DOI:
10.1039/D4DT03509G
(Paper)
Dalton Trans., 2025,
54, 4049-4060
Immobilization of palladium(II) acetate on a polymer-anchored Schiff base as a new heterogeneous catalyst for three-component one-Pot Mizoroki–Heck coupling via an in situ witting reaction†
Received
21st December 2024
, Accepted 2nd February 2025
First published on 10th February 2025
Abstract
In this work, 4-((4-methylbenzyl)oxy)benzaldehyde resin-supported Pd(II)-Schiff base PS@Pd(OAc)2 was synthesized. FT-IR, XPS, TGA, ICP-MS, and powder XRD established the structure of PS@Pd(OAc)2. The morphology and distribution of elements on PS@Pd(OAc)2 were determined using SEM, TEM, and elemental mapping analysis. The heterogenized PS@Pd(OAc)2 catalyst was found to be efficient in promoting three-component Mizoroki–Heck coupling via an in situ Witting reaction, where (E)-1,2-diphenylethenes were efficiently synthesized from the Witting salt. In this one-pot reaction, this catalyst exhibits a distinct synergistic effect. The significant mass-transfer limitation results from a nonsignificant distribution of active sites amalgamated with the crumpling of catalysts, which facilitates the smooth, easy movement of the reactants and products toward the well-spaced active catalytic sites on the catalyst's surface. These characteristics increase the catalytic activity of PS@Pd(OAc)2. Moreover, the catalyst was found to be quite robust for this reaction with very little metal leaching; thus, it can be efficiently recycled. Hence, multiple uses were established, and its reusability was proven in this important reaction.
1. Introduction
The Heck reaction is a palladium-catalyzed cross-coupling between aryl halides and olefins, emerging as a crucial method for the formation of carbon–carbon bonds in organic synthesis. Since its independent discovery by Mizoroki and Heck, this transformation has been extensively applied across fields such as pharmaceuticals, agrochemicals, and materials science.1,2 Palladium salts, such as Pd(OAc)2 and PdCl2, have played a crucial role as catalysts in this reaction, offering a cost-effective, efficient, and versatile approach to accessing diverse molecular architectures.3 The Heck reaction catalytic cycle includes several key steps, namely oxidative addition of the aryl halide to the palladium(0) species, migratory insertion of the alkene, β-hydride elimination to give the coupled product, and regeneration of the active palladium species.4,5 Palladium salts serve as precursors to the active catalytic species, which can be generated in situ under suitable conditions, eliminating the need for complex pre-synthesized catalysts. This simplicity makes palladium salts particularly attractive for large-scale industrial applications and academic research alike.6,7 Among the most commonly employed palladium salts, Pd(OAc)2 is notable for its solubility and ease of handling, often enabling ligand-free reaction conditions. Such ligand-free systems align with green chemistry principles by reducing waste and simplifying purification processes. Similarly, PdCl2 has demonstrated robust catalytic performance, particularly in aqueous and solvent-free conditions, addressing sustainability concerns while maintaining high efficiency.8–10 Palladium salts are useful not only for simple substrates but also for complex substrates, such as sterically hindered olefins and multifunctionalized aryl halides. For example, regio- and stereoselective Heck reactions have been accomplished with Pd(OAc)2 to access structurally diverse molecules with excellent control over product configuration. All these developments have expanded the reaction's applicability to previously challenging synthetic targets, furthering the versatility of palladium salts as catalysts.11,12 The Wittig reaction, introduced by Georg Wittig in 1954, is a pivotal method for the synthesis of alkenes through the reaction of a phosphonium ylide with a carbonyl compound. This transformation produces an alkene along with triphenylphosphine oxide as a byproduct and has become an indispensable tool in organic synthesis due to its versatility and high selectivity. Its ability to form carbon–carbon double bonds under mild reaction conditions has made it widely applicable in the synthesis of natural products, pharmaceuticals, and advanced materials.13,14 A key strength of the Wittig reaction lies in its stereoselectivity, which allows for the formation of E- or Z-alkenes, depending on the ylide and reaction conditions. Stabilized ylides, containing electron-withdrawing groups such as esters or ketones, generally favor the formation of E-alkenes. Mechanistically, the reaction proceeds through the formation of a betaine intermediate, followed by cyclization to yield an oxaphosphetane. The collapse of this oxaphosphetane generates the desired alkene and triphenylphosphine oxide, driven by the formation of a strong P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond.15–17 Variations, such as the Schlosser modification, enable precise control of reaction conditions to enhance product stereochemistry and expand the reaction. These variations have been extensively applied in the synthesis of structurally complex molecules and high-value alkenes.18,19 Recent advancements have led to the development of a strategy that integrates the Heck reaction with an in situ Wittig reaction. This approach involves the direct generation of the alkene from a phosphonium ylide and a carbonyl compound, enabling the two key reactions to occur in a single reaction vessel. This one-pot methodology not only streamlines the synthetic process but also enhances atom economy and minimizes the need for isolating intermediate species.20–23 The in situ generation of the alkene via the Wittig reaction allows for immediate coupling with aryl or vinyl halides in the presence of a palladium catalyst.24 This tandem strategy provides a more efficient and selective route for synthesizing conjugated systems, styrenes, and functionalized dienes, all under mild reaction conditions. By eliminating the need for isolation and purification of the intermediate alkene, this approach significantly reduces the overall reaction time and increases product yield, offering an attractive alternative to traditional methods.25 A significant advancement in this field is the application of heterogeneous polymer-supported Pd(II) catalysts. These catalysts offer several advantages over traditional soluble Pd salts, including improved catalyst stability, recyclability, and ease of separation from the reaction mixture. The use of polymer-supported Pd(II) catalysts in the Heck reaction, coupled with an in situ Wittig reaction, significantly reduces catalyst leaching, a common issue when using Pd salts, thereby providing a more sustainable and efficient pathway for C–C bond formation.26 The combination of polymer-supported Pd(II) catalysts with an in situ Wittig reaction facilitates a one-pot, streamlined process for the synthesis of complex conjugated systems, styrenes, and functionalized dienes. These catalysts exhibit high stability under reaction conditions and enable straightforward catalyst recovery, presenting an attractive alternative to Pd salts, which typically require the use of excess palladium to achieve satisfactory yields and may suffer from catalyst deactivation during the reaction.27–30 By enhancing the stability and minimizing leaching of the palladium species, polymer-supported catalysts significantly improve reaction efficiency and product yields while reducing environmental impact.31 Moreover, the polymer support provides enhanced control over reaction conditions, including solvent polarity, temperature, and catalyst loading, which can be optimized to improve both the Wittig and Heck reactions in the tandem process. This precise control contributes to higher stereoselectivity and regioselectivity in the formation of alkenes. In contrast, Pd salts often exhibit inconsistent selectivity due to catalyst loss or deactivation, particularly when less reactive substrates are employed.29,30 The integration of polymer-supported Pd(II) catalysts with an in situ Wittig–Heck reaction has led to broader substrate scope, higher yields, and enhanced selectivity, establishing this methodology as a promising approach for the efficient and scalable synthesis of a wide range of alkenes and conjugated systems.
O bond.15–17 Variations, such as the Schlosser modification, enable precise control of reaction conditions to enhance product stereochemistry and expand the reaction. These variations have been extensively applied in the synthesis of structurally complex molecules and high-value alkenes.18,19 Recent advancements have led to the development of a strategy that integrates the Heck reaction with an in situ Wittig reaction. This approach involves the direct generation of the alkene from a phosphonium ylide and a carbonyl compound, enabling the two key reactions to occur in a single reaction vessel. This one-pot methodology not only streamlines the synthetic process but also enhances atom economy and minimizes the need for isolating intermediate species.20–23 The in situ generation of the alkene via the Wittig reaction allows for immediate coupling with aryl or vinyl halides in the presence of a palladium catalyst.24 This tandem strategy provides a more efficient and selective route for synthesizing conjugated systems, styrenes, and functionalized dienes, all under mild reaction conditions. By eliminating the need for isolation and purification of the intermediate alkene, this approach significantly reduces the overall reaction time and increases product yield, offering an attractive alternative to traditional methods.25 A significant advancement in this field is the application of heterogeneous polymer-supported Pd(II) catalysts. These catalysts offer several advantages over traditional soluble Pd salts, including improved catalyst stability, recyclability, and ease of separation from the reaction mixture. The use of polymer-supported Pd(II) catalysts in the Heck reaction, coupled with an in situ Wittig reaction, significantly reduces catalyst leaching, a common issue when using Pd salts, thereby providing a more sustainable and efficient pathway for C–C bond formation.26 The combination of polymer-supported Pd(II) catalysts with an in situ Wittig reaction facilitates a one-pot, streamlined process for the synthesis of complex conjugated systems, styrenes, and functionalized dienes. These catalysts exhibit high stability under reaction conditions and enable straightforward catalyst recovery, presenting an attractive alternative to Pd salts, which typically require the use of excess palladium to achieve satisfactory yields and may suffer from catalyst deactivation during the reaction.27–30 By enhancing the stability and minimizing leaching of the palladium species, polymer-supported catalysts significantly improve reaction efficiency and product yields while reducing environmental impact.31 Moreover, the polymer support provides enhanced control over reaction conditions, including solvent polarity, temperature, and catalyst loading, which can be optimized to improve both the Wittig and Heck reactions in the tandem process. This precise control contributes to higher stereoselectivity and regioselectivity in the formation of alkenes. In contrast, Pd salts often exhibit inconsistent selectivity due to catalyst loss or deactivation, particularly when less reactive substrates are employed.29,30 The integration of polymer-supported Pd(II) catalysts with an in situ Wittig–Heck reaction has led to broader substrate scope, higher yields, and enhanced selectivity, establishing this methodology as a promising approach for the efficient and scalable synthesis of a wide range of alkenes and conjugated systems.
2. Results and discussion
2.1. Synthesis of PS-H1L
A 100 mL oven-dried round-bottom flask was charged with 2.0 g of 4-benzyloxybenzaldehyde polystyrene resin beads in 20 mL of absolute alcohol. The mixture was allowed to swell at room temperature for 1 hour. Then, 10 mL of 1,2-diaminocyclohexane was introduced, and 2–3 drops of glacial acetic acid were added. The reaction mixture was stirred at 85 °C for 24 h. After completion of the reaction, solid product was isolated by filtration and sequentially washed with MeOH, THF, DCM, and CH3CN to remove unreacted 1,2-diaminocyclohexane and other impurities. The material was dried 2–3 times under vacuum to afford PS-H1L as a light-yellow powder (Scheme 1).
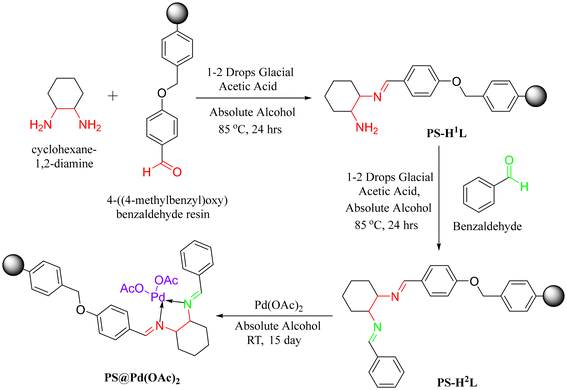 |
| | Scheme 1 Synthesis of PS-H1L, PS-H2L, and PS@Pd(OAc)2. | |
2.2. Synthesis of PS-H2L
A 100 mL oven-dried round-bottom flask was charged with 2.0 g of PS-H1L in 20 mL of absolute alcohol. Then, 10 mL of benzaldehyde and 2–3 drops of glacial acetic acid were added. The reaction mixture was stirred at 85 °C for 24 h. After completion of the reaction, solid product PS-H2L was isolated by filtration and washed sequentially with MeOH, THF, DCM, and CH3CN to remove unreacted benzaldehyde and other impurities. The material was dried under the vacuum 2–3 times to afford PS-H2L as a yellow powder.
2.3. Synthesis of PS@Pd(OAc)2
A 100 mL oven-dried round-bottom flask was charged with 2.0 g of PS-H2L in 20 mL of THF. Then, 500 mg of Pd(OAc)2 was added, and the reaction mixture was stirred under a nitrogen atmosphere at room temperature for 15 days. After completion of the reaction, the solid product, PS@Pd(OAc)2, was isolated by filtration and washed sequentially with MeOH, DCM, and absolute alcohol to remove excess Pd(OAc)2 and other impurities. The material was dried under vacuum 5–6 times to afford PS@Pd(OAc)2 as a dark grey solid.
3. Results and discussion
3.1. Synthesis and characterization of PS@Pd(OAc)2
The IR spectra presented in Fig. 1 demonstrate the chemical changes across three samples: PS-H1L, PS-H2L, and PS@Pd(OAc)2. Each spectrum reveals key functional group vibrations, indicating structural modifications as the polymer supports are modified and metal-loaded. Fig. 1 shows the N–H symmetric and asymmetric stretching vibrations of 1,2-diaminocyclohexane at 3352 cm−1 and 3290 cm−1, respectively, and the N–H bending mode at 1527 cm−1. This indicates the presence of the second amine group remains free in the polymer structure PS-H1L. A strong peak at 1624 cm−1 corresponds to the CN stretching vibration, which is present in both PS-H1L and PS-H2L. This peak indicates the formation of imine bonds, typically resulting from condensation between aldehydes and amines, confirming the structural backbone of the polymer. The peak remains sharp in the metal-loaded spectrum PS@Pd(OAc)2, suggesting that imine groups are retained after palladium incorporation. The CC aromatic stretching is observed at 1485 cm−1, confirming the aromatic nature of the polymer backbone. The C–O–C stretching vibration at 1149 cm−1 indicates the presence of ether linkages that are already incorporated in the structures of PS-H1L, PS-H2L, and PS@Pd(OAc)2. In the PS@Pd(OAc)2 spectrum, the appearance of a new peak at 507 cm−1 corresponds to the Pd–N coordination bond. This peak confirms that palladium has successfully coordinated with nitrogen atoms from the polymer ligand, which is essential for catalytic activity. Furthermore, the C–C stretching vibration is observed at 820 cm−1, indicating alkyl or aromatic groups present within the polymer matrix. Overall, the IR spectra reveal the successful synthesis and chemical transformations within the polymer system. The retention of imine groups (CN) and the appearance of the Pd–N coordination band confirm the successful loading of palladium onto the polymer support, making it a potential candidate for catalytic applications.
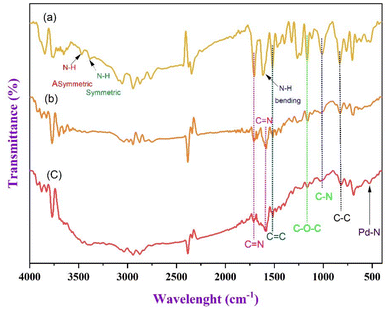 |
| | Fig. 1 FT-IR spectra of (a) PS-H1L, (b) PS-H2L, and (c) PS@Pd(OAc)2. | |
Fig. 2 shows the SEM images of PS-H1L (Fig. 2A), PS-H2L (Fig. 2B), and PS@Pd(OAc)2 (Fig. 2C) exhibits noticeable differences in surface morphology and porosity. In images A and B, both PS-H1L and PS-H2L exhibit smooth, spherical particles with well-defined shapes and minimal surface irregularities. Their morphology suggests low porosity, as the surfaces appear compact and non-porous, with no visible cracks or openings. However, PS@Pd(OAc)2 (Fig. 2C) presents a distinct contrast, showing particles with surface disruptions, including cracks and fractures. This indicates a significant increase in porosity, likely due to the deposition of Pd(OAc)2, which alters the material's surface texture and increases surface area.32–34 Thus, while PS-H1L and PS-H2L maintain smooth, non-porous surfaces, PS@Pd(OAc)2 demonstrates a more irregular and porous structure, resulting from the modifications during Pd(OAc)2 deposition.
 |
| | Fig. 2 FESEM images of (A) PS-H1L, (B) PS-H2L, and (C) PS@Pd(OAc)2. | |
Fig. 3 depicts HRTEM images of PS-H1L (Fig. 3A), PS-H2L (Fig. 3B), and PS@Pd(OAc)2 (Fig. 3C), which can be compared based on surface characteristics, morphology, and porosity. PS-H1L (Fig. 3A) shows a relatively smooth surface with well-defined, dispersed spherical particles, indicating higher porosity and less aggregation. In contrast, PS-H2L (Fig. 3B) displays a rougher surface with more clustered and overlapping spherical particles, suggesting lower porosity due to increased aggregation and particle fusion. Conversely, PS@Pd(OAc)2 (Fig. 3C) presents a denser and more irregular structure with darker regions, likely due to the deposition of Pd(OAc)2, resulting in a less porous, compact morphology. This palladium incorporation leads to a denser surface with fewer visible spaces between particles compared to the other two samples.35–37 Overall, PS-H1L (Fig. 3A) has the most porous and uniform structure, PS-H2L (Fig. 3B) is more aggregated with reduced porosity, and PS@Pd(OAc)2 (Fig. 3C) is the densest with minimal porosity due to the palladium complex coating.
 |
| | Fig. 3 HRTEM images of (a) PS-H1L, (b) PS-H2L, and (c) PS@Pd(OAc)2. | |
Fig. 4 shows the powder X-ray diffraction (PXRD) patterns for PS-H2L and PS@Pd(OAc)2. The (a) PS-H2L sample (red curve) differs from the (b) PS@Pd(OAc)2 sample (black curve). The red curve shows a broad peak between 15° and 30° (2θ), indicating the polymer is mostly amorphous. In contrast, the black curve for PS@Pd(OAc)2 shows sharp peaks at 40°, 46°, and 68°, corresponding to the Pd(111), Pd(200), and Pd(220) planes, which indicates the presence of palladium in a crystalline form.38,39 The inclusion of Pd(II) ions into the polymer beads leads to partial crystallinity, showing a strong interaction between the metal and the Schiff base. The moderate intensity of the palladium peaks suggests that the palladium is finely distributed, which is beneficial for catalytic applications.
 |
| | Fig. 4 Powder XRD analysis of (a) PS-H2L and (b) PS@Pd(OAc)2. | |
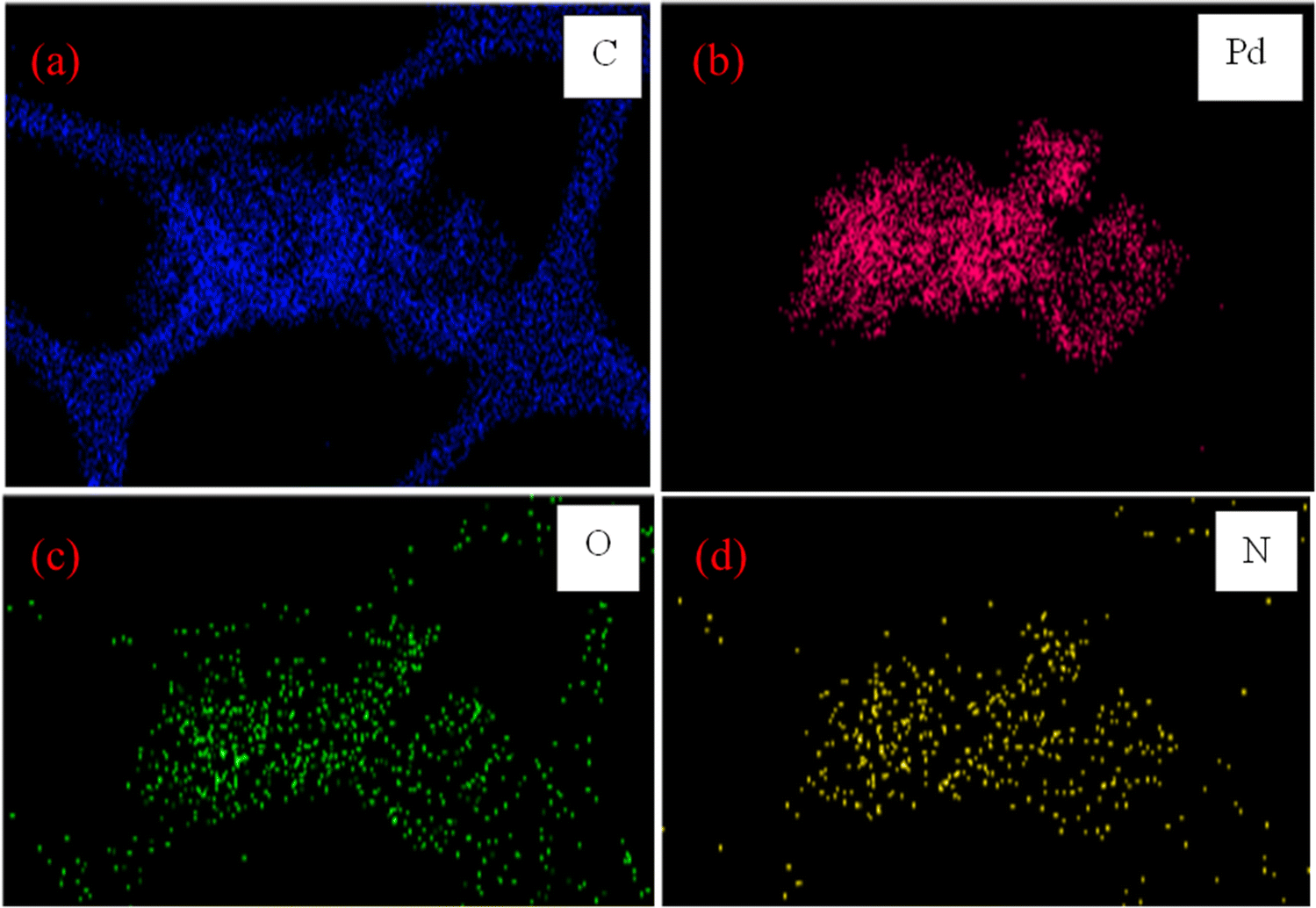
The elemental mapping analysis in Fig. 5 shows the distribution of four elements in the PS@Pd(OAc)2 catalyst using different colors. The blue map (C Kα1_2) indicates a high carbon content, showing that the polystyrene beads are the main supporting structure. The red map (Pd Kα1) shows the localization of palladium (Pd), highlighting the regions where Pd(II) species are immobilized. The green map (O Kα1) indicates the presence of oxygen, likely from functional groups that help stabilize Pd. The yellow map (N Kα1_2) shows nitrogen-containing functional groups, which also contribute to Pd stabilization. The colors reveal that Pd is not uniformly distributed but is confined to specific active sites in the polymer. In addition, ICP-MS analysis of the catalyst indicates that the amount of Pd was found to be 8.7%.
 |
| | Fig. 5 Elemental mapping analysis images of PS@Pd(OAc)2 for key elements (a) C, (b) Pd, (c) O, and (d) N. | |
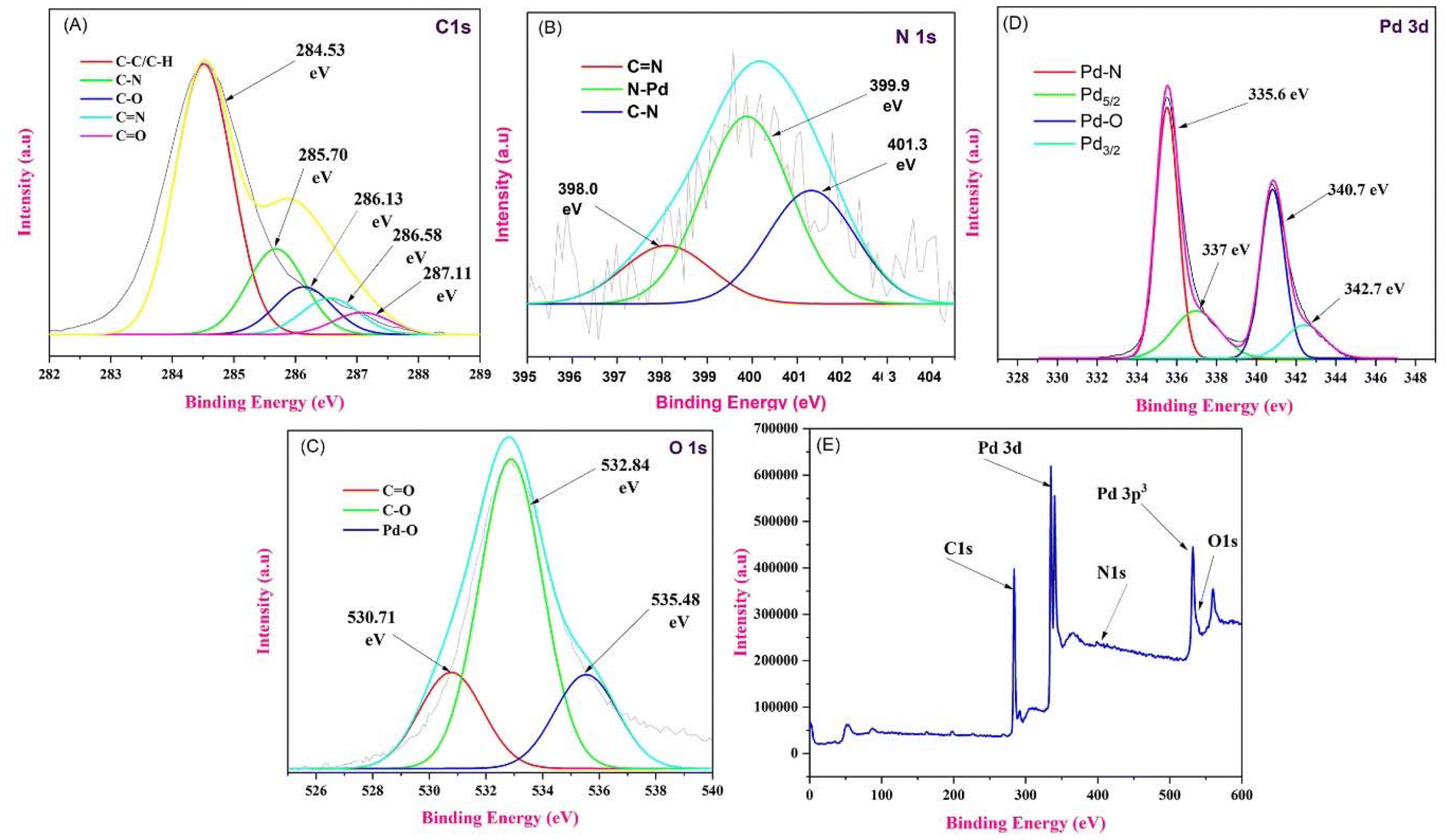
X-ray photoelectron spectroscopy (XPS) measurements were performed to investigate the existence of C 1s, O 1s, N 1s, and Pd 3d states in the PS@Pd(OAc)2 catalyst. In Fig. 6, the XPS spectra of Pd 3d are shown, revealing two main doublets identified through Gaussian fitting, indicating the presence of two distinct Pd states. Compared to the Pd0 metal state with a binding energy (Eb) of ∼335.2 eV, the low-energy doublet observed here (Eb(Pd 3d5/2) ∼335.7 eV and Pd 3d3/2 ∼341 eV) is shifted to higher binding energies. This shift can be attributed to oxidized Pd clusters or single atoms. Another Pd 3d doublet, with binding energies of ∼337 eV for Pd 3d5/2 and ∼342.7 eV for Pd 3d3/2, corresponds to Pd2+ ions incorporated into the catalyst structure.40 These XPS results confirm that the primary state of Pd in the catalyst is Pd2+, with contributions from both oxidized single atoms and clusters. In the C 1s spectrum of PS@Pd(OAc)2, different peaks correspond to various carbon bonding states. The red curve at 284.53 eV represents C–C/C–H bonds, the green curve at 285.70 eV corresponds to C–N bonds, the blue curve at 286.13 eV is attributed to C–O bonds, the cyan curve at 286.58 eV represents CN bonds, and the magenta curve at 287.11 eV indicates CO bonds. In the N 1s spectrum, the red curve, with a peak at 398.0 eV, is assigned to CN bonds, indicating nitrogen in a double bond with carbon. The green curve, peaking at 399.9 eV, corresponds to N–Pd interactions, suggesting the coordination of nitrogen with palladium. Finally, the blue curve, centered at 401.3 eV, is associated with C–N bonds, indicative of nitrogen in a single bond with carbon. The O 1s spectrum was deconvoluted into distinct peaks, each representing different chemical states of oxygen on the material's surface. The red curve, located at a binding energy of 530.71 eV, corresponds to oxygen in a carbonyl group (CO), indicating the presence of such functional groups on the surface. The green curve, with a binding energy of 532.84 eV, represents the C–O bond, typically associated with hydroxyl or ether groups. Finally, the blue curve, at 535.48 eV, is attributed to Pd–O bonds, suggesting an interaction between palladium and oxygen on the surface. This analysis provides insight into the different oxygen species present, highlighting the interaction between oxygen and palladium, along with the presence of carbon-containing functional groups.41–43
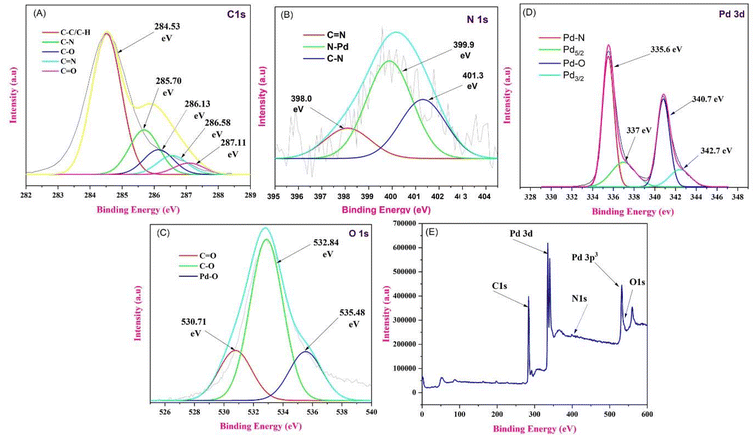 |
| | Fig. 6 (A) C 1s scan XPS spectrum of PS@Pd(OAc)2. (B) N 1s scan XPS spectrum of PS@Pd(OAc)2. (C) O 1s scan XPS spectrum of PS@Pd(OAc)2. (D) Pd 3d scan XPS spectrum of PS@Pd(OAc)2 and (E) full-scan XPS spectrum of PS@Pd(OAc)2. | |
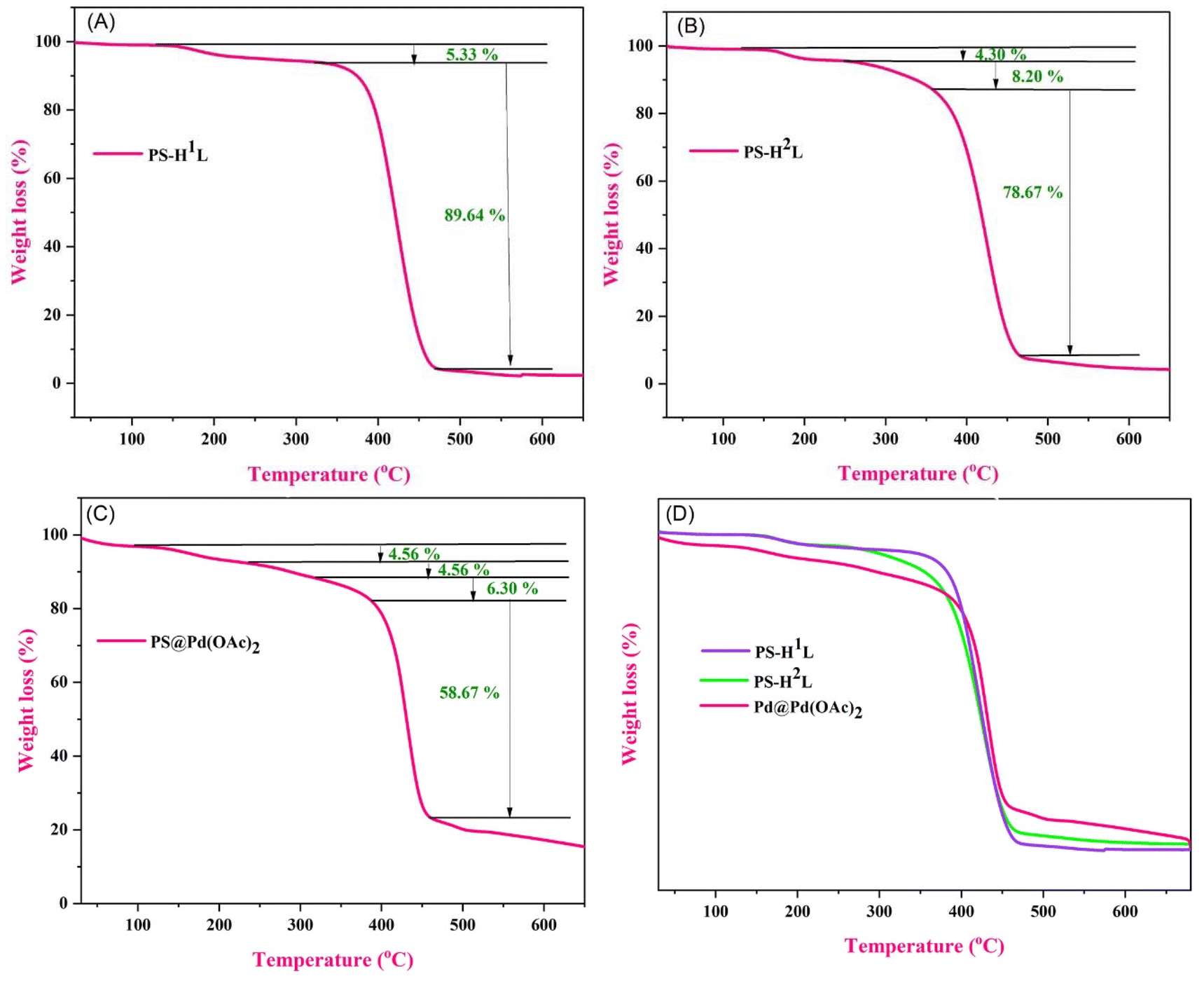
In the thermogravimetric analysis (TGA) curves, the initial small mass losses observed in all three figures are likely due to the evaporation of moisture or other volatile components, a common occurrence in TGA analysis. In PS-H1L (Fig. 7A), the first 5.33% weight loss can be attributed to moisture, followed by a major 89.64% decomposition at higher temperatures, indicating a relatively simple single-stage thermal degradation. In PS-H2L (Fig. 7B), the initial 4.30% loss is also likely from moisture, with further weight losses of 8.20% and 78.67% indicating a more complex multi-stage decomposition process. For PS@Pd(OAc)2 (Fig. 7C), the first two small losses of 4.56% each could be moisture-related, with subsequent 6.30% and 58.67% losses showing a more gradual decomposition pattern, likely influenced by the presence of Pd(OAc)2, which enhances its thermal stability. Overall, PS-H1L has the simplest thermal profile, PS-H2L exhibits multiple stages of decomposition, and PS@Pd(OAc)2 shows smaller, gradual losses, indicating higher stability due to the palladium acetate content.44
 |
| | Fig. 7 TGA spectra of (A) PS-H1L, (B) PS-H2L, and (C) PS@Pd(OAc)2. (D) Comparison spectra between PS-H1L, PS-H2L, and PS@Pd(OAc)2. | |
In Fig. 8, the derivative thermogravimetric analysis (DTGA) curves for PS-H2L, PS-H1L, and PS@Pd(OAc)2 exhibit similar thermal behavior, with major weight loss events occurring between 420 °C and 435 °C, indicating decomposition in this temperature range. The PS-H2L sample (Fig. 8B) exhibits decomposition at 425.68 °C, maintaining stability before and after this temperature, while PS@Pd(OAc)2 (Fig. 8C) decomposes slightly later at 433.15 °C. The increased thermal stability in the PS@Pd(OAc)2 sample is likely due to the presence of palladium acetate, which shifts the decomposition to a higher temperature compared to the others. Conversely, PS-H1L (Fig. 8A) decomposes earlier at 423 °C, making it the least thermally stable among the three. When comparing the materials, PS@Pd(OAc)2 stands out as the most stable due to its enhanced decomposition temperature, followed by PS-H2L, and finally PS-H1L, which decomposes at the lowest temperature. This comparison indicates that while all three materials are stable up to around 420 °C, the addition of palladium acetate notably improves the thermal stability, and PS-H1L is the least resistant to thermal degradation.45–48
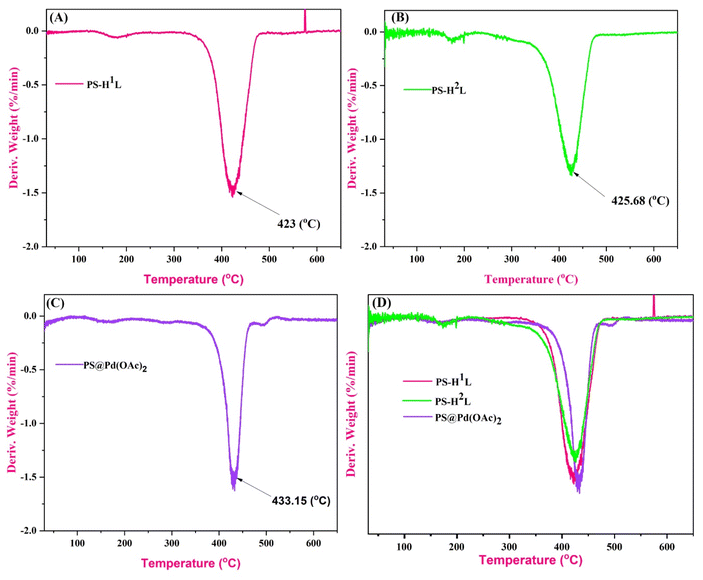 |
| | Fig. 8 DTGA spectra of (A) PS-H1L, (B) PS-H2L, and (C) PS@Pd(OAc)2. (D) Comparison spectra between PS-H1L, PS-H2L, and PS@Pd(OAc)2. | |
4. Experimental section
4.1. General
All chemicals were acquired from Merck (Sigma-Aldrich), TCI, AVRA, and SRL Chemical Company. 1H NMR and 13C NMR spectra were recorded on Bruker Avance neo 400 spectrometers. Chemical shifts are reported in ppm relative to TMS as an internal standard. The reactions were monitored using TLC on silica gel 60 F254 plates. IR spectra were recorded on a Bruker spectrometer as KBr pellets. The morphology of the catalyst was studied by field-emission scanning electron microscopy (FESEM, Model-JSM7610F PLUS), and TEM images were taken using a TEM microscope (JEOL JEM 2100 PLUS). The elemental composition was determined by elemental mapping using TEM (JEOL JEM 2100 PLUS). TG-DTA measurements were performed on an SII EXTRAR600TG-DTA instrument.
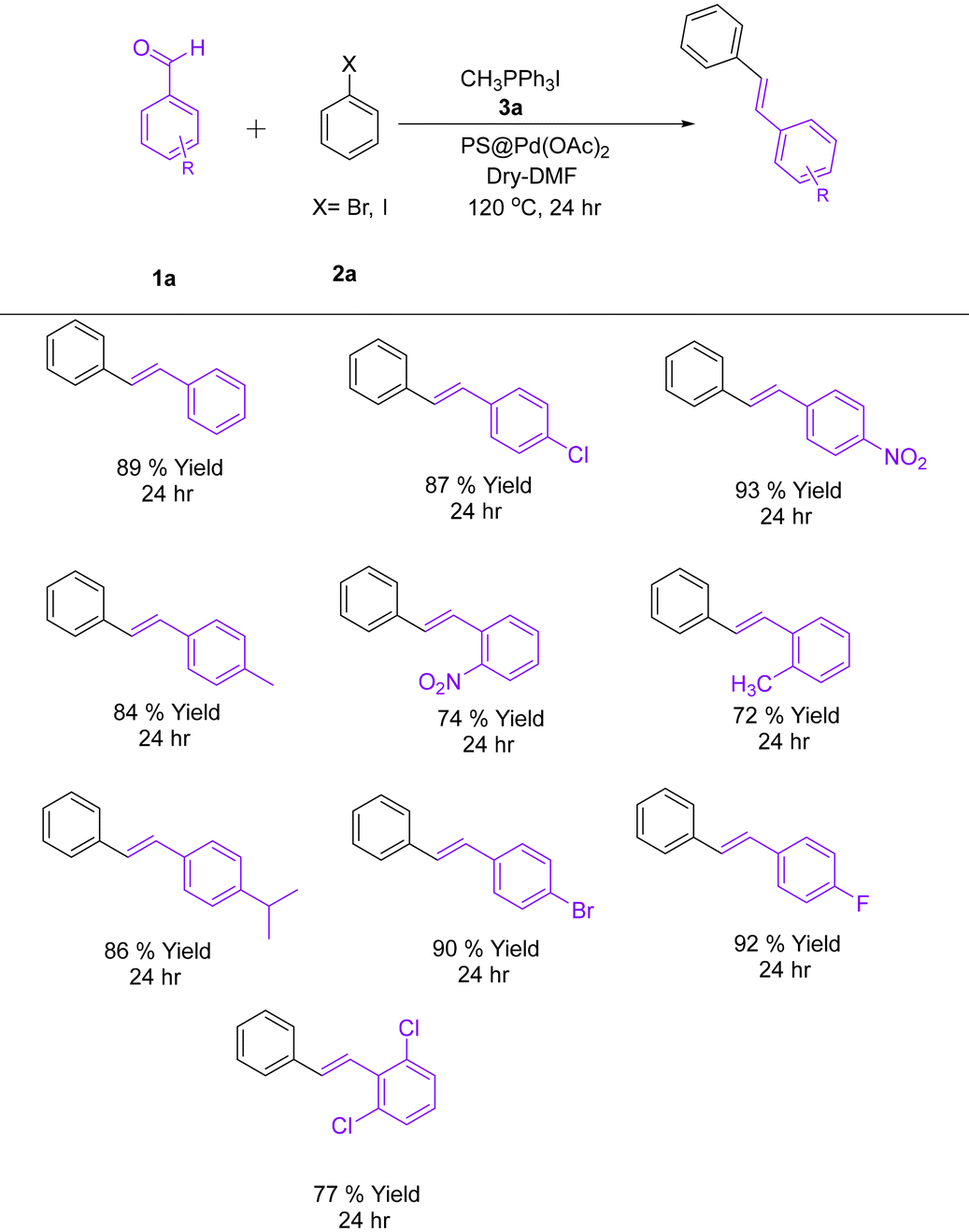
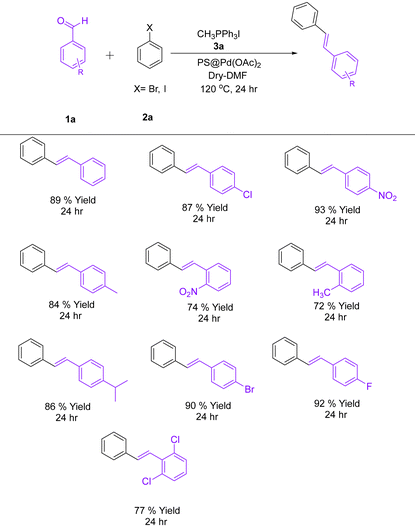
4.2. General procedure for the synthesis of (E)-1,2-diphenylethene derivatives
A 25 mL oven-dried round-bottom flask was used that was equipped with a magnetic stir bar. To it, benzaldehyde (2.0 mmol, 1.0 equiv.), Wittig salt (3.0 mmol, 1.5 equiv.), iodobenzene (2.4 mmol, 1.2 equiv.), and PS@Pd(OAc)2 (2.0 mol%) were added. To this, potassium carbonate (6.0 mmol, 3.0 equiv.) was added as the base. The reaction mixture was dissolved in 5 mL of anhydrous DMF and stirred at 120 °C under a nitrogen atmosphere for 24 h. Upon completion, as monitored using thin-layer chromatography, the reaction mixture was cooled to room temperature. The mixture was diluted with ethyl acetate and filtered to remove the polymer-supported catalyst. The filtrate was washed sequentially with brine water. The organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo (Table 1). The crude product was purified using column chromatography on silica gel to give the desired product (Scheme 2).
 |
| | Scheme 2 Mechanism of Mizoroki–Heck coupling via an in situ Witting reaction. | |
Table 1 Optimization of the reaction conditions for the synthesis of (E)-1,2-diphenylethene
|

|
| Entry |
Catalyst |
Solvent |
Base |
Temperature (°C) |
Time |
Yielda (%) |
| Reaction conditions: 1a (2.0 mmol, 1.0 equiv.), 2a (2.4 mmol, 1.2 equiv.), 3a (3.0 mmol, 1.5 equiv.), PS@Pd(OAc)2 in mol%, base (6.0 mmol, 3.0 equiv.), solvent (5 mL), 24 h. Isolated yields of 4a. All reactions were performed under a nitrogen atmosphere. |
| 1 |
0.5 mol% |
DMF |
K2CO3 |
100 |
24 h |
35 |
| 2 |
1.0 mol% |
DMF |
K2CO3 |
100 |
24 h |
59 |
| 3 |
1.5 mol% |
DMF |
K2CO3 |
100 |
24 h |
67 |
| 4 |
2.0 mol% |
DMF |
K2CO3 |
100 |
24 h |
68 |
| 5 |
2.5 mol% |
DMF |
K2CO3 |
100 |
24 h |
68 |
| 6 |
2.0 mol% |
DMF |
K2CO3 |
100 |
06 h |
25 |
| 7 |
2.0 mol% |
DMF |
K2CO3 |
100 |
12 h |
54 |
| 8 |
2.0 mol% |
DMF |
K2CO3 |
100 |
18 h |
72 |
| 9 |
2.0 mol% |
DMF |
K2CO3 |
100 |
24 h |
79 |
| 10 |
2.0 mol% |
DMF |
K2CO3 |
100 |
30 h |
79 |
| 11 |
2.0 mol% |
DMF |
K2CO3 |
50 |
24 h |
Trace |
| 12 |
2.0 mol% |
DMF |
K2CO3 |
80 |
24 h |
54 |
| 13 |
2.0 mol% |
DMF |
K2CO3 |
120 |
24 h |
81 |
| 14 |
2.0 mol% |
DMF |
K2CO3 |
140 |
24 h |
81 |
| 15 |
2.0 mol% |
1,4-Dioxane |
K2CO3 |
120 |
24 h |
47 |
| 16 |
2.0 mol% |
K2CO3 |
120 |
24 h |
89 |
| 17 |
2.0 mol% |
Dry-DMF |
K2CO3 |
120 |
24 h |
52 |
| 18 |
2.0 mol% |
Toluene |
K2CO3 |
120 |
24 h |
62 |
| 19 |
2.0 mol% |
DMSO |
K2CO3 |
120 |
24 h |
85 |
| 20 |
2.0 mol% |
DMA |
NaOAc |
120 |
24 h |
77 |
| 21 |
2.0 mol% |
Dry-DMF |
Na2CO3 |
120 |
24 h |
84 |
The Heck coupling via an in situ Wittig reaction proceeds through a series of well-defined mechanistic steps. The reaction begins with the oxidative addition of an alkyl halide (species A) to the palladium(0) catalyst. In this step, the palladium(0) species reacts with the carbon–halogen bond of the alkyl halide, resulting in the formation of a palladium(II)–alkyl complex (species B). This step is critical as it activates the halide for further transformations and increases the oxidation state of palladium from 0 to +2. Concurrently, a phosphonium ylide, derived from the deprotonation of a phosphonium salt, reacts with an aldehyde or ketone (species C) present in the reaction system. This Wittig reaction forms an alkene intermediate (species D) along with triphenylphosphine oxide as a byproduct. The formation of the alkene intermediate in situ is an essential feature of the reaction, as it directly participates in the subsequent Heck coupling step. The alkene intermediate (species D) undergoes coordination to the palladium(II)–alkyl complex, forming a π-complex (species E). This coordination step is followed by migratory insertion, where the double bond of the alkene inserts into the palladium–carbon bond, generating a new palladium–alkyl intermediate (species F). This step effectively links the alkene to the palladium center, allowing for the formation of the carbon–carbon bond. In the final stage of the reaction, β-hydride elimination occurs, leading to the formation of the coupled product (species G) and regenerating the palladium(0) catalyst. The regenerated catalyst re-enters the catalytic cycle, ensuring its continuous activity. Throughout the process, a base is employed to neutralize the hydroiodic acid (HI) generated during oxidative addition and to assist in maintaining the palladium catalyst in its active form. This stepwise combination of the Wittig reaction and the Heck coupling reaction in a single pot enhances the overall efficiency of the synthesis, reduces the need for intermediate isolation, and promotes atom economy. Each step in the mechanism is carefully designed to ensure smooth progression and high yields of the desired product (Table 2).
Table 2 Synthesis of (E)-1,2-diphenylethene derivatives
| Reaction conditions: 1a (2.0 mmol, 1.0 equiv.), 2a (2.4 mmol, 1.2 equiv.), 3a (3.0 mmol, 1.5 equiv.), PS@Pd(OAc)2 containing Pd 2.0 mol%, K2CO3 (6.0 mmol, 3.0 equiv.), dry-DMF (5 mL), temperature: 120 °C, 24 h. All reactions were performed under a nitrogen atmosphere. |
|
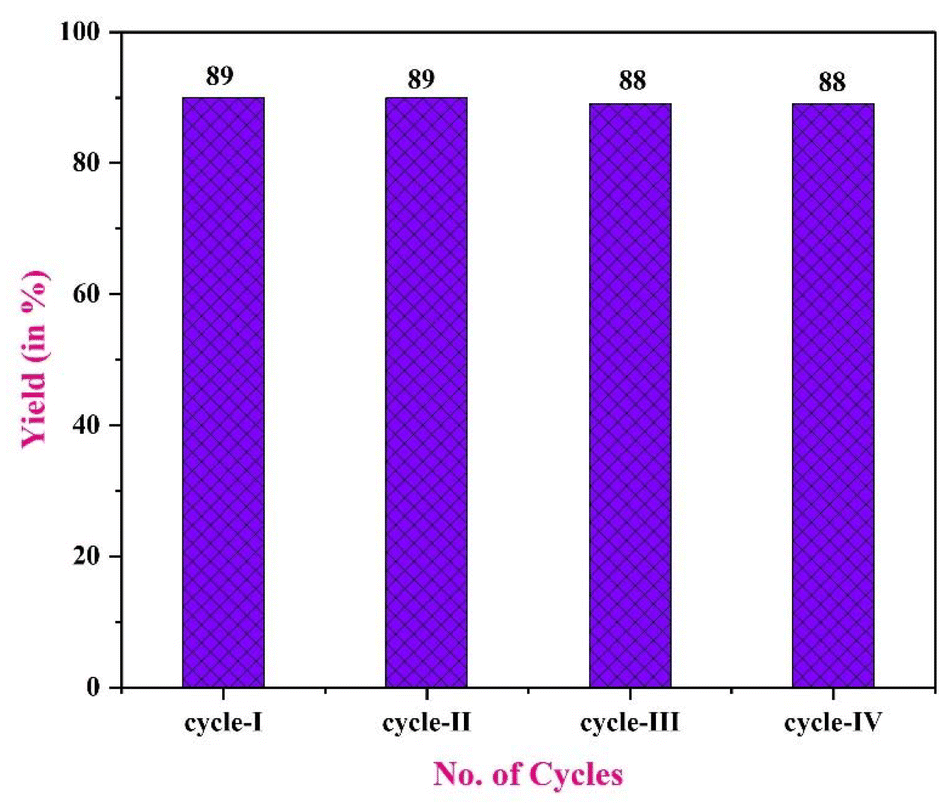
5. Recycling study of PS@Pd(OAc)2
The polymer-bound Pd(II) complex demonstrates excellent stability and recyclability, making it a promising catalyst for sustainable chemical transformations. This was shown through a series of experiments in which the catalyst was separated by filtration, washed, and reused across multiple reaction cycles. Even after four cycles, the catalyst maintained minimal loss in activity, as shown in Fig. 9, indicating the catalytic efficacy. This highlights its prolonged usability without significant performance decline. The strong immobilization of Pd(II) onto the polymer support effectively prevents leaching and preserves the integrity of the active sites, ensuring consistent catalytic performance over extended use. These findings confirm the robust nature of the catalyst, making it suitable for long-term applications where recyclability, recoverability, and sustained activity are essential. Therefore, the polymer-bound Pd(II) complex emerges as a sustainable and efficient catalytic system for continuous chemical processes.49–51
 |
| | Fig. 9 Recycling study of PS@Pd(OAc)2. | |
6. Heterogeneity test
The recyclability of the catalyst was examined through a systematic analysis of metal ion leaching. In Set-I, the reaction was allowed to proceed for 6 h; however, after this period, the catalyst was removed and the reaction continued for an additional 24 h, resulting in a low yield of 34%. This outcome suggests that the removal of the catalyst significantly impacted reaction efficiency. In Set-II, the catalyst was retained for the entire 24 h, achieving a much higher yield of 89%. These findings highlight the necessity of the catalyst's continuous presence for optimal performance. Additionally, after the 6 h reaction in Set-I, an ICP-MS analysis of the reaction mixture revealed no leaching of Pd, confirming that Pd(OAc)2 remained fully immobilized and did not dissolve into the solution. This underscores the stability of the catalyst and its critical role in maintaining efficiency. This study thus provides critical insights into catalytic behavior and reaction optimization (Table 3).
Table 3 Leaching study of the catalyst
| No |
Experiment |
Time (h) |
Conditions |
Yield (%) |
| 1 |
Set-I |
6 |
The catalyst was removed and the reaction continued for 24 hours |
34 |
| 2 |
Set-II |
24 |
The reaction continued with the catalyst for all 24 hours |
89 |
7. Conclusion
This study successfully demonstrates the efficacy of a heterogeneous polymer-supported Pd metal catalyst in facilitating the Mizoroki–Heck coupling reaction via an in situ Wittig reaction pathway. This novel approach offers significant advantages over traditional methodologies employing direct Pd metal. The polymer-supported catalyst exhibits remarkable stability under reaction conditions, enabling consistent catalytic performance over multiple cycles, and thereby highlighting its recyclability. Furthermore, the system achieves superior product yields with enhanced reaction efficiency. These findings underscore the potential of polymer-supported Pd catalysts as a sustainable and robust alternative in the development of high-performance catalytic systems.
Data availability
The characterization data that support the findings of this study, including SEM, TEM, elemental mapping, TGA, ICP-MS, IR, NMR, and HRMS, are available to the readers.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
The authors are thankful to the Department of Chemistry, the Maharaja Sayajirao University of Baroda, Vadodara, for providing necessary laboratory facilities and DST-FIST for providing NMR facilities. The authors also thank the funding agency Science and Engineering Research Board, Department of Science and Technology (award no: EEQ/2022/000473), and Research and Development Cell, the Maharaja Sayajirao University of Baroda (award no: RDC/Dir./2023-24/19/17), for financially supporting the corresponding author.
References
- F. Saleem, G. K. Rao, A. Kumar, S. Kumar, M. P. Singh and A. K. Singh, RSC Adv., 2014, 4, 56102–56111 RSC.
- G. Z. Wang, R. Shang, W. M. Cheng and Y. Fu, J. Am. Chem. Soc., 2017, 139, 18307–18312 CrossRef CAS PubMed.
- A. F. Littke and G. C. Fu, J. Am. Chem. Soc., 2001, 123, 6989–7000 CrossRef CAS PubMed.
- G. S. Lee, D. Kim and S. H. Hong, Nat. Commun., 2021, 12, 991 CrossRef CAS PubMed.
- T. Shirai, Y. Migitera, R. Nakajima and T. Kumamoto, J. Org. Chem., 2024, 89, 2787–2793 CrossRef CAS PubMed.
-
D. A. Watson, Legacy of Richard Heck, American Chemical Society, 2016, DOI:10.1021/acs.organomet.6b00261, preprint.
- A. Kamal, V. Srinivasulu, B. N. Seshadri, N. Markandeya, A. Alarifi and N. Shankaraiah, Green Chem., 2012, 14, 2513–2522 RSC.
- J. B. Liu, F. J. Chen, N. Liu and J. Hu, RSC Adv., 2015, 5, 45843–45846 RSC.
- A. K. Danodia, R. K. Saunthwal, M. Patel, R. K. Tiwari and A. K. Verma, Org. Biomol. Chem., 2016, 14, 6487–6496 RSC.
- K. N. Sharma, N. Satrawala, A. K. Srivastava, M. Ali and R. K. Joshi, Org. Biomol. Chem., 2019, 17, 8969–8976 RSC.
- J. Yu, Z. Hong, X. Yang, Y. Jiang, Z. Jiang and W. Su, Beilstein J. Org. Chem., 2018, 14, 786–795 CrossRef CAS PubMed.
- C. Wang, G. Xiao, T. Guo, Y. Ding, X. Wu and T. P. Loh, J. Am. Chem. Soc., 2018, 140, 9332–9336 CrossRef CAS PubMed.
- K. Zhang, L. Q. Lu and W. J. Xiao, Chem. Commun., 2019, 55, 8716–8721 RSC.
-
P. A. Byrne, Investigation of Reactions Involving Pentacoordinate Intermediates, 2012, pp. 1–56 Search PubMed.
- W. Yan, D. Wang, J. Feng, P. Li, D. Zhao and R. Wang, Org. Lett., 2012, 14, 2512–2515 CrossRef CAS PubMed.
-
M. M. Heravi, M. Ghanbarian, V. Zadsirjan and B. Alimadadi Jani, Advances in Kumada–Tamao–Corriu cross-coupling reaction: an update, Springer-Verlag Wien, 2019, DOI:10.1007/s00706-019-02465-9.
- P. Miao, R. Li, X. Lin, L. Rao and Z. Sun, Green Chem., 2021, 23, 1638–1641 RSC.
- Z. X. Deng, Y. Zheng, Z. Z. Xie, Y. H. Gao, J. A. Xiao, S. Q. Xie, H. Y. Xiang, X. Q. Chen and H. Yang, Org. Lett., 2020, 22, 488–492 CrossRef CAS PubMed.
- K. Ayub and R. Ludwig, RSC Adv., 2016, 6, 23448–23458 RSC.
- H. A. Patel, V. J. Bhanvadia, H. M. Mande, S. S. Zade and A. L. Patel, Org. Biomol. Chem., 2019, 17, 9467–9478 RSC.
- J. F. Bai, H. Sasagawa, T. Yurino, T. Kano and K. Maruoka, Chem. Commun., 2017, 53, 8203–8206 RSC.
- H. J. Xu, Y. Q. Zhao and X. F. Zhou, J. Org. Chem., 2011, 76, 8036–8041 CrossRef CAS PubMed.
- H. Wu, J. Yang, J. Šečkute and N. K. Devaraj, Angew. Chem., Int. Ed., 2014, 53, 5805–5809 CrossRef CAS PubMed.
- H. A. Patel, A. L. Patel and A. V. Bedekar, Appl. Organomet. Chem., 2015, 29, 1–6 CrossRef CAS.
- M. Winkler, L. Montero De Espinosa, C. Barner-Kowollik and M. A. R. Meier, Chem. Sci., 2012, 3, 2607–2615 RSC.
-
A. Koranne, S. Turakhia, V. K. Jha, S. Gupta, R. Ravi, A. Mishra, A. K. Aggarwal, C. K. Jha, N. Dheer and A. K. Jha, The Mizoroki–Heck reaction between in situ generated alkenes and aryl halides: cross-coupling route to substituted olefins, Royal Society of Chemistry, 2023, 10.1039/d3ra03533f.
- L. Yu, Y. Huang, Z. Wei, Y. Ding, C. Su and Q. Xu, J. Org. Chem., 2015, 80, 8677–8683 CrossRef CAS PubMed.
- L. Huang, Z. Wang and J. Tan, React. Chem. Eng., 2020, 5, 921–934 RSC.
- A. Ruiu, W. S. J. Li, M. Senila, C. Bouilhac, D. Foix, B. Bauer-Siebenlist, K. Seaudeau-Pirouley, T. Jänisch, S. Böringer and P. Lacroix-Desmazes, Molecules, 2023, 28, 6342 CrossRef CAS PubMed.
- A. M. Fiore, G. Romanazzi, C. Leonelli, P. Mastrorilli and M. M. Dell’anna, Catalysts, 2022, 12, 506 CrossRef CAS.
- L. C. Lee, J. He, J. Q. Yu and C. W. Jones, ACS Catal., 2016, 6, 5245–5250 CrossRef CAS.
- W. Mansour, M. Fettouhi, Q. Saleem and B. El Ali, Appl. Organomet. Chem., 2021, 35, e6195 CrossRef CAS.
- Q. Yang, Y. Zhang, W. Zeng, Z. C. Duan, X. Sang and D. Wang, Green Chem., 2019, 21, 5683–5690 RSC.
- I. C. Howard, C. Hammond and A. Buchard, Polym. Chem., 2019, 10, 5894–5904 RSC.
- R. Kurane, P. Bansode, S. Khanapure, R. Salunkhe and G. Rashinkar, Res. Chem. Intermed., 2016, 42, 7807–7821 CrossRef CAS.
- C. Zhang, Y. Wang, X. Zhang, R. Wang, L. Kou, R. Li and C. Fan, RSC Adv., 2020, 10, 37644–37656 RSC.
- J. J. Boruah and S. P. Das, RSC Adv., 2018, 8, 34491–34504 RSC.
- J. Mondal, R. Gomes, A. Modak and A. Bhaumik, Recyclable Catal., 2013, 1 DOI:10.2478/recat-2013-0001.
- B. Qi, L. Di, W. Xu and X. Zhang, J. Mater. Chem. A, 2014, 2, 11885–11890 RSC.
- X. Wang, J. Chen, J. Zeng, Q. Wang, Z. Li, R. Qin, C. Wu, Z. Xie and L. Zheng, Nanoscale, 2017, 9, 6643–6648 RSC.
- A. Brzęczek-Szafran, M. Gwóźdź, B. Gaida, M. Krzywiecki, M. Pawlyta, A. Blacha-Grzechnik, A. Kolanowska, A. Chrobok and D. Janas, Sci. Rep., 2024, 14, 11106 CrossRef PubMed.
-
H. Wei, X. Y. Yang, H. C. van der Mei and H. J. Busscher, X-Ray Photoelectron Spectroscopy on Microbial Cell Surfaces: A Forgotten Method for the Characterization of Microorganisms Encapsulated With Surface-Engineered Shells, Frontiers Media S.A., 2021, DOI:10.3389/fchem.2021.666159.
- B. Liu, J. Z. Li, X. F. Gong, Y. L. Zhang, Q. Y. Zhou, J. J. Cai, Z. G. Liu, X. L. Sui and Z. B. Wang, Ionics, 2020, 26, 1913–1922 CrossRef CAS.
- N. Charef, L. Arrar, A. Ourari, R. M. Zalloum and M. S. Mubarak, J. Macromol. Sci., Part A: Pure Appl. Chem., 2010, 47, 177–184 CrossRef CAS.
- S. Nanaki, R. M. Eleftheriou, P. Barmpalexis, M. Kostoglou, E. Karavas and D. Bikiaris, Sci, 2019, 1, 48 CrossRef.
- A. Valkov, M. Zinigrad, A. Sobolev and M. Nisnevitch, Int. J. Mol. Sci., 2020, 21, 3512 CrossRef CAS PubMed.
- H. Memon and Y. Wei, J. Appl. Polym. Sci., 2020, 137, e49541 CrossRef.
- H. Memon, Y. Wei and C. Zhu, Mater. Today Commun., 2021, 29, 102814 CrossRef CAS.
- S. U. Raut, S. A. Deshmukh, S. H. Barange and P. R. Bhagat, Catal. Today, 2023, 408, 81–91 CrossRef CAS.
- D. V. Bhuse and P. R. Bhagat, ChemistrySelect, 2022, 7, e202200328 CrossRef CAS.
- S. U. Raut, K. R. Balinge, S. A. Deshmukh, S. H. Barange, B. C. Mataghare and P. R. Bhagat, Catal. Sci. Technol., 2022, 12, 5917–5931 RSC.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*