
Nucleic acid-binding bis-acridine orange dyes with improved properties for bioimaging and PCR applications†
Olesia
Kulyk
a,
Alexander
Krivoshey
a,
Olga
Kolosova
 a,
Ivanna
Prylutska
b,
Tudor
Vasiliu
cd,
Razvan
Puf
c,
Francesca
Mocci
e,
Aatto
Laaksonen
cfgh,
Sergiy
Perepelytsya
i,
Dmytro
Kobzev
aj,
Rostyslav
Svoiakov
a,
Zenoviy
Tkachuk
b and
Anatoliy
Tatarets‡
*a
a,
Ivanna
Prylutska
b,
Tudor
Vasiliu
cd,
Razvan
Puf
c,
Francesca
Mocci
e,
Aatto
Laaksonen
cfgh,
Sergiy
Perepelytsya
i,
Dmytro
Kobzev
aj,
Rostyslav
Svoiakov
a,
Zenoviy
Tkachuk
b and
Anatoliy
Tatarets‡
*a
aInstitute of Functional Materials Chemistry of State Scientific Institution “Institute for Single Crystals” of National Academy of Sciences of Ukraine, Kharkiv, 61072, Ukraine. E-mail: tatarets@isc.kh.ua; Tel: +38 097 7566276
bInstitute of Molecular Biology and Genetics of the National Academy of Sciences of Ukraine, Kyiv, 03143, Ukraine
cCenter of Advanced Research in Bionanoconjugates and Biopolymers, “Petru Poni” Institute of Macromolecular Chemistry, Iasi, 700487, Romania
dThe Research Institute of the University of Bucharest (ICUB), Bucharest, 050663, Romania
eDipartimento di Scienze Chimiche e Geologiche, Università di Cagliari, Cagliari, 09042, Italy
fDepartment of Materials and Environmental Chemistry, Division of Physical Chemistry, Arrhenius Laboratory, Stockholm University, Stockholm, 106 91, Sweden
gState Key Laboratory of Materials-Oriented and Chemical Engineering, Nanjing Tech University, Nanjing, 210009, P. R. China
hDepartment of Engineering Sciences and Mathematics, Division of Energy Science, Luleå University of Technology, Luleå, 97187, Sweden
iBogolyubov Institute for Theoretical Physics of the National Academy of Sciences of Ukraine, Kyiv, 03143, Ukraine
jInstitute for Experimental Molecular Imaging, Center for Biohybrid Medical Systems, RWTH Aachen University Clinic, Aachen, 52074, Germany
First published on 23rd October 2024
Abstract
Understanding the intricate interactions of molecular dyes with nucleic acids is pivotal for advancing medical and biochemical applications. In this work, we present a comprehensive study of the interplay between a novel series of bis-acridine orange (BAO) dyes and double-stranded DNA (dsDNA). These BAO dyes were intentionally designed as two acridine orange units connected by neutral linkers featuring a 2,5-disubstituted thiophene moiety. Comparative analysis of BAO compounds with the widely utilized DNA-binding dye EvaGreen (EG) was carried out for fibroblast staining and qPCR analysis. The results show that BAO dyes outperform EG by supporting PCR amplification over a broader concentration range (0.5–5.0 μM). Furthermore, they exhibit an exceptional capability to generate consistent DNA melting curves regardless of DNA concentration fluctuations. Molecular dynamics simulations showed that BAO dyes when interacting with dsDNA unfold from the stacked conformation to the elongated one. The difference in the energy between the conformations is shown to be concomitant with fluorescence enhancement. This study enriches our understanding of the intricate interplay between innovative BAO dyes and dsDNA, fostering their applications in medical and biochemical research, particularly in qPCR methodologies and bioimaging techniques.
1. Introduction
A large variety of ligands able to bind to DNA have an important role in medicine, both for therapeutic and diagnostics purposes. The interactions between drug molecules and DNA play a crucial role in pharmacology, particularly in the development of drugs for combating diseases such as cancer and infections.1 In diagnostic applications, there are molecules specifically designed to bind to DNA and act as reporters enabling the detection of biological events involving DNA.2 They are engineered to bind to DNA in specific ways resulting in the production of a signal that reflects the binding. Among the most important reporters are fluorescent dyes emitting light when they interact with DNA.3Two techniques involving dyes commonly used in molecular biology to detect and analyze nucleic acid-containing materials are histological staining and the real-time quantitative polymerase chain reaction (qPCR). In staining, either a single dye is used to highlight a specific tissue component, or a mixture of dyes can be used to visualize and highlight different objects with different colors. In nucleic acid staining, it is common to use fluorescent dyes that bind to DNA or RNA and emit when exposed to light of a certain wavelength.4,5 Among the most used fluorescent molecules that stain DNA are ethidium bromide,6SYBR Green (SG),7DAPI,8Hoechst,9 and acridine orange (AO) dyes.6
qPCR is a powerful tool of molecular biology,10 used for medical diagnostics,11 genotyping,12 DNA cloning,13 and forensic DNA analysis.14 qPCR consists of a series of repeated thermal cycles leading to an exponential amplification of the target DNA sequence in a biological sample.15,16 In qPCR, the accumulation of the amplification product (amplicon) is monitored and quantified as the reaction progresses in real time, while in conventional PCR, the results are evaluated only at the end of the reaction.15 Analysis of the amplicons can be carried out using fluorogenic dyes that substantially increase their fluorescence upon binding to DNA,17,18 or fluorescently labeled oligonucleotides (hybridization probes).19 The advantage of hybridization probes is their high sensitivity and specificity to DNA sequences. However, the complexity of their production significantly increases the cost of the analysis. In contrast, the use of fluorogenic dyes provides a simpler and less expensive DNA detection.
To date, a number of dyes suitable for qPCR applications have been developed,17 but most of them suffer from one or several drawbacks such as PCR inhibition, photobleaching, cytotoxicity, genotoxicity, poor thermal and/or hydrolytic stability, insufficient specificity to double-stranded DNA (dsDNA), and/or unsatisfactory brightness of dye-DNA complexes. Among the best commercially available qPCR dyes are SG7 and EvaGreen (EG).20 These dyes are highly specific dsDNA binding dyes with absorption and emission maxima in the presence of dsDNA at approximately 500 and 530 nm, respectively, that make them compatible with practical qPCR systems. SG, however, is insufficiently stable and degrades under routine PCR conditions,21 and also inhibits PCR at concentrations above 0.34 μM, while for EG, the limiting concentration is almost 4 times higher (1.33 μM).22 The use of these dyes at higher concentrations is limited since they can interfere with the amplification through the deactivation of Taq-polymerase or undesirable binding of a dye to primers, dimers and non-specific PCR products, or due to the capability of dyes to alter the ion composition in the reaction mixture.23 On the other hand, the high dye concentration eliminates the dye redistribution problem24 allowing the generation of stronger and more robust fluorescent signals for higher resolution melt curve analysis and single-base-pair mismatch discrimination.25 Hence, dsDNA binding dyes which can be used at higher, saturating concentrations without inhibiting the PCR reaction are beneficial for accurate quantitation and reliable melt curve analysis. Moreover, high concentrations of dyes can help to detect DNA fragments in the presence of quenching substances.26
Recently, we developed a series of bis-acridine orange (BAO) dyes,27 demonstrated their applicability for qPCR assays, and found that BAO-1 (Fig. 1), consisting of two acridine orange moieties bound by a linker containing a 2,5-disubstituted thiophene cycle as the central core, exhibited the best performance without qPCR inhibition at dye concentrations up to 1.75 μM. Moreover, BAO-1 demonstrated good stability, instrument compatibility, and no cytotoxic or genotoxic effects at concentrations well above the qPCR working concentration. Importantly, BAO-1 outperformed the widely used qPCR dye SYBR Green I, showcasing its potential as a highly effective alternative in qPCR assays.27
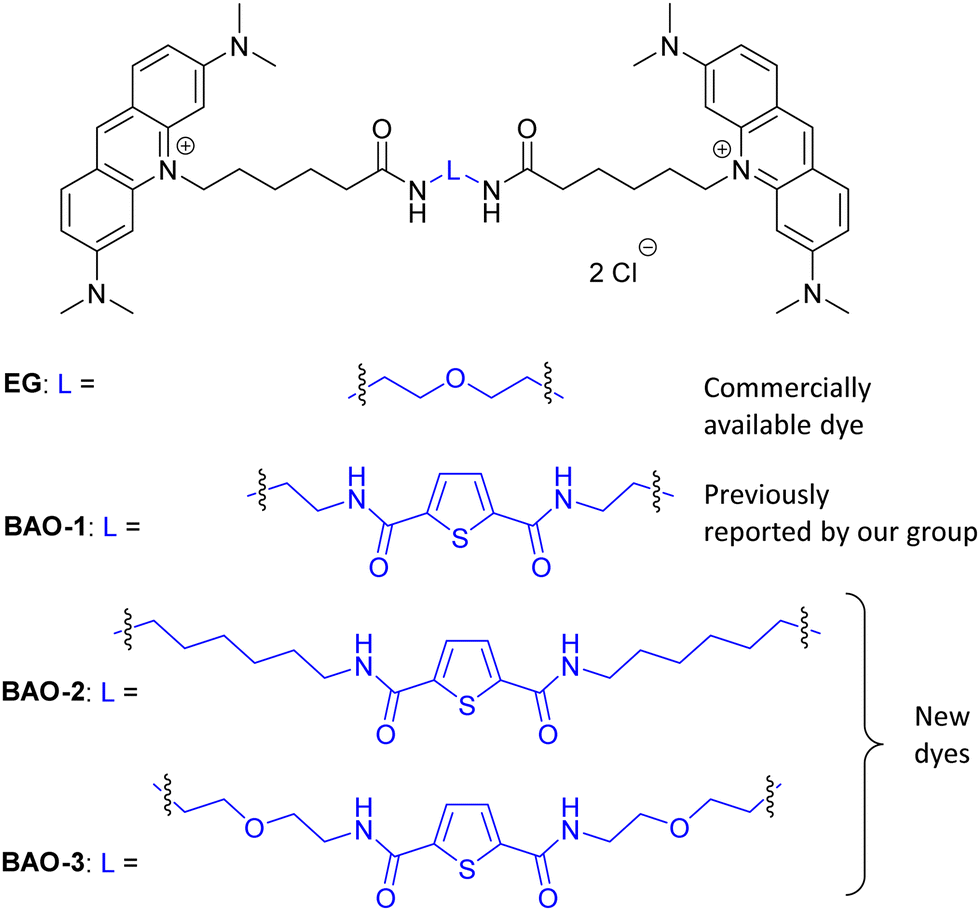 | ||
| Fig. 1 Chemical structures of commercial EG and novel BAO dyes. | ||
In this work, in order to further study the applicability of the BAO type dyes in dsDNA research, we: (1) compared the properties of BAO-1 with EG that has a similar molecular structure (Fig. 1); (2) varied hydrophilicity and linker flexibility of the BAO type dyes by variation of the bridge length and oxygen substitution in the bridge (BAO-2 and BAO-3; Fig. 1); and (3) extended the use of the dyes for DNA sensing applications.
To explain the advantages of the BAO dyes over EG, we studied the interactions of the ligands with dsDNA at the atomistic level by means of molecular dynamics simulations. To verify the conformational preferences of the dyes, and the effect of their interaction with dsDNA on their optical properties, we used umbrella sampling simulations to study the evolution from a compact conformation of the dye characterized by the stacking of the aromatic moieties to an extended conformation and checked the energy difference between these two states. Furthermore, we investigated the spectral properties of the dyes free in solutions and upon interaction with dsDNA to further study the performance of the dyes in qPCR analyses and cell imaging.
2. Materials and methods
2.1. Synthesis and characterization
Detailed information regarding the starting materials, synthesis, purification protocols, and spectral measurements can be found in the ESI.† The synthesis of BAO-1, BAO-2, and BAO-3 dyes (Fig. 1) followed a procedure similar to our previously reported method (Section S1, ESI†).27 The structures of all synthesized compounds were confirmed using 1H and 13C NMR spectroscopy, as well as mass spectrometry (Sections S6–S8, ESI†). Experimental procedures and details on the measurement of absorption, fluorescence excitation and emission spectra, fluorescence lifetimes, binding constants, brightness, and stability tests are provided in Section S2 of the ESI.†2.2. Cell preparation, cytotoxicity assay and confocal microscopy
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. The cells were used at passages 3–4. Cultured fibroblasts formed a 100% confluent monolayer in culture flasks.
:2. The cells were used at passages 3–4. Cultured fibroblasts formed a 100% confluent monolayer in culture flasks.
All experiments were carried out following the European Convention for the Protection of Vertebrate Animals used for Experimental and other Scientific Purposes (EST 123), Directive 2010/63/EU for the protection of animals used for scientific purposes, and Recommendation 2007/526/EC. The study was approved by the Commission on Ethics and Bioethics (Kharkiv National Medical University, Kharkiv, Ukraine).
The fibroblasts were seeded in 96-well cell culture plates (SPL, Republic of Korea) at a concentration of 1 × 104 cells per well and incubated for 24 h in a CO2-incubator (Thermo Fisher Scientific, USA) at 37 °C and in an atmosphere of 5% CO2. The stock solutions of 250 μM EG or BAO dyes in DMSO were prepared and aliquots of 6 μL, 12 μL, or 24 μL were added to 1 mL of cultured fibroblasts to the final dye concentrations: 1.5 μM, 3.0 μM, or 6.0 μM, respectively. Thereafter, fibroblasts treated with these dyes were additionally incubated for 24 h. For the control sample, the fibroblasts were incubated for 24 h without the tested dye (negative control) or with the corresponding concentrations (0.6%, 1.2%, 2.4%) of DMSO. The area covered by fibroblasts on a growth surface was then estimated for each sample. Then the medium was removed and 0.1 mL of culture medium with 15 μL of MTT at a concentration of 5 mg mL−1 was added. The resulting samples were incubated in a CO2-incubator for 3 h at 37 °C and 5% CO2. Then the medium was discarded and 0.1 mL of DMSO with 10% sodium dodecyl sulfate (SDS) was added to dissolve the formazan, followed by incubation for 60 min at 37 °C. Absorbance was measured using a UV/Vis microplate reader SM600 at 570 nm.
2.3. PCR and post-PCR experiments
Two-step reverse transcription quantitative PCR (RT-qPCR) was used to perform the experiments.Solutions of the dyes BAO-1, BAO-2, BAO-3, and EG (cDye ∼ 10 μM) in nuclease-free water (Thermo Scientific, USA) were prepared by diluting stock solutions of the dyes (1 mM in DMSO). The final dye concentration was varied (0.25–5.0 μM) in a total reaction volume of 20 μL. The complementary DNA (cDNA) obtained from human leukocyte RNA by a reverse transcription using the Maxima First Strand cDNA Synthesis Kit for RT-qPCR (Thermo Scientific, USA) was used as a template. Three sets of forward and reverse primers, i.e. STAT4, STAT6 and HLA-B, were used in the experiments. Their sequences were as follows: STAT4 forward primer – CAGTGAAAGCCATCTCGGAGGA, STAT4 reverse primer – TGTAGTCTCGCAGGATGTCAGC (amplicon length – 140 bp); STAT6 forward primer – CCTTGGAGAACAGCATTCCTGG, STAT6 reverse primer – GCACTTCTCCTCTGTGACAGAC (amplicon length – 116 bp); HLA-B forward primer – CAGGGCTCTGATGTGTCTC, HLA-B reverse primer – TCAGTCCCTCACAAGACAG (amplicon length – 63 bp).
A reaction mixture for qPCR included the following components: HOT FIREPol® DNA Polymerase – 0.025 U μL−1, HOT FIREPol® 10 × Buffer B1 – 1X, MgCl2 – 1.5 mM, dNTP mix – 200 μM each (all were purchased from Solis BioDyne), 1 mM solution of a studied dye in DMSO that was brought to the final concentration by nuclease-free water for PCR, mix of forward and reverse primers – 0.3 μM each, cDNA – 12 ng and nuclease-free water for PCR.
A negative control (NTC) was prepared for each of the dyes to determine the possible contamination or background fluorescence. The NTC reaction mixture included all components, except the template.
The analysis was performed on CFX96 Touch real-time PCR detection system C1000 Touch (Bio-Rad) according to the standard protocol: initial activation – 95 °C, 12 min; denaturation – 95 °C, 15 s; annealing – 60 °C, 20 s; elongation – 72 °C, 20 s. Fluorescence measurements were acquired for the SYBR channel.
In qPCR the cycle threshold (Ct) value was defined as the number of cycles required for the fluorescent signal to cross the threshold. The fluorescence intensities of dyes (end-point fluorescence intensities) were expressed in relative fluorescence units (RFU). The values of Ct and the end-point fluorescence intensity were presented as an average from all experiments with the SD error. The Ct value was used as an indicator of the inhibitory effect on PCR efficiency since significant decreases in efficiency would result in increased Ct values. The threshold concentrations were considered as the highest possible dye concentrations without causing significant Ct delay and nonspecific product formation. The average Ct values were plotted against the dye concentrations for each dye and PCR amplicons used. A linear relationship was observed where the slope of the trend line indicates the degree of inhibition. In addition, the average end-point fluorescence intensity and the amplicons melting temperature under given PCR conditions were compared. Only one typical amplicon melting curve for each concentration of investigated dye was presented to simplify the data visualization.
All qPCR experiments were repeated twice and run in triplicates, followed by post-PCR product melting curve analysis to confirm product specificity. The melt curve protocol was as follows: heating for 10 s at 95 °C, cooling to 62 °C and then heating from 62 °C to 95 °C with 0.2 °C increments per each 10 s.
2.4. Methods and models for molecular dynamics (MD) simulations
The initial dye structures were built with Avogadro software36 and parameterized with AmberTools 18 package, using the general AMBER force field (GAFF2).37 The partial atomic charges were obtained by RESP38 using Gaussian software at the B3LYP/6-31G(d,p) level of theory39 using the same protocol as described in our previous works.40,41
For each system, prior to the MD simulations, an energy minimization process was performed to remove faulty contacts between atoms, followed by a 10 ns NPT MD simulation (298 K, 1 bar) in which a force constant of 10 kJ mol−1 nm−2 was applied to DNA's atoms to avoid deformations of the DNA structure as a consequence of a solvating surrounding far from equilibrium.
The GROMACS 202042 software package was used to perform all the NPT simulations with the temperatures set at 298 K, by using a Nosé–Hoover thermostat43 with a time constant of 4 ps. The pressure was controlled by a Parrinello–Rahman barostat and set to 1 bar and isotropic pressure coupling with a constant time of 10 ps.44 The time step was 2 fs. And the LINCS algorithm45 was used to constrain the length of all covalent bonds involving hydrogen atoms. The Particle Mesh Ewald (PME) algorithm was used for the long-range electrostatic interactions.46 The length of each simulation trajectory was 500 ns for the BAO-1, BAO-2, and BAO-3 dyes, and 1000 ns for EG. The main details of the simulation box and MD trajectory length are reported in Table 1.
| Dye | Number of atoms | Number of water molecules | Number of ions (Na+/Cl−) | Box size (nm × nm × nm) | Simulation length |
|---|---|---|---|---|---|
| BAO-1 | 71250 |
23181 |
106/66 | 9 × 9 × 9 | 500 ns |
| BAO-2 | 71259 |
23178 |
106/66 | 9 × 9 × 9 | 500 ns |
| BAO-3 | 71234 |
23171 |
106/66 | 9 × 9 × 9 | 500 ns |
| EG | 71254 |
23187 |
106/66 | 9 × 9 × 9 | 1000 ns |
The umbrella method involves pulling the dye over a range of windows (23–26 windows depending on the dye's structure), at a pull rate of 0.1 nm per window, from 0.4 nm to 2.7–3 nm. Each window was simulated for 40 ns to allow a large sampling of the conformational space and to reach a detailed energy profile.
3. Results and discussion
3.1. Spectral properties of dimeric dyes
The absorption, fluorescence excitation and emission maxima (λmax), molar absorptivities (ε), absorption bands half-widths (FWHM), Stokes shifts, fluorescence quantum yields (ΦF), mean fluorescence lifetimes (τmean), and brightness (B) of the BAO dyes and EG measured free in aqueous and methanol solutions (cDye ∼ 1 μM), and in non-covalent complexes with dsDNA (cDNA = 100 μg mL−1) are summarized in Table 2. All the absorption, excitation and emission spectra are given in Fig. S1 (ESI†). As shown in Table 2 and Fig. S1 (ESI†), dimeric dyes EG, BAO-1, BAO-2, and BAO-3 exhibit similar absorption and emission maxima, differing by no more than 4 nm in all measured media.| Dye | Media | λ max (Abs), nm | Abs. FWHM, cm−1 | λ max (Exc), nm | λ max (Em), nm | Stokes shift, cm−1 | ε, M−1 cm−1 | Φ F, % | τ mean, ns | B (488nm), M−1 cm−1 | K b × 104, M−1 |
|---|---|---|---|---|---|---|---|---|---|---|---|

|
Tris buffer | 472 | 2030 | 496 | 524 | 1080 | 7020020 |
1.8 | 1.40 | 830 | — |
| MeOH | 495 | 2000 | 495 | 522 | 1040 | 90100 |
19 | 1.65 | 15200 |
— | |
| dsDNA | 501 | 1990 | 502 | 528 | 980 | 80800 |
44 | 5.41 | 25100 |
3.1 | |

|
Tris buffer | 476 | 2130 | 499 | 524 | 960 | 80100 |
2.5 | 1.33 | 1500 | — |
| MeOH | 496 | 1990 | 496 | 521 | 970 | 107600 |
24 | 1.56 | 22600 |
— | |
| dsDNA | 504 | 1800 | 505 | 527 | 830 | 85300 |
54 | 4.67 | 27900 |
5.9 | |
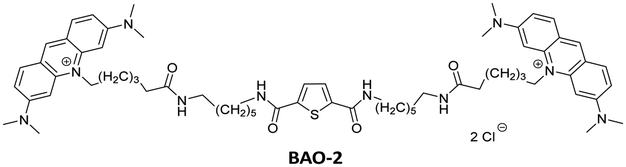
|
Tris buffer | 475 | 1940 | 497 | 525 | 1070 | 80300 |
2.2 | 1.30 | 1200 | — |
| MeOH | 495 | 1810 | 496 | 521 | 970 | 107200 |
29 | 1.61 | 27000 |
— | |
| dsDNA | 502 | 2110 | 503 | 527 | 900 | 85100 |
39 | 4.54 | 24600 |
2.5 | |
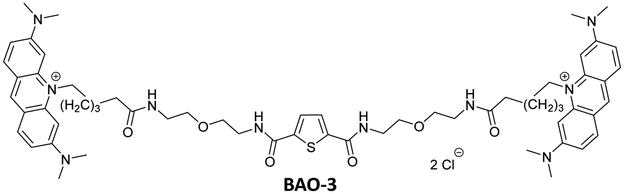
|
Tris buffer | 475 | 1950 | 499 | 525 | 990 | 80000 |
2.0 | 1.26 | 1000 | — |
| MeOH | 496 | 1860 | 495 | 522 | 1050 | 107100 |
25 | 1.61 | 22600 |
— | |
| dsDNA | 503 | 1920 | 504 | 527 | 870 | 84900 |
46 | 4.76 | 24600 |
1.9 |
In Tris buffer, the absorption spectrum contains two main bands: the major maxima lie in the region of 472–476 nm for all dimeric dyes. We assume that they correspond to the intramolecular aggregated forms of the dimeric dyes while the shoulder band at ∼495 nm could be related to non-aggregated forms. A similar trend was observed for AO earlier: the peak at 491 nm is a characteristic of the AO in the monomeric state but the π-conjugated system of AO favors the formation of dimers in aqueous media resulting in a shoulder band at about 470 nm.49 The ratio of the intensities of these two bands is slightly different for the dyes under study but in each case the intensity of the band at ∼475 nm is 1.4–1.7 times higher than that at ∼495 nm evidencing the predominance of the aggregated form of BAO dyes and EG in Tris buffer. Meanwhile, in the fluorescence excitation spectra of all these dyes measured in Tris buffer and detected at 530 nm, the excitation band at ∼498 nm becomes predominant (Fig. S1 (ESI†), Table 2). This indicates that the non-aggregated form fluoresces while the aggregated form does not contribute to the fluorescence signal in the green region (at about 530 nm). In Tris buffer, the fluorescence maxima for EG and BAO dyes reside in the narrow region of 524–525 nm (Fig. S1 (ESI†)). The fluorescence quantum yields are very low (1.8–2.5%) again indicating that the molecules exist preferably in the aggregated form. The fluorescence lifetimes (FLTs) in Tris buffer for all dimeric dyes differ insignificantly and are in the range of 1.26–1.91 ns (Table 2 and Table S1 (ESI†)).
Interestingly, the fluorescence spectra of dimeric dyes exhibit the red band with the maxima approximately at 630 nm. In the case of AO, the red emission appears only when interacting with RNA or single-stranded DNA due to aggregation of AO molecules.50 In our experiments, the most pronounced red fluorescence at ∼630 nm in Tris buffer is observed for BAO-2. The fluorescence excitation spectrum of BAO-2 registered at 630 nm (dashed blue line, Fig. S1 (ESI†)) shows the predominance of a band at 475 nm related to the aggregated form of BAO-2. Moreover, the shape of this excitation spectrum well coincides with the absorption spectrum of BAO-2 in Tris buffer. It is worth mentioning that the fluorescence excitation spectra of BAO-1 and BAO-3 detected at 630 nm do not show distinct peaks at 475 nm indicating that their stacked forms do not fluoresce.
When the aqueous medium is replaced by the less polar methanol, the intramolecular aggregates of BAO dyes and EG are destroyed. As a result, the absorption band at ∼475 nm drops down and the band at ∼495 nm increases. The fluorescence intensity as well as quantum yields also increase. The degree of dimer dissociation for dimeric dyes in methanol is different than in water and there is a correlation between quantum yields of the dyes in methanol and absorption bands half-widths (Table 2). A narrower absorption band and higher fluorescence quantum yield are evidence of weaker aggregation. The quantum yields increase in the order of EG < BAO-1 < BAO-3 < BAO-2. Meanwhile, the absorption bands half-widths decrease in the opposite order from 2000 cm−1 to 1810 cm−1. Thus, the BAO-2 dye is less prone to form intramolecular aggregates in methanol than the other dyes.
In dsDNA solution, a more pronounced increase in the quantum yield of EG and BAO dyes occurs as a result of the dye–dsDNA interaction while the overall shape of the absorption spectra remains the same as in methanol (Fig. S1 (ESI†), Table 2). The quantum yields increase in the order of BAO-2 < EG < BAO-3 < BAO-1, while the absorption bands half-widths change in the opposite order: BAO-2 > EG > BAO-3 > BAO-1. FLTs of the dye–dsDNA complexes also increase 3.49–3.86 times, while in methanol they increase only slightly (1.17–1.36 times). Thus, the effect of signal amplification upon binding to dsDNA is more pronounced for fluorescence lifetimes than for quantum yields. The enhanced quantum yields as well as the FLTs upon binding to dsDNA compared to those in methanol can be explained by more pronounced destruction of intramolecular dye aggregates due to the additional fixation of AO units within DNA strands. Thus, BAO-1 shows the weakest tendency to form aggregates among other dimeric dyes while BAO-2 turned out to be the most aggregated dye. This finding suggests that the nature of the linker connecting two AO moieties in BAO dyes affects their binding to dsDNA that, in turn, leads to different fluorescence enhancement.
Complexation of the dimeric dyes with dsDNA leads to long-wavelength shifts in absorption and fluorescence spectra. It is known that the binding of a positively charged dye to a negatively charged counterion can lead to ion pair-like species, which cause band splitting.51 Depending on the symmetry of the resulting ion pair complex, only the transition corresponding to the long-wavelength part of the split band leading to the bathochromic shift can be allowed.49 So, the red shift in the spectra of EG and BAO dyes can be attributed to the electrostatic interactions between negatively charged phosphate groups of dsDNA and amino groups of positively charged AO fragments.
The binding constants (Kb) between the studied dyes and dsDNA were determined from the plot oflog(Y/(1 − Y)) vs. log[DNA] (Fig. S2 (ESI†)) according to eqn (S5) (ESI†) by observing the changes in the fluorescence intensity with increasing concentration of dsDNA. For the EG and BAO dyes, the Hill coefficients are close to 1, suggesting that the binding of one molecule of a dye does not influence the binding of other molecules (absence of cooperativity). The binding constants were found to increase in the order of BAO-3 < BAO-2 < EG < BAO-1 (Table 2).
One of the key parameters that reflect the applicability and sensitivity of a dye for fluorescent applications is its brightness, which is expressed as quantum yield multiplied by molar absorptivity at a specific wavelength.52 All the studied dyes show almost the same brightness (24600 − 27900 M−1 cm−1) in the presence of dsDNA (Table 2). Only the dye BAO-1 which has the highest brightness in the dsDNA solution as compared to the others can be emphasized.
Another important parameter for a qPCR dye is its photo-, thermal and chemical stability. We evaluated these characteristics for the new BAO dyes and compared them with those of commercial PCR dyes SYBR Green I and EG. The results are shown in Fig. S3 and S4 (ESI†). As shown in the graphs, all dimeric dyes demonstrate comparable stability and significantly outperform SYBR Green I in all tests.
As a result of our spectral study, we conclude that EG and the new dyes BAO-1, BAO-2 and BAO-3 exhibit almost the same values of absorption and emission maxima, molar absorptivities, Stokes shifts, and stability. The quantum yields and brightness of all the dyes in dsDNA solution are essentially increased as an expected result of the dye–dsDNA interaction and this makes them suitable for imaging and sensing of nucleic acids. However, the values of quantum yields are not markedly different from one dimeric dye to another. Therefore, EG and all new BAO dyes were investigated further for nucleic acid sensing in fluorescence imaging and qPCR experiments.
3.2. Cytotoxicity and cell imaging studies
Before advancing to cell staining and qPCR experiments, all the dyes underwent safety evaluation through an MTT assay using cultured rat skin fibroblasts. The stock solutions of dyes in DMSO were prepared and added to cultured fibroblasts so that the final dye concentrations were 1.5 μM, 3.0 μM, or 6.0 μM and 0.6%, 1.2%, or 2.4% of DMSO respectively. Untreated fibroblasts were used as a negative control (NC). Also, as a control, cultured cells containing only the corresponding quantity of DMSO and no dye were used. We found that incubating fibroblasts with various aliquots of stock solutions of EG and BAO dyes in DMSO did not impact the cells' metabolic activity, indicating no cytotoxic effect on cell viability (Fig. S5 (ESI†)). This was confirmed by the absence of a statistically significant decrease in absorbance values between the control samples treated with the same concentration of DMSO and the fibroblasts treated with the dyes under study. The obtained results reaffirm the safety of the dyes and allow us to confidently proceed with cell staining and qPCR experiments.It is known that acridine orange is a cell-permeable dye which is used to differentiate DNA- and RNA-containing cellular compartments, as well as for distinguishing cell death by apoptosis and necrosis. However, acridine orange stains both dead and live cells thus complicating the interpretation of results.53
In our work, we employed BAO dyes to stain both live and dead fibroblasts. To simulate cell death accompanied by increased membrane permeability, we treated fibroblasts with ice cold ethyl alcohol for 1 min. The results of the staining were compared with AO and well-known EG dyes.
As expected, AO stained both live and dead cells (Fig. 2). In contrast, neither BAO dyes nor EG displayed detectable signals in live cells, indicating that these dyes cannot penetrate the cell membrane. Upon membrane disruption, the BAO and EG dyes entered the cells and stained the cytoplasm and nuclei. In line with AO imaging studies,53,54BAO dyes provided diffuse staining of the cytoplasm and nuclear bodies due to the abundance of RNA in both green and red channels (Fig. 2). The nucleus displayed a distinct green hue related to DNA presence, and red punctate structures associated with the nuclear bodies (Fig. 2). The merged images revealed the overlap of the signal from the red fluorescence channel with the green one (Fig. 2). This overlap appeared as greenish-orange or orange regions at lower dye concentrations and reddish-orange or red regions at higher ones. The increase in fluorescence intensity in the red channel was attributed to the formation of insoluble dye–RNA complexes upon exceeding a certain dye-to-RNA ratio, as was shown previously in the case of AO.53,54
 | ||
| Fig. 2 Laser scanning confocal microscopy images of live and dead fibroblast cells incubated for 30 minutes with different concentrations of AO, EG and BAO dyes without washing. The laser excitation wavelength was 488 nm, and the fluorescence was recorded in green (detection range was set between 508–550 nm) and red (583–700 nm) channels. Images represent an overlay of both emission channels. (Individual images collected in green and red channels are represented in Fig. S6 and S7 (ESI†), respectively.) | ||
In the merged images of cells stained with dimeric BAO-1, BAO-3, and EG dyes at a concentration of 3.0 μM, dye–RNA complexes are represented by orange spots in the cytoplasm and nuclear bodies while dye–DNA complexes are evident as green spots in the nuclei (Fig. 2). At a dye concentration of 6.0 μM, the emission of dye–RNA complexes turned to red.
Interestingly, BAO-2 preferentially stained RNA at all investigated concentrations. As shown in Fig. 2, the emission of its dye–RNA complexes appeared as orange spots in the cytoplasm and nuclear bodies at 1.5 μM and 3.0 μM dye concentrations, turning into bright red areas at higher concentrations.
We further investigated the ratio between the fluorescence intensities obtained in the green (Fig. S6 (ESI†)) and red (Fig. S7 (ESI†)) channels. As shown in Fig. 3, AO stained DNA (green channel) more markedly than RNA (red channel) in dead fibroblasts, and the RNA-to-DNA ratio, i.e. the ratio between the dye–RNA and dye–dsDNA fluorescence intensities, decreased from 0.42 to 0.28 with increased AO concentration from 1.5 μM to 6.0 μM, respectively, which is consistent with the literature.53 In contrast, the RNA-to-DNA ratio remained almost unchanged and independent on concentration for BAO-1, EG, and BAO-3. The staining of fibroblasts with BAO-2, however, differed. At a 1.5 μM dye concentration the fluorescence of the dye–RNA complexes was 6 times higher than that of the dye–dsDNA complexes, while at 6.0 μM dye concentration the dye–RNA complexes fluorescence increased to 10, thus indicating a higher specificity of the BAO-2 dye to RNA.
 | ||
| Fig. 3 Average fluorescence intensities of the dead fibroblast cells stained with AO, EG or BAO dyes in green and red channels. The final concentrations of the dyes in staining solution were 1.5 μM, 3.0 μM, or 6.0 μM for EG or BAO dyes and twice higher (3.0 μM, 6.0 μM, 12.0 μM) for AO to get the same quantity of chromophore units during cell staining. Data are shown as the mean ± SEM. | ||
The average fluorescence intensity (image brightness) was quantified as an average integrated density of the nuclei in the green channel and the cells in the red channel. The image brightness of AO in complexes with dsDNA and RNA was measured to be in the range of 2.4 × 105 – 3.1 × 105 a.u. and 0.7 × 105 – 1.3 × 105 a.u., respectively. Both dye–dsDNA and dye–RNA complexes in the presence of BAO-3 or EG demonstrated higher brightness (3.6 × 105 – 5.1 × 105 a.u.) while the brightness of nucleic acids complexes with BAO-1 was approximately 2.5 times lower. Thus, BAO-1, BAO-3, and EG can be utilized to equally stain DNA and RNA in dead cells. Surprisingly, the RNA complexes with BAO-2 at 6.0 μM dye concentration provided an extremely bright signal in the red channel (1.4 × 106 a.u.) compared to the signal of its dye–dsDNA complexes in the green channel (1.2 × 105 a.u.). Therefore, BAO-2 might be useful for specific RNA staining of dead cells.
To sum up, EG and BAO dyes can be used to differentiate live and dead cells and, therefore, to investigate cell injury and cell death. Notably, EG and BAO-3 allow obtaining brighter cell images both in red and green channels while BAO-2 is superior for visualization of RNA-rich compartments.
3.3. qPCR experiments
We compared the influence of the thiophene-containing linker in the dye structures on the qPCR efficiency of EG and BAO dyes. The PCR inhibition effect of the BAO dyes and commercial EG was investigated using real-time qPCR with constant concentration of DNA and varying dye concentrations. Specifically, BAO-1 was studied at 0.5, 1.0, 2.0, 3.0 and 4.0 μM, BAO-2 and BAO-3 were used at concentrations of 0.5, 1.0, 2.0, 3.0, 4.0, and 5.0 μM while EG was used at four concentrations (0.25, 0.5, 1.0, and 2.5 μM) varied in a total reaction volume of 20 μL.There are three key parameters describing the performance of the dyes in qPCR analysis. First, the cycle threshold value (Ct), which is the number of cycles required to reach the threshold for declaring a positive test result. The higher Ct the higher the degree of qPCR inhibition. Second, the slope of the trendline calculated from a linear fit of the plot of Ct value vs. dye concentration. A positive slope indicates increased qPCR inhibition, which should be avoided in qPCR assays. Third, the high fluorescence intensity of dyes at the end-point of qPCR ensures the robustness of the signal upon analysis of the desired amplicons.
Thus, upon amplification of the STAT4 gene (Fig. 4a), BAO-2 and BAO-3 demonstrated the least pronounced qPCR inhibition as evidenced by the trendline with a negative slope. When BAO-1 was used at concentrations of 0.5 and 1.0 μM, the Ct values were similar to those of BAO-3. But with the increase in the dye concentration to 4.0 μM, the linear growth of the Ct value to 34.9 ± 0.5 was observed for BAO-1. In contrast to BAO-2 and BAO-3, the slope of the trendline for EG and BAO-1 was positive indicating a higher degree of PCR inhibition in the presence of these dyes. When comparing EG with BAO-1, the use of EG at a concentration of 1.5 μM increased the Ct value to 32.1 ± 0.1, while BAO-1 reached approximately the same Ct value (31.7 ± 0.3) only at a twice higher concentration. Moreover, the slope of the trendline for EG was ∼2 times higher than for BAO-1. Based on these results, EG shows the most pronounced inhibiting effect on PCR among all studied dyes. A similar trend was observed during the amplification of the STAT6 and HLA-B genes (Fig. S8a and c (ESI†)).
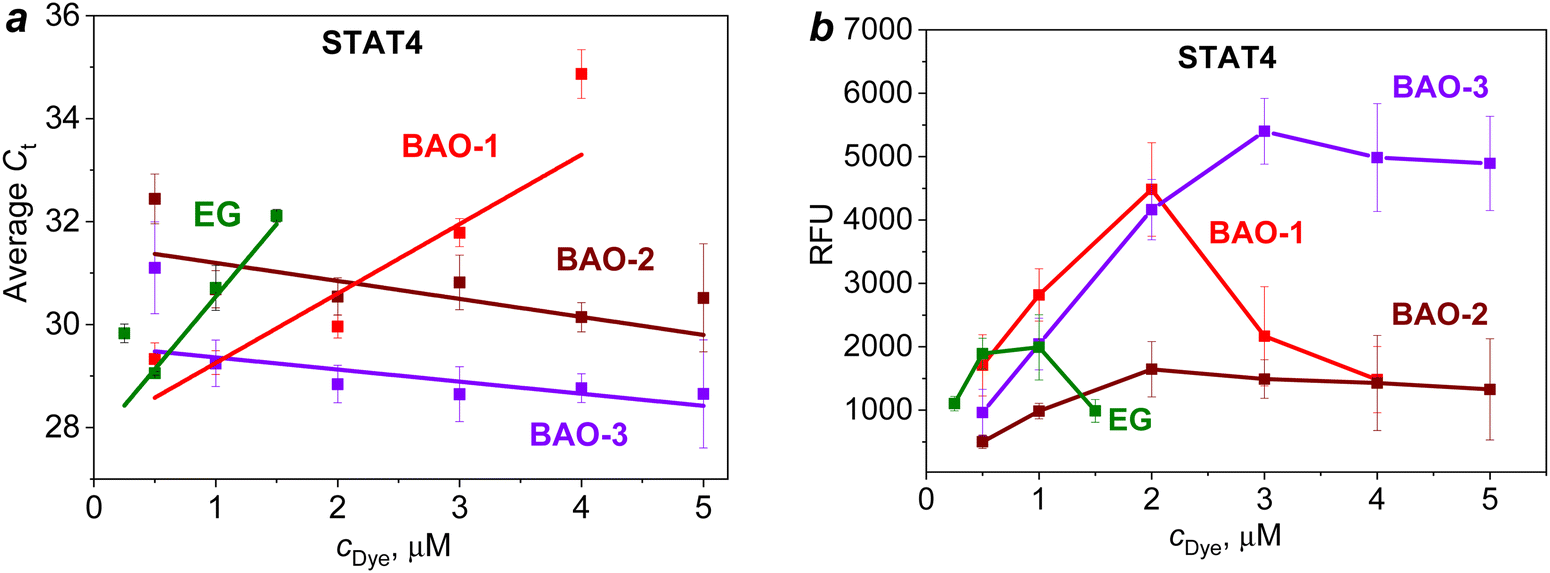 | ||
| Fig. 4 The dependence of the average cycle threshold (Ct) values (a) and end-point fluorescence intensity in relative fluorescence units (RFU) (b) on dye concentration upon the amplification of STAT4 gene on CFX96 Touch Real-Time PCR Detection System C1000 Touch (Bio-Rad). Only points without significant PCR inhibition were included in the linear fits. The slope of the trendline from the fit is as follows: EG: 2.82 ± 0.72, BAO-1: 1.35 ± 0.34, BAO-2: −0.35 ± 0.17, and BAO-3: −0.24 ± 0.15. | ||
Among the studied dyes, BAO-3 did not show concentration-dependent inhibition of amplification within the concentration range of 0.5–5.0 μM. When BAO-3 was used, the amplicon length did not affect the amplification. However, in the presence of BAO-2 the inhibitory degree on short amplicon HLA-B was higher when compared to longer amplicons. A comparison of the average Ct values for the same concentrations of BAO-2 and BAO-3 shows that BAO-3 gives a lower Ct level than BAO-2. Despite the low inhibitory effect, the use of the lowest and highest concentrations of BAO-2 and BAO-3, i.e. 0.5 and 5.0 μM, led to a large difference between the repeats (∼2 cycles). Comparing BAO-2 and BAO-3 with commercial EG, the former dyes can be used in a wide concentration range from 0.5 to 5.0 μM without significant Ct delays while the optimal concentration of EG under applied conditions was comparatively low (0.25–0.50 μM).
In the next step, the end-point fluorescence intensity of all the dyes was investigated (Fig. S8b and d (ESI†) and Fig. 4b). Upon amplification of the STAT4 gene, BAO-1 and BAO-3 demonstrated high end-point fluorescence intensity which at optimal concentrations (0.5–2.0 μM) exceeded that of BAO-2 and EG (Fig. 4b). However, the use of BAO-1 at concentrations of 3.0–5.0 μM resulted in a sharp decrease in the end-point fluorescence intensity. This is explained by the ability of BAO-1 to inhibit amplification at high dye concentrations. For BAO-3, a linear increase in the end-point fluorescence intensity was observed making this dye suitable for use at higher concentrations yet maintaining the same end-point fluorescence intensity without a PCR inhibition effect.
To discriminate specific products predetermined by primers from reaction artifacts, the post-PCR melt curve analysis was carried out. It was revealed that the concentration of the studied dyes can affect the melting temperature (Fig. S9–S11 (ESI†)). BAO-1 caused the most pronounced concentration-dependent change showing the average melting point shift of 2 °C between the lowest and highest concentrations. In addition, the use of BAO-1 at concentrations of 4.0–5.0 μM during the amplification of STAT6 gene promoted the formation of non-specific products with lower melting temperatures (Fig. S10d (ESI†)). Meanwhile, an increase in the concentrations of BAO-3 and EG caused the temperature shift of ∼1 °C while for BAO-2 it is only 0.5 °C (Fig. S9–S11 (ESI†)).
Thus, elongation of the linker connecting AO moieties in BAO-2 and BAO-3 dyes leads to a decrease in qPCR inhibition by the dye. At the same time, the introduction of an oxygen atom into the linker in the case of BAO-3 leads to an increase in end-point fluorescence intensity. The highest dye concentrations at which qPCR is not inhibited under applied conditions are up to 2.0 μM for BAO-1, 3.0 μM for BAO-2, 4.0 μM for BAO-3, and 0.5 μM for EG.
Analyzing the qPCR performance of the studied dyes, we found a correlation between the binding constants for dye–dsDNA complexes (Section 3.1) and qPCR inhibition. Specifically, BAO-3 with the lowest binding constant (1.9 × 104 M−1) exhibited the least level of qPCR inhibition at relatively high dye concentrations. In contrast, EG and other BAO dyes with higher binding constants (up to 5.9 × 104 M−1) inhibited qPCR thus leading to the formation of non-specific products and negatively impacting melt curve analysis at the same dye concentrations. Apparently, for optimal qPCR performance, an ideal PCR dye should exhibit sufficient affinity to form a dye–dsDNA complex but not so high as to cause qPCR inhibition and complicate post-PCR analysis. This observation aligns well with previously reported data.17
Among studied dyes, BAO-3 exhibits the lowest degree of qPCR inhibition and outperforms commercial EG in terms of the end-point fluorescence intensity providing a robust PCR signal and a strong sharp DNA melting peak during post-PCR analysis.
Despite minor differences in the spectral properties of the studied dimeric dyes (Section 3.1), they show different behavior in qPCR experiments. To further clarify the correlation between the structure of BAO dyes and their properties upon binding to dsDNA we carried out molecular dynamic simulations.
3.4. Molecular modeling of the interaction of BAO dyes with dsDNA and RNA
The interaction of bis-acridine orange dyes with the DNA double helix can proceed according to various possible scenarios: external binding to the phosphate groups and/or to the minor and major grooves, mono- and bis-intercalation (Fig. 5). Due to the electrostatic positive charge on acridine orange moieties of BAO dyes, their interaction with the phosphate groups of the double helix backbone from the outside of the dsDNA macromolecule is very probable (Fig. 5a). The negatively charged phosphate groups of dsDNA are exposed to the solution and the interaction of BAO dyes should start with the substitution of counterions from the ionic atmosphere around the double helix and binding to the phosphate groups. Then the dyes can penetrate in the internal regions of the dsDNA macromolecule, i.e. the minor and major grooves, and adopt compact or elongated conformations that are stabilized by the interaction with dsDNA atoms (Fig. 5b and c). In principle, when the dyes are localized in the grooves of dsDNA, the intercalation of the acridine orange moieties between nucleotide bases can occur (Fig. 5d). Previous studies indicated that the fluorescence enhancement of similar bis-acridine dyes bound to dsDNA was not due to intercalation.55,56 Thus, the intercalation of BAO dyes will not be considered in the present study. | ||
| Fig. 5 Possible scenarios of the interaction of BAO dyes with the DNA double helix: electrostatic interaction of BAO dyes with the phosphate groups from outside the double helix (a); localization of BAO dyes in the minor groove (b) and in the major groove (c) of the double helix; intercalation of acridine orange moieties between the nucleotide bases (d) and (e). | ||
MD simulations were carried out to explain the better performance of BAO-1, BAO-2, and BAO-3 dyes under applied qPCR conditions compared to the commercial EG to which they differ only in the structure of the linker.
As detailed in the Materials and methods section, the simulation box comprised a 22 base-pair (bp) dsDNA fragment, 23000 water molecules, and one dye molecule.
The formation process of the EG–dsDNA complex and the variety of conformations that EG dye can adopt in the complex are depicted in Fig. 6.
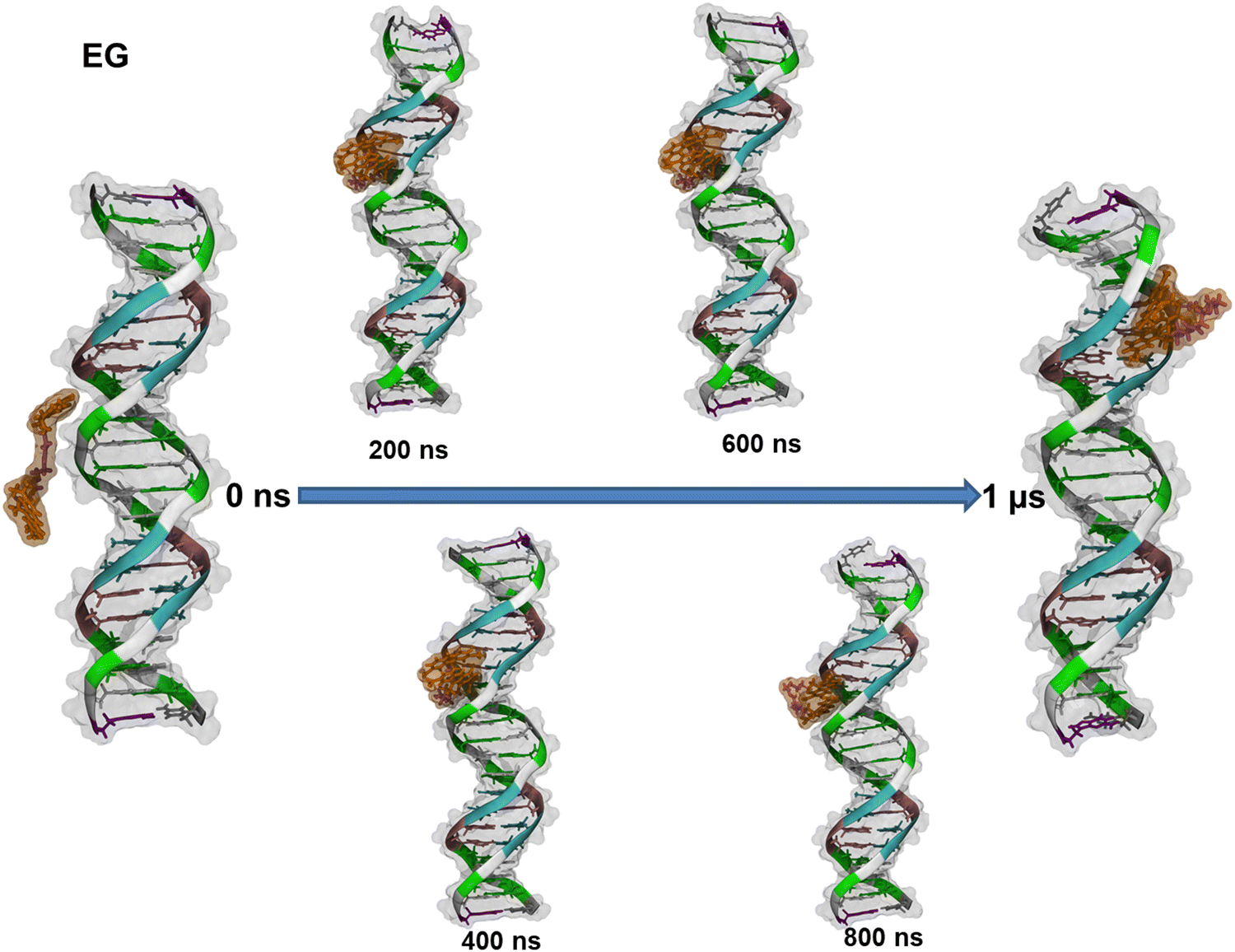 | ||
| Fig. 6 Evolution of the complex between EG and the dsDNA over 1 μs. Color code: guanine in green, cytosine in white, thymine in cyan and adenine in pink. The AO moieties of the dye are orange and the linker between them is purple. In addition to the cartoon and tube representations, the van der Waals radii of all atoms are depicted as a transparent surface (in grey for the dsDNA). | ||
We found that EG enters the minor groove of dsDNA with one AO fragment at the beginning and remains there for the entire simulation. Furthermore, visual inspection reveals that after 250 ns the EG dye adopts a folded conformation. Similar behavior was observed during the formation of the complex of BAO-1 with dsDNA (Fig. S12 (ESI†)). Similarly to EG, BAO-1 enters the minor groove of dsDNA and adopts a folded conformation. However, this conformation differs from the conformation of EG. Unlike EG, that has two AO moieties stacked and the linker is not involved in the stacking, the BAO dyes form a 3-components stack with the two AO components on each side of the central thiophene ring (Fig. 7).
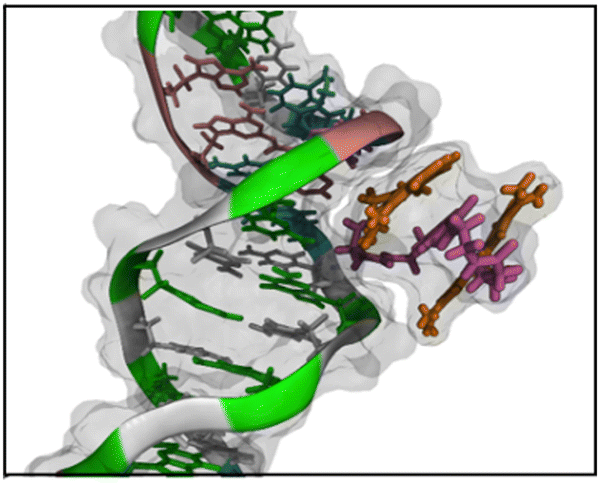 | ||
| Fig. 7 Highlight of the 3-components stacked conformation of BAO-3 dyes; a similar conformation is observed for BAO-1 and BAO-2. Color code: guanine in green, cytosine in white, thymine in cyan and adenine in pink. The AO moieties of the dye are orange and the linker between them is purple. In addition to the cartoon and tube representations of the molecules, the van der Waals radii of all atoms are depicted as a transparent grey surface. | ||
While the BAO-1 dye interacts with dsDNA similarly to EG entering the minor groove of dsDNA and forming a stable complex with dsDNA, the BAO-2 and BAO-3 dyes behave differently. As shown in Fig. 8, initially both BAO-2 and BAO-3 behave similarly to BAO-1 with the dye adopting the 3-components stacked conformation and entering the minor groove of dsDNA. However, by the end of the simulation, the dyes are partially stretched inside the minor groove, suggesting that the 3-components stacked conformation of BAO-2 and BAO-3 is less stable than the same conformation of BAO-1.
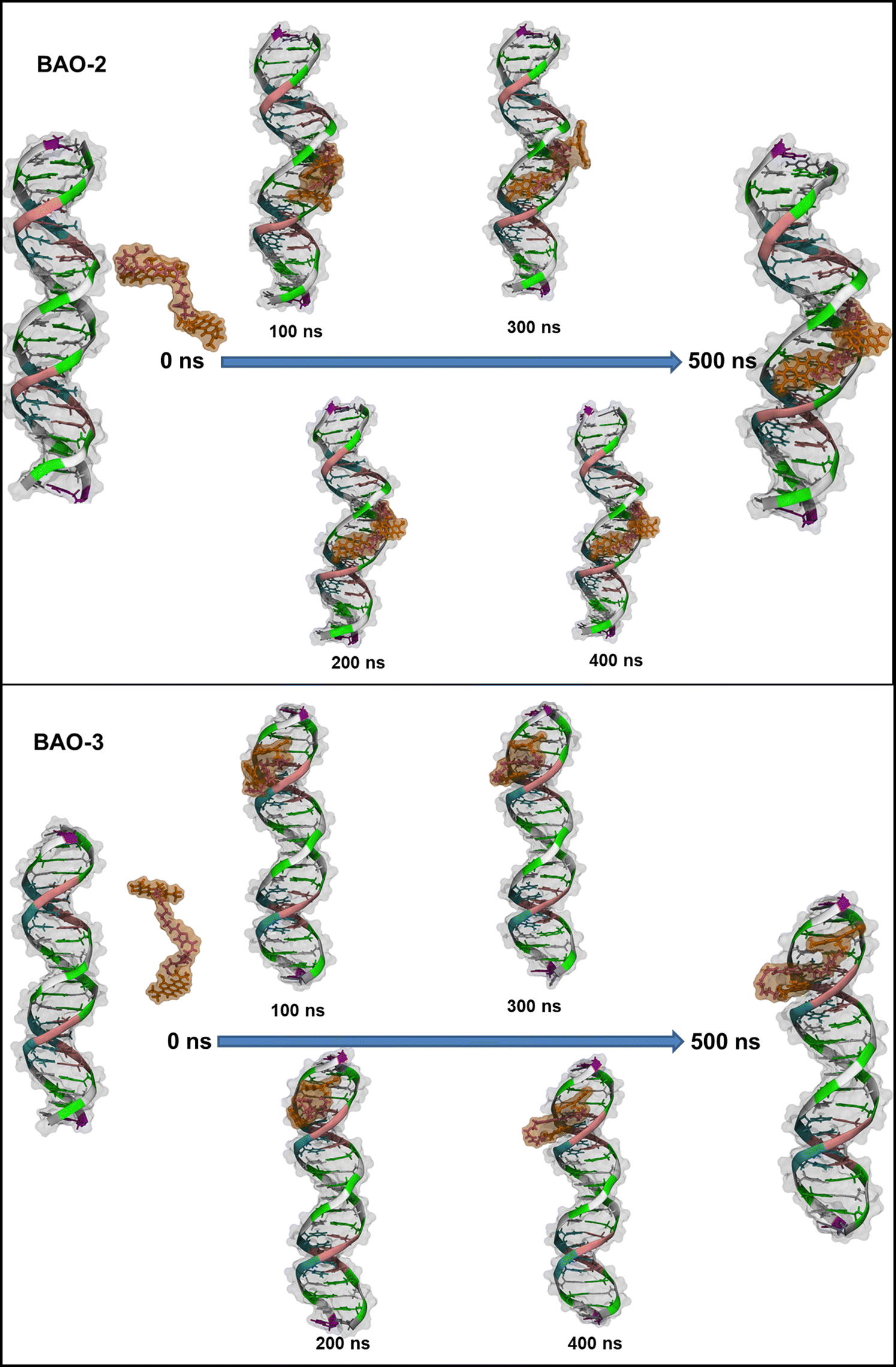 | ||
| Fig. 8 Formation of the complex between BAO-2 and BAO-3 with dsDNA over 500 ns. Color code: guanine in green, cytosine in white, thymine in cyan and adenine in pink. The AO moieties of the dye are orange and the linker between them is purple. In addition to the cartoon and tube representations of the molecules, the van der Waals radii of all atoms are depicted as a transparent surface (in grey for the dsDNA, orange for the AO and purple for the linker). | ||
Considering that the only noticeable difference between EG, BAO-1 and BAO-2, BAO-3 is the stability of the stacked conformation when BAO-2 and BAO-3 interact with dsDNA, we hypothesized that the improved efficiency in the qPCR test is due to their ability to change from the stacked conformation to a stretched one. To test this, we performed four umbrella sampling (US) simulations. In each US we used each dye in a stacked conformation as an initial point and then gradually increased the distance between the AO parts, while monitoring the changes in energy of the simulated system. Fig. 9 depicts the graphs obtained from these simulations with inlets depicting the most representative structures of the dyes taken at certain distances between the AO moieties. In addition, for better viewing, all inlets are presented in full size in Fig. S13–S18 (ESI†).
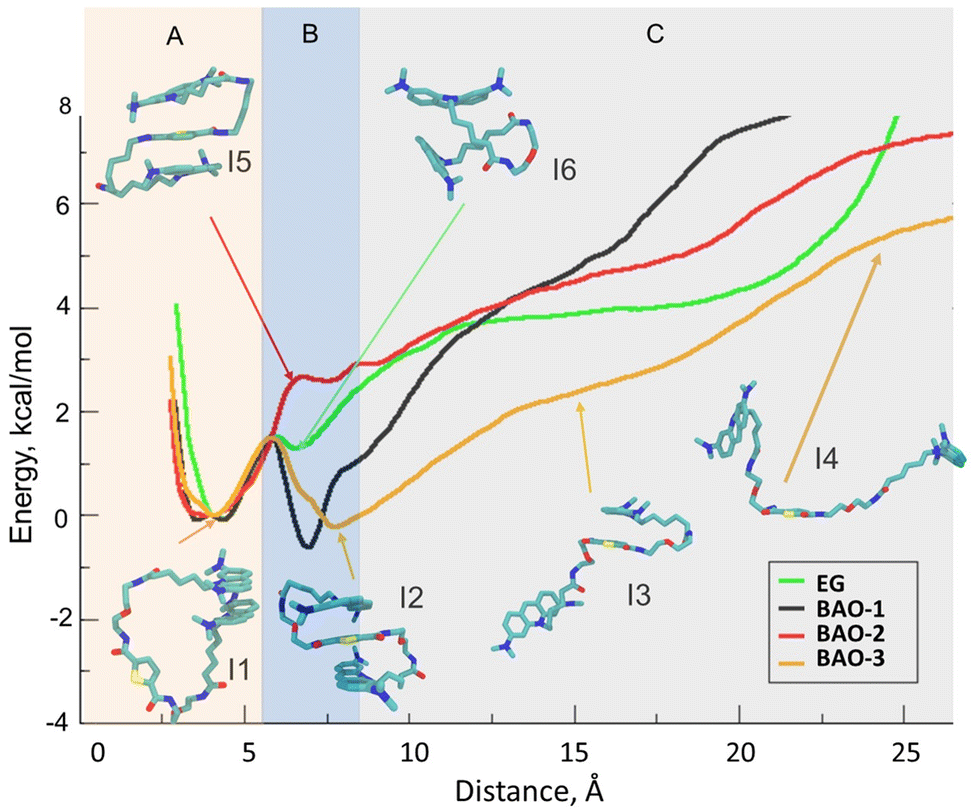 | ||
| Fig. 9 Potential of mean force (PMF) at varying distance between the two AO moieties. Color code: BAO-1, black; BAO-2, red; BAO-3, orange; EG, green. Inset code: I1, an example of AO stacking in BAO-3; I2, a 3-components stacked conformation of BAO-3; I3, stacking of one AO moiety with thiophene in BAO-3; I4, stretched conformation of BAO-3; I5, a 3-components stacked conformation of BAO-2; I6, perpendicular conformation of EG. Region A corresponds to the I1-type conformation, where the AO moieties are stacked. Region B roughly corresponds to the 3-components stacked conformation for the BAO dyes and to the I6 conformation of the EG dye. Region C corresponds to the conformations, where the AO moieties are not interacting with each other. | ||
As shown in Fig. 9, EG has an energy minimum at the distance of 4 Å between the AO parts, which corresponds to the closed conformation with the two AO moieties stacked to each other. This minimum at the distance between the AO moieties of 4 Å is present in all dyes, and the stacked conformation is depicted in the inset I1 in Fig. 9 for dye BAO-3. At larger distances, in the case of EG, the energy increases with the distance between the two AO moieties, except for a small local minimum at 6–7 Å which corresponds to the conformation depicted in inset I6 in Fig. 9 that shows the two AO moieties stacked perpendicularly to each other. While all studied dyes have an energy minimum at the distance of 4 Å (Region A, Fig. 9), there are important differences in subsequent sections of the curves (Regions B and C) for each dye. For BAO-1 and BAO-3, there is a second energy minimum at the distance of ca. 6 and 8 Å between the AO parts which corresponds to the 3-components stacked conformation. Moreover, this second energy minimum has a lower energy value than at 4 Å which indicates that these dyes prefer the 3-components stacked conformation over the closed one. Although BAO-2 can adopt the 3-components stacked conformation like the BAO-1 and BAO-3 dyes, this conformation does not correspond to the energy minimum (as for BAO-1 and BAO-3), but to the maximum. In the region corresponding to the 3-components stacked conformation, i.e. 6–7 Å (Region B, Fig. 9), the energy for EG and BAO-2 are similar; however, as mentioned above, this value corresponds to a local maximum for BAO-2 while EG has a local minimum. As can be seen in insets I5 and I6 in Fig. 9, although the energies for EG and BAO-2 are similar, the corresponding conformations are essentially different. The lack of a local minimum at the distance of 6–8 Å is attributed to the fact that dye BAO-2 has two alkyl moieties in the linker that are pushed to the side, into water, as depicted in inset I5 of Fig. 9, when the molecule adopts the 3-components stacked conformation. So, any decrease in energy gained from the 3-components stacked conformation is compensated by the displacement of the hydrophobic hydrocarbon chains of the linker toward water. In the case of BAO-3, it can be clearly seen that the difference in energy between the stacked and unstacked conformation is smaller when compared with the other dyes, which means that the BAO-3 dye can unstack more easily than the other dyes. This difference is attributed to the presence in the linker of an oxygen atom that increases the hydrophilicity of the dye. Also, as depicted in inset I3 of Fig. 9 the dye can take a semi-stacked conformation, where one AO moiety is stacked with the thiophene and the other one resides in water. Visual inspection of the umbrella sampling trajectories clearly revealed how already at 8 Å one of the AO moieties is not stacked anymore to the other aromatic rings. Considering that the stacking quenches the fluorescence, the observed differences in energies could explain why BAO-3 provides a higher rate of end-point fluorescence intensity than the others in qPCR experiments since it has a higher probability of being unstacked upon binding with dsDNA.
When examining the simulations of the dyes with RNA (Fig. S19–S22, ESI†), a different behavior of the EG and BAO dyes is observed. Unlike simulations with the dsDNA where the BAO dyes remain in the 3-components stacked conformation, in the simulations with the RNA, the BAO dyes adopt the 2-components stacked conformation similar to EG. Visual inspection of the simulation trajectories revealed that this difference arises due to an interaction of the thiophene moiety with the nucleic bases of the RNA, as seen in Fig. S23 (ESI†). This effect does not occur in the case of the dsDNA because, when dsDNA is in the B-DNA canonical structure, the nucleic bases are tightly stacked with each other thus preventing the thiophene from finding any free base to interact with. These findings are consistent with the literature data on AO,50 verifying that the red emission observed upon interaction with RNA is due to the aggregation of AO molecules.
4. Conclusions
To conclude, we present here a series of novel dimeric BAO dyes containing two acridine orange moieties connected by various linkers, each featuring a 2,5-disubstituted thiophene cycle as the central core. The nucleic acid sensing properties of BAO dyes were compared to those of a widely used commercial DNA-binding dye, EG. All the dyes demonstrated almost the same spectral and photophysical properties in the aqueous and methanol media, as well as upon binding to dsDNA, while BAO dyes exhibited markedly improved characteristics in qPCR and cell imaging experiments as compared to EG. Additionally, EG and BAO dyes exhibited comparable photostability, and thermal and chemical stability, significantly outperforming SYBR Green I.All the investigated dyes showed a significant increase in fluorescence quantum yield, lifetime and brightness upon binding to dsDNA, and can therefore be used for imaging and sensing of nucleic acids. BAO-3 exhibited selectivity for both dsDNA and RNA when applied to fibroblast staining, providing good quality fluorescence microscopy images with high brightness and contrast. A similar trend was observed for EG. Surprisingly, BAO-2 at high concentration facilitated highly specific and exceptionally bright RNA visualization in dead fibroblast cells. Hence, BAO-2 and BAO-3 are the most promising candidates for imaging applications including the investigation of cell damage, cell death, and a wide array of other biomedical uses.
The qPCR efficiency of BAO-2 and BAO-3 dyes was compared to that of BAO-1 and EG. The elongation of the linker joining AO moieties in BAO-2 and BAO-3 dyes enhanced the qPCR performance. Furthermore, the introduction of an oxygen atom into the linker in the case of BAO-3 resulted in an increased end-point fluorescence intensity. Thus, BAO-3 outperformed the other studied dyes and demonstrated the following advantages over commercial EG: it can be used in qPCR over a broader range of dye concentrations (0.5–5 μM) without causing qPCR inhibition and provides more reliable and consistently shaped DNA melting curves that are less affected by dye concentration.
To explain the improved qPCR performance of BAO-3 and to gain deeper insight into the binding mechanism of BAO dyes to dsDNA, we used molecular dynamics simulations. It is known that the fluorescence of EG in the presence of dsDNA is related to the existence of an equilibrium between stacked and unstacked conformations of the EG molecule. Our umbrella sampling simulations demonstrated that the transition from the stacked to the unstacked conformation requires less energy for the BAO-1 and BAO-3 dyes than for EG. This means that, compared to EG, the equilibrium of BAO-3 is shifted towards the unstacked conformation, thus explaining stronger binding to dsDNA and better performance of BAO-3 in qPCR. In addition, the simulations indicate that the dyes preferentially bind to the minor groove and reveal a major difference between the EG and BAO dyes. In the stacked conformation, the EG has two AO moieties stacked, while in the case of the BAO dyes, the stacked structure is represented by the 3-component stacked conformation, with the thiophene ring inserted between two stacked AO moieties. The PMF analysis showed that the 3-component stacked conformation is more favorable from an energetic point for BAO-1 and BAO-3.
We believe that the studied BAO dyes can further extend the scope of using dsDNA-specific dyes in biomedical and diagnostic applications.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
A. T. and A. K. thank National Research Foundation of Ukraine [project No. 2021.01/0414] for the financial support of the experiments on dye synthesis, spectral investigation and fluorescence microscopy. A. L. acknowledges the financial support from the Swedish Research Council and from Kempe Foundation. A. L., T. V. and R. P. acknowledge financial support from European Union's Horizon Europe Research and Innovation Programme [No. 101086667], project BioMat4CAST (BioMat4CAST - “Petru Poni” Institute of Macromolecular Chemistry Multi-Scale in Silico Laboratory for Complex and Smart Biomaterials). The computations were enabled by resources provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) and the Swedish National Infrastructure for Computing (SNIC) at PDC partially funded by the Swedish Research Council [No. 2019-03865]. F. M., A. L. and S. P. acknowledge the role of COST Action CA21101 – Confined Molecular Systems: From a New Generation of Materials to the Stars (COSY), supported by COST (European Cooperation in Science and Technology). S. P. thanks the NAS of Ukraine for the partial support of the research [project No. 0120U100855]. The authors thank Dr I. F. Kovalenko and Dr V. Yu. Prokopyuk (Research Institute of Experimental and Clinical Medicine of Kharkiv National Medical University, Ukraine) for conducting the fluorescence imaging and cytotoxicity assessment. The authors are grateful to Dr V. I. Musatov, Dr M. B. Kaidash and Mrs O. Yu. Stepanenko (Institute of Functional Materials Chemistry of SSI “Institute for Single Crystals” of NAS of Ukraine, Kharkiv, Ukraine) for performing analyses by NMR spectroscopy, mass-spectrometry and HPLC, respectively.References
- K. Watanabe and N. Seki, Int. J. Mol. Sci., 2024, 25, 752 CrossRef CAS PubMed.
- S. A. Yoon, S. Y. Park, Y. Cha, L. Gopala and M. H. Lee, Strategies of Detecting Bacteria Using Fluorescence-Based Dyes, Front. Chem., 2021, 9, 743923 CrossRef CAS PubMed.
- A. Ligasová and K. Koberna, Molecules, 2021, 26, 5515 CrossRef PubMed.
- Y. Wang, R. Zhou, W. Liu, C. Liu and P. Wu, Chin. Chem. Lett., 2020, 31, 2950–2954 CrossRef CAS.
- H.-A. Bennett, A. McAdorey and H. Yan, J. Fluoresc., 2024, 34, 1193–1205 CrossRef CAS PubMed.
- S. Nafisi, A. A. Saboury, N. Keramat, J.-F. Neault and H.-A. Tajmir-Riahi, J. Mol. Struct., 2007, 827, 35–43 CrossRef CAS.
- A. I. Dragan, R. Pavlovic, J. B. McGivney, J. R. Casas-Finet, E. S. Bishop, R. J. Strouse, M. A. Schenerman and C. D. Geddes, J. Fluoresc., 2012, 22, 1189–1199 CrossRef CAS PubMed.
- K. Munyenyembe, C. Timmons, A. K. M. Weiner, L. A. Katz and Y. Yan, Eur. J. Protistol., 2021, 81, 125840 CrossRef PubMed.
- J. Bucevičius, G. Lukinavičius and R. Gerasimaitė, Chemosensors, 2018, 6, 18 CrossRef.
- H. Zhu, H. Zhang, Y. Xu, S. Laššáková, M. Korabečná and P. Neužil, Biotechniques, 2020, 69, 317–325 CrossRef CAS PubMed.
- T. Kadja, C. Liu, Y. Sun and V. P. Chodavarapu, Sensors, 2022, 22, 2320 CrossRef CAS PubMed.
- P. W. Chomczynski, K. M. Vires, M. Rymaszewski and J. A. Heiny, PLoS One, 2022, 17, e0267348 CrossRef CAS PubMed.
- K. Wang, Y. Niu, Q. Wang, H. Liu, Y. Jin and S. Zhang, PeerJ, 2017, 5, e3260 CrossRef PubMed.
- D. Jordan and D. Mills, Front. Ecol. Evol., 2021, 9, 1–7 CrossRef.
- M. Jalali, J. Zaborowska and M. Jalali, in Basic Science Methods for Clinical Researchers, Elsevier, 2017, pp. 1–18 Search PubMed.
- Quantitative Real-Time PCR, ed., R. Biassoni and A. Raso, Springer New York, New York, 2020, vol. 2065 Search PubMed.
- H. Gudnason, M. Dufva, D. D. Bang and A. Wolff, Nucleic Acids Res., 2007, 35, e127 CrossRef PubMed.
- F. Domahidy, B. Kovács, L. Cseri, G. Katona, B. Rózsa, Z. Mucsi and E. Kovács, ChemPhotoChem, 2024, e202400080 CrossRef CAS.
- B. Juskowiak, Anal. Bioanal. Chem., 2011, 399, 3157–3176 CrossRef CAS PubMed.
- L. C. T. Shoute and G. R. Loppnow, Phys. Chem. Chem. Phys., 2018, 20, 4772–4780 RSC.
- A. Karsai, S. Müller, S. Platz and M.-T. Hauser, Biotechniques, 2002, 32, 790–796 CrossRef CAS PubMed.
- F. Mao, W.-Y. Leung and X. Xin, BMC Biotechnol., 2007, 7, 76 CrossRef PubMed.
- M. Sidstedt, P. Rådström and J. Hedman, Anal. Bioanal. Chem., 2020, 412, 2009–2023 CrossRef CAS PubMed.
- P. T. Monis, S. Giglio and C. P. Saint, Anal. Biochem., 2005, 340, 24–34 CrossRef CAS PubMed.
- G. H. Reed, J. O. Kent and C. T. Wittwer, Pharmacogenomics, 2007, 8, 597–608 CrossRef CAS PubMed.
- L. Jansson, M. Koliana, M. Sidstedt and J. Hedman, Biotechnol. Rep., 2017, 14, 34–37 CrossRef PubMed.
- O. G. Kulyk, O. S. Kolosova, R. P. Svoiakov, D. V. Kobzev, I. V. Hovor, I. M. Kraievska, E. V. Sanin, A. I. Krivoshey, Z. Y. Tkachuk and A. L. Tatarets, Dyes Pigm., 2022, 200, 110148 CrossRef CAS.
- Y. Chang, K. Guo, Q. Li, C. Li, Z. Guo and H. Li, Cell. Physiol. Biochem., 2016, 39, 157–171 CrossRef CAS PubMed.
- T. Vasiliu, F. Mocci, A. Laaksonen, L. D. V. Engelbrecht and S. Perepelytsya, Caging polycations: Effect of increasing confinement on the modes of interaction of spermidine3+ with DNA double helices, Front. Chem., 2022, 10, 836994 CrossRef PubMed.
- S. Perepelytsya, J. Uličný, A. Laaksonen and F. Mocci, Nucleic Acids Res., 2019, 47, 6084–6097 CrossRef CAS PubMed.
- D. A. Case, T. E. Cheatham, T. Darden, H. Gohlke, R. Luo, K. M. Merz, A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS PubMed.
- I. Ivani, P. D. Dans, A. Noy, A. Pérez, I. Faustino, A. Hospital, J. Walther, P. Andrio, R. Goñi, A. Balaceanu, G. Portella, F. Battistini, J. L. Gelpí, C. González, M. Vendruscolo, C. A. Laughton, S. A. Harris, D. A. Case and M. Orozco, Nat. Methods, 2016, 13, 55–58 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- P. Mark and L. Nilsson, J. Phys. Chem. A, 2001, 105, 9954–9960 CrossRef CAS.
- I. S. Joung and T. E. Cheatham, J. Phys. Chem. B, 2009, 113, 13279–13290 CrossRef CAS PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- L. Huang and B. Roux, J. Chem. Theory Comput., 2013, 9, 3543–3556 CrossRef CAS PubMed.
- F.-Y. Dupradeau, A. Pigache, T. Zaffran, C. Savineau, R. Lelong, N. Grivel, D. Lelong, W. Rosanski and P. Cieplak, Phys. Chem. Chem. Phys., 2010, 12, 7821 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, et al.Gaussian 09, Revision A.02, Gaussian Inc. Wallingford CT, Wallingford CT, 2009, 34 Search PubMed.
- T. Vasiliu, B. F. Craciun, A. Neamtu, L. Clima, D. L. Isac, S. S. Maier, M. Pinteala, F. Mocci and A. Laaksonen, Biomater. Sci., 2021, 9, 6623–6640 RSC.
- Z. Zhang, T. Vasiliu, F. Li, A. Laaksonen, X. Zhang, F. Mocci and X. Ji, J. CO2 Util., 2022, 60, 101978 CrossRef CAS.
- GROMACS 2020 DOI:10.5281/zenodo.3562495.
- D. J. Evans and B. L. Holian, J. Chem. Phys., 1985, 83, 4069–4074 CrossRef CAS.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- M. Zgarbová, M. Otyepka, J. Šponer, A. Mládek, P. Banáš, T. E. Cheatham and P. Jurečka, J. Chem. Theory Comput., 2011, 7, 2886–2902 CrossRef PubMed.
- G. M. Torrie and J. P. Valleau, J. Comput. Phys., 1977, 23, 187–199 CrossRef.
- M. I. Garcia Fernandez, D. Ceccarelli and U. Muscatello, Anal. Biochem., 2004, 328, 174–180 CrossRef CAS PubMed.
- S. Ichimura, Biopolymers, 1975, 14, 1033–1047 CrossRef CAS PubMed.
- F. Feichtmayr and J. Schlag, Ber. Bunsenges. Phys. Chem., 1964, 68, 95–102 CrossRef CAS.
- A. H. Ashoka, I. O. Aparin, A. Reisch and A. S. Klymchenko, Chem. Soc. Rev., 2023, 52, 4525–4548 RSC.
- J. R. Plemel, A. V. Caprariello, M. B. Keough, T. J. Henry, S. Tsutsui, T. H. Chu, G. J. Schenk, R. Klaver, V. W. Yong and P. K. Stys, J. Cell Biol., 2017, 216, 1163–1181 CrossRef CAS PubMed.
- G. Lober, J. Lumin., 1981, 22, 221–265 CrossRef.
- S. Takenaka, Y. Sakakibara, H. Ueyama and T. Nojima, Nucleic Acids Res. Suppl., 2001, 2003(3), 151–152 Search PubMed.
- K. Mizuki, Y. Sakakibara, H. Ueyama, T. Nojima, M. Waki and S. Takenaka, Org. Biomol. Chem., 2005, 3, 578–580 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01775g |
| ‡ Present Address: Dr Anatoliy Tatarets, Institute of Functional Materials Chemistry of State Scientific Institution “Institute for Single Crystals” of National Academy of Sciences of Ukraine, 60 Nauky Ave., 61072 Kharkiv, Ukraine. |
| This journal is © The Royal Society of Chemistry 2024 |