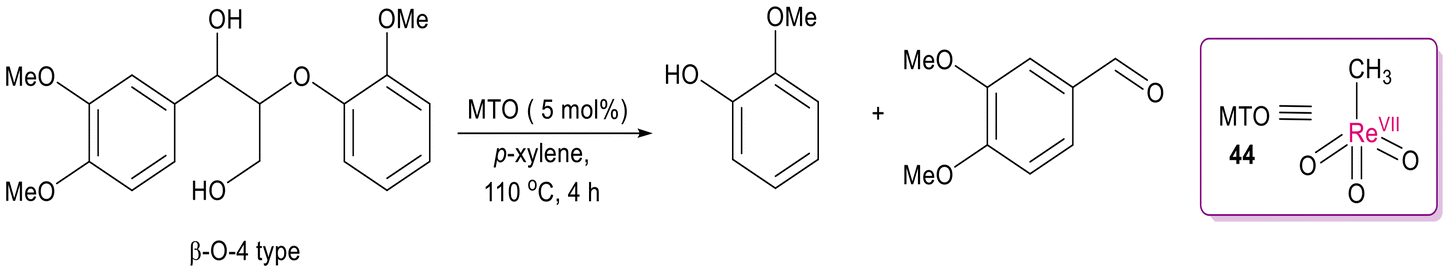
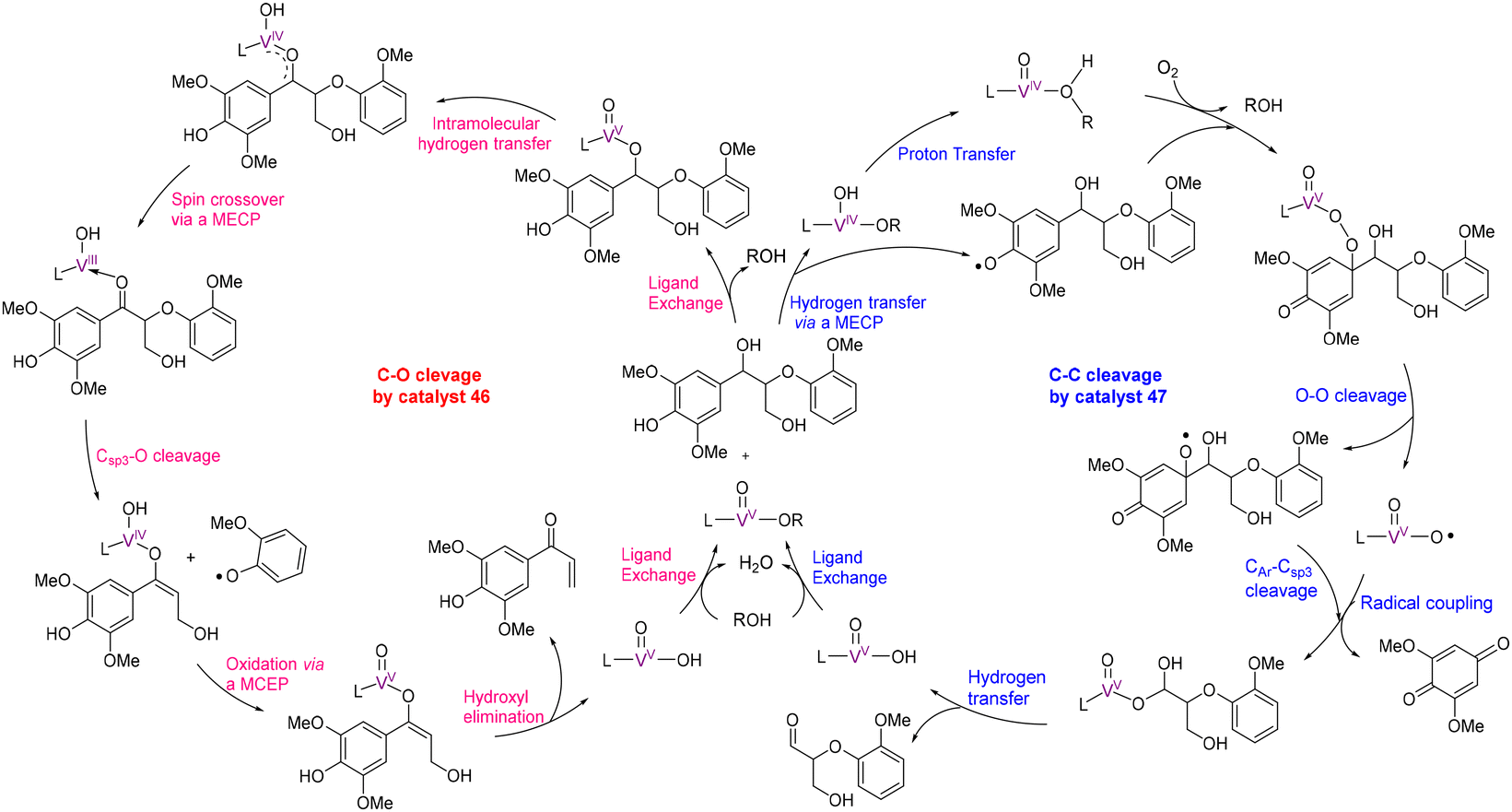
DOI:
10.1039/D4DT02555E
(Perspective)
Dalton Trans., 2025,
54, 3977-4012
Depolymerization by transition metal complexes: strategic approaches to convert polymeric waste into feedstocks
Received
8th September 2024
, Accepted 23rd December 2024
First published on 30th December 2024
Abstract
At present, plastic pollution is a global environmental catastrophe and a major threat to mankind. Moreover, the increasing manufacture of various plastic products is causing rapid depletion of precious resources. Thus, transforming plastic waste into feedstock, which can maintain a circular economy, has emerged as a significant technique for waste management and carbon resource conservation. Furthermore, the urgent development of effective depolymerization methods is vital to save our planet from man-made plastic pollution. Among various chemical depolymerization techniques developed thus far, cleavage of the polymeric skeleton by transition metal complexes is a highly emerging, effective and exciting strategy. In this context, herein, we have summarized mechanistic approaches for cleaving various polymeric bonds using organometallic catalysts. The recently developed strategies, catalyst design and mechanisms for depolymerization of synthetic and natural polymers with polar (C–N, C–O, C–Cl, and Si–O) and non-polar (C–C) skeletal bonds are systematically discussed in detail.
 Suman Dolai | Suman Dolai pursued studies in Chemistry and earned a master's degree in Chemistry from the Indian Institute of Technology Guwahati in 2018. Continuing his love for the subject, he began pursuing Ph.D. in 2019 in Inorganic Chemistry at the Indian Institute of Technology Kharagpur under the supervision of Dr Sanjib K. Patra. Suman is currently working on polymerization and depolymerization chemistry by developing heterobimetallic catalysts for sustainable chemistry. |
 Chinmoy K. Behera | Chinmoy Kumar Behera earned his M.Sc. degree in Chemistry from Maharaja Sriram Chandra Bhanjadeo University in 2018. Driven by his growing interest in chemistry, Chinmoy started his doctoral research journey in Inorganic Chemistry at IIT Kharagpur in 2019 under the guidance of Dr Sanjib K. Patra. His research focuses on developing controlled living polymerization to synthesize smart metallopolymers with diverse applications. |
 Sanjib K. Patra | Sanjib K Patra received his Ph.D. from the Indian Institute of Technology Kanpur in 2007. After completing postdoctoral research as a Royal Society postdoctoral fellow and Marie Curie postdoctoral fellow at the University of Bristol under the supervision of Prof. Ian Manners, he joined as faculty member at the Department of Chemistry, Indian Institute of Technology Kharagpur, in 2011. Currently his research interest includes developing functional wire-like organometallic complexes, developing smart and multifunctional side-chain metallopolymers, tailoring multifunctional terpyridyl conjugates and metallogels, and designing monometallic and bimetallic transition metal complexes as sustainable photo/redox catalysts for useful transformations such as functionalization of atmospheric CO2 and degradation of waste polymers to generate useful feedstock. |
1. Introduction
The twentieth century was a revolutionary period that offered us plastics, which soon became our daily requirement. Although plastics were initially a boon, they have presently become an environmental threat. Since the last decade, approximately 6.3 billion tonnes of plastic waste has been generated in ecosystems, which is a constant menace to society.1 Furthermore, despite many commitments from different countries regarding the mitigation of plastic wastes, Rochman and coworkers estimated that annual plastic waste may reach 53 million metric tons by 2030.2 Alternatively, highly developed industries aim to supply plastics according to the global demand. The use of single-use plastics is one of the main reasons for the accumulation of plastic waste. Thus, considering the current scenario, industries need to develop efficient protocols to achieve a circular economy by managing this plastic waste.3 Governments of different countries have already set a number of ambitious commitments to control this current concern of plastic hazard.4,5 Urgent attention to effective depolymerization is crucial to save our planet from this global crisis. Hence, this topic is highly significant to convey the importance of its research to scientific communities.
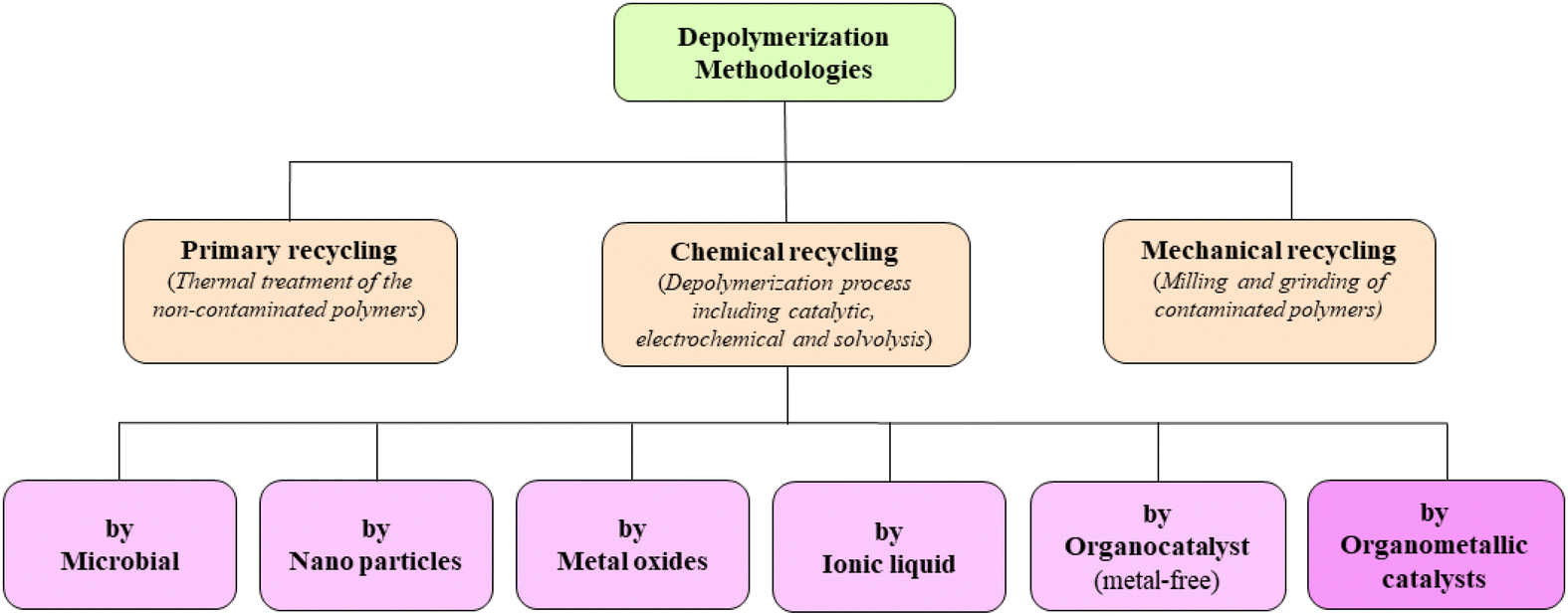
Besides environmental pollution, another serious concern related to the excessive usage of plastic materials is the depletion of precious carbon resources. Transforming plastic waste into feedstock will align with the circular economy, which should be the most effective protocol for waste management and carbon resource conservation. Thus, used plastic materials may be considered valuable waste, offering an inexpensive source of raw materials or value-added products in the chemical and pharmaceutical sectors.6–8 Multiple methodologies have been explored since 2000 and continue to be developed. As illustrated in the flowchart given in Fig. 1, mainly three methodologies are followed to recycle or reuse polymeric materials. Generally, recycling centers use primary recycling methods, which are based on melting and re-moulding clean and pure used-plastic materials. Although it is the least expensive method, it is limited to specific types of polymers and requires clean, often singly used and well-separated plastic materials.9,10 Alternatively, mechanical recycling, involving pre-treatment and palletization of contaminated polymers, compromises product quality and durability with each recycling step. In contrast, chemical recycling, specifically the depolymerization method involves bond cleavage to generate monomers or low molecular weight oligomers. However, the depolymerization process faces inherent challenges due to robust skeletal bonds in the entangled polymeric structure. The resulting monomers after successful depolymerization can be reused to produce polymers with improved quality and durability. Chemical depolymerization surpasses mechanical processes, offering a cost-effective recycling method by appropriate catalytic reactions, notably with transition metal and organometallic catalysts.
 |
| | Fig. 1 Classifications of conventional depolymerization methodologies. | |
Depolymerization by catalysts is one of the subways to cleave the robust polymeric bonds, and also an effective method to convert waste to reusable resources. Judiciously designed organometallic catalysts and metal-free organocatalysts are efficient in activating and breaking the polar and non-polar bonds of small molecules. Inspired by this exciting chemistry, in recent times, multiple research groups have attempted to cleave the robust bonds in the polymeric skeleton. Catalysts, especially transition metal- and organometallic-based catalysts, have emerged as effective tools in breaking polymeric skeletal bonds. These catalysts not only contribute to breaking polymers efficiently but also offer “closing the loop of polymer making and breaking”. Some recent reviews covered the depolymerization catalysts focusing on specific methodologies11,12 and discrete information13–16 about the circular economy. However, although organometallic catalysts demonstrate superior efficiency and selectivity in depolymerization, achieving the highest depolymerization efficiency is still the goal to date. Also, despite its drawbacks such as catalyst deactivation and contamination barriers, catalytic depolymerization represents a promising avenue for sustainable recycling under mild conditions.
Herein, in this review, we focus our discussion on the strategic development of well-defined organometallic catalysts, emphasizing the design and mechanism for cleaving robust nonpolar and polar bonds in diverse polymeric structures. The bond-breaking processes are categorized and the role of ligands, catalyst design and probable mechanisms are systematically discussed and presented. The coordination spheres of these catalysts exert a multifaceted influence, enhancing their catalytic properties. Coordinated ligands govern their electronic and steric effects, fine-tuning catalytic reactions for better control and selectivity. Ongoing research aims to transform these catalytic processes into recycling technologies to mitigate existing barriers. Thus, this review comprehensively addresses the catalyst design, recent developments, strategies and mechanisms in depolymerization reactions of robust polymers, with classification of the skeletal bond polarity. Also, detailed strategies for the cleavage of the non-polar and polar polymeric bonds of synthetic polymers and cleavage of the C–C and C–O bonds in biopolymers are systematically discussed.
2. Strategies for cleavage of polymeric bonds by organometallic complexes
To realize effective depolymerization through the cleavage of polymeric skeletal bonds by catalysts, one must understand the nature of the bonds to be cleaved. Thus, Tong and co-workers calculated the bond dissociation enthalpies (BDE) for model compounds corresponding to six commonly used polymers (M1–M6) using density functional theory, as shown in Fig. 2.17 According to the results, the thermal stability of the four types of polyolefins follows the trend of PVC < PS < PP < PE. The cleavage of polymeric bonds is thermodynamically more challenging compared to other small molecules because of the possible existence of various intramolecular and intermolecular noncovalent interactions such as hydrogen bonding, π–π interaction, van der Waals interaction and ionic interaction.18 Therefore, the strategically induced non-covalent interaction in functional polymers, which modulates their mechanical, physiochemical and biochemical properties, creates further complexity in breaking the polymeric chain. Hence, to cleave the highly stable bonds in diverse polymeric systems, defining specific strategies are crucial. Our discussion categorizes and emphasizes the designed organometallic catalysts to break the polar and non-polar polymeric bonds.
 |
| | Fig. 2 Bond dissociation enthalpies (BDE, kJ mol−1) of homonuclear and heteronuclear bonds in model compounds of commonly used polymers.17 | |
2.1 Strategies to break down non-polar polymers
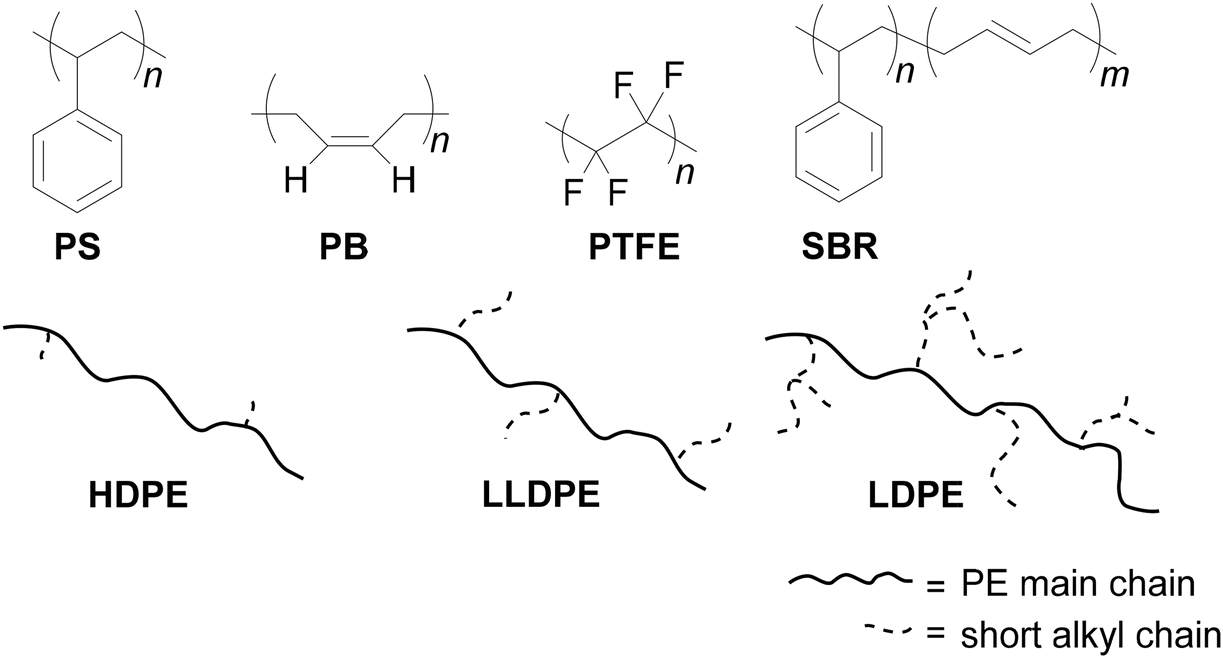
Polyolefins are hydrocarbon polymers having non-polar bonds (such as C–C and C–H), which are chemically robust given that there is no site for nucleophilic or electrophilic attack, making them resistant to degradation (Fig. 3). Examples include PE (polyethylene), PB (polybutadiene), PS (polystyrene), PTFE (polytetrafluoroethylene) and related hydrocarbon homo and copolymers. Notably, PE became commercially significant very quickly after its discovery by Fawcett and Gibson in 1933 at Imperial Chemical Industries (ICI).19 The production of polyolefins is the highest among the categories of polymers. For example, 65% of the total 278 million tons of polymers produced in 2018 was only polyolefins, which included 47.3 million tons of HDPE, 33.4 million tons of LDPE, 22.2 million tons of LLDPE, and 76 million tons of PP.20,21 The production of polyolefins continues to increase each year. Thus, this exponential accumulation of polyolefins waste needs serious attention. The degradation of this type of chemically stable polymeric waste to monomers, oligomers or value-added useful chemicals is highly challenging but essential for maintaining a circular economy and waste management. In the following subsections, we discuss the ground-breaking strategies for activating non-polar skeletal bonds (C–C and C–H) to produce valuable feedstock.
 |
| | Fig. 3 Chemical structure of commonly used polymers containing non-polar bonds. PS: polystyrene, PB: polybutadiene, PTFE: polytetrafluoroethylene, SBR: styrene butadiene rubber, HDPE: high density polyethylene, LDPE: low-density polyethylene, and LLDPE: linear low-density polyethylene. | |
2.1.1. Depolymerization of polyolefins by tandem dehydrogenation and olefin metathesis.
Herein, we discuss the recent progress in cleaving hydrocarbon-based polymeric skeletal bonds via the strategic development of transition metal-based catalysis. We restrict our discussion to the advancement of homogeneous and well-defined heterogeneous transition metal catalytic systems in this perspective. In general, catalytic methods for the depolymerization of polyolefins begin with C–H bond activation and can proceed through a number of chemical intermediates. These methods include a carbocation intermediate for catalytic cracking by mesoporous materials and nanozeolite catalysts.22 Other cracking methods have also been reported, involving oxidation by HNO3 for the conversion of LDPE to various dicarboxylic acids, HOOC(CH2)xCOOH (x = 1–5), or by a mixture of NO and O2 gases for the degradation of polystyrene to useful small molecules such as benzoic acid and nitrobenzoic acid.23,24 These cracking processes can also proceed through adsorbed alkyne intermediates for hydrogenolysis by using well-dispersed Pt nanoparticles supported on SrTiO3 perovskite nanocuboid metal surfaces.25–27 However, these non-transition metal or nanoparticle-based catalytic systems will not be included for further discussion in our perspective.
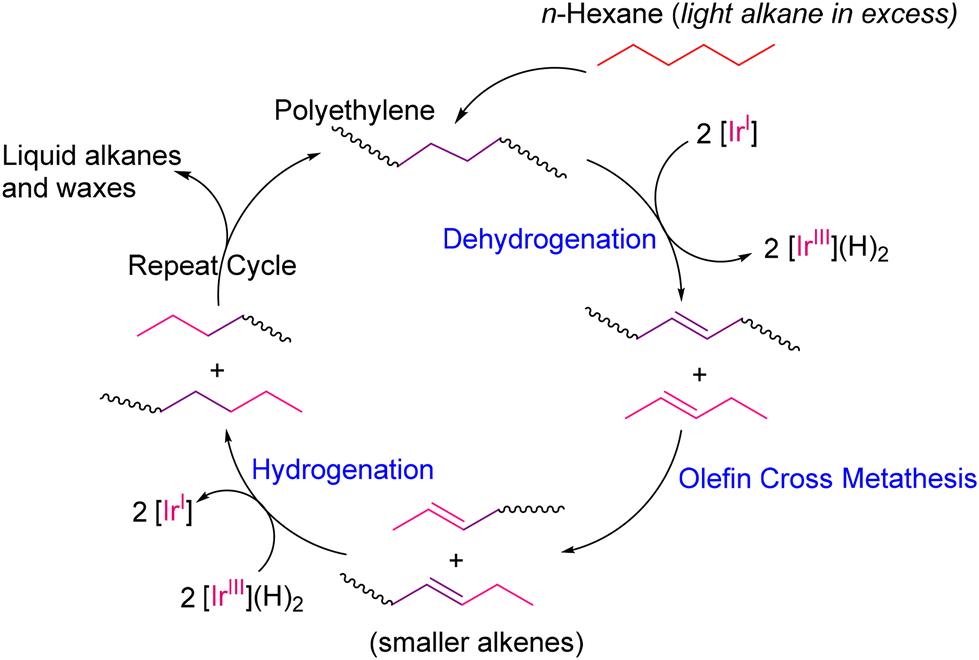
Tandem dehydrogenation and olefin cross metathesis (TDOCM) is one of the most effective protocols to recently emerge for the depolymerization of polyethylene, as first pioneered by the group of Goldman and Brookhart.28 TDOCM proceeds via a olefin-intermediate process (OIP) through the dehydrogenation of alkanes, as proposed by Beckham and co-workers (Fig. 4).29 The dehydrogenated olefin intermediate again is involved in the olefin metathesis reaction to form different hydrocarbons with reduced chain lengths. Thereby, dehydrogenation and cross metathesis work in a tandem manner in the TDOCM method to activate the non-polar C–C bonds, and finally degrade into useful resources.
 |
| | Fig. 4 Schematic free energy diagram of olefin-intermediate process (OIP) for activating non-polar C–C bonds: strategy for TDOCM.28,29 | |
TDOCM starts with the very widely used dehydrogenation strategy of polyolefin as the first step by a suitable dehydrogenation catalyst. Various groups reported the use of Ir(I)-based homogenous catalysts for alkane dehydrogenation, which was essentially the alkane activation step.30 In this case, several groups successfully obtained the dehydrogenated products by Ir(III) complexes in presence of a hydrogen acceptor or sacrificial acceptor (alkenes such as norbornene, tert-butylethylene and 1-decene) to produce alkanes.31–39 This strategy was used by Goldman, where they first introduced an iPrPCP-Ir(III)(H)2 (iPrPCP = 2,6-bis[di(isopropyl)-phosphinomethyl]phenyl)-based pincer catalyst (1) for the selective dehydrogenation of n-alkanes (with a TON value of 97) in the presence of a hydrogen acceptor (alkene), as illustrated in Scheme 1.40 This catalyst produced α-olefins as the major product from the linear alkanes. However, α-olefins are the thermodynamically least stable among the corresponding regioisomers and any method for their formation must likely begin with the activation of a strong C–H bond. The mechanism consists of two consecutive catalytic cycles, as follows (i) transfer dehydrogenation and (ii) isomerization. Additionally, the transfer dehydrogenation cycle consists of two microscopic reversal steps, namely (i) oxidative addition of C–H bond of alkane and (ii) β-hydrogen elimination. They observed that the isomer distribution of the products was only dependent on the concentration and nature of the hydrogen acceptor. Moreover, they also proposed that the competition between the hydrogen acceptor alkene and 1-octene towards the insertion reaction in the Ir–H bond of catalyst 1 is a major factor in determining the isomer distribution and fraction of 1-octene product. Additionally, they also demonstrated that the bulkiness of the ligand played a significant role in controlling the rate of the reaction and regioselectivity of the alkene products by varying the pincer ligand to tBuPCP (2,6-bis[di(tert-butyl)-phosphinomethyl]phenyl) from iPrPCP. Many experimental and theoretical works have been reported for the further modification of this family of dehydrogenation catalysts to investigate the effect of the steric bulk of PCP-based pincer ligands on the catalytic efficiency in the transfer dehydrogenation reaction of alkanes.41–43
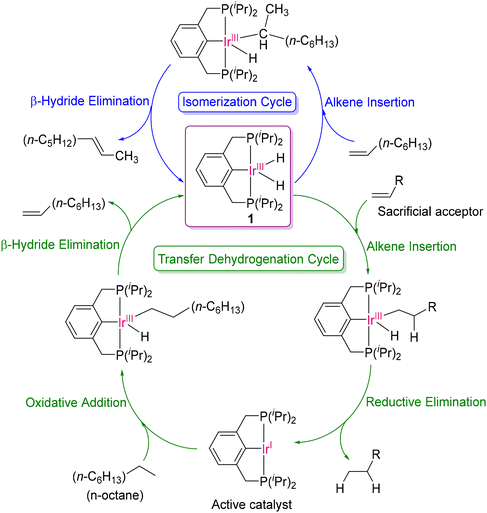 |
| | Scheme 1 Concurrent cycle of isomerisation and transfer dehydrogenation of n-octane with the PCP-Ir(III) Brookhart catalyst.40 | |
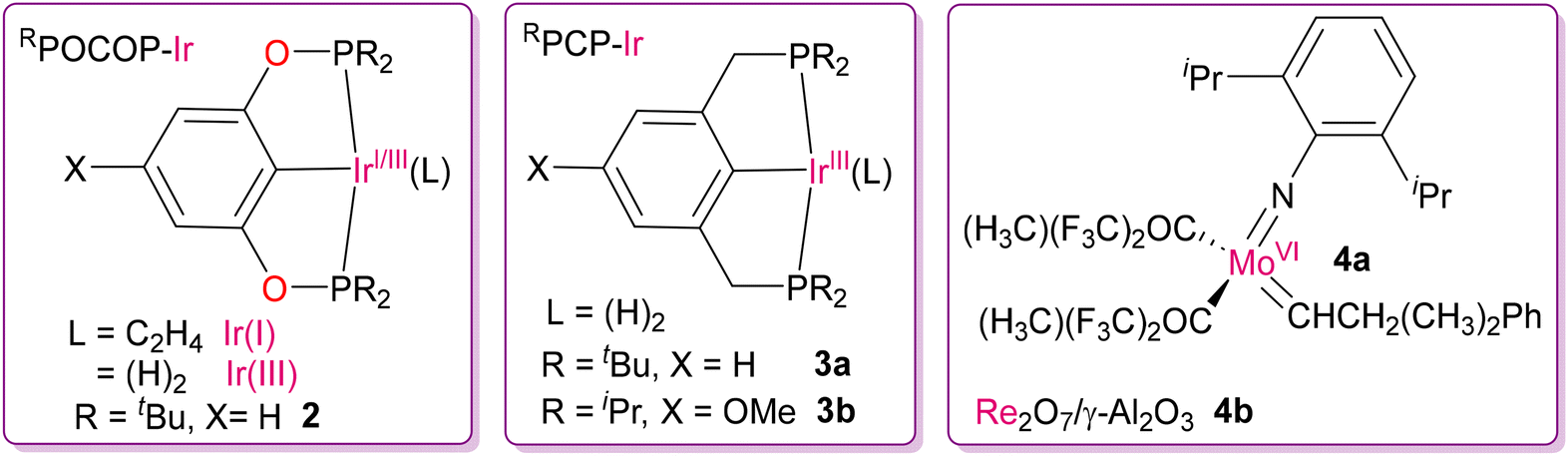
The development of highly active and exciting category of dehydrogenation catalysts to obtain regioselective isomeric products of α-olefins motivated researchers to utilize these catalytic systems for the depolymerization of hydrocarbon polymers. One of the first pioneering works in this field of depolymerization was reported in 2006 by the Goldman and Brookhart group utilizing the TDOCM strategy.44 They successfully demonstrated the inter-conversion of alkanes to new hydrocarbon products with a lower molecular weight by homogeneous and heterogeneous catalysts at moderate temperatures via a tandem combination of two independent catalytic process, where one is dehydrogenation and other is alkene metathesis. They judiciously designed homogeneous RPOCOP-Ir(I/III) (2) and RPCP-Ir(III) (3a and 3b) catalysts, with bis(phosphinite) and bis(phosphine) pincer ligands respectively, for the dehydrogenation of hexanes, as shown in Fig. 5.45 It was obvious that tert-butylethylene (TBE) was necessary as a sacrificial hydrogen acceptor for the Ir(III) catalyst precursors for converting into the active form of Ir(I) dehydrogenating catalysts. As an alkene-metathesis catalyst, they used a Schrock-type imido-alkylidene complex of Mo(VI) (4a).46 The Schrock-type catalyst was preferred over the widely used Grubbs catalyst given that the latter reacts with the Ir(III/I) catalyst and deactivates the dehydrogenating ability.44 In the TDOCM process, the hydrogen atoms are eliminated during dehydrogenation, and then undergo re-addition into the olefinic intermediates, yielding a rearranged mixture of alkanes via metathesis followed by hydrogenation. After the formation of 1-hexene through dehydrogenation at the terminal position of n-hexane, there are two possible pathways for the next steps (direct isomerization or metathesis followed by isomerization) to form a new pair of alkenes, as shown in Scheme 2. Lastly, alkene cross-metathesis and subsequent hydrogenation by [Ir(III)(H)2] species yielded a new pair of hydrocarbons, n-pentane and n-heptane, completing the catalytic cycle. With the help of NMR spectroscopic studies (31P NMR and 1H NMR) they tried to gain insight into the mechanism by detecting the resting states as well as the extent of alkane metathesis by this combination of catalysts. It was revealed that although the initial major resting states were either Ir–H2 or Ir–(C2H4), at the later stage of the reaction (4 days) they identified Ir-(1-hexene) species as the major resting state in solution. This was because of the instability and degradation of the alkene metathesis catalyst under the reaction conditions (125 °C), resulting in an increase in the concentration of 1-hexene, and the dehydrogenating catalysts were still active. This problem of catalyst deactivation at high temperature was solved to some extent by using a stable and heterogeneous catalyst for metathesis. The highest activity and greater stability were observed by replacing the Mo(VI) catalyst (4a) with a supported Re2O7/γ-Al2O3 metathesis catalyst (4b) at 175 °C for 4 to 5 days. In their study, catalysts 3b in combination with 4b showed the best result while attempting the metathesis of n-decane using tert-butylethylene as the acceptor, yielding alkane products ranging from C2–C28 over the course of 3 h, as monitored by GC. In addition to alkane disproportionation (self-metathesis), comproportionation (cross-metathesis) of low and high molecular weight alkanes was also observed, leading to the production of alkanes with intermediate molecular weights.
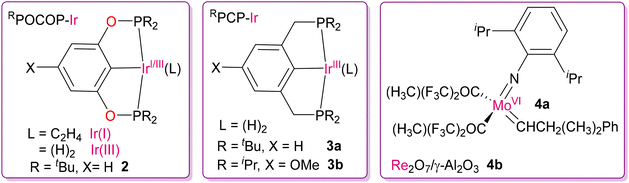 |
| | Fig. 5 Ir(I/III)-based catalysts for the selective dehydrogenation of saturated C–C bonds, along with Mo(VI)- and Re(VII)-based metathesis catalysts.44 | |
 |
| | Scheme 2 Two possible pathways for alkane metathesis via dehydrogenation and cross alkene-metathesis to reduce the chain length.44 | |
Furthermore, the same group also developed a heterogeneous dehydrogenating system of homogenous analogues of PNP-Ir(I) or POCOP-Ir(I) catalysts, exhibiting higher productivities and longer catalytic lifetimes compared to its corresponding homogeneous system.47 The idea was to install basic groups such as –OMe, –NMe2, –OK, and –OP(tBu)2 at the para-position of the POCOP and PCP ligand moiety, which can be strongly adsorbed on γ-alumina through a Lewis acid-Lewis base interaction to get the modified catalysts 5 and 6a–6c, respectively, as shown in Fig. 6. In the case of the alkene metathesis reaction, they screened two long-lived heterogeneous catalysts, which were Re2O7/γ-Al2O3 and MoO3/CoO/γ-Al2O3. Using this combination of heterogeneous catalytic systems and following TDOCM approach, they optimized the alkane metathesis reaction using a two-pot reaction device operating at two different temperatures for the two different catalysts (220 °C for dehydrogenation and 50 °C for alkene metathesis catalyst) considering the thermal stability of the individual catalytic systems. These two separate catalysts could be physically recovered and were partially recyclable for multiple cycles to achieve a better turnover number as high as 6900.
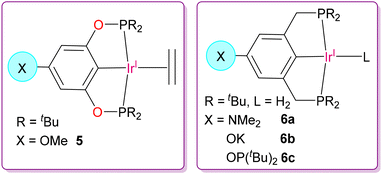 |
| | Fig. 6 Chemical structures of Ir(I) catalysts of POCOP and PCP pincer ligands functionalized with para-substituted basic groups for investigation as alumina-supported dehydrogenating catalysts.47 | |
In 2016, TDOCM was employed to depolymerize a real polyethylene sample to produce fuel and waxes under mild conditions in a regulated manner.48 Guan, Huang and co-workers used Ir(I/III) catalysts (2, 3a and 3b) for dehydrogenation in combination with the well-known heterogeneous olefin metathesis catalyst 4b (Re2O7/γ-Al2O3) to depolymerize PE to liquid alkanes and waxy solids, utilizing n-octane or n-hexane as both the solvent and co-reactant. As shown in Scheme 3, the PCP-Ir(I) catalyst first converts PE and the light alkane (n-hexane) to unsaturated species (alkenes). Subsequently, the alkenes are modified to smaller alkenes through cross metathesis reaction of a long alkene and a small alkene (hexene) by the olefin metathesis catalyst. Through the repetition of this process, the chain length of PE gradually decreases, ultimately leading to the depolymerization of PE. Finally, saturated alkanes are produced by hydrogenating the generated alkenes by the hydrogenating catalyst, Ir(III)–H2. Numerous types of plastic samples, including HDPE, LDPE, and LLDPE of high molecular weights of up to millions, could be degraded to low molecular weight oils and waxes within a day at 175 °C in the presence of excess low-cost light alkanes such as petroleum ether. According to the mechanism, it was understood that the efficiency of shortening the PE chain depends on the position of the double bond of the alkene generated from PE. Degradation is faster when the internal double bond is in the middle of the polymer chain. The POCOP-Ir(I) catalyst (2) was found to be more effective due to its higher regioselectivity for the generation of internal alkenes in comparison to the PCP-Ir(I) catalysts (3a and 3b), which mainly produced terminal alkenes.
 |
| | Scheme 3 General mechanism of tandem dehydrogenation and olefin cross metathesis (TDOCM) for degradation of the PE sample to produce liquid alkanes and waxes.48 | |
Following the successful dehydrogenation of poly(α-olefins) using catalysts 3a and 3b by Goldman and Coates in 2005,49 the deconstruction of post-consumer HDPE into telechelic macro-monomers with possible upcycling opportunity has been reported recently in 2022 by Delferro, Coates and co-workers.50 By utilizing a POCOP-Ir(I) catalyst (6C), unsaturation was induced in HDPE by catalytic dehydrogenation without an alkene acceptor. The partly unsaturated HDPE was converted into telechelic macro-monomers by cross metathesis with 2-hydroxyethyl acrylate by a Hoveyda–Grubbs 2nd generation (HG2) catalyst. Moreover, further functionalization and chemical modification of the macro-monomers are possible for repolymerization to access new polymers. In this way, the waste polyethylene materials can enter the loop of the chemical recycling process.
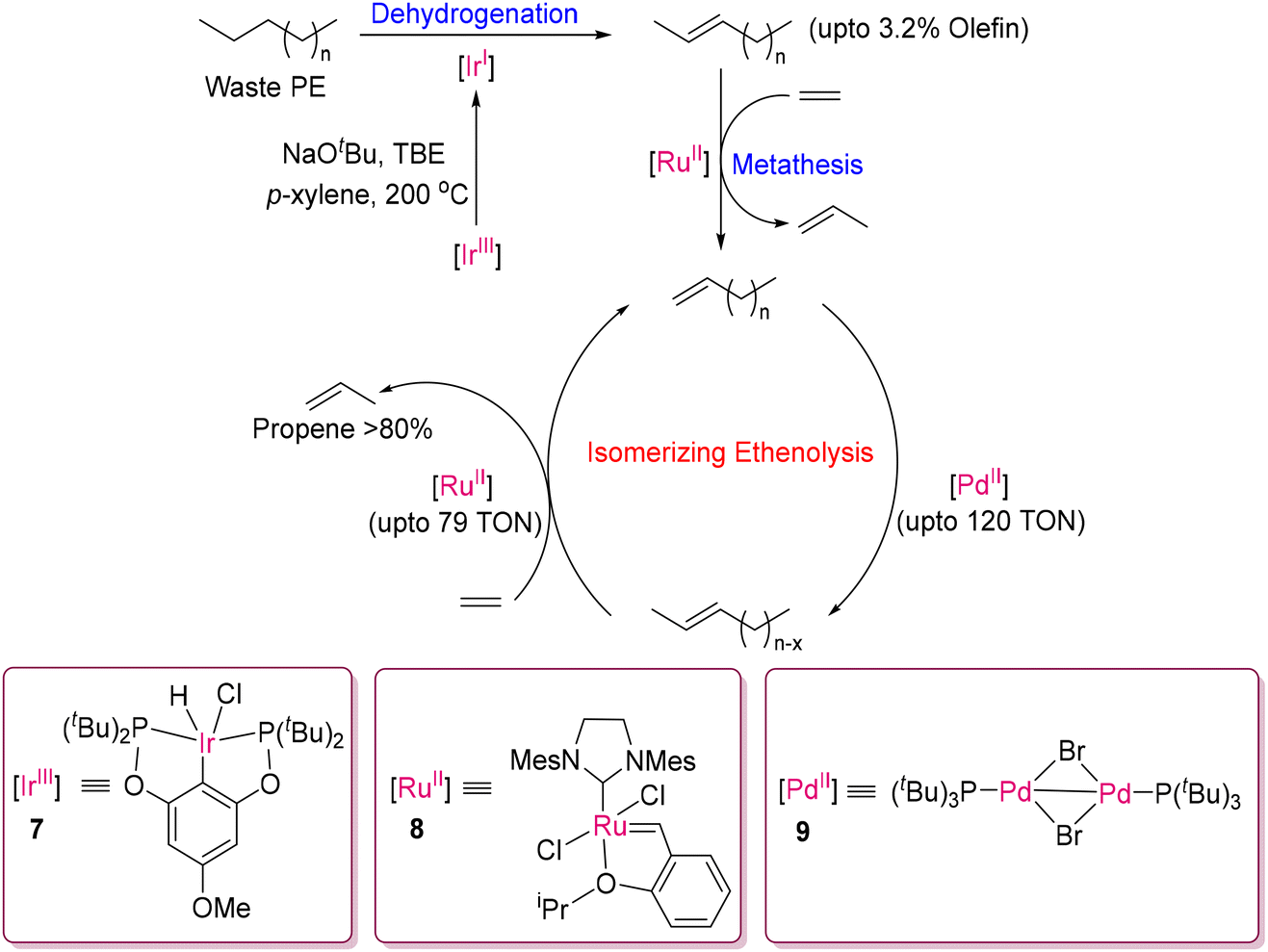
Recently, Hartwig and co-workers introduced a modified TDOCM process to produce propene in up to 80%, yield as shown in Scheme 4.51 In this modified method, a tandem process involving the partial dehydrogenation of polyethylene and isomerizing ethenolysis of the desaturated chain was employed strategically. Waste polyethylene was first dehydrogenated by the tBuPOCOP-Ir(I) dehydrogenation catalyst (which is formed from 7in situ) in the presence of tert-butylethylene as a sacrificial hydrogen acceptor to produce predominantly internal alkenes, which can then undergo ethenolysis through olefin metathesis by HG-2 catalyst (8) to form a wide range of alkene fragments.51 Furthermore, the combination of a dinuclear Pd(II) isomerization catalyst (9), furnishing exclusively internal alkenes, and a Ru(II) metathesis catalyst (8) converts the unsaturated fragmented PE to propylene (up to 87% yield) via cross olefin metathesis with ethylene. This strategy, designated as a dehydrogenation and isomerizing ethenolysis (DIE) process, is an exciting and very promising approach to convert waste PE to industrially important monomer feedstock. Scott, Guironnet, and co-workers independently developed another method involving TDOCM by incorporating a tandem catalytic pathway involving ethenolysis, dehydrogenation and isomerization processes.52 This approach utilizes distinct homogeneous catalysts tailored for each process based on Ru(II) (10), Ir(I/III) (5) and Pd(II) (9) catalysts, respectively. Additionally, methyltrioxorhenium (MTO) supported on activated chlorinated alumina (Cl–Al2O3) (11) acted as both an ethenolysis and isomerization catalyst, as shown in Fig. 7. The compatibility of these catalysts was screened with low molecular weight PE (Mn = 3000 Da), achieving an impressive 94% yield of propylene. This method demonstrates a potential approach by combining multiple catalytic processes to efficiently convert polymeric substrates into valuable olefins. Very recently, Hartwig and co-workers reported an elegant approach for converting PE and PP to propylene and isobutylene using tungsten oxide supported on silica (WO3/SiO2) and sodium supported on γ-alumina (Na/γ-Al2O3).53 This approach was found to be remarkably efficient with a conversion yield of more than 90% at 320 °C, and without the dehydrogenation of the starting polyolefins.
 |
| | Scheme 4 Formation of propene of up to 80% yield from waste PE via a modified TDOCM process developed by Hartwig's group.51 | |
 |
| | Fig. 7 Chemical structures of the catalysts used by Scott, Guironnet, and co-workers for degradation of PE to propylene through the TDOCM approach.52 | |
Thus, TDOCM catalyst systems can play a vital role in deconstructing waste hydrocarbon polymers, with future enhancements in efficiency and selectivity. The challenges in catalyst recovery suggest a potential shift to heterogeneous catalysis and industrial applicability. This strategy, coupled with well-designed transition metal-based catalysts, is expected to shape the future of depolymerization processes.
2.1.2. Depolymerization by C–C bond cleavage applying the principle of microscopic reversibility.
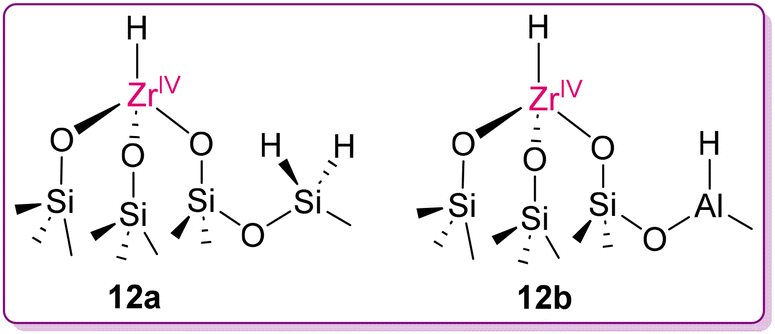
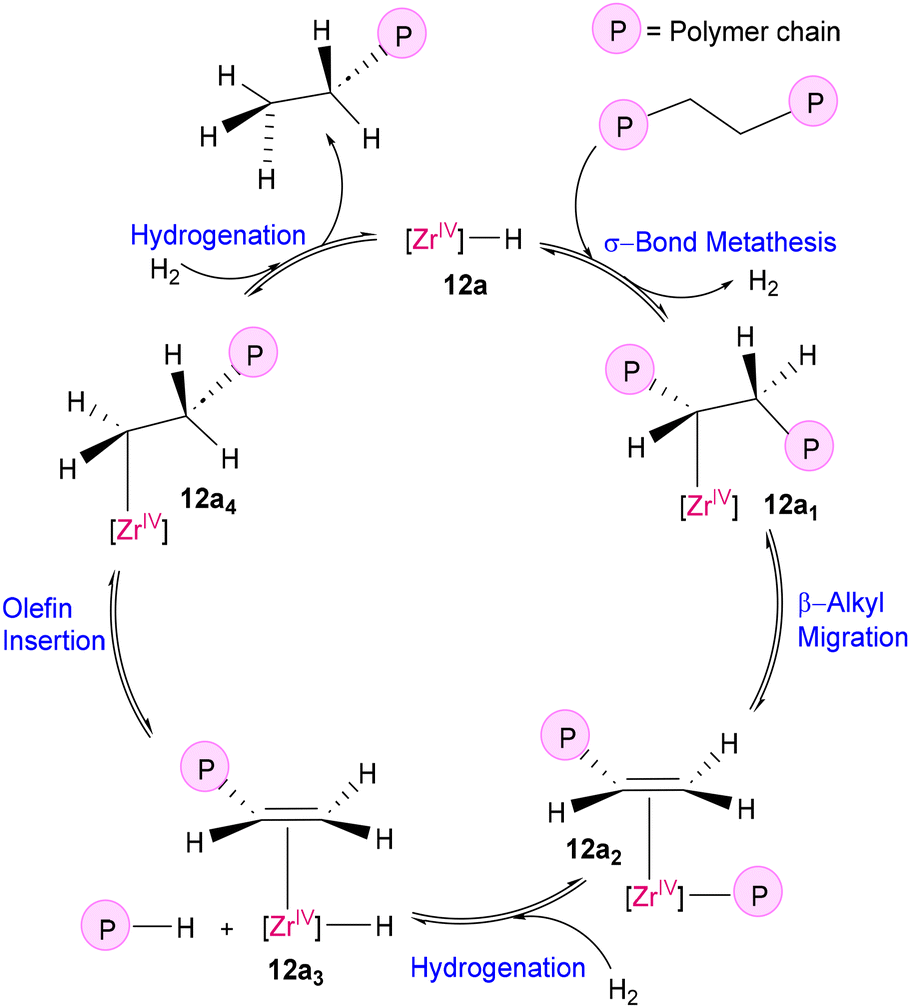
The principle of microscopic reversibility suggests that any forward and reverse molecular process occur at the same rate at equilibrium. Applying this principle to the mechanistic aspect, polymerization catalysts can also act as depolymerization catalysts. In Ziegler–Natta-type polymerization, the key step involves the insertion of an olefinic bond into a metal–alkyl bond. This step can be considered the microscopic opposite of β-alkyl transfer, unveiling the interconnection between polymerization and depolymerization processes, as illustrated in Fig. 8.54 Although this step is theoretically reversible from a mechanistic perspective, it is thermodynamically difficult at moderate temperature, stimulating researchers to find solutions to overcome this limitation. In this line of work, Dufaud and Basset successfully activated and cleaved the C–C bonds of chemically stable PE using a highly electrophilic zirconium monohydride catalyst supported to silica-alumina, [(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiO)3ZrH] (12a and 12b) using H2 (105 Pa) at 150 °C (Fig. 9).55 The heterogeneous catalyst was synthesized by the condensation of ZrNp4 (Np = neopentyl) with the surface silanol groups of a partially dehydroxylated silica-alumina gel, followed by hydrogenolysis with H2, with the simultaneous generation of silicon dihydride (12a) and aluminium hydride (12b), as proposed by them. As illustrated in Scheme 5, the proposed mechanism involves the facile electrophilic activation of C–H bonds of alkanes through σ-bond metathesis at the d0 metal center of a highly electrophilic catalyst [(SiO)3ZrH] (12a), followed by β-alkyl migration to form alkyl-olefin-Zr(IV) species (12a1), which upon hydrogenation releases degraded alkanes and forms an alkene-ZrH species (12a3). Insertion of the coordinated alkene in the Zr–H bond again generates an alkyl–Zr(IV) species (12a4), which subsequently undergoes hydrogenolysis, liberating another alkane fragment with regeneration of the original catalyst.
SiO)3ZrH] (12a and 12b) using H2 (105 Pa) at 150 °C (Fig. 9).55 The heterogeneous catalyst was synthesized by the condensation of ZrNp4 (Np = neopentyl) with the surface silanol groups of a partially dehydroxylated silica-alumina gel, followed by hydrogenolysis with H2, with the simultaneous generation of silicon dihydride (12a) and aluminium hydride (12b), as proposed by them. As illustrated in Scheme 5, the proposed mechanism involves the facile electrophilic activation of C–H bonds of alkanes through σ-bond metathesis at the d0 metal center of a highly electrophilic catalyst [(SiO)3ZrH] (12a), followed by β-alkyl migration to form alkyl-olefin-Zr(IV) species (12a1), which upon hydrogenation releases degraded alkanes and forms an alkene-ZrH species (12a3). Insertion of the coordinated alkene in the Zr–H bond again generates an alkyl–Zr(IV) species (12a4), which subsequently undergoes hydrogenolysis, liberating another alkane fragment with regeneration of the original catalyst.
 |
| | Fig. 8 Insertion of olefin and microscopic reverse of the β-alkyl transfer reaction in Ziegler–Natta-type polymerization.54 | |
 |
| | Fig. 9 Predicted chemical structure of the immobilised Zr–H catalyst in the partially dehydroxylated silica–alumina surface.55 | |
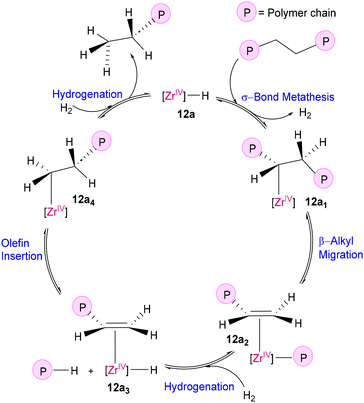 |
| | Scheme 5 Plausible mechanism of the hydrogenolysis of polyethylene by the immobilized Zr(IV)–H catalyst (12a).55 | |
Schwartz's reagent, [Cp2ZrHCl] (13a), has also been widely employed for depolymerization following the same approach of microreversibility.56 The Schwartz hydrozirconation is a well-known reaction in modern synthetic chemistry for the functionalization of terminal or single internal alkenes or alkyne bonds, and converting to terminal organozirconium species by alkyl rearrangement of internal hydrozirconation. Internal alkene bonds have been shown to be less reactive to Schwartz's reagent than the terminal double bonds given that the internal hydrozirconated complexes are typically unstable due to possible retro-hydrozirconation or β-H elimination. In 2013, for the first time, Tang and co-workers thoughtfully applied Schwartz's reagent for chain-scission reaction in polymers with alkene bonds such as 1,4-polybutadiene (PB), styrene–butadiene and polyisoprene.57 The key step is C–C bond activation, leading to cleavage through β-alkyl elimination of the metal–alkyl complex.58 Very recently, Veige and co-workers also followed the same strategy to degrade PB using ansa-metallocene complexes (vide infra).59 In Tang's strategy, involving the carboalumination of olefin, the Cp2ZrCl2(13a)/iBu3Al catalyst combination was preferred. The in situ generation of the catalytically active species “Zr(IV)–H” followed by hydrozirconation and transmetalation was proposed for the chain-scission reaction of PB, as shown in Scheme 6. After the hydrozirconation of the PB backbone, β-alkyl removal causes chain cleavage, which then releases the hydrocarbon chain as well as a Zr(IV)-end capped chain. Lastly, the chain-transfer reaction leads to the formation organo-aluminium end groups, which can be further functionalized to achieve valuable telechelic polymers. It should also be noted that the performance critically depends on activation or transmetalation steps, as is evidenced by the trend obtained by varying the [Al]/[Zr] ratio. A high excess of iBu3Al inhibits the Zr(IV) catalyst by forming an undesirable Al–Zr bimetallic complex.
 |
| | Scheme 6 Degradation mechanism of PB using [Cp2ZrClH]. For clarity only one of two β-alkyl elimination events is depicted.56,57 | |
The very recent work of Veige and co-workers59 involving the systematic high-throughput experimentation (HTE) screening of twenty-eight polymerization catalyst precursors belonging to the families of unbridged-metallocenes, ansa-metallocenes, and hemi-and post-metallocenes of group 4 metals (Ti, Zr, Hf) for the degradation of cis-1,4-polybutadiene (PB), following reaction conditions similar to Tang and co-workers, revealed important structure–activity correlations. The HTE setup consisted of a 96-well high-pressure reactor fitted with glass vials and individual magnetic stirrers mounted in a Freeslate Extended Core Module robotic platform. The accelerating accumulation of plastics in the environment requires rapid screening methods, and thus the HTE protocol may be more useful than the much slower one-at-a-time catalyst design approach. The efficiency of a catalyst was quantified by the number of chain scissoring events (ηCSE). The most remarkable observation in their work is the degradation activity at least to some extent by all the molecular catalysts. This implies that C–C bond cleavage, through β-alkyl elimination, is a fundamentally accessible reaction for degradation from a microreversibility viewpoint. Some of the representative non-bridged metallocene (13a and b), ansa-metallocene (14–16), and hemi-metallocene (17a–b)-based catalysts of group 4 metals (Fig. 10) show promising chain cleaving efficiency. The simplest catalyst in this family, Cp2ZrCl2 (13a), displayed moderate but highly promising degradation efficiency with an ηCSE value of 4.1. A subtle balance between electronic and steric effects dictates the degradation performance. For example, catalyst 13b with indenyl carbocyclic ligands, which are bulkier but less electron donating than Cp, improved the ηCSE to 8.5. The 16-electron ansa-metallocene catalysts such as 14–16 also showed an improved degradation efficiency. In general, Zr(IV) complexes showed superiority over Ti/Hf analogues having the same ligand moieties, as also observed while comparing the ηCSE values of 21.0 and 8.7 imparted by 15b and 16, respectively. The Ti(IV) hemi-metallocene catalyst 17a with pentamethylcyclopentadienyl and amidinate ligands showed a poor performance in PB degradation (ηCSE = 2.0), but to a great surprise, the other hemi-metallocene catalyst 17b having a combination of phosphinimide and less bulky and less electron-donating substituted cyclopentadienyl ligand (Cp-C6F5) showed the highest ηCSE of 35.7. This study certainly sets a platform to use the already developed libraries of catalysts for olefin polymerization as future and efficient catalysts for depolymerization to convert waste hydrocarbon polymers to fuel or reusable monomers.
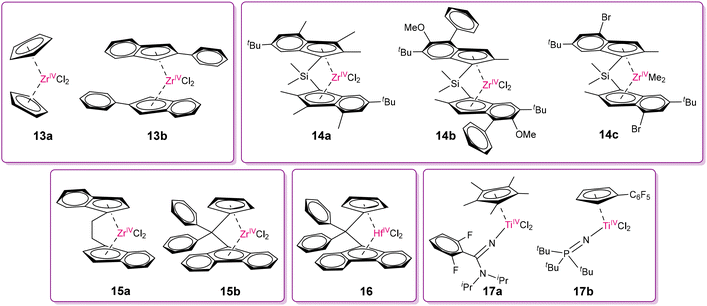 |
| | Fig. 10 Non-bridged metallocene (13a–b), ansa-metallocene (14–16) and hemi-metallocene (17a–b) Zr(IV)/Ti(IV)/Hf(IV)-based catalysts showing promising efficiency in PB degradation.59 | |
2.1.3. Other recent emerging strategies of depolymerization.
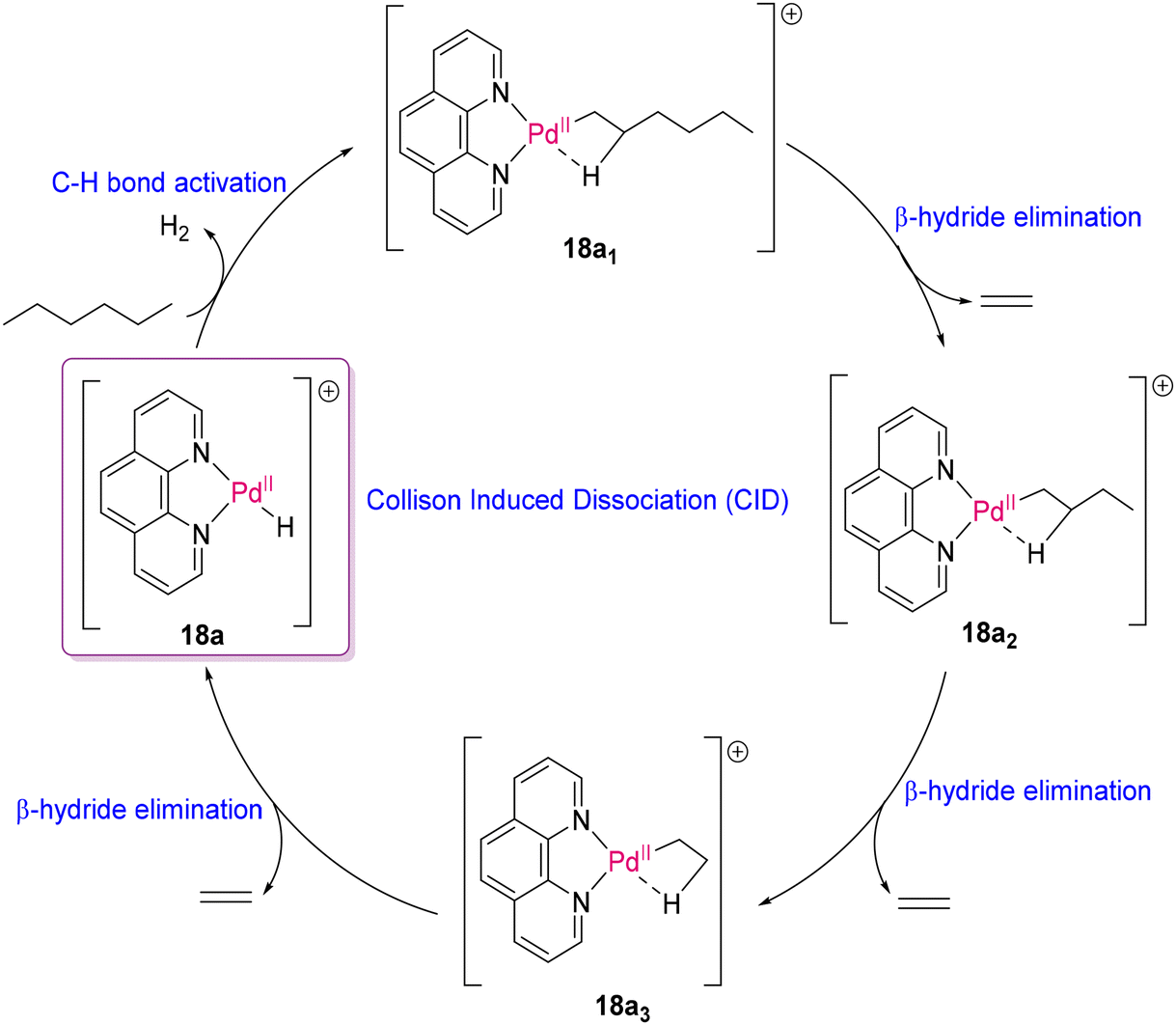
In parallel to TDOCM and reverse polymerization strategies, many groups also explored late transition metal complexes for the cleavage of non-polar polymers. In 2020, Ryzhov, O'Hair and co-workers reported that the cationic complexes [(phen)M(R)]+ (phen = 1,10-phenanthroline; M = Ni, Pd, Pt; R = –H, –Et) could catalyze the deoxygenation reaction of fatty acids60 and acceptorless dehydrogenation of ethane.61 Logically, this category of catalysts should also have potential to depolymerize polyolefins through C–H bond activation. In the following year, the same group demonstrated the degradation of n-hexane as a model linear alkyl chain system using [(phen)Pd(R)]+ catalysts (18a, R = H; 18b, R = CH3) by means of multistage mass spectrometry (MSn) experiments in a linear ion trap mass spectrometer and DFT studies.62 The formation of the Pd(II)-alkyl α-complex [(phen)Pd(C6H13)]+ (18a1) through C–H activation at the α-position of n-hexane, and then fragmentation of coordinated hexyl to the step-wise extrusion of ethylene through collision-induced dissociation (CID) via the formation of Pd(II) species (18a1–18a3) were proposed, as shown in Scheme 7. Gas phase experiments and DFT calculations also supported C–H activation at the β- and γ-sites together with the α-position of n-hexane. DFT studies also revealed the possibility of interconversion to other isomers by the “chain-walking” mechanism from α- to β- and β- to γ-hexyl complexes of Pd(II) associated with low energy barriers, and subsequent degradation of hexyl group.
 |
| | Scheme 7 Proposed mechanism for fragmentation of n-hexane by the Pd(II)–H catalyst.62 | |
Jones and co-workers developed an unorthodox approach for the in situ degradation of crosslinked polybutadiene networks embedded with a metathesis catalyst by applying a thermally induced alkene metathesis reaction to get liquid hydrocarbon fargments.63 The well-known Hoveyda–Grubbs alkene metathesis catalyst HG2 (19a) and an analogous but less efficient Schiff base Ru(II) catalyst, commercially known as HeatMet (19b), bearing a chelating ester moiety developed by Grela and coworkers64 were compared for the metathetic depolymerization of crosslinked PB materials. As shown in Scheme 8, the uniformly distributed Ru(II) catalyst (19a or 19b) was directly incorporated into the hydroxyl-terminated polybutadiene (HTPB) and PB, followed by crosslinking by utilizing chain-end urethane formed via alcohol-isocyanate or main-chain thiol–ene chemistry, using a triphenyl bismuth (TPBi) catalyst and diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide (TPO) photoinitiator, respectively. It was noticed that with a low catalyst loading (0.08% relative to the moles of alkene bond in polymers), HG2 facilitated rapid in situ depolymerization under ambient conditions (25 °C), whether HeatMet was able to depolymerize only after increasing the temperature to 100 °C, but to a relatively lower extent compared to the performance of HG2 (at 25 °C). There is no doubt that this innovative approach through the crosslinking and incorporation of a latent metathesis catalyst may open a new gateway to develop smart and green PB rubbers, which can be easily upcycled.
 |
| | Scheme 8 Latent Ru(II)-based metathesis catalysts for depolymerization of smart PB based polymers.63 | |
2.2. Hydrogenation methods for depolymerizing polar skeletal bonds
Various commercial polymers, having polar skeletal bonds of heteroatoms, such as polycarbonate (PC), polylactide (PLA), polyethylene terephthalate (PET), polycaprolactone (PCL), polydioxanone (PDO), polyethylene glycol (PEG) and polydimethylsiloxanes (PDMS) have widespread applications in daily life (Fig. 11). However, their high global consumption and extensive use have led to substantial waste generation, necessitating the development of efficient methods for depolymerization to maintain a circular economy and for greener waste management. The catalytic cleavage of polar bonds by transition metal complexes offers a promising avenue for developing efficient depolymerization chemistry. Among the various depolymerization methods, metal complex-catalyzed cleavage provides distinct advantages such as mild conditions, higher catalytic efficiency, and better product selectivity. The discussion in this section will elucidate the depolymerization strategy and its mechanism for different polar polymeric structures though cleavage of C–O, C–N, C–Cl and Si–O bonds by transition metal-based catalysts. Strategies such as direct hydrogenation, transfer hydrogenation and hydrosilylation reactions have been explored, yielding valuable depolymerized products by various research groups worldwide. The strategies of breaking polymeric bonds will be systematically discussed in the following sections by categorizing the catalysts based on the position of the metals in the periodic table. From a commercial perspective, hydrogenation plays a crucial role in producing fine chemicals and pharmaceuticals, showcasing its significance in organic synthesis.65–68 The hydrogenation process mostly involves the use of H2 gas under high-pressure conditions. Hence, the transfer hydrogen (TH) strategy involving hydrogen donors is always advantageous due its easy accessibility, affordability, and simpler and safer reaction setup.
 |
| | Fig. 11 Widely used polymeric skeletons with polar skeletal bonds. PC: polycarbonate, PC-BPA: polycarbonate-bisphenol-A, PET: polyethylene terephthalate, PCL: polycaprolactone, PDO: polydioxanone, PA: polylactic acid, PES: (polyethylene succinate), PEG: polyethylene glycol, PVC: polyvinylchloride, PU: polyurethane, and PDMS: polydimethylsiloxanes. | |
2.2.1 Group 9 metal complexes.
Group 9 metal complexes of Ir(I/III) or Rh(I/III) are very effective catalysts for the transfer hydrogenation of unsaturated substrates by hydrogen donors (such as cyclohexene and 2-propanol).69 In addition, these metal complexes are also active catalysts towards direct hydrogenation and hydrosilylation reactions.
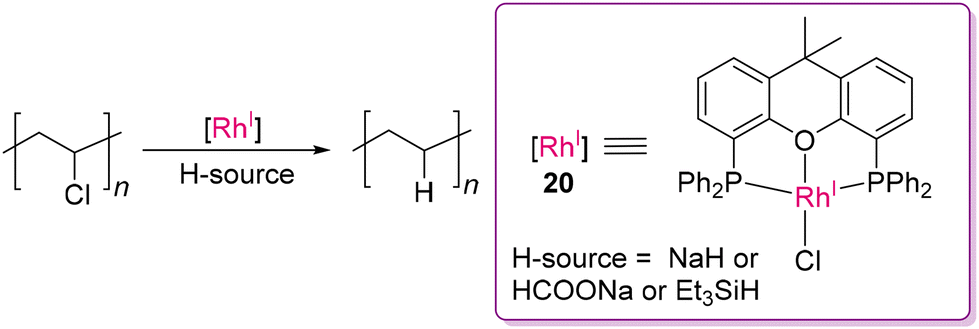
The depolymerization of PVC is challenging due to the presence of the C–Cl bond. One of the trending approaches to address the challenges of PVC depolymerization is plasticizer-mediated electro-dechlorination, enabling the conversion of PVC into valuable chloro-arene compounds.70 Alternatively, hydrodechlorination has also emerged as a promising technique to remove chlorine atoms from PVC, transforming it into polyethylene, which then can be depolymerized by the established methods (vide supra). Notably, Fieser and co-workers developed an Rh(I) complex (20) based on the xantphos ligand as a selective hydrochlorinating catalyst, which in presence of hydride donors, activated the C(sp3)–Cl bond by transfer hydrogenation to form polyethylene, as shown in Scheme 9.71,72 Hydrodechlorination serves as an indirect method for depolymerizing PVC, offering a cleaner, potentially less hazardous approach for recycling and repurposing PVC waste.71–74
 |
| | Scheme 9 Hydrodechlorination of PVC by the Rh(I) complex based on the xantphos ligand.71,72 | |
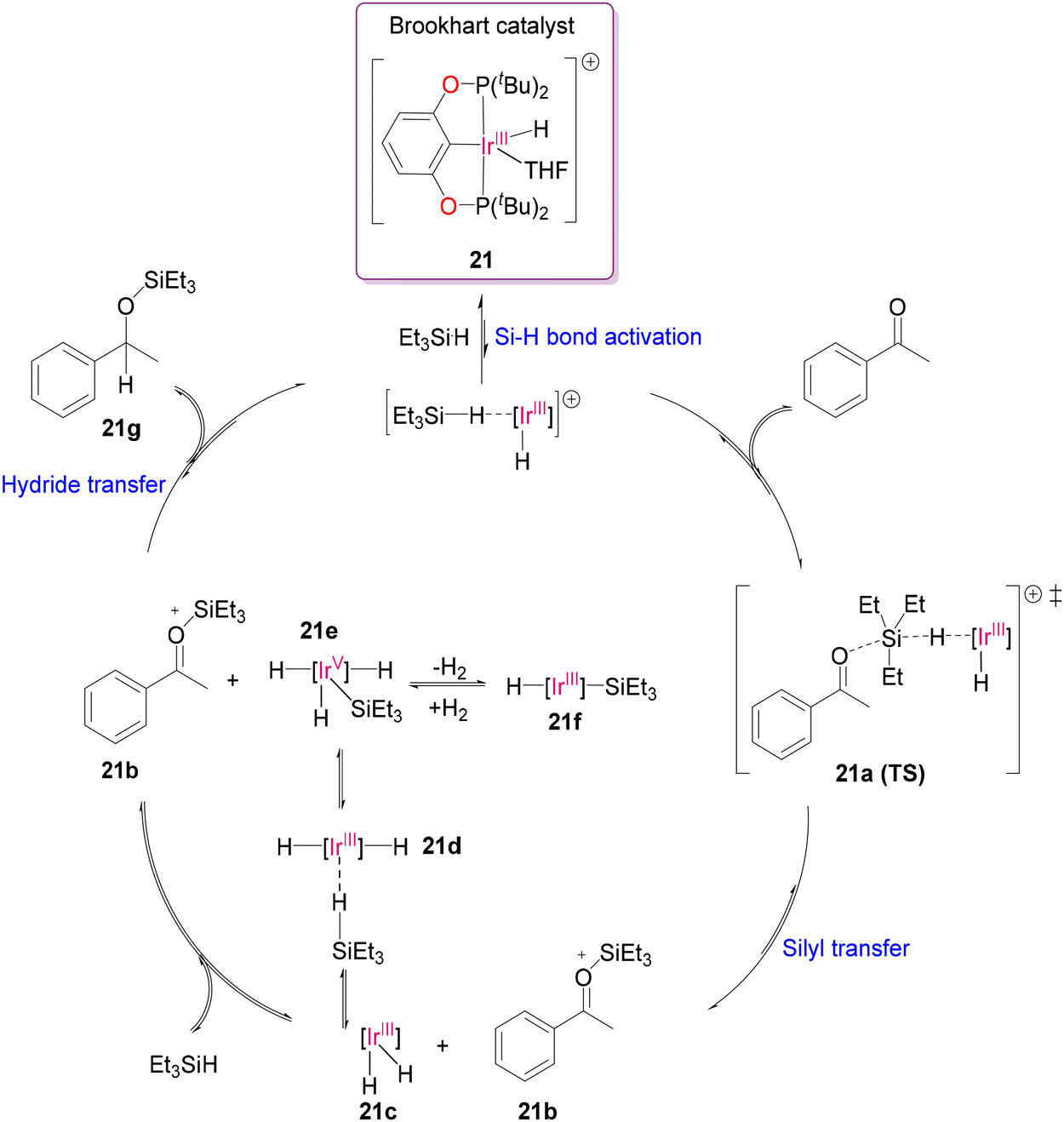
Classical aminolysis, transesterification or hydrolysis reactions have been widely used in the catalytic depolymerization of polyesters75 and polycarbonates.76 Brookhart and other research groups successfully demonstrated the functionalization of various functional groups such as carbonyl and carboxyl groups as well as ethers by utilization of hydrosilylation reaction catalysed by Ir(III) complexes.77–81 In 2015, Cantat and co-worker successfully used the commercially available B(C6F5)3 and [Ph3C+B(C6F5)4−] as Lewis acid catalysts and hydrosilanes as reductants for the metal-free reductive depolymerization of waste polyesters, polycarbonates and polyethers.82 Later, they explored the cationic pincer complex, [Ir(POCOP)H(THF)][B(C6F5)4] (POCOP = 1,3-(tBu2PO)2C6H3) (21), also known as Brookhart's Ir(III) catalyst (Scheme 10), to overcome the limitations associated with the above-mentioned Lewis acid catalysts such as solubility, high loading amount and poor selectivity.83 The Ir(III) catalyst was utilised to crack PCL, PDO, PLA, PES, PET and PPC in the presence of silane through the hydrosilylation protocol. Although the sequence of reactivity for the reduction of carbonyl groups typically follows the order of ketones > esters > carbonates,84 Brookhart's Ir(III) catalyst exhibits distinct reactivity, prioritizing the reduction of monofunctional carbonate (such as PPC) over lactide monomer (such as PLA) when catalyzed with 1 mol% of catalyst 21 and excess Et3SiH. This selective hydrosilylation of PPC over PLA indicates a specific reactivity trend. Oestreich and co-workers proposed a modified mechanism with the small molecule acetophenone, involving two catalytic cycles with an Si(IV) center (Scheme 11).81 The reversible coordination of silane to the cationic Brookhart's catalyst [Ir(III)–H] followed by nucleophilic substitution with the carbonyl oxygen form the silyl carboxonium product (21b) and the dihydride-Ir(III) species (21c) through a transition state (21a) via Si–H bond activation. The trihydride-Ir(V) species (21e) species is believed to be the most likely hydride transfer agent to form 21f as a silane-mediated hydrosilylated product. Repetition of the whole process with another oxygen atom of the carbonyl carbon produces the depolymerized product, as shown in Scheme 11. By analyzing the mechanism critically, they confirmed the formation of Ir(V) species, which was stabilized by the judiciously designed ligand moiety with σ-donor ligands, enhancing the hydride donor ability. Thus, regulating the hydricity on the Ir-centre plays a crucial role in successfully cleaving the C–O bond in the PET polymer to form various silyl derivatives.81,85,86
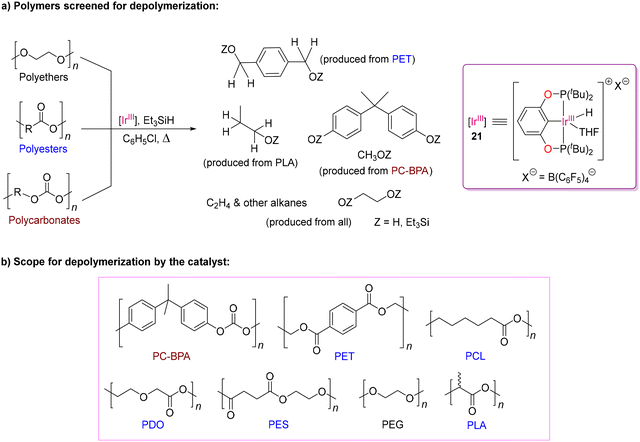 |
| | Scheme 10 Cleavage of C–O bonds in polymers by the cationic pincer Ir(III) complex.83 | |
 |
| | Scheme 11 Proposed mechanism using the POCOP-Ir(III) catalyst and Et3SiH for cleaving C–O bond in acetophenone (as a model carbonyl compound).81 | |
The development of methods for the successful depolymerization of various polar polymers by the (POCOP)-Ir(III) catalyst prompted researchers to explore the catalytic efficiency of Ir(III) catalysts for the depolymerization of chemically resistant polyurethane (PU). PU is a highly valued polymer, which is widely utilized in many common products, including insulation materials, shoe bottoms, mattresses, and sophisticated sections of medical devices, aircraft, spacecraft, etc. In recent years, Skrydstrup and co-workers successfully attempted to deconstruct PU with an Ir(III) catalyst, as shown in Scheme 12.87 In the work by this group, [Ir(H)2(Cl)-[HN(C2H4PiPr2)2]] (22), named (iPrMACHO)-Ir(III), was utilized. Not only (MACHO)-Ir(III) but also the Fe(II), Mn(I) and Ru(II) metal centers with MACHO ligand were used for the efficient hydrogenation of PU. Flexible solid, flexible foamed, rigid solid, and rigid foamed samples based on PU were examined. Aromatic amines and a polyol fraction were isolated in a high yield. The reaction condition was applied using 2 wt% (iPrMACHO)-Ir(III) catalyst, 2 equiv. of KtOBu per catalyst at 150–180 °C under 30 bar H2. They could isolate different types of formanilides with the aniline fractions. Their finding suggested that formanilides may serve as intermediates in the hydrogenation of PU, where the breakage of the carbon–nitrogen bond is preceded by the initial reduction of the carbon–oxygen bond. MeOH, formed through the formanilide intermediate, was identified and quantified by GC-mass spectrometry from the depolymerized crude product mixtures of PU samples. The plausible mechanism of the MACHO-based metal complexes for the hydrogenation of polar bonds is presented in the next sub-section for the ease of discussion (vide infra). From the viewpoint of sustainable chemistry, the authors also mentioned that green solvents such as isopropyl alcohol and less expensive (iPrMACHO)-Mn(I) complex can also be utilized with slightly increased temperature and base loading for some selected PU samples. It is noteworthy to mention that hydrogen gas at high pressure is efficient for large-scale depolymerization but involves higher energy cost, safety risks, and moderate selectivity. Alternatively, silanes offer better selectivity and milder reaction conditions, though they are more expensive and generate silicon-based byproducts.
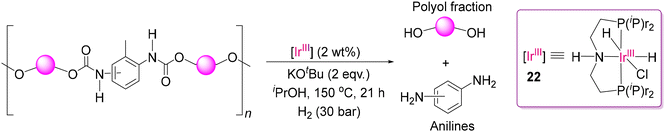 |
| | Scheme 12 Degradation of polyurethane by the (iPrMACHO)-Ir(III) catalyst in the presence of high-pressure H2.87 | |
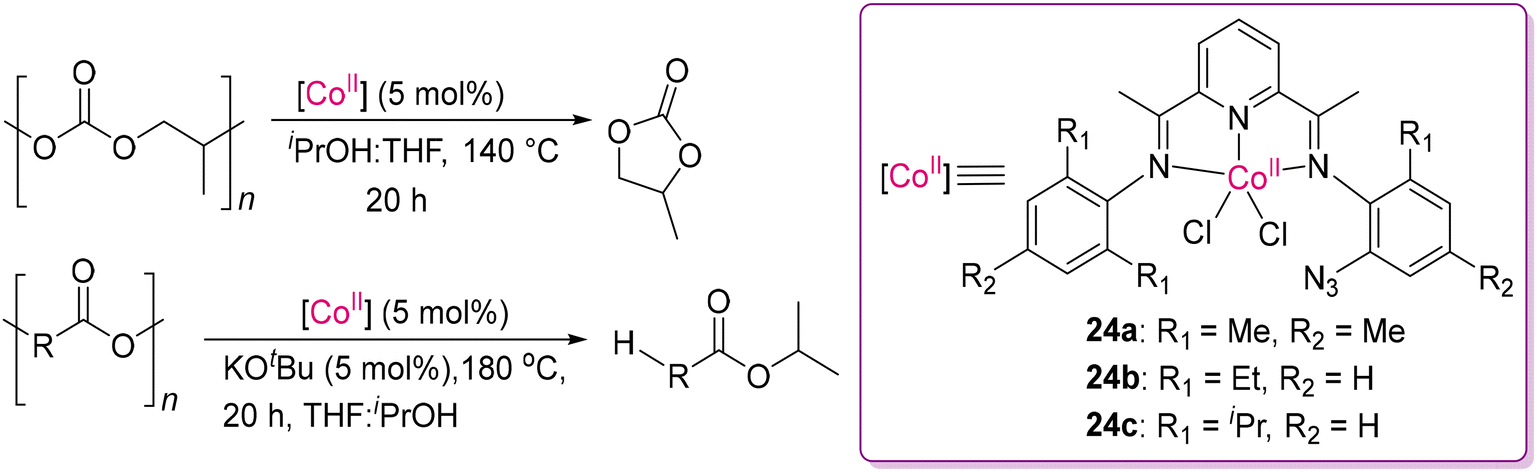
It is always desirable to replace the expensive 4d and 5d-transition metal catalysts by the more economic 3d-transition metal congeners. In this direction, Sundararaju and co-workers successfully demonstrated phosphine-free, air- and moisture-stable, high valent Co(III) complexes with appropriate ligands, showing catalytic activity towards hydrogenative bond cleavage of carbonates into the corresponding diols/alcohols.88 However, it is challenging to functionalize the least reactive substrates such as carbonate and carbamate/urea of the carbonyl family due to the low electrophilicity of the carbonyl bonds.89,90 Thus, to overcome this problem, they developed a novel [Cp*Co-(pyridine-2-carboxylate)I] catalyst (23b) and its analogues (23c–23e) by varying the anionic N,O-chelating ligands from [Cp*Co(CO)(I)2] (23a), as shown in Scheme 13. A wide variety of carbonates, including symmetrical, unsymmetrical acyclic carbonates and cyclic carbonates, were hydrogenated under a low-catalyst loading. Monomeric compounds were obtained efficiently via the hydrogenative depolymerization of carbonate-based polymers, showing its practical utility sustainable chemistry. They examined cobalt complexes (23a–23e) and observed that complex 23c was the most effective, providing the highest yield of the corresponding diols. The catalysts were also effective in the presence of isopropanol as a transfer hydrogenating agent, replacing the harsh condition of high-pressure hydrogen gas, making this process more economically viable. It is noteworthy to mention that using Co(II) complexes with triphos-based ligands, similar types of hydrogenation products were obtained from small molecular organic cyclic and acyclic carbonates, as reported by Beller and co-workers.91 A recent development was reported by Fieser and co-workers using air-stable Co(II) catalysts (24a–24c) with pyridine diimine-based ligands, demonstrating an effective route for the catalytic cyclodepolymerization (CDP) of polypropylene carbonates (PPC), producing cyclic carbonates as valuable feedstocks, as shown in Scheme 14.92 Moreover, this study successfully demonstrated a proof of concept of mixed recycling stream for a mixture of PPC and PBPAC (poly(bisphenol A carbonate)), leading to the full conversion of the respective monomers through CDP and solvolytic depolymerization, respectively, while the solvolysis condition was strictly followed. However, when the CDP condition (without base or iPrOH) was followed, only selective CDP for PPC was observed, ensuring that the other polymer PBPAC remained undisturbed.
 |
| | Scheme 13 Phosphine-free Co(III) catalysts for conversion of acyclic carbonates to useful diols.88 | |
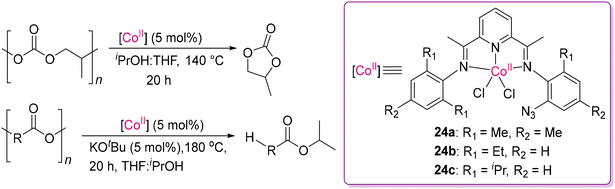 |
| | Scheme 14 Cyclodepolymerization and solvolytic depolymerization of polycarbonates and polyesters by CoII catalysts.92 | |
2.2.2 Group 8 metal complexes.
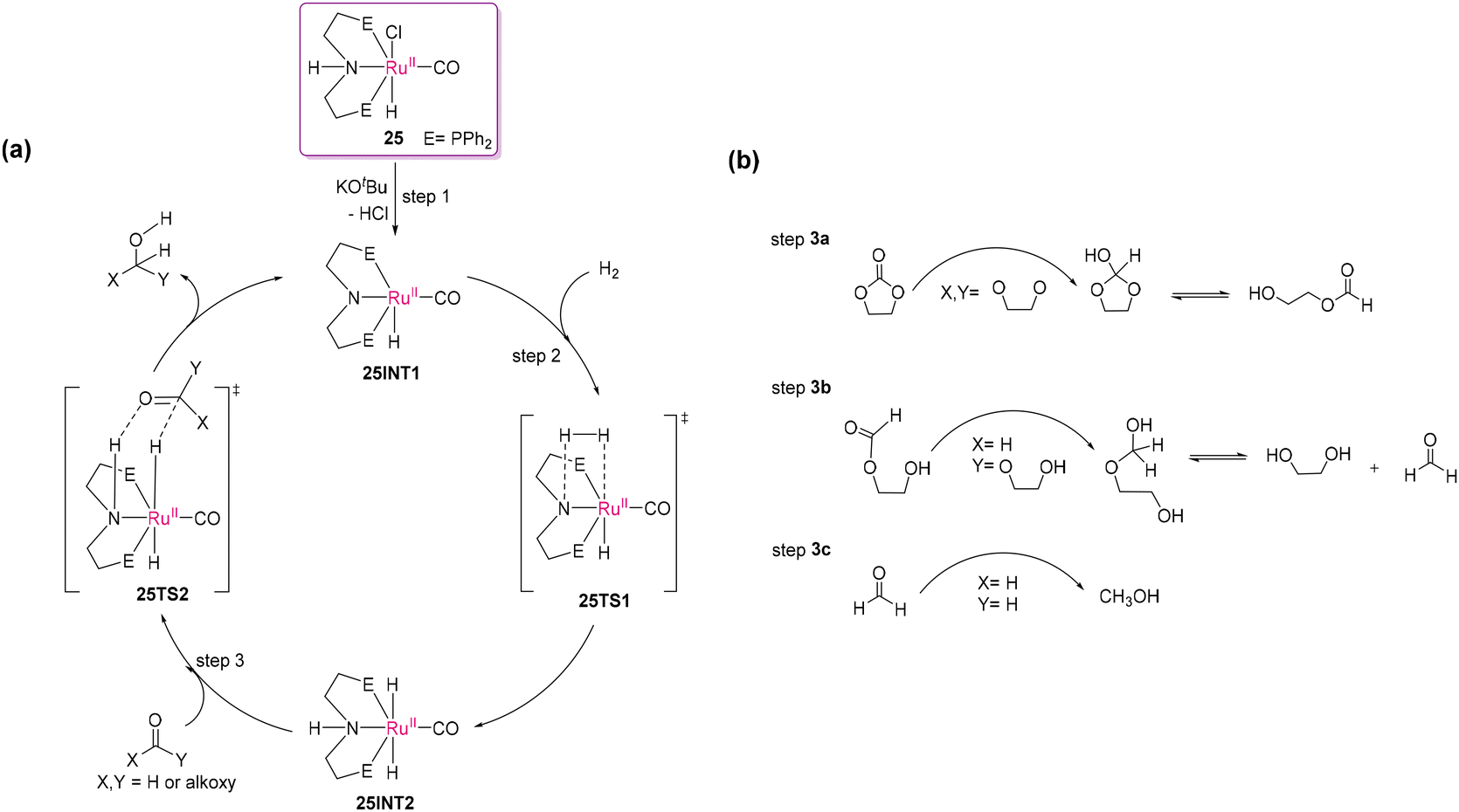
The ground-breaking development of depolymerization began with group 8 metal complexes. In this subsection, we account for the continuous evolution of hydrogenative bond cleavage of polar polymeric bonds. It is crucial to note that the initial impetus for bond cleavage reactions in polymeric carbonates emerged from the activation chemistry of the CO2 molecule for utilization as a valuable C1 feedstock. However, the functionalization of the chemically stable CO2 poses a significant challenge, prompting the exploration for innovative catalytic processes in both academia and industry.93–95 In this line of research, it is essential to highlight the work by Milstein and co-workers, which was the first report on an indirect method for converting CO2 into methanol by hydrogenating carbonates or carbamates employing a pincer-type [(PNN)RuII(CO)(H)(Cl)] catalyst (PNN = 2-(di-tert-butylphosphinomethyl)-6-(diethylaminomethyl)pyridine).96 This idea greatly inspired to many research groups working in this field. Notably, Ding and co-workers developed a highly efficient method for the catalytic hydrogenation of cyclic carbonates to generate methanol and the corresponding diols by using the [(PNP)RuII(CO)(H)(Cl)] (PNP = HN(CH2CH2PPh2)2) pincer complex (25) as the catalyst under relatively mild conditions, as depicted in Scheme 15.97 Ru(II) complexes with different PNP-based ligands and varying substituents at the phosphorus center as isopropyl, tert-butyl, cyclohexyl, adamantyl were screened; however, the best catalytic efficiency was observed for the phenyl-substituted PNP ligand. This process provides a facile approach for the simultaneous production of two important bulk chemicals, namely methanol and ethylene glycol from ethylene carbonate, which is industrially produced by reacting ethylene oxide with CO2. Moreover, this catalytic system also provides a potential process for the utilization of waste poly(propylene carbonate) as a resource of 1,2-propyleneglycol and methanol through hydrogenative depolymerization. A possible mechanism was proposed, as shown in Scheme 16, in which the N⋯H interaction between the ligand and the substrate plays a crucial role in facilitating the reduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in carbonate through secondary coordination sphere interaction. The catalytic cycle starts with the activation of the Ru(II) catalyst (25), resulting in the formation of Ru(II)-amido complex 25INT1 with 16-valence electrons (step 1). Interaction with H2 forms Ru(II)-dihydride (25INT2) through a four-membered transition state (25TS1), followed by heterolytic cleavage of the H–H bond (step 2). The regeneration of 25INT1 (step 3) occurs via the nucleophilic addition of 25INT2 to the carbonyl group in the substrate. This strategy involves the utilization of the N–H proton of the ligand moiety, which plays a key role in all the transfer hydrogenation steps. Three independent cycles may produce different hydrogenated products (steps 3a–3c), demonstrating the stepwise hydrogenation by the Ru(II) catalyst. Ethylene carbonate is first converted into 1,3-dioxolan-2-ol, which is in equilibrium with 2-hydroxyethylformate (step 3a). Further reduction of 2-hydroxyethylformate produces 2-(hydroxymethoxy)ethanol, in equilibrium with formaldehyde and glycol (step 3b). The final step (3c) generates formaldehyde to methanol, completing the reduction process. This comprehensive mechanism showcases the intricate steps involved in hydrogenation and offers insights into selective bond cleavage in polymeric scaffolds using suitably designed Ru(II) complexes.
O bond in carbonate through secondary coordination sphere interaction. The catalytic cycle starts with the activation of the Ru(II) catalyst (25), resulting in the formation of Ru(II)-amido complex 25INT1 with 16-valence electrons (step 1). Interaction with H2 forms Ru(II)-dihydride (25INT2) through a four-membered transition state (25TS1), followed by heterolytic cleavage of the H–H bond (step 2). The regeneration of 25INT1 (step 3) occurs via the nucleophilic addition of 25INT2 to the carbonyl group in the substrate. This strategy involves the utilization of the N–H proton of the ligand moiety, which plays a key role in all the transfer hydrogenation steps. Three independent cycles may produce different hydrogenated products (steps 3a–3c), demonstrating the stepwise hydrogenation by the Ru(II) catalyst. Ethylene carbonate is first converted into 1,3-dioxolan-2-ol, which is in equilibrium with 2-hydroxyethylformate (step 3a). Further reduction of 2-hydroxyethylformate produces 2-(hydroxymethoxy)ethanol, in equilibrium with formaldehyde and glycol (step 3b). The final step (3c) generates formaldehyde to methanol, completing the reduction process. This comprehensive mechanism showcases the intricate steps involved in hydrogenation and offers insights into selective bond cleavage in polymeric scaffolds using suitably designed Ru(II) complexes.
 |
| | Scheme 15 Depolymerization of poly(propylene carbonate) by the (PNP)-Ru(II) pincer complex.97 | |
 |
| | Scheme 16 (a) Mechanism of activation of the catalyst and degradation of the C–O bond in cyclic and acyclic polymers. (b) Stepwise degradation the cyclic carbonate to methanol.97 | |
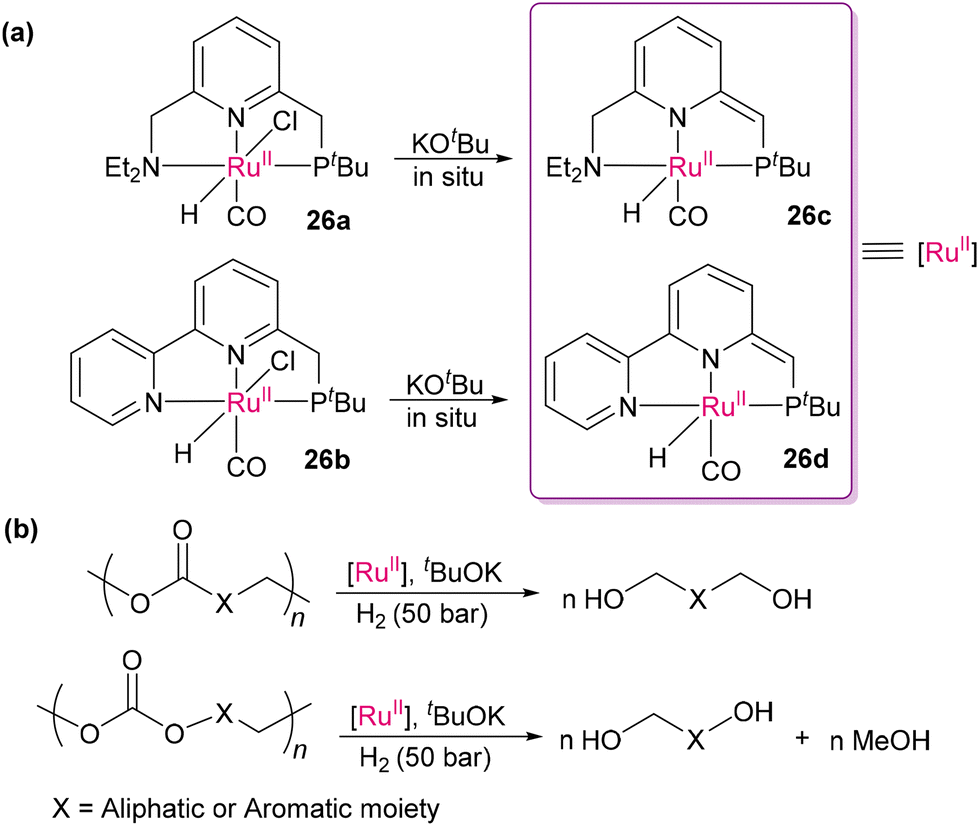
Following the promising approach by Milstein's group using [(PNN)Ru(CO)(H)(Cl)],96 Robertson and co-workers utilized PNN-Ru(II)-based catalysts (PNN = (2-(di-tert-butylphosphinomethyl)-6-(diethylaminomethyl)pyridine) or 6-di-tert-butylphosphinomethyl-2,2′-bipyridine) (26a and b) for the hydrogenative depolymerization of polyesters and polycarbonates into diols, glycols, and methanol, offering an effective approach for recycling waste plastics (Scheme 17).98 This method offers great efficiency for various polymers of the polyester and polycarbonate families. The proposed mechanism is similar to that of Milstein's system,96 where metal–ligand cooperation and aromatization–dearomatization (through 26c2–26c3 intermediates) of the heteroaromatic PNN pincer ligands play a crucial role in activating robust substrates (such as dimethyl carbonate as the model skeletal fragment of polyester and polycarbonate), as presented in Scheme 18.96 Although the hemilability of PNN ligands brings additional catalytic activities towards robust polar bonds, the use of high-pressure H2 makes these hydrogenation processes harsh on the bulk scale, which is considered a limitation.
 |
| | Scheme 17 (a) In situ generation of catalytically active complexes (26c and 26d) with a de-aromatized ligand from the precatalysts PNN-Ru(II) (26a and 26b). (b) Hydrogenative cleavage of robust C–O bonds in polyesters (producing diols) and polycarbonates (producing glycols and methanol).98 | |
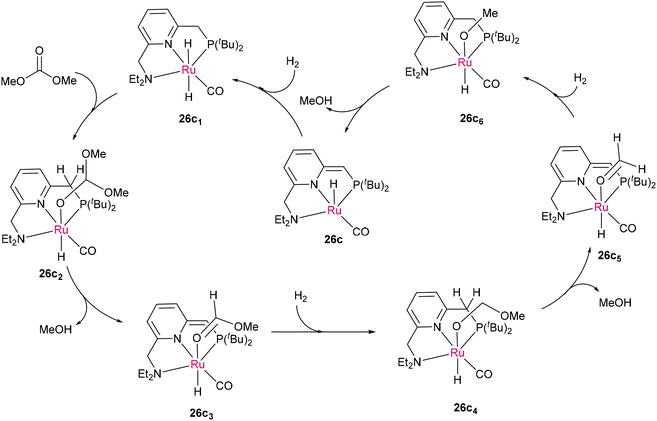 |
| | Scheme 18 Hydrogenation of dimethyl carbonate through methyl formate to methanol by the PNN-Ru(II) pincer complex.96 | |
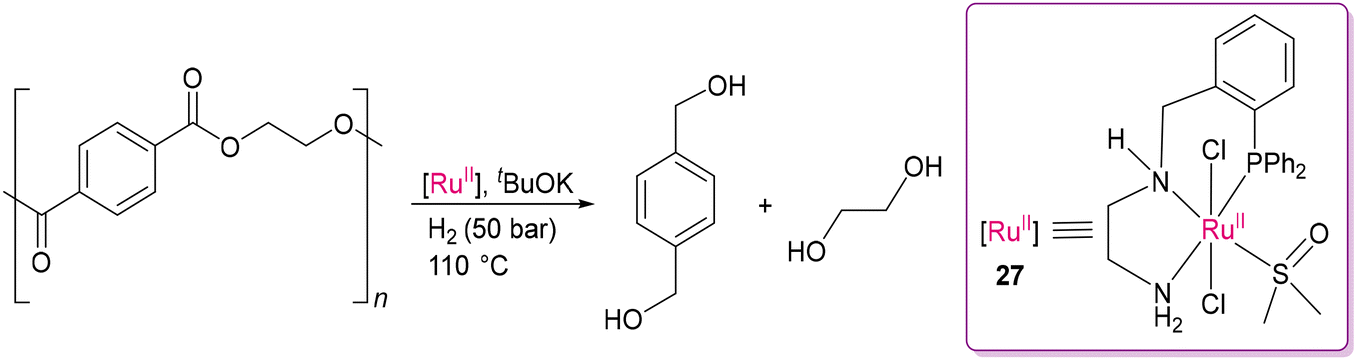
In 2015, Clarke and co-workers reported several chiral and achiral phosphine-diamine, phosphine-amino-alcohol, phosphine-amino-amide-based Ru(II) catalysts for the hydrogenation of a range of esters. The meridional geometry imposed by the tridentate ligands with labile amide/amino/alcohol donor sites, together with weakly coordinated DMSO is beneficial for activating the H–H bond and concomitantly C–O bond cleavage in the depolymerization of polyester samples. Notably, the Ru(II) complex of N′-(2-(diphenylphosphino)benzyl)ethane-1,2-diamine as a PNN pincer ligand with N–H substituent in the P^NH^NH2 tridentate ligand (27) was found to be substantially more efficient than the other catalysts (Scheme 19).99 The optimal catalytic conditions included 1–2 mol% of 27, a suitable base (tBuOK), and 50 bar of H2 at 110 °C in toluene-anisole solvent. The continuous proton transfer process in the presence of molecular hydrogen is crucial for breaking C–O bonds. This study emphasizes the importance of ligand selection and labile ancillary ligands around the metal center for effective depolymerization.100 As evidenced by the kinetic studies, the ligand N–H group in their catalysts dramatically enhances H–H activation, as also observed for Noyori's catalyst in asymmetric hydrogenation.101
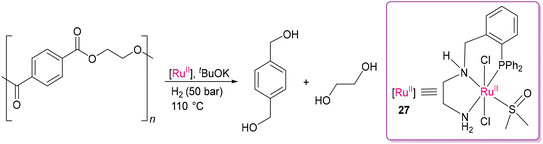 |
| | Scheme 19 Cleavage of robust C–O bonds in polyesters with the Ru(II)-PNN pincer complex.99 | |
Contemporary to Clarke's report, Klankermayer and co-workers developed Ru(II) complexes, [Ru(triphos)(tmm)] (triphos = (1,1,1-tri(diphenylphosphinomethyl)ethane), tmm = trimethylenemethane) (28a) and [Ru(triphos-xyl)tmm] (triphos-xyl = 1,1,1-tri(bis(3,5-dimethylphenyl)phosphinomethyl)ethane) (28b), for the transformation of polyester and polycarbonate scaffolds (Scheme 20).102,103 Scrutinizing the design of the catalyst, it can be claimed that the labile nature of the tmm and π-acceptor triphos ligands plays a crucial role in the formation of dihydride Ru(II) active catalytic species. The tmm ligand can be eliminated either by molecular hydrogen or a catalytic amount of acid to form the catalytically active species. The dihydride Ru(II) species (28a1) was proposed to be the active species for hydrogenation, as also supported by DFT calculation, together with two neutral dihydride of Ru(II) dimer complexes, while treating 28a with H2.104 The presence, and also an optimum quantity of the acid additive, HNTf2, significantly influences catalyst activation and reactivity. However, excessive HNTf2 can lead to reduced conversion and noticeable side reactions.104 The proposed mechanism from DFT calculation considering the neutral dihydride Ru(II) active species (28a1) is illustrated in Scheme 21.104 Coordination of the carbonyl group (28a2) of methyl benzoate, followed by migratory insertion in the Ru–H bond forms the mono-hydride complex (28a3). The neutral mixed classical/non-classical hydride complex 28a4 was proposed as a result of the coordination of an H2 molecule with 28a3, which is necessary for the hydrogenolysis of the Ru–O bond. The conventional dihydride Ru(II) species (28a5) is produced as a result of proton transfer to the coordinated oxygen, with the consequent formation of hemiacetal of benzyl alcohol as the primary product, which is then converted to benzaldehyde and methanol. The catalytic cycle involves the repeated hydrogenation of benzaldehyde to form benzyl alcohol as the final product. This proposed mechanism is applicable for converting polyester and polycarbonate into polyol and methanol.
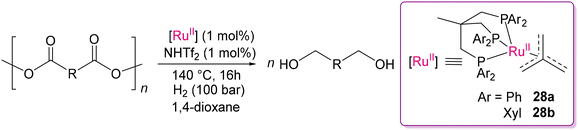 |
| | Scheme 20 Hydrogenolysis of the selected polyester and polycarbonate materials using [Ru(triphos)tmm] or [Ru(triphos-xyl)tmm] and HNTf2 in the presence of H2 gas.102,103 | |
 |
| | Scheme 21 Catalytic pathway by the in situ-synthesized PPP-Ru(II) dihydride complex to cleave a small ester molecule (methyl benzoate) as a proof of principle to degrade the polyester.104 | |
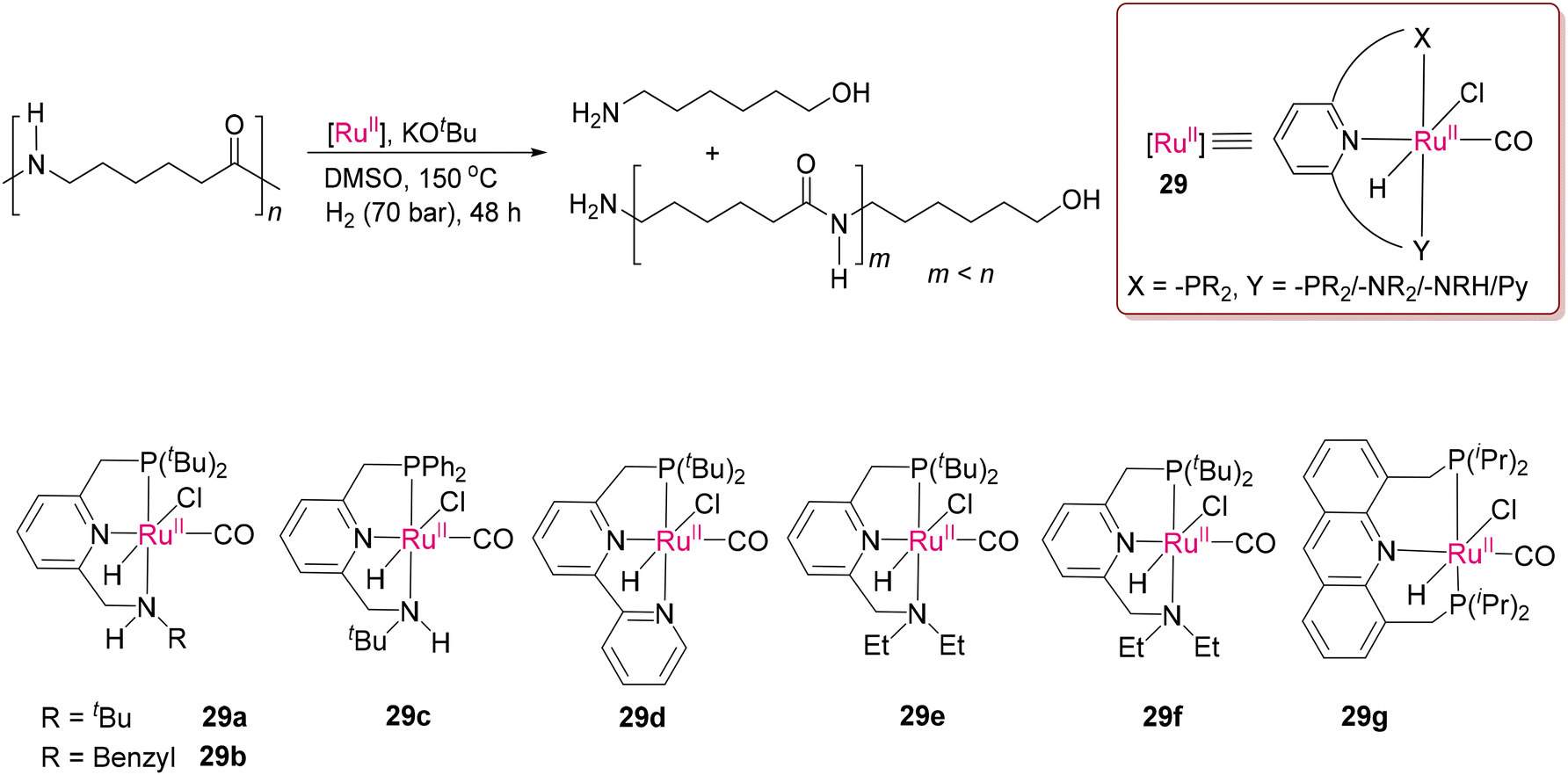
Given that it is one of the strongest bonds to cleave, if the C–N bond of amide is present in the polymer backbone, the level of difficulty is multiplied by several folds. The robustness associated with polymeric bonds and poor solubility of nylon samples in common organic solvents make depolymerization highly challenging through homogenous catalysis. Recently, Milstein and co-workers successfully cleaved the chemically robust C–N bonds in nylons by various Ru(II) catalysts (29a–29g) having PNN or PNP pincer ligands, as presented in Scheme 22.105 The “metal–ligand cooperation” (MLC) with the polymer substrates serves as an operating tool to cleave the strong polyamide bonds through catalytic hydrogenation in the presence of high-pressure condition, and more importantly using low toxic aprotic solvents such as DMSO, avoiding acid medium,106 given that the PNN-Ru(II) pincer catalysts undergo deactivation in acid medium. As proposed, hydrogenation occurs through concerted steps (Scheme 23), as supported by DFT calculations. The step involving H–H bond activation is achieved by the deprotonated and dearomatized form of the coordinated pincer ligand (29a1) through deprotonation of the benzylic position of the P-arm. Subsequent formation of the energetically favoured ruthenium dihydride species 29a2 was proposed. Amide hydrogenation happens by proton transfer from the N–H group of 29a2 through a key transition state involving interaction with the –NH group of a ligand similar to Noyori's mechanism.107 Thereafter, thermodynamically favoured C–N bond dissociation was predicted from the hemiaminal (hydrogenated product of amide).108–110
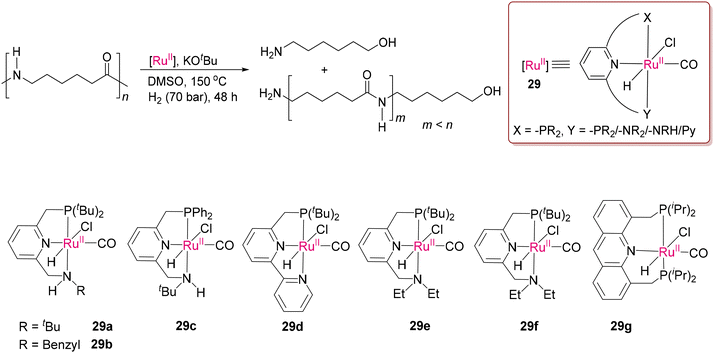 |
| | Scheme 22 PNN/PNP-Ru(II) catalysts (29a–29g) for breaking the robust C–N bonds in polyamides reported by Milstein and co-workers.105 | |
 |
| | Scheme 23 Proposed mechanism of C–N bond cleavage in a model compound (N-ethylpropionamide) by PNP-Ru(II) complex 29a, as supported by DFT calculations (other high energy dearomatized canonical species of 29a1 are omitted for simplification).105 | |
Inspired by the successful hydrogenation of organic carbonates, carbamates and formates, Enthaler and co-workers explored the MACHO-Ru(II)-BH (30) and Milstein's versatile PNN-Ru(II) catalysts (29e) for the hydrogenative depolymerization of poly(bisphenol carbonate), as illustrated in Scheme 24.111,112 Catalyst 30 was synthesized from bis[(2-diphenylphosphino)-ethyl]ammonium chloride and [Ru(II)(CO)Cl(H)(PPh3)3] in refluxing toluene in basic medium, followed by reduction by NaBH4.113 They successfully converted the poly(bisphenol A carbonate) samples to methanol and bisphenol using both catalysts.
 |
| | Scheme 24 Cleavage of polymeric bonds in poly(bisphenol A carbonate) by PNP- and MACHO-based Ru(II) complexes to produce methanol as primary feedstock.111,112 | |
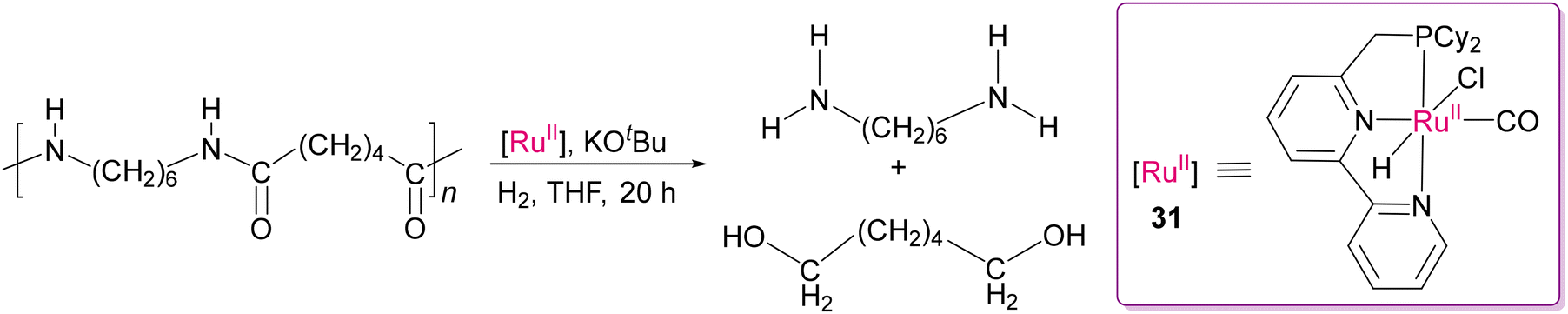
Milstein's group successfully realized the depolymerization of robust polyamides using DMSO as the solvent at elevated temperature (vide supra),97 but this reaction condition limits the turnover number due to the deactivation of the Ru(II) catalyst and possible unwanted decomposition of the solvent. Schaub and group114 addressed this problem by switching to THF as the solvent and replacing the tert-butyl group (in 29d) by cyclohexyl group at the P-arm of the pincer ligand in the PNN-Ru(II) complex (31), as shown in Scheme 25. This simple but effective modification increased the yield of hydrogenated products of polyamides, showcasing an improved system for the hydrogenative depolymerization of polyamide 66 and polyurethane samples.
 |
| | Scheme 25 Depolymerization of polyamides via cleavage of C–N bonds by PNN-Ru(II) complex (31) in the THF medium developed by Schaub.114 | |
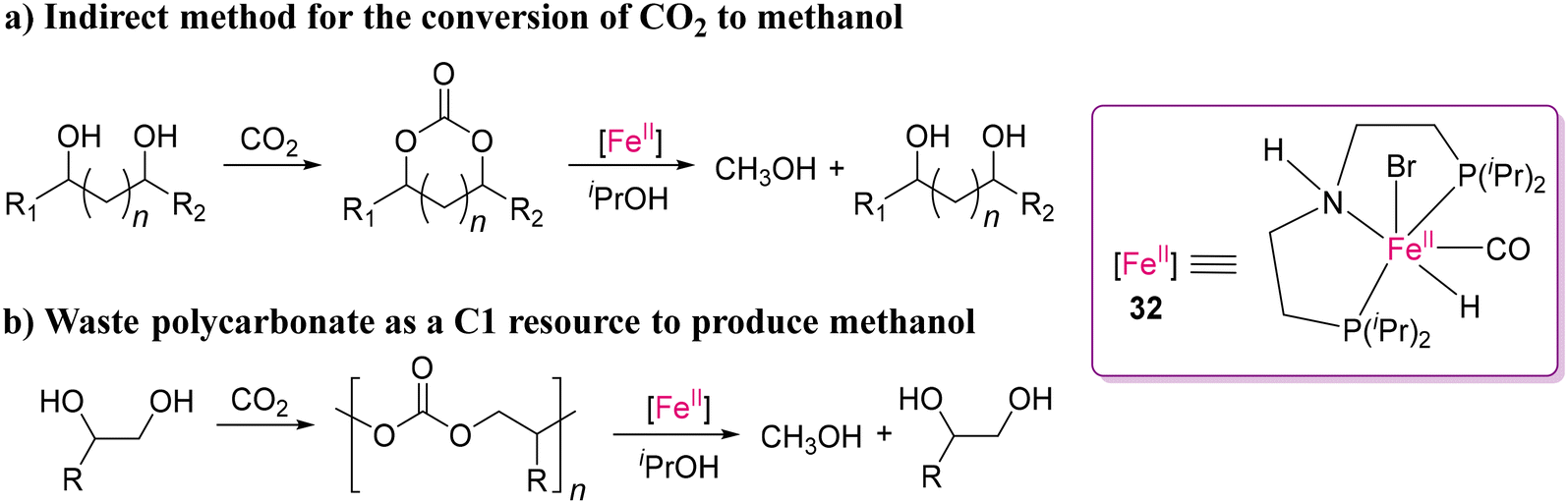
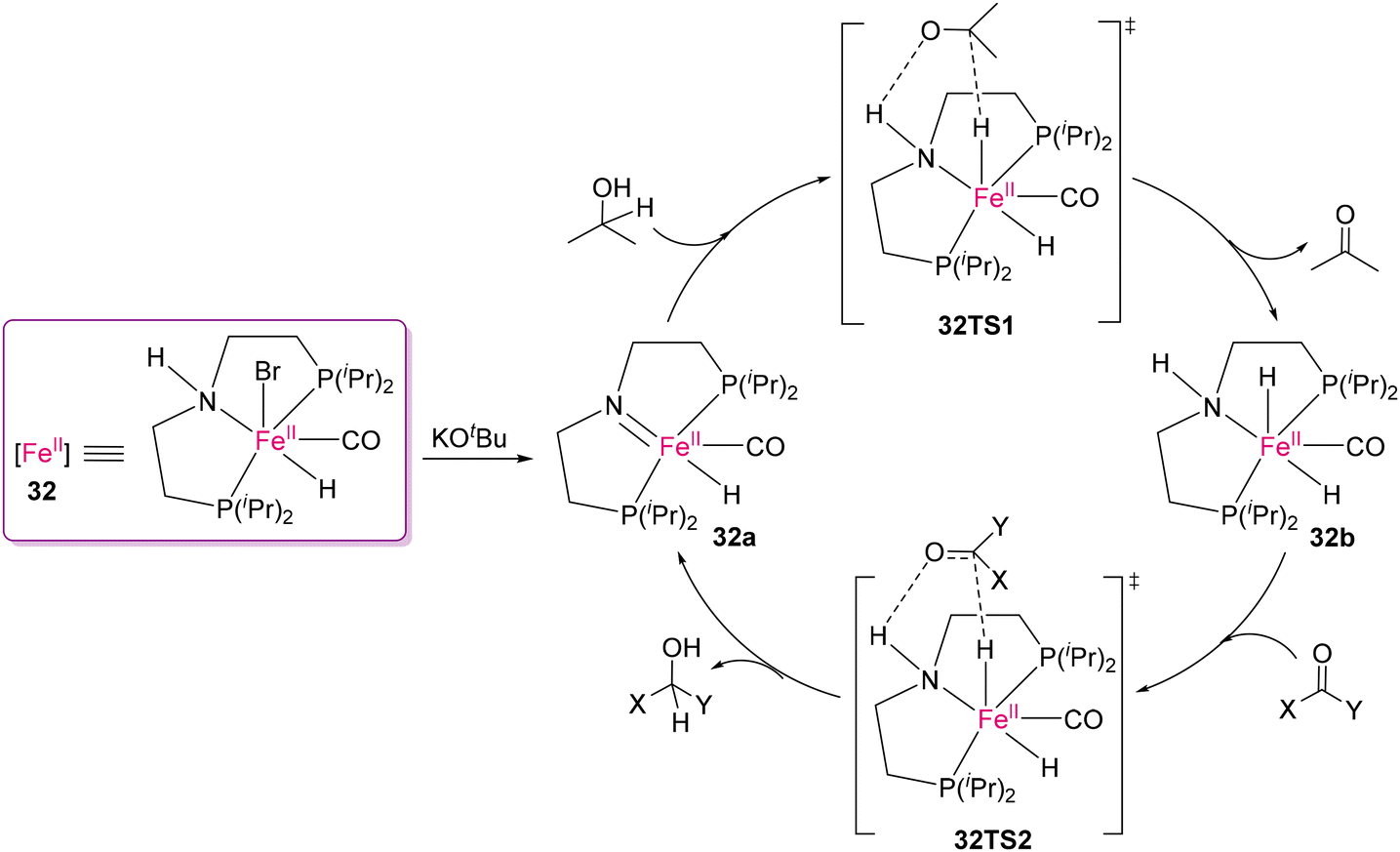
Although noble metal complexes have been used owing to their efficient selectivity and efficiency, earth abundant metals are the obvious choice for bulk scale conversion owing to their affordability. To date, the successful use of Fe(II)-based catalysts for cleaving polar bonds by transfer hydrogenation in polymeric scaffolds is limited. It is important to emphasize that transfer hydrogenation utilizing easily accessible and non-toxic hydrogen donors such as isopropanol is an appealing alternative to traditional hydrogenation processes.68,115 Notably, the transfer hydrogenation of molecular cyclic carbonates and ketones using ruthenium(II) catalysts was successfully demonstrated by Hong's116 and Vries's117 groups. An exciting and pioneering work was reported by Werner and co-workers on the transfer hydrogenation of organic carbonates using the MACHO-Fe(II) catalyst (32) for converting polycarbonates into value-added products, as shown in Scheme 26.118 Several cyclic carbonates as well as polycarbonates were transformed into their corresponding diols and MeOH by catalyst 32 using isopropanol as the hydrogen donor and solvent. Most importantly, this process can also be used for conversion of CO2 to methanol through the formation of carbonates from diol compounds. They proposed a catalytic cycle, where the catalytically active 16e amido-complex 32a is formed by the deprotonation of Fe(II) complex 32 (Scheme 27). Following the deprotonation, dehydrogenation of iPrOH results in the formation of 18e complex 32b and acetone via the 32TS1 transition state. In the last step, 32a is regenerated by the transfer of hydrogen from complex 32b to the CO group of a carbonyl substrate through 32TS2.
 |
| | Scheme 26 Conversion of polycarbonates into methanol and polyols by the Fe(II) catalyst using isopropanol as a hydrogen donor.118 | |
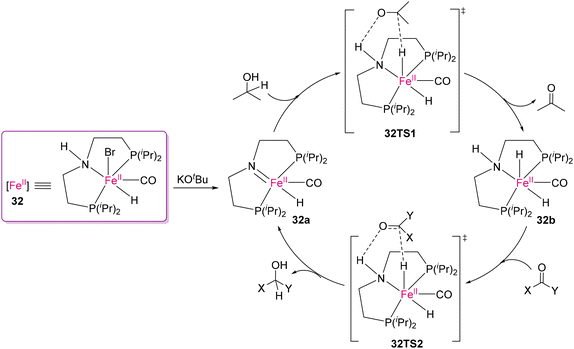 |
| | Scheme 27 Proposed mechanism of the base-promoted transfer hydrogenation of carbonates by the PNP-Fe(II) catalyst.116 | |
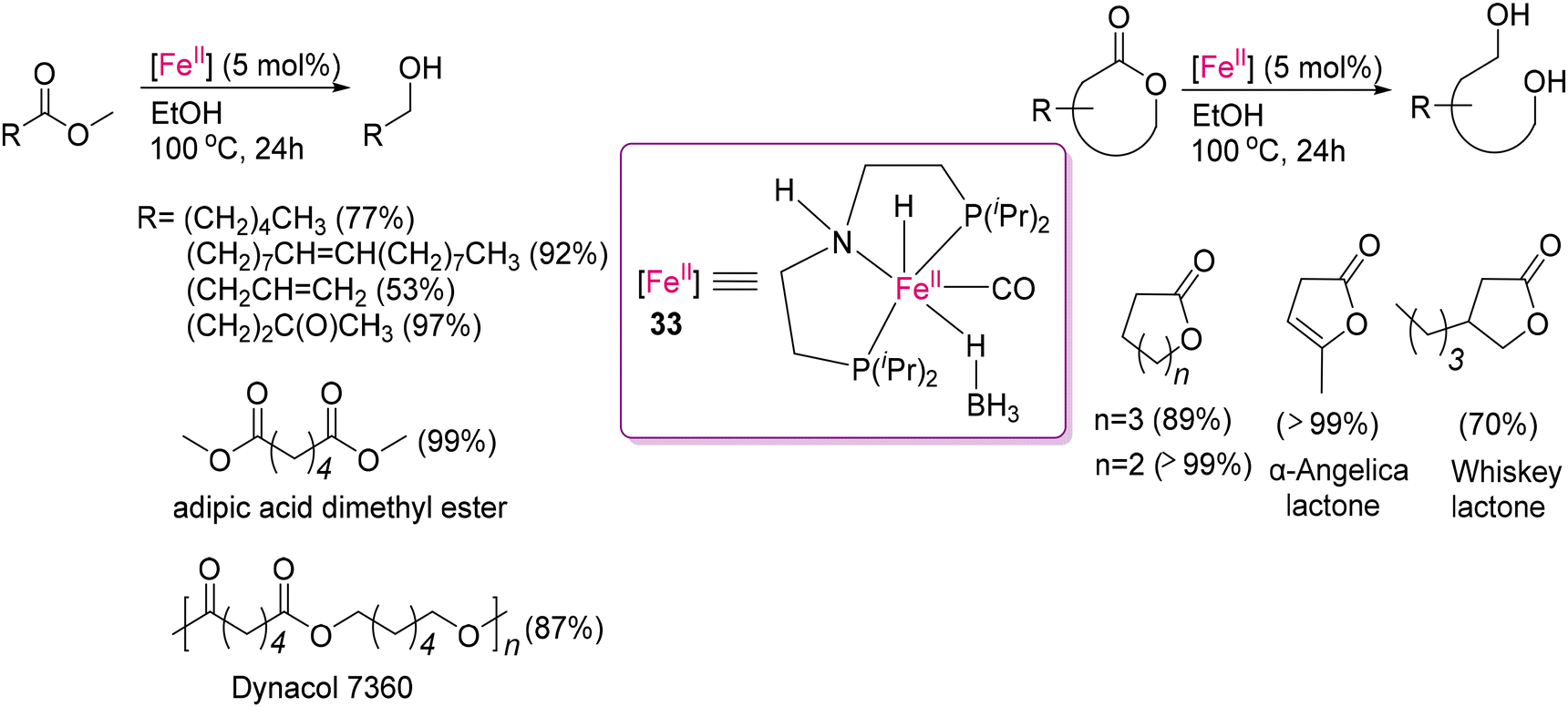
At the same time, Vries and co-workers reported the Fe(II)-MACHO-BH (33)-catalysed base-free transfer hydrogenation of esters, as shown in Scheme 28, to achieve the depolymerization of aromatic esters, aliphatic esters and lactones, including important renewable substrates such as methyl oleate and α-Angelica lactone.119 Instead of isopropanol, they used ethanol as a hydrogen donor, which is more environmentally acceptable and renewable. They suggested a mechanism involving transfer hydrogenation, similar to that reported before by the same group,117 and also by Beller and co-workers.120,121 The key feature of Vries's strategy is the mild activation step through the outer sphere mechanism, eliminating the coordinated BH3 from complex 33, as illustrated in Scheme 29. Unlike the other activation steps (using strong base), the coordinated BH3 easily reacts with alcohol to form the corresponding borates and the active catalytic species 33a. They postulated a low energy barrier, allowing equilibrium between the Fe(II) amine (33a) and amido-complexes (33b), which is responsible for the hydrogenation of the carbonyl substrates and polymeric samples to produce the depolymerised fragments.
 |
| | Scheme 28 PNP-Fe(II) complex for depolymerization of acyclic and cyclic polycarbonates (conversion yield in parentheses).119 | |
 |
| | Scheme 29 Transfer hydrogenation of carbonyl compounds by the MACHO-Fe(II)-BH complex under base-free conditions.120 | |
2.2.3 Group 6 transition metal complexes.
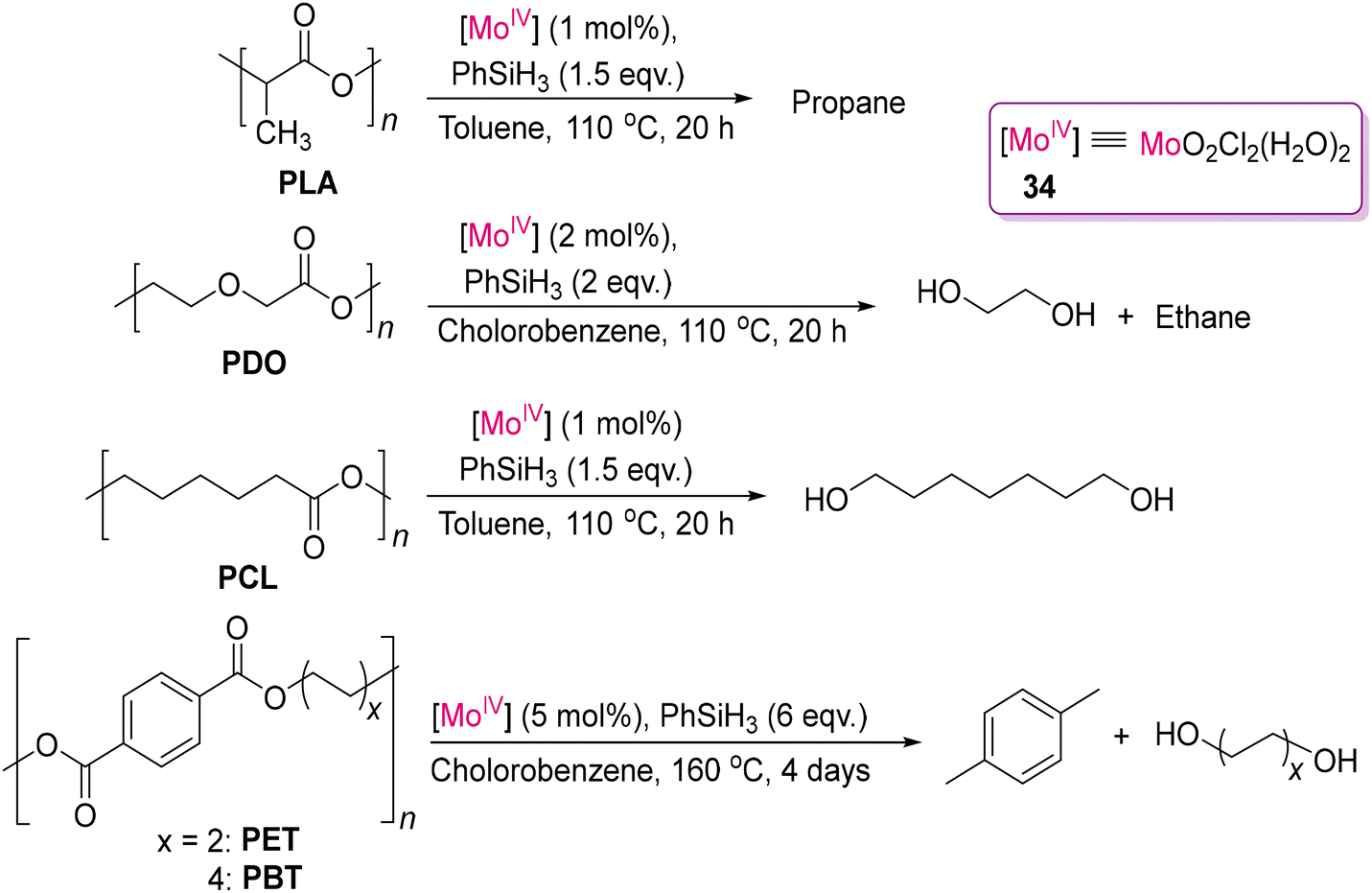
Fernandes and co-workers reported the reductive depolymerization of plastic waste (such as PCL, PLA, PDO, PET and PBT samples) into value-added compounds and fuels using silanes as reducing agents catalysed by MoO2Cl2(H2O)2 (34), as shown in Scheme 30.122 This catalyst is air-stable, easy to synthesize and economic compared to other noble metal catalysts. It can be easily obtained by extraction from a hydrochloric acid solution of Na2MoO4 with diethyl ether. The choice of silanes was found to be crucial for achieving the best efficiency. The reductive depolymerization of various samples of PCL, PDO, PET and PBT yielded the corresponding diols. However, in the case of PLA, the highly demanding propane as fuel was isolated instead of 1,2-propanediol.
 |
| | Scheme 30 Reductive depolymerization by the Mo(IV) catalyst reported by Fernandes and co-workers.122 | |
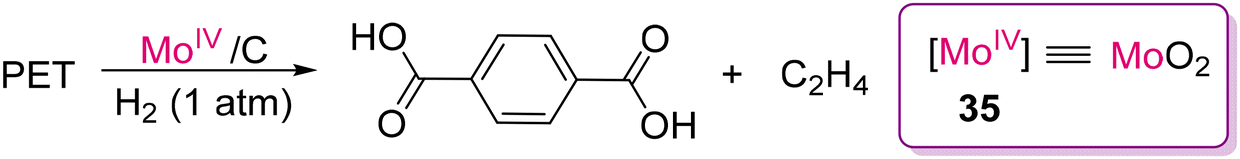
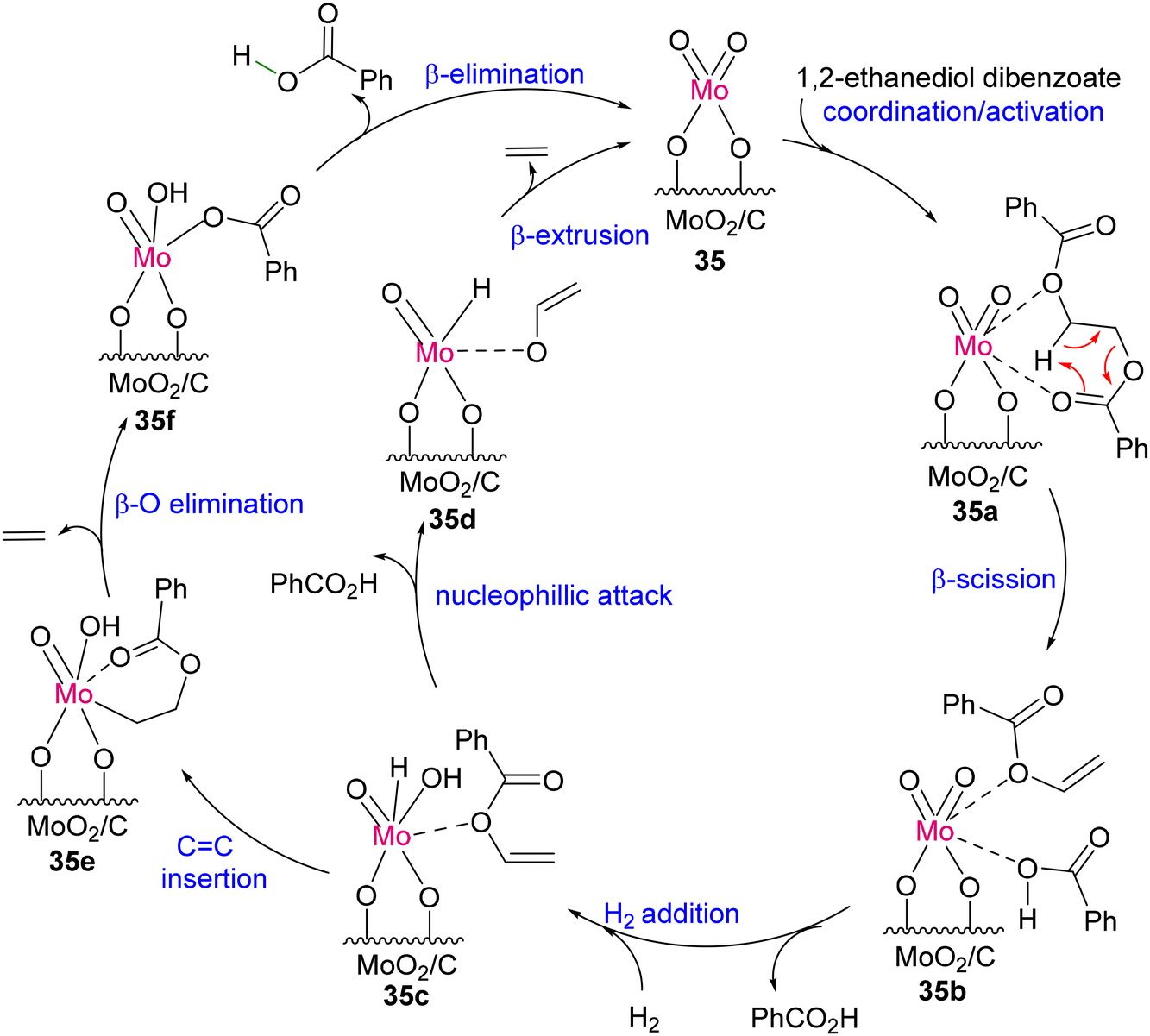
Around the same time, Tobin J. Marks and co-workers developed an earth-abundant, air- and moisture-stable single-site Mo-dioxo complex grafted on carbon (MoO2/C; 35), which could act as an efficient and reusable catalyst for the depolymerization of PET samples under 1 atmospheric pressure of H2. The conversion afforded terephthalic acid and ethylene selectively in high yield, as shown in Scheme 31.123 Mechanistic insight was established with a model compound of 1,2-ethanediol dibenzoate. As proposed, 35 forms a hexacoordinated Mo-dioxo complex (35a) with 1,2-ethanediol dibenzoate (Scheme 32). As a result of β-scission, benzoic acid and vinyl benzoate are formed together with the Mo-dioxo species of 35b. The addition of H–H across the MoO bond forms a species with both Mo–H and Mo–OH (35c), which can react as a nucleophile to vinyl benzoate to generate 35d and benzoic acid. The β-extrusion of 35d yields ethylene and completes the catalytic cycle. Another reaction pathway was predicted considering CC insertion into Mo–H of 35c, forming Mo–alkyl species 35e followed by β-oxygen elimination to form ethylene and Mo–benzoate species 35f. Liberation of benzoic acid through elimination reaction ends the catalytic cycle.
 |
| | Scheme 31 Controlled and efficient catalysis shown by MoO2/C to degrade PET.123 | |
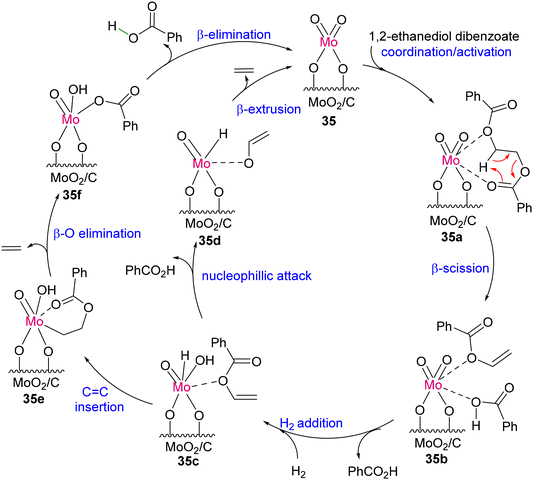 |
| | Scheme 32 Proposed catalytic pathway for PET degradation by the heterogeneous C/MoO2 catalyst.123 | |
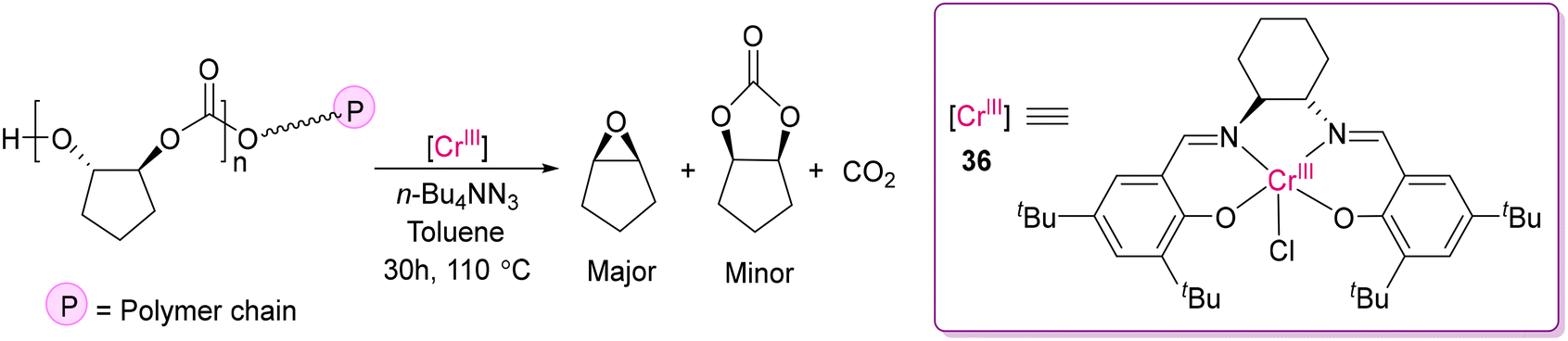
Although 3d transition metal complexes have great potential for versatile catalytic applications, in the field of depolymerization, their application is limited to date. Darensbourg and co-workers investigated the depolymerization of poly(cyclopentene carbonate) by (salen)Cr(III)Cl (salen = (R,R′)-(N,N′-bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexenediimine)) in the presence of n-Bu4NN3 (Scheme 33).124 While monitoring the depolymerization of polycarbonates by the [(salen)Cr(III)Cl] catalyst (36) by 1H NMR, it was realized that N3− played a pivotal role in making this depolymerization efficient by minimizing the time of the reaction and favouring the acid–base reaction between [Cr]–N3− and HO-P (P = polymer chain), forming [Cr]–OP + HN3. The formation of cis-cyclopentene oxide together with CO2 as the major recycled monomer over cis-cyclopentene carbonate was explained as a result of the low energy barrier of intramolecular nucleophilic displacement of carbonate-terminated polymer chain to the epoxide monomer.
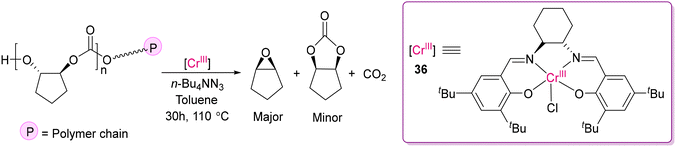 |
| | Scheme 33 Depolymerization of polycarbonates by the [(salen)Cr(III)Cl] catalyst in the presence of azide.124 | |
2.2.4. Group 7 and 12 transition metal complexes.
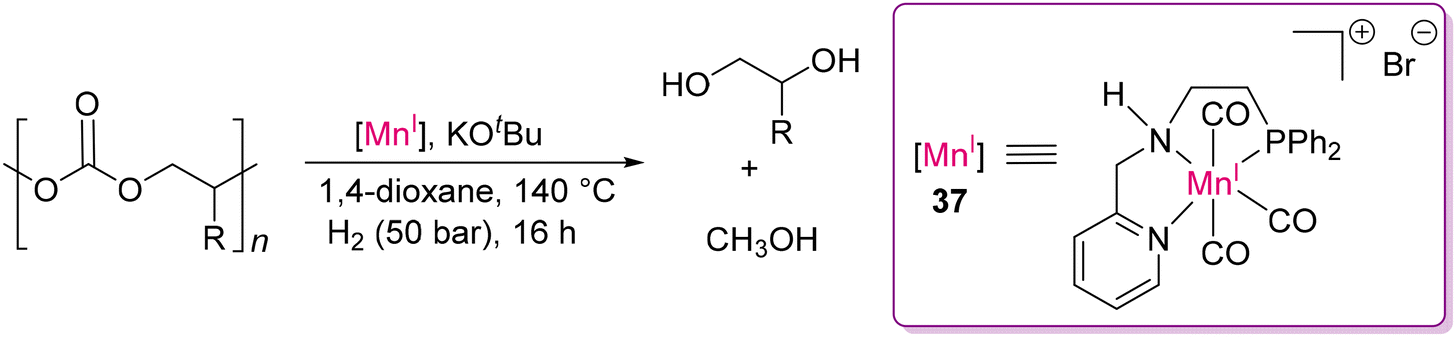
Modern industrial chemistry relies heavily on the hydrogenation of polar bonds, which is catalyzed by various transition metal-based catalysts, most of which are based on precious noble metals, as already been discussed vide supra.83 However, there is high interest and demand to replace noble metal catalysts with earth abundant metals to make the process sustainable. In this direction, Rueping and co-workers developed an Mn(I) catalyst (37) with a PyCH2NH(CH2)2PPh2 (PNN) ligand to hydrogenate poly(propylene carbonate).125
This group reported that 1 mol% of catalyst 37 could hydrogenate poly(propylene carbonate), producing methanol with almost full conversion (Scheme 34). According to the proposed mechanism, the amine proton (N–H) of the ligand plays a vital role in H–H and substrate activation through ligand metal cooperation, as discussed for Milstein's PNP-Ru(II) catalyst (vide supra).105
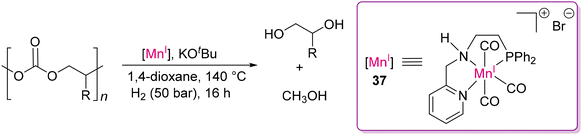 |
| | Scheme 34 Depolymerization of poly(propylene carbonate) by the PNN-Mn(I) catalyst.125 | |
Considering economic and sustainability aspects, considerable efforts have been devoted to developing Zn(II) catalysts for depolymerizing different polar polymers. Recently, Enthaler and co-workers explored the use of Zn(II) catalysts in the depolymerization of chemically stable polyethers including polysiloxanes to produce compounds as potential precursor materials in polymer chemistry.126–129 Explicitly, due to the intrinsic properties of the Si–O skeletal bond, the reduction of polysiloxanes is extremely difficult, and only a few processes involving high-temperature (>200 °C) or less environmentally friendly conditions have been reported to date.130–137 As shown by Enthaler, the Lewis acidic Zn(OTf)2 (38) can cleave the Si–O bond of inorganic silicone polymers in the presence of benzoyl fluoride under mild conditions to form a stable Si–F bond, as shown in Scheme 35.126 They proposed that the activation of stable Si–O occurs by a significant decrease in the bond-dissociation energy through the coordination of an oxygen atom to the Lewis acidic Zn(II) centre, as supported by theoretical calculations, which then facilitates reaction with benzoyl fluoride and consequent cleavage of Si–O to form the Si–F bond. Through a repetitive bond cleaving process, the polymer can be gradually converted into low-molecular-weight fragments such as R2SiF2 and FR2Si-O-SiR2F. These compounds can be reused after appropriate functionalization to produce new polymers. Thus, their successful investigation with simple and affordable Zn(II) catalysts in combination with benzoyl fluoride for the depolymerization of end-of-life polysiloxanes can be a breakthrough finding.
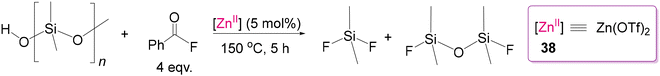 |
| | Scheme 35 Cleavage of Si–O bonds in silicones by Zn(OTf)2 in the presence of benzoyl fluoride.126 | |
Wood, Jones and co-workers reported the use of an imino-monophenolate Zn(II) complex (39) for the degradation of end-of-life PLA through methanolysis reaction to obtain methyl lactate, as illustrated in Scheme 36.138,139 These complexes have the advantage of easy synthetic method and straightforward large-scale production. Calhorda, Dagorne, Avilés and co-workers also demonstrated Zn(II) complexes with N-heterocyclic carbene ligand, which was capable of the production and degradation of PLA.140 Sobota's group showed the degradation of PLA through alcoholysis under solvothermal condition by Ca(II)- and Mg(II)-alkoxides synthesised in situ.141
 |
| | Scheme 36 Zn(II) Lewis acid catalyst for methanolysis of polylactic acid.138,139 | |
Very recently, Williams and co-workers designed an exciting category of homo- and hetero-bimetallic complexes (40a–40c) with macrocyclic ligand based on earth abundant metal ions for taking the advantage of the metal–metal cooperative effect for successful application in the depolymerization of poly(cyclohexene carbonate) under relatively milder reaction conditions with the highest TOF value of 8100 h−1 for the Mg(II)–Co(II) system (40b) (Scheme 37).142–144
 |
| | Scheme 37 Depolymerization of polycarbonates by homo- and hetero-binuclear catalysts reported by Williams’ group.142–144 | |
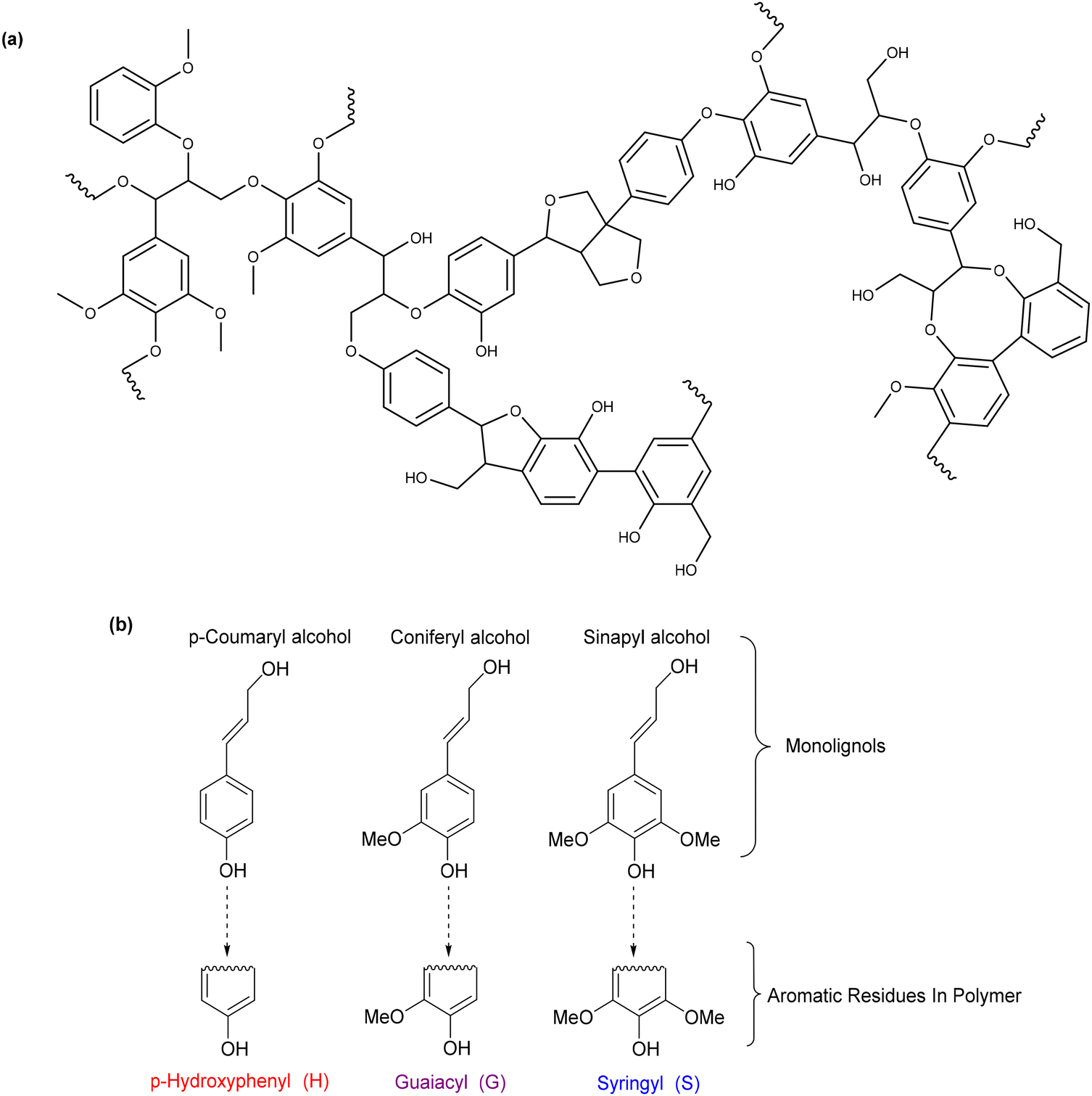
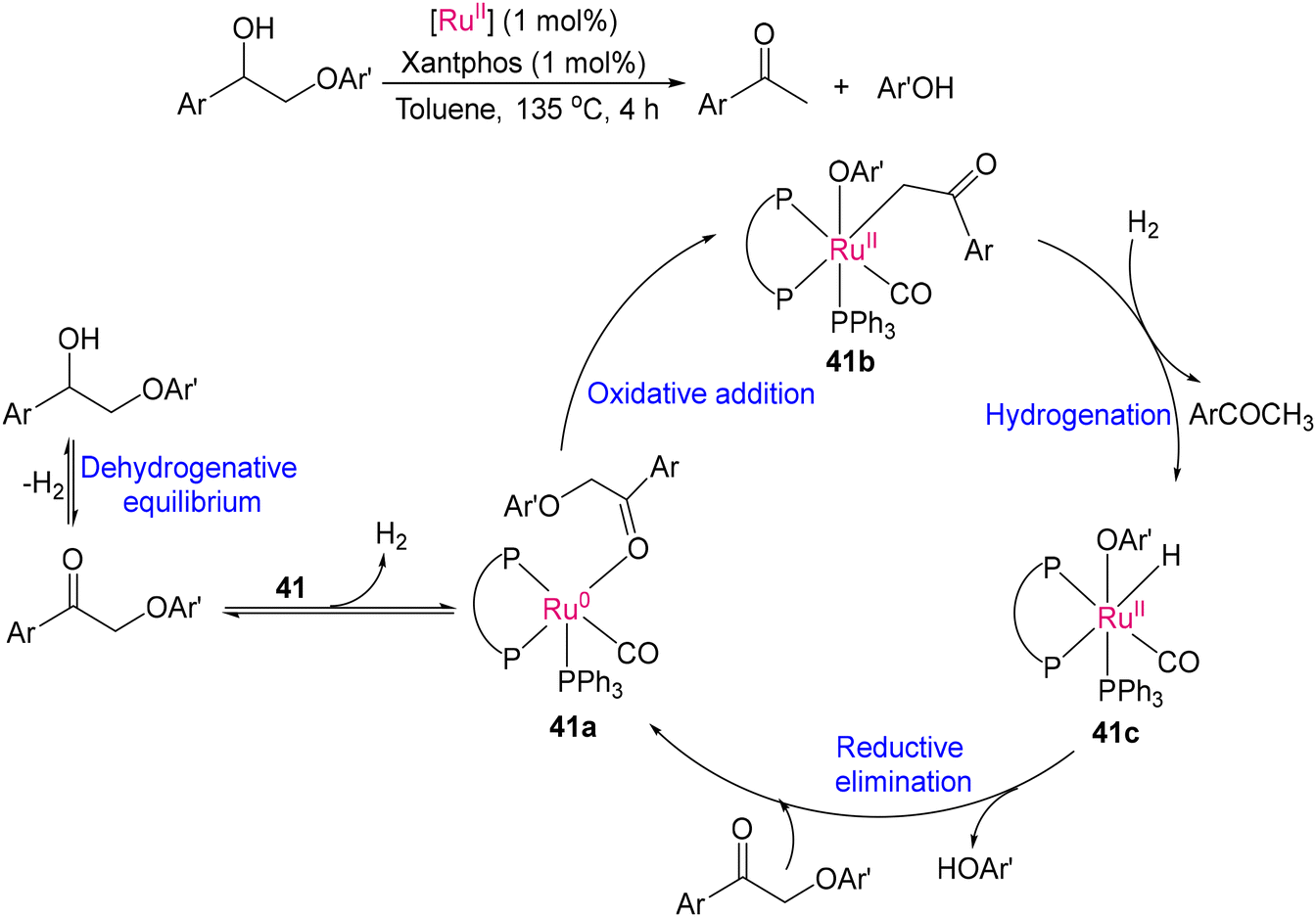
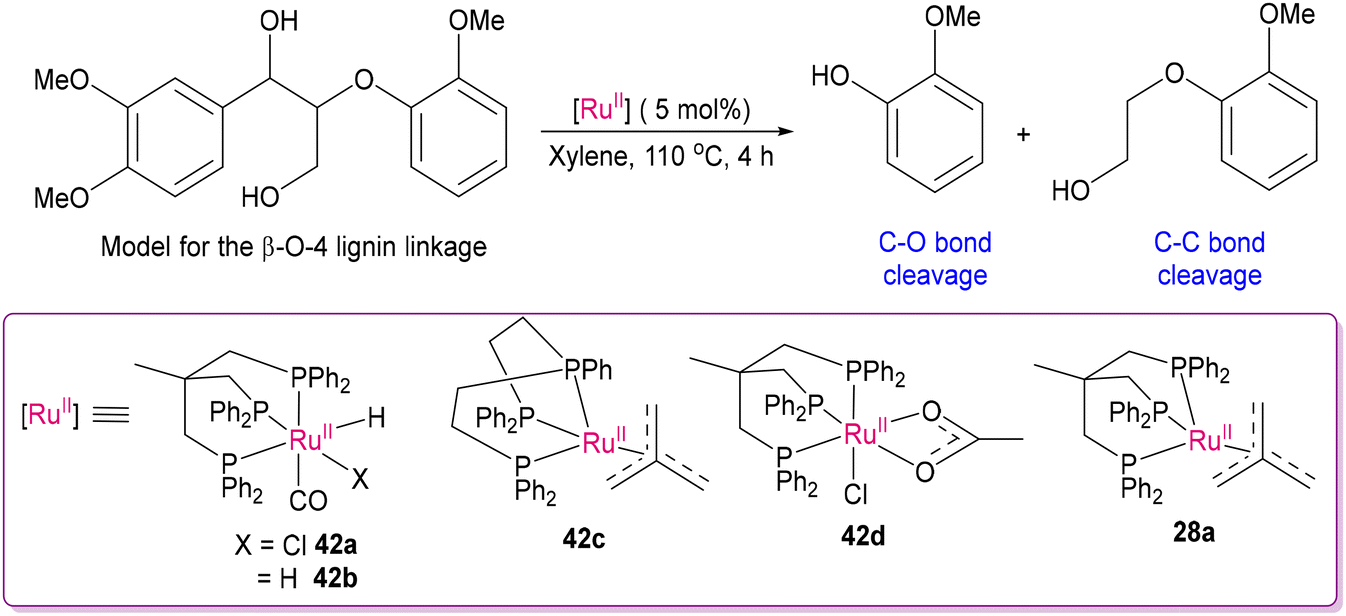
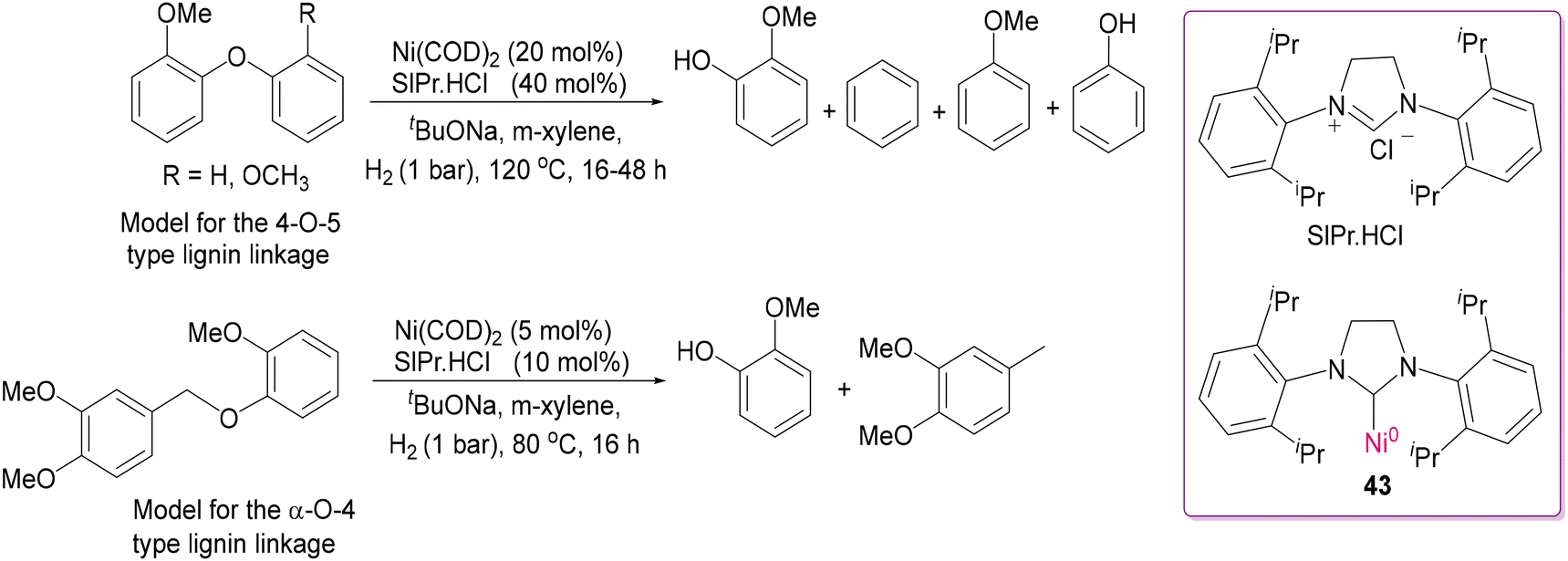
2.3 Deconstructing biopolymers: cleaving C–O and C–C bonds
In the previous sections, we discussed several depolymerization strategies for synthetic polymers with polar and non-polar bonds. However, modified approaches are required to break the similar types of bonds present in the complex interlinked network of biopolymers. Biopolymers such as lignin have huge potential as a renewable feedstock and may serve as a sustainable source of aromatic compounds that can advantageously replace petrochemicals in the long run.145 However, it remains a challenge to depolymerize lignin, given that the C–C and C–O bonds are distributed in a complex network. Lignin, derived from aromatic monolignols, is mainly comprised of aromatic subunits such as p-hydroxyphenyl (H), guaiacyl (G), and syringyl (S) linked via C–O or C–C bonds (Fig. 12). The diversity in lignin structures arises due to the difference in the ratio of monolignols and the way in which these monolignols are covalently linked with each other. For example, hardwood lignin is rich in G and S units with trace H units, while softwood lignin is predominantly G with minimal H. Grass-based lignin exhibits nearly equal proportions of G and S units and minimal H units but greater than the hard and softwood lignins.146 Attempts to isolate native lignin from plant tissues following the developed delignification process often lead to structural changes due to condensation reactions. Thus, to overcome this challenge, technical lignin is preferred for depolymerization and functionalization. Technical lignins are byproducts of conventional pulping processes such as kraft, soda, organosolv, hydrolysis, and sulfite processes.147 Research on technical lignin has become an important area for developing sustainable processes to produce high-value chemicals and fuels.16 However, during the delignification process, it creates structurally accessible and non-accessible parts, which often generate complexity in the depolymerization process, even in technical lignin. The major types of linkages in lignin includes β-O-4, α-O-4, 4-O-5, β-1, α-1 and 5–5, as shown in Fig. 13. The bond dissociation energies (BDE) of the C–O and C–C bonds in lignin model compounds for the six prevalent linkages were theoretically calculated using density functional theory by Huang's and Beckham groups.148,149 In these complex systems, the relationship between the bond length and BDE is not always straightforward like that in small molecules. In these various types of linkages in lignin, the average BDE follows the order of Cα–O < Cβ–O < Cα–Cβ (β-1) < Cα–Cβ (β-O-4) < C4–O < O–C5 < Cα–C1 < C5–C5. Among them, the average BDE values of Cα–O for the α-O-4 linkage and Cβ–O for the β-O-4 linkage are the lowest in energy. A good correlation between the bond length and BDE for the C–C linkage exists, but in the case of C–O linkages, this correlation is not observed. Certainly, the depolymerization of lignin is an intriguing process that is complicated by the structural intricacy and recalcitrance present in these aromatic biopolymers having a random network through strong C–C and C–O bonds. The most recurring type is the β-O-4 linkage, which typically makes up about 50% of all the linkages, and therefore it has been the focus of most depolymerization strategies. According to the above discussion, it is very clear that the depolymerization of lignin is a challenging task, which needs careful design of catalysts.
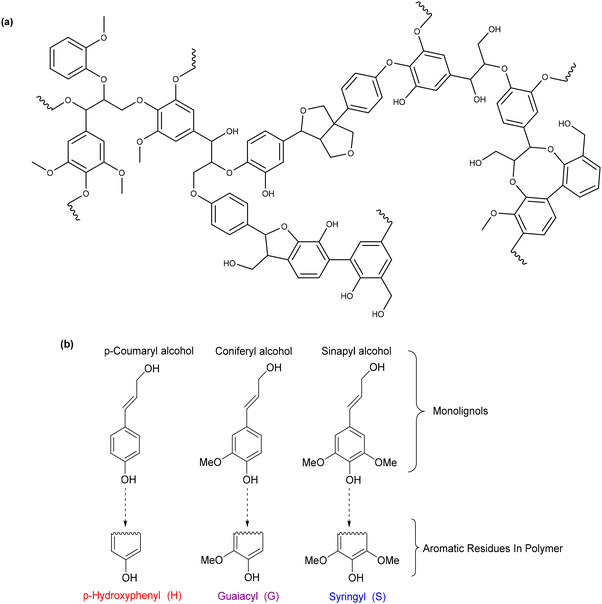 |
| | Fig. 12 (a) Representative interlinked network chemical structures of lignin and (b) aromatic subunits of ‘true lignin’.146 | |
 |
| | Fig. 13 (a) Six prevalent linkages in technical lignin related model compounds. (b) Structural accessibility of technical lignin for the catalyst.148 | |
In 2010, one of the pioneering works by Ellman and coworkers was to successfully cleave C–O bonds (β-O-4) by an Ru(II) catalyst (Scheme 38).150 They used [RuH2(CO)(PPh3)3] and 9,9-dimethyl-bis(diphenylphosphino)xanthene (Xantphos) with the in situ formation of the catalyst, [RuH2(CO)(PPh3)(Xantphos)] (41), to perform tandem catalysis for the essential hydrogen shuttling and C–O bond activation. The catalytic system was successfully applied to a polyether resembling isolated lignin of similar molecular weight with almost 99% conversion. The mechanism involves Ru(II)-catalyzed dehydrogenative equilibrium between a benzylic alcohol and the corresponding aryl ketone.151 Reductive elimination of the Ru(II) catalyst (41) to Ru(0) complex (41a), followed by C–O bond activation of the coordinated aryl ketone moiety through oxidative addition reaction results in the formation of the Ru(II)-enolate (41b). Hydrogenation of 41b to Ru(II)-alkoxide species (41c) and subsequent reductive elimination lead to the formation of phenol, thus completing the catalytic cycle, as shown in Scheme 39.
 |
| | Scheme 38 Ru(II)-catalyzed C–O bond cleavage in lignin-related polymers reported by Ellman and co-workers.150 | |
 |
| | Scheme 39 Proposed mechanism of Ru(II)-catalyzed C–O bond cleavage in a model lignin.151 | |
Klankermayer and coworkers reported a series of slightly modified Ru(II) catalysts with TRIPHOS chelating phosphine (42) and trimethylenemethane (TMM) ligands (28a), showing comparable C–O and C–C cleavage in a 1,3-diol lignin model substrate with β-O-4 linkage, forming guaiacol and other alcoholic products, as shown in Scheme 40.152 Catalysts 42b and 42c were found to be efficient catalysts for C–O cleavage,104 whereas the other catalysts (28a, 42a and 42d) were selective for catalyzing redox-neutral C–C cleavage through a dehydrogenation-initiated retro-aldol and intramolecular hydrogen transfer mechanism. Although both the C–C and C–O bond cleavage pathways require the presence of a secondary hydroxyl group, the presence of a primary alcohol is essential to access the C–C bond cleavage, as proposed by Klankermayer and co-workers. Thus, the selection of the catalyst plays a crucial role in determining the cleavage pathway and the fragmented yields from a particular lignin substrate.
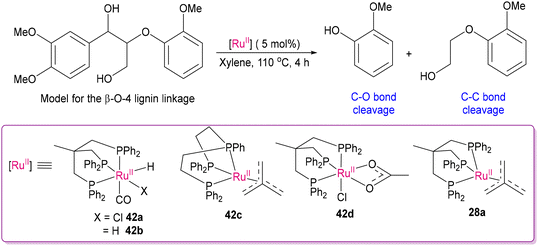 |
| | Scheme 40 Cleaving of C–O and C–C bonds by Ru(II)-Triphos/TMM catalysts.152 | |
The catalytic C–O bond cleavage of aromatic compounds by NiCl2(PPh3)2 in the presence of Grignard reagents was first reported by Wenkert and coworkers, forming biaryl compounds through the cross-coupling of aryl ethers.153 Extending this reactivity to the hydrogenolysis of the C–O bond is a difficult task, given that hydrogen is less reactive than the carbon nucleophile R−M+, where M is a main group metal. However, in 2011, Hartwig and co-workers developed the Ni(0)–NHC catalyst (43), generated in situ from Ni(COD)2 and SIPr·HCl ligand, enabling the selective hydrogenation and cleavage of C–O bonds by H2 in α-O-4 and 4-O-5 lignin model systems, as illustrated in Scheme 41.154 The Ni(0) catalyst system could efficiently catalyze the reductive cleavage of aromatic C–O bonds in non-activated aryl–aryl or aryl–alkyl ethers.
 |
| | Scheme 41 Ni(0)-NHC-catalyzed selective hydrogenolysis of aryl ethers resembling 4-O-5 and β-O-4 lignin linkages.154 | |
In this direction, Kühn and co-workers reported an efficient and selective approach to cleave C–O bonds of several β-hydroxy ethers with catalytic amounts of MeReO3 (MTO) (44), as shown in Scheme 42.155 They further revealed that MeReO2 (MDO), spectroscopically detected as its hexyne adduct, was the catalytically active species formed via the reduction of MTO by the secondary alcohols present in the model lignin. The catalyst efficiently cleaves the model lignin of β-O-4 linkage to guaiacol (as major product) and various other multiple fragments. However, the range of lignin model compounds that can be cleaved by this protocol is limited to having a β-hydroxy ether moiety, restricting its wider applicability.
 |
| | Scheme 42 MeReO3-mediated C–O cleavage of a model lignin compound.155 | |
Further, Stephenson and co-workers developed a two-step photo-catalytic lignin degradation strategy involving milder conditions that overcomes the limitations of traditional methods.156 In contrast to the conventional approaches requiring elevated temperatures, which cannot tolerate functional groups such as phenols and γ-alcohols, this novel method is efficient even at ambient temperatures and compatible with the functional groups present in native and processed lignins. The photocatalytic cleavage involves the use of [4-AcNH-TEMPO][BF4] (Bobbitt's salt), which acts as a mediator for benzylic oxidation, followed by chemoselective visible-light-mediated reductive C−O bond cleavage using the photocatalyst [Ir(ppy)2(dtbbpy)][PF6] (ppy = 2-phenylpyridine, dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine) (45), as shown in Scheme 43.157 The mechanism of the reductive Cα–O bond cleavage is based on the well-established reductive quenching process of 45, as previously established by this group for reductive dehalogenation. When visible light is absorbed by the photocatalyst, it generates an excited state, [Ir]3+*, via metal-to-ligand charge transfer. This excited state accepts an electron from the amine or amine-formate complex and generates reactive [Ir]2+ species. Being a strong reductant with a reduction potential of −1.51 V (vs. SCE), it generates a radical anion through single electron transfer to the benzylic ketone or aliphatic aldehyde, which then undergoes fragmentation to generate an alkoxy anion and the corresponding Cα-radical. Subsequent protonation of the alkoxy anion and H-atom abstraction by Cα-radical yields the degraded products of the model lignin.
 |
| | Scheme 43 Two-step catalytic degradation of model lignin using an Ir(III) photocatalyst.157 | |
In 2013, Toste and co-workers demonstrated the exceptional performance of a vanadyl(V)-based complex of a Schiff base ligand (46) as a catalyst for C(sp3)–O cleavage in nonphenolic β-O-4 dimeric/trimeric lignin models and organosolv lignins derived from Miscanthus giganteus (Scheme 44).158 From the organosolv lignins, several useful degraded products such as vanillin, syringic acid, syringaldehyde, 4-hydroxybenzaldehyde and vanillic acid were detected. The same group also reported that a bis-(8-oxyquinolate) vanadium(V) complex (47) enabled selective Ar–C(sp3) cleavage in phenolic substrates of a dimeric lignin model system. Later, in 2016, Fu and co-workers proposed two possible mechanistic cycles for the cleavage of C(sp3)–OAr and Ar–C(sp3) bonds by theoretical and experimental studies, as illustrated in Scheme 45.159 The catalytic cycle involves stepwise oxidation and reduction of the vanadyl species switching between the various accessible common oxidation states as VV–VIV–VIII–VIV–VV. The cycle of C(sp3)–O cleavage by catalyst 46 starts with ligand exchange in the VV-OR catalyst with the benzylic hydroxyl group of the lignin model compound, followed by intramolecular hydrogen transfer to generate the VIV intermediate, which then isomerizes to the VIII intermediate through a minimum energy spin crossover point (MECP). Successively, C(sp3)–OAr cleavage occurs at the γ-position (with respect to the vanadium center), generating the VIV intermediate, which then undergoes oxidation via second spin crossover, forming VV species. Finally, hydroxyl group elimination and ligand exchange regenerate the active VV catalyst to complete the catalytic cycle. Interestingly, the bis(8-oxyquinolate)vanadium complex (47) selectively cleaves the Ar–C(sp3) of the phenolic β-O-4 lignin model compounds. Here, unlike C(sp3)–OAr cleavage, the catalytic cycle starts with a spin-crossover-involved intermolecular hydrogen transfer, generating a phenoxyl radical and VIV–OH from VVO. The VIV intermediate undergoes proton transfer (via a proton shuttle mechanism) to generate an alcohol-ligated VIVO intermediate, and then reacts with O2 and releases the alcohol and the phenoxyl radical to form a superoxo-VVO intermediate. Stepwise O–O (of superoxo) and Ar–C(sp3) cleavage, followed radical coupling generates a gem-diolate VV intermediate and 2,6-dimethoxy-1,4-benzoquinone. Subsequently, the VV intermediate undergoes intramolecular proton transfer and ligand exchange to regenerate the active VV catalyst. Dehydration of β-hydroxyaldehyde, as produced in this cycle, could also be accomplished by the same catalytic system. The energetic span model suggests that the selectivity of the two catalysts towards C(sp3)–OAr and Ar–C(sp3) cleavage by changing the ligand systems arises due to the difference in T1-HOMO separation/charge dispersion effects and the turnover frequency (TOF)-determining transition state (TDTS) involving the vanadyl species of variable oxidation states.
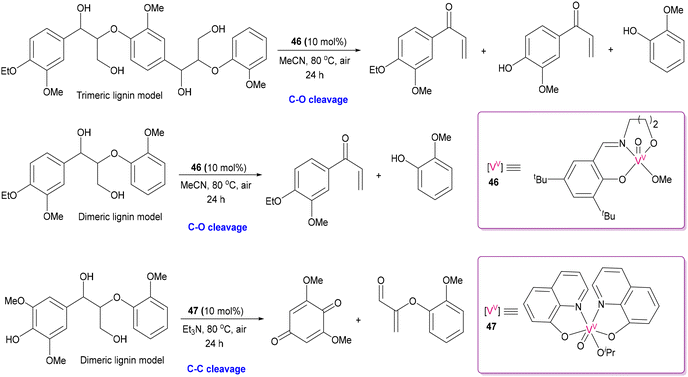 |
| | Scheme 44 Vanadium-catalyzed degradation of dimeric and trimeric lignin model systems.158 | |
 |
| | Scheme 45 Plausible mechanism for vanadium(V)-catalyzed degradation of a dimeric lignin model substrate.159 | |
In recent times, Riisager and coworkers systematically screened various transition metal-based catalysts for the oxidative depolymerization of technical lignin from softwood for converting to bio-oil as renewable stock and value-added aromatic monomers under mild conditions in alkaline aqueous medium using molecular oxygen as the primary oxidant.160 The best-performing system was the VO(acac)2–Cu(OAc)2 (V–Cu) catalyst, resulting a bio-oil yield of ca. 50% and a total increase in aromatic monomers of 27% by weight. The mechanistic details of the depolymerization process for the lignin system by this category of catalyst systems have not been clearly reported. However, the combined effect of V–Cu catalysts, as also observed in lignin model compounds and lignin in previous reports,161–163 is responsible for depolymerization via the combination of C–C and C–O bond cleavage processes.
Following a different but efficient strategy, Cantat and co-workers successfully extended the application of Brookhart's cationic Ir(III) catalyst, 21 (vide supra), for the depolymerization of softwood and hardwood lignin under hydrosilylation conditions, as shown in Scheme 46.164 Notably, the same group reported that the [B(C6F5)3] Lewis acid in the presence of hydrosilanes could act as an efficient catalyst system for the reductive depolymerization of softwood and hardwood lignin through C–O cleavage.147 However, the instability associated with [B(C6F5)3] under the reaction condition limits its applicability in the direct reductive polymerization of lignin substrates, stimulating the development of stable but efficient catalyst systems. The pioneering work by Brookhart showed that the cationic Ir(III) complex (21) could be an efficient catalyst to cleave the C(sp3)–O bond in alkylethers under hydrosilylation conditions to yield silylether and alkane.77 The similar chemical behavior of the Lewis acid B(C6F5)3 and the Ir(III) complex (21) led Cantat and co-workers to compare the stabilities and activities for the reduction of C(sp3)–O linkages for both model molecules and lignin matrices.164 Although less active than B(C6F5)3, complex 21, being more stable, was found to be an effective catalyst in presence of Et3SiH for the reductive depolymerization of various softwood and hardwood lignin to monoaromatics. For example, catalyst 21 with a loading of 10 wt% (ca. 1.3 mol% per aromatic unit) and 300 wt% of Et3SiH at 70 °C successfully depolymerized industrial pine lignin to the tris-silylated primary alcohol with 120 wt% yield. However, a minimum quantity of catalyst is required (≥5 wt% of 21) for depolymerization given that common impurities (such as residual carbohydrates and lipids present in lignin) deactivate and poison the catalyst system. Brookhart and co-workers elucidated the mechanism in full detail for the cleavage of C(sp3)–O by Ir(III) catalyst 21 (vide supra).77
 |
| | Scheme 46 Depolymerization of true lignin using the Brookhart catalyst.164 | |
In this section, we only highlighted the most efficient catalytic systems employing transition metal complexes for the depolymerization of lignin through C–O and C–C cleavage of less BDE motifs (β-O-4, α-O-4 and 4-O-5). However, the same strategy can be translated to the real bio-derived lignin by further modification of the catalysts. For further reading on the valorization of lignin by catalysts, readers can refer to the reviews published by Stephenson165 and Ragauskas.166
3. Conclusion
This comprehensive summary highlights the recent development and strategies for the depolymerization of polymeric waste by transition metal-based catalysts. The judicious design of organometallic catalysts directed by the reactivity of the metal centre and suitable ligand selection for breaking the nonpolar and polar skeletal bonds was strategically discussed. In this context, we provide a compilation of organometallic catalysts that exhibit notable efficacy in depolymerizing polymeric waste by elucidating the ligand and coordination effects in the catalytic reactions and the mechanistic approach facilitating the cleavage of chemical bonds. Organometallic catalysts offer high efficiency, milder condition, selectivity, reduced catalyst-loading and reusability, offering a sustainable solution to the challenge of reusing plastic waste, specifically the chemically robust hydrocarbon polymers with non-polar skeletal bonds. This perspective highlights that Ir(III) and Ru(II) organometallic catalysts demonstrate significant potential for depolymerizing both polar and nonpolar polymers through different mechanisms. However, it is desirable that cost-effective and more affordable transition metal complexes, particularly based on Fe(II), Co(II), and Zn(II), are the future for catalyzing bulk depolymerization processes. Hence, there is no doubt that depolymerization using transition metal-based catalysts holds promising future prospect for converting plastic waste to its monomeric form, valuable materials, fragmented hydrocarbons as fuels and other feedstock, not only for reducing its environmental impact but also promoting chemical upcycling. However, understanding the mechanism of depolymerization the catalysis reaction is crucial for designing next-generation catalysts that are both efficient and economically feasible. Ongoing research aims to fully unlock the potential of transition metal complexes as depolymerization catalysts, paving the way for a more sustainable and circular approach to polymer waste management.
Data availability
No primary research results, software or code has been included and no new data were generated or analysed as part of this submitted review for publication as perspective (Invited) in Dalton Transaction.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
SKP acknowledges the support from SERB, Govt. India (CRG/2019/005907). SD and CKB acknowledge IIT Kharagpur and UGC India, respectively, for doctoral fellowship.
References
- L. Jia, S. Evans and S. van der Linden, Nat. Commun., 2019, 10, 9–11 CrossRef
 .
.
- S. B. Borrelle, J. Ringma, K. L. Law, C. C. Monnahan, L. Lebreton, A. McGivern, E. Murphy, J. Jambeck, G. H. Leonard, M. A. Hilleary, M. Eriksen, H. P. Possingham, H. De Frond, L. R. Gerber, B. Polidoro, A. Tahir, M. Bernard, N. Mallos, M. Barnes and C. M. Rochman, Science, 2020, 369, 1515–1518 CrossRef CAS PubMed .
- W. W. Y. Lau, Y. Shiran, R. M. Bailey, E. Cook, M. R. Stuchtey, J. Koskella, C. A. Velis, L. Godfrey, J. Boucher, M. B. Murphy, R. C. Thompson, E. Jankowska, A. Castillo Castillo, T. D. Pilditch, B. Dixon, L. Koerselman, E. Kosior, E. Favoino, J. Gutberlet, S. Baulch, M. E. Atreya, D. Fischer, K. K. He, M. M. Petit, U. R. Sumaila, E. Neil, M. V. Bernhofen, K. Lawrence and J. E. Palardy, Science, 2020, 369, 1455–1461 CrossRef CAS PubMed .
-
B. C. Howard, S. Gibbens, E. Zachos and L. Parker, A Running List of Action on Plastic Pollution, 2018 Search PubMed .
- UN resolution pledges to plastic reduction by 2030.
- A. R. Rahimi and J. M. Garciá, Nat. Rev. Chem., 2017, 1, 1–11 CrossRef .
- M. Hong and E. Y. X. Chen, Green Chem., 2017, 19, 3692–3706 RSC .
- H. Tang, N. Li, G. Li, A. Wang, Y. Cong, G. Xu, X. Wang and T. Zhang, Green Chem., 2019, 21, 2709–2719 RSC .
- J. M. García, Chem, 2016, 1, 813–815 Search PubMed .
-
D. S. Achilias, L. Andriotis, L. A. Koutsidis, D. A. Louka, N. P. Nianias, P. Siafaka, L. Tsagkalias and G. Tsintzou in Material Recycling – Trends and Perspectives, InTech, 2012 Search PubMed .
- C. Wang and O. El-Sepelgy, Curr. Opin. Green Sustainable Chem., 2021, 32, 100547 CrossRef .
- A. C. Fernandes, Green Chem., 2021, 23, 7330–7360 RSC .
- I. A. Ignatyev, W. Thielemans and B. Vander Beke, ChemSusChem, 2014, 7, 1579–1593 CrossRef PubMed .
- J. Aguado, D. P. Serrano and J. M. Escola, Ind. Eng. Chem. Res., 2008, 47, 7982–7992 CrossRef .
- S. Kumar, A. K. Panda and R. K. Singh, Resour., Conserv. Recycl., 2011, 55, 893–910 CrossRef .
- S. Guadix-Montero and M. Sankar, Top. Catal., 2018, 61, 183–198 CrossRef .
- J. B. Huang, G. S. Zeng, X. S. Li, X. C. Cheng and H. Tong, IOP Conf. Ser. Earth Environ. Sci., 2018, 167, 012029 CrossRef .
- J. Chen, Q. Peng, X. Peng, H. Zhang and H. Zeng, Chem. Rev., 2022, 122, 14594–14678 CrossRef CAS .
-
F. Ullmann, polymers and Plastics: Products and Processes, Wiely VCH, 2016, p. 937 Search PubMed .
- A. H. Westlie, E. Y. X. Chen, C. M. Holland, S. S. Stahl, M. Doyle, S. R. Trenor and K. M. Knauer, Macromol. Rapid Commun., 2022, 43, 1–15 CrossRef .
- Anonymous Polyethylene August 2020 World Analysis, IHS Markit 2021.
- D. P. Serrano, J. Aguado and J. M. Escola, ACS Catal., 2012, 2, 1924–1941 CrossRef .
- A. Pifer and A. Sen, Angew. Chem., Int. Ed., 1998, 37, 3306–3308 CrossRef .
- E. Bäckström, K. Odelius and M. Hakkarainen, Ind. Eng. Chem. Res., 2017, 56, 14814–14821 CrossRef .
- G. Celik, R. M. Kennedy, R. A. Hackler, M. Ferrandon, A. Tennakoon, S. Patnaik, A. M. Lapointe, S. C. Ammal, A. Heyden, F. A. Perras, M. Pruski, S. L. Scott, K. R. Poeppelmeier, A. D. Sadow and M. Delferro, ACS Cent. Sci., 2019, 5, 1795–1803 CrossRef PubMed .
- D. D. Hibbitts, D. W. Flaherty and E. Iglesia, ACS Catal., 2016, 6, 469–482 CrossRef .
- J. H. Sinfelt, Adv. Catal., 1973, 23, 91–119 Search PubMed .
- M. C. Haibach, S. Kundu, M. Brookhart and A. S. Goldman, Acc. Chem. Res., 2012, 45, 947–958 CrossRef PubMed .
- L. D. Ellis, S. V. Orski, G. A. Kenlaw, A. G. Norman, K. L. Beers, Y. Román-Leshkov and G. T. Beckham, ACS Sustainable Chem. Eng., 2021, 9, 623–628 CrossRef PubMed .
- G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681–703 CrossRef PubMed .
- J. A. Maguire, A. Petrillo and A. S. Goldman, J. Am. Chem. Soc., 1992, 114, 9492–9498 CrossRef .
- J. Belli and C. M. Jensen, Organometallics, 1996, 15, 1532–1534 CrossRef CAS .
- G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681–703 CrossRef CAS PubMed .
- M. J. Burk and R. H. Crabtree, J. Am. Chem. Soc., 1987, 109, 8025–8032 CrossRef CAS .
- T. Aoki and R. H. Crabtree, Organometallics, 1993, 12, 294–298 CrossRef CAS .
- T. Fujii, Y. Higashino and Y. Saito, J. Chem. Soc., Dalton Trans., 1993, 517–520 RSC .
- K. Nomura and Y. Saito, J. Chem. Soc., Chem. Commun., 1988, 161–162 RSC .
- J. A. Maguire, W. T. Boese and A. S. Goldman, J. Am. Chem. Soc., 1989, 111, 7088–7093 CrossRef .
- J. A. Maguire and A. S. Goldman, J. Am. Chem. Soc., 1991, 113, 6706–6708 CrossRef .
- F. Liu, E. B. Pak, B. Singh, C. M. Jensen and A. S. Goldman, J. Am. Chem. Soc., 1999, 121, 4086–4087 CrossRef .
- S. Kundu, Y. Choliy, G. Zhuo, R. Ahuja, T. J. Emge, R. Warmuth, M. Brookhart, K. Krogh-Jespersen and A. S. Goldman, Organometallics, 2009, 28, 5432–5444 CrossRef .
- R. Ahuja, B. Punji, M. Findlater, C. Supplee, W. Schinski, M. Brookhart and A. S. Goldman, Nat. Chem., 2011, 3, 167–171 CrossRef .
- B. Punji, T. J. Emge and A. S. Goldman, Organometallics, 2010, 29, 2702–2709 CrossRef .
- A. S. Goldman, A. H Roy, Z. Huang, R. Ahuja, W. Schinski and M. Brookhart, Science, 2006, 312, 257–261 CrossRef PubMed .
- I. Göttker-Schnetmann, P. White and M. Brookhart, J. Am. Chem. Soc., 2004, 126, 1804–1811 CrossRef .
- R. H. Grubbs, Angew. Chem., Int. Ed., 2006, 45, 3760–3765 CrossRef PubMed .
- Z. Huang, E. Rolfe, E. C. Carson, M. Brookhart, A. S. Goldman, S. H. El-Khalafy and A. H. Roy MacArthur, Adv. Synth. Catal., 2010, 352, 125–135 CrossRef .
- X. Jia, C. Qin, T. Friedberger, Z. Guan and Z. Huang, Sci. Adv., 2016, 2, 1–8 Search PubMed .
- A. Ray, K. Zhu, Y. V. Kissin, A. E. Cherian, G. W. Coates and A. S. Goldman, Chem. Commun., 2005, 2, 3388–3390 RSC .
- A. Arroyave, S. Cui, J. C. Lopez, A. L. Kocen, A. M. Lapointe, M. Delferro and G. W. Coates, J. Am. Chem. Soc., 2022, 144, 23280–23285 CrossRef PubMed .
- R. J. Conk, S. Hanna, J. X. Shi, J. Yang, N. R. Ciccia, L. Qi, B. J. Bloomer, S. Heuvel, T. Wills, J. Su, A. T. Bell and J. F. Hartwig, Science, 2022, 377, 1561–1566 CrossRef PubMed .
- N. M. Wang, G. Strong, V. Dasilva, L. Gao, R. Huacuja, I. A. Konstantinov, M. S. Rosen, A. J. Nett, S. Ewart, R. Geyer, S. L. Scott and D. Guironnet, J. Am. Chem. Soc., 2022, 144, 18526–18531 CrossRef PubMed .
- R. J. Conk, J. F. Stahler, J. X. Shi, J. Yang, N. G. Lefton, J. N. Brunn, A. T. Bell and J. F. Hartwig, Science, 2024, 1327, 1322–1327 CrossRef .
-
H. Sinn and W. Kaminsky, in Advances in Organometallic Chemistry, Elsevier, 1980, vol. 18, pp. 99–149 Search PubMed .
- V. Dufaud and J. M. Basset, Angew. Chem., Int. Ed., 1998, 37, 806–810 CrossRef .
- D. W. Hart and J. Schwartz, J. Am. Chem. Soc., 1974, 96, 8115–8116 CrossRef .
- J. Zheng, Y. Lin, F. Liu, H. Tan, Y. Wang and T. Tang, Chem. – Eur. J., 2013, 19, 541–548 CrossRef PubMed .
- C. H. Jun, Chem. Soc. Rev., 2004, 33, 610–618 RSC .
- F. D. Cannavacciuolo, R. Yadav, A. Esper, A. Vittoria, G. Antinucci, F. Zaccaria, R. Cipullo, P. H. M. Budzelaar, V. Busico, G. P. Goryunov, D. V. Uborsky, A. Z. Voskoboynikov, K. Searles, C. Ehm and A. S. Veige, Angew. Chem., Int. Ed., 2022, 61, e202202258 CrossRef PubMed .
- K. Parker, G. K. Weragoda, V. Pho, A. J. Canty, A. Polyzos, R. A. J. O'Hair and V. Ryzhov, ChemCatChem, 2020, 12, 5476–5485 CrossRef .
- K. Parker, G. K. Weragoda, A. J. Canty, A. Polyzos, V. Ryzhov and R. A. J. O'Hair, Organometallics, 2020, 39, 4027–4036 CrossRef .
- K. Parker, G. K. Weragoda, A. J. Canty, V. Ryzhov and R. A. J. O'Hair, Organometallics, 2021, 40, 857–868 CrossRef .
- J. A. Herman, M. E. Seazzu, L. G. Hughes, D. R. Wheeler, C. M. Washburn and B. H. Jones, ACS Appl. Polym. Mater., 2019, 1, 2177–2188 CrossRef .
- S. J. Czarnocki, I. Czeluśniak, T. K. Olszewski, M. Malinska, K. Woźniak and K. Grela, ACS Catal., 2017, 7, 4115–4121 CrossRef .
-
L. Cervený, Catalytic hydrogenation, Elsevier, 1986 Search PubMed .
-
J. G. de Vries and C. J. Elsevier, Handbook of homogeneous hydrogenation, WeinheimWiley-VCH, 2007 Search PubMed .
-
P. G. Andersson and I. J. Munslow, Modern reduction methods, John Wiley & Sons, 2008 Search PubMed .
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef PubMed .
- A. T. Normand and K. J. Cavell, Eur. J. Inorg. Chem., 2008, 2008, 2781–2800 CrossRef .
- D. E. Fagnani, D. Kim, S. I. Camarero, J. F. Alfaro and A. J. McNeil, Nat. Chem., 2023, 15, 222–229 CrossRef PubMed .
- S. Bac, M. E. Fieser and S. Mallikarjun Sharada, Phys. Chem. Chem. Phys., 2022, 24, 3518–3522 RSC .
- N. G. Bush, M. K. Assefa, S. Bac, S. Mallikarjun Sharada and M. E. Fieser, Mater. Horiz., 2023, 10, 2047–2052 RSC .
- S. C. Kosloski-Oh, Z. A. Wood, Y. Manjarrez, J. P. De Los Rios and M. E. Fieser, Mater. Horiz., 2021, 8, 1084–1129 RSC .
- R. K. Jha, B. J. Neyhouse, M. S. Young, D. E. Fagnani and A. J. McNeil, Chem. Sci., 2024, 15, 5802–5813 RSC .
- F. Gardea, J. M. Garcia, D. J. Boday, K. M. Bajjuri, M. Naraghi and J. L. Hedrick, Macromol. Chem. Phys., 2014, 215, 2260–2267 CrossRef .
- E. Quaranta, D. Sgherza and G. Tartaro, Green Chem., 2017, 19, 5422–5434 RSC .
- S. Park and M. Brookhart, Organometallics, 2010, 29, 6057–6064 CrossRef CAS .
- S. Park, D. Bézier and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 11404–11407 CrossRef CAS .
- I. S. Kim, M. Y. Ngai and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 14891–14899 CrossRef CAS .
- S. Park and M. Brookhart, Chem. Commun., 2011, 47, 3643–3645 RSC .
- T. T. Metsänen, P. Hrobárik, H. F. T. Klare, M. Kaupp and M. Oestreich, J. Am. Chem. Soc., 2014, 136, 6912–6915 CrossRef PubMed .
- E. Feghali and T. Cantat, ChemSusChem, 2015, 8, 980–984 CrossRef CAS .
- L. Monsigny, J. C. Berthet and T. Cantat, ACS Sustainable Chem. Eng., 2018, 6, 10481–10488 CrossRef CAS .
- P. A. Dub and T. Ikariya, ACS Catal., 2012, 2, 1718–1741 CrossRef CAS .
- J. Yang, P. S. White and M. Brookhart, J. Am. Chem. Soc., 2008, 130, 17509–17518 CrossRef CAS PubMed .
- T. Robert and M. Oestreich, Angew. Chem., Int. Ed., 2013, 52, 5216–5218 CrossRef CAS PubMed .
- L. Gausas, S. K. Kristensen, H. Sun, A. Ahrens, B. S. Donslund, A. T. Lindhardt and T. Skrydstrup, JACS Au, 2021, 1, 517–524 CrossRef CAS PubMed .
- P. Dahiya, M. K. Gangwar and B. Sundararaju, ChemCatChem, 2021, 13, 934–939 CrossRef CAS .
- J. Pritchard, G. A. Filonenko, R. Van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2015, 44, 3808–3833 RSC .
- E. Balaraman, Y. Ben-David and D. Milstein, Angew. Chemie, 2011, 123, 11906–11909 CrossRef .
- F. Ferretti, F. K. Scharnagl, A. Dall'Anese, R. Jackstell, S. Dastgir and M. Beller, Catal. Sci. Technol., 2019, 9, 3548–3553 RSC .
- K. D. Knight and M. E. Fieser, Inorg. Chem. Front., 2023, 11, 298–309 RSC .
- R. Noyori, Nat. Chem., 2009, 1, 5–6 CrossRef CAS PubMed .
-
M. Aresta, Carbon dioxide as chemical feedstock, John Wiley & Sons, 2010 Search PubMed .
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed .
- E. Balaraman, C. Gunanathan, J. Zhang, L. J. W. Shimon and D. Milstein, Nat. Chem., 2011, 3, 609–614 CrossRef CAS PubMed .
- L. Zhang, Z. Han, X. Zhao, Z. Wang and K. Ding, Angew. Chemie, 2015, 127, 6284–6287 CrossRef .
- E. M. Krall, T. W. Klein, R. J. Andersen, D. S. Reader, B. C. Dauphinais, S. P. McIlrath, A. A. Fischer, M. J. Carney and N. J. Robertson, Chem. Commun., 2014, 50, 4884–4887 RSC .
- J. A. Fuentes, S. M. Smith, M. T. Scharbert, I. Carpenter, D. B. Cordes, A. M. Z. Slawin and M. L. Clarke, Chem. – Eur. J., 2015, 21, 10851–10869 CrossRef CAS PubMed .
- S. D. Phillips, J. A. Fuentes and M. L. Clarke, Chem. – Eur. J., 2010, 16, 8002–8005 CrossRef CAS .
- P. A. Dub, N. J. Henson, R. L. Martin and J. C. Gordon, J. Am. Chem. Soc., 2014, 136, 3505–3521 CrossRef CAS PubMed .
- S. Westhues, M. Meuresch and J. Klankermayer, Angew. Chem., Int. Ed., 2016, 55, 12841–12844 CrossRef CAS .
- M. Meuresch, S. Westhues, W. Leitner and J. Klankermayer, Angew. Chem., Int. Ed., 2016, 55, 1392–1395 CrossRef CAS PubMed .
- T. Vom Stein, M. Meuresch, D. Limper, M. Schmitz, M. Hölscher, J. Coetzee, D. J. Cole-Hamilton, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2014, 136, 13217–13225 CrossRef CAS .
- A. Kumar, N. Von Wolff, M. Rauch, Y. Q. Zou, G. Shmul, Y. Ben-David, G. Leitus, L. Avram and D. Milstein, J. Am. Chem. Soc., 2020, 142, 14267–14275 CrossRef CAS .
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. Robert McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 1–24 CrossRef .
- R. Noyori, M. Yamakawa and S. Hashiguchi, J. Org. Chem., 2001, 66, 7931–7944 CrossRef CAS .
- A. Kumar, N. A. Espinosa-Jalapa, G. Leitus, Y. Diskin-Posner, L. Avram and D. Milstein, Angew. Chem., Int. Ed., 2017, 56, 14992–14996 CrossRef CAS PubMed .
- A. Kumar, T. Janes, N. A. Espinosa-Jalapa and D. Milstein, Angew. Chem., Int. Ed., 2018, 57, 12076–12080 CrossRef CAS PubMed .
- N. A. Espinosa-Jalapa, A. Kumar, G. Leitus, Y. Diskin-Posner and D. Milstein, J. Am. Chem. Soc., 2017, 139, 11722–11725 CrossRef CAS PubMed .
- T. O. Kindler, C. Alberti, J. Sundermeier and S. Enthaler, ChemistryOpen, 2019, 8, 1410–1412 CrossRef CAS PubMed .
- C. Alberti, S. Eckelt and S. Enthaler, ChemistrySelect, 2019, 4, 12268–12271 CrossRef CAS .
-
Q. Yao, in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, 2015, pp. 1–3 Search PubMed .
- W. Zhou, P. Neumann, M. Al Batal, F. Rominger, A. S. K. Hashmi and T. Schaub, ChemSusChem, 2021, 14, 4176–4180 CrossRef CAS PubMed .
- G. A. Filonenko, R. Van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2018, 47, 1459–1483 RSC .
- S. H. Kim and S. H. Hong, ACS Catal., 2014, 4, 3630–3636 CrossRef CAS .
- R. A. Farrar-Tobar, Z. Wei, H. Jiao, S. Hinze and J. G. de Vries, Chem. – Eur. J., 2018, 24, 2725–2734 CrossRef CAS PubMed .
- X. Liu, J. G. De Vries and T. Werner, Green Chem., 2019, 21, 5248–5255 RSC .
- R. A. Farrar-Tobar, B. Wozniak, A. Savini, S. Hinze, S. Tin and J. G. de Vries, Angew. Chem., Int. Ed., 2019, 58, 1129–1133 CrossRef CAS PubMed .
- C. Bornschein, S. Werkmeister, B. Wendt, H. Jiao, E. Alberico, W. Baumann, H. Junge, K. Junge and M. Beller, Nat. Commun., 2014, 5, 411 Search PubMed .
- S. Werkmeister, K. Junge, B. Wendt, E. Alberico, H. Jiao, W. Baumann, H. Junge, F. Gallou and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 8722–8726 CrossRef CAS PubMed .
- B. F. S. Nunes, M. C. Oliveira and A. C. Fernandes, Green Chem., 2020, 22, 2419–2425 RSC .
- Y. Kratish, J. Li, S. Liu, Y. Gao and T. J. Marks, Angew. Chem., Int. Ed., 2020, 59, 19857–19861 CrossRef CAS PubMed .
- D. J. Darensbourg, S. H. Wei, A. D. Yeung and W. C. Ellis, Macromolecules, 2013, 46, 5850–5855 CrossRef CAS .
- V. Zubar, Y. Lebedev, L. M. Azofra, L. Cavallo, O. El-Sepelgy and M. Rueping, Angew. Chemie, 2018, 130, 13627–13631 CrossRef .
- S. Enthaler, Angew. Chemie, 2014, 126, 2754–2759 CrossRef .
- S. Enthaler and M. Weidauer, Chem. – Eur. J., 2012, 18, 1910–1913 CrossRef CAS PubMed .
- M. Weidauer, B. Heyder, D. Woelki, M. Tschiersch, A. Köhler-Krützfeldt and S. Enthaler, Resour. Technol., 2015, 1, 73–79 Search PubMed .
- S. Enthaler, Eur. J. Lipid Sci. Technol., 2013, 115, 239–245 CrossRef CAS .
- L. H. Sommer and G. R. Ansul, J. Am. Chem. Soc., 1955, 77, 2482–2485 CrossRef CAS .
- A. W. Apblett and A. R. Barron, Organometallics, 1990, 9, 2137–2141 CrossRef CAS .
- R. Mulhaupt, J. Calabrese and S. D. Ittel, Organometallics, 1991, 10, 3403–3406 CrossRef CAS .
- C. N. McMahon, S. G. Bott, L. B. Alemany, H. W. Roesky and A. R. Barron, Organometallics, 1999, 18, 5395–5408 CrossRef CAS .
- C.-L. Chang, H. S.-J. Lee and C.-K. Chen, J. Polym. Res., 2005, 12, 433–438 CrossRef CAS .
- M. Okamoto, K. Miyazaki, A. Kado and E. Suzuki, Chem. Commun., 2001, 1838–1839 RSC .
- T. Fouquet, J. Bour, V. Toniazzo, D. Ruch and L. Charles, Rapid Commun. Mass Spectrom., 2012, 26, 2057–2067 CrossRef CAS PubMed .
- M. G. Voronkov, A. A. Trukhina, L. I. Belousova, G. A. Kuznetsova and N. N. Vlasova, Russ. J. Org. Chem., 2007, 43, 501–506 CrossRef CAS .
- L. A. Román-Ramírez, P. McKeown, M. D. Jones and J. Wood, ACS Catal., 2019, 9, 409–416 CrossRef .
- L. A. Román-Ramírez, P. McKeown, C. Shah, J. Abraham, M. D. Jones and J. Wood, Ind. Eng. Chem. Res., 2020, 59, 11149–11156 CrossRef .
- C. Fliedel, D. Vila-Viçosa, M. J. Calhorda, S. Dagorne and T. Avilés, ChemCatChem, 2014, 6, 1357–1367 CrossRef CAS .
- R. Petrus, D. Bykowski and P. Sobota, ACS Catal., 2016, 6, 5222–5235 CrossRef CAS .
- F. N. Singer, A. C. Deacy, T. M. McGuire, C. K. Williams and A. Buchard, Angew. Chem., Int. Ed., 2022, 61, e202201785 CrossRef CAS PubMed .
- T. M. McGuire, A. C. Deacy, A. Buchard and C. K. Williams, J. Am. Chem. Soc., 2022, 144, 18444–18449 CrossRef CAS PubMed .
- M. L. Smith, T. M. McGuire, A. Buchard and C. K. Williams, ACS Catal., 2023, 13, 15770–15778 CrossRef CAS PubMed .
- C. M. Welker, V. K. Balasubramanian, C. Petti, K. M. Rai, S. De Bolt and V. Mendu, Energies, 2015, 8, 7654–7676 CrossRef CAS .
- K. Ninomiya, K. Ochiai, M. Eguchi, K. Kuroda, Y. Tsuge, C. Ogino, T. Taima and K. Takahashi, Ind. Crops Prod., 2018, 111, 457–461 CrossRef CAS .
- E. Feghali, G. Carrot, P. Thuéry, C. Genre and T. Cantat, Energy Environ. Sci., 2015, 8, 2734–2743 RSC .
- J.-B. Huang, W. U. Shu-Bin, C. Hao, M. Lei, J.-J. Liang and H. Tong, J. Fuel Chem. Technol., 2015, 43, 429–436 CrossRef CAS .
- S. Kim, S. C. Chmely, M. R. Nimlos, Y. J. Bomble, T. D. Foust, R. S. Paton and G. T. Beckham, J. Phys. Chem. Lett., 2011, 2, 2846–2852 CrossRef CAS .
- J. M. Nichols, L. M. Bishop, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2010, 132, 16725–16725 CrossRef CAS .
- N. A. Owston, A. J. Parker and J. M. J. Williams, Chem. Commun., 2002, 8, 624–625 Search PubMed .
- T. vom Stein, T. den Hartog, J. Buendia, S. Stoychev, J. Mottweiler, C. Bolm, J. Klankermayer and W. Leitner, Angew. Chemie, 2015, 127, 5957–5961 CrossRef .
- E. Wenkert, E. L. Michelotti and C. S. Swindell, J. Am. Chem. Soc., 1979, 101, 2246–2247 CrossRef CAS .
- A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS PubMed .
- R. G. Harms, I. I. E. Markovits, M. Drees, H. C. Mult, W. A. Herrmann, M. Cokoja and F. E. Kühn, ChemSusChem, 2014, 7, 429–434 CrossRef CAS PubMed .
- J. D. Nguyen, E. M. D'amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS PubMed .
- J. D. Nguyen, B. S. Matsuura and C. R. J. Stephenson, J. Am. Chem. Soc., 2014, 136, 1218–1221 CrossRef CAS PubMed .
- J. M. W. Chan, S. Bauer, H. Sorek, S. Sreekumar, K. Wang and F. D. Toste, ACS Catal., 2013, 3, 1369–1377 CrossRef CAS .
- Y. Y. Jiang, L. Yan, H. Z. Yu, Q. Zhang and Y. Fu, ACS Catal., 2016, 6, 4399–4410 CrossRef CAS .
- F. Walch, O. Y. Abdelaziz, S. Meier, S. Bjelić, C. P. Hulteberg and A. Riisager, Catal. Sci. Technol., 2021, 11, 1843–1853 RSC .
- J. Mottweiler, M. Puche, C. Räuber, T. Schmidt, P. Concepción, A. Corma and C. Bolm, ChemSusChem, 2015, 8, 2106–2113 CrossRef CAS PubMed .
- T. Rinesch, J. Mottweiler, M. Puche, P. Concepción, A. Corma and C. Bolm, ACS Sustainable Chem. Eng., 2017, 5, 9818–9825 CrossRef CAS .
- C. Mattsson, S.-I. Andersson, T. Belkheiri, L.-E. Åmand, L. Olausson, L. Vamling and H. Theliander, Biomass Bioenergy, 2016, 95, 364–377 CrossRef CAS .
- L. Monsigny, E. Feghali, J. C. Berthet and T. Cantat, Green Chem., 2018, 20, 1981–1986 RSC .
- M. D. Kärkäs, B. S. Matsuura, T. M. Monos, G. Magallanes and C. R. J. Stephenson, Org. Biomol. Chem., 2016, 14, 1853–1914 RSC .
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed .
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Chinmoy K.
Behera
,
Chinmoy K.
Behera