
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cerium(III) yldiide complexes with divergent CO reactivity†
Alexander J. Gremilliona,
Hemant Kumara,
Iker Del Rosalb,
Steven P. Kelley a,
Laurent Maron*b,
Viktoria H. Gessner*c and
Justin R. Walensky*a
a,
Laurent Maron*b,
Viktoria H. Gessner*c and
Justin R. Walensky*a
aDepartment of Chemistry, University of Missouri, Columbia, MO 65211, USA. E-mail: walenskyj@missouri.edu
bUniversité de Toulouse and CNRS, INSA, UPS, CNRS, UMR 5212, Toulouse 31077, France. E-mail: Laurent.maron@irsamc.ups-tlse.fr
cFaculty of Chemistry and Biochemistry, Ruhr-University Bochum, 44801 Bochum, Germany. E-mail: viktoria.gessner@rub.de
First published on 12th December 2024
Abstract
Synthesis of cerium yldiide complexes and their reactivity with CO is demonstrated. In the case of the sulphur-tethered yldiide, the ketenyl complex is formed with release of PPh3, while Ph3PCCO is formed along with a sulfinato ligand in the case of the tosyl-substituted yldiide. Computational analysis shows that this diverging reactivity is due to the stability of the two isomers in the first step of each mechanism.
Metalated ylides, also known as yldiides (Fig. 1), have emerged as an interesting class of reagents and ligands, with applications ranging from the generation of highly efficient phosphines for applications in transition metal catalysis to the stabilization of unusual bonding situations.1–3 In addition, yldiides have demonstrated remarkable reactivity with small molecules, affording rare moieties and reagents.4,5 For example, exchange of the phosphine group in yldiides with CO produces ketenyl anions, which can be further functionalized with other substrates to ketenes as well as other carbonyl-containing compounds. Ketenyl anions have often been proposed as intermediates and although there has been a resurgence in recent years,6–9 they remain a rare functional moiety,10–13 making their structural characterization crucial for gaining further insights into their reactivity patterns.
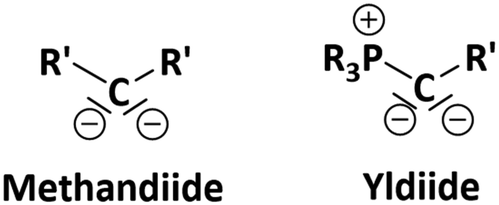 | ||
| Fig. 1 ChemDraw showing methandiide versus yldiide ligands. | ||
The selectivity of the PPh3/CO exchange in yldiides was found to depend on the nature of the alkali metal.14 This dependency was attributed to the different coordination chemistry of the metal cations in the decisive intermediates, but no intermediate structures could be identified. The metal dependency suggests that carbonylation in the coordinating sphere of other metals in the periodic table could offer an additional means for selectivity control and might provide further insights into the underlying mechanism. Given the similarities in bonding between the s-block metals and lanthanides, we turned our attention toward the rare earth metals.
Small molecule activation with organolanthanide complexes has been an active area of interest due to their ability to even transform relatively challenging molecules such as CO.15 While ketene-type ligands are known in f element chemistry, these are rare and have always been stabilized by a carboxylate group forming a ketene carboxylate.16–21 We selected cerium as first test system, as Ce(III) typically forms coloured compounds, often facilitating reaction monitoring. Additionally, due to its high oxophilicity, we speculated that it would result in further interactions with the carbonyl oxygen. Herein, we report the synthesis of two cerium(III) yldiide complexes, [(C5Me5)2Ce{C(PPh3)(R)}], R = P(S)Ph2, SO2(4-CH3C6H4), from the salt metathesis reactions of [(C5Me5)2CeCl2K(THF)] with the corresponding potassium yldiides. These yldiides react with CO to form, in the case of R = SPPh2, a ketenyl anion with concomitant release of PPh3, or a sulfinato complex with release of Bestmann's ylide, Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCO (Scheme 1).
CCO (Scheme 1).
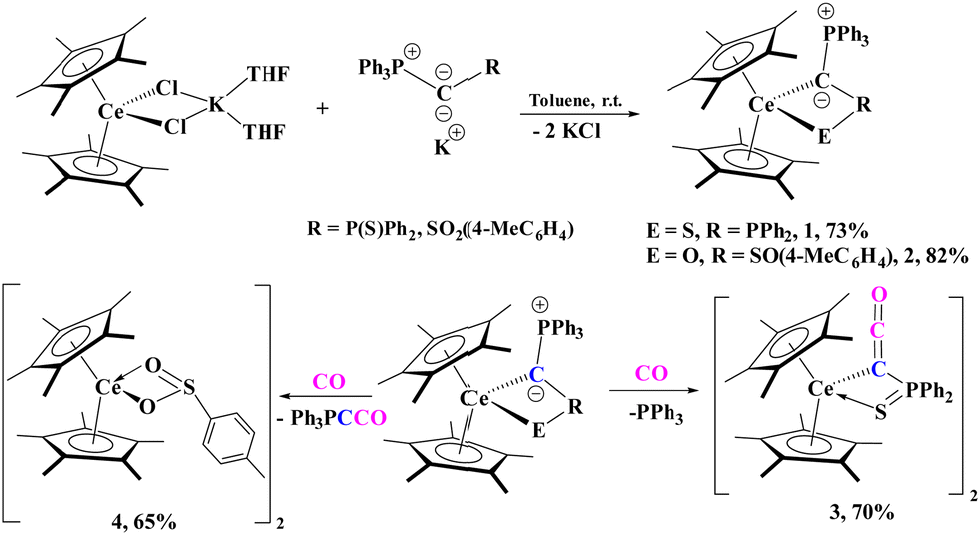 | ||
| Scheme 1 Synthesis of cerium metalated yldiide complexes 1 and 2 (top) and divergent reactivity with CO (bottom). | ||
Treatment of [(C5Me5)2CeCl2K(THF)2] with [K{C(PPh3)(R)}], yields the corresponding cerium(III) yldiides, [(C5Me5)2Ce{C(PPh3)(R)}] (R = P(S)Ph2, 1; SO2(4-CH3C6H4), 2), with a colour change from yellow to bright red/pink observed in both reactions. Both 1 and 2 can be isolated in good crystalline yields of 73% and 82%, respectively.
The solid-state structure of 1 and 2 were determined by X-ray crystallography (Fig. 2), each being monomeric and having pseudo-tetrahedral geometries with two (C5Me5)1− ligands and a bidentate yldiide coordinated through the ylidic carbon and either sulphur (in 1) or oxygen (in 2). Organocerium(III) compounds with Ce–C (σ bonds) are relatively rare,22–32 and the Ce–C (ylidic) bond lengths in 1 and 2 are long at 2.623(2) and 2.597(3) Å, respectively. These distances can be compared to Ce(III)–C (alkyl) complexes such as the 2.50(2)–2.54(2) Å in [Ce(tBu4)][Li(THF)4],33 2.600(2)–2.614(2) Å in Ce(CH2Ph)3(THF)3, 2.577(4) Å or 2.584(4) Å in [(C5tBu3H2)2Ce(CH2Ph)] (two molecules in the asymmetric unit),34 and 2.535(5) Å in [(C5Me5)2Ce{C(H)(SiMe3)2}]. A few Ce(III)–C(aryl) bonds have been reported at distances of 2.5574(17) Å and 2.563(3) Å in [(C5Me4SiMe3)2Ce(κ2-ortho-oxazoline)] and [Li(DME)3][(C5Me5)2Ce(biphenyl)],22 respectively. The Ce–S distance in 1 is 2.8775(6) Å, much longer than cerium(III)–thiolate distances in [Ce(SMes*)3], Mes* = 2,4,6-tBu3C6H2, which range from 2.703(4)–2.744(4) Å,35 but similar to the 2.8567(17)–3.0381(19) Å in [Ce(THF)3(SC6F5)3]2,36 but this is also a dimer in the solid-state so Ce–S distances are expectedly longer.
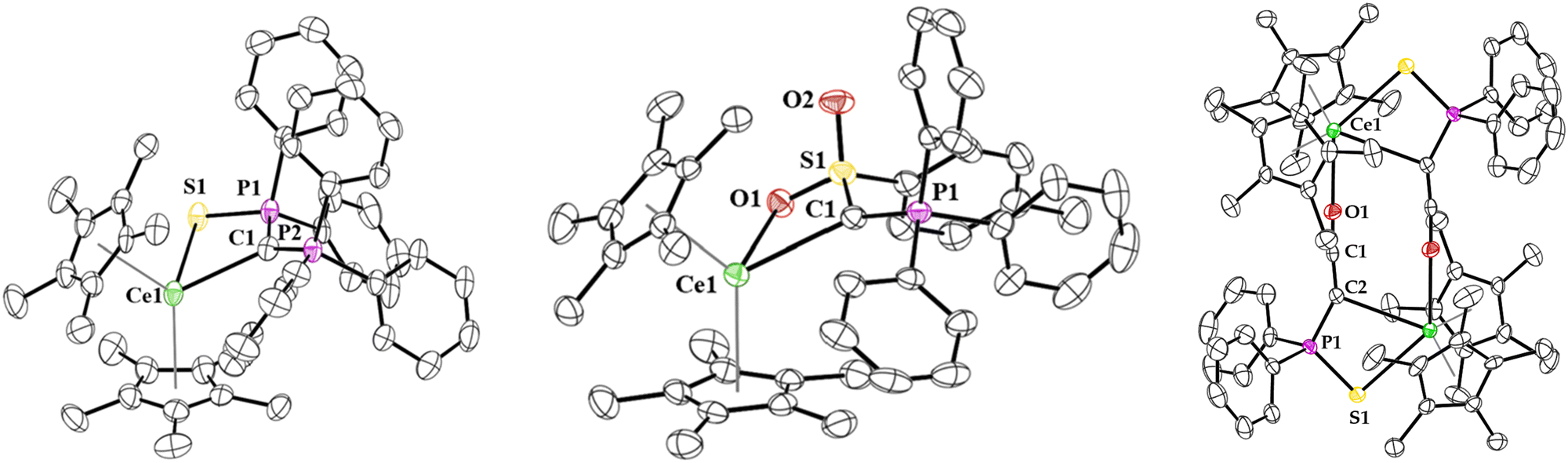 | ||
| Fig. 2 Molecular structure of 1 (left), 2 (middle), and 3 (right) shown at the 50% probability level. The hydrogen atoms have been omitted for clarity. | ||
The 1H NMR and UV-vis spectra of 1 and 2 show similar features as expected given the similar coordination environment. In 1, two 31P NMR resonances are observed at −25.4 and −96.6 ppm, while the 31P NMR signal for 2 is seen at 2.86 ppm. Unfortunately, similar to other Ce(III) complexes,30 we also could not observe the ylidic carbon in the 13C NMR spectrum.
With 1 and 2 in hand, treatment with 1 atm CO was conducted with each. Both reactions require approx. 12 hours for completion at room temperature. In the case of 1, the growth of a signal corresponding to PPh3 (−5.31 ppm, Fig. S18, ESI†) in the 31P NMR spectrum as well as another resonance at 21 ppm for the Ce complex was observed. The solid-state structure of 1 with CO revealed a ketenyl product, [(C5Me5)2Ce{C(CO)(P(S)Ph2)}]2 (3) which forms a dimer in the solid-state (Fig. 2) in 70% crystalline yield. Further, the infrared spectrum of 3 showed a characteristic CCO stretching vibration at 2088 cm−1.37 The Ce-C(ketenyl) distance in 3 is 2.851(6) Å, which is longer than the sum of the covalent radii of 2.77 Å, presumably due to the dimeric structure in the solid-state. Therefore, these Ce-C bonds in 1, 2, and 3 are some of the longest reported. The Ce–S1 distance of 2.988(6) Å and Ce–O1 distance of 2.531(4) Å are in the expected range for a dimer. The C1–C2 distance of 1.241(6) Å is longer than the one observed in the potassium ketenyl (1.178(8) Å), while the C1–O1 distance of 1.206(5) Å is slightly shorter (cf. 1.248(8) Å).5
In contrast to 1, the carbonylation of 2 does not result in the formation of PPh3 but in a compound characterized by a signal at 2.86 ppm in the 31P NMR spectrum. X-ray crystallography of crystals obtained from the reaction mixture revealed the formation of complex [(C5Me5)2Ce(μ:κ1-O2S-4-MeC6H4)]2 (4) with a bridging sulfinato ligand (Fig. S24, ESI†). This is consistent with the 31P NMR chemical shift at 2.86 ppm which is assigned to Bestmann's ylide, Ph3PCCO.38 Similar reactivity has been observed with the lithium yldiide producing mostly Ph3PCCO, while the Na and K form yielded the ketenyl anion as the major product.8 In nearly all carbonylation reactions of the alkali metal yldiides, both the phosphaketenyl, Ph3PCCO, and ketenyl moieties were observed, while we only observed formation of 4 in the case of Ce, which we could isolate in a 65% yield. We note that the UV-vis spectra for 4 does not display a band around 540 nm which is observed in both 1 and 2, which suggests that this absorption is due to the yldiide ligand.
To gain insights into the reaction of complexes 1 and 2 with CO density functional theory (DFT) calculations (B3PW91) calculations were carried out. For clarity, structures of all computed intermediates and transition states along with the charges and WBI are available in the ESI† (Fig. S25 and S26). In the case of the complex 2 (Fig. 3, left), the formation of a ketenyl complex (not experimentally observed) was also considered for comparison. For the formation of both complexes, the reaction of 2 begins in the same way that is the CO insertion into the Ce–C bond. This readily occurs with a low activation barrier (4.6 kcal mol−1) in line with a facile [2σ+2π] metathesis reaction. Following the intrinsic reaction coordinate, it yields a thermodynamically stable ketenyl type intermediate (−11.9 kcal mol−1). This intermediate possesses two isomers, shown in the middle of Fig. 3. In these isomers, the ketenyl moiety binds to the metal through the C1 carbon atom (Int1) or through both the C1 carbon and oxygen atom (Int1′). Although kinetically facile (barrier of 7.5 kcal mol−1), the O-bonded isomer Int1′ is less stable than the non-bonded one Int1 by 5.3 kcal mol−1. Interestingly, the two competing reactions start from the most stable intermediate Int1 (blue and black in Fig. 3, left). As can be seen, the two pathways (S–C versus P–C bond breaking) are in kinetic competition (barrier difference of 0.7 kcal mol−1 favouring the formation of the sulfinato complex 4). However, there is a clear thermodynamic preference for the formation of 4, which confirms its exclusive formation in the experiment. The similar barriers for the two reactions are explained by the Wiberg bond indexes (WBI) in the Int1 that are similar for the P–C and S–C bonds (0.98–0.99).
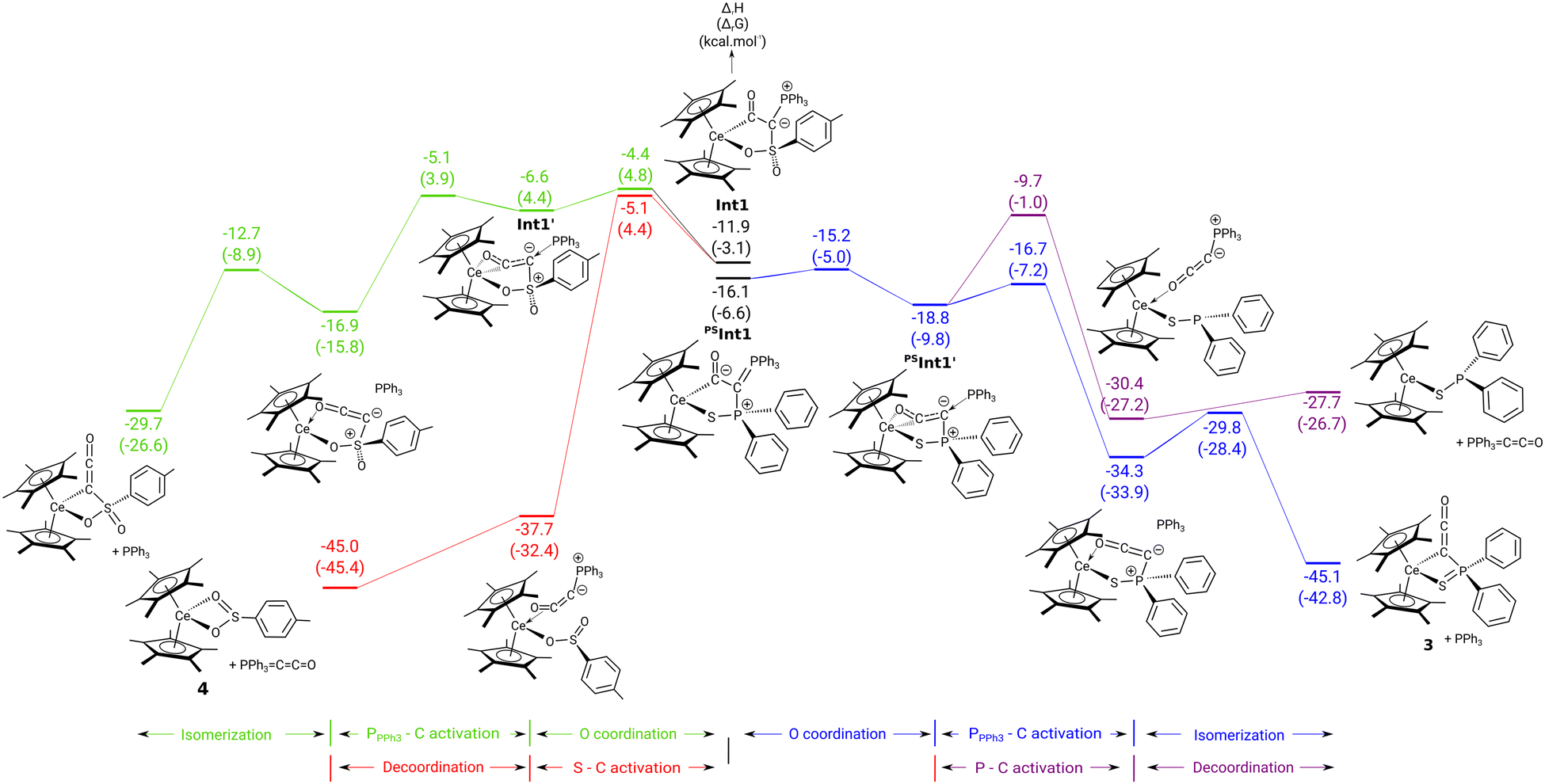 | ||
| Fig. 3 Computed enthalpy (kcal mol−1) profile for the transformation of complex 1 to 3 (right) and 2 to 4 (left) upon CO addition. | ||
The situation is different for the reaction of 1 with CO (Fig. 3, right). In this case, the CO-bonded isomer of the ketenyl intermediate (PSInt1′) is energetically favoured (−18.8 vs. −16.1 kcal mol−1) with a low activation barrier of isomerization (0.9 kcal mol−1). As observed for 2, the two competing reactions (P–C vs. S–C activation) start from the most stable isomer PSInt′. In case of the thiophosphinoyl system, the P–C bond breaking pathway (black lines in Fig. 3) is predicted to be both kinetically and thermodynamically favoured over the S–C bond cleavage (blue lines on Fig. 3, right). This can be attributed to the already strong P–C bond activation in the O-bonded intermediate PSInt1′ reflected by a low WBI(P–C) of only 0.59 similar to the one found in the transition state. P–C cleavage finally yields the ketenyl complex 3.
The difference of reactivity between 1 and 2 is therefore attributed to the difference of stability of the two isomers after CO insertion. In the case of 1, the presence of the weak sulphur donor atom makes the O-bonded isomer the most stable, while the stronger oxygen donor in 2 inverts the situation, making the C-bonded isomer is more stable.
In summary, we have isolated Ce(III) complexes with two different yldiide ligands, each exhibiting long cerium–carbon bonds. These distances are reminiscent of sterically crowded complexes with f elements, indicating no double bond character. Both complexes display divergent reactivity when treated with CO, one forming a rare ketenyl complex along with PPh3, while the other one eliminates the ylide moiety, forming Ph3PCCO and a cerium sulfinato complex. Calculations reveal that this diverging reactivity is due two competing coordination modes in the intermediate ketene complex, which depend on the binding strength of the second donor (sulfonyl or thiophosphinoyl). The stronger coordination of the sulfonyl group prevents the binding via the oxygen of the ketene moiety and thus results in the elimination of Ph3PCCO. These findings explain why both the alkali metal yldiides previously reported and the cerium complexes presented here show divergent reactivity with CO. This adds to the limited organocerium(III) chemistry, which we are continuing to investigate with these unusual metalated ylide ligands.
This project was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC-2033-390677874-RESOLV. J. R. W. gratefully acknowledge the Alexander von Humboldt Foundation for the award of a research fellowship and the U.S. Department of Energy, Office of Basic Energy Sciences, Heavy Element Program under Award DE-SC0021273. L. M. is a senior member of the Institut Universitaire de France and acknowledge the HPCs CALcul en Midi-Pyrénées (CALMIP-EOS-grant 1415).
Data availability
The dataset supporting this article have been uploaded as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- L. T. Scharf and V. H. Gessner, Inorg. Chem., 2017, 56, 8599 CrossRef CAS PubMed.
- L. T. Scharf, D. M. Andrada, G. Frenking and V. H. Gessner, Chem. – Eur. J., 2017, 23, 4422 CrossRef CAS PubMed.
- M. Jorges, A. Gremillion, D. Knyszek, S. P. Kelley, J. R. Walensky and V. H. Gessner, Chem. Commun., 2024, 23, 3190 RSC.
- M. Jörges, F. Krischer and V. H. Gessner, Science, 2022, 378, 1331 CrossRef PubMed.
- F. Krischer and V. H. Gessner, JACS Au, 2024, 4, 1709 CrossRef CAS PubMed.
- S. A. Gonsales, I. Ghiviriga, K. A. Abboud and A. S. Veige, Dalton Trans., 2016, 45, 15783–15785 RSC.
- J. A. Buss, G. A. Bailey, J. Oppenheim, D. G. VanderVelde, W. A. I. Goddard and T. Agapie, J. Am. Chem. Soc., 2019, 141, 15664–15674 CrossRef CAS PubMed.
- R. Wei, X.-F. Wang, D. A. Ruiz and L. L. Liu, Angew. Chem., Int. Ed., 2023, 62, 41 Search PubMed.
- A. Lachguar, I. Del Rosal, L. Maron, E. Jeanneau, L. Veyre, C. Thieuleux and C. Camp, J. Am. Chem. Soc., 2024, 146, 18306–18319 CrossRef CAS PubMed.
- R. P. Woodbury, N. R. Long and M. W. Rathke, J. Org. Chem., 1978, 43, 376 CrossRef CAS.
- G. Erker and R. Hock, Angew. Chem., Int. Ed. Engl., 1989, 28, 179 CrossRef.
- H. Kai, K. Iwamoto, N. Chatani and S. Murai, J. Am. Chem. Soc., 1996, 118, 7634 CrossRef CAS.
- A. M. Turner, et al., Astrophys. J., 2020, 896, 88 CrossRef CAS.
- F. Krischer, M. Jörges, T.-F. Leung, H. Darmandeh and V. H. Gessner, Angew. Chem., Int. Ed., 2023, 62, 41 CrossRef PubMed.
- N. Mahieu, J. Piątkowski, T. Simler and G. Nocton, Chem. Sci., 2023, 14, 443–457 RSC.
- W. J. Evans, J. W. Grate, L. A. Hughes, H. Zhang and J. L. Atwood, J. Am. Chem. Soc., 1985, 107, 3728 CrossRef CAS.
- W. J. Evans, D. S. Lee, J. W. Ziller and N. Kaltsoyannis, J. Am. Chem. Soc., 2006, 128, 14176 CrossRef CAS PubMed.
- M. Fang, et al., J. Am. Chem. Soc., 2012, 134, 6064 CrossRef CAS PubMed.
- A. J. Ryan, J. W. Ziller and W. J. Evans, Chem. Sci., 2020, 11, 2006 RSC.
- T. Simler, K. N. McCabe, L. Maron and G. Nocton, Chem. Sci., 2022, 13, 7449 RSC.
- R. J. Ward, I. del Rosal, S. P. Kelley, L. Maron and J. R. Walensky, Chem. Sci., 2023, 14, 2024 RSC.
- H. J. Heeres, J. Renkema, M. Booij, A. Meetsma and J. H. Teuben, Organometallics, 1988, 7, 2495 CrossRef CAS.
- M. Zimmermann and R. Anwander, Chem. Rev., 1988, 110, 6194 CrossRef PubMed.
- T. Berger, J. Lebon, C. Maichle-Mössmer and R. Anwander, Angew. Chem., Int. Ed., 2021, 60, 15622 CrossRef CAS PubMed.
- A. J. Wooles, D. P. Mills, W. Lewis, A. J. Blake and S. T. Liddle, Dalton Trans., 2009, 500 Search PubMed.
- W. J. Evans, J. M. Perotti and J. W. Ziller, J. Am. Chem. Soc., 2005, 127, 3894 CrossRef CAS PubMed.
- E. L. Werkema and R. A. Andersen, J. Am. Chem. Soc., 2008, 130, 7153 CrossRef CAS PubMed.
- G. B. Panetti, et al., Nat. Commun., 2021, 12, 1713 CrossRef CAS PubMed.
- D. Gu, C. Yi and W. Ren, Inorg. Chem., 2019, 58, 9260 CrossRef CAS PubMed.
- P. L. Arnold, et al., Chem. Sci., 2018, 9, 8035 RSC.
- I. J. Casely, S. T. Liddle, A. J. Blake, C. Wilson and P. L. Arnold, Chem. Commun., 2007, 5037 RSC.
- P. Pandey, et al., Organometallics, 2023, 42, 1267 CrossRef CAS.
- E. L. Werkema, R. A. Andersen, L. Maron and O. Eisenstein, Dalton Trans., 2010, 39, 6648 RSC.
- M. Roger, et al., J. Am. Chem. Soc., 2006, 128, 8790 CrossRef CAS PubMed.
- J. H. Melman, C. Rohde, T. J. Emge and J. G. Brennan, Inorg. Chem., 2002, 41, 28 CrossRef CAS PubMed.
- A. Brar, D. K. Unruh, A. J. Aquino and C. Krempner, Chem. Commun., 2019, 55, 3513–3516 RSC.
- J. Frey and Z. Rappoport, J. Am. Chem. Soc., 1996, 118, 5169–5181 CrossRef CAS.
- R. Schobert, Org. Synth., 2005, 82, 140 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2362374–2362377 and 2394687. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc04562a |
| This journal is © The Royal Society of Chemistry 2025 |