DOI:
10.1039/D4BM01101E
(Paper)
Biomater. Sci., 2025, Advance Article
Rapid in situ forming PEG hydrogels for mucosal drug delivery†
Received
20th August 2024
, Accepted 27th March 2025
First published on 3rd April 2025
Abstract
In situ gelling polymeric biomaterials have proven useful as drug delivery vehicles to enable sustained release at the sites of disease or injury. However, if delivered to mucosal tissues, such as the eyes, nose, and gastrointestinal and cervicovaginal tracts, these gels must also possess the ability to adhere to an epithelium coated in mucus. Towards this end, we report a new rapid in situ gelling polyethylene glycol-based hydrogel. Unlike other chemistries that enable rapid gel formation via irreversible covalent bonds, we use a bio-reducible linker allowing the gels to be naturally degraded over several days once administered. We identified a set of 6 lead formulations, which rapidly transform into disulfide-linked PEG hydrogels in 30 seconds or less. These rapidly forming PEG hydrogels were also able to conform and adhere to mucosal tissues via PEG-mucin entanglements and hydrogen bonding. Controlled release of protein-based cargoes from the PEG gels was achieved over several hours, whereas 40 nm nanoparticle-based cargos were retained over 24 hours. We also found that these rapid in situ forming PEG gels were well tolerated by mammalian cells and were retained in the nasal cavity of mice for up to 1 week. These studies support further testing and development of rapid in situ forming PEG gels for drug delivery to improve therapeutic retention and efficacy at mucosal sites.
Introduction
Mucosal tissues such as the eyes, stomach, nose, lungs, and vagina are often sites of inflammatory and infectious diseases, making them a desirable site for therapeutic delivery. Moreover, the mucosa may also be useful as a non-invasive route for drug delivery to local and distal sites as these tissues possess a large surface area that is highly vascularized and immunologically active.1–3 However, mucosal tissues possess natural defense mechanisms to facilitate the rapid clearance of potentially infectious or irritating materials from these sites, which can lead to reduced drug bioavailability when locally administered.4–7 This has motivated the use of formulations with enhanced viscosity (e.g., ointments, creams and gels) to act as a local drug depot for sustained delivery.8–10 Thus, biomaterial systems have been sought to provide these benefits for delivery of existing drugs and others under clinical development.11
Delivery of therapeutic-loaded gels to the cervicovaginal, ocular, nasal, and rectal routes has often necessitated the design of in situ forming hydrogels that can be administered in a liquid form and rapidly transform upon administration to act as a local drug depot.7,12–14 This can be achieved with thermosensitive polymers such as Pluronic and/or the use of click chemistry including dibenzocyclooctyne (DBCO)-azide and norbornene-tetrazine for rapid hydrogel assembly.15–19 A drawback of the latter approach using click chemistry is these gels are not reversible and contain crosslinks which will remain intact such that these gels will not be naturally degraded once they have delivered a therapeutic payload. Sulfhydryl crosslinked hydrogel biomaterials can also be constructed using maleimide or vinyl sulfone as reactive groups.20–22 While highly stable, sulfhydryl crosslinked gels typically form on the order of minutes which is less than ideal for in situ gelation. As an alternative, sulfide-dipyridyl disulfide reactions can also occur rapidly but, in general, they are underexplored for hydrogel formulations used in drug delivery. These crosslinks can also be reversed under reducing conditions and are likely to degrade over time once administered to biological tissues. Moreover, this cross-linking mirrors that found with mucus gels and prior work has shown that hydrogel attachment to mucosal tissues may be facilitated via reactions with available cysteines.23–25
In this work, we designed hydrogel biomaterials that form rapidly through thiol-pyridyl disulfide exchange. This material is composed of polyethylene glycol (PEG), a widely used polymer, and forms nearly instantaneously upon mixing under physiological conditions. Since the prepared hydrogels have thiol and pyridyl sulfides, we hypothesized that adhesion of the PEG hydrogel to mucosal tissues may be favored through direct reactions with thiol groups on mucin biopolymers. To determine if the PEG hydrogels are potentially useful for drug delivery applications, we assessed the release rates of different types of cargoes encapsulated within the PEG gels and evaluated their biocompatibility. This study establishes a new simple approach for formulating rapid in situ forming PEG gels, which could be used in mucosal drug delivery applications.
Materials and methods
Materials
4-Arm PEG-SH (PEG-4SH) 10 kDa and 20 kDa were purchased from Laysan Bio. 4-Arm PEG-OPSS (PEG-4OPSS) 10 kDa and 20 kDa were purchased from Creative PEGWorks. Dithiothreitol (DTT) and L-glutathione reduced were obtained from Sigma-Aldrich. Tetramethylrhodamine labeled BSA (TRITC-BSA) and cyanine 5 conjugated IgG (Cy5-IgG) were purchased from Protein Mods. Resazurin reagent and calcein AM were purchased from Biotium. Propidium iodide (Invitrogen), FluoSpheres and cell culture reagents such as DMEM and fetal bovine serum were obtained from Thermo Fisher Scientific.
Hydrogel synthesis
4-Arm thiol terminated PEG (PEG-4SH) dissolved in phosphate buffered saline (PBS) (>0.5% w/v) is mixed with equal volumes of 4-arm orthopyridyl disulfide terminated polyethylene glycol (PEG-4OPSS) dissolved in PBS (>0.5% w/v) for crosslinking and rapid gelation. The tube inversion method was used to measure gelation time at a body temperature of 37 °C. Briefly, polymer solutions of 100 μL each were mixed in Eppendorf tubes placed on a heat block at 37 °C. The tubes were taken out and inverted to see gelation of the polymers every 5 s. Time was noted when the solution stopped running on the walls of the tubes. It was repeated for all formulations of different polymer weight percentages and performed in triplicate.
Equilibrium swelling
100 μL of cylindrical gels were made by mixing the polymer solutions in equal volumes in tip cutoff 1 mL syringes. Gels were then pushed out of the syringes into 5 mL of PBS and incubated at 37 °C for 24 hours to reach equilibrium swelling. After 24 hours, gels were taken out of PBS and weighed to measure the mass of the swollen network. Gels were then freeze-dried to measure the mass of the dried network. The equilibrium swelling ratio was calculated using the below equation.
Degradation
Degradation of PEG hydrogels was measured in PBS, PBS + dithiothreitol (DTT) and PBS + glutathione. To measure degradation in PBS, 100 μL cylindrical gels were prepared and placed in 5 mL of PBS at 37 °C as described previously. Gels were weighed before immersion in PBS and at different time points after immersion in PBS. Percentage mass remaining was calculated using the below equation. To measure degradation in reducing agents, 200 μL of 100 mM DTT or 100 mM glutathione in PBS was added to 100 μL gels prepared in Eppendorf tubes and incubated at 37 °C. The tubes were inverted every minute for DTT and every ∼10 hours for glutathione to check gel degradation. Time was noted when the entire gel transitioned to the liquid state.
Scanning electron microscopy
Gels were prepared as described in the equilibrium swelling section. Briefly, gels composed of 2% w/v PEG-4SH and 2% w/v PEG-4OPSS using 20 kDa polymers were incubated in PBS at 37 °C for 24 hours to achieve equilibrium swelling. Following incubation, the gels were freeze-dried, coated with carbon, and imaged using a Hitachi SU-70 Schottky field emission gun scanning electron microscope.
Fourier transform infrared spectroscopy
To confirm crosslinking between the polymers, Fourier-transform infrared (FTIR) spectroscopy was performed using a Bruker VERTEX 70 FT-IR spectrometer equipped with a Platinum-ATR accessory and a diamond crystal plate. Spectra were collected for individual 10 kDa and 20 kDa polymers of PEG-4SH and PEG-4OPSS, as well as for freeze-dried hydrogels composed of 2% w/v PEG-4SH and 2% w/v PEG-4OPSS synthesized using 10 kDa and 20 kDa polymers.
Bulk rheology
To confirm successful formation of PEG hydrogels and to understand polymer weight percentages on mechanical properties, bulk rheological measurements were performed using an ARES G2 rheometer (TA instruments). PEG gel precursor solutions were loaded onto a 25 mm diameter parallel plate at a gap of 1000 μm at 37 °C. The solution was allowed to gel and equilibrate to 37 °C for 5 min. A humidity chamber was used to prevent solvent evaporation and consequent hydrogel drying. To determine the linear viscoelastic region of the fully formed gel, a strain sweep measurement was performed at 0.1–10% strain at a frequency of 1 rad s−1. To determine the elastic modulus, G′(ω), and viscous modulus, G′′(ω), a frequency sweep measurement is conducted within the linear viscoelastic region of the gel, at 10% strain amplitude and angular frequencies from 0.1 to 100 radians per s. To elucidate the interaction between PEG solution and mucins, a flow sweep was performed from a shear rate of 0.01 to 100 s−1 using 40 nm cone-plate geometry.
Self-healing
100 μL square gels were formed in 3D printed square molds. Two gels were pushed out of the molds and were brought together for 10 min before being held vertically to observe self-healing. One of the gels was kept transparent while rhodamine B was added to the other gel to aid in visualization. The response of the hydrogel to the application and the removal of shear were examined in shear-induced failure and recovery experiments using oscillatory time sweeps at a frequency of 10 rad s−1. Alternating high and low strain cycles were applied every 2 minutes, with high strains of 200%, 500%, and 1000% for the failure phase and a low strain of 1% for the recovery phase. The rheometer parameters were consistent with those described previously.
Particle tracking microrheology
Particle tracking microrheology was performed as described in our previous work.26 Briefly, solutions were prepared with 1 μL of ∼0.002% w/v suspension of 100 nm fluorescent muco-inert nanoparticles and added into a 25 μL solution of PEG-4SH and PEG-4OPSS prior to gelation in a custom microscopy chamber, sealed with a cover slip, and equilibrated for 30 min at room temperature before imaging. Ten-second movies at 30 ms temporal resolution were acquired with a high-speed CMOS camera equipped on an inverted confocal microscope with a 63×/1.4 NA oil objective. Movies were analyzed using custom written tracking software in MATLAB to extract 2D x, y-coordinates of MIP centroids over time. From these trajectories, time-averaged mean squared displacement (MSD; 〈Δr2(τ)〉) as a function of lag time, τ, is calculated as 〈Δr2(τ)〉 = 〈[x(t + τ) − x(t)]2〉 + 〈[y(t + τ) − y(t)]2〉, where x(t) and y(t) are the spatial coordinates of particles as a function of time t. Using the generalized Stokes–Einstein relation, measured MSD values were used to compute the viscoelastic properties of the hydrogels (e.g., G′(ω), G′′(ω), micro-viscosity). Hydrogel network pore size, ξ, was estimated based on MSD using the equation, ξ ≈ (〈Δr2(τ)〉)1/2 + a.27 All measurements described were performed in n = 3 PEG gel preparations per formulation.
Drug release
0.2% w/v TRITC labeled bovine serum albumin (TRITC-BSA) or Cy5 labeled IgG (Cy5-IgG) was added to 2% w/v 4-arm PEG-SH solution and mixed with equal volumes of 3% w/v of 4-arm PEG-OPSS in tip cut-off syringes to make 100 μL cylindrical hydrogels. Resulting gels comprised of 0.1% w/v TRITC-BSA/Cy5-IgG, 1% w/v 4-arm PEG-SH and 1.5% w/v 4-arm PEG-OPSS were immersed in 1 mL of PBS and incubated at 37 °C. For nanoparticle loading, 2% v/v FluoSpheres were encapsulated. Supernatants were collected at each time point and replaced with a fresh buffer. To examine protein release, fluorescence of the collected supernatants was measured by UV/Vis spectroscopy using a Tecan Spark multimode microplate reader. Fluorescence Ex/Em wavelengths of 545/575 were used for TRITC-BSA quantification and Ex/Em wavelengths of 650/670 were used for Cy5-IgG quantification.
Mucoadhesion
Mucoadhesive strength of the gels was measured using a pull-apart adhesion test.28 Square sections of 10 mm fresh porcine intestine were cut and 100 μL of PEG gel precursor solutions were applied on the luminal side of the tissue. It was allowed to set for 5 min and the apical side of the intestinal tissue was superglued to the clamps of a dynamic mechanical analyzer (TA Instruments, DMA Q800). Samples were initially isothermally compressed at a force of 1 N for 5 minutes to ensure the superglue dries and is pulled at a rate of 0.5 N min−1 until failure. The adhesion strength of each sample was recorded and replicated three times. To visualize mucoadhesion, a solution of 1% w/v PEG-4SH, 2% w/v PEG-4OPSS, and 0.01% w/v rhodamine B (200 μL total) was added onto a small section of the intestinal tissue obtained from Animal Biotech Industries and allowed to set for 2 min before being held vertically to visualize and take photographs.
Biocompatibility
Biocompatibility of PEG polymers and PEG gels was evaluated on HEK 293T cells. Briefly, HEK 293T cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin–streptomycin. Cells were seeded onto a 96 well plate at a density of 20 000 cells per well. After allowing cell adherence overnight, cells were treated with 20 μL solution of polymers dissolved in PBS (pH 7.4) in the concentration range of 1%–4% w/v or 20 μL of PEG-4SH and PEG-4OPSS gels and incubated at 37 °C, 5% CO2 for 24 hours. Following the treatment, cell culture supernatant containing polymers or gels was removed and replaced with resazurin containing media. Cells with the resazurin reagent were incubated at 37 °C, 5% CO2 for 3 hours. 100 μL of the cell culture supernatant containing resazurin was transferred to a 96-well black plate and fluorescence was measured at Ex/Em wavelengths of 570/585 nm. Cell viability was measured relative to the cells grown in media without any treatment. For live/dead staining, 1 × 105 cells were seeded in an 8-well chambered cell culture slide. After allowing cell adherence overnight, 50 μL of 2% PEG-4SH and 2% PEG-4OPSS gels were added to each well and incubated at 37 °C, 5% CO2 for 24 hours. Following the treatment, live cell staining was performed using 1 μM calcein AM and 500 nM propidium iodide and cells were imaged using a Zeiss confocal microscope.
In vivo gel retention
All experimental procedures conducted in mice were performed in accordance with the standards established by the US Animal Welfare Acts, set forth in the NIH guidelines as well as the Policy and Procedures Manual of the University of Maryland Institutional Animal Care and Use Committee (IACUC). These procedures were approved under the IACUC protocol # R-MAY-22-25 at the University of Maryland. Female BALB/c mice (6–8 weeks old, Charles River Laboratories) were used for the study. To prepare the hydrogel formulation, a 2% w/v PEG-4SH 20 kDa solution was mixed with near-infrared-labeled 100 nm polystyrene nanoparticles (excitation: 715 nm, emission: 755 nm). The PEG-4SH/nanoparticle solution and a 2% w/v PEG-4OPSS 20 kDa solution were loaded into a Twin-Syringe Delivery System (M-System, MedMix) for simultaneous mixing and delivery. The final concentration of nanoparticles in the gel was 1.5 μg μL−1, which was approximately 2 × 1010 particles per 10 μL. Mice were fully anesthetized using isoflurane and positioned on their stomachs during administration. To minimize the risk of nasal blockage, 10 μL of the combined liquid solution was carefully administered dropwise into a single nostril of each mouse (n = 3). To evaluate whether the hydrogel enhances cargo retention, a control group of mice (n = 3) received an intranasal dose of nanoparticles suspended in PBS under identical conditions. Fluorescence signals from the hydrogel-encapsulated nanoparticles were measured at 0, 2, 5, and 48 hours, as well as at 7 and 15 days post-administration using the IVIS Spectrum Imaging System. Background signal was recorded from untreated control mice (n = 1). Throughout the study period, body weight was monitored to assess the overall health of the animals. At the end of the study, mice were euthanized, and nasal and lung tissues were harvested for ex vivo fluorescence imaging to evaluate nanoparticle retention and distribution.
Results and discussion
Formulation of rapid in situ forming PEG hydrogels
In this study, we developed a new method for the rapid formation of polyethylene glycol (PEG)-based hydrogels under physiological conditions of 37 °C and at a pH of 7.4. These gels are also formed via bio-reducible disulfide linkers by combining thiol terminated 4-arm PEG (PEG-SH) and OPSS terminated 4-arm PEG (PEG-OPSS). An overview of the crosslinking mechanism for 4-arm PEG-SH and 4-arm PEG-OPSS is depicted in Fig. 1. After being dissolved in phosphate buffered solution (PBS), we observed gel formation within 30 seconds through rapid crosslinking via di-sulfide bond formation. Table 1 summarizes the time to gel as well as degradation time under reducing conditions for 6 lead formulations. Although a wide range of weight percentages were tested and found to rapidly form gels as noted in Table S1,† lead formulations were selected, which retained stability in PBS for ≥24 hours.
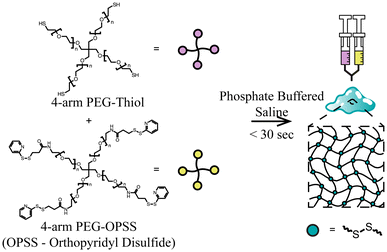 |
| | Fig. 1 Schematic illustration of the preparation of rapid in situ forming PEG hydrogels. | |
Table 1 Gelation time and degradation time of rapid forming PEG gels
| Formulation number |
4-Arm PEG-SH (%w/v) |
4-Arm PEG-OPSS (%w/v) |
Gelation time |
Degradation time in DTT |
Degradation time in GSH |
| 10 kDa PEG |
| F1 |
1 |
1.5 |
∼15 s |
∼8 min |
∼30 h |
| F3 |
1 |
2 |
∼5 s |
∼9 min |
∼60 h |
| F5 |
2 |
2 |
∼5 s |
∼6 min |
∼60 h |
| |
| 20 kDa PEG |
| F2 |
1 |
1.5 |
∼30 s |
∼3 min |
∼30 h |
| F4 |
1 |
2 |
∼20 s |
∼5 min |
∼30 h |
| F6 |
2 |
2 |
∼15 s |
∼7 min |
∼40 h |
The disulfide reducing agent glutathione (GSH) is present throughout the tissues in the body at a μM to mM concentration range.29 Thus, the degradation rate of PEG gels once administered in vivo is likely to be affected by the presence of GSH. Further, external disulfide reducing agents such as dithiothreitol (DTT) could be used for removal of the gel from the site of application as needed. We tested the effect of these compounds on our PEG gels in vitro using 100 mM DTT and GSH individually. As indicated in Table 1, DTT degrades gels in under 10 minutes, whereas glutathione degrades in 1 to 3 days.
Characterization of rapid forming PEG gels. The effects of polymer weight percentage and polymer molecular weight on the mechanical strength of hydrogels were studied and are shown in Fig. 2A. Formulations F1 to F5 have a storage modulus ranging from ∼400 to 800 Pa. Formulation F6 containing 20 kDa sized 2% w/v 4-arm PEG-SH and 2% w/v 4-arm PEG-OPSS possessed a significantly higher storage modulus of 1712 ± 267 Pa, indicative of an increased elasticity in comparison with other formulations tested.
 |
| | Fig. 2 Physical characterization of rapid in situ forming PEG gels. (A) Storage modulus (G′) for each gel at a frequency of 1 rad s−1. (n = 3), ****p < 0.0001 for one-way ANOVA with Tukey's multiple comparison test. (B) Estimated pore size for particle tracking microrheology using densely PEGylated 100 nm nanoparticles as probes. *p < 0.05, ***p < 0.001, ****p < 0.0001 for the Kruskal–Wallis test. (C) Equilibrium swelling ratio after immersion in PBS. (n = 3), *p < 0.05 for the Kruskal–Wallis test. (D) Degradation profile of PEG gels after immersion in PBS for 30 hours. (E) Dissolution of PEG gels after 10 minutes in PBS or under reducing conditions in DTT. SEM images of 20 kDa PEG gels at ×200 (F) and ×450 (G) magnifications. Data are expressed as mean ± SD. | |
We next evaluated the pore size of each formulation using particle tracking microrheology (Fig. 2B). We hypothesized that increasing PEG-SH/PEG-OPSS concentrations or molecular weights would lead to decreases in pore size which may change the release kinetics of encapsulated cargoes. Interestingly, we found only a weak dependence on the total PEG concentration with a relatively small range of pore sizes observed across formulation conditions. We did observe changes in the pore size as a function of PEG MW, but a consistent trend was not observed. Total available functional groups for crosslinking, flexibility of polymer chain to facilitate disulfide bond formation and steric hindrance due to the bulkiness of OPSS reactive groups compared to SH could be reasons for the inconsistent trends.
Swelling and degradation dictate the rate of release of therapeutics and biodegradability, which are vital for use in biomedical applications. Fig. 2C shows the equilibrium swelling ratio for each formulation. F6 with the highest storage modulus also has the highest swelling ratio and is 2-fold higher than the rest of the formulations. For degradation studies (Fig. 2D), PEG gels were immersed in PBS volume that is 50 times the volume of the gel. An initial increase in gel mass was observed due to swelling followed by slow degradation. Gels made of 10 kDa polymers, F1, F3, and F5 showed degradation up to 30% of the initial swollen mass in 30 days, whereas gels made of 20 kDa polymers, F2, F4, and F6 degraded up to 15% of the initial swollen mass in the same time frame. Longer retention and slow degradation of PEG gels can be due to the stability of ether linkages of PEG polymer and susceptibility to degradation due to auto-oxidation.30 Fig. 2E further illustrates that the linkers within the PEG gels are reducible as immersion of the rhodamine-loaded PEG gel in PBS alone shows rhodamine release but no degradation of the gel. However, the addition of DTT shows complete degradation of the gel within 10 minutes. Scanning electron microscopy images (Fig. 2F and G) of the freeze-dried hydrogels of formulation F6 made with 20 kDa polymers reveal the porous structure of the hydrogel network. FTIR spectra, as shown in Fig. S7,† confirm successful crosslinking between PEG-4SH and PEG-4OPSS. The spectra also show consistent peaks across hydrogels, regardless of the polymer molecular weight, indicating that crosslinking is independent of polymer size.
Self-healing of rapid in situ forming PEG hydrogels. To assess the functional resilience of the rapidly forming PEG hydrogels, their self-healing properties were investigated. When two gels of formulation F2 (1% w/v 4-arm PEG-SH, 20 kDa, crosslinked with 1.5% w/v 4-arm PEG-OPSS, 20 kDa) are brought into contact, they autonomously heal at the interface through disulfide bond reformation and hydrogen bonding as shown in Fig. 3A. The self-healing behavior was further assessed via a cyclic time sweep test with alternating low/high strains using formulation F6. Strain levels of 200%, 500%, and 1000% which are beyond the linear viscoelastic region shown in Fig. 3B were applied for 2 minutes to induce material failure, followed by a 2 minute recovery phase under a low strain of 1%. The hydrogels exhibited self-healing across all tested strains, with varying efficiencies. At 200% strain, the hydrogels achieved near-complete recovery, regaining a substantial portion of their original mechanical strength (Fig. 3C). Furthermore, the responses were rapid and repeatable. Although self-healing persisted at 500% strain, recovery efficiency declined, resulting in only partial restoration of mechanical properties (Fig. 3D). Under extreme deformation at 1000% strain, the hydrogels retained limited self-repair capacity, with markedly reduced mechanical recovery (Fig. 3E). These findings underscore the ability of PEG hydrogels to autonomously repair damage, minimizing the risk of detachment at the application site and preventing therapeutic cargo leakage. This self-healing capability further highlights their potential for use in dynamic physiological environments, such as localized delivery to the cardiac tissue, where repeated deformation and recovery are critical for sustained functionality.
 |
| | Fig. 3 Self-healing of rapid in situ forming PEG gels. (A) Photographs of the self-healing behavior of PEG gels. (B) Amplitude sweep demonstrating a linear viscoelastic region below 100% strain. Cyclic strains of 1% (low, unshaded area) and 200% (C), 500% (D), and 1000% (E) (high, shaded area) at a frequency of 10 rad s−1. | |
Mucoadhesive properties of rapid in situ forming PEG hydrogels. We next evaluated the ability of these PEG gels to adhere to mucosal tissues. Prior work has shown that PEG-based biomaterials can adhere to mucus-coated tissues through a combination of PEG-mucin entanglement and hydrogen bonding (Fig. 4A).31–33 We hypothesized that our gels may also be able to form disulfide bonds with cysteine-rich domains of mucins via PEG-OPSS and/or PEG-SH, leading to increases in mucoadhesion. To test this, we applied PEG-4OPSS and PEG-4SH solutions to the surface of pig intestine where a uniform layer of PEG gel was instantaneously formed (Fig. 4B). Pull-apart tests were then performed to measure their mucoadhesive strength immediately and 24 hours after application (Fig. 4C and D). We also included for comparison tissues treated with 2% w/v 4-arm PEG-DBCO and 2% w/v 4-arm PEG-azide as a control for PEG gels without OPSS/SH groups to form disulfide bonds and 4% w/v chitosan solution, which is known to possess mucoadhesive properties. All formulations tested were found to adhere to pig intestine with mucoadhesive strengths >600 Pa. Although statistically non-significant, the 4% w/v chitosan possessed greater mucoadhesive strength than all PEG gel formulations, likely due to the net-positively charged chitosan adhering to net-negatively charged mucin chains. However, PEG-DBCO and PEG-azide gels showed comparable mucoadhesive strength to PEG-OPSS and PEG-SH gels, indicating our gels may not be able to access the cysteines on the mucosal tissue. Therefore, adhesion is likely mediated by entanglement during sol–gel transition and hydrogen bonding. This is further confirmed through flow sweep measurements, which showed no change in viscosity when mucin solutions were mixed with PEG-4SH and PEG-4OPSS, indicating no covalent bond formation (Fig. S2C†).
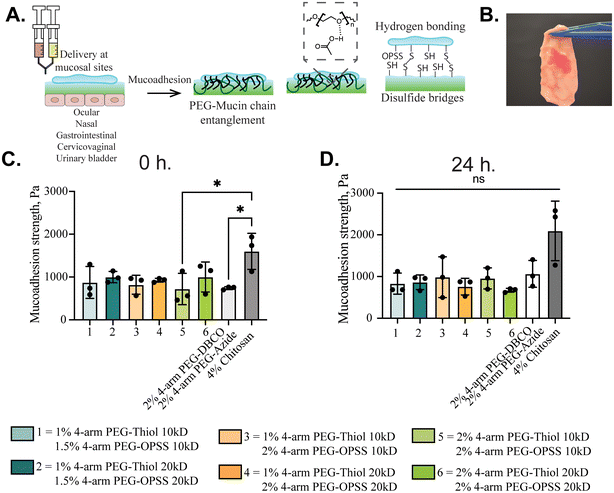 |
| | Fig. 4 Mucoadhesive properties of rapid in situ forming PEG hydrogels. (A) Schematic illustration of potential mechanisms enabling PEG gel adhesion to mucosal tissues. (B) Image of PEG gels following application to porcine intestinal tissue. (C) Mucoadhesive strength as measured by a pull-apart test for PEG gels and 4% w/v chitosan immediately. (n = 3), *p < 0.05 for one-way ANOVA with Tukey's multiple comparison test. (D) 24 hours (n = 3), non-significant (ns) as per the Kruskal–Wallis test after application to porcine intestinal tissue. Data are expressed as mean ± SD. | |
Release of cargoes from rapid forming PEG gels. We characterized the release kinetics of different model cargos, including bovine serum albumin (BSA), immunoglobulin (IgG), and 40 nm nanoparticles (NP), from PEG gels over 24 hours. We found that BSA and IgG were released in roughly 4 hours and 10 hours, respectively, whereas 40 nm NPs were retained within the gel for at least 24 hours (Fig. 5). This is likely explained by the difference in cargo size with BSA and IgG being ∼4–5 times smaller than 40 nm NP. These data indicate that the release of protein therapeutics would likely be driven by diffusion out of the gel, whereas the delivery of encapsulated nanoparticles is more likely to be driven by gel degradation. Furthermore, the polymer weight percentage and swelling ratio have no effect on BSA release, a minimal effect on IgG release, and the highest effect on 20 nm nanoparticles, exhibiting increased release in formulation F6 due to a higher swelling ratio (Fig. S5†).
 |
| | Fig. 5 Release kinetics of model therapeutic cargoes from rapid forming PEG hydrogels. Cumulative mass release profiles of bovine serum albumin (BSA), immunoglobulin (IgG), and 40 nm nanoparticles (NPs) from PEG hydrogels with either 10 kDa or 20 kDa PEG-SH and PEG-OPSS over 24 hours (n = 3). Data are expressed as mean ± SD. | |
Biocompatibility of rapid forming PEG gels. To ensure the gel precursor components and the PEG gel itself are well-tolerated by mammalian cells, we conducted biocompatibility studies using HEK-293 cells (Fig. 6). First, we treated HEK-293 cells with either 4-arm PEG-SH or PEG-OPSS at concentrations up to 4% w/v (Fig. 6A). We found that 10 kDa 4-arm PEG-SH was well-tolerated at all concentrations tested. Although statistically significant differences were observed with other treatments, more than 80% cell viability was observed except for 10 kDa 4-arm PEG OPSS at 4% w/v concentration. We next treated HEK-293 cells with PEG gels and no toxicity was observed for all lead formulations tested (Fig. 6B). These data suggest that PEG gels are generally safe for use as an injectable biomaterial. However, acute exposure to 10 kDa 4-arm PEG formulations may pose some toxicity concerns, and thus, 20 kDa PEG formulations may be better suited for use in future drug delivery applications.
 |
| | Fig. 6 Biocompatibility of rapid forming PEG gels. Viability of HEK-293 cells following treatment with (A) 4-arm PEG solutions (n = 5) and (B) PEG hydrogels (n = 4). (C) Live/dead fluorescence staining following treatment with PEG hydrogels. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 for one-way ANOVA with Dunnett's multiple comparison test. Data are expressed as mean ± SD. | |
In vivo nasal retention of rapid in situ forming PEG gels. To evaluate the in vivo retention of the rapid-forming PEG hydrogel, we assessed its ability to prolong the residence time of a model cargo (nanoparticles) in the nasal cavity of mice. Materials deposited in the nasal mucosa were subject to rapid clearance every 15 to 20 minutes through mucociliary clearance, posing a significant challenge for effective nasal drug delivery. To test whether the hydrogel could enhance retention, we encapsulated near-infrared (NIR)-emitting 100 nm polystyrene nanoparticles (NPs) within the gel and administered them intranasally. Since polystyrene NPs are non-biodegradable, the presence of a fluorescence signal directly correlates with gel retention. As shown in Fig. 7A and B, NPs administered in PBS (without hydrogel) were cleared from the nasal cavity within 5 hours and completely undetectable by 48 hours. In contrast, NPs delivered using the hydrogel vehicle exhibited near-complete retention up to 48 hours. By day 15, fluorescence signals returned to baseline levels, consistent with the complete degradation of the hydrogel and clearance of NPs. Additionally, body weight monitoring throughout the study (Fig. 7C) showed a steady increase, indicating no adverse effects from the hydrogel treatment. These results demonstrate that the rapid in situ forming PEG hydrogel significantly enhances cargo retention in the nasal cavity and provides an ideal formulation strategy for sustained drug release at mucosal sites.
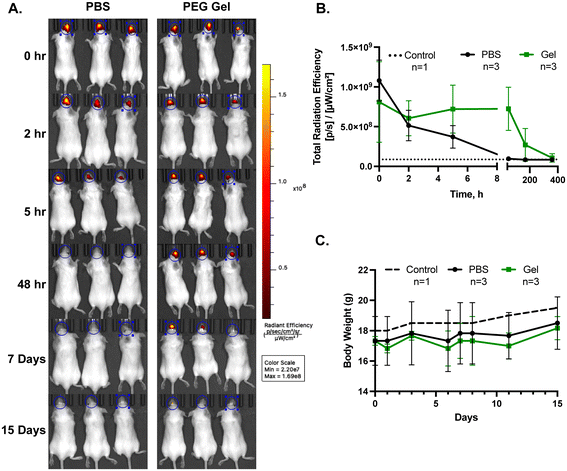 |
| | Fig. 7 In vivo nasal retention of rapid forming PEG gels. (A) IVIS images of mice at different time points following administration with either PBS or rapid forming PEG hydrogels loaded with near infrared labeled, 100 nm polystyrene nanoparticles. (B) Quantification of total radiation efficiency from IVIS images, used to assess the in vivo retention of PEG hydrogels over time. (C) Body weight of mice monitored throughout the study to evaluate the overall health and tolerability of PEG hydrogels. Data are expressed as mean ± SD. | |
Conclusion
We have developed a rapid in situ forming PEG hydrogel capable of adhering to mucosal tissues. Unlike other chemistries used in previous work, we could form bio-reducible disulfide-linked PEG gels that are able to be degraded over time (days to weeks) upon administration. Both protein- and nanoparticle-based therapeutics may be encapsulated into the gel for extended release at mucosal sites. Future work will focus on further development of PEG gels as a novel sprayable and/or injectable in situ forming hydrogel for the delivery of therapeutics to mucosal tissues.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
T. Y. and G. A. D. have a pending patent based on the hydrogel formulation described in this manuscript.
Acknowledgements
This study was supported by the NIH (R21 EB030834 to GAD and R01 GM141132 to PK) and a UM Ventures Medical Device Development Fund grant to GAD.
References
- J. R. McGhee, J. Mestecky, M. T. Dertzbaugh, J. H. Eldridge, M. Hirasawa and H. Kiyono, The mucosal immune system: from fundamental concepts to vaccine development, Vaccine, 1992, 10, 75–88, DOI:10.1016/0264-410x(92)90021-b.
- E. A. Naumova, T. Dierkes, J. Sprang and W. H. Arnold, The oral mucosal surface and blood vessels, Head Face Med., 2013, 9, 8, DOI:10.1186/1746-160X-9-8.
- L.-A. Keller, O. Merkel and A. Popp, Intranasal drug delivery: opportunities and toxicologic challenges during drug development, Drug Delivery Transl. Res., 2022, 12, 735–757, DOI:10.1007/s13346-020-00891-5.
- N. A. Peppas and J. J. Sahlin, Hydrogels as mucoadhesive and bioadhesive materials: a review, Biomaterials, 1996, 17, 1553–1561, DOI:10.1016/0142-9612(95)00307-X.
- T. Yeruva, S. Yang, S. Doski and G. A. Duncan, Hydrogels for Mucosal Drug Delivery, ACS Appl. Bio Mater., 2023, 6, 1684–1700, DOI:10.1021/acsabm.3c00050.
- X. Murgia, B. Loretz, O. Hartwig, M. Hittinger and C.-M. Lehr, The role of mucus on drug transport and its potential to affect therapeutic outcomes, Adv. Drug Delivery Rev., 2018, 124, 82–97, DOI:10.1016/j.addr.2017.10.009.
- S. R. Van Tomme, G. Storm and W. E. Hennink, In situ gelling hydrogels for pharmaceutical and biomedical applications, Int. J. Pharm., 2008, 355, 1–18, DOI:10.1016/j.ijpharm.2008.01.057.
- A. Bak, M. Ashford and D. J. Brayden, Local delivery of macromolecules to treat diseases associated with the colon, Adv. Drug Delivery Rev., 2018, 136–137, 2–27, DOI:10.1016/j.addr.2018.10.009.
- I. Seah, X. J. Loh and X. Su, A topical gel for extended ocular drug release, Nat. Biomed. Eng., 2020, 4, 1024–1025, DOI:10.1038/s41551-020-00645-1.
- M. S. Roberts, H. S. Cheruvu, S. E. Mangion, A. Alinaghi, H. A. E. Benson, Y. Mohammed, A. Holmes, J. Van Der Hoek, M. Pastore and J. E. Grice, Topical drug delivery: History, percutaneous absorption, and product development, Adv. Drug Delivery Rev., 2021, 177, 113929, DOI:10.1016/j.addr.2021.113929.
- A. Mandal, J. R. Clegg, A. C. Anselmo and S. Mitragotri, Hydrogels in the clinic, Bioeng. Transl. Med., 2020, 5, e10158, DOI:10.1002/btm2.10158.
- M. Agrawal, S. Saraf, S. Saraf, S. K. Dubey, A. Puri, U. Gupta, P. Kesharwani, V. Ravichandiran, P. Kumar, V. G. M. Naidu, U. S. Murty, Ajazuddin and A. Alexander, Stimuli-responsive In situ gelling system for nose-to-brain drug delivery, J. Controlled Release, 2020, 327, 235–265, DOI:10.1016/j.jconrel.2020.07.044.
- M. H. Asfour, S. H. Abd El-Alim, G. E. A. Awad and A. A. Kassem, Chitosan/β-glycerophosphate in situ forming thermo-sensitive hydrogel for improved ocular delivery of moxifloxacin hydrochloride, Eur. J. Pharm. Sci., 2021, 167, 106041, DOI:10.1016/j.ejps.2021.106041.
- Y. C. Kim, M. D. Shin, S. F. Hackett, H. T. Hsueh, R. Lima e Silva, A. Date, H. Han, B.-J. Kim, A. Xiao, Y. Kim, L. Ogunnaike, N. M. Anders, A. Hemingway, P. He, A. S. Jun, P. J. McDonnell, C. Eberhart, I. Pitha, D. J. Zack, P. A. Campochiaro, J. Hanes and L. M. Ensign, Gelling hypotonic polymer solution for extended topical drug delivery to the eye, Nat. Biomed. Eng., 2020, 4, 1053–1062, DOI:10.1038/s41551-020-00606-8.
- K. S. Anseth and H.-A. Klok, Click
Chemistry in Biomaterials, Nanomedicine, and Drug Delivery, Biomacromolecules, 2016, 17, 1–3, DOI:10.1021/acs.biomac.5b01660.
- H. Zhan, H. de Jong and D. W. P. M. Löwik, Comparison of Bioorthogonally Cross-Linked Hydrogels for in Situ Cell Encapsulation, ACS Appl. Bio Mater., 2019, 2, 2862–2871, DOI:10.1021/acsabm.9b00253.
- T. Yeruva and C. H. Lee, Enzyme Responsive Delivery of Anti-Retroviral Peptide via Smart Hydrogel, AAPS PharmSciTech, 2022, 23, 234, DOI:10.1208/s12249-022-02391-w.
- C. Bahou, R. J. Spears, A. M. Ramírez Rosales, L. N. C. Rochet, L. J. Barber, K. S. Stankevich, J. F. Miranda, T. C. Butcher, A. M. Kerrigan, V. K. Lazarov, W. Grey, V. Chudasama and C. D. Spicer, Hydrogel Cross-Linking via Thiol-Reactive Pyridazinediones, Biomacromolecules, 2023, 24, 4646–4652, DOI:10.1021/acs.biomac.3c00290.
- H. T. Hoang, S.-H. Jo, Q.-T. Phan, H. Park, S.-H. Park, C.-W. Oh and K. T. Lim, Dual pH-/thermo-responsive chitosan-based hydrogels prepared using “click” chemistry for colon-targeted drug delivery applications, Carbohydr. Polym., 2021, 260, 117812, DOI:10.1016/j.carbpol.2021.117812.
- T. S. Hebner, B. E. Kirkpatrick, B. D. Fairbanks, C. N. Bowman, K. S. Anseth and D. S. W. Benoit, Radical-Mediated Degradation of Thiol–Maleimide Hydrogels, Adv. Sci., 2024, 11, 2402191, DOI:10.1002/advs.202402191.
- J. I. Paez, A. Farrukh, R. Valbuena-Mendoza, M. K. Włodarczyk-Biegun and A. del Campo, Thiol-Methylsulfone-Based Hydrogels for 3D Cell Encapsulation, ACS Appl. Mater. Interfaces, 2020, 12, 8062–8072, DOI:10.1021/acsami.0c00709.
- J. Yu, X. Xu, F. Yao, Z. Luo, L. Jin, B. Xie, S. Shi, H. Ma, X. Li and H. Chen, In situ covalently cross-linked PEG hydrogel for ocular drug delivery applications, Int. J. Pharm., 2014, 470, 151–157, DOI:10.1016/j.ijpharm.2014.04.053.
- A. Bernkop-Schnürch, Thiomers: A new generation of mucoadhesive polymers, Adv. Drug Delivery Rev., 2005, 57, 1569–1582, DOI:10.1016/j.addr.2005.07.002.
- R. P. Brannigan and V. V. Khutoryanskiy, Progress and Current Trends in the Synthesis of Novel Polymers with Enhanced Mucoadhesive Properties, Macromol. Biosci., 2019, 19, 1900194, DOI:10.1002/mabi.201900194.
- V. Grabovac and D. Guggi, A. Bernkop-Schnürch, Comparison of the mucoadhesive properties of various polymers, Adv. Drug Delivery Rev., 2005, 57, 1713–1723, DOI:10.1016/j.addr.2005.07.006.
- K. Joyner, D. Song, R. F. Hawkins, R. D. Silcott and G. A. Duncan, A rational approach to form disulfide linked mucin hydrogels, Soft Matter, 2019, 15, 9632–9639, 10.1039/c9sm01715a.
- K. Joyner, S. Yang and G. A. Duncan, Microrheology for biomaterial design, APL Bioeng., 2020, 4, 041508, DOI:10.1063/5.0013707.
- J. L. Daristotle, M. Erdi, L. W. Lau, S. T. Zaki, P. Srinivasan, M. Balabhadrapatruni, O. B. Ayyub, A. D. Sandler and P. Kofinas, Biodegradable, Tissue Adhesive Polyester Blends for Safe, Complete Wound Healing, ACS Biomater. Sci. Eng., 2021, 7, 3908–3916, DOI:10.1021/acsbiomaterials.1c00865.
- G. Wu, J. R. Lupton, N. D. Turner, Y.-Z. Fang and S. Yang, Glutathione Metabolism and Its Implications for Health, J. Nutr., 2004, 134, 489–492, DOI:10.1093/jn/134.3.489.
- M. E. Payne, O. O. Kareem, K. Williams-Pavlantos, C. Wesdemiotis and S. M. Grayson, Mass spectrometry investigation into the oxidative degradation of poly(ethylene glycol), Polym. Degrad. Stab., 2021, 183, 109388, DOI:10.1016/j.polymdegradstab.2020.109388.
- L. Serra, J. Doménech and N. A. Peppas, Design of poly(ethylene glycol)-tethered copolymers as novel mucoadhesive drug delivery systems, Eur. J. Pharm. Biopharm., 2006, 63, 11–18, DOI:10.1016/j.ejpb.2005.10.011.
- N. V. Efremova, Y. Huang, N. A. Peppas and D. E. Leckband, Direct Measurement of Interactions between Tethered Poly(ethylene glycol) Chains and Adsorbed Mucin Layers, Langmuir, 2002, 18, 836–845, DOI:10.1021/la011303p.
- P. Schattling, E. Taipaleenmäki, Y. Zhang and B. Städler, A Polymer Chemistry Point of View on Mucoadhesion and Mucopenetration, Macromol. Biosci., 2017, 17, 1700060, DOI:10.1002/mabi.201700060.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Robert J. Morris III
*a,
Robert J. Morris III







