
Tunable optical and thermodynamic properties: the ignition of energetic metal complexes with different crystal fields†
Kun
Wang
 *ab,
Xiaoqi
Yang
a,
Peipei
Zhang
a,
Shu
Bu
a,
Longjiu
Cheng
a and
Jianguo
Zhang
b
*ab,
Xiaoqi
Yang
a,
Peipei
Zhang
a,
Shu
Bu
a,
Longjiu
Cheng
a and
Jianguo
Zhang
b
aDepartment of Chemistry, Key Laboratory of Functional Inorganic Materials of Anhui Province, Anhui University, Hefei, Anhui 230601, P. R. China. E-mail: wangkun@ahu.edu.cn
bState Key Laboratory of Explosion Science and Technology, Beijing Institute of Technology, Beijing 100081, P. R. China
First published on 8th March 2025
Abstract
Primary explosives are generally composed of a metal, energetic ligands and oxyacid anions, and are exploded by optical or thermal initiation pathways to provide energy. Nowadays, near-infrared (NIR) laser initiation is a safe way to ignite explosives to effectively avoid electromagnetic interference. Therefore, low NIR laser sensitivity and high thermal stability are the preconditions for applicable laser-initiated explosives. Here, metallic 1,5-diaminotetrazole perchlorate complexes, [M(DAT)6](ClO4)2 (M2+ = Cr2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+ and Zn2+), have been selected to understand the laser sensitivity and thermal stability of octahedral energetic complexes using time-dependent density-functional theory methods (TD-DFT) and Car–Parrinello molecular dynamics (CPMD) methods. Compared with the energetic complexes [M(1-AT)x](NO3)2 (x = 2 or 3), which adopt a tetragonal/square crystal field, the optical properties are obviously influenced by the coordination field. In a departure from traditional viewpoints, the thermal safety is reflected not only by the stability of the thermal ignition stage, but also by the performance of the stable deflagration stage and the deflagration to detonation transition (DDT) stage. This work helps to enhance the NIR sensitivity of complexes by tuning the crystal field and to improve the thermal safety based on the metal–ligand interactions, which is valuable for the exploration and design of stable laser-ignited energetic materials.
1. Introduction
Nitrogen-rich metal complexes, that are highly prone to explode via a rapid decomposition accompanied by an intensive release of energy and the formation of high-temperature gaseous reaction products, are broadly used in various military, industrial, and civilian applications.1–3 Energetic metal complexes (EMC) have gained attention worldwide due to their different thermodynamic or optical properties, which are directly related to their metal centers.4,5Recently, laser initiation technology that has strong anti-interference ability and safety has been developing quickly. The laser initiation process is generally characterized by metal-to-ligand charge transfer (MLCT) or ligand-to-metal charge transfer (LMCT) and a strong absorption intensity in the near-infrared (NIR) region, and is limited by currently developed NIR laser igniter technology.6–8 One crucial factor in determining the thermal stability and laser initiation threshold of a primary explosive is the interaction between the metal center and the energetic ligand,9 where the sensitivity can be regulated by altering the metal center.10 For example, alkali metals coordinated with the ligand 4,4′,5,5′-tetranitro-2,2′-biimidazole (TNBI) possess shorter onset times than alkaline earth metal-containing complexes, which indicates that the initiation delay times have a definite relationship with the metal ion type.6 Fe/Cu complexes of the ligands 1-amino-5H-tetrazole (1AT) and 2-amino-5H-tetrazole (2-AT) are both are more sensitive to mechanical stimuli than the corresponding Mn/Zn complexes.11 Sifain et al. demonstrated that the conjugation of the ligand is an important factor that leads to low energy charge transfer.12 Peng et al. synthesized Co/Cd/Ni/Cu complexes coordinated with 5,5′-azotetrazole-1,1′-diol (H2AzTO), where a closed-shell Cd(II) complex has the highest thermal stability, whilst the Cu(II) complex has the highest heat of detonation.13 Liu et al. prepared Cu-coordinated 1,2-bis(3-nitroamino-1,2,4-triazol-5-yl)ethane (BNATE), a metal quadridentate chelate, with excellent photosensitivity and a lower laser-initiation threshold.14 Similarly, Cu(II)-coordinated 4,5-ditetrazolyl-1,2,3-triazole (H3BTT)15 and thiazole-4-carbohydrazide with perchloric acid both appear to have excellent thermal stability and photosensitivity.16 Based on our previous studies, the coordination field of the metal center affects the light absorption in the NIR region, which can be enhanced by the magnetism exchange interaction between the metal or by the Jahn–Teller effect.17 Not only do the photophysical properties affect the stability and thermal initiation pathway, but the coordination pathway of the metal center also affects the stability and thermal initiation pathway.18–20 In order to understand how the optical properties are affected by different crystal fields, seven [M(DAT)6](ClO4)2 octahedral complexes (M2+ = Cr2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+ and Zn2+) were selected and compared with our previously studied [M(1-AT)x](NO3)2 (M2+ = Cr2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+ and Zn2+, x = 2 or 3) complexes. The thermal safeties of the primary explosives were evaluated again based on the metal–ligand relationship.
In this paper, the crystalline [Mn/Co/Zn(DAT)6](ClO4)2 complexes (CCDC-704380/783306/742791) were obtained from experiments.10,18,21,22 In the present study, reasonable crystal structures are required to evaluate the effect of the coordination field on the thermal and optical properties. Herein, DAT is characterized as having a weak ligand field which will not change the electron configuration of the metal center by coordination. Generally, the 4th row transition metal complexes appear to have similar coordination environments and the same symmetries.3,4,6,8,10,11 Therefore, for the unknown crystal structures of [Cr/Fe/Ni/Cu(DAT)6](ClO4)2, it is effective to directly substitute the metal centers17,19 of the known crystal structures with one from an unknown structure. This is similar to the phenomenon for the known [Mn/Co/Zn(DAT)6](ClO4)2 complexes, all of which belong to the trigonal syngony, where, based on their crystal structures, the spatial arrangements are known to be completely the same.10 Therefore, it is reasonable to expect that all of the predicted [Cr/Fe/Co/Ni(DAT)6](ClO4)2 complexes are also isostructural.
In the present study, based on the [Zn(DAT)6](ClO4)2 crystal obtained by experiment, we first substituted the metal center with Cr/Fe/Ni/Cu to construct the DAT series of EMCs to help us understand how the optical/thermal properties of these complexes are influenced by the metal–ligand–anion relationship. Secondly, we optimized the crystal structures of the seven [M(DAT)6](ClO4)2 (M2+ = Cr2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+ and Zn2+) complexes to obtain the electronic structures and UV-vis-NIR (ultraviolet-visible-near-infrared) spectra used to predict which EMCs are possible NIR sensitive candidates. Thirdly, the gas-phase structures of the [M(DAT)6](ClO4)2 complexes were abstracted from the crystal structures to understand their electronic excitation properties using time-dependent density-functional theory (TD-DFT) methods to conduct natural transition orbital (NTO) analysis and obtain the hole–electron distribution, and inter-fragment charge transition (IFCT).23,24 Fourthly, all the optical properties have been compared with that of a series of tetragonal (distorted octahedron) [M(1-AT)x](NO3)2 complexes in order to understand the effects of different crystal fields. Limited by the known synthesized energetic complexes, it is difficult to find complexes with the same ligands and anions. Thus, the comparison between [M(DAT)6](ClO4)2 and [M(1-AT)x](NO3)2 was chosen as both the 1-AT and DAT ligands are derivates of aminotetrazoles with closely related structures. Moreover, based on experimental results of the thermal stability and optical sensitivities, we can understand the effect that the coordination field has on the thermal/optical properties of the complexes. Finally, the Car–Parrinello molecular dynamics (CPMD) method has been applied to simulate the thermal initiation of the seven [M(DAT)6](ClO4)2 complexes to understand their thermodynamic stabilities. The comparison between the previously studied [M(1-AT)x](NO3)2 (x = 2 or 3) complexes, with a distorted octahedral coordination field,17 and the [M(DAT)6](ClO4)2 (M2+ = Cr2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+ and Zn2+) standard octahedral complexes, gives us clues as to how the optical and thermodynamic properties are influenced by the coordination field of the metal center. The flowchart of our calculations with the detail of the methods used has been summarized in Fig. 1. The detailed descriptions and the tests of the calculations have been summarized in the Methods section and ESI.†
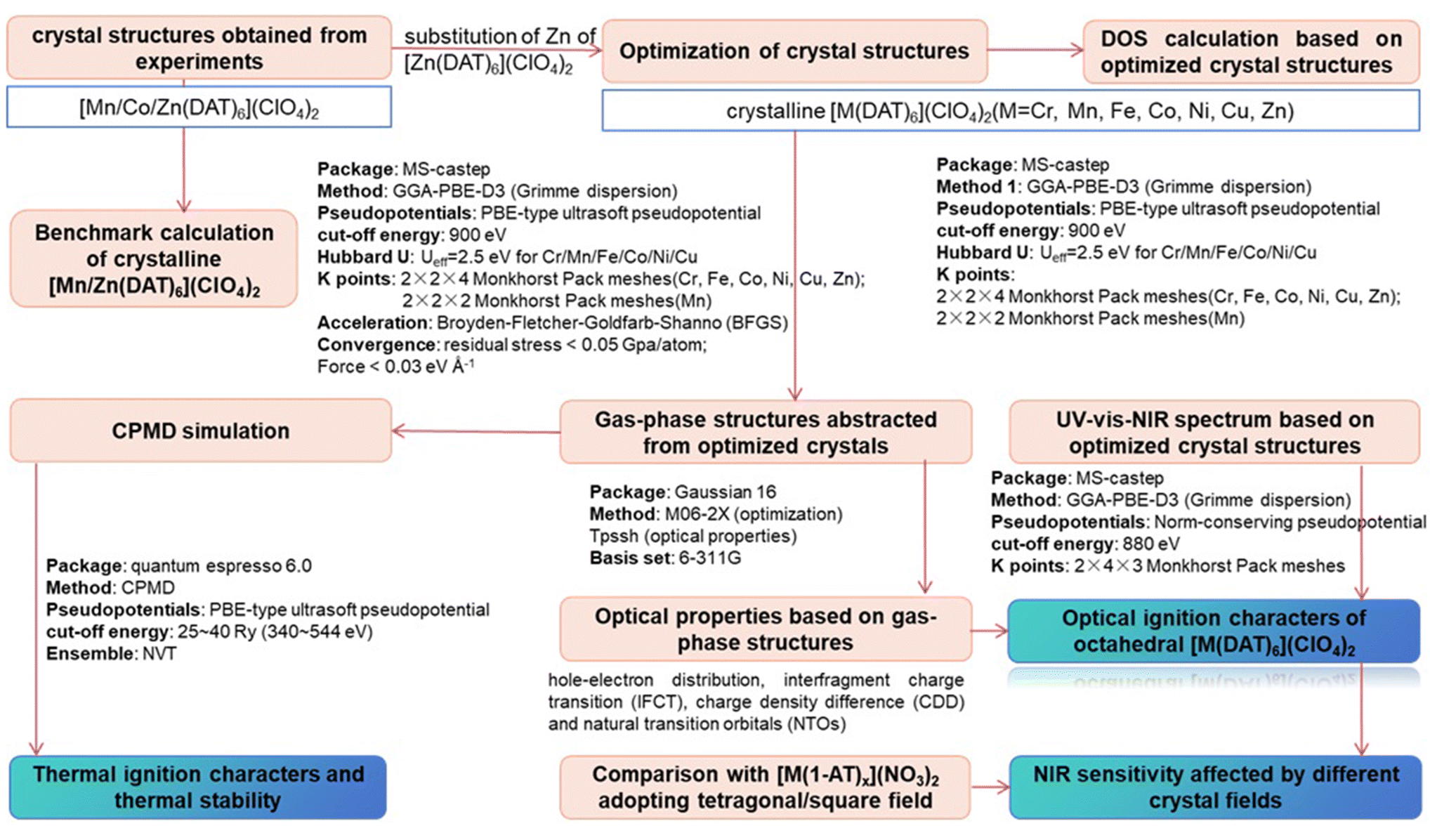 | ||
| Fig. 1 The flowchart of our calculations with the details of the methods used. | ||
2. Computational methods
The seven crystal structures of [M(DAT)6](ClO4)2 (M2+ = Cr2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+ and Zn2+) were studied using the CASTEP code.25 Calculations were performed using the generalized-gradient approximation (GGA) for the Perdew, Burke, and Ernzerhof (PBE) exchange–correlation functional within density-functional theory for crystals.26 The core electrons are described by a PBE type ultrasoft pseudopotential27,28 with a valence state of 3d44s2 for Cr, 3d54s2 for Mn, 3d64s2 for Fe, 3d74s2 for Co, 3d84s2 for Ni, 3d94s2 for Cu, 3d104s2 for Zn, 2s22p2 for C, 2s22p3 for N, 2s22p4 for O, 3s23p5 for Cl and 1s1 for H. During optimization, the cutoff energy was set as 900 eV based on the tests. We have implemented the Hubbard U model in the optimization to obtain accurate electronic structures, where the effective U parameters have been set as 2.5 eV for Cr/Fe/Co/Ni/Cu to account for errors in exchange correlations on their 3d orbitals. With the exception of the 2 × 2 × 2 Monkhorst–Pack meshes29 for [Mn(DAT)6](ClO4)2, 2 × 2 × 4 k-points are used to describe the Brillouin zones for the Cr/Fe/Co/Ni/Cu/Zn crystals based on the benchmark calculation, where the details are summarized in section 1 of the ESI.† The different k-points of [Mn(DAT)6](ClO4)2 arise because its unit cell has Brillion zones that are different to those of the other six crystals. The Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm30 was applied in the structural relaxation of the atomic position until the residual force of the atom and stress were less than 0.03 eV Å−1 and 0.05 GPa. For the molecular structures abstracted from the crystal structures, the M06-2X method31 and 6-311G basis set were selected to optimize the gas-phase structure, and to describe the electronic structures of the ground state and excited state, based on previous benchmark calculations.12,32 A total of 70 singlet excited states have been studied to give the complete UV-vis-NIR spectra. All gas-phase calculations were performed using the Gaussian 16 software package.33 In order to further explore the electronic excitation characteristics of all the complexes, natural transition orbital (NTO) analysis, the hole–electron distribution and inter-fragment charge transition (IFCT) were analysed using the Multiwfn software34,35 and VMD software.36To understand the role of the metal center in the thermal ignition pathways of [M(DAT)6](ClO4)2, the CPMD method was applied with the GGA-PBE method in the Quantum Espresso 6.0 package.37 Each optimized molecular structure was put in a 15 × 15 × 15 Å box. The kinetic energy cutoff of 25–40 Ry (340.0–544 eV) was set for the molecular dynamics, with an effective electron mass of 500–700 a.u. In the simulations, the time step was set as 4.0 a.u (about 0.1 fs) for the seven complexes. The nose thermostat38 and the NVT ensemble were employed with an oscillation frequency of 550–750 THz. The external temperature directly increased to 2500 K and the simulation time was set as 2 ps (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps) for the seven complexes to understand the mechanisms of the initial decompositions and DDT processes. The flowchart of our calculations with the details of the methods used for each step has been summarized in Fig. 1.
000 steps) for the seven complexes to understand the mechanisms of the initial decompositions and DDT processes. The flowchart of our calculations with the details of the methods used for each step has been summarized in Fig. 1.
3. Results and analysis
3.1 Optimized structures
The optimized crystalline and molecular structures of [M(DAT)6](ClO4)2 (M2+ = Cr2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+ and Zn2+) are shown in Fig. 2. Both the crystal and gas-phase structural parameters and details are summarized in ESI section 2 and 3.†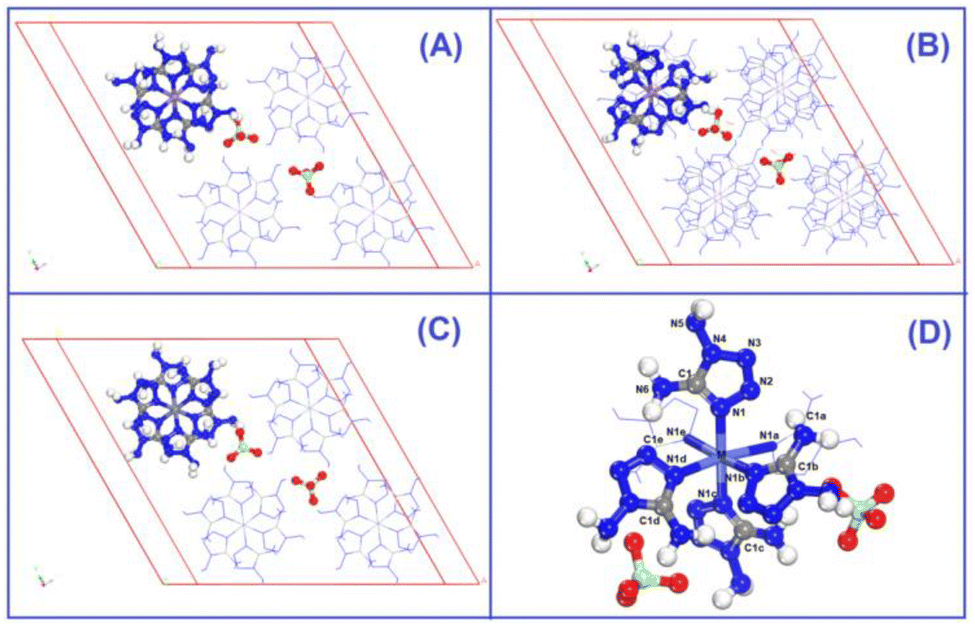 | ||
| Fig. 2 The optimized crystal structures of (A) [Co(DAT)6](ClO4)2, (B) [Mn(DAT)6](ClO4)2, and (C) [Zn(DAT)6](ClO4)2 and the predicted crystalline complexes obtained by the substitution of Zn with Cr/Fe/Ni/Cu. (D) The optimized gas-phase structure of the seven [M(DAT)6](ClO4)2 complexes. | ||
The crystal structures shown in Fig. 2A and C all belong to the trigonal syngony with P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) symmetry, except for [Mn(DAT)6](ClO4)2, which has Pc1 symmetry. In all of the seven crystals, the metal center adopts sp3d2 hybridization and coordinates six DAT ligands through their N atoms to form an octahedral structure where the Nh–M–Nv angle is close to 90° between the horizontal and vertical direction. All the C and N atoms of the DAT ligand adopt the sp2 hybridization in the formation of a 5-center-5-electron delocalized π bond, where the N1 atom (labelled in Fig. 2D) with the most negative charge forms an M–N coordination bond. Based on the Mulliken charge analysis of this coordination bond, Cr/Zn/Cu appear to have the most positive charge of the metals and correspond with the largest negative charges located on the coordinated N atoms. Therefore, the Cr/Cu/Zn complexes appear to have strong Coulomb interactions between the metal and the N atom, with strongly polarized atom charges. The electron population analysis shows that the metal–ligand interaction in the Mn/Fe/Co/Ni complexes is mainly maintained by covalent bonds with large populations, where the Ni–N bond appears to have the shortest length and largest electron population value, indicating that it has the strongest covalency. We considered the molecular structures based on the crystals shown in Fig. 2D. For the Cr/Mn/Fe/Co molecules, the high-spin states are more stable than the low-spin ones at the M062X/def2tzvp level of theory. The energies of the high-spin and low-spin states for all the complexes are compared in Table S4.† Based on the NPA (natural population analysis) of the atomic charge and the population analysis of the gas-phase structures, the Cr/Cu/Zn–N bonds display lower WBI (Wiberg bond index) and larger charge polarization than the Mn/Fe/Co/Ni–N bonds, which is consistent with the crystal structures.
symmetry, except for [Mn(DAT)6](ClO4)2, which has Pc1 symmetry. In all of the seven crystals, the metal center adopts sp3d2 hybridization and coordinates six DAT ligands through their N atoms to form an octahedral structure where the Nh–M–Nv angle is close to 90° between the horizontal and vertical direction. All the C and N atoms of the DAT ligand adopt the sp2 hybridization in the formation of a 5-center-5-electron delocalized π bond, where the N1 atom (labelled in Fig. 2D) with the most negative charge forms an M–N coordination bond. Based on the Mulliken charge analysis of this coordination bond, Cr/Zn/Cu appear to have the most positive charge of the metals and correspond with the largest negative charges located on the coordinated N atoms. Therefore, the Cr/Cu/Zn complexes appear to have strong Coulomb interactions between the metal and the N atom, with strongly polarized atom charges. The electron population analysis shows that the metal–ligand interaction in the Mn/Fe/Co/Ni complexes is mainly maintained by covalent bonds with large populations, where the Ni–N bond appears to have the shortest length and largest electron population value, indicating that it has the strongest covalency. We considered the molecular structures based on the crystals shown in Fig. 2D. For the Cr/Mn/Fe/Co molecules, the high-spin states are more stable than the low-spin ones at the M062X/def2tzvp level of theory. The energies of the high-spin and low-spin states for all the complexes are compared in Table S4.† Based on the NPA (natural population analysis) of the atomic charge and the population analysis of the gas-phase structures, the Cr/Cu/Zn–N bonds display lower WBI (Wiberg bond index) and larger charge polarization than the Mn/Fe/Co/Ni–N bonds, which is consistent with the crystal structures.
Being classic water-free energetic complexes, the seven [M(DAT)6](ClO4)2 complexes adopt similar octahedral structures, where the 1,5-diaminotetrazole (DAT) ligand is a representative energetic ligand with a conjugated structure,19,39 but exhibit completely different explosion performances. Based on experiment,10,18,21,22 [Co(DAT)6](ClO4)2 is known to be sensitive to both impact and friction stimuli, with an impact sensitivity (IS) of 0.6 J and a friction sensitivity (FS) of 9 N, which is completely different from the Mn/Zn-DAT compounds,8,40 where Zn-DAT has almost no IS but quite a large FS. The band gaps of the Cr/Fe/Cu complexes are zero, indicating that they are electronic conductors due to their active β-d electrons, as shown in Fig. 3A. The other four complexes have band gaps that follow the sequence Co < Ni < Mn < Zn, which is positively correlated with the known IS values (Co < Mn < Zn). The theoretical level of the optimization, density of states (DOS) and partial density of states (pDOS) are summarized in ESI Fig. S1–S7.† The energy range of the valence bands of all the complexes, −12–−9 eV, is mainly composed of the H 1s, C/N 2s and 2p states, corresponding to N–H bonds, N–N bonds and C–N bonds. The energy bands in the range of −6 to −3 eV are mainly composed of the 2p states and the hybrid sp2 orbitals of the C and N atoms, which mainly correspond to the N–N bonds and C–N bonds. The electrons of the metals distribute around the Fermi level, composed of the 3d states and slightly composed of the 4s/4p states. We have summarized the contribution of the 3d states of the metal centers in the DOS of the seven complexes shown in Fig. 3A. The inner d electrons of the metals and the N sp electrons occupy the bands from −3 eV to the Fermi level, indicating that the M–N and N–N bonds of the tetrazole ring are the most active bonds, corresponding to the hot spot in the dissociation. To understand the activity of metal center, we calculated the d-band center for each transition metal (Fig. 3B). The empty d band for Cr/Mn appears to result in a positive d band center. For the other five metal complexes, the value of the d band center follows the sequence of Zn < Cu < Fe < Ni < Co. Based on the d band center theory, a higher d band center corresponds to a stronger absorption ability with respect to the fragments in the dissociation.
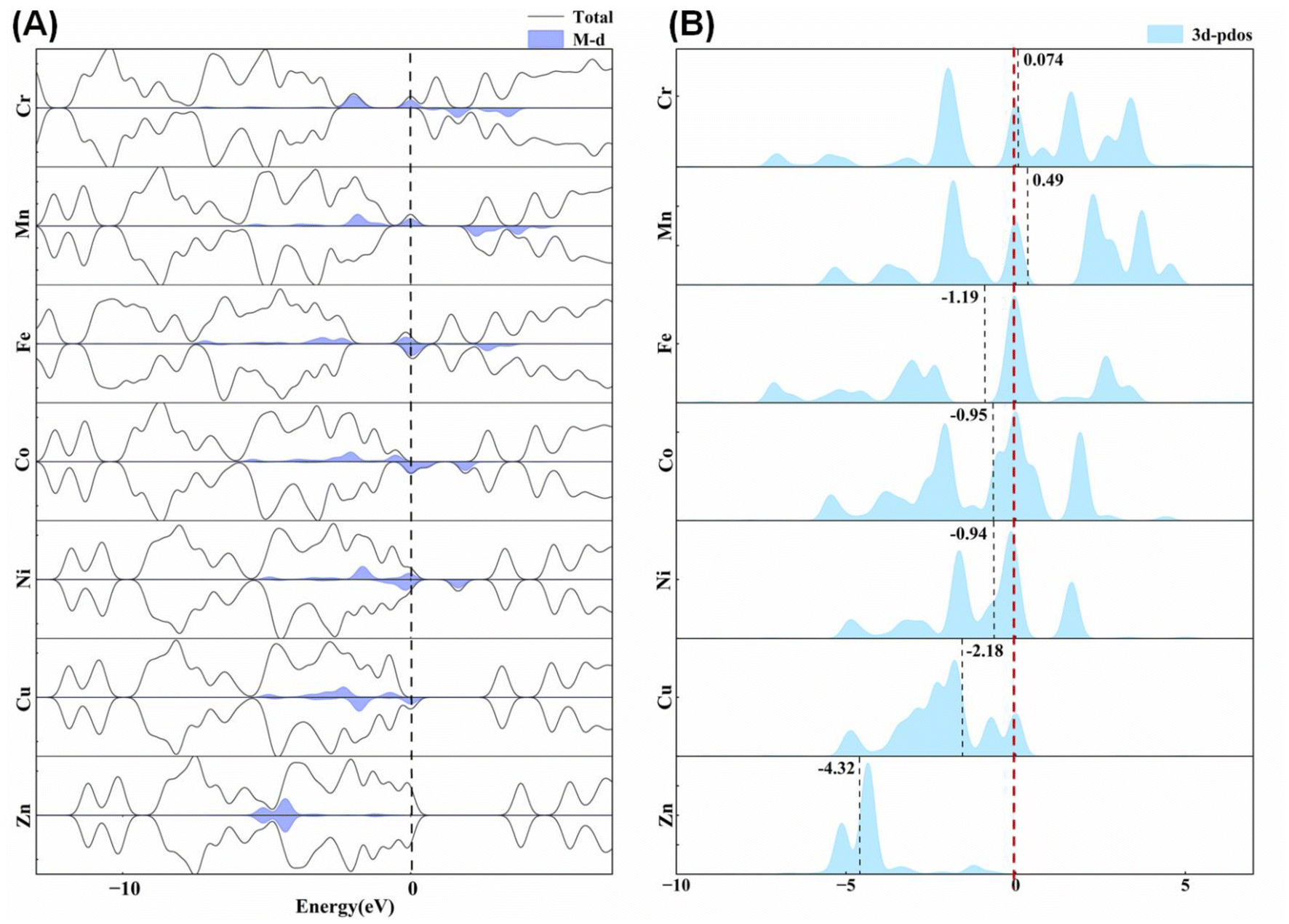 | ||
| Fig. 3 (A) The total density of states (DOS) and the role of the 3d states of the metal centers. (B) The d-band centers of the metal centers of the seven M(DAT)6(ClO4)2 complexes. | ||
Hirshfeld surfaces41 and two-dimensional fingerprint plots were applied to analyze the molecular interactions in each crystal structure (Fig. 4). Obviously, all the surfaces shown in Fig. 4A and B are hillocky and humpy, corresponding to the irregular coordination environments. The red dots show that the distance between the molecules is less than the van der Waals distance, indicating the presence of strong intermolecular interactions. Most of the red dots at the surface edges are related to the strong intermolecular hydrogen bonds of the type Cl–O⋯H between the ClO4− species and the ligands. The seven complexes all have similarly shaped fingerprint plots (Fig. 4C), where the hydrogen bonds, including the N–H⋯H–N bonds and Cl–O⋯H bonds, make the largest contribution to maintaining the structural stability of the crystal. The DAT ligands interact with each other through different types of hydrogen bonds, leading to a layered conformation.
 | ||
| Fig. 4 (A) Molecular structures; (B) Hirshfeld surfaces; (C) 2D fingerprint plots and (D) percentages of the weak intermolecular actions of the seven [M(DAT)6](ClO4)2 complexes. | ||
In order to understand the role of the metal center in the stabilization of the six crystals, we have calculated the metal stabilization energies (MSE) based on the definition [M2+ + (DAT)6](ClO4−)2 → [M(DAT)6](ClO4)2, where the energies of the reactants and products are calculated in the corresponding unit cell to ensure the same environment. The MSE of the seven crystals follows the sequence Fe > Ni > Mn > Co > Zn > Cu > Cr. Based on the analysis of coordination bonds, the ionic-type metal–ligand interactions (Cr/Cu/Zn) exhibit a weaker metal stabilization effect than the covalent type interactions (Mn/Fe/Co/Ni). The different stabilization effects of the metal centers result in different detonation pathways.
3.2 Optical properties of [M(DAT)6](ClO4)2
Based on the optimized crystal structures, the UV-vis-NIR spectra in the range of 900–1200 nm (corresponding to 1.03–1.38 eV) in the solid state is summarized in Fig. 5. This analysis was conducted because the most widely applied wavelengths for laser ignition are in the in the near-infrared (NIR) region,6–8 for example, the most practical and available wavelength for optical initiation is that of Nd:YAG lasers (1064 nm). The strength of absorption (cm−1) of the seven crystals at 900 nm follows the sequence of Zn(DAT)6(ClO4)2 (0) ≈ Mn(DAT)6(ClO4)2 (0) ≈ Ni(DAT)6(ClO4)2 (0.01) ≈ Co(DAT)6(ClO4)2 (0.05) < Cr(DAT)6(ClO4)2 (154.07) < Fe(DAT)6(ClO4)2 (487.05) < Cu(DAT)6(ClO4)2 (1413.94), which is also the sequence in the range of 900–1200 nm. Therefore, the Cr/Fe/Cu complexes exhibit excellent potential to be applied as the laser-ignited explosives. | ||
| Fig. 5 UV-vis-NIR spectra of [M(DAT)6](ClO4)2 (M = Cr, Mn, Fe, Co, Ni, Cu, Zn) in the NIR region of 900–1200 nm. | ||
In order to understand the detailed excitation modes of the complexes, we abstracted the two neighboring molecules of [M(DAT)6](ClO4)2, based on the optimized crystals, in order to confirm the excitation properties at the tpssh/def2tzvp level of theory in Gaussian 16. The absorption spectra of the gas-phase states is consistent with that of the solid states (ESI Fig. S23†), indicating that the abstracted structures are reasonable. The energy gap and oscillator strengths of the molecular [M(DAT)6](ClO4)2 complexes are summarized in ESI Tables S20–S26.† The greater the number of absorption bands and the larger the oscillator strengths are, the higher the possibility a complex has to be ignited with an NIR laser.
Based on the spectra in Fig. 5, we first analysed the hole–electron distribution of the charge transition in the NIR area. The dominant natural transition orbitals (NTOs) of the hole–electron pairs in the NIR region along with the excitation mode and the oscillator strength (OS) are listed in Table 2. All the detailed results about the optical properties of all the complexes are summarized in ESI section 5.† The spectra in Fig. 5 indicate that [Cu(DAT)6](ClO4)2 exhibits the most optical activity in the NIR region. There is only one absorption band located in the NIR region, which mainly corresponds to the charge transfer from the metal to the ligand (S0 → S8) with the largest OS value of 0.16. For [Fe(DAT)6](ClO4)2, it is the second most NIR sensitive complex based on our calculations, with four absorption bands in the required area. The hole–electron pairs in Table 2 indicate that the MLCT of S0 → S10 and LMCT of S0 → S11 have the largest probabilities of NIR light absorption, with OS values of 0.06 and 0.04, respectively. [Cr(DAT)6](ClO4)2 is the third most NIR sensitive complex and has three bands in this region. The largest OS value of the Cr complex is only 0.05, which is the lowest of the Cu/Fe/Cr complexes. Therefore, absorption in the NIR region is the only the precondition for a laser-ignited explosive. However, the optical sensitivity is determined by the probability of absorption in the NIR region, as reflected by the OS value.
3.3 The effect of the crystal field on the NIR sensitivity: comparison of [M(DAT)6](ClO4)2 and [M(1-AT)x](NO3)2
For the previously studied metal complexes with 1-amino-5H-tetrazole (1-AT) ligands,17 which adopt a tetragonal (distorted octahedron) coordination field in their Cr/Mn/Fe/Co/Cu/Zn complexes and a square field for the Ni complex (Fig. 6), the strength of absorption in the NIR region follows the sequence [Zn(1-AT)3](NO3)2 ≈ [Co(1-AT)3](NO3)2 < [Cr(1-AT)3](NO3)2 < [Ni(1-AT)2](NO3)2 < [Mn(1-AT)3](NO3)2 < [Cu(1-AT)3](NO3)2 < [Fe(1-AT)3](NO3)2. The absorption properties of [Mn/Cr(DAT)6](ClO4)2, standard octahedral complexes, are completely different from those of [Mn/Cr(NO3)2](1-AT)3. For the hexa-coordinated complex, no absorption in the NIR area can be found for [Mn(DAT)6](ClO4)2 whilst [Cr(DAT)6](ClO4)2 exhibits a strong absorption in the NIR area. But, based on DFT calculations, for the tetragonal field complexes, [Mn(1-AT)3](NO3)2 exhibits a strong absorption and only a slight absorption can be found for [Cr(1-AT)3](NO3)2 in the NIR region. In the meantime, to understand the effect of the ligands (DAT and 1-AT) and ions (ClO4− and NO3−) on absorption, we calculated the absorption spectra of 1-AT, DAT, NO3− and ClO4−, all of which are compared in ESI Fig. S22.† Without a metal, none of the ligands and anions show absorption in the NIR area. Moreover, no absorption signals for 1-AT and DAT can be found in the range of 400–600 nm. Therefore, the differences in the optical properties of the complexes are mainly caused by the crystal of for each metal complex. | ||
| Fig. 6 (A) The sketch map of the splitting of the d orbitals in the octahedral field, tetragonal field and square field. (B) The energy and components of the HOMO and LUMO of each metal cation. (C) Schematic representation of the tetrahedral structures: [M(1-AT)x](NO3)2 (M = Cr, Mn, Fe, Co, Cu, Zn), square structures: [Ni(1-AT)2](NO3)2, and octahedral structures: [M(DAT)6](ClO4)2 (M = Cr, Mn, Fe, Co, Ni, Cu, Zn). | ||
Based on previous research,17 effective charge transfer between a ligand and a metal center, such as LMCT and MLCT, is the determining factor for accelerating light absorption in the NIR region. However, both MLCT and LMCT are affected by the coordination field. For all transition metals, five generated d orbitals (in a spherical field) will split into different crystal fields, as has been summarized in Fig. 6. With respect to light absorption, the d electrons can be excited between the split d orbitals or between the metal and ligands. Regardless of the excitation pathways of the d electrons, the activity of the d electrons is the determining factor in the excitation.
All the metals of the seven [M(DAT)6](ClO4)2 complexes and seven [M(1-AT)x](NO3)2 complexes adopt high spin states. But the crystal fields of the complexes are different from each other. The splitting energy of the crystal field of the octahedral field, tetragonal field and square field is 10 Dq, shown as Δ0 in Fig. 6A. The activity of the d electrons during the excitation can be evaluated using the crystal field stabilization energy (CFSE) with the electron configuration. The energy and components of the HOMO and LUMO of each metal cation have been summarized in Fig. 6B. We have summarized the electronegativity, coordination field, the corresponding electron configuration of the d → d excitation based on the spectroscopic term, the number of single electrons and the CFSE in Table 3. Cr2+ exhibits low electronegativity (the second lowest of the seven metal cations), suggesting that it can easily donate its valence electrons during excitation. Given that the ligand and anions in the vis-NIR region show similar absorption effects (ESI Fig. S22†), the different optical properties of [Cr(1-AT)3](NO3)2 and [Cr(DAT)6](ClO4)2 are mainly caused by the different crystal fields. As shown in Table 3, Cr2+ has four unpaired electrons in both [Cr(1-AT)3](NO3)2 and [Cr(DAT)6](ClO4)2. Unlike in [Cr(1-AT)3](NO3)2, an electron occupies the higher energy dz2 orbital in [Cr(DAT)6](ClO4)2 without the splitting of the eg orbitals, where the higher CFSE (−6 Dq vs. −9.14 Dq) indicates that the d electrons in the octahedral field are more active than those in the tetragonal field. Moreover, the asymmetrical distribution of high-spin electron in the generated dx2−y2 and dz2 orbitals, results in an obvious Jahn–Teller effect in the octahedral field. It is known that the Jahn–Teller effect broadens the absorption bands of d–d transition.17 However, Jahn–Teller effect is weakened by the splitting of the dx2−y2 and dz2 orbitals in [Cr(1-AT)3](NO3)2. Therefore, for [Cr(DAT)6](ClO4)2, the lower CFSE and stronger Jahn–Teller effect both accelerate the MLCT, corresponding to the three strong excitation modes (S0 to S14, S15 and S20) shown in Table 2. For the Mn complexes, there are five unpaired 3d electrons in the orbitals of both [Mn(DAT)6](ClO4)2 and [Mn(1-AT)3](NO3)2 with the same CFSE. However, the activity of Mn2+ is strongly affected by the crystal field. In the octahedral field, d–d excitation is completely forbidden based on the selection rules of the Mn2+ spectrum. Therefore, the charge transfer between the metal and ligands/anions requires higher energy in an octahedral field, with no absorption signal in the NIR region. From the NIR region to the visible region, the first possible excitation mode is S0 → S82 with a wavelength of 534.86 nm, where the corresponding oscillator strength is 0.02. However, with the splitting of three T2g orbitals in the tetragonal field of Mn(NO3)2(1-AT)3, the possible 4E → 4T2 excitation mode based on d–d interactions improve the activity of the d electrons and leads to MLCT (3d electron → π* orbital of 1-AT) in the NIR region.
In the Ni complexes, the Ni2+ of [Ni(DAT)6](ClO4)2 in an octahedral field generally exhibits a lower CFSE (−12 Dq) than in a square field (−4.28 Dq for [Ni(1-AT)2](NO3)2), suggesting that [Ni(DAT)6](ClO4)2 has less active d electrons. In the NIR region, the first possible excitation mode is S0 → S93 for [Ni(DAT)6](ClO4)2 with a wavelength of 579.31 nm and an oscillator strength of 0.01. But in [Ni(1-AT)2](NO3)2, with the splitting of the T2g and Eg orbitals, a greater number of d–d excitation modes, such as the as 1A1g → 1A2g, 1A1g → 1B1g and 1A1g → 1Eg modes, improves the activity of the d electrons. Additionally, the low LUMO energy of Ni2+ in the square field enhances the activity of the d orbitals with respect to light, especially in the CT process of NO3− → Ni2+.
For both the Fe and Cu complexes, the d electrons appear to have quite high activities with respect to the NIR region in both the octahedral and tetragonal fields. For [Fe(DAT)6](ClO4)2, with an Fe2+ ion with the lowest LUMO energy level and weakest electronegativity of the seven metal cations, it is easy to complete MLCT and LMCT via light absorption. In Table 3, both LMCT and MLCT are obviously seen with oscillator strengths of 0.04–0.06. The CFSE of [Fe(DAT)6](ClO4)2 is larger than that of Fe(NO3)2(1-AT)3, suggesting that it has d electrons with high activity. However, based on Table 3, there are no exchange interactions between the neighbouring high-spin Fe2+ ion of [Fe(DAT)6](ClO4)2 in the possible excitation of 5T2g → 5Eg in the NIR region, which is different from the excitation of 3T2 → 3E in Fe(NO3)2(1-AT)3. Therefore, the absorption range of [Fe(DAT)6](ClO4)2 cannot be broadened without M–M exchange interactions. For the Cu complexes, the lower CFSE and strong Jahn–Teller effect of [Cu(DAT)6](ClO4)2 indicate that its d electrons have high activity, leading to the strongest absorption in the NIR region. Although both [Fe/Cr(DAT)6](ClO4)2 have more absorption bands than [Cu(DAT)6](ClO4)2, [Cu(DAT)6](ClO4)2 exhibits the highest probability of the only absorption in the NIR region with an oscillator strength of 0.16. The strongly electronegative Cu2+ exhibits strong MLCT in the excitation. The Cu complexes in both crystal fields have similar d electron behaviors, in which the Jahn–Teller effect broadens the absorption bands of the d–d transition (2Eg → 2T2g) and improves its sensitivity to NIR absorption.
The Co and Zn complexes in both crystal fields show similar properties. In both the [Co/Zn(DAT)6](ClO4)2 and [Co/Zn(1-AT)3](NO3)2 complexes, the distribution of the d electrons in Co2+ and Zn2+ are similar. For the Co complexes, high-spin Co2+, with the 4F spectral term, splits into A1g, T1g and T2g levels in an octahedral field, and T2 and E levels in a tetragonal field. Therefore, for both complexes, the energy for local MC excitation is lower than 1.03 eV. Additionally, with a high electronegativity and low HOMO energy, the energy required for MLCT is higher than 1.38 eV. In [Co(DAT)6](ClO4)2, the lowest MLCT is S0 → S44, with a wavelength of 731.74 nm and an oscillator strength of 0.01. As for the Zn complexes in the two different crystal fields, Zn2+ is the only closed-shell species of the seven complexes. In both the octahedral and tetragonal field, d–d excitation is completely forbidden. The zero oscillator strength of the excitation suggests that the Zn complex has low optical sensitivity.
With respect to the metal centers, enhancing the activity of the d electrons of the metal is a pathway to improve the NIR laser sensitivity, which can be realized by decreasing the CFSE of each metal or increasing the possibility of d–d excitation based on split spectroscopic terms, both of which are determined by the crystal fields. The CFSE can be tuned by controlling the configuration of the d electrons, based on the control of the oxidation number in the formation of the complex. The possibility of d–d excitation can be tuned by the crystal field. Generally, for high-spin configurations of energetic metal complexes, the octahedral crystal field exhibits a lower CFSE than the tetrahedral field, but higher than a square field. But for the half-closed shell or closed-shell metal cations, such as Mn2+ or Zn2+, accelerating the splitting of the spectral terms of the d orbitals is an effective way to improve the activity of the d electrons. Additionally, Jahn–Teller effects and exchange interaction between neighboring high-spin electrons will further improve the activity of the d electrons. Finally, based on the high activity of the d electrons, tuning the ligands is an effective way to decrease the excitation energy between the metal and ligands and to increase the NIR sensitivity. This can be achieved by coordination of ligands with strong e-donor characters with low electronegativity metals like Cr/Mn/Fe/Co, or the coordination of ligands with strong electron-withdrawing character with high electronegativity metals like Ni/Cu/Zn.
3.4 The thermal stability of crystalline M(DAT)6(ClO4)2
In the simulation, the ClO4− anions move first, caused by the ionic interaction between [M(DAT)6]2+ and (ClO4)−. Then the decomposition is triggered by the cleavage of the covalent bond. Based on the population analysis of each crystal (Table 1), the M–N bond exhibits the weakest covalency. Therefore, all the ignition pathways are triggered by the cleavage of the weakest coordinative M–N bond. The initial decomposition has been summarized in Scheme 1, where the M–N bonds are the hot spot of explosion. For the seven M–N bonds, Mn–N appears to have the strongest stability with a bond dissociation energy (BDE) of 64.52 kcal mol−1 which breaks at 0.41 ps. Fe–N is the weakest bond and cleaves at 0.15 ps with a BDE of 31.66 kcal mol−1. The thermal stability of the complexes during the initiation stage follows the sequence [Mn(DAT)6](ClO4)2 > [Cu(DAT)6](ClO4)2 > [Cr(DAT)6](ClO4)2 ≈ [Co(DAT)6](ClO4)2 > [Zn(DAT)6](ClO4)2 > [Ni(DAT)6](ClO4)2 > [Fe(DAT)6](ClO4)2, which is consistent with the experimental results.10 Based on experiment, [Zn(DAT)6](ClO4)2 starts to melt at 170.5 °C, which is lower than [Mn/Co(DAT)6](ClO4)2 (larger than 200 °C). Therefore, the thermal stability of the initiation stage is determined by the covalency of the coordinative M–N bonds, where a higher covalency of the M–N bond suggests a larger BDE, leading to a higher stability of initiation.
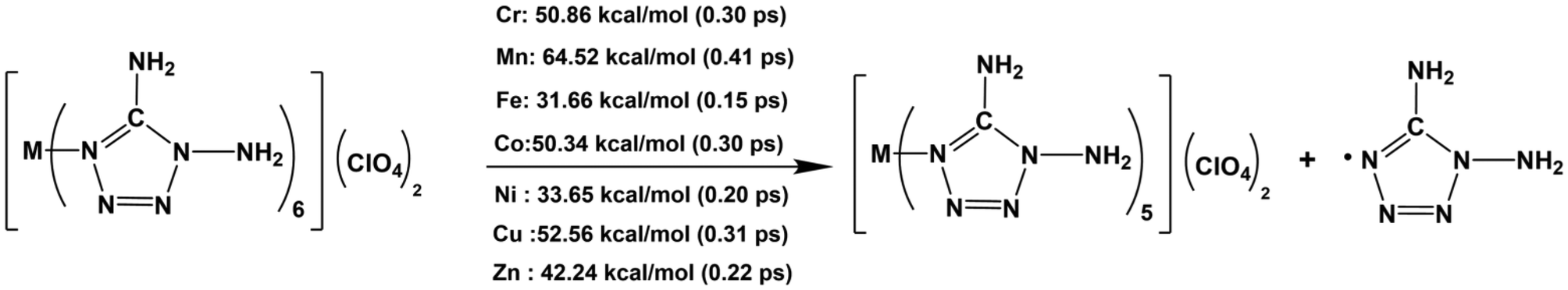 | ||
| Scheme 1 The initial decomposition of [M(DAT)6](ClO4)2 and the initial decomposition times. | ||
| Cr | Mn | Fe | Co | Ni | Cu | Zn | |
|---|---|---|---|---|---|---|---|
| a The IS and FS values are obtained from experimental results.10,18,21,22 | |||||||
| Symmetry |
P |
Pc1 |
P |
P |
P |
P |
P |
| M–N1(D/P) | 2.24/0.13 | 2.28/0.19 | 2.23/0.19 | 2.20/0.20 | 2.17/0.22 | 2.20/0.15 | 2.22/0.21 |
| N1–N2(D/P) | 1.38/0.72 | 1.38/0.68 | 1.38/0.68 | 1.38/0.60 | 1.38/0.68 | 1.38/0.68 | 1.38/0.68 |
| C1–N1(D/P) | 1.35/1.01 | 1.35/1.01 | 1.35/1.00 | 1.35/1.00 | 1.35/1.01 | 1.35/1.01 | 1.35/1.00 |
| N–M–N (°) | 88.09 | 95.20 | 89.05 | 91.67 | 87.86 | 88.01 | 87.71 |
| AC (N1/M) | −0.13/0.40 | −0.11/0.32 | −0.06/0.27 | −0.10/0.24 | −0.09/0.20 | −0.12/0.38 | −0.12/0.40 |
| MSE (eV) | −11.80 | −15.01 | −17.81 | −14.46 | −15.55 | −12.00 | −13.62 |
| Band gap (eV) | 0 | 2.15 | 0 | 0.51 | 1.63 | 0 | 3.59 |
| IS (J)a | — | 3.6 | — | 0.6 | — | — | — |
| FS (N)a | — | 252 | — | 9 | — | — | >360 |
| Hole | Electron | |
|---|---|---|
| [Cr(DAT) 6 ](ClO 4 ) 2 |
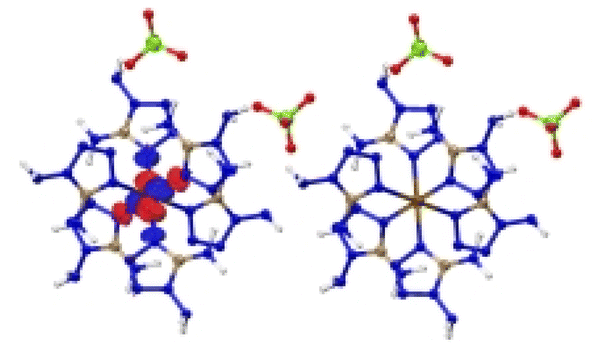
|
|
| Excitation: S0 → S14 | ||
| ΔE(S0 → S14) = 1.164 eV | ||
| OS = 0.04 | ||
| CT modes (ratio): MLCT (99%) | ||
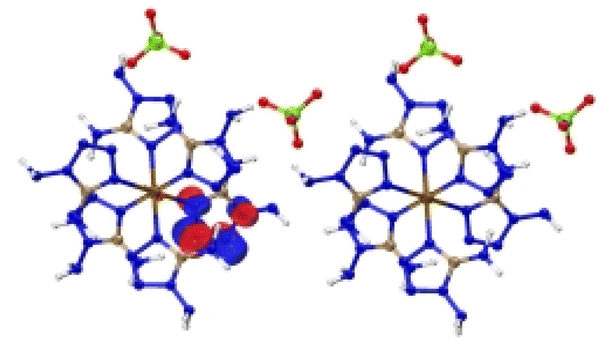
| [Cr(DAT) 6 ](ClO 4 ) 2 |

|
|
| Excitation: S0 → S15 | ||
| ΔE(S0 → S15) = 1.168 eV | ||
| OS = 0.05 | ||
| CT modes (ratio): MLCT (99%) | ||
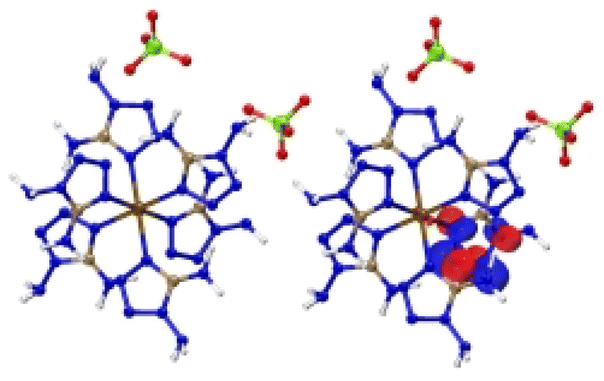
| [Cr(DAT) 6 ](ClO 4 ) 2 |

|
|
| Excitation: S0 → S20 | ||
| ΔE(S0 → S20) = 1.258 eV | ||
| OS = 0.02 | ||
| CT modes (ratio): MLCT (99%) | ||
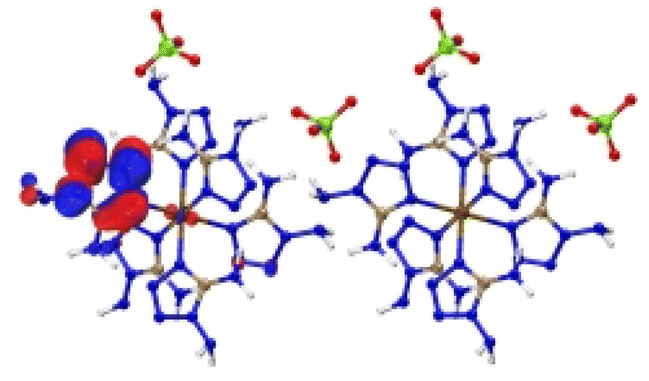
| [Fe(DAT) 6 ](ClO 4 ) 2 |

|
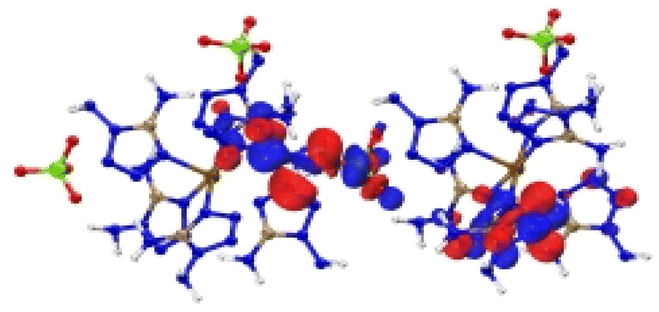
|
| Excitation: S0 → S10 | ||
| ΔE(S0 → S10) = 1.091 eV | ||
| OS = 0.06 | ||
| CT modes (ratio): LLCT, MLCT (99%) | ||
| [Fe(DAT) 6 ](ClO 4 ) 2 |
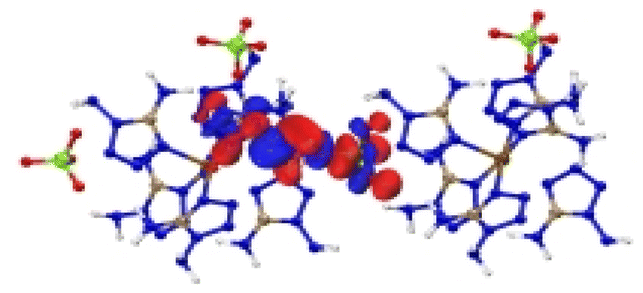
|

|
| Excitation: S0 → S11 | ||
| ΔE(S0 → S11) = 1.207 eV | ||
| OS = 0.04 | ||
| CT modes (ratio): LLCT, LMCT (97%) | ||
| [Fe(DAT) 6 ](ClO 4 ) 2 |
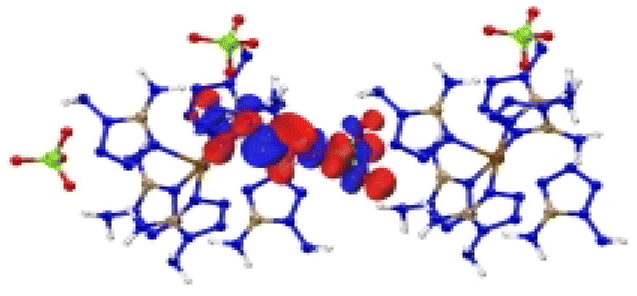
|

|
| Excitation: S0 → S13 | ||
| ΔE(S0 → S13) = 1.253 eV | ||
| OS = 0.04 | ||
| CT modes (ratio): LLCT, LMCT (90%) | ||
| [Cu(DAT) 6 ](ClO 4 ) 2 |
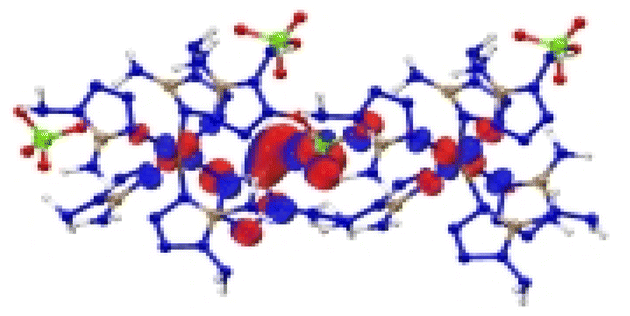
|

|
| Excitation: S0 → S8 | ||
| ΔE(S0 → S8) = 1.126 eV | ||
| OS = 0.16 | ||
| CT modes (ratio): LLCT, MLCT (99%) |
| Metal | X | Ligand | Coordination field | Electron configuration | CFSE (S) | ||
|---|---|---|---|---|---|---|---|
| Cr2+ | 8.54 | DAT | Octahedral | (t2g)3(eg)1 |
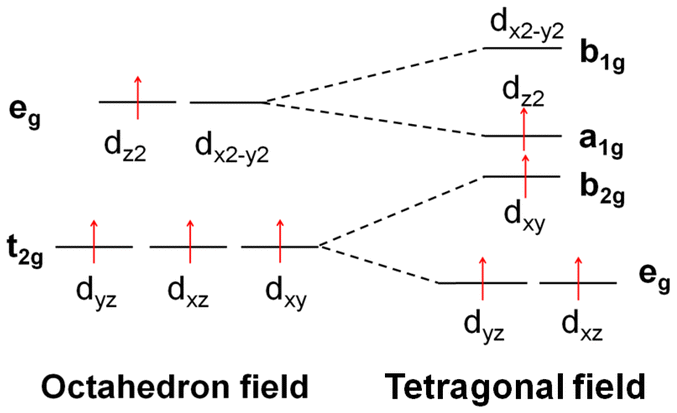
|
−6 Dq (4) | |
| 1-AT | Pyramidal | (eg)2(a1g)1(b2g)1 | −9.14 Dq (4) | ||||
| 5Eg → 5T2g | 5E → 5T2 | ||||||
| Mn2+ | 8.88 | DAT | Octahedral | (t2g)3(eg)2 |
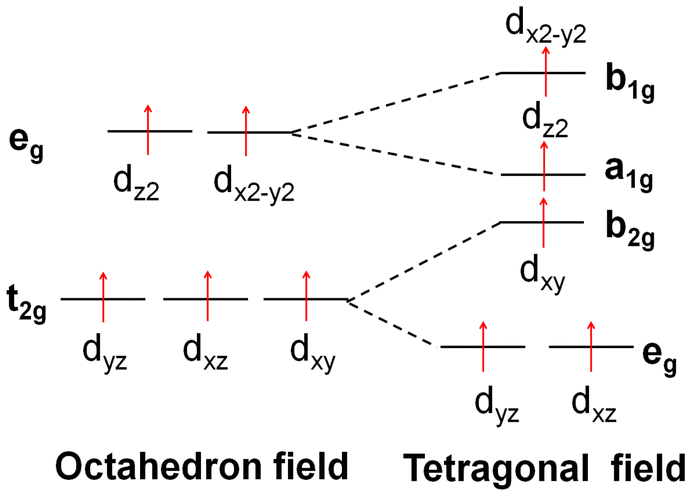
|
0 Dq (5) | |
| 1-AT | Pyramidal | (eg)2(a1g)1(b2g)1(b1g)1 | 0 Dq (5) | ||||
| Forbidden (d → d) | 4E → 4T2 | ||||||
| Fe2+ | 8.43 | DAT | Octahedral | (t2g)4(eg)2 |

|
−4 Dq (4) | |
| 1-AT | Pyramidal | (eg)3(a1g)1(b2g) 1(b1g)1 | −4.57 Dq (4) | ||||
| 5T2g → 5Eg | 3T2 → 3E | ||||||
| Co2+ | 9.10 | DAT | Octahedral | (t2g)5(eg)2 |

|
−8 Dq (3) | |
| 1-AT | Pyramidal | (eg)4(b2g)1(a1g)2 | −9.14 Dq (3) | ||||
| 4T2g → 4A2g; 4T1g → 4A2g | 2T2 → 2E | ||||||
| Ni2+ | 9.60 | DAT | Octahedral | (t2g)6(eg)2 |

|
−12 Dq (2) | |
| 1-AT | Square | (eg)4(a1g)2(b2g)2 | −4.28 Dq (2) | ||||
| 3T1g → 3T2g; 3T2g → 3A2g | 1A1g → 1A2g; 1A1g → 1B1g;1A1g → 1Eg | ||||||
| Cu2+ | 10.28 | DAT | Octahedral | (t2g)6(eg)3 |
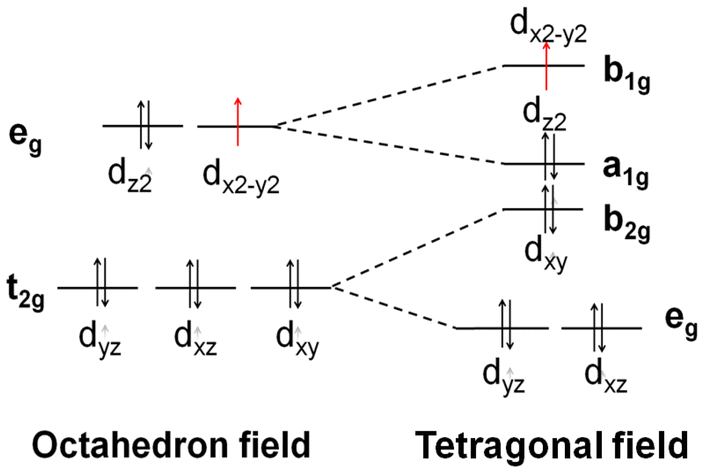
|
−6 Dq (1) | |
| 1-AT | Pyramidal | (eg)4(b2g)2(a1g)2(b1g)1 | −9.14 Dq (1) | ||||
| 2Eg → 2T2g | 2E → 2T2 | ||||||
| Zn2+ | 10.38 | DAT | Octahedral | (t2g)6(eg)4 |

|
0 | |
| 1-AT | Pyramidal | (eg)4(b2g)2(a1g)2(b1g)2 | 0 | ||||
| Forbidden (d → d) | Forbidden (d → d) | ||||||
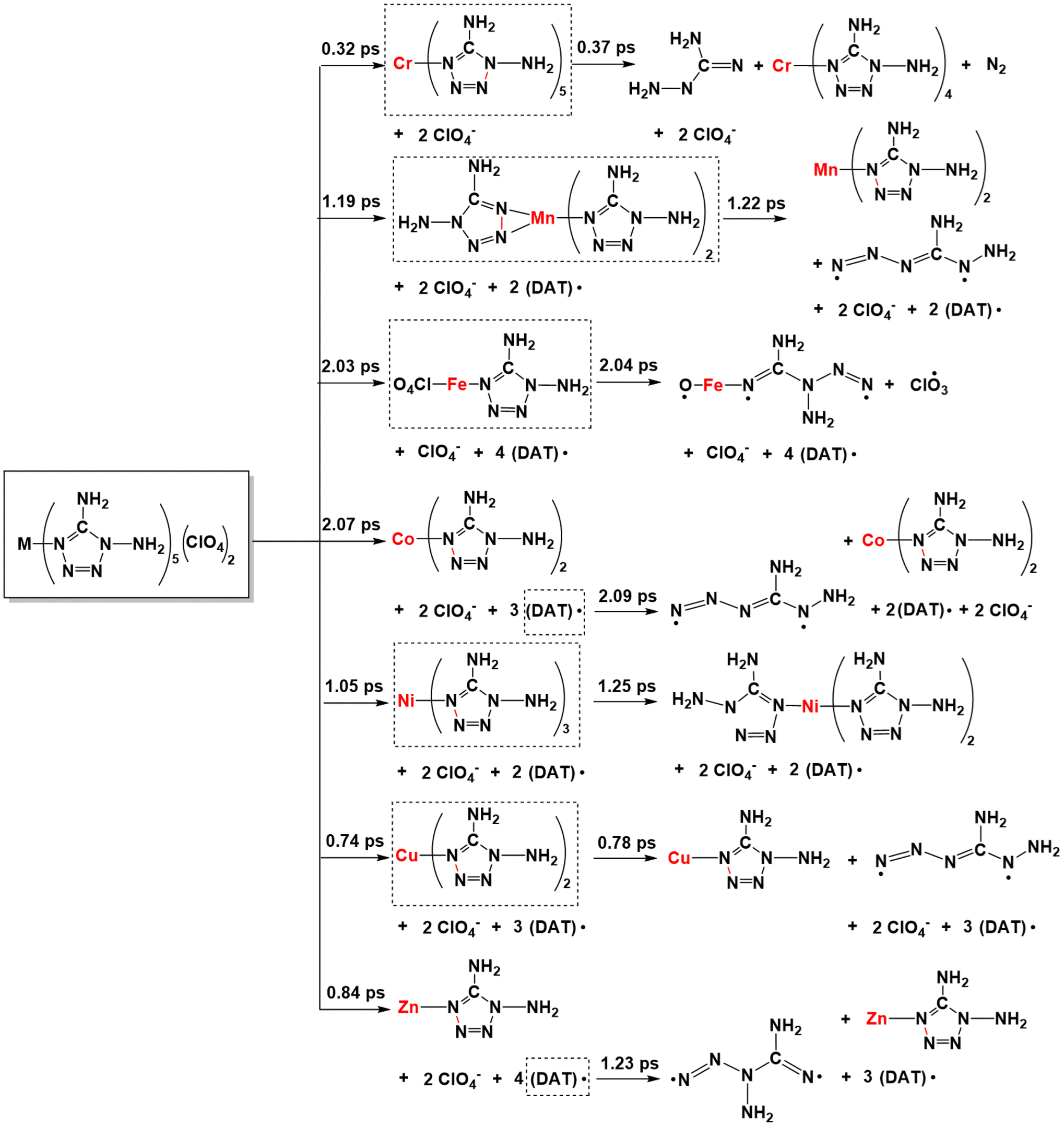 | ||
| Scheme 2 The transition pathways from deflagration to detonation of [M(DAT)6](ClO4)2. | ||
With the generation of more and more leaving groups, the seven systems gradually destabilized in response to simulated thermal stimuli, where the key intermediates are shown in boxes in Scheme 2. From [Cr(DAT)5](ClO4)2, the cleavage of the Cr–N bond requires 50.86 kcal mol−1 at 0.32 ps, followed by the N–N bond cleavage of the tetrazole ring at 0.37 ps. Only 9.24 kcal mol−1 is required for the ring-opening step. For [Mn(DAT)5](ClO4)2, the ring-opening step requires 7.99 kcal mol−1 accompanied by the dissociation of the fourth DAT radical after the formation of [Mn(DAT)5](ClO4)2. A similar ring-opening pathway can be found in the Co (2.09 ps), Cu (0.78 ps) and Zn (1.23 ps) complexes, with energies of 12.30, 12.29 and 1.91 kcal mol−1, respectively. Fe2+, with the highest metal stabilization energy, participates in the cleavage of the FeO–ClO3 bond and Fe–(N)DAT bond at 2.04 ps with an energy of 234.93 kcal mol−1. Without Cl–O bond cleavage, Ni2+, with the second highest MSE, also takes part in the ring-opening step at 1.25 ps with an energy of 23.55 kcal mol−1. As for the trajectory from the [M(DAT)5](ClO4)2 complexes, the larger the d-band center of the metal center, the more groups there are connected with it. In the chain-growth stage, a metal center with a large stabilization energy, such as the Fe and Ni systems, indicates a better ability to control the ligands in the dissociation, where the ring-opening step is from the metal-connected DAT ligand with a large positive enthalpy. The metals with lower MSE exhibit a weak stabilization effect, leading to a lower decomposition energy in the chain-growth stage and a longer DDT period. Therefore, the ring-opening step starts from the dissociative DAT radical, without the participation of the metals, such as the Cr/Mn/Co/Cu/Zn complexes. The typical snapshots of the ignition and chain-growth steps are summarized in ESI section 4.2.†
From the generation of the destabilized intermediates to the continuous explosive chain-growth, the interval is, with the exception of the Zn complex, short, generally less than 0.05 ps. Here, based on the further decomposition of [Mn(DAT)5](ClO4), we can understand that the stable deflagration period is from the beginning to the moment that the destabilized intermediates are generated. Meanwhile, the DDT process is from the generation of the destabilized structures until the moment of ring-opening. In the seven systems, the [Co(DAT)6](ClO4)2 (2.07 ps) and [Fe(DAT)6](ClO4)2 (2.03 ps) complexes have the longest stable deflagration periods. The deflagration period of the heating process follows the sequence of [Co(DAT)6](ClO4)2 (2.07 ps) > [Fe(DAT)6](ClO4)2 (2.03 ps) > [Mn(DAT)6](ClO4)2 (1.19 ps) > [Ni(DAT)6](ClO4)2 (1.05 ps) > [Zn(DAT)6](ClO4)2 (0.84 ps) > [Cu(DAT)6](ClO4)2 (0.74 ps) > [Cr(DAT)6](ClO4)2 (0.32 ps), which is similar to the sequence of MSE as we discussed before. Therefore, it is reasonable to conclude that a higher MSE generally corresponds to a longer stable deflagration period. Based on experiment,10 the temperature of the exothermic decomposition of Mn/Co/Zn starts at 200.4 °C, 213.8 °C and 185.3 °C, respectively, which is completely consistent with the sequence of the heating process period (stable deflagration period) of each complex. Moreover, following the stable deflagration period, the DDT period is affected by the stability of the destabilized intermediates (boxes with dashed lines in Scheme 2) and follows the sequence [Zn(DAT)6](ClO4)2 (0.39 ps) > [Ni(DAT)6](ClO4)2 (0.20 ps) > [Cr(DAT)6](ClO4)2 (0.05 ps) > [Cu(DAT)6](ClO4)2 (0.04 ps) > [Mn(DAT)6](ClO4)2 (0.03 ps) > [Co(DAT)6](ClO4)2 (0.02 ps) > [Fe(DAT)6](ClO4)2 (0.01 ps). Thus, the stable deflagration and DDT periods are continuous stages affected by the different structural properties of the complexes. To evaluate the safety of primary explosives, stable thermal-ignition properties and short DDT periods are the most important factors to be considered. Therefore, [Mn/Cu/Cr/Co(DAT)6](ClO4)2 exhibit excellent thermal stability and explosive properties.
4. Conclusions
In summary, we have clarified how the coordination field affects the optical properties of [M(DAT)6](ClO4)2 complexes by comparison with a series of [M(1-AT)x](NO3)2 (x = 2 or 3) complexes. Additionally, we have investigated how the metal–ligand interactions in the [M(DAT)6](ClO4)2 complexes affect their thermal ignition and explosion properties. All the metals adopt high-spin configurations when forming crystals, where the intermolecular N–H⋯H–N hydrogen bonds and Cl–O⋯H bonds contribute most to maintain the structural stability of the crystal.With regards the relationship between the crystal field and the optical properties, in order to improve the NIR sensitivity of an energetic complex, firstly, it is an efficient strategy to decrease the CFSE of the metal center or to increase the possible ways to achieve d–d excitation, both of which are determined by the crystal field and tuned by the oxidation number of the metal of each complex. Secondly, Jahn–Teller effects and exchange interactions between neighbouring high-spin electrons will further improve the activity of the d electrons and redshift the absorption wavelength. Finally, it is also effective to combine ligands with strong electron-donating character with low electronegativity metals like Cr/Mn/Fe/Co, or to combine ligands with strong electron-withdrawing character with high electronegativity metals like Ni/Cu/Zn. In the simulations, the optical sensitivity is determined by the probability of absorption in the NIR region, as reflected by the OS value. Based on their strong absorptions in the 900–1200 nm region, the [Cr/Fe/Cu(DAT)6](ClO4)2 complexes have the greatest potential to be applied as laser-ignited explosives.
Different from the traditional way of evaluating thermal stability, the initial decomposition of each complex is split into the thermal ignition step, a stable deflagration stage and DDT step, which are continuous but independent stages affected by the different structural properties of the complexes. Firstly, a stable coordinative bond with a high BDE is associated with the stability of the thermal-ignition stage. Secondly, a higher metal stabilization energy generally corresponds with a longer stable deflagration period. A larger d-band center corresponds to better controllability of the fragments in the chain-growth stage. [Mn/Cu/Cr/Co(DAT)6](ClO4)2 satisfy the safety requirements of primary explosives, where having stable thermal-ignition properties and short DDT periods are the most important factors to be considered.
Therefore, [Cu/Cr(DAT)6](ClO4)2 appears to be both NIR sensitive and thermally stable based on this research. Additionally, this work is helpful for establishing the metal-determined mechanisms of the optical and thermal initiation of energetic complexes, and for exploring the most appropriate pathway for the ignition of energetic materials.
Author contributions
K. W. and J. Z. designed the research; X. Y., P. Z., S. B., L. C. and K. W. analysed the data; X. Y., S. B., and K. W. performed the research; X. Y., S. B. and K. W. wrote the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was made possible as a result of generous grants from the National Natural Science Foundation of China (21701001), the opening project of the State Key Laboratory of Explosion Science and Safety Protection (Beijing Institute of Technology, KFJJ24-15M) and the Natural Science Research Project of Anhui Province (KJ2020ZD04). The calculations were carried out at the Hefei Advanced Computing Center. We are also thankful for the help and discussions of Prof. John E. McGrady (Department of Chemistry, Inorganic Chemistry Laboratory, University of Oxford).References
- Y. Yang, X. Li, Y. Sun, J. Tian, H. Liu, B. Wu and J. Wang, Preparation and characterization of HMX/EVA/hBNNSs micro-composites with improved thermal stability and reduced sensitivity, Def. Technol., 2021, 17, 650–656 CrossRef.
- S. Zhou, J. Chen, X. Li, F. Jia, D. Zhang, L. Xie, B. Li and L. Jiang, Catalytic pyrolysis of 5-Amino-1H-Tetrazole with copper and its oxide: A deep insight into the combustion mechanism for high nitrogen compound, Fuel, 2023, 334, 126764 Search PubMed.
- G. Tao, T. Brendan and J. M. Shreeve, Energetic nitrogen-rich Cu(II) and Cd(II) 5, 5′-azobis (tetrazolate) complexes, Inorg. Chem., 2009, 48, 9918–9923 Search PubMed.
- C. Lei, H. Yang, Q. Zhang and G. Cheng, Series of azido and fused-tetrazole explosives: combining good thermal stability and low sensitivity, ACS Appl. Mater. Interfaces, 2022, 14, 39091–39097 CrossRef CAS PubMed.
- J. A. Bjorgaard, A. E. Sifain, T. Nelson, T. W. Myers, J. M. Veauthier, D. E. Chavez, R. J. Scharff and S. Tretiak, Two-photon absorption in conjugated energetic molecules, J. Phys. Chem. A, 2016, 120, 4455–4464 CrossRef CAS PubMed.
- Y. Wang, S. Xu, H. Li, H. Ma, Y. Zhang and Z. Guo, Laser ignition of energetic complexes: impact of metal ion on laser initiation ability, New J. Chem., 2021, 45, 12705–12710 RSC.
- M. E. Casida, Time-dependent density functional response theory for molecules, in Recent Advances In Density Functional Methods: (Part I), 1995, pp. 155–192 Search PubMed.
- N. Szimhardt, M. H. Wurzenberger, L. Zeisel, M. S. Gruhne, M. Lommel, T. M. Klapötke and J. Stierstorfer, 1-AminoTriazole Transition–Metal Complexes as Laser–Ignitable and Lead–Free Primary Explosives, Chem. – Eur. J., 2019, 25, 1963–1974 CrossRef CAS PubMed.
- M. Joas, T. M. Klapötke, J. Stierstorfer and N. Szimhardt, Synthesis and Characterization of Various Photosensitive Copper(II) Complexes with 5-(1-Methylhydrazinyl)–1H–tetrazole as Ligand and Perchlorate, Nitrate, Dinitramide, and Chloride as Anions, Chem. – Eur. J., 2013, 19, 9995–10003 CrossRef CAS PubMed.
- K. Wang, D. Zeng, J. G. Zhang, Y. Cui, T. Zhang, Z. Li and X. Jin, Controllable explosion: fine-tuning the sensitivity of high-energy complexes, Dalton Trans., 2015, 44, 12497–12501 RSC.
- N. Szimhardt, M. H. Wurzenberger, L. Zeisel, M. S. Gruhne, M. Lommel and J. Stierstorfer, Maximization of the energy capability level in transition metal complexes through application of 1-amino-and 2-amino-5 H-tetrazole ligands, J. Mater. Chem. A, 2018, 6, 16257–16272 RSC.
- A. E. Sifain, J. A. Bjorgaard, T. W. Myers, J. M. Veauthier, D. E. Chavez, O. V. Prezhdo, R. J. Scharff and S. Tretiak, Photoactive excited states in explosive Fe(II) tetrazine complexes: A time-dependent density functional theory study, J. Phys. Chem. C, 2016, 120, 28762–28773 CrossRef CAS.
- J. Zhang, B. Jin, X. Li, W. Hao, T. Huang, B. Lei, Z. Guo, J. Shen and R. Peng, Study of H2AzTO-based energetic metal-organic frameworks for catalyzing the thermal decomposition of ammonium perchlorate, Chem. Eng. J., 2021, 404, 126287 Search PubMed.
- Q. Zhang, R. Lv, Y. Wang, Z. Luo, P. Pan, T. Wang and Q. Liu, Energetic copper(II) quadridentate chelate: a novel green laser-sensitive primary explosive, Inorg. Chem. Front., 2024, 11, 3168–3177 RSC.
- L. Luo, H. Zhou, Q. Liu, H. Deng, W. Hao, J. Guo, T. Huang, B. Jin and R. Peng, Assessment of the thermal stability, catalytic behavior, and laser ignitability of energetic coordination polymer [Cu (HBTT)(H2O)] n, Energ. Mater. Front., 2021, 2, 186–192 CrossRef CAS.
- C. Zhang, T. Wang, Z. Lu, Z. Yi, M. Xu, Y. Li, Q. Yu, Z. Li and J. Zhang, A new sulfur-containing laser-sensitive primary explosive based on thiazole-4-carbohydrazide, Energ. Mater. Front., 2024, 5, 191–198 CrossRef CAS.
- S. Bu, J. Zhang and K. Wang, Metal-Determined Explosive Characteristics of M (NO3) 2 (1-AT) x of Thermal and Laser Ignition (M2+= Cr2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+, and Zn2+; x = 2 or 3), Inorg. Chem., 2024, 63, 3212–3220 CrossRef CAS PubMed.
- S. Qi, Z. Li, Z. Zhou, Y. Cui, G. Zhang, T. Zhang, J. Zhang and L. Yang, Crystal structure, thermal decomposition behaviours and sensitivity properties of a novel energetic compound [Co (DAT)6](ClO4)2, Chin. J. Chem., 2011, 29, 59–64 CrossRef CAS.
- P. Zhang, J. Zhang, B. Shu, X. Huang, L. Shi, L. Cheng, J. Zhang and K. Wang, Metal-Tuned Energetic Bactericidal Characteristics: Synthesis and Theoretical Study of [M(atrz)(IO3)2]n (M = Mn, Fe, Co, Ni, Cu, and Zn), J. Phys. Chem. C, 2024, 128, 2240–2246 CrossRef CAS.
- X. He, P. Wu, X. Huang, C. Dai, C. Li, L. Cheng, T. Zhang, J. Zhang and K. Wang, Exoergic pathways triggered by O/H radicals in different metallic carbohydrazide perchlorates (M2+= Mn2+, Fe2+, Co2+, Ni2+, Zn2+ and Cd2+), Phys. Chem. Chem. Phys., 2022, 24, 10877–10886 RSC.
- Y. Shu, J. Zhang and T. Zhang, Synthesis, Crystal Structure, Thermal Behavior and Sensitivity Properties of New Energetic Compound [Mn(DAT)6](ClO4)2, Chem. J. Chin. Univ., 2009, 30, 1935–1939 Search PubMed.
- J. Shang, J. Zhang, Y. Cui, T. Zhang, Y. Shu and L. Yang, Synthesis, crystal structure, and properties of an energetic compound Zn (1, 5-diaminotetrazole)6(ClO4)2, Acta Chim. Sin., 2010, 3, 233–238 Search PubMed.
- C. V. Caillie and R. D. Amos, Geometric derivatives of excitation energies using SCF and DFT, Chem. Phys. Lett., 1999, 308, 249–255 CrossRef.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS.
- M. D. Segall, P. J. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. Clark and M. Payne, First-principles simulation: ideas, illustrations and the CASTEP code, J. Phys.: Condens. Matter., 2002, 14, 2717 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- T. D. Kühne, M. Krack, F. R. Mohamed and M. Parrinello, Efficient and accurate Car-Parrinello-like approach to Born-Oppenheimer molecular dynamics, Phys. Rev. Lett., 2007, 98, 066401 CrossRef PubMed.
- R. Car and M. Parrinello, Unified approach for molecular dynamics and density-functional theory, Phys. Rev. Lett., 1985, 55, 2471 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B, 1976, 13, 5188 CrossRef.
- T. H. Fischer and J. Almlof, General methods for geometry and wave function optimization, J. Phys. Chem., 1992, 96, 9768–9774 CrossRef CAS.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS.
- J. Yang, G. Zhang, J. Zhang, D. Chen and Q. Zhang, New perspectives on the laser initiation for metal tetrazine complexes: a theoretical study, Phys. Chem. Chem. Phys., 2022, 24, 305–312 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. Scuseria, M. Robb and J. Cheeseman, Gaussian 16, Wallingford, 2016, vol. 1572 Search PubMed.
- T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- T. Lu and F. Chen, Quantitative analysis of molecular surface based on improved Marching Tetrahedra algorithm, J. Mol. Graphics, 2012, 38, 314–323 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: visual molecular dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni and I. Dabo, QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- S. Nosé, A unified formulation of the constant temperature molecular dynamics methods, J. Phys. Chem., 1984, 81, 511–519 CrossRef.
- R. J. C. Gálvez, G. Holl, K. Karaghiosoff, T. M. Klapötke, K. Löhnwitz, P. Mayer, H. Nöth, K. Polborn, C. J. Rohbogner and M. Suter, Derivatives of 1, 5-diamino-1 H-tetrazole: a new family of energetic heterocyclic-based salts, Inorg. Chem., 2005, 44, 4237–4253 CrossRef PubMed.
- T. Kon'kova, Y. N. Matyushin, E. Miroshnichenko and A. Vorob'ev, The thermochemical properties of transition metal complex salts with 1, 5-diaminotetrazole, Russ. J. Phys. Chem. B, 2009, 3, 987–989 CrossRef.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces, Chem. Commun., 2007, 37, 3814–3816 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional calculation methods (section 1), parameters of both gas-phase and crystal structures (section 2 and 3), trajectories of CPMD simulation (section 4) and charge transferring analysis in excitation of the optical characters (section 5). See DOI: https://doi.org/10.1039/d4dt03513e |
| This journal is © The Royal Society of Chemistry 2025 |