
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpression of hyperconjugative stereoelectronic interactions in borazines†
Vivek Chandrakant
Wakchaure
 a,
Jacopo
Dosso
b,
Martina
Crosta
a,
Hanspeter
Kählig
a,
Benjamin D.
Ward
b and
Davide
Bonifazi
*a
a,
Jacopo
Dosso
b,
Martina
Crosta
a,
Hanspeter
Kählig
a,
Benjamin D.
Ward
b and
Davide
Bonifazi
*a
aInstitute of Organic Chemistry, Faculty of Chemistry, University of Vienna, Währinger Strasse 38, 1090, Vienna, Austria. E-mail: davide.bonifazi@univie.ac.at
bSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, UK
First published on 4th December 2024
Abstract
This paper discusses hyperconjugative stereoelectronic effects in borazines. A series of alkyl-substituted borazines were synthesized and analysed by NMR spectroscopy and X-ray diffraction. Supported by NBO analyses, the significant decreases in 1JCH coupling constant for the CH groups adjacent to the boron atoms are consistent with the presence of 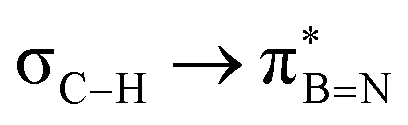 and
and 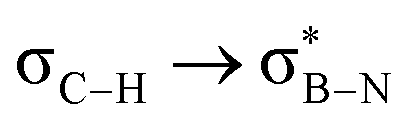 interactions. These interactions lower the electrophilicity of boron atoms, enhancing moisture stability and establishing these molecules as valuable scaffolds in synthetic chemistry and materials science.
interactions. These interactions lower the electrophilicity of boron atoms, enhancing moisture stability and establishing these molecules as valuable scaffolds in synthetic chemistry and materials science.
The significance of BN-doped molecular scaffolds has recently surged due to their broad applications in optoelectronic devices,1–4 thermal management materials,5 and catalysis.6 Among these, hexasubstituted borazine is an attractive option for BN-based architectures.7 The borazine core's susceptibility to hydrolysis in water has hindered its widespread use.8 To prevent borazine hydrolytic decomposition, bulky aryl groups can be added to the boron atom, where ortho-substituents create steric hindrance shielding B-atoms from water.9 In another architecture, amino groups were used to stabilize the borazine ring within a hybrid cyclomatrix polymer.9f In search of other approaches preventing hydrolysis, we noticed that B,B′,B′′-trialkyl-N,N′,N′′-triphenyl borazines (alkyl moieties being i- and n-propyl, i- and n-butyl) exhibit remarkable counterintuitive moisture resistance when compared to its aryl-substituted congeners.10 A comprehensive understanding of the factors affecting the stability of these molecules is still lacking. It is possible that hyperconjugative stereoelectronic interactions occur between the electron-donating alkyl groups and the electrophilic borazine ring (Fig. 1b),11 similar to what is seen in trialkyl boranes.9h,i With this hypothesis in mind, in this paper, we address the presence of such hyperconjugative effects through NMR spectroscopy and X-ray investigations of a series of B-alkyl borazine derivatives.12 Theoretical modeling further supported these studies by elucidating the molecular orbitals involved in those interactions.13 Three types of alkyl groups to be attached to the boron centres were chosen: –CH3, –CH2–, and –CH–. While molecules 1a–b bear methyl substituents, 2a–e and 3a–b feature methylene (as –nBu, –nHex, –nOct, benzyl and (trimethylsilyl)methyl substituents) and methine (as –iPr and cyclohexyl substituents) substitutions on the boron atoms (Fig. 1a), respectively.
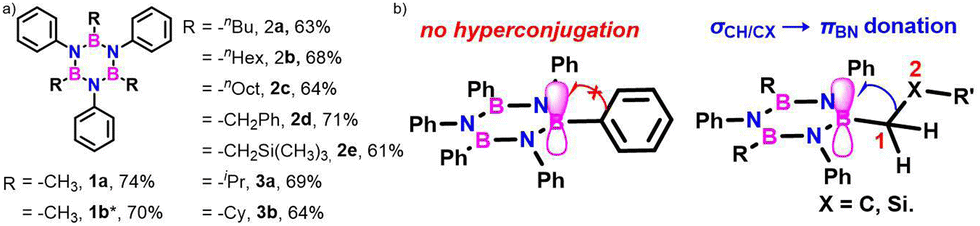 | ||
| Fig. 1 (a) Alkyl borazines studied in this work (*2′,6′-dimethyl-[1,1′-biphenyl]-4-amine used for the synthesis). (b) Stereoelectronic interactions in B-aryl and B-alkyl borazines. | ||
The synthesis for these alkyl borazines have been carried out by adapting known protocols.9c Intermediate trichloroborazole (BNCl) was prepared by refluxing aniline with BCl3 in toluene (Scheme S1, ESI†). The reaction of BNCl with the respective organoalkyl-lithium/Grignard nucleophiles in THF gave the final product in good yields (61–74%, Fig. 1a and Scheme S2, ESI†). The structures of all alkyl borazines were characterized by 1H and 13C NMR spectroscopy, and with high-resolution mass spectrometry (Fig. S1–S48, ESI†) and selected structures by single-crystal X-ray diffraction analysis. All B,B′,B′′-trialkyl borazine derivatives exhibited low reactivity towards moisture, allowing them to be handled in the air without precautions and purified through column chromatography. Thermogravimetric analysis (TGA) indicated that all derivatives remain stable above 200 °C (Fig. S49–S51, ESI†).
X-ray diffraction investigations of selected borazine derivatives were first attempted to shed further light on their structural properties (Fig. 2a–f and Tables S1–S6 and Fig. S52–S57, ESI†). In all derivatives, the central borazine ring displays a nearly flat geometry, with average ∠N–B–N and ∠(B–N–B) angles of 116.2(4)° and 123.7(4)°, respectively, and with typical B–C1 bond lengths of 1.575(14) Å (Fig. 2).11c For molecules 2a, 3a, and 3b, the C1–C2 bond lengths (average value ∼1.526(4) Å) fall within the expected range for corresponding single bonds.11a The average torsional angles ∠(N–B–C1–H/C2/Si), describing the relative orientation of the B–C–H/C/Si bonds to the borazine ring, are ∼90° for all borazines (Fig. 2). This perpendicular arrangement of the C–H/C/Si bonds is to be expected when in the presence of a hyperconjugation. Moreover, the bond angle ∠(B–C1–C2) for 2a, 3a and 3b is found to be 113(2)°, 116(3)° and 118(6)°, respectively. Similarly, the bond angle ∠(B–C1–Si) is 118(2)° for molecule 2e (Fig. 2). These ∠(B–C1–H/C2/Si) angles are wider than those between C(sp3), again suggesting that a hyperconjugative interaction is at play.11a
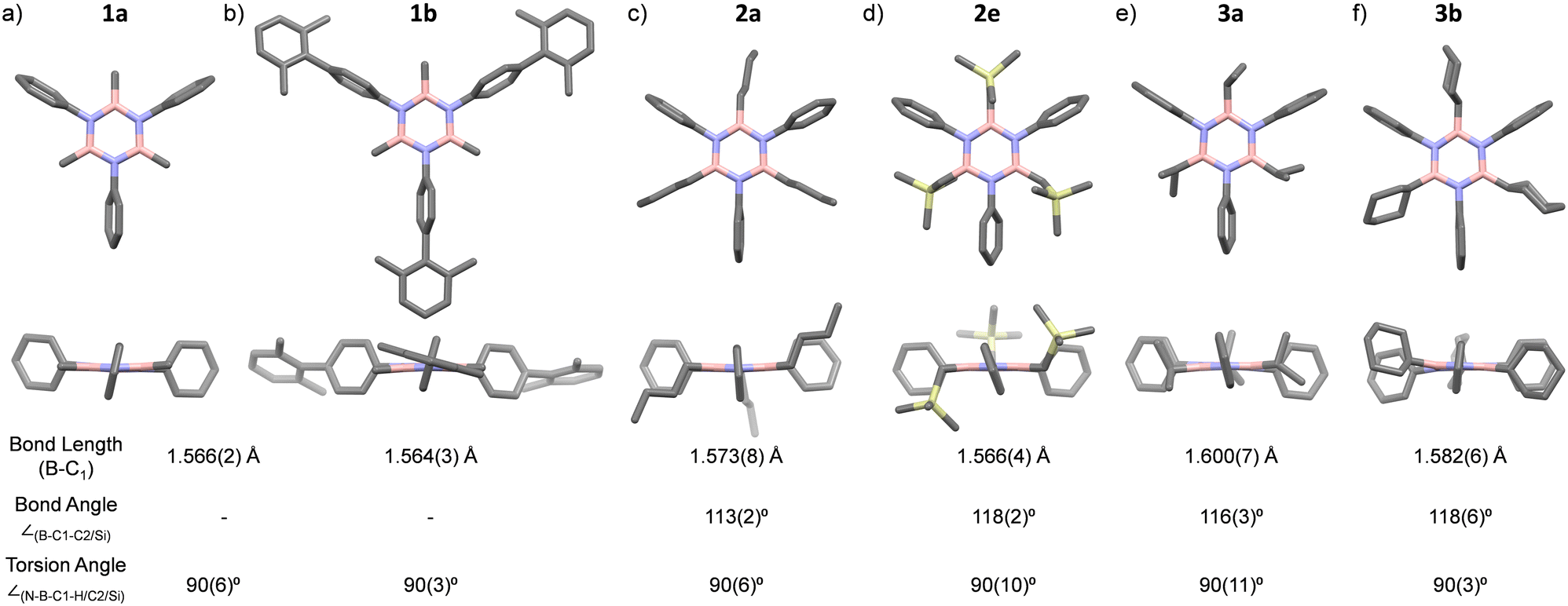 | ||
Fig. 2 Single-crystal X-ray structures of borazines and relevant structural parameters (a) 1a, C2/c, (b) 1b, C2/c, (c) 2a, Cc, (d) 2e, P21/c, (e) 3a, P![[1 with combining overline]](https://www.rsc.org/images/entities/char_0031_0305.gif) , and (f) 3b, Pna21. Grey: C, pink: B, yellow: Si, and blue: N. The torsion angles are the average of the values measured for C–H/C/Si bonds that display an orientation that is close to that of perpendicularity. , and (f) 3b, Pna21. Grey: C, pink: B, yellow: Si, and blue: N. The torsion angles are the average of the values measured for C–H/C/Si bonds that display an orientation that is close to that of perpendicularity. | ||
Considering that one-bond NMR spin–spin coupling constants are experimental probes for determining any stereoelectronic interactions (Perlin effect),14 we tackled the determination of the 1H–13C coupling constants (JCH) of the C–H bonds connected to the B-atoms by analysis of either the carbon satellite signals in the 1H NMR spectra or the doublet cross peaks in proton-coupled Heteronuclear single quantum coherence (HSQC) experiments (Fig. 3). For 1a and 1b, the experimental 1JCH value for the B–Me groups is 116.4 and 116.6 Hz (Fig. 3a), respectively, which is lower than that of the Me group in the same molecule (2JCH = 126.6 Hz, Fig. S8 and S13, ESI†). Similarly, the 1JCH values for molecules 2a–e are within the 112 Hz to 118 Hz range, which is also lower than the other JCH values (Fig. 3b and Fig. S18, S23, S28, S33 and S38, ESI†). Further, by examining the 29Si NMR spectrum of molecule 2e, we could determine the coupling constants 1JCSi for –CH2– and –CH3 groups attached to the Si-atom. The results indicate that 1JCSi of –CH2– exhibits a value of ∼12.3 Hz lower than that measured with –CH3 (Fig. S38, ESI†). This suggests that the C–Si bond experiences an elongation that one could attribute to a hyperconjugation. For 3a and 3b, the 1JCH values are 116.1 and 112.3 Hz, respectively (Fig. 3c and Fig. S43 and S48, ESI†). Notably, these couplings were the lowest among the series. These observations are in line with the X-ray-measured ∠(B–C1–C2) angle values (Fig. 2), confirming strong hyperconjugative interactions between the B(pz) orbital and the σC–H, σC–C and σC–Si bonds of the relevant substituents.
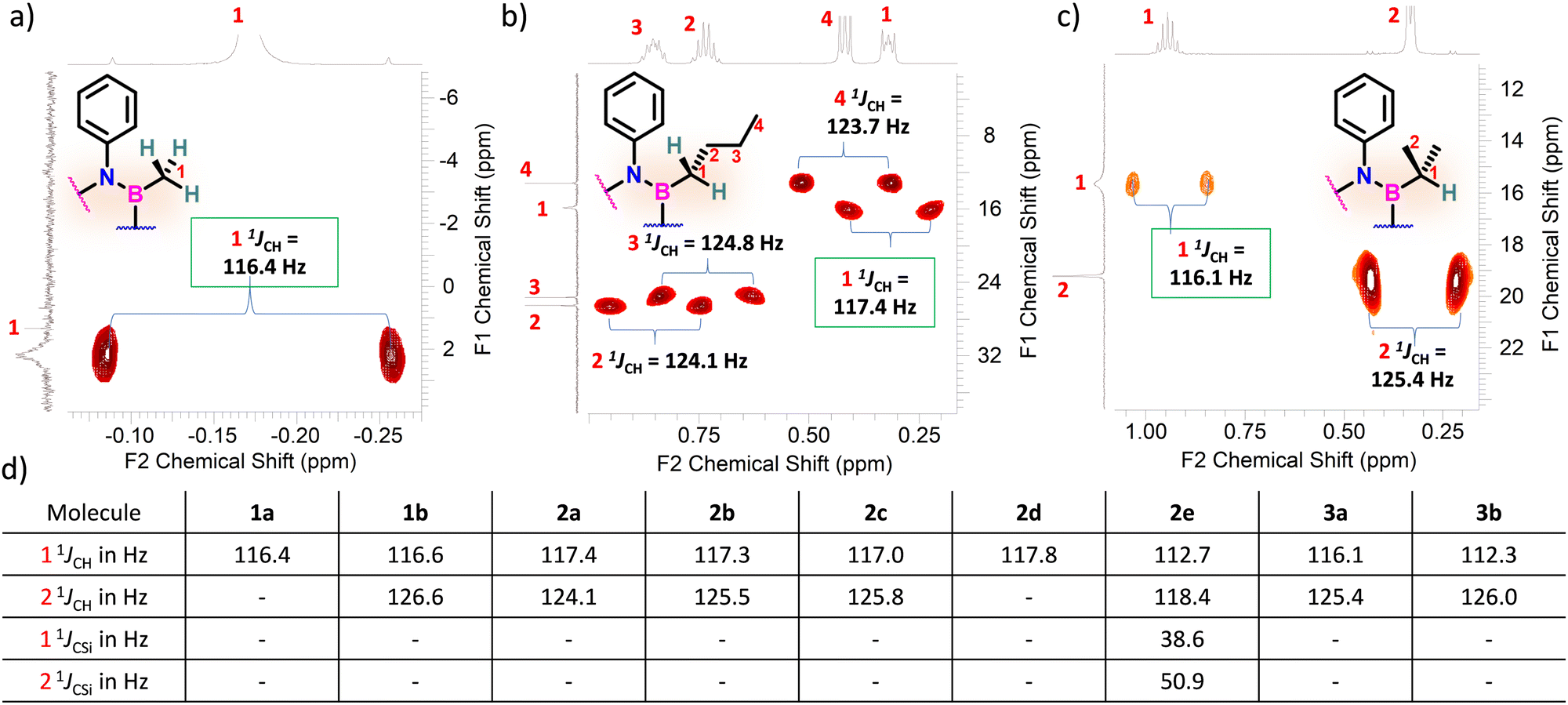 | ||
| Fig. 3 Excerpts of the HSQC spectra displaying the relevant 1JCH coupling constants for (a) 1a, (b) 2a, and (c) 3a and. (d) 1 1JCH, 2 1JCH, 1 1JCSi and 2 1JCSi values for each alkyl borazines. | ||
DFT calculations were employed to explore hyperconjugation, with bonding analysed via natural bonding orbital (NBO) analyses.13c The findings revealed weak interactions between the σC–H and σC–C orbitals and both the 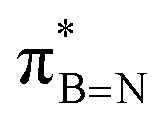 and
and 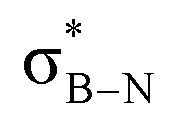 orbitals for all derivatives. The σC–H orbital, positioned perpendicularly to the ring, exhibited significant overlap with the π* orbital. For 1a and 1b, the optimised structure contains a C–H lying orthogonal to the borazine plane (Fig. 2a and b) and two that are at the midpoint between the orthogonal and parallel planes (
orbitals for all derivatives. The σC–H orbital, positioned perpendicularly to the ring, exhibited significant overlap with the π* orbital. For 1a and 1b, the optimised structure contains a C–H lying orthogonal to the borazine plane (Fig. 2a and b) and two that are at the midpoint between the orthogonal and parallel planes (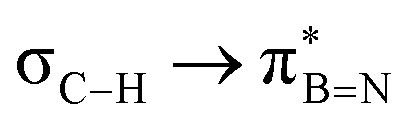 and
and 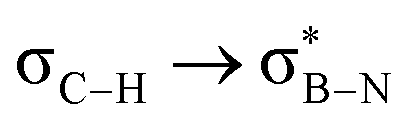 in Fig. 4a and Fig. S58, ESI†). Since the overlap between σC–H and the given unoccupied orbitals depends on the orientation of the substituent, the stabilisation energy (E2) associated with these interactions was computationally probed as a function of the angle between the proximal σC–H/C/Si orbital and the borazine ring upon rotation about the B–C bond (molecule 1a in Fig. 4). As expected, the
in Fig. 4a and Fig. S58, ESI†). Since the overlap between σC–H and the given unoccupied orbitals depends on the orientation of the substituent, the stabilisation energy (E2) associated with these interactions was computationally probed as a function of the angle between the proximal σC–H/C/Si orbital and the borazine ring upon rotation about the B–C bond (molecule 1a in Fig. 4). As expected, the 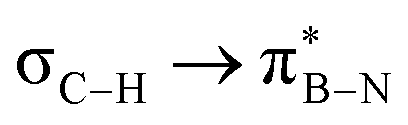 interaction is the strongest, 6.5 kcal mol−1, when the C–H bond lies perpendicular to the BN ring as this orientation gives the most significant orbital overlap between the σC–H and
interaction is the strongest, 6.5 kcal mol−1, when the C–H bond lies perpendicular to the BN ring as this orientation gives the most significant orbital overlap between the σC–H and 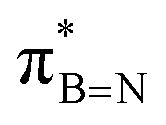 orbitals. As the Me group is rotated, E2 decreases, and no interaction is detected with ∠(N–B–C1–H) values <20°. Conversely, the
orbitals. As the Me group is rotated, E2 decreases, and no interaction is detected with ∠(N–B–C1–H) values <20°. Conversely, the 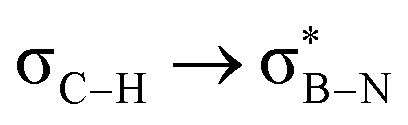 interaction gives the strongest stabilisation, 3.51 kcal mol−1, when the C–H bond lies in the same plane as the BN ring. The effect is not as pronounced as the
interaction gives the strongest stabilisation, 3.51 kcal mol−1, when the C–H bond lies in the same plane as the BN ring. The effect is not as pronounced as the 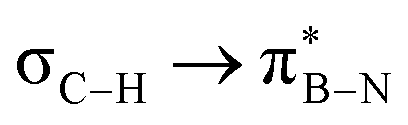 , but it contributes conferring an additional stabilisation. Similarly, NBO analysis shows both
, but it contributes conferring an additional stabilisation. Similarly, NBO analysis shows both 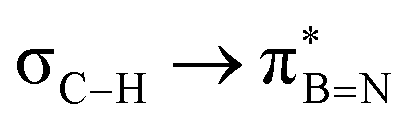 and
and 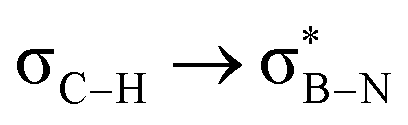 interactions in the case of –CH2– and –CH– (3a–b) substitutions (Fig. S59–S67, ESI†). Also, the stabilisation energies were probed for proximal σC–C/Si bonds (Fig. S61, ESI†).
interactions in the case of –CH2– and –CH– (3a–b) substitutions (Fig. S59–S67, ESI†). Also, the stabilisation energies were probed for proximal σC–C/Si bonds (Fig. S61, ESI†).
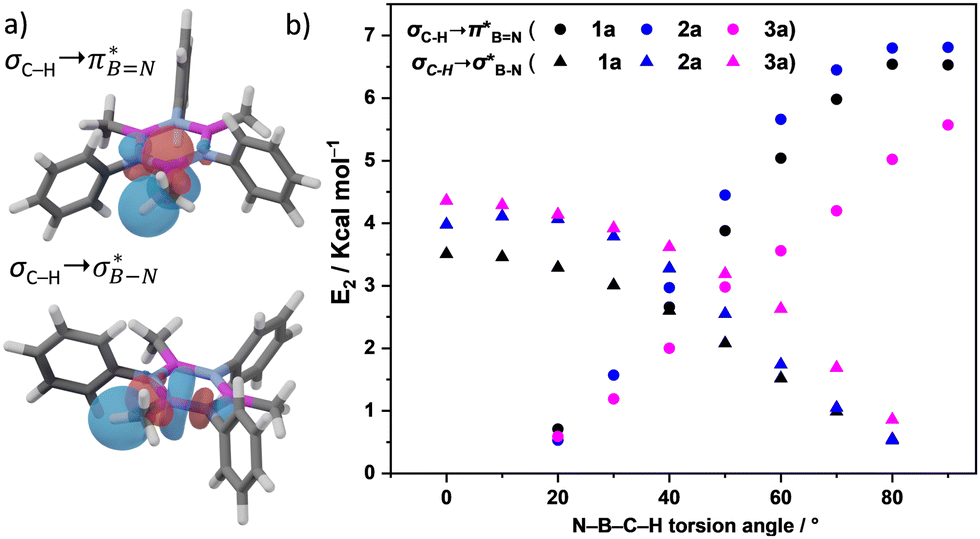 | ||
Fig. 4 (a) NBO for 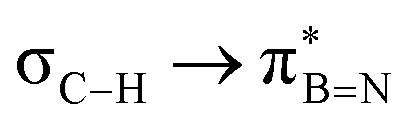 and and 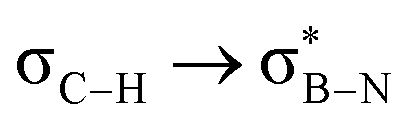 1a (PBE/def2-TZVP, orbitals with only one B-atom are shown), (b) hyperconjugative energies upon rotation of the B-alkyl group. 1a (PBE/def2-TZVP, orbitals with only one B-atom are shown), (b) hyperconjugative energies upon rotation of the B-alkyl group. | ||
Interactions between the σC–C and BN ring orbitals are significant when sterically demanding alkyl groups prevent the C–H group from lying perpendicular to the ring, and 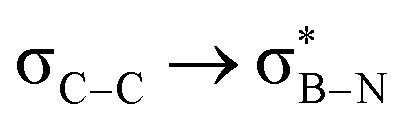 interactions are present in 2a–e and 3a–b (Fig. S60–S66, ESI†) but offered less of a stabilising effect than those involving σC–H (Fig. 4b). Notably,
interactions are present in 2a–e and 3a–b (Fig. S60–S66, ESI†) but offered less of a stabilising effect than those involving σC–H (Fig. 4b). Notably, 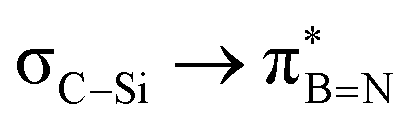 and
and 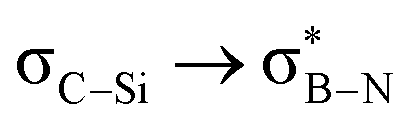 interactions in 2e displayed stronger stabilization than those with σC–C (Fig. S61, ESI†). The maximum E2 value is observed at the torsion angles ∠(N–B–C1–H/C2/Si) of ∼90° and 0° for
interactions in 2e displayed stronger stabilization than those with σC–C (Fig. S61, ESI†). The maximum E2 value is observed at the torsion angles ∠(N–B–C1–H/C2/Si) of ∼90° and 0° for 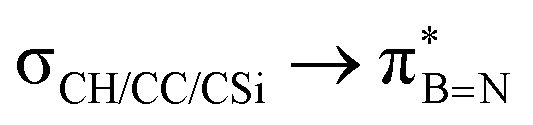 and
and 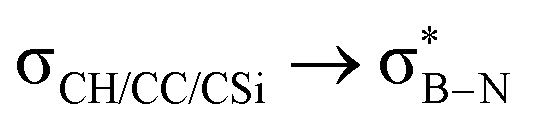 interactions, respectively (Table 1). We evaluate energy changes for 1a using a different basis set and calculation function (Fig. S67, ESI†), confirming the reliability of the data.
interactions, respectively (Table 1). We evaluate energy changes for 1a using a different basis set and calculation function (Fig. S67, ESI†), confirming the reliability of the data.
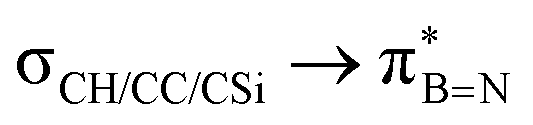 and 0° for
and 0° for 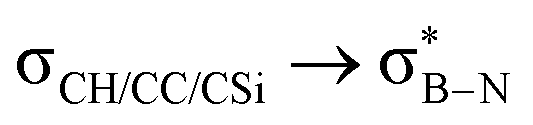 (PBE/def2-TZVP)
(PBE/def2-TZVP)
| Molecule | E 2/kcal mol−1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1a | 1b | 2a | 2b | 2c | 2d | 2e | 3a | 3b | |
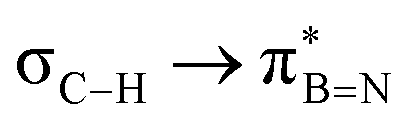
|
6.53 | 6.48 | 6.81 | 6.40 | 6.40 | 5.70 | 5.28 | 5.57 | 4.70 |
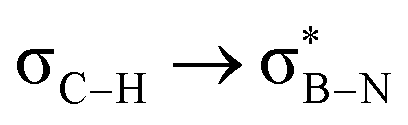
|
3.51 | 3.52 | 3.98 | 3.99 | 3.99 | 3.96 | 4.37 | 4.36 | 4.39 |
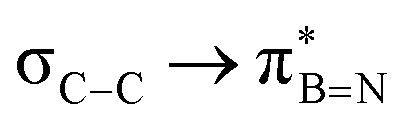
|
— | — | 3.83 | 3.87 | 3.80 | 2.66 | — | 3.85 | 3.47 |
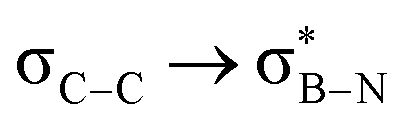
|
— | — | 1.28 | 1.28 | 1.28 | 1.04 | — | 1.52 | 1.41 |
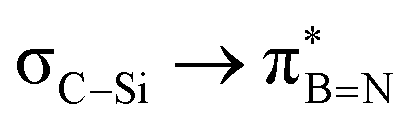
|
— | — | — | — | — | — | 7.94 | — | — |
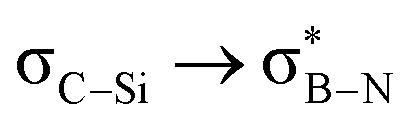
|
— | — | — | — | — | — | 2.22 | — | — |
Further, we calculated the NMR coupling constant using the GIAO method.15 The results (Table S7, ESI†) show that the difference ΔJ between 1JC1H and 1JC2H is 6 to 10 Hz, which is in agreement with the experimental values. Similarly, the calculated ΔJ between 1JC1Si and 1JC2Si in 2e is 12 Hz, matching the experimental data (Table S8, ESI†). We examined the isodesmic reactions with borazine 3, which has Me and Ph moieties on the N and B atoms (ESI,† Section S7.2.3). Our findings revealed higher values for enthalpy (+18.5 kcal mol−1) and Gibbs free energy (+17.4 kcal mol−1) compared to congener 1a, highlighting the significant impact of hyperconjugation. NBO interactions in Me- and Et-substituted benzene congeners (4Me and 4Et) yielded 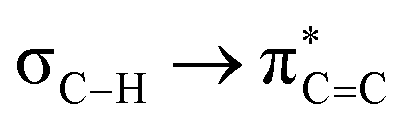 values of 5.1 and 3.4 kcal mol−1. These values indicate that 4Me and 4Et exhibit reduced hyperconjugation than their BN counterparts, as evidenced by their higher 1JC1H and 1JC2H values than those of 1a and 2b, Table S9, ESI†).
values of 5.1 and 3.4 kcal mol−1. These values indicate that 4Me and 4Et exhibit reduced hyperconjugation than their BN counterparts, as evidenced by their higher 1JC1H and 1JC2H values than those of 1a and 2b, Table S9, ESI†).
As a proof of concept exploiting the hyperconjugation to stabilize borazine derivatives, we successfully prepared cross-linked borazine-based polymers9f using either 1,6-dilithium hexane or 1,8-dilithium octane as nucleophiles (Fig. 5a). The insoluble nature of the obtained materials in common organic solvents suggests the presence of a crosslinked structure. Also, it exhibits hydrolytic stability. Solid-state 13C NMR spectra (Fig. 5b and Fig. S68–S70, ESI†) distinctly displayed aromatic and aliphatic signals corresponding to the phenyl ring and alkyl chain on the borazine structure. Scanning electron microscope (SEM) analyses revealed the morphology of P1 and P2 polymers, showing aggregates and macroscopic porosity (Fig. 5c and Fig. S71 and S72, ESI†). TGA analysis indicated approximately 50% weight loss (up to 500 °C), which matches the alkyl moieties’ loss (Fig. S73, ESI†).
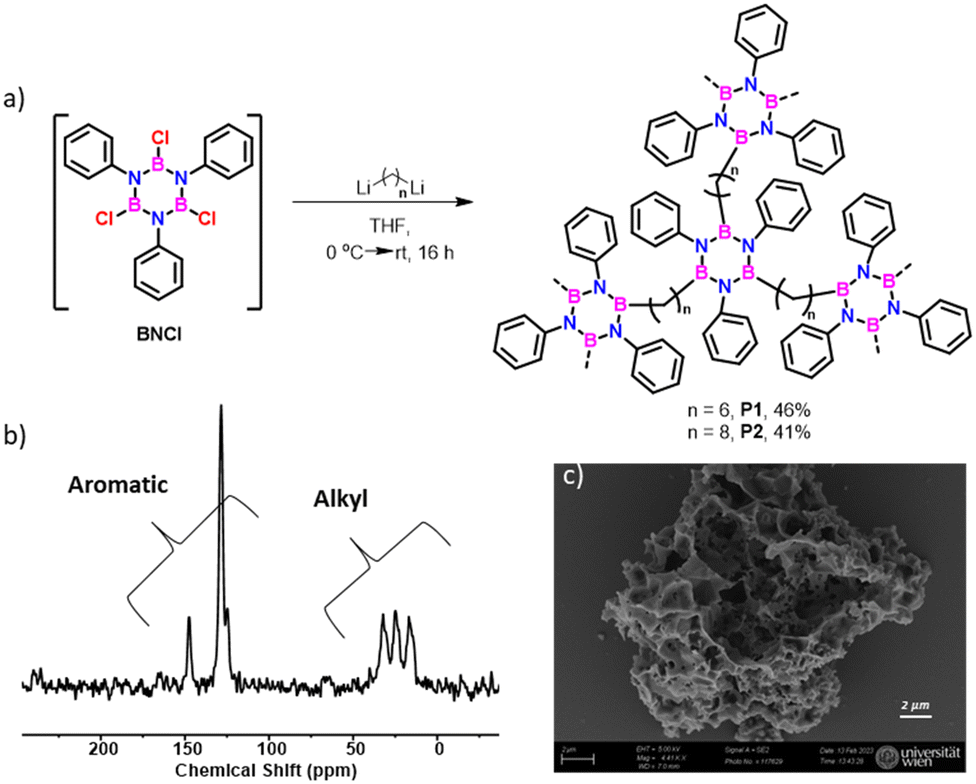 | ||
| Fig. 5 (a) Synthesis of cross-linked polymers P1 and P2, (b) solid-state 1H → 13C cross-polarization and magic angle spinning (CPMAS) NMR spectrum of P1, and (c) SEM images of P1 (see CPMAS NMR spectrum for reference borazine 2a in Fig. S68, ESI†). | ||
In conclusion, our studies into alkyl borazines using NMR spectroscopy have revealed key insights into stereoelectronic interactions. The findings confirm the existence of interactions between the C1–H, C1–C2, and C1–Si bonds of the B-alkyl group with the 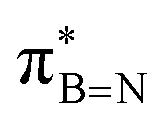 orbital of the BN ring. Computational analyses provide a nuanced understanding of the interplay between the σC–H/C/Si orbitals and both the
orbital of the BN ring. Computational analyses provide a nuanced understanding of the interplay between the σC–H/C/Si orbitals and both the 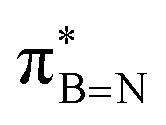 and
and 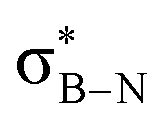 orbitals. As proof of principle, we have developed stable crosslinked borazine polymers that demonstrate moisture resistance. Our findings enhance the knowledge of borazines and expand their applications, offering exciting opportunities for using inorganic benzene in new materials and advanced chemistry.
orbitals. As proof of principle, we have developed stable crosslinked borazine polymers that demonstrate moisture resistance. Our findings enhance the knowledge of borazines and expand their applications, offering exciting opportunities for using inorganic benzene in new materials and advanced chemistry.
D. B. acknowledges the EU (MSCA-ITN-ETN, STiBNite, no. 956923 & HORIZON-IA, DecoChrom, no. 760973) and the University of Vienna for funding; we acknowledge access to the HPC service (ARCCA) at Cardiff University, we thank Dr D. Romito (Vienna) and Dr P. K. Mondal (Elettra Trieste) for help in some X-ray analyses.
Data availability
The data supporting this article are in the ESI,† while the original data are available from the authors upon request.Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) I. H. T. Sham, C. C. Kwok, C. M. Che and N. Zhu, Chem. Commun., 2005, 3547–3549 RSC; (b) D. Bonifazi, F. Fasano, M. M. Lorenzo-Garcia, D. Marinelli, H. Oubaha and J. Tasseroul, Chem. Commun., 2015, 51, 15222–15236 RSC; (c) D. Marchionni, S. Basak, A. N. Khodadadi, A. Marrocchi and L. Vaccaro, Adv. Funct. Mater., 2023, 33, 2303635 CrossRef CAS; (d) I. Neogi and A. M. Szpilman, Synthesis, 2022, 1877–1907 CrossRef CAS.
- (a) Z. Huang, S. Wang, R. D. Dewhurst, N. V. Ignat‘ev, M. Finze and H. Braunschweig, Angew. Chem., Int. Ed., 2020, 59, 8800–8816 CrossRef CAS PubMed; (b) S. Madayanad Suresh, D. Hall, D. Beljonne, Y. Olivier and E. Zysman-Colman, Adv. Funct. Mater., 2020, 30, 1908677 CrossRef CAS.
- (a) J. Dosso, J. Tasseroul, F. Fasano, D. Marinelli, N. Biot, A. Fermi and D. Bonifazi, Angew. Chem., Int. Ed., 2017, 56, 4483–4487 CrossRef CAS PubMed; (b) S. Oda and T. Hatakeyama, Bull. Chem. Soc. Jpn., 2021, 94, 950–960 CrossRef CAS; (c) J. Kashida, Y. Shoji, Y. Ikabata, H. Taka, H. Sakai, T. Hasobe, H. Nakai and T. Fukushima, Angew. Chem., Int. Ed., 2021, 60, 23812–23818 CrossRef CAS PubMed.
- (a) S. Wang, Coord. Chem. Rev., 2001, 215, 79–98 CrossRef CAS; (b) Y.-L. Rao and S. Wang, Inorg. Chem., 2011, 50, 12263–12274 CrossRef CAS PubMed.
- (a) M. R. H. Mazumder, L. D. Mathews, S. Mateti, N. V. Salim, J. Parameswaranpillai, P. Govindaraj and N. Hameed, Appl. Mater. Today, 2022, 29, 101672 CrossRef; (b) Y. Luo, L. Zou, J. Qiao, J. Zhang, K. Liu, H. Wu, P. Lin and Y. Chen, ACS Appl. Energy Mater., 2022, 5, 11591–11603 CrossRef CAS.
- A. K. Thakur, K. Kurtyka, M. Majumder, X. Yang, H. Q. Ta, A. Bachmatiuk, L. Liu, B. Trzebicka and M. H. Rummeli, Adv. Mater. Interfaces, 2022, 9, 2101964 CrossRef CAS.
- (a) A. Wakamiya, T. Ide and S. Yamaguchi, J. Am. Chem. Soc., 2005, 127, 14859–14866 CrossRef CAS PubMed; (b) S. Kervyn, N. Kalashnyk, M. Riello, B. Moreton, J. Tasseroul, J. Wouters, T. S. Jones, A. De Vita, G. Costantini and D. Bonifazi, Angew. Chem., Int. Ed., 2013, 52, 7410–7414 CrossRef CAS PubMed; (c) J. Dosso, T. Battisti, B. D. Ward, N. Demitri, C. Hughes, A. P. Williams, K. D. M. Harris and D. Bonifazi, Chem. – Eur. J., 2020, 26, 6608–6621 CrossRef CAS PubMed.
- T. Yoshizaki, H. Watanabe and T. Nakagawa, Inorg. Chem., 1968, 7, 422–429 CrossRef CAS.
- (a) K. Nagasawa, Inorg. Chem., 1966, 5, 442–445 CrossRef CAS; (b) S. Kervyn, O. Fenwick, F. Di Stasio, Y. S. Shin, J. Wouters, G. Accorsi, S. Osella, D. Beljonne, F. Cacialli and D. Bonifazi, Chem. – Eur. J., 2013, 19, 7771–7779 CrossRef CAS PubMed; (c) D. Marinelli, F. Fasano, B. Najjari, N. Demitri and D. Bonifazi, J. Am. Chem. Soc., 2017, 139, 5503–5519 CrossRef CAS PubMed; (d) N. Kalashnyk, P. Ganesh Nagaswaran, S. Kervyn, M. Riello, B. Moreton, T. S. Jones, A. De Vita, D. Bonifazi and G. Costantini, Chem. – Eur. J., 2014, 20, 11856–11862 CrossRef CAS PubMed; (e) J. Dosso, D. Marinelli, N. Demitri and D. Bonifazi, ACS Omega, 2019, 4, 9343–9351 CrossRef CAS PubMed; (f) N. A. Riensch, A. Deniz, S. Kühl, L. Müller, A. Adams, A. Pich and H. Helten, Polym. Chem., 2017, 8, 5264 RSC; (g) J. R. Sanzone, C. T. Hu and K. A. Woerpel, J. Am. Chem. Soc., 2017, 139, 8404–8407 CrossRef CAS PubMed; (h) A. R. Cowley, A. J. Downs, S. Marchant, V. A. Macrae, R. A. Taylor and S. Parsons, Organometallics, 2005, 24, 5702–5709 CrossRef CAS; (i) B. Wrackmeyer and O. L. Tok, Z. Naturforsch. B, 2005, 60, 259–264 CrossRef CAS.
- (a) S. J. Groszos and S. F. Stafiej, J. Am. Chem. Soc., 1958, 80, 1357–1360 CrossRef CAS; (b) J. H. Smalley and S. F. Stafiej, J. Am. Chem. Soc., 1959, 81, 582–586 CrossRef CAS; (c) R. J. Brotherton and A. L. McCloskey, Adv. Chem., 1964, 42, 131–138 CAS; (d) I. B. Atkinson, D. C. Blundell and D. B. Clapp, J. Inorg. Nucl. Chem., 1972, 34, 3037–3041 CrossRef CAS.
- (a) A. H. Maulitz, P. Stellberg and R. Boese, J. Mol. Struct.: THEOCHEM, 1995, 338, 331–340 CrossRef; (b) I. V. Alabugin, Stereoelectronic Effects: A Bridge Between Structure and Reactivity, Wiley, Hoboken, 2016 CrossRef; (c) R. Boese, A. H. Maulitz and P. Stellberg, Chem. Ber., 1994, 127, 1887–1889 CrossRef CAS.
- (a) G. Cuevas and E. Juaristi, J. Am. Chem. Soc., 2002, 124, 13088–13096 CrossRef CAS PubMed; (b) R. Boese, D. Blaser, N. Niederprum, M. Nusse, W. A. Brett, P. V. R. Schleyer, M. Buhl and N. J. R. V. E. Hommes, Angew. Chem., Int. Ed. Engl., 1992, 31, 314–316 CrossRef.
- (a) I. V. Alabugin, J. Org. Chem., 2000, 65, 3910–3919 CrossRef CAS PubMed; (b) I. V. Alabugin and T. A. Zeidan, J. Am. Chem. Soc., 2002, 124, 3175–3185 CrossRef CAS PubMed; (c) I. V. Alabugin, G. P. Gomes and M. Abdo, Comput. Mol. Sci., 2018, 9, e1389 CrossRef; (d) Y. Mo, H. Jiao and P. V. R. Schleyer, J. Org. Chem., 2004, 69, 3493–3499 CrossRef CAS PubMed; (e) F. Weinhold and C. R. Landis, Valency and bonding: a natural bond orbital donor-acceptor perspective, Cambridge University Press, Cambridge, UK, 2005 Search PubMed.
- (a) A. S. Perlin and B. Casu, Tetrahedron Lett., 1969, 10, 2921–2924 CrossRef; (b) S. Wolfe, B. M. Pinto, V. Varma and R. Y. N. Leung, Can. J. Chem., 1990, 68, 1051–1062 CrossRef CAS; (c) E. Juaristi and G. Cuevas, Acc. Chem. Res., 2007, 40, 961–970 CrossRef CAS PubMed.
- X. S. Monfort and O. Eisenstein, Polyhedron, 2006, 25, 339–348 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2261150, 2261151, 2280127, 2280128, 2284380 and 2307378. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc05188b |
| This journal is © The Royal Society of Chemistry 2025 |