
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTerpolymerization reactions of epoxides, CO2, and the third monomers toward sustainable CO2-based polymers with controllable chemical and physical properties
Koichi
Nakaoka
and
Tadashi
Ema
 *
*
Division of Applied Chemistry, Graduate School of Natural Science and Technology, Okayama University, Tsushima, Okayama 700-8530, Japan. E-mail: ema@cc.okayama-u.ac.jp
First published on 27th November 2024
Abstract
Carbon dioxide (CO2) serves as a cheap, abundant, and renewable C1 building block for the synthesis of organic compounds and polymers. Selective and efficient CO2 fixation processes are still challenging because of the kinetic and thermodynamic stability of CO2. Among various CO2 fixation processes, the ring-opening copolymerization (ROCOP) of epoxides and CO2 gives aliphatic polycarbonates with high atom economy, although the chemical and physical properties of the resulting polycarbonates are not necessarily satisfactory. Introducing the third monomers into this ROCOP system provides new terpolymers, and the thermal, optical, mechanical or degradation properties can be added or tuned by incorporating new polymer backbones derived from the third monomers at the expense of the CO2 content. Here we review the terpolymerization reactions of epoxides, CO2, and the third monomers such as cyclic anhydrides, lactones, lactides, heteroallenes, and olefins. The development of catalysts and the control of the polymer structures are described together with the chemical and physical properties of the resulting polymers.
 Koichi Nakaoka | Koichi Nakaoka was born in Osaka, Japan in 1988. He received his bachelor's degree in 2011 and master's degree in 2013 from Osaka University under the supervision of Professor Akio Baba. He joined Asahi KASEI in 2013 and was promoted to a team leader in 2020. He obtained his doctor's degree from Okayama University under the supervision of Professor Tadashi Ema in 2024. He moved to PPG Japan in 2024. His research interest is in the design and synthesis of sustainable materials based on green chemistry. |
 Tadashi Ema | Tadashi Ema was born in Gifu, Japan in 1966. He received his bachelor's degree from Kyoto University in 1989, and he obtained his master's degree in 1991 and doctor's degree in 1994 from Kyoto University under the supervision of Professor Hisanobu Ogoshi. He was appointed as an Assistant Professor at Okayama University in 1994 and was promoted to an Associate Professor in 2002 and a Full Professor in 2012. His research interest is in the design and synthesis of unique molecular catalysts for sustainable organic synthesis. |
1. Introduction
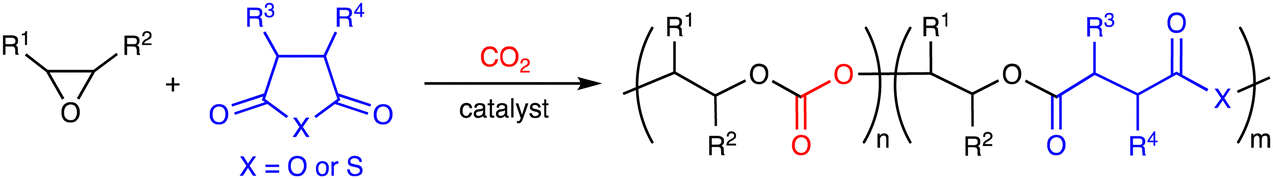
Carbon dioxide (CO2) is a cheap, abundant, and renewable C1 building block, while it is a greenhouse gas causing severe damage to the global environment. The effective utilization of CO2 as well as the mitigation of CO2 emission is required to realize sustainable carbon-neutral societies based on resource circulation.1 On the other hand, the accumulation of plastics in the environment and living organisms is also a big problem to be solved. More environmentally benign plastics should be developed and used. The creation of CO2-based polymers will contribute to finding the solution to these global issues. Because CO2 is the most oxidized form of carbon with high stability, the effective and efficient conversion of CO2 into useful compounds and polymers is still challenging. The thermodynamic stability of CO2 can be overcome by using high-energy chemicals, heat, or electric current, while the kinetic stability of CO2 can be overcome by lowering activation barriers using catalysts.Ring-opening copolymerization (ROCOP) of epoxides and CO2 can produce aliphatic polycarbonates in a highly atom-economical manner (Scheme 1).2,3 Because the ROCOP of epoxides and CO2 occasionally competes with the ring-opening polymerization (ROP) of epoxides to give polyethers (Scheme 1), selective catalysts for the ROCOP of epoxides and CO2 are needed, and much progress has been made. On the other hand, the chemical or physical properties of the resultant aliphatic CO2-based polycarbonates are rather limited. When the polycarbonates have unsatisfactory properties, there is an option of employing the third monomer at the expense of the CO2 content.4 The terpolymerization strategy may be quite effective and even essential for the creation of more useful and practical polymers. The terpolymerization reactions of epoxides, CO2, and the third monomers, such as cyclic anhydrides,5 lactones,6 lactides,7 heteroallenes,8,9 and olefins,10 are effective strategies for the development of new CO2-based polymers (Scheme 2). The thermal, optical, mechanical, adhesive, or degradation properties can be added or tuned by not only incorporating different polymer backbones derived from the third monomers but also controlling the sequence structures such as block, gradient, and random structures.11 Controlling these factors is important for the modulation of heat resistance, mechanical strength, elasticity, or biodegradability, leading to the creation of functional materials such as heat-resistant resins, tough plastics, elastomers, adhesives, and environmentally benign plastics. Two catalytic cycles, which may be switchable, need to proceed, and the development of the selective and controllable catalysts for terpolymerization reactions is challenging and important for the further advancement of CO2-based polymers.
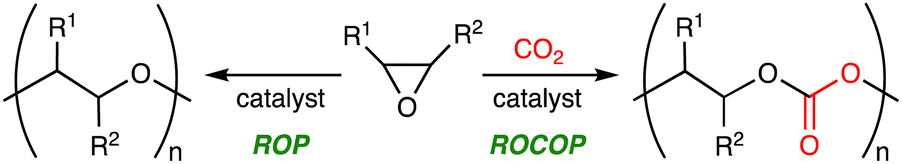 | ||
| Scheme 1 ROCOP of epoxides and CO2 and ROP of epoxides. | ||
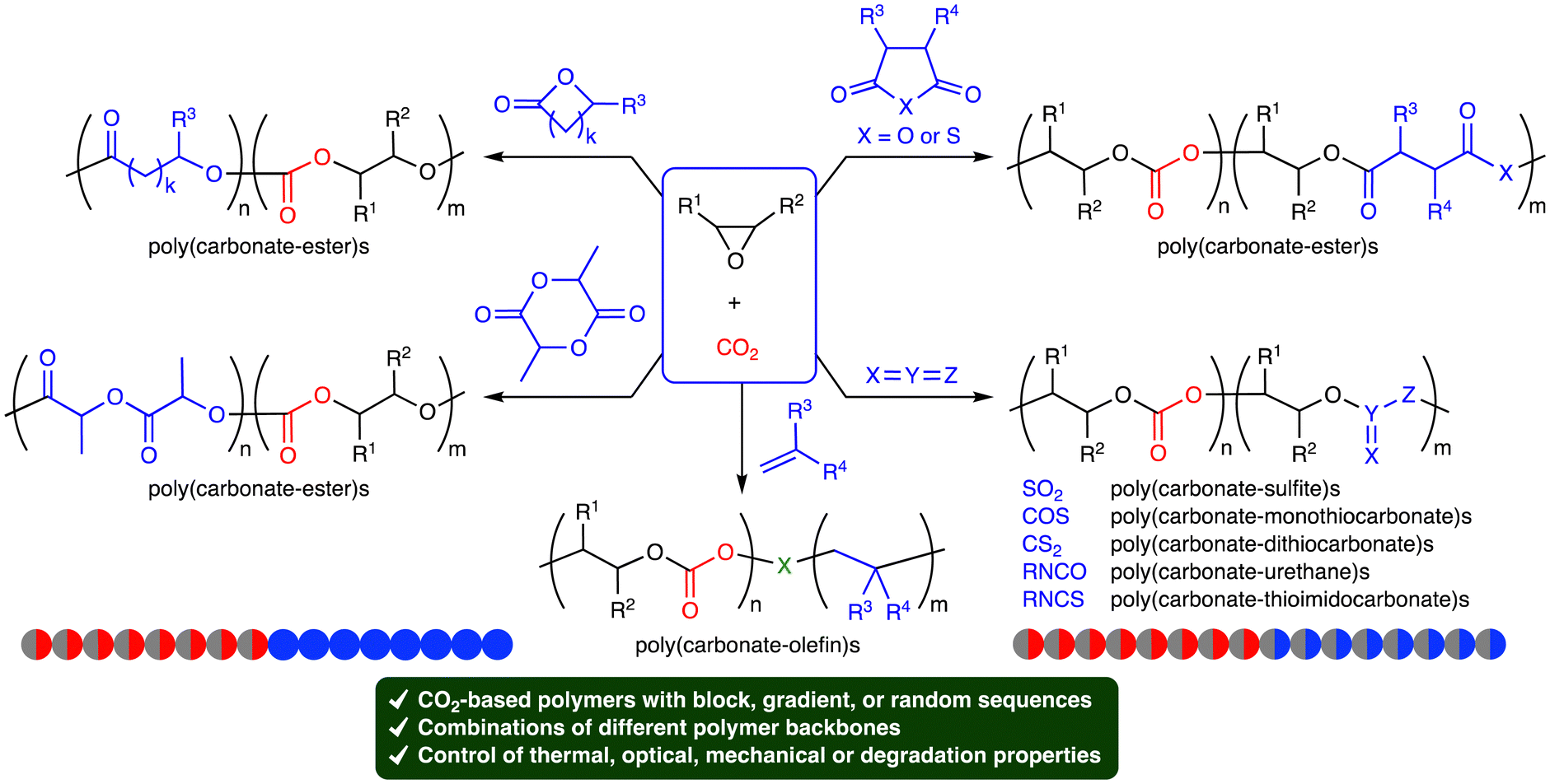 | ||
| Scheme 2 Terpolymerization reactions of epoxides, CO2, and the third monomers. | ||
Here we provide an overview of the terpolymerization reactions of epoxides, CO2, and the third monomers for the synthesis of CO2-based polymers made up of different polymer backbones, which are suitable for the improvement or control of chemical and physical properties. The terpolymerization reactions of two different epoxides and CO2 or epoxides, CO2, and cyclic carbonates are not included herein, although the resulting polycarbonates often have excellent properties.12,13 Polymerization reactions with the stepwise feeding of different monomers such as epoxides and lactones are also excluded, whereas terpolymerization reactions with gas switching, for example, between CO2 and N2 are introduced herein. The reactions, catalysts, and chemical or physical properties of the resulting CO2-based polymers are concisely summarized one by one. Herein, one will find the beneficial effects, potential, and challenging aspects of the catalytic terpolymerization strategy to create new CO2-based polymers with excellent chemical and physical properties.
2. Terpolymerization reactions of epoxides, CO2, and cyclic anhydrides
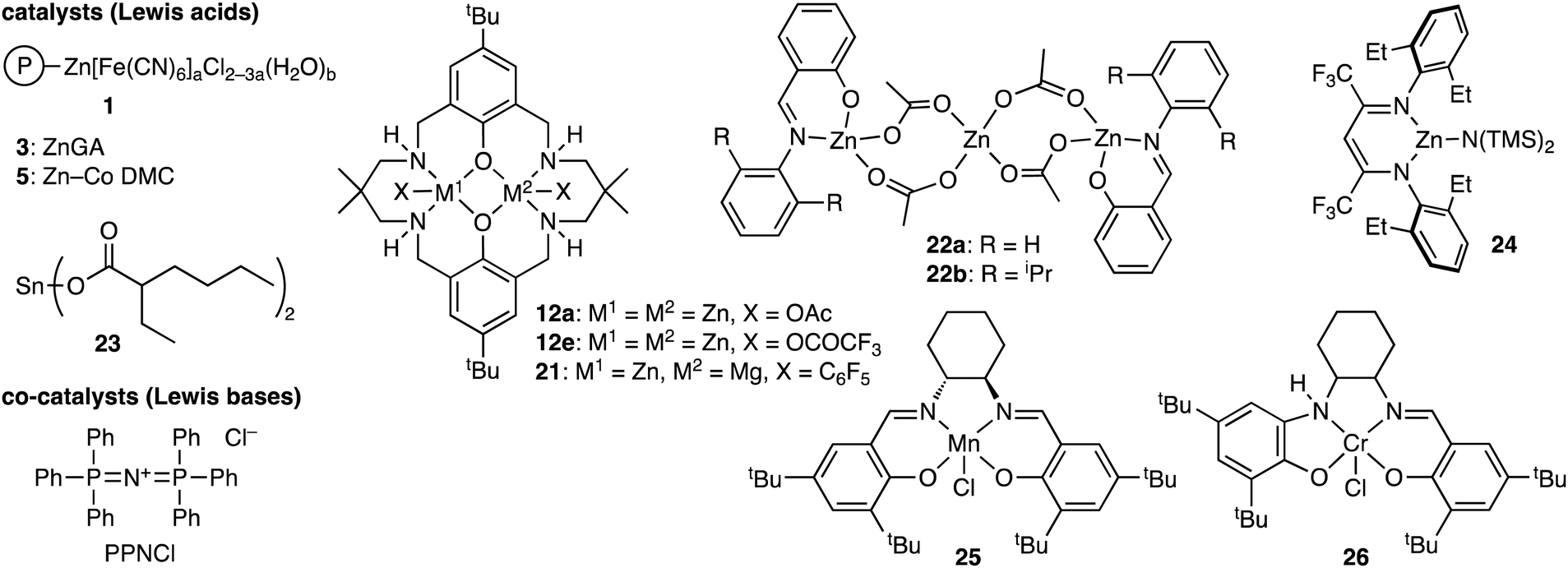
The terpolymerization reactions of epoxides, CO2, and cyclic anhydrides give poly(carbonate-ester)s (Scheme 3a). Because the carbonate and ester linkages show different biodegradabilities, it is possible to adjust the degradability of the poly(carbonate-ester)s by regulating the ratio of the polyester linkage to the polycarbonate linkage. In addition, the chemical and physical properties of the terpolymers can be altered by changing the structures and proportions of the monomers. The representative catalytic cycles, catalysts, and reaction conditions for the terpolymerization reactions of epoxides, CO2, and cyclic anhydrides are shown in Scheme 3b, Fig. 1 and Table 1, respectively.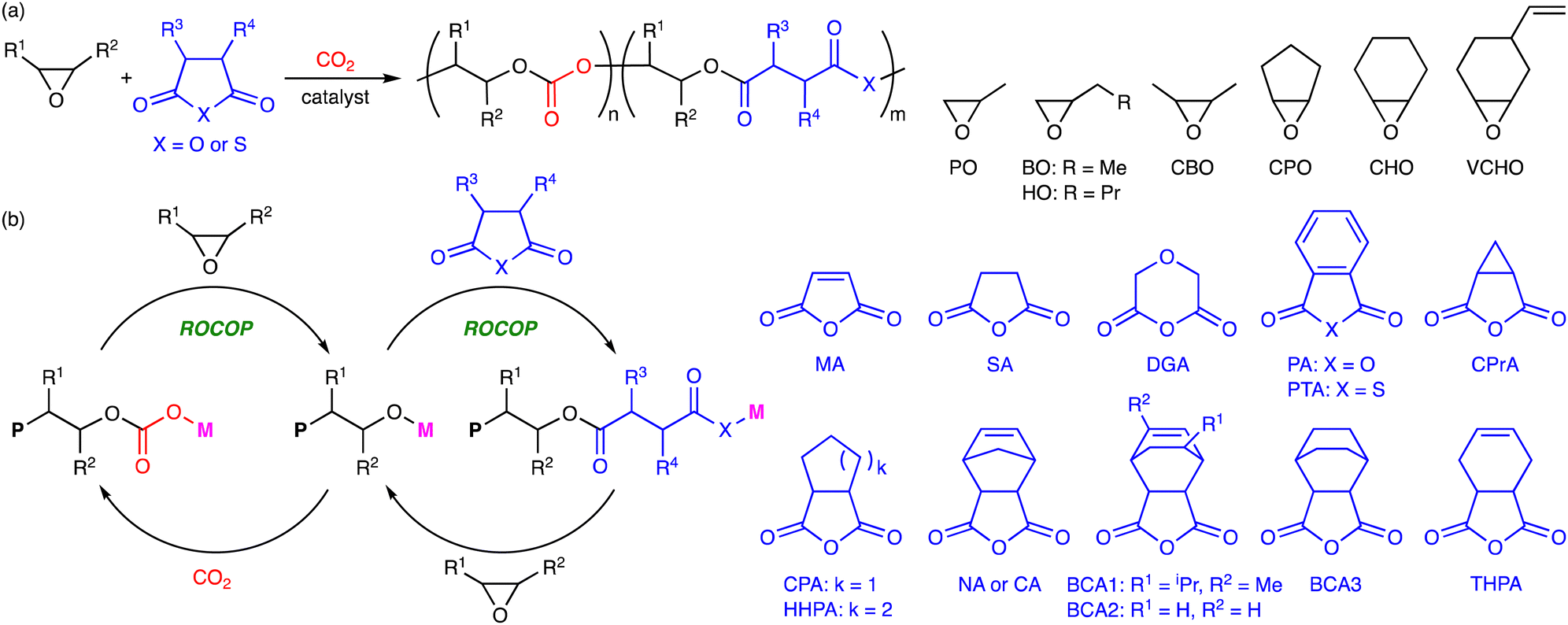 | ||
| Scheme 3 (a) Representative reactions and (b) catalytic cycles. P simply represents a part of a polymer chain that may differ in each step, while M represents a Lewis acid catalyst. | ||
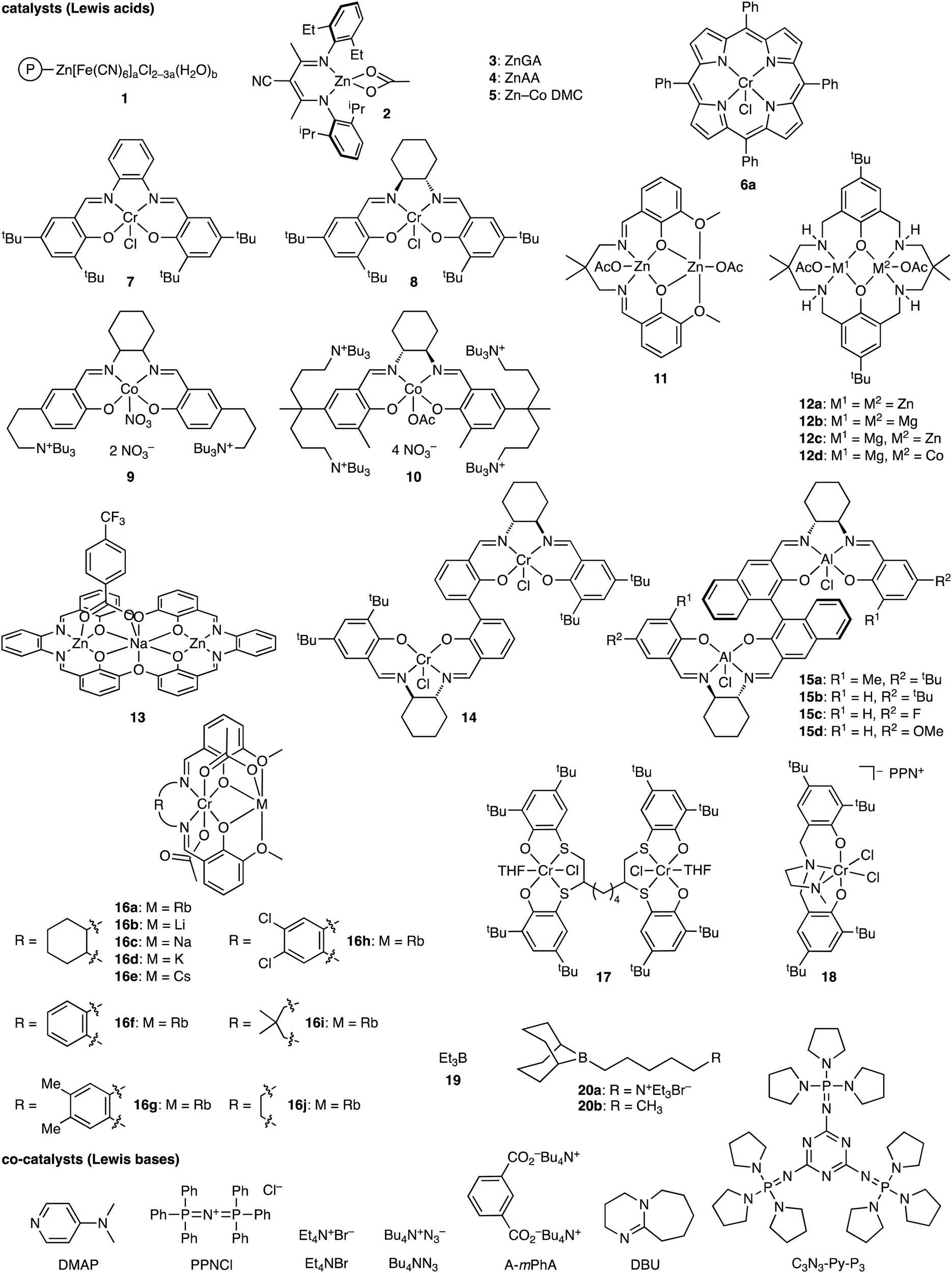 | ||
| Fig. 1 Catalysts and co-catalysts for the terpolymerization reactions of epoxides, CO2, and cyclic anhydrides. | ||
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Epoxide | Anhydride | CO2 (MPa) | Cat. | Co-cat. | T (°C) | M n (kg mol−1) | Sequenceb | Ref. |
| a Maximum value. b Sequence structures of polymers. R: random, G: gradient, B: block. | |||||||||
| 1 | PO | MA | 3.5–4 | 1 | — | 50–80 | 78.1 | R | 5a |
| 2 | CHO, VCHO | DGA, SA | 0.34–5.5 | 2 | — | 50–55 | 37.0 | B | 5b |
| 3 | PO | MA | 5.2 | 3 | — | 60 | 67.1 | R | 5c |
| 4 | PO | PA | 0.5–5.0 | 3 | — | 75 | 221 | R | 5h |
| 5 | PO | MA | 2.5–5.0 | 4 | — | 75 | 152 | G | 5k |
| 6 | CHO | MA | 1.0–4.0 | 5 | — | 75–90 | 14.1 | R, B | 5d |
| 7 | CHO | SA, CPrA, CPA, PA | 5.0 | 6a, 7 | DMAP | 80 | 16 | B | 5e |
| 8 | PO | SA | 4.0–5.0 | 6a | PPNCl | 25 | — | B | 5g |
| 9 | CHO | PA | 3.4 | 8 | PPNCl | 80 | — | B | 5f |
| 10 | PO | NA | 3.0 | 9 | — | 60 | 94.4 | B | 5i |
| 11 | PO | PA | 3.5 | 10 | — | 80 | 381 | G, B | 5j |
| 12 | CHO | PA | 3.0 | 11 | — | 100 | 7.1 | B | 5l |
| 13 | CHO | PA | 0.10 | 12a | — | 100 | — | B | 5m |
| 14 | CHO | BCA1–3, CA, THPA | 0.10 | 12a–b | — | 100 | 11.8 | B | 5n |
| 15 | CHO | PA, BCA1 | 0.10–2.0 | 12 | — | 100 | 19.4 | R, B | 5q |
| 16 | CHO | PA | 0.10 | 13 | — | 90–100 | 4.06 | B | 5r |
| 17 | CHO | PA | 1.0 | 14 | PPNCl | 80 | 10.2 | B | 5o |
| 18 | CBO, CPO, CHO | PA | 0.5–2.0 | 15 | PPNCl | 0–60 | 30.0 | R | 5w |
| 19 | CHO | PTA | 0.4 | 16 | — | 100 | 16.9 | R | 5y |
| 20 | PO, CHO, VCHO, HO | PA | 2.0 | 17 | PPNCl | 45–80 | 10.3 | B | 5z |
| 21 | CHO | PA | 4.0 | 18 | — | 100, 60 | 17.6 | B | 5aa |
| 22 | PO | PA | 0.5–3.0 | 19 | PPNCl | 60–100 | 58.3 | R, B | 5s |
| 23 | EO | PA | 1.0 | 19 | A-mPhA | 40–45 | 273 | G | 5v |
| 24 | CHO, PO, BO | PA | 1.0 | 19 | PPNCl | 80 | 29.8 | B | 5t |
| 25 | CHO | PA | 1.0 | 19 | C3N3-Py-P3 | 80 | 15.5 | R, G, B | 5x |
| 26 | CHO, PO | SA, PA | 1.0 | 19 | Bu4NN3, PPNCl | 60–80 | 22.7 | R, G, B | 5u |
| 27 | PO, CHO | PA, SA, HHPA | 2.0 | 19 | DBU | 60 | 12.0 | B | 5ab |
| 28 | CHO | PA | 0.15–4.0 | 20a | — | 60–120 | 62.3 | G, B | 5ac |
In 2006, Huang and co-workers reported the terpolymerization of propylene oxide (PO), CO2, and maleic anhydride (MA) by using polymer-supported bimetallic (PBM) complex 1 as a catalyst.5a The viscosity, glass transition temperature (Tg), and decomposition temperature of the terpolymers were much higher than those of poly(propylene carbonate) (PPC). The terpolymers also showed a higher degradability than PPC, and their degradation rates increased with increasing content of MA in the terpolymers.
Coates and co-workers developed an excellent homogeneous catalytic system using β-diiminate (BDI) ZnII complex 2, which enabled the first block terpolymerization of cyclohexene oxide (CHO) or 4-vinyl-1-cyclohexene-1,2-epoxide (VCHO), CO2, and diglycolic anhydride (DGA) or succinic anhydride (SA) in a one-step procedure.5b Diblock terpolymers with very little tapering were obtained. This discovery led to the development of several other selective catalysts.
Meng and co-workers found that the ZnII glutarate catalyst (ZnGA) 3 prepared from zinc oxide and glutaric acid acted as a good catalyst for the terpolymerization of PO, CO2, and MA.5c The Tg values of the terpolymers increased with increasing molecular weights. The decomposition temperatures of the terpolymers were enhanced in proportion to the MA content because the double bonds derived from MA readily underwent crosslinking at high temperature. Tensile tests also indicated that the mechanical properties of the terpolymers increased with increasing molecular weight. The same group also reported the terpolymerization of PO, CO2, and phthalic anhydride (PA) using ZnGA 3.5h In this system, CO2 was much more reactive than PA. The incorporation of a small amount of PA enhanced the thermal properties of the terpolymers. They also reported the terpolymerization of PO, CO2, and MA using ZnII adipate (ZnAA) 4, and the sulfonation of the resulting terpolymers afforded new biodegradable surfactants.5k The terpolymer with comparable hydrophilic and hydrophobic segments exhibited the best surface activity.
Zhang and co-workers achieved the terpolymerization of CHO, CO2, and MA using heterogeneous Zn–Co double metal cyanide (DMC) 5 to obtain poly(ester-carbonate) with a low content of ether units.5d THF used as a solvent dramatically inhibited the polyether formation owing to the coordination of THF to the ZnII center. Moreover, the polycarbonate chain containing a small amount of the polyester linkage showed a slightly higher decomposition temperature than the fully alternating polycarbonate.
Duchateau and co-workers achieved the terpolymerization of CHO, CO2, and cyclic anhydride using Cr(TPP)Cl 6a or Cr(salophen)Cl 7 as a catalyst and 4-(N,N-dimethylamino)pyridine (DMAP) as a co-catalyst.5e SA, cyclopropane-1,2-dicarboxylic acid anhydride (CPrA), cyclopentane-1,2-dicarboxylic acid anhydride (CPA) or PA was used as an anhydride monomer. Both 6a and 7 in combination with DMAP afforded poly(ester-carbonate)s with completely alternating structures because the presence of CO2 suppressed the formation of the polyether linkage associated with the homopolymerization of CHO. Chisholm and co-workers reported the terpolymerization of PO, CO2, and SA using 6a and bis(triphenylphosphine)iminium chloride (PPNCl) as a catalyst and a co-catalyst, respectively, to give poly(ester-b-carbonate).5g The preferential reactivity of SA over CO2 was ascribed to the higher concentration of SA relative to CO2 dissolved in the reaction mixture.
Darensbourg and co-workers studied the kinetics of the terpolymerization of CHO, CO2, and PA using Cr(salen)Cl 8 and PPNCl as a catalyst and a co-catalyst, respectively, to afford diblock copolymers with very little tapering.5f The polyester formation was much faster than the polycarbonate formation. In the latter case, the epoxide ring opening was much slower, and CO2 insertion into the metal–alkoxide bond was highly reversible.
Liu and co-workers performed the terpolymerization of PO, CO2, and norbornene anhydride (NA) using bifunctional Co(salen)NO3 tethering two quaternary ammonium salts 9 as a catalyst.5i The reaction afforded diblock copolymers containing reactive norbornene moieties, which could be applied to the thiol–ene click reaction. Lee and co-workers reported that bifunctional Co(salen)OAc tethering four quaternary ammonium salts 10 showed a high catalytic activity of 2.2 kg-polymer per g-catalyst for the terpolymerization of PO, CO2, and PA.5j The Tg value of the terpolymer was higher than that of the PO/CO2 alternating copolymer, PPC, and lower than that of the PO/PA alternating copolymer.
Williams and co-workers used dinuclear ZnII complex 11 for the terpolymerization of CHO, CO2, and PA. The polyester was initially produced selectively, and poly(cyclohexene carbonate) (PCHC) was then formed, delivering poly(ester-b-carbonate)s in a one-step manner.5l They also studied the block-selective copolymerization from a mixture of CHO and PA under 1 atm CO2 using dinuclear ZnII complex 12a to obtain poly(ester-b-carbonate)s.5m CHO/PA copolymerization proceeded first until the complete consumption of PA, and CHO/CO2 copolymerization then occurred. DFT calculations indicated that PA insertion into CHO was more favorable than CO2 insertion into CHO. The same group also used sterically congested tricyclic anhydrides (BCA1–3), carbic anhydride (CA), and 1,2,3,6-tetrahydrophthalic anhydride (THPA) as the third monomers in the terpolymerization, and both catalysts 12a and 12b showed excellent catalytic performances to synthesize block copolymers from bio-derived anhydride BCA1, CHO, and CO2.5n Interestingly, ZnII-based catalyst 12a initially catalyzed CHO/BCA1 polymerization, after which alternating CHO/CO2 polymerization occurred to give poly(ester-b-carbonate)s. In sharp contrast, more active MgII-based catalyst 12b formed the polycarbonate block, and after the removal of CO2, CHO/BCA1 copolymerization proceeded to give poly(carbonate-b-ester)s. This unusual selectivity of 12b was ascribed to the rigidity and steric hindrance of the tricyclic anhydrides. DSC analysis demonstrated that the Tg values of the polymers could be controlled by the carbonate/ester block ratios. Later, different block structures were realized with a series of dinuclear catalysts 12.5q ZnIIZnII complex 12a yielded poly(ester-b-carbonate)s, MgIIMgII complex 12b or MgIICoII complex 12d gave poly(carbonate-b-ester)s, and MgIIZnII complex 12c furnished random copolymers from CHO, CO2, and BCA1. The most active and selective MgIICoII catalyst 12d also delivered triblock (ABA), pentablock (BABAB), and heptablock (ABABABA) polymers by switching the atmosphere between CO2 and N2. They also reported that trinuclear ZnIINaIZnII catalyst 13 catalyzed CHO/PA ROCOP, CHO/CO2 ROCOP, and CHO ROP from mixtures of CHO and PA by using the atmosphere of CO2 or N2.5r
Liu, Lu, and co-workers reported the terpolymerization of CHO, CO2, and PA with dinuclear CrIII complex 14. PA was consumed completely within 2 h to give polyesters, and then the polymerization entered the second phase, leading to the formation of poly(ester-b-carbonate)s without forming the polyether segments.5o More recently, they achieved the asymmetric terpolymerization reactions of meso-epoxides, CO2, and PA using chiral bimetallic AlIII complex 15, giving terpolymers with a random distribution of carbonate and ester segments, which differed from that observed for tapered or gradient terpolymers.5w All the catalysts exhibited good catalytic activities to afford enantioenriched terpolymers with high enantioselectivity and narrow polydispersity. Especially, catalyst 15b gave the terpolymer with the highest molecular weight and enantiomeric purity. Various epoxides such as CHO, cyclopentene oxide (CPO), and cis-2,3-butene oxide (CBO) were reactive, and the terpolymerization of the epoxides, CO2, and PA proceeded with excellent enantioselectivities of up to 99% ee.
Plajer and co-workers reported the terpolymerization of CHO, CO2, and phthalic thioanhydride (PTA) forming random poly(ester-thioester-carbonate) by employing heterobimetallic CrIII-based catalysts 16.5y The ratios of the ester, thioester, and carbonate linkages varied, depending on the structures of the catalysts (alkali metals and backbones). Interestingly, the ratios of the thioester to ester linkages were always lower than 1, implying the loss of the S atoms and the enrichment of the O atoms in the polymer backbones, which is due to the O/S exchange at the chain end followed by the release of thiirane (cyclohexene sulfide). The terpolymers degraded into oligomers under UV irradiation as a result of the selective cleavage of the thioester linkages. The quaterpolymerization of CHO, CO2, PA, and PTA was also achieved, where the terpolymerization of CHO, CO2, and PA occurred until PA was consumed, and the subsequent terpolymerization of CHO, CO2, and PTA proceeded to produce poly(ester-thioester-carbonate) with some tapering.
Capacchione and co-workers developed dinuclear CrIII complexes, among which 17 was the most active for the terpolymerization reactions of epoxides, CO2, and PA as well as the copolymerization reactions of epoxides and CO2.5z The 1H and 13C NMR spectra showed no polycarbonate sequences in the polyester segments, suggesting the formation of diblock poly(ester-b-carbonate) copolymers. It is considered that after the faster reaction of the epoxides with PA to fully consume PA, the carbonate block was elongated by the slower reaction of the epoxides with CO2. Kozak and co-workers conducted two-step polymerization with CrIII ate complex 18 to synthesize block copolymers.5aa Poly(ester-b-carbonate) was obtained by the ROCOP of CHO and PA under N2 at 100 °C followed by the ROCOP of CHO and CO2 at 60 °C.
Metal-free catalysts have also been developed since the discovery of ROCOP of epoxides/CO2 with Et3B (19) and onium salts by Gnanou, Feng, and co-workers.3b Xiao, Meng, and co-workers performed the terpolymerization of PO, CO2, and PA using 19 and PPNCl.5s A random poly(ester-carbonate) segment formed first, and a long PPC-enriched segment grew after the full consumption of PA, which afforded poly[(ester-carbonate)-b-carbonate]. The terpolymer with an aromatic polyester content of 43 mol% displayed a Tg value of 47 °C, which was higher than that of PPC (38 °C). The terpolymers exhibited tensile strengths of up to 37.6 MPa, which was much higher than that of PPC (9 MPa, Mn = 28 kg mol−1). The terpolymers also exhibited satisfactory degradation behaviors under the standard composting conditions. They also reported the quaterpolymerization of CHO, PO, CO2, and PA using 19 and PPNCl to obtain polymers with an Mn of up to 77.7 kg mol−1.5p They succeeded in changing Tg values between 86 and 116 °C and enhancing tensile strengths to 54.8 MPa by modulating the monomer ratios. They also reported that gradient terpolymers were successfully synthesized by the terpolymerization of ethylene oxides (EO), CO2, and PA using 19 and the tetrabutylammonium salt of m-phthalic acid (A-mPhA).5v Thermal gravimetric analysis (TGA) indicated that the terpolymers had two fast decomposition stages: the first weight loss peak corresponded to the decomposition of the poly(ethylene carbonate) (PEC) sequence, and the second weight loss peak was assigned to the thermal degradation of the EO/PA polyester. The terpolymers had a higher thermal stability than PEC. Liu, Kang, Li, and co-workers achieved the terpolymerization of CHO, CO2, and PA with 19 and PPNCl to deliver diblock copolymers with little tapering in a one-pot one-step manner.5t Furthermore, the ROCOP of CHO and PA and the ROCOP of CHO and CO2 were done in a sequential one-pot manner by changing the atmosphere from N2 to CO2 to synthesize well-defined diblock poly(ester-b-carbonate)s. Other epoxides such as PO and 1,2-butylene oxide (BO) were also used to construct diblock poly(ester-b-carbonate)s. All materials showed single Tg values between the Tg values for the corresponding polyester and polycarbonate homopolymers. Liu, Zhong, Li, and co-workers reported the chemoselective terpolymerization of CHO, CO2, and PA using 19 and phosphazene C3N3-Py-P3 as binary organocatalysts.5x The terpolymers with block, tapered, or random structures could be selectively synthesized by changing the molar ratio of C3N3-Py-P3 to 19. DFT calculations clarified the effect of the molar ratio of C3N3-Py-P3 to 19 on the chemoselectivity. Gnanou, Feng, and co-workers demonstrated the terpolymerization of PO or CHO, CO2, and SA or PA in the presence of 19 and PPNCl or tetrabutylammonium azide (Bu4NN3).5u Only single glass transition temperature was observed for all the terpolymers, and the Tg value could be tuned by varying the ester contents. Wang and co-workers found a combination of 19 and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) showing the self-switchable polymerization of PO, PA, and CO2 to afford poly(propylene phthalate)-b-poly(propylene carbonate) with linear and hyperbranched architectures.5ab The linear and hyperbranched topologies were realized by employing benzyl alcohol and 1,2,4-benzene tricarboxylic anhydride as a polymerizable chain transfer agent, respectively, and the effect of the polymeric topology on viscosity, glass transition temperature, and thermal stability was revealed. For example, the Tg values of the linear and hyperbranched polymers were 50.1 °C and 44.2 °C, respectively.
Yang, Wu, and co-workers observed that bifunctional organoborane 20a with quaternary ammonium bromide produced gradient terpolymers from CHO, CO2, and PA, while the corresponding binary catalyst, 20b, and Et4NBr, afforded diblock terpolymers with tapering segments.5ac DFT calculations rationalized the experimental results; in the case of 20a, synergistic electrostatic interactions operate in the transition state, leading to the more competitive formation of the polyester and polycarbonate linkages. The gradient terpolymers with different polyester contents ranging from 5 mol% to 64 mol% had Tg values in the range of 114 °C to 128 °C, although the gradient terpolymers had a decomposition temperature (Td5%) at 280 °C, which was lower than that of the diblock terpolymers (298 °C).
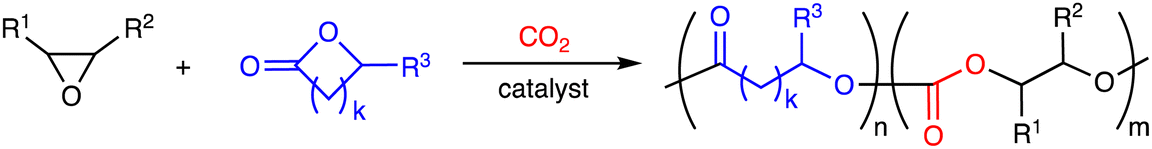
3. Terpolymerization reactions of epoxides, CO2, and lactones
Polymers containing the carbonate and ester linkages can also be obtained by the terpolymerization reactions of epoxides, CO2, and lactones, where the ROCOP and ROP mechanisms are involved (Scheme 4a). This approach is accessible to polymers that cannot be synthesized by the terpolymerization reactions of epoxides, CO2, and cyclic anhydrides. The representative catalytic cycles, catalysts, and reaction conditions for the terpolymerization reactions of epoxides, CO2, and lactones are shown in Scheme 4b, Fig. 2 and Table 2, respectively.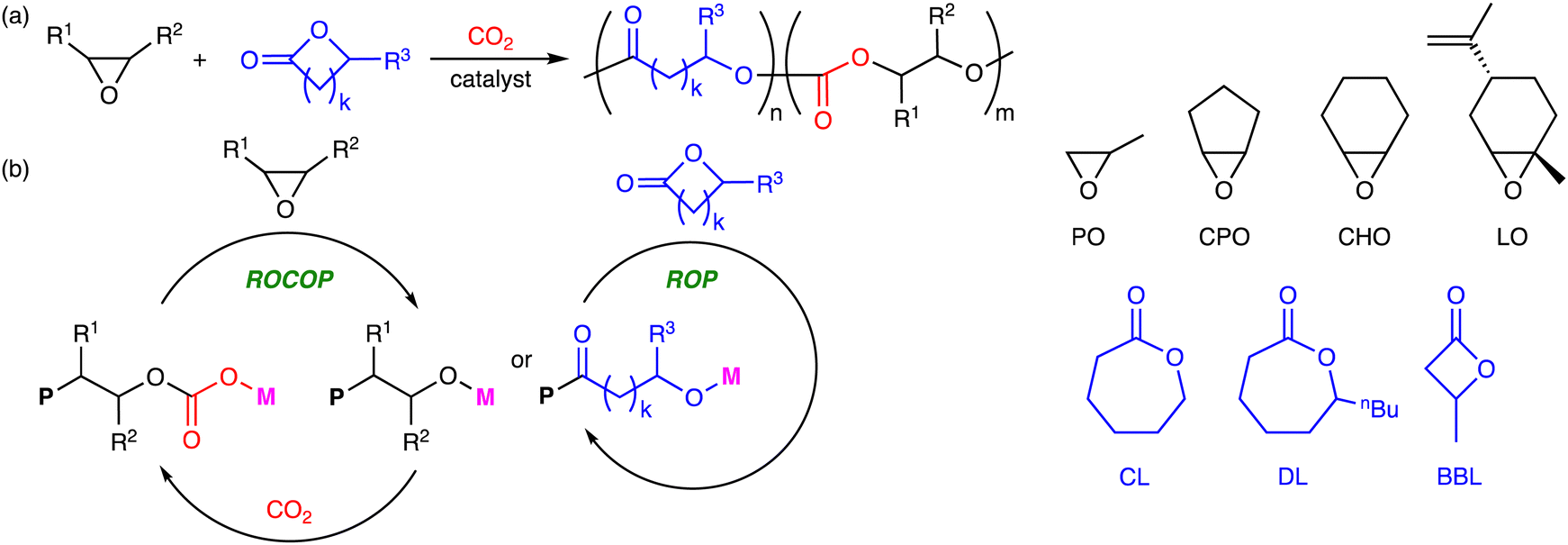 | ||
| Scheme 4 (a) Representative reactions and (b) catalytic cycles. P simply represents a part of a polymer chain that may differ in each step, while M represents a Lewis acid catalyst. | ||
 | ||
| Fig. 2 Catalysts and co-catalysts for the terpolymerization reactions of epoxides, CO2, and lactones. | ||
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Epoxide | Lactone | CO2 (MPa) | Cat. | Co-cat. | T (°C) | M n (kg mol−1) | Sequenceb | Ref. |
| a Maximum value. b Sequence structures of polymers. R: random, G: gradient, B: block, N/A: data not available. | |||||||||
| 1 | PO | CL | 2.8 | 3 | — | 60 | 275 | B | 6a,6b |
| 2 | PO | CL | 4.0 | 1 | — | 50–90 | 73.6 | N/A | 6c |
| 3 | CHO | CL | 0.10 | 12a | — | 80 | 4.81 | B | 6d |
| 4 | CHO | CL | 0.10 | 12e | — | 80 | 13.8 | B | 6e |
| 5 | CHO | DL | 2.0 | 21 | — | 80 | 71.9 | B | 6k |
| 6 | CHO | CL | 5.0 | 22 | — | 70–100 | 234.3 | G | 6f |
| 7 | CHO | CL | 4.0 | 5, 23 | — | 100 | 35.2 | B | 6g |
| 8 | CHO, CPO | BBL | 0.1–5.0 | 24 | — | 50–60 | 174 | R, G, B | 6i |
| 9 | CHO, CPO, LO | BBL | 0.3–4.0 | 24 | — | 40–60 | 233 | R, B | 6j |
| 10 | CHO | CL | 1.5 | 25 | PPNCl | 80 | 41.8 | B | 6l |
| 11 | CHO | CL | 1.0–4.0 | 26 | PPNCl | 60–100 | 67.1 | G | 6m |
In 2003, Ree and co-workers carried out the terpolymerization of PO, CO2, and ε-caprolactone (CL) using ZnGA 3 as a catalyst to produce poly(propylene carbonate-caprolactone)s, PPC-PCL, with improved biodegradability.6a,6b All the terpolymers showed a single Tg originating from the polycarbonate blocks and a single Tm originating from the polyester blocks. The terpolymers underwent enzymatic degradation in the presence of a lipase in a phosphate buffer (pH 7.0) at 37 °C for 10 days. Huang and co-workers reported the terpolymerization of PO, CO2, and CL with PBM complex 1.6c The incorporation of CL into the polymer skeletons increased the viscosity, glass transition temperature, and the degradation rate of the terpolymers.
Williams and co-workers reported the selective synthesis of poly(cyclohexene carbonate-caprolactone)s, PCHC-PCL, from CHO, CO2, and CL using dinuclear ZnII complex 12a,6d which also catalyzed the terpolymerization reactions of epoxides, CO2, and cyclic anhydrides as mentioned above. This is the first example of a single catalyst that not only promoted two distinct polymerization reactions but also switched them by the addition of exogeneous switching reagents, epoxide or CO2. The catalytic mechanism was elucidated by experiments and DFT calculations.6h The same terpolymerization was catalyzed by dinuclear ZnII complex 12e, and ABA block polymers, PCL-b-PCHC-b-PCL, were prepared in one pot.6e The ROCOP of CHO and CO2 occurred first to produce polycarbonates, and after removal of CO2, the subsequent ROP of CL on both sides of the polymer chains produced the triblock copolymers. These polymers showed controllable Tg values from −54 °C to 34 °C, depending on the block compositions. The one-pot switchable catalysis (reverse procedure) was also applied to the synthesis of PCHC-b-PDL-b-PCHC triblock polymers from CHO, CO2, and ε-decalactone (DL) with heterodinuclear ZnIIMgII catalyst 21.6k The block polymers showed not only excellent mechanical properties and thermal stability but also good degradation behaviors upon gentle heating under acidic conditions.
Xiao, Meng, and co-workers reported the terpolymerization of CHO, CO2, and CL with Schiff base trinuclear ZnII complexes 22.6f22b was more active than 22a. In this terpolymerization system, CL was much more reactive than the CHO/CO2 pair, and the resulting terpolymers had high molecular weights.
Zhang and co-workers achieved the highly efficient terpolymerization of CHO, CO2, and CL with binary catalysts, Zn–Co DMC 5 and stannous octoate (23).6g The ROCOP of CHO and CO2 with 5 and the ROP of CL with 23 occurred independently with matched polymerization rates to give PCHC and PCL, respectively, and the two independently growing chains underwent the cross-chain exchange reaction to produce multiblock copolymers, which are the first examples of biodegradable polycarbonate-polyester multiblock copolymers. In addition, improved chemical and physical properties were confirmed in glass transition temperature, melting temperature, and tensile tests. For example, the film material of the multiblock copolymers showed an elongation at break of 22.8%, which was much higher than those of PCHC (3.3%) and a PCHC/PCL blend (1.8%).
Rieger and co-workers achieved the terpolymerization reactions of epoxides, CO2, and rac-β-butyrolactone (BBL), which is less reactive than CL or lactide.6i Catalyst 24 could switch between the ROP of BBL and the ROCOP of epoxides and CO2, and the following three terpolymerization procedures were established. The first procedure provided the block structures by the ROP of BBL in the beginning followed by the addition of 40 bar CO2 initiating the ROCOP of epoxides and CO2. The second one was the reverse procedure starting from the ROCOP of epoxides and CO2 followed by the ROP of BBL after complete CO2 release. In the third procedure, lowering the CO2 pressure to 3 bar afforded random terpolymers composed of epoxides, CO2, and BBL. Not only CHO and CPO but also limonene oxide (LO), a bio-based monomer, was successfully used for the terpolymerization.6j Although the terpolymers with a block structure showed two Tg values because of the phase separation, only one Tg was observed for those with a random structure. As for mechanical behaviors, the Young's modulus of each of the block and random polymers was smaller than that of PCHC because of the incorporation of soft poly(3-hydroxybutyrate) segments.
Pang and co-workers synthesized AB-type diblock copolymers and ABA-type triblock copolymers by the terpolymerization of CHO, CO2, and CL using Mn(salen)Cl 25.6l In this catalyst system, cyclic anhydrides were also used as monomers, and the one-pot switchable copolymerization of anhydrides, epoxides, CO2, and CL delivered AB, ABA, and ABC-type block copolymers. They also reported a facile method for the synthesis of gradient poly(carbonate-ester), PCHC-g-PCL, from a mixture of CHO, CO2, and CL with CrIII complex 26.6m Compared with a symmetrical Cr(salen)Cl complex, this catalyst had an exceptional ability to synthesize biodegradable terpolymers with gradient character. The polymerization initially produced a PCHC segment, which then changed to a PCL-enriched segment by a cross-propagation reaction, and the chain configurations could be modulated by adjusting the reaction conditions. When the proportion of the PCL in the terpolymers was increased, a melting peak appeared and the degradation temperature increased.
4. Terpolymerization reactions of epoxides, CO2, and lactides
Polylactide (PLA), which can be derived from renewable resources, has a Tg of 60 °C, high crystallinity, and biodegradability, but the brittleness of PLA restricts potential applicability. On the other hand, poly(carbonate-ester)s synthesized by the terpolymerization reactions of epoxides, CO2, and lactides (Scheme 5a) may have excellent properties originating from both PLA and polycarbonates. The representative catalytic cycles, catalysts, and reaction conditions for the terpolymerization reactions of epoxides, CO2, and lactides are shown in Scheme 5b, Fig. 3 and Table 3, respectively. | ||
| Scheme 5 (a) Representative reactions and (b) catalytic cycles. P simply represents a part of a polymer chain that may differ in each step, while M represents a Lewis acid catalyst. | ||
 | ||
| Fig. 3 Catalysts and co-catalysts for the terpolymerization reactions of epoxides, CO2, and lactides. | ||
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Epoxide | Lactide | CO2 (MPa) | Cat. | Co-cat. | T (°C) | M n (kg mol−1) | Sequenceb | Ref. |
| a Maximum value. b Sequence structures of polymers. R: random, G: gradient, B: block, N/A: data not available. c Stereochemistry of lactide not indicated. | |||||||||
| 1 | CHO | rac, L | 4.0 | 27 | — | 90 | 41.6 | R | 7a |
| 2 | PO | rac | 3.5 | 1 | — | 70 | 14.3 | N/A | 7b |
| 3 | PO | L | 4.0 | 28 | — | 60–80 | 156 | R, B | 7c |
| 4 | PO | L | 5.0 | 4 | — | 60–90 | 132 | B | 7e |
| 5 | PO | L | CO2 : PO = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
4 | — | 70 | 68 | B | 7d |
| 6 | PO | rac | 0.1–4.0 | 6 | PPNCl | 23–60 | 14.7 | B | 7f |
| 7 | VCHO | rac | 0.10–4.0 | 6c | PPNCl | 25–70 | 8.2 | B | 7k |
| 8 | PO | L | 2.0 | 29a | DBU | rt, 40 | 7.15 | B | 7g |
| 9 | PO, SO, CHO | L | 2.0 | 29b–e, 30a–b | PPNCl | 60 | 13.6 | B | 7h |
| 10 | PO | L | 3.0 | 31 | — | 25–60 | 15.3 | B | 7i |
| 11 | PO, EPI, GMA | —c | 1.0–4.0 | 32 | PPNCl | 60 | 49.3 | B | 7n |
| 12 | PO, BO | L | 0.20–2.0 | 30b, 3 | PPNCl | 60 | 698 | B | 7l |
| 13 | PO, BO | L | 0.20–2.0 | 30 | PPNCl, O2 | 60 | 28.7 | B | 7m |
| 14 | PO | rac | 3.5 | 33 | TEC, TBC, MOC, BBC | 55 | 11.3 | N/A | 7j |
| 15 | CHO | L | 0.5–4.0 | 34 | — | 60 | 9.1 | R, B | 7o |
| 16 | CHO | L | 0.5–0 | 35 | — | 45 | 8.6 | B | 7p |
In 2006, Kröger, Döring, and co-workers reported the first example of the terpolymerization of CHO, CO2, and lactides with ZnII complexes 27 for the synthesis of poly(cyclohexene carbonate-lactide)s whose composition was adjusted by monomer feeding.7a The Tg values of the terpolymers fell between those of pure PLA and pure PCHC and increased with increasing the PCHC content, indicating the random structures of the terpolymers.
Liu and co-workers synthesized terpolymers from PO, CO2, and rac-lactides by using PBM 1 as a catalyst.7b The terpolymers showed a much higher hydrolytic degradability in tris buffer (pH 7.4) over 10 weeks than PPC. Wang and co-workers conducted the terpolymerization of PO, CO2, and L-lactide using Y(CCl3CO2)3-ZnEt2-glycerin ternary catalyst 28.7c The resulting terpolymers showed a decomposition temperature Td of up to 242.9 °C, which was much higher than that of pure PPC (197.1 °C), and the elongation at break of the terpolymers reached 40.5%, which was three times larger than that of pure PPC.
Wang, Meng, and co-workers reported the synthesis of carbonate-ester terpolymers with a long L-lactide-rich sequence via the terpolymerization of PO, CO2, and L-lactide using ZnAA 4.7e The thermal stability of the terpolymers was greatly improved; the decomposition temperature (Td5%) of the terpolymers was modulated from 235.9 °C to 258.6 °C, which was much higher than that for pure PPC (219.9 °C) and close to that for pure PLA (257.0 °C). This is because the zipper-like depolymerization reaction of PPC was suppressed by the L-lactide-rich sequence; the activation energy for PLA decomposition (280 kJ mol−1) is much higher than that for PPC decomposition (80 kJ mol−1). Regarding the mechanical properties, all terpolymers exhibited greater tensile strengths than PPC; the tensile strength of the terpolymers increased from 27.1 MPa to 49.5 MPa upon increasing the crystalline PLA segment. The same catalyst system was also reported by Sakharov and co-workers.7d
The one-pot regioselective and stereoselective terpolymerization of PO, CO2, and rac-lactide was reported by Xie and co-workers.7f Interestingly, M(TPP)Cl 6 and PPNCl catalyzed the polymerization to produce PPC–PLA–PPO (poly(propylene oxide)) multiblock copolymers with good regioregularity (head-to-tail structure) in the PPC block and good stereoregularity (isotacticity) in the PLA block despite the use of the achiral catalyst and racemic starting materials. They also reported the terpolymerization of VCHO, CO2, and rac-lactide with 6c.7k The glass transition temperature of the resulting polymers could be adjusted by changing the molar ratio of the monomers, reaction temperature, CO2 pressure, and the amount of the catalyst.
Pang, Chen, and co-workers developed a switchable system with 29a and DBU to achieve chemoselective block copolymerization between lactide ROP and PO/CO2 ROCOP in one pot.7g This catalyst system is based on the reversible adsorption of CO2 onto DBU and the chain shuttling polymerization using i-PrOH as a chain shuttling reagent. Pang and co-workers also devised a ternary catalyst system comprising the CoII complex (29b, 29c, or 30a), the CoIII complex (29d, 29e, or 30b), and PPNCl, which produced poly(ester-carbonate)s with no polyether structures from a mixture of PO, CO2, and L-lactide.7h The resultant polymers were biodegradable, and the Tg values were tunable between 38 °C and 52 °C, which contrasts with pure PPC having a Tg value of 35–40 °C. The mechanistic investigation revealed that the CoIII and CoII complexes catalyzed PO/CO2 ROCOP and lactide ROP, respectively, and the combined polymers were formed via the transfer of the growing chains. Styrene oxide (SO) and CHO were also reactive in this copolymerization. They also developed trinuclear CoIII complex 31 for the terpolymerization of PO, CO2, and L-lactide to produce multiblock copolymers.7i The lactide ROP and PO/CO2 ROCOP were separately catalyzed by 31 without any co-catalyst to give the terpolymers in one pot. Recently, Pang, Chen, and co-workers developed an electrochemically controlled switchable copolymerization system with redox-active heteronuclear complex 32 for the synthesis of multi-block copolymers.7n When the catalyst was in a reduced state, the ROP of lactide proceeded (ON), and the ROCOP of epoxide and CO2 did not occur (OFF). In contrast, when the catalyst underwent the electrochemical oxidation, the ROCOP of epoxide and CO2 proceeded (ON), and the ROP of lactide did not occur (OFF). The electrochemical switching between the active (ON) and dormant (OFF) states of the catalyst realized the synthesis of the multi-block copolymers.
Pang, Deng, and co-workers reported a heterogeneous ternary catalyst system composed of dinuclear CoIII complex 30b, ZnGA 3, and PPNCl for the terpolymerization of epoxides, CO2, and L-lactide.7l Interestingly, 30b could not catalyze the PO/CO2 ROCOP and the lactide ROP simultaneously because the strong coordination of CO2 prevented the latter, while 3 was also inactive for the ROP of lactide. The intermolecular cooperation between the two metal catalysts realized the terpolymerization, forming PLA/PPC multiblock copolymers with high molecular weights of up to 698 kg mol−1. Several homogeneous ternary catalyst systems (TCSs) were also developed.7m TCS I was composed of 30a, 30b, and PPNCl, while TCS IIIa consisted of 30b, 30c, and PPNCl, and TCS IIIb comprised 30b, O2, and PPNCl. TCS IIIa and TCS IIIb showed a higher catalytic activity than TCS I.
Lin, Zhu, and co-workers reported Zn–Fe DMC 33 and quaternary ammonium salts for the terpolymerization of PO, CO2, and lactide.7j The catalyst systems were synthesized from ZnBr2, K3Fe(CN)6, and quaternary ammonium salts via ball grinding, and the combination of Zn–Fe DMC and triethylmethylammonium chloride (TEC) showed the highest catalytic activity.
Castro-Osma, Lara-Sánchez, and co-workers developed bimetallic InIII complex 34 for the terpolymerization of CHO, CO2, and L-lactide.7o Terpolymers were obtained without any co-catalyst, and the degree of CO2 incorporation could be modulated by changing the CO2 pressure and the monomer amounts. The terpolymers exhibited single Tg values between those of pure PLA and pure PCHC, which suggested the formation of random copolymers, and the Tg values increased as the ratio of CHO to L-lactide increased. The terpolymer with the highest ratio of CHO to L-lactide showed good thermal stability (a Td5% of up to 244 °C) because of the higher PCHC content.
Wang, Xia, Li, and co-workers employed bifunctional organoboron 35 for the one-pot two-step synthesis of poly(carbonate-b-ester).7p When a mixture of CHO, L-lactide, and 35 was subjected to CO2 (5 bar) at 45 °C for 1 h, the ROCOP of CHO and CO2 proceeded selectively to form PCHC in 99% conversion. The release of CO2 allowed the ROP of L-lactide to produce the PCHC-b-PLA block polymer. In contrast, this type of block polymer could not be obtained when a similar experiment was done at a higher CO2 pressure (20 bar); the ROP of L-lactide did not proceed probably because the polymer end was capped with CO2 to form a carbonate anion, a weak nucleophile for the ring-opening of L-lactide.
5. Terpolymerization reactions of epoxides, CO2, and heteroallenes
Heteroallenes such as CO2, sulfur dioxide (SO2), carbonyl sulfide (COS), and carbon disulfide (CS2), isocyanates (RNCO), and isothiocyanates (RNCS) are important reagents for organic synthesis. The ROCOP of epoxides and heteroallenes can incorporate new linkages into the main chains of the polymers, which may improve their chemical and physical properties. Therefore, the terpolymerization reactions of epoxides, CO2, and heteroallenes provide new opportunities to create new CO2-based polymers (Scheme 6a), even if they are challenging because of the difference in reactivity between CO2 and heteroallenes.8 In fact, there are only several reports on this type of terpolymerization. The representative catalytic cycles, catalysts, and reaction conditions for the terpolymerization reactions of epoxides, CO2, and heteroallenes are shown in Scheme 6b, Fig. 4 and Table 4, respectively. | ||
| Scheme 6 (a) Representative reactions and (b) catalytic cycles. P simply represents a part of a polymer chain that may differ in each step, while M represents a Lewis acid catalyst. | ||
 | ||
| Fig. 4 Catalysts and co-catalysts for the terpolymerization reactions of epoxides, CO2, and heteroallenes. | ||
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Epoxide | Heteroallene | CO2 (MPa) | Cat. | Co-cat. | T (°C) | M n (kg mol−1) | Sequenceb | Ref. |
| a Maximum value. b Sequence structures of polymers. R: random, G: gradient, B: block. | |||||||||
| 1 | CHO | SO2 | SO2 : CO2 = 1:4–16 |
8 | —, PPNCl | 40–90 | 9.99 | B | 9a |
| 2 | CHO | COS | 0.6–3.0 | 36, 37 | PPNCl | 25–80 | 50.2 | R, G | 9b |
| 3 | CHO | CS2 | 0.4 | 16a | — | 100 | 12.7 | R | 9c |
| 4 | CHO | RNCO | 0.1 | 38 | — | 80–100 | 7.4 | R | 9d |
| 5 | CHO | RNCS | 0.5–2.0 | 38 | — | 80–100 | 183 | G, B | 9d |

In 2019, Jia, Shan, and co-workers achieved the terpolymerization of CHO, CO2, and SO2 with CrIII(salen)Cl 8 to synthesize poly(carbonate-sulfite)s.9a Interestingly, no nucleophilic co-catalyst was needed to promote this terpolymerization because SO2 acted as a nucleophile as well as a reactant. TGA indicated that the first weight loss appeared at 211.4–242.7 °C, corresponding to the degradation of the sulfite units. The second weight loss appeared at 242.7–327.7 °C, corresponding to the decomposition of the carbonate and ether units.
Ren, Darensbourg, and co-workers synthesized terpolymers with tunable randomly distributed sulfur atoms from CHO, CO2, and COS using 36 or 37 as a catalyst.9b By changing the CO2 pressure, random polycarbonates with different COS contents in the terpolymers could be prepared, and the polymers showed good optical properties such as the Abbe number as high as 48.6 and a refractive index of 1.501. The optical parameters of the terpolymers exceeded those of homopolymer blends or diblock copolymers with equal sulfur compositions.
Neale, Plajer, and co-workers reported the terpolymerization of CHO, CO2, and CS2 with heterobimetallic catalyst 16a.9c The mechanism of O/S scrambling is proposed to rationalize the oxygen enrichment in the terpolymers. For example, the alkoxide chain end attacks the adjacent dithiocarbonate linkage (–OC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S)S–) to generate the thiolate chain end and the monothiocarbonate linkage (–OC(S)O–), which is followed by the release of thiirane, as supported by DFT calculations. Although the terpolymers had Tg values and refractive indexes that were similar to those of PCHC, the sulfur-containing terpolymers were susceptible to oxidative and photolytic cleavage. The terpolymers degraded into oligomers under aqueous H2O2 or UV irradiation owing to the selective degradation of the dithiocarbonate units, which is in contrast to the corresponding all-oxygen CHO/CO2 copolymers being fully intact under the same conditions.
S)S–) to generate the thiolate chain end and the monothiocarbonate linkage (–OC(S)O–), which is followed by the release of thiirane, as supported by DFT calculations. Although the terpolymers had Tg values and refractive indexes that were similar to those of PCHC, the sulfur-containing terpolymers were susceptible to oxidative and photolytic cleavage. The terpolymers degraded into oligomers under aqueous H2O2 or UV irradiation owing to the selective degradation of the dithiocarbonate units, which is in contrast to the corresponding all-oxygen CHO/CO2 copolymers being fully intact under the same conditions.
Ema and co-workers achieved the terpolymerization of CHO, CO2, and aryl isothiocyanates to produce poly(carbonate-thioimidocarbonate)s with gradient character, and the ratio of the polythioimidocarbonate to the polycarbonate units in the terpolymers could be controlled by the CO2 pressure.9d When the scope of aryl isothiocyanates was explored, p-tolyl isothiocyanate was modestly incorporated into the terpolymer, while aryl isothiocyanates with electron-withdrawing groups showed higher reactivity to give terpolymers with higher polythioimidocarbonate contents. On the other hand, the terpolymerization of CHO, CO2, and aryl isocyanates furnished poly(carbonate-urethane)s with random sequences. It should be noted that these terpolymerization reactions were catalyzed by a single catalyst, bifunctional AlIII porphyrin 38. This fact clearly demonstrates that cooperative catalysis with the metal center and the quaternary ammonium salts of bifunctional AlIII porphyrins is effective for polymer synthesis.14,15 Poly(carbonate-thioimidocarbonate)s underwent partial degradation upon acid treatment or UV irradiation to give polycarbonates, while poly(carbonate-urethane)s were stable under the same conditions.
6. Terpolymerization reactions of epoxides, CO2, and olefins
The terpolymerization reactions of epoxides, CO2, and olefins have been achieved by ingenious strategies. In 2018, Xie, Wang, and co-workers reported the unprecedented terpolymerization of epoxides, CO2, and olefins using AlIII porphyrin 39 as a catalyst (Scheme 7a).10a Well-defined CO2-based diblock copolymers were obtained in one step by the simultaneous ROCOP of epoxides and CO2 and the reversible addition–fragmentation chain transfer (RAFT) polymerization of olefin monomers using TTC-COOH and 2,2′-azobis(isobutyronitrile) (AIBN) as a chain transfer agent with the trithiocarbonate (TTC) group and a radical initiator, respectively. More recently, Yue, Ren, and co-workers achieved the terpolymerization reactions of epoxides, CO2, and acrylate esters with dinuclear CoIII catalyst 40 (Scheme 7b).10b In the presence of CO2, various polycarbonate-b-polyacrylates, which had a wide range of Mn and Tg values, were obtained successfully. In contrast, in the absence of CO2, the copolymerization of epoxides and acrylates proceeded in a sequence-controlled manner, delivering polyether-b-polyacrylates; the polyether formation occurred first to consume most of the epoxides, and the non-catalytic anionic polymerization of acrylate esters took place from the alkoxide ends of the growing polymers (not shown). Several examples of stepwise terpolymerization reactions have also been reported,16 although they are not described herein. | ||
| Scheme 7 Terpolymerization reactions of epoxides, CO2, and olefins. | ||
7. Conclusions and outlook
The mitigation of CO2 emission is crucially important because one of the most urgent issues is global warming caused by greenhouse gases such as CO2, while the effective and efficient utilization of CO2 is also considerably important from the viewpoint of resource circulation aimed at carbon neutrality. In addition, the accumulation of plastics in the environment and living organisms is also a big problem to be solved. More environmentally benign and sustainable plastics should be created and used. The creation of CO2-based polymers can contribute to finding the solution to these global issues. Among various CO2-based polymers, aliphatic polycarbonates synthesized by the copolymerization of epoxides and CO2 are fascinating from the viewpoint of high CO2 contents and good chemical and physical properties, and biomass-based epoxides can also be employed. Nevertheless, synthetic efficiencies and polymer functions are often insufficient for practical use. The incorporation of the third monomer may be effective for the improvement of the latter (polymer functions), although the former (synthetic efficiencies) may become more difficult to achieve. Here we provided an overview of the terpolymerization reactions of epoxides, CO2, and the third monomers such as cyclic anhydrides, lactones, lactides, heteroallenes, and olefins. They present promising routes to new CO2-based polymers with altered chemical and physical properties. Especially, we focused on the catalysts/cocatalysts, reaction conditions, and tunable polymer properties. The incorporation of various monomers not only improves the thermal and mechanical properties of the terpolymers but also adjusts the degradation rates. In addition, the precise control of polymer sequences enhances the ability to tailor the properties of materials for specific applications. Further studies and efforts are required for the further improvement of synthetic efficiencies and the exploration of new CO2-based polymers. The terpolymerization reactions of epoxides, CO2, and the third monomers will contribute significantly to the development of environmentally benign and sustainable polymers.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Notes and references
- Recent reviews: (a) J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed; (b) C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. M. Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS PubMed; (c) C. Kim, C.-J. Yoo, H.-S. Oh, B. K. Min and U. Lee, J. CO2 Util., 2022, 65, 102239 CrossRef CAS; (d) P. Challa, G. Paleti, V. R. Madduluri, S. B. Gadamani, R. Pothu, D. R. Burri, R. Boddula, V. Perugopu and S. R. R. Kamaraju, Catal. Surv. Asia, 2022, 26, 80–91 CrossRef; (e) L.-Q. Qiu, X. Yao, Y.-K. Zhang, H.-R. Li and L.-N. He, J. Org. Chem., 2023, 88, 4942–4964 CrossRef CAS PubMed.
- Reviews: (a) M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC; (b) S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460–1479 CrossRef CAS; (c) X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC; (d) M. Taherimehr and P. P. Pescarmona, J. Appl. Polym. Sci., 2014, 131, 41141 CrossRef; (e) D. J. Darensbourg and A. D. Yeung, Polym. Chem., 2014, 5, 3949–3962 RSC; (f) G. Trott, P. K. Saini and C. K. Williams, Phil. Trans. R. Soc., A, 2016, 374, 20150085 CrossRef PubMed; (g) S. J. Poland and D. J. Darensbourg, Green Chem., 2017, 19, 4990–5011 RSC; (h) Y. Wang and D. J. Darensbourg, Coord. Chem. Rev., 2018, 372, 85–100 CrossRef CAS; (i) C. M. Kozak, K. Ambrose and T. S. Anderson, Coord. Chem. Rev., 2018, 376, 565–587 CrossRef CAS; (j) M. Scharfenberg, J. Hilf and H. Frey, Adv. Funct. Mater., 2018, 28, 1704302 CrossRef; (k) D. J. Darensbourg, Green Chem., 2019, 21, 2214–2223 RSC; (l) A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, Green Chem., 2019, 21, 406–448 RSC; (m) V. Paradiso, V. Capaccio, D. H. Lamparelli and C. Capacchione, Catalysts, 2020, 10, 825 CrossRef CAS; (n) J. Huang, J. C. Worch, A. P. Dove and O. Coulembier, ChemSusChem, 2020, 13, 469–487 CrossRef CAS PubMed; (o) G. A. Bhat and D. J. Darensbourg, Green Chem., 2022, 24, 5007–5034 RSC; (p) C. A. L. Lidston, S. M. Severson, B. A. Abel and G. W. Coates, ACS Catal., 2022, 12, 11037–11070 CrossRef CAS; (q) F. Siragusa, C. Detrembleur and B. Grignard, Polym. Chem., 2023, 14, 1164–1183 RSC; (r) G. A. Bhat and D. J. Darensbourg, Coord. Chem. Rev., 2023, 492, 215277 CrossRef CAS.
- Selected examples: (a) S. Inoue, H. Koinuma and T. Tsuruta, J. Polym. Sci., Part B: Polym. Lett., 1969, 7, 287–292 CrossRef CAS; (b) D. Zhang, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, J. Am. Chem. Soc., 2016, 138, 11117–11120 CrossRef CAS PubMed; (c) M. Reiter, S. Vagin, A. Kronast, C. Jandl and B. Rieger, Chem. Sci., 2017, 8, 1876–1882 RSC; (d) Z. Huang, Y. Wang, N. Zhang, L. Zhang and D. J. Darensbourg, Macromolecules, 2018, 51, 9122–9130 CrossRef CAS; (e) H. Nagae, R. Aoki, S. Akutagawa, J. Kleemann, R. Tagawa, T. Schindler, G. Choi, T. P. Spaniol, H. Tsurugi, J. Okuda and K. Mashima, Angew. Chem., Int. Ed., 2018, 57, 2492–2496 CrossRef CAS; (f) R. Duan, C. Hu, Z. Sun, H. Zhang, X. Pang and X. Chen, Green Chem., 2019, 21, 4723–4731 RSC; (g) N. G. Patil, S. K. Boopathi, P. Alagi, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2019, 52, 2431–2438 CrossRef CAS; (h) H. Asaba, T. Iwasaki, M. Hatazawa, J. Deng, H. Nagae, K. Mashima and K. Nozaki, Inorg. Chem., 2020, 59, 7928–7933 CrossRef CAS; (i) W. Lindeboom, D. A. X. Fraser, C. B. Durr and C. K. Williams, Chem. Eur. J., 2021, 27, 12224–12231 CrossRef CAS; (j) R. Zhang, Q. Kuang, H. Cao, S. Liu, X. Chen, X. Wang and F. Wang, CCS Chem., 2022, 5, 750–760 CrossRef; (k) G.-W. Yang, C.-K. Xu, R. Xie, Y.-Y. Zhang, C. Lu, H. Qi, L. Yang, Y. Wang and G.-P. Wu, Nat. Synth., 2022, 1, 892–901 CrossRef; (l) Y.-N. Li, H.-H. Yang and X.-B. Lu, J. Polym. Sci., 2022, 60, 2078–2085 CrossRef CAS; (m) H. Nagae, S. Matsushiro, J. Okuda and K. Mashima, Chem. Sci., 2023, 14, 8262–8268 RSC.
- Reviews on terpolymerizations: (a) J. Liang, S. Ye, S. Wang, M. Xiao and Y. Meng, Polym. J., 2021, 53, 3–27 CrossRef; (b) D. H. Lamparelli and C. Capacchione, Catalysts, 2021, 11, 961 CrossRef CAS.
- (a) Y. Liu, K. Huang, D. Peng and H. Wu, Polymer, 2006, 47, 8453–8461 CrossRef CAS; (b) R. C. Jeske, J. M. Rowley and G. W. Coates, Angew. Chem., Int. Ed., 2008, 47, 6041–6044 CrossRef CAS PubMed; (c) P. F. Song, M. Xiao, F. G. Du, S. J. Wang, L. Q. Gan, G. Q. Liu and Y. Z. Meng, J. Appl. Polym. Sci., 2008, 109, 4121–4129 CrossRef CAS; (d) X.-K. Sun, X.-H. Zhang, S. Chen, B.-Y. Du, Q. Wang, Z.-Q. Fan and G.-R. Qi, Polymer, 2010, 51, 5719–5725 CrossRef CAS; (e) S. Huijser, E. H. Nejad, R. Sablong, C. de Jong, C. E. Koning and R. Duchateau, Macromolecules, 2011, 44, 1132–1139 CrossRef CAS; (f) D. J. Darensbourg, R. R. Poland and C. Escobedo, Macromolecules, 2012, 45, 2242–2248 CrossRef CAS; (g) A. Bernard, C. Chatterjee and M. H. Chisholm, Polymer, 2013, 54, 2639–2646 CrossRef CAS; (h) Y. Liu, M. Xiao, S. Wang, L. Xia, D. Hang, G. Cui and Y. Meng, RSC Adv., 2014, 4, 9503–9508 RSC; (i) Z. Duan, X. Wang, Q. Gao, L. Zhang, B. Liu and I. Kim, J. Polym. Sci., 2014, 52, 789–795 CrossRef CAS; (j) J. Y. Jeon, S. C. Eo, J. K. Varghese and B. Y. Lee, Beilstein J. Org. Chem., 2014, 10, 1787–1795 CrossRef PubMed; (k) Y. Liu, K. Deng, S. Wang, M. Xiao, D. Han and Y. Meng, Polym. Chem., 2015, 6, 2076–2083 RSC; (l) A. Thevenon, J. A. Garden, A. J. P. White and C. K. Williams, Inorg. Chem., 2015, 54, 11906–11915 CrossRef CAS; (m) C. Romain, Y. Zhu, P. Dingwall, S. Paul, H. S. Rzepa, A. Buchard and C. K. Williams, J. Am. Chem. Soc., 2016, 138, 4120–4131 CrossRef CAS PubMed; (n) P. K. Saini, G. Fiorani, R. T. Mathers and C. K. Williams, Chem. Eur. J., 2017, 23, 4260–4265 CrossRef CAS; (o) Y. Liu, J.-Z. Guo, H.-W. Lu, H.-B. Wang and X.-B. Lu, Macromolecules, 2018, 51, 771–778 CrossRef CAS; (p) S. Ye, W. Wang, J. Liang, S. Wang, M. Xiao and Y. Meng, ACS Sustainable Chem. Eng., 2020, 8, 17860–17867 CrossRef CAS; (q) G. Rosetto, A. C. Deacy and C. K. Williams, Chem. Sci., 2021, 12, 12315–12325 RSC; (r) A. J. Plajer and C. K. Williams, Angew. Chem., Int. Ed., 2021, 60, 13372–13379 CrossRef CAS; (s) J. Liang, S. Ye, W. Wang, C. Fan, S. Wang, D. Han, W. Liu, Y. Cui, L. Hao, M. Xiao and Y. Meng, J. CO2 Util., 2021, 49, 101558 CrossRef CAS; (t) J. Zhang, L. Wang, S. Liu, X. Kang and Z. Li, Macromolecules, 2021, 54, 763–772 CrossRef CAS; (u) V. K. Chidara, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2021, 54, 2711–2719 CrossRef CAS; (v) S. Ye, Y. Ren, J. Liang, S. Wang, S. Huang, D. Han, Z. Huang, W. Liu, M. Xiao and Y. Meng, J. CO2 Util., 2022, 65, 102223 CrossRef CAS; (w) G.-H. He, B.-H. Ren, S. Wang, Y. Liu and X.-B. Lu, Angew. Chem., Int. Ed., 2023, 62, e202304943 CrossRef CAS; (x) Y. Ma, X. You, J. Zhang, X. Wang, X. Kou, S. Liu, R. Zhong and Z. Li, Angew. Chem., Int. Ed., 2023, 62, e202303315 CrossRef CAS PubMed; (y) M. R. Stühler, C. Gallizioli, S. M. Rupf and A. J. Plajer, Polym. Chem., 2023, 14, 4848–4855 RSC; (z) F. Niknam, A. Denk, A. Buonerba, B. Rieger, A. Grassi and C. Capacchione, Catal. Sci. Technol., 2023, 13, 4684–4692 RSC; (a a) C. W. Vos, J. Beament and C. M. Kozak, Polym. Chem., 2023, 14, 5083–5093 RSC; (a b) M. Zhao, S. Zhu, G. Zhang, Y. Wang, Y. Liao, J. Xu, X. Zhou and X. Xie, Macromolecules, 2023, 56, 2379–2387 CrossRef CAS; (a c) R. Xie, Y. Wang, S. Li, B. Li, J. Xu, J. Liu, Y. He, G.-W. Yang and G.-P. Wu, Angew. Chem., Int. Ed., 2024, 63, e202404207 CrossRef CAS.
- (a) Y. Hwang, J. Jung, M. Ree and H. Kim, Macromolecules, 2003, 36, 8210–8212 CrossRef CAS; (b) Y. Hwang, H. Kim and M. Ree, Macromol. Symp., 2005, 224, 227–237 CrossRef CAS; (c) S. Liu, H. Xiao, K. Huang, L. Lu and Q. Huang, Polym. Bull., 2006, 56, 53–62 CrossRef CAS; (d) C. Romain and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 1607–1610 CrossRef CAS; (e) S. Paul, C. Romain, J. Shaw and C. K. Williams, Macromolecules, 2015, 48, 6047–6056 CrossRef CAS; (f) Y. Xu, S. Wang, L. Lin, M. Xiao and Y. Meng, Polym. Chem., 2015, 6, 1533–1540 RSC; (g) Y. Li, J. Hong, R. Wei, Y. Zhang, Z. Tong, X. Zhang, B. Du, J. Xu and Z. Fan, Chem. Sci., 2015, 6, 1530–1536 RSC; (h) C. Romain, Y. Zhu, P. Dingwall, S. Paul, H. S. Rzepa, A. Buchard and C. K. Williams, J. Am. Chem. Soc., 2016, 138, 4120–4131 CrossRef CAS PubMed; (i) S. Kernbichl, M. Reiter, F. Adams, S. Vagin and B. Rieger, J. Am. Chem. Soc., 2017, 139, 6787–6790 CrossRef CAS PubMed; (j) S. Kernbichl, M. Reiter, J. Mock and B. Rieger, Macromolecules, 2019, 52, 8476–8483 CrossRef CAS; (k) G. S. Sulley, G. L. Gregory, T. T. D. Chen, L. P. Carrodeguas, G. Trott, A. Santmarti, K.-Y. Lee, N. J. Terrill and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 4367–4378 CrossRef CAS PubMed; (l) Z. Yang, C. Hu, F. Cui, X. Pang, Y. Huang, Y. Zhou and X. Chen, Angew. Chem., Int. Ed., 2022, 61, e202117533 CrossRef CAS PubMed; (m) Y. Zhou, Z. Gao, C. Hu, S. Meng, R. Duan, Z. Sun and X. Pang, Macromolecules, 2022, 55, 9951–9959 CrossRef CAS.
- (a) M. Kröger, C. Folli, O. Walter and M. Döring, Adv. Synth. Catal., 2006, 348, 1908–1918 CrossRef; (b) S. Liu, J. Wang, K. Huang, Y. Liu and W. Wu, Polym. Bull., 2011, 66, 327–340 CrossRef CAS; (c) L. Gu, Y. Qin, Y. Gao, X. Wang and F. Wang, Chin. J. Chem., 2012, 30, 2121–2125 CrossRef CAS; (d) Z. N. Nysenko, E. E. Said-Galiev, M. I. Buzin, Y. E. Belevtsev, M. M. Ilin, G. G. Nikiforova and A. M. Sakharov, Mendeleev Commun., 2014, 24, 236–238 CrossRef CAS; (e) L. Tang, W. Luo, M. Xiao, S. Wang and Y. Meng, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1734–1741 CrossRef CAS; (f) D. Xie, Z. Yang, L. Wu, C. Zhang and M. H. Chisholm, Polym. Int., 2018, 67, 883–893 CrossRef CAS; (g) C. Hu, R. Duan, S. Yang, X. Pang and X. Chen, Macromolecules, 2018, 51, 4699–4704 CrossRef CAS; (h) X. Li, C. Hu, X. Pang, R. Duan and X. Chen, Catal. Sci. Technol., 2018, 8, 6452–6457 RSC; (i) R. Duan, C. Hu, Z. Sun, H. Zhang, X. Pang and X. Chen, Green Chem., 2019, 21, 4723–4731 RSC; (j) N. Liu, C. Gu, M. Chen, J. Zhang, W. Yang, A. Zhan, K. Zhang, Q. Lin and L. Zhu, ChemistrySelect, 2020, 5, 2388–2394 CrossRef CAS; (k) Y. Chen, W. Wang, D. Xie, L. Wu and C. Zhang, J. Polym. Sci., 2021, 59, 1528–1539 CrossRef CAS; (l) X. Li, R.-L. Duan, C.-Y. Hu, X. Pang and M.-X. Deng, Polym. Chem., 2021, 12, 1700–1706 RSC; (m) X. Li, C.-Y. Hu, R.-L. Duan, Z.-Z. Liang, X. Pang and M.-X. Deng, Polym. Chem., 2021, 12, 3124–3131 RSC; (n) Y. Huang, C. Hu, X. Pang, Y. Zhou, R. Duan, Z. Sun and X. Chen, Angew. Chem., Int. Ed., 2022, 61, e202202660 CrossRef CAS PubMed; (o) M. Martínez de Sarasa Buchaca, F. de la Cruz-Martínez, L. F. Sánchez-Barba, J. Tejeda, A. M. Rodríguez, J. A. Castro-Osma and A. Lara-Sánchez, Dalton Trans., 2023, 52, 3482–3492 RSC; (p) Y. Li, X. Kou, X. Wang, L. Xia and Z. Li, Polym. Chem., 2024, 15, 2476–2481 RSC.
- Z. Li, R. J. Mayer, A. R. Ofial and H. Mayr, J. Am. Chem. Soc., 2020, 142, 8383–8402 CrossRef CAS PubMed.
- (a) Y. Zhi, Y. Miao, W. Zhao, J. Wang, Y. Zheng, H. Su, Q. Jia and S. Shan, Polymer, 2019, 165, 11–18 CrossRef CAS; (b) T.-J. Yue, B.-H. Ren, W.-J. Zhang, X.-B. Lu, W.-M. Ren and D. J. Darensbourg, Angew. Chem., Int. Ed., 2021, 60, 4315–4321 CrossRef CAS PubMed; (c) J. Stephan, M. R. Stühler, S. M. Rupf, S. Neale and A. J. Plajer, Cell Rep. Phys. Sci., 2023, 4, 101510 CrossRef CAS; (d) K. Nakaoka, S. Muranaka, I. Yamamoto and T. Ema, Polym. Chem., 2024, 15, 707–713 RSC.
- (a) Y. Wang, Y. Zhao, Y. Ye, H. Peng, X. Zhou, X. Xie, X. Wang and F. Wang, Angew. Chem., Int. Ed., 2018, 57, 3593–3597 CrossRef CAS PubMed; (b) X.-Y. Fu, T.-J. Yue, B.-H. Ren, H. Wang, W.-M. Ren and X.-B. Lu, Angew. Chem., Int. Ed., 2024, 63, e202401926 CrossRef CAS PubMed.
- (a) Y.-Y. Zhang, G.-P. Wu and D. J. Darensbourg, Trends Chem., 2020, 2, 750–763 CrossRef CAS; (b) A. C. Deacy, G. L. Gregory, G. S. Sulley, T. T. D. Chen and C. K. Williams, J. Am. Chem. Soc., 2021, 143, 10021–10040 CrossRef CAS PubMed.
- Selected examples: (a) D. J. Darensbourg and Y. Wang, Polym. Chem., 2015, 6, 1768–1776 RSC; (b) F. W. Shaarani and J. J. Bou, Sci. Total Environ., 2017, 598, 931–936 CrossRef CAS PubMed; (c) W. Mo, C. Zhuo, H. Cao, S. Liu, X. Wang and F. Wang, Macromol. Chem. Phys., 2022, 223, 2100403 CrossRef CAS; (d) M. Sengoden, G. A. Bhat and D. J. Darensbourg, Macromolecules, 2023, 56, 2362–2369 CrossRef CAS; (e) S. Tang, H. Suo, R. Qu, H. Tang, M. Sun and Y. Qin, Polymers, 2023, 15, 748 CrossRef CAS PubMed; (f) W. Mo, C. Zhuo, S. Liu, X. Wang and F. Wang, Polym. Chem., 2023, 14, 152–160 RSC.
- Selected examples: (a) Z. N. Nysenko, E. E. Said-Galiev, G. G. Nikiforova, M. I. Buzin, A. A. Glazkov, M. M. Ilin, G. A. Belyaev, V. V. Rusak and A. M. Sakharov, Russ. Chem. Bull., 2022, 71, 1770–1776 CrossRef CAS; (b) Z. Gao, B. Gao, S. Meng, Z. Yang, Z. Liang, Z. Sun, Y. Zhou and X. Pang, Macromolecules, 2023, 56, 2062–2069 CrossRef CAS.
- (a) J. Deng, M. Ratanasak, Y. Sako, H. Tokuda, C. Maeda, J. Hasegawa, K. Nozaki and T. Ema, Chem. Sci., 2020, 11, 5669–5675 RSC; (b) C. Maeda, H. Inoue, A. Ichiki, T. Okihara and T. Ema, ACS Catal., 2022, 12, 13042–13049 CrossRef CAS; (c) C. Maeda, K. Kawabata, K. Niki, Y. Sako, T. Okihara and T. Ema, Polym. Chem., 2023, 14, 4338–4343 RSC.
- T. Ema, Bull. Chem. Soc. Jpn., 2023, 96, 693–701 CrossRef CAS.
- Selected examples: (a) Y.-Y. Zhang, G.-W. Yang and G.-P. Wu, Macromolecules, 2018, 51, 3640–3646 CrossRef CAS; (b) H.-J. Zhou, G.-W. Yang, Y.-Y. Zhang, Z.-K. Xu and G.-P. Wu, ACS Nano, 2018, 12, 11471–11480 CrossRef CAS PubMed; (c) Y. Zhao, Y. Wang, X. Zhou, Z. Xue, X. Wang, X. Xie and R. Poli, Angew. Chem., Int. Ed., 2019, 58, 14311–14318 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |