
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe influence of size, metal loading and oxygen vacancies on the catalytic performance of Au/CeO2−x in the sunlight-powered reverse water gas shift reaction†
Jordi
Volders
 abc,
Sander
Bossier
d,
Sander
Stulens
abc,
Bjorn
Joos
abc,
Thomas
Vranken
abc,
Francesc
Sastre
e,
Jan
D'Haen
b,
Ken
Elen
abc,
Marcel A.
Verheijen
fg,
Pegie
Cool
d,
An
Hardy
abc,
Pascal
Buskens
*ae and
Marlies K.
Van Bael
*abc
abc,
Sander
Bossier
d,
Sander
Stulens
abc,
Bjorn
Joos
abc,
Thomas
Vranken
abc,
Francesc
Sastre
e,
Jan
D'Haen
b,
Ken
Elen
abc,
Marcel A.
Verheijen
fg,
Pegie
Cool
d,
An
Hardy
abc,
Pascal
Buskens
*ae and
Marlies K.
Van Bael
*abc
aInstitute for Materials Research (imo-imomec), DESINe Group, Hasselt University, Agoralaan Building D, Diepenbeek 3590, Belgium. E-mail: marlies.vanbael@uhasselt.be
bIMOMEC Associated Laboratory, IMEC vzw, Wetenschapspark 1, Diepenbeek 3590, Belgium
cEnergyVille, Thor Park 8320, Genk 3600, Belgium
dLaboratory of Adsorption and Catalysis, Department of Chemistry, University of Antwerp, Universiteitsplein 1, Wilrijk B-2610, Belgium
eThe Netherlands Organisation for Applied Scientific Research (TNO), Eindhoven, The Netherlands. E-mail: pascal.buskens@tno.nl
fDepartment of Applied Physics, Eindhoven University of Technology, Eindhoven 5600 MB, The Netherlands
gEurofins Materials Science, High Tech Campus 11, Eindhoven 5656 AE, The Netherlands
First published on 10th December 2024
Abstract
This study reports the conversion of CO2 and H2 to CO and H2O at low temperature and low pressure (up to 203 °C, p = 3.5 bar) using plasmonic Au/CeO2−x photocatalysts, with mildly concentrated sunlight as the sole energy source (up to 9 kW m−2). Systematic catalytic studies were carried out by varying the CeO2−x particle size, Au particle size and loading, and the concentration of oxygen vacancies. Upon illumination, all Au/CeO2−x catalysts showed a CO production of up to 2.6 ± 0.2 mmol CO per gAu per h (104 ± 8 μmol CO per gcat per h), while the supports without Au did not show any activity. We determined that both photothermal and non-thermal effects contribute to the light-driven reverse water-gas shift reaction catalysed by plasmonic Au/CeO2−x. A photothermal contribution was found from the exponential relationship between the CO production and the solar irradiance. In the dark, all Au/CeO2−x photocatalysts and supports without Au produced CH4 instead of CO with ≥97% selectivity, indicating a significant non-thermal contribution in light. A linear dependence of catalytic activity on the accessible interface area between CeO2−x and Au was found, which is in line with an associative formate-mediated reaction mechanism occurring at the metal–support interface. Tuning the VO content through thermal treatments yielded decreased photocatalytic activity for oxidised samples, identifying them as pre-catalysts. The stability of the Au/CeO2−x photocatalysts was evaluated, demonstrating that the catalytic performance was affected by adsorption of H2O as a reaction product, which could be fully restored upon heating in vacuo.
1 Introduction
To this day a large majority of the world's total energy supply is provided by fossil fuels, giving rise to extensive CO2 emissions.1 In view of the transition to a circular, net zero emission economy, CO2 should not be seen as a waste product but as a feedstock for conversion into various carbon-based chemicals and fuels (carbon capture and utilisation, CCU).2–8 For instance, syngas (a mixture of CO and H2) could be produced from CO2 and subsequently used to synthesise long-chain hydrocarbons via the Fischer–Tropsch process as well as various platform chemicals such as methanol, acetic acid and aldehydes.9–15 Syngas can be obtained by CO2 reduction in the reverse water gas shift (rWGS) reaction (eqn (1)).13,16 The rWGS reaction is endothermic and competes with the exothermic methanation process (Sabatier reaction, eqn (2)). To predominantly produce CO, conventionally a high temperature of >600 °C is required.13,17,18| CO2 + H2 ⇌ CO + H2O ΔH298K = 41.2 kJ mol−1 | (1) |
| CO2 + 4H2 ⇌ CH4 + 2H2O ΔH298K = −165.0 kJ mol−1 | (2) |
Photocatalysis, which utilises the energy of (sun)light to carry out the rWGS reaction at near-ambient conditions, is a promising alternative. Traditional photocatalysts are semiconducting materials, with ZnO and TiO2 as common examples, in which light-generated electron–hole pairs drive reactions.19–21 A challenge for these photocatalysts is that the majority of stable semiconductors have a bandgap energy exceeding 3 eV, which absorbs only UV wavelengths, excluding the majority of the solar spectrum. Possible ways to mediate this challenge are bandgap engineering and the use of cocatalysts.21 Plasmonic nanoparticles (NPs), which are able to harvest a broad range of the solar spectrum, could act as cocatalysts in tandem with a semiconductor.7,22–33 For metal NPs that are much smaller than the wavelength of incident light, the free electron gas of the metal exhibits an oscillation with a frequency equal to that of incident light, also known as the localised surface plasmon resonance (LSPR). This LSPR can activate reactants through several pathways. As a first effect, heat will be generated in the local vicinity of the plasmonic NP through the Joule effect, allowing for enhanced reaction kinetics. Secondly, light can be re-emitted at the same frequency as the incident light concentrated at specific positions at the NP surface, dependent on particle morphology. This effect is known as near-field enhancement which can strongly benefit photo-activated processes or generate additional charge carriers in the semiconductor. In a third pathway, the energy of the LSPR can be used to promote an electron to a higher energy level, referred to as a hot electron. When the plasmonic NP is in junction with a semiconductor, hot electrons can be injected into the conduction band over the Schottky barrier, increasing the charge carrier lifetime and thus their probability of participating in a reaction.34–36 Finally, the conventional generation of electron–hole pairs in the semiconductor by UV excitation can also occur. The local heat generation is defined as a photothermal contribution, while the near-field enhancement, hot electron generation and injection and bandgap transition combined are referred to as non-thermal contributors.35,37–41
CeO2 is a semiconductor with a face-centred cubic (FCC) fluorite crystal structure which is rarely fully stoichiometric and exhibits relatively large amounts of oxygen vacancies (VOs) in its lattice. Every VO results in two reduced Ce3+ sites while retaining the fluorite structure and can readily be reversed into stoichiometric CeO2.42–48 The VOs allow for increased adsorption of CO2 and have been shown to act as catalytic sites for rWGS on supported CeO2−x, for both redox and associative reaction mechanisms. In the redox mechanism, the reaction occurs at a redox site of the catalyst, specifically at VO sites of the support material as commonly catalytic metals for rWGS do not readily change valence states. In the associative mechanism, H2 is dissociated at the catalytic metal and will react with CO2 adsorbed on the support to result in carbon-containing intermediates (e.g. formates, carbonates), which are subsequently converted to CO.18,49–56 Additionally, it has been demonstrated that deposition of metal NPs, e.g. Au, on the surface of CeO2−x can significantly decrease the required temperature to form VOs at the surface.57–62 Due to these unique properties, CeO2−x has been studied as a support52,63–70 and photocatalyst in various applications like selective organic reactions,71–75 hydrogen evolution reaction76 and pollutant degradation.77–81
Several supported plasmonic materials have been investigated as photocatalysts for sunlight-driven rWGS: Ag,82 Al,83 Co,84 Cu,85,86 Fe,87,88 Mo,89 Ni,90 Pt,91–94 Pd (ref. 91, 95–97) and Ru.98 In addition, Au has been widely reported as a plasmonic photocatalyst due to its strong LSPR effect and high selectivity towards rWGS. An overview of publications reporting supported Au plasmonic photocatalysts can be found in Table S1.†25,27,30,85,99–103 Production rates are either normalized by the mass of the photocatalyst including the support, denoted as gcat−1, or by the mass of only the plasmonic metal (denoted as gAu−1). For supported Au plasmonic photocatalysts, no specific correlations between plasmonic Au size, loading and activity have been reported. In addition, the Au/CeO2−x catalytic system for light-driven plasmonic rWGS is not yet fully understood: no correlation between photocatalytic activity and key structural properties has been reported. Upadhye et al. reported an Au/CeO2−x photocatalyst with a CO production rate of 85.02 mmol gcat−1 h−1, which was a significant enhancement compared to the Au/Al2O3 photocatalyst with an insulating support. They also found the largest rate enhancement between dark and light experiments for Au/CeO2−x compared to Au/TiO2 and Au/Al2O3. For Au/TiO2 they proposed a carboxyl-mediated associative mechanism at the metal–support interface where the LSPR assists in dissociation of the carboxyl intermediate or H2O formation.85 Lu et al. reported an Au/CeO2−x photocatalyst with a CO production rate of approximately 7 mmol gcat−1 h−1; they attributed the activity difference between light and dark experimental conditions to the plasmon-associated dissociation of H2 on the Au surface.103
In our study, we prepared and characterized a series of Au/CeO2−x catalysts and focused on the influence of key structural properties of Au and CeO2−x, more specifically particle size, Au loading and oxygen vacancies, on the catalytic performance of Au/CeO2−x in the sunlight-powered rWGS reaction. By the obtained results, we gained insights on the impact of photothermal heating and the nature of non-thermal effects contributing to the reaction.
2 Results and discussion
2.1 Characterisation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water mixture (1:2 for L-HT and 3:1 for S-HT), then transferred to a Teflon-lined autoclave and left in an oven at 160 °C (20 h for L-HT, 14 h for S-HT). Finally, the solvent was evaporated in ambient air and a powder was obtained. A sample list with a summary of characterisation is available in Table S2.† Particle sizing with dynamic light scattering (DLS) (Fig. S1c and h†) of the as-synthesised dispersion yielded a particle size of 376 ± 6 nm and 53 ± 3 nm for L-HT and S-HT, respectively, with a confidence interval of 98%. Particle size distributions from transmission electron microscopy (TEM) analyses (Fig. S1a, b, d and e†) yielded particle sizes of 200 ± 7 nm and 35.5 ± 0.7 nm for L-HT and S-HT, respectively, with a confidence interval of 98%. The hydrodynamic diameter obtained by DLS is likely a larger value than particle sizes found in TEM analysis due to the presence of the electric double layer, organic residuals and agglomeration. Thermogravimetric analysis (TGA) was performed (Fig. S1i†) and a weight loss of 68% and 72% was observed for L-HT and S-HT below 450 °C, indicating a large amount of organic material still present in the powders. This was confirmed by attenuated total reflection Fourier-transform infrared (ATR-FTIR) spectroscopy (Fig. S1g†), where characteristic CH and CH2 stretch vibrations (around 2900 cm−1) originating from the PVP surfactant are present in both L-HT and S-HT samples. X-ray diffraction (XRD) (Fig. 1c) analyses of both samples exhibited the characteristic fluorite structure of CeO2 (JCPDS 65-5923). Additional peaks can be attributed to the LaB6 internal standard (JCPDS 06-0401) which was added as a reference for accurate peak position determination.
:water mixture (1:2 for L-HT and 3:1 for S-HT), then transferred to a Teflon-lined autoclave and left in an oven at 160 °C (20 h for L-HT, 14 h for S-HT). Finally, the solvent was evaporated in ambient air and a powder was obtained. A sample list with a summary of characterisation is available in Table S2.† Particle sizing with dynamic light scattering (DLS) (Fig. S1c and h†) of the as-synthesised dispersion yielded a particle size of 376 ± 6 nm and 53 ± 3 nm for L-HT and S-HT, respectively, with a confidence interval of 98%. Particle size distributions from transmission electron microscopy (TEM) analyses (Fig. S1a, b, d and e†) yielded particle sizes of 200 ± 7 nm and 35.5 ± 0.7 nm for L-HT and S-HT, respectively, with a confidence interval of 98%. The hydrodynamic diameter obtained by DLS is likely a larger value than particle sizes found in TEM analysis due to the presence of the electric double layer, organic residuals and agglomeration. Thermogravimetric analysis (TGA) was performed (Fig. S1i†) and a weight loss of 68% and 72% was observed for L-HT and S-HT below 450 °C, indicating a large amount of organic material still present in the powders. This was confirmed by attenuated total reflection Fourier-transform infrared (ATR-FTIR) spectroscopy (Fig. S1g†), where characteristic CH and CH2 stretch vibrations (around 2900 cm−1) originating from the PVP surfactant are present in both L-HT and S-HT samples. X-ray diffraction (XRD) (Fig. 1c) analyses of both samples exhibited the characteristic fluorite structure of CeO2 (JCPDS 65-5923). Additional peaks can be attributed to the LaB6 internal standard (JCPDS 06-0401) which was added as a reference for accurate peak position determination.
 | ||
| Fig. 1 (a) Particle size distributions of L CeO2−x support from TEM analysis with lognormal fit. (b) Representative TEM image of L CeO2−x particles. (c) XRD diffractograms of L-HT, S-HT, L and S CeO2−x particles with LaB6 as internal standard. Intensity is normalised on the wt% of sample in the sample/LaB6 mixture. Graphs have been stacked for visual clarity. The * denotes an impurity peak of the Al sample stage in the L sample. (d) Particle size distributions of S CeO2−x support from TEM analysis with lognormal fit. (e) Representative TEM image of S CeO2−x particles. (f) H2-TPR profiles of S and L CeO2−x particles; graphs have been stacked for visual clarity. | ||
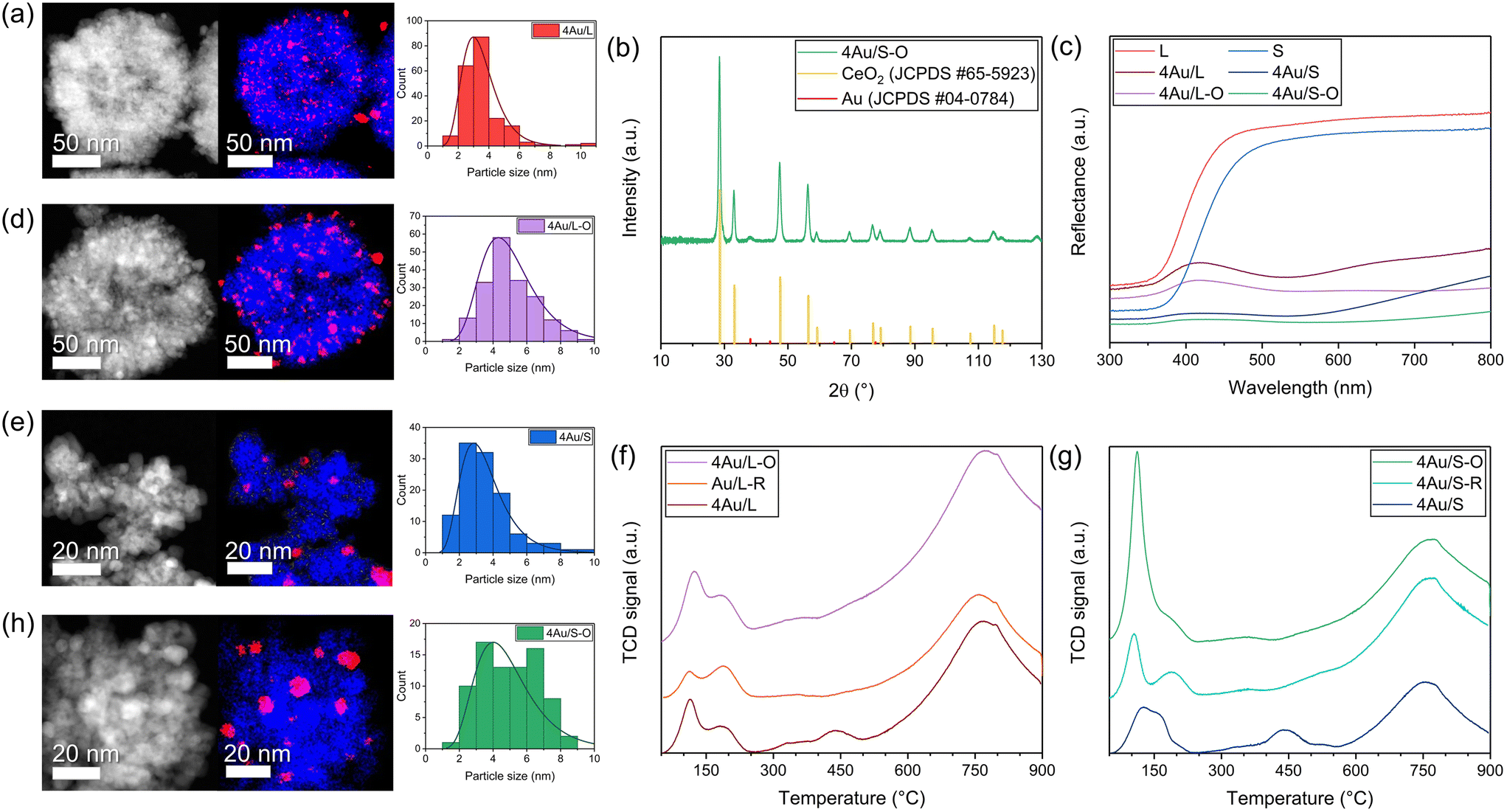 | ||
| Fig. 2 Representative HAADF-STEM image, EDX mapping in STEM mode and Au particle size distribution from TEM analysis with lognormal fit of (a) 4Au/L, (d) 4Au/L-O, (e) 4Au/S and (h) 4Au/S-O Au/CeO2−x samples. CeO2−x is coloured blue and Au is coloured red in EDX mapping, lower magnification shown for (a) and (d) to illustrate CeO2−x size particle difference. (b) XRD diffractogram of 4Au/S-O. (c) Reflective diffuse UV-vis spectra of L, 4Au/L, 4Au/L-O, S, 4Au/S and 4Au/S-O samples; graphs have been stacked for visual clarity. H2-TPR profiles of (f) 4Au/L-O, 4Au/L-R and 4Au/L and (g) 4Au/S-O, 4Au/S-R and 4Au/S; graphs have been stacked for visual clarity. | ||
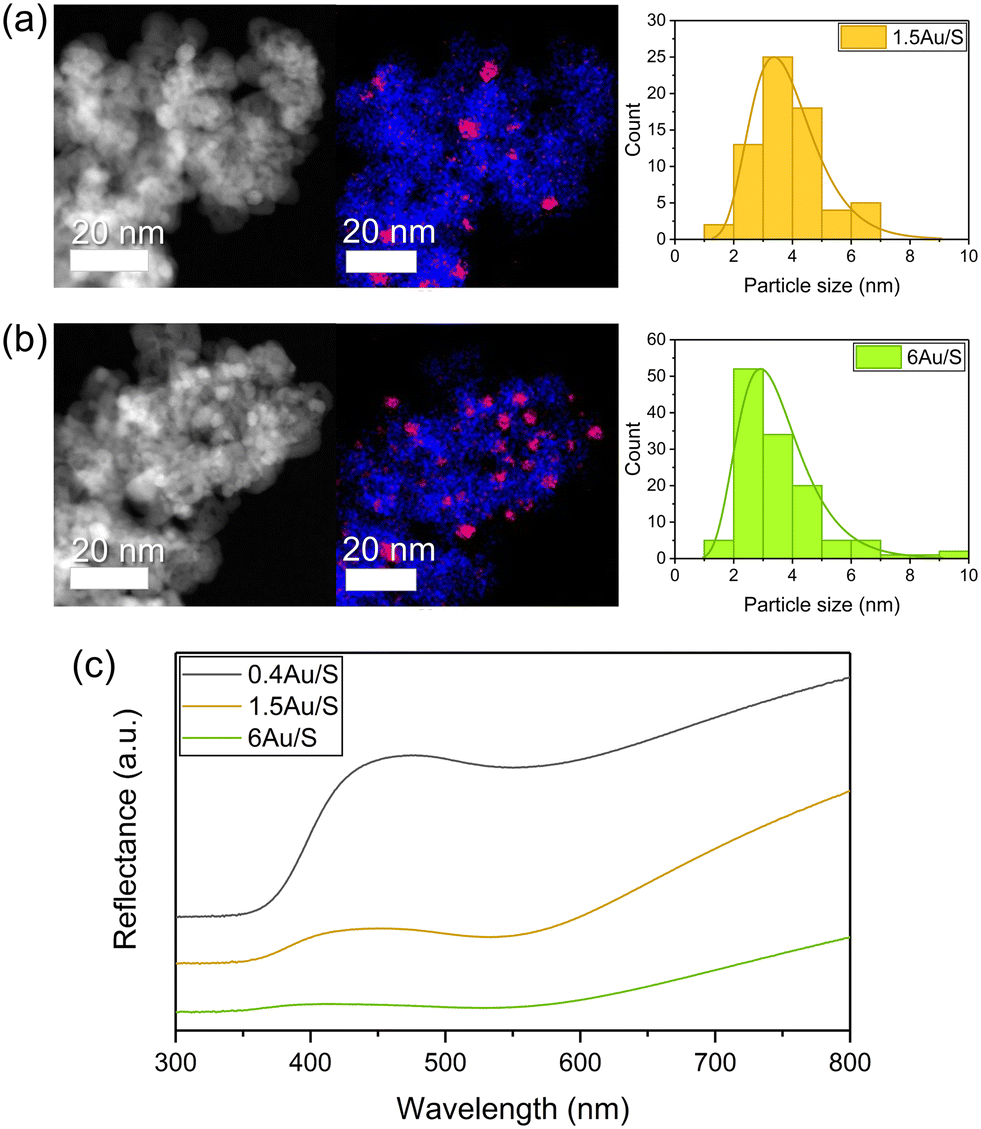 | ||
| Fig. 3 Representative HAADF-STEM image, EDX mapping in STEM mode and Au particle size distribution from TEM analysis with lognormal fit of (a) 1.5Au/S and (b) 6Au/S. CeO2−x is coloured blue and Au is coloured red in EDX mapping. (c) Diffuse reflective UV-vis spectra of 0.4Au/S, 1.5Au/S and 6Au/S; graphs have been stacked for visual clarity. | ||
2.2 Catalysis
:8 ml min−1:4 ml min−1 H2:CO2:N2 at 3.5 bar. No other products besides CH4 and CO were detected, and no reactivity was observed for bare L and S supports upon illumination, while they solely produced CH4 when externally heated to 180 °C without illumination. Confidence intervals of 98% are shown for catalyst activity. A summary of catalyst activity and selectivity is shown in Table S5.†
Accurate temperature measurements are essential for differentiating between thermal and non-thermal contributions. The catalyst temperature (Fig. 4c and S5a†) was measured with a thermocouple in the reactor vessel, a thermocouple at the bottom of the catalyst bed and fibre Bragg grating (FBG)-based fibre optic sensors inside the catalyst bed, as reported in a previous work (Fig. 4d and e).26 A temperature difference of almost 100 °C is observed between the FBG result at the top of the catalyst surface compared to the thermocouple at the bottom of the catalyst bed. Furthermore, the presence of photothermal contributions in the reaction was investigated by studying the influence of light variation on the CO production, where the irradiance was varied between 6 kW m−2 and 9 kW m−2 (AM1.5) (6 and 9 suns). In order to eliminate the contribution of the semiconductor to the reaction, a UV cutoff filter (<395 nm) was mounted in the light path during additional experiments. All catalysts possess selectivity towards CO of more than 98%. An exponential relationship (Fig. 4a and b) is observed for 4Au/L-O, 4Au/S-O, 4Au/L-R and 4Au/S-R, confirming the presence of a photothermal contribution in all catalysts. As heat generated by plasmonic particles is linearly dependent on the incident light intensity, an exponential increase in activity is expected with light variation for photothermal processes, following Arrhenius' law.35 No significant production (refer to Fig. S6a† for a simplified representation) difference can be observed between large (4Au/L-O and 4Au/L-R) and small (4Au/S-O and 4Au/S-R) CeO2−x support samples, indicating that there is no direct significant impact of CeO2−x surface area on the photocatalytic activity. From this it can be deduced that the pathway for the sunlight-driven rWGS reaction over Au/CeO2−x is presumably not progressing through a redox mechanism, but instead through an associative mechanism, which aligns with reports in the literature.52,101 Using the temperature at the catalyst surface from FBG, it was possible to calculate the apparent activation energy for the light-powered reaction using the Arrhenius equation (Fig. S7a–c†). Apparent activation energies of 1.2 kJ mol−1, 1.0 kJ mol−1, 1.2 kJ mol−1 and 0.8 kJ mol−1 were obtained for 4Au/L-O, 4Au/S-O, 4Au/L-R and 4Au/S-R, respectively. When making use of the UV cutoff filter, apparent activation energies of 1.0 kJ mol−1 and 1.7 kJ mol−1 were found for 4Au/S-O and 4Au/S-R, respectively. The apparent activation energy values obtained above are all very low compared to previously reported photocatalytic apparent activation energies of 146 kJ mol−1 for Au/CeO2 (ref. 103) and 29 kJ mol−1 for Au/TiO2 in the same reactor setup,27 so an exceptionally active photocatalyst with very fast CO production would be expected. This, however, was not the case. A possible explanation could be that only a small fraction of the catalyst material is producing a significant amount of CO, while the majority of the catalyst material is not active, possibly amongst others due to limited light penetration into the catalyst bed (small skin depth). This can be demonstrated by examining the temperature depth profile within the catalyst bed during reaction (Fig. S5b†). The temperature of the 4Au/S-R photocatalyst upon 9 kW m−2 illumination is 203.3 °C at the surface, 197.8 °C at 0.15 mm depth, 193.3 °C at 0.30 mm depth and only 171.9 °C at 0.50 mm depth. Then, following Arrhenius' equation, about half CO production rate is expected at 0.30 mm depth and <10% CO production rate is expected at 0.50 mm depth. This was further confirmed by studying CO production (as a function of illuminated area) in relation to catalyst loading in the reactor (Fig. S8†). No homogeneous spread of catalyst over the holder could be achieved with catalyst loadings <50 mg, and from 60 mg catalyst loading onwards a plateau in CO production is reached. This confirms that the vast majority of photocatalytic activity takes place at the surface of the photocatalyst bed.
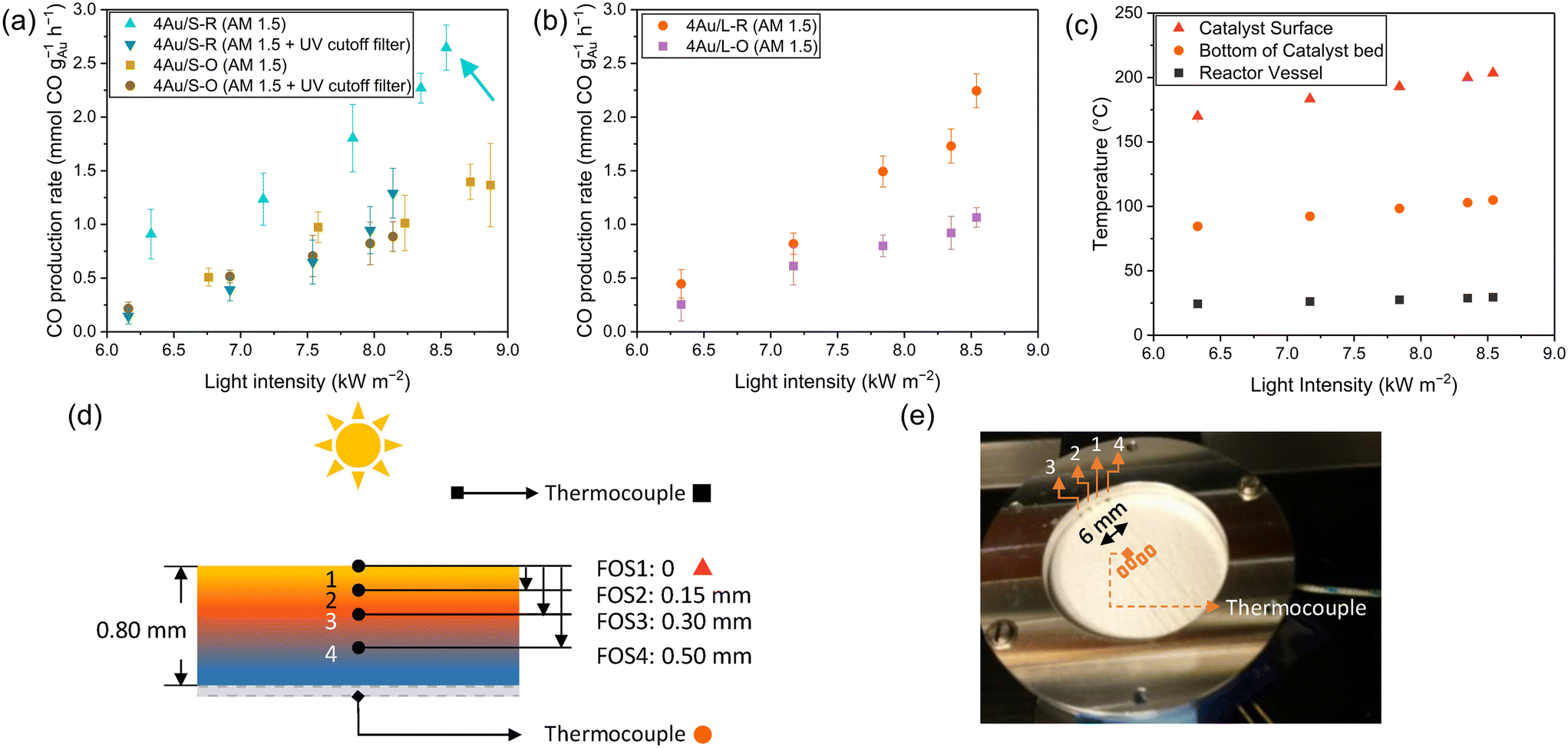 | ||
| Fig. 4 (a) CO production of 4Au/S-R and 4Au/S-O under varied illumination using AM1.5 filter and AM1.5 + UV cutoff filter, (b) 4Au/L-R and 4Au/L-O under varied illumination using AM1.5 filter, (c) temperature of reactor vessel, bottom of the catalyst bed and top of the catalyst of 4Au/S-R irradiance variation experiments. Schematic (d) and photo (e) of the FBG-based fibre optic sensors inside the catalyst bed and thermocouples in the reactor; the figure has been adapted from a previously published work.26 Reaction conditions: mixture of CO2:H2:N2 (8 ml min−1:8 ml min−1:4 ml min−1) at 3.5 bar pressure, 200 mg Au/CeO2−x photocatalyst, varied irradiance from solar simulator (AM1.5 with and without UV cut-off filter). | ||
Another observation is a significant decrease in CO production for 4Au/S-R (Fig. 4a) when making use of the UV cutoff filter, paired with a significant increase in apparent activation energy, indicating a substantial contribution of CeO2−x light absorption above bandgap. This difference in apparent activation energy could indicate a different rate-determining step in the catalytic cycle. This was not observed for 4Au/S-O under the same reaction conditions, where no impact of UV contribution was obtained. Wang et al.101 recently reported that the plasmonic rWGS reaction over Au/Al2O3 is driven by an associative mechanism with both carboxylic and formate intermediates and VOs acting as active sites. For smaller Au NPs (4.3 nm and 6.7 nm) the mechanism was found to be mainly dominated by mono- and bridged formate intermediates. The rate-determining step was identified as the decomposition of HCOO* into CO* and OH* species, resulting in a cumulation of the formate intermediates on active sites during reaction. Applying this mechanism to our results, it can be interpreted that for 4Au/S-R the rate-determining step is similarly the dissociation of the formate intermediates, with an accumulation of these formate intermediates at the VO sites. A reasonable explanation for the lower apparent activation energy upon UV illumination stems from additional charge carriers being generated in the semiconductor which aid in dissociation of the formate intermediate to form CO. In contrast, 4Au/S-O is in fact a precatalyst, as the active sites for catalysis, which are VOs at the catalyst surface, still need to be generated, which lowers overall catalytic activity. No significant impact of UV irradiation is observed, indicating that UV-generated charge carriers generated in the CeO2−x do not significantly contribute to the formation of oxygen vacancies compared to the LSPR effect.
Compared to Au/TiO2 catalysts under similar reaction conditions and without external heating, our catalysts are outperformed in activity by Au/TiO2 with small Au nanoparticles (1.6 nm).25,27 The CO selectivity is similarly high (≥98%). For Au/TiO2 catalysts with larger Au nanoparticles (6 nm, 16 nm) that activity is in the same range as for the Au/CeO2−x catalysts reported in this manuscript.30 Direct comparison with Au/Al2O3 and Au/SiO2 is not possible, since the catalysts reported in the respective studies were both illuminated and externally heated to temperatures between 300 °C and 400 °C.85,100–102 In a previous study on Au/CeO2, Lu et al. reported a higher activity, albeit at a significantly higher irradiance of 32.0 kW m−2.103
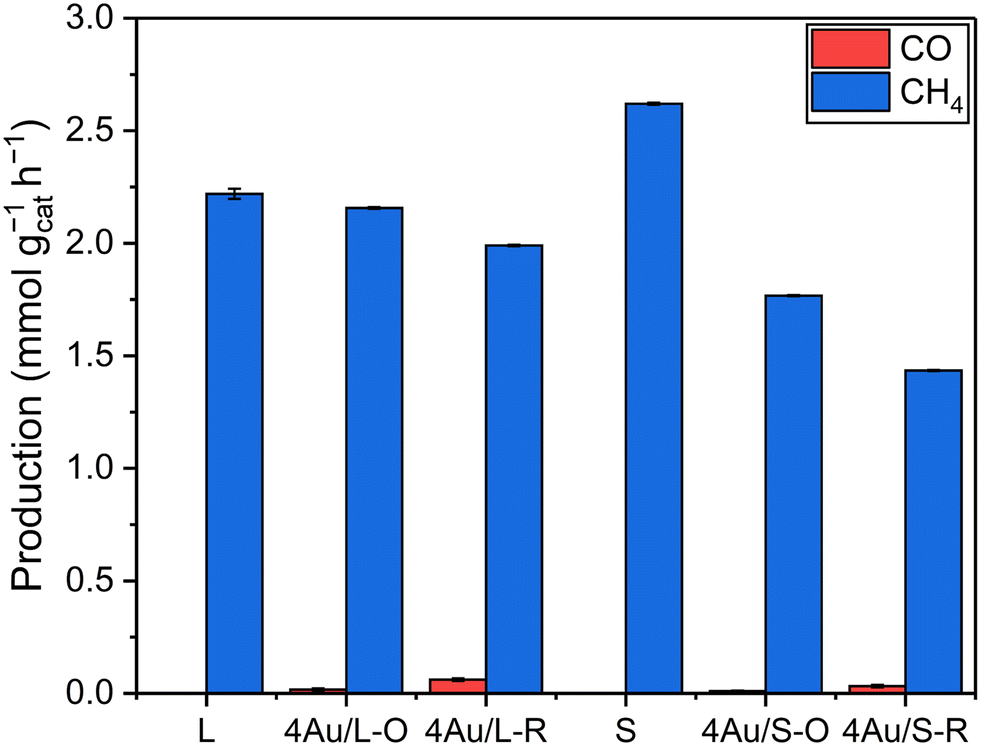 | ||
| Fig. 5 CO and CH4 production rates of L, 4Au/L-O, 4Au/L-R, S, 4Au/S-O and 4Au/S-R in the dark, thermally heated to 180 °C and no illumination. Reaction conditions: mixture of CO2:H2:N2 (8 ml min−1:8 ml min−1:4 ml min−1) at 3.5 bar pressure, 200 mg Au/CeO2−x photocatalyst, no illumination and reactor heating to 180 °C. | ||
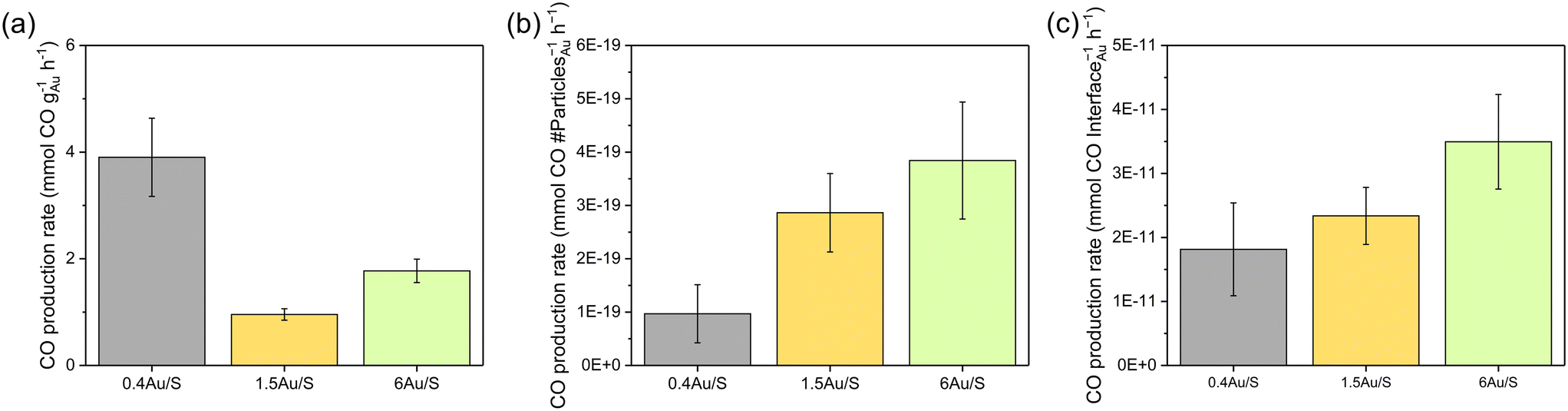 | ||
| Fig. 6 CO production of 0.4Au/S, 1.5Au/S and 6.0 Au/S normalised on (a) mass of Au, (b) circumference of Au particles and (c) number of contact points (amount of Au particles). Reaction conditions: mixture of CO2:H2:N2 (8 ml min−1:8 ml min−1:4 ml min−1) at 3.5 bar pressure, 200 mg Au/CeO2−x photocatalyst, 8.54 kW m−2 illumination from solar simulator (AM1.5). | ||
The calculated values (Table S6†) represent catalytic activity normalised on the interface area between metal and support (scales linearly with the sum of circumferences of the base of all hemispherical Au NPs) (Fig. 6c) and normalised on the number of Au particles (Fig. 6b). The total volume of Au in the sample was considered. When normalising the CO production with respect to the weight of Au (Fig. 6a), significantly different values are obtained for the samples (0.4Au/S, 1.5Au/S and 6Au/S), which is also the case when normalising with respect to the number of Au particles.
This indicates that the Au weight or the number of Au particles are not directly determining the CO production rate. However, when the CO production rate is normalised on the interface area between metal and support, no significant differences were observed. This indicates that the CO production rate may scale directly with the number of active sites, which aligns with the associative mechanism reported above. As such, the CO production rate is expected to increase linearly with the number of active sites as it will increase the dissociation of formate intermediates. As a verification, the same calculations were performed for 4Au/L-O, 4 Au/L-R, 4Au/S-O and 4Au/S-R (Fig. S6b†) and similarly no significant differences between samples could be observed, indicating that the difference in CO activity between reduced and oxidised samples could be attributed to Au particle growth in oxidised samples. These observations underline careful consideration when choosing a plasmonic metal deposition method on semiconducting supports for the sunlight-driven rWGS, as the activity of the photocatalyst is strongly dependent on the interfacial sites.
The stability of CO production of the 4Au/S-R catalyst was investigated over 38 h (Fig. 7a). Initially a CO production rate of 2.42 ± 0.06 mmol CO per gAu per h was obtained, and after 38 h 1.59 ± 0.05 mmol CO per gAu per h was observed, resulting in an activity loss of 34%. This loss in activity could be fully attributed to H2O accumulation on the surface at VO sites, effectively poisoning the active sites for rWGS. The reactor was left at 90 °C under vacuum for 2 h and then allowed to cool, allowing the catalyst to fully regenerate. A CO production rate of 2.53 ± 0.09 mmol CO per gAu per h was found upon illumination. Similar results were found for 4Au/S-O over 15 h (Fig. S10†), with an initial CO production rate of 1.87 ± 0.06 mmol CO per gAu per h and a CO production rate of 0.84 ± 0.06 mmol CO per gAu per h after 15 h with an activity loss of 55%. The catalyst could similarly be regenerated under vacuum at 90 °C for 2 h and a CO production rate of 1.7 ± 0.1 mmol CO per gAu per h upon illumination. On–off experiments were carried out to probe catalyst stability with a fluctuating irradiance (Fig. 7b). The lamp was turned on for 1 h and then turned off for 1 h per cycle. 4 of these cycles were performed during one day. The catalyst remained under dark conditions at room temperature overnight. The following day, 4 more cycles were performed. Similar losses of CO production due to accumulation of H2O at the catalyst surface were observed. In practical application, the catalyst may be regenerated, e.g. by thermal treatment or in vacuo. Furthermore, for equilibrium-limited reactions such as the rWGS process, adsorption of water as a reaction product increases the conversion by shifting the equilibrium according to Le Chatelier's principle. Sorbents and/or membrane technology are applied to achieve this and facilitate CO2 capture and utilization.108
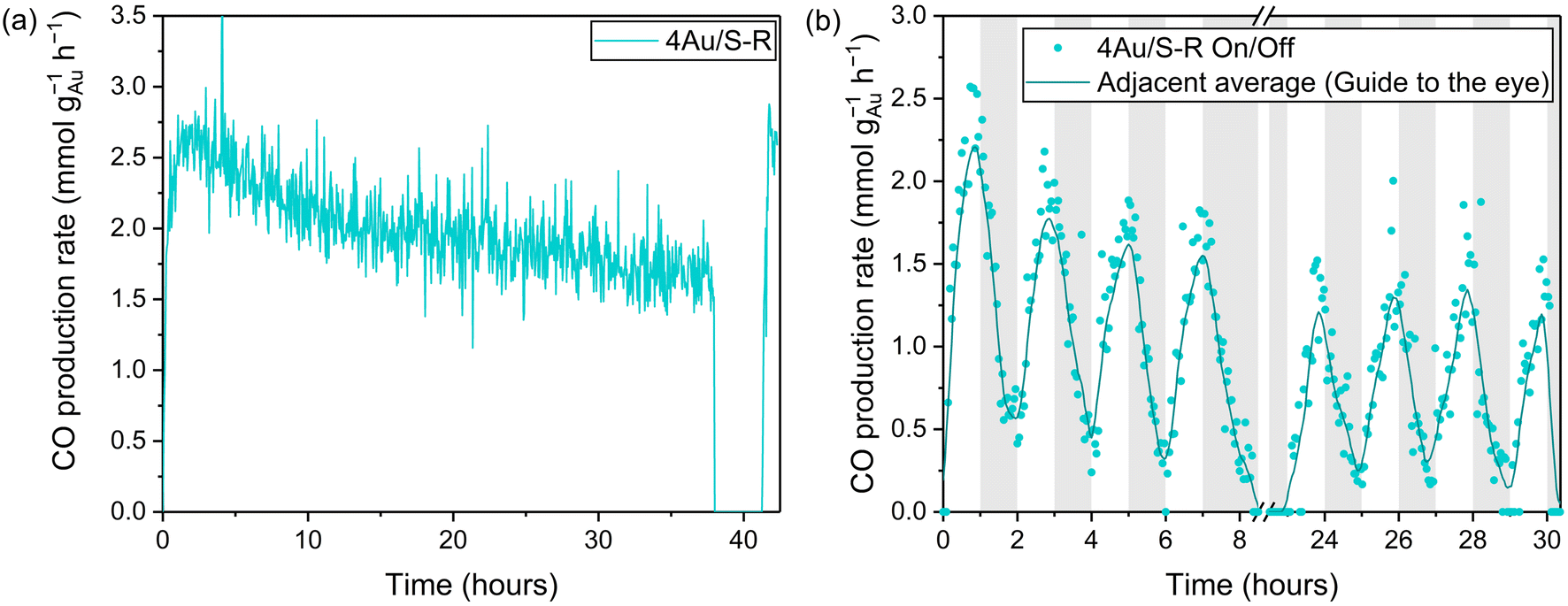 | ||
| Fig. 7 (a) CO production of 4Au/S-R over 38 h, CO production after regeneration in vacuum at 90 °C has been appended at 40 h, and (b) CO production of 4Au/S-R during on–off experiments, the lamp was turned on–off every hour. Reaction conditions: mixture of CO2:H2:N2 (8 ml min−1:8 ml min−1:4 ml min−1) at 3.5 bar pressure, 200 mg Au/CeO2−x photocatalyst, 8.54 kW m−2 illumination from solar simulator (AM1.5). | ||
 | ||
| Fig. 8 Proposed associative monobridged formate-mediated reaction mechanism for the plasmonic-driven rWGS reaction over Au/CeO2−x upon illumination. In fully oxidised catalysts, the rate-determining step (blue 1) is the plasmonic-driven generation of vacancies near Au NPs. For catalysts with a reducing treatment, the rate-determining step (blue 2) is proposed to be the dissociation of the formate intermediate. Atoms are labelled as Au (gold), C (black), H (white), O (red). | ||
3 Conclusion
We successfully produced CeO2−x support materials by hydrothermal synthesis and decorated them with Au nanoparticles on their surface. For systematic catalytic studies, we produced materials with varying CeO2−x particle size, Au particle size and loading, and concentration of oxygen vacancies. In addition to conventional compositional and structural characterization, we determined the H2 uptake of these materials as a function of temperature to shed light on the concentration of oxygen vacancies before and after catalysis. We determined that both photothermal and non-thermal effects contributed to the light-driven rWGS reaction catalysed by plasmonic Au/CeO2−x. Upon illumination, all Au/CeO2−x catalysts tested in this study produced CO with a selectivity of ≥98% at low catalyst surface temperatures (up to 203 °C). The supports without Au were inactive. The exponential relationship between the CO production rate and the irradiance confirms the contribution of photothermal heating. For all tested catalysts, low apparent activation energies were obtained (0.8–1.7 kJ mol−1). The contrasting low overall activity of the tested catalysts (up to 2.6 ± 0.2 mmol g−1 h−1) is amongst others due to the limited light penetration depth. UV light only contributed to the performance of reduced catalysts. Their oxidized counterparts remained unaffected. The latter is likely due to the fact that oxidized Au/CeO2 is merely a precatalyst, for which the formation of oxygen vacancies to form the actual catalyst is rate determining. For the reduced catalysts, we assume that the decomposition of formate intermediates is rate determining (associative mechanism). In the dark, all Au/CeO2−x catalysts and the supports without Au formed CH4 instead of CO. The latter indicates that plasmon-induced desorption of CO plays a vital role in preventing further reduction to CH4. Systematic studies with varying Au loading and particle size show a linear dependence of the catalytic activity on the accessible interfacial area between CeO2−x and Au. This observation is in line with the proposed associative mechanism for which the rate-determining decomposition of formate takes place at the metal–support interface. An assessment of the catalyst stability in steady-state and fluctuating operating conditions demonstrated that the catalytic performance was affected by adsorption of H2O as reaction product, which could be reversed upon heating in vacuo. For spent catalysts, we observed that the concentration of oxygen vacancies substantially increased for oxidized Au/CeO2, whilst it remained similar for their reduced counterparts. This confirms the formation of active catalyst by reduction of the oxidized precatalyst. Structural changes were not observed for spent catalysts, indicating good stability under the applied reaction conditions.Data availability
All data are available in the manuscript and the submitted ESI† file. For raw data, please contact the corresponding authors.Author contributions
Jordi Volders: conceptualisation, data curation, formal analysis, investigation, methodology, visualisation, writing – original draft, writing – review & editing. Sander Bossier: investigation, writing – review & editing. Sander Stulens: investigation, visualisation, writing – review & editing. Bjorn Joos: investigation, writing – review & editing. Thomas Vranken: methodology, resources, writing – review & editing. Francesc Sastre: formal analysis, investigation, methodology, resources, writing – review & editing. Jan D'Haen: investigation, visualisation, writing – review & editing. Ken Elen: conceptualisation, data curation, funding acquisition, methodology, supervision, writing – review & editing. Marcel A. Verheijen: funding acquisition, investigation, methodology, resources, visualisation, writing – review & editing. Pegie Cool: funding acquisition, methodology, supervision, writing – review & editing. An Hardy: conceptualisation, data curation, funding acquisition, methodology, supervision, writing – review & editing. Pascal Buskens: conceptualisation, data curation, funding acquisition, methodology, supervision, writing – review & editing. Marlies K. Van Bael: conceptualisation, data curation, funding acquisition, methodology, supervision, writing – review & editing. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement 101015960 (SPOTLIGHT), the EC Interreg Flanders-The Netherlands project FOTON and the Research Foundation Flanders (FWO-Vlaanderen) under project No. G0A4924N (PICaSSo). Solliance and the Dutch province of Noord-Brabant are acknowledged for funding the TEM facility. We would like to thank Robbe Jacops for ICP-OES measurements, Pieter De Meyer for TGA analyses and Naomi Billiet and Nele Debusschere for XRD measurements.References
- IEA, Key World Energy Statistics 2021, https://www.iea.org/reports/key-world-energy-statistics-2021, (accessed 25/06/2024).
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment, Chem. Rev., 2018, 118, 434–504 CrossRef CAS.
- J. H. Clark, T. J. Farmer, L. Herrero-Davila and J. Sherwood, Circular economy design considerations for research and process development in the chemical sciences, Green Chem., 2016, 18, 3914–3934 RSC.
- L. Guo, J. Sun, Q. Ge and N. Tsubaki, Recent advances in direct catalytic hydrogenation of carbon dioxide to valuable C2+ hydrocarbons, J. Mater. Chem. A, 2018, 6, 23244–23262 RSC.
- A. Rafiee, K. Rajab Khalilpour, D. Milani and M. Panahi, Trends in CO2 conversion and utilization: A review from process systems perspective, J. Environ. Chem. Eng., 2018, 6, 5771–5794 CrossRef CAS.
- A. I. Osman, M. Hefny, M. I. A. Abdel Maksoud, A. M. Elgarahy and D. W. Rooney, Recent advances in carbon capture storage and utilisation technologies: a review, Environ. Chem. Lett., 2021, 19, 797–849 CrossRef CAS.
- F. Sastre, A. V. Puga, L. Liu, A. Corma and H. García, Complete Photocatalytic Reduction of CO2 to Methane by H2 under Solar Light Irradiation, J. Am. Chem. Soc., 2014, 136, 6798–6801 CrossRef CAS.
- F. Sastre, A. Corma and H. García, Visible-Light Photocatalytic Conversion of Carbon Monoxide to Methane by Nickel(II) Oxide, Angew. Chem., Int. Ed., 2013, 52, 12983–12987 CrossRef CAS.
- R. Franke, D. Selent and A. Börner, Applied Hydroformylation, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS.
- W. Gao, R. Gao, Y. Zhao, M. Peng, C. Song, M. Li, S. Li, J. Liu, W. Li, Y. Deng, M. Zhang, J. Xie, G. Hu, Z. Zhang, R. Long, X.-D. Wen and D. Ma, Photo-Driven Syngas Conversion to Lower Olefins over Oxygen-Decorated Fe5C2 Catalyst, Chem, 2018, 4, 2917–2928 CAS.
- G. J. Sunley and D. J. Watson, High productivity methanol carbonylation catalysis using iridium: The Cativa™ process for the manufacture of acetic acid, Catal. Today, 2000, 58, 293–307 CrossRef CAS.
- J. R. Rostrup-Nielsen, New aspects of syngas production and use, Catal. Today, 2000, 63, 159–164 CrossRef CAS.
- P. Kaiser, R. B. Unde, C. Kern and A. Jess, Production of Liquid Hydrocarbons with CO2 as Carbon Source based on Reverse Water-Gas Shift and Fischer-Tropsch Synthesis, Chem. Ing. Tech., 2013, 85, 489–499 CrossRef CAS.
- R. Detz, M. Beerse, N. Meulendijks, P. Buskens and B. van der Zwaan, Towards the Use of Renewable Syngas for the Decarbonization of Industry, ChemSusChem, 2024, e202400059 CrossRef CAS.
- C. Xavier Silva, J. Moncada Botero, F. Sastre, J. van den Ham, P. Buskens, N. Meulendijks and R. J. Detz, Techno-economic analysis for the sunlight-powered reverse water gas shift process: Scenarios, costs, and comparative insights, Sustain. Energy Technol. Assessments, 2024, 65, 103768 CrossRef.
- P. Frontera, A. Macario, M. Ferraro and P. Antonucci, Supported Catalysts for CO2 Methanation: A Review, Catalysts, 2017, 7, 59 CrossRef.
- X. Su, X. Yang, B. Zhao and Y. Huang, Designing of highly selective and high-temperature endurable RWGS heterogeneous catalysts: recent advances and the future directions, J. Energy Chem., 2017, 26, 854–867 CrossRef.
- Y. A. Daza and J. N. Kuhn, CO2 conversion by reverse water gas shift catalysis: comparison of catalysts, mechanisms and their consequences for CO2 conversion to liquid fuels, RSC Adv., 2016, 6, 49675–49691 RSC.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Cocatalysts for Selective Photoreduction of CO2 into Solar Fuels, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed.
- J. Low, J. Yu, M. Jaroniec, S. Wageh and A. A. Al-Ghamdi, Heterojunction Photocatalysts, Adv. Mater., 2017, 29, 1601694 CrossRef PubMed.
- M. D. Khan, I. Fareed, M. ul Hassan Farooq, M. Akram, S. ur Rehman, Z. Ali, Z. Tariq, M. Irshad, C. Li and F. K. Butt, Methodologies for enriched photocatalytic CO2 reduction: an overview, Int. J. Environ. Sci. Technol., 2024, 21, 3489–3526 CrossRef CAS.
- D. Burova, J. Rohlfs, F. Sastre, P. M. Molina, N. Meulendijks, M. A. Verheijen, A.-S. Kelchtermans, K. Elen, A. Hardy, M. K. Van Bael and P. Buskens, Comparing the Performance of Supported Ru Nanocatalysts Prepared by Chemical Reduction of RuCl3 and Thermal Decomposition of Ru3(CO)12 in the Sunlight-Powered Sabatier Reaction, Catalysts, 2022, 12, 284 CrossRef CAS.
- R. Grote, R. Habets, J. Rohlfs, F. Sastre, N. Meulendijks, M. Xu, M. A. Verheijen, K. Elen, A. Hardy, M. K. Van Bael, T. den Hartog and P. Buskens, Collective photothermal effect of Al2O3-supported spheroidal plasmonic Ru nanoparticle catalysts in the sunlight-powered Sabatier reaction, ChemCatChem, 2020, 12, 5618–5622 CrossRef CAS.
- J. Rohlfs, K. W. Bossers, N. Meulendijks, F. Valega Mackenzie, M. Xu, M. A. Verheijen, P. Buskens and F. Sastre, Continuous-Flow Sunlight-Powered CO2 Methanation Catalyzed by γ-Al2O3-Supported Plasmonic Ru Nanorods, Catalysts, 2022, 12, 126 CrossRef CAS.
- P. Martínez Molina, K. W. Bossers, J. D. Wienk, J. Rohlfs, N. Meulendijks, M. A. Verheijen, P. Buskens and F. Sastre, Sunlight Powered Continuous Flow Reverse Water Gas Shift Process Using a Plasmonic Au/TiO2 Nanocatalyst, Chem. – Asian J., 2023, 18, e202300405 CrossRef PubMed.
- M. Xu, T. den Hartog, L. Cheng, M. Wolfs, R. Habets, J. Rohlfs, J. van den Ham, N. Meulendijks, F. Sastre and P. Buskens, Using Fiber Bragg Grating Sensors to Quantify Temperature Non-Uniformities in Plasmonic Catalyst Beds under Illumination, ChemPhotoChem, 2022, 6, e202100289 CrossRef CAS.
- P. Martínez Molina, N. Meulendijks, M. Xu, M. A. Verheijen, T. den Hartog, P. Buskens and F. Sastre, Low Temperature Sunlight-Powered Reduction of CO2 to CO Using a Plasmonic Au/TiO2 Nanocatalyst, ChemCatChem, 2021, 13, 4507–4513 CrossRef.
- J. H. A. Schuurmans, T. M. Masson, S. D. A. Zondag, P. Buskens and T. Noël, Solar-Driven Continuous CO2 Reduction to CO and CH4 using Heterogeneous Photothermal Catalysts: Recent Progress and Remaining Challenges, ChemSusChem, 2024, 17, e202301405 CrossRef CAS PubMed.
- F. Sastre, C. Versluis, N. Meulendijks, J. Rodríguez-Fernández, J. Sweelssen, K. Elen, M. K. Van Bael, T. den Hartog, M. A. Verheijen and P. Buskens, Sunlight-Fueled, Low-Temperature Ru-Catalyzed Conversion of CO2 and H2 to CH4 with a High Photon-to-Methane Efficiency, ACS Omega, 2019, 4, 7369–7377 CrossRef CAS.
- J. Volders, K. Elen, A. Raes, R. Ninakanti, A.-S. Kelchtermans, F. Sastre, A. Hardy, P. Cool, S. W. Verbruggen, P. Buskens and M. K. Van Bael, Sunlight-Powered Reverse Water Gas Shift Reaction Catalysed by Plasmonic Au/TiO2 Nanocatalysts: Effects of Au Particle Size on the Activity and Selectivity, Nanomaterials, 2022, 12, 4153 CrossRef CAS PubMed.
- A. Moores and F. Goettmann, The plasmon band in noble metal nanoparticles: an introduction to theory and applications, New J. Chem., 2006, 30, 1121–1132 RSC.
- A. Gellé and A. Moores, Plasmonic nanoparticles: Photocatalysts with a bright future, Curr. Opin. Green Sustainable Chem., 2019, 15, 60–66 CrossRef.
- R.-G. Ciocarlan, N. Blommaerts, S. Lenaerts, P. Cool and S. W. Verbruggen, Recent Trends in Plasmon-Assisted Photocatalytic CO2 Reduction, ChemSusChem, 2023, 16, e202201647 CrossRef CAS.
- G. Baffou and R. Quidant, Nanoplasmonics for chemistry, Chem. Soc. Rev., 2014, 43, 3898–3907 RSC.
- G. Baffou, I. Bordacchini, A. Baldi and R. Quidant, Simple experimental procedures to distinguish photothermal from hot-carrier processes in plasmonics, Light: Sci. Appl., 2020, 9, 108 CrossRef.
- R. Asapu, N. Claes, R.-G. Ciocarlan, M. Minjauw, C. Detavernier, P. Cool, S. Bals and S. W. Verbruggen, Electron Transfer and Near-Field Mechanisms in Plasmonic Gold-Nanoparticle-Modified TiO2 Photocatalytic Systems, ACS Appl. Nano Mater., 2019, 2, 4067–4074 CrossRef CAS.
- G. Baffou, F. Cichos and R. Quidant, Applications and challenges of thermoplasmonics, Nat. Mater., 2020, 19, 946–958 CrossRef CAS.
- Y. Sivan, J. H. Baraban and Y. Dubi, Experimental practices required to isolate thermal effects in plasmonic photo-catalysis: lessons from recent experiments, OSA Continuum, 2020, 3, 483–497 CrossRef.
- Y. Dubi and Y. Sivan, “Hot” electrons in metallic nanostructures—non-thermal carriers or heating?, Light: Sci. Appl., 2019, 8, 89 CrossRef PubMed.
- Y. Dubi, I. W. Un and Y. Sivan, Thermal effects – an alternative mechanism for plasmon-assisted photocatalysis, Chem. Sci., 2020, 11, 5017–5027 RSC.
- R. Kamarudheen, G. J. W. Aalbers, R. F. Hamans, L. P. J. Kamp and A. Baldi, Distinguishing Among All Possible Activation Mechanisms of a Plasmon-Driven Chemical Reaction, ACS Energy Lett., 2020, 5, 2605–2613 CrossRef CAS.
- A. Trovarelli, Catalytic Properties of Ceria and CeO2-Containing Materials, Catal. Rev.: Sci. Eng., 1996, 38, 439–520 CrossRef CAS.
- A. Trovarelli, C. de Leitenburg and G. Dolcetti, CO and CO2 hydrogenation under transient conditions over Rh–CeO2: novel positive effects of metal–support interaction on catalytic activity and selectivity, J. Chem. Soc., Chem. Commun., 1991, 472–473, 10.1039/C39910000472.
- P. Ebrahimi, A. Kumar and M. Khraisheh, A Review of CeO2 Supported Catalysts for CO2 Reduction to CO through the Reverse Water Gas Shift Reaction, Catalysts, 2022, 12, 1101 CrossRef CAS.
- M. Boaro, S. Colussi and A. Trovarelli, Ceria-Based Materials in Hydrogenation and Reforming Reactions for CO2 Valorization, Front. Chem., 2019, 7, 28, DOI:10.3389/fchem.2019.00028.
- A. E. Herzog, T. J. Michael, A. D. Dunkelberger, M. D. Johannes, D. R. Rolison, P. A. DeSario and T. G. Novak, Nanostructured CeO2 photocatalysts: optimizing surface chemistry, morphology, and visible-light absorption, Nanoscale, 2024, 16, 9659–9679, 10.1039/D4NR00676C.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Fundamentals and Catalytic Applications of CeO2-Based Materials, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed.
- M. Melchionna and P. Fornasiero, The role of ceria-based nanostructured materials in energy applications, Mater. Today, 2014, 17, 349–357 CrossRef CAS.
- A. Goguet, F. C. Meunier, D. Tibiletti, J. P. Breen and R. Burch, Spectrokinetic Investigation of Reverse Water-Gas-Shift Reaction Intermediates over a Pt/CeO2 Catalyst, J. Phys. Chem. B, 2004, 108, 20240–20246 CrossRef CAS.
- L. Lin, S. Yao, Z. Liu, F. Zhang, N. Li, D. Vovchok, A. Martínez-Arias, R. Castañeda, J. Lin, S. D. Senanayake, D. Su, D. Ma and J. A. Rodriguez, In Situ Characterization of Cu/CeO2 Nanocatalysts for CO2 Hydrogenation: Morphological Effects of Nanostructured Ceria on the Catalytic Activity, J. Phys. Chem. C, 2018, 122, 12934–12943 CrossRef CAS.
- M. Li, T. H. My Pham, Y. Ko, K. Zhao, L. Zhong, W. Luo and A. Züttel, Support-Dependent Cu–In Bimetallic Catalysts for Tailoring the Activity of Reverse Water Gas Shift Reaction, ACS Sustainable Chem. Eng., 2022, 10, 1524–1535 CrossRef CAS.
- M. Ziemba, J. Weyel and C. Hess, Elucidating the mechanism of the reverse water–gas shift reaction over Au/CeO2 catalysts using operando and transient spectroscopies, Appl. Catal., B, 2022, 301, 120825 CrossRef CAS.
- M. Zhu, Q. Ge and X. Zhu, Catalytic Reduction of CO2 to CO via Reverse Water Gas Shift Reaction: Recent Advances in the Design of Active and Selective Supported Metal Catalysts, Trans. Tianjin Univ., 2020, 26, 172–187 CrossRef CAS.
- E. Pahija, C. Panaritis, S. Gusarov, J. Shadbahr, F. Bensebaa, G. Patience and D. C. Boffito, Experimental and Computational Synergistic Design of Cu and Fe Catalysts for the Reverse Water–Gas Shift: A Review, ACS Catal., 2022, 12, 6887–6905 CrossRef CAS.
- L. F. Bobadilla, J. L. Santos, S. Ivanova, J. A. Odriozola and A. Urakawa, Unravelling the Role of Oxygen Vacancies in the Mechanism of the Reverse Water–Gas Shift Reaction by Operando DRIFTS and Ultraviolet–Visible Spectroscopy, ACS Catal., 2018, 8, 7455–7467 CrossRef CAS.
- H. Dong, M. Jung, Y. Zhang, S. Wang and S. Ding, Supported noble metal-based catalysts for thermal CO2 hydrogenation to CO, Mol. Catal., 2024, 560, 114133 CrossRef CAS.
- A. Vourros, I. Garagounis, V. Kyriakou, S. A. C. Carabineiro, F. J. Maldonado-Hódar, G. E. Marnellos and M. Konsolakis, Carbon dioxide hydrogenation over supported Au nanoparticles: Effect of the support, J. CO2 Util., 2017, 19, 247–256 CrossRef CAS.
- S.-Y. Lai, Y. Qiu and S. Wang, Effects of the structure of ceria on the activity of gold/ceria catalysts for the oxidation of carbon monoxide and benzene, J. Catal., 2006, 237, 303–313 CrossRef CAS.
- Q. Fu, S. Kudriavtseva, H. Saltsburg and M. Flytzani-Stephanopoulos, Gold–ceria catalysts for low-temperature water-gas shift reaction, Chem. Eng. J., 2003, 93, 41–53 CrossRef CAS.
- U. R. Pillai and S. Deevi, Highly active gold-ceria catalyst for the room temperature oxidation of carbon monoxide, Appl. Catal., A, 2006, 299, 266–273 CrossRef CAS.
- S. A. C. Carabineiro, S. S. T. Bastos, J. J. M. Órfão, M. F. R. Pereira, J. J. Delgado and J. L. Figueiredo, Exotemplated ceria catalysts with gold for CO oxidation, Appl. Catal., A, 2010, 381, 150–160 CrossRef CAS.
- L. G. A. van de Water, S. K. Wilkinson, R. A. P. Smith and M. J. Watson, Understanding methanol synthesis from CO/H2 feeds over Cu/CeO2 catalysts, J. Catal., 2018, 364, 57–68 CrossRef CAS.
- X. Zhu, X. Qu, X. Li, J. Liu, J. Liu, B. Zhu and C. Shi, Selective reduction of carbon dioxide to carbon monoxide over Au/CeO2 catalyst and identification of reaction intermediate, Chin. J. Catal., 2016, 37, 2053–2058 CrossRef CAS.
- S. Li, Y. Xu, Y. Chen, W. Li, L. Lin, M. Li, Y. Deng, X. Wang, B. Ge, C. Yang, S. Yao, J. Xie, Y. Li, X. Liu and D. Ma, Tuning the Selectivity of Catalytic Carbon Dioxide Hydrogenation over Iridium/Cerium Oxide Catalysts with a Strong Metal–Support Interaction, Angew. Chem., Int. Ed., 2017, 56, 10761–10765 CrossRef CAS PubMed.
- G. Kim, S. Shin, Y. Choi, J. Kim, G. Kim, K.-J. Kim and H. Lee, Gas-Permeable Iron-Doped Ceria Shell on Rh Nanoparticles with High Activity and Durability, JACS Au, 2022, 2, 1115–1122 CrossRef CAS.
- S. Li, Y. Xu, H. Wang, B. Teng, Q. Liu, Q. Li, L. Xu, X. Liu and J. Lu, Tuning the CO2 Hydrogenation Selectivity of Rhodium Single-Atom Catalysts on Zirconium Dioxide with Alkali Ions, Angew. Chem., Int. Ed., 2023, 62, e202218167 CrossRef CAS PubMed.
- S. Mine, T. Yamaguchi, K. W. Ting, Z. Maeno, S. M. A. H. Siddiki, K. Oshima, S. Satokawa, K.-i. Shimizu and T. Toyao, Reverse water-gas shift reaction over Pt/MoOx/TiO2: reverse Mars–van Krevelen mechanism via redox of supported MoOx, Catal. Sci. Technol., 2021, 11, 4172–4180 RSC.
- A. S. Malik, H. Bali, F. Czirok, Á. Szamosvölgyi, G. Halasi, A. Efremova, B. Šmíd, A. Sápi, Á. Kukovecz and Z. Kónya, Turning CO2 to CH4 and CO over CeO2 and MCF-17 supported Pt, Ru and Rh nanoclusters – Influence of nanostructure morphology, supporting materials and operating conditions, Fuel, 2022, 326, 124994 CrossRef CAS.
- L. C. Wang, D. Widmann and R. J. Behm, Reactive removal of surface oxygen by H2, CO and CO/H2 on a Au/CeO2 catalyst and its relevance to the preferential CO oxidation (PROX) and reverse water gas shift (RWGS) reaction, Catal. Sci. Technol., 2015, 5, 925–941 RSC.
- L. C. Wang, M. Tahvildar Khazaneh, D. Widmann and R. J. Behm, TAP reactor studies of the oxidizing capability of CO2 on a Au/CeO2 catalyst – A first step toward identifying a redox mechanism in the Reverse Water–Gas Shift reaction, J. Catal., 2013, 302, 20–30 CrossRef CAS.
- Y. Guo and H. Thérien-Aubin, Nanofibrous Photocatalytic Membranes Based on Tailored Anisotropic Gold/Ceria Nanoparticles, ACS Appl. Mater. Interfaces, 2021, 13, 37578–37588 CrossRef CAS.
- B. Li, B. Zhang, S. Nie, L. Shao and L. Hu, Optimization of plasmon-induced photocatalysis in electrospun Au/CeO2 hybrid nanofibers for selective oxidation of benzyl alcohol, J. Catal., 2017, 348, 256–264 CrossRef CAS.
- E. I. García-López, Z. Abbasi, F. Parrino, V. La Parola, L. F. Liotta and G. Marcì, Au/CeO2 Photocatalyst for the Selective Oxidation of Aromatic Alcohols in Water under UV, Visible and Solar Irradiation, Catalysts, 2021, 11, 1467 CrossRef.
- W. Zhao, Q. Dong, C. Sun, D. Xia, H. Huang, G. Yang, G. Wang and D. Y. C. Leung, A novel Au/g-C3N4 nanosheets/CeO2 hollow nanospheres plasmonic heterojunction photocatalysts for the photocatalytic reduction of hexavalentchromium and oxidation of oxytetracycline hydrochloride, Chem. Eng. J., 2021, 409, 128185 CrossRef CAS.
- A. Taratayko, Y. Larichev, V. Zaikovskii, N. Mikheeva and G. Mamontov, Ag–CeO2/SBA-15 composite prepared from Pluronic P123@SBA-15 hybrid as catalyst for room-temperature reduction of 4-nitrophenol, Catal. Today, 2021, 375, 576–584 CrossRef CAS.
- D. V. Dao, T. T. D. Nguyen, P. Uthirakumar, Y.-H. Cho, G.-C. Kim, J.-K. Yang, D.-T. Tran, T. D. Le, H. Choi, H. Y. Kim, Y.-T. Yu and I.-H. Lee, Insightful understanding of hot-carrier generation and transfer in plasmonic Au@CeO2 core–shell photocatalysts for light-driven hydrogen evolution improvement, Appl. Catal., B, 2021, 286, 119947 CrossRef CAS.
- J.-H. Wang, M. Chen, Z.-J. Luo, L. Ma, Y.-F. Zhang, K. Chen, L. Zhou and Q.-Q. Wang, Ceria-Coated Gold Nanorods for Plasmon-Enhanced Near-Infrared Photocatalytic and Photoelectrochemical Performances, J. Phys. Chem. C, 2016, 120, 14805–14812 CrossRef CAS.
- A. A. Fauzi, A. A. Jalil, N. S. Hassan, F. F. A. Aziz, M. S. Azami, I. Hussain, R. Saravanan and D. V. N. Vo, A critical review on relationship of CeO2-based photocatalyst towards mechanistic degradation of organic pollutant, Chemosphere, 2022, 286, 131651 CrossRef CAS PubMed.
- S. Ali, S. Basak, S. Sikdar and M. Roy, Synergetic effects of green synthesized CeO2 nanorod-like catalyst for degradation of organic pollutants to reduce water pollution, Environ. Nanotechnol., Monit. Manage., 2021, 16, 100539 CAS.
- J. Shen, J. Shen, W. Zhang, X. Yu, H. Tang, M. Zhang, Zulfiqar and Q. Liu, Built-in electric field induced CeO2/Ti3C2-MXene Schottky-junction for coupled photocatalytic tetracycline degradation and CO2 reduction, Ceram. Int., 2019, 45, 24146–24153 CrossRef CAS.
- M. K. Kesarla, M. O. Fuentez-Torres, M. A. Alcudia-Ramos, F. Ortiz-Chi, C. G. Espinosa-González, M. Aleman, J. G. Torres-Torres and S. Godavarthi, Synthesis of g-C3N4/N-doped CeO2 composite for photocatalytic degradation of an herbicide, J. Mater. Res. Technol., 2019, 8, 1628–1635 CrossRef CAS.
- M. Tahir, B. Tahir, N. A. S. Amin and Z. Y. Zakaria, Photo-induced reduction of CO2 to CO with hydrogen over plasmonic Ag-NPs/TiO2 NWs core/shell hetero-junction under UV and visible light, J. CO2 Util., 2017, 18, 250–260 CrossRef CAS.
- H. Robatjazi, H. Zhao, D. F. Swearer, N. J. Hogan, L. Zhou, A. Alabastri, M. J. McClain, P. Nordlander and N. J. Halas, Plasmon-induced selective carbon dioxide conversion on earth-abundant aluminum-cuprous oxide antenna-reactor nanoparticles, Nat. Commun., 2017, 8, 27 CrossRef.
- S. Ning, H. Xu, Y. Qi, L. Song, Q. Zhang, S. Ouyang and J. Ye, Microstructure Induced Thermodynamic and Kinetic Modulation to Enhance CO2 Photothermal Reduction: A Case of Atomic-Scale Dispersed Co–N Species Anchored Co@C Hybrid, ACS Catal., 2020, 10, 4726–4736 CrossRef CAS.
- A. A. Upadhye, I. Ro, X. Zeng, H. J. Kim, I. Tejedor, M. A. Anderson, J. A. Dumesic and G. W. Huber, Plasmon-enhanced reverse water gas shift reaction over oxide supported Au catalysts, Catal. Sci. Technol., 2015, 5, 2590–2601 RSC.
- Z. Yang, M. Zeng, K. Wang, X. Yue, X. Chen, W. Dai and X. Fu, Visible light-assisted thermal catalytic reverse water gas reaction over Cu-CeO2: The synergistic of hot electrons and oxygen vacancies induced by LSPR effect, Fuel, 2022, 315, 123186 CrossRef CAS.
- H. Zhang, T. Wang, J. Wang, H. Liu, T. D. Dao, M. Li, G. Liu, X. Meng, K. Chang, L. Shi, T. Nagao and J. Ye, Surface-Plasmon-Enhanced Photodriven CO2 Reduction Catalyzed by Metal–Organic-Framework-Derived Iron Nanoparticles Encapsulated by Ultrathin Carbon Layers, Adv. Mater., 2016, 28, 3703–3710 CrossRef CAS.
- J. Zhao, Q. Yang, R. Shi, I. N. Waterhouse Geoffrey, X. Zhang, W. Li-Zhu, T. Chen-Ho and T. Zhang, FeO–CeO2 nanocomposites: an efficient and highly selective catalyst system for photothermal CO2 reduction to CO, NPG Asia Mater., 2020, 12, 5, DOI:10.1038/s41427-019-0171-5.
- X. Wan, Y. Li, Y. Chen, J. Ma, Y.-A. Liu, E.-D. Zhao, Y. Gu, Y. Zhao, Y. Cui, R. Li, D. Liu, R. Long, K. M. Liew and Y. Xiong, A nonmetallic plasmonic catalyst for photothermal CO2 flow conversion with high activity, selectivity and durability, Nat. Commun., 2024, 15, 1273 CrossRef CAS PubMed.
- J. Yu, A. Muhetaer, X. Gao, Z. Zhang, Y. Yang, Q. Li, L. Chen, H. Liu and D. Xu, Highly Active Hydrogen-rich Photothermal Reverse Water Gas Shift Reaction on Ni/LaInO3 Perovskite Catalysts with Near-unity Selectivity, Angew. Chem., Int. Ed., 2023, 62, e202303135 CrossRef CAS.
- N. Blommaerts, N. Hoeven, D. Arenas Esteban, R. Campos, M. Mertens, R. Borah, A. Glisenti, K. De Wael, S. Bals, S. Lenaerts, S. W. Verbruggen and P. Cool, Tuning the turnover frequency and selectivity of photocatalytic CO2 reduction to CO and methane using platinum and palladium nanoparticles on Ti-Beta zeolites, Chem. Eng. J., 2021, 410, 128234 CrossRef CAS.
- H. Ge, Y. Kuwahara, K. Kusu, H. Kobayashi and H. Yamashita, Enhanced visible-NIR absorption and oxygen vacancy generation of Pt/HxMoWOy by H-spillover to facilitate photothermal catalytic CO2 hydrogenation, J. Mater. Chem. A, 2022, 10, 10854–10864 RSC.
- H. Ge, Y. Kuwahara, K. Kusu and H. Yamashita, Plasmon-induced catalytic CO2 hydrogenation by a nano-sheet Pt/HxMoO3−y hybrid with abundant surface oxygen vacancies, J. Mater. Chem. A, 2021, 9, 13898–13907 RSC.
- Z. Li, Q. Ul Hassan, W. Zhang, L. Zhu, J. Gao, X. Shi, Y. Huang, P. Liu and G. Zhu, Promotion of dual-reaction pathway in CO2 reduction over Pt0/SrTiO3–δ: Experimental and theoretical verification, Chin. J. Catal., 2023, 46, 113–124 CrossRef CAS.
- J. Jia, H. Wang, Z. Lu, P. G. O'Brien, M. Ghoussoub, P. Duchesne, Z. Zheng, P. Li, Q. Qiao, L. Wang, A. Gu, A. A. Jelle, Y. Dong, Q. Wang, K. K. Ghuman, T. Wood, C. Qian, Y. Shao, C. Qiu, M. Ye, Y. Zhu, Z.-H. Lu, P. Zhang, A. S. Helmy, C. V. Singh, N. P. Kherani, D. D. Perovic and G. A. Ozin, Photothermal Catalyst Engineering: Hydrogenation of Gaseous CO2 with High Activity and Tailored Selectivity, Adv. Sci., 2017, 4, 1700252 CrossRef.
- J. Jia, P. G. O'Brien, L. He, Q. Qiao, T. Fei, L. M. Reyes, T. E. Burrow, Y. Dong, K. Liao, M. Varela, S. J. Pennycook, M. Hmadeh, A. S. Helmy, N. P. Kherani, D. D. Perovic and G. A. Ozin, Visible and Near-Infrared Photothermal Catalyzed Hydrogenation of Gaseous CO2 over Nanostructured Pd@Nb2O5, Adv. Sci., 2016, 3, 1600189 CrossRef.
- Z. Zhu, X. Hu, X. An, M. Xiao, L. Zhang, C. Li and L. He, Photothermal Catalytic CO2 Hydrogenation with High Activity and Tailored Selectivity Over Monodispersed Pd-Ni Nanoalloys, Chem. – Asian J., 2022, 17, e202200993 CrossRef CAS.
- Z. Wu, J. Shen, C. Li, C. Zhang, K. Feng, Z. Wang, X. Wang, D. M. Meira, M. Cai, D. Zhang, S. Wang, M. Chu, J. Chen, Y. Xi, L. Zhang, T.-K. Sham, A. Genest, G. Rupprechter, X. Zhang and L. He, Mo2TiC2 MXene-Supported Ru Clusters for Efficient Photothermal Reverse Water–Gas Shift, ACS Nano, 2023, 17, 1550–1559 CrossRef CAS.
- B. Tahir, M. Tahir and N. A. S. Amin, Photocatalytic CO2 conversion over Au/TiO2 nanostructures for dynamic production of clean fuels in a monolith photoreactor, Clean Technol. Environ. Policy, 2016, 18, 2147–2160 CrossRef CAS.
- X. Zhang, X. Li, D. Zhang, N. Q. Su, W. Yang, H. O. Everitt and J. Liu, Product selectivity in plasmonic photocatalysis for carbon dioxide hydrogenation, Nat. Commun., 2017, 8, 14542 CrossRef CAS.
- K. Wang, S. Shao, Y. Liu, M. Cao, J. Yu, C. H. Lau, Y. Zheng and X. Fan, DRIFTS-SSITKA-MS investigations on the mechanism of plasmon preferentially enhanced CO2 hydrogenation over Au/γ-Al2O3, Appl. Catal., B, 2023, 328, 122531 CrossRef CAS.
- I. Ro, R. Carrasquillo-Flores, J. A. Dumesic and G. W. Huber, Intrinsic kinetics of plasmon-enhanced reverse water gas shift on Au and Au–Mo interfacial sites supported on silica, Appl. Catal., A, 2016, 521, 182–189 CrossRef CAS.
- B. Lu, F. Quan, Z. Sun, F. Jia and L. Zhang, Photothermal reverse-water-gas-shift over Au/CeO2 with high yield and selectivity in CO2 conversion, Catal. Commun., 2019, 129, 105724 CrossRef CAS.
- C. Li, Y. Sakata, T. Arai, K. Domen, K.-i. Maruya and T. Onishi, Carbon monoxide and carbon dioxide adsorption on cerium oxide studied by Fourier-transform infrared spectroscopy. Part 1.—Formation of carbonate species on dehydroxylated CeO2, at room temperature, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 929–943 RSC.
- S. Loridant, Raman spectroscopy as a powerful tool to characterize ceria-based catalysts, Catal. Today, 2021, 373, 98–111 CrossRef CAS.
- B. Chen, B. Wang, Y. Sun, X. Wang, M. Fu, J. Wu, L. Chen, Y. Tan and D. Ye, Plasma-Assisted Surface Interactions of Pt/CeO2 Catalyst for Enhanced Toluene Catalytic Oxidation, Catalysts, 2019, 9, 2 CrossRef.
- R. Schmitt, A. Nenning, O. Kraynis, R. Korobko, A. I. Frenkel, I. Lubomirsky, S. M. Haile and J. L. M. Rupp, A review of defect structure and chemistry in ceria and its solid solutions, Chem. Soc. Rev., 2020, 49, 554–592 RSC.
- J. Boon, Sorption-enhanced reactions as enablers for CO2 capture and utilisation, Curr. Opin. Chem. Eng., 2023, 40, 100919 CrossRef.
- W. Chen, G. Qian, Y. Wan, D. Chen, X. Zhou, W. Yuan and X. Duan, Mesokinetics as a Tool Bridging the Microscopic-to-Macroscopic Transition to Rationalize Catalyst Design, Acc. Chem. Res., 2022, 55, 3230–3241 CrossRef CAS PubMed.
- W. Chen, W. Fu, X. Duan, B. Chen, G. Qian, R. Si, X. Zhou, W. Yuan and D. Chen, Taming Electrons in Pt/C Catalysts to Boost the Mesokinetics of Hydrogen Production, Engineering, 2022, 14, 124–133 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy01194e |
| This journal is © The Royal Society of Chemistry 2025 |