.
- A. Bánréti, M. Sass and Y. Graba, Autophagy, 2013, 9, 819–829 CrossRef PubMed.
- M. Del Carlo, D. Schwartz, E. A. Erickson and R. F. Loeser, Free Radicals Biol. Med., 2007, 42, 1350–1358 CrossRef CAS PubMed.
- E.-H. Hong, S.-J. Lee, J.-S. Kim, K.-H. Lee, H.-D. Um, J.-H. Kim, S.-J. Kim, J.-I. Kim and S.-G. Hwang, J. Biol. Chem., 2010, 285, 1283–1295 CrossRef CAS PubMed.
- X. Chevalier, P. Goupille, A. D. Beaulieu, F. X. Burch, W. G. Bensen, T. Conrozier, D. Loeuille, A. J. Kivitz, D. Silver and B. E. Appleton, Arthritis Rheum., 2009, 61, 344–352 CrossRef CAS PubMed.
- M. A. Karsdal, M. Michaelis, C. Ladel, A. S. Siebuhr, A. R. Bihlet, J. R. Andersen, H. Guehring, C. Christiansen, A. C. Bay-Jensen and V. B. Kraus, Osteoarthritis Cartilage, 2016, 24, 2013–2021 CrossRef CAS PubMed.
- E. F. Ekman, J. S. Gimbel, A. E. Bello, M. D. Smith, D. S. Keller, K. M. Annis, M. T. Brown, C. R. West and K. M. Verburg, J. Rheumatol., 2014, 41, 2249–2259 CrossRef CAS PubMed.
- O. H. Jeon, C. Kim, R. M. Laberge, M. Demaria, S. Rathod, A. P. Vasserot, J. W. Chung, D. H. Kim, Y. Poon, N. David, D. J. Baker, J. M. van Deursen, J. Campisi and J. H. Elisseeff, Nat. Med., 2017, 23, 775–781 CrossRef CAS PubMed.
- C. H. Evans, V. B. Kraus and L. A. Setton, Nat. Rev. Rheumatol., 2014, 10, 11–22 CrossRef CAS PubMed.
- G. S. M. John, V. K. Vuttaradhi, S. Takeuchi, R. S. Pitani, G. Venkatraman and S. K. Rayala, J. Nanobiotechnol., 2020, 18, 74 CrossRef CAS PubMed.
- T. Y. Yang, J. P. Chen, K. W. Ku, S. H. Fu and B. R. Hsu, Transplant. Proc., 2005, 37, 1828–1829 CrossRef CAS PubMed.
- F. Danhier, E. Ansorena, J. M. Silva, R. Coco, A. Le Breton and V. Preat, J. Controlled Release, 2012, 161, 505–522 CrossRef CAS PubMed.
- H. K. Makadia and S. J. Siegel, Polymers, 2011, 3, 1377–1397 CrossRef CAS PubMed.
- A. Singh, R. Agarwal, C. A. Diaz-Ruiz, N. J. Willett, P. Wang, L. A. Lee, Q. Wang, R. E. Guldberg and A. J. Garcia, Adv. Healthcare Mater., 2014, 3, 1562–1567 CrossRef CAS PubMed , 1525.
- J. E. Blecha, M. O. Anderson, J. M. Chow, C. C. Guevarra, C. Pender, C. Penaranda, M. Zavodovskaya, J. F. Youngren and C. E. Berkman, Bioorg. Med. Chem. Lett., 2007, 17, 4026–4029 CrossRef CAS PubMed.
- T. Shen, J. Shi, N. Wang, X. Yu, C. Zhang, J. Li, L. Wei, C. Ma, X. Zhao, M. Lian, C. Jiang and D. Zhu, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2015, 309, L449–L462 CrossRef CAS PubMed.
- K. Chen, Y. Yan, C. Li, J. Yuan, F. Wang, P. Huang, N. Qian, J. Qi, H. Zhou, Q. Zhou, L. Deng, C. He and L. Guo, Cell Death Dis., 2017, 8, e3109 CrossRef CAS PubMed.
- S. Barth, D. Glick and K. F. Macleod, J. Pathol., 2010, 221, 117–124 CrossRef CAS PubMed.
- K. V. Greco, A. J. Iqbal, L. Rattazzi, G. Nalesso, N. Moradi-Bidhendi, A. R. Moore, M. B. Goldring, F. Dell’Accio and M. Perretti, Biochem. Pharmacol, 2011, 82, 1919–1929 CrossRef CAS PubMed.
- H. Fang, L. Huang, I. Welch, C. Norley, D. W. Holdsworth, F. Beier and D. Cai, Sci. Rep., 2018, 8, 2855 CrossRef PubMed.
- S. S. Glasson, T. J. Blanchet and E. A. Morris, Osteoarthritis Cartilage, 2007, 15, 1061–1069 CrossRef CAS PubMed.
- H. Huang, J. D. Skelly, D. C. Ayers and J. Song, Sci. Rep., 2017, 7, 42294 CrossRef CAS PubMed.
- S. Mahr, J. Menard, V. Krenn and B. Müller, Ann. Rheum. Dis., 2003, 62, 1234–1237 CrossRef CAS PubMed.
- H. L. Ma, T. J. Blanchet, D. Peluso, B. Hopkins, E. A. Morris and S. S. Glasson, Osteoarthritis Cartilage, 2007, 15, 695–700 CrossRef PubMed.
- B. He, M. Christin, S. Mouchbahani-Constance, A. Davidova and R. Sharif-Naeini, Osteoarthritis Cartilage, 2017, 25, 2091–2099 CrossRef CAS PubMed.
- M. Wang, E. R. Sampson, H. Jin, J. Li, Q. H. Ke, H.-J. Im and D. Chen, Arthritis Res. Ther., 2013, 15, R5 CrossRef CAS PubMed.
- H. Li, D. Wang, Y. Yuan and J. Min, Arthritis Res. Ther., 2017, 19, 248 CrossRef PubMed.
- A. G. Juby, K. Skeith and P. Davis, Clin. Rheumatol., 2005, 24, 535–538 CrossRef PubMed.
- M. Hou, Y. Zhang, X. Zhou, T. Liu, H. Yang, X. Chen, F. He and X. Zhu, Cell Death Dis., 2021, 12, 483 CrossRef CAS PubMed.
- J. P. Pelletier, J. P. Caron, C. Evans, P. D. Robbins, H. I. Georgescu, D. Jovanovic, J. C. Fernandes and J. Martel-Pelletier, Arthritis Rheum., 1997, 40, 1012–1019 CrossRef CAS PubMed.
- G. S. Mala John, S. Takeuchi, G. Venkatraman and S. K. Rayala, Curr. Cancer Drug Targets, 2020, 20, 86–103 CrossRef CAS PubMed.
- E. H. Hong, S. J. Lee, J. S. Kim, K. H. Lee, H. D. Um, J. H. Kim, S. J. Kim, J. I. Kim and S. G. Hwang, J. Biol. Chem., 2010, 285, 1283–1295 CrossRef CAS PubMed.
- A. P. Evan and K. D. Gardner, Kidney Int., 1979, 15, 7–19 CrossRef CAS PubMed.
- B. C. Geiger, S. Wang, R. F. Padera, Jr., A. J. Grodzinsky and P. T. Hammond, Sci. Transl. Med., 2018, 10, 469 Search PubMed.
- T. Aigner, A. Sachse, P. M. Gebhard and H. I. Roach, Adv. Drug Delivery Rev., 2006, 58, 128–149 CrossRef CAS PubMed.
- J. Martel-Pelletier, A. J. Barr, F. M. Cicuttini, P. G. Conaghan, C. Cooper, M. B. Goldring, S. R. Goldring, G. Jones, A. J. Teichtahl and J. P. Pelletier, Nat. Rev. Dis. Primers, 2016, 2, 16072 CrossRef PubMed.
- M. Arra, G. Swarnkar, K. Ke, J. E. Otero, J. Ying, X. Duan, T. Maruyama, M. F. Rai, R. J. O’Keefe, G. Mbalaviele, J. Shen and Y. Abu-Amer, Nat. Commun., 2020, 11, 3427 CrossRef CAS PubMed.
- D. M. Findlay, Rheumatology, 2007, 46, 1763–1768 CrossRef CAS PubMed.
- T. Malinin and E. A. Ouellette, Osteoarthritis Cartilage, 2000, 8, 483–491 CrossRef CAS PubMed.
- B. Carames, N. Taniguchi, D. Seino, F. J. Blanco, D. D’Lima and M. Lotz, Arthritis Rheum., 2012, 64, 1182–1192 CrossRef CAS PubMed.
- S. Y. Kim, T.-B. Kim, K.-A. Moon, T. J. Kim, D. Shin, Y. S. Cho, H.-B. Moon and K.-Y. Lee, Exp. Mol. Med., 2008, 40, 461–476 CrossRef CAS PubMed.
- A. Mobasheri, C. Matta, R. Zakany and G. Musumeci, Maturitas, 2015, 80, 237–244 CrossRef CAS PubMed.
- M. J. Daniels, J. B. Nourse, H. Kim, V. Sainati, M. Schiavina, M. G. Murrali, B. Pan, J. J. Ferrie, C. M. Haney, R. Moons, N. S. Gould, A. Natalello, R. Grandori, F. Sobott, E. J. Petersson, E. Rhoades, R. Pierattelli, I. Felli, V. N. Uversky, K. A. Caldwell, G. A. Caldwell, E. S. Krol and H. Ischiropoulos, Sci. Rep., 2019, 9, 2937 CrossRef PubMed.
- F. Debacq-Chainiaux, J. D. Erusalimsky, J. Campisi and O. Toussaint, Nat. Protoc., 2009, 4, 1798–1806 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01342e |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |