
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Two-step continuous flow-driven synthesis of 1,1-cyclopropane aminoketones†
Viktoria Velichko a,
Davide Moia,
Francesco Soddua,
Roberto Scipionea,
Enrico Poddab,
Alberto Luridianaa,
Dario Cambie*c,
Francesco Secci*a and
Maria Chiara Cabuaa
a,
Davide Moia,
Francesco Soddua,
Roberto Scipionea,
Enrico Poddab,
Alberto Luridianaa,
Dario Cambie*c,
Francesco Secci*a and
Maria Chiara Cabuaa
aDipartimento di Scienze Chimiche e Geologiche, Università degli Studi di Cagliari, Complesso Universitario di Monserrato, 09042, Monserrato (Cagliari), Italy. E-mail: fsecci@unica.it
bCentro Servizi d’Ateneo per la Ricerca CeSAR, 09042, Monserrato (Cagliari), Italy
cMax Planck Institute of Colloids and Interfaces, Biomolecular Systems Department, Am Mühlenberg 1, 14476 Potsdam, Germany
First published on 23rd December 2024
Abstract
The continuous flow telescoped synthesis of 1,1-cyclopropane aminoketones was achieved by optimizing the photocyclization of 1,2-diketones to 2-hydroxycylobutanones (HCBs) and their reaction with aryl- and alkylamines, via tandem condensation C4–C3-ring contraction reaction. With the achieved operational conditions, we were able to obtain a library of cyclopropylamines with good chemical yields, high productivity, and short residence times.
Aminocyclopropanes have consistently captured the attention of the organic chemistry community due to their crucial roles in the synthesis of biologically active molecules with applications in drug discovery and in total synthesis.1 In particular, constrained amino derivatives have been proposed as therapeutic agents including MAO inhibitors,2 opioid antagonists,3 anticancer,4 carboxypeptidase,5a and protease inhibitors,5b and anabolic,5c and antidepressant drugs6 (Scheme 1a). In recent years, interest in nitrogen-decorated strained carbocyclic derivatives has expanded. This is mainly due to the possibility of triggering cyclopropylamine ring-opening reactions through the generation of chemo- or photoinduced radical species (oxidative cleavage) or by nucleophilic ring-opening of cyclopropanes functionalized with EDG-groups (polar reactions). This reactivity is often enhanced in push–pull systems, where donor amine groups and EWG-groups coexist.7 In particular, research has looked into ring-opening and ring expansion rearrangements,8 and metal-catalysed and light-induced [3+2]-annulation strategies,9 for converting three membered ring compounds into synthetically valuable building blocks.10 Conventional approaches pointing at the construction of cyclopropylamines have involved Curtius11a and Hofmann rearrangement,11b Kulinkovich–Szymoniak reaction,12 and carbene transfer of diazo compounds.13 More recently Rh,14 Ru15 and organo-Zn mediated cyclopropanation,16 and Cu-catalysed three-component cyclopropane-alkenyl amination17 have been reported. Furthermore, organocatalytic,18 electrochemical19 and metal-free redox-neutral strategies have been unlocked for the preparation of these compounds.20
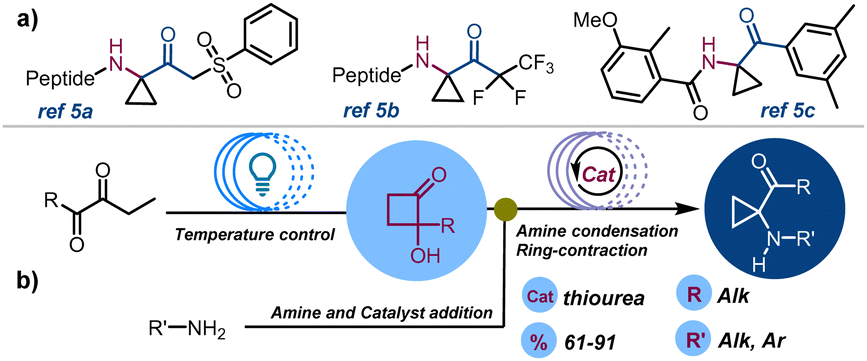 | ||
| Scheme 1 (a) Selected 1,1-cyclopropane aminoketone bioactive compounds; (b) this work: two-step synthesis of 1,1-cyclopropane aminoketones from 1,2-diketones under continuous flow conditions. | ||
Despite these advancements, 1,1-cyclopropane-aminoketones still represent a relatively underdeveloped class of compounds, hindered by challenging synthetic methods and limited functionalization options.21 In 2020, a new synthetic batch methodology has been proposed, enabling access to cyclopropyl aminoketones through the condensation of primary and secondary amines to 2-hydroxycyclobutanones (HCBs) followed by α-iminol rearrangement.22 However, this method requires extended reaction times (48–72 h) and entails the multi-step preparation of starting materials. Herein, we report our investigations around the development of a continuous-flow cyclopropane aminoketone two-step synthesis.23 This seamless process combines the swift and secure photocyclization of 1,2-diketones to access HCBs and the addition of aryl- and alkylamines, streamlining and elevating the synthesis of high-value organic compounds (Scheme 1).
We initiated our investigation by optimizing the continuous-flow type II Norrish–Yang (NY II) photocyclization of 1,2-diketones 1 to HCBs 2 by revisiting our previously reported procedure.24 In that approach, a 0.1 M solution of diketone was pumped into a custom-made flow reactor exposed to blue LEDs, producing a panel of differently substituted HCBs in good yields within a 30 minute residence time (tres). However, to standardize and enhance this synthetic protocol, to more efficient, and commercially available light sources, we substituted the LED light with Kessil lamps, using a thermostatic bath to control the temperature (see ESI,† Section S2). Indeed, earlier observations indicated that the starting diketones 1 and HCBs 2 could undergo degradative processes at high temperatures. Additionally, the photoclosure reaction itself can generate secondary products due to the competition between NY-I (fragmentation)25 and NY-II (cyclization), leading to reaction yield erosion. To prove our hypotheses, diketone 1a (0.1 M, ACN) was reacted in a 2 mL custom-made reactor (R1) placed in a thermostatic bath (19–21 °C) and exposed to light (427 nm). The reactor was connected to a syringe pump via the inlet and to a collection container via the outlet. Using this set-up, tres screening indicated that the best reaction conversion could be achieved within 30 minutes, producing the compound 2a in 81% yield (P: 18.2 mg h−1) as reported in Table 1 (entry 1). However, in 15 minutes tres 2a was produced in 79% yield and fairly higher productivity (P: 36.1 mg mL−1, entry 3). As a comparison, the same stock solution was pumped into a Vapourtec UV-150 photoreactor (5 mL) setting the temperature at 19 °C. Shorter time reaction (tres = 1 min) led to 2a in 79% yield (flow rate 5 mL min−1). This marked a 15-times acceleration compared to our previous continuous flow approach, demonstrating a high level of productivity (entry 7 also refers to the ESI†).
|
|||
|---|---|---|---|
| Entry | tres (min) | Yielda 2a (%) | Productivity (mg h−1) |
| a Yields were determined by qNMR using 1,2,4,5-tetrachloro-3-nitrobenzene (TCN) as an internal standard.b Reaction conditions: hexane-2,3-dione (1a, 0.1 M in ACN), FEP tube (ϕ i.d.: 0.8 mm, 2 mL vol.) at 19–21 °C, syringe pump, Kessil PR 427 nm.c Reaction conditions: Vapourtec UV-150 photoflow reactor (ϕ i.d.: 0.8 mm, 5 mL volume), 440 nm, 60 W. | |||
| 1 | 30 | 81b | 18.5 |
| 2 | 20 | 80b | 27.4 |
| 3 | 15 | 79b | 36.1 |
| 4 | 10 | 69b | 47.2 |
| 5 | 5 | 51b | 69.8 |
| 6 | 5 | 85c | 116.4 |
| 7 | 1 | 79c | 541.0 |

Using the conditions reported in Table 1 (entry 3) we were also able to successfully synthesize other HCB derivatives. The photocyclization of pentane-2,3-dione 1b allowed us to obtain the corresponding cyclobutanone 2b in 88% yield. While 5-methylhexane-2,3-dione 1c afforded 2c in 90% yield.26
Building on these results, we focused our efforts on the second phase of this investigation concerning the synthesis of cyclopropane-derivatives 4 under continuous flow conditions. The reaction between the amine 3a and 1a was selected as a model transformation to study the α-iminol C4–C3 ring-contraction reaction22 (Scheme 1b).
A 2 mL coil reactor (R2) maintained at 22–24 °C was connected via a Y-mixer to two pumps dispensing HCB 2a (0.1 M in ACN) and 3a (0.1 M in ACN). Using this initial configuration, the target compound 4a was successfully obtained in a 41% yield (Table 2, entry 1).
|
||||
|---|---|---|---|---|
| Entry | tres (min) | Cat. (mol%) | Temp. (°C) | Yield of 2aa (%) |
| Reactions were performed in a 2 mL coil reactor at the reported temperatures.a Yields were determined by qNMR using TCN as an internal standard.b Reactions were carried out on a R-series (R4) Vapourtec module as described in the ESI. | ||||
| 1 | 15 | — | 24 | 41 |
| 2 | 30 | — | 24 | 83 |
| 3 | 15 | I (10) | 24 | 82 |
| 4 | 15 | II (2.5) | 24 | 80 |
| 5 | 15 | III (20) | 24 | 77 |
| 6 | 30 | II (2.5) | 24 | 89 |
| 7 | 180 | I (10) | 24 | 90 |
| 8 | 180 | II (2.5) | 24 | 90 |
| 9 | 15 | II (2.5) | 50b | 89 |
| 10 | 5 | — | 125b | 89 |
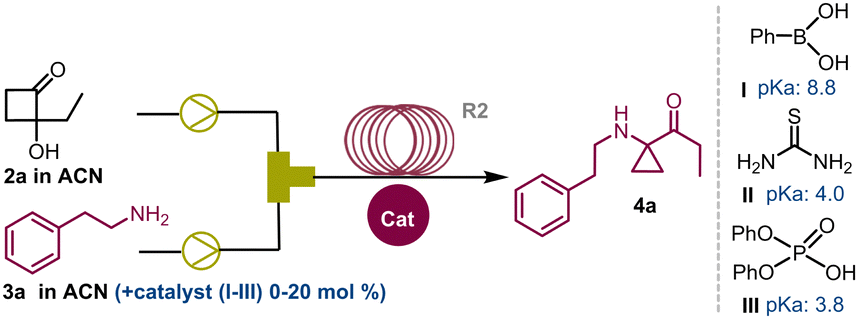
However, attempts to increase the yields by simply modifying the reaction times were unsuccessful (see ESI,† Section S2). Subsequently, we tested a panel of acid catalysts to expedite the reaction without altering other parameters. Optimal outcomes were achieved by employing 10 mol% of phenylboronic acid I or 2.5 mol% of thiourea II, which yielded 4a in 82% (entry 3) and 80% yield (entry 4), respectively. On the other hand, diphenyl phosphate III proved less effective, affording compound 4a in 77% yield at 20 mol% loading (entry 5). Moreover, extending the residence time (tres) from 15 to 180 minutes allowed us to achieve a 90% yield of 4a in reactions in the presence of catalysts I and II (entries 7 and 8). Other reaction conditions were investigated by using a Vapourtec system (see ESI,† Section S3). Reactions conducted at 125 °C and in the absence of catalysts were performed with 5 minute tres, affording the compound 4a in 89% yield (entry 10). To do this, a back pressure regulator system (BPR) was connected at the inlet of R2 to address pressure issues arising from the ACN boiling point (5.2 bar, ESI† Section S3.2). Finally, carrying out these reactions with a stock solution of amine and cat. II (2.5 mol%) at 50 °C, the desired cyclopropyl adduct 4a was obtained in 89% yield (entry 9). Further implementations of the continuous flow setup and the exploration of other reaction conditions (see ESI†) did not show any improvement. Overall, in a cost–benefit balance, the reaction conditions summarised in entry 5 (Table 2) were deemed as the best compromise between productivity and reproducibility. With these results in hand, the scope and the limitations of this synthetic strategy were explored. Reactions carried out with both aliphatic and aromatic amines 3a–z and HCBs 1a–c (also including the active pharmaceutical ingredients tyramine 3b, tryptamine 3c, and procaine 3v) delivered the corresponding reaction products in good to high yields (61–91%), showing high functional group tolerance and good degree of purity. However, the synthesis of compounds 4u and 4v required modifications to the flow conditions. Enamine 3u was reacted with 1a and 1b in toluene (1.0 M) at 80 °C, yielding the corresponding compounds in 25% and 27% yield, respectively (see ESI,† Section S5.1).
Very interestingly, derivatives 4y and 4z, observed by 1H NMR analysis of the corresponding crude mixtures, spontaneously transformed into crystalline imines 5y and 5z upon isolation from the reaction medium, as confirmed by XRD analysis (Scheme 2; see also ESI,† Section S8). Subsequently, it was observed that this behaviour is common to all the aliphatic cyclopropylamines having a geminal dimethylamino-system on the carbocyclic unit that we tested (ESI†). Beyond this observation, this represents the first example of crystalline structures of 2-hydroxycyclobutyl imine to date. At this point, following the systematic optimization of each reaction step, the focus was on merging the two flow reaction processes to achieve a telescoped continuous-flow, two-step synthesis of 1,1-cyclopropane aminoketones 4 from compound 1 (Scheme 3). To perform this, a 4 mL FEP coil reactor (R1), maintained at 19 °C and exposed to blue light (427 nm), was connected to a syringe pump feeding a 0.1 M stock solution of 1a in ACN (flow rate: 0.27 mL min−1). The reactor outlet was connected via a Y-mixer with a stream of amine 3a (0.1 M in ACN) containing 2.5 mol% of thiourea using a second syringe pump and a back pressure regulator (BPR, pressure = 5.2 bar). The nucleophilic addition of 3a to HCB 1a occurred in a second 4 mL tubing reactor (R2) maintained at 22–24 °C and connected directly to a collection container (flow rate: 0.54 mL min−1). Experiments carried out with this configuration enabled compound 4a to be obtained in 80% yield over two-steps (overall tres over two-steps: 30 min; P: 208 mg h−1).
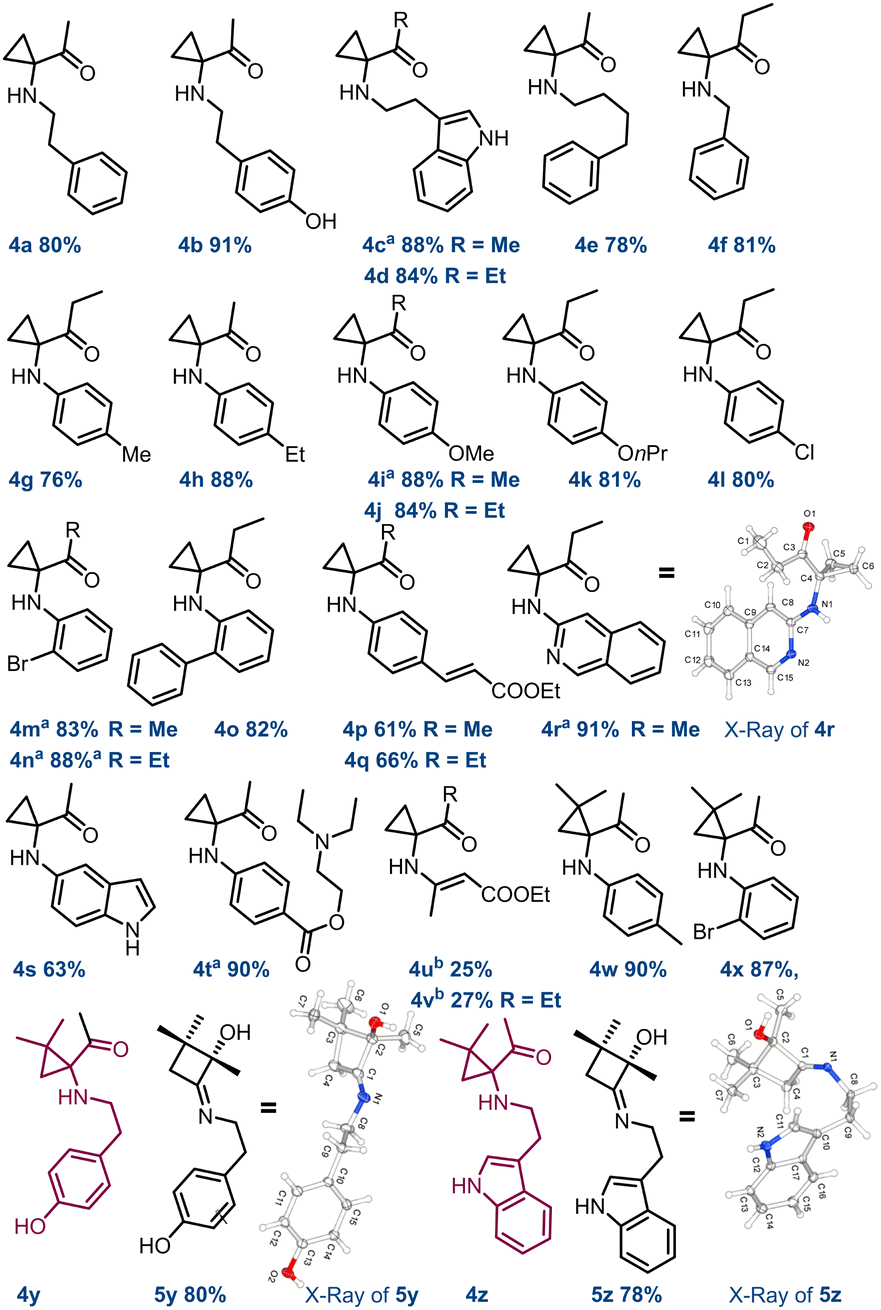 | ||
| Scheme 2 Continuous flow synthesis of 1,1-cyclopropane aminoketone substrate scope. Yields of compounds 4c, 4i, 4m–n, 4r, and 4t were determined by qNMR using 1,3,5-trimethoxybenzene as an internal standard. aCompounds 4c, 4i, 4m–n, 4r, and 4t were purified after reduction with NaBH4 in THF at 0 °C; breaction carried out in toluene (1.0 M) at 80 °C. 4r CCDC 2373555, 5y CCDC 2373554, 5z CCDC 2373556.† | ||
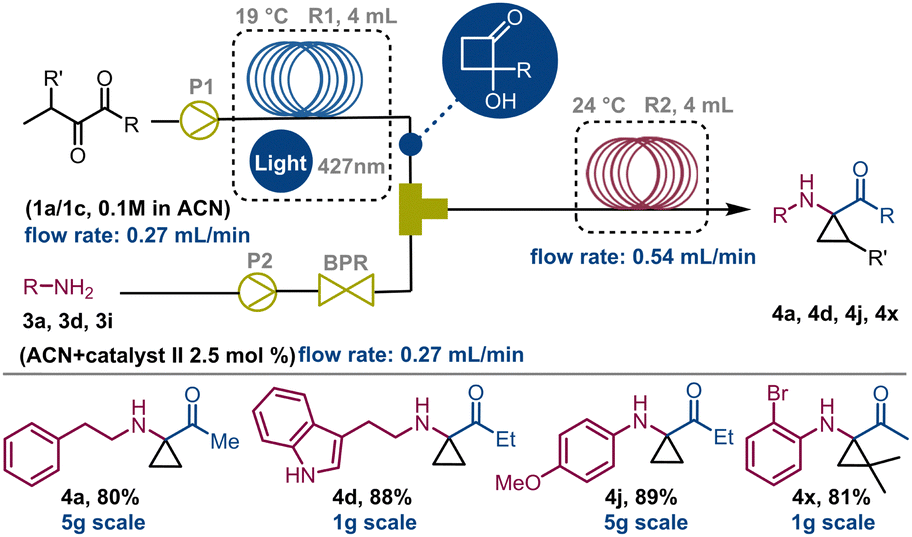 | ||
| Scheme 3 Telescoped synthesis of 1,1-cyclopropane aminoketones under continuous flow conditions. | ||
This process was also evaluated in the telescoped synthesis of derivatives 4d (88%), 4j (89%) and 4x (81%). Nevertheless, performing these processes on a multigram scale we were able to obtain approximately 5g of compounds 4a and 4j over 12 hours, and 1g of 4d and 4x after about 2 hours (see ESI,† Section S3). These values are aligned with those obtained for the individual transformations (steps 1 and 2) confirming the efficiency of the entire designed process. To demonstrate the synthetic utility of these derivatives, compound 4n, once reduced to 6n with NaBH4 before purification, was submitted to selective acylation to produce derivative 7 in 75% yield. Also, the Pd-induced tandem ring-opening-intramolecular cyclization reaction of 6n allowed access to quinoline 8 in 70% yield, probably via adduct A (Scheme 4a). On the other hand, stirring 6c in THF at 50 °C in the presence of SiO2, compound 9 was produced in 81% yield. As in the previous case, it is proposed that cyclopropane ring-opening triggers an iminium-like equilibrium, leading to the formation of an α-imino-ketone adduct. Once protonated, this adduct can be susceptible of nucleophilic attack by the indole moiety, leading to the tricyclic compound 9 (Scheme 4b).
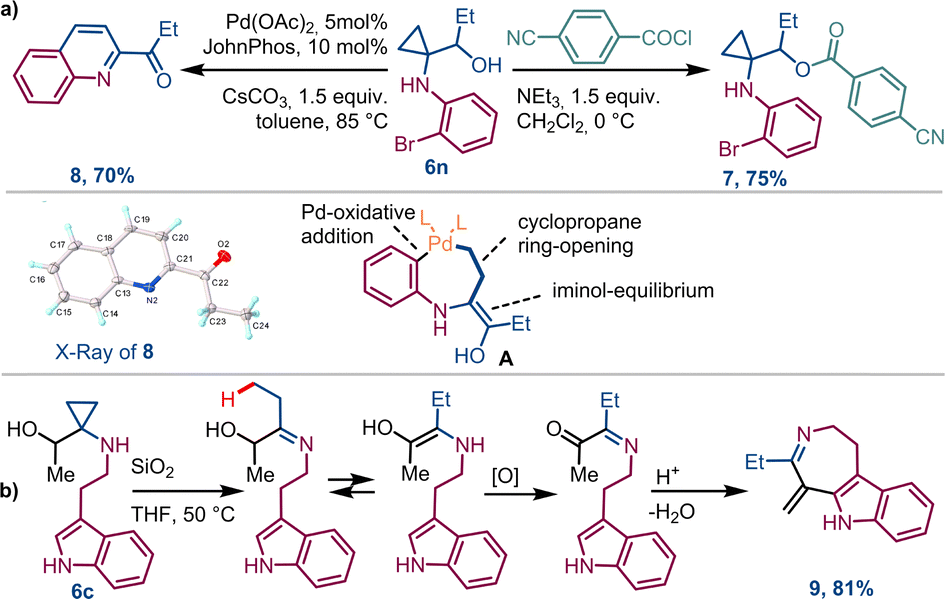 | ||
| Scheme 4 Post functionalization of 1,1-cyclopropane aminoketones 4n and 4c via alcohols 6n (a) and 6c (b). CCDC deposition number of 8: 2387960. | ||
In summary, we have developed and optimized two continuous flow processes, involving (a) the photocyclization of 1,2-diketones to access HCBs and (b) the reaction between a panel of amines and HCBs to obtain cyclopropane aminoketones. The two established protocols were subsequently merged to design a telescoped 2-step continuous-flow process, enabling the direct transformation of diketones into the corresponding cyclopropylamines in good yields, eliminating intermediate purifications, shortening the reaction times (30 minutes vs. 48 hours for batch reactions), and maximizing the process productivity. Studies dedicated to understanding the reaction mechanism leading to the formation of compounds 8 and 9 are currently underway in our laboratories.
Open Access funding provided by the Max Planck Society.
Data availability
The data supporting this article have been included in the ESI.† Crystallographic data for 4r, 5y, 5z and 8 has been deposited at the CCDC [CCDC numbers: 2373554, 2373555, 2373556, 2387960] and can be obtained from [https://www.ccdc.cam.ac.uk/structures/?gad_source=1&gclid=Cj0KCQjwn9y1BhC2ARIsAG5IY4fF7OAulkNmvDvTV06QberBmhXhiavEOu9tw1Vlb87_36Wo_5ULGsaAu4lEALw_wcB].Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) T. T. Talele, J. Med. Chem., 2016, 59, 8712 CrossRef; (b) Z. Casar, Synthesis, 2020, 1315 CrossRef.
- J. Mills, R. W. Kattau, I. H. Slater and R. W. Fuller, J. Med. Chem., 1968, 11, 95 CrossRef.
- S. Dongbang, B. Pedersen and J. A. Ellman, Chem. Sci., 2019, 10, 535 RSC.
- M. Pieroni, G. Annunziato, E. Azzali, P. Dessanti, C. Mercurio, G. Meroni, P. Trifiro, P. Vianello, M. Villa, C. Beato, M. Varasi and G. Costantino, Eur. J. Med. Chem., 2015, 92, 377 CrossRef PubMed.
- (a) S. Husbaonds, C. A. Sucking and C. J. Sucking, Tetrahedron, 1994, 50, 9729 CrossRef; (b) A. Johansson, A. Poliakov, E. Akerblom, K. Wiklund, G. Lindeberg, S. Winiwarter, U. H. Danielson, B. Samuelsson and A. Hallberg, Bioorg. Med. Chem., 2003, 11, 2551 CrossRef; (c) A. Garcia, E. G. Mata, C. M. Tice, R. E. Horman, E. Nicolas, F. Albericio and L. E. Michelotti, J. Comb. Chem., 2005, 7, 843 CrossRef.
- N. W. Johnson, J. Kasparec, W. H. Miller, M. B. Rouse, D. Suarez and X. Tian, WO2012135113A2, 2012.
- (a) O. O. Sokolova and J. F. Bower, Chem. Rev., 2021, 121, 80 CrossRef PubMed; (b) M.-M. Wang, T. V. T. Nguyen and J. Waser, Chem. Soc. Rev., 2022, 51, 7344 RSC; (c) M. A. Cavitt, L. H. Phun and S. France, Chem. Soc. Rev., 2014, 43, 804 RSC.
- (a) J. Lee, U. Js, S. C. Blackstock and J. K. Cha, J. Am. Chem. Soc., 1997, 119, 10241 CrossRef; (b) S. O. Nilsson Lill, R. R. Naredla, M. E. Zielinski, L. Knoecer and D. A. Klumpp, J. Org. Chem., 2013, 78(17), 8922 CrossRef PubMed; (c) P.-C. Xu, S. Qian, X. Meng, Y. Zheng and S. Huang, Org. Lett., 2024, 26, 2806 CrossRef PubMed.
- (a) S. Maity, M. Zhu, R. S. Shinabery and N. Zhen, Angew. Chem., Int. Ed., 2012, 51, 222 CrossRef; (b) D. H. White, A. Noble, K. I. Booker-Milburn and V. K. Aggarwal, Org. Lett., 2021, 23, 3038 CrossRef PubMed; (c) Y. Dai, S. Liang, G. Zeng, H. Huang, X. Zhao, S. Cao and Z. Jiang, Chem. Sci., 2022, 13, 3787 RSC.
- F. Brackmann and A. de Meijere, Chem. Rev., 2007, 107, 4493 CrossRef.
- (a) J. A. Miller, E. J. Hennessy, W. J. Marshall, M. A. Scialdone and S. T. Nguyen, J. Org. Chem., 2003, 68, 7884 CrossRef PubMed; (b) J. Huang, Y. Geng, Y. Wang and J. Xu, Ind. Eng. Chem. Res., 2019, 58, 16389 CrossRef.
- P. Bertus and J. Szymoniak, Synlett, 2007, 1346 CrossRef.
- (a) V. K. Aggarwal, J. de Vicente Roger and V. Bonnert, Org. Lett., 2001, 3, 2785 CrossRef PubMed; (b) L. A. Adams, V. K. Aggarwal, R. V. Bonnert, B. Bressel, R. J. Cox, J. Shepherd, J. de Vicente, M. Walter, W. G. Whittingham and C. L. Winn, J. Org. Chem., 2003, 68, 9433 CrossRef PubMed.
- (a) Y. Zhu, T. Zhou, H. Zhang, J. He, H. Li, M. Lang, J. Wang and S. Peng, J. Org. Chem., 2022, 87, 1074 CrossRef PubMed; (b) H.-F. Tu, A. Jeandin and M. G. Suero, J. Am. Chem. Soc., 2022, 144, 16737 CrossRef.
- S. Chanthamath, D. T. Nguyen, K. Shibatomi and S. Iwasa, Org. Lett., 2013, 15, 772 CrossRef.
- (a) M. S. West, L. R. Mills, T. R. McDonald, J. B. Lee, D. Ensan and S. A. L. Rousseaux, Org. Lett., 2019, 21, 8409 CrossRef PubMed; (b) M. S. West, J. E. Pia and S. A. L. Rousseaux, Org. Lett., 2022, 24, 5869 CrossRef PubMed.
- (a) Y. Zhang, Y. Li, W. Zhou, M. Zhang, Q. Zhang, R. Jia and J. Zhao, Chem. Commun., 2020, 56, 12250 RSC; (b) R. Sakae, N. Matsuda, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 1228 CrossRef PubMed.
- C. D. Papageorgiou, M. A. Cubillo de Dios, S. V. Ley and M. J. Gaunt, Angew. Chem., Int. Ed., 2004, 43, 4641 CrossRef.
- T. Cantin, A. B. Charette, T. Poisson and P. Jubault, Synthesis, 2023, 2943 Search PubMed.
- D. Kim, H.-J. Jeon, Y. Kwak, S. J. Lee, T.-G. Nam, J. H. Yu, H. An and K. B. Hong, RSC Adv., 2024, 14, 831 RSC.
- (a) M. C. Schroeder and J. S. Kiely, J. Heterocycl. Chem., 1988, 25, 1769 CrossRef; (b) A. Johansson, A. Poliakov, E. Akerblom, K. Wiklund, G. Lindeberg, S. Winiwarter, U. H. Danielson, B. Samuelsson and A. Hallberg, Bioorg. Med. Chem., 2003, 11, 2551 CrossRef PubMed; (c) L. A. T. Allen, R.-C. Raclea, P. Nathoa and P. J. Parsona, Org. Biomol. Chem., 2021, 18, 498 RSC.
- L. Serusi, F. Soddu, F. Cuccu, G. Peretti, A. Luridiana, F. Secci, P. Caboni, D. J. Aitken and A. Frongia, Adv. Synth. Catal., 2020, 365, 4159 CrossRef.
- (a) L. Capaldo, Z. Wen and T. Noël, Chem. Sci., 2023, 14, 4230 RSC; (b) F. M. Akwi and P. Watts, Chem. Commun., 2018, 54, 13894 RSC; (c) D. Moi, M. C. Cabua, V. Velichko, A. Cocco, R. Mocci, S. Porcu, A. Chiappone, M. Piras, S. Bianco, F. Pesciaioli and F. Secci, Molecules, 2022, 27, 7943 CrossRef.
- (a) F. Secci, S. Porcu, A. Luridiana, A. Frongia and P. C. Ricci, Org. Biomol. Chem., 2020, 18, 3684 RSC; (b) V. Velichko, D. Cambie and F. Secci, React. Chem. Eng., 2024, 9, 1721 RSC.
- M. Oelgemöller and N. Hoffmann, Org. Biomol. Chem., 2016, 14, 7392 RSC.
- (a) F. Turnu, A. Luridiana, A. Cocco, S. Porcu, A. Frongia, G. Sarais and F. Secci, Org. Lett., 2019, 21, 7329 CrossRef PubMed; (b) S. Porcu, A. Luridiana, A. Martis, A. Frongia, G. Sarais, D. J. Aitken, T. Boddaert, R. Guillot and F. Secci, Chem. Commun., 2018, 54, 13547 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, and 1H and 13C NMR spectra. CCDC 2373554–2373556, 2387960. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc04089a |
| This journal is © The Royal Society of Chemistry 2025 |