
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insight into the influence of Re and Cl on Ag catalysts in ethylene epoxidation†
Claudia J.
Keijzer
 a,
Pim T.
Weide
a,
Kristiaan H.
Helfferich
a,
Justyna
Zieciak
b,
Marco
de Ridder
b,
Remco
Dalebout
a,
Tracy L.
Lohr
c,
John R.
Lockemeyer
d,
Peter
van den Brink
b and
Petra E.
de Jongh
*a
a,
Pim T.
Weide
a,
Kristiaan H.
Helfferich
a,
Justyna
Zieciak
b,
Marco
de Ridder
b,
Remco
Dalebout
a,
Tracy L.
Lohr
c,
John R.
Lockemeyer
d,
Peter
van den Brink
b and
Petra E.
de Jongh
*a
aMaterials Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, The Netherlands. E-mail: P.E.deJongh@uu.nl
bShell Global Solutions International, Amsterdam, The Netherlands
cShell Catalysts & Technologies, Houston, Texas, USA
dShell Global Solutions US Inc. Houston, Texas, USA
First published on 6th November 2024
Abstract
Commercial ethylene epoxidation catalysts consist of α-alumina supported Ag particles and usually contain a mixture of promoters. High selectivity catalysts typically include a small amount of rhenium species. We studied a series of Ag catalysts promoted with Re loadings up to 4 at% (Re/(Re + Ag)), which is intentionally higher than in optimized commercial catalysts to facilitate characterization and to amplify the influence on catalysis. Sequential impregnation brought Re and Ag in such close contact that they formed a new characterized phase of AgReO4. Chemisorption experiments showed that both ReOx and AgReO4 species act as a reversible reservoir for O2. Ethylene epoxidation was performed without and with the industrially crucial ethyl chloride promoter in the feed. Without the chloride (Cl), the ethylene oxide selectivity increased when Re was present, whereas the combination of Re and Cl decreased the ethylene oxide selectivity at higher Re loadings. Systematic ethylene oxide isomerization experiments revealed that Re and Cl individually inhibit the isomerization on the Ag surface. However, Re and Cl combined increased the isomerization, which can be explained by the surface becoming overly electrophilic. This hence shows the importance of studying promoters both individually and combined.
1. Introduction
Ethylene oxide (EO) is the primary building block for poly(ethylene glycol), which is used for making a myriad of chemicals such as antifreeze agents, insulating materials, and pharmaceuticals.1–3 EO is produced by epoxidation of ethylene, which is catalyzed by Ag particles on α-Al2O3.4,5 Ag displays unique activity and selectivity towards ethylene oxide compared to other metals due to its moderate oxidizing behavior.6 During ethylene epoxidation conditions, the Ag surface is partially oxidized with either electrophilic (weakly bound) or nucleophilic (strongly bound) oxygen.7 It is generally believed that electrophilic oxygen promotes the selective oxidation of ethylene to ethylene oxide, whereas nucleophilic oxygen results in total combustion.8–10 Ethylene oxide can isomerize to acetaldehyde, which is catalyzed by support hydroxyl groups, hence an inert support material such as α-alumina is used to suppress this side-reaction.11 Ag itself was also found to isomerize EO to acetaldehyde.12 Acetaldehyde subsequently oxidizes to CO2 and water on the Ag surface. The selectivity towards ethylene oxide depends on the ratio between the various reaction rates and the ratio between active surface and support sites.11,13With CO2 as main side-product (0.2–0.3 Mt per Mt EO),2 it is of great importance to understand how the EO selectivity is influenced, and hence optimized. In industry, ethylene epoxidation runs at relatively low ethylene conversions (7–15%),3 since the EO selectivity inversely depends on the ethylene conversion due to subsequent side reactions.14 Another approach to increase selectivity is to introduce promoters to the catalyst. Gaseous organochlorides such as ethyl or vinyl chloride decompose to Cl species on the Ag surface, blocking oxygen vacant sites which suppresses the overall activity.15 In addition, Cl was found to increase the ratio between the concentration of electrophilic oxygen and nucleophilic oxygen on the Ag surface, which is beneficial for the selective oxidation pathway.7
Besides gaseous chloride compounds, a variety of solid promoters are typically used.16,17 In general, catalysts classified as “high activity” in industrial terminology contain alkali promoters and so-called “high selectivity” catalysts contain a mixture of rhenium and alkali species.3 The mechanism behind Re promotion in ethylene oxide catalysts has been a topic of debate,18–22 but literature addresses Re in combination with Cs and chloride,18,19,22 or only Re without the industrially used chloride promoter.21 In the latter, it was found that Re2O7 species block Ag sites with high oxygen affinities, inhibiting the formation of nucleophilic oxygen and increasing the EO selectivity.21 In another study with chloride in the feed, Re–Ag catalysts were found to be less active and less selective,18 while Re2O7 increased the amount of electrophilic oxygen. Dellamorte et al. described morphological changes within Ag catalysts with 25 ppm Re and mentioned the use of vinyl chloride in the feed.23 However, catalytic data and a clear understanding of the effect of Re is limited.
In addition, “high selectivity” catalysts contain low Re loadings (35–900 ppm of the total catalyst weight5,17). As a consequence, the effect of Re on (structural) properties of Ag catalysts is difficult to investigate. An intermediate [Ag(μ-ethylenediamine)][ReO4] phase, precipitated from a precursor solution, has been investigated in an earlier study which demonstrated that Re and Ag species can have a close proximity during the preparation of commercial catalysts.20 In this work, we study Ag catalysts with 0–4 at% Re loadings. Using X-ray diffraction (XRD), the formation of AgReO4 is studied, confirming the presence of Re7+ species, but not as the earlier believed Re2O7. Re-promoted Ag catalysts were tested without and with varying amounts of ethyl chloride in the feed to compare with literature. Re in absence of Cl inhibited the ethylene conversion and hence indirectly increased the EO selectivity, whereas Re and Cl together at high concentrations resulted in an overall decrease in selectivity. In addition to ethylene epoxidation experiments, we separately investigated EO isomerization behavior with and without chloride, which is crucial to understand EO selectivity.
2. Materials and methods
2.1 Catalyst preparation
Re-promoted Ag catalysts supported on α-alumina were prepared with sequential incipient wetness impregnation. Prior to the first impregnation, 8 m2 g−1 α-alumina extrudates (BASF, Al-4196, pore volume of 0.45 cm3 g−1) were crushed and sieved to a fraction <212 μm and dried in vacuum at 200 °C for 2 h. Ammonium perrhenate (NH4ReO4, ≥99%, Aldrich Chemistry and Fisher) was dissolved in MilliQ® water, obtaining solutions of 0.016, 0.08 and 0.16 M. The dried α-alumina was impregnated with one of these solutions up to 90% of its pore volume, aiming for final Re loadings of 0.4, 2 and 4 at% compared to the Ag loading (Re/(Ag + Re)). The impregnated material was dried in stagnant air at 60 °C for 20 h. After 15, 30 and 45 min of drying, all materials were mixed thoroughly to promote a homogeneous distribution. The dried samples were transferred to a U-shaped borosilicate reactor and calcined for 2 h at 500 °C (5 °C min−1 ramp) in 25% O2 in N2 (gas hourly space velocity (GHSV) of 7000 h−1).The Ag deposition was based on a procedure described elsewhere.24 Silver oxalate was used as Ag precursor, which was synthesized by adding an aqueous silver nitrate (≥99.0%, Sigma-Aldrich) to an aqueous solution of oxalic acid (≥99.0%, Sigma-Aldrich) in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mol ratio. Silver oxalate precipitated immediately and was washed three times in MilliQ® water and once in ethanol, after which it was dried in air at room temperature. Please note that silver oxalate is shock-sensitive, and should therefore be handled with great care.
:1 mol ratio. Silver oxalate precipitated immediately and was washed three times in MilliQ® water and once in ethanol, after which it was dried in air at room temperature. Please note that silver oxalate is shock-sensitive, and should therefore be handled with great care.
For the deposition of Ag, the (Re-promoted) α-alumina samples were dried in vacuum at 200 °C for 2 h. Silver oxalate was dissolved in a MilliQ®/ethylene diamine (99%, Sigma-Aldrich) solution (4:1 mol ratio). The dried powder was impregnated up to 90% of its pore volume with the Ag2C2O4/MilliQ®/ethylene diamine solution, aiming for a Ag loading of 15 wt%. The pore volume of the Re-promoted α-alumina powder was assumed to be similar as the pure α-alumina. After impregnation, the material was subjected to a similar drying and calcination procedure as described for the Re-impregnation, except that the heating temperature was 215 °C.
AgReO4 was deposited on α-alumina via co-impregnation. Silver oxalate and ammonium perrhenate (Ag:Re mol ratio of 1:1) were dissolved in a MilliQ®/ethylene diamine solution (4:1 mol ratio). Dried α-alumina powder was impregnated with this solution, up to 90% of its pore volume, aiming for a AgReO4 loading of 15 wt%. A similar drying and calcination procedure as for the Ag deposition was performed after impregnation. Afterwards, a second heating step was performed for 2 h at 500 °C (5 °C min−1 ramp) in 25% O2 in N2 (GHSV of 7000 h−1).
2.2 Characterization
Scanning electron microscopy (SEM) was used to determine the Ag particle size of the samples. A FEI Helios G3 UC microscope was operated at 5–10 kV in immersion mode. Double-sided carbon adhesive tabs (Electron Microscopy Sciences) were used to attach the sample powder to the SEM holder, upon which a 7.5 nm PdPt layer was sputtered to create conductivity. For each sample, the diameter of at least 200 Ag particles was measured using ImageJ software. Surface averaged particle diameters (dp,s) with corresponding standard deviations (σp,s) were calculated using eqn (1), with n the number of counted silver particles and di the diameter of particle i. | (1) |
Crystalline phases within the samples were determined with X-ray diffraction (XRD) using a Bruker D2 Phaser equipped with a Co Kα source (λ = 0.1789 nm) and operated at 30 kV and 10 mA, under constant rotation of 15 rpm. Diffractograms were measured between 20–80° 2θ with a step size of 0.020° 2θ and 1 s per step. The measured diffractograms were analyzed with Bruker TOPAS V5 software and fitted with theoretical diffractograms from the PDF-4+ 2016 database. The following PDF cards were used: 04-004-2852 (α-Al2O3), 04-003-5319 (Ag), 04-014-4906 (AgReO4) and 04-004-1280 (ReO2).25
O2 chemisorption was used to determine the O2 uptake of the catalysts, using a Micromeritics ASAP 2020 apparatus. 100–200 mg of sample was loaded in a U-shaped quartz reactor between two layers of quartz wool. The sample was evacuated at 100 °C for 30 min with a heating ramp of 10 °C min−1. Thereafter, the sample was flushed with O2 for 10 min at 100 °C, before increasing the temperature to 215 °C (10 °C min−1). This step was followed by an evacuation step and a treatment with H2 for 60 min, in order to clean the surface. After another evacuation step of 30 min, the O2 chemisorption experiment was performed with an equilibration time of 10 s.
X-ray photoelectron spectroscopy (XPS) analysis was conducted utilizing the advanced Nexsa G2 Surface Analysis System, featuring an X-ray photoelectron spectrometer equipped with a hemispherical energy analyzer and a monochromatic Al Kα source. Operating at 12 keV, the monochromatic Al Kα source underwent optimization, employing a pass energy of 250 eV for the survey scan and 50 eV for the subsequent high-resolution scan. The X-ray spot size was set at 400 μm during the analyses. In preparation for XPS measurements, all samples were processed into pressed powders and affixed to a stainless steel stub using 3 M 666 double-sided tape. A charge neutralizer was used to minimize charging of the sample surfaces. The spectra referenced to the Al 2p peak with a binding energy of 74.4 eV. Further details on the XPS interpretation are listed in section A of the ESI.†
Inductively coupled plasma (ICP) analysis was performed at Mikroanalytisches Laboratorium Kolbe to determine the weight loading of Re and Ag within the catalysts.
2.3 Ethylene epoxidation and ethylene oxide isomerization experiments
Prior to testing the catalysts in the ethylene epoxidation reaction, the catalysts were sieved to a 90–150 μm sieve fraction. In a typical catalytic run, 100 mg catalyst was diluted with 500 mg SiC (212–425 μm), which had been washed in nitric acid (65%, AnalaR Normapur®, 10 mL gSiC−1) and calcined at 800 °C to remove organic and inorganic impurities. The diluted catalyst was loaded in a quartz reactor (4 mm internal diameter) between two layers of quartz wool. The experiments were performed at 215 °C, with 7.5 vol% ethylene, 7.5 vol% oxygen, and ethyl chloride concentrations of 0–3 ppm in balance helium, with total gas flows ranging from 16–66 mL min−1. A fixed ratio was chosen, not optimizing the process conditions for each experiment, as the exact ratio is not critical for the selectivity as shown earlier.26 Reaction products were analyzed with an online Interscience Compact GC, equipped with a Porabond Q column and Molsieve 5 Å column. At room temperature, the reactant concentrations in the feed were evaluated to guarantee the feed composition.Ethylene conversion and ethylene oxide selectivity were determined using eqn (2) and (3), respectively. Average conversions and selectivities were calculated after the catalysts had equilibrated at each EC concentration for 15–20 h. The carbon mass balance was calculated using eqn (4), and was 100 ± 3% for each datapoint.
 | (2) |
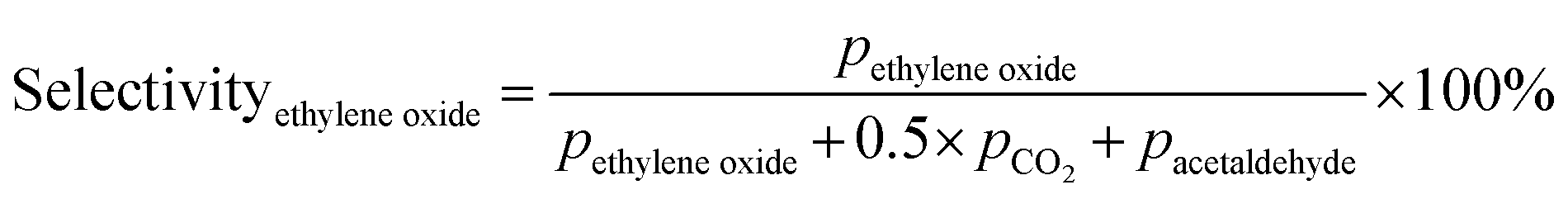 | (3) |
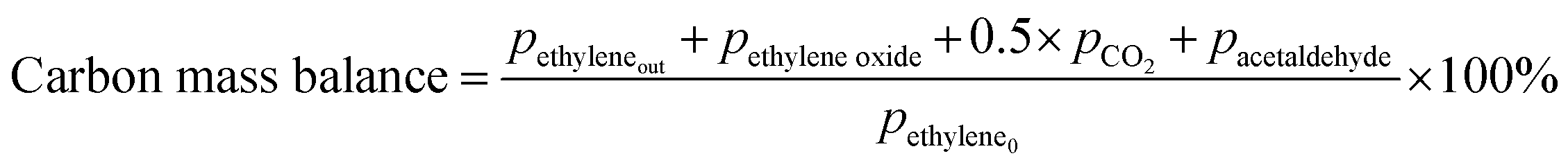 | (4) |
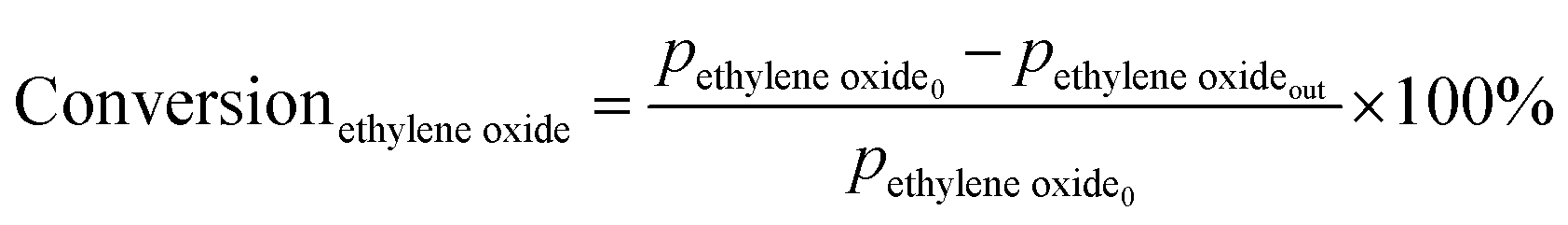 | (5) |
 | (6) |
 | (7) |
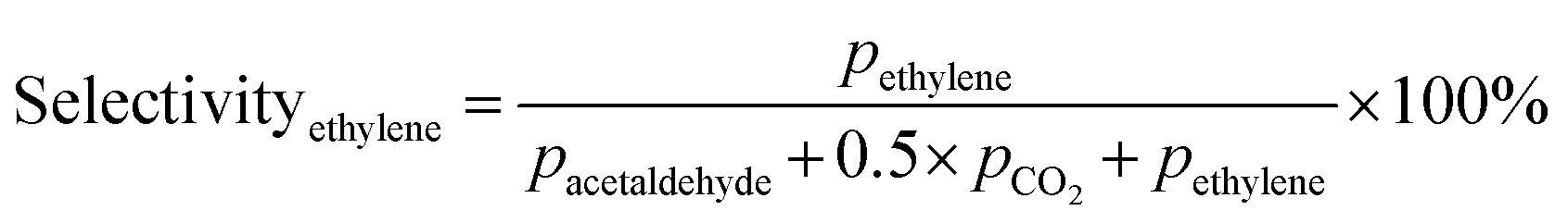 | (8) |
3. Results and discussion
3.1 Catalyst structure
The oxidation state of Re on α-alumina prior to Ag deposition was determined with XPS, as no crystalline ReOx phases within the 4Re sample could be detected with XRD (Fig. S3†). Rhenium can have several oxidation states varying from −3 to +7.27Fig. 1 shows the spectra of Re-promoted α-alumina. The sample names refer to the final Re loading after silver deposition in at% (Re/(Re + Ag)). For all samples, the most dominant Re 4f5/2 and 4f7/2 peaks are around 49 and 46 eV, which is typical for an oxidation state of +7. A minor mismatch between the Re7+ fits and the spectra at lower binding energies suggests the presence of Re6+, with 4f5/2 and 4f7/2 peaks around 47 and 44 eV, respectively. Zoomed-in spectra are shown in Fig. S1.† The Re6+/Re7+ ratio increases from 0.08 to 0.23 when the Re loading decreases from 4 to 0.4 at% (Table S1†), which might be explained by an increasing surface-to-bulk ratio. Nevertheless, Re7+ remains the predominant phase in these samples. Re7+ corresponds to the Re2O7 stoichiometry, which is also reported in literature after calcination of ReOx above 500 °C.28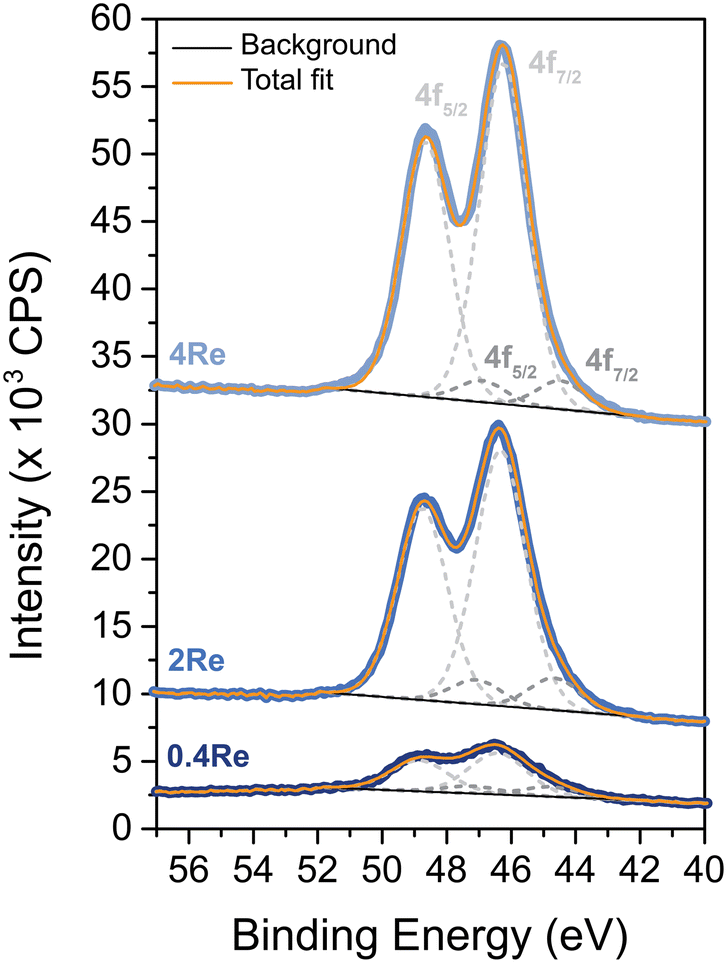 | ||
| Fig. 1 X-ray photoelectron spectra of the Re 4f region of re on α-alumina after calcination at 500 °C. Sample names refer to the final Re loading after Ag deposition in at% (Re/(Re + Ag)). Light grey fits correspond to Re7+ and dark grey fits correspond to Re6+. | ||
Once Ag particles were deposited on the (Re-promoted) α-alumina, SEM was performed. Fig. 2 shows the alumina-supported silver catalysts with rhenium loadings of 0 to 4 at% compared to silver. The silver particles are depicted as white to light grey spheres on the darker grey α-alumina support. Re at such low loadings could not be distinguished from the Ag. The surface averaged particle diameters of the (Re-promoted) Ag catalysts are summarized in Table 1, together with the theoretical silver and rhenium loadings and the experimental loadings measured with ICP and XPS. The particle diameters of all catalysts are 70–80 nm, but increasing the Re loading (above 0.4Re–Ag) slightly narrowed the particle diameter distributions. In literature, Re was reported to cause a trimodal Ag particle size distribution, or to have no effect on particle size at all.18,22,23 In our present study, relatively high loadings of Re were deposited prior to the Ag, which might have increased the concentration of anchoring sites for the Ag precursor. Since the Ag loading and particle sizes are similar, differences in catalyst performance can be ascribed to the influence of Re rather than to Ag particle size effects or a different surface ratio between Ag and α-alumina.13 The experimental loadings determined with ICP were as expected, whereas the Re loading determined with XPS was higher than the ICP measurements. As XPS is a surface sensitive technique, this means that the Re is preferentially located at the surface rather than in the bulk of the material.
 | ||
| Fig. 2 SEM images of Ag (A) and Re-promoted Ag catalysts, with 0.4 at% Re (B), 2 at% Re (C), and 4 at% Re (D). Histograms of particle diameters are shown as insets. For each catalyst more than 200 Ag particles were measured. | ||
| Catalyst | Ag particle diameter dp,s (nm) | Ag loading (wt%) | Re/(Re + Ag) (at%) | |||
|---|---|---|---|---|---|---|
| Theoretical | ICP | Theoretical | Bulk, ICP | Surface, XPS | ||
| Ag | 79 ± 26 | 14.8 | 14.5 | 0 | 0 | 0 |
| 0.4Re–Ag | 80 ± 27 | 14.6 | 14.6 | 0.39 | 0.37 | 3.07 |
| 2Re–Ag | 71 ± 23 | 15.9 | 14.8 | 2.08 | 2.22 | 9.67 |
| 4Re–Ag | 72 ± 15 | 14.7 | 14.3 | 3.87 | 3.55 | 15.27 |
To determine the crystalline phases in the samples, X-ray diffraction was performed. Fig. 3 shows the diffractograms of the unpromoted and Re-promoted Ag catalysts, with reference diffractograms of α-Al2O3 and Ag at the bottom of the Figure. The diffractograms of the Ag and 0.4Re–Ag catalysts only show Ag and α-Al2O3 peaks. For 2Re–Ag and 4Re–Ag, however, a small diffraction line around 32° 2θ was detected, which belongs to AgReO4 which is a thermodynamically stable phase under these conditions (Fig. S14†). Previously, an intermediate phase [Ag(μ-ethylenediamine)][ReO4] had been identified during the preparation of ethylene epoxidation catalysts,20 where Ag and Re precursors were dissolved in the same solution. In another study, AgReO4 colloids were synthesized in solution.29 We show that also sequential impregnation can bring Re and Ag in such close contact that they form a single crystalline phase upon calcination.
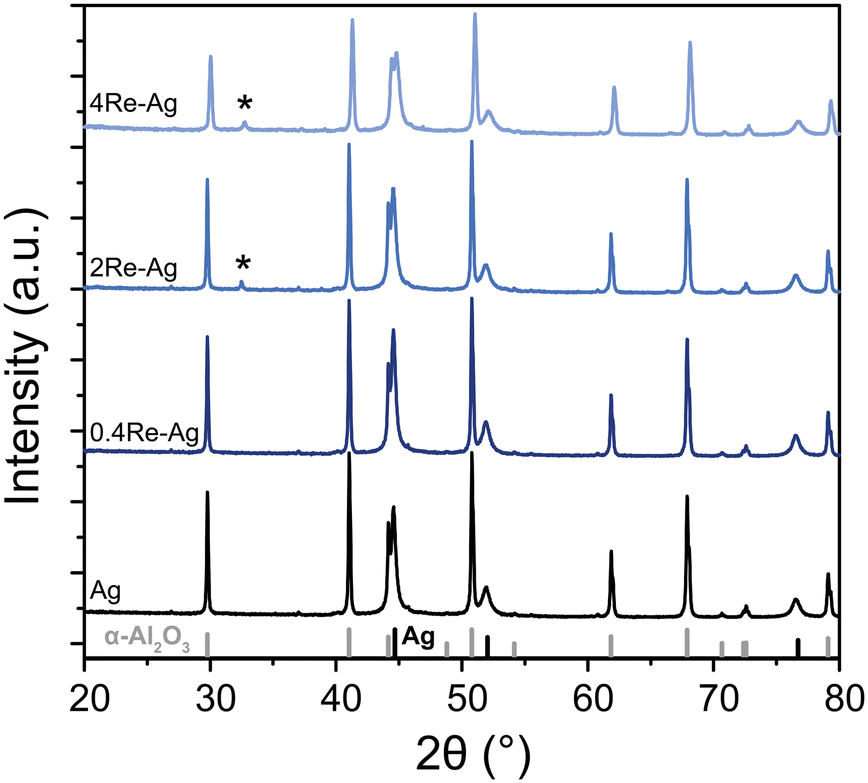 | ||
| Fig. 3 X-ray diffractograms of the Ag and Re-promoted Ag catalysts. Theoretical stick diffraction patterns of α-Al2O3 (grey) and Ag (black) are depicted below the diffractograms as reference. The asterisk (*) indicates AgReO4. | ||
The Re in the samples containing Ag (and thus AgReO4) has a predominant oxidation state of +7 according to XPS (Fig. S1 and Table S1†). In literature, a similar oxidation state has been reported, but it was attributed to Re2O7 rather than to AgReO4.18 Prolonging the calcination time from 2 to 12 h increased the amount of crystalline AgReO4 from 0.5 to 0.8 wt% in the sample (Fig. S4 and Table S3†). Re2O7 has a relatively low melting temperature of 297 °C,30 which might explain its mobility on the α-Al2O3 support and contact with the Ag surface during calcination. In patent literature, Re-promoted Ag catalysts are conditioned for several hours in an O2-containing feed without ethylene.31,32 It is hence expected that AgReO4 species also form in commercial catalysts with less Re, although in a lower concentration and hence more difficult to detect.
Industrial Ag catalysts contain low Re loadings (35–900 ppm of the total catalyst weight5,17), and consequently the influence of Re on structural properties is difficult to investigate. By increasing the Re loading, characterization becomes feasible, and effects on catalysis are amplified, which can give insight into the working mechanism of Re as a promoter. Therefore, once AgReO4 was detected in the Re-promoted Ag catalysts, 15 wt% AgReO4 was deposited on α-alumina to further investigate the properties of this phase. After calcination treatments at 215 °C and 500 °C, X-ray diffractograms, SEM, and STEM-EDX images were collected (Fig. 4). At 215 °C no crystalline AgReO4 had formed, whereas at 500 °C intense diffraction lines due to AgReO4 are visible. Calculated crystallite sizes are ca. 70 nm, which is also in the upper limit of the XRD due to experimental line broadening, whereas the AgReO4 particles visible in SEM are 100–500 nm and are shaped irregularly (Fig. 4B) which implies that these particles contain multiple crystalline domains. STEM-EDX maps of Al–Re (Fig. 4C) and Al–Ag (Fig. 4D) confirm that Ag and Re are in close proximity, but also small Ag particles are present on the α-Al2O3 support. In contrast, XRD does not show crystalline Ag peaks, possibly due to the detection limit, which suggests that the bulk is mostly AgReO4. AgReO4 colloids have been reported earlier,29 but to our knowledge have never been deposited on a support material.
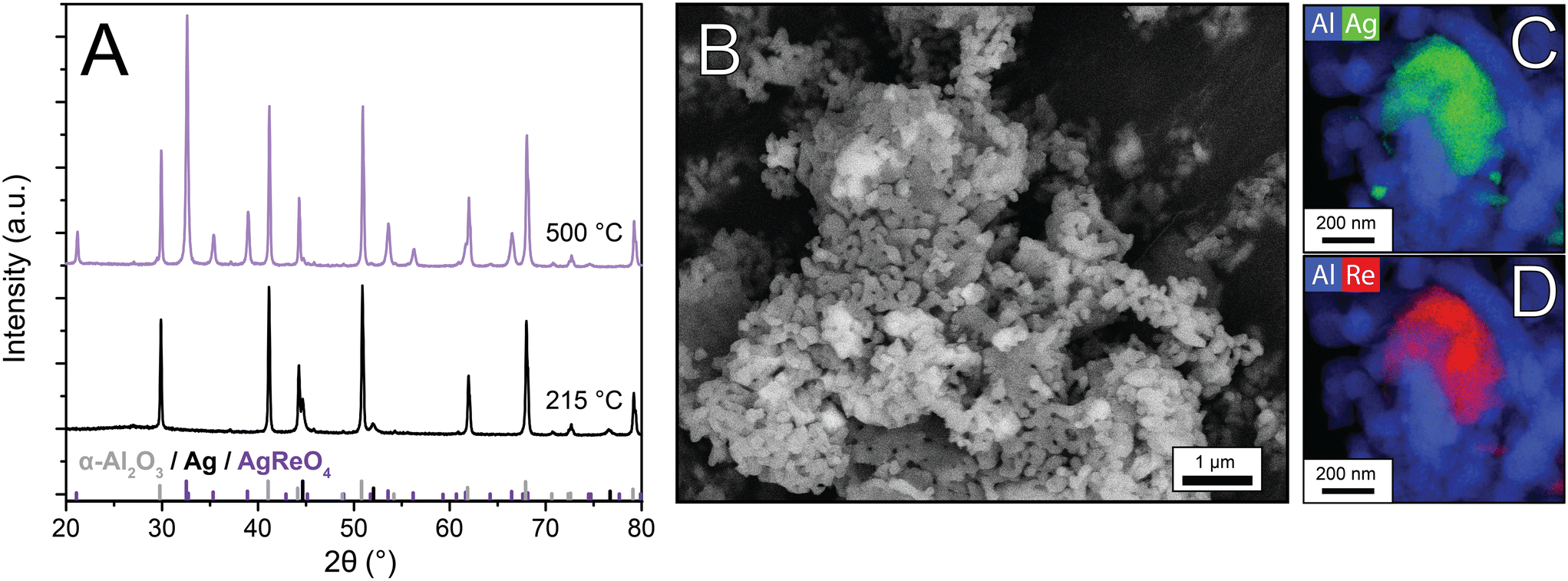 | ||
| Fig. 4 X-ray diffractograms of the AgReO4 samples calcined at 215 and 500 °C, with theoretical stick diffraction patterns of α-Al2O3, Ag and AgReO4 below (A), SEM image of AgReO4/α-Al2O3 calcined at 500 °C (B) and STEM-EDX maps of a zoomed in AgReO4/α-Al2O3 particle (C and D). | ||
3.2 Effect of Re on O2 uptake
During ethylene epoxidation, both the adsorption of oxygen on the silver surface and the subsequent reaction of ethylene with the adsorbed oxygen can be rate-limiting steps.33,34 Hence the adsorption of oxygen is an important parameter to consider.35 Re is a redox-active element with oxidation states between −3 to +7,27 and in the case of pure rhenium oxides this corresponds to an oxygen/rhenium ratio varying between 0 and 3.5. The effect of Re on O2 uptake was investigated using O2 chemisorption at 215 °C, which is the same temperature used for catalytic testing. Table 2 summarizes the O2 uptake of the (Re-promoted) catalysts, prior to and after Ag deposition. It should be noted that the samples were reduced at 215 °C in H2 for 60 min prior to the O2 chemisorption analysis to clean the surface, following a protocol developed to characterize the active silver surface.36 O2 isotherms are shown in Fig. S6 and S7.† For comparison, the fraction of crystalline AgReO4 in the samples determined with XRD is also listed.| Sample | Crystalline AgReO4a (wt%) | Total O2 uptake (μmolO2 gsample−1) | Total O2 uptake – O2 uptake by Agb (μmolO2 gsample−1) |
|---|---|---|---|
| a Crystalline fraction of the sample, determined with XRD after 2 h calcination at 215 °C except for sample AgReO4 which was subsequently calcined at 500 °C. b The O2 uptake of Ag was subtracted from the total O2 uptake of the Re–Ag catalysts, assuming the O2 uptake by Ag was similar in these catalysts. For the Re and AgReO4 samples no crystalline Ag was present, hence no subtraction was performed. | |||
| 2Re | 0 | 18.6 | 18.6 |
| 4Re | 0 | 37.6 | 37.6 |
| Ag | 0 | 16.3 | 0 |
| 0.4Re–Ag | 0 | 17.0 | 0.7 |
| 2Re–Ag | 0.28 ± 0.02 | 45.5 | 29.2 |
| 4Re–Ag | 0.48 ± 0.03 | 74.5 | 58.2 |
| AgReO4 | 10.78 ± 0.05 | 171.8 | 171.8 |
The 2Re and 4Re samples both contain Re2O7 and show significant O2 uptake, which can be explained by the reoxidation of the Re-containing samples after the treatment in H2 to clean the surface. In case of 2Re–Ag and 4Re–Ag, the O2 uptake is higher than of the individual Ag and Re samples combined. The formation and/or presence of AgReO4 in these samples might explain the increased O2 uptake. 0.4Re–Ag has a similar uptake as the Ag sample (Fig. S7†) and no detectable AgReO4 with XRD. The AgReO4 sample showed the highest O2 uptake of 172 μmolO2 gsample−1 with a corresponding 10.78 wt% crystalline AgReO4. It is clear from these experiments that both ReOx and AgReO4 species are a reversible reservoir for O.
Repeating the measurement on the AgReO4 sample resulted in a lower O2 uptake of 107 μmol gsample−1 (Fig. S8†). During the chemisorption experiments the samples are treated in H2 at 215 °C to clean the surface, and according to XRD this resulted in a decrease of crystalline AgReO4 from 10.8 to 6.6 wt% and the formation of 1.3 wt% Ag (Fig. S6 and Table S3†), which explains the lower O2 uptake. XPS measurements were conducted after subjecting the AgReO4, 4Re–Ag and 4Re samples to similar gas treatments used for the chemisorption experiments. After reduction, the Re was reduced, containing Re in oxidation states down to 0, but after re-oxidation almost all Re7+ was regained for the AgReO4 and 4Re–Ag samples (Fig. S2 and Table S2†). AgReO4 shows interesting behavior in these reduction and oxidation cycles, which might be relevant for other reactions that require redox-active sites, and for the role of Re as a promoter for the Ag-catalyzed ethylene epoxidation.
3.3 Ethylene epoxidation
The Re-promoted Ag catalysts were tested in the ethylene epoxidation reaction. In this reaction, a chloride promotor is typically co-fed in the reactant gas feed to increase the ethylene oxide selectivity by modifying the nature of the active silver sites.3,7 Depending on the composition of solid promotors (alkali only or rhenium with alkali), different chloride concentrations show optimal activity and selectivity results, which is often not taken into account in academic studies but essential to compare different promoter compositions.3 Therefore, the catalysts were evaluated with varying chloride levels. Fig. 5A shows the ethylene conversion versus ethyl chloride concentration. The effect of rhenium is most pronounced without ethyl chloride, which is in line with literature:37 the conversion decreases with an order of magnitude upon increasing Re, and AgReO4 does not show a significant catalytic activity. Raw catalytic data are shown in Fig. S9.† In literature, Ag catalysts promoted with small amounts of Re (and no Cl) also have a decreased activity compared to Ag-only catalysts.21 Possibly, certain Ag sites are blocked by dispersed [ReO4]− species. In the AgReO4 sample with crystalline AgReO4 it might be that no metallic Ag remains due to the 1:1 ratio between Ag and Re. The crystalline AgReO4 loading is unaltered after catalysis with EC (Table S3†), which shows that this phase is stable in ethylene epoxidation reaction conditions. The particle diameters for all Re-promoted Ag catalysts increased from 70–80 nm to approximately 90 nm after ethylene epoxidation (Fig. S18†), which implies that Re promotion did not significantly affect the stability of the Ag particles.
 | ||
| Fig. 5 Ethylene conversion (A) and EO selectivity (B) as a function of the ethyl chloride concentration, at 215 °C and in a total gas flow of 66 mL min−1. (C) EO selectivity as a function of ethylene conversion at different EC concentrations, assigned with different symbols. | ||
Ethylene conversion decreases for all catalysts upon increasing the chloride concentration. In literature, catalysts promoted with both Re and Cs are reported to show an increase in activity with increasing chloride concentrations.3 The origin of this increase in activity with both Cs and Re is still under debate: it is speculated that [ReO4]− serves as molecular spacer for Cs-species and thereby preventing the formation of CsCl.38 The Re loadings in our study are intentionally higher than in industrial catalysts to study the influence of Re and Cl on ethylene epoxidation, without Cs. Catalytic tests between 0–1 ppm ethyl chloride (EC) were performed to investigate if the conversion would show an increase within this lower EC range (Fig. S9–S11†). The Re–Ag catalysts did not show an increase in conversion between 0–1 ppm EC (Fig. S12†), and hence with such high Re loadings and without Cs behave differently than the earlier reported Re–Cs promoted Ag catalysts.
Fig. 5B shows the EO selectivity as a function of EC concentration. Rhenium increases the ethylene oxide selectivity at 0 ppm EC (albeit, at a lower conversion). For ethylene epoxidation, a decrease in conversion typically causes an increase in selectivity due to the limitation of secondary reactions.24 This effect is illustrated in Fig. 5C which displays EO selectivity vs. conversion plots of the catalysts. Interestingly, without the chloride the Re-promoted catalysts show similar selectivities of ca. 35%, while 0.4Re–Ag shows a much higher conversion compared to 2Re–Ag and 4Re–Ag. Upon increasing the chloride concentration the Ag and 0.4Re–Ag catalysts show similar selectivities at relatively similar conversions, which is in line with literature,37 but the selectivity decreases with increasing Re loading (Fig. 5C). In this research, we deliberately chose higher Re loadings compared to the optimized Re loadings used in industry (35–900 ppm of the total catalyst weight),5,17 and 0.4Re–Ag is the only catalyst that reaches the upper limit of these low Re loadings (ca. 900 ppm). Therefore, it should be noted that the influence of Re on catalysis is intentionally magnified to provide insights into the mechanism of Re promoted silver catalysts, but does not represent the optimum composition for effective catalysts.
3.4 Ethylene oxide isomerization as measure for total selectivity
EO isomerization is an important factor influencing EO selectivity.11,14 Inert supports limit isomerization, but EO can also isomerize and combust on the Ag surface.12 We performed dedicated EO isomerization experiments to understand the origin of the changes in EO selectivity during ethylene epoxidation by Re and EC. Fig. 6 shows the EO isomerization data without O2, and additionally with O2 in the feed to approach catalytic reaction conditions at 0 ppm EC. Without oxygen the Ag, 0.4Re–Ag and 4Re–Ag catalysts have similar EO conversions, whereas 4Re and AgReO4 show remarkably high EO conversions of 50 to 100%, respectively. Re2O7 is a Lewis acid,39 which might explain the high isomerization activity of 4Re. Calculations of equilibrium concentrations show that AgReO4 and Re2O7 reduce to Ag, ReO2 and ReO3 during EO isomerization without O2, but are thermodynamically stable in the presence of O2 (Fig. S14†). XRD indeed showed that the amount of crystalline AgReO4 in the AgReO4 sample decreased from ca. 10 to 5 wt% during EO isomerization and that Ag and ReO2 species formed (Fig. S6 and Table S3†). | ||
| Fig. 6 Ethylene oxide (EO) conversion without and with oxygen (A), and corresponding product selectivities without (B) and with oxygen (C). Experiments were performed with 0.15 vol% ethylene oxide (EO), 0.075 vol% acetaldehyde (AA), 7.5 vol% O2 in He at 215 °C. No ethyl chloride has been added in these experiments. | ||
Strikingly, introducing O2 in the feed did not change the EO conversions of the 0.4Re–Ag and 4Re–Ag catalysts. As these conversions are similar to the α-Al2O3 (ca. 20%), this suggests that the only sites active in EO isomerization are the support surface groups. Moreover, it seems that no separate ReOx species are present in the 0.4Re–Ag and 4Re–Ag samples, as these would have increased the EO conversion as shown for the 4Re sample. In contrast, the unpromoted Ag catalyst now has an EO conversion of ca. 70%. These EO conversion trends are inversely related with the selectivity trends from Fig. 5 at 0 ppm EC, where 0.4Re–Ag and 4Re–g have similarly increased EO selectivities compared to the Ag catalyst (albeit, at lower conversions). A small amount of Re thus inhibits all EO isomerization on the Ag surface without chloride in the feed, which shows the value of Re in commercial catalysts.
Product selectivities during EO isomerization experiments give further insight into the mechanism of Re-promoted Ag catalysts. Without O2 in the feed, mostly acetaldehyde is formed (Fig. 6B), which is the direct product of isomerization and its formation does not require oxygen. Strikingly, ethylene was detected during tests with 4Re, 4Re–Ag and AgReO4. Stacked-bed experiments confirmed that ethylene was formed from ethylene oxide and not from acetaldehyde in the gas feed (Fig. S15†). It is known that Re7+ species catalyze the deoxygenation of epoxides to alkenes.40 Trace amounts of O2 and CO2 were detected in the feed (Fig. S16†). In the case of the Ag catalyst this might be caused by the release of weakly adsorbed O2, resulting in EO combustion. 4Re, 4Re–Ag and AgReO4 form slightly more O2, but these samples also display high O2 uptakes at 215 °C as determined with O2 chemisorption (Table 2), which underestimates the release of O2 upon ethylene formation.
With oxygen in the feed (Fig. 6C), CO2 is formed by most of the catalysts, which is the result of the total oxidation of acetaldehyde and/or EO. Compared to the isomerization without oxygen, less ethylene is formed for the 4Re and AgReO4 samples. When increasing the Re loading from 0.4 to 4 at% the CO2 selectivity decreases drastically to 8%. Not only does a small amount of Re inhibit the EO conversion to acetaldehyde, but it also inhibits acetaldehyde (or EO) combustion on the Ag surface and decreases the amount of CO2 emitted.
The EO isomerization results are in line with the selectivity trends without ethyl chloride (EC) in the feed. With EC, the ethylene oxide selectivity decreases with increased Re loading (Fig. 5). As EC is an industrially used promotor, EO isomerization tests were also performed after stabilizing the Ag and 4Re–Ag catalysts with the chloride during catalysis (Fig. 7). In earlier EO isomerization studies no ethyl chloride was co-fed and only (alkali promoted) Ag catalysts or supports were investigated.41–43 Interestingly, EC decreases the EO conversion of the Ag catalyst from 75 to 10%. We tested another Ag catalyst supported on α-alumina from a different batch, which displayed similarly low EO conversions after stabilization with EC (Fig. S17†). This contradicts an earlier study on Cl2-promoted Ag(111) crystals where Cl2 was reported to promote EO isomerization.44 It is unclear, however, whether in these previous studies these Ag(111) crystals were stabilized with Cl2 during ethylene epoxidation for a prolonged period, which is required for a meaningful evaluation of the catalysts' performance and can take up at least 10–20 h.3,13 To our knowledge, this is the first time that (Re-promoted) Ag catalysts have been systematically studied under industrially relevant isomerization conditions including the gaseous chloride promoter.
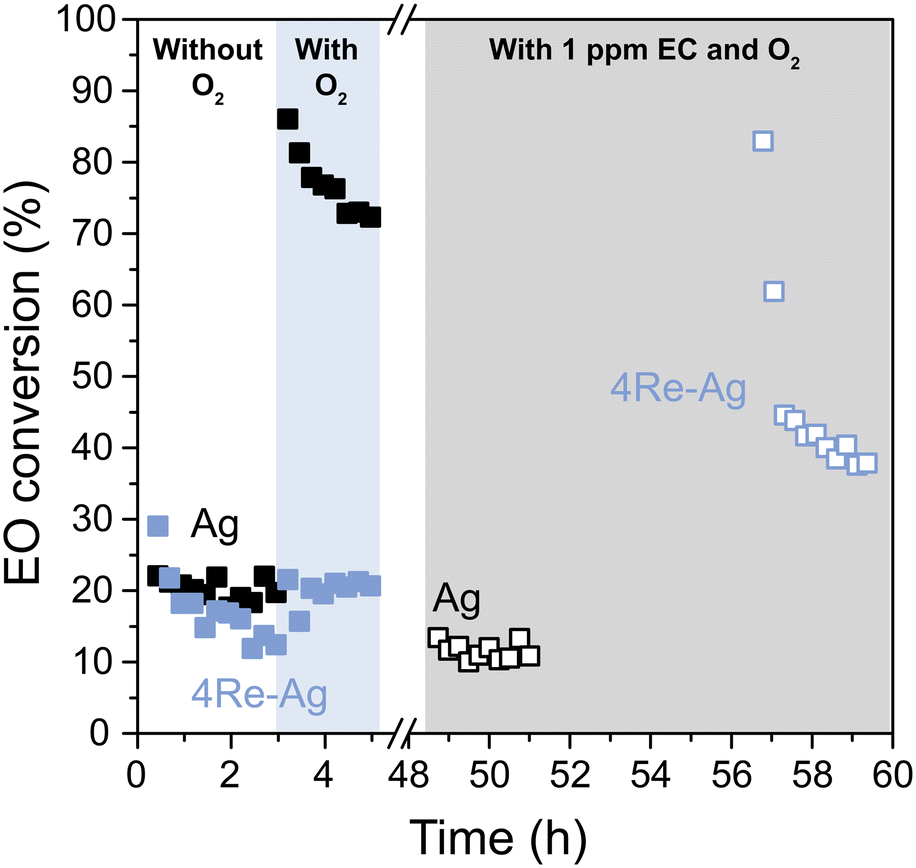 | ||
| Fig. 7 EO isomerization activity of the Ag catalyst (black) and 4Re–Ag catalyst (blue), without (closed symbols) and with ethyl chloride (open symbols) in the feed after ethylene epoxidation had taken place for ca. 50 h at 215 °C. | ||
During ethylene epoxidation, the Ag surface is partially oxidized with either electrophilic or nucleophilic oxygen.7 Electrophilic oxygen promotes the formation of ethylene oxide, whereas nucleophilic oxygen results in total combustion.8–10 EC is known to increase the concentration of electrophilic oxygen species on the silver surface and decrease the nucleophilic oxygen.7 As nucleophilic oxygen is more prone to attack the C–H bond of EO and hence to catalyze its combustion, a reduction in nucleophilic oxygen species explains a decrease in EO conversion. In contrast, the 4Re–Ag catalyst shows an increased EO conversion, which is in line with the decreased EO selectivity from Fig. 5 with EC. Possibly, electrophilic O species are promoted by the chloride, while a surplus of AgReO4 make the O species overly electrophilic. This can activate the C–O bond from ethylene oxide which hence increases the EO conversion. It is clear that by studying AgReO4 separately, it is not merely a spectator phase but influences pathways of the ethylene epoxidation mechanism. In addition, we show that for both the unmodified and EC-modified experiments, the degree of EO isomerization accounts for the difference in EO selectivity for all catalysts which emphasizes the importance of separately understanding isomerization behavior and investigating catalysts after stabilization with the chloride.
4. Conclusions
Crystalline AgReO4 formed in Re–Ag catalysts, which has not been identified before. Previously reported Re7+ species likely originate from this AgReO4 phase, rather than from separate Re2O7 species, as was assumed so far. Both ReOx and AgReO4 species are reservoirs for O2. The catalysts were tested without and with the industrially relevant ethyl chloride promoter in the feed and in all cases, Re decreased the activity due to [ReO4]− blocking active silver sites. Without chloride, Re increased the EO selectivity while lowering the conversion. The combination of chloride and Re resulted in similar conversions and selectivities for 0.4Re–Ag and Ag, but the 2Re–Ag and 4Re–Ag showed a decreased EO selectivity compared to Ag at equivalent conversions. Including the industrially relevant ethyl chloride, we show for the first time that these changes in selectivity relate to the catalysts' EO isomerization behavior. This shows that the effects of chloride and Re are not limited to the selective oxidation pathway, but should also be considered for the subsequent isomerization reaction which influences the EO selectivity.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
CJK was funded at the Advanced Research Center for Chemical Building Blocks consortium (project number 2021.032.A.UU), which is co-founded and co-financed by the Dutch Research Council (NWO) and the Netherlands Ministry of Economic Affairs and Climate Policy. SEM and STEM-EDX measurements were performed at the Electron Microscopy Centre at Utrecht University. The authors would like to thank Eric Hellebrand and Chris Schneijdenberg (SEM) for technical support. Dennis Becker (Bruker AXS) is acknowledged for his help with resolving an error in the PDF+ database. Julie Cao and Litao Bai (Shell) are acknowledged for setting up and conducting the XPS measurements.References
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm and F. R. Wurm, et al., Polymerization of ethylene oxide, propylene oxide, and other alkylene oxides: Synthesis, novel polymer architectures, and bioconjugation, Chem. Rev., 2016, 116(4), 2170–2243, DOI:10.1021/acs.chemrev.5b00441.
- W. H. Faveere, S. Van Praet, B. Vermeeren, K. N. R. Dumoleijn, K. Moonen and E. Taarning, et al., Toward Replacing Ethylene Oxide in a Sustainable World: Glycolaldehyde as a Bio-Based C2 Platform Molecule, Angew. Chem., Int. Ed., 2021, 60(22), 12204–12223, DOI:10.1002/anie.202009811.
- J. R. Lockemeyer and T. L. Lohr, Ethylene Oxide Catalysis Under Commercial Conditions – A Guide for Researchers, ChemCatChem, 2023, 15, e202201511, DOI:10.1002/cctc.202201511.
- G. H. Law and H. C. Chitwood, Process of making olefin oxides, US 2279469, 1942.
- A. M. Lauritzen, Ethylene oxide catalyst and process for preparing the catalyst, US 4761394, 1988.
- M. O. Özbek, I. Onal and R. A. Van Santen, Why silver is the unique catalyst for ethylene epoxidation, J. Catal., 2011, 284(2), 230–235, DOI:10.1016/j.jcat.2011.08.004.
- T. C. R. Rocha, M. Hävecker, A. Knop-Gericke and R. Schlögl, Promoters in heterogeneous catalysis: The role of Cl on ethylene epoxidation over Ag, J. Catal., 2014, 312, 12–16, DOI:10.1016/j.jcat.2014.01.002.
- V. I. Bukhtiyarov, A. I. Nizovskii, H. Bluhm, M. Hävecker, E. Kleimenov and A. Knop-Gericke, et al., Combined in-situ XPS and PTRMS study of ethylene epoxidation over silver, J. Catal., 2006, 238(2), 260–269, DOI:10.1016/j.jcat.2005.11.043.
- M. O. Özbek and R. A. Van Santen, The mechanism of ethylene epoxidation catalysis, Catal. Lett., 2013, 143, 131–141, DOI:10.1007/s10562-012-0957-3.
- T. E. Jones, R. Wyrwich, S. Böcklein, E. A. Carbonio, M. T. Greiner and A. Y. Klyushin, et al., The Selective Species in Ethylene Epoxidation on Silver, ACS Catal., 2018, 8(5), 3844–3852, DOI:10.1021/acscatal.8b00660.
- J. E. van den Reijen, W. C. Versluis, S. Kanungo, M. F. D'Angelo, K. P. de Jong and P. E. de Jongh, From qualitative to quantitative understanding of support effects on the selectivity in silver catalyzed ethylene epoxidation, Catal. Today, 2019, 338, 31–39, DOI:10.1016/j.cattod.2019.04.049.
- J. H. Miller, A. Joshi, X. Li and A. Bhan, Catalytic degradation of ethylene oxide over Ag/α-Al2O3, J. Catal., 2020, 389, 714–720, DOI:10.1016/j.jcat.2020.07.008.
- C. J. Keijzer, L. C. J. Smulders, D. Wezendonk, J. W. de Rijk and P. E. de Jongh, Influence of Ag particle size and Ag : Al2O3 surface ratio in catalysts for the chloride-promoted ethylene epoxidation, Catal. Today, 2024, 428, 114447, DOI:10.1016/j.cattod.2023.114447.
- T. Pu, H. Tian, M. E. Ford, S. Rangarajan and I. E. Wachs, Overview of Selective Oxidation of Ethylene to Ethylene Oxide by Ag Catalysts, ACS Catal., 2019, 10727–10750, DOI:10.1021/acscatal.9b03443.
- M. O. Özbek, I. Önal and R. A. Van Santen, Chlorine and Caesium Promotion of Silver Ethylene Epoxidation Catalysts, ChemCatChem, 2013, 5(2), 443–451, DOI:10.1002/cctc.201200690.
- J. R. Lockemeyer, Process for preparing a catalyst, the catalyst, and a use of the catalyst, US 2005/0222442 A1, 2005.
- M. Matusz, Process for the production of an olefin oxide, a 1,2-diol, a 1,2-diol ether, a 1,2-carbonate, or an alkanolamine, US 2009/0281345 A1, 2009.
- W. Diao, C. D. Digiulio, M. T. Schaal, S. Ma and J. R. Monnier, An investigation on the role of Re as a promoter in Ag-Cs-Re/α-Al2O3 high-selectivity, ethylene epoxidation catalysts, J. Catal., 2015, 322, 14–23, DOI:10.1016/j.jcat.2014.11.007.
- M. A. Salaev, Computational insights into promoting effects of alkali metals, Re, and Cl for silver catalysts of ethylene epoxidation, Mol. Catal., 2021, 507, 111574, DOI:10.1016/j.mcat.2021.111574.
- D. Badea, A. Karpov, M. Jansen and J. Bruns, [Ag(μ-en)][ReO4]: An Intermediate during Preparation of the Ethylene Epoxidation Catalyst, Z. Anorg. Allg. Chem., 2022, 648, e202200267, DOI:10.1002/zaac.202200267.
- J. C. Dellamorte, J. Lauterbach and M. A. Barteau, Rhenium promotion of Ag and Cu-Ag bimetallic catalysts for ethylene epoxidation, Catal. Today, 2007, 120(2), 182–185, DOI:10.1016/j.cattod.2006.07.051.
- D. Ren, G. Cheng, J. Li, J. Li, W. Dai and X. X. Sun, et al. Effect of Rhenium Loading Sequence on Selectivity of Ag–Cs Catalyst for Ethylene Epoxidation, Catal. Lett., 2017, 147(12), 2920–2928, DOI:10.1007/s10562-017-2211-5.
- J. C. Dellamorte, J. Lauterbach and M. A. Barteau, Promoter-induced morphological changes of Ag catalysts for ethylene epoxidation, Ind. Eng. Chem. Res., 2009, 48(13), 5943–5953, DOI:10.1021/ie801627k.
- J. E. van den Reijen, S. Kanungo, T. A. J. Welling, M. Versluijs-Helder, T. A. Nijhuis and K. P. de Jong, et al., Preparation and particle size effects of Ag/a-Al2O3 catalysts for ethylene epoxidation, J. Catal., 2017, 356, 65–74, DOI:10.1016/j.jcat.2017.10.001.
- S. Gates-Rector and T. Blanton, The Powder Diffraction File: a quality materials characterization database, Powder Diffr., 2019, 34(4), 352–360 CrossRef CAS.
- J. T. Jankowiak and M. A. Barteau, Ethylene epoxidation over silver and copper-silver bimetallic catalysts: II. Cs and Cl promotion, J. Catal., 2005, 236(2), 379–386, DOI:10.1016/j.jcat.2005.10.017.
- H. Jin, X. Liu, P. An, C. Tang, H. Yu and Q. Zhang, et al., Dynamic rhenium dopant boosts ruthenium oxide for durable oxygen evolution, Nat. Commun., 2023, 14, 354, DOI:10.1038/s41467-023-35913-6.
- J. Okal, L. Kępiński, L. Krajczyk and W. Tylus, Oxidation and redispersion of a low-loaded Re/γ-Al2O3 catalyst, J. Catal., 2003, 219(2), 362–371, DOI:10.1016/S0021-9517(03)00165-9.
- L. Brütsch and C. Feldmann, Synthesis and Morphology of AgReO4 Plates, Rods, and Stars, Z. Anorg. Allg. Chem., 2017, 643(12), 789–792, DOI:10.1002/zaac.201700072.
- S. Lwin and I. E. Wachs, Olefin metathesis by supported metal oxide catalysts, ACS Catal., 2014, 4(8), 2505–2520, DOI:10.1021/cs500528h.
- M. Matusz, R. L. Paddock and R. C. Yeates, Methods for conditioning an ethylene epoxidation catalyst and associated methods for the production of ethylene oxide, WO 2018/102377 A1, 2018.
- M. Matusz, R. L. Paddock, R. C. Yeates and J. R. Lockemeyer, Methods for conditioning an ethylene epoxidation catalyst and associated methods for the production of ethylene oxide, EP 3548471 B1, 2021.
- S. Linic and M. A. Barteau, Control of ethylene epoxidation selectivity by surface oxametallacycles, J. Am. Chem. Soc., 2003, 125(14), 4034–4035, DOI:10.1021/ja029076g.
- C. Stegelmann, N. C. Schiødt, C. T. Campbell and P. Stoltze, Microkinetic modeling of ethylene oxidation over silver, J. Catal., 2004, 221(2), 630–649, DOI:10.1016/j.jcat.2003.10.004.
- M. Huš and A. Hellman, Dipole effect on ethylene epoxidation: Influence of alkali metals and chlorine, J. Catal., 2018, 363, 18–25, DOI:10.1016/j.jcat.2018.04.008.
- S. R. Seyedmonir, D. Strohmayer, G. Geoffroy and A. Vannice, Characterization of Supported Silver Catalysts, J. Catal., 1984, 436, 424–436 CrossRef.
- A. Hwang, J. Klaucke, C. Lizandara-Pueyo, A. Karpov and E. Iglesia, Roles of Re and Cs Promoters and Organochlorine Moderators in the Synthesis of Ethylene Oxide on Ag-based Catalysts, ChemCatChem, 2023, 202301369, DOI:10.1002/cctc.202301369.
- B. W. J. Chen, B. Wang, M. B. Sullivan, A. Borgna and J. Zhang, Unraveling the Synergistic Effect of Re and Cs Promoters on Ethylene Epoxidation over Silver Catalysts with Machine Learning-Accelerated First-Principles Simulations, ACS Catal., 2022, 12(4), 2540–2551, DOI:10.1021/acscatal.1c05419.
- T. J. Korstanje, E. F. De Waard, J. T. B. H. Jastrzebski and R. J. M. Klein Gebbink, Rhenium-catalyzed dehydration of nonbenzylic and terpene alcohols to olefins, ACS Catal., 2012, 2(10), 2173–2181, DOI:10.1021/cs300455w.
- T. Nakagiri, M. Murai and K. Takai, Stereospecific Deoxygenation of Aliphatic Epoxides to Alkenes under Rhenium Catalysis, Org. Lett., 2015, 17(13), 3346–3349, DOI:10.1021/acs.orglett.5b01583.
- B. T. Egelske, W. Xiong, H. Zhou and J. R. Monnier, Effects of the method of active site characterization for determining structure-sensitivity in Ag-catalyzed ethylene epoxidation, J. Catal., 2022, 410, 221–235, DOI:10.1016/j.jcat.2022.03.021.
- N. Macleod, J. M. Keel and R. M. Lambert, The effects of catalyst aging under industrial conditions: ethylene oxide conversion over Ag-Cs/α-Al2O3 catalysts, Catal. Lett., 2003, 86(1–3), 51–56, DOI:10.1023/A:1022602807322.
- D. A. Bulushev, E. A. Paukshtis, Y. N. Nogin and B. S. Bal'zhinimaev, Transient response and infrared studies of ethylene oxide reactions on silver catalysts and supports, Appl. Catal., A, 1995, 123(2), 301–322, DOI:10.1016/0926-860X(94)00221-5.
- S. A. Tan, R. B. Grant and R. M. Lambert, Secondary Chemistry in the Selective Oxidation of Ethylene: Effect of Cl and Cs Promoters on the Adsorption, Isomerisation, and Combustion of Ethylene Oxide on Ag(111), J. Catal., 1987, 106, 54–64 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00858h |
| This journal is © The Royal Society of Chemistry 2025 |